 Xuejia Tang
Xuejia Tang Chenghai Zuo3†
Chenghai Zuo3†
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 08 July 2021
Sec. Cancer Molecular Targets and Therapeutics
Volume 11 - 2021 | https://doi.org/10.3389/fonc.2021.701291
Glioblastoma (GBM) remains the most lethal and common primary brain tumor, even after treatment with multiple therapies, such as surgical resection, chemotherapy, and radiation. Although great advances in medical development and improvements in therapeutic methods of GBM have led to a certain extension of the median survival time of patients, prognosis remains poor. The primary cause of its dismal outcomes is the high rate of tumor recurrence, which is closely related to its resistance to standard therapies. During the last decade, glioblastoma stem cells (GSCs) have been successfully isolated from GBM, and it has been demonstrated that these cells are likely to play an indispensable role in the formation, maintenance, and recurrence of GBM tumors, indicating that GSCs are a crucial target for treatment. Herein, we summarize the current knowledge regarding GSCs, their related signaling pathways, resistance mechanisms, crosstalk linking mechanisms, and microenvironment or niche. Subsequently, we present a framework of targeted therapy for GSCs based on direct strategies, including blockade of the pathways necessary to overcome resistance or prevent their function, promotion of GSC differentiation, virotherapy, and indirect strategies, including targeting the perivascular, hypoxic, and immune niches of the GSCs. In summary, targeting GSCs provides a tremendous opportunity for revolutionary approaches to improve the prognosis and therapy of GBM, despite a variety of challenges.
Malignant gliomas are the most frequent and lethal cancers originating in the central nervous system (1), and include three main high-grade glioma phenotypes based on genome-wide expression profiling and DNA methylation analysis in recent clinical assessments: proneural, mesenchymal, and classical (2, 3). Glioblastoma (GBM), also known as glioblastoma multiforme, is one of the most biologically aggressive subtypes due to its high malignancy and genetic heterogeneity (4, 5). The standard therapeutic approaches for GBM include maximal surgical resection, radiotherapy, chemotherapy (with various chemotherapeutic agents), and other innovative therapies combined with conventional approaches (6–8). Despite this, only 5.5% of patients between the ages of 55 and 64 survive for five years from diagnosis, and life expectancy has only increased from approximately 6 to 12–18 months after maximal standard treatments. This dismal prognosis is associated with the high rate of relapse and resistance to current standard therapies (9, 10). Overall, treatment of GBM is still challenging compared to other solid tumors, despite the great advances in medical development and improvements in therapeutic methods. Therefore, there is an urgent need to identify novel therapeutic targets to optimize the therapeutic approaches to GBM treatment.
Disease management and standard therapies are the absence of work due to the considerable intratumoral phenotype, intrinsic heterogeneity, and extensive infiltration of tumor cells throughout the brain (11). Thus far, several studies have identified the presence of stem cell-like cells in solid tumors, termed cancer stem cells (CSCs). These cells have high plasticity and the ability to proliferation, self-renew, and give rise to pathognomonic heterogeneous cancer cells that comprise the tumor (1, 12–14). A study on a pure CSC tumor model found that CSCs form the basis of tumorigenesis and continue to proliferate through self-renewal and segregation into different tumor cells (15). Simultaneously, numerous studies have shown that the highly malignant CSC subpopulation of tumor cells shows more aggressive potential compared to non-CSCs, despite the fact that the differences between CSCs and non-CSCs are not fully clear (16–18). A huge amount of evidence has shown that malignant CSC subpopulation of tumor cells in GBM are probably linked to malignant relapse, resistance to standard therapies due to their characteristic abilities to self-renewal, differentiation, growing and progression (12, 19–25). Therefore, CSCs causes of tumor characteristics such as proliferation, maintenance, malignant relapse, and metastasis in GBM (26). The major reasons for this resistance to chemo- and radiotherapy are their assumed quiescence, high capacity for extensive DNA repair, a higher mitochondrial reserve and their location in hypoxic niches (2, 27, 28). In order to combat therapeutic resistance of CSCs, it is necessary to understand both the mechanisms of inherent resistance and the surrounding niche of CSCs, which might be perceived as an interesting alternative to targeting therapy for GBM. It is also crucial to further understand the characteristics of these cells. First, CSCs must be identified prospectively from a variety of tumor cells. Common biomarkers, including CD133, CD44, and CD24, have been used to identify and enrich CSCs; however, several studies on CSCs have failed to confirm their reproducibility and accuracy owing to the genetic heterogeneity of CSCs, and the lack of universally informative biomarkers (29, 30). Furthermore, with the aim of treating this highly malignant disease, identification and blockade of CSC signaling pathways, such as the Notch, sonic hedgehog (SHH) and Wnt signaling pathways (31), which are closely correlated with tumor characteristics including proliferation, maintenance, malignant relapse, and metastasis in GBM, will provide vital targets for GBM treatment.
In contrast to the bulk tumor populations, the unique properties of CSCs suggest that targeting the stemness of CSCs in GBM and developing targeted CSC therapies could offer an unprecedented therapeutic opportunity for GBM (Figure 1). This is important, as the refinement of the current standard therapy techniques for GBM is essential to improve the poor disease outcome. However, targeting CSCs for GBM therapeutics remains difficult, owing to the slow cycling of CSCs, the high expression of drug export proteins, and having no potential ability in CSCs to expressing the oncoproteins that could be targeted by the new generation of smart drugs, such as Gleevec and Iressa (32). In order to gain a better understanding of the characteristics of CSCs and CSCs-targeted therapeutic approaches to overcome therapeutic challenges, including the resistance and tumor recurrence in GBM, we summarize the current knowledge regarding glioblastoma stem cells (GSCs), their related signaling pathways, resistance mechanisms, crosstalk linking mechanisms, and microenvironment or niche. Subsequently, we present a framework of targeted therapy for GSCs based on direct strategies, including blockade of the pathways necessary to overcome resistance or prevent their function, promotion of GSC differentiation, virotherapy, and indirect strategies, including targeting the perivascular, hypoxic, and immune niches of the GSCs. In summary, targeting GSCs provides a tremendous opportunity for revolutionary approaches to improve the prognosis and therapy of GBM, despite a variety of challenges. We believe that this review could help to guide the future of GBM research and therapy.
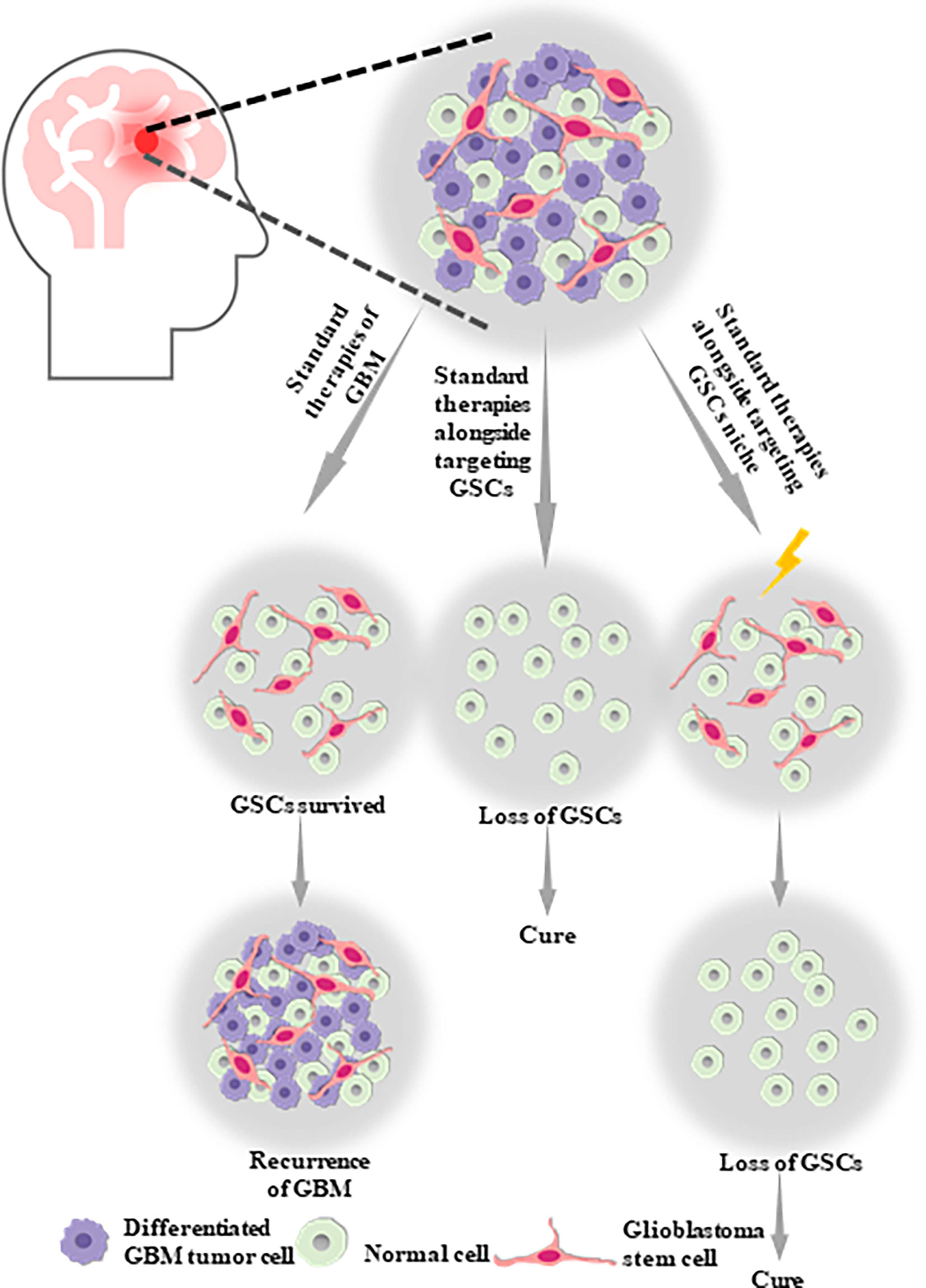
Figure 1 Glioblastoma stem cells (GSCs) are likely to be one of the potential causes of tumor proliferation, maintenance, malignant recurrence and metastasis in GBM, which aid in the self-renew, multilineage differentiation and proliferation of GSCs. Therefore, standard therapies alongside targeting GSCs or targeting GSCs niche might be the optimal therapy strategy for GBM.
In the 1960s, James Till and Ernest McCulloch first discovered stem cells after performing modified spleen colony formation assays in vivo (33). The crucial capacity for undergoing asymmetric division via self-renewal of stem cells have been described, which could generate two daughter cells with different cell fates that is a true stem cell and the other one is a progenitor with a high cycle ability despite a limited number of cell cycles, and simultaneously the loss of this balanced process could induce a cancer-like state (34). Recently, stem cells as undifferentiated cells have been clearly defined that have the ability to undergo asymmetric division, malignant proliferation, self-renewal, plasticity, differentiation into progenitor cells or regeneration of injured tissue (35). The cell cultures of GBM have been verified to show capable of forming neurospheres in vitro with high expression of biomarkers like CD133 and Nestin (36), and these sphere-forming cells derived from human GBM into mice could also grow tumor in vivo (20). The same cell culture conditions could been used as for these normal neural stem cells (NSCs) (37), which have the ability to produce neural and glial cells of the nervous system, are one of the primary stem cell types in the nervous system, and the transformation from NSCs to GSCs has been demonstrated to be a possible factor in the formation of glioma (38). There are at least two neurogenic regions of the adult mammalian brain where NSCs are identified: the subventricular zone (SVZ) of the lateral ventricles and the subgranular zone (SGZ) of the hippocampus in the dentate gyrus. Stem cells near the SVZ or SGZ are always in a state of quiescence or active mitosis, as shown in several murine models (39–41). In addition, NSCs situated in the SVZ could also undergo asymmetric division as they are able to orient their division and their mitotic spindle acting as a molecular trigger for both daughter cells in the microenvironment (42–44). The malignant proliferation of NSCs has been proven to be one of the likely origins and identifying characteristics of cells that induce tumorigenesis, including proliferation, maintenance, malignant relapse, and metastasis in GBM due to the genetic mutations in oncogenes accumulation in NSCs, which may give rise to the dedifferentiation in normal brain cells and induce tumor cells (45–47).
These tumor cells possessing stem cell-like features seem to be responsible for tumor progression and tumor relapse in various solid tumors including GBM. The authentic origin of CSCs is a problem that has been extensively investigated, but some controversy about this question remains, due to the incomplete and inconsistent experimental data (48–51).
Interestingly, the tumor-propagating cells in GBM named GSCs have properties similar to those of normal NSCs, including the enhanced ability to self-renew and undergo multilineage segregation into different tumor cells (52, 53). In addition to these similar stem cell properties, the common pathways between GSCs and NSCs, such as Notch, bone morphogenic proteins(BMPs), Wnt, NF-κB, platelet-derived growth factor (PDGF), epidermal growth factor (EGF), SHH, and transforming growth factor–b (TGF-b) are closely linked to the development of the nervous system (36, 54, 55). Among these, PDGF signaling is also important for the transformation of normal NSCs into GSCs, facilitating the formation and proliferation of GBM (56–58). EGFR signaling has been demonstrated to play a pivotal role in regulating normal NSC expansion, migration, differentiation, and survival in neural development (59–62), inducing tumorigenic potential (63, 64). The similar gene expression profiles between NSCs and GSCs also support the hypothesis that CSCs are the other malignant variants of NSCs (65, 66). Furthermore, GSCs and NSCs express semblable neuronal or glial markers, such as CD133, CD15, integrin-α6, L1CAM and (67). In contrast to the remaining isolated cells which lack these markers, CSCs with common NSC markers have undergone orthotopic tumor formation in nude mice, and the orthotopic tumors are closely related to human GBM (68). In summary, there is plenty of evidence to indicate the common properties, signaling pathways, gene expression, and biomarkers between NSCs and GSCs, however, it is still difficult to confirm whether GSCs are derived from malignant mutation of NSCs, which could induce tumor formation, or whether they are derived from other mature cells with the ability to self-renew and differentiate into various tumor cells. A reliable approach to targeting GSCs for GBM treatment would only selectively eliminate GSCs after identifying and isolating GSCs from other NSCs. In regards to this problem, further studies on the relationship between NSCs and GSCs are needed.
Specific and precise criteria play an indispensable role in identifying and isolating GSCs from other cells (69). GSCs have been isolated from solid tumors and classified as one of the first cell types (23). Conversely, GSCs are effective at initiating tumor xenografts in vivo, owing to the formation of heterogeneous tumors that resemble the original parent tumor. On the contrary, GSCs have been demonstrated to self-renew and form neurospheres when grown in vitro, which could be used to assess the proliferation of GSCs via the frequencies of GSCs in tumors (70, 71). Consequently, GSCs cultured in vivo or in vitro play an important role in the pathogenesis and development of GBM, because of the greater reliability and physiological relevance of the model.
The use of various specific surface markers or molecular mediators such as CD133, CD90, CD44, L1CAM, A2B5, and GPD1 is a common method to identify GSCs and define lineage-specific subpopulations within the tumor, although the cause of their reproducibility and accuracy is still unclear (29, 30, 72). However, the GSC niche as a potential marker source may cause epigenetic changes and variations in CSC phenotypes via microenvironmental signals in vivo or in vitro (31). The recognition of GSCs as therapeutically targetable remains a major challenge to overcome. Furthermore, some stem cells might additionally express ABC transporters binding with ATP, which are able to pump the fluorescent dye HOECHST-33342 out of the cell and to recognize unlabeled side population (SP) highly enriched in stem cells in neural tissues (67), which could be an alternative approach for identifying GSCs. Another approach to identify a subpopulation of GSCs is the observation of autofluorescence around 520 nm under laser excitation at 488 nm, owing to the autofluorescence properties and distinctive morphology of GSCs (73). In summary, various strategies for the isolation and identification of GSCs have been produced, and have facilitated efforts to isolate and identify GSCs via new and more effective technology, which may require a thorough insight into both the essential molecular and morphogenic processes regarding how GSCs are associated with tumor initiation or tumor proliferation in GBM. To improve the diagnostic and therapeutic prediction value, it is important to develop methods that are more effective to isolate and identify GSCs in GBM.
The isolation and identification of GSCs in GBM may contribute to a better understanding of the mechanisms underlying tumorigenesis and novel therapeutic strategies for GBM. It may be an ideal and effective approach to sort and target GSC populations via GSC markers to distinguish the expression of stem cell surface markers in GSCs, and are functionally correlated with the maintenance of GSCs, providing a powerful tool to investigate the tumorigenic process in the cerebral nervous system and promote the diagnosis, and treatment of GBM. To date, a great number of advances have been made in understanding GSC markers in the last several years. Here, we focus on several putative GSC markers, including CD133 (PROM-1), Nestin, LGR5, B23 (NPM1), and GPD1.
CD133, as the initial isolation marker of GSCs, is still the best validated marker. CD133, which belongs to the Pentaspan transmembrane glycoprotein family member, is also known as prominin-1 (PROM-1) or AC133 (74). It is a membrane bound glycoprotein encoded by the PROM1 gene and the chemical structure of CD133 involves a single-chain polypeptide of 865 amino acids with 5-TM regions, an extracellular N-terminus and a cytoplasmic C-terminus (75). CD133 was first discovered in human hematopoietic progenitor cells and was later described in mouse tissue (76). With the aim of enriching and identifying tumor-propagating and tumor-initiating cells, CD133 has become an alternative tool in some experiments. For example, CD133+ cells in GBM, identified by flow cytometry, have been considered tumor-initiating cells and have been demonstrated to be responsible for the proliferation of tumor cells, as well as resistance to radiation therapy based on immune-deficient NOD/SCID mouse xenograft models (77). However, there are still some controversies regarding CD133+ cells. Some studies have questioned the idea that CD133 as a stem cell surface marker is a common and useful tool for identifying and defining GSCs, including the findings that CD133+ cells are differentiated, while CD133− cells also have the ability to initiate tumors in some cases (78), indicating that CD133 might not be a reliable GSC informative marker. There are still some debates regarding the definitive function of CD133 in GBM, but it is clear that the expression of CD133 may change due to several interactions with the tumor microenvironment, and may play a possible role in cell differentiation and the epithelial to mesenchymal transition (79). Given the limited knowledge regarding the definitive role of CD133 and its expression across the differentiation spectrum of GSCs, it is necessary to have a thorough insight into the function of CD133 in GBM.
The neuroepithelial stem cell protein commonly known as nestin, first described as an antigen of rat-401 against embryonic spinal cord and later identified as a class VI intermediate filament protein (80), has been shown to be expressed in neuroepithelial stem cells, and is highly expressed in several types of human malignancies, including higher grade GBM. Nestin has been shown to be strongly correlated with lower cancer patient survival, while some researchers hold the opposing view that there is no connection between Nestin expression and poor prognosis in GBM (78, 81–85). Nestin, as another putative marker for the GSC phenotype, probably plays a significant role in aggressive growth metastasis and self-renewal capacity of CSCs, organizing the cytoskeleton, cell signaling, organogenesis, and cell metabolism, and represents the proliferation, migration, and multi-differentiated characteristics of multi-lineage progenitor cells, and it is thus a more suitable target molecule to identify GSCs in GBM than CD133 (86, 87). Zhang et al. previously showed that there have been considerable refinements in clinical prognostic accuracy by adding a putative marker for the GSCs Nestin into CD133 (88). Although a definitive description of Nestin in GSCs remains elusive, its role in transformed cells, especially GSCs, its involvement in forming the cytoskeleton and the thorough mechanisms underlying this relationship between Nestin expression and miscellaneous capacity of GSCs is known. Thus, a combined detection of Nestin and CD133 co-expression may be a potential indicator of the biological invasion of GBM.
Leucine-rich repeat-containing G protein-coupled receptor 5 (LGR5) was first reported as a marker of intestinal stem cells and also serves as a novel functional marker of GSCs. LGR5 belongs to the seven-transmembrane receptor subclass of the G protein-coupled receptor family, is closely correlated with the Wnt signaling pathway and is also known as Gpr49 (89). LGR5 contributes to tumor formation, proliferation, and aggressiveness (90–94). In 2018, Zhang et al.demonstrated that LGR5 has a variety of roles, including promoting GSC epithelial-mesenchymal transition (EMT) by activating the Wnt/β-catenin pathway in vitro and in vivo, and should be an effective indicator of prognosis in GBM, along with predicting glioma recurrence (95). Therefore, targeting LGR5 in GSCs may facilitate the treatment of GBM with ideal therapeutic approaches to promote the further understanding of the novel functional marker of GSCs.
B23, also known as nucleophosmin or NPM1, is both a chaperone of nucleic acids and a nucleolar protein shuttling between the nucleoli, nucleoplasm, and cytoplasm, and is involved in various functions, including centrosome duplication, ribosome maturation and export, intracellular transport, chromatin remodeling (core and linker histone binding), apoptosis, and mRNA splicing in diverse cellular processes, and plays an important role in the cellular response to different stress stimuli and cell cycle control (96, 97). It has been found that B23 is overexpressed at both the mRNA and protein levels in hematologic malignancies and other types of cancer compared with the normal brain, especially in gliomas (98). Consequently, B23 is considered a promising therapeutic target for GBM treatment.
GPD1, also named as GPD-C, GPDH-C, or HTGTI, is one of three isoenzymes of human glycerol-3-phosphate dehydrogenase that are involved in catalyzing the conversion of dihydroxyacetone phosphate (DHAP) with NADH and glycerol-3-phosphate (G3P) with NAD+, which is critically implicated in the transport of reducing equivalents across the mitochondrial membrane and triacylglycerol synthesis (99–101). Scientists have demonstrated that aberrant GPD1 expression is found in dormant GSCs, but not in NSCs in vivo, and its expression could aggravate the progression of GBM due to its significance for GSC proliferation and maintenance, furnishing dormant GSCs with functional correlation as a therapeutic target (102). Therefore, loss of GPD1 could result in impairment of GSC maintenance pathways in GBM, prolonging patient survival. Furthermore, it is better to shed light on the crosstalk between GPD1 and edema in GBM, as edema occurs in patients suffering from human GBM (103).
Although growing putative GSC markers like CD133, CD90, CD15, A2B5, ALDH1, Label-retention, Nestin, proteasome activity, and ABC transporters (78, 104) have been used to recognize GSCs, and a considerable number of studies have been conducted on these markers to provide compelling and informative evidence that these putative GSC markers are powerful tools to investigate the tumorigenic process in the cerebral nervous system and promote the diagnosis and treatment of GBM (Table 1). However, the exact mechanisms and functionalities of these putative markers in GSCs have not been thoroughly elucidated. Given this problem, it is crucial to have a thorough and wide insight into identifying both specific markers of GSCs and the molecular mechanisms for the development of improved and tailored targeting of GSC treatments for GBM, especially paying closer attention to the various ideas of GSC isolation and identification, such as the concomitant use of different stem cell markers, instead of a single marker.
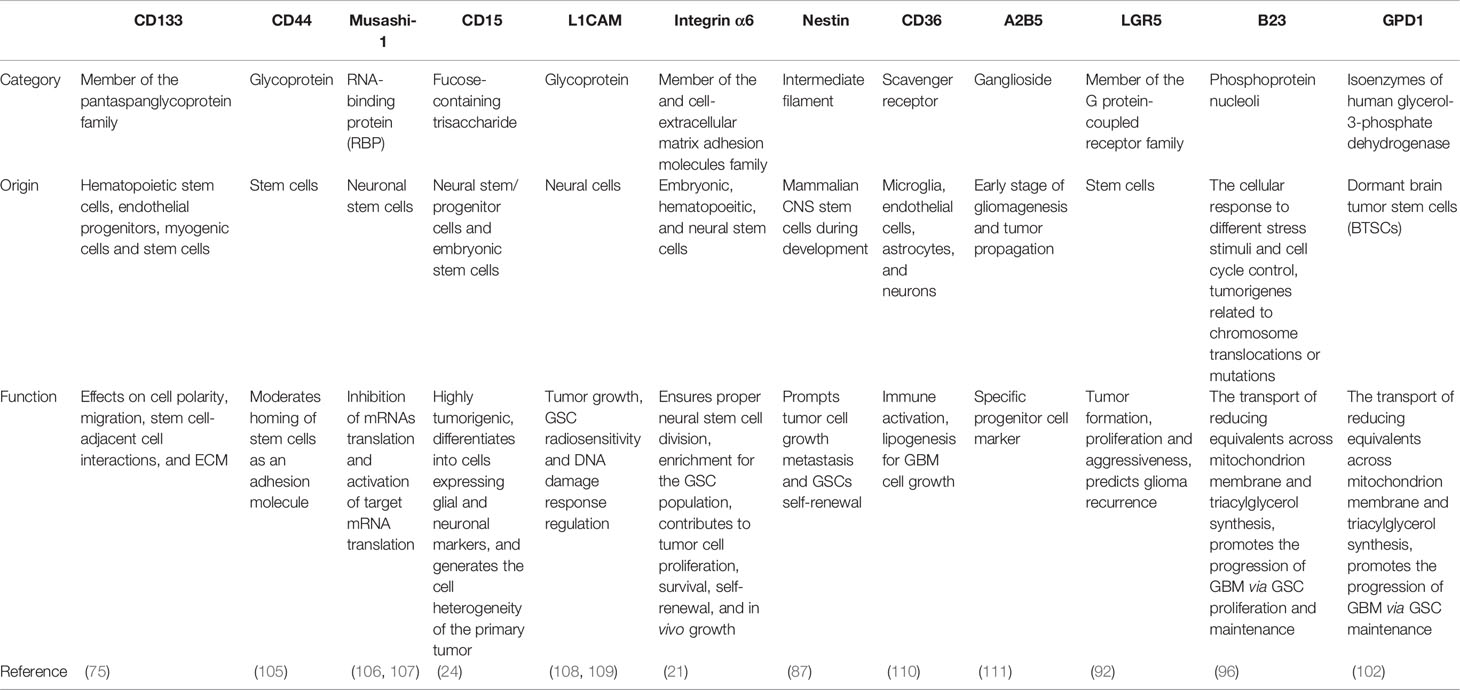
Table 1 Common markers and novel biomarkers on GSCs.
Niches, clearly defined as the tumor microenvironment (TME) composed of extracellular matrix (ECM) and a complex tissue of cells including astrocytes, macrophages, pericytes, fibroblasts, and endothelial cells play a significant role in stem cells that not only an anatomic structural unit surrounding stem cells, but also a functional unit that provides complex and dynamic interactions with stem cells (13, 112). In the human brain, stem cells, including normal NSCs, are enriched in specific regions that consist of endothelial and ependymal cells to maintain their stem cell properties (14). The relationship between TME and stem cells is synergistic and codependent like the ‘‘seed-and-soil’’ relationship rather than passive relationship, which are adapted in GSCs, as they also require a specific niche to support their stem-like characteristics, including division other than self-renewal (113), high proliferation (114), and multidirectional differentiation into various tumor cells. In addition, multilineage and inherent crosstalk between GSCs and microenvironment with various tumor components such as the (ECM), cellular compartments such as cancer-associated fibroblasts, immune cells, differentiated neural cells, etc., and the blood–brain barrier (BBB) through tumor-derived pericytes is facilitated to form an ideal microenvironment that could enhance the invasive tumor-properties, or resistance to chemotherapy and radiotherapy (79, 115). The chemo- and radio- resistance of GBM is thus responsible for an isolated properties of GSCs, along with the intrinsic dependance on the synergistic interaction between these tumor cells and TME. These properties of the GSCs niche could be conducive to GBM heterogeneity, plasticity, and malignancy, determining the fate of GSCs.
There are three major GSC niches, including the perivascular niche via angiogenic pathways, the perinecrotic or hypoxic niche via inducing hypoxia, and the immune niche. The perivascular niche is surrounded by a mass of blood vessels feeding the tumor with abnormal structure and function. The synergistic and codependent relationship between the GBM perivascular niche and GSCs can contribute to the progression of tumor angiogenesis (116). There are a great number of CSCs under hypoxic conditions, which indicates a positive player in maintenance of CSCs through supporting the critical stem cell traits of multipotency, self-renewal, and tumorigenicity (117). HIFs(HIF-1α and HIF-2α) in the hypoxic niche are upregulated via the expression of HIF-1α and HIF-2α isoforms in GBM and the roles of both HIF-1α and HIF-2α seem to be overlapping with 75% homologies between HIF-1α and HIF-2α (118). In addition, it has been found that tumor aggression increased via acting on the GSCs and the infiltration of a large number of immune cells in the GBM immune niche, showing that the hypoxic response along with inflammation are overlapping despite the immune privilege in the normal human brain (119). The hypoxic environments could induce synthesis of HIF-1α with the protein stabilized through engaging the T-cell receptor (120), enhance the lytic ability of CD8+ T lymphocytes (121) as well as interferon-gamma secretion by CD4+ T cells (122), impair cytotoxic T lymphocyte (CTL) development, proliferation and expression of inflammatory cytokines (121), and also recruit immunosuppressive cells via GSCs signaling including tumor-associated macrophages (TAMs), myeloid-derived suppressor cells (MDSCs), and Tregs that could promote angiogenesis, inhibit the immune response by secreting chemokines and growth factors such as vascular endothelial growth factor (VEGF), transforming growth factor-β1 (TGF-β1), neurotensin, SDF1, and soluble colony-stimulating factor 1 (sCSF-1) and expressing surface molecules that engage inhibitory molecules on effector immune cells (123), ultimately leading to the formation of an immunosuppressive microenvironment. In a word, targeting of the GSC niche, particularly the perivascular niche, the hypoxic niche, and the immune niche, is still one of the crucial strategies to solve resistance to current standard therapy for GBM and improve the poor disease prognosis of GBM.
A sequence of signaling pathways and receptors upregulated by GSCs involved in tumor proliferation, maintenance, and resistance to chemotherapy and radiotherapy, could improve the stem-like features and aberrant cell survival, ultimately giving rise to oncogenesis in the brain (36). It is thus crucial to acquire a thorough insight into the pivotal signaling pathways and receptors responsible for GSC maintenance, including Notch, Wnt, SHH and Receptor Tyrosine Kinase (RTK) pathways, to understand the stem cell properties of GSCs and facilitate the development of improved and tailored targeting GSCs treatments for GBM.
It has been previously confirmed that the Notch signaling pathway is critically associated with stem cell fate determination, proliferation, maintenance of cell quiescence, metastasis, and modulation of differentiation of both normal NSCs and GSCs (124). Via mutual effects with ligands on a considerable number of cells, the Notch pathway, including the four Notch receptors (Notch 1–Notch 4) and five ligands (Jagged-1 and Jagged-2, and Delta-like-1, Delta-like-3, and Delta-like-4), can regulate the interaction between cells and neighboring cells covering a short range (125, 126). The Notch signaling pathway is activated by sequential proteolytic cleavage reactions, causing release and nuclear translocation of the intracellular domains of Notch receptors (NICDs) and a series of transcription-dependent pathways, activated by the Notch signaling pathway are essential (127). Inhibitors of the γ-secretase (GSIs) complex which participates in regulating the last proteolytic step for release of NICDs, such as RO4929097 (29), play a significant role in blocking the Notch signaling pathway both in vitro and in vivo, owing to its ability to activating the Notch signaling pathway (128).
The Wnt/β-catenin signaling pathway is critically implicated in modulating the differentiation and proliferation of normal neural cells such as NSCs or cells of the astroglial lineage (129–131), as well as the self-renewal, differentiation, and expansion of GSCs (132–134). The nuclear localization of stabilized β-catenin is able to abnormally activate the β-catenin signaling pathway of GSCs, and is intrinsically correlated to tumorigenesis, growth invasion in GBM (135, 136), and the expression of MGMT, which is responsible for the resistance to TMZ (137). However, it remains highly difficult to target the Wnt/β-catenin signaling pathway owing to the serious side effects of this inhibition, as this pathway is critically related to a considerable number of physiological processes in human organs including the brain (138).
It has been well demonstrated that normal NSC fate determination, proliferation, maintenance, differentiation, and ventral patterning in the special and pivotal region called SVZ is intrinsically dependent on the SHH pathway in the adult brain and organogenesis, such as neural progenitor modulation during embryonic development (36). The SHH signaling pathway is critically implicated in the oncogenesis of GBM and the self-renewal of GSCs, as the activation of the SHH signaling pathway upregulates the drug efflux Pglycoprotein (ABCB1), ABC transporter ABCG2, multi-drug resistance-associated protein-1(ABCC1/MRP1), MGMT, B cell-specific Moloney murine leukemia virus integration site 1 (BMI1) (31, 139–141). Furthermore, the loss of P53 causes up-regulation of the potent transcription factor Nanog, which is closely correlated with SHH signaling pathway activity and the ability to regulate GSC properties, eventually contributing to chemo-resistance to TMZ in GSCs via regulating the expression of the TMZ resistance marker MGMT (141) and maintaining the self-renewal, differentiation, and expansion of GSCs based on recent studies (71).
RTK pathways are transmembrane proteins composed of a unique extracellular ligand-binding domain, a transmembrane helix, an intracellular tyrosine kinase domain, and a series of tyrosine residues (142, 143). RTK pathways include some of the most extensively studied pathways in oncology; here, we will focus on several important growth factor receptors in GBM and the targeted strategies to inhibit them.
The epidermal growth factor receptor (EGFR) is a transmembrane RTK that is critically responsible for the modulation of stem cell expansion, metastasis, differentiation, and survival in the human brain (144). In the case of heterodimers or homodimers formed by binding to ligands consisting of EGF or transforming growth factor-α (TGF-α), EGFR causes autophosphorylation of its C-terminal tail and activation of downstream signaling via its docking site of the SRC homology domain (145). It has been found that the over-expression of EGFR in humans accounts for 40%–60% of primary GBM tumors, especially in the classical subtype (146), however, the over-expression of EGFR in GBM is intrinsically implicated in gain-of-function missense mutations and in-frame deletions in the extracellular domain, rather than responsiveness to EGFR inhibitors (147, 148). The transactivation of β-catenin enables EGFR activation, leading to GSC expansion, metastasis, differentiation, tumor formation (149), and subsequently the human oncogenic EGFR is over-expressed, enhancing the self-renewal ability of GSCs, and further contributing to tumor- initiation or tumor- proliferation in GBM (36, 63, 64). Furthermore, in a GSCs marker study conducted by Song et al. (150), in 2021, SH3KBP1, a promising therapeutic target in GBM patients, was demonstrated to have the capacity to activate and modulate EGFR signaling. There are three kinds of EGFR tyrosine kinase inhibitors (TKIs) in total, including first-generation reversible small-molecule TKIs, which target EGFR and its co-receptor HER2, such as Erlotinib and Gefitinib (151), second-generation TKIs which bind irreversibly to EGFR, such as Afatinib, Neratinib and Dacomitinib (138) and third-generation irreversible inhibitors such as TKIs AZD9291 (Osimertinib), which have excellent blood–brain barrier penetration and have been shown to be effective in preclinical tests (152).
The PDGF family of receptors, one of the main deregulated RTK pathways in GBM, is comprised of two categories of receptors by different genes encoding PDGFRα, which is mainly responsible for developing oligodendrocytes (153) and PDGFRβ, which is important for blood vessel formation (154) during embryonic development. It has been found that the activation of PDGFR-signaling via a PDGF-nitric oxide (NO)-ID4-regulatory circuit (155) in humans accounts for tumorigenesis in 30% of GBM patients, and the overexpression or alterations of PDGF ligands are commonly found in GBM patients, especially in a proneural subtype of GBM, which contributes to the development of GBM and the function of GSCs, such as the self-renewal and tumor-initiating capacity to different extents among PDGFRα and PDGFRβ (155). Among several PDGF inhibitors, such as imatinib, tandutinib, AG1433, and nilotinib (138, 156), the small molecule TKI imatinib (also known as Gleevec; Glivec; STI-571) has been demonstrated to enable the growth of GBM xenografts in vivo to inhibit and the radiosensitivity of human GSCs in vitro to increase (157), but it has no significant therapeutic effect on recurrent GBM (156).
Three RTKs, VEGFR1, VEGFR2, and VEGFR3, which are encoded by the genes FLT1, KDR, and FLT4, constitute the VEGF receptor (VEGFR) family (158–160), a pivotal regulator of vasculogenesis, angiogenesis, and lymphangiogenesis (VEGFR3) in the normal body (161). GBM is one of various tumors which highly expresses VEGF and its receptors, resulting in highly vascularized tumors and increased microvasculature compared to other normal tissues (162), and is an important target in glioblastoma aberrant VEGFR2 signaling, which is an important pathway affecting survival, proliferation, migration, and vessel permeability in tumor cells (163). Furthermore, it has recently been demonstrated that human cartilage glycoprotein-39 or chitinase-like protein-1 (YKL-40) may be effective targets, as they are able to upregulate VEGF expression and induce new tumor vasculature (164). There are a wide variety of VEGFR inhibitors (TKIs) with different treatment effects, including atalanib (PTK787), mainly against VEGFR2, PDGFR, and c-kit (165), sorafenib (166), tivozanib (167), pazopanib combined with lapatinib (168), Cediranib (169), and SU1498 (170).
Therapeutic targeting of GSC pathways and receptors, such as Notch, Wnt, SHH, EGFR, PDGFR, and VEGFR presented in this review, which are critically implicated in tumor cell proliferation, maintenance, and resistance to current therapies, is of significant interest as it provides reliable and physiologically relevant trials to block these stem cells via the inhibition of pathways and receptors. Among these, EGFR, PDGFR, and VEGFR inhibitors have shown less advantages for their limitations compared to GSC pathways, such as Notch, Wnt, and SHH pathways, suggesting that targeting GSC pathways might become a future direction and an excellent solution in GBM therapy.
Multilineage differentiation both in vitro and in vivo is an important property in NSCs, and NSCs share the ability to promote cell differentiation in common with GSCs (68). In addition, GSCs have been proven to become inherently more sensitive to therapy, less capable of engraftment, and enable apoptosis to be directly induced in some settings after differentiating into more terminal glioblastoma cells (2). Hence, the promotion of GSC differentiation might be a key player in the therapeutic strategies for GBM. The bone morphogenic proteins (BMPs) involved in promoting normal neural precursor differentiation into astrocytes and post-transcriptional modification using miRNA, have been demonstrated to play a positive role in the stem cell niche of the adult brain, increase in GSC differentiation, and suppression of glioblastoma tumorigenicity in vivo, leading to the elimination of GSCs and sensitization of GBM to chemotherapeutics (2, 171). Recently, the overexpression of miR-128 or miR-302a has been shown to promote GSC differentiation, enhance senescence mediated by axitinib treatment, and further impair GSC proliferation (172). Furthermore, graphene oxide (GO), a new carbon material, has been reported to have potential for use in GBM treatment, as GO could decrease the expression of stem cell markers such as SOX2 and CD133, and increase the expression of differentiation-related markers such as GFAP and β-III tubulin, ultimately inducing the differentiation of GSCs (173). Sulindac, a non-steroidal anti-inflammatory drug (NSAID), is capable of inducing GSC differentiation and sensitizing them to oxidative stress (174).In the future, novel approaches for the promotion of GSC differentiation and potent anti-GBM agents such as GO and Sulindac, which could be used to suppress GSCs and become useful for future clinical applications, are urgently needed.
As strictly intracellular organisms, viruses can replicate inside host cells by hijacking the cellular machinery. Among these various targeting GSC strategies, virotherapy has shown therapeutic value in terms of preventing GBM recurrence; here, we will shed light on oncolytic virotherapy (OV), which has been extensively studied. Oncolytic virotherapy possesses lytic properties and is capable of achieving tumor cell lysis through intra-neoplastic virus replication (175). Several approaches for delivering OV with stem cells against recurrent GBM have been studied, including intra-arterial delivery of allogeneic bone marrow-derived human mesenchymal stem cells loaded with the oncolytic adenovirus DNX-2401 (BM-hMSCs-DNX2401), and injection of NSCs to deliver an oncolytic adenovirus into newly diagnosed GBM (4). Furthermore, this might be an ideal strategy to overcome the limitations of oncolytic viral vectors, such as limited biodistribution, dismal replication, and negligible transduction of neighboring tumor cells after intracranial injection to study some vectors that possess the capacity to specifically target the small population of GSCs with resistance to chemotherapy and radiotherapy, in addition to more globally targeting the bulk tumor populations.
There are three major methods of indirect targeting of the GSC niche, including targeting of: the perivascular niche via angiogenic pathways, the perinecrotic or hypoxic niche via inducing hypoxia, and the immune niche (Figure 2), which is still one of the crucial strategies to solve resistance to current standard therapy for GBM and improve the poor disease prognosis of GBM.
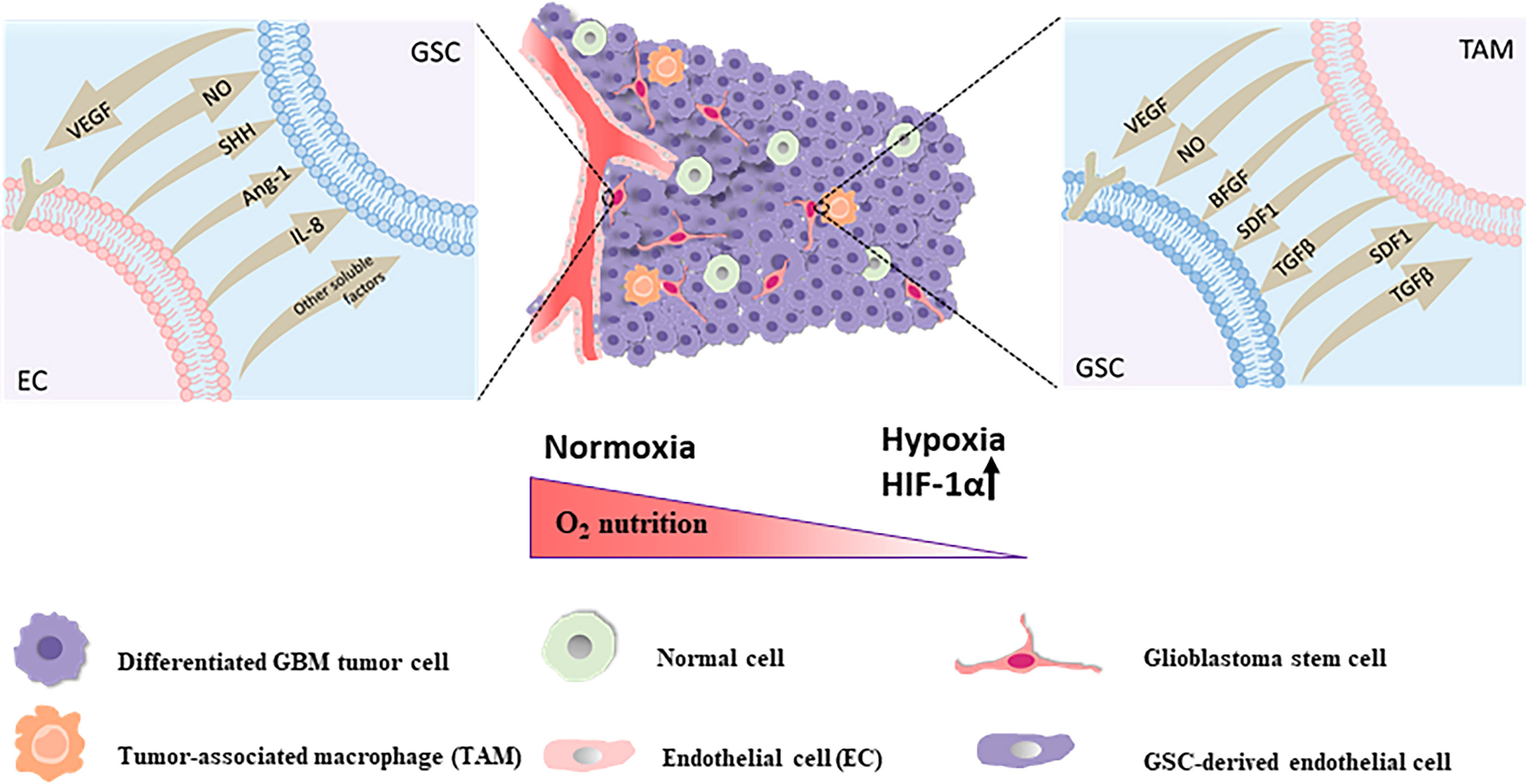
Figure 2 The perivascular niche The self-renewal and maintenance of neighboring GSCs within the perivascular maintenance niche could been promoted via secretion of endothelial-derived diffusible signals including nitric oxide (NO), SHH, angiopoietin-1 (Ang-1), IL-8 and other soluble factors. GSCs are capable of stimulating the proliferation of endothelial cells and the sprouting of new vessels via secretion of VEGF and SDF-1 in the local tumor environment for sustainability and expansion of the vascular maintenance niche. The hypoxic niche With the constant invasion of GSCs, activation of hypoxia-related factors might take an active role in stemcell maintenance and one of the main factors is the hypoxia response Hypoxia Inducible Factor 1α (HIF-1α). The immune niche On the one hand, for TAMs, GSCs could chemo-attract and recruit TAMs to the tumor site, prompt the growth of macrophages and induce the polarization of TAMs into the immunosuppressive M2 phenotype through secreting chemokines and growth factors like VEGF, transforming growth factor-β (TGF-β), SDF1 and soluble colony-stimulating factor 1 (sCSF-1). On the other hand, the accumulation of these pro-tumorigenic TAMs in tumor could secrete cytokines and signaling molecules such as basic fibroblast growth factor (bFGF), TGFβ, SDF1, VEGF, and nitric oxide (NO), contributing to tumor progression and GSCs maintenance in turn.
The perivascular niche is surrounded by a mass of blood vessels feeding the tumor with abnormal structure and function, including poor and irregular formation (leaky and friable blood vessels), hypoxia, loss of hierarchy, and an impaired blood–brain barrier (BBB). This vasculature is distinct from the normal blood vessels in humans, which are formed primarily through vasculogenesis and angiogenesis, and transport gases, nutrients, and waste products for the human body (176, 177). The synergistic and codependent relationship between the GBM perivascular niche and GSCs has been extensively proven. For example, it is known that GBM mediates the perivascular region to maintain GSCs survival, proliferation, and migration (178), simultaneously GSCs act as a regulator of the cancer-specific vasculature, infiltrating the tumor and thus contributing to the progression of tumor angiogenesis (116). High levels of angiogenic factors produced by GSCs in the human body are conducive to the abnormal formation of tumor blood vessels under hypoxic microenvironments, including vascular endothelial growth factor (VEGF), SDF-1, PDGF, and fibroblast growth factor (FGF), providing beneficial conditions for GSC survival and expansion (178–180). Consequently, this might become an ideal approach for GBM therapy by indirectly targeting the perivascular niche of GSCs and interfering in the aberrant vascular proliferation via the inhibitors angiogenic factors in tumor and simultaneously combined with other treatments that might inhibit progression of tumor malignancy or GSC differentiation, such as in combination with immunotherapeutics, especially those designed to repolarize macrophages (181) to overcome the challenge that malignant progression in GBM is prompted by increasing the invasion and metastasis of tumor cells after the blockage of angiogenesis that could shrink the original tumor (14).
Hypoxia as one of the diagnostic hallmarks of GBM, with median oxygen saturation levels of <2% in necrotic regions, compared to the normal median oxygen concentrations of approximately 5% in physiological tissues, owing to vigorous metabolic activity and plentiful oxygen consumption in heterogeneous tumor cells, along with chaotic and poor functioning blood vessels (182) that are able to maintain and promote the intrinsic cellular function of GSCs involved in self-renewal and differentiation, as well as enhance angiogenesis, tumor aggression, and chemo- and radiotherapy resistance in GBM, which are the main reasons for GBM patient death (183, 184). It has been demonstrated that one of the main regulators of the hypoxia response Hypoxia Inducible Factor-1α (HIF-1α) and Vascular Endothelial Growth Factor A (VEGFA) staining and tumor vascularity are critically implicated in worse progression-free survival and are responsible for lower patient survival rates of GBM based on dynamic contrast-enhanced MRI analyses (185, 186), while another hypoxia inducible factor (HIF) family HIF-2α indicates an important role of maintaining GSC survival and proliferation, as blockage of HIF-2α via the gene silencing technology or others would significantly compromise the intrinsic cellular features of GSCs (26, 187). Moreover, VEGFA is one of the main factors prompting the invasion of GSCs and polarizing immune cells into an immunosuppressive phenotype, causing treatment resistance to both standard and modern approaches via tumor-associated macrophage M2 polarization, increased regulatory T cells, and higher rates of PD-1+ CD8+ T cells (188–191). Bevacizumab, a humanized monoclonal antibody and inhibitor of VEGFA, is thus a promising anti-VEGF agent to create persistent normalization through pruning vessels and overcoming the recurrence of hypoxia and the emergence of resistance in GBM (192).
In GBM mouse model systems, scientists found that tumor aggression increased and the infiltration of a large number of immune cells, such as TAMs and other CD11b+ myeloid cells emerged in the brain after treating recurrent GBM patients with bevacizumab or the angiokinase inhibitor cediranib, despite the immune privilege in the normal human brain (119, 193–195). The direct interaction of GSCs with these immune cells could contribute to resistance of antiangiogenic therapy by generating VEGF-independent angiogenesis and immunosuppression, which leads to the development of tumors lacking the ability to respond to VEGF inhibition (196, 197). TAMs are the most prevalent tumor-infiltrating inflammatory cells in GBM (188, 189). A considerable number of infiltrating TAMs in the GSC niche, including the blood-derived macrophages and resident microglia (198), indicates a pivotal player in GBM tumor progression and GSC maintenance by a pleiotrophin–PTPRZ1 signaling axis (199), and is also positively linked to the poor prognosis of GBM and the high malignancy grade of glioma (200, 201). The relationship between the GBM immune niche and GSCs is also synergistic and interactive. On the one hand, for TAMs, GSCs could facilitate chemo-attraction and recruit TAMs to the tumor site upon the hypoxic conditions, prompt the growth of macrophages, and induce the polarization of TAMs into the immunosuppressive M2 phenotype by secreting chemokines and growth factors such as VEGF, TGF-β1, neurotensin, SDF1, and sCSF-1 (202–206). Additionally, for T lymphocytes, GSCs play a direct role in inhibiting the proliferation and activation of T cells and inducing T cell apoptosis, which may be mediated via the inhibitory, co-stimulatory molecule B7-H1, soluble factors such as galectin-3, the T cell chemokine attractants VEGF, chemokine (C-C motif) ligand 2 (CCL2), and prostaglandin E2 (202, 207–210), leading to silencing of the immune response and escape immune surveillance in GBM. Contrarily, the accumulation of these pro-tumorigenic TAMs in tumors could trigger the secretion of high levels of pro-inflammatory cytokines, such as RAGE, COX2, and NF-kB (211, 212), contributing to tumor progression and GSC maintenance. Accordingly, targeting innate immune cells and receptors correlated with their secreta, such as macrophage colony stimulation factor receptor (213), have been shown to increase tumor sensitivity to anti-GBM therapy, and could become a promising therapeutic target for GBM.
Here, we shed light on the knowledge of GSCs that are consistent with the general definition of CSCs, their resistance mechanism, vital signal pathways, the crosstalk between GSCs and their niche, and describe how this can prompt the persistence and progression of these stem cells. We also provide a framework for targeting strategies to GSCs consisting of direct and indirect strategies with some constructive opinions presented towards different targeting strategies in the review. Targeting GSCs provides a tremendous opportunity for revolutionary approaches, and a wide variety of reliable therapeutic strategies have been identified that are being clinically translated to improve the prognosis and therapy of GBM after several decades of stem cell therapy experiments, despite the need to simultaneously meet a variety of challenges, such as how to target GSCs effectively while avoiding impairing normal NSCs and progenitor cells. Accordingly, more authentically useful approaches for the isolation and identification of GSCs, further laboratory and clinical investigations regarding crosstalk between GBM and GSCs are needed to gain insights into maintenance, therapy resistance, and recurrence in tumors, as well as the origin, properties, and progression of GSCs. In the future, efficient design of clinical trials, novel approaches for targeting GSCs, and potent anti-GBM agents that could be used to suppress GSCs and become useful for future clinical applications are urgently needed.
XT, RT, and YH conceived the article. XT compiled the review and prepared the draft of the manuscript. PF, RT, YH, ZZ, GL, and YQ reviewed and edited the manuscript. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Tasaki T, Fujita M, Okuda T, Yoneshige A, Kato A. MET Expressed in Glioma Stem Cells Is a Potent Therapeutic Target for Glioblastoma Multiform. Anticancer Res (2016) 36(7):3571.
2. Auffinger B, Spencer D, Pytel P, Ahmed AU, Lesniak MS. The Role of Glioma Stem Cells in Chemotherapy Resistance and Glioblastoma Multiforme Recurrence. Expert Rev Neurother (2015) 15(7):741–52. doi: 10.1586/14737175.2015.1051968
3. Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, et al. Integrated Genomic Analysis Identifies Clinically Relevant Subtypes of Glioblastoma Characterized by Abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell (2010) 17(1):98–110. doi: 10.1016/j.ccr.2009.12.020
4. Wen PY, Weller M, Lee EQ, Alexander BM, Barnholtz-Sloan JS, Barthel FP, et al. Glioblastoma in Adults: A Society for Neuro-Oncology (SNO) and European Society of Neuro-Oncology (EANO) Consensus Review on Current Management and Future Directions. Neuro Oncol (2020) 22(8):1073–113. doi: 10.1093/neuonc/noaa106
5. Villa C, Miquel C, Mosses D, Bernier M, Di Stefano D. The 2016 World Health Organization Classification of Tumours of the Central Nervous System. Presse Med (2018) 47(11-12 Pt 2):e187–200. doi: 10.1016/j.lpm.2018.04.015
6. Khan I, Mahfooz S, Hatiboglu MA. Herbal Medicine for Glioblastoma: Current and Future Prospect. Med= Chem (2020) 16:1–22. doi: 10.2174/1573406416666200130100833
7. Khan I, Mahfooz S, Elbasan E, Karaçam B, Oztanir M, Hatiboglu M. Targeting Glioblastoma: The Current State of Different Therapeutic Approache. Curr Neuropharmacol (2021) 19. doi: 10.2174/1570159X19666210113152108
8. Fernandes C, Costa A, Osório L, Lago RC, Linhares P, Carvalho B, et al. Current Standards of Care in Glioblastoma Therap. In: De Vleeschouwer S, editor. Glioblastom. Brisbane (AU: Codon Publications (2017).
9. Arbab AS, Rashid MH, Angara K, Borin TF, Lin P-C, Jain M, et al. Major Challenges and Potential Microenvironment-Targeted Therapies in Glioblastom. Int J Mol Sci (2017) 18(12):2732. doi: 10.3390/ijms18122732
10. Alphandéry E. Glioblastoma Treatments: An Account of Recent Industrial Development. Front Pharmacol (2018) 9:879. doi: 10.3389/fphar.2018.00879
11. Volovetz J, Berezovsky AD, Alban T, Chen Y, Lauko A, Aranjuez GF, et al. Identifying Conserved Molecular Targets Required for Cell Migration of Glioblastoma Cancer Stem Cells. Cell Death Dis (2020) 11(2):152. doi: 10.1038/s41419-020-2342-2
12. Chen J, Li Y, Yu TS, McKay RM, Burns DK, Kernie SG, et al. A Restricted Cell Population Propagates Glioblastoma Growth After Chemotherapy. Nature (2012) 488(7412):522–6. doi: 10.1038/nature11287
13. Binello E, Germano IM. Targeting Glioma Stem Cells: A Novel Framework for Brain Tumors. Cancer Sci (2011) 102(11):1958–66. doi: 10.1111/j.1349-7006.2011.02064.x
14. Chen J, McKay RM, Parada LF. Malignant Glioma: Lessons From Genomics, Mouse Models, and Stem Cells. Cell (2012) 149(1):36–47. doi: 10.1016/j.cell.2012.03.009
15. Sundar SJ, Hsieh JK, Manjila S, Lathia JD, Sloan A. The Role of Cancer Stem Cells in Glioblastoma. Neurosurg Focus (2014) 37(6):E6. doi: 10.3171/2014.9.FOCUS14494
16. Cheng L, Wu Q, Guryanova OA, Huang Z, Huang Q, Rich JN, et al. Elevated Invasive Potential of Glioblastoma Stem Cells. Biochem Biophys Res Commun (2011) 406(4):643–8. doi: 10.1016/j.bbrc.2011.02.123
17. Grzybicki DM. Enhanced Invasion In Vitro and the Distribution Patterns In Vivo of CD133+ Glioma Stem Cells. Yearbook Pathol Lab Med (2012) 2012:224–6. doi: 10.1016/j.ypat.2011.11.133
18. Inoue A, Takahashi H, Harada H, Kohno S, Ohue S, Kobayashi K, et al. Cancer Stem-Like Cells of Glioblastoma Characteristically Express MMP-13 and Display Highly Invasive Activity. Int J Oncol (2010) 37(5):1121–31. doi: 10.3892/ijo_00000764
19. Beier D, Hau P, Proescholdt M, Lohmeier A, Wischhusen J, Oefner PJ, et al. CD133(+) and CD133(-) Glioblastoma-Derived Cancer Stem Cells Show Differential Growth Characteristics and Molecular Profiles. Cancer Res (2007) 67(9):4010–5. doi: 10.1158/0008-5472.can-06-4180
20. Galli R, Binda E, Orfanelli U, Cipelletti B, Gritti A, De Vitis S, et al. Isolation and Characterization of Tumorigenic, Stem-Like Neural Precursors From Human Glioblastoma. Cancer Res (2004) 64(19):7011–21. doi: 10.1158/0008-5472.can-04-1364
21. Lathia JD, Gallagher J, Heddleston JM, Wang J, Eyler CE, Macswords J, et al. Integrin Alpha 6 Regulates Glioblastoma Stem Cells. Cell Stem Cell (2010) 6(5):421–32. doi: 10.1016/j.stem.2010.02.018
22. Ogden AT, Waziri AE, Lochhead RA, Fusco D, Lopez K, Ellis JA, et al. Identification of A2B5+CD133- Tumor-Initiating Cells in Adult Human Gliomas. Neurosurgery (2008) 62(2):505–14. doi: 10.1227/01.neu.0000316019.28421.95
23. Singh SK, Hawkins C, Clarke ID, Squire JA, Dirks PB. Identification of Human Brain Tumor Initiating Cells. Nature (2004) 432(7015):396–401. doi: 10.1038/nature03128
24. Son MJ, Woolard K, Nam DH, Lee J, Fine HA. SSEA-1 is an Enrichment Marker for Tumor-Initiating Cells in Human Glioblastoma. Cell Stem Cell (2009) 4(5):440–52. doi: 10.1016/j.stem.2009.03.003
25. Haas TL, Sciuto MR, Brunetto L, Valvo C, Signore M, Fiori ME, et al. Integrin α7 Is a Functional Marker and Potential Therapeutic Target in Glioblastom. Cell Stem Cell (2017) 21(1):35–50. doi: 10.1016/j.stem.2017.04.009
26. Wang J, Ma Y, Cooper MK. Cancer Stem Cells in Glioma: Challenges and Opportunities. Trans Cancer Res (2013) 2(5):429–41. doi: 10.3978/j.issn.2218-676X.2013.08.01
27. Vlashi E, Lagadec C, Vergnes L, Matsutani T, Masui K, Poulou M, et al. Metabolic State of Glioma Stem Cells and Nontumorigenic Cells. Proc Of Natl Acad Of Sci Of USA (2011) 108(38):16062–7. doi: 10.1073/pnas.1106704108
28. Soeda A, Park M, Lee D, Mintz A, Androutsellis-Theotokis A, McKay RD, et al. Hypoxia Promotes Expansion of the CD133-Positive Glioma Stem Cells Through Activation of HIF-1α. Oncogene (2009) 28(45):3949–59. doi: 10.1038/onc.2009.252
29. Dantas-Barbosa C, Bergthold G, Daudigeos-Dubus E, Blockus H, Boylan JF, Ferreira C, et al. Inhibition of the NOTCH Pathway Using Gamma-Secretase Inhibitor RO4929097 has Limited Antitumor Activity in Established Glial Tumors. Anti-Cancer Drugs (2015) 26(3):272–83. doi: 10.1097/CAD.0000000000000190
30. Hiddingh L, Tannous BA, Teng J, Tops B, Jeuken J, Hulleman E, et al. EFEMP1 Induces γ-Secretase/Notch-Mediated Temozolomide Resistance in Glioblastoma. Oncotarget (2014) 5(2):363–74. doi: 10.18632/oncotarget.1620
31. Sharifzad F, Ghavami S, Verdi J, Mardpour S, Mollapour Sisakht M, Azizi Z, et al. Glioblastoma Cancer Stem Cell Biology: Potential Theranostic Targets. Drug Resist Updates: Rev Commentaries Antimicrobial Anticancer Chemother (2019) 42:35–45. doi: 10.1016/j.drup.2018.03.003
32. Stiles CD, Rowitch DH. Glioma Stem Cells: A Midterm Exam. Neuron (2008) 58(6):832–46. doi: 10.1016/j.neuron.2008.05.031
33. Siminovitch L, McCulloch EA, Till JE. The Distribution of Colony-Forming Cells Among Spleen Colonies. J Cell Comp Physiol (1963) 62:327–36. doi: 10.1002/jcp.1030620313
34. Caussinus E, Hirth F. Asymmetric Stem Cell Division in Development and Cancer. Prog Mol Subcellular Biol (2007) 45:205–25. doi: 10.1007/978-3-540-69161-7_9
35. Zakrzewski W, Dobrzynski M, Szymonowicz M, Rybak Z. Stem Cells: Past, Present, and Future. Stem Cell Res Ther (2019) 10(1):68. doi: 10.1186/s13287-019-1165-5
36. Liebelt BD, Shingu T, Zhou X, Ren J, Shin SA, Hu J. Glioma Stem Cells: Signaling, Microenvironment, and Therap. Stem Cells Int (2016) 2016:7849890. doi: 10.1155/2016/7849890
37. Xie Y, Bergstrom T, Jiang Y, Johansson P, Marinescu VD, Lindberg N, et al. The Human Glioblastoma Cell Culture Resource: Validated Cell Models Representing All Molecular Subtype. EBioMedicine (2015) 2(10):1351–63. doi: 10.1016/j.ebiom.2015.08.026
38. Alcantara Llaguno S, Chen J, Kwon CH, Jackson EL, Li Y, Burns DK, et al. Malignant Astrocytomas Originate From Neural Stem/Progenitor Cells in a Somatic Tumor Suppressor Mouse Model. Cancer Cell (2009) 15(1):45–56. doi: 10.1016/j.ccr.2008.12.006
39. Bayin NS, Modrek AS, Placantonakis DG. Brain Tumor Stem Cells. Mol Pathol Nervous Syst Tumors: Biol Stratification Targeted Therapies (2015) 8:23–34. doi: 10.1007/978-1-4939-1830-0_2
40. Byer SJ, Brossier NM, Peavler LT, Eckert JM, Watkins S, Roth KA, et al. Malignant Peripheral Nerve Sheath Tumor Invasion Requires Aberrantly Expressed EGF Receptors and is Variably Enhanced by Multiple EGF Family Ligands. J Neuropathol Exp Neurol (2013) 72(3):219–33. doi: 10.1097/NEN.0b013e3182859939
41. Chugh R, Wathen JK, Maki RG, Benjamin RS, Patel SR, Meyers PA, et al. Phase II Multicenter Trial of Imatinib in 10 Histologic Subtypes of Sarcoma Using a Bayesian Hierarchical Statistical Model. J Clin Oncol: Off J Am Soc Clin Oncol (2009) 27(19):3148–53. doi: 10.1200/jco.2008.20.5054
42. Lathia JD, Hitomi M, Gallagher J, Gadani SP, Adkins J, Vasanji A, et al. Distribution of CD133 Reveals Glioma Stem Cells Self-Renew Through Symmetric and Asymmetric Cell Divisions. Cell Death Dis (2011) 2:e200. doi: 10.1038/cddis.2011.80
43. Schmidtstraßburger U. Neural Stem Cells in Development and Agin. Cancer Sci (2012) 110(2):499–508. doi: 10.1159/000338027
44. Little JN, Dwyer ND. P53 Deletion Rescues Apoptosis and Microcephaly in a Kif20b Mouse Mutant. Hum Mol Gene (2018) 28(3):434-47. doi :10.1093/hmg/ddy350
45. Friedmann-Morvinski D, Verma IM. Dedifferentiation and Reprogramming: Origins of Cancer Stem Cells. EMBO Rep (2014) 15(3):244–53. doi: 10.1002/embr.201338254
46. Campos B, Olsen LR, Urup T, Poulsen HS. A Comprehensive Profile of Recurrent Glioblastoma. Oncogene (2016) 35(45):5819–25. doi: 10.1038/onc.2016.85
47. Lee JH, Lee JE, Kahng JY, Kim SH, Park JS, Yoon SJ, et al. Human Glioblastoma Arises From Subventricular Zone Cells With Low-Level Driver Mutations. Nature (2018) 560(7717):243–7. doi: 10.1038/s41586-018-0389-3
48. Rycaj K, Tang DG. Cell-Of-Origin of Cancer Versus Cancer Stem Cells: Assays and Interpretation. Cancer Res (2015) 75(19):4003–11. doi: 10.1158/0008-5472.CAN-15-0798
49. Friedmann-Morvinski D, Bushong EA, Ke E, Soda Y, Marumoto T, Singer O, et al. Dedifferentiation of Neurons and Astrocytes by Oncogenes can Induce Gliomas in Mice. Science (2012) 338(6110):1080–4. doi: 10.1126/science.1226929
50. Nair N, Calle AS, Zahra MH, Prieto-Vila M, Oo AKK, Hurley L, et al. A Cancer Stem Cell Model as the Point of Origin of Cancer-Associated Fibroblasts in Tumor Microenvironment. Sci Rep (2017) 7(1):6838. doi: 10.1038/s41598-017-07144-5
51. Carvalho LS, Gonçalves N, Fonseca NA, Moreira JN. Cancer Stem Cells and Nucleolin as Drivers of Carcinogenesi. Pharmaceuticals (2021) 14(1):60. doi: 10.3390/ph14010060
52. Hu B, Wang Q, Wang YA, Hua S, Sauve CG, Ong D, et al. Epigenetic Activation of WNT5A Drives Glioblastoma Stem Cell Differentiation and Invasive Growt. Cell (2016) 167(5):1281–95 e18. doi: 10.1016/j.cell.2016.10.039
53. Hu J, Ho AL, Yuan L, Hu B, Hua S, Hwang SS, et al. From the Cover: Neutralization of Terminal Differentiation in Gliomagenesis. Proc Natl Acad Sci USA (2013) 110(36):14520–7. doi: 10.1073/pnas.1308610110
54. Godlewski J, Nowicki MO, Bronisz A, Williams S, Otsuki A, Nuovo G, et al. Targeting of the Bmi-1 Oncogene/Stem Cell Renewal Factor by microRNA-128 Inhibits Glioma Proliferation and Self-Renewal. Cancer Res (2008) 68(22):9125–30. doi: 10.1158/0008-5472.CAN-08-2629
55. Silber J, Lim DA, Petritsch C, Persson AI, Maunakea AK, Yu M, et al. miR-124 and miR-137 Inhibit Proliferation of Glioblastoma Multiforme Cells and Induce Differentiation of Brain Tumor Stem Cells. BMC Med (2008) 6:14. doi: 10.1186/1741-7015-6-14
56. Huse JT, Holland EC. Genetically Engineered Mouse Models of Brain Cancer and the Promise of Preclinical Testing. Brain Pathol (2009) 19(1):132–43. doi: 10.1111/j.1750-3639.2008.00234.x
57. Jackson EL, Garcia-Verdugo JM, Gil-Perotin S, Roy M, Quinones-Hinojosa A, VandenBerg S, et al. PDGFR Alpha-Positive B Cells are Neural Stem Cells in the Adult SVZ That Form Glioma-Like Growths in Response to Increased PDGF Signaling. Neuron (2006) 51(2):187–99. doi: 10.1016/j.neuron.2006.06.012
58. Leder K, Pitter K, LaPlant Q, Hambardzumyan D, Ross BD, Chan TA, et al. Mathematical Modeling of PDGF-Driven Glioblastoma Reveals Optimized Radiation Dosing Schedule. Cell (2014) 156(3):603–16. doi: 10.1016/j.cell.2013.12.029
59. Ayuso-Sacido A, Moliterno JA, Kratovac S, Kapoor GS, O’Rourke DM, Holland EC, et al. Activated EGFR Signaling Increases Proliferation, Survival, and Migration and Blocks Neuronal Differentiation in Post-Natal Neural Stem Cells. J Neuro-Oncol (2010) 97(3):323–37. doi: 10.1007/s11060-009-0035-x
60. Lindberg OR, Persson A, Brederlau A, Shabro A, Kuhn HG. EGF-Induced Expansion of Migratory Cells in the Rostral Migratory Stream. PloS One (2012) 7(9):e46380. doi: 10.1371/journal.pone.0046380
61. Sanalkumar R, Vidyanand S, Lalitha Indulekha C, James J. Neuronal vs. Glial Fate of Embryonic Stem Cell-Derived Neural Progenitors (ES-NPs) is Determined by FGF2/EGF During Proliferation. J Mol Neurosci: MN (2010) 42(1):17–27. doi: 10.1007/s12031-010-9335-z
62. Wang J, Yu RK. Interaction of Ganglioside GD3 With an EGF Receptor Sustains the Self-Renewal Ability of Mouse Neural Stem Cells In Vitro. Proc Natl Acad Sci USA (2013) 110(47):19137–42. doi: 10.1073/pnas.1307224110
63. Ayuso-Sacido A, Graham C, Greenfield JP, Boockvar JA. The Duality of Epidermal Growth Factor Receptor (EGFR) Signaling and Neural Stem Cell Phenotype: Cell Enhancer or Cell Transformer? Curr Stem Cell Res Ther (2006) 1(3):387–94. doi: 10.2174/157488806778226849
64. Nicholas MK, Lukas RV, Jafri NF, Faoro L, Salgia R. Epidermal Growth Factor Receptor - Mediated Signal Transduction in the Development and Therapy of Gliomas. Clin Cancer Res: An Off J Am Assoc Cancer Res (2006) 12(24):7261–70. doi: 10.1158/1078-0432.CCR-06-0874
65. Jackson EL, Alvarez-Buylla A. Characterization of Adult Neural Stem Cells and Their Relation to Brain Tumors. Cells Tissues Organs (2008) 188(1-2):212–24. doi: 10.1159/000114541
66. Zhu Y, Guignard F, Zhao D, Liu L, Burns DK, Mason RP, et al. Early Inactivation of P53 Tumor Suppressor Gene Cooperating With NF1 Loss Induces Malignant Astrocytoma. Cancer Cell (2005) 8(2):119–30. doi: 10.1016/j.ccr.2005.07.004
67. Brescia P, Richichi C, Pelicci G. Current Strategies for Identification of Glioma Stem Cells: Adequate or Unsatisfactory? J Oncol (2012) 2012:376894. doi: 10.1155/2012/376894
68. Dirks PB. Brain Tumor Stem Cells: Bringing Order to the Chaos of Brain Cancer. J Clin Oncol: Off J Am Soc Clin Oncol (2008) 26(17):2916–24. doi: 10.1200/JCO.2008.17.6792
69. Ablett MP, Singh JK, Clarke RB. Stem Cells in Breast Tumours: Are They Ready for the Clinic? Eur J Cancer (2012) 48(14):2104–16. doi: 10.1016/j.ejca.2012.03.019
70. Azari H, Millette S, Ansari S, Rahman M, Deleyrolle LP, Reynolds BA. Isolation and Expansion of Human Glioblastoma Multiforme Tumor Cells Using the Neurosphere Assay. J Vis Exp (2011) 56):e3633. doi: 10.3791/3633
71. Abou-Antoun TJ, Hale JS, Lathia JD, Dombrowski SM. Brain Cancer Stem Cells in Adults and Children: Cell Biology and Therapeutic Implication. Neurotherapeutics (2017) 14(2):372–84. doi: 10.1007/s13311-017-0524-0
72. Flavahan WA, Drier Y, Liau BB, Gillespie SM, Venteicher AS, Stemmer-Rachamimov AO, et al. Insulator Dysfunction and Oncogene Activation in IDH Mutant Gliomas. Nature (2016) 529(7584):110–4. doi: 10.1038/nature16490
73. Clement V, Marino D, Cudalbu C, Hamou MF, Mlynarik V, de Tribolet N, et al. Marker-Independent Identification of Glioma-Initiating Cells. Nat Methods (2010) 7(3):224–8. doi: 10.1038/nmeth.1430
74. Xu YY, Gao P, Sun Y, Duan YR. Development of Targeted Therapies in Treatment of Glioblastoma. Cancer Biol Med (2015) 12(3):223–37. doi: 10.7497/j.issn.2095-3941.2015.0020
75. Xu HS, Qin XL, Zong HL, He XG, Cao L. Cancer Stem Cell Markers in Glioblastoma - An Update. Eur Rev Med Pharmacol Sci (2017) 21(14):3207–11.
76. Weigmann A, Corbeil D, Hellwig A, Huttner WB. Prominin, a Novel Microvilli-Specific Polytopic Membrane Protein of the Apical Surface of Epithelial Cells, is Targeted to Plasmalemmal Protrusions of Non-Epithelial Cells. Proc Natl Acad Sci USA (1997) 94(23):12425–30. doi: 10.1073/pnas.94.23.12425
77. Kang MK, Kang SK. Tumorigenesis of Chemotherapeutic Drug-Resistant Cancer Stem-Like Cells in Brain Glioma. Stem Cells Dev (2007) 16(5):837–47. doi: 10.1089/scd.2007.0006
78. Ludwig K, Kornblum HI. Molecular Markers in Glioma. J Neuro-Oncol (2017) 134(3):505–12. doi: 10.1007/s11060-017-2379-y
79. Ruiz-Garcia H, Alvarado-Estrada K, Schiapparelli P, Quinones-Hinojosa A, Trifiletti DM. Engineering Three-Dimensional Tumor Models to Study Glioma Cancer Stem Cells and Tumor Microenvironmen. Front Cell Neurosci (2020) 14:558381. doi: 10.3389/fncel.2020.558381
80. Klein WM, Wu BP, Zhao S, Wu H, Klein-Szanto AJ, Tahan SR. Increased Expression of Stem Cell Markers in Malignant Melanoma. Modern Pathol: An Off J United States Can Acad Pathol Inc (2007) 20(1):102–7. doi: 10.1038/modpathol.3800720
81. Krupkova O Jr., Loja T, Zambo I, Veselska R. Nestin Expression in Human Tumors and Tumor Cell Lines. Neoplasma (2010) 57(4):291–8. doi: 10.4149/neo_2010_04_291
82. Khan IS, Ehtesham M. Targeting Glioblastoma Cancer Stem Cells: The Next Great Hope? Neurosurg Focus (2014) 37(6):E7. doi: 10.3171/2014.9.focus14509
83. Singh SK, Clarke ID, Terasaki M, Bonn VE, Hawkins C, Squire J, et al. Identification of a Cancer Stem Cell in Human Brain Tumors. Cancer Res (2003) 63(18):5821–8. doi: 10.1002/cncr.11592
84. Wilson RJ, Thomas CD, Fox R, Roy DB, Kunin WE. Spatial Patterns in Species Distributions Reveal Biodiversity Change. Nature (2004) 432(7015):393–6. doi: 10.1038/nature03031
85. Hatanpaa KJ, Hu T, Vemireddy V, Foong C, Raisanen JM, Oliver D, et al. High Expression of the Stem Cell Marker Nestin is an Adverse Prognostic Factor in WHO Grade II-III Astrocytomas and Oligoastrocytomas. J Neuro-Oncol (2014) 117(1):183–9. doi: 10.1007/s11060-014-1376-7
86. Ehrmann J, Kolar Z, Mokry J. Nestin as a Diagnostic and Prognostic Marker: Immunohistochemical Analysis of its Expression in Different Tumours. J Clin Pathol (2005) 58(2):222–3. doi: 10.1136/jcp.2004.021238
87. Neradil J, Veselska R. Nestin as a Marker of Cancer Stem Cells. Cancer Sci (2015) 106(7):803–11. doi: 10.1111/cas.12691
88. Zhang M, Song T, Yang L, Chen R, Wu L, Yang Z, et al. Nestin and CD133: Valuable Stem Cell-Specific Markers for Determining Clinical Outcome of Glioma Patients. J Exp Clin Cancer Res: CR (2008) 27:85. doi: 10.1186/1756-9966-27-85
89. Barker N, Huch M, Kujala P, van de Wetering M, Snippert HJ, van Es JH, et al. Lgr5(+ve) Stem Cells Drive Self-Renewal in the Stomach and Build Long-Lived Gastric Units In Vitro. Cell Stem Cell (2010) 6(1):25–36. doi: 10.1016/j.stem.2009.11.013
90. Choi YJ, Kim N, Lee HS, Park SM, Park JH, Yoon H, et al. Expression of Leucine-Rich Repeat-Containing G-Protein Coupled Receptor 5 and CD44: Potential Implications for Gastric Cancer Stem Cell Marke. J Cancer Prev (2016) 21(4):279–87. doi: 10.15430/JCP.2016.21.4.279
91. Takahashi H, Ishii H, Nishida N, Takemasa I, Mizushima T, Ikeda M, et al. Significance of Lgr5(+ve) Cancer Stem Cells in the Colon and Rectum. Ann Surg Oncol (2011) 18(4):1166–74. doi: 10.1245/s10434-010-1373-9
92. Parry PV, Engh JA. Knockdown of LGR5 Suppresses the Proliferation of Glioma Cells In Vitro and In Vivo. Neurosurgery (2014) 74(2):N14–5. doi: 10.1227/01.neu.0000442976.61335.f6
93. Chen X, Wei B, Han X, Zheng Z, Huang J, Liu J, et al. LGR5 is Required for the Maintenance of Spheroid-Derived Colon Cancer Stem Cells. Int J Mol Med (2014) 34(1):35–42. doi: 10.3892/ijmm.2014.1752
94. Xi HQ, Cai AZ, Wu XS, Cui JX, Shen WS, Bian SB, et al. Leucine-Rich Repeat-Containing G-Protein-Coupled Receptor 5 is Associated With Invasion, Metastasis, and Could be a Potential Therapeutic Target in Human Gastric Cancer. Br J Cancer (2014) 110(8):2011–20. doi: 10.1038/bjc.2014.112
95. Zhang J, Cai H, Sun L, Zhan P, Chen M, Zhang F, et al. LGR5, a Novel Functional Glioma Stem Cell Marker, Promotes EMT by Activating the Wnt/beta-Catenin Pathway and Predicts Poor Survival of Glioma Patients. J Exp Clin Cancer Res: CR (2018) 37(1):225. doi: 10.1186/s13046-018-0864-6
96. Cela I, Di Matteo A. Nucleophosmin in Its Interaction With Ligand. Int J Mol Sci (2020) 21(14):E4885. doi: 10.3390/ijms21144885
97. Holmberg Olausson K, Elsir T, Moazemi Goudarzi K, Nistér M, Lindström MS. NPM1 Histone Chaperone is Upregulated in Glioblastoma to Promote Cell Survival and Maintain Nucleolar Shape. Sci Rep (2015) 5:16495. doi: 10.1038/srep16495
98. Chen J, Sun J, Yang L, Yan Y, Shi W, Shi J, et al. Upregulation of B23 Promotes Tumor Cell Proliferation and Predicts Poor Prognosis in Glioma. Biochem Biophys Res Commun (2015) 466(1):124–30. doi: 10.1016/j.bbrc.2015.08.118
99. Ou X, Ji C, Han X, Zhao X, Li X, Mao Y, et al. Crystal Structures of Human Glycerol 3-Phosphate Dehydrogenase 1 (GPD1). J Mol Biol (2006) 357(3):858–69. doi: 10.1016/j.jmb.2005.12.074
100. Menaya J, González-Manchón C, Parrilla R, Ayuso MS. Molecular Cloning, Sequencing and Expression of a cDNA Encoding a Human Liver NAD-Dependent Alpha-Glycerol-3-Phosphate Dehydrogenase. Biochim Biophys Acta (1995) 1262(1):91–4. doi: 10.1016/0167-4781(95)00069-s
101. Mráček T, Drahota Z, Houštěk J. The Function and the Role of the Mitochondrial Glycerol-3-Phosphate Dehydrogenase in Mammalian Tissues. Biochim Biophys Acta (2013) 1827(3):401–10. doi: 10.1016/j.bbabio.2012.11.014
102. Rusu P, Shao C, Neuerburg A, Acikgoz AA, Wu Y, Zou P, et al. GPD1 Specifically Marks Dormant Glioma Stem Cells With a Distinct Metabolic Profil. Cell Stem Cell (2019) 25(2):241–57 e8. doi: 10.1016/j.stem.2019.06.004
103. Kaal EC, Vecht CJ. The Management of Brain Edema in Brain Tumors. Curr Opin Oncol (2004) 16(6):593–600. doi: 10.1097/01.cco.0000142076.52721.b3
104. He J, Liu Y, Zhu T, Zhu J, Dimeco F, Vescovi AL, et al. CD90 is Identified as a Candidate Marker for Cancer Stem Cells in Primary High-Grade Gliomas Using Tissue Microarrays. Mol Cell Proteomics: MCP (2012) 11(6):M111 010744. doi: 10.1074/mcp.M111.010744
105. Gu Q, Chen X, Zhou L, Liu X. Exosome EpCAM Promotes the Metastasis of Glioma by Targeting the CD44 Signaling Molecule on the Surface of Glioma Cells. Adv Clin Exp Med: Off Organ Wroclaw Med Univ (2020) 29(11):1277–82. doi: 10.17219/acem/126051
106. Cragle CE, MacNicol MC, Byrum SD, Hardy LL, Mackintosh SG, Richardson WA, et al. Musashi Interaction With Poly(A)-Binding Protein is Required for Activation of Target mRNA Translation. J Biol Chem (2019) 294(28):10969–86. doi: 10.1074/jbc.RA119.007220
107. Yarmishyn AA, Yang YP, Lu KH, Chen YC, Chien Y, Chou SJ, et al. Musashi-1 Promotes Cancer Stem Cell Properties of Glioblastoma Cells via Upregulation of YTHDF1. Cancer Cell Int (2020) 20(1):597. doi: 10.1186/s12935-020-01696-9
108. Maness PF, Schachner M. Neural Recognition Molecules of the Immunoglobulin Superfamily: Signaling Transducers of Axon Guidance and Neuronal Migration. Nat Neurosci (2007) 10(1):19–26. doi: 10.1038/nn1827
109. Jackson M, Hassiotou F, Nowak A. Glioblastoma Stem-Like Cells: At the Root of Tumor Recurrence and a Therapeutic Target. Carcinogenesis (2015) 36(2):177–85. doi: 10.1093/carcin/bgu243
110. Hale JS, Otvos B, Sinyuk M, Alvarado AG, Hitomi M, Stoltz K, et al. Cancer Stem Cell-Specific Scavenger Receptor CD36 Drives Glioblastoma Progression. Stem Cells (Dayton Ohio) (2014) 32(7):1746–58. doi: 10.1002/stem.1716
111. Auvergne R, Wu C, Connell A, Au S, Cornwell A, Osipovitch M, et al. PAR1 Inhibition Suppresses the Self-Renewal and Growth of A2B5-Defined Glioma Progenitor Cells and Their Derived Gliomas In Vivo. Oncogene (2016) 35(29):3817–28. doi: 10.1038/onc.2015.452
112. Scheres B. Stem-Cell Niches: Nursery Rhymes Across Kingdoms. Nat Rev Mol Cell Biol (2007) 8(5):345–54. doi: 10.1038/nrm2164
113. Man J, Shoemake J, Zhou W, Fang X, Wu Q, Rizzo A, et al. Sema3C Promotes the Survival and Tumorigenicity of Glioma Stem Cells Through Rac1 Activation. Cell Rep (2014) 9(5):1812–26. doi: 10.1016/j.celrep.2014.10.055
114. Fan X, Khaki L, Zhu TS, Soules ME, Talsma CE, Gul N, et al. NOTCH Pathway Blockade Depletes CD133-Positive Glioblastoma Cells and Inhibits Growth of Tumor Neurospheres and Xenografts. Stem Cells (Dayton Ohio) (2010) 28(1):5–16. doi: 10.1002/stem.254
115. Cheng L, Huang Z, Zhou W, Wu Q, Donnola S, Liu JK, et al. Glioblastoma Stem Cells Generate Vascular Pericytes to Support Vessel Function and Tumor Growth. Cell (2013) 153(1):139–52. doi: 10.1016/j.cell.2013.02.021
116. Jhaveri N, Chen TC, Hofman FM. Tumor Vasculature and Glioma Stem Cells: Contributions to Glioma Progression. Cancer Lett (2016) 380(2):545–51. doi: 10.1016/j.canlet.2014.12.028
117. Colwell N, Larion M, Giles AJ, Seldomridge AN, Sizdahkhani S, Gilbert MR, et al. Hypoxia in the Glioblastoma Microenvironment: Shaping the Phenotype of Cancer Stem-Like Cells. Neuro Oncol (2017) 19(7):887–96. doi: 10.1093/neuonc/now258
118. Renfrow JJ, Soike MH, Debinski W, Ramkissoon SH, Mott RT, Frenkel MB, et al. Hypoxia-Inducible Factor 2α: A Novel Target in Gliomas. Future Med Chem (2018) 10(18):2227–36. doi: 10.4155/fmc-2018-0163
119. Lu-Emerson C, Duda DG, Emblem KE, Taylor JW, Gerstner ER, Loeffler JS, et al. Lessons From Anti-Vascular Endothelial Growth Factor and Anti-Vascular Endothelial Growth Factor Receptor Trials in Patients With Glioblastoma. J Clin Oncol: Off J Am Soc Clin Oncol (2015) 33(10):1197–213. doi: 10.1200/jco.2014.55.9575
120. Nakamura H, Makino Y, Okamoto K, Poellinger L, Ohnuma K, Morimoto C, et al. TCR Engagement Increases Hypoxia-Inducible Factor-1 Alpha Protein Synthesis via Rapamycin-Sensitive Pathway Under Hypoxic Conditions in Human Peripheral T Cells. J Immunol (Baltimore Md: 1950) (2005) 174(12):7592–9. doi: 10.4049/jimmunol.174.12.7592
121. Doedens AL, Phan AT, Stradner MH, Fujimoto JK, Nguyen JV, Yang E, et al. Hypoxia-Inducible Factors Enhance the Effector Responses of CD8(+) T Cells to Persistent Antigen. Nat Immunol (2013) 14(11):1173–82. doi: 10.1038/ni.2714
122. Roman J, Rangasamy T, Guo J, Sugunan S, Meednu N, Packirisamy G, et al. T-Cell Activation Under Hypoxic Conditions Enhances IFN-Gamma Secretion. Am J Respir Cell Mol Biol (2010) 42(1):123–8. doi: 10.1165/rcmb.2008-0139OC
123. Nizet V, Johnson RS. Interdependence of Hypoxic and Innate Immune Responses. Nat Rev Immunol (2009) 9(9):609–17. doi: 10.1038/nri2607
124. Saito N, Aoki K, Hirai N, Fujita S, Iwabuchi S. Abstract 774: Notch Pathway Activation Predicts Resistance to Bevacizumab Therapy in Glioblastoma. Cancer Res (2017) 77(13 Supplement):774–. doi: 10.1158/1538-7445.AM2017-774
125. Lai EC. Notch Signaling: Control of Cell Communication and Cell Fate. Dev (Cambridge England) (2004) 131(5):965–73. doi: 10.1242/dev.01074
126. Wu J, Bresnick EH. Bare Rudiments of Notch Signaling: How Receptor Levels are Regulated. Trends Biochem Sci (2007) 32(10):477–85. doi: 10.1016/j.tibs.2007.09.002
127. Wang J, Wakeman TP, Lathia JD, Hjelmeland AB, Wang X-F, White RR, et al. Notch Promotes Radioresistance of Glioma Stem Cells. Stem Cells (Dayton Ohio) (2009) 28:17–28. doi: 10.1002/stem.261. N/A-N/A.
128. Huppert SS, Le A, Schroeter EH, Mumm JS, Saxena MT, Milner LA, et al. Embryonic Lethality in Mice Homozygous for a Processing-Deficient Allele of Notch1. Nature (2000) 405(6789):966–70. doi: 10.1038/35016111
129. Paina S, Garzotto D, Demarchis S, Marino M, Moiana A, Conti L, et al. Wnt5a is a Transcriptional Target of Dlx Homeogenes and Promotes Differentiation of Interneuron Progenitors In Vitro and In Vivo. J Neurosci (2011) 31(7):2675–87. doi: 10.1523/JNEUROSCI.3110-10.2011
130. Pei Y, Brun SN, Markant SL, Lento W, Gibson P, Taketo MM, et al. WNT Signaling Increases Proliferation and Impairs Differentiation of Stem Cells in the Developing Cerebellum. Dev (Cambridge England) (2012) 139(10):1724. doi: 10.1242/dev.050104
131. Mariachiara Z, Patricia G, Sihana Z, Marzia C, Di IP, Francesco C, et al. The Role of Wnt Signal in Glioblastoma Development and Progression: A Possible New Pharmacological Target for the Therapy of This Tumo. Genes (2018) 9(2):105. doi: 10.3390/genes9020105
132. Grigoryan T, Wend P, Klaus A, Birchmeier W. Deciphering the Function of Canonical Wnt Signals in Development and Disease: Conditional Loss- and Gain-of-Function Mutations of β-Catenin in Mice. Genes Dev (2008) 22(17):2308–41. doi: 10.1101/gad.1686208
133. Munji RN, Choe Y, Li G, Siegenthaler JA, Pleasure SJ. Wnt Signaling Regulates Neuronal Differentiation of Cortical Intermediate Progenitor. J Neurosci Off J Soc Neurosci (2011) 31(5):1676–87. doi: 10.1523/JNEUROSCI.5404-10.2011
134. Binda E, Visioli A, Giani F, Trivieri N, Palumbo O, Restelli S, et al. Wnt5a Drives an Invasive Phenotype in Human Glioblastoma Stem-Like Cell. Cancer Res (2017) 77(4):996–1007. doi: 10.1158/0008-5472.can-16-1693
135. Atkins RJ, Dimou J, Paradiso L, Morokoff AP, Kaye AH, Drummond KJ, et al. Regulation of Glycogen Synthase Kinase-3 Beta (GSK-3β) by the Akt Pathway in Gliomas. J Clin Neurosci (2012) 19(11):1558–63. doi: 10.1016/j.jocn.2012.07.002
136. Hatten ME, Roussel MF. Development and Cancer of the Cerebellum. Trends Neurosci (2011) 34(3):134–42. doi: 10.1016/j.tins.2011.01.002
137. Lee Y, Lee J-K, Ahn SH, Lee J, Nam D-H. WNT Signaling in Glioblastoma and Therapeutic Opportunities. Lab Invest (2015) 96(2):137–50. doi: 10.1038/labinvest.2015.140
138. El-Khayat SM, Arafat WO. Therapeutic Strategies of Recurrent Glioblastoma and its Molecular Pathways ‘Lock Up the Beast’. Ecancermedicalscience (2021) 15:1176. doi: 10.3332/ecancer.2021.1176
139. Wee B, Pietras A, Ozawa T, Bazzoli E, Podlaha O, Antczak C, et al. ABCG2 Regulates Self-Renewal and Stem Cell Marker Expression But Not Tumorigenicity or Radiation Resistance of Glioma Cells. Sci Rep (2016) 6:25956. doi: 10.1038/srep25956
140. Shahi MH, Farheen S, Mariyath M, Castresana JS. Potential Role of Shh-Gli1-BMI1 Signaling Pathway Nexus in Glioma Chemoresistance. Tumour Biol: J Int Soc Oncodevelopmental Biol Med (2016) 37(11):15107–14. doi: 10.1007/s13277-016-5365-7
141. Hombach-Klonisch S, Mehrpour M, Shojaei S, Harlos C, Ghavami S. Glioblastoma and Chemoresistance to Alkylating Agents: Involvement of Apoptosis, Autophagy, and Unfolded Protein Respons. Pharmacol Ther (2017) 184:13–41. doi: 10.1016/j.pharmthera.2017.10.017
142. Lemmon MA, Schlessinger J. Cell Signaling by Receptor Tyrosine Kinases. Cell (2010) 141(7):1117–34. doi: 10.1016/j.cell.2010.06.011
143. van der Geer P, Hunter T, Lindberg RA. Receptor Protein-Tyrosine Kinases and Their Signal Transduction Pathways. Annu Rev Cell Biol (1994) 10:251–337. doi: 10.1146/annurev.cb.10.110194.001343
144. Arteaga CL, Engelman JA. ERBB Receptors: From Oncogene Discovery to Basic Science to Mechanism-Based Cancer Therapeutics. Cancer Cell (2014) 25(3):282–303. doi: 10.1016/j.ccr.2014.02.025
145. Singh B, Coffey RJ. Trafficking of Epidermal Growth Factor Receptor Ligands in Polarized Epithelial Cells. Annu Rev Physiol (2014) 76:275–300. doi: 10.1146/annurev-physiol-021113-170406
146. Nguyen HS, Shabani S, Awad AJ, Kaushal M. Molecular Markers of Therapy-Resistant Glioblastoma and Potential Strategy to Combat Resistanc. Int J Mol Sci (2018) 19(6):1765. doi: 10.3390/ijms19061765
147. Vivanco I, Robins HI, Rohle D, Campos C, Grommes C, Nghiemphu PL, et al. Differential Sensitivity of Glioma- Versus Lung Cancer-Specific EGFR Mutations to EGFR Kinase Inhibitors. Cancer Discov (2012) 2(5):458–71. doi: 10.1158/2159-8290.cd-11-0284
148. Lee JC, Vivanco I, Beroukhim R, Huang JH, Feng WL, DeBiasi RM, et al. Epidermal Growth Factor Receptor Activation in Glioblastoma Through Novel Missense Mutations in the Extracellular Domain. PloS Med (2006) 3(12):e485. doi: 10.1371/journal.pmed.0030485
149. Yang W, Xia Y, Ji H, Zheng Y, Liang J, Huang W, et al. Corrigendum: Nuclear PKM2 Regulates β-Catenin Transactivation Upon EGFR Activation. Nature (2017) 550(7674):142. doi: 10.1038/nature24008
150. Song H, Wang Y, Shi C, Lu J, Yuan T, Wang X. SH3KBP1 Promotes Glioblastoma Tumorigenesis by Activating EGFR Signaling. Front Oncol (2020) 10:583984. doi: 10.3389/fonc.2020.583984
151. Krawczyk P, Kowalski DM, Ramlau R, Kalinka-Warzocha E, Winiarczyk K, Stencel K, et al. Comparison of the Effectiveness of Erlotinib, Gefitinib, and Afatinib for Treatment of non-Small Cell Lung Cancer in Patients With Common and Rare EGFR Gene Mutations. Oncol Lett (2017) 13(6):4433–44. doi: 10.3892/ol.2017.5980
152. Liu X, Chen X, Shi L, Shan Q, Cao Q, Yue C, et al. The Third-Generation EGFR Inhibitor AZD9291 Overcomes Primary Resistance by Continuously Blocking ERK Signaling in Glioblastoma. J Exp Clin Cancer Res: CR (2019) 38(1):219. doi: 10.1186/s13046-019-1235-7
153. Fruttiger M, Karlsson L, Hall AC, Abramsson A, Calver AR, Boström H, et al. Defective Oligodendrocyte Development and Severe Hypomyelination in PDGF-A Knockout Mice. Dev (Cambridge England) (1999) 126(3):457–67. doi: 10.1007/s004290050219
154. Hellström M, Kalén M, Lindahl P, Abramsson A, Betsholtz C. Role of PDGF-B and PDGFR-Beta in Recruitment of Vascular Smooth Muscle Cells and Pericytes During Embryonic Blood Vessel Formation in the Mouse. Dev (Cambridge England) (1999) 126(14):3047–55. doi: 10.1007/s004290050265
155. Eun K, Jeon HM, Kim SO, Choi SH, Lee SY, Jin X, et al. A Cell-Autonomous Positive-Signaling Circuit Associated With the PDGF-NO-ID4-Regulatory Axis in Glioblastoma Cells. Biochem Biophys Res Commun (2017) 486(2):564–70. doi: 10.1016/j.bbrc.2017.03.089
156. Tilak M, Holborn J, New LA, Lalonde J, Jones N. Receptor Tyrosine Kinase Signaling and Targeting in Glioblastoma Multiform. Int J Mol Sci (2021) 22(4):1831. doi: 10.3390/ijms22041831
157. Oertel S, Krempien R, Lindel K, Zabel A, Milker-Zabel S, Bischof M, et al. Human Glioblastoma and Carcinoma Xenograft Tumors Treated by Combined Radiation and Imatinib (Gleeve). Strahlentherapie Und Onkol: Organ Der Deutschen Rontgengesellschaft (2006) 182(7):400–7. doi: 10.1007/s00066-006-1445-8
158. Matthews W, Jordan CT, Gavin M, Jenkins NA, Copeland NG, Lemischka IR. A Receptor Tyrosine Kinase cDNA Isolated From a Population of Enriched Primitive Hematopoietic Cells and Exhibiting Close Genetic Linkage to C-Kit. Proc Natl Acad Sci USA (1991) 88(20):9026–30. doi: 10.1073/pnas.88.20.9026
159. Galland F, Karamysheva A, Pebusque MJ, Borg JP, Rottapel R, Dubreuil P, et al. The FLT4 Gene Encodes a Transmembrane Tyrosine Kinase Related to the Vascular Endothelial Growth Factor Receptor. Oncogene (1993) 8(5):1233–40. doi: 10.1038/ng0593-98
160. Shibuya M, Yamaguchi S, Yamane A, Ikeda T, Tojo A, Matsushime H, et al. Nucleotide Sequence and Expression of a Novel Human Receptor-Type Tyrosine Kinase Gene (Flt) Closely Related to the Fms Family. Oncogene (1990) 5(4):519–24. doi: 10.1016/0027-5107(90)90097-N
161. Shibuya M. Vascular Endothelial Growth Factor (VEGF) and Its Receptor (VEGFR) Signaling in Angiogenesis: A Crucial Target for Anti- and Pro-Angiogenic Therapie. Genes Cancer (2011) 2(12):1097–105. doi: 10.1177/1947601911423031
162. Loureiro L, Neder L, Callegaro-Filho D, Koch L, Malheiros S. The Immunohistochemical Landscape of the VEGF Family and its Receptors in Glioblastomas. Surg Exp Pathol (2020) 3(1):1–8. doi: 10.1186/s42047-020-00060-5
163. Guarnaccia L, Navone SE, Trombetta E, Cordiglieri C, Cherubini A, Crisà FM, et al. Angiogenesis in Human Brain Tumors: Screening of Drug Response Through a Patient-Specific Cell Platform for Personalized Therapy. Scientific Reports (2018) 8(1):8748. doi: 10.1038/s41598-018-27116-7
164. Batista KMP, Eulate-Beramendi SA, Pińa K, Figueira PR, Canal AF, Chasin JMA, et al. Mesenchymal/proangiogenic Factor YKL-40 Related to Glioblastomas and its Relationship With the Subventricular Zone. Folia Neuropathol (2017) 55(1):14–22. doi: 10.5114/fn.2017.66709
165. Gerstner ER, Eichler AF, Plotkin SR, Drappatz J, Doyle CL, Xu L, et al. Phase I Trial With Biomarker Studies of Vatalanib (PTK787) in Patients With Newly Diagnosed Glioblastoma Treated With Enzyme Inducing Anti-Epileptic Drugs and Standard Radiation and Temozolomide. J Neuro-Oncol (2011) 103(2):325–32. doi: 10.1007/s11060-010-0390-7
166. Lee EQ, Kuhn J, Lamborn KR, Abrey L, DeAngelis LM, Lieberman F, et al. Phase I/II Study of Sorafenib in Combination With Temsirolimus for Recurrent Glioblastoma or Gliosarcoma: North American Brain Tumor Consortium Study 05-02. Neuro Oncol (2012) 14(12):1511–8. doi: 10.1093/neuonc/nos264
167. Kalpathy-Cramer J, Chandra V, Da X, Ou Y, Emblem KE, Muzikansky A, et al. Phase II Study of Tivozanib, an Oral VEGFR Inhibitor, in Patients With Recurrent Glioblastoma. J Neuro-Oncol (2017) 131(3):603–10. doi: 10.1007/s11060-016-2332-5
168. Reardon DA, Groves MD, Wen PY, Nabors L, Mikkelsen T, Rosenfeld S, et al. A Phase I/II Trial of Pazopanib in Combination With Lapatinib in Adult Patients With Relapsed Malignant Glioma. Clin Cancer Res: An Off J Am Assoc Cancer Res (2013) 19(4):900–8. doi: 10.1158/1078-0432.ccr-12-1707
169. Batchelor TT, Mulholland P, Neyns B, Nabors LB, Campone M, Wick A, et al. Phase III Randomized Trial Comparing the Efficacy of Cediranib as Monotherapy, and in Combination With Lomustine, Versus Lomustine Alone in Patients With Recurrent Glioblastoma. J Clin Oncol: Off J Am Soc Clin Oncol (2013) 31(26):3212–8. doi: 10.1200/jco.2012.47.2464
170. Popescu AM, Alexandru O, Brindusa C, Purcaru SO, Tache DE, Tataranu LG, et al. Targeting the VEGF and PDGF Signaling Pathway in Glioblastoma Treatment. Int J Clin Exp Pathol (2015) 8(7):7825–37.
171. Ciechomska IA, Gielniewski B, Wojtas B, Kaminska B. EGFR/FOXO3a/BIM Signaling Pathway Determines Chemosensitivity of BMP4-Differentiated Glioma Stem Cells to Temozolomide. Experimental and Mol Med (2020) 52(8):1326–40. doi: 10.1038/s12276-020-0479-9
172. Cardoso AM, Morais CM, Pena F, Marante T, Cunha PP, Jurado AS, et al. Differentiation of Glioblastoma Stem Cells Promoted by miR-128 or miR-302a Overexpression Enhances Senescence-Associated Cytotoxicity of Axitinib. Hum Mol Genet (2021) 30(3-4):160–71. doi: 10.1093/hmg/ddab011
173. Wang X, Zhou W, Li X, Ren J, Ji G, Du J, et al. Graphene Oxide Suppresses the Growth and Malignancy of Glioblastoma Stem Cell-Like Spheroids via Epigenetic Mechanisms. J Res Trans Med (2020) 18(1):200. doi: 10.1186/s12967-020-02359-z
174. Allani SK, Weissbach H, Lopez Toledano MA. Sulindac Induces Differentiation of Glioblastoma Stem Cells Making Them More Sensitive to Oxidative Stress. Neoplasma (2018) 65(3):376–88. doi: 10.4149/neo_2018_170404N245
175. Dey M, Ulasov IV, Tyler MA, Sonabend AM, Lesniak MS. Cancer Stem Cells: The Final Frontier for Glioma Virotherapy. Stem Cell Rev Rep (2011) 7(1):119–29. doi: 10.1007/s12015-010-9132-7
176. Zhu TS, Costello MA, Talsma CE, Flack CG, Crowley JG, Hamm LL, et al. Endothelial Cells Create a Stem Cell Niche in Glioblastoma by Providing NOTCH Ligands That Nurture Self-Renewal of Cancer Stem-Like Cells. Cancer Res (2011) 71(18):6061–72. doi: 10.1158/0008-5472.can-10-4269
177. Jain RK, di Tomaso E, Duda DG, Loeffler JS, Sorensen AG, Batchelor TT. Angiogenesis in Brain Tumours. Nat Rev Neurosci (2007) 8(8):610–22. doi: 10.1038/nrn2175
178. Calabrese C, Poppleton H, Kocak M, Hogg TL, Fuller C, Hamner B, et al. A Perivascular Niche for Brain Tumor Stem Cells. Cancer Cell (2007) 11(1):69–82. doi: 10.1016/j.ccr.2006.11.020
179. Farahani E, Patra HK, Jangamreddy JR, Rashedi I, Kawalec M, Rao Pariti RK, et al. Cell Adhesion Molecules and Their Relation to (Cancer) Cell Stemness. Carcinogenesis (2014) 35(4):747–59. doi: 10.1093/carcin/bgu045
180. Plaks V, Kong N, Werb Z. The Cancer Stem Cell Niche: How Essential is the Niche in Regulating Stemness of Tumor Cells? Cell Stem Cell (2015) 16(3):225–38. doi: 10.1016/j.stem.2015.02.015
181. Wirsching HG, Roth P. Weller M. A Vasculature-Centric Approach to Developing Novel Treatment Options for Glioblastoma. Expert Opin Ther Targets (2021) 25(2):87–100. doi: 10.1080/14728222.2021.1881062
182. Leone RD, Powell JD. Metabolism of Immune Cells in Cancer. Nat Rev Cancer (2020) 20(9):516–31. doi: 10.1038/s41568-020-0273-y
183. Codrici E, Enciu AM, Popescu ID, Mihai S, Tanase C. Glioma Stem Cells and Their Microenvironments: Providers of Challenging Therapeutic Target. Stem Cells Int (2016) 2016:5728438. doi: 10.1155/2016/5728438
184. Chedeville AL, Madureira PA. The Role of Hypoxia in Glioblastoma Radiotherapy Resistance. Cancers (2021) 13(3):542. doi: 10.3390/cancers13030542
185. Jensen RL, Mumert ML, Gillespie DL, Kinney AY, Schabel MC, Salzman KL. Preoperative Dynamic Contrast-Enhanced MRI Correlates With Molecular Markers of Hypoxia and Vascularity in Specific Areas of Intratumoral Microenvironment and is Predictive of Patient Outcome. Neuro Oncol (2014) 16(2):280–91. doi: 10.1093/neuonc/not148
186. Clara CA, Marie SK, de Almeida JR, Wakamatsu A, Oba-Shinjo SM, Uno M, et al. Angiogenesis and Expression of PDGF-C, VEGF, CD105 and HIF-1α in Human Glioblastoma. Neuropathol: Off J Japanese Soc Neuropathol (2014) 34(4):343–52. doi: 10.1111/neup.12111
187. Nusblat LM, Tanna S, Roth CM. Gene Silencing of HIF-2alpha Disrupts Glioblastoma Stem Cell Phenotype. Cancer Drug Resist (2020) 3(2):199–208. doi: 10.20517/cdr.2019.96
188. Escribese MM, Casas M, Corbí AL. Influence of Low Oxygen Tensions on Macrophage Polarization. Immunobiology (2012) 217(12):1233–40. doi: 10.1016/j.imbio.2012.07.002
189. Tamura R, Ohara K, Sasaki H, Morimoto Y, Kosugi K, Yoshida K, et al. Difference in Immunosuppressive Cells Between Peritumoral Area and Tumor Core in Glioblastoma. World Neurosurg (2018) 120:e601–e10. doi: 10.1016/j.wneu.2018.08.133
190. Tamura R, Tanaka T. The Role of Vascular Endothelial Growth Factor in the Hypoxic and Immunosuppressive Tumor Microenvironment: Perspectives for Therapeutic Implications. Med Oncol (2019) 37(1):2. doi: 10.1007/s12032-019-1329-2
191. Tamura R, Tanaka T, Ohara K, Miyake K, Morimoto Y, Yamamoto Y, et al. Persistent Restoration to the Immunosupportive Tumor Microenvironment in Glioblastoma by Bevacizumab. Cancer Sci (2019) 110(2):499–508. doi: 10.1111/cas.13889
192. Hambardzumyan D, Bergers G. Glioblastoma: Defining Tumor Niche. Trends Cancer (2015) 1(4):252–65. doi: 10.1016/j.trecan.2015.10.009
193. Gerstner ER, Chen PJ, Wen PY, Jain RK, Batchelor TT, Sorensen G. Infiltrative Patterns of Glioblastoma Spread Detected via Diffusion MRI After Treatment With Cediranib. Neuro Oncol (2010) 12(5):466–72. doi: 10.1093/neuonc/nop051
194. Gerstner ER, Frosch MP, Batchelor TT. Diffusion Magnetic Resonance Imaging Detects Pathologically Confirmed, Nonenhancing Tumor Progression in a Patient With Recurrent Glioblastoma Receiving Bevacizumab. J Clin Oncol: Off J Am Soc Clin Oncol (2010) 28(6):e91–3. doi: 10.1200/jco.2009.25.0233
195. Piao Y, Liang J, Holmes L, Zurita AJ, Henry V, Heymach JV, et al. Glioblastoma Resistance to Anti-VEGF Therapy is Associated With Myeloid Cell Infiltration, Stem Cell Accumulation, and a Mesenchymal Phenotype. Neuro Oncol (2012) 14(11):1379–92. doi: 10.1093/neuonc/nos158
196. Rivera LB, Bergers G. Intertwined Regulation of Angiogenesis and Immunity by Myeloid Cells. Trends Immunol (2015) 36(4):240–9. doi: 10.1016/j.it.2015.02.005
197. Rivera LB, Meyronet D, Hervieu V, Frederick MJ, Bergsland E, Bergers G. Intratumoral Myeloid Cells Regulate Responsiveness and Resistance to Antiangiogenic Therapy. Cell Rep (2015) 11(4):577–91. doi: 10.1016/j.celrep.2015.03.055
198. Gieryng A, Pszczolkowska D, Walentynowicz KA, Rajan WD, Kaminska B. Immune Microenvironment of Gliomas. Lab Invest (2017) 97(5):498. doi: 10.1038/labinvest.2017.19
199. Cui Q, Shi H, Ye P, Li L, Qu Q, Sun G, et al. M(6)A RNA Methylation Regulates the Self-Renewal and Tumorigenesis of Glioblastoma Stem Cell. Cell Rep (2017) 18(11):2622–34. doi: 10.1016/j.celrep.2017.02.059
200. Bingle L, Brown NJ, Lewis CE. The Role of Tumour-Associated Macrophages in Tumour Progression: Implications for New Anticancer Therapies. J Pathol (2002) 196(3):254–65. doi: 10.1002/path.1027
201. Roggendorf W, Strupp S, Paulus W. Distribution and Characterization of Microglia/Macrophages in Human Brain Tumors. Acta Neuropathol (1996) 92(3):288–93. doi: 10.1007/s004010050520
202. Filatova A, Acker T, Garvalov BK. The Cancer Stem Cell Niche(s): The Crosstalk Between Glioma Stem Cells and Their Microenvironment. Biochim Biophys Acta (2013) 1830(2):2496–508. doi: 10.1016/j.bbagen.2012.10.008
203. Wang SC, Hong JH, Hsueh C, Chiang CS. Tumor-Secreted SDF-1 Promotes Glioma Invasiveness and TAM Tropism Toward Hypoxia in a Murine Astrocytoma Model. Lab Investigation J Tech Methods Pathol (2012) 92(1):151–62. doi: 10.1038/labinvest.2011.128
204. Yi L, Xiao H, Xu M, Ye X, Hu J, Li F, et al. Glioma-Initiating Cells: A Predominant Role in Microglia/Macrophages Tropism to Glioma. J Neuroimmunol (2011) 232(1-2):75–82. doi: 10.1016/j.jneuroim.2010.10.011
205. Gabrusiewicz K, Ellert-Miklaszewska A, Lipko M, Sielska M, Frankowska M, Kaminska B. Characteristics of the Alternative Phenotype of Microglia/Macrophages and its Modulation in Experimental Gliomas. PloS One (2011) 6(8):e23902. doi: 10.1371/journal.pone.0023902
206. Wu A, Wei J, Kong LY, Wang Y, Priebe W, Qiao W, et al. Glioma Cancer Stem Cells Induce Immunosuppressive Macrophages/Microglia. Neuro Oncol (2010) 12(11):1113–25. doi: 10.1093/neuonc/noq082
207. Audia A, Conroy S, Glass R, Bhat KPL. The Impact of the Tumor Microenvironment on the Properties of Glioma Stem-Like Cell. Front Oncol (2017) 7:143. doi: 10.3389/fonc.2017.00143
208. Wei J, Wu A, Kong LY, Wang Y, Fuller G, Fokt I, et al. Hypoxia Potentiates Glioma-Mediated Immunosuppression. PloS One (2011) 6(1):e16195. doi: 10.1371/journal.pone.0016195
209. Wei J, Barr J, Kong LY, Wang Y, Wu A, Sharma AK, et al. Glioblastoma Cancer-Initiating Cells Inhibit T-Cell Proliferation and Effector Responses by the Signal Transducers and Activators of Transcription 3 Pathway. Mol Cancer Ther (2010) 9(1):67–78. doi: 10.1158/1535-7163.mct-09-0734
210. Jordan JT, Sun W, Hussain SF, DeAngulo G, Prabhu SS, Heimberger AB. Preferential Migration of Regulatory T Cells Mediated by Glioma-Secreted Chemokines can be Blocked With Chemotherapy. Cancer Immunol Immunother: CII (2008) 57(1):123–31. doi: 10.1007/s00262-007-0336-x
211. Liu HL, Wang YN, Feng SY. Brain Tumors: Cancer Stem-Like Cells Interact With Tumor Microenvironment. World J Stem Cells (2020) 12(12):1439–54. doi: 10.4252/wjsc.v12.i12.1439
212. Aderetti DA, Hira VVV, Molenaar RJ, van Noorden CJF. The Hypoxic Peri-Arteriolar Glioma Stem Cell Niche, an Integrated Concept of Five Types of Niches in Human Glioblastoma. Biochim Biophys Acta Rev Cancer (2018) 1869(2):346–54. doi: 10.1016/j.bbcan.2018.04.008
Keywords: glioblastoma, glioblastoma stem cells, biomarkers, signal pathways, targeted therapy
Citation: Tang X, Zuo C, Fang P, Liu G, Qiu Y, Huang Y and Tang R (2021) Targeting Glioblastoma Stem Cells: A Review on Biomarkers, Signal Pathways and Targeted Therapy. Front. Oncol. 11:701291. doi: 10.3389/fonc.2021.701291
Received: 01 May 2021; Accepted: 25 June 2021;
Published: 08 July 2021.
Edited by:
Zhi Sheng, Virginia Tech, United StatesReviewed by:
Bernard Rogister, University of Liège, BelgiumCopyright © 2021 Tang, Zuo, Fang, Liu, Qiu, Huang and Tang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yi Huang, eWlod2FuZ0AxMjYuY29t; Rongrui Tang, dGFuZ3JyXzIwMThAMTYzLmNvbQ==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.