 Aditya A. Mohan
Aditya A. Mohan William H. Tomaszewski
William H. Tomaszewski Aden P. Haskell-MendozaKelly M. HotchkissKirit SinghJessica L. ReedyPeter E. Fecci
Aden P. Haskell-MendozaKelly M. HotchkissKirit SinghJessica L. ReedyPeter E. Fecci John H. Sampson
John H. Sampson Mustafa Khasraw*
Mustafa Khasraw*- Preston Robert Tisch Brain Tumor Center at Duke, Department of Neurosurgery, Duke University Medical Center, Durham, NC, United States
We have only recently begun to understand how cancer metabolism affects antitumor responses and immunotherapy outcomes. Certain immunometabolic targets have been actively pursued in other tumor types, however, glioblastoma research has been slow to exploit the therapeutic vulnerabilities of immunometabolism. In this review, we highlight the pathways that are most relevant to glioblastoma and focus on how these immunometabolic pathways influence tumor growth and immune suppression. We discuss hypoxia, glycolysis, tryptophan metabolism, arginine metabolism, 2-Hydroxyglutarate (2HG) metabolism, adenosine metabolism, and altered phospholipid metabolism, in order to provide an analysis and overview of the field of glioblastoma immunometabolism.
Introduction
Advances in immunotherapies have revolutionized cancer care, yet unfortunately, they have been largely unsuccessful in managing glioblastoma. One of the primary obstacles in treating glioblastoma with immunotherapy has been overcoming the heterogeneous and immunosuppressive tumor microenvironment (TME) that is, at least in part, regulated by tumor metabolism. Since 1927, when Otto Warburg et al. first described tumor’s preferential use of glycolysis to generate adenosine triphosphate (ATP) (1), there has been a burgeoning interest in understanding tumor metabolism and how it influences tumor growth. However, it is only recently that our understanding of tumor metabolism has extended beyond the confines of the tumor cell membrane and that we have begun to understand how tumor metabolism affects noncancerous cells such as tumor-infiltrating immune cells.
While glioblastoma cells are metabolically distinct from noncancerous tissue in the brain, certain metabolic similarities exist between glioblastoma and proliferating immune cells. These similarities include an upregulation of glucose utilization, glycolysis, fatty acid oxidization, amino acid metabolism, and nucleotide synthesis. As such, glioblastoma cells can induce immunosuppression by outcompeting immune cells for critical nutrients. In addition to contending with immune cells for metabolites, certain glioblastoma cells can also avail distinctive metabolic pathways to produce unique metabolites such as 2-Hydroxyglutarate (2HG) and extracellular adenosine, which can directly suppress the immune system. While there are multiple mechanisms by which tumors can alter their metabolism and influence the immune system, we have utilized large-scale omics analysis to selectively highlight pathways that are critical to glioblastoma pathogenesis (2–5).
In this review, we outline how altered metabolic pathways in glioblastoma contribute to immunosuppression and discuss approaches to target these phenomena, in order to improve future immunotherapy outcomes.
Hypoxia
Hypoxia is a key feature of tumor growth and describes a condition in which the oxygen demand within an organism, cell, or tissue exceeds the available supply, typically described as < 10 mmHg O2 (6). Hypoxia is frequently found in solid tumors, including glioblastoma, due to rapid tumor growth, ultimately outstripping vascular supply and therefore, preventing O2 diffusion (6, 7). In gliomas, these hypoxic changes can be visualized on MR imaging, with high grade lesions displaying prominent ring-shaped contrast-enhancement with a hypointense center. Histological analysis of these regions often reveals highly anaplastic cells surrounding a necrotic tumor core, termed pseudopalisading necrosis (7, 8). The most important transcription factors in the cellular response to low pO2 are the hypoxia-inducible factors (HIFs). HIF-mediated signaling plays a role in vasculogenesis, tumor and cancer stem-like cell proliferation, and immunosuppression within the tumor microenvironment (TME). This family consists of a constitutively expressed β subunit (HIF1β), and at least three tightly regulated α subunits, HIF1α, HIF2α, and HIF3α. In normoxic conditions, the heterodimeric protein’s α subunit is rapidly degraded by the proteasome. Hypoxic conditions stabilize the α subunit and allow it to translocate to the nucleus, dimerize with the β subunit and induce transcription of hypoxia response genes (6, 7). In glioblastoma, it has been demonstrated that cells in the perivascular niche and necrotic areas upregulate the expression of HIF2α, and that this expression colocalizes with the stem cell markers CD133 and Olig2 (9, 10). Hypoxia increases growth and proliferation of glioma cells and glioma stem-cells, and strongly induces HIF2α, as well as stem genes. Ectopic expression of non-degradable HIF2α induced a stem-like phenotype in glioma cells and enhanced tumorigenicity in vivo (11). Hypoxia induced by the antibody bevacizumab that targets the vascular endothelial growth factor (VEGF), has also been shown to induce autophagy-related genes, resulting in a resistance mechanism to anti-VEGF therapy that could be abrogated by autophagy inhibition (12). In the past two decades, many basic investigations focusing on targeting hypoxia to increase the efficacy of VEGF inhibition were initiated, including inhibiting autophagy with chloroquine, or attempting to prevent HIF1α synthesis with mTOR inhibitors such as temsirolimus and everolimus (13).
Hypoxic changes in the glioma microenvironment may also modulate key immune effector molecules (Figure 1). Hypoxia has been further shown to induce T cell exhaustion through mitochondrial fragmentation and decreased oxidative phosphorylation, among other mechanisms (14). Furthermore, under hypoxic conditions, glioma cells secrete interleukins IL-6 and IL-8, which serve as autocrine proliferative signals and localize to perinecrotic regions with many pseudopalisading glioblastoma cells (15, 16). IL-6 signaling also plays a role in maintaining the tumor stem cell niche and stimulating angiogenesis (15, 17). Finally, IL-6 has been shown to induce upregulation of the programmed death-ligand 1 (PD-L1) on tumor-infiltrating and circulating myeloid cells (18). Additionally, glioma cells and proliferating endothelial cells in the hypoxic perivascular niche respond to HIF1α and VEGF signaling by upregulation of the chemokine receptor CXCR4, allowing for increased migration (19).
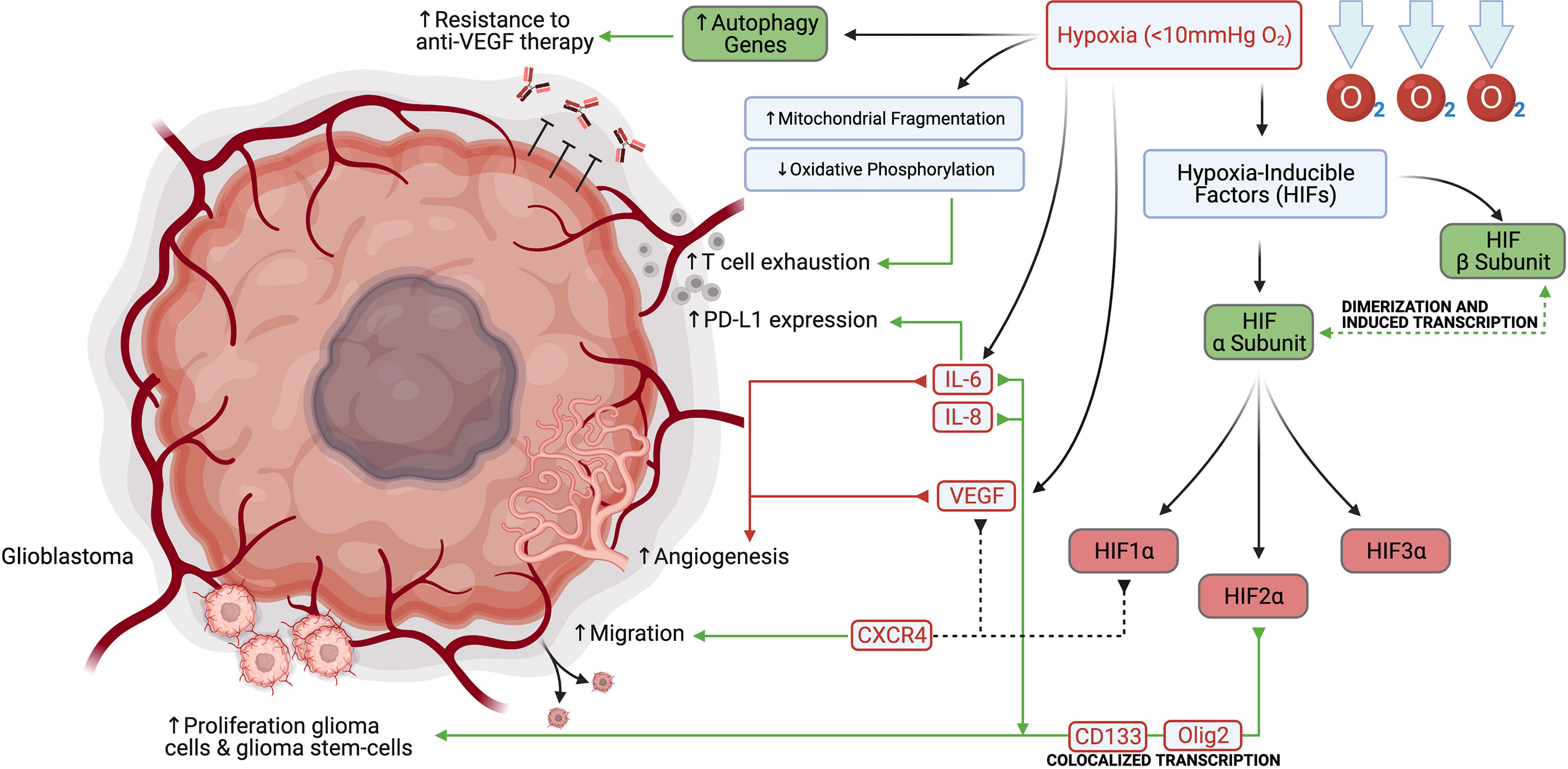
Figure 1 Hypoxia promotes glioblastoma and glioblastoma stem-cell proliferation, migration, angiogenesis, PD-L1 expression, and resistance to anti-VEGF therapy.
A small-molecule HIF2α inhibitor PT2385, which was later improved to the second-generation inhibitor PT2977 (now known as ‘MK-6482’), has also been shown to block the transcription of HIF2α-responsive genes, including VEGFA, CCND1 and the glucose transporter–encoding gene SLC2A1, and both molecules demonstrated on-target antitumor activity in mouse xenograft models of renal cell cancer (20). PT2385 showed promising preliminary promising activity in early phase development. This is also studied in glioblastoma and in a combination study with nivolumab (21). MK-6482 is nearly identical to PT2385, but with a more favourable pharmacokinetic profile and is also undergoing evaluation in early phase trials (20).
Glycolysis
Glycolysis is the primary metabolic pathway that provides energy and involves the breakdown of glucose to form the high energy molecules ATP and NADH. The brain is an energy demanding organ with about 25% of the body’s glucose consumption being devoted to brain function (22). Despite the brain’s high energy demand, it has relatively low levels of glucose when compared to plasma (23). Glucose transporter 1 (GLUT1) is responsible for shuttling glucose into the brain, as well as driving it into cells (24). Neurons, oligodendrocytes, astrocytes, and tumor cells are especially dependent on glucose for survival and energy production (25, 26). Neurons additionally express Glucose transporter 3 (GLUT3), which is five-fold more efficient at transporting glucose than GLUT1 (24). The PI3k-Akt-mTOR pathway is primarily responsible for fulfilling the energy demands of transformed cells, neurons, and glia (27, 28).
Aerobic glycolysis (Warburg’s effect) is a hallmark of cancer and is a process through which cancer cells produce lactate after undergoing glucose-mediated oxidative phosphorylation (29). Like other tumors, glioblastoma highly expresses GLUT1 and its energy demands are greater than that of normal brain cells (30, 31). Transformed, neuronal, and glial cells have high energy demands in a low glucose environment, and act as a sink that depletes glucose which limits immune cell anti-tumor effector functions. Gliomas can further recruit and maintain immunosuppressive immune populations such as pro-tumor mononuclear phagocytes which also undergo glycolysis and deplete available glucose, among other nutrients including L-arginine and L-cysteine, from the tumor microenvironment (32, 33). Blockage of the Akt-mTOR pathway via administration of Akt inhibitors in low glucose environments has been shown to inhibit growth of glioma cells (34). Additionally, high expression of a glycolysis related gene signature was associated with cancer progression, adhesion, proliferation, angiogenesis, and drug resistance, further demonstrating the important role of glycolysis in glioblastoma (35).
Immune cells, and in particular effector T cells, are dependent on glycolysis to support their proliferation and effector functions (36) (Figure 2). TCR signaling in T cells results in PI3k-Akt-mTOR signaling, which further increases glucose requirements (37). Low glucose availability is a known driver of the exhaustion phenotype in T cells (38). Recent research suggests that exhausted T cells exist on a continuum from a precursor exhausted state, that are responsive to checkpoint blockade, to a terminally exhausted state, which are refractory to checkpoint blockade therapy (39). Precursor exhausted T cells have been shown to have reduced expression of glycolysis related genes in relation to naïve and effector T cells (40). Interestingly the PD1/PDL1 receptor ligand pair has divergent functions in T cells and the tumor. Ligation of PD1 on T cells reduces glucose uptake, where increased expression of PDL1 on tumor cells improves tumor glycolysis (41–43). Reinforcing exhaustion in T cells via inhibitory signals and reducing glucose uptake is a synergistic way that the tumor cells maintain the immunosuppressive microenvironment. Furthermore, lactate accumulation in the glioblastoma TME is in of itself a potent immunosuppressive agent. Lactate has been shown to polarize macrophages towards an M2 phenotype, impair lymphocyte proliferation, activation, and degranulation (44). Broad pharmaceutical inhibition of glycolysis may not result in a net anti-tumor effect, as it has pro-tumor effects in glioma cells, but is also important for anti-tumor effects in T cells. Drugs that disrupt glycolysis preferentially in the tumor or bolster glucose uptake specifically in T cells would be attractive methods of leveraging metabolism to provide an anti-tumor effect.
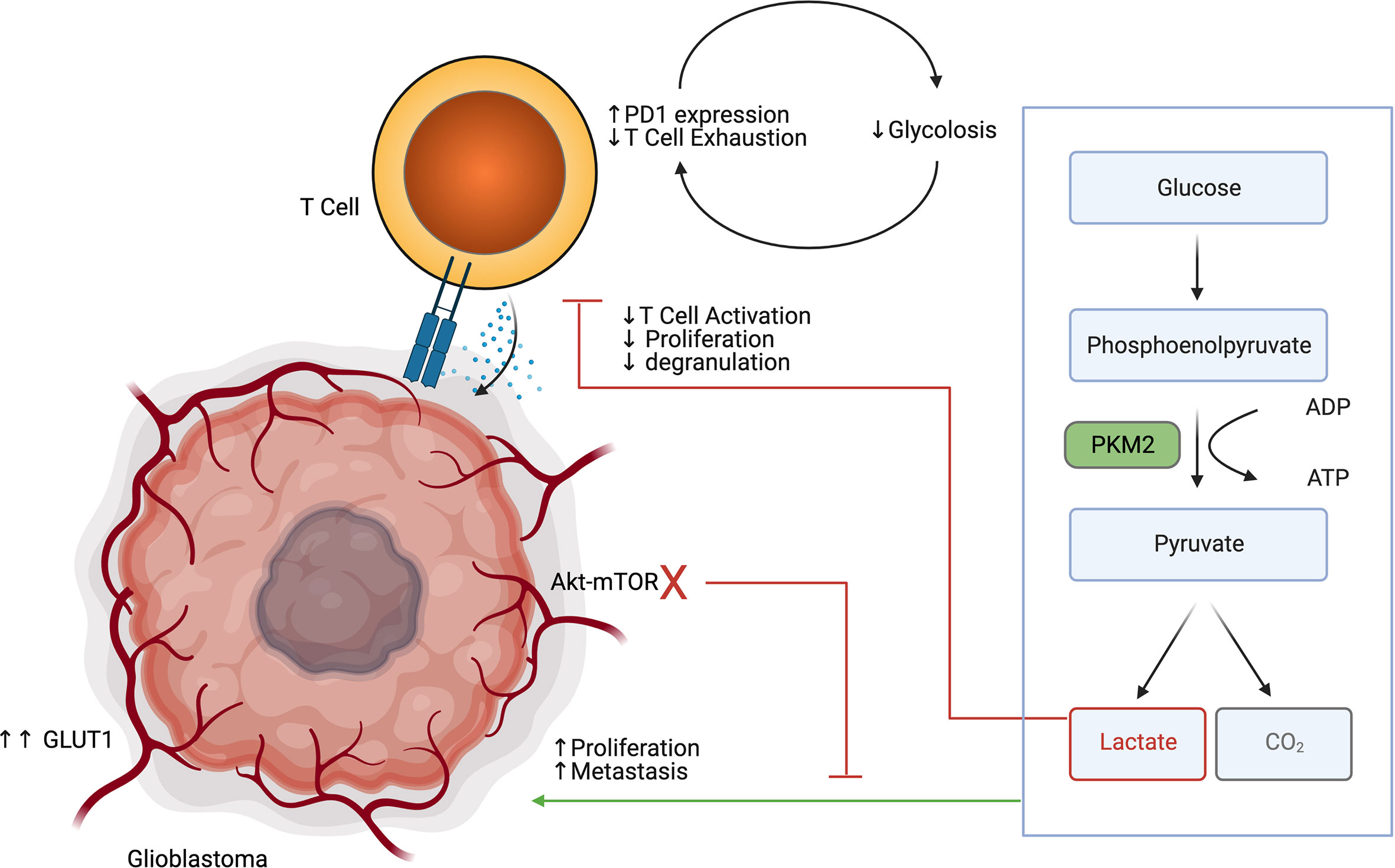
Figure 2 Glioblastoma glycolysis drives tumor progression while inhibiting T cell glycolysis, activation, proliferation and degranulation.
Tryptophan Metabolism
The tryptophan catabolism is among the most characterized immunometabolic pathways in glioblastoma since it contributes to both tumor progression and immune evasion. Tryptophan, the least abundant amino acid, can be incorporated into proteins, modified to produce serotonin, or metabolized to produce kynurenines (Kyn). Briefly, tryptophan is metabolized by indoleamine 2,3-dioxygenase (IDO)1/2 and tryptophan 2,3-dioxygenase (TDO), another important enzyme of the kynurenine pathway, to produce N‐formyl kynurenine, which is converted to kynurenine by arylformamidase (AFMID). Kynurenine is further metabolized through various pathways to produce metabolites including kynurenic acid, anthranilic acid, 3‐hydroxykynurenine, xanthurenic acid, quinolinic acid, picolinic acid, and nicotinamide‐adenine‐mononucleotide (45). Although difficult to control each of these metabolites’ pathways individually, it has been possible to inhibit their production through upstream IDO1/2 and TDO inhibition. IDO1/2 expression are typically found in peripheral tissues, while TDO expression is associated with hepatic tryptophan metabolism. Although recent evidence suggests that there may be value in specifically targeting TDO in the context of glioblastoma, tryptophan metabolism in glioblastoma has primarily been explored in the setting of IDO1/2.
Kynurenines and quinolinic acid have been previously described to be able to drive neoplastic proliferation through Wnt/β‐catenin signaling (46). Kynurenines further influence tumorigenesis in glioblastoma by modulating DNA repair enzyme, polymerase kappa, thereby preventing DNA damage and allowing genomic instability to propagate leading to tumor heterogeneity (47). Kynurenines and quinolinic acid may also promote cell proliferation in a fibroblast growth factor‐1 (FGF‐1) dependent manner (48). Lastly, nicotinamide‐adenine‐mononucleotide can be converted to NAD+, which confers tumor cells’ resistance to oxidative stress. Although the mechanism remains largely unclear, IDO expression may play a role in tumor angiogenesis and metastasis through control of IFNg and IL-6 (49). Mondal et al. found that in vivo IDO inhibition reduced metastasis and neovascularization (50). In patients with glioblastoma, IDO expression was strongly associated with shortened overall survival (51).
IDO1/2 mediated depletion of tryptophan was initially thought of as an ancient innate immune mechanism to prevent the growth of microorganisms while reducing inflammation and autoimmunity in areas such as the brain (52). In fact, IDO1/2 expression is significantly increased in inflammatory tissues due to IFNg, TGF-β, and PGE2 signaling to potently inhibit active inflammation (Figure 3). Wainwright et al. were among the first to demonstrate that glioblastoma cells significantly upregulated the expression of IDO1 and suggested that IDO1 may also contribute to tumor progression by promoting an immunosuppressive phenotype (53).
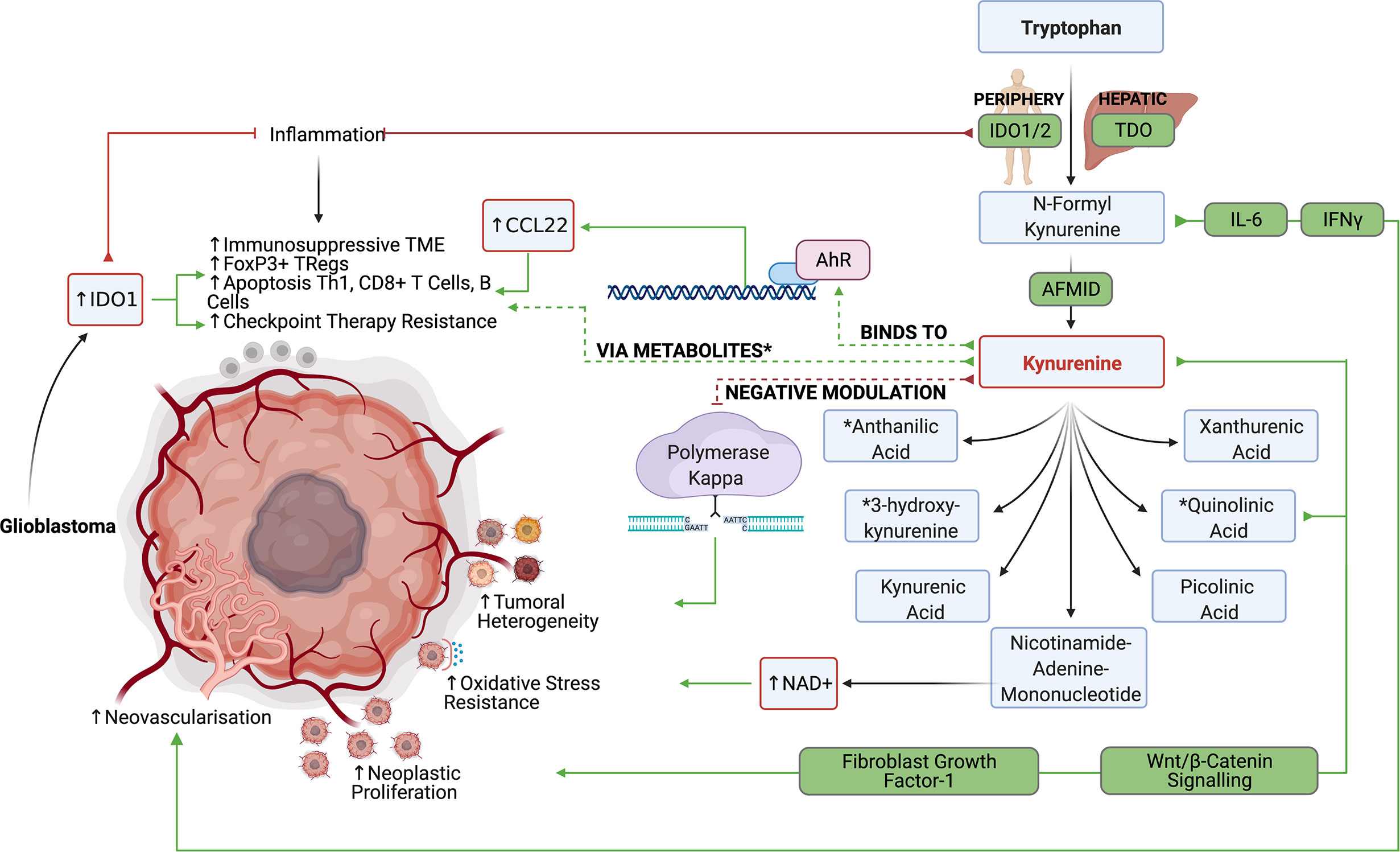
Figure 3 The IDO1/2 and TDO pathway allow the production of various tryptophan metabolites, which suppress anti-glioblastoma lymphocyte responses while promoting tumor growth.
In both clinical studies and preclinical murine glioma models, tumors with high expression levels of IDO1 were infiltrated with more FoxP3+ regulatory T cells (53). It has been suggested that tryptophan metabolism can induce Treg differentiation based on the kynurenines’ ability to bind to cytosolic ligand-activated transcription factor AhR (54). Kynurenine-driven AhR activation also induces the production of CCL22 to recruit Tregs into the glioblastoma TME (55). In addition to inducing suppressive T cell populations, tryptophan metabolism may blunt antitumor CD4 and CD8 responses through various mechanisms. Opitz et al. found that TDO derived kynurenines reduced the proliferation of both CD4+ and CD8+ T cells and reduced LCA+ CD8+ T cell infiltration in human gliomas with high TDO expression (56). Furthermore, depletion of tryptophan in the TME leads to an accumulation of unbound tryptophan–tRNA in T cells which activates the GCN2 mediated stress response and inhibits RNA transcription and protein synthesis in T cells leading to cell cycle arrest and apoptosis (57). Although currently contested, some evidence suggests that tryptophan deprivation may also inhibit the mTOR pathway in T cells to inhibit effector T cell functions. Additionally, metabolites like quinolinic acid, 3-hydoxyanthranilic acid, and 3‐hydroxykynurenine have been shown to induce apoptosis in Th1 helper cells, CD8+ Effector T cells, B cells, while sparing immunosuppressive Th2 helper cells.
IDO inhibitors such as 1-L-MT, IDO-IN-2, Navoximod (GDC-0919), IDO-IN-1, Linrodostat, coptisine chloride, PF-06840003, and TDO inhibitors such as 680C91 have recently been developed (58). While IDO inhibitors such as navoximod did not improve antitumor responses in preclinical glioblastoma models, Kesarwani et al. found that navoximod synergistically improved antitumor responses when combined with RT and immune checkpoint blockade (59). Hanihara et al. and Li et al. similarly found that while 1-L-MT did not improve antitumor immunity on its own, 1-L-MT significantly synergized with temozolomide administration and radiation therapy (60, 61). Wainwright et al. found that IDO inhibitors particularly synergized with PD1 and CTLA4 blockade in the mice (62, 63). Interestingly, advanced age is associated with an increase of brain IDO expression and this is not reversed by IDO enzyme inhibitor treatment (64). It remains to be seen if targeting IDO will translate into clinical benefit in cancer and in gliomas.
Arginine Metabolism
Arginine is yet another amino acid substrate that is actively metabolized by tumor cell to promote tumor progression and immunosuppression. L-arginine is critical in the urea cycle and is a modulator of immune function and tumor metabolism. L-arginine is utilized as a substrate for both Arginase 1 (ARG1) and cytokine inducible nitric oxide synthase (iNOS). ARG1 converts L-arginine to urea and ornithine, which is further utilized in the urea cycle. iNOS converts L-arginine to citrulline and nitric oxide (NO), which is important for directing anti-tumor functions in immune cells (65).
Depletion of arginine has been identified as a successful treatment strategy in cancers that are deficient in aspects of arginine metabolism and are reliant on exogenous sources (Figure 4). This approach has been successful in Leukemia, where transformed cells were found to be deficient in asparagine synthase and were not capable of producing asparagine. This left the tumors vulnerable to treatment with L-asparaginase which depleted asparagine (66). Similarly the function arginosuccinate synthase 1 (ASS1) is defective in some tumors, which makes them dependent on exogenous arginine (66). In glioblastoma, there seems to be an abundance of arginine transporters, which is evidenced by a notable accumulation of byproducts of arginine metabolism (67, 68). This suggests that arginine metabolism is functional, and may be sensitive to targeted depletion. A recent study that utilized a pegylated recombinant human ARG1, depleted arginine, and induced cytotoxicity in glioma cells (69). Similarly, selective iNOS inhibitors 1400W and S-MIU have recently been shown to reduce tumor growth in a EGFRvIII mutant overexpressing U87 glioblastoma model (70). How arginine fosters immunosuppression in the TME is also an area of research that seeks to elucidate the tumor promoting effects of arginine metabolism.
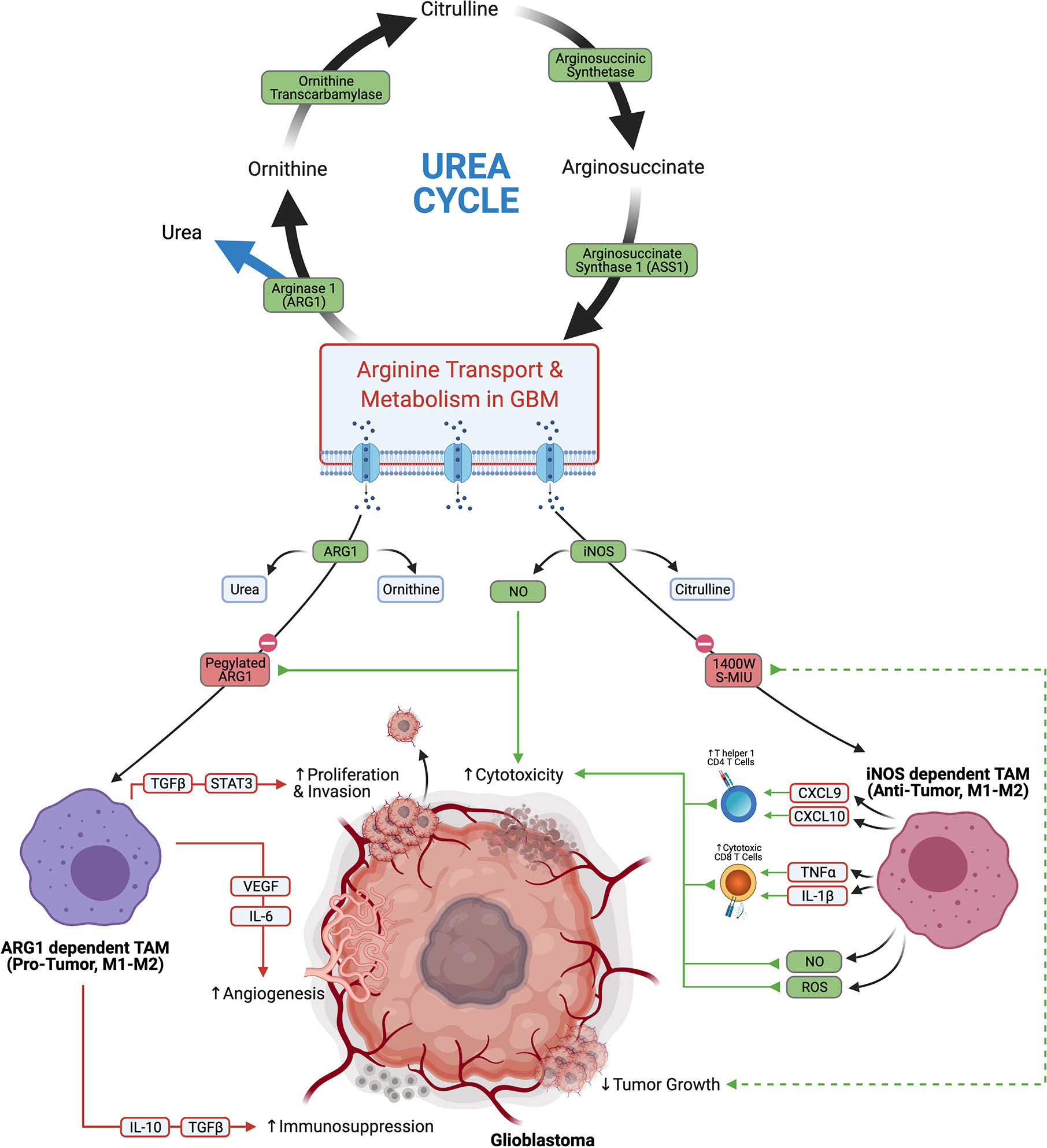
Figure 4 Glioblastoma arginine metabolism via iNOS or Arg1 polarizes the tumor associated macrophages towards anti-tumor tumor or pro-tumor phenotypes, respectively.
Macrophages are a large component of the TME, constituting up to 30% of the tumor by weight (71, 72), and the divergent functions of arginine metabolism are best appreciated in this immune cell subset. Macrophages are a highly plastic cell type, which can adopt either pro- or anti-tumor function depending on their environmental cues. Macrophage polarization has traditionally been thought of to exist on a continuum from M1 to M2 phenotypes which confer inflammatory/anti-tumor phenotypes and repair/pro-tumor phenotypes, respectively. Recent research suggests that the M1-M2 dichotomy is likely an oversimplification (73), which is underscored by the numerous reports that tumor associated macrophages (TAMs) in glioblastoma express a mixture of M1 and M2 related genes (74, 75). TAMs which primarily metabolize arginine via iNOS are considered more anti-tumor. iNOS dependent anti-tumor TAMs skew the TME towards cytotoxicity through stimulating Th1 responses via secretion of CXCL9 and CXCL10, inducing cytotoxic CD8s through TNFα and IL1β, and direct killing of tumor cells through nitric oxide (NO) and Reactive Oxygen Species (ROS). Conversely TAMs which metabolize arginine primarily through ARG1 are thought to have more pro-tumor activity. ARG1 dependent pro-tumor TAMs stimulate angiogenesis through VEGF and IL6, promote invasion and proliferation via TGFβ and STAT3, and support immunosuppression through IL-10 and TGFβ (65). TAMs in glioblastoma are considered to be pro-tumor overall, and their accumulation correlates with worse prognostic outcomes (76). Due to the highly plastic nature of TAMs and their abundance in the glioblastoma TME, they are an attractive target for repolarization from a pro-tumor to an anti-tumor phenotype. Finding ways to selectively shift arginine metabolism in TAMs towards iNOS presents an interesting treatment modality that could potentially skew a large portion of the TME to an overall anti-tumor effect.
2-Hydroxyglutarate Metabolism
2HG production represents a unique immune-metabolomic pathway found in many cancer cells, including low-grade gliomas and secondary glioblastoma. Within low-grade gliomas and secondary glioblastoma, 2HG is often produced due to mutations in the catalytic domains of isocitrate dehydrogenase isoform 1 (IDH1) and isocitrate dehydrogenase isoform 2 (IDH2). The most common mutations in IDH1 include R132H, R132C, R132L, R132S, and R100Q, while the most common mutations in IDH2 include R140Q, R140G, R140W, R140L, R172K, R172G, R172M, R172Q, R172T, R172S (77). While IDH1 is found in the cytoplasm of cells, IDH2 is found in the mitochondrial matrix. Despite their differences in cellular sub-localization, both wildtype IDH1 and wildtype IDH2 catalyze the decarboxylation of isocitrate using NADP+ and Mg2+ as cofactors and produce α-Ketoglutaric acid and CO2 (78). Wildtype IDH1 and wildtype IDH2 are normally also able to catalyze the reverse reaction by reducing α-Ketoglutaric acid into isocitrate using NADPH as a cofactor. α-Ketoglutaric acid also acts as a substrate for an alternative reduction reaction that incompletely reduces α-Ketoglutaric acid into 2-Hydroxyglutarate instead of isocitrate in an NADPH driven manner. Somatic missense mutations of arginine in IDH1 and IDH2 lead to impaired oxidative carboxylation and favor the incomplete reduction of α-Ketoglutaric acid into 2-Hydroxyglutarate (79). While 2HG is produced by other enzymes including hydroxyacid-oxoacid transhydrogenase (80), human phosphoglycerate dehydrogenase (81), lactate dehydrogenase (82), and l-malate dehydrogenase (83), it is believed that IDH1/2 mutations are almost exclusively what drive 2HG overaccumulation in low-grade glioma and secondary glioblastoma. In grade II/III gliomas carrying IDH1/2 mutations, 2HG concentrations have been found between 1 and ∼30 mM (77). The overaccumulation of 2HG can both promote gliomagenesis while inhibiting anti-tumor immunity.
The oncogenic process can be mediated through epigenetic regulation, 2HG, inhibition of DNA repair enzymes, promotion of autophagy, and promotion of invasiveness. 2HG exerts control over cellular epigenetics by favoring the hypermethylation of various genes by inhibiting α-Ketoglutaric acid-dependent dioxygenases such as Tet methylcytosine dioxygenases (TETs) (84). Koivunen et al. demonstrated that TET2, in particular, was inhibited by 2HG (85). TET2 inhibition was demonstrated to decrease tumor cell differentiation and promote tumorigenesis in the setting of glioblastoma by Garcia et al. (86) Perhaps most notably, 2HG was shown to increase methylation of histone lysines and c-Myc binding at the promoter of the telomerase reverse transcriptase (TERT) gene encouraging tumor transformation and immortalization (87). 2HG was also shown by Chen et al. to inhibit the AlkB family of DNA repair enzymes such as ALKBH2 and ALKBH3 (88). 2HG further promotes autophagy and cell survival by indirectly controlling mTORC1 and mTORC2 signaling. 2HG does this by activity inhibiting KDM4A, which allows DEPTOR to activate the mTORC1/2 pathway (78). 2HG may also have a role in destabilizing the basement membrane of glioblastoma cells through the inhibition of collagen stabilizing enzymes such as PLOD1, PLOD3, P4HA1, and PHA3 (79). In addition to IDH1/2 mutations helping produce 2HG, these mutations also favor the consumption of NADPH instead of their production. The depletion of NADPH helps induce cellular dysregulation and impairs cellular defense against reactive oxygen species (77).
While 2HG has been shown to influence tumor growth, 2HG has also been demonstrated to modulate anti-tumor immunity (Figure 5). Bunse et al. demonstrated that 2HG produced by tumors can be transported into immune cells via SLC13A3, which generally impairs immune function (89). Within T cells, 2HG was shown to inhibit T cell activity by inhibiting enzymes such as ornithine decarboxylase, transcription factors such as NF-κB p65, and the NFAT pathway through NFATC1 (89). It was demonstrated that T-cells treated with 2HG producing astrocytes demonstrated decreased production of IFN-γ and IL-2 upon activation (89). 2HG also directly inhibits T cell activation by inhibiting the steps that lead to calcium influx, such as early ATP-dependent TCR signaling events, c-Jun N-terminal kinase (JNK), and PLC-γ1(Y783) phosphorylation (89). Of note, the effect of 2HG on the suppression of T-cell activation was most prominent in CD4+ T cells. 2HG was also found by Kohanbasch et al. to reduce the expression of STAT1 in DC cells, thereby inhibiting the secretion of CXCL10 in the glioblastoma TME (90). This represents yet another mechanism by which 2HG may suppress T cell activity. Interestingly, IDH1/2 mutant tumors are generally infiltrated by T cells expressing less PD-1 than those T cells found in IDH1/2 wildtype tumors (89). This may be due to 2HG inhibiting NFAT translocation, which is necessary for inducing PD-1 expression. IDH1/2 mutant tumors are also generally infiltrated by less immunosuppressive M2 Macrophages (91). Amankulor et al. suggest that the decreased immunosuppressive cell infiltration in IDH1/2 mutant tumors may play a role in controlling the growth of secondary glioblastoma (92).
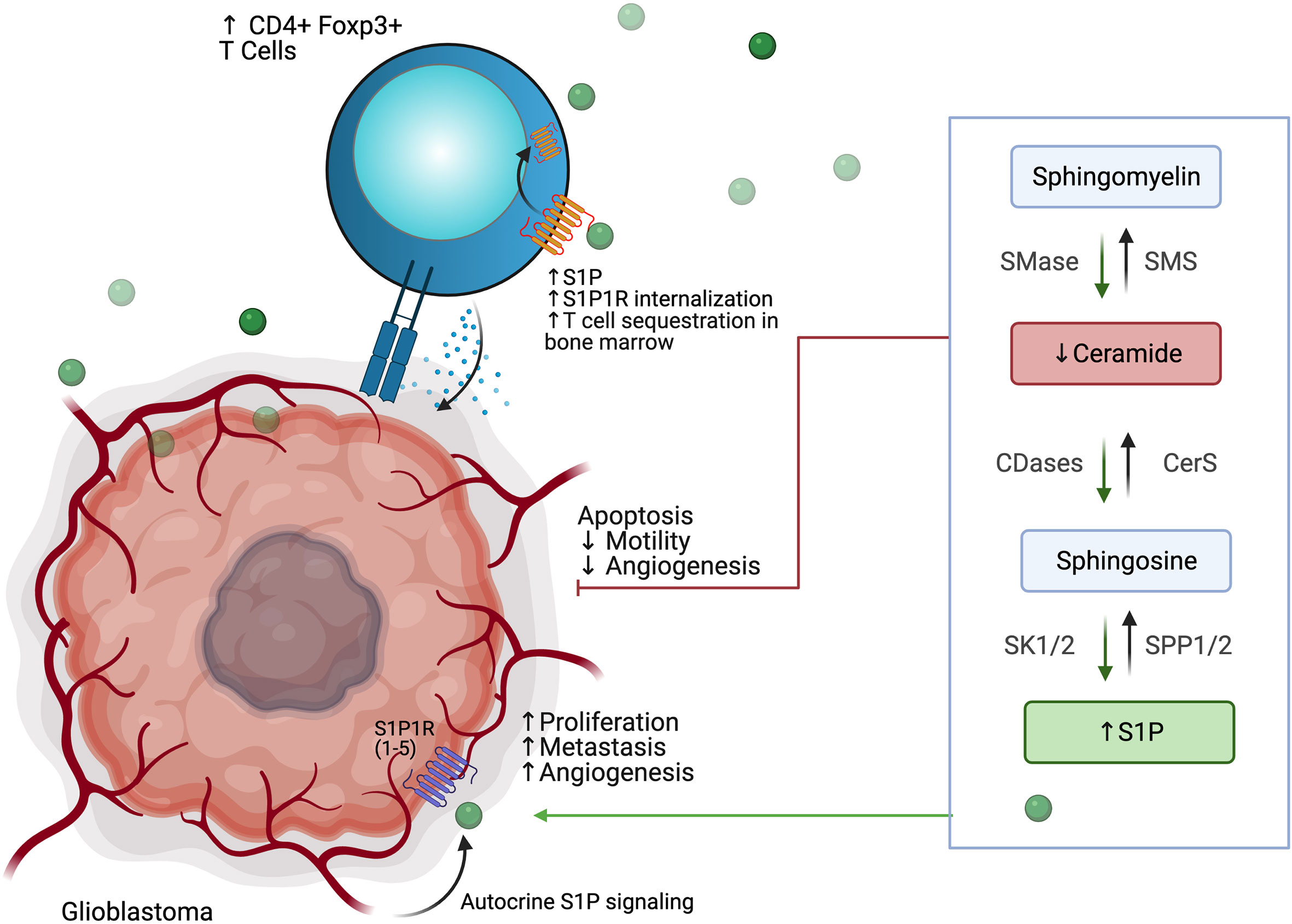
Figure 5 2 HG produced by IDH1/2 mutant secondary glioblastoma promotes tumor survival while impairing T cell activation and degranulation.
Given the immunometabolic importance of IDH1/2 mutations, IDH mutation-specific inhibitors have been developed. IDH1 mutations have been targeted through molecules such as Ivosidenib, BAY-1436032, and AG-5198 (93). IDH2 mutations have been targeted through molecules such as Enasidenib, AGI-6780, and GI-6780 (93). IDH1 and IDH2 mutations have been co-targeted though molecules such as AG-881, which is currently being evaluated in the phase 3 INDIGO trial (NCT04164901) in patients with residual or recurrent Grade 2 glioma with an IDH1 or IDH2 mutation (93). Since IDH mutations may help anti-tumor immunity by decreasing PD1 expression and decreasing immunosuppressive cell infiltration, there may rationale to combine IDH mutation inhibitors with other immunostimulatory therapies such as checkpoint blockade. Bunse et al. demonstrated that BAY1436032 and PD-1 therapy increased overall survival in a murine IDH1 R132H model (89). Similarly, Kadiyala et al. found that 2HG inhibition, IR treatment, temozolomide, and anti-PDL1 administration led to complete tumor regression in 60% of mice bearing IDH1 mutant gliomas (94). In addition to small molecule inhibition, Platten et al. recently demonstrated that the IDH1 R132H mutant pathway could also be targeted by a peptide vaccine approach in newly diagnosed gliomas (95). While IDH inhibitors and vaccines represent one of the remarkable success stories in low grade IDH mutated gliomas, they are unfortunately not a therapeutic option in IDH wild type glioblastoma.
Adenosine Metabolism
In normal physiology, adenosine and ATP are found in the cytosol of tissues, while these metabolites’ extracellular levels are rarely observable (96). In certain pathologies such as gliomagenesis, intracellular adenosine can be secreted via bidirectional equilibrating nucleoside transporters (97), and ATP is released extracellularly via plasma membrane disruption or ATP efflux induced by hypoxia or inflammation and mediated by ABC transporters, anion channels, connexins, pannexins, and receptors like P2X7R (98). Once in the extracellular environment, ATP is degraded by enzymes such as CD73 and CD39 to produce adenosine. While the CD73 and CD39 mediated pathway of adenosine production are the most relevant to glioblastoma, extracellular adenosine may also be generated via ecto-phosphodiesterase/pyrophosphatase family proteins, nicotinamide adenine dinucleotide glycohydrolases, prostatic acid phosphatase, and alkaline phosphatase (99, 100). Extracellular adenosine is regulated either by cellular uptake or extracellular adenosine deaminase enzymes. The extracellular adenosine that remains can signal through high-affinity A2a and low-affinity A2b receptors expressed on tumors, tumor-associated cells, and immune cells. Blocking this adenosine signaling represents an intriguing target to modulating anti-tumor responses.
The adenosine metabolism pathway is of particular importance in glioblastoma and low-grade gliomas. Ott et al. recently found that in patients with gliomas, the A2aR/CD73/CD39 pathway was most frequently expressed (101). In the hypoxic glioblastoma TME setting, there is an increased expression of HIF1a in tumor tissue leading to increased expression of CD39 and CD73 on tumor cells, immune cells, stromal cells, and endothelial cell, leading to increased extracellular adenosine. This extracellular adenosine can signal through adenosine receptors to improve tumor survival, stimulate tumor cell proliferation, and induce tumor cell invasion and angiogenesis. Adenosine can improve tumor cell survival through the AKT and ERK pathways, inhibiting caspase pathway activation, upregulation of Bcl2 family antiapoptotic genes and downregulation of P53 (102, 103). Adenosine has been shown to induce tumor cell proliferation through various pathways including but not limited to AKT, ERK, JNK, and P38, ERa, and upregulation of cyclin proteins (104–106). Adenosine has been shown to increase the expression of matrix metalloproteases (MMPs) and FXYD5, which disrupt the tumor’s ECM and drive invasiveness by reducing cell adhesion, respectively (107). Lastly, adenosine signaling can help drive angiogenesis by inducing increased VEGF, IL8, angiopoietin 2, and erythropoietin (100).
In addition to regulating tumor growth, adenosine signaling has a multifaceted role in controlling anti-tumor immunity (Figure 6). While extracellular ATP functions as danger associated molecular pattern (DAMP) that can stimulate both innate and adaptive immunity, extracellular adenosine serves to dampen the immune system (108). Within T cells, adenosine signaling through receptors such as A2aR inhibits MAP kinase, protein kinase C, NFkB, and NFAT pathways and inhibit proximal TCR signaling (109, 110). T cells treated with adenosine demonstrated decreased IL-2, TNFa, and INFg production, decreased CD28 expression, and increased expression of PD1, CTLA4, and LAG3 (108, 111, 112). Adenosine signaling was also shown to help generate Tregs by increasing the expression of FoxP3 in CD4 T cells (113). These Tregs were found to upregulate CD39 and CD73, creating a positive feedback loop in the TME (113). Adenosine signaling similarly blunted NK cell target cell killing, proliferation, and the production of IFNg and TNFa (114–116). DC cells treated with adenosine exhibited decreased expression of TNFa and IL12 while increasing their expression of immunosuppressive factors including IL-5, IL-6, IL-10, TGFβ, arginase, and IDO, and PD-L2 (117, 118). In macrophages, adenosine signaling via A2aR and A2bR were shown to induce M2 polarization (119) and blunted the secretion of neutrophil chemoattractants (111). Adenosine signaling also directly impaired neutrophil function finding their ability to adhere, degranulate, phagocytose, and produce TNFa and superoxide (120).
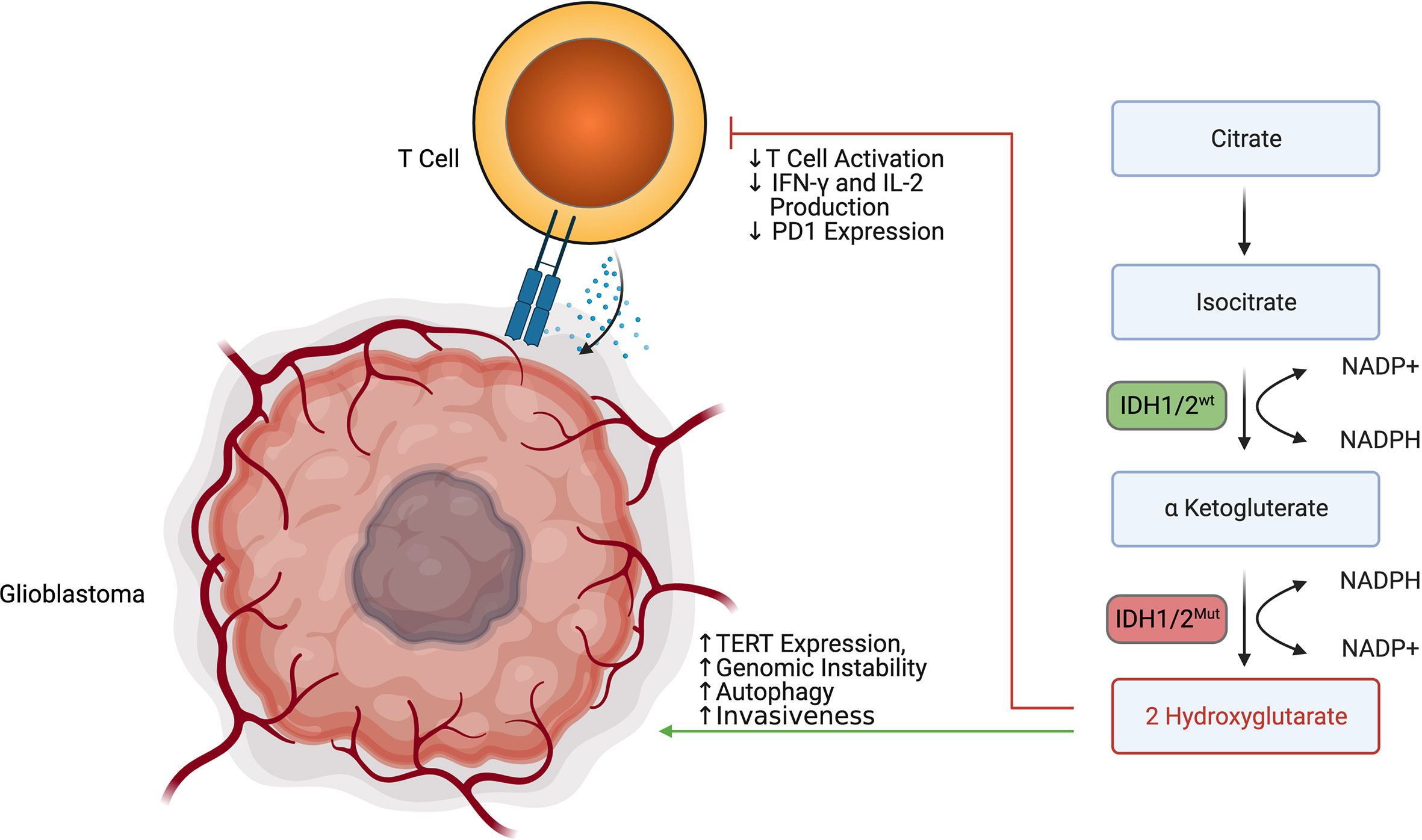
Figure 6 Glioblastoma cells control the amount of extracellular adenosine in the glioblastoma immune microenvironment and use adenosine to decrease T cell degranulation and increase T cell exhaustion.
Sitkovsky et al. were one of the first to demonstrate enhanced anti-tumor immunity in A2aR knockout mice and demonstrated that A2aR could be inhibited using pharmacologic blockade (121, 122). In preclinical models, the immunosuppressive effect of adenosine signaling has been blunted using anti-CD73 and anti-CD39 blockade (123). In multiple murine tumor models, A2aR inhibition synergized with anti-PD1, anti-TIM3, and anti-CTLA4 antibodies to improve survival and reduce tumor metastasis (124–126). Inhibitors of this pathway such as Istradefylline, SCH-442416, Preladenant, BAY-545, Ciforadenant (CPI-444), Imaradenant (AZD4635), SCH58261, AB928 and AB680 have emerged and many of them are in clinical development (123). Targeting the adenosine signaling pathway in glioblastoma remains an active area of research.
Altered Phospholipid Metabolism
Sphingolipids are important structural components of the cell membrane that play a role in membrane fluidity and integrity. Many sphingolipids are also highly bioactive and play roles in a variety of cellular processes. Sphingosine, the first discovered sphingolipid, is induced by cellular stressors, including chemotherapy and radiation, and functions in cytoskeletal reorganization, cell cycle regulation, senescence and apoptosis. Since then, many other sphingomyelins have been identified, including ceramide, a molecule involved in regulation of apoptosis and is believed to be the central hub of sphingolipid metabolism, as well as sphingosine-1-phosphate, or S1P, which has roles in promoting survival, migration, and inflammation (127, 128).
Sphingolipid synthesis occurs de novo via condensation of serine and palmitate to 3-keto-dyhydrosphingosine, which via several intermediate steps involving ceramide synthases (CERS1-6) is converted to ceramide, a molecule well-identified as a pro-apoptotic signal (128, 129). Ceramide consists of an 18-carbon sphingosine long-chain base that contains an amide-linkage to a fatty acyl chain of variable carbon number; synthesis by CERS1-6 is the rate-limiting step of de novo ceramide synthesis and each enzyme is responsible for ceramides of specific fatty acyl chain length. Ceramides can also be synthesized via salvage following breakdown of complex sphingolipids such as sphingomyelins, via the subcellularly-localized sphingomyelinases (acid, neutral, and alkaline SMases), and cerebrosides, via glucosylceramidase and galactosylceramidase. Ceramide breakdown via ceramidases leads to sphingosine formation, which may be recycled or phosphorylated by the sphingosine kinases SK1 and Sk2 to form S1P. S1P is a ligand for the five G-protein coupled receptors S1PR1-5 and is normally rapidly metabolized via S1P phosphatase (SGPP) and S1P lyase 1 (SPL). Activation of S1PRs results in cellular proliferation and further production of S1P to promote cell motility and survival. This tight linkage of interconnected pathways for the rapid synthesis and breakdown of ceramide (pro-apoptotic) and S1P (pro-survival) has given rise to the “sphingolipid rheostat” model, in which the balance of these two biomolecules plays an important and potentially targetable role in normal cellular function and oncogenesis (127–131).
Derangement of the sphingolipid rheostat is implicated in the pathogenesis of glioblastoma (Figure 7). Analysis of human glioma tissue revealed significantly lower ceramide levels in high grade tumors relative lower grade tumors, and relative to peritumoral brain tissue (132). This difference was most dramatic for the C18 ceramide. Likewise, S1P levels in glioma tissues were higher than in normal gray matter; glioma stem-like cells have also been shown to secrete S1P as an autocrine, resulting in proliferation and increased expression stemness markers (132, 133). Taken together, this S1P/ceramide shift represents a common, targetable feature of malignancy even with regard for the heterogeneity displayed within and between glioblastomas. In gliomas, this shift is likely due to multiple alterations in sphingolipid synthesis, including increased expression of SK1 (134–136), deletion of chromosomal regions containing SPL (137), and downregulation of S1P phosphatase 2 (138), resulting in increased S1P levels; as well as inhibition of ceramide synthase (139) and increased expression of ceramidases (134). The activity of SMases in glioblastoma is still being elucidated, but it has been shown that acid SMase may sensitize glioma cells to chemotherapy and radiation by increasing metabolism of sphingomyelin to ceramide and consequent apoptosis in the context of p53-deficiency; conversely, neutral SMase may be involved in increasing ceramide production in p53-wildtype cells (129). Increased understanding of the proapoptotic role of SMases in glioblastoma may yield new therapeutic targets.
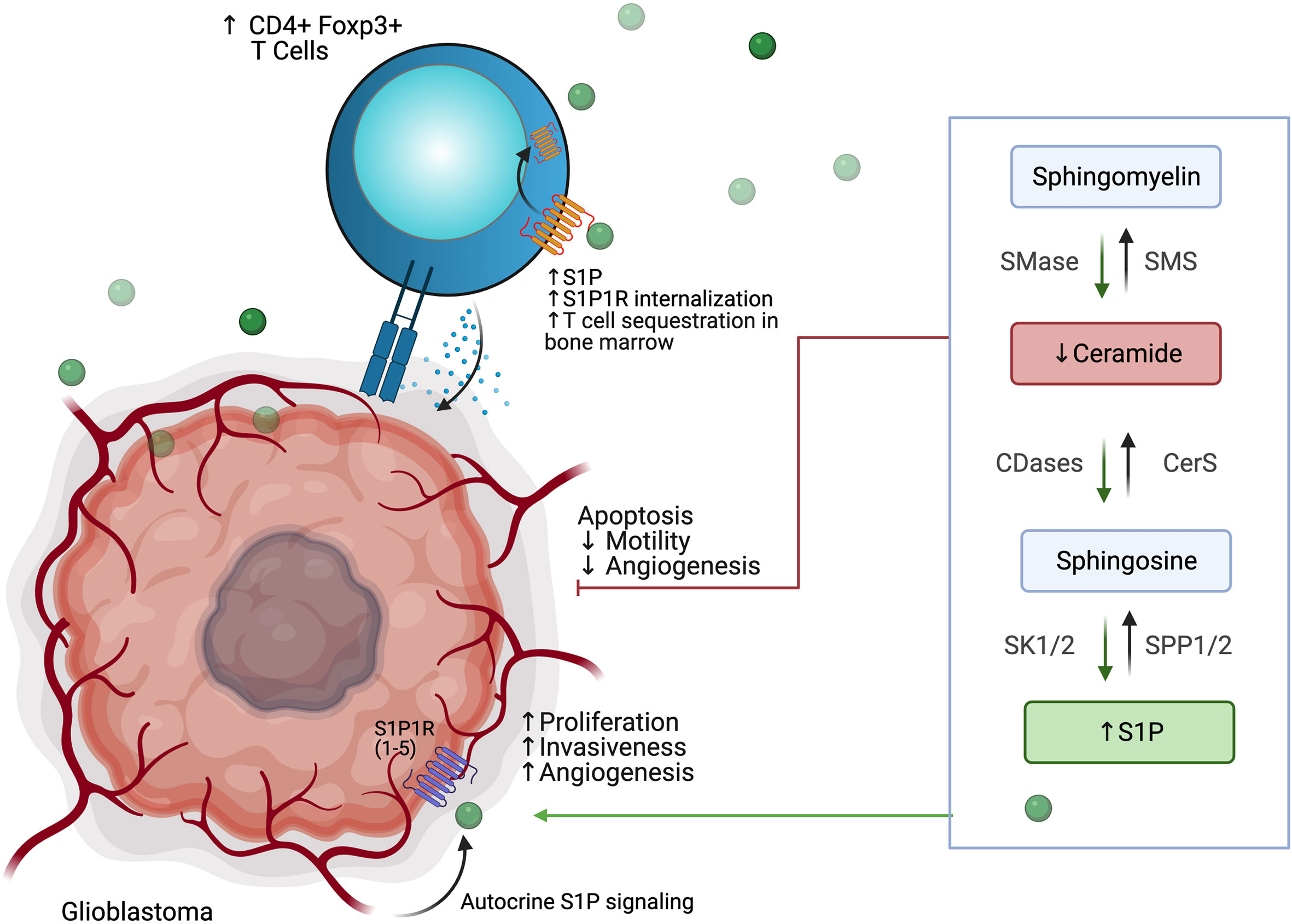
Figure 7 Glioblastoma cells use altered phospholipid metabolism to induce tumor proliferation, invasiveness, and angiogenesis while sequestering T cell populations away from the tumor microenvironment.
Interactions of glioblastoma with surrounding neuronal, glial, and immune cells in the TME are continuing to be appreciated. As discussed above, S1P plays an autocrine role in gliomas, and has been shown to be constitutively secreted in rodent glioma cells and human glioblastoma cell lines, likely due to SK1 activity (134, 140, 141). Increased SK1 activity, in turn, has been shown to be induced by microenvironmental IL-1 and HIF-2α activity (142, 143). S1P is also capable of acting as a chemoattractant for innate and adaptive immune cells (144, 145). Increased glioblastoma-derived S1P may thus promote formation of TAMs. TAMs, in turn, may also contribute S1P to the TME and increase SK1 activity (130). TAMs also produce NO, which has been shown to decrease acid SMase activity in glioma cells, resulting in therapeutic resistance (146). Despite the breakdown of the blood-brain barrier (BBB) in glioblastoma, there is often a paucity of T-cells in the TME or within the tumor (so-called tumor-infiltrating lymphocytes, or TILs). Those cells that are present are often the CD4+ CD25+ FoxP3+ population of immunosuppressive regulatory T-cells, termed Tregs (147). In addition to their role in apoptosis, global alterations of SMase expression modulate differentiation of T cell populations, with acid SMase activation linked to increased numbers of CD4+ Th1 and Th17 cells, while mice deficient in acid SMase exhibit increased Tregs (148, 149). Recently, it has been shown that glioblastomas may cause sequestration of T cells in bone marrow via T-cell internalization of S1PR1, and enforced expression of S1PR1 in combination with T-cell activation via 4-1BB agonism can increase survival in vivo (144). 4-1BB agonism has also been shown to rescue the poor efficacy of PD-1 blockade in glioblastoma in vivo; translation of these results to clinical trials is greatly anticipated (150). Blockade of S1PR1 is commonly employed using the sphingosine analog fingolimod to reduce immune trafficking in multiple sclerosis; fingolimod may also inhibit ceramide synthases, SK1, and SPL and was proposed as a possible therapeutic for glioblastoma (144, 151). A small trial of fingolimod was initiated with the aim of assessing whether sequestration of lymphocytes via S1PR1 antagonism could reduce post-chemoradiation lymphopenia in glioblastoma patients, but results have not yet been published (NCT02490930). Interestingly, Baeyens et al. recently found that monocytes in the lymph node may also produce S1P and influence T cell differentiation and T cell residence time in the lymph node (152). Their work suggests an alternative mechanism by which drugs that target S1P signaling can influence the glioblastoma TME and immune populations.
Additionally, an emerging mediator of aberrant phospholipid metabolism in glioblastoma is polymerase I and transcript release factor (PTRF), also known as Cavin1. PTRF was originally discovered to be involved in dissociation of RNA polymerase I-rRNA-DNA ternary complexes during transcription (153, 154). Through its colocalization with caveolin1 (Cav1) on the plasma membrane, it has been subsequently identified as essential for the formation of caveolae, cell-membrane infoldings that are 50-100 nm in diameter and function in cell signaling, lipid metabolism, and endocytosis (155, 156). Indeed, mutations in PTRF cause congenital generalized lipodystrophies in humans, providing further evidence for its role in lipid metabolism (157, 158). PTRF has also been shown to play a role in oncogenesis, as reduced PTRF expression in prostate and lung cancer is associated with progressive disease (45).
There is increasing evidence of a role for PTRF in the growth and progression of glioblastoma. PTRF has been shown to be upregulated in chemoresistant glioma cells and in human tumor tissues, with increasing PTRF expression correlating glioma grade and with tumor recurrence (155, 159). Huang et al. showed that EGFRvIII, an EGFR mutant with constitutively active tyrosine kinase activity present in ~25% of glioblastoma patients, drove PTRF upregulation (159). Blockade of PI3K and AKT reduced PTRF expression, showing a role for PTRF in EGFR-driven gliomagenesis even in the absence of the EGFRvIII mutation. This overexpression of PTRF in gliomas results in increased secretion of exosomes, cell growth, and aberrant methylation (159).
Interestingly, in silico analyses have suggested that PTRF expression is negatively correlated with the presence of cytotoxic lymphocytes intratumorally (160). Yi et al. recently showed that overexpression of PTRF in primary glioblastoma cells results in accumulation of lysophosphatidylcholine (LPC) species and decreased phosphatidylcholine (PC), resulting in increased membrane fluidity, endocytosis, and levels of the protein cytoplasmic phospholipase A2 (cPLA2), which provides fatty acids for mitochondrial fatty acid oxidation. In vivo, PTRF overexpression resulted in increased tumor growth and shorter survival. The authors found that intratumoral interferon gamma (IFN-γ) and granzyme B (GzmB) were decreased, with decreased numbers of CD8+ TILs, providing evidence for the role of abnormal phospholipid synthesis in glioblastoma immunosuppression. Strikingly, inhibition of cPLA2 restored IFN-γ and GzmB levels and resulted in increased TIL accumulation (161). Future investigations of cPLA2 in combination with existing immune activating therapies such as checkpoint blockade or CAR-T cells are warranted.
Discussion
Several metabolic pathways are implicated in maintaining immunosuppression and glioblastoma outgrowth in the TME. These aspects have the potential to be exploited therapeutically but also for the development of diagnostic tools, including imaging tools such as MR spectroscopy for 2HG (162), hyperpolarized [1-13C] lactate (163), intratumoral acidity using pH-weighted amine chemical exchange saturation transfer (CEST) MRI (164) and amino acid PET tracers like 18F-fluoroethyltyrosine (FET) (165).
Targeting metabolic pathways has potential for conditioning the TME to become more responsive to frontline immunotherapies that have succeeded in more immunogenic cancers, as well as providing the opportunity to expand the limited treatment modalities that are currently approved. As experimental tools mature, our ability to better appreciate the heterogeneity between and within tumors advances. Many of the pathways mentioned have attracted study in more immunogenic cancers while preclinical data is sparse for glioblastoma models.
While there are a number of ongoing clinical trials exploring glioblastoma immunotherapies from the perspective of checkpoint blockade, there are relatively fewer trials pursuing immunometabolism modulation. For example, despite the extensive characterization of pathways like adenosine metabolism, glioblastoma research has yet to pursue A2aR inhibitors in the clinic. While the tryptophan metabolism pathway is by far the most clinically explored in IDH1 wildtype glioblastoma with 2 studies completed (NCT02052648, NCT02502708) and two studies recruiting (NCT04047706, NCT04049669), other pathways have unfortunately been less pursued. With the exception of one study pursuing arginine metabolism (NCT04587830) many of the other pathways analyzed in this review have not yet been explored in the clinic.
Metabolic mechanisms in glioma, and their interactions with the TME, and immune cells are helpful to develop precision medicine approaches. The presence of infiltrating immune cells in the TME presents a challenge but also a potential for therapeutic targets. Effector CD8+ T-cells express high levels of co-stimulatory and co-inhibitory molecules with a preferential accumulation of regulatory T cells (Tregs) in CNS tumors (166). The immunosuppressive environment of brain tumors has been highlighted in gliomas and other CNS tumors (144). Tregs play an essential role in ameliorating auto-immunity, but in the setting of brain TME, their anti-inflammatory activity creates a more permissive environment for tumor progression (167).
While targeting the IDH metabolic pathway with IDH inhibitors, and also more recently the IDH antigen, has demonstrated encouraging preliminary results in IDH mutated gliomas (95), glioblastoma or IDH wild type gliomas lack a uniformly expressed tumor specific antigen and are highly heterogenous. Research focus on targeting the metabolism in IDH wildtype glioblastoma, investigation of the role of metabolic pathways in glioblastoma, developing an appreciation for their differing activities across tumor types, and an increased willingness to explore these pathways in glioblastoma without first waiting for exploration in other tumors, should allow for selective and targeted treatment options and should inspire hope to treat patients with glioblastoma with immunotherapy.
Conclusions
The field of immunometabolism represents a unique opportunity with emerging data supporting further research to fully understand mechanisms of resistance and to find potential synergy between immunometabolic pathways as well as other immunotherapy modalities. In addition to the pathways outlined, there remain other unknown metabolic aspects to discover to improve available therapies for patients with glioblastoma.
Author Contributions
MK, AM, and JR conceived of and designed the work. AM, MK, WT, and AH-M drafted, and subsequently all authors revised the manuscript. KH and KS developed the figures, which were revised with input from all authors. All authors contributed to the article and approved the submitted version.
Conflict of Interest
PF reports consulting for Monteris Medical. JS has an equity interest in Istari Oncology, which has licensed intellectual property from Duke related to the use of poliovirus and D2C7 in the treatment of glioblastoma. JS is an inventor on patents related to PEP-CMV DC vaccine with tetanus, as well as poliovirus vaccine and D2C7 in the treatment of glioblastoma. JS has an equity interest in Annias Immunotherapeutics, which has licensed intellectual property from Duke related to the use of the pepCMV vaccine in the treatment of glioblastoma. MK reports advisory roles for Janssen, AbbVie, and Jackson Laboratory for Genomic Medicine, and research funding from AbbVie, Bristol-Myers Squibb, and Specialized Therapeutics.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Warburg O, Wind F, Negelein E. The Metabolism of Tumors in the Body. J Gen Physiol (1927) 8:519–30. doi: 10.1085/jgp.8.6.519
2. Heiland DH, Worner J, Gerrit Haaker J, Delev D, Pompe N, Mercas B, et al. The Integrative Metabolomic-Transcriptomic Landscape of Glioblastome Multiforme. Oncotarget (2017) 8:49178–90. doi: 10.18632/oncotarget.16544
3. Gadaleta F, Bessonov K, Van Steen K. Integration of Gene Expression and Methylation to Unravel Biological Networks in Glioblastoma Patients. Genet Epidemiol (2017) 41:136–44. doi: 10.1002/gepi.22028
4. Kesarwani P, Prabhu A, Kant S, Chinnaiyan P. Metabolic Remodeling Contributes Towards an Immune-Suppressive Phenotype in Glioblastoma. Cancer Immunol Immunother (2019) 68:1107–20. doi: 10.1007/s00262-019-02347-3
5. Dentro SC, Leshchiner I, Haase K, Tarabichi M, Wintersinger J, Deshwar AG, et al. Characterizing Genetic Intra-Tumor Heterogeneity Across 2,658 Human Cancer Genomes. bioRxiv (2020) 312041. doi: 10.1101/312041
6. Jawhari S, Ratinaud MH, Verdier M. Glioblastoma, Hypoxia and Autophagy: A Survival-Prone ‘Menage-a-Trois’. Cell Death Dis (2016) 7:e2434. doi: 10.1038/cddis.2016.318
7. Monteiro AR, Hill R, Pilkington GJ, Madureira PA. The Role of Hypoxia in Glioblastoma Invasion. Cells (2017) 6(4):45. doi: 10.3390/cells6040045
8. Rong Y, Durden DL, Van Meir EG, Brat DJ. ‘Pseudopalisading’ Necrosis in Glioblastoma: A Familiar Morphologic Feature That Links Vascular Pathology, Hypoxia, and Angiogenesis. J Neuropathol Exp Neurol (2006) 65:529–39. doi: 10.1097/00005072-200606000-00001
9. Seidel S, Garvalov BK, Wirta V, von Stechow L, Schanzer A, Meletis K, et al. A Hypoxic Niche Regulates Glioblastoma Stem Cells Through Hypoxia Inducible Factor 2 Alpha. Brain (2010) 133:983–95. doi: 10.1093/brain/awq042
10. Li Z, Bao S, Wu Q, Wang H, Eyler C, Sathornsumetee S, et al. Hypoxia-Inducible Factors Regulate Tumorigenic Capacity of Glioma Stem Cells. Cancer Cell (2009) 15:501–13. doi: 10.1016/j.ccr.2009.03.018
11. Heddleston JM, Li Z, McLendon RE, Hjelmeland AB, Rich JN. The Hypoxic Microenvironment Maintains Glioblastoma Stem Cells and Promotes Reprogramming Towards a Cancer Stem Cell Phenotype. Cell Cycle (2009) 8:3274–84. doi: 10.4161/cc.8.20.9701
12. Hu YL, DeLay M, Jahangiri A, Molinaro AM, Rose SD, Carbonell WS, et al. Hypoxia-Induced Autophagy Promotes Tumor Cell Survival and Adaptation to Antiangiogenic Treatment in Glioblastoma. Cancer Res (2012) 72:1773–83. doi: 10.1158/0008-5472.CAN-11-3831
13. Rapisarda A, Melillo G. Overcoming Disappointing Results With Antiangiogenic Therapy by Targeting Hypoxia. Nat Rev Clin Oncol (2012) 9:378–90. doi: 10.1038/nrclinonc.2012.64
14. Liu YN, Yang JF, Huang DJ, Ni HH, Zhang CX, Zhang L, et al. Hypoxia Induces Mitochondrial Defect That Promotes T Cell Exhaustion in Tumor Microenvironment Through MYC-Regulated Pathways. Front Immunol (2020) 11:1906. doi: 10.3389/fimmu.2020.01906
15. Brat DJ, Bellail AC, Van Meir EG. The Role of Interleukin-8 and its Receptors in Gliomagenesis and Tumoral Angiogenesis. Neuro Oncol (2005) 7:122–33. doi: 10.1215/S1152851704001061
16. Rolhion C, Penault-Llorca F, Kemeny JL, Lemaire JJ, Jullien C, Labit-Bouvier C, et al. Interleukin-6 Overexpression as a Marker of Malignancy in Human Gliomas. J Neurosurg (2001) 94:97–101. doi: 10.3171/jns.2001.94.1.0097
17. Ma J, Benitez JA, Li J, Miki S, Ponte de Albuquerque C, Galatro T, et al. Inhibition of Nuclear PTEN Tyrosine Phosphorylation Enhances Glioma Radiation Sensitivity Through Attenuated DNA Repair. Cancer Cell (2019) 35:504–18.e507. doi: 10.1016/j.ccell.2019.01.020
18. Lamano JB, Lamano JB, Li YD, DiDomenico JD, Choy W, Veliceasa D, et al. Glioblastoma-Derived IL6 Induces Immunosuppressive Peripheral Myeloid Cell PD-L1 and Promotes Tumor Growth. Clin Cancer Res (2019) 25:3643–57. doi: 10.1158/1078-0432.CCR-18-2402
19. Zagzag D, Lukyanov Y, Lan L, Ali MA, Esencay M, Mendez O, et al. Hypoxia-Inducible Factor 1 and VEGF Upregulate CXCR4 in Glioblastoma: Implications for Angiogenesis and Glioma Cell Invasion. Lab Invest (2006) 86:1221–32. doi: 10.1038/labinvest.3700482
20. Choueiri TK, Kaelin WG. Targeting the HIF2–VEGF Axis in Renal Cell Carcinoma. Nat Med (2020) 26:1519–30. doi: 10.1038/s41591-020-1093-z
21. Strowd RE, Ellingson BM, Wen PY, Ahluwalia MS, Piotrowski AF, Desai AS, et al. Safety and Activity of a First-in-Class Oral HIF2-alpha Inhibitor, PT2385, in Patients With First Recurrent Glioblastoma (GBM). J Clin Oncol (2019) 37:2027–7. doi: 10.1200/JCO.2019.37.15_suppl.2027
22. Mergenthaler P, Lindauer U, Dienel GA, Meisel A. Sugar for the Brain: The Role of Glucose in Physiological and Pathological Brain Function. Trends Neurosci (2013) 36:587–97. doi: 10.1016/j.tins.2013.07.001
23. Flavahan WA, Wu Q, Hitomi M, Rahim N, Kim Y, Sloan AE, et al. Brain Tumor Initiating Cells Adapt to Restricted Nutrition Through Preferential Glucose Uptake. Nat Neurosci (2013) 16:1373–82. doi: 10.1038/nn.3510
24. Vannucci SJ, Maher F, Simpson IA. Glucose Transporter Proteins in Brain: Delivery of Glucose to Neurons and Glia. Glia (1997) 21:2–21. doi: 10.1002/(SICI)1098-1136(199709)21:1<2::AID-GLIA2>3.0.CO;2-C
25. Vaughn AE, Deshmukh M. Glucose Metabolism Inhibits Apoptosis in Neurons and Cancer Cells by Redox Inactivation of Cytochrome C. Nat Cell Biol (2008) 10:1477–83. doi: 10.1038/ncb1807
26. Amaral AI, Hadera MG, Tavares JM, Kotter MR, Sonnewald U. Characterization of Glucose-Related Metabolic Pathways in Differentiated Rat Oligodendrocyte Lineage Cells. Glia (2016) 64:21–34. doi: 10.1002/glia.22900
27. Agostini M, Romeo F, Inoue S, Niklison-Chirou MV, Elia AJ, Dinsdale D, et al. Metabolic Reprogramming During Neuronal Differentiation. Cell Death Differ (2016) 23:1502–14. doi: 10.1038/cdd.2016.36
28. Masui K, Tanaka K, Akhavan D, Babic I, Gini B, Matsutani T, et al. mTOR Complex 2 Controls Glycolytic Metabolism in Glioblastoma Through FoxO Acetylation and Upregulation of c-Myc. Cell Metab (2013) 18:726–39. doi: 10.1016/j.cmet.2013.09.013
29. Ganapathy-Kanniappan S, Geschwind JF. Tumor Glycolysis as a Target for Cancer Therapy: Progress and Prospects. Mol Cancer (2013) 12:152. doi: 10.1186/1476-4598-12-152
30. Guo H, Nan Y, Zhen Y, Zhang Y, Guo L, Yu K, et al. miRNA-451 Inhibits Glioma Cell Proliferation and Invasion by Downregulating Glucose Transporter 1. Tumour Biol (2016) 37:13751–61. doi: 10.1007/s13277-016-5219-3
31. Spence AM, Muzi M, Graham MM, O’Sullivan F, Link JM, Lewellen TK, et al. 2-[18f]Fluoro-2-Deoxyglucose and Glucose Uptake in Malignant Gliomas Before and After Radiotherapy. Correlation with Outcome. Clin Cancer Res (2002) 8:971–9.
32. Won WJ, Deshane JS, Leavenworth JW, Oliva CR, Griguer CE. Metabolic and Functional Reprogramming of Myeloid-Derived Suppressor Cells and Their Therapeutic Control in Glioblastoma. Cell Stress (2019) 3:47–65. doi: 10.15698/cst2019.02.176
33. Raber P, Ochoa AC, Rodriguez PC. Metabolism of L-arginine by Myeloid-Derived Suppressor Cells in Cancer: Mechanisms of T Cell Suppression and Therapeutic Perspectives. Immunol Invest (2012) 41:614–34. doi: 10.3109/08820139.2012.680634
34. Hou X, Liu Y, Liu H, Chen X, Liu M, Che H, et al. PERK Silence Inhibits Glioma Cell Growth Under Low Glucose Stress by Blockage of p-AKT and Subsequent HK2’s Mitochondria Translocation. Sci Rep (2015) 5:9065. doi: 10.1038/srep09065
35. Zhang C, Wang M, Ji F, Peng Y, Wang B, Zhao J, et al. A Novel Glucose Metabolism-Related Gene Signature for Overall Survival Prediction in Patients With Glioblastoma. BioMed Res Int (2021) 2021:8872977–8872977. doi: 10.1155/2021/8872977
36. Ho PC, Bihuniak JD, Macintyre AN, Staron M, Liu X, Amezquita R, et al. Phosphoenolpyruvate Is a Metabolic Checkpoint of Anti-Tumor T Cell Responses. Cell (2015) 162:1217–28. doi: 10.1016/j.cell.2015.08.012
37. Siska PJ, Rathmell JC. T Cell Metabolic Fitness in Antitumor Immunity. Trends Immunol (2015) 36:257–64. doi: 10.1016/j.it.2015.02.007
38. Siska PJ, van der Windt GJ, Kishton RJ, Cohen S, Eisner W, MacIver NJ, et al. Suppression of Glut1 and Glucose Metabolism by Decreased Akt/mTORC1 Signaling Drives T Cell Impairment in B Cell Leukemia. J Immunol (Baltimore Md 1950) (2016) 197:2532–40. doi: 10.4049/jimmunol.1502464
39. Kallies A, Zehn D, Utzschneider DT. Precursor Exhausted T Cells: Key to Successful Immunotherapy? Nat Rev Immunol (2020) 20:128–36. doi: 10.1038/s41577-019-0223-7
40. Bengsch B, Johnson AL, Kurachi M, Odorizzi PM, Pauken KE, Attanasio J, et al. Bioenergetic Insufficiencies Due to Metabolic Alterations Regulated by the Inhibitory Receptor PD-1 Are an Early Driver of CD8(+) T Cell Exhaustion. Immunity (2016) 45:358–73. doi: 10.1016/j.immuni.2016.07.008
41. Patsoukis N, Bardhan K, Chatterjee P, Sari D, Liu B, Bell LN, et al. PD-1 Alters T-Cell Metabolic Reprogramming by Inhibiting Glycolysis and Promoting Lipolysis and Fatty Acid Oxidation. Nat Commun (2015) 6:6692. doi: 10.1038/ncomms7692
42. Chang CH, Qiu J, O’Sullivan D, Buck MD, Noguchi T, Curtis JD, et al. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell (2015) 162:1229–41. doi: 10.1016/j.cell.2015.08.016
43. Parsa AT, Waldron JS, Panner A, Crane CA, Parney IF, Barry JJ, et al. Loss of Tumor Suppressor PTEN Function Increases B7-H1 Expression and Immunoresistance in Glioma. Nat Med (2007) 13:84–8. doi: 10.1038/nm1517
44. de la Cruz-Lopez KG, Castro-Munoz LJ, Reyes-Hernandez DO, Garcia-Carranca A, Manzo-Merino J. Lactate in the Regulation of Tumor Microenvironment and Therapeutic Approaches. Front Oncol (2019) 9:1143. doi: 10.3389/fonc.2019.01143
45. Badawy AA. Kynurenine Pathway of Tryptophan Metabolism: Regulatory and Functional Aspects. Int J Tryptophan Res (2017) 10:1178646917691938. doi: 10.1177/1178646917691938
46. Thaker AI, Rao MS, Bishnupuri KS, Kerr TA, Foster L, Marinshaw JM, et al. IDO1 Metabolites Activate Beta-Catenin Signaling to Promote Cancer Cell Proliferation and Colon Tumorigenesis in Mice. Gastroenterology (2013) 145:416–25.e411-414. doi: 10.1053/j.gastro.2013.05.002
47. Bostian AC, Eoff RL. Aberrant Kynurenine Signaling Modulates DNA Replication Stress Factors and Promotes Genomic Instability in Gliomas. Chem Res Toxicol (2016) 29:1369–80. doi: 10.1021/acs.chemrestox.6b00255
48. Di Serio C, Cozzi A, Angeli I, Doria L, Micucci I, Pellerito S, et al. Kynurenic Acid Inhibits the Release of the Neurotrophic Fibroblast Growth Factor (FGF)-1 and Enhances Proliferation of Glia Cells, In Vitro. Cell Mol Neurobiol (2005) 25:981–93. doi: 10.1007/s10571-005-8469-y
49. Munn DH, Mellor AL. Indoleamine 2,3 Dioxygenase and Metabolic Control of Immune Responses. Trends Immunol (2013) 34:137–43. doi: 10.1016/j.it.2012.10.001
50. Mondal A, Smith C, DuHadaway JB, Sutanto-Ward E, Prendergast GC, Bravo-Nuevo A, et al. IDO1 Is an Integral Mediator of Inflammatory Neovascularization. EBioMedicine (2016) 14:74–82. doi: 10.1016/j.ebiom.2016.11.013
51. Mitsuka K, Kawataki T, Satoh E, Asahara T, Horikoshi T, Kinouchi H. Expression of Indoleamine 2,3-Dioxygenase and Correlation With Pathological Malignancy in Gliomas. Neurosurgery (2013) 72:1031–1038; discussion 1038-1039. doi: 10.1227/NEU.0b013e31828cf945
52. Yuasa HJ, Ball HJ, Ho YF, Austin CJ, Whittington CM, Belov K, et al. Characterization and Evolution of Vertebrate Indoleamine 2, 3-Dioxygenases IDOs From Monotremes and Marsupials. Comp Biochem Physiol B Biochem Mol Biol (2009) 153(2):137–44. doi: 10.1016/j.cbpb.2009.02.002
53. Wainwright DA, Balyasnikova IV, Chang AL, Ahmed AU, Moon KS, Auffinger B, et al. IDO Expression in Brain Tumors Increases the Recruitment of Regulatory T Cells and Negatively Impacts Survival. Clin Cancer Res (2012) 18:6110–21. doi: 10.1158/1078-0432.CCR-12-2130
54. Vogel CF, Goth SR, Dong B, Pessah IN, Matsumura F. Aryl Hydrocarbon Receptor Signaling Mediates Expression of Indoleamine 2,3-Dioxygenase. Biochem Biophys Res Commun (2008) 375:331–5. doi: 10.1016/j.bbrc.2008.07.156
55. Wainwright DA, Dey M, Chang A, Lesniak MS. Targeting Tregs in Malignant Brain Cancer: Overcoming IDO. Front Immunol (2013) 4:116. doi: 10.3389/fimmu.2013.00116
56. Opitz CA, Litzenburger UM, Sahm F, Ott M, Tritschler I, Trump S, et al. An Endogenous Tumour-Promoting Ligand of the Human Aryl Hydrocarbon Receptor. Nature (2011) 478:197–203. doi: 10.1038/nature10491
57. Munn DH, Sharma MD, Baban B, Harding HP, Zhang Y, Ron D, et al. GCN2 Kinase in T Cells Mediates Proliferative Arrest and Anergy Induction in Response to Indoleamine 2,3-Dioxygenase. Immunity (2005) 22:633–42. doi: 10.1016/j.immuni.2005.03.013
58. Prendergast GC, Malachowski WJ, Mondal A, Scherle P, Muller AJ. Indoleamine 2,3-Dioxygenase and Its Therapeutic Inhibition in Cancer. Int Rev Cell Mol Biol (2018) 336:175–203. doi: 10.1016/bs.ircmb.2017.07.004
59. Kesarwani P, Prabhu A, Kant S, Kumar P, Graham SF, Buelow KL, et al. Tryptophan Metabolism Contributes to Radiation-Induced Immune Checkpoint Reactivation in Glioblastoma. Clin Cancer Res (2018) 24:3632–43. doi: 10.1158/1078-0432.CCR-18-0041
60. Hanihara M, Kawataki T, Oh-Oka K, Mitsuka K, Nakao A, Kinouchi H. Synergistic Antitumor Effect With Indoleamine 2,3-Dioxygenase Inhibition and Temozolomide in a Murine Glioma Model. J Neurosurg (2016) 124:1594–601. doi: 10.3171/2015.5.JNS141901
61. Li M, Bolduc AR, Hoda MN, Gamble DN, Dolisca SB, Bolduc AK, et al. The Indoleamine 2,3-Dioxygenase Pathway Controls Complement-Dependent Enhancement of Chemo-Radiation Therapy Against Murine Glioblastoma. J Immunother Cancer (2014) 2:21. doi: 10.1186/2051-1426-2-21
62. Correction: Durable Therapeutic Efficacy Utilizing Combinatorial Blockade Against IDO, CTLA-4, and PD-L1 in Mice With Brain Tumors. Clin Cancer Res (2015) 21:662. doi: 10.1158/1078-0432.CCR-14-3211
63. Wainwright DA, Chang AL, Dey M, Balyasnikova IV, Kim CK, Tobias A, et al. Durable Therapeutic Efficacy Utilizing Combinatorial Blockade Against IDO, CTLA-4, and PD-L1 in Mice With Brain Tumors. Clin Cancer Res (2014) 20:5290–301. doi: 10.1158/1078-0432.CCR-14-0514
64. Ladomersky E, Zhai L, Lauing KL, Bell A, Xu J, Kocherginsky M, et al. Advanced Age Increases Immunosuppression in the Brain and Decreases Immunotherapeutic Efficacy in Subjects With Glioblastoma. Clin Cancer Res (2020) 26:5232–45. doi: 10.1158/1078-0432.ccr-19-3874
65. Rath M, Müller I, Kropf P, Closs EI, Munder M. Metabolism Via Arginase or Nitric Oxide Synthase: Two Competing Arginine Pathways in Macrophages. Front Immunol (2014) 5:532. doi: 10.3389/fimmu.2014.00532
66. Phillips MM, Sheaff MT, Szlosarek PW. Targeting Arginine-Dependent Cancers With Arginine-Degrading Enzymes: Opportunities and Challenges. Cancer Res Treat (2013) 45:251–62. doi: 10.4143/crt.2013.45.4.251
67. Kobayashi K, Ohnishi A, Promsuk J, Shimizu S, Kanai Y, Shiokawa Y, et al. Enhanced Tumor Growth Elicited by L-Type Amino Acid Transporter 1 in Human Malignant Glioma Cells. Neurosurgery (2008) 62:493–503; discussion 503-494. doi: 10.1227/01.neu.0000316018.51292.19
68. Chinnaiyan P, Kensicki E, Bloom G, Prabhu A, Sarcar B, Kahali S, et al. The Metabolomic Signature of Malignant Glioma Reflects Accelerated Anabolic Metabolism. Cancer Res (2012) 72:5878–88. doi: 10.1158/0008-5472.Can-12-1572-t
69. Khoury O, Ghazale N, Stone E, El-Sibai M, Frankel AE, Abi-Habib RJ. Human Recombinant Arginase I (Co)-PEG5000 [HuArgI (Co)-PEG5000]-induced Arginine Depletion Is Selectively Cytotoxic to Human Glioblastoma Cells. J Neurooncol (2015) 122:75–85. doi: 10.1007/s11060-014-1698-5
70. Jahani-Asl A, Bonni A. iNOS: A Potential Therapeutic Target for Malignant Glioma. Curr Mol Med (2013) 13:1241–9. doi: 10.2174/1566524011313080002
71. Morantz RA, Wood GW, Foster M, Clark M, Gollahon K. Macrophages in Experimental and Human Brain Tumors. Part 2: Studies of the Macrophage Content of Human Brain Tumors. J Neurosurg (1979) 50:305–11. doi: 10.3171/jns.1979.50.3.0305
72. Rossi ML, Hughes JT, Esiri MM, Coakham HB, Brownell DB. Immunohistological Study of Mononuclear Cell Infiltrate in Malignant Gliomas. Acta Neuropathol (1987) 74:269–77. doi: 10.1007/bf00688191
73. Ginhoux F, Schultze JL, Murray PJ, Ochando J, Biswas SK. New Insights Into the Multidimensional Concept of Macrophage Ontogeny, Activation and Function. Nat Immunol (2016) 17:34–40. doi: 10.1038/ni.3324
74. Müller S, Kohanbash G, Liu SJ, Alvarado B, Carrera D, Bhaduri A, et al. Single-Cell Profiling of Human Gliomas Reveals Macrophage Ontogeny as a Basis for Regional Differences in Macrophage Activation in the Tumor Microenvironment. Genome Biol (2017) 18:234–4. doi: 10.1186/s13059-017-1362-4
75. Chen Z, Hambardzumyan D. Immune Microenvironment in Glioblastoma Subtypes. Front Immunol (2018) 9:1004. doi: 10.3389/fimmu.2018.01004
76. Hambardzumyan D, Gutmann DH, Kettenmann H. The Role of Microglia and Macrophages in Glioma Maintenance and Progression. Nat Neurosci (2016) 19:20–7. doi: 10.1038/nn.4185
77. Cairns RA, Mak TW. Oncogenic Isocitrate Dehydrogenase Mutations: Mechanisms, Models, and Clinical Opportunities. Cancer Discov (2013) 3:730–41. doi: 10.1158/2159-8290.CD-13-0083
78. Jezek P. 2-Hydroxyglutarate in Cancer Cells. Antioxid Redox Signal (2020) 33:903–26. doi: 10.1089/ars.2019.7902
79. Gagné LM, Boulay K, Topisirovic I, Huot ME, Mallette FA. Oncogenic Activities of IDH1/2 Mutations: From Epigenetics to Cellular Signaling. Trends Cell Biol (2017) 27:738–52. doi: 10.1016/j.tcb.2017.06.002
80. Struys EA, Verhoeven NM, Ten Brink HJ, Wickenhagen WV, Gibson KM, Jakobs C. Kinetic Characterization of Human Hydroxyacid-Oxoacid Transhydrogenase: Relevance to D-2-Hydroxyglutaric and Gamma-Hydroxybutyric Acidurias. J Inherit Metab Dis (2005) 28:921–30. doi: 10.1007/s10545-005-0114-x
81. Fan J, Teng X, Liu L, Mattaini KR, Looper RE, Vander Heiden MG, et al. Human Phosphoglycerate Dehydrogenase Produces the Oncometabolite D-2-Hydroxyglutarate. ACS Chem Biol (2015) 10:510–6. doi: 10.1021/cb500683c
82. Intlekofer AM, Dematteo RG, Venneti S, Finley LW, Lu C, Judkins AR, et al. Hypoxia Induces Production of L-2-Hydroxyglutarate. Cell Metab (2015) 22:304–11. doi: 10.1016/j.cmet.2015.06.023
83. Rzem R, Vincent MF, Van Schaftingen E, Veiga-da-Cunha M. L-2-Hydroxyglutaric Aciduria, a Defect of Metabolite Repair. J Inherit Metab Dis (2007) 30:681–9. doi: 10.1007/s10545-007-0487-0
84. Dang L, Su SM. Isocitrate Dehydrogenase Mutation and (R)-2-Hydroxyglutarate: From Basic Discovery to Therapeutics Development. Annu Rev Biochem (2017) 86:305–31. doi: 10.1146/annurev-biochem-061516-044732
85. Koivunen P, Lee S, Duncan CG, Lopez G, Lu G, Ramkissoon S, et al. Transformation by the (R)-Enantiomer of 2-Hydroxyglutarate Linked to EGLN Activation. Nature (2012) 483:484–8. doi: 10.1038/nature10898
86. Garcia MG, Carella A, Urdinguio RG, Bayon GF, Lopez V, Tejedor JR, et al. Epigenetic Dysregulation of TET2 in Human Glioblastoma. Oncotarget (2018) 9:25922–34. doi: 10.18632/oncotarget.25406
87. Ohka F, Ito M, Ranjit M, Senga T, Motomura A, Motomura K, et al. Quantitative Metabolome Analysis Profiles Activation of Glutaminolysis in Glioma With IDH1 Mutation. Tumour Biol (2014) 35:5911–20. doi: 10.1007/s13277-014-1784-5
88. Chen F, Bian K, Tang Q, Fedeles BI, Singh V, Humulock ZT, et al. Oncometabolites D- and L-2-Hydroxyglutarate Inhibit the AlkB Family DNA Repair Enzymes Under Physiological Conditions. Chem Res Toxicol (2017) 30:1102–10. doi: 10.1021/acs.chemrestox.7b00009
89. Bunse L, Pusch S, Bunse T, Sahm F, Sanghvi K, Friedrich M, et al. Suppression of Antitumor T Cell Immunity by the Oncometabolite (R)-2-Hydroxyglutarate. Nat Med (2018) 24:1192–203. doi: 10.1038/s41591-018-0095-6
90. Kohanbash G, Carrera DA, Shrivastav S, Ahn BJ, Jahan N, Mazor T, et al. Isocitrate Dehydrogenase Mutations Suppress STAT1 and CD8+ T Cell Accumulation in Gliomas. J Clin Invest (2017) 127:1425–37. doi: 10.1172/JCI90644
91. Komohara Y, Ohnishi K, Kuratsu J, Takeya M. Possible Involvement of the M2 Anti-Inflammatory Macrophage Phenotype in Growth of Human Gliomas. J Pathol (2008) 216:15–24. doi: 10.1002/path.2370
92. Amankulor NM, Kim Y, Arora S, Kargl J, Szulzewsky F, Hanke M, et al. Mutant IDH1 Regulates the Tumor-Associated Immune System in Gliomas. Genes Dev (2017) 31:774–86. doi: 10.1101/gad.294991.116
93. Golub D, Iyengar N, Dogra S, Wong T, Bready D, Tang K, et al. Mutant Isocitrate Dehydrogenase Inhibitors as Targeted Cancer Therapeutics. Front Oncol (2019) 9:417. doi: 10.3389/fonc.2019.00417
94. Kadiyala P, Carney SV, Gauss JC, Garcia-Fabiani MB, Haase S, Alghamri MS, et al. Inhibition of 2-Hydroxyglutarate Elicits Metabolic Reprogramming and Mutant IDH1 Glioma Immunity in Mice. J Clin Invest (2021) 131(4):e139542. doi: 10.1172/JCI139542
95. Platten M, Bunse L, Wick A, Bunse T, Le Cornet L, Harting I, et al. A Vaccine Targeting Mutant IDH1 in Newly Diagnosed Glioma. Nature (2021) 592:463–8. doi: 10.1038/s41586-021-03363-z
96. Falzoni S, Donvito G, Di Virgilio F. Detecting Adenosine Triphosphate in the Pericellular Space. Interface Focus (2013) 3:20120101. doi: 10.1098/rsfs.2012.0101
97. Pastor-Anglada M, Perez-Torras S. Who Is Who in Adenosine Transport. Front Pharmacol (2018) 9:627. doi: 10.3389/fphar.2018.00627
98. Sabirov RZ, Okada Y. ATP Release Via Anion Channels. Purinergic Signal (2005) 1:311–28. doi: 10.1007/s11302-005-1557-0
99. Zimmermann H, Zebisch M, Strater N. Cellular Function and Molecular Structure of Ecto-Nucleotidases. Purinergic Signal (2012) 8:437–502. doi: 10.1007/s11302-012-9309-4
100. Vigano S, Alatzoglou D, Irving M, Menetrier-Caux C, Caux C, Romero P, et al. Targeting Adenosine in Cancer Immunotherapy to Enhance T-Cell Function. Front Immunol (2019) 10:925. doi: 10.3389/fimmu.2019.00925
101. Ott M, Tomaszowski KH, Marisetty A, Kong LY, Wei J, Duna M, et al. Profiling of Patients With Glioma Reveals the Dominant Immunosuppressive Axis Is Refractory to Immune Function Restoration. JCI Insight (2020) 5(17):e134386. doi: 10.1172/jci.insight.134386
102. Merighi S, Benini A, Mirandola P, Gessi S, Varani K, Leung E, et al. Hypoxia Inhibits Paclitaxel-Induced Apoptosis Through Adenosine-Mediated Phosphorylation of Bad in Glioblastoma Cells. Mol Pharmacol (2007) 72:162–72. doi: 10.1124/mol.106.031849
103. Kim H, Kang JW, Lee S, Choi WJ, Jeong LS, Yang Y, et al. A3 Adenosine Receptor Antagonist, Truncated Thio-Cl-IB-MECA, Induces Apoptosis in T24 Human Bladder Cancer Cells. Anticancer Res (2010) 30:2823–30.
104. Gessi S, Bencivenni S, Battistello E, Vincenzi F, Colotta V, Catarzi D, et al. Inhibition of A2A Adenosine Receptor Signaling in Cancer Cells Proliferation by the Novel Antagonist TP455. Front Pharmacol (2017) 8:888. doi: 10.3389/fphar.2017.00888
105. Lin Z, Yin P, Reierstad S, O’Halloran M, Coon VJ, Pearson EK, et al. Adenosine A1 Receptor, a Target and Regulator of Estrogen Receptoralpha Action, Mediates the Proliferative Effects of Estradiol in Breast Cancer. Oncogene (2010) 29:1114–22. doi: 10.1038/onc.2009.409
106. Zhou Y, Chu X, Deng F, Tong L, Tong G, Yi Y, et al. The Adenosine A2b Receptor Promotes Tumor Progression of Bladder Urothelial Carcinoma by Enhancing MAPK Signaling Pathway. Oncotarget (2017) 8:48755–68. doi: 10.18632/oncotarget.17835
107. Mittal D, Sinha D, Barkauskas D, Young A, Kalimutho M, Stannard K, et al. Adenosine 2B Receptor Expression on Cancer Cells Promotes Metastasis. Cancer Res (2016) 76:4372–82. doi: 10.1158/0008-5472.CAN-16-0544
108. Allard B, Longhi MS, Robson SC, Stagg J. The Ectonucleotidases CD39 and CD73: Novel Checkpoint Inhibitor Targets. Immunol Rev (2017) 276:121–44. doi: 10.1111/imr.12528
109. Linnemann C, Schildberg FA, Schurich A, Diehl L, Hegenbarth SI, Endl E, et al. Adenosine Regulates CD8 T-Cell Priming by Inhibition of Membrane-Proximal T-Cell Receptor Signalling. Immunology (2009) 128:e728–737. doi: 10.1111/j.1365-2567.2009.03075.x
110. Jimenez JL, Punzon C, Navarro J, Munoz-Fernandez MA, Fresno M. Phosphodiesterase 4 Inhibitors Prevent Cytokine Secretion by T Lymphocytes by Inhibiting Nuclear Factor-KappaB and Nuclear Factor of Activated T Cells Activation. J Pharmacol Exp Ther (2001) 299:753–9.
111. Cekic C, Linden J. Purinergic Regulation of the Immune System. Nat Rev Immunol (2016) 16:177–92. doi: 10.1038/nri.2016.4
112. Allard D, Turcotte M, Stagg J. Targeting A2 Adenosine Receptors in Cancer. Immunol Cell Biol (2017) 95:333–9. doi: 10.1038/icb.2017.8
113. Deaglio S, Dwyer KM, Gao W, Friedman D, Usheva A, Erat A, et al. Adenosine Generation Catalyzed by CD39 and CD73 Expressed on Regulatory T Cells Mediates Immune Suppression. J Exp Med (2007) 204:1257–65. doi: 10.1084/jem.20062512
114. Hatfield SM, Kjaergaard J, Lukashev D, Schreiber TH, Belikoff B, Abbott R, et al. Immunological Mechanisms of the Antitumor Effects of Supplemental Oxygenation. Sci Transl Med (2015) 7:277ra230. doi: 10.1126/scitranslmed.aaa1260
115. Miller JS, Cervenka T, Lund J, Okazaki IJ, Moss J. Purine Metabolites Suppress Proliferation of Human NK Cells Through a Lineage-Specific Purine Receptor. J Immunol (Baltimore Md 1950) (1999) 162:7376–82.
116. Beavis PA, Divisekera U, Paget C, Chow MT, John LB, Devaud C, et al. Blockade of A2A Receptors Potently Suppresses the Metastasis of CD73+ Tumors. Proc Natl Acad Sci U S A (2013) 110:14711–6. doi: 10.1073/pnas.1308209110
117. Milo R, Jorgensen P, Moran U, Weber G, Springer M. BioNumbers–the Database of Key Numbers in Molecular and Cell Biology. Nucleic Acids Res (2010) 38:D750–3. doi: 10.1093/nar/gkp889
118. Li L, Huang L, Ye H, Song SP, Bajwa A, Lee SJ, et al. Dendritic Cells Tolerized With Adenosine A(2)AR Agonist Attenuate Acute Kidney Injury. J Clin Invest (2012) 122:3931–42. doi: 10.1172/JCI63170
119. Csoka B, Selmeczy Z, Koscso B, Nemeth ZH, Pacher P, Murray PJ, et al. Adenosine Promotes Alternative Macrophage Activation Via A2A and A2B Receptors. FASEB J (2012) 26:376–86. doi: 10.1096/fj.11-190934
120. Yago T, Tsukamoto H, Liu Z, Wang Y, Thompson LF, McEver RP. Multi-Inhibitory Effects of A2A Adenosine Receptor Signaling on Neutrophil Adhesion Under Flow. J Immunol (Baltimore Md 1950) (2015) 195:3880–9. doi: 10.4049/jimmunol.1500775
121. Ohta A, Gorelik E, Prasad SJ, Ronchese F, Lukashev D, Wong MK, et al. A2A Adenosine Receptor Protects Tumors From Antitumor T Cells. Proc Natl Acad Sci U S A (2006) 103:13132–7. doi: 10.1073/pnas.0605251103
122. Ohta A, Sitkovsky M. Role of G-Protein-Coupled Adenosine Receptors in Downregulation of Inflammation and Protection From Tissue Damage. Nature (2001) 414:916–20. doi: 10.1038/414916a
123. Leone RD, Emens LA. Targeting Adenosine for Cancer Immunotherapy. J Immunother Cancer (2018) 6:57. doi: 10.1186/s40425-018-0360-8
124. Beavis PA, Milenkovski N, Henderson MA, John LB, Allard B, Loi S, et al. Adenosine Receptor 2A Blockade Increases the Efficacy of Anti-PD-1 Through Enhanced Antitumor T-Cell Responses. Cancer Immunol Res (2015) 3:506–17. doi: 10.1158/2326-6066.CIR-14-0211
125. Mittal D, Young A, Stannard K, Yong M, Teng MW, Allard B, et al. Antimetastatic Effects of Blocking PD-1 and the Adenosine A2A Receptor. Cancer Res (2014) 74:3652–8. doi: 10.1158/0008-5472.CAN-14-0957
126. Iannone R, Miele L, Maiolino P, Pinto A, Morello S. Adenosine Limits the Therapeutic Effectiveness of Anti-CTLA4 mAb in a Mouse Melanoma Model. Am J Cancer Res (2014) 4:172–81.
127. Ogretmen B. Sphingolipid Metabolism in Cancer Signalling and Therapy. Nat Rev Cancer (2018) 18:33–50. doi: 10.1038/nrc.2017.96
128. Hannun YA, Obeid LM. Principles of Bioactive Lipid Signalling: Lessons From Sphingolipids. Nat Rev Mol Cell Biol (2008) 9:139–50. doi: 10.1038/nrm2329
129. Hawkins CC, Ali T, Ramanadham S, Hjelmeland AB. Sphingolipid Metabolism in Glioblastoma and Metastatic Brain Tumors: A Review of Sphingomyelinases and Sphingosine-1-Phosphate. Biomolecules (2020) 10(10):1357. doi: 10.3390/biom10101357
130. Riboni L, Abdel Hadi L, Navone SE, Guarnaccia L, Campanella R, Marfia G. Sphingosine-1-Phosphate in the Tumor Microenvironment: A Signaling Hub Regulating Cancer Hallmarks. Cells (2020) 9(2):337. doi: 10.3390/cells9020337
131. Tea MN, Poonnoose SI, Pitson SM. Targeting the Sphingolipid System as a Therapeutic Direction for Glioblastoma. Cancers (Basel) (2020) 12(1):111. doi: 10.3390/cancers12010111
132. Riboni L, Campanella R, Bassi R, Villani R, Gaini SM, Martinelli-Boneschi F, et al. Ceramide Levels are Inversely Associated With Malignant Progression of Human Glial Tumors. Glia (2002) 39:105–13. doi: 10.1002/glia.10087
133. Marfia G, Campanella R, Navone SE, Di Vito C, Riccitelli E, Hadi LA, et al. Autocrine/Paracrine Sphingosine-1-Phosphate Fuels Proliferative and Stemness Qualities of Glioblastoma Stem Cells. Glia (2014) 62:1968–81. doi: 10.1002/glia.22718
134. Abuhusain HJ, Matin A, Qiao Q, Shen H, Kain N, Day BW, et al. A Metabolic Shift Favoring Sphingosine 1-Phosphate at the Expense of Ceramide Controls Glioblastoma Angiogenesis. J Biol Chem (2013) 288:37355–64. doi: 10.1074/jbc.M113.494740
135. Li J, Guan HY, Gong LY, Song LB, Zhang N, Wu J, et al. Clinical Significance of Sphingosine Kinase-1 Expression in Human Astrocytomas Progression and Overall Patient Survival. Clin Cancer Res (2008) 14:6996–7003. doi: 10.1158/1078-0432.CCR-08-0754
136. Van Brocklyn JR, Jackson CA, Pearl DK, Kotur MS, Snyder PJ, Prior TW. Sphingosine Kinase-1 Expression Correlates With Poor Survival of Patients With Glioblastoma Multiforme: Roles of Sphingosine Kinase Isoforms in Growth of Glioblastoma Cell Lines. J Neuropathol Exp Neurol (2005) 64:695–705. doi: 10.1097/01.jnen.0000175329.59092.2c
137. Steck PA, Ligon AH, Cheong P, Yung WK, Pershouse MA. Two Tumor Suppressive Loci on Chromosome 10 Involved in Human Glioblastomas. Genes Chromosomes Cancer (1995) 12:255–61. doi: 10.1002/gcc.2870120404
138. Ogawa C, Kihara A, Gokoh M, Igarashi Y. Identification and Characterization of a Novel Human Sphingosine-1-Phosphate Phosphohydrolase, hSPP2. J Biol Chem (2003) 278:1268–72. doi: 10.1074/jbc.M209514200
139. Jensen SA, Calvert AE, Volpert G, Kouri FM, Hurley LA, Luciano JP, et al. Bcl2L13 Is a Ceramide Synthase Inhibitor in Glioblastoma. Proc Natl Acad Sci U S A (2014) 111:5682–7. doi: 10.1073/pnas.1316700111
140. Riccitelli E, Giussani P, Di Vito C, Condomitti G, Tringali C, Caroli M, et al. Extracellular Sphingosine-1-Phosphate: A Novel Actor in Human Glioblastoma Stem Cell Survival. PloS One (2013) 8:e68229. doi: 10.1371/journal.pone.0068229
141. Edsall LC, Cuvillier O, Twitty S, Spiegel S, Milstien S. Sphingosine Kinase Expression Regulates Apoptosis and Caspase Activation in PC12 Cells. J Neurochem (2001) 76:1573–84. doi: 10.1046/j.1471-4159.2001.00164.x
142. Bouquerel P, Gstalder C, Muller D, Laurent J, Brizuela L, Sabbadini RA, et al. Essential Role for SphK1/S1P Signaling to Regulate Hypoxia-Inducible Factor 2alpha Expression and Activity in Cancer. Oncogenesis (2016) 5:e209. doi: 10.1038/oncsis.2016.13
143. Paugh BS, Bryan L, Paugh SW, Wilczynska KM, Alvarez SM, Singh SK, et al. Interleukin-1 Regulates the Expression of Sphingosine Kinase 1 in Glioblastoma Cells. J Biol Chem (2009) 284:3408–17. doi: 10.1074/jbc.M807170200
144. Chongsathidkiet P, Jackson C, Koyama S, Loebel F, Cui X, Farber SH, et al. Sequestration of T Cells in Bone Marrow in the Setting of Glioblastoma and Other Intracranial Tumors. Nat Med (2018) 24:1459–68. doi: 10.1038/s41591-018-0135-2
145. Gude DR, Alvarez SE, Paugh SW, Mitra P, Yu J, Griffiths R, et al. Apoptosis Induces Expression of Sphingosine Kinase 1 to Release Sphingosine-1-Phosphate as a “Come-and-Get-Me” Signal. FASEB J (2008) 22:2629–38. doi: 10.1096/fj.08-107169
146. Perrotta C, Cervia D, Di Renzo I, Moscheni C, Bassi MT, Campana L, et al. Nitric Oxide Generated by Tumor-Associated Macrophages Is Responsible for Cancer Resistance to Cisplatin and Correlated With Syntaxin 4 and Acid Sphingomyelinase Inhibition. Front Immunol (2018) 9:1186. doi: 10.3389/fimmu.2018.01186
147. Fecci PE, Mitchell DA, Whitesides JF, Xie W, Friedman AH, Archer GE, et al. Increased Regulatory T-Cell Fraction Amidst a Diminished CD4 Compartment Explains Cellular Immune Defects in Patients With Malignant Glioma. Cancer Res (2006) 66:3294–302. doi: 10.1158/0008-5472.CAN-05-3773
148. Bai A, Guo Y. Acid Sphingomyelinase Mediates Human CD4(+) T-Cell Signaling: Potential Roles in T-Cell Responses and Diseases. Cell Death Dis (2017) 8:e2963. doi: 10.1038/cddis.2017.360
149. Zhou Y, Salker MS, Walker B, Munzer P, Borst O, Gawaz M, et al. Acid Sphingomyelinase (ASM) Is a Negative Regulator of Regulatory T Cell (Treg) Development. Cell Physiol Biochem (2016) 39:985–95. doi: 10.1159/000447806
150. Woroniecka KI, Rhodin KE, Dechant C, Cui X, Chongsathidkiet P, Wilkinson D, et al. 4-1BB Agonism Averts TIL Exhaustion and Licenses PD-1 Blockade in Glioblastoma and Other Intracranial Cancers. Clin Cancer Res (2020) 26:1349–58. doi: 10.1158/1078-0432.CCR-19-1068
151. Pitman MR, Woodcock JM, Lopez AF, Pitson SM. Molecular Targets of FTY720 (Fingolimod). Curr Mol Med (2012) 12:1207–19. doi: 10.2174/156652412803833599
152. Baeyens A, Bracero S, Chaluvadi VS, Khodadadi-Jamayran A, Cammer M, Schwab SR, et al. Monocyte-Derived S1P in the Lymph Node Regulates Immune Responses. Nature (2021) 592:290–5. doi: 10.1038/s41586-021-03227-6
153. Jansa P, Mason SW, Hoffmann-Rohrer U, Grummt I. Cloning and Functional Characterization of PTRF, a Novel Protein Which Induces Dissociation of Paused Ternary Transcription Complexes. EMBO J (1998) 17:2855–64. doi: 10.1093/emboj/17.10.2855
154. Mason SW, Sander EE, Grummt I. Identification of a Transcript Release Activity Acting on Ternary Transcription Complexes Containing Murine RNA Polymerase I. EMBO J (1997) 16:163–72. doi: 10.1093/emboj/16.1.163
155. Wang X, Liu T, Bai Y, Liao H, Qiu S, Chang Z, et al. Polymerase I and Transcript Release Factor Acts as an Essential Modulator of Glioblastoma Chemoresistance. PloS One (2014) 9:e93439. doi: 10.1371/journal.pone.0093439
156. Hill MM, Bastiani M, Luetterforst R, Kirkham M, Kirkham A, Nixon SJ, et al. PTRF-Cavin, a Conserved Cytoplasmic Protein Required for Caveola Formation and Function. Cell (2008) 132:113–24. doi: 10.1016/j.cell.2007.11.042
157. Patni N, Garg A. Congenital Generalized Lipodystrophies–New Insights Into Metabolic Dysfunction. Nat Rev Endocrinol (2015) 11:522–34. doi: 10.1038/nrendo.2015.123
158. Hayashi YK, Matsuda C, Ogawa M, Goto K, Tominaga K, Mitsuhashi S, et al. Human PTRF Mutations Cause Secondary Deficiency of Caveolins Resulting in Muscular Dystrophy With Generalized Lipodystrophy. J Clin Invest (2009) 119:2623–33. doi: 10.1172/JCI38660
159. Huang K, Fang C, Yi K, Liu X, Qi H, Tan Y, et al. The Role of PTRF/Cavin1 as a Biomarker in Both Glioma and Serum Exosomes. Theranostics (2018) 8:1540–57. doi: 10.7150/thno.22952
160. Guo Q, Guan GF, Cheng W, Zou CY, Zhu C, Cheng P, et al. Integrated Profiling Identifies Caveolae-Associated Protein 1 as a Prognostic Biomarker of Malignancy in Glioblastoma Patients. CNS Neurosci Ther (2019) 25:343–54. doi: 10.1111/cns.13072
161. Yi K, Zhan Q, Wang Q, Tan Y, Fang C, Wang Y, et al. PTRF/Cavin-1 Remodels Phospholipid Metabolism to Promote Tumor Proliferation and Suppress Immune Responses in Glioblastoma by Stabilizing cPLA2. Neuro Oncol (2021) 23:387–99. doi: 10.1093/neuonc/noaa255
162. Chen W, Peng Y, Jiang X, Zhao J, Zhao H, Zhu Y. Isomers Identification of 2-Hydroxyglutarate Acid Disodium Salt (2HG) by Terahertz Time-Domain Spectroscopy. Sci Rep (2017) 7:12166. doi: 10.1038/s41598-017-11527-z
163. Chaumeil MM, Radoul M, Najac C, Eriksson P, Viswanath P, Blough MD, et al. Hyperpolarized (13)C MR Imaging Detects No Lactate Production in Mutant IDH1 Gliomas: Implications for Diagnosis and Response Monitoring. NeuroImage Clin (2016) 12:180–9. doi: 10.1016/j.nicl.2016.06.018
164. Wang Y-L, Yao J, Chakhoyan A, Raymond C, Salamon N, Liau LM, et al. Association Between Tumor Acidity and Hypervascularity in Human Gliomas Using pH-weighted Amine Chemical Exchange Saturation Transfer Echo-Planar Imaging and Dynamic Susceptibility Contrast Perfusion MRI at 3T. Am J Neuroradiol (2019) 40:979–86. doi: 10.3174/ajnr.A6063
165. Hayes AR, Jayamanne D, Hsiao E, Schembri GP, Bailey DL, Roach PJ, et al. Utilizing 18F-Fluoroethyltyrosine (FET) Positron Emission Tomography (PET) to Define Suspected Nonenhancing Tumor for Radiation Therapy Planning of Glioblastoma. Pract Radiat Oncol (2018) 8:230–8. doi: 10.1016/j.prro.2018.01.006
166. Friebel E, Kapolou K, Unger S, Núñez NG, Utz S, Rushing EJ, et al. Single-Cell Mapping of Human Brain Cancer Reveals Tumor-Specific Instruction of Tissue-Invading Leukocytes. Cell (2020) 181:1626–42.e1620. doi: 10.1016/j.cell.2020.04.055
Keywords: glioblastoma, immunotherapy, metabolism, immunometabolism, tryptophan, arginine, 2HG, adenosine
Citation: Mohan AA, Tomaszewski WH, Haskell-Mendoza AP, Hotchkiss KM, Singh K, Reedy JL, Fecci PE, Sampson JH and Khasraw M (2021) Targeting Immunometabolism in Glioblastoma. Front. Oncol. 11:696402. doi: 10.3389/fonc.2021.696402
Received: 16 April 2021; Accepted: 26 May 2021;
Published: 16 June 2021.
Edited by:
Terrance Johns, University of Western Australia, AustraliaReviewed by:
Lukas Bunse, German Cancer Research Center (DKFZ), GermanySerena Pellegatta, Fondazione Istituto Neurologio Carlo Besta (IRCCS), Italy
Copyright © 2021 Mohan, Tomaszewski, Haskell-Mendoza, Hotchkiss, Singh, Reedy, Fecci, Sampson and Khasraw. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mustafa Khasraw, bXVzdGFmYS5raGFzcmF3QGR1a2UuZWR1