 Marion Grard1,2†Camille Chatelain1,2†Tiphaine Delaunay1,2
Marion Grard1,2†Camille Chatelain1,2†Tiphaine Delaunay1,2 Elvire Pons-Tostivint1,2,3
Elvire Pons-Tostivint1,2,3 Jaafar Bennouna1,2,3
Jaafar Bennouna1,2,3 Jean-François Fonteneau1,2*
Jean-François Fonteneau1,2*- 1Université de Nantes, Inserm, CRCINA, Nantes, France
- 2Labex IGO, Immunology Graft Oncology, Nantes, France
- 3CHU de Nantes, oncologie thoracique et digestive, Université de Nantes, Nantes, France
Homozygous deletion (HD) of the tumor suppressor gene CDKN2A is the most frequent genetic alteration in malignant pleural mesothelioma and is also frequent in non-small cell lung cancers. This HD is often accompanied by the HD of the type I interferons (IFN I) genes that are located closed to the CDKN2A gene on the p21.3 region of chromosome 9. IFN I genes encode sixteen cytokines (IFN-α, IFN-β…) that are implicated in cellular antiviral and antitumor defense and in the induction of the immune response. In this review, we discuss the potential influence of IFN I genes HD on thoracic cancers therapy and speak in favor of better taking these HD into account in patients monitoring.
Frequent Homozygous Co-Deletion of the CDKN2A Tumor Suppressor Gene and the IFN I Genes in Thoracic Cancers
Non-small cell lung cancer (NSCLC) is the most common cause of cancer death worldwide often due to long-term tobacco smoking. Malignant pleural mesothelioma (MPM) is a rare cancer that is mainly due to asbestos exposure. As other cancers, some genomic alterations are found in NSCLC and MPM tumor cells, especially in locus containing tumor suppressor genes. These alterations are in part responsible for the disease.
In MPM cells, the most frequent genomic alteration is the homologous deletion (HD) in the p21.3 region of chromosome 9 (1–6). These HDs are variable in length but they mainly overlap at the level of the cyclin-dependent kinase inhibitor 2A (CDKN2A) tumor suppressor gene located in this region (Figures 1A, B). Fluorescence in situ hybridization (FISH) studies reported that CDKN2A gene HDs are found in 60 to 80% of patients (3–6). Copy number alteration study from The Cancer Genome Atlas (TCGA) reported a lower frequency of 44% of patients with CDKN2A gene HD in MPM (Figure 1B) (7). However, TCGA study is performed on tumor biopsies that often contain non-malignant cells. These non-malignant cells may mask CDKN2A gene HD that are only present in tumor cells. Thus, some patients with CDKN2A gene HD were probably not detected in the TCGA study.
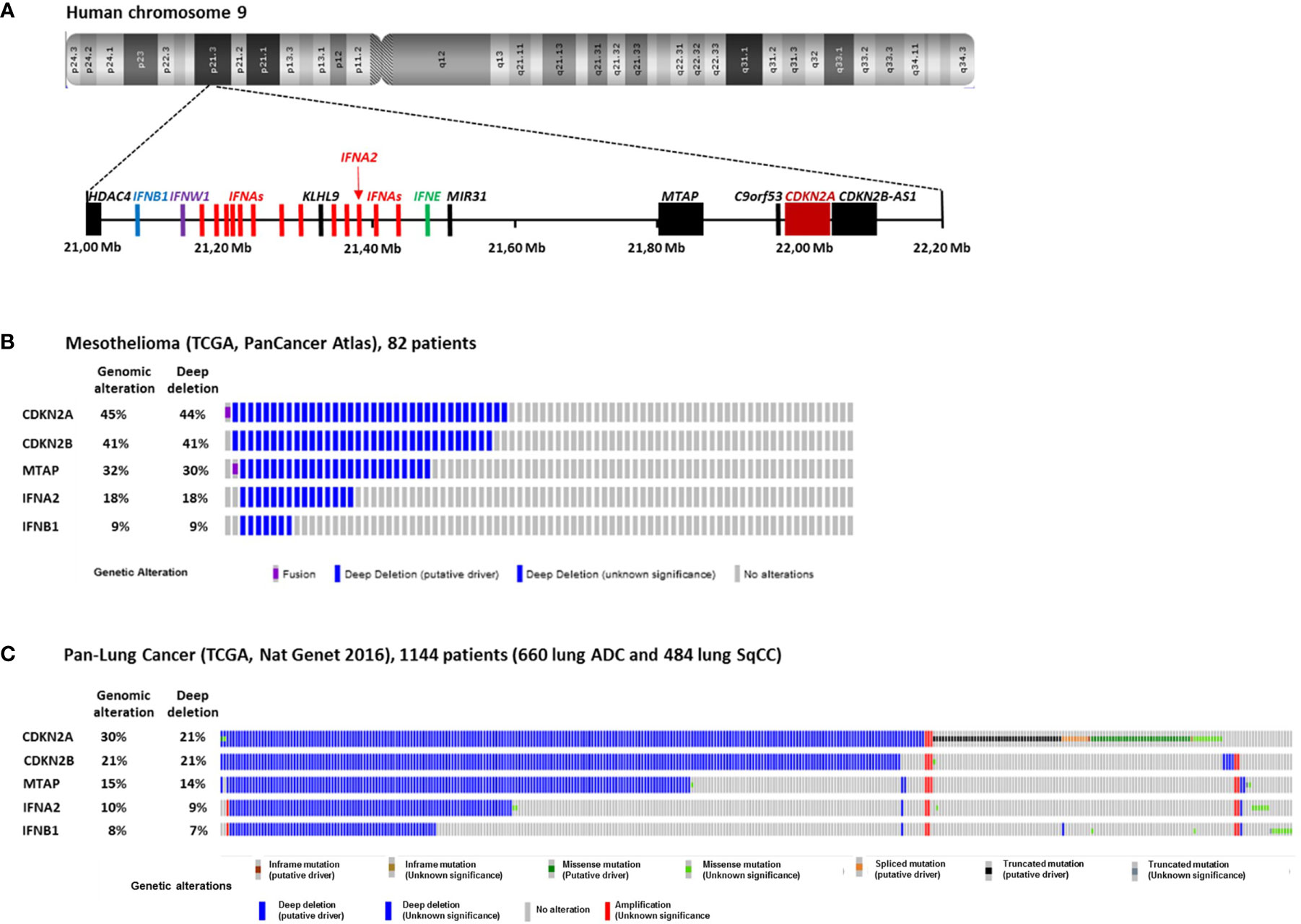
Figure 1 Homozygous Deletions in the p21.3 region of chromosome 9 in MPM and NSCLC. (A) Schematic representation of genes present in the p21.3 region of chromosome 9 between positions 21,000,000 and 22,200,000 drawn from UCSC Genome Browser (https://genome.ucsc.edu/). (B) Oncoprint representation of CDKN2A, CDKN2B, MTAP, IFNA2 and IFNB1 genomic alterations found in tumor samples of 82 MPM patients. Oncoprint was performed with cbioportals website (http://www.cbioportal.org/) using TCGA Pancancer atlas data. (C) Oncoprint representation of CDKN2A, CDKN2B, MTAP, IFNA2 and IFNB1 genomic alterations found in tumor samples of 1144 NSCLC patients. Oncoprint was performed with cbioportals website (http://www.cbioportal.org/) using TCGA Pan lung cancer data. Only the patients with at least one genomic alteration in the five genes are shown. ADC, adenocarcinoma; SqCC, squamous cell carcinoma.
In NSCLC, CDKN2A gene HDs were also identified in the 1990s (8–10). They were then found more frequently in a subset of patient with intact retinoblastoma (rb) pathway (11). FISH studies on 85 and 19 NSCLC patients reported CDKN2A gene HD in 21 and 29% of patients respectively (12, 13), and FISH study on 31 squamous cell carcinoma (SqCC) patients reported them in 16% (14). TCGA study on 1144 NSCLC patients (660 lung adenocarcinoma and 484 lung SqCC) reported CDKN2A gene HD in 21% of patients (Figure 1C) (15).
The CDKN2A gene encodes several proteins, notably p16INK4a and p14arf that are implicated in the regulation of the cell cycle. p16INK4a binds to cyclin-dependent kinase 4 and 6 (CDK4/6) and inhibits its capacity with cyclin D1 to phosphorylate rb protein and the translocation of the transcription factor E2F from the cytoplasm to the nucleus (16). In absence of p16INK4a, E2F translocates to the nucleus and allows the transition from G1 phase to S phase of the cell cycle. p14arf also acts as a tumor suppressor via the p53 pathway and its absence favors the entry in the cell cycle.
Close to CDKN2A gene, CDKN2B and MTAP are two other genes that are often co-deleted with CDKN2A in MPM (Figure 1B) and NSCLC (Figure 1C). CDKN2B encode the p15Ink4b protein that interacts with CDK4/6 and inhibits its activation by cyclin D and thus acts as a tumor suppressor (17). MTAP encodes the S-methyl-5’-thioadenosine phosphorylase (MTAP) implicated in the polyamine metabolism (18).
Further downstream from CDKN2A and MTAP in the p21.3 region of human chromosome 9, a cluster of 16 genes encodes the type I interferons (IFN I): IFN-β, IFN-ε, IFN-ω and 13 IFN-α (Figure 1A) (19). IFNB1 is the furthest gene from CDKN2A. In the 1990s, IFN I genes HDs were identified in a fraction of NSCLC and MPM patients with CDKN2A gene HD (1, 2, 8, 20). We recently reported that in 78 short-term–cultured MPM cell lines, 57 (73%) and 18 (23%) cell lines harbors CDKN2A and IFNBI genes HD respectively, whereas in TCGA study performed on 82 patients, these percentage where smaller, probably due to non-malignant cells contamination (44 and 9%) (Figure 1B) (21). Thus, about 10 to 20% of mesothelioma patients present HD of all the IFN I genes. In NSCLC, the TCGA study on 1,144 patients reported 21 and 7% of patients with CDKN2A and IFNBI gene HDs respectively (Figure 1C). Interestingly, NSCLC patients with IFN I and CDKN2A gene HDs have a significantly worst disease free survival than patients with only CDKN2A gene HD (22), suggesting a tumor suppressor role for IFN I genes in this cancer.
The Type I Interferon Response in Thoracic Cancers
Type I interferon (IFN I) response is key in antiviral immune response (Figure 2). The IFN I response allows infected and immune cells to report via IFN-α and -β secretion the presence of the virus to neighboring cells and to the immune system via the IFN-α/-β receptor (IFNAR) which is expressed by virtually all somatic cells (23). The presence of viral genome or intermediaries of its replication is detected by cytoplasmic pattern recognition receptors (PRR) and lead to the production of IFN I via two main pathways: the stimulator of interferon genes protein (STING) pathway for DNA viruses and the mitochondrial antiviral-signaling protein (MAVS) pathway for RNA viruses (23, 24). IFN I are also produced by immune cells notably via Toll like receptor (TLR) activation, especially plasmacytoid dendritic cells (pDC) (25). IFN-β is expressed by all nucleated cells in response to infection, whereas the IFN-α are mainly produced by immune cells. Among IFN-α, IFN-α2 was the first cytokines to be approved in clinics for cancer treatment in 1986 and is the most studied (26).
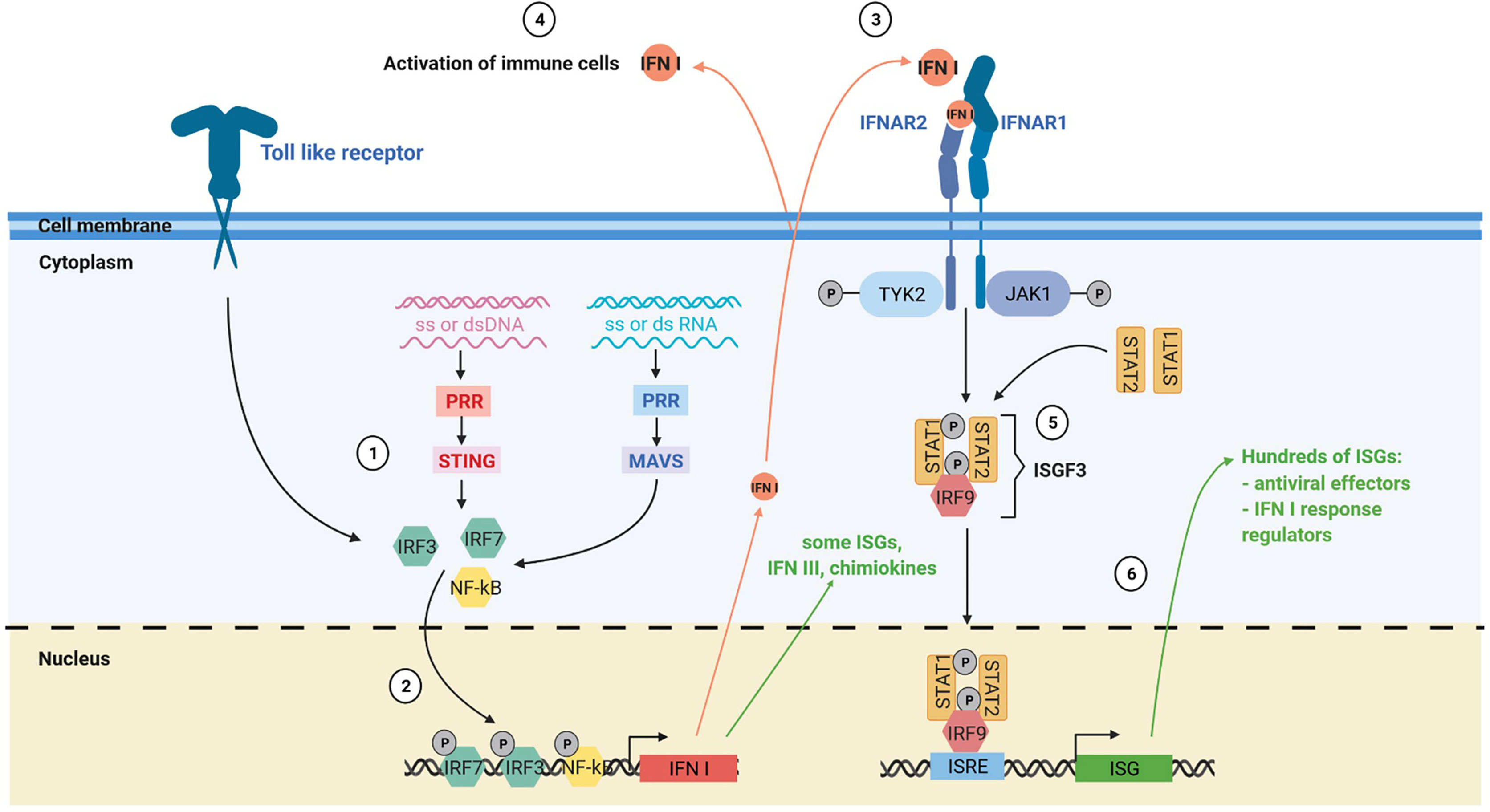
Figure 2 The IFN I response. (1) The IFN I response is triggered by different stimuli such as ssDNA, dsDNA, ssRNA and dsRNA via the toll like receptors, the MAVS and the STING pathway. (2) Activation of these pathways induces the nuclear translocation of transcription factors such as IRF3, IRF7 and NF-kB that trigger IFN I production and expression of some interferon stimulated genes (ISGs). (3) Secreted IFN I trigger IFNAR signaling on neighboring cells, notably immune cells. (4) IFN I and other soluble factors of the IFN I response activate the immune response. (5) Activation of IFNAR signaling by IFN I leads to the formation of the interferon stimulated gene factor 3 complex. (6) This complex translocates to the nucleus and activates numerous ISGs with antiviral, immunomodulatory and regulatory functions. PRR, pattern recognition receptors.
Cells exposed to IFN I express hundreds of IFN-stimulated genes (ISGs). Many ISGs encode proteins that induce a state of anti-viral resistance. These antiviral proteins act by blocking the different stages of the viral cycle, from the entry of the virus, through the inhibition of its replication, to the release of its progeny by the infected cell (23, 27). The IFN I also play a crucial role in the induction and the regulation of the antiviral adaptive immune response, notably by favoring antigen cross-priming by dendritic cells (28, 29). However, during chronic infection, prolonged IFN I response can have deleterious effects by inducing immune dysfunctions (30).
IFN I response is often induced during cancer development and treatments. Several pathways are involved in this induction. Presence of mitochondrial or nuclear DNA in the cytoplasm of tumor cells can induce the secretion of IFN I via the STING pathway (31–39). Expression of endogenous retrovirus (ERV) under the form of dsRNA due to epigenetic deregulation in tumor cells can also trigger the expression of IFN I via the MAVS pathway (35, 40–44). Non-malignant cells from the tumor microenvironment, such as phagocytic cells notably dendritic cells (DC), can also produce IFN I via activation of the STING pathway after engulfment of dead tumor cells. This occurs due to accumulation of DNA from engulfed dead tumor cells in DC cytoplasm (39, 45–47). A recent analysis of 31 cancer types in TCGA database by Liu et al. shows that, lung adenocarcinoma, MPM and SqCC are the 3rd, 6th and 8th respectively in the intensity of an IFN I signature based on the expression of 38 ISGs (35). Globally, IFN I signature correlates with the degree of immune cells infiltration, but some tumors with an interferon signature and with no immune cells infiltration were also found.
IFN I signaling is able to modulate the expressions of hundreds of genes (27). This signaling pathway also induces expression of noncoding RNAs including long noncoding RNAs, miRNA and ERV RNA (27, 41, 48). Due to its potential toxicity and inflammatory effects, IFN I production and signaling are tightly regulated by numerous positive and negative regulators, many of which are ISGs (49). Thus, induction of IFN I response in tumors has multiple complex effects that are rather unfavorable to tumor development. The IFN I can restrict tumor growth by reducing proliferation of tumor cells, inducing their apoptosis, limiting their migratory capacity and inhibiting angiogenesis (50, 51). Furthermore, they increase antigen presentation by HLA molecules, stimulate the innate and adaptive antitumor immune response and inhibit CD4+ regulatory T cells (30, 50, 52–56). IFN I were shown to play an important role in tumor immuno-editing in mouse models of chemically-induced and transposable tumors (57). They signal tumor cells to the immune system. They are necessary for the priming of anti-tumor T cell response as the abrogation of IFN I signaling in CD8+ dendritic cells blocks their capacity to cross-present antigens in mouse (54, 55). They also participate in the activation of anti-tumor NK cell response (56). Induction of anti-tumor NK and T cell responses lead to the secretions of the type II interferon-gamma (IFN-γ) that will further shapes the immunogenicity and the immunosuppressive microenvironment of the tumor with the IFN I (58). Indeed, IFN I are also involved in setting up the immunosuppressive tumor environment by inducing the expression of numerous inhibitory molecules such as PD1 and PDL1 that block CD8+ T cell cytotoxicity (30, 50, 58). IFN I can also induce Indoleamine 2,3-dioxygenase (IDO) expression that reduces locally the amount of tryptophan needed for T cell functions and favors their differentiation in Treg (59). Thus, the IFN I in tumors play a dual role by stimulating the innate and adaptive immune response and inducing feedback mechanisms to control its magnitude.
IFN I in NSCLC modulates numerous pathways implicated in proliferation, survival and apoptosis of tumor cells including JAK/STAT, Src kinases, Vav proto-oncogene, PTEN/PI3-K/AKT, Crk proteins and MAP kinase signaling pathways (51). Several recent studies reported that a constitutive activation of the IFN I response in lung tumors correlates with tumor inflammation and immune checkpoint inhibitors efficacy (60–62). Furthermore, DNA damages and dysfunctions of the DNA damage response are inducers of the IFN I response and have been linked to immune checkpoint efficiency (37, 63). The renewed interest in IFN I response also comes from the observation that they are necessary for the radiotherapy abscopal response (64–67), and that they participate in the induction of the antitumor immune response by chemotherapy (68). This is also due to the identification of new potential immune checkpoint such as ADAR and Trex1 which are ISG that functions as negative regulators of the IFN I response by inactivating the nucleic acids that stimulate this response (35, 40, 65, 66). By blocking ADAR or Trex function, IFN I response is amplified which promotes the antitumor immune response. Thus, there is still a great interest to adapt treatments or find new therapeutic strategies to activate the IFN I response locally especially in cold tumors with no or low immune cells infiltrate.
Potential Consequences of IFN I Genes HD for Thoracic Cancers Therapy
Given the central role of IFN I response in tumor immune surveillance, the frequent loss of all copies of IFN I genes that accompanied CDKN2A gene HD in tumor cells likely plays a role in tumor immune escape. In NSCLC, like in other cancers, patients with only CDKN2A gene HD have a longer survival compared to patients with IFN I and CDKN2A genes HD (22). Thus, IFN I genes act as tumor suppressors genes in malignant cells. Beside this study of Ye et al., nothing is known on the prognostic value and immunotherapy biomarker potential of IFN I genes HD in NSCLC, MPM and other cancers. These deletions were described as early as the mid-1990s and yet they have not been well documented. It may be because they were discovered in studies focusing on the CDKN2A tumor suppressor gene HD. Furthermore, reports that IFN I treatment in NSCLC and MPM has limited clinical benefit at that time, may have decrease the interest of studying IFN I gene HD. Finally, most studies on the role of IFN I on antitumor immune response were performed with IFNARko mouse models that are easier to obtain than mouse models ko for all IFN I genes. These IFNARko models are instrumental to understand the role of IFN I signaling on tumor cells and the different subtypes of immune cells but are less suitable to study the source of the IFN I production.
Questions arise regarding presence of IFN I genes HD in tumor cells. There are several potential cellular sources of IFN I secretion in tumors, that are basically tumor and immune cells. Thus, the first question is the role of IFN I production by tumor cells and if absence of this production is compensated by other cellular sources. Several recent studies suggest that triggering of the tumor cells IFN I production play a role in the induction of the anti-tumor immune response.
Kitajima et al. reported that the lack of response to immune checkpoint blockade (ICB) of patients with KRAS-LKB1–mutant lung cancers is due to the inhibition of STING expression via the loss of LKB1 (62). They show that KRAS-LKB1–mutant tumor cells are not able to sense cytoplasmic dsDNA via the STING pathway, and to produce IFN I in response. In consequence, T cells infiltration and PD-L1 expression in KRAS-LKB1-mutant tumors is reduced and ICB therapy is ineffective.
Demaria’s team showed that triggering of tumor cell IFN I response is necessary for induction of anti-tumor immune response by radiotherapy (64, 65). They first reported that abscopal response in mouse is abrogated when cancer cells in the irradiated tumor do not express cGAS/STING or overexpress the exonuclease Trex1 (65). Irradiation induces the presence of cytoplasmic DNA that triggers the IFN-β production via the STING pathway and lead to the expression of the ISG Trex1. This ISG is an exonuclease that degrades DNA in the cytoplasm and thus decreases IFN I response by tumor cells and triggering of the antitumor immune response. By inactivating Trex1 in tumor cells, the IFN I response induced by irradiation is increased, as well as the antitumor immune response. Thus, tumor cell IFN I response is essential for abscopal effect of radiation in that mouse model. Demaria’s team then showed that combination of radiotherapy and anti-CTLA4 blockade in NSCLC patients that have failed anti-CTLA4 alone or in combination with chemotherapy, induced IFN-β in the blood and an antitumor T cell response in responding patients (64).
These studies highlight the important role on the antitumor immune response of triggering the IFN I response via the STING pathway in tumor cells. However, IFN I response in tumor cells can also be induced by the sensing of endogenous dsRNA via the MAVS pathway and that also plays a role in the stimulation of the antitumor immune response (35, 40–44). Best evidences come from the study of an ISG, the adenosine deaminase acting on RNA (ADAR) that acts on the MAVS pathway like Trex1 does on the STING pathway (35, 40). The ADAR protein, by converting A to I, disrupts the normal A:U pairing which destabilizes the dsRNA into ssRNA. dsRNA edited by ADAR are no longer able to trigger the IFN I response by the dsRNA cytoplasmic sensor Mda5. Thus like Trex1, ADAR inactivates the stimuli at the origin of the IFN I production by tumor cells. In a mouse model, Ishizuka et al. reported that loss of ADAR1 in tumor cells overcomes the resistance to immune checkpoint inhibitors by increasing the IFN I response via the MAVS pathway and, thus, the inflammation of the tumor microenvironment (40). This results was confirmed by Liu et al., that show that both, the MAVS and the STING pathway are needed to maintain the IFN I response in tumor cells that have lost ADAR (35).
Altogether these studies on the STING and the MAVS pathway show that triggering of the IFN I response in tumor cells is central to inflame the microenvironment. However, it does not establish clearly that IFN I production by tumor cells is required. Indeed, in these studies, the MAVS or the STING pathway is inactivated. This inactivation impairs IFN I production and also the expression of lots of other genes. Indeed, when the MAVS or the STING pathway are triggered, activated transcription factors such as IRF3 and NF-kB not only induce IFN I production, but also the expression of many other genes with many being ISG (Figure 2). In MPM cell lines that have lost IFN I genes, exposition to attenuated measles virus still resulted in the induction of expression of a small subset of genes (21). Among these genes, some may play a role in the inflammation of the microenvironment, such as the chemokines CCL5, CXCL10 and CXCL11, or the type III interferons. Thus, triggering of the IFN I response in tumor cells that have lost IFN I genes may still conserve a certain capacity to inflame the microenvironment. By studying patients with IFN I genes HD tumors, we would better understand the IFN I contribution of tumor cells versus non-malignant cells in cancer development and therapies. We would also better define the contribution of IFN I versus other cytokines/chemokines induced by the triggering of the MAVS or the STING pathway.
IFN I genes HD may also be interesting for new cancer therapies such as antitumor virotherapy using oncolytic replicative viruses. We studied the replication and oncolytic activity of the attenuated Schwarz strain of measles virus (MV) on 22 human MPM cell lines and four types of healthy cell (fibroblasts, mesothelial, endothelial and lung epithelial cells) (69). We found that the healthy cells and seven MPM cell lines were resistant to MV replication due to a protective functional IFN I response. The 15 others MPM cell lines were permissive to MV replication and lysis due to a defective IFN I response. Among these 15 cell lines, 11 were unable to produce IFN I when exposed to MV. We showed later that eight of these 11 cell lines have lost both copies of the IFN I genes (21). The three others cells line have at least one copy of IFN I genes but are not able to produce IFN I in response to the virus suggesting another type of defects of the IFN I response in these MPM cells lines (69). These 11 MV-sensitive MPM cell lines that are unable to produce IFN I in response to MV become MV-resistant if exposed to exogenous IFN I, suggesting that IFNAR signaling is functional in these cell lines. The four other MV-sensitive MPM cell lines were able to produce IFN I in response to MV, but unable to control viral replication suggesting a defect of the IFN I response in IFNAR signaling. This defective IFNAR signaling has been previously reported in tumor cells of some patients with MPM (70). It has been associated to mark decrease of IFNAR, IRF9 and PKR expression and to tumor sensitivity to an oncolytic vesicular stomatitis virus. These studies illustrate the diversity of defects found in the IFN I response from one patients to another in MPM. Such converging selection of tumor cells with a deficient IFN I response highlights the tumor suppressive role of this response.
Other questions are still pending regarding IFN I genes HD. These HD are diverse in length (Figure 2). For some patients, only a part of IFN I genes are lost and IFNB1 gene that encodes IFN-β is preserved (Figure 1). Consequences of these partial losses are also to define. During tumor development, do these HD appear concomitantly to CDKN2A HD or do they appear later conferring an additional advantage to the tumor variant that carries them? The best techniques for detecting IFN I genes HD is probably FISH assay that can be performed cost effectively on paraffin-embedded tissue and allow to identify homozygous and hemizygous deletions at the single cell level (71). It can also be performed by polymerase chain reaction-based techniques or whole exome sequencing on tumor biopsies, but large amount of non-malignant cells in the biopsy may hide the deletions.
With the success of cancer immunotherapy and recent advances in understanding the IFN I tumor suppressor role, IFN I genes HD should be studied and taken into account in the monitoring of MPM and NSCLC patients. This would likely lead to new strategies and improvements of immunotherapy.
Author Contributions
Literature review: MG, CC, and JFF. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by “La Ligue Régionale Grand Ouest contre le Cancer” (CSIRGO: CD16, CD22, CD44, CD49, CD72, CD79 and CD85), “La Ligue Nationale contre le Cancer”, “L’association ARSMESO44 “, “La fondation ARC”, L’Agence Nationale pour la Recherche (ANR-16-CE18-0016), and “LabEX IGO program supported by the National Research Agency via the investment of the future program ANR-11-LABX-0016-01”. TD was supported by a grant from Ligue contre le Cancer.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Dr. Didier Jean for his insightful discussion. Figure 2 was created with BioRender.com.
References
1. Cheng JQ, Jhanwar SC, Klein WM, Bell DW, Lee WC, Altomare DA, et al. p16 Alterations and Deletion Mapping of 9p21-p22 in Malignant Mesothelioma. Cancer Res (1994) 54(21):5547–51.
2. Xio S, Li D, Vijg J, Sugarbaker DJ, Corson JM, Fletcher JA. Codeletion of p15 and p16 in Primary Malignant Mesothelioma. Oncogene (1995) 11(3):511–5.
3. Cheng YY, Yuen ML, Rath EM, Johnson B, Zhuang L, Yu TK, et al. CDKN2A and MTAP Are Useful Biomarkers Detectable by Droplet Digital PCR in Malignant Pleural Mesothelioma: A Potential Alternative Method in Diagnosis Compared to Fluorescence in Situ Hybridisation. Front Oncol (2020) 10:579327. doi: 10.3389/fonc.2020.579327
4. Illei PB, Rusch VW, Zakowski MF, Ladanyi M. Homozygous Deletion of CDKN2A and Codeletion of the Methylthioadenosine Phosphorylase Gene in the Majority of Pleural Mesotheliomas. Clin Cancer Res (2003) 9(6):2108–13.
5. Hwang HC, Sheffield BS, Rodriguez S, Thompson K, Tse CH, Gown AM, et al. Utility of BAP1 Immunohistochemistry and p16 (Cdkn2a) FISH in the Diagnosis of Malignant Mesothelioma in Effusion Cytology Specimens. Am J Surg Pathol (2016) 40(1):120–6. doi: 10.1097/PAS.0000000000000529
6. Marshall K, Jackson S, Jones J, Holme J, Lyons J, Barrett E, et al. Homozygous Deletion of CDKN2A in Malignant Mesothelioma: Diagnostic Utility, Patient Characteristics and Survival in a UK Mesothelioma Centre. Lung Cancer (2020) 150:195–200. doi: 10.1016/j.lungcan.2020.10.020
7. Hmeljak J, Sanchez-Vega F, Hoadley KA, Shih J, Stewart C, Heiman D, et al. Integrative Molecular Characterization of Malignant Pleural Mesothelioma. Cancer Discovery (2018) 8(12):1548–65. doi: 10.1158/2159-8290.CD-18-0804
8. Dreyling MH, Bohlander SK, Adeyanju MO, Olopade OI. Detection of CDKN2 Deletions in Tumor Cell Lines and Primary Glioma by Interphase Fluorescence in Situ Hybridization. Cancer Res (1995) 55(5):984–8.
9. Kamb A, Gruis NA, Weaver-Feldhaus J, Liu Q, Harshman K, Tavtigian SV, et al. A Cell Cycle Regulator Potentially Involved in Genesis of Many Tumor Types. Science (1994) 264(5157):436–40. doi: 10.1126/science.8153634
10. Nobori T, Miura K, Wu DJ, Lois A, Takabayashi K, Carson DA. Deletions of the Cyclin-Dependent Kinase-4 Inhibitor Gene in Multiple Human Cancers. Nature (1994) 368(6473):753–6. doi: 10.1038/368753a0
11. Shapiro GI, Edwards CD, Kobzik L, Godleski J, Richards W, Sugarbaker DJ, et al. Reciprocal Rb Inactivation and p16INK4 Expression in Primary Lung Cancers and Cell Lines. Cancer Res (1995) 55(3):505–9.
12. Okami K, Cairns P, Westra WH, Linn JF, Ahrendt SA, Wu L, et al. Detailed Deletion Mapping at Chromosome 9p21 in Non-Small Cell Lung Cancer by Microsatellite Analysis and Fluorescence in Situ Hybridization. Int J Cancer (1997) 74(6):588–92. doi: 10.1002/(sici)1097-0215(19971219)74:6<588::aid-ijc5>3.0.co;2-q
13. Panani AD, Maliaga K, Babanaraki A, Bellenis I. Numerical Abnormalities of Chromosome 9 and p16CDKN2A Gene Deletion Detected by FISH in non-Small Cell Lung Cancer. Anticancer Res (2009) 29(11):4483–7.
14. Dessy E, Rossi E, Berenzi A, Tironi A, Benetti A, Grigolato P. Chromosome 9 Instability and Alterations of p16 Gene in Squamous Cell Carcinoma of the Lung and in Adjacent Normal Bronchi: FISH and Immunohistochemical Study. Histopathology (2008) 52(4):475–82. doi: 10.1111/j.1365-2559.2008.02969.x
15. Campbell JD, Alexandrov A, Kim J, Wala J, Berger AH, Pedamallu CS, et al. Distinct Patterns of Somatic Genome Alterations in Lung Adenocarcinomas and Squamous Cell Carcinomas. Nat Genet (2016) 48(6):607–16. doi: 10.1038/ng.3564
16. Burkhart DL, Sage J. Cellular Mechanisms of Tumour Suppression by the Retinoblastoma Gene. Nat Rev Cancer (2008) 8(9):671–82. doi: 10.1038/nrc2399
17. Kim WY, Sharpless NE. The Regulation of INK4/ARF in Cancer and Aging. Cell (2006) 127(2):265–75. doi: 10.1016/j.cell.2006.10.003
18. Urso L, Cavallari I, Sharova E, Ciccarese F, Pasello G, Ciminale V. Metabolic Rewiring and Redox Alterations in Malignant Pleural Mesothelioma. Br J Cancer (2020) 122(1):52–61. doi: 10.1038/s41416-019-0661-9
19. Diaz MO. The Human Type I Interferon Gene Cluster. Semin Virol (1995) 6(3):143–9. doi: 10.1006/smvy.1995.0019
20. Mead LJ, Gillespie MT, Irving LB, Campbell LJ. Homozygous and Hemizygous Deletions of 9p Centromeric to the Interferon Genes in Lung Cancer. Cancer Res (1994) 54(9):2307–9.
21. Delaunay T, Achard C, Boisgerault N, Grard M, Petithomme T, Chatelain C, et al. Frequent Homozygous Deletions of Type I Interferon Genes in Pleural Mesothelioma Confer Sensitivity to Oncolytic Measles Virus. J Thorac Oncol (2020) 15(5):827–42. doi: 10.1016/j.jtho.2019.12.128
22. Ye Z, Dong H, Li Y, Ma T, Huang H, Leong HS, et al. Prevalent Homozygous Deletions of Type I Interferon and Defensin Genes in Human Cancers Associate With Immunotherapy Resistance. Clin Cancer Res (2018) 24(14):3299–308. doi: 10.1158/1078-0432.CCR-17-3008
23. Schneider WM, Chevillotte MD, Rice CM. Interferon-Stimulated Genes: A Complex Web of Host Defenses. Annu Rev Immunol (2014) 32:513–45. doi: 10.1146/annurev-immunol-032713-120231
24. Iwasaki A. A Virological View of Innate Immune Recognition. Annu Rev Microbiol (2012) 66:177–96. doi: 10.1146/annurev-micro-092611-150203
25. Brubaker SW, Bonham KS, Zanoni I, Kagan JC. Innate Immune Pattern Recognition: A Cell Biological Perspective. Annu Rev Immunol (2015) 33:257–90. doi: 10.1146/annurev-immunol-032414-112240
26. Paul F, Pellegrini S, Uze G. Ifna2: The Prototypic Human Alpha Interferon. Gene (2015) 567(2):132–7. doi: 10.1016/j.gene.2015.04.087
27. Schoggins JW. Interferon-Stimulated Genes: What Do They All do? Annu Rev Virol (2019) 6(1):567–84. doi: 10.1146/annurev-virology-092818-015756
28. Crouse J, Kalinke U, Oxenius A. Regulation of Antiviral T Cell Responses by Type I Interferons. Nat Rev Immunol (2015) 15(4):231–42. doi: 10.1038/nri3806
29. Schiavoni G, Mattei F, Gabriele L. Type I Interferons as Stimulators of DC-Mediated Cross-Priming: Impact on Anti-Tumor Response. Front Immunol (2013) 4:483. doi: 10.3389/fimmu.2013.00483
30. Snell LM, McGaha TL, Brooks DG. Type I Interferon in Chronic Virus Infection and Cancer. Trends Immunol (2017) 38(8):542–57. doi: 10.1016/j.it.2017.05.005
31. Mackenzie KJ, Carroll P, Martin CA, Murina O, Fluteau A, Simpson DJ, et al. cGAS Surveillance of Micronuclei Links Genome Instability to Innate Immunity. Nature (2017) 548(7668):461–5. doi: 10.1038/nature23449
32. McArthur K, Whitehead LW, Heddleston JM, Li L, Padman BS, Oorschot V, et al. BAK/BAX Macropores Facilitate Mitochondrial Herniation and mtDNA Efflux During Apoptosis. Science (2018) 359(6378):eaao6047. doi: 10.1126/science.aao6047
33. Harding SM, Benci JL, Irianto J, Discher DE, Minn AJ, Greenberg RA. Mitotic Progression Following DNA Damage Enables Pattern Recognition Within Micronuclei. Nature (2017) 548(7668):466–70. doi: 10.1038/nature23470
34. Marcus A, Mao AJ, Lensink-Vasan M, Wang L, Vance RE, Raulet DH. Tumor-Derived Cgamp Triggers a STING-Mediated Interferon Response in Non-Tumor Cells to Activate the NK Cell Response. Immunity (2018) 49(4):754–63.e4. doi: 10.1016/j.immuni.2018.09.016
35. Liu H, Golji J, Brodeur LK, Chung FS, Chen JT, deBeaumont RS, et al. Tumor-Derived IFN Triggers Chronic Pathway Agonism and Sensitivity to ADAR Loss. Nat Med (2019) 25(1):95–102. doi: 10.1038/s41591-018-0302-5
36. Vanpouille-Box C, Demaria S, Formenti SC, Galluzzi L. Cytosolic DNA Sensing in Organismal Tumor Control. Cancer Cell (2018) 34(3):361–78. doi: 10.1016/j.ccell.2018.05.013
37. Mouw KW, Goldberg MS, Konstantinopoulos PA, D’Andrea AD. Dna Damage and Repair Biomarkers of Immunotherapy Response. Cancer Discovery (2017) 7(7):675–93. doi: 10.1158/2159-8290.CD-17-0226
38. Yamazaki T, Kirchmair A, Sato A, Buque A, Rybstein M, Petroni G, et al. Mitochondrial DNA Drives Abscopal Responses to Radiation That Are Inhibited by Autophagy. Nat Immunol (2020) 21(10):1160–71. doi: 10.1038/s41590-020-0751-0
39. Ahn J, Xia T, Konno H, Konno K, Ruiz P, Barber GN. Inflammation-Driven Carcinogenesis is Mediated Through STING. Nat Commun (2014) 5:5166. doi: 10.1038/ncomms6166
40. Ishizuka JJ, Manguso RT, Cheruiyot CK, Bi K, Panda A, Iracheta-Vellve A, et al. Loss of ADAR1 in Tumours Overcomes Resistance to Immune Checkpoint Blockade. Nature (2019) 565(7737):43–8. doi: 10.1038/s41586-018-0768-9
41. Canadas I, Thummalapalli R, Kim JW, Kitajima S, Jenkins RW, Christensen CL, et al. Tumor Innate Immunity Primed by Specific Interferon-Stimulated Endogenous Retroviruses. Nat Med (2018) 24(8):1143–50. doi: 10.1038/s41591-018-0116-5
42. Panda A, de Cubas AA, Stein M, Riedlinger G, Kra J, Mayer T, et al. Endogenous Retrovirus Expression is Associated With Response to Immune Checkpoint Blockade in Clear Cell Renal Cell Carcinoma. JCI Insight (2020) 5(11):e137569. doi: 10.1172/jci.insight.121522
43. Roulois D, Loo Yau H, Singhania R, Wang Y, Danesh A, Shen SY, et al. Dna-Demethylating Agents Target Colorectal Cancer Cells by Inducing Viral Mimicry by Endogenous Transcripts. Cell (2015) 162(5):961–73. doi: 10.1016/j.cell.2015.07.056
44. Sun S, Frontini F, Qi W, Hariharan A, Ronner M, Wipplinger M, et al. Endogenous Retrovirus Expression Activates Type-I Interferon Signaling in an Experimental Mouse Model of Mesothelioma Development. Cancer Lett (2021) 507:26–38. doi: 10.1016/j.canlet.2021.03.004
45. Woo SR, Fuertes MB, Corrales L, Spranger S, Furdyna MJ, Leung MY, et al. STING-Dependent Cytosolic DNA Sensing Mediates Innate Immune Recognition of Immunogenic Tumors. Immunity (2014) 41(5):830–42. doi: 10.1016/j.immuni.2014.10.017
46. Barber GN. STING: Infection, Inflammation and Cancer. Nat Rev Immunol (2015) 15(12):760–70. doi: 10.1038/nri3921
47. Xu MM, Pu Y, Han D, Shi Y, Cao X, Liang H, et al. Dendritic Cells But Not Macrophages Sense Tumor Mitochondrial DNA for Cross-priming Through Signal Regulatory Protein Alpha Signaling. Immunity (2017) 47(2):363–73.e5. doi: 10.1016/j.immuni.2017.07.016
48. Suarez B, Prats-Mari L, Unfried JP, Fortes P. LncRNAs in the Type I Interferon Antiviral Response. Int J Mol Sci (2020) 21(17):6447. doi: 10.3390/ijms21176447
49. Arimoto KI, Miyauchi S, Stoner SA, Fan JB, Zhang DE. Negative Regulation of Type I IFN Signaling. J Leukoc Biol (2018) 103:1099–116. doi: 10.1002/JLB.2MIR0817-342R
50. Zhou L, Zhang Y, Wang Y, Zhang M, Sun W, Dai T, et al. A Dual Role of Type I Interferons in Antitumor Immunity. Adv Biosyst (2020) 4(11):e1900237. doi: 10.1002/adbi.201900237
51. Galani V, Kastamoulas M, Varouktsi A, Lampri E, Mitselou A, Arvanitis DL. Ifns-Signaling Effects on Lung Cancer: An Up-to-Date Pathways-Specific Review. Clin Exp Med (2017) 17(3):281–9. doi: 10.1007/s10238-016-0432-3
52. Zitvogel L, Galluzzi L, Kepp O, Smyth MJ, Kroemer G. Type I Interferons in Anticancer Immunity. Nat Rev Immunol (2015) 15(7):405–14. doi: 10.1038/nri3845
53. Vidal P. Interferon Alpha in Cancer Immunoediting: From Elimination to Escape. Scand J Immunol (2020) 91(5):e12863. doi: 10.1111/sji.12863
54. Diamond MS, Kinder M, Matsushita H, Mashayekhi M, Dunn GP, Archambault JM, et al. Type I Interferon is Selectively Required by Dendritic Cells for Immune Rejection of Tumors. J Exp Med (2011) 208(10):1989–2003. doi: 10.1084/jem.20101158
55. Fuertes MB, Kacha AK, Kline J, Woo SR, Kranz DM, Murphy KM, et al. Host Type I IFN Signals are Required for Antitumor CD8+ T Cell Responses Through CD8{alpha}+ Dendritic Cells. J Exp Med (2011) 208(10):2005–16. doi: 10.1084/jem.20101159
56. Swann JB, Hayakawa Y, Zerafa N, Sheehan KC, Scott B, Schreiber RD, et al. Type I IFN Contributes to NK Cell Homeostasis, Activation, and Antitumor Function. J Immunol (2007) 178(12):7540–9. doi: 10.4049/jimmunol.178.12.7540
57. Dunn GP, Bruce AT, Sheehan KC, Shankaran V, Uppaluri R, Bui JD, et al. A Critical Function for Type I Interferons in Cancer Immunoediting. Nat Immunol (2005) 6(7):722–9. doi: 10.1038/ni1213
58. Benci JL, Xu B, Qiu Y, Wu TJ, Dada H, Twyman-Saint Victor C, et al. Tumor Interferon Signaling Regulates a Multigenic Resistance Program to Immune Checkpoint Blockade. Cell (2016) 167(6):1540–54.e12. doi: 10.1016/j.cell.2016.11.022
59. Lemos H, Mohamed E, Huang L, Ou R, Pacholczyk G, Arbab AS, et al. Sting Promotes the Growth of Tumors Characterized by Low Antigenicity Via IDO Activation. Cancer Res (2016) 76(8):2076–81. doi: 10.1158/0008-5472.CAN-15-1456
60. Della Corte CM, Sen T, Gay CM, Ramkumar K, Diao L, Cardnell RJ, et al. Sting Pathway Expression Identifies Nsclc With an Immune-Responsive Phenotype. J Thorac Oncol (2020) 15(5):777–91. doi: 10.1016/j.jtho.2020.01.009
61. Thompson JC, Hwang WT, Davis C, Deshpande C, Jeffries S, Rajpurohit Y, et al. Gene Signatures of Tumor Inflammation and Epithelial-to-Mesenchymal Transition (EMT) Predict Responses to Immune Checkpoint Blockade in Lung Cancer With High Accuracy. Lung Cancer (2020) 139:1–8. doi: 10.1016/j.lungcan.2019.10.012
62. Kitajima S, Ivanova E, Guo S, Yoshida R, Campisi M, Sundararaman SK, et al. Suppression of STING Associated With LKB1 Loss in KRAS-Driven Lung Cancer. Cancer Discovery (2019) 9(1):34–45. doi: 10.1158/2159-8290.CD-18-0689
63. Ricciuti B, Recondo G, Spurr LF, Li YY, Lamberti G, Venkatraman D, et al. Impact of DNA Damage Response and Repair (Ddr) Gene Mutations on Efficacy of PD-(L)1 Immune Checkpoint Inhibition in Non-Small Cell Lung Cancer. Clin Cancer Res (2020) 26(15):4135–42. doi: 10.1158/1078-0432.CCR-19-3529
64. Formenti SC, Rudqvist NP, Golden E, Cooper B, Wennerberg E, Lhuillier C, et al. Radiotherapy Induces Responses of Lung Cancer to CTLA-4 Blockade. Nat Med (2018) 24(12):1845–51. doi: 10.1038/s41591-018-0232-2
65. Vanpouille-Box C, Alard A, Aryankalayil MJ, Sarfraz Y, Diamond JM, Schneider RJ, et al. DNA Exonuclease Trex1 Regulates Radiotherapy-Induced Tumour Immunogenicity. Nat Commun (2017) 8:15618. doi: 10.1038/ncomms15618
66. Diamond JM, Vanpouille-Box C, Spada S, Rudqvist NP, Chapman JR, Ueberheide BM, et al. Exosomes Shuttle Trex1-Sensitive IFN-Stimulatory dsDNA From Irradiated Cancer Cells to Dcs. Cancer Immunol Res (2018) 6(8):910–20. doi: 10.1158/2326-6066.CIR-17-0581
67. Skoulidis F, Goldberg ME, Greenawalt DM, Hellmann MD, Awad MM, Gainor JF, et al. Stk11/Lkb1 Mutations and PD-1 Inhibitor Resistance in KRAS-Mutant Lung Adenocarcinoma. Cancer Discov (2018) 8(7):822–35. doi: 10.1158/2159-8290.CD-18-0099
68. Yum S, Li M, Chen ZJ. Old Dogs, New Trick: Classic Cancer Therapies Activate Cgas. Cell Res (2020) 30(8):639–48. doi: 10.1038/s41422-020-0346-1
69. Achard C, Boisgerault N, Delaunay T, Roulois D, Nedellec S, Royer PJ, et al. Sensitivity of Pleural Mesothelioma to Oncolytic Measles Virus Depends on Defects of the Type I Interferon Response. Oncotarget (2015) 6(42):44892–904. doi: 10.18632/oncotarget.6285
70. Saloura V, Wang LC, Fridlender ZG, Sun J, Cheng G, Kapoor V, et al. Evaluation of an Attenuated Vesicular Stomatitis Virus Vector Expressing Interferon-Beta for Use in Malignant Pleural Mesothelioma: Heterogeneity in Interferon Responsiveness Defines Potential Efficacy. Hum Gene Ther (2010) 21(1):51–64. doi: 10.1089/hum.2009.088
71. Husain AN, Colby TV, Ordonez NG, Allen TC, Attanoos RL, Beasley MB, et al. Guidelines for Pathologic Diagnosis of Malignant Mesothelioma 2017 Update of the Consensus Statement From the International Mesothelioma Interest Group. Arch Pathol Lab Med (2018) 142(1):89–108. doi: 10.5858/arpa.2017-0124-RA
Keywords: lung cancer, mesothelioma, type I interferon, CDKN2A (p16), homozygous deletion, immunotherapy, STING
Citation: Grard M, Chatelain C, Delaunay T, Pons-Tostivint E, Bennouna J and Fonteneau J-F (2021) Homozygous Co-Deletion of Type I Interferons and CDKN2A Genes in Thoracic Cancers: Potential Consequences for Therapy. Front. Oncol. 11:695770. doi: 10.3389/fonc.2021.695770
Received: 15 April 2021; Accepted: 07 June 2021;
Published: 24 June 2021.
Edited by:
Emanuela Felley-Bosco, University of Zurich, SwitzerlandReviewed by:
Yoshitaka Sekido, Aichi Cancer Center, JapanSteven Albelda, University of Pennsylvania, United States
Copyright © 2021 Grard, Chatelain, Delaunay, Pons-Tostivint, Bennouna and Fonteneau. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jean-François Fonteneau, amVhbi1mcmFuY29pcy5mb250ZW5lYXVAaW5zZXJtLmZy
†These authors have contributed equally to this work and share first authorship