 Alessandra Romano1†
Alessandra Romano1† Paola Storti2†
Paola Storti2† Valentina Marchica2Grazia Scandura1Laura Notarfranchi2Luisa Craviotto2,3
Valentina Marchica2Grazia Scandura1Laura Notarfranchi2Luisa Craviotto2,3 Francesco Di Raimondo1,4*
Francesco Di Raimondo1,4* Nicola Giuliani3*
Nicola Giuliani3*- 1Department of Surgery and Medical Specialties, University of Catania, Catania, Italy
- 2Department of Medicine and Surgery, University of Parma, Parma, Italy
- 3Azienda Ospedaliero-Universitaria di Parma, Parma, Italy
- 4U.O.C. Ematologia, A.O.U. Policlinico–San Marco, Catania, Italy
Monoclonal antibodies (mAbs) directed against antigen-specific of multiple myeloma (MM) cells have Fc-dependent immune effector mechanisms, such as complement-dependent cytotoxicity (CDC), antibody-dependent cellular cytotoxicity (ADCC), and antibody-dependent cellular phagocytosis (ADCP), but the choice of the antigen is crucial for the development of effective immuno-therapy in MM. Recently new immunotherapeutic options in MM patients have been developed against different myeloma-related antigens as drug conjugate-antibody, bispecific T-cell engagers (BiTEs) and chimeric antigen receptor (CAR)-T cells. In this review, we will highlight the mechanism of action of immuno-therapy currently available in clinical practice to target CD38, SLAMF7, and BCMA, focusing on the biological role of the targets and on mechanisms of actions of the different immunotherapeutic approaches underlying their advantages and disadvantages with critical review of the literature data.
Introduction
Multiple Myeloma (MM) is the second most frequent hematological neoplasm, due to uncontrolled proliferation of neoplastic plasma cells (PCs) in and out the bone marrow (BM), surrounded by a permissive and protective tumor microenvironment (TME) (1, 2). The cross talk between MM cells and their surrounding TME has been a major obstacle for the development of immunotherapy. However, thanks to increasing body of evidence about the molecular arms of MM/TME interaction and the introduction of multiple novel agents (3), median patient survival prolonged from 3 to 8–10 years (4). MM PCs are strictly dependent on BM microenvironment cells and they express different molecules on the surface as receptors and adhesion molecules that exploit the function of crosstalk and adhesion with the BM microenvironment (5). Some of these molecules, such as Cluster of Differentiation 38 (CD38), signaling lymphocyte activation molecule family member 7 (SLAMF7), and B cell maturation antigen (BCMA), are highly expressed by MM PCs characterizing them as good target for novel therapeutic strategies as monoclonal antibodies (6–8).
Monoclonal antibodies (mAbs) are a group of agents with immune-based mechanism of actions that in recent years have changed the management of newly diagnosed and relapsed/refractory MM (RRMM) (6, 9). Moreover, the development of a new generation of mAbs, including antibody-drug conjugates (ADCs) and bispecific antibodies (bsAbs) has the potential to additional improve the clinical outcome of MM patients (6, 10). Isotype dictates mAbs activity (11), and most anti-MM mAbs are IgG antibodies. The IgG subclass, allotype, and glycosylation pattern are the main factors involved in the interaction strength of the IgG-Fc domain with Fc engaging molecules, including the classical IgG-Fc receptors (FcγR), the neonatal Fc-receptor (FcRn), the Tripartite motif-containing protein 21 (TRIM21), the first component of the classical complement cascade (C1), the Fc-receptor-like receptors (FcRL4/5). The effector potential strength of the interaction between IgG mAbs and Fc engaging molecules will not be described, being out of scope of this manuscript. Several extensive and updated reviews are available about this topic (12, 13).
This review will describe main therapeutic targets in MM cells and the BM microenvironment and the mAbs in use in the anti-MM therapy focusing on their mechanism of actions and strategies to improve their efficacy.
CD38
Target Definition
Human Cyclic ADP ribose hydrolase, also known as CD38, is a 43.7 kDa type II transmembrane glycoprotein, encoded by CD38 gene located on chromosome 4 (14). E.L. Reinherz, S. Schlossman and colleagues, first identified this surface molecule in 1980 during their analysis of the human lymphocyte surface using mAbs in search of the T-cell receptor (15). Therefore, at the beginning, it was considered a marker of T cells; afterwards, it was exploited as a phenotypic marker to recognize and classify T and B leukemia (16).
Physiological Expression and Function of CD38
This molecule is widely expressed in lymphoid and myeloid lineages (14, 17–19). Resting natural killer (NKs) cells and monocytes express it at low levels, as well as other cell types belonging to the hematopoietic lineage (Table 1): erythrocytes, platelets, and dendritic cells (DCs) (16, 20–22). Moreover, CD38 is expressed by different T cells subtypes T cell precursors as CD4+CD8+ double‐positive thymocytes (19). Within the circulating pool, CD38 is expressed by CD4+/CD45RA+ naive T cells as well as by subset of CD4+CD25+FoxP3+ regulatory T cells (Tregs) and by a subset of memory T cells (19, 23). CD38 is also a marker of activated T cells (19). Among CD8 T cells, CD38 is strongly expressed during chronic infection. CD38 is also expressed at high levels by peripheral blood mononuclear cells upon in vitro and in vivo activation (24). Subsequentially, CD38 expression is also modified during different stages of B cell differentiation. It is present at high levels on BM B cell precursors (immature or transitional) and is downregulated in mature B cells and is expressed at high level in terminally differentiated PC (19). Moreover, CD38 is expressed at high levels in a subset of B regulatory (Bregs) CD19+CD24hi cells and on IL-10-producing plasmablast with regulatory functions, on the other hand memory B cell population show a low expression of CD38 (CD24hiCD38loCD27+) (25). CD38 is also expressed on pathological cells such as chronic lymphocytic leukemia cells, where a presence of a major clone CD38+ positive is correlated with an unfavorable prognosis, and on MM cells (26). Analysis of CD38 distribution within MM bone niche revealed that only PCs express CD38 at high levels (27). Nevertheless, some studies demonstrated that CD38 expression is highly heterogeneous on MM cells, without a difference between newly diagnosed and relapsed/refractory MM patients (28). Moreover, in the MM bone microenvironment, CD38 decreases during osteoblasts (OBs) differentiation (29) and recently has been demonstrated that CD38 is expressed on the surface of early osteoclasts (OCs) progenitors but it is lost during in-vitro differentiation toward OCs (27).
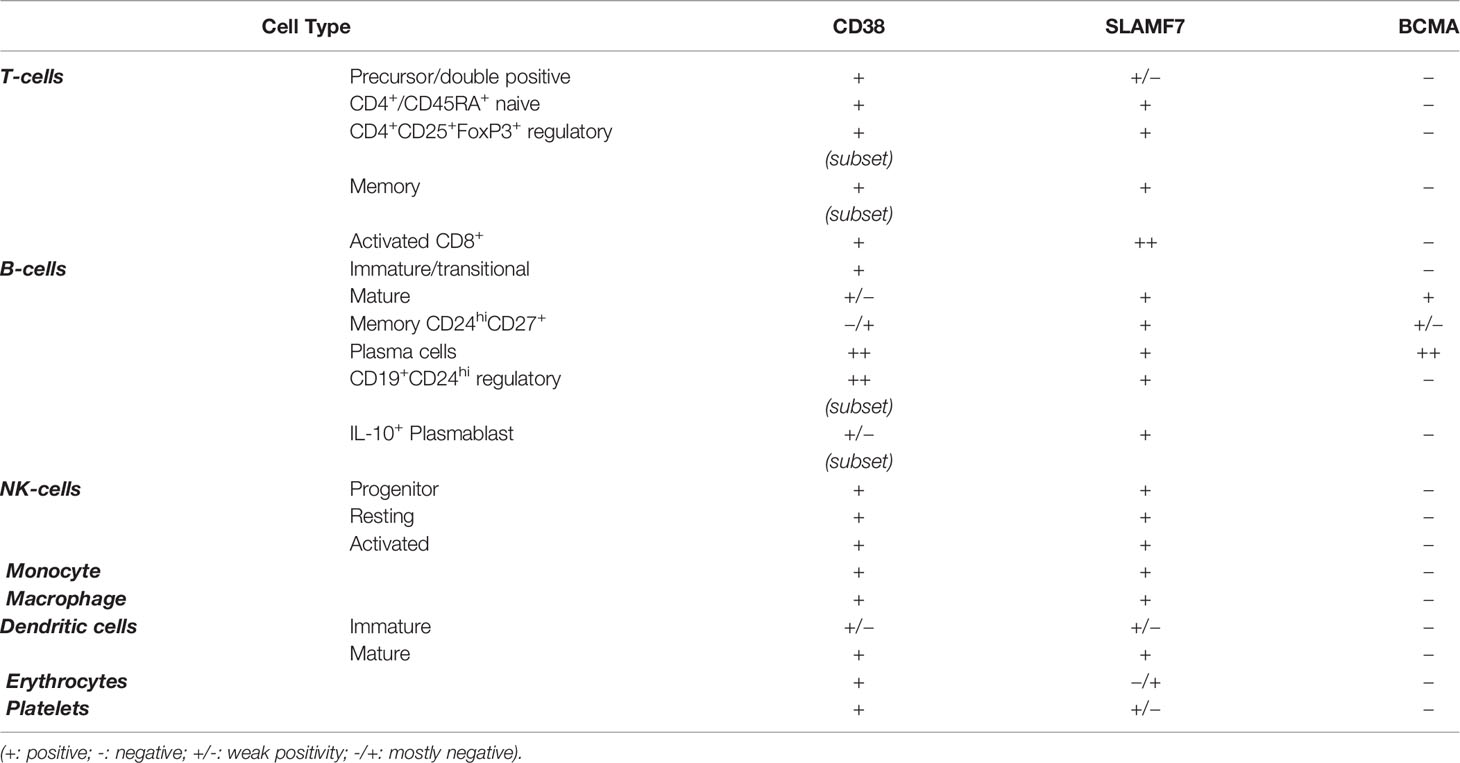
Table 1 Expression of CD38, SLAMF7, and BCMA in cells circulating in peripheral blood.
CD38 has a dual function of receptor and enzyme. As receptor, it regulates cellular adhesion, signal transduction, and calcium signaling. CD38 interacts with hyaluronic acid and the non-substrate ligand CD31, which is constitutively expressed by endothelial cells, leading to the activation of NF-kB, ZAP-70, and ERK1/2 pathways (26, 30). It has been generally known as a receptor despite a very short cytoplasmic tail that led to an inability to transduce the signal (6). Indeed, to act as a receptor, CD38 needs to be redirected to lineage-depended receptors of the cell membrane: BCR/CD19/CD21 in B cells, CD3/TCR in T cells, and CD16/CD61 in NK cells (26, 31).
The extracellular domain of CD38 acts as an ectoenzyme that, depending on the pH, is involved in the catabolism of nicotinamide adenine dinucleotide (NAD+) and nicotinamide adenine dinucleotide phosphate (NADP+) generating calcium signaling molecules, such as adenosine (ADO), that have immunosuppressive functions (32, 33).
All these data indicate multiple roles of CD38 in MM, becoming one of the most attractive antibody targets of the immunotherapeutic approaches to inhibit MM cell growth and survival and revert immunosuppression in MM patients.
Monoclonal Antibodies Anti-CD38: In Vitro Molecular Rationale for Use
Several anti-CD38 antibodies have been developed in the last decade with different mechanisms of action. CD38-targeting antibodies such as daratumumab (DARA), MOR202, and isatuximab (ISA), have high single agent activity in heavily pretreated MM patients by pleiotropic mechanisms of actions (Table 2).
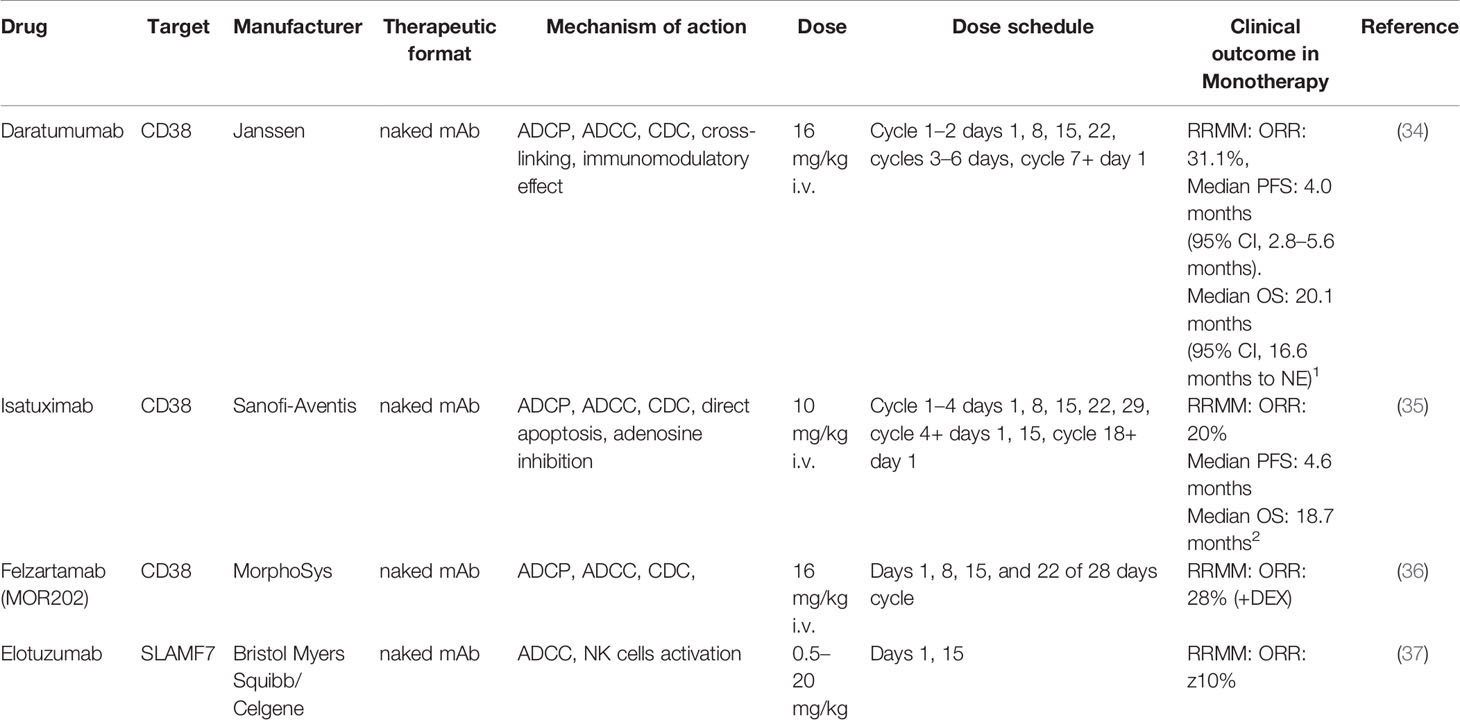
Table 2 Monoclonal antibodies against CD38 and SLAMF7 (Major clinical trials with published data).
DARA is the first anti-CD38 mAb approved in MM therapy. It is a fully human immunoglobulin G1 kappa (IgG1ƙ) mAb (38) that binds two sequences of a unique CD38 epitope outside the catalytic domain (33, 39). It entered in clinical trial in 2015, for use as a monotherapy in the treatment of MM patients who had received at least three previous lines of therapy (40), now the use has been expanded to newly diagnosed MM.
ISA is a humanized IgG1k mAb, which binds to a specific 23-amino acid discontinuous epitope that include a part of the CD38 catalytic site and it has been selected due to its multiple mechanisms of actions (33, 41). ISA is now approved in combination with pomalidomide and dexamethasone, for the treatment of MM patients with who have received at least two prior therapies (42).
Anti-CD38 antibodies exert their anti-MM activity by different mechanisms of action including classical FC-dependent immune effector mechanisms, namely the antibody-dependent cell-mediated cytotoxicity (ADCC) and antibody-dependent cellular phagocytosis (ADCP) and the complement-dependent cytotoxicity (CDC), and direct and immunomodulatory effects (43) (Figure 1).
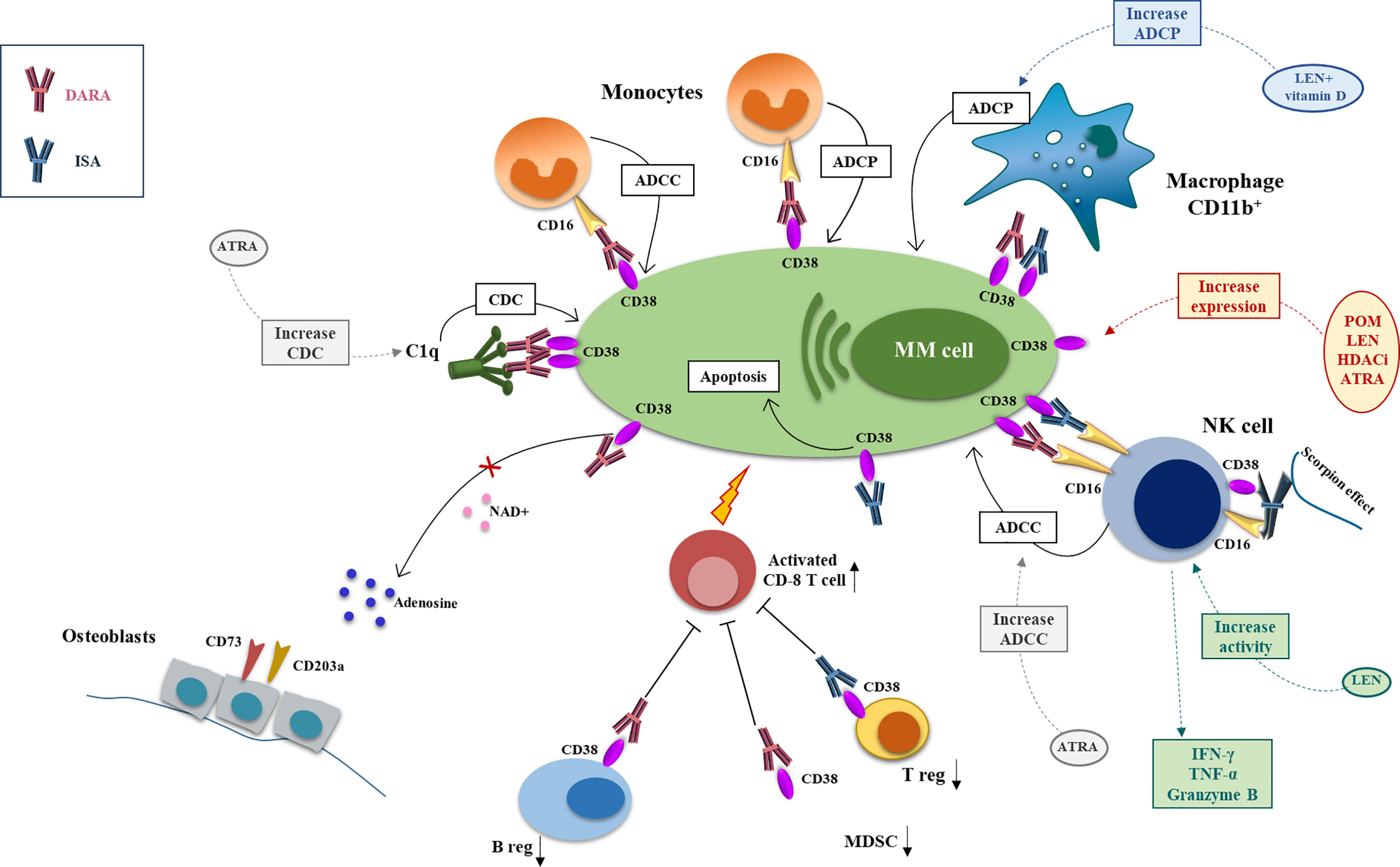
Figure 1 Mechanism of action and major drug combination of anti-CD38 mAbs, daratutmumab, and isatuximab. The anti-CD38 mAbs exert their antimyeloma activity through different mechanisms of actions that can be potentiate by different anti-MM drugs. CDC is activated by engagement of the C1q by DARA and initiates the classical complement cascade and the recognition of MM cells by phagocytic cells and the production of the anaphylatoxins. This mechanism can be increased by ATRA. ADCC involves NK cell and monocytes that through CD16 recognize the anti-CD38 mAbs on MM cell surface and activate the cytotoxic process. ISA can activate directly the NK cells through the scorpion effect. NK cell activity can be boosted by ATRA and LEN. ADCP is carried by CD16+ monocytes and CD11b+ macrophage; LEN+ vitamin D can enhance anti-CD38 mAbs-mediated macrophages phagocytic activity. ISA can also have a direct anti-MM effect inducing MM cell apoptosis. DARA has also an immunomodulatory function downregulating the immunosuppressor ADO, diminishing Breg and MDSCs and activating CD8+ T cells. ISA exerts its immunomodulating potential downregulating Treg (DARA, daratumumab; ISA, isatuximab; CDC, complement depend cytotoxicity; ADCC, antibody depend cytotoxicity; ADCP, antibody depend phagocytosis; ATRA, all-trans retinoic acid; LEN, lenalidomide).
Antibody-Dependent Cell-Mediated Cytotoxicity (ADCC)
Anti-CD38 antibodies can bind the Fc gamma receptors (FcγRs) (44) on the immune effector cells inducing the ADCC (40).
The binding with the Fc fragment of the anti-CD38 mAbs produces the intracellular phosphorylation of the tyrosine-based activating motifs of the FcγRs that leads in the lysis of MM cells (45). In particular the cell types FcγRs-expressing that are mainly involved in the ADCC-anti-CD38 mAbs mediated are NK cells which express CD32 and CD16, monocytes expressing CD16 and macrophages CD64+ (45).
NK cells are probably the main mediator of ADCC by mAbs. In vitro and ex-vivo data demonstrated that, DARA, by its binding with CD16, induces NK cells activation through the induction of STAT1 phosphorylation and activation of NF-kB p65 (46). Activated NK produced pro-inflammatory cytokines, as interferon gamma (IFN-γ) and tumor necrosis factor alpha (TNF-α) that lead to the recruitment of immune cells and MM cells killing (45, 46). Recent ex vivo data report the important role of the BM adaptive NK cell, characterized by a lower expression of CD38 and high expression of NKG2C, an activating NK receptor, in the response to DARA treatment of newly diagnosed MM patients (47). In particular, this NK subset sorted from BM of MM patients have higher ADCC capacity to kill MM cell coated with DARA compared to the conventional NK cell and adaptive NK cell frequencies is correlated with DARA response ex vivo (47).
Moreover, DARA could also enhance CD38+ NK cell apoptosis through a fratricide NK-to-NK ADCC without the involvement of tumor cells (46). This mechanism could be the basis of the rapid depletion of NK cell in MM patients after DARA treatment. Finally, also CD14+CD16+ monocytes can induce ADCC against MM cell coated by DARA (43, 48, 49).
On the other hand, in vitro data support that ISA induces ADCC by NK cells more efficiently against MM cells with higher density of CD38 on the surface, leading to the production of INF-γ and TNF-α (50). Moreover, it is hypothesized that ISA can directly activate NK cells through the cross-link of CD38 and CD16 on their surface (the scorpion effect) and activated NK cells can kill CD38low and CD38− target cells (50–52). Finally, Moreno et al. suggest that the depletion of NK cells after ISA treatment could be imputed to an exhaustion of these cells due to the higher ISA-mediated activation, rather than a fratricide mechanism (51).
Antibody-Dependent Cellular Phagocytosis (ADCP)
Phagocytosis contributes to the anti-MM activity of the anti-CD38 mAbs, as well (43). In vitro studies have demonstrated that DARA-coated MM cells are rapidly engulfed by tumor-associated macrophages (53). Recently, it has been demonstrated in an ex-vivo assay that the CD16+ subset of monocytes is essential in DARA MM cells-killing activity and the inhibition of the anti-phagocytic signal CD47-SIRPα significantly improves the DARA effect mediated by CD16+ monocytes (49).
On the other hand, Moreno et al. demonstrated that in vitro ISA triggers ADCP by CD11b+ macrophages only on MM cells that present a high level of CD38 molecules on the surface and the ability of ISA to induces ADPC also in NOD/scid/γc−/− (NSG) mice (51).
Complement-Dependent Cytotoxicity (CDC)
The Fc tail of the anti-CD38 mAbs engage the C1q molecule and initiates the classical complement cascade, leading the deposition of C3b on MM cell surface inducing the CDC, the recognition by phagocytic cells and the production of the anaphylatoxins C3a and C5a (40, 45). This effect could be imputed to a mechanism recently described by different groups on anti-CD20 mAbs: the establishments of non-covalent interactions between the mAbs Fc tails resulting in the formation of antigen dimerization that adjuvate the constitution of antibody hexamers after antigen binding on cells that recruit and activated C1 (54–56). DARA is the most effective inducer of CDC, while ISA can induce CDC only in a few MM samples with high expression of CD38 on PCs (51). The in vitro CDC induction by both DARA and ISA is reduced in presence of high level of inhibitory complement regulatory proteins CD59 and CD55 on MM cells (28, 50).
Immunomodulatory Effects
As CD38 is expressed on several immune cells, anti-CD38 mAbs have also immunomodulatory effects. DARA treatment reduced CD19+C24+CD38+ Bregs in MM patients and in vitro generated MDSCs (CD11b+CD14−HLA-DR−CD33+CD15+CD38+) causing a modification of the antitumor response (57). Moreover, DARA induces CD4+ and CD8+ T cells expansion in MM patients and in particular the effector memory CD8+ T cells concomitant with a decrease of naïve T cells subset (57). Indeed, the reduction of immunosuppressive cells could lead to an increase in T-cell numbers, T-cell clonality, as well as T-cell activity contain higher levels of granzyme B after exposure to DARA (57, 58).
Like DARA, ISA reduces T regulatory cells (Tregs) and blocks the production of immune inhibitory cytokines like interleukin (IL)-10 (59). Recently it has been demonstrated that ISA also depletes CD38hi B lymphocyte precursors and NK cells (51).
Finally, DARA treatment possibly modulates the enzymatic activity of CD38 by reducing the ADO levels. The axis CD38/CD203a/CD73 converts NAD+ to ADO: NAD+ reduction leads to the development of exhausted T cells and adenosine has an immunosuppressive effect on NK and CD8+ cells (60, 61). Van de Donk et al. showed that DARA reduces CD38 cyclase activity, increasing NAD+ levels and decreasing ADO levels (58). Indeed, targeting CD38 with anti-CD38 mAbs could restore the immune functions.
Direct Effects
ISA was selected initially based on its in vitro ability to directly induce MM cell death independently of effector cells and independently of Fc fragment binding to FcRs by binding the CD38 activating the classical caspase, lysosome death pathways, lysosomal membrane permeabilization, and cathepsin hydrolase release (62). Moreover, it is reported that ISA could induce reactive oxygen species production and promote to MM cell death (62). In contrast with these data, a recent paper reported no direct killing activity on ISA on MM cell in vitro (51).
In contrast, DARA did not show a direct killing effect on MM cells (43).
Mechanisms to Potentiate the Effects of Anti-CD38 mAbs in MM
Several pre-clinical studies indicate that the effects of anti-CD38 may be potentiated with other drugs and compounds. A synergistic effect between DARA and lenalidomide (LEN) in the induction of ADCC cytotoxicity has been previously demonstrated (63). Indeed, it is known that LEN stimulates NK cell increasing their production of IFN-γ, TNF-α, and granzyme B (64). Interestingly, an up-regulation of DARA-dependent ADCC was described in peripheral blood mononucleated cells (PBMCs) isolated from MM patients during or just after LEN treatment, thus further supporting the potential benefits from this combination (63).
Other studies showed that LEN enhances DARA-induced MM cell lysis by an increased frequency of CD3−CD56+ NK cells, with no alterations of T cell and monocyte compartments, even in patients refractory to LEN (65). Consistently, data obtained in humanized mice engrafted with MM cells from LEN refractory patients confirmed the capacity of LEN to potentiate the DARA effect (65). Accordingly, Van der Veer et al. (66) showed that the synergism between DARA and LEN/bortezomib treatment was more prominent in CD138+ CD38+ cells of MM patients refractory to LEN (66).
More recently, it has been also suggested that also vitamin D can potentiate the synergism between LEN and anti-CD38 mAbs combination mediated by the increase of the ADCP (67) due to LEN ability to induce CYP27B1 expression in macrophages (68). DARA-LEN synergism could be also due to the decrease of the frequency of inhibitory T cell populations induced by LEN (69). Indeed, it has been demonstrated that LEN up-regulates CD38 expression on Tregs and increases the fraction of CD38-high Tregs sensitizing this population to the anti-CD38, ISA (59).
Our group previously demonstrated that LEN and pomalidomide (POM) up-regulate CD38 expression by MM cells (70). This finding was recently confirmed by others (71): showing that the activity of DARA in combination with LEN was correlated by the increased CD38 surface expression by MM cells but not by NK cells by LEN (71). Lastly, Jiang et al. (62) showed that POM, enhances anti-CD38 mAbs effect both by the direct killing of MM cells, and by the indirect cytotoxicity effect (62). Interestingly it has been also reported that POM synergized with ISA in CD38-high MM cells with mutated p53 (62) supporting the use of POM and ISA combination in this type of high-risk MM patients.
Panobinostat is a pan-HDACi able to increase the expression of CD38 by PCs but not T cells (72) and consequently to potentiate the effect of DARA (72). Ricolinostat also increases CD38 expression on the surface of MM cells and it augments the ADCC by DARA against MM cell lines but not CDC effect (73).
Other drugs could be also used to increase the efficacy of anti-CD38 mAbs through the modulation of CD38 expression by MM cells and/or the effector cells. Different studies showed that agents such all-trans-Retinoic acid (ATRA) can be used to improve the effect of DARA and to overcome its resistance by increasing the expression of CD38 in MM cell (28). Nijhof et al. showed that treatment with ATRA significantly increased the expression of CD38 enhancing DARA-induced ADCC and CDC (28). The mechanism involving the modulation of CD38 by ATRA can be explained by the presence of a retinoic acid responsive element located in the first intron of the CD38 gene (74). Interestingly, treatment with ATRA also reduced the expression of CD55 and CD59 in MM cells (75). These studies clearly provide the rationale to design clinical trial with ATRA and DARA in refractory MM patients.
Bispecific Antibodies Against CD38
An antibody containing two different antigen-binding sites within one molecule is known as a bispecific antibody (BsAb). In particular, Bispecific T-cell engaging (BiTE) antibodies are a new class of drugs that can bind both a specific antigen on the surface of the tumor cells and the CD3e chain on T cells (76). BsAbs that target CD38 are in developing the last years and some of them are under evaluation in Phase I studies.
AMG424 is a novel CD38/CD3 BiTE, and recently it has been reported that AMG 424 can kill cancer cells expressing high and low levels of CD38 in vitro and increases T-cell proliferation, but with attenuated cytokine release (77). However, since CD38 is expressed in normal immune cells and non-hematopoietic tissues, it is associated with off-target toxicity (77). A phase 1 first-in-human trial (NCT03445663) of the drug in patients with R/R MM started in July 2018.
This year, it has been published a new BiTe against CD38: Bi38-3. Bi38-3 is made of two single-chain variable fragments anti-human CD38 and CD3e; it activates T-cell-mediated lysis of CD38+ MM cells in vitro, ex vivo, and in vivo. Moreover, it has been reported that it has no toxicity on B, T, and NK cells in vitro (78). Furthermore, Bi38-3 triggers the killing of MM cells from resistant patients and, since it recognizes a specific epitope on the Fc region of CD38, could be efficient also in patients after daratumumab therapy (78).
SLAMF7/CS1/CD319
Target Definition
The signaling lymphocyte SLAMF7, also known as CRACC or CD319 is encoded by SLAMF7 gene present on chromosome 1 at locus 1q23-24 (79, 80), is a 66kDa glycoprotein member of the SLAM superfamily. The SLAM family include several related CD2 subset of the immunoglobulin superfamily of receptors expressed on the surface of a wide variety of hematopoietic cells, including CD150, CD48, CD244, CD229, CD84 NK-T-B-antigen (NTB-A), also known as SF2000 in human or Ly108 in mouse, CD352, CD319, B lymphocyte activator macrophage expressed (BLAME, Slamf8), and SF2001 (CD84H, Slamf9) (79).
Like most SLAM receptors, SLAMF7/CS1 is a self-ligand, which exert activating or inhibitory influences on cells of the immune system depending on cellular context and the availability of effector proteins (81, 82).
SLAMF7 contains a membrane proximal C-type Ig fold and a membrane distal V-type Ig, a cytoplasmic region including two immunoreceptor tyrosine-based switch motifs (ITSM). The phosphorylation of tyrosine-based motifs in SLAMF7 induces downstream molecules activation including PLCγ1, PLCγ2, and PI3K kinases, regulating a variety of cell functions. SLAM receptors triggered by homo- or heterotypic cell-cell interactions control the activation and differentiation of a wide variety of immune cells and the interplay between innate and adaptive immune response. Downstream signaling is mediated by recruitment of small cytoplasmic adapter proteins, namely SH2D1A/SAP and/or SH2D1B/EAT-2. In humans, SLAMF7/CS1 has two splice variants, constitutively expressed on NK cells, namely a long-form CS1-L and a short form CS1-S, which lacks an ITSM motif required for NK cells activation described above (83). CS1L mediates NK cell activation through a SH2D1B/EAT-2 dependent, SH2D1A/SAP-independent extracellular signal- regulated ERK-mediated pathway. Thus, SLAMF7 can act as an activator if it can bind EAT-2, otherwise it is an inhibitor of downstream signaling (82).
Physiological Expression and Function of SLAMF7
SLAMF7, first described in NK cells (84, 85) and macrophages, is involved in numerous functions, including PCs survival, cell adhesion, NK cell– and CD8 T cell–mediated cytotoxicity (79). Several other hematopoietic cells express SLAMF7, including myeloid cells, activated T cells, most B cells, including antibody-producing PCs (81).
In NK cells, SLAMF7 is usually a positive regulator of NK cell activation, as consequence of the binding with the SAP family adaptor Ewing’s sarcoma-associated transcript 2 (EAT-2) via phosphorylated tyrosine 281 (Y281) in its cytoplasmic segment, thereby triggering activating signals involving phospholipase C-γ (PLC-γ) (86, 87) to induce polarization of cytotoxic granules (86). In the absence of EAT-2, or in excess of another adapter protein SAP (87), SLAMF7 recruits SHIP-1 and mediates inhibitory effects, as found in NK cells derived from EAT-2-deficient mice, and in normal activated T cells (81) and MM PCs (82), which lack EAT-2.
Expression and Function of SLAMF7/CS1 in Multiple Myeloma
Primary myeloma cells and human myeloma cell lines express higher levels of SLAMF7 than the normal or reactive counterpart, as consequence of genetic derangements (Table 1). In particular, cancer cells carrying the translocation t(4; 14) seem to have a greater expression of SLAMF7 and the in vitro inhibition of the expression of SLAMF7 in these cells is able to reduce the formation of colonies and to induce apoptosis and arrest of cells in G1, thus indicating an important role of this receptor in the proliferation of MM cells (88). The promoter region of SLAMF7/CS1 can bind the identity marker of PCs Blimp-1 (B-lymphocyte-induced maturation protein-1), which is dysregulated in MM, to enhance SLAMF7/CS1 transcription (89). This finding could explain why increased expression of SLAMF7 has been reported also in other B-cell disorders, like chronic lymphocytic leukemia and diffuse large B cell lymphoma (90). High levels of soluble SLAMF7 (sCS1) (91, 92) and increased mRNA of SLAMF7 in purified PCs have been documented in patients affected by monoclonal gammopathies in the entire spectrum, from monoclonal gammopathy of unknown significance (MGUS) through smoldering-, active-, and relapsed-refractory MM relapsed patients (93) and in autoimmune diseases, like systemic lupus erythematosus (94), or systemic infections in response to IFN-⍺ stimulation (80).
In 199 newly diagnosed MM patients, the amount of sCS1 was positively associated to active MM and not appreciated in healthy or stage I MM patients (95). Increased sCS1 was associated to most aggressive presentation, in both newly diagnosed and relapsed MM-patients, associated to lower probability to achieve deep response and reduced progression free survival, even if it could not be shown as an emerging independent prognostic factor (91).
Monoclonal Antibodies Anti-SLAMF7: In Vitro Molecular Rationale for Use of Elotuzumab
Elotuzumab is a humanized IgG1 mAb directed selectively against SLAMF7, unable to induce direct or complement-mediated lysis of MM cells, as shown in vitro and by absence of clinical activity as single agent (82, 96) (Table 2). In MM cells, high SLAMF7 expression is not able to induce neither proliferation nor apoptosis, due to lack of both EAT-2 (required to activate downstream signaling) and SHIP-2 (required to inhibit of downstream signaling) (82). Targeting SLAMF7/CS1 in vitro inhibited cell viability of human MM cell lines co-cultured with bone-marrow stromal cells (BMSCs) in a dose-dependent fashion, overcoming the stimulatory and protective effects of microenviroment on MM growth and survival (95), implying that its efficacy occurred through an indirect mechanism.
Elotuzumab can induce SLAMF7 expression in NK cells and acting as self-ligand can amplify its molecular effect (97). Favoring the homotypic SLAMF7-SLAMF7 interaction between NK and MM cells, elotuzumab further promotes natural cytotoxicity in a CD16-independent manner (85). Indeed, an Fc mutant form of Elotuzumab, unable to bind CD16, could promote cytotoxicity of SLAMF7+ target cells by NK cells derived from healthy donors, in particular when these cells were previously exposed to IL2. Therefore, it is likely that additional effects of Elotuzumab on immune system are based on involvement of other SLAMF7+ cells such as CD8+ lymphocytes (98), dendritic cells, activated monocytes, and dendritic plasmacytoid cells (99), as shown by reduced efficacy in CD8+ T- cells-depleted mice (98). Elotuzumab may inhibit the plasmocytoid dendritic cells that play an important role within the microenvironment that support MM cells growth and survival (99).
In Vivo Effects of Elotuzumab in MM and Strategies to Improve Clinical Efficacy of Immunotherapy Addressed Against SLAMF7
In vivo, most activity of elotuzumab can be attributed to NK-cells engagement via two main mechanisms. First, ADCC triggering, via engagement of FCγRIIIa/CD16a (93, 95). Second, a direct binding of elotuzumab’s Fab domain with the SLAMF7 receptor enhances EAT-2 recruitment to promote NK cells activation (81, 82, 84, 85, 87, 97, 100, 101). For this reason, differently from daratumumab, another mAb used in the same setting of relapsed/refractory (RRMM) patients, elotuzumab does not affect viability of NK cells (102), explaining the increasing interest for this molecule in the emerging adoptive CAR NK-cell therapies (103) (Figure 2).
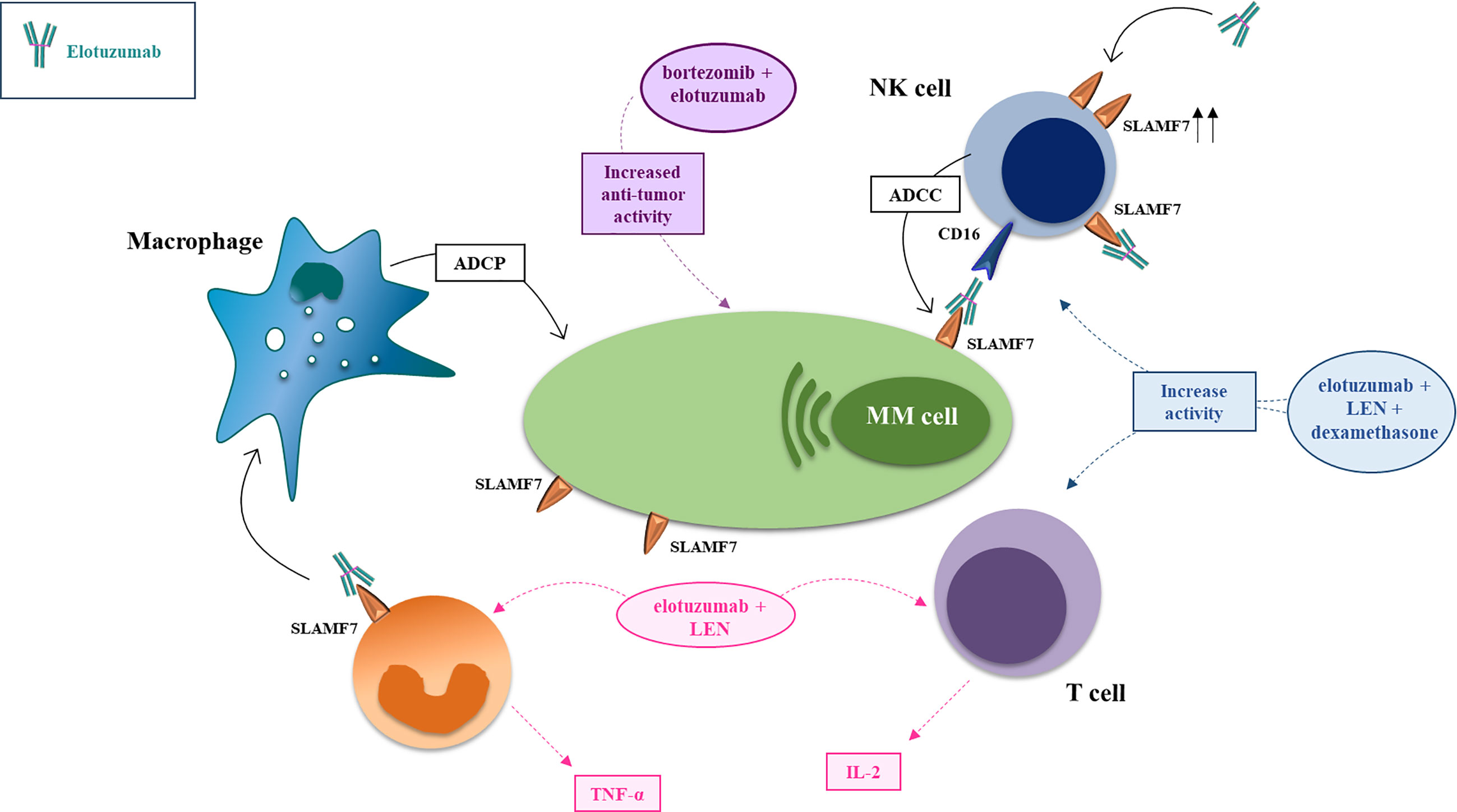
Figure 2 Mechanism of action and major drug combination of the anti-SLAMF7 mAb elotuzumab. The anti-SLAMF7 mAb elotuzumab exerts anti-MM effects via several indirect mechanisms: (i) promoting macrophage-mediated antibody-dependent cellular phagocytosis (ADCP) engaging co-stimulatory signaling to enhance ADCP in macrophages expressing both SLAMF7 and EAT-2; (ii) facilitating NK cell-mediated antibody-dependent cellular cytotoxicity (ADCC) of myeloma cells through Fc-dependent interactions with CD16 (FcγRIIIA); (iii) enhancing co-stimulatory signaling in NK cells, thereby potentiating natural cytotoxicity of myeloma cells, via simultaneous engagement of ITAM-linked activating receptors on NK cells (e.g. NKp46 or CD16) with ligands on myeloma cells; (iv) tagging myeloma cells for cell recognition; (v) elimination of immunosuppressive CD8+CD28−CD57+ Tregs which overexpress SLAMF7. In combination with proteasome inhibitors (e.g. bortezomib, carfilzomib) or immunomodulators (e.g. lenalidomide, pomalidomide), elotuzumab enhances anti-tumor effects via activation of T-cells and NK-cells.
To increase the cytotoxic effect of elotuzumab several strategies have been explored. In general, the combination with drugs able to induce SLAMF7 expression, recruit NK-cells and promoting ADCC is been extensively evalauted and it is highly recommended. In the pivotal studies, Van Rhee et al. treated SCID-human xenograft mice and documented that antitumor activity was enhanced if the MM cells were pretreated with bortezomib, even if pretreatment with bortezomib did not affect SLAMF7 expression (96).
In mice, the combination of lenalidomide and elotuzumab was very effective in reducing tumor volume and increasing the infiltration of NK cells into the tumor microenvironment, an effect enhanced by IL-2 secreted by T cells and TNF-alpha produced by monocytes (101) and macrophages (104). According to observation that Elotuzumab is able to reduce tumor burden and prolong survival in a MM model with SCID-beige mouse lacking B-, T-cells and with a reduced NK function, a further mechanism of action has been recently proposed. Elotuzumab could recruit monocytes, promote the infiltration of M1-polarized tumor-associated macrophages with enhanced ADCP of MM cells through engagement of the Fcγ receptor (104).
Immunomodulatory drugs (IMiDs) look like the ideal backbone to combine with Elotuzumab for their direct and indirect effects on both T- and NK-cells function (64, 105, 106), as shown in the relapsed/refractory setting by trials ELOQUENT-2, which tested efficacy and safety of elotuzumab combined to lenalidomide and dexamethasone (107, 108) and ELOQUENT-3 study, which tested efficacy and safety of elotuzumab combined to pomalidomide and dexamethasone (109). Unfortunately, the ELOQUENT-1 trial, which evaluated elotuzumab combined to lenalidomide and dexamethasone in the setting of newly diagnosed, transplantation-ineligible MM patients, failed to demonstrate additive clinical activity of elotuzumab (110).
Elotuzumab can be combined with other mAbs to increase the activity of effector cells, like the checkpoint inhibitor pembrolizumab (111) to promote tumor-infiltrating NK and CD8+ T-cell activation, intratumoral cytokine and chemokine release (98).
Elotuzumab has ben used to arm an anti-CD3 (OKT3) antibody to develop a bispecific antibody-armed activated T cell to induce adaptive cellular and humoral immune responses in MM patients, to mediate MM cytotoxicity independently from major histocompatibility complex. Ex vivo arming unarmed activated T cells avoids the need to administer large quantities (mg/kg) of bispecific antibody to reduce adverse events, like cytokine release syndrome. This strategy utilizes humoral antibody targeting by ATC. Secretion of Th1 cytokines upon binding of the effector cells to the myeloma cells not only augments tumoricidal activity directed at the malignant B cells, but may increase local cytokine and chemokine secretion that leads to shifting the tumor microenvironment to recruit endogenous immune effectors and induce an endogenous immune response (112). The targeting domain derived from elotuzumab has been used to develop T cells expressing an SLAMF7 CAR, with promising activity in preclinical models in vitro and in vivo, leading to ongoing phase 1/2a clinical trials CARAMBA and MELANI-01 (113).
Finally, a SLAMF7-targeted mAb ahs been conjugated with a payload drug (e.g. DM1, DM4, SN38, MMAE, MMAF) through a linker (e.g. SMCC, SPDB, MC-Vc-PAB). Azintuxizumab vedotin (ABBV-838) was the first-in-class antibody-drug conjugate (ADC) in which a SLAMF7-targeted mAb was linked to monomethyl auristatin E (MMAE) via a cathepsin B-cleavable peptide linker. Two phase I clinical trials have been started but in June 2017 AbbVie decided to terminate the phase-I/Ib trial NCT02462525 for insufficient clinical activity (114), with only 10% of overall response rate (115).
The BAFF-APRIL-BCMA System
Target Definition
The persistence of normal and neoplastic plasma cells (PCs) depends on survival factors provided in the bone marrow as consequence of direct contact to mesenchymal stromal cells (116) or the B-Cell Maturation Antigen (BCMA) triggering, induced by its two ligands, namely, B-cell-Activating Factor (BAFF; BLys and CD257) (117) and A PRoliferation-Inducing Ligand (APRIL; CD256), that are respectively produced mainly by macrophages (118) and osteoclasts (119, 120).
BCMA (also referred as TNFRSF17, CD269) is a transmembrane glycoprotein belonging to the tumor necrosis factor superfamily, selectively induced during B-cell differentiation into plasmablasts and bone marrow PCs (121), neoplastic PCs (120, 122), while it is nearly absent on naive and memory B cells (121, 122) CD34 stem cells, and other normal tissue cells (123) (Table 1). In normal and neoplastic mouse plasma cells, and in the human MM cell line MM1.s, the BCMA expression is under control of the master plasma cell gene IRF4 (124, 125), even if the post-translational regulation of BCMA can be largely compensated for reduced transcription, by mechanisms still under investigation (124).
Following stimulation with APRIL or BAFF, BCMA becomes a trimer, eliciting a signaling cascade involved in the activation of MAP kinases and the induction of anti-apoptotic proteins, such as Bcl-2, Bcl-XL, and the antiapoptotic protein myeloid cell leukemia 1 (MCL-1) (126).
The BAFF-APRIL-BCMA System Regulates Plasma Cells Homeostasis
BAFF is required for homeostasis and maintaining normal B-cell development, and survival of malignant B- and PCs, by increasing the levels of the pro-survival molecules B cell lymphoma 2 (Bcl-2) and Bcl-x and by decreasing the levels of the proapoptotic molecule Bcl-2-homologous antagonist/killer (Bak) (127, 128). BCMAko mice have shorter survival of long-lived bone marrow PCs compared to wild-type controls while maintaining a normal phenotype (129). BAFF binds mainly the BAFF Receptor (BAFF-R), which triggers naïve B cell survival and maturation. During B-cell development BAFF-R is first expressed on immature B cells with the highest expression levels on transitional and mature B cells and decreased levels on germinal center B cells, while BCMA and TACI (Transmembrane activator and CAML interactor) are expressed in a more restricted manner and support the survival of PCs. BAFF/BCMA binding activates NF-kB and the MAPK8/JNK signaling pathways, to sustain long-term humoral immunity, survival, and proliferation to regulate B cell antibody responses, isotype switching, and homeostasis (130). While BAFF is required for B cell homeostasis, the excessive production of BAFF is detrimental to the host. Transgenic mice overexpressing BAFF suffer from increased production of autoantibodies and symptoms of autoimmune diseases (130–132).
APRIL binds to BCMA with higher affinity interaction than BAFF to prevent activation of the endoplasmic reticulum (ER)-associated Casp12 contributing to maintenance of long-lived PCs in the niche (116). APRIL can also bind heparin sulfate proteoglycans to potentiate TACI and BCMA activation through its multimerization (133, 134), but the underlying molecular mechanisms are still largely unknown.
BAFF and APRIL are equally potent in inducing bone marrow plasma cell survival. TACI mediates the BAFF- and APRIL-induced generation of PCs and T cell-independent immunoglobulin isotype switching and secretion, whereas the function of BCMA is restricted to the maintenance of PCs and antigen presentation by B cells, through the activation of AKT, MAPK, and via NF-kB (120).
BCMA is shed from the surface of PCs via γ-secretase–mediated cleavage, with consequent releases of soluble BCMA (sBCMA). sBCMA acts as a decoy neutralizing APRIL (135) and sequesters B-cell activating factor BAFF (136), thereby preventing it from performing its signaling to stimulate normal B-cell and plasma cell development, resulting in reduced polyclonal antibody levels (136).
The BAFF-APRIL-BCMA System in Multiple Myeloma
BCMA is detectable in malignant PCs throughout the duration of the disease, with progressive increased expression from monoclonal gammopathy of uncertain significance (MGUS) to smoldering myeloma to active MM, with the highest levels correlated to the worst prognosis (137). In MM patients, BCMA mRNA is upregulated in PCs, and in CD138− progenitor cells responsible for recurrences (138). Conversely, a downregulation of BCMA reduces the viability and formation of myeloma colonies (120). In vivo, BCMA-overexpressing tumors increased neo-angiogenesis and transcription of genes crucial for osteoclast activation, adhesion, and angiogenesis/metastasis, as well as genes mediating immune inhibition including programmed death ligand 1 (PDL1), transforming growth factor β (TGF- β), and interleukin 10 (IL-10) to orchestrate the complex interplay between myeloma and microenvironment cells (120).
Soluble BCMA (sBCMA) is higher in supernatants of mononuclear cell cultures of MM-BM than the marrow of healthy subjects (135), progressively increased from healthy, MGUS, and active MM, and higher in patients with progressive disease, associated to reduced overall survival (139). In mice, sBCMA levels correlated with the change in tumor volume in response to melphalan or cyclophosphamide with bortezomib (139). In vivo, sBCMA correlated with the percentage of bone marrow PCs, even in patients with non-secretory myeloma and with the depth of response to treatment. Patients with levels above the median had shorter progression free survival and overall survival (140). Normalization of sBCMA during any treatment was predictor of increased overall response rate, overall survival (141), and achievement of complete response (141). Currently, sBCMA (139, 140) and sTACI are investigated as novel biomarker of disease activity in B-cell disorders with prognostic value (142), with two main advantages: independence from renal kidney and shorter half-life (24–36 h) than monoclonal components IgG (21 days) and IgA (7 days), or free light chains (140, 141).
Based on the above-mentioned circuitry, BCMA has been recently evaluated as highly selective antigen for neoplastic PCs, representing that tumor associated antigen ideal for the development of target therapy (143). Moreover, due to lack of expression on B-cell precursors, a rapid recovery of cell B immunity could be expected upon discontinuation of anti BCMA treatment.
How to Target the BAFF-APRIL-BCMA System in Multiple Myeloma: Tabalumab
Tabalumab (LY 2127399) is an-anti BAFF human mAb developed by Eli Lilly and Company, designed for the treatment of autoimmune diseases and B cell malignancies (144). In MM, two phase II studies, conducted in US and Japanese cohorts, failed to show any clinical improvement in progression free survival (145–147), probably for high BAFF concentrations in RRMM patients or the induction of compensatory pathways via APRIL/BCMA engagement (148–151), suggesting the need to combine tabalumab with anti‐APRIL antibody or TACI‐Fc fusion protein, a potent inhibitor of both BAFF and APRIL, to augment clinical efficacy in RRMM.
How to Target BCMA in Multiple Myeloma: The Antibodies Drug Conjugates
Currently, the development of immunotherapy against BCMA is directed towards three approaches: antibodies drug conjugates (ADCs), the bispecific antibodies, and the CAR-T cells, that have been recently described in a comprehensive review (Tables 3 and 4) (163).
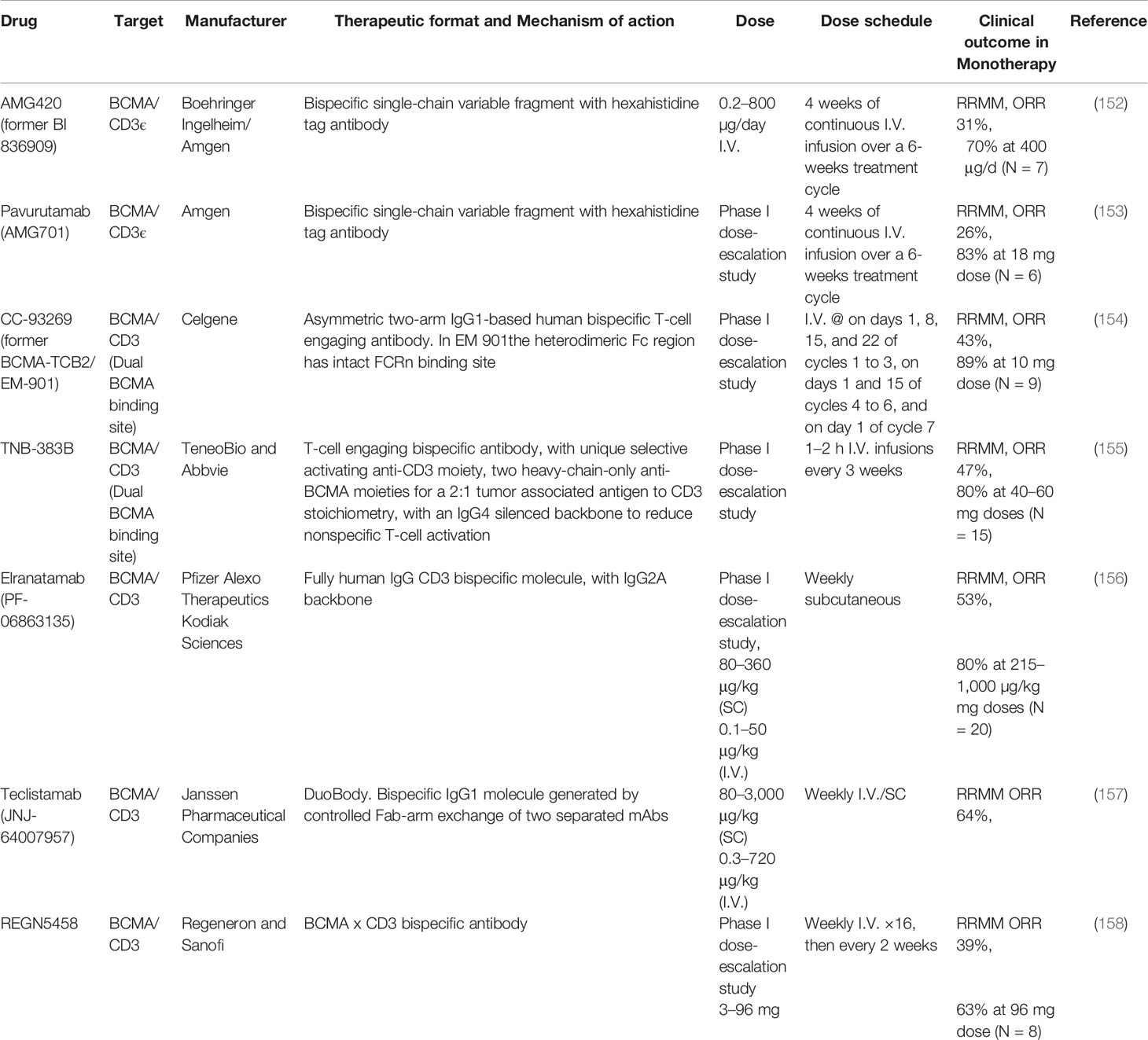
Table 3 BsAbs against BCMA in clinical development (Major clinical trials with published data).
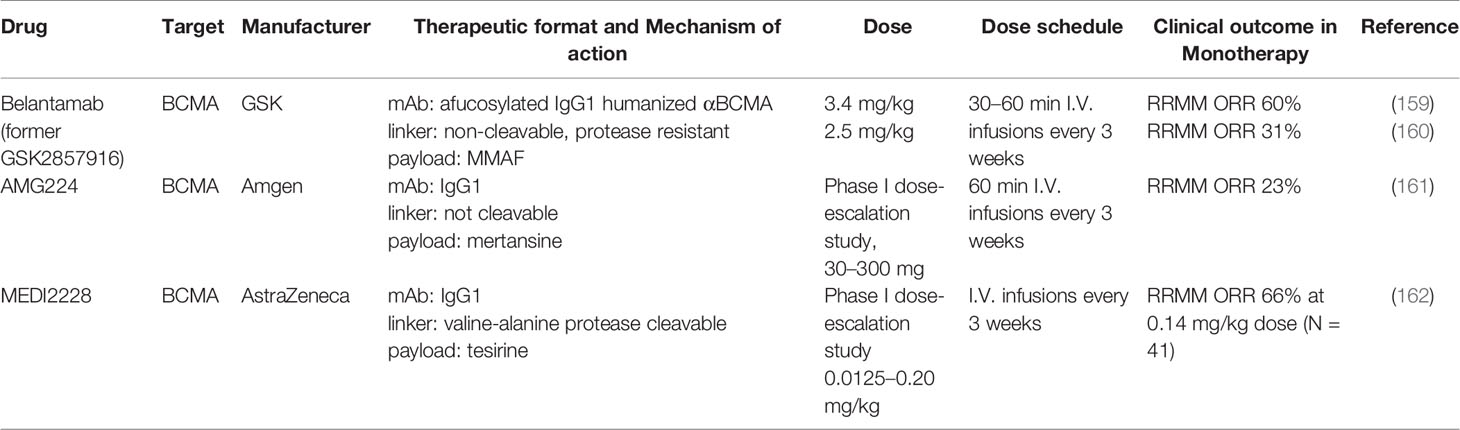
Table 4 ADCs against BCMA in clinical development.
ADC technologies combine mAbs (generally IgG1 due to the availability of multiple lysines required for optimal conjugation), selective for the antigen on the target cell, with toxic payloads (generally targeting microtubules or DNA duplication) (164, 165) and a linker between the antibody and the cytotoxic agent, as extensively described in excellent reviews (166). Based on ADC design, linkers can be stable in serum or in the circulation, thus after the initial internalization antigen/ADC complex is followed by its complete degradation in the lysosome (166). Otherwise, linkers could be cleaved only under certain specific conditions to ensure drug delivery. For example, the hydrazine linkers are susceptible to acidic conditions, the disulfide linkers to reducing equivalents (glutathione), and the peptide linkers to proteases (164, 165, 167, 168).
The first anti-BCMA mAb cSG1 was initially evaluated by Seattle Genetics in 2007, developed both as a naked mAb (not further tested in large clinical trials) as well as with a drug conjugate (ADC) (143). Alone, or in combination with bortezomib or lenalidomide, cSG1was able to induce cytotoxicity of myeloma cells in vitro, even in the presence of BMSCs, and to reduce the migratory capacity of MM cells through the inhibition of NFkB (120).
The first-in-class anti-BCMA ADC investigated in clinical trials is Belantamab Mafodotin (GSK2857916). The Belantamab Mafodotin platform has three peculiarities: i) afucosylated IgG1to ensure the highest affinity for the FCγRIIIa/CD16a receptor of the effector cells to mediate ADCC; ii) a protease non-cleavable linker, to avoid serum degradation and release of the payload outside the cell of interest. The linker is cleaved inside the cell; iii) a powerful cytotoxic agent as payload, monomethyl auristatin (MMAF), designed to be much more active when actively delivered inside cells with a mAb, compared to treatment in the untargeted form. Thanks to its peculiar structure, Belantamab Mafodotin has different mechanisms of action:
1) induces the arrest of MM cells in G2/M phase resulting in apoptosis
2) induces a powerful ADCC via binding of the defucosylated Fc fragment of NK and PBMC cells
3) induces ADCP via binding of the defucosylated Fc fragment of macrophages
4) competes with BAFF and APRIL, reducing their signal of activation of NFkB
5) reduces activity of BCMA+ dendritic plasmacytoid cells which support proliferation and drug resistance of MM cells (169).
Belantamab Mafodotin was first tested in both disseminated and subcutaneous human MM xenograft mouse models where it was shown to induce a complete eradication of the neoplasm without inducing weight loss of the mice, thus confirming the absence of toxicity (170). Subsequently, it was investigated in phase I (171) and phase II trials (160), with encouraging about 30% of overall response in penta-refractory patients and now is being tested in combination with lenalidomide and pomalidomide for patients with relapsed/refractory MM (143), as recently described in several recent comprehensive reviews (172–180). The most common grade 3–4 adverse events include: keratopathy (181, 182), thrombocytopenia, and anemia (160). Blurred vision, keratitis, dry eye, and microcystic epithelial damage are typically associated to ADCs due to o non-specific ADC uptake into actively dividing basal epithelial limbal stem cells residing in the basal epithelial layer of the cornea (183).
Future developments to improve drug-induced toxicities include the combination of Belantamab Mafodotin with both immunomodulatory drugs and proteasome inhibitors, extending dosing intervals (i.e. every 4–6 week dosing versus every 3 week dosing) (179), together with the clinical studies involving other anti-BCMA ADCs with several promising different payloads (184).
Other BCMA-targeting ADCs include AMG-224, CC-99712, SG1-auristatin, MEDI228, and HDP-101, as summarized in very recent comprehensive reviews (172, 185–187).
AMG 224 is an antihuman BCMA IgG1 antibody conjugated with mertansine, an antitubulin maytansinoid, through a non-cleavable linker. In the dose escalation NCT02561962 phase 1 trial, 40 patients received intravenous AMG 224 every 3 weeks at prespecified doses of 30–300 mg in a 3 + 3 design, with no mandated pre-medications. The objective response rate (ORR) for the study was 23%, including six responses in dose escalation and three responses in the dose expansion. In the dose escalation cohort, the most common AEs include thrombocytopenia, fatigue, nausea, AST increase, and anemia (161).
In MEDI2228 a fully human BCMA-binding IgG1 antibody is conjugated to DNA-damaging agent pyrrolobenzodiazepine (PBD) via a protease-cleavable linker, showing higher clinical activity than to a monomethyl auristatin F (MMAF) analog, also in the presence of high levels of sBCMA, due to induced DNA damage responses (DDR) and synergized with multiple DDR-inhibitors. A phase 1 dose-escalation/-expansion study of MEDI2228 as monotherapy in relapsed/refractory patients is currently ongoing (NCT03489525).
In HDP-101 a BCMA-specific antibody is conjugated to the RNA polymerase inhibitor amanitin, a synthetic derivative belonging to the amatoxin family, identified more than 40 years ago in the mushroom Amanita phalloides. These substances, responsible for severe hepatotoxicity secondary to the ingestion of these fungi, bind with high affinity to RNA Polymerase II, thus reducing transcription and protein synthesis and are effective against both rapidly dividing and resting cells. Based on preclinical studies, HDP-101 has large clinical activity in models with a knockout of tumor suppressor TP53 and knockdown of RNA polymerase POLR2A, which mimics the deletion of 17p in a subtype of high-risk MM patients. Preclinical data have shown that HDP-101 has significant anti-tumor activity both in vitro and on xenograft models and results of the clinical study are expected in 2021 (188).
How to Target BCMA in Multiple Myeloma: The Bispecific Antibodies and Beyond
A further challenge of immunotherapy is retargeting the effector cells (NK-cells, macrophages, T-cells) to provide rapid activation, robust and durable cytotoxic responses, and potentially generate immunologic memory (189). The engagement of CD3 (part of the T-cell receptor) induces both proliferation of CD4 and CD8 T-cells and cytotoxic activity by CD8 and in part CD4 cells against the target. The engagement of CD3 is the major proliferation signal, even though there may be additional indirect mechanism of proliferation induction by cytokines. Thus, the interaction between the patient’s own T lymphocytes and the tumor cells expressing a specific antigen could be facilitated, to eliminate cancer without genetic alteration of the T cells or need for ex vivo expansion/manipulation, providing off-the-shelf immuno-oncotherapy (189, 190). In this scenario, the class of bispecific antibodies (bsAbs), also known as dual-targeting molecules, includes antibodies or derived proteins engineered to have multiple binding sites, each with a unique antigen specificity, to different epitopes, to physically bridge two or more cells.
There are two major factors which could affect pharmacokinetics (191) of bsAbs immunotherapy: i) the binding to FcRn (neonatal Fc receptor), which in turn mediates the long half-life of IgG molecules in vivo and it is involved in transcytosis from the vascular space out into tissue compartments; ii) the potential higher immunogenicity of anti-drug antibodies due to the presence of non-natural structural motifs (192). To this end, Fc mutations have been heavily employed in new generation mAbs to modify interaction with FC-γ-receptors, to increase or decrease CDC, ADCC, and ADCP. Mutations to modify FcRn binding are also attempted by different groups to modify pharmacokinetics, but these have not reached the clinic (191). ADC hapten-like structure across eight molecules tested in 11 phase I–II clinical trials do not appear to increase patient immune responses beyond those generally observed for mAb biotherapeutics (193), but data lack in MM setting. It is still under investigation if larger molecules could hard penetrate the tumor, especially when extramedullary bulky masses are present.
Two formats of bsAbs have been extensively studied in MM: BiTE (Bispecific T-cell engager, developed by Amgen, Thousand Oaks, CA, USA) (10, 184, 190, 194) and DuoBody (developed by Genmab A/S, Copenhagen, Denmark). In the BiTE molecules, binding domains are two single‐chain variable fragment (scFv) regions, arised from mAbs, joined by a flexible peptide linker: one, to recognize tumor‐expressed antigens, and another to engage effector T-cells. The second scFv binding domain is always specific for CD3, the invariable part of the T‐cell receptor complex. When a BiTE molecule engages both a cytotoxic T cell and a tumor cell, the T cells start to proliferate, increasing overall numbers of effector cells and strengthening the potency of BiTE therapy (195). Once the cytolytic synapse has occurred, the T cells release perforin and granzyme B, thus inducing the apoptosis of the tumor cells. Furthermore, the activation of lymphocytes induces the release of cytokines that amplify the immunological response by involving other immune cells and induce a proliferation of T cells (189, 190, 196).
AMG 420 (formerly BI 836909) was the first anti-BCMA BiTE used in relapsed/refractory MM patients. AMG 420 is a bispecific single-chain variable fragment consisting of two linked single-chain variable fragments (scFvs) (197). The BCMA scFv is positioned N-terminally, and the CD3ϵscFvC-terminally followed by a hexahistidine (His6tag) (197). In vitro experiments have documented that both T lymphocyte subpopulations (CD4+ and CD8+) contribute to the antibody-induced lysis of MM cells, associated to autologous T-cell activation, documented by increased secretion of IFNγ, IL-2, IL-6, IL-10, and TNFα in a dose dependent manner, in T-cells obtained from both newly diagnosed and RRMM patients (Figure 3). The maximum cytolytic activity was reached between 16 and 24 h, greater in presence of peripheral blood mononuclear cells (PBMCs), suggesting the engagement of other blood cells. The cytolytic activity of AMG420 was not affected by the co-culture of MM cells with stromal cells, that usually confer drug resistance, or in presence of soluble APRIL and BCMA, which could interfere with or bind the antibody. The same encouraging results have been obtained in vivo, in both mouse xenograft models with the insertion of human T cells and in cynomolgus monkeys, where a dose-dependent decrease of bone marrow PCs could be documented as well (197). Clinical application of AMG420 was very promising in RRMM setting, with a response rate of 70% at the maximum tolerated dose, including for half patients the achievement of MRD-negative complete response (152). However, further clinical development has been stopped, due to the short half-life of AMG420, requiring a continuous infusion of 4 weeks, and the high rate of infections, mainly due to the requirement of a catheter for i.v. injection.
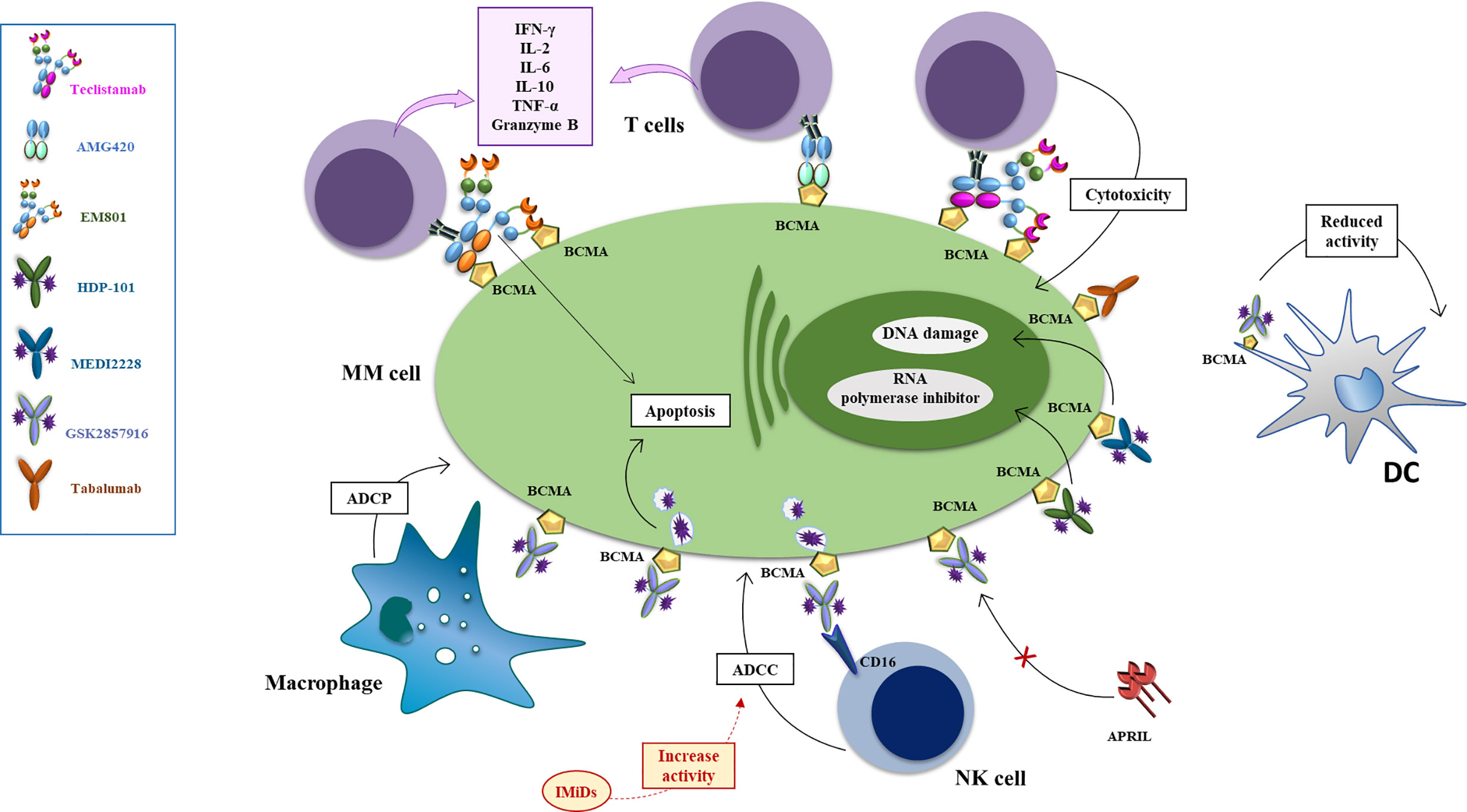
Figure 3 Mechanisms of action of anti-BCMA mAb, antibody drug conjugates and bispecific antibodies. The antibody drug conjugate Belantamab mafodotin exerts anti-MM effect via several mechanisms: i. inducing ADCP via binding of the defucosylated Fc fragment of macrophages the arrest of MM cells in G2/M phase resulting in apoptosis; ii. inducing a powerful ADCC via binding of the defucosylated Fc fragment of NK and PBMC cells (an effect enhanced by combination with lenalidomide) iii. competing with BAFF and APRIL, reducing their signal of activation of NFkB in MM cells (an effect enhanced by combination with bortezomib) iv. reducing activity of BCMA+dendritic plasmacytoid cells which support proliferation and drug resistance of MM cells. Upon binding with MM cells via BCMA, MEDI2228 releases pyrrolobenzodiazepine to promote DNA damage and cell death, while HDP-101 releases the RNA polymerase inhibitor amanitin, to reduce transcription and protein synthesis, resulting in apoptosis of both rapidly dividing and resting cells. AMG 224 is an antihuman BCMA IgG1 antibody conjugated with mertansine, to inhibit the assembly of microtubules with consequent cell death. Tabalumab (LY 2127399) is an-anti BAFF human naked monoclonal antibody that neutralizes the membrane-bound and soluble forms of this factor, reducing their signal of activation of NFkB in MM cells. Bispecific monoclonal antibodies can simultaneously bind to two different types of antigen to engage effector cells against neoplastic cells. EM-801 and AMG-420 are two examples of BCMA/CD3 bispecific T-cell engager. Teclistamab is a BCMA/CD3 DuoBody.
In the attempt to improve drug manageability and prolong its clearance, new bsAbs have been engineered to have a Fc moiety which makes them more like a complete antibody (198). Among them, AMG701 has a mean elimination half-life of 112 h (4.7 days) and it is able to induce a potent T-cell-dependent cellular toxicity against BCMA positive MM cell lines, together with a dose-dependent T-cell activation a cytokine secretion. In vivo, it was able to inhibit growth of tumor xenografts, prolong survival of an orthotopic mouse xenograft model and deplete MM cells in cynomolgus monkeys (199). In RRMM patients, AMG 701 potently induced autologous cell lysis, T-cell proliferation, and differentiation leading to higher CD8/CD4 ratios, acting synergistically with lenalidomide and pomalidomide to prevent myeloma relapse in vivo (200). The interim analysis of the Phase 1 dose escalation trial (NCT03287908) presented at ASH 2020 (153) provided encouraging signs of activity as a single agent in heavily pre-treated MM patients. AMG 701 was given to 85 R/R MM patients who had received at least three prior lines of therapy, and a median of six lines. The response rate was 36% at doses of 3–18 mg with responses lasting up to 26 months in one patient. Six of seven patients, who were tested for minimal residual disease (MRD), were MRD-negative. In the most recent evaluable cohort, there was an 83% ORR, with 4/5 responders being triple refractory (153).
EM801 is an asymmetric IgG1 bsAb first developed by EngMab AG with Celgene. EM801 incorporates bivalent binding to BCMA, a head-to-tail fusion of BCMA- and CD3ε-binding Fab domains and an engineered Fc region, with completely abolished binding to FcγRs and C1q, carrying a heterodimeric Fc region with intact FcRn binding. The molecular structure enables prolonged half-life to approximately 4 days, thus allowing the possibility of intravenous or subcutaneous administration once a week (201). In vitro, EM801 induces a strong, dose dependent bond between T lymphocytes and MM cells with consequent activation of T cells, documented by the hyper-expression of CD25 and CD69 and release of granzyme B and inflammatory cytokines such as IFNγ, TNFα, and IL-2 (Figure 3). At an E:T ratio of 2:1, 5:1, and 10:1 EM801 induces a killing of MM cells of 60, 70, and 80% respectively and the depletion of a subpopulation of T lymphocytes (CD4+ or CD8+) not significantly reduces the cytocidal effect of the antibody. Experiments conducted on primary MM cells have documented that EM801 was able to induce mortality in 77 and 83% primary samples obtained respectively from newly diagnosed and RRMM patients, without toxicity on microenvironmental cells. In vivo, anticancer activity has been documented in both mouse models and in cynomolgus monkeys, where a single administration induced a significant reduction of BCMA+ cells after only 24 h. A clinical study documented almost a 90% overall response rate, including 44.4% sCR/CR and more than 90% of MRD negativity achievement, with a good toxicity profile (184).
The further development of CC-93269 (former EM901), a human IgG1-based T-cell engager that binds to BCMA and CD3 epsilon in a 2 + 1 format, is promising as well, as shown by Dr. Costa at ASH 2019 (154). In a dose-escalation trial of 30 patients with relapsed or refractory disease, the 10-mg dose of CC-93269 induced responses in 89% of patients, including complete or stringent complete remission in 44%. Minimal residual disease negativity was achieved by 92% of responders. Although cytokine-release syndrome occurred in about three-quarters of patients, cases were mostly grade 1 or 2, and it tapered off after the first dose.
TNB-383B and TNB-384B have been developed by Tenebio based on in silico analysis of heavy chain only/fixed light chain antibody sequences (202). TNB-383B is a BCMA x CD3 bispecific T-cell redirecting antibody incorporating an activating a unique anti-CD3 moiety (selective in the -383B platform, pan-T-cell activator in the -384B platform), two heavy-chain-only anti-BCMA moieties for a 2:1 tumor associated antigen to CD3 stoichiometry, and a silenced human IgG4 Fc tail (203). The bivalent BCMA binding reduces APRIL competition, conferring high specificity and avidity to the anti-BCMA moieties. Differently from other pan-T cell activating T-bsAbs that can overstimulate T cells, TNB-383B preferentially activates effector over regulatory T-cells and uncouple cytokine release from anti-tumor activity, induce PC lysis, regardless of very high or low E:T ratio (203), reducing the CRS risk, as shown in further clinical development (155, 202, 203). Data presented at last 2020 ASH meeting showed a favorable safety profile in patients with R/R MM and achieved an overall response rate of 80% at doses ≥40 mg every 3 weeks. The most common adverse events were cytokine release syndrome, fatigue, headache, anemia, infection, and nausea. TNB-383B was well tolerated at doses up to 40 mg, without the need for step/split dosing. A preliminary ORR of 52% (12/23) was observed at doses ≥5.4 mg, including deep (6 PR/3 VGPR/3 CR) and durable (up to 24 weeks) responses despite dosing only every 3 weeks (155).
PF-06863135 (Elranatamab) is an anti-BCMA x anti-CD3 BsAb that consists of targeting arms within an IgG2a Fc backbone, given with a weekly subcutaneous (SC) infusion to allow higher doses than intravenous administration without increasing adverse events (156, 204). Recent update about its safety and efficacy in RRMM patients has been reported at ASH 2020 by Dr. Lesokhin. Responses were achieved with SC dosing of Elranatamab in 6 of 8 (75%) patients at the two highest dose levels evaluated. However, the enrollment of the phase-2 trial MagnetisMM-3 has been paused in May 2021 in US due to peripheral neuropathy.
Novel mechanisms of drug resistance are emerging, like the loss or reduction of BCMA antigen, requiring alternative antigens to target. A promising antigen is the G-protein-coupled receptor class 5 member D (GPRC5D), expressed selectively at high levels in MM cells and associated to inferior outcome in MM patients, independently from BCMA expression, even if its function and ligand is still largely known (205).
Talquetamab (JNJ-64407564) is a GPRC5D x CD3 DuoBody able, in vitro, to induce cytotoxicity independently from the number of BCMA receptors or the amount of sBCMA, and, in vivo, to recruit T cells at the tumor site, without affecting humoral immunity due to lack of expression on B memory cells. Robust preclinical data provided the rationale for the ongoing phase NCT03399799I clinical trial (122).
Cevostamab (BFCR4350A) is a new bispecific antibody developed by Roche that simultaneously binds to the CD3 protein on immune T-cells and a portion of the Fc receptor-like protein 5 (FcRH5), a protein receptor found in nearly all myeloma cells, more highly expressed in cell carrying 1q21 abnormalities (206). Preliminary data discussed at ASH 2020 showed ORR of about 53%, irrespective of target expression level in patients. Deep and durable responses were observed in patients with high-risk cytogenetics, triple-class refractory disease, and/or prior exposure to anti-CD38 monoclonal antibodies, CAR T cells, or antibody-drug conjugates (207). In peripheral blood, Cevostamab induced robust CD8+ T-cell activation and proliferation and IFN-γ induction at active doses (3.6 mg), up to 20-fold higher than at baseline. CD8+ tumor-infiltrating T-cell levels were higher on treatment in responders than in non-responders, and T-cell expansion by end of the first cycle was more pronounced in responders than in non-responders, irrespective of baseline CD8+ T-cell levels (208).
The recent discovery that, under physiological conditions, IgG4 can engage Fab-arms exchange (209), has prompted the technology of DuoBody platform (195, 210, 211). In Duobodies, introducing matched mutations at the CH3 interfaces creates a IgG1 bispecific antibody favoring and then stabilizing Fab arm exchange, for the generation of stable bispecific IgG1 antibodies in which heavy and light chain homodimers from two different antibodies form a single heterodimeric bispecific antibody (211–214).
Teclistamab (JNJ-64007957) is a DuoBody bsAb that induces T cell-mediated cytotoxicity against BCMA-expressing myeloma cells, independently from the amount of sBCMA, APRIL, or BAFF (Pillarisetti et al., 2020). Teclistamab has shown to be highly active in vitro on immortalized and primary myeloma cells, obtained also from daratumumab-refractory patients (215). Teclistamab is currently being evaluated in a Phase 2 clinical study for the treatment of relapsed or refractory multiple myeloma (NCT04557098) and is also being explored in combination studies (NCT04586426, NCT04108195). At ASCO 2020 congress, Dr. Usmani presented excellent preliminary results from the ongoing study of weekly teclistamab in RRMM (NCT03145181), with manageable safety across all doses explored and 78% overall response rate (102). However, the efficacy of Teclistamab was inversely related to the PDL-1 expression on RRMM cells, thus ongoing trials are investigating the possibility of overcoming this resistance using a combination with PDL1 inhibitors (216). Additional partners for combination therapy include γ-secretase inhibitors which potentiate Teclistamab killing capacity by elevating BCMA surface expression (215).
HPN217 is a tri-specific T cell activating construct (TriTAC) consisting of three binding domains: an N-terminal single domain antibody (sdAb) that binds to human BCMA, a middle sdAb that binds to human serum albumin (HSA), and a C-terminal single chain Fv (scFv) that binds to CD3ϵ of the T cell receptor (TCR) complex (217). The in vitro pharmacological activity of HPN217 was evaluated by T cell-dependent cellular cytotoxicity (TDCC) assays. In co-cultures of T cells from normal human or cynomolgus monkey donors, target tumor cells, and HSA, HPN217 mediated dose-dependent and BCMA-dependent cytotoxicity with EC50 values ranging from 0.05 to 0.7 nM. Killing was dependent on expression of BCMA on target tumor cells.
Non-clinical in vivo properties of HPN217 were evaluated in xenograft models and a single dose pharmacokinetic (PK) study in cynomolgus monkeys. HPN217 mediated dose-dependent growth suppression against the RPMI-8226 MM model and Jeko-1 mantle cell lymphoma model expressing relatively low levels of 5,600 and 2,200 copies of BCMA per cell, respectively. Serum half-life, volume of distribution, and clearance appeared to be independent of dose. HPN217 was demonstrated to be stable and remained intact up to 3 weeks in vivo as demonstrated by a functional ligand binding assay using recombinant CD3ϵ and BCMA, respectively, to capture and detect HPN217. Importantly, serum samples collected 1 week after dosing were as potent as stock HPN217 in MM tumor cell killing in TDCC assays.
RO7297089 is a potent therapeutic agent in vitro and selectively kills BCMA expressing MM-PCs by activating innate immunity, via ADCC and ADCP, with low incidence of acute cytokine release. In a 1-month repeat-dose study in cynomolgus monkeys, RO7297089 was well tolerated, and there were no test article-related adverse effects at up to 50 mg/kg, with no significant cytokine release. RO7297089 represents a novel and promising MOA with a favorable safety profile, distinct from the T cell-based BCMA-targeting modalities in the clinic (218, 219).
CTX‐4419, is a first‐in‐class NKp30xBCMA bispecific, able to induce cytokine production, NK-cell proliferation, and potent tumor cell killing of target cells, independently from high, intermediate, or low BCMA expression. Differently BCMA‐IgG1 mAbs can activate NK cells in the absence of CD16A engagement (220).
2A9-MICA consists of human MICA extracellular region and a single-chain antibody fragment (scFv) that targets BCMA generated by phage display technology. In vitro, 2A9-MICA activated NK cell-mediated cytotoxicity and induced NK cells to kill BCMA-positive human myeloma cells. Moreover, in BCMA-positive, MM-bearing nude mice, 2A9-MICA specifically targeted tumor tissue, where it effectively recruited immune cells and inhibited tumor tissue growth showed superior antitumor activity (221).
Blocking PD-1/PDl-1 Axis by mAbs in MM
Several experimental pre-clinical data indicate that PD-L1/PD-1 blockade by mAbs provided promising anti-MM effects. In vitro PD-L1/PD-1 blockade overcame BM MSC-mediated MM growth and directly enhanced NK and T cell mediated anti-MM responses (222, 223). MM cells by PD-L1 expression inhibit the activity of CTLs, acquiring a proliferative advantage which results in immune evasion and resistance to anti-myeloma drugs (224). In vivo PD-L1 blockade prolonged mice survival after stem-cell transplantation (225–228) as well as PD-1 blockade also prolonged the survival in disseminated myeloma-bearing mice (228, 229), by mainly acting on CD4+ or CD8+ T cells (229). In these models, PD-1 expression on both CD8+ and CD4+ T cells was higher in mice with advanced MM as compared to non-tumor bearing ones.
mAbs targeting the PD-1/PD-L1 axis can be divided into two different groups: (i) those against the PD-1 receptor and (ii) those against the ligands (PD-L1/PD-L2). Nivolumab, pembrolizumab, and pidilizumab are the main anti-PD-1 mAbs used whereas anti-PD-L1 mAbs are durvalumab and atezolizumab. However, despite promising pre-clinical data, the use of mAbs antiPD-1/PD-L1 mAbs as single agents did not show a significant clinical effect in relapsed refractory MM. On the other hand, the phase III trial evaluating lenalidomide and dexamethasone in combination with pembrolizumab in patients with MM presented unexpected safety findings and was discontinued. Accordingly, the other clinical trials anti PD-1/PD-L1 mAbs in combination with IMiDs have been interrupted. Actually the identification of the best MM patients candidate to the treatment with PD-1/PD-L1 blockade is still be unknown.
Conclusions: Challenges and Perspectives of Immunotherapy in Multiple Myeloma
Immunotherapy is revolutionizing the therapeutic scenario of both newly diagnosed and refractory-relapsed MM patients. Novel challenges are emerging on how to choose the target and the therapeutic format as summarized in Table 5, the best sequential approach, timing, and patients’ characteristics.
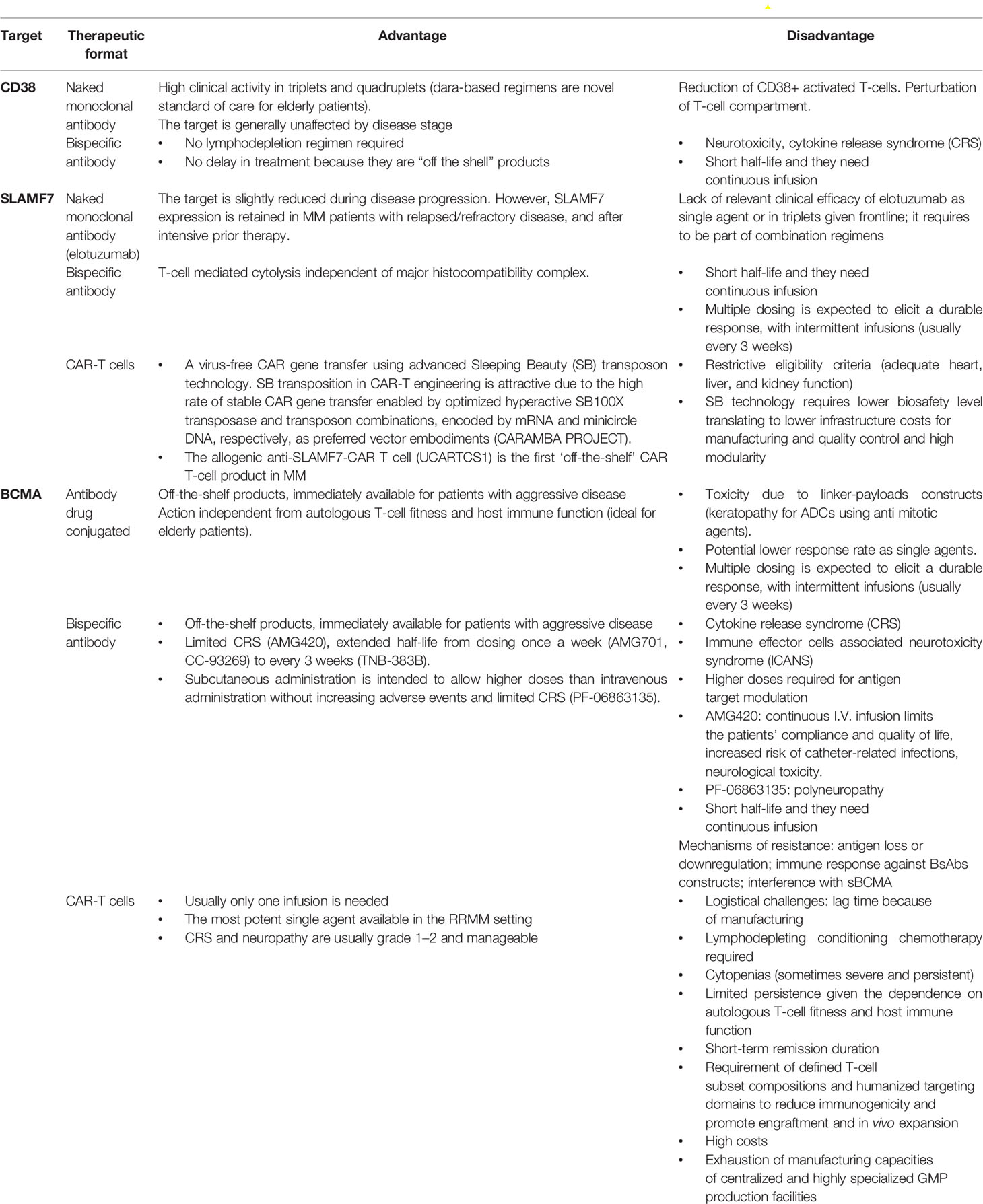
Table 5 Vantages and Disadvantage of monoclonal antibodies, Bispecific antibodies, Antibody drug conjugated and CAR-T cells.
CAR-T cell therapy against BCMA is one of the most powerful single-agent for RRMM patients (with ORR range 50–90% across the studies), but it is affected by logistical constrains, with up to 20% drop-out rate in the manufacturing time (4–7 weeks) for complications associated with disease progression (230). To overcome these limitations, reduce the high costs and face with exhaustion of manufacturing capacities of centralized and highly specialized production facilities, some technological improvements are ongoing, including virus-free gene transfer, automated point-of-care production and allogeneic cell products to provide off-the-shelf CAR-T cells products (113, 231, 232).
The limited persistence and lack of survival plateau is an additional limitation of CAR-T cells in MM. Some authors suggest to identify upfront (e.g. high risk patients with extramedullary presentation or adverse cytogenetics or biallelic TP53 inactivation) or early during the treatment (e.g. after induction or at first minimal residual disease detection after autologous stem cell transplantation) those patients with the highest chance of benefiting from T-cells redirecting therapies with the final goal of cure (231) or achievement of persistence of minimal residual disease negativity (233). However, T-cell function looks like compromised since the asymptomatic phase of disease (234–236), arising the question about how to improve the efficacy of current immunotherapy approaches and their toxicity profile. For example, T cell proliferation decreased in presence of mature neutrophils (234, 236–238). The cytotoxic potential of T cells engaged by EM801 increased notably with the depletion of mature neutrophils (236), arising the question if immunotherapy should be adapted to an extensive immune profiling not limited to T-cells only.
In immunotherapy of both solid and hematological cancers, there is an increasing evidence about the prominent role of antibody’s constant region, much of which is mediated through interaction of the Fc with FcγRs, that could be engineered to modify their pharmacokinetics and pharmacodynamics. Neutrophils could positively affect the activity of several mAbs, in vitro and in vivo, via the recognition of IgA‐opsonized tumor targets by FcαRI/CD64, as recently reviewed (239). However, the mechanisms of neutrophil‐induced tumor killing are still under debate and the role of neutrophils, either positive or negative, is far from clear.
Reducing progressively the costs, potential toxicity, and therapy complexity is the major strength of off-the-shelf immunotherapeutic strategies like bispecific antibodies and ADCs, which function is largely independent from autologous T-cell fitness and host immune function. Cytokine release syndrome and immune effector cells associated neurotoxicity syndrome can be managed at lower dosages or changing the format to reduce non-specific T-cell activation (as in TNB-383B).
While clinical trials are not yet mature for a direct comparison of several classes of agents and we still need larger series and further confirmation, the favorable safety profile of the first-in-class belantamab mafodotin, makes it a potential great choice for the elderly patients (232), especially when the therapeutic goal is not disease eradication but long-term control of disease, with repeated infusions. The most relevant challenge for the ADCs development is to reduce toxicity related to linker-payloads constructs, like keratopathy associated to ADCs with anti-mitotic agents and neuropathy (240).
The mechanisms involved in the acquired resistance to anti-CD38 mAbs are not fully understood but could involve the downregulation of the CD38 on cell surface, NK and T cells number and exhaustion, overexpression of complement inhibitory proteins, and expression of inhibitory pathways as CD47-SIRPα (45, 49). The therapy in combination with immunomodulatory drugs seems to potentiate the effect of the anti-CD38 mAbs compared with single-agent treatment, increasing the activity of NK cells and on macrophage (9). These mechanisms lead to reach significant response even in relapsed/refractory MM patients. Other drugs (HDACs and ATRA) could overcome the resistance to anti-CD38 mAbs increasing the expression of the CD38 molecule (28, 72, 73).
In contrast to BCMA, CD38 and SLAMF7 antigens show stable expression levels throughout the successive lines of MM treatments, making their contemporary dual targeting an emerging therapeutic option. Hemibodies are a pair of complementary antibody fragments that redirect T cells against cancer-defining antigen combinations. Hemibodies addressing CD38 and SLAMF7 recruit T cells for the exquisite elimination of dual antigen positive multiple myeloma cells while leaving single antigen positive bystanders unharmed. Differently from T-cell redirecting therapies targeting only CD38 and SLAMF7 targeting hemibodies do not induce massive cytokine release and T cell fratricide reactions, translating into very low off-tumor toxicity in clinical settings (241).
The unique mechanisms of action of monoclonal antibodies make them a perfect component to be used alone or in combination with present therapeutic treatments, which could improve the efficacy of the treatment and probably overcome resistance (114). In the upcoming years, a robust selection of patients, based on both genomic and immune profiling to test respectively the clonal architecture and the host immune fitness, combined to multi-target immunotherapy could induce a further major paradigm shift to offer long-term control of disease and hopefully cure to most of MM patients.
Author Contributions
Conceptualization: FR and NG. PS and AR, writing the paper; FR and NG—writing and editing. VM prepared the figures. LN and GS prepared the tables. LC revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported in part by two grants from the Associazione Italiana per la Ricerca sul Cancro (AIRC) IG2017 n. 20299 (NG), IG n.22131 (FR), Ministero della Salute Italiana PE-2016-02361261 and Università degli Studi di Catania, “Fondi di ateneo 2020-2022, Università di Catania, linea Open Access.”
Conflict of Interest
NG received research funding and honoraria from Amgen, Bristol Mayers Squibb, Celgene, Millenium Pharmaceutical, and Janssen Pharmaceutical. AR and FR received research funding and honoraria from Amgen.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Associazione Italiana contro Leucemie, Linfomi e Mielomi ONLUS, ParmAIL and AIL Catania, and FON.CA.NE.SA. (Fondazione Catanese per lo Studio delle Malattie Neoplastiche del Sangue) for the support.
References
1. Anderson KC. Multiple Myeloma. Hematol Oncol Clin North Am (2014) 28(5):xi–xii. doi: 10.1016/j.hoc.2014.08.001
2. Romano A, Conticello C, Cavalli M, Vetro C, La Fauci A, Parrinello NL, et al. Immunological Dysregulation in Multiple Myeloma Microenvironment. BioMed Res Int (2014) 2014:198539. doi: 10.1155/2014/198539
3. Romano A, Conticello C, Cavalli M, Vetro C, Di Raimondo C, Di Martina V, et al. Salvage Therapy of Multiple Myeloma: The New Generation Drugs. BioMed Res Int (2014) 2014:456037. doi: 10.1155/2014/456037
4. Gulla A, Anderson KC. Multiple Myeloma: The (R)Evolution of Current Therapy and a Glance Into Future. Haematologica (2020) 105(10):2358–67. doi: 10.3324/haematol.2020.247015
5. De Raeve HR, Vanderkerken K. The Role of the Bone Marrow Microenvironment in Multiple Myeloma. Histol Histopathol (2005) 20(4):1227–50. doi: 10.14670/HH-20.1227
6. Giuliani N, Accardi F, Marchica V, Dalla Palma B, Storti P, Toscani D, et al. Novel Targets for the Treatment of Relapsing Multiple Myeloma. Expert Rev Hematol (2019) 12(7):481–96. doi: 10.1080/17474086.2019.1624158
7. Chillemi A, Zaccarello G, Quarona V, Lazzaretti M, Martella E, Giuliani N, et al. CD38 and Bone Marrow Microenvironment. Front Biosci (2014) 19:152–62. doi: 10.2741/4201
8. Jung SH, Lee HJ, Vo MC, Kim HJ, Lee JJ. Immunotherapy for the Treatment of Multiple Myeloma. Crit Rev Oncol Hematol (2017) 111:87–93. doi: 10.1016/j.critrevonc.2017.01.011
9. Musto P, La Rocca F. Monoclonal Antibodies in Relapsed/Refractory Myeloma: Updated Evidence From Clinical Trials, Real-Life Studies, and Meta-Analyses. Expert Rev Hematol (2020) 13(4):331–49. doi: 10.1080/17474086.2020.1740084
10. Dalla Palma B, Marchica V, Catarozzo MT, Giuliani N, Accardi F. Monoclonal and Bispecific Anti-BCMA Antibodies in Multiple Myeloma. J Clin Med (2020) 9(9):3022. doi: 10.3390/jcm9093022
11. Beers SA, Glennie MJ, White AL. Influence of Immunoglobulin Isotype on Therapeutic Antibody Function. Blood (2016) 127(9):1097–101. doi: 10.1182/blood-2015-09-625343
12. de Taeye SW, Rispens T, Vidarsson G. The Ligands for Human IgG and Their Effector Functions. Antibodies (Basel) (2019) 8(2):30. doi: 10.3390/antib8020030
13. Bournazos S, Gupta A, Ravetch JV. The Role of IgG Fc Receptors in Antibody-Dependent Enhancement. Nat Rev Immunol (2020) 20(10):633–43. doi: 10.1038/s41577-020-00410-0
14. Deaglio S, Morra M, Mallone R, Ausiello CM, Prager E, Garbarino G, et al. Human CD38 (ADP-Ribosyl Cyclase) Is a Counter-Receptor of CD31, an Ig Superfamily Member. J Immunol (1998) 160(1):395–402.
15. Reinherz EL, Kung PC, Goldstein G, Levey RH, Schlossman SF. Discrete Stages of Human Intrathymic Differentiation: Analysis of Normal Thymocytes and Leukemic Lymphoblasts of T-Cell Lineage. Proc Natl Acad Sci United States America (1980) 77(3):1588–92. doi: 10.1073/pnas.77.3.1588
16. van de Donk NW, Janmaat ML, Mutis T, Lammerts van Bueren JJ, Ahmadi T, Sasser AK, et al. Monoclonal Antibodies Targeting CD38 in Hematological Malignancies and Beyond. Immunol Rev (2016) 270(1):95–112. doi: 10.1111/imr.12389
17. Deaglio S, Aydin S, Vaisitti T, Bergui L, Malavasi F. CD38 at the Junction Between Prognostic Marker and Therapeutic Target. Trends Mol Med (2008) 14(5):210–8. doi: 10.1016/j.molmed.2008.02.005
18. Deaglio S, Mallone R, Baj G, Arnulfo A, Surico N, Dianzani U, et al. CD38/CD31, a Receptor/Ligand System Ruling Adhesion and Signaling in Human Leukocytes. Chem Immunol (2000) 75:99–120. doi: 10.1159/000058765
19. Malavasi F, Deaglio S, Funaro A, Ferrero E, Horenstein AL, Ortolan E, et al. Evolution and Function of the ADP Ribosyl Cyclase/CD38 Gene Family in Physiology and Pathology. Physiol Rev (2008) 88(3):841–86. doi: 10.1152/physrev.00035.2007
20. Lee HC, Aarhus R. Wide Distribution of an Enzyme That Catalyzes the Hydrolysis of Cyclic ADP-Ribose. Biochim Biophys Acta (BBA) - Protein Structure Mol Enzymol (1993) 1164(1):68–74. doi: 10.1016/0167-4838(93)90113-6
21. Lee HC, Zocchi E, Guida L, Franco L, Benatti U, De Flora A. Production and Hydrolysis of Cyclic ADP-Ribose at the Outer Surface of Human Erythrocytes. Biochem Biophys Res Commun (1993) 191(2):639–45. doi: 10.1006/bbrc.1993.1265
22. Ramaschi G, Torti M, Festetics ET, Sinigaglia F, Malavasi F, Balduini C. Expression of Cyclic ADP-Ribose-Synthetizing CD38 Molecule on Human Platelet Membrane. Blood (1996) 87(6):2308–13. doi: 10.1182/blood.V87.6.2308.bloodjournal8762308
23. Sandoval-Montes C, Santos-Argumedo L. CD38 Is Expressed Selectively During the Activation of a Subset of Mature T Cells With Reduced Proliferation But Improved Potential to Produce Cytokines. J Leuk Biol (2005) 77(4):513–21. doi: 10.1189/jlb.0404262
24. Malavasi F, Funaro A, Alessio M, Demonte LB, Ausiello CM, Dianzani U, et al. Cd38 - a Multilineage Cell Activation Molecule With a Split Personality. Int J Clin Lab Res (1992) 22(2):73–80. doi: 10.1007/Bf02591400
25. Oleinika K, Mauri C, Salama AD. Effector and Regulatory B Cells in Immune-Mediated Kidney Disease. Nat Rev Nephrol (2019) 15(1):11–26. doi: 10.1038/s41581-018-0074-7
26. Malavasi F, Deaglio S, Damle R, Cutrona G, Ferrarini M, Chiorazzi N. CD38 and Chronic Lymphocytic Leukemia: A Decade Later. Blood (2011) 118(13):3470–8. doi: 10.1182/blood-2011-06-275610
27. Costa F, Toscani D, Chillemi A, Quarona V, Bolzoni M, Marchica V, et al. Expression of CD38 in Myeloma Bone Niche: A Rational Basis for the Use of Anti-CD38 Immunotherapy to Inhibit Osteoclast Formation. Oncotarget (2017) 8(34):56598–611. doi: 10.18632/oncotarget.17896
28. Nijhof IS, Groen RW, Lokhorst HM, van Kessel B, Bloem AC, van Velzen J, et al. Upregulation of CD38 Expression on Multiple Myeloma Cells by All-Trans Retinoic Acid Improves the Efficacy of Daratumumab. Leukemia (2015) 29(10):2039–49. doi: 10.1038/leu.2015.123
29. Quarona V, Ferri V, Chillemi A, Bolzoni M, Mancini C, Zaccarello G, et al. Unraveling the Contribution of Ectoenzymes to Myeloma Life and Survival in the Bone Marrow Niche. Ann New York Acad Sci (2015) 1335:10–22. doi: 10.1111/nyas.12485
30. Watt SM, Gschmeissner SE, Bates PA. PECAM-1: Its Expression and Function as a Cell Adhesion Molecule on Hemopoietic and Endothelial Cells. Leuk Lymphoma (1995) 17(3-4):229–44. doi: 10.3109/10428199509056827
31. Morandi F, Airoldi I, Marimpietri D, Bracci C, Faini AC, Gramignoli R. CD38, a Receptor With Multifunctional Activities: From Modulatory Functions on Regulatory Cell Subsets and Extracellular Vesicles, to a Target for Therapeutic Strategies. Cells (2019) 8(12):1527. doi: 10.3390/cells8121527
32. Aarhus R, Graeff RM, Dickey DM, Walseth TF, Lee HC. ADP-Ribosyl Cyclase and CD38 Catalyze the Synthesis of a Calcium-Mobilizing Metabolite From NADP. J Biol Chem (1995) 270(51):30327–33. doi: 10.1074/jbc.270.51.30327
33. Horenstein ALF, Faini AC, Morandi F, Bracci C, Lanza F, Giuliani N, et al. The Circular Life of Human CD38: From Basic Science to Clinics and Back. Molecules (2020) 25:4844. doi: 10.3390/molecules25204844
34. Usmani SZ, Weiss BM, Plesner T, Bahlis NJ, Belch A, Lonial S, et al. Clinical Efficacy of Daratumumab Monotherapy in Patients With Heavily Pretreated Relapsed or Refractory Multiple Myeloma. Blood (2016) 128(1):37–44. doi: 10.1182/blood-2016-03-705210
35. Mikhael J, Richter J, Vij R, Cole C, Zonder J, Kaufman JL, et al. A Dose-Finding Phase 2 Study of Single Agent Isatuximab (Anti-CD38 mAb) in Relapsed/Refractory Multiple Myeloma. Leukemia (2020) 34(12):3298–309. doi: 10.1038/s41375-020-0857-2
36. Raab MS, Engelhardt M, Blank A, Goldschmidt H, Agis H, Blau IW, et al. MOR202, a Novel Anti-CD38 Monoclonal Antibody, in Patients With Relapsed or Refractory Multiple Myeloma: A First-in-Human, Multicentre, Phase 1-2a Trial. Lancet Haematol (2020) 7(5):e381–e94. doi: 10.1016/S2352-3026(19)30249-2
37. Zonder JA, Mohrbacher AF, Singhal S, van Rhee F, Bensinger WI, Ding H, et al. A Phase 1, Multicenter, Open-Label, Dose Escalation Study of Elotuzumab in Patients With Advanced Multiple Myeloma. Blood (2012) 120(3):552–9. doi: 10.1182/blood-2011-06-360552
38. de Weers M, Tai YT, van der Veer MS, Bakker JM, Vink T, Jacobs DC, et al. Daratumumab, a Novel Therapeutic Human CD38 Monoclonal Antibody, Induces Killing of Multiple Myeloma and Other Hematological Tumors. J Immunol (2011) 186(3):1840–8. doi: 10.4049/jimmunol.1003032
39. Noort WA, Groen RWJ, Raymakers R, Aalders L, Hofhuis FM, Van Kessel B, et al. Daratumumab, a Novel Therapeutic Human CD38 Monoclonal Antibody, Induces Killing of Refractory Patient-Derived Multiple Myeloma Cells, Growing in a Novel Humanized Mouse MM Model. Blood (2012) 120(21):940–. doi: 10.1182/blood.V120.21.940.940
40. van de Donk N, Richardson PG, Malavasi F. CD38 Antibodies in Multiple Myeloma: Back to the Future. Blood (2018) 131(1):13–29. doi: 10.1182/blood-2017-06-740944
41. Deckert J, Wetzel M-C, Bartle LM, Skaletskaya A, Goldmacher VS, Vallée F, et al. SAR650984, A Novel Humanized CD38-Targeting Antibody, Demonstrates Potent Antitumor Activity in Models of Multiple Myeloma and Other CD38<sup<+</sup< Hematologic Malignancies. Clin Cancer Res (2014) 20(17):4574. doi: 10.1158/1078-0432.CCR-14-0695
42. Attal M, Richardson PG, Rajkumar SV. Isatuximab Plus Pomalidomide and Low-Dose Dexamethasone Versus Pomalidomide and Low-Dose Dexamethasone in Patients With Relapsed and Refractory Multiple Myeloma (ICARIA-MM): A Randomised, Multicentre, Open-Label, Phase 3 Study (Vol 394, Pg 2096, 2019). Lancet (2019) 394(10214):2072–. doi: 10.1016/S0140-6736(19)32556-5
43. van de Donk N, Usmani SZ. CD38 Antibodies in Multiple Myeloma: Mechanisms of Action and Modes of Resistance. Front Immunol (2018) 9:2134. doi: 10.3389/fimmu.2018.02134
44. Junker F, Gordon J, Qureshi O. Fc Gamma Receptors and Their Role in Antigen Uptake, Presentation, and T Cell Activation. Front Immunol (2020) 11:1393(1393). doi: 10.3389/fimmu.2020.01393
45. Saltarella I, Desantis V, Melaccio A, Solimando AG, Lamanuzzi A, Ria R, et al. Mechanisms of Resistance to Anti-CD38 Daratumumab in Multiple Myeloma. Cells (2020) 9(1):167. doi: 10.3390/cells9010167
46. Wang Y, Zhang Y, Hughes T, Zhang J, Caligiuri MA, Benson DM, et al. Fratricide of NK Cells in Daratumumab Therapy for Multiple Myeloma Overcome by Ex Vivo-Expanded Autologous NK Cells. Clin Cancer Res: Off J Am Assoc Cancer Res (2018) 24(16):4006–17. doi: 10.1158/1078-0432.CCR-17-3117
47. Cho H, Kim KH, Lee H, Kim CG, Chung H, Choi YS, et al. Adaptive Natural Killer Cells Facilitate Effector Functions of Daratumumab in Multiple Myeloma. Clin Cancer Res: Off J Am Assoc Cancer Res (2021) 27(10):2947–58. doi: 10.1158/1078-0432.CCR-20-3418
48. Yeap WH, Wong KL, Shimasaki N, Teo EC, Quek JK, Yong HX, et al. CD16 Is Indispensable for Antibody-Dependent Cellular Cytotoxicity by Human Monocytes. Sci Rep (2016) 6:34310. doi: 10.1038/srep34310
49. Storti P, Vescovini R, Costa F, Marchica V, Toscani D, Dalla Palma B, et al. CD14(+) CD16(+) Monocytes Are Involved in Daratumumab-Mediated Myeloma Cells Killing and in Anti-CD47 Therapeutic Strategy. Br J Haematol (2020) 190:430–6. doi: 10.1111/bjh.16548
50. Zhu C, Song Z, Wang A, Srinivasan S, Yang G, Greco R, et al. Isatuximab Acts Through Fc-Dependent, Independent, and Direct Pathways to Kill Multiple Myeloma Cells. Front Immunol (2020) 11:1771. doi: 10.3389/fimmu.2020.01771
51. Moreno L, Perez C, Zabaleta A, Manrique I, Alignani D, Ajona D, et al. The Mechanism of Action of the Anti-CD38 Monoclonal Antibody Isatuximab in Multiple Myeloma. Clin Cancer Res: Off J Am Assoc Cancer Res (2019) 25(10):3176–87. doi: 10.1158/1078-0432.CCR-18-1597
52. Malavasi F, Faini AC. Mechanism of Action of a New Anti-CD38 Antibody: Enhancing Myeloma Immunotherapy. Clin Cancer Res: Off J Am Assoc Cancer Res (2019) 25(10):2946–8. doi: 10.1158/1078-0432.CCR-19-0260
53. Overdijk MB, Verploegen S, Bogels M, van Egmond M, Lammerts van Bueren JJ, Mutis T, et al. Antibody-Mediated Phagocytosis Contributes to the Anti-Tumor Activity of the Therapeutic Antibody Daratumumab in Lymphoma and Multiple Myeloma. mAbs (2015) 7(2):311–21. doi: 10.1080/19420862.2015.1007813
54. Diebolder CA, Beurskens FJ, de Jong RN, Koning RI, Strumane K, Lindorfer MA, et al. Complement Is Activated by IgG Hexamers Assembled at the Cell Surface. Science (2014) 343(6176):1260–3. doi: 10.1126/science.1248943
55. Rougé L, Chiang N, Steffek M, Kugel C, Croll TI, Tam C, et al. Structure of CD20 in Complex With the Therapeutic Monoclonal Antibody Rituximab. Science (2020) 367:1224–30. doi: 10.1126/science.aaz9356
56. Kumar A, Planchais C, Fronzes R, Mouquet H, Reyes N. Binding Mechanisms of Therapeutic Antibodies to Human CD20. Science (2020) 369:793–9. doi: 10.1126/science.abb8008
57. Krejcik J, Casneuf T, Nijhof IS, Verbist B, Bald J, Plesner T, et al. Daratumumab Depletes CD38+ Immune Regulatory Cells, Promotes T-Cell Expansion, and Skews T-Cell Repertoire in Multiple Myeloma. Blood (2016) 128(3):384–94. doi: 10.1182/blood-2015-12-687749
58. van de Donk N. Immunomodulatory Effects of CD38-Targeting Antibodies. Immunol Lett (2018) 199:16–22. doi: 10.1016/j.imlet.2018.04.005
59. Feng X, Zhang L, Acharya C, An G, Wen K, Qiu L, et al. Targeting CD38 Suppresses Induction and Function of T Regulatory Cells to Mitigate Immunosuppression in Multiple Myeloma. Clin Cancer Res: Off J Am Assoc Cancer Res (2017) 23(15):4290–300. doi: 10.1158/1078-0432.ccr-16-3192
60. Horenstein AL, Chillemi A, Quarona V, Zito A, Roato I, Morandi F, et al. NAD(+)-Metabolizing Ectoenzymes in Remodeling Tumor-Host Interactions: The Human Myeloma Model. Cells (2015) 4(3):520–37. doi: 10.3390/cells4030520
61. Vijayan D, Young A, Teng MWL, Smyth MJ. Targeting Immunosuppressive Adenosine in Cancer. Nat Rev Cancer (2017) 17(12):709–24. doi: 10.1038/nrc.2017.86
62. Jiang H, Acharya C, An G, Zhong M, Feng X, Wang L, et al. SAR650984 Directly Induces Multiple Myeloma Cell Death via Lysosomal-Associated and Apoptotic Pathways, Which Is Further Enhanced by Pomalidomide. Leukemia (2016) 30(2):399–408. doi: 10.1038/leu.2015.240
63. van der Veer MS, de Weers M, van Kessel B, Bakker JM, Wittebol S, Parren PW, et al. Towards Effective Immunotherapy of Myeloma: Enhanced Elimination of Myeloma Cells by Combination of Lenalidomide With the Human CD38 Monoclonal Antibody Daratumumab. Haematologica (2011) 96(2):284–90. doi: 10.3324/haematol.2010.030759
64. Hayashi T, Hideshima T, Akiyama M, Podar K, Yasui H, Raje N, et al. Molecular Mechanisms Whereby Immunomodulatory Drugs Activate Natural Killer Cells: Clinical Application. Br J Haematol (2005) 128(2):192–203. doi: 10.1111/j.1365-2141.2004.05286.x
65. Nijhof IS, Groen RW, Noort WA, van Kessel B, de Jong-Korlaar R, Bakker J, et al. Preclinical Evidence for the Therapeutic Potential of CD38-Targeted Immuno-Chemotherapy in Multiple Myeloma Patients Refractory to Lenalidomide and Bortezomib. Clin Cancer Res: Off J Am Assoc Cancer Res (2015) 21(12):2802–10. doi: 10.1158/1078-0432.ccr-14-1813
66. van der Veer MS, de Weers M, van Kessel B, Bakker JM, Wittebol S, Parren PW, et al. The Therapeutic Human CD38 Antibody Daratumumab Improves the Anti-Myeloma Effect of Newly Emerging Multi-Drug Therapies. Blood Cancer J (2011) 1(10):e41. doi: 10.1038/bcj.2011.42
67. Endell J, Samuelsson C, Boxhammer R, Strauss S, Steidl S. Effect of MOR202, a Human CD38 Antibody, in Combination With Lenalidomide and Bortezomib, on Bone Lysis and Tumor Load in a Physiologic Model of Myeloma. J Clin Oncol (2011) 29(15_suppl):8078–. doi: 10.1200/jco.2011.29.15_suppl.8078
68. Busch L, Mougiakakos D, Buttner-Herold M, Muller MJ, Volmer DA, Bach C, et al. Lenalidomide Enhances MOR202-Dependent Macrophage-Mediated Effector Functions via the Vitamin D Pathway. Leukemia (2018) 32(11):2445–58. doi: 10.1038/s41375-018-0114-0
69. Gorgun G, Calabrese E, Soydan E, Hideshima T, Perrone G, Bandi M, et al. Immunomodulatory Effects of Lenalidomide and Pomalidomide on Interaction of Tumor and Bone Marrow Accessory Cells in Multiple Myeloma. Blood (2010) 116(17):3227–37. doi: 10.1182/blood-2010-04-279893
70. Bolzoni M, Storti P, Bonomini S, Todoerti K, Guasco D, Toscani D, et al. Immunomodulatory Drugs Lenalidomide and Pomalidomide Inhibit Multiple Myeloma-Induced Osteoclast Formation and the RANKL/OPG Ratio in the Myeloma Microenvironment Targeting the Expression of Adhesion Molecules. Exp Hematol (2013) 41(4):387–97 e1. doi: 10.1016/j.exphem.2012.11.005
71. Fedele PL, Willis SN, Liao Y, Low MS, Rautela J, Segal DH, et al. IMiDs Prime Myeloma Cells for Daratumumab-Mediated Cytotoxicity Through Loss of Ikaros and Aiolos. Blood (2018) 132(20):2166–78. doi: 10.1182/blood-2018-05-850727
72. Garcia-Guerrero E, Gogishvili T, Danhof S, Schreder M, Pallaud C, Perez-Simon JA, et al. Panobinostat Induces CD38 Upregulation and Augments the Antimyeloma Efficacy of Daratumumab. Blood (2017) 129(25):3386–8. doi: 10.1182/blood-2017-03-770776
73. Garcia-Guerrero E, Gotz R, Doose S, Sauer M, Rodriguez-Gil A, Nerreter T, et al. Upregulation of CD38 Expression on Multiple Myeloma Cells by Novel HDAC6 Inhibitors Is a Class Effect and Augments the Efficacy of Daratumumab. Leukemia (2020) 35(1):201–4. doi: 10.1038/s41375-020-0840-y
74. Kishimoto H, Hoshino S, Ohori M, Kontani K, Nishina H, Suzawa M, et al. Molecular Mechanism of Human CD38 Gene Expression by Retinoic Acid. Identification of Retinoic Acid Response Element in the First Intron. J Biol Chem (1998) 273(25):15429–34. doi: 10.1074/jbc.273.25.15429
75. Nijhof IS, Casneuf T, van Velzen J, van Kessel B, Axel AE, Syed K, et al. CD38 Expression and Complement Inhibitors Affect Response and Resistance to Daratumumab Therapy in Myeloma. Blood (2016) 128(7):959–70. doi: 10.1182/blood-2016-03-703439
76. Bannas P, Hambach J, Koch-Nolte F. Nanobodies and Nanobody-Based Human Heavy Chain Antibodies As Antitumor Therapeutics. Front Immunol (2017) 8:1603. doi: 10.3389/fimmu.2017.01603
77. Zuch de Zafra CL, Fajardo F, Zhong W, Bernett MJ, Muchhal US, Moore GL, et al. Targeting Multiple Myeloma With AMG 424, a Novel Anti-CD38/CD3 Bispecific T-Cell-Recruiting Antibody Optimized for Cytotoxicity and Cytokine Release. Clin Cancer Res: Off J Am Assoc Cancer Res (2019) 25(13):3921–33. doi: 10.1158/1078-0432.CCR-18-2752
78. Fayon M, Martinez-Cingolani C, Abecassis A, Roders N, Nelson E, Choisy C, et al. Bi38-3 Is a Novel CD38/CD3 Bispecific T-Cell Engager With Low Toxicity for the Treatment of Multiple Myeloma. Haematologica (2021) 106(4):1193–7. doi: 10.3324/haematol.2019.242453
79. Cannons JL, Tangye SG, Schwartzberg PL. SLAM Family Receptors and SAP Adaptors in Immunity. Annu Rev Immunol (2011) 29:665–705. doi: 10.1146/annurev-immunol-030409-101302
80. Buller CW, Mathew PA, Mathew SO. Roles of NK Cell Receptors 2b4 (CD244), CS1 (CD319), and LLT1 (CLEC2D) in Cancer. Cancers (Basel) (2020) 12(7):1755. doi: 10.3390/cancers12071755
81. Cruz-Munoz ME, Dong Z, Shi X, Zhang S, Veillette A. Influence of CRACC, a SLAM Family Receptor Coupled to the Adaptor EAT-2, on Natural Killer Cell Function. Nat Immunol (2009) 10(3):297–305. doi: 10.1038/ni.1693
82. Guo H, Cruz-Munoz ME, Wu N, Robbins M, Veillette A. Immune Cell Inhibition by SLAMF7 Is Mediated by a Mechanism Requiring src Kinases, CD45, and SHIP-1 That Is Defective in Multiple Myeloma Cells. Mol Cell Biol (2015) 35(1):41–51. doi: 10.1128/mcb.01107-14
83. Lee JK, Boles KS, Mathew PA. Molecular and Functional Characterization of a CS1 (CRACC) Splice Variant Expressed in Human NK Cells That Does Not Contain Immunoreceptor Tyrosine-Based Switch Motifs. Eur J Immunol (2004) 34(10):2791–9. doi: 10.1002/eji.200424917
84. Campbell KS, Cohen AD, Pazina T. Mechanisms of NK Cell Activation and Clinical Activity of the Therapeutic SLAMF7 Antibody, Elotuzumab in Multiple Myeloma. Front Immunol (2018) 9:2551. doi: 10.3389/fimmu.2018.02551
85. Pazina T, James AM, Colby KB, Yang Y, Gale A, Jhatakia A, et al. Enhanced SLAMF7 Homotypic Interactions by Elotuzumab Improves NK Cell Killing of Multiple Myeloma. Cancer Immunol Res (2019) 7(10):1633–46. doi: 10.1158/2326-6066.Cir-18-0579
86. Pérez-Quintero LA, Roncagalli R, Guo H, Latour S, Davidson D, Veillette A. EAT-2, a SAP-Like Adaptor, Controls NK Cell Activation Through Phospholipase Cγ, Ca++, and Erk, Leading to Granule Polarization. J Exp Med (2014) 211(4):727–42. doi: 10.1084/jem.20132038
87. Wilson TJ, Garner LI, Metcalfe C, King E, Margraf S, Brown MH. Fine Specificity and Molecular Competition in SLAM Family Receptor Signalling. PloS One (2014) 9(3):e92184. doi: 10.1371/journal.pone.0092184
88. Xie Z, Gunaratne J, Cheong LL, Liu SC, Koh TL, Huang G, et al. Plasma Membrane Proteomics Identifies Biomarkers Associated With MMSET Overexpression in T(4;14) Multiple Myeloma. Oncotarget (2013) 4(7):1008–18. doi: 10.18632/oncotarget.1049
89. Kim JR, Mathew SO, Mathew PA. Blimp-1/PRDM1 Regulates the Transcription of Human CS1 (SLAMF7) Gene in NK and B Cells. Immunobiology (2016) 221(1):31–9. doi: 10.1016/j.imbio.2015.08.005
90. Chen J, Zhong M-C, Guo H, Davidson D, Mishel S, Lu Y, et al. SLAMF7 Is Critical for Phagocytosis of Haematopoietic Tumour Cells. Via Mac-1 Integrin Nat (2017) 544(7651):493–7. doi: 10.1038/nature22076
91. Ishibashi M, Soeda S, Sasaki M, Handa H, Imai Y, Tanaka N, et al. Clinical Impact of Serum Soluble SLAMF7 in Multiple Myeloma. Oncotarget (2018) 9(78):34784–93. doi: 10.18632/oncotarget.26196
92. Kikuchi J, Hori M, Iha H, Toyama-Sorimachi N, Hagiwara S, Kuroda Y, et al. Soluble SLAMF7 Promotes the Growth of Myeloma Cells via Homophilic Interaction With Surface SLAMF7. Leukemia (2020) 34(1):180–95. doi: 10.1038/s41375-019-0525-6
93. Hsi ED, Steinle R, Balasa B, Szmania S, Draksharapu A, Shum BP, et al. CS1, a Potential New Therapeutic Antibody Target for the Treatment of Multiple Myeloma. Clin Cancer Res: Off J Am Assoc Cancer Res (2008) 14(9):2775–84. doi: 10.1158/1078-0432.CCR-07-4246
94. Malaer JD, Mathew PA. CS1 (SLAMF7, CD319) Is an Effective Immunotherapeutic Target for Multiple Myeloma. Am J Cancer Res (2017) 7(8):1637–41.
95. Tai YT, Dillon M, Song W, Leiba M, Li XF, Burger P, et al. Anti-CS1 Humanized Monoclonal Antibody HuLuc63 Inhibits Myeloma Cell Adhesion and Induces Antibody-Dependent Cellular Cytotoxicity in the Bone Marrow Milieu. Blood (2008) 112(4):1329–37. doi: 10.1182/blood-2007-08-107292
96. van Rhee F, Szmania SM, Dillon M, van Abbema AM, Li X, Stone MK, et al. Combinatorial Efficacy of Anti-CS1 Monoclonal Antibody Elotuzumab (HuLuc63) and Bortezomib Against Multiple Myeloma. Mol Cancer Ther (2009) 8(9):2616–24. doi: 10.1158/1535-7163.Mct-09-0483
97. Collins SM, Bakan CE, Swartzel GD, Hofmeister CC, Efebera YA, Kwon H, et al. Elotuzumab Directly Enhances NK Cell Cytotoxicity Against Myeloma via CS1 Ligation: Evidence for Augmented NK Cell Function Complementing ADCC. Cancer Immunol Immunother (2013) 62(12):1841–9. doi: 10.1007/s00262-013-1493-8
98. Bezman NA, Jhatakia A, Kearney AY, Brender T, Maurer M, Henning K, et al. PD-1 Blockade Enhances Elotuzumab Efficacy in Mouse Tumor Models. Blood Adv (2017) 1(12):753–65. doi: 10.1182/bloodadvances.2017004382
99. Ritchie D, Colonna M. Mechanisms of Action and Clinical Development of Elotuzumab. Clin Transl Sci (2018) 11(3):261–6. doi: 10.1111/cts.12532
100. Veillette A, Guo H. CS1, a SLAM Family Receptor Involved in Immune Regulation, Is a Therapeutic Target in Multiple Myeloma. Crit Rev Oncol Hematol (2013) 88(1):168–77. doi: 10.1016/j.critrevonc.2013.04.003
101. Balasa B, Yun R, Belmar NA, Fox M, Chao DT, Robbins MD, et al. Elotuzumab Enhances Natural Killer Cell Activation and Myeloma Cell Killing Through Interleukin-2 and TNF-α Pathways. Cancer Immunol Immunother (2015) 64(1):61–73. doi: 10.1007/s00262-014-1610-3
102. Wang X, Walter M, Urak R, Weng L, Huynh C, Lim L, et al. Lenalidomide Enhances the Function of CS1 Chimeric Antigen Receptor-Redirected T Cells Against Multiple Myeloma. Clin Cancer Res (2018) 24(1):106–19. doi: 10.1158/1078-0432.Ccr-17-0344
103. Shah UA, Mailankody S. CAR T and CAR NK Cells in Multiple Myeloma: Expanding the Targets. Best Pract Res Clin Haematol (2020) 33(1):101141. doi: 10.1016/j.beha.2020.101141
104. Kurdi AT, Glavey SV, Bezman NA, Jhatakia A, Guerriero JL, Manier S, et al. Antibody-Dependent Cellular Phagocytosis by Macrophages Is a Novel Mechanism of Action of Elotuzumab. Mol Cancer Ther (2018) 17(7):1454–63. doi: 10.1158/1535-7163.MCT-17-0998
105. Davies FE, Raje N, Hideshima T, Lentzsch S, Young G, Tai YT, et al. Thalidomide and Immunomodulatory Derivatives Augment Natural Killer Cell Cytotoxicity in Multiple Myeloma. Blood (2001) 98(1):210–6. doi: 10.1182/blood.v98.1.210
106. Lagrue K, Carisey A, Morgan DJ, Chopra R, Davis DM. Lenalidomide Augments Actin Remodeling and Lowers NK-Cell Activation Thresholds. Blood (2015) 126(1):50–60. doi: 10.1182/blood-2015-01-625004
107. Lonial S, Dimopoulos M, Palumbo A, White D, Grosicki S, Spicka I, et al. Elotuzumab Therapy for Relapsed or Refractory Multiple Myeloma. New Engl J Med (2015) 373(7):621–31. doi: 10.1056/NEJMoa1505654
108. Dimopoulos MA, Lonial S, White D, Moreau P, Weisel K, San-Miguel J, et al. Elotuzumab, Lenalidomide, and Dexamethasone in RRMM: Final Overall Survival Results From the Phase 3 Randomized ELOQUENT-2 Study. Blood Cancer J (2020) 10(9):91. doi: 10.1038/s41408-020-00357-4
109. Dimopoulos MA, Dytfeld D, Grosicki S, Moreau P, Takezako N, Hori M, et al. Elotuzumab Plus Pomalidomide and Dexamethasone for Multiple Myeloma. New Engl J Med (2018) 379(19):1811–22. doi: 10.1056/NEJMoa1805762
110. Dimopoulos MA, Facon T, Richardson PGG, Orlowski RZ, San-Miguel JF, Lonial S, et al. ELOQUENT-1: A Phase III, Randomized, Open-Label Trial of Lenalidomide/Dexamethasone With or Without Elotuzumab in Subjects With Previously Untreated Multiple Myeloma (CA204-006). J Clin Oncol (2012) 30(15_suppl):TPS8113–TPS. doi: 10.1200/jco.2012.30.15_suppl.tps8113
111. Ghobrial I, Cruz CH, Garfall A, Shah N, Munshi N, Kaufman J, et al. Immunotherapy in Multiple Myeloma: Accelerating on the Path to the Patient. Clin Lymphoma Myeloma Leuk (2019) 19(6):332–44. doi: 10.1016/j.clml.2019.02.004
112. Lum LG, Thakur A, Elhakiem A, Alameer L, Dinning E, Huang M. Anti-CS1 × Anti-CD3 Bispecific Antibody (BiAb)-Armed Anti-CD3 Activated T Cells (CS1-BATs) Kill CS1(+) Myeloma Cells and Release Type-1 Cytokines. Front Oncol (2020) 10:544. doi: 10.3389/fonc.2020.00544
113. Prommersberger S, Reiser M, Beckmann J, Danhof S, Amberger M, Quade-Lyssy P, et al. CARAMBA: A First-in-Human Clinical Trial With SLAMF7 CAR-T Cells Prepared by Virus-Free Sleeping Beauty Gene Transfer to Treat Multiple Myeloma. Gene Ther (2021). doi: 10.1038/s41434-021-00254-w
114. Abramson HN. Monoclonal Antibodies for the Treatment of Multiple Myeloma: An Update. Int J Mol Sci (2018) 19(12):3924. doi: 10.3390/ijms19123924
115. Vij R, Nath R, Afar DEH, Mateos MV, Berdeja JG, Raab MS, et al. First-In-Human Phase I Study of ABBV-838, an Antibody-Drug Conjugate Targeting SLAMF7/CS1 in Patients With Relapsed and Refractory Multiple Myeloma. Clin Cancer Res (2020) 26(10):2308–17. doi: 10.1158/1078-0432.Ccr-19-1431
116. Cornelis R, Hahne S, Taddeo A, Petkau G, Malko D, Durek P, et al. Stromal Cell-Contact Dependent PI3K and APRIL Induced NF-κB Signaling Prevent Mitochondrial- and ER Stress Induced Death of Memory Plasma Cells. Cell Rep (2020) 32(5):107982. doi: 10.1016/j.celrep.2020.107982
117. Benson MJ, Dillon SR, Castigli E, Geha RS, Xu S, Lam KP, et al. Cutting Edge: The Dependence of Plasma Cells and Independence of Memory B Cells on BAFF and APRIL. J Immunol (2008) 180(6):3655–9. doi: 10.4049/jimmunol.180.6.3655
118. Balázs M, Martin F, Zhou T, Kearney J. Blood Dendritic Cells Interact With Splenic Marginal Zone B Cells to Initiate T-Independent Immune Responses. Immunity (2002) 17(3):341–52. doi: 10.1016/s1074-7613(02)00389-8
119. An G, Acharya C, Feng X, Wen K, Zhong M, Zhang L, et al. Osteoclasts Promote Immune Suppressive Microenvironment in Multiple Myeloma: Therapeutic Implication. Blood (2016) 128(12):1590–603. doi: 10.1182/blood-2016-03-707547
120. Tai YT, Acharya C, An G, Moschetta M, Zhong MY, Feng X, et al. APRIL and BCMA Promote Human Multiple Myeloma Growth and Immunosuppression in the Bone Marrow Microenvironment. Blood (2016) 127(25):3225–36. doi: 10.1182/blood-2016-01-691162
121. Laabi Y, Gras MP, Brouet JC, Berger R, Larsen CJ, Tsapis A. The BCMA Gene, Preferentially Expressed During B Lymphoid Maturation, Is Bidirectionally Transcribed. Nucleic Acids Res (1994) 22(7):1147–54. doi: 10.1093/nar/22.7.1147
122. Dogan A, Siegel D, Tran N, Fu A, Fowler J, Belani R, et al. B-Cell Maturation Antigen Expression Across Hematologic Cancers: A Systematic Literature Review. Blood Cancer J (2020) 10(6):73. doi: 10.1038/s41408-020-0337-y
123. Cho SF, Anderson KC, Tai YT. Targeting B Cell Maturation Antigen (BCMA) in Multiple Myeloma: Potential Uses of BCMA-Based Immunotherapy. Front Immunol (2018) 9:1821. doi: 10.3389/fimmu.2018.01821
124. Low MSY, Brodie EJ, Fedele PL, Liao Y, Grigoriadis G, Strasser A, et al. IRF4 Activity Is Required in Established Plasma Cells to Regulate Gene Transcription and Mitochondrial Homeostasis. Cell Rep (2019) 29(9):2634–45.e5. doi: 10.1016/j.celrep.2019.10.097
125. Fedele PL, Liao Y, Gong JN, Yao Y, van Delft MF, Low MSY, et al. The Transcription Factor IRF4 Represses Proapoptotic BMF and BIM to Licence Multiple Myeloma Survival. Leukemia (2020). doi: 10.1038/s41375-020-01078-0
126. Peperzak V, Vikström I, Walker J, Glaser SP, LePage M, Coquery CM, et al. Mcl-1 Is Essential for the Survival of Plasma Cells. Nat Immunol (2013) 14(3):290–7. doi: 10.1038/ni.2527
127. Schiemann B, Gommerman JL, Vora K, Cachero TG, Shulga-Morskaya S, Dobles M, et al. An Essential Role for BAFF in the Normal Development of B Cells Through a BCMA-Independent Pathway. Science (2001) 293(5537):2111–4. doi: 10.1126/science.1061964
128. Sakai J, Akkoyunlu M. The Role of BAFF System Molecules in Host Response to Pathogens. Clin Microbiol Rev (2017) 30(4):991–1014. doi: 10.1128/cmr.00046-17
129. O’Connor BP, Raman VS, Erickson LD, Cook WJ, Weaver LK, Ahonen C, et al. BCMA Is Essential for the Survival of Long-Lived Bone Marrow Plasma Cells. J Exp Med (2004) 199(1):91–8. doi: 10.1084/jem.20031330
130. Bossen C, Cachero TG, Tardivel A, Ingold K, Willen L, Dobles M, et al. TACI, Unlike BAFF-R, Is Solely Activated by Oligomeric BAFF and APRIL to Support Survival of Activated B Cells and Plasmablasts. Blood (2008) 111(3):1004–12. doi: 10.1182/blood-2007-09-110874
131. Groom J, Kalled SL, Cutler AH, Olson C, Woodcock SA, Schneider P, et al. Association of BAFF/BLyS Overexpression and Altered B Cell Differentiation With Sjögren’s Syndrome. J Clin Invest (2002) 109(1):59–68. doi: 10.1172/jci14121
132. Mackay F, Woodcock SA, Lawton P, Ambrose C, Baetscher M, Schneider P, et al. Mice Transgenic for BAFF Develop Lymphocytic Disorders Along With Autoimmune Manifestations. J Exp Med (1999) 190(11):1697–710. doi: 10.1084/jem.190.11.1697
133. Huard B, McKee T, Bosshard C, Durual S, Matthes T, Myit S, et al. APRIL Secreted by Neutrophils Binds to Heparan Sulfate Proteoglycans to Create Plasma Cell Niches in Human Mucosa. J Clin Invest (2008) 118(8):2887–95. doi: 10.1172/jci33760
134. Kimberley FC, Van Bostelen L, Cameron K, Hardenberg G, Marquart JA, Hahne M, et al. The Proteoglycan (Heparan Sulfate Proteoglycan) Binding Domain of APRIL Serves as a Platform for Ligand Multimerization Andcross-Linking. FASEB J (2009) 23(5):1584–95. doi: 10.1096/fj.08-124669
135. Laurent SA, Hoffmann FS, Kuhn PH, Cheng Q, Chu Y, Schmidt-Supprian M, et al. γ-Secretase Directly Sheds the Survival Receptor BCMA From Plasma Cells. Nat Commun (2015) 6:7333. doi: 10.1038/ncomms8333
136. Sanchez E, Gillespie A, Tang G, Ferros M, Harutyunyan NM, Vardanyan S, et al. Soluble B-Cell Maturation Antigen Mediates Tumor-Induced Immune Deficiency in Multiple Myeloma. Clin Cancer Res (2016) 22(13):3383–97. doi: 10.1158/1078-0432.Ccr-15-2224
137. Lee L, Bounds D, Paterson J, Herledan G, Sully K, Seestaller-Wehr LM, et al. Evaluation of B Cell Maturation Antigen as a Target for Antibody Drug Conjugate Mediated Cytotoxicity in Multiple Myeloma. Br J Haematol (2016) 174(6):911–22. doi: 10.1111/bjh.14145
138. Kinneer K, Flynn M, Thomas SB, Meekin J, Varkey R, Xiao X, et al. Preclinical Assessment of an Antibody-PBD Conjugate That Targets BCMA on Multiple Myeloma and Myeloma Progenitor Cells. Leukemia (2019) 33(3):766–71. doi: 10.1038/s41375-018-0278-7
139. Sanchez E, Li M, Kitto A, Li J, Wang CS, Kirk DT, et al. Serum B-Cell Maturation Antigen Is Elevated in Multiple Myeloma and Correlates With Disease Status and Survival. Br J Haematol (2012) 158(6):727–38. doi: 10.1111/j.1365-2141.2012.09241.x
140. Ghermezi M, Li M, Vardanyan S, Harutyunyan NM, Gottlieb J, Berenson A, et al. Serum B-Cell Maturation Antigen: A Novel Biomarker to Predict Outcomes for Multiple Myeloma Patients. Haematologica (2017) 102(4):785–95. doi: 10.3324/haematol.2016.150896
141. Jew S, Chang T, Bujarski S, Soof C, Chen H, Safaie T, et al. Normalization of Serum B-Cell Maturation Antigen Levels Predicts Overall Survival Among Multiple Myeloma Patients Starting Treatment. Br J Haematol (2020) 92:272–80. doi: 10.1111/bjh.16752
142. Thaler FS, Laurent SA, Huber M, Mulazzani M, Dreyling M, Ködel U, et al. Soluble TACI and Soluble BCMA as Biomarkers in Primary Central Nervous System Lymphoma. Neuro Oncol (2017) 19(12):1618–27. doi: 10.1093/neuonc/nox097
143. Gavriatopoulou M, Ntanasis-Stathopoulos I, Dimopoulos MA, Terpos E. Anti-BCMA Antibodies in the Future Management of Multiple Myeloma. Expert Rev Anticancer Ther (2019) 19(4):319–26. doi: 10.1080/14737140.2019.1586539
144. Zagouri F, Terpos E, Kastritis E, Dimopoulos MA. Emerging Antibodies for the Treatment of Multiple Myeloma. Expert Opin Emerg Drugs (2016) 21(2):225–37. doi: 10.1080/14728214.2016.1186644
145. Iida S, Ogiya D, Abe Y, Taniwaki M, Asou H, Maeda K, et al. Dose-Escalation Study of Tabalumab With Bortezomib and Dexamethasone in Japanese Patients With Multiple Myeloma. Cancer Sci (2016) 107(9):1281–9. doi: 10.1111/cas.13000
146. Raje NS, Faber EA, Richardson PG, Schiller G, Hohl RJ, Cohen AD, et al. Phase 1 Study of Tabalumab, a Human Anti-B-Cell Activating Factor Antibody, and Bortezomib in Patients With Relapsed/Refractory Multiple Myeloma. Clin Cancer Res (2016) 22(23):5688–95. doi: 10.1158/1078-0432.Ccr-16-0201
147. Raje NS, Moreau P, Terpos E, Benboubker L, Grząśko N, Holstein SA, et al. Phase 2 Study of Tabalumab, a Human Anti-B-Cell Activating Factor Antibody, With Bortezomib and Dexamethasone in Patients With Previously Treated Multiple Myeloma. Br J Haematol (2017) 176(5):783–95. doi: 10.1111/bjh.14483
148. Coquery CM, Wade NS, Loo WM, Kinchen JM, Cox KM, Jiang C, et al. Neutrophils Contribute to Excess Serum BAFF Levels and Promote CD4+ T Cell and B Cell Responses in Lupus-Prone Mice. PloS One (2014) 9(7):e102284. doi: 10.1371/journal.pone.0102284
149. Moisini I, Davidson A. BAFF: A Local and Systemic Target in Autoimmune Diseases. Clin Exp Immunol (2009) 158(2):155–63. doi: 10.1111/j.1365-2249.2009.04007.x
150. Moreaux J, Legouffe E, Jourdan E, Quittet P, Rème T, Lugagne C, et al. BAFF and APRIL Protect Myeloma Cells From Apoptosis Induced by Interleukin 6 Deprivation and Dexamethasone. Blood (2004) 103(8):3148–57. doi: 10.1182/blood-2003-06-1984
151. Salazar-Camarena DC, Ortiz-Lazareno PC, Cruz A, Oregon-Romero E, Machado-Contreras JR, Muñoz-Valle JF, et al. Association of BAFF, APRIL Serum Levels, BAFF-R, TACI and BCMA Expression on Peripheral B-Cell Subsets With Clinical Manifestations in Systemic Lupus Erythematosus. Lupus (2016) 25(6):582–92. doi: 10.1177/0961203315608254
152. Topp MS, Duell J, Zugmaier G, Attal M, Moreau P, Langer C, et al. Anti-B-Cell Maturation Antigen BiTE Molecule AMG 420 Induces Responses in Multiple Myeloma. J Clin Oncol (2020) 38(8):775–83. doi: 10.1200/jco.19.02657
153. Harrison SJ, Minnema MC, Lee HC, Spencer A, Kapoor P, Madduri D, et al. A Phase 1 First in Human (FIH) Study of AMG 701, an Anti-B-Cell Maturation Antigen (BCMA) Half-Life Extended (HLE) BiTE® (Bispecific T-Cell Engager) Molecule, in Relapsed/Refractory (RR) Multiple Myeloma (MM). Blood (2020) 136(Supplement 1):28–9. doi: 10.1182/blood-2020-134063
154. Costa LJ, Wong SW, Bermúdez A, de la Rubia J, Mateos M-V, Ocio EM, et al. First Clinical Study of the B-Cell Maturation Antigen (BCMA) 2 + 1 T Cell Engager (TCE) CC-93269 in Patients (Pts) With Relapsed/Refractory Multiple Myeloma (RRMM): Interim Results of a Phase 1 Multicenter Trial. Blood (2019) 134(Supplement_1):143–. doi: 10.1182/blood-2019-122895
155. Rodriguez C, D’Souza A, Shah N, Voorhees PM, Buelow B, Vij R, et al. Initial Results of a Phase I Study of TNB-383B, a BCMA X CD3 Bispecific T-Cell Redirecting Antibody, in Relapsed/Refractory Multiple Myeloma. Blood (2020) 136(Supplement 1):43–4. doi: 10.1182/blood-2020-139893
156. Lesokhin AM, Levy MY, Dalovisio AP, Bahlis NJ, Solh M, Sebag M, et al. Preliminary Safety, Efficacy, Pharmacokinetics, and Pharmacodynamics of Subcutaneously (SC) Administered PF-06863135, a B-Cell Maturation Antigen (BCMA)-CD3 Bispecific Antibody, in Patients With Relapsed/Refractory Multiple Myeloma (RRMM). Blood (2020) 136(Supplement 1):8–9. doi: 10.1182/blood-2020-133355
157. Garfall AL, Usmani SZ, Mateos M-V, Nahi H, van de Donk NWCJ, San-Miguel JF, et al. Updated Phase 1 Results of Teclistamab, a B-Cell Maturation Antigen (BCMA) X CD3 Bispecific Antibody, in Relapsed and/or Refractory Multiple Myeloma (RRMM). Blood (2020) 136(Supplement 1):27–. doi: 10.1182/blood-2020-138831
158. Madduri D, Rosko A, Brayer J, Zonder J, Bensinger WI, Li J, et al. REGN5458, a BCMA X CD3 Bispecific Monoclonal Antibody, Induces Deep and Durable Responses in Patients With Relapsed/Refractory Multiple Myeloma (RRMM). Blood (2020) 136(Supplement 1):41–2. doi: 10.1182/blood-2020-139192
159. Trudel S, Lendvai N, Popat R, Voorhees PM, Reeves B, Libby EN, et al. Antibody-Drug Conjugate, GSK2857916, in Relapsed/Refractory Multiple Myeloma: An Update on Safety and Efficacy From Dose Expansion Phase I Study. Blood Cancer J (2019) 9(4):37. doi: 10.1038/s41408-019-0196-6
160. Lonial S, Lee HC, Badros A, Trudel S, Nooka AK, Chari A, et al. Belantamab Mafodotin for Relapsed or Refractory Multiple Myeloma (DREAMM-2): A Two-Arm, Randomised, Open-Label, Phase 2 Study. Lancet Oncol (2020) 21(2):207–21. doi: 10.1016/s1470-2045(19)30788-0
161. Lee HC, Raje NS, Landgren O, Upreti VV, Wang J, Avilion AA, et al. Phase 1 Study of the Anti-BCMA Antibody-Drug Conjugate AMG 224 in Patients With Relapsed/Refractory Multiple Myeloma. Leukemia (2021) 35(1):255–8. doi: 10.1038/s41375-020-0834-9
162. Kumar SK, Migkou M, Bhutani M, Spencer A, Ailawadhi S, Kalff A, et al. Phase 1, First-In-Human Study of MEDI2228, a BCMA-Targeted ADC in Patients With Relapsed/Refractory Multiple Myeloma. Blood (2020) 136(Supplement 1):26–7. doi: 10.1182/blood-2020-136375
163. D’Agostino M, Raje N. Anti-BCMA CAR T-Cell Therapy in Multiple Myeloma: Can We do Better? Leukemia (2020) 34(1):21–34. doi: 10.1038/s41375-019-0669-4
164. Chalouni C, Doll S. Fate of Antibody-Drug Conjugates in Cancer Cells. J Exp Clin Cancer Res (2018) 37(1):20. doi: 10.1186/s13046-017-0667-1
165. Yaghoubi S, Karimi MH, Lotfinia M, Gharibi T, Mahi-Birjand M, Kavi E, et al. Potential Drugs Used in the Antibody-Drug Conjugate (ADC) Architecture for Cancer Therapy. J Cell Physiol (2020) 235(1):31–64. doi: 10.1002/jcp.28967
166. Hasan M, Alam S, Poddar SK. Antibody-Drug Conjugates: A Review on the Epitome of Targeted Anti- Cancer Therapy. Curr Clin Pharmacol (2018) 13(4):236–51. doi: 10.2174/1574884712666180802095521
167. Joubert N, Beck A, Dumontet C, Denevault-Sabourin C. Antibody-Drug Conjugates: The Last Decade. Pharm (Basel) (2020) 13(9):245. doi: 10.3390/ph13090245
168. Erickson HK, Park PU, Widdison WC, Kovtun YV, Garrett LM, Hoffman K, et al. Antibody-Maytansinoid Conjugates Are Activated in Targeted Cancer Cells by Lysosomal Degradation and Linker-Dependent Intracellular Processing. Cancer Res (2006) 66(8):4426–33. doi: 10.1158/0008-5472.Can-05-4489
169. Chauhan D, Singh AV, Brahmandam M, Carrasco R, Bandi M, Hideshima T, et al. Functional Interaction of Plasmacytoid Dendritic Cells With Multiple Myeloma Cells: A Therapeutic Target. Cancer Cell (2009) 16(4):309–23. doi: 10.1016/j.ccr.2009.08.019
170. Tai YT, Mayes PA, Acharya C, Zhong MY, Cea M, Cagnetta A, et al. Novel Anti-B-Cell Maturation Antigen Antibody-Drug Conjugate (GSK2857916) Selectively Induces Killing of Multiple Myeloma. Blood (2014) 123(20):3128–38. doi: 10.1182/blood-2013-10-535088
171. Trudel S, Lendvai N, Popat R, Voorhees PM, Reeves B, Libby EN, et al. Targeting B-Cell Maturation Antigen With GSK2857916 Antibody-Drug Conjugate in Relapsed or Refractory Multiple Myeloma (BMA117159): A Dose Escalation and Expansion Phase 1 Trial. Lancet Oncol (2018) 19(12):1641–53. doi: 10.1016/s1470-2045(18)30576-x
172. Demel I, Bago JR, Hajek R, Jelinek T. Focus on Monoclonal Antibodies Targeting B-Cell Maturation Antigen (BCMA) in Multiple Myeloma: Update 2021. Br J Haematol (2021) 193(4):705–22. doi: 10.1111/bjh.17235
173. Chen Y, Nagarajan C, Tan MS, Martinelli G, Cerchione C. BCMA-Targeting Approaches for Treatment of Multiple Myeloma. Panminerva Med (2021) 63(1):28–36. doi: 10.23736/s0031-0808.20.04121-x
174. Fang Y, Hou J. Immunotherapeutic Strategies Targeting B Cell Maturation Antigen in Multiple Myeloma. Mil Med Res (2021) 8(1):9. doi: 10.1186/s40779-021-00302-x
175. Hosoya H, Sidana S. Antibody-Based Treatment Approaches in Multiple Myeloma. Curr Hematol Malig Rep (2021) 16(2):183–91. doi: 10.1007/s11899-021-00624-6
176. Jullien M, Touzeau C, Moreau P. Monoclonal Antibodies as an Addition to Current Myeloma Therapy Strategies. Expert Rev Anticancer Ther (2021) 21(1):33–43. doi: 10.1080/14737140.2021.1837627
177. Maples KT, Johnson C, Lonial S. Antibody Treatment in Multiple Myeloma. Clin Adv Hematol Oncol (2021) 19(3):166–74.
178. Martino M, Paviglianiti A. An Update on B-Cell Maturation Antigen-Targeted Therapies in Multiple Myeloma. Expert Opin Biol Ther (2021), 13:1–10. doi: 10.1080/14712598.2021.1872540
179. Sanchez L, Dardac A, Madduri D, Richard S, Richter J. B-Cell Maturation Antigen (BCMA) in Multiple Myeloma: The New Frontier of Targeted Therapies. Ther Adv Hematol (2021) 12:2040620721989585. doi: 10.1177/2040620721989585
180. Vozella F, Fazio F, Lapietra G, Petrucci MT, Martinelli G, Cerchione C. Monoclonal Antibodies in Multiple Myeloma. Panminerva Med (2021) 63(1):21–7. doi: 10.23736/s0031-0808.20.04149-x
181. Farooq AV, Degli Esposti S, Popat R, Thulasi P, Lonial S, Nooka AK, et al. Corneal Epithelial Findings in Patients With Multiple Myeloma Treated With Antibody–Drug Conjugate Belantamab Mafodotin in the Pivotal, Randomized, DREAMM-2 Study. Ophthalmol Ther (2020) 9(4):889–911. doi: 10.1007/s40123-020-00280-8
182. Popat R, Warcel D, O’Nions J, Cowley A, Smith S, Tucker WR, et al. Characterization of Response and Corneal Events With Extended Follow-Up After Belantamab Mafodotin (GSK2857916) Monotherapy for Patients With Relapsed Multiple Myeloma: A Case Series From the First-Time-in-Human Clinical Trial. Haematologica (2020) 105(5):e261–e3. doi: 10.3324/haematol.2019.235937
183. Zhao H, Atkinson J, Gulesserian S, Zeng Z, Nater J, Ou J, et al. Modulation of Macropinocytosis-Mediated Internalization Decreases Ocular Toxicity of Antibody-Drug Conjugates. Cancer Res (2018) 78(8):2115–26. doi: 10.1158/0008-5472.Can-17-3202
184. Shah N, Chari A, Scott E, Mezzi K. Usmani SZ. B-Cell Maturation Antigen (BCMA) in Multiple Myeloma: Rationale for Targeting and Current Therapeutic Approaches. Leukemia (2020) 34(4):985–1005. doi: 10.1038/s41375-020-0734-z
185. Bruins WSC, Zweegman S, Mutis T, van de Donk N. Targeted Therapy With Immunoconjugates for Multiple Myeloma. Front Immunol (2020) 11:1155. doi: 10.3389/fimmu.2020.01155
186. Lancman G, Richter J, Chari A. Bispecifics, Trispecifics, and Other Novel Immune Treatments in Myeloma. Hematol Am Soc Hematol Educ Program (2020) 2020(1):264–71. doi: 10.1182/hematology.2020000110
187. Barilà G, Rizzi R, Zambello R, Musto P. Drug Conjugated and Bispecific Antibodies for Multiple Myeloma: Improving Immunotherapies Off the Shelf. Pharm (Basel) (2021) 14(1):40. doi: 10.3390/ph14010040
188. Pahl A, Lutz C, Hechler T. Amanitins and Their Development as a Payload for Antibody-Drug Conjugates. Drug Discovery Today Technol (2018) 30:85–9. doi: 10.1016/j.ddtec.2018.08.005
189. Huehls AM, Coupet TA, Sentman CL. Bispecific T-Cell Engagers for Cancer Immunotherapy. Immunol Cell Biol (2015) 93(3):290–6. doi: 10.1038/icb.2014.93
190. Einsele H, Borghaei H, Orlowski RZ, Subklewe M, Roboz GJ, Zugmaier G, et al. The BiTE (Bispecific T-Cell Engager) Platform: Development and Future Potential of a Targeted Immuno-Oncology Therapy Across Tumor Types. Cancer (2020) 126(14):3192–201. doi: 10.1002/cncr.32909
191. Regazzi M, Golay J, Molinaro M. Monoclonal Antibody Monitoring: Clinically Relevant Aspects, A Systematic Critical Review. Ther Drug Monit (2020) 42(1):45–56. doi: 10.1097/ftd.0000000000000681
192. Golay J, Regazzi M. Key Features Defining the Disposition of Bispecific Antibodies and Their Efficacy In Vivo. Ther Drug Monit (2020) 42(1):57–63. doi: 10.1097/ftd.0000000000000668
193. Carrasco-Triguero M, Dere RC, Milojic-Blair M, Saad OM, Nazzal D, Hong K, et al. Immunogenicity of Antibody–Drug Conjugates: Observations Across 8 Molecules in 11 Clinical Trials. Bioanalysis (2019) 11(17):1555–68. doi: 10.4155/bio-2018-0259
194. Caraccio C, Krishna S, Phillips DJ, Schürch CM. Bispecific Antibodies for Multiple Myeloma: A Review of Targets, Drugs, Clinical Trials, and Future Directions. Front Immunol (2020) 11:501. doi: 10.3389/fimmu.2020.00501
195. Gramer MJ, van den Bremer ET, van Kampen MD, Kundu A, Kopfmann P, Etter E, et al. Production of Stable Bispecific IgG1 by Controlled Fab-Arm Exchange: Scalability From Bench to Large-Scale Manufacturing by Application of Standard Approaches. MAbs (2013) 5(6):962–73. doi: 10.4161/mabs.26233
196. Ross SL, Sherman M, McElroy PL, Lofgren JA, Moody G, Baeuerle PA, et al. Bispecific T Cell Engager (BiTE®) Antibody Constructs can Mediate Bystander Tumor Cell Killing. PloS One (2017) 12(8):e0183390. doi: 10.1371/journal.pone.0183390
197. Hipp S, Tai YT, Blanset D, Deegen P, Wahl J, Thomas O, et al. A Novel BCMA/CD3 Bispecific T-Cell Engager for the Treatment of Multiple Myeloma Induces Selective Lysis. Vitro Vivo Leuk (2017) 31(8):1743–51. doi: 10.1038/leu.2016.388
198. Chen Y, Nagarajan C, Tan MS, Martinelli G, Cerchione C. BCMA-Targeting Approaches for Treatment of Multiple Myeloma. Panminerva Med (2020) 63(1):28–36. doi: 10.23736/s0031-0808.20.04121-x
199. Goldstein RL, Goyos A, Li CM, Deegen P, Bogner P, Sternjak A, et al. AMG 701 Induces Cytotoxicity of Multiple Myeloma Cells and Depletes Plasma Cells in Cynomolgus Monkeys. Blood Adv (2020) 4(17):4180–94. doi: 10.1182/bloodadvances.2020002565
200. Cho S-F, Lin L, Xing L, Li Y, Wen K, Yu T, et al. The Immunomodulatory Drugs Lenalidomide and Pomalidomide Enhance the Potency of AMG 701 in Multiple Myeloma Preclinical Models. Blood Adv (2020) 4(17):4195–207. doi: 10.1182/bloodadvances.2020002524
201. Seckinger A, Delgado JA, Moser S, Moreno L, Neuber B, Grab A, et al. Target Expression, Generation, Preclinical Activity, and Pharmacokinetics of the BCMA-T Cell Bispecific Antibody EM801 for Multiple Myeloma Treatment. Cancer Cell (2017) 31(3):396–410. doi: 10.1016/j.ccell.2017.02.002
202. Buelow B, Choudry P, Clarke S, Dang K, Davison L, Force Aldred S, et al. Pre-Clinical Development of TNB-383B, a Fully Human T-Cell Engaging Bispecific Antibody Targeting BCMA for the Treatment of Multiple Myeloma. J Clin Oncol (2018) 36(15_suppl):8034–. doi: 10.1200/JCO.2018.36.15_suppl.8034
203. Foureau DM, Bhutani M, Robinson M, Guo F, Pham D, Buelow B, et al. Ex Vivo Efficacy of BCMA-Bispecific Antibody TNB-383B in Relapsed/Refractory Multiple Myeloma. eJHaem (2020) 1(1):113–21. doi: 10.1002/jha2.69
204. Raje NS, Jakubowiak A, Gasparetto C, Cornell RF, Krupka HI, Navarro D, et al. Safety, Clinical Activity, Pharmacokinetics, and Pharmacodynamics From a Phase I Study of PF-06863135, a B-Cell Maturation Antigen (BCMA)-CD3 Bispecific Antibody, in Patients With Relapsed/Refractory Multiple Myeloma (RRMM). Blood (2019) 134(Supplement_1):1869–. doi: 10.1182/blood-2019-121805
205. Atamaniuk J, Gleiss A, Porpaczy E, Kainz B, Grunt TW, Raderer M, et al. Overexpression of G Protein-Coupled Receptor 5D in the Bone Marrow Is Associated With Poor Prognosis in Patients With Multiple Myeloma. Eur J Clin Invest (2012) 42(9):953–60. doi: 10.1111/j.1365-2362.2012.02679.x
206. Li J, Stagg NJ, Johnston J, Harris MJ, Menzies SA, DiCara D, et al. Membrane-Proximal Epitope Facilitates Efficient T Cell Synapse Formation by Anti-FcRH5/CD3 and Is a Requirement for Myeloma Cell Killing. Cancer Cell (2017) 31(3):383–95. doi: 10.1016/j.ccell.2017.02.001
207. Cohen AD, Harrison SJ, Krishnan A, Fonseca R, Forsberg PA, Spencer A, et al. Initial Clinical Activity and Safety of BFCR4350A, a FcRH5/CD3 T-Cell-Engaging Bispecific Antibody, in Relapsed/Refractory Multiple Myeloma. Blood (2020) 136(Supplement 1):42–3. doi: 10.1182/blood-2020-136985
208. Nakamura R, Lear S, Wilson D, Koeppen H, Vaze A, Trudel S, et al. Early Pharmacodynamic Changes in T-Cell Activation, Proliferation, and Cytokine Production Confirm the Mode of Action of BFCR4350A, a FcRH5/CD3 T-Cell-Engaging Bispecific Antibody, in Patients With Relapsed/Refractory Multiple Myeloma. Blood (2020) 136(Supplement 1):14–5. doi: 10.1182/blood-2020-136980
209. van der Neut Kolfschoten M, Schuurman J, Losen M, Bleeker WK, Martínez-Martínez P, Vermeulen E, et al. Anti-Inflammatory Activity of Human IgG4 Antibodies by Dynamic Fab Arm Exchange. Science (2007) 317(5844):1554–7. doi: 10.1126/science.1144603
210. Schuurman J, Graus YF, Labrijn AF, Ruuls S, Parren PW. Opening the Door to Innovation. MAbs (2014) 6(4):812–9. doi: 10.4161/mabs.29004
211. Labrijn AF, Janmaat ML, Reichert JM, Parren P. Bispecific Antibodies: A Mechanistic Review of the Pipeline. Nat Rev Drug Discovery (2019) 18(8):585–608. doi: 10.1038/s41573-019-0028-1
212. Labrijn AF, Buijsse AO, van den Bremer ET, Verwilligen AY, Bleeker WK, Thorpe SJ, et al. Therapeutic IgG4 Antibodies Engage in Fab-Arm Exchange With Endogenous Human IgG4 in vivo. Nat Biotechnol (2009) 27(8):767–71. doi: 10.1038/nbt.1553
213. Labrijn AF, Meesters JI, de Goeij BE, van den Bremer ET, Neijssen J, van Kampen MD, et al. Efficient Generation of Stable Bispecific IgG1 by Controlled Fab-Arm Exchange. Proc Natl Acad Sci USA (2013) 110(13):5145–50. doi: 10.1073/pnas.1220145110
214. Labrijn AF, Meesters JI, Priem P, de Jong RN, van den Bremer ET, van Kampen MD, et al. Controlled Fab-Arm Exchange for the Generation of Stable Bispecific Igg1. Nat Protoc (2014) 9(10):2450–63. doi: 10.1038/nprot.2014.169
215. Pillarisetti K, Powers G, Luistro L, Babich A, Baldwin E, Li Y, et al. Teclistamab Is an Active T Cell-Redirecting Bispecific Antibody Against B-Cell Maturation Antigen for Multiple Myeloma. Blood Adv (2020) 4(18):4538–49. doi: 10.1182/bloodadvances.2020002393
216. Frerichs KA, Broekmans MEC, Marin Soto JA, van Kessel B, Heymans MW, Holthof LC, et al. Preclinical Activity of JNJ-7957, a Novel BCMA×CD3 Bispecific Antibody for the Treatment of Multiple Myeloma, Is Potentiated by Daratumumab. Clin Cancer Res (2020) 26(9):2203–15. doi: 10.1158/1078-0432.Ccr-19-2299
217. Law C-L, Aaron W, Austin R, Barath M, Callihan E, Evans T, et al. Preclinical and Nonclinical Characterization of HPN217: A Tri-Specific T Cell Activating Construct (TriTAC) Targeting B Cell Maturation Antigen (BCMA) for the Treatment of Multiple Myeloma. Blood (2018) 132(Supplement 1):3225–. doi: 10.1182/blood-2018-99-113921
218. Gantke T, Reusch U, Kellner C, Ellwanger K, Fucek I, Weichel M, et al. AFM26 Is a Novel, Highly Potent BCMA/CD16A-Directed Bispecific Antibody for High Affinity NK-Cell Engagement in Multiple Myeloma. J Clin Oncol (2017) 35(15_suppl):8045–. doi: 10.1200/JCO.2017.35.15_suppl.8045
219. Ross T, Reusch U, Wingert S, Haneke T, Klausz K, Otte A-K, et al. Preclinical Characterization of AFM26, a Novel B Cell Maturation Antigen (BCMA)-Directed Tetravalent Bispecific Antibody for High Affinity Retargeting of NK Cells Against Myeloma. Blood (2018) 132(Supplement 1):1927–. doi: 10.1182/blood-2018-99-118970
220. Watkins-Yoon J, Guzman W, Oliphant A, Haserlat S, Leung A, Chottin C, et al. CTX-8573, an Innate-Cell Engager Targeting BCMA, Is a Highly Potent Multispecific Antibody for the Treatment of Multiple Myeloma. Blood (2019) 134(Supplement_1):3182–. doi: 10.1182/blood-2019-128749
221. Wang Y, Li H, Xu W, Pan M, Qiao C, Cai J, et al. BCMA-Targeting Bispecific Antibody That Simultaneously Stimulates NKG2D-Enhanced Efficacy Against Multiple Myeloma. J Immunother (2020) 43(6):175–88. doi: 10.1097/cji.0000000000000320
222. Gorgun G, Samur MK, Cowens KB, Paula S, Bianchi G, Anderson JE, et al. Lenalidomide Enhances Immune Checkpoint Blockade-Induced Immune Response in Multiple Myeloma. Clin Cancer Res: Off J Am Assoc Cancer Res (2015) 21(20):4607–18. doi: 10.1158/1078-0432.CCR-15-0200
223. Benson DM Jr., Bakan CE, Mishra A, Hofmeister CC, Efebera Y, Becknell B, et al. The PD-1/PD-L1 Axis Modulates the Natural Killer Cell Versus Multiple Myeloma Effect: A Therapeutic Target for CT-011, a Novel Monoclonal Anti-PD-1 Antibody. Blood (2010) 116(13):2286–94. doi: 10.1182/blood-2010-02-271874
224. Tamura H, Ishibashi M, Yamashita T, Tanosaki S, Okuyama N, Kondo A, et al. Marrow Stromal Cells Induce B7-H1 Expression on Myeloma Cells, Generating Aggressive Characteristics in Multiple Myeloma. Leukemia (2013) 27(2):464–72. doi: 10.1038/leu.2012.213
225. Motzer RJ, Escudier B, McDermott DF, George S, Hammers HJ, Srinivas S, et al. Nivolumab Versus Everolimus in Advanced Renal-Cell Carcinoma. New Engl J Med (2015) 373(19):1803–13. doi: 10.1056/NEJMoa1510665
226. Ansell SM, Lesokhin AM, Borrello I, Halwani A, Scott EC, Gutierrez M, et al. PD-1 Blockade With Nivolumab in Relapsed or Refractory Hodgkin’s Lymphoma. New Engl J Med (2015) 372(4):311–9. doi: 10.1056/NEJMoa1411087
227. Rosenblatt J, Glotzbecker B, Mills H, Vasir B, Tzachanis D, Levine JD, et al. PD-1 Blockade by CT-011, Anti-PD-1 Antibody, Enhances Ex Vivo T-Cell Responses to Autologous Dendritic Cell/Myeloma Fusion Vaccine. J Immunother (2011) 34(5):409–18. doi: 10.1097/CJI.0b013e31821ca6ce
228. Hallett WH, Jing W, Drobyski WR, Johnson BD. Immunosuppressive Effects of Multiple Myeloma Are Overcome by PD-L1 Blockade. Biol Blood Marrow Transplant: J Am Soc Blood Marrow Transplant (2011) 17(8):1133–45. doi: 10.1016/j.bbmt.2011.03.011
229. Kearl TJ, Jing W, Gershan JA, Johnson BD. Programmed Death Receptor-1/Programmed Death Receptor Ligand-1 Blockade After Transient Lymphodepletion to Treat Myeloma. J Immunol (2013) 190(11):5620–8. doi: 10.4049/jimmunol.1202005
230. Rodríguez-Otero P, Prósper F, Alfonso A, Paiva B, San Miguel JF. CAR T-Cells in Multiple Myeloma Are Ready for Prime Time. J Clin Med (2020) 9(11):3577. doi: 10.3390/jcm9113577
231. Rasche L, Hudecek M, Einsele H. What Is the Future of Immunotherapy in Multiple Myeloma? Blood (2020) 136(22):2491–7. doi: 10.1182/blood.2019004176
232. Mohan M, Hari P, Dhakal B. Immunotherapy in Multiple Myeloma—Time for a Second Major Paradigm Shift. JCO Oncol Pract (2021) 18:OP2100032. doi: 10.1200/op.21.00032 OP.21.00032.
233. Romano A, Palumbo GA, Parrinello NL, Conticello C, Martello M, Terragna C. Minimal Residual Disease Assessment Within the Bone Marrow of Multiple Myeloma: A Review of Caveats, Clinical Significance and Future Perspectives. Front Oncol (2019) 9:699. doi: 10.3389/fonc.2019.00699
234. Romano A, Parrinello NL, Simeon V, Puglisi F, La Cava P, Bellofiore C, et al. High-Density Neutrophils in MGUS and Multiple Myeloma Are Dysfunctional and Immune-Suppressive Due to Increased STAT3 Downstream Signaling. Sci Rep (2020) 10(1):1983. doi: 10.1038/s41598-020-58859-x
235. Zavidij O, Haradhvala NJ, Mouhieddine TH, Sklavenitis-Pistofidis R, Cai S, Reidy M, et al. Single-Cell RNA Sequencing Reveals Compromised Immune Microenvironment in Precursor Stages of Multiple Myeloma. Nat Cancer (2020) 1(5):493–506. doi: 10.1038/s43018-020-0053-3
236. Perez C, Botta C, Zabaleta A, Puig N, Cedena MT, Goicoechea I, et al. Immunogenomic Identification and Characterization of Granulocytic Myeloid-Derived Suppressor Cells in Multiple Myeloma. Blood (2020) 136(2):199–209. doi: 10.1182/blood.2019004537
237. Giallongo C, Tibullo D, Parrinello NL, La Cava P, Di Rosa M, Bramanti V, et al. Granulocyte-Like Myeloid Derived Suppressor Cells (G-MDSC) Are Increased in Multiple Myeloma and Are Driven by Dysfunctional Mesenchymal Stem Cells (MSC). Oncotarget (2016) 7(52):85764–75. doi: 10.18632/oncotarget.7969
238. Romano A, Parrinello NL, La Cava P, Tibullo D, Giallongo C, Camiolo G, et al. PMN-MDSC and Arginase Are Increased in Myeloma and may Contribute to Resistance to Therapy. Expert Rev Mol Diagn (2018) 18(7):675–83. doi: 10.1080/14737159.2018.1470929
239. Heemskerk N, van Egmond M. Monoclonal Antibody-Mediated Killing of Tumour Cells by Neutrophils. Eur J Clin Invest (2018) 48 Suppl 2(Suppl Suppl 2):e12962. doi: 10.1111/eci.12962
240. Zhao P, Zhang Y, Li W, Jeanty C, Xiang G, Dong Y. Recent Advances of Antibody Drug Conjugates for Clinical Applications. Acta Pharm Sin B (2020) 10(9):1589–600. doi: 10.1016/j.apsb.2020.04.012
Keywords: monoclonal antibodies, multiple myeloma, CD38, SLAMF7, BCMA, antibody-drug conjugate, bispecific antibodies
Citation: Romano A, Storti P, Marchica V, Scandura G, Notarfranchi L, Craviotto L, Di Raimondo F and Giuliani N (2021) Mechanisms of Action of the New Antibodies in Use in Multiple Myeloma. Front. Oncol. 11:684561. doi: 10.3389/fonc.2021.684561
Received: 23 March 2021; Accepted: 09 June 2021;
Published: 08 July 2021.
Edited by:
María-Victoria Mateos, University Hospital of Salamanca, SpainReviewed by:
Pooja Arora, University of Delhi, IndiaTijana Martinov, Fred Hutchinson Cancer Research Center, United States
Copyright © 2021 Romano, Storti, Marchica, Scandura, Notarfranchi, Craviotto, Di Raimondo and Giuliani. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nicola Giuliani, bmljb2xhLmdpdWxpYW5pQHVuaXByLml0; Francesco Di Raimondo, ZGlyYWltb25AdW5pY3QuaXQ=
†These authors have contributed equally to this work and share first authorship