 Jun Wei
Jun Wei Eli Gilboa2
Eli Gilboa2 George A. Calin
George A. Calin Amy B. Heimberger
Amy B. Heimberger
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 31 August 2021
Sec. Cancer Immunity and Immunotherapy
Volume 11 - 2021 | https://doi.org/10.3389/fonc.2021.682129
This article is part of the Research Topic Impact of the Glioma Microenvironment on Antitumor Immunity View all 14 articles
Glioblastomas are heterogeneous and have a poor prognosis. Glioblastoma cells interact with their neighbors to form a tumor-permissive and immunosuppressive microenvironment. Short noncoding RNAs are relevant mediators of the dynamic crosstalk among cancer, stromal, and immune cells in establishing the glioblastoma microenvironment. In addition to the ease of combinatorial strategies that are capable of multimodal modulation for both reversing immune suppression and enhancing antitumor immunity, their small size provides an opportunity to overcome the limitations of blood-brain-barrier (BBB) permeability. To enhance glioblastoma delivery, these RNAs have been conjugated with various molecules or packed within delivery vehicles for enhanced tissue-specific delivery and increased payload. Here, we focus on the role of RNA therapeutics by appraising which types of nucleotides are most effective in immune modulation, lead therapeutic candidates, and clarify how to optimize delivery of the therapeutic RNAs and their conjugates specifically to the glioblastoma microenvironment.
Glioblastoma cells would not survive without close connection and dependence on their adjacent molecular and cellular components. Multilevel and complicated communication between glioblastoma cells and nonmalignant cells promotes a permissive microenvironment for gliomagenesis. The glioblastoma microenvironment can be taken as a local niche comprising glioma cells, immune cells, parenchymal cells, and their associated molecular factors and subcellular vesicles. Microglia, a major type of parenchymal cells, contribute significantly to the brain tumor mass and immunosuppressive microenvironment. In addition, these cells can secrete a number of factors together with glioblastoma cells and nonneoplastic astrocytes that have an effect on glioblastoma progression.
Heterogenous molecular factors contribute to the complexity of the glioblastoma microenvironment and emphasize the importance of local niche influence to the tumor cells. An important contributor to these molecular factors is short noncoding RNAs (ncRNAs) including small noncoding microRNAs (miRNAs) and small interfering RNAs (siRNAs) carried by extracellular vehicles (EVs). miRNAs account for a large portion of the human transcriptome in the glioblastoma cells and their surrounding cells, and these miRNAs regulate numerous hallmarks of glioblastoma such as proliferation, invasion, immune escape and resistance to treatment (1). The development of various RNA agents including siRNA, antisense oligonucleotides (ASO), and aptamers for specific gene targeting and knockdown offers enormous therapeutic potential (2). In this manuscript, we discuss the current state of knowledge of miRNA immune regulatory functions and the impact of immunomodulatory miRNAs on the glioblastoma microenvironment. Next, we will review possible use of miRNAs, their analogue siRNAs, and aptamers as antiglioblastoma therapeutics. Finally, we will discuss potential RNA nucleotide therapeutics targeting immunomodulatory pathways and survey effective strategies for delivery to the glioblastoma microenvironment.
Our understanding of gene expression modulation evolved upon the discovery of the role of miRNA as an epigenetic regulator. miRNAs at 21–23 nucleotide long can suppress target gene expression by binding to the 3′ untranslated regions (UTRs) of mRNA with partial complementarity base-pairing (3). At present, around 2,500 human miRNAs have been identified that act in this capacity (4). miRNA and mRNA interactions can be complex: a mRNA can be modulated by multiple miRNAs and a miRNA can target many different mRNAs. Thus, it is not surprising that miRNAs play important roles in glioblastoma initiation and progression. miRNAs have been indicated as critical regulators of glioblastoma stem cell maintenance (5), epigenetic regulation (6), tumorigenesis (7), oncogenic pathways, and migration (8, 9). Furthermore, miRNA is involved in regulating radio- and chemotherapy resistance and sensitivity and may serve as biomarkers for diagnosis and outcome (10, 11).
Upregulated miRNAs in glioblastoma cells can act as oncogenes (oncomiRs) and silence onco-suppressor genes. A prototypical example and one of the first oncomiRs identified is miR-21 which is involved in malignant processes by targeting genes important for proliferation, cell survival, invasion, and treatment resistance (12). Other upregulated miRNAs such as the miR-17-92 cluster, miR-10b, and miR-15b have been investigated in preclinical studies and shown to be indispensable for tumor initiation. As such, oncomiR blockade in glioma cells could activate numerous tumor suppressor genes (13) and also restore immune surveillance of the glioblastoma (14, 15). Liu et al., for example, demonstrated that miR-340-5p suppression in glioblastoma cell enhanced M2 macrophage polarization and macrophage recruitment to the glioblastoma microenvironment (16), suggesting restoration of miR-340-5p could be a potential strategy to reverse immune suppression mediated by M2 macrophages.
In contrast, some miRNAs may function as tumor suppressors and could be therapeutically reconstituted. For instance, the miR-1, miR-7, miR-34a, miR-124, miR-128, miR-138, and miR-181 family are a group of suppressor miRNAs inhibiting glioblastoma progression when they are overexpressed (8, 17, 18). These miRNAs not only directly target oncogenic/tumor suppressor pathways in glioma cells but also exert broad regulatory effects on the immune system. Our group found that miR-124 inhibited multiple targets in the signal transducer and activator of transcription 3 (STAT3) signaling pathway and reversed immune dysfunction of T cells induced by glioblastoma stem cells (GSCs) (19). We also showed that miR-138 can target multiple immune checkpoint molecules such as CTLA-4 and programmed cell death protein 1 (PD-1) to inhibit tumor-infiltrating Tregs. In vivo administration of miR-138 suppressed tumor development and significantly prolonged survival time of immune-competent glioma-bearing mice, but not immune-deficient mice (20), indicating the pivotal role of miR-138 in immunological tumor surveillance.
One of the key mechanisms for intercellular communication in the tumor microenvironment are exosomes that contain a wide variety of miRNAs (12–16, 21). Exosomal miR-21 has been shown to be an important mediator of immune cell reprogramming by glioblastoma cells to create a niche favorable for cancer progression (14). miR-1246 has been identified as the most enriched miRNA in glioblastoma-derived exosomes and mediates glioblastoma-induced protumorigenic macrophage formation by targeting TERF2IP and subsequently activating the STAT3 pathway (22). miR-214-5p, another glioblastoma-derived exosomal miRNA, mediates proinflammatory responses by targeting CXCR5 in primary microglia upon lipopolysaccharide stimulation (23). Furthermore, exosomal miR-29a and miR-92a from glioblastoma cells promotes the proliferation and immunosuppressive phenotype of glioblastoma-infiltrating macrophages (GIMs) by targeting protein kinase cAMP-dependent type I regulatory subunit alpha and high-mobility group box transcription factor 1 (24). Given this is an emerging area of investigation, it is likely that almost every key pathway and mechanism elucidated for gliomagenesis will have a network of miRNA control.
Secondary to an immunosuppressive microenvironment, glioblastoma patients are deficient in antitumor immunity leading to malignant progression and resistance to treatment. GIMs, the most frequent infiltrating immune cell subset (25), originate from peripheral blood monocytes in response to tumor-derived chemokines. Upon entry into the glioblastoma, the macrophages adopt a M0/M2 phenotype with the capacity to promote tumor cell invasion and exert immune suppression through factors like tumor growth factor-β (TGFβ) (26, 27). We have shown that miR-142-3p, by modulating the TGFβ signaling, inhibits the M2 phenotype of GIMs, and systemic in vivo administration of miR-142-3p induces antiglioma immune function (28). Ishii et al. reported that exogenous expression of miR-130a and miR-145 in myeloid-derived suppressor cells (MDSCs) decreased tumor metastasis through downregulation of TGFβ receptor II (TβRII) and related immune suppressive cytokines (29). Reciprocally, GIM-derived exosomal miR-22-3p, miR-27a-3p, and miR-221-3p can promote the proneural-to-mesenchymal transition of GSCs by simultaneously targeting CHD7 (30). miR-21 is also enriched in GIM-derived exosomes and mediates temozolomide (TMZ) resistance for which the STAT3 inhibitor pacritinib could overcome this resistance by downregulating miR-21 (31). Moreover, the downregulation of miR-21 promotes M1 macrophage polarization (32). As such, miR-21 deregulation is an operational mechanism in both GIMs and glioblastoma cells that modulates multiple oncogenic molecules, and signaling pathway such as STAT3 and anti-miR-21 strategies could be therapeutically developed.
In addition to the crosstalk between glioblastoma and immune cells, astrocytes also contribute to tumor growth, invasion, and immune suppression (33). For example, miR-19a is transferred from astrocytes to metastatic cancer cells in the central nervous system (CNS). Zhang et al. have demonstrated that exosomal transfer of miR-19a downregulates PTEN and subsequently promotes cytokine chemokine ligand 2 (CCL2) secretion. CCL2 recruits macrophages that contribute to immune resistance (34). Another astrocyte-associated miRNA, miR-10b, is overexpressed in gliomas and brain metastasis, and this miRNA can induce astrocyte transformation (13). Targeting astrocytes and their associated miRNAs is an emerging strategy for potentially treating CNS tumors.
Oligodendrocytes have important protecting roles because they produce myelin for neuron protection. Being a major cell population in the glioblastoma microenvironment, oligodendrocytes interact with astrocytes and microglia and participate in the formation of a tumor permissive niche (35, 36). Oligodendrocyte progenitor cells (OPCs) are the largest proliferating population in the CNS, and together with GIMs, are enriched at the infiltrating edge (37). Several miRNAs such as miR-219, miR-129-2, and miR-338 have higher expression at the infiltrating edge and are involved in oligodendrocyte differentiation (38). Negative regulators of oligodendrocyte differentiation such as SOX6, HES5, PDGFRA, and ZFP238 are suppressed by these miRNAs (39). One of the important functions of miR-219 is to mediate OPC differentiation to oligodendrocytes (40). Additionally, miR-219 indirectly promotes receptor tyrosine kinase signaling activity by targeting and inhibiting epidermal growth factor receptor expression. Thus, the evidence points to the involvement of oligodendrocyte-associated miRNAs in the glioblastoma microenvironment and warrants further study to determine their exact targets mediating the crosstalk of the OPCs and tumor cells.
Glioblastoma is highly vascularized, and its invasion and outgrowth rely on a nutrient supply by acquiring new blood vessel formation (41). Endothelial cell proliferation from the tumor is a direct measure of its malignancy. GSC-associated exosomes are capable of inducing angiogenesis of endothelia cells mediated by miR-21 (42). Some endothelial cell-associated miRNAs such as miR-145-5p and miR-5096 can transfer between human microvascular endothelial cells (HMECs) and glioblastoma cells through gap junctions. In the process, miR-145-5p is downregulated in early gliomagenesis and acts as a tumor suppressor when passing from HMECs into glioma cells, whereas miR-5096 is transferred from glioma cells into HMECs and promotes angiogenesis (43). Finally, the miR-221/222 cluster has also been shown to enhance angiogenesis and silencing attenuates angiogenesis by inhibiting the JAK/STAT pathway (44). All these targetable miRNAs associated with the various cell lineages in the glioblastoma microenvironment are depicted in Figure 1.
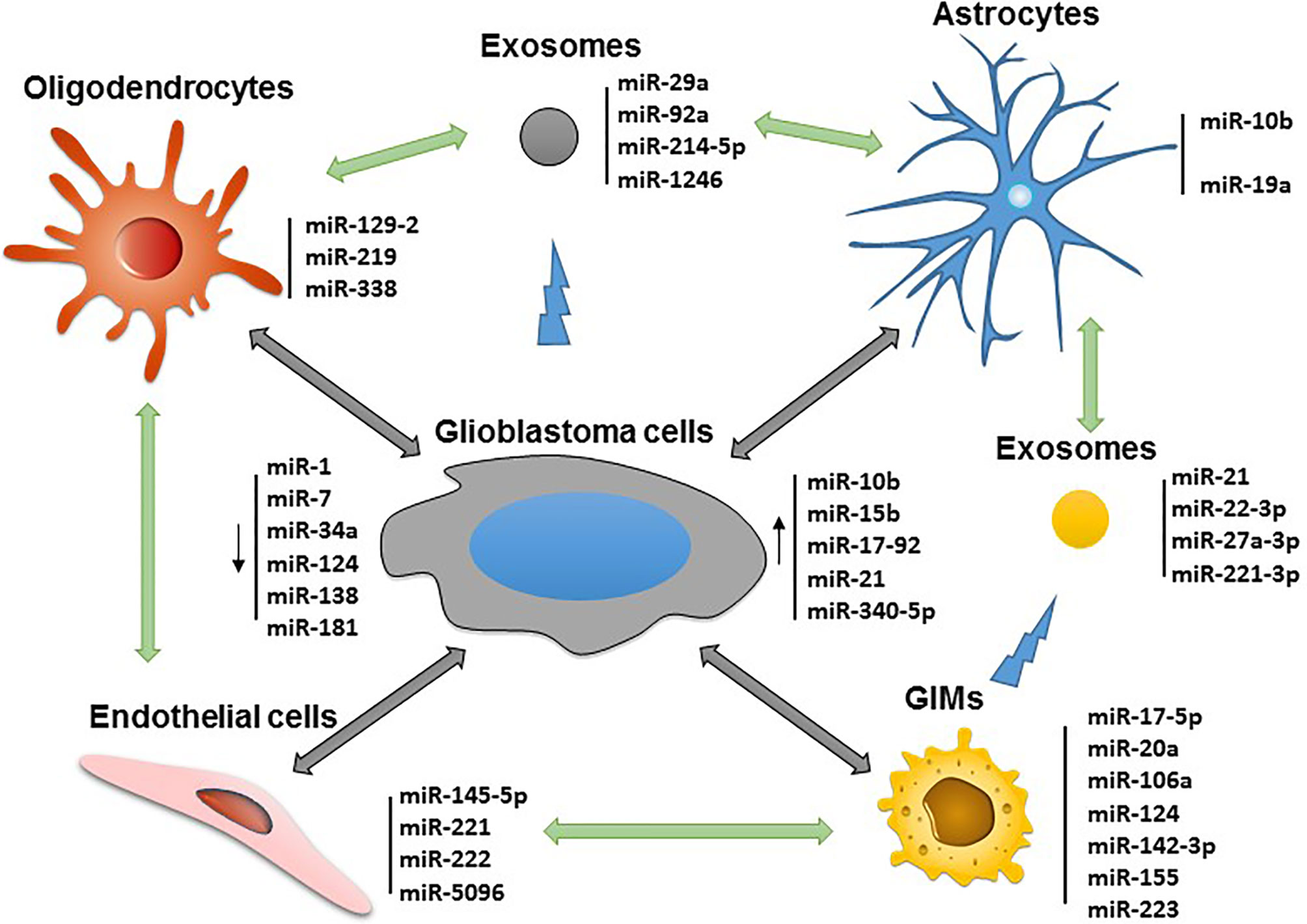
Figure 1 Potential miRNAs that can be targeted for immune modulation and therapeutic application in the glioblastoma microenvironment.
In addition to the use of miRNAs, siRNA, which are 21–27 base pair double-stranded oligonucleotides, are another treatment modality for inhibiting protein synthesis at the posttranscriptional level. Although both miRNA and siRNA strategies are capable of BBB penetration, miRNA has the therapeutic advantage of target networks which would be beneficial in complex heterogenous cancers such as glioblastoma. However, miRNA off target effects remains a substantial concern. In contrast, siRNA approaches have much greater specificity that is counterbalanced by tumor plasticity and escape mechanisms.
Abnormal vasculature is enriched in glioblastoma as a consequence of upregulated angiogenic factors such as VEGF. Increased VEGF causes new blood vessels to form within the tumor via angiogenesis and the associated proliferation of endothelial cells (45). The resulting vascular networks display increased vessel permeability and enlarged vessel size that results in plasma leakage into the tumor tissue and disruption to the BBB. Together, these abnormalities induce inflammation, cerebral edema, and increased interstitial pressure. Thus, antiangiogenic VEGF treatments have been extensively investigated including monoclonal antibodies such as bevacizumab and small molecules targeting its receptor VEGFR (46). These agents have limitations based on short half-life, limited efficacy in patient overall survival improvement, and systemic toxicity. Some siRNA formulations targeting VEGF have been evaluated in early stage clinical trials, but this have not been advanced further (47, 48).
T cells are present in the glioblastoma microenvironment, although at lower frequencies than GIMs (25). They have a profoundly exhausted phenotype characterized by expression of multiple immune checkpoint ligands (49, 50) likely accounting for their inability to control tumor growth. The lack of effective T-cell response is also highlighted by the ineffectiveness of checkpoint blockade immunotherapy in glioblastoma. Nonetheless, anti-PD-1 therapy achieves potent antiglioma activity in mouse glioma activity possibly through the depletion of PD-1+ macrophages and proinflammatory polarization in the glioblastoma microenvironment (51). A RNAi specific to the PD-1/PD-L1 pathway was delivered by a hemagglutinating virus of Japan-envelope—a nonreplicating viral vector that was capable of inhibiting immune suppression and eliciting antiglioma immune responses (52). Similarly, we showed that miR-138 could downregulate both CTLA-4 and PD-1 to inhibit tumor-infiltrating regulatory T cells (Tregs) and in vivo administration induced tumor reduction and prolonged the survival of immune syngeneic glioma-bearing mice (20).
Neuronal activity is involved in glioblastoma growth and progression (53). In the normal brain microenvironment, neurons are strong mitogenic signalers stimulating the growth of neural and oligodendrocyte precursor cells—an important consideration in the role of stem/progenitor cells in glioblastoma (54). Elegant studies of neuronal activity conducted by Venkatesh et al. in xenograft glioma mouse models show that presynaptic and postsynaptic function is disrupted in the presence of glioma with microenvironmental neurolignin 3 (NLGN3) being hijacked to induce signaling through the PI3K/PTEN/AKT/mTOR pathway (55). Neurons and OPCs produce NLGN3 by cleavage of ADAM10 sheddase, so the inhibition of this enzyme blocks NLGN3 secretion into the tumor microenvironment and suppresses glioma outgrowth in preclinical models. Therefore, siRNAs targeting NLGN3/ADAM10 are promising for treating glioblastoma by modulating the interaction between neuronal cells and tumor cells (56).
Growth differentiation factor (GDF15) is highly expressed in glioblastoma as a secreted cytokine participate in regulating tumor cell proliferation and immunosuppression (57). GDF15 promotes GSC stemness by activating the leukemia inhibitor factor–STAT3 pathway (58). Thus, it represents a potential therapeutic target in glioblastoma treatment by siRNA targeting GDF15 or its cognate receptor GFRAL (the GDNF family receptor alpha like) (59).
Temozolomide (TMZ) is the standard-of-care for glioblastoma and the other brain tumors, but many patients show limited response due to unmethylated O6-methylguanine-DNA methyltransferase (MGMT). Efforts to inhibit MGMT activity by systemic delivery of a siRNA have been made to silence the TMZ resistance gene MGMT. Wang et al. developed a MGMT siRNA nanoparticle that when combined with TMZ was found to reduce tumor growth and significantly extending survival in a GSC xenograft model relative to TMZ monotherapy (60). Like MGMT, other DNA damage response mediators such as ataxia-telangiectasia-mutated, ataxia-terlangiectasia-Rad3-related, DNA-dependent protein kinase, and poly-ADP-ribose polymerase could also be targeted with a siRNAs, which represent novel strategies for overcoming chemotherapy resistance (61–64).
The immunosuppressive potency of the glioblastoma microenvironment may be a function of tumor invasiveness and epithelial-mesenchymal transition (EMT), in which c-MET plays a key role. c-MET was recently shown to mediate EMT via activation of Wnt/β-catenin signaling (65). MiR-128-3p targeting c-MET inhibited glioblastoma migration and invasion and enhanced TMZ therapeutic efficacy in vivo (66). Other miRNAs such as miR-34a, miR-144-3p, and miR-562 have been reported to exert activity against glioblastoma proliferation and invasion by also targeting c-MET (66–68). Hence, these therapeutic miRNAs and c-MET siRNAs could be used to treat glioblastoma by means of suppressing tumor invasion and EMT.
GIM mediate immunosuppression is mediated by the chitinase-3-like-1 (CHI3L1)/Gal3-PI3K/ATK/mTOR axis. Chen et al. showed that inhibiting CHI3L1 complexes reversed GIM immunosuppression and delayed tumor progression (69). In other cancer models, genetic ablation of CHI3L1 in vivo reduced macrophage recruitment and increased effector T-cell infiltration in the tumor (70). Thus, siRNA targeting CHI3L1 is another potential therapy that could be beneficial to glioblastoma patients.
Overactivation of TGFβ signaling plays a critical role in reprogramming the glioblastoma to be immune suppressive and mediates immune escape and treatment resistance. In the glioblastoma microenvironment, a variety of dysfunctional cellular components and their interaction such as tumor, T, myeloid and NK cells are governed by the TGFβ signaling pathway. Consequently, the anti-TGFβ latency-associated peptide antibody can enhance antitumor immune responses in murine glioblastoma models (71); miR-142-3p targeting the TGFβR1 on M2 macrophages results in glioblastoma growth inhibition (28), and targeting the TGFβ-integrin axis improves NK cell antiglioblastoma activity (72). Anti-TGFβ RNA therapeutic represents a promising treatment avenue to be investigated in glioblastoma patients. A recent study showed that antisense oligonucleotides specifically targeting TGFβ1 and TGFβ2 exerts strong antiglioblastoma activity in vitro and in vivo (73).
S100A gene family members can modulate EMT, GSC stemness, and immune cell infiltration and are candidate therapeutic targets for glioblastoma patients (74). S100A4 is the most studied as a central player controlling EMT, stemness, and neutrophil infiltration. Its depletion downregulates the glioblastoma progression and treatment resistance (75, 76). Several microRNAs such as miR-124 (77), miR-149-3p (78), and miR-520c (79) have been identified as targeting S100A4, and their mimics resulted in antiglioblastoma activity in vivo. Therefore, these suppressor miRNAs and siRNAs targeting S100A4 could also be explored for therapeutic activity in glioblastoma patients.
Antisense oligonucleotides (ASOs) bind sequences specifically by Watson-Crick base pairing to the target RNA and regulate protein expression through RNase H mediated degradation and ribosome blockage by steric hindrance (80, 81). Significant advancement of oligonucleotide chemistry and numerous delivery platforms enhance ASO development and clinical application. Recently, the FDA has approved a few nucleic acid-based drugs, which stimulates greater interest in ASO therapeutic development. Currently, a variety of ASO drugs are being tested in clinical trials to treat cancer, infectious, and neurodegenerative diseases (82). Some ASO drugs target oncomiRs that promote tumorigenesis and metastasis. For example, RG-012, an anti-miR-21 ASO for Alport syndrome is being evaluated to ascertain if it decreases the rate of progression of renal fibrosis (83). Cobomarsen (MRG-106) is a miR-155 inhibitory ASO presently in phase II trials treating T cell leukemia and lymphoma (84). The anti-miR-21 and anti-miR-155 ASO strategy have been tested in preclinical models and showed potent anti-glioma efficacy (85, 86). Therefore, these anti–oncomiR ASOs can potentially be applied for the treatment of glioblastoma patients that have miR–21 and miR–155 dysregulation.
Aptamers are short oligonucleotides that possess a 3–D distinct structure for their target recognition and binding. Systematic Evolution of Ligands by EXponential enrichment (SELEX) is the most used screening approach to identify specific aptamers binding a target with high affinity and selectivity. Their relatively small molecular weight (one–tenth that of monoclonal antibody) makes them more accessible to the glioblastoma microenvironment (87). In addition, aptamers are called chemical antibodies since they can be derived completely using chemical synthesis. Moreover, the low immunogenicity and long shelf life are advantageous features of aptamers for clinical application. Oligonucleotide aptamers are synthesized and assembled with cell–free automation enabling rapid and cost–effective production with minimal variation between batches. However, for clinical utility, they require further chemical modification for improving their in vivo half–life because of fast renal excretion and nuclease degradation. These modifications should have minimal effect on the affinity and specificity of the aptamers, and simultaneously improve their stability. Some promising modifications include inverted thymidine capping on the terminals, two hydroxyl group modifications in the ribose ring, the phosphodiester bond replacement and PEGylation (88). De La Fuente et al. identified several human and mouse specific RNA aptamers using tumor–associated myeloid cells as the targets via SELEX. These aptamers were specific to tumor–associated MDSCs in several cancer lineages including glioblastoma and had high binding affinity—highlighting their application as therapeutics targeting to the tumor microenvironment (89). Conjugating these MDSC–specific aptamers to a tumoricidal agent could significantly improve their potency.
Nucleotide aptamers and siRNAs share the same nucleic acid units, so base pair annealing, or covalent linkage can create an aptamer–siRNA chimera. These chimeras have great advantages over protein and cellular products: single component simplicity, small size, and easy manufacturing (90, 91). They also have less immunogenicity because the human immune system does not recognize nucleic acids as foreign molecules (92). Additionally, the siRNA portion in the chimeras can still be recognized and processed by Dicer with no compromised efficacy, resulting in their target mRNA degradation and protein depletion (93). The first aptamer–siRNA chimera designated PSMA aptamer–Plk1 siRNA was constructed in 2006. Since then, a number of aptamer–siRNA chimeras have been made with improved stability, targetting specificity and in vivo efficacy (94). A list of the examples for targeted RNAi potentially applicable to the glioblastoma and its associated microenvironment are presented in Table 1.

Table 1 Summary of aptamer–siRNA chimeras potentially applicable to the glioblastoma.
In spite of significant successes of immune checkpoint blockade (ICB) in treating cancer patients, thus far, this immunotherapy approach has minimal efficacy for the vast majority of glioblastoma patients secondary to a wide variety of mechanisms such as mutations in the antigen presentation pathway and the IFN–γ signaling pathway (106). Protein tyrosine phosphatase (PTPN2) has been identified by in vivo CRISPR screening as a new target–mediating resistance to ICB immunotherapy. Knockdown of PTPN2 results in enhanced ICB therapeutic efficacy by promoting antigen presentation and IFN–γ signaling in tumor cells (107). PTPN2 is overexpressed in glioblastoma and its expression associates with IDH wild–type expression and the mesenchymal subtype that indicates a worse prognosis. Furthermore, there is an inverse relationship between PTPN2 and an inflammatory response in glioblastoma (108). Thus, we believe that a PTPN2 siRNA–tumor–specific aptamer therapeutic may provide an important strategy to overcome ICB resistance of glioblastoma patients.
Another target mediating ICB resistance is A–to–I editing of interferon–inducible RNA species (ADAR1) that encodes an adenosine deaminase that inhibits the sensing of endogenous double–stranded RNAs (dsRNAs), and subsequently hinders antitumor immunity. Thus, ADAR1 inhibition can improve patient responses when combined with PD–1 blockade by overcoming the resistance mechanism of nonresponding to endogenous dsRNAs (109). ADAR1 is found highly active in glioma tissues and cells and essential for the maintenance of gliomagenesis (110), so we propose that ADAR1 can be a potential target inhibited by tumor cell specific aptamer–ADAR1 siRNA conjugates.
One of the major challenges in developing effective cancer immunotherapy is to identify tumor–specific and immunogenic neoantigens to stimulate a robust and durable immune response. There are several approaches to induce neoantigens in tumor cells in situ by aptamer–siRNAs specific to unique pathways that trigger the expression of neoantigens. The first pathway is nonsense–mediated messenger RNA decay (NMD), which is a highly conserved surveillance mechanism in mammal cells that prevents the translation of mRNAs with a premature stopping codon. NMD inhibition using tumor–specific oligonucleotide aptamer–targeted delivery of siRNAs to NMD–associated molecules such as SMG1 and UPF2 results in the expression of de novo antigens encoded by the premature stopping codon–containing mRNAs and their immune–mediated tumor rejection in metastatic and subcutaneous tumor models (111). This strategy is readily applicable to human glioblastoma as NMD pathway is important for gliomagenesis detection (112) and SMG1 mRNA expression is present in glioma cells (TCGA_GBM data). The second one is to target the transporter associated with antigen processing (TAP). Genetic ablation results in drastically enhanced presentation of new MHC class I–restricted epitopes independent of TAP. These induced new antigen epitopes form MHC–peptide complexes for engaging and activating CD8+ T cells capable of killing TAP–deficient tumor cells. Administration of TAP siRNAs conjugated to a tumor–targeting nucleolin aptamer (AS1411) has been shown to exert antitumor activity in multiple mouse tumor models (113). Both TAP and nucleolin are expressed in gliomas, making AS1411 and TAP siRNA conjugates an appealing candidate of the RNA–based immune therapy for treating glioblastoma patients with preclinical efforts underway by our group.
STAT3 has been shown to be a signaling hub in tumor cells as well as tumor–associated immune cells (114, 115). In the glioblastoma microenvironment, STAT3 is persistently activated in glioma cells, myeloid, and T cells and promotes tumor cell survival, proliferation, invasion, and immunosuppression (26, 116, 117). Due to CTLA4 upregulation on tumor–infiltrating CD8+ T cells, a CTLA4‐targeting aptamer STAT3 siRNA chimera was created that triggers CD8+ T cell reactivation in the tumor microenvironment. Additionally, this chimera inhibits tumor–infiltrating Tregs and shows significant antitumor efficacy in multiple primary and metastatic tumor models (95). The Yu group has generated a DNA aptamer CpG1668–STAT3 siRNA chimera linked by a C3 carbon chain, which preserved the immunostimulatory properties of CpG1668 and at the same time does not interfere with Dicer processing of siRNA, thereby contributing to synergistic antitumor effects (118). Other aptamers have been tested including: (1) a STAT3 siRNA that successfully induces antitumor effects in glioblastoma when conjugated with a PDR3 aptamer against PDGFRα (104), and (2) a PDGFRβ–specific aptamer–siRNA chimera designated Gin4.T–STAT3 that could efficiently antagonize STAT3 in PDGFRβ+ GBM xenografts (103, 119).
Two major cell components in the glioblastoma microenvironment responsible for tumor escape from immune surveillance include GSCs and GIMs (26, 116, 120). Osteopontin (OPN), a key molecule–mediating immune suppression in this setting, is highly expressed in both GSCs and GIMs. It is a secreted phosphoprotein chemokine that also operates intracellularly with both forms playing important roles in tumor growth and metastasis (121). Our data indicate that highly expressed OPN in the glioblastoma microenvironment is indispensable for macrophage infiltration. We have further shown that both tumor–derived OPN and nontumor–derived OPN are essential for glioblastoma development. A deficiency of OPN in either glioblastoma cells or immune cells results in a marked reduction in numbers of immune suppressive M2 macrophages and enhanced T–cell effector function (87). As such, OPN is an attractive therapeutic target specific to the glioblastoma microenvironment. Interestingly, periostin sharing the same RGD functional motif with OPN is secreted from GSCs and correlates with GIM infiltration in human glioblastoma. Periostin depletion diminishes the tumor supportive M2 type of GIMs in xenografts (122). CCL2, another chemokine highly enriched in the glioma microenvironment, is important for attracting both CCR2+Ly–6C+ monocytic MDSCs and CCR4+ Tregs. CD163+ GIMs are a major source producing CCL2 in the glioblastoma microenvironment (123). GIM–derived CCL8 contributes to the stemness maintenance and invasion of glioblastoma cells through ERK1/2 pathway and its blockade significantly decreases invasion of glioma cells (124). Boeck et al. have also shown that IL–33 is another important chemokine–mediating GIM infiltration since its expression correlates with GIM density in human and mouse glioma tissues. Furthermore, both intracellular and secreted isoforms of IL–33 upregulate other chemokines that collectively recruit and transform peripheral innate immune cells to create an immunosuppressive environment (125). Bispecific aptamer siRNA conjugates to the aforementioned chemokines are plausible strategies and could be developed.
Bispecific aptamers, composed of two aptamers, exhibit concurrent binding to two different entities such as antigens. Absence of costimulatory signal in the tumor microenvironment results T–cell energy (126). Accordingly, an aptamer specific to 4–1BB receptor has been developed to target and activate tumor–infiltrating T cells (127). Pastor et al. generated the first bispecific aptamers consisting of a bivalent 4–1BB aptamer and a tumor–specific PSMA aptamer, enhancing the conjugate delivery to the tumor niche and activation of costimulatory responses. Profound antitumor activity of a 4–1BB–PSMA aptamer chimera has been observed in multiple immune competent mouse models including colon cancer and melanoma lung metastasis when administered systemically (128). A similar strategy could be considered for glioblastoma but would require the selection of a subset of patients that express a given tumor antigen. Other bispecific aptamers have been engineered to specifically target CD28–expressing T cells in multidrug resistance–associated protein 1 melanoma that triggered prolonged survival of tumor–bearing mice. The associated immune mechanism included reactivation of tumor–infiltrating T cells via CD28 costimulation by CD28 aptamer binding and crosslinking (95). Schrand et al. synthesized another bivalent aptamer by fusing an agonistic 4–1BB aptamer with an aptamer specific to VEGF and showed effective targeting of the stroma in the tumor microenvironment. This 4–1BB–VEGF aptamer was capable of inducing the activation and expansion of CD8+ effector T cells and promoting T memory cell differentiation that prevented tumor recurrence across cancer lineages (129). Since VEGF expression is a common feature of glioblastoma, a strategy in which T cells are activated and expanded may induce glioma cell–specific killing (130), but this will require preclinical testing. A list of the examples for bispecific aptamers potentially applicable to the glioblastoma and its associated microenvironment is presented in Table 2.
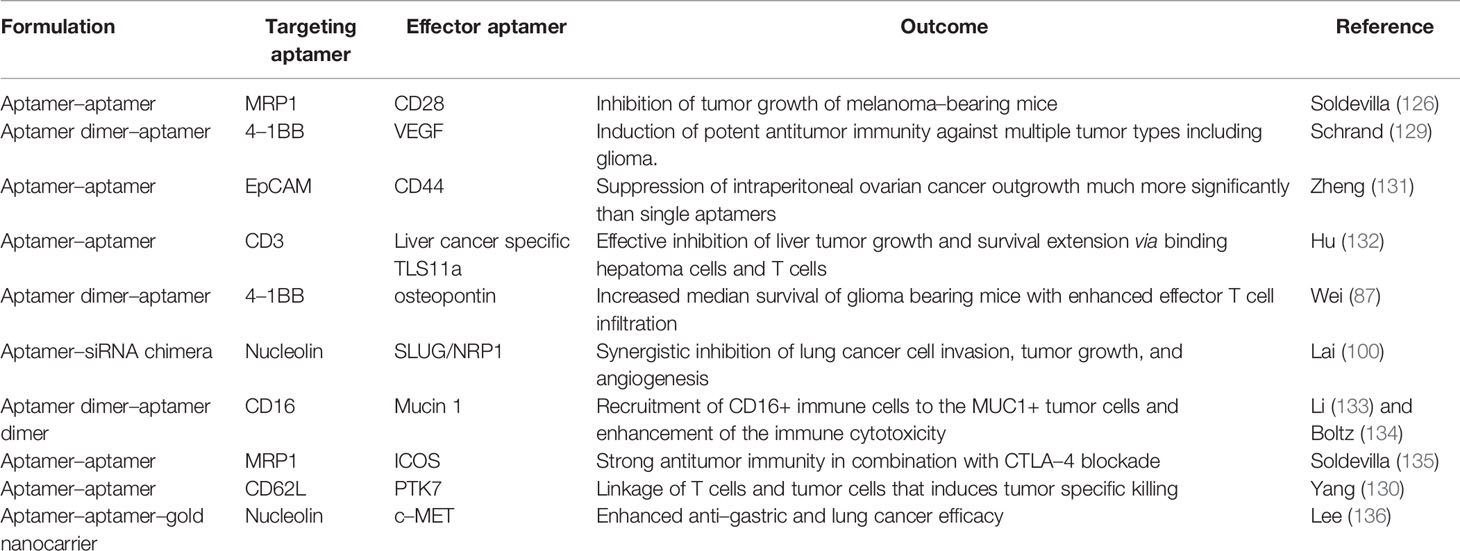
Table 2 Summary of bispecific aptamers applicable to the glioblastoma treatment.
Aptamer–siRNA chimeras and bispecific aptamers are one of the most efficient strategies for target delivery modules because of their high specificity and binding affinity, fully automated synthesis, and great potential for clinical application. Nonetheless, a major obstacle needs to be overcome for delivery of siRNAs into the cytoplasm of the targeted tumor and/or immune cells. One hurdle for the efficient delivery of aptamer–siRNA chimeras to the cytoplasm is the negative charge of nucleotides and endosomal degradation. A strategy that could overcome this limitation is to embed the cell–specific aptamers into siRNA encapsulating nanoparticles, which improve the delivery efficacy of naked siRNAs passing through the cellular barriers.
Many cell types in the glioblastoma microenvironment interact with each other through microvesicles and exosomes (137). As a natural system of miRNA delivery, these vesicles can be secreted from genetically engineered miRNA overexpressing cells or generated from exosomes transfected with miRNAs (138, 139). Glioblastoma cells or stem cells, for example, could be genetically modified to express exogenous tumor suppressor miRNAs and the elaborated exosomes with the tumor–suppressor miRNA used as a therapeutic product. Proof–of–principal preclinical studies using this type of strategy have been shown to reduce the tumor burden and have potential clinical utility (140). Although there are other contents in the exosomes such as other RNA and protein molecules, this composition can be altered by the cell status and signaling stimuli (141). As such, these natural vesicles may be an excellent RNAi carrier system (142). Continuing efforts are warranted to improve capacity of these exosome nanocarriers for passing the BBB and efficient delivery of their siRNA cargo to the glioblastoma microenvironment.
Other types of natural modifications include conjugation to chitosan, a natural polysaccharide composed of repeating N–acetyl–glucosamine and glucosamine units. The cationic charge of chitosan can rapidly form complexes with negatively charged nucleic acids. Furthermore, highly reactive amino and hydroxyl groups of chitosan allow for chemical modification and linkage of cognate ligands (143). Noh et al. demonstrated that systemic administration of EGFL6 siRNA—chitosan nanoparticles were delivered to endothelial ovarian cancer cells and markedly inhibited tumor progression (144).
For effective glioblastoma therapy, nanoparticles that are delivered systemically must have tumor and/or immune specificity with little measurable side effects. More recently, RNA nanocarriers have gained attention as a versatile natural platform of nanoassembly and construction. The three–strand packaging RNA complex in the polyhedra bacteriophage phi29 is self–assembled and highly dynamic. This unique feature is utilized to generate a variety of RNA nanoparticles with a wide range of specific sizes and shapes. The pRNA–3WJ motif is a three–RNA–strand scaffold that is capable of targeting intracranial gliomas in mice (145).
Lipid–formulated nanoparticles have intensively been utilized in laboratory studies and clinical trials for RNA therapeutics delivery because of the ease of manufacturing and high biocompatibility (146). Sun et al. developed a novel liposome system simultaneously delivering survivin siRNA and paclitaxel to the glioblastoma. Specifically, a CD133–specific RNA aptamer and a low–density lipoprotein receptor–related protein were integrated into the exterior membrane of the liposomes, resulting in dual targeting ability to bind glioblastoma cells and endothelial cells in the tumor microenvironment. This lipid nanoformulation could enrich in the tumor niche via efficiently binding to the low–density lipoprotein receptor expressing endothelial cells, and selectively induce apoptosis of CD133+ GSCs as well as endothelial stroma cells (147). Another similar liposome siRNA delivery carrier has been developed to treat breast cancer by means of CD44 aptamer targeting. These liposomes were found to efficiently inhibit CD44+ tumor outgrowth in vivo (148). CD44 is also found overexpressed in glioma cells and a common GSC surface marker (149). As such, an anti–CD44 aptamer–equipped siRNA liposome delivery system may be applicable to glioblastoma.
A critical determinant of successful delivery of RNA is the prevention of nuclease degradation. Polymeric nanocarriers have been extensively utilized for the protection of miRNAs and siRNAs due to their positive charge (150). Recently, cationic polymers were broadly used to form stable complexes with negatively charged RNA. Among them, polyethyleneimine is the most commonly used for nucleic acid delivery, but its clinical application is hampered by its inherent toxicity. One alternative approach is to use hybrid polymers such as PEI–chitosan hybrid nanocarriers that show an improved safety profile (151). Another strategy is to link polyethyleneimine polyplexes with brain–targeting rabies virus glycoprotein. These nanoparticles have shown both effective brain tumor targeting and low toxicity (152).
Despite enormous and continuous efforts for developing new and combinational treatment strategies for glioblastoma, there has been minimal improvement in survival. The brain tumor microenvironment is a key driver that promotes and regulates tumor initiation and progress and mediates the treatment resistance in both primary and metastatic brain malignancies. Molecular dissection into the protumorigenic functions of single elements of the brain tumor microenvironment has resulted in the discovery of a number of promising noncoding RNA therapeutic targets. These RNA therapeutics include miRNAs, siRNAs, and aptamers and are emerging as a novel avenue to treat brain cancer patients. Since miRNAs can act upon multiple targets and pathways regulating immune suppression and chemoresistance, they may be more effective in treating the malignancies such as glioblastoma that are highly heterogeneous. Nonetheless, one major challenge remains for clinical application for treating glioblastoma is the ability of an agent to cross the BBB. This issue is alleviated by their small molecular weight and compact size, which can be further enhanced with nanocarrier–specific targeted delivery. Increasingly sophisticated nanoparticle systems, also relying on targeting moieties for BBB penetration and/or improved target cell transfection efficacy, may provide an avenue toward clinical application. On the other hand, it should be noted that too complicated systems based on multiple components may prohibit drug approval and transition into the clinic.
The unique characteristics of aptamers make them highly attractive for targeted therapy of glioblastoma and the other malignancies. A given aptamer can be conjugated to another aptamer, siRNA or miRNA, which leads to the production of multimodal chimeric therapeutics with novel functions enabling simultaneous targeting of numerous molecules and cell subsets in the glioblastoma microenvironment. Their small size and simple structure may have key advantages superior for passing through the BBB and gaining access to the brain tumor when administered systemically.
JW provided ideas for the project and wrote the initial draft. ABH, GAC and EG completed the final version. All authors contributed to the article and approved the submitted version.
Funding was provided by the Ben and Catherine Ivy Foundation, the MD Anderson Cancer Center Provost Fund and NIH grant CA120813.
GC is one of the scientific founders of Ithax Pharmaceuticals.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Jethwa K, Wei J, McEnery K, Heimberger AB. miRNA–Mediated Immune Regulation and Immunotherapeutic Potential in Glioblastoma. Clin Invest (2011) 1:1637–50. doi: 10.4155/cli.11.159
2. Pastor F, Berraondo P, Etxeberria I, Frederick J, Sahin U, Gilboa E, et al. An RNA Toolbox for Cancer Immunotherapy. Nature reviews. Drug Discov (2018) 17:751–67. doi: 10.1038/nrd.2018.132
3. Rupaimoole R, Calin GA, Lopez–Berestein G, Sood AK. miRNA Deregulation in Cancer Cells and the Tumor Microenvironment. Cancer Discovery (2016) 6:235–46. doi: 10.1158/2159-8290.CD-15-0893
4. Acunzo M, Romano G, Wernicke D, Croce CM. MicroRNA and Cancer–a Brief Overview. Adv Biol Regul (2015) 57:1–9. doi: 10.1016/j.jbior.2014.09.013
5. Katsushima K, Kondo Y. Non–Coding RNAs as Epigenetic Regulator of Glioma Stem–Like Cell Differentiation. Front Genet (2014) 5:14. doi: 10.3389/fgene.2014.00014
6. Kreth S, Thon N, Kreth FW. Epigenetics in Human Gliomas. Cancer Lett (2014) 342:185–92. doi: 10.1016/j.canlet.2012.04.008
7. Nikaki A, Piperi C, Papavassiliou AG. Role of microRNAs in Gliomagenesis: Targeting miRNAs in Glioblastoma Multiforme Therapy. Expert Opin Invest Drugs (2012) 21:1475–88. doi: 10.1517/13543784.2012.710199
8. Bronisz A, Wang Y, Nowicki MO, Peruzzi P, Ansari K, Ogawa D, et al. Extracellular Vesicles Modulate the Glioblastoma Microenvironment via a Tumor Suppression Signaling Network Directed by miR–1. Cancer Res (2014) 74:738–50. doi: 10.1158/0008-5472.CAN-13-2650
9. Godlewski J, Bronisz A, Nowicki MO, Chiocca EA, Lawler S. microRNA–451: A Conditional Switch Controlling Glioma Cell Proliferation and Migration. Cell Cycle (Georgetown Tex) (2010) 9:2742–8. doi: 10.4161/cc.9.14.12248
10. Hermansen SK, Kristensen BW. MicroRNA Biomarkers in Glioblastoma. J Neuro–Oncol (2013) 114:13–23. doi: 10.1007/s11060-013-1155-x
11. Anfossi S, Babayan A, Pantel K, Calin GA. Clinical Utility of Circulating non–Coding RNAs – an Update. Nature Reviews. Clin Oncol (2018) 15:541–63. doi: 10.1038/s41571-018-0035-x
12. Møller HG, Rasmussen AP, Andersen HH, Johnsen KB, Henriksen M, Duroux M. A Systematic Review of microRNA in Glioblastoma Multiforme: Micro–Modulators in the Mesenchymal Mode of Migration and Invasion. Mol Neurobiol (2013) 47:131–44. doi: 10.1007/s12035-012-8349-7
13. El Fatimy R, Subramanian S, Uhlmann EJ, Krichevsky AM. Genome Editing Reveals Glioblastoma Addiction to MicroRNA–10b. Mol Ther (2017) 25:368–78. doi: 10.1016/j.ymthe.2016.11.004
14. Abels ER, Maas SLN, Nieland L, Wei Z, Cheah PS, Tai E, et al. Glioblastoma–Associated Microglia Reprogramming Is Mediated by Functional Transfer of Extracellular miR–21. Cell Rep (2019) 28:3105–19.e7. doi: 10.1016/j.celrep.2019.08.036
15. Guo X, Qiu W, Liu Q, Qian M, Wang S, Zhang Z, et al. Immunosuppressive Effects of Hypoxia–Induced Glioma Exosomes Through Myeloid–Derived Suppressor Cells via the miR–10a/Rora and miR–21/Pten Pathways. Oncogene (2018) 37:4239–59. doi: 10.1038/s41388-018-0261-9
16. Liu Y, Li X, Zhang Y, Wang H, Rong X, Peng J, et al. An miR–340–5p–Macrophage Feedback Loop Modulates the Progression and Tumor Microenvironment of Glioblastoma Multiforme. Oncogene (2019) 38:7399–415. doi: 10.1038/s41388-019-0952-x
17. Yang L, Ma Y, Xin Y, Han R, Li R, Hao X. Role of the microRNA 181 Family in Glioma Development. Mol Med Rep (2018) 17:322–9. doi: 10.3892/mmr.2017.7895
18. Banelli B, Forlani A, Allemanni G, Morabito A, Pistillo MP, Romani M. MicroRNA in Glioblastoma: An Overview. Int J Genomics (2017) 2017:7639084. doi: 10.1155/2017/7639084
19. Wei J, Wang F, Kong LY, Xu S, Doucette T, Ferguson SD, et al. miR–124 Inhibits STAT3 Signaling to Enhance T Cell–Mediated Immune Clearance of Glioma. Cancer Res (2013) 73:3913–26. doi: 10.1158/0008-5472.CAN-12-4318
20. Wei J, Nduom EK, Kong LY, Hashimoto Y, Xu S, Gabrusiewicz K, et al. MiR–138 Exerts Anti–Glioma Efficacy by Targeting Immune Checkpoints. Neuro–oncology (2016) 18:639–48. doi: 10.1093/neuonc/nov292
21. Anfossi S, Fu X, Nagvekar R, Calin GA. MicroRNAs, Regulatory Messengers Inside and Outside Cancer Cells. Adv Exp Med Biol (2018) 1056:87–108. doi: 10.1007/978-3-319-74470-4_6
22. Qian M, Wang S, Guo X, Wang J, Zhang Z, Qiu W, et al. Hypoxic Glioma–Derived Exosomes Deliver microRNA–1246 to Induce M2 Macrophage Polarization by Targeting TERF2IP via the STAT3 and NF–κb Pathways. Oncogene (2020) 39:428–42. doi: 10.1038/s41388-019-0996-y
23. Yang JK, Liu HJ, Wang Y, Li C, Yang JP, Yang L, et al. Exosomal miR–214–5p Released From Glioblastoma Cells Modulates Inflammatory Response of Microglia After Lipopolysaccharide Stimulation Through Targeting Cxcr5. CNS Neurol Disord Drug Targets (2019) 18:78–87. doi: 10.2174/1871527317666181105112009
24. Guo X, Qiu W, Wang J, Liu Q, Qian M, Wang S, et al. Glioma Exosomes Mediate the Expansion and Function of Myeloid–Derived Suppressor Cells Through microRNA–29a/Hbp1 and microRNA–92a/Prkar1a Pathways. Int J Cancer (2019) 144:3111–26. doi: 10.1002/ijc.32052
25. Hussain SF, Yang D, Suki D, Aldape K, Grimm E, Heimberger AB. The Role of Human Glioma–Infiltrating Microglia/Macrophages in Mediating Antitumor Immune Responses. Neuro–Oncology (2006) 8:261–79. doi: 10.1215/15228517-2006-008
26. Wu A, Wei J, Kong LY, Wang Y, Priebe W, Qiao W, et al. Glioma Cancer Stem Cells Induce Immunosuppressive Macrophages/Microglia. Neuro–Oncology (2010) 12:1113–25. doi: 10.1093/neuonc/noq082
27. Gabrusiewicz K, Rodriguez B, Wei J, Hashimoto Y, Healy LM, Maiti SN, et al. Glioblastoma–Infiltrated Innate Immune Cells Resemble M0 Macrophage Phenotype. JCI Insight (2016) 1(2):e85841. doi: 10.1172/jci.insight.85841
28. Xu S, Wei J, Wang F, Kong LY, Ling XY, Nduom E, et al. Effect of miR––3p on the M2 Macrophage and Therapeutic Efficacy Against Murine Glioblastoma. J Natl Cancer Inst (2014) 106:dju162. doi: 10.1093/jnci/dju162
29. Ishii H, Vodnala SK, Achyut BR, So JY, Hollander MC, Greten TF, et al. miR–130a and miR–145 Reprogram Gr–1(+)CD11b(+) Myeloid Cells and Inhibit Tumor Metastasis Through Improved Host Immunity. Nat Commun (2018) 9:2611. doi: 10.1038/s41467-018-05023-9
30. Zhang Z, Xu J, Chen Z, Wang H, Xue H, Yang C, et al. Transfer of MicroRNA via Macrophage–Derived Extracellular Vesicles Promotes Proneural–To–Mesenchymal Transition in Glioma Stem Cells. Cancer Immunol Res (2020) 8:966–81. doi: 10.1158/2326-6066.CIR-19-0759
31. Chuang HY, Su YK, Liu HW, Chen CH, Chiu SC, Cho DY, et al. Preclinical Evidence of STAT3 Inhibitor Pacritinib Overcoming Temozolomide Resistance via Downregulating miR––Enriched Exosomes From M2 Glioblastoma–Associated Macrophages. J Clin Med (2019) 8:959. doi: 10.20944/preprints201905.0374.v1
32. Xi J, Huang Q, Wang L, Ma X, Deng Q, Kumar M, et al. miR–21 Depletion in Macrophages Promotes Tumoricidal Polarization and Enhances PD–1 Immunotherapy. Oncogene (2018) 37:3151–65. doi: 10.1038/s41388-018-0178-3
33. Brandao M, Simon T, Critchley G, Giamas G. Astrocytes, the Rising Stars of the Glioblastoma Microenvironment. Glia (2019) 67:779–90. doi: 10.1002/glia.23520
34. Zhang L, Zhang S, Yao J, Lowery FJ, Zhang Q, Huang WC, et al. Microenvironment–Induced PTEN Loss by Exosomal microRNA Primes Brain Metastasis Outgrowth. Nature (2015) 527:100–4. doi: 10.1038/nature15376
35. Peferoen L, Kipp M, van der Valk P, van Noort JM, Amor S. Oligodendrocyte–Microglia Cross–Talk in the Central Nervous System. Immunology (2014) 141:302–13. doi: 10.1111/imm.12163
36. Hide T, Shibahara I, Kumabe T. Novel Concept of the Border Niche: Glioblastoma Cells Use Oligodendrocytes Progenitor Cells (GAOs) and Microglia to Acquire Stem Cell–Like Features. Brain Tumor Pathol (2019) 36:63–73. doi: 10.1007/s10014-019-00341-2
37. Hide T, Komohara Y, Miyasato Y, Nakamura H, Makino K, Takeya M, et al. Oligodendrocyte Progenitor Cells and Macrophages/Microglia Produce Glioma Stem Cell Niches at the Tumor Border. EBioMedicine (2018) 30:94–104. doi: 10.1016/j.ebiom.2018.02.024
38. Hide T, Komohara Y. Oligodendrocyte Progenitor Cells in the Tumor Microenvironment. Adv Exp Med Biol (2020) 1234:107–22. doi: 10.1007/978-3-030-37184-5_8
39. Zhao X, He X, Han X, Yu Y, Ye F, Chen Y, et al. MicroRNA–Mediated Control of Oligodendrocyte Differentiation. Neuron (2010) 65:612–26. doi: 10.1016/j.neuron.2010.02.018
40. Galloway DA, Moore CS. miRNAs As Emerging Regulators of Oligodendrocyte Development and Differentiation. Front Cell Dev Biol (2016) 4:59. doi: 10.3389/fcell.2016.00059
41. Dubois LG, Campanati L, Righy C, D’Andrea–Meira I, Spohr TC, Porto–Carreiro I, et al. Gliomas and the Vascular Fragility of the Blood Brain Barrier. Front Cell Neurosci (2014) 8:418. doi: 10.3389/fncel.2014.00418
42. Sun X, Ma X, Wang J, Zhao Y, Wang Y, Bihl JC, et al. Glioma Stem Cells–Derived Exosomes Promote the Angiogenic Ability of Endothelial Cells Through miR–21/VEGF Signal. Oncotarget (2017) 8:36137–48. doi: 10.18632/oncotarget.16661
43. Lu Y, Chopp M, Zheng X, Katakowski M, Buller B, Jiang F. MiR–145 Reduces ADAM17 Expression and Inhibits In Vitro Migration and Invasion of Glioma Cells. Oncol Rep (2013) 29:67–72. doi: 10.3892/or.2012.2084
44. Xu CH, Liu Y, Xiao LM, Chen LK, Zheng SY, Zeng EM, et al. Silencing microRNA–221/222 Cluster Suppresses Glioblastoma Angiogenesis by Suppressor of Cytokine Signaling––Dependent JAK/STAT Pathway. J Cell Physiol (2019) 234:22272–84. doi: 10.1002/jcp.28794
45. Weathers SP, de Groot J. VEGF Manipulation in Glioblastoma. Oncol (Williston Park NY) (2015) 29:720–7.
46. Kochar AS, Madhavan M, Manjila S, Scoco A, Belle VK, Geertman RT. Contemporary Updates on Clinical Trials of Antiangiogenic Agents in the Treatment of Glioblastoma Multiforme. Asian J Neurosurg (2018) 13:546–54. doi: 10.4103/ajns.AJNS_266_16
47. Tabernero J, Shapiro GI, LoRusso PM, Cervantes A, Schwartz GK, Weiss GJ, et al. 3rd, First–In–Humans Trial of an RNA Interference Therapeutic Targeting VEGF and KSP in Cancer Patients With Liver Involvement. Cancer Discovery (2013) 3:406–17. doi: 10.1158/2159-8290.CD-12-0429
48. Kaiser PK, Symons RC, Shah SM, Quinlan EJ, Tabandeh H, Do DV, et al. RNAi–Based Treatment for Neovascular Age–Related Macular Degeneration by Sirna–027. Am J Ophthalmol (2010) 150:33–39.e2. doi: 10.1016/j.ajo.2010.02.006
49. Ott M, Tomaszowski KH, Marisetty A, Kong LY, Wei J, Duna M, et al. Profiling of Patients With Glioma Reveals the Dominant Immunosuppressive Axis Is Refractory to Immune Function Restoration. JCI Insight (2020) 5(17):e134386. doi: 10.1172/jci.insight.134386
50. Woroniecka K, Chongsathidkiet P, Rhodin K, Kemeny H, Dechant C, Farber SH, et al. T–Cell Exhaustion Signatures Vary With Tumor Type and Are Severe in Glioblastoma. Clin Cancer Res (2018) 24:4175–86. doi: 10.1158/1078-0432.CCR-17-1846
51. Rao G, Latha K, Ott M, Sabbagh A, Marisetty A, Ling X, et al. Anti–PD–1 Induces M1 Polarization in the Glioma Microenvironment and Exerts Therapeutic Efficacy in the Absence of CD8 Cytotoxic T Cells. Clin Cancer Res (2020) 26:4699–712. doi: 10.1158/1078-0432.CCR-19-4110
52. Sugii N, Matsuda M, Okumura G, Shibuya A, Ishikawa E, Kaneda Y, et al. Hemagglutinating Virus of Japan–Envelope Containing Programmed Cell Death–Ligand 1 siRNA Inhibits Immunosuppressive Activities and Elicits Antitumor Immune Responses in Glioma. Cancer Sci (2020) 112:81–90. doi: 10.1111/cas.14721
53. Venkatesh HS, Johung TB, Caretti V, Noll A, Tang Y, Nagaraja S, et al. Neuronal Activity Promotes Glioma Growth Through Neuroligin–3 Secretion. Cell (2015) 161:803–16. doi: 10.1016/j.cell.2015.04.012
54. Lan X, Jörg DJ, Cavalli FMG, Richards LM, Nguyen LV, Vanner RJ, et al. Fate Mapping of Human Glioblastoma Reveals an Invariant Stem Cell Hierarchy. Nature (2017) 549:227–32. doi: 10.1038/nature23666
55. Venkatesh HS, Tam LT, Woo PJ, Lennon J, Nagaraja S, Gillespie SM, et al. Targeting Neuronal Activity–Regulated Neuroligin–3 Dependency in High–Grade Glioma. Nature (2017) 549:533–7. doi: 10.1038/nature24014
56. Kohutek ZA, diPierro CG, Redpath GT, Hussaini IM. ADAM––Mediated N–Cadherin Cleavage Is Protein Kinase C–Alpha Dependent and Promotes Glioblastoma Cell Migration. J Neurosci (2009) 29:4605–15. doi: 10.1523/JNEUROSCI.5126-08.2009
57. Roth P, Junker M, Tritschler I, Mittelbronn M, Dombrowski Y, Breit SN, et al. GDF–15 Contributes to Proliferation and Immune Escape of Malignant Gliomas. Clin Cancer Res (2010) 16:3851–9. doi: 10.1158/1078-0432.CCR-10-0705
58. Zhu S, Yang N, Guan Y, Wang X, Zang G, Lv X, et al. GDF15 Promotes Glioma Stem Cell–Like Phenotype via Regulation of ERK1/2–C–Fos–LIF Signaling. Cell Death Discov (2021) 7:3. doi: 10.1038/s41420-020-00395-8
59. Suriben R, Chen M, Higbee J, Oeffinger J, Ventura R, Li B, et al. Antibody–Mediated Inhibition of GDF15–GFRAL Activity Reverses Cancer Cachexia in Mice. Nat Med (2020) 26:1264–70. doi: 10.1038/s41591-020-0945-x
60. Wang K, Kievit FM, Chiarelli PA, Stephen ZR, Lin G, Silber JR, et al. siRNA Nanoparticle Suppresses Drug–Resistant Gene and Prolongs Survival in an Orthotopic Glioblastoma Xenograft Mouse Model. Adv Funct Mater (2021) 31. doi: 10.1002/adfm.202007166
61. Karlin J, Allen J, Ahmad SF, Hughes G, Sheridan V, Odedra R, et al. Orally Bioavailable and Blood–Brain Barrier–Penetrating ATM Inhibitor (AZ32) Radiosensitizes Intracranial Gliomas in Mice. Mol Cancer Ther (2018) 17:1637–47. doi: 10.1158/1535-7163.MCT-17-0975
62. Wengner AM, Siemeister G, Lücking U, Lefranc J, Wortmann L, Lienau P, et al. The Novel ATR Inhibitor BAY 1895344 Is Efficacious as Monotherapy and Combined With DNA Damage–Inducing or Repair–Compromising Therapies in Preclinical Cancer Models. Mol Cancer Ther (2020) 19:26–38. doi: 10.1158/1535-7163.MCT-19-0019
63. Mohiuddin IS, Kang MH. DNA–PK as an Emerging Therapeutic Target in Cancer. Front Oncol (2019) 9:635. doi: 10.3389/fonc.2019.00635
64. Higuchi F, Nagashima H, Ning J, Koerner MVA, Wakimoto H, Cahill DP. Restoration of Temozolomide Sensitivity by PARP Inhibitors in Mismatch Repair Deficient Glioblastoma is Independent of Base Excision Repair. Clin Cancer Res (2020) 26:1690–9. doi: 10.1158/1078-0432.CCR-19-2000
65. Huang M, Zhang D, Wu JY, Xing K, Yeo E, Li C, et al. Wnt–Mediated Endothelial Transformation Into Mesenchymal Stem Cell–Like Cells Induces Chemoresistance in Glioblastoma. Sci Transl Med (2020) 12:eaay7522. doi: 10.1126/scitranslmed.aay7522
66. Zhao C, Guo R, Guan F, Ma S, Li M, Wu J, et al. MicroRNA––3p Enhances the Chemosensitivity of Temozolomide in Glioblastoma by Targeting C–Met and EMT. Sci Rep (2020) 10:9471. doi: 10.1038/s41598-020-65331-3
67. Lan F, Yu H, Hu M, Xia T, Yue X. miR––3p Exerts Anti–Tumor Effects in Glioblastoma by Targeting C–Met. J Neurochem (2015) 135:274–86. doi: 10.1111/jnc.13272
68. Nie X, Su Z, Yan R, Yan A, Qiu S, Zhou Y. MicroRNA–562 Negatively Regulated C–MET/AKT Pathway in the Growth of Glioblastoma Cells. OncoTargets Ther (2019) 12:41–9. doi: 10.2147/OTT.S186701
69. Chen A, Jiang Y, Li Z, Wu L, Santiago U, Zou H, et al. Chitinase––Like–1 Protein Complexes Modulate Macrophage–Mediated Immune Suppression in Glioblastoma. J Clin Invest (2021) 131:e147552. doi: 10.1172/JCI147552
70. Cohen N, Shani O, Raz Y, Sharon Y, Hoffman D, Abramovitz L, et al. Fibroblasts Drive an Immunosuppressive and Growth–Promoting Microenvironment in Breast Cancer via Secretion of Chitinase 3–Like 1. Oncogene (2017) 36:4457–68. doi: 10.1038/onc.2017.65
71. Gabriely G, da Cunha AP, Rezende RM, Kenyon B, Madi A, Vandeventer T, et al. Targeting Latency–Associated Peptide Promotes Antitumor Immunity. Sci Immunol (2017) 2:eaaj1738. doi: 10.1126/sciimmunol.aaj1738
72. Shaim H, Shanley M, Basar R, Daher M, Gumin J, Zamler DB, et al. Targeting the αv Integrin/TGF–β Axis Improves Natural Killer Cell Function Against Glioblastoma Stem Cells. J Clin Invest (2021) 131:e142116. doi: 10.1172/JCI142116
73. Papachristodoulou A, Silginer M, Weller M, Schneider H, Hasenbach K, Janicot M, et al. Therapeutic Targeting of Tgfβ Ligands in Glioblastoma Using Novel Antisense Oligonucleotides Reduces the Growth of Experimental Gliomas. Clin Cancer Res (2019) 25:7189–201. doi: 10.1158/1078-0432.CCR-17-3024
74. Zhang Y, Yang X, Zhu XL, Bai H, Wang ZZ, Zhang JJ, et al. S100A Gene Family: Immune–Related Prognostic Biomarkers and Therapeutic Targets for Low–Grade Glioma. Aging (2021) 13:15459–78. doi: 10.18632/aging.203103
75. Chow KH, Park HJ, George J, Yamamoto K, Gallup AD, Graber JH, et al. S100A4 Is a Biomarker and Regulator of Glioma Stem Cells That Is Critical for Mesenchymal Transition in Glioblastoma. Cancer Res (2017) 77:5360–73. doi: 10.1158/0008-5472.CAN-17-1294
76. Liang J, Piao Y, Holmes L, Fuller GN, Henry V, Tiao N, et al. Neutrophils Promote the Malignant Glioma Phenotype Through S100A4. Clin Cancer Res (2014) 20:187–98. doi: 10.1158/1078-0432.CCR-13-1279
77. Choe N, Kwon DH, Shin S, Kim YS, Kim YK, Kim J, et al. The microRNA miR–124 Inhibits Vascular Smooth Muscle Cell Proliferation by Targeting S100 Calcium–Binding Protein A4 (S100a4). FEBS Lett (2017) 591:1041–52. doi: 10.1002/1873-3468.12606
78. Yang D, Du G, Xu A, Xi X, Li D. Expression of miR––3p Inhibits Proliferation, Migration, and Invasion of Bladder Cancer by Targeting S100A4. Am J Cancer Res (2017) 7:2209–19.
79. Mudduluru G, Ilm K, Fuchs S, Stein U. Epigenetic Silencing of miR–520c Leads to Induced S100A4 Expression and its Mediated Colorectal Cancer Progression. Oncotarget (2017) 8:21081–94. doi: 10.18632/oncotarget.15499
80. Ward AJ, Norrbom M, Chun S, Bennett CF, Rigo F. Nonsense–Mediated Decay as a Terminating Mechanism for Antisense Oligonucleotides. Nucleic Acids Res (2014) 42:5871–9. doi: 10.1093/nar/gku184
81. Rinaldi C, Wood MJA. Antisense Oligonucleotides: The Next Frontier for Treatment of Neurological Disorders. Nature Reviews. Neurology (2018) 14:9–21. doi: 10.1172/JCI142116
82. Dhuri K, Bechtold C, Quijano E, Pham H, Gupta A, Vikram A, et al. Antisense Oligonucleotides: An Emerging Area in Drug Discovery and Development. J Clin Med (2020) 9:2004. doi: 10.3390/jcm9062004
83. Shah MY, Ferrajoli A, Sood AK, Lopez–Berestein G, Calin GA. microRNA Therapeutics in Cancer – An Emerging Concept. EBioMedicine (2016) 12:34–42. doi: 10.1016/j.ebiom.2016.09.017
84. Seto AG, Beatty X, Lynch JM, Hermreck M, Tetzlaff M, Duvic M, et al. Cobomarsen, an Oligonucleotide Inhibitor of miR–155, Co–Ordinately Regulates Multiple Survival Pathways to Reduce Cellular Proliferation and Survival in Cutaneous T–Cell Lymphoma. Br J Haematol (2018) 183:428–44. doi: 10.1111/bjh.15547
85. Oh B, Song H, Lee D, Oh J, Kim G, Ihm SH, et al. Anti–Cancer Effect of R3V6 Peptide–Mediated Delivery of an anti–microRNA–21 Antisense–Oligodeoxynucleotide in a Glioblastoma Animal Model. J Drug Target (2017) 25:132–9. doi: 10.1080/1061186X.2016.1207648
86. Milani R, Brognara E, Fabbri E, Manicardi A, Corradini R, Finotti A, et al. Targeting Mir−155−5p and Mir−221−3p by Peptide Nucleic Acids Induces Caspase−3 Activation and Apoptosis in Temozolomide−Resistant T98G Glioma Cells. Int J Oncol (2019) 55:59–68. doi: 10.3892/ijo.2019.4810
87. Wei J, Marisetty A, Schrand B, Gabrusiewicz K, Hashimoto Y, Ott M, et al. Osteopontin Mediates Glioblastoma–Associated Macrophage Infiltration and is a Potential Therapeutic Target. J Clin Invest (2019) 129:137–49. doi: 10.1172/JCI121266
88. Ni S, Yao H, Wang L, Lu J, Jiang F, Lu A, et al. Chemical Modifications of Nucleic Acid Aptamers for Therapeutic Purposes. Int J Mol Sci (2017) 18:1683. doi: 10.3390/ijms18081683
89. De La Fuente A, Zilio S, Caroli J, Van Simaeys D, Mazza EMC, Ince TA, et al. Aptamers Against Mouse and Human Tumor–Infiltrating Myeloid Cells as Reagents for Targeted Chemotherapy. Sci Transl Med (2020) 12:eaav9760. doi: 10.1126/scitranslmed.aav9760
90. Cerchia L, de Franciscis V. Targeting Cancer Cells With Nucleic Acid Aptamers. Trends Biotechnol (2010) 28:517–25. doi: 10.1016/j.tibtech.2010.07.005
91. Zhou J, Rossi J. Aptamers as Targeted Therapeutics: Current Potential and Challenges. Nature reviews. Drug Discov (2017) 16:181–202. doi: 10.1038/nrd.2016.199
92. Song KM, Lee S, Ban C. Aptamers and Their Biological Applications. Sensors (Basel Switzerland) (2012) 12:612–31. doi: 10.3390/s120100612
93. McNamara JO 2nd, Andrechek ER, Wang Y, Viles KD, Rempel RE, Gilboa E, et al. Cell Type–Specific Delivery of siRNAs With aptamer–siRNA Chimeras. Nat Biotechnol (2006) 24:1005–15. doi: 10.1038/nbt1223
94. Kruspe S, Giangrande PH. Aptamer–siRNA Chimeras: Discovery, Progress, and Future Prospects. Biomedicines (2017) 5:45. doi: 10.3390/biomedicines5030045
95. Herrmann A, Priceman SJ, Swiderski P, Kujawski M, Xin H, Cherryholmes GA, et al. CTLA4 Aptamer Delivers STAT3 siRNA to Tumor–Associated and Malignant T Cells. J Clin Invest (2014) 124:2977–87. doi: 10.1172/JCI73174
96. Gilboa–Geffen A, Hamar P, Le MT, Wheeler LA, Trifonova R, Petrocca F, et al. Gene Knockdown by EpCAM Aptamer–siRNA Chimeras Suppresses Epithelial Breast Cancers and Their Tumor–Initiating Cells. Mol Cancer Ther (2015) 14:2279–91. doi: 10.1158/1535-7163.MCT-15-0201-T
97. Berezhnoy A, Castro I, Levay A, Malek TR, Gilboa E. Aptamer–Targeted Inhibition of mTOR in T Cells Enhances Antitumor Immunity. J Clin Invest (2014) 124:188–97. doi: 10.1172/JCI69856
98. Hussain AF, Tur MK, Barth S. An aptamer–siRNA Chimera Silences the Eukaryotic Elongation Factor 2 Gene and Induces Apoptosis in Cancers Expressing αvβ3 Integrin. Nucleic Acid Ther (2013) 23:203–12. doi: 10.1089/nat.2012.0408
99. Rajagopalan A, Berezhnoy A, Schrand B, Puplampu–Dove Y, Gilboa E. Aptamer–Targeted Attenuation of IL–2 Signaling in CD8(+) T Cells Enhances Antitumor Immunity. Mol Ther (2017) 25:54–61. doi: 10.1016/j.ymthe.2016.10.021
100. Lai WY, Wang WY, Chang YC, Chang CJ, Yang PC, Peck K. Synergistic Inhibition of Lung Cancer Cell Invasion, Tumor Growth and Angiogenesis Using aptamer–siRNA Chimeras. Biomaterials (2014) 35:2905–14. doi: 10.1016/j.biomaterials.2013.12.054
101. Wang T, Gantier MP, Xiang D, Bean AG, Bruce M, Zhou SF, et al. EpCAM Aptamer–Mediated Survivin Silencing Sensitized Cancer Stem Cells to Doxorubicin in a Breast Cancer Model. Theranostics (2015) 5:1456–72. doi: 10.7150/thno.11692
102. Subramanian N, Kanwar JR, Kanwar RK, Krishnakumar S. Targeting Cancer Cells Using LNA–Modified Aptamer–siRNA Chimeras. Nucleic Acid Ther (2015) 25:317–22. doi: 10.1089/nat.2015.0550
103. Esposito CL, Nuzzo S, Catuogno S, Romano S, de Nigris F, de Franciscis V. STAT3 Gene Silencing by Aptamer–siRNA Chimera as Selective Therapeutic for Glioblastoma. Mol Ther Nucleic Acids (2018) 10:398–411. doi: 10.1016/j.omtn.2017.12.021
104. Yoon S, Wu X, Armstrong B, Habib N, Rossi JJ. An RNA Aptamer Targeting the Receptor Tyrosine Kinase Pdgfrα Induces Anti–Tumor Effects Through STAT3 and P53 in Glioblastoma. Mol Ther Nucleic Acids (2019) 14:131–41. doi: 10.1016/j.omtn.2018.11.012
105. Rehmani H, Li Y, Li T, Padia R, Calbay O, Jin L, et al. Addiction to Protein Kinase Cι Due to PRKCI Gene Amplification can be Exploited for an Aptamer–Based Targeted Therapy in Ovarian Cancer. Signal Transduct Target Ther (2020) 5:140. doi: 10.1038/s41392-020-0197-8
106. Kalbasi A, Ribas A. Tumour–Intrinsic Resistance to Immune Checkpoint Blockade. Nature reviews. Immunology (2020) 20:25–39. doi: 10.1038/s41577-019-0218-4
107. Manguso RT, Pope HW, Zimmer MD, Brown FD, Yates KB, Miller BC, et al. In Vivo CRISPR Screening Identifies Ptpn2 as a Cancer Immunotherapy Target. Nature (2017) 547:413–8. doi: 10.1038/nature23270
108. Wang PF, Cai HQ, Zhang CB, Li YM, Liu X, Wan JH, et al. Molecular and Clinical Characterization of PTPN2 Expression From RNA–Seq Data of 996 Brain Gliomas. J Neuroinflamm (2018) 15:145. doi: 10.1186/s12974-018-1187-4
109. Ishizuka JJ, Manguso RT, Cheruiyot CK, Bi K, Panda A, Iracheta–Vellve A, et al. Loss of ADAR1 in Tumours Overcomes Resistance to Immune Checkpoint Blockade. Nature (2019) 565:43–8. doi: 10.1038/s41586-018-0768-9
110. Yang B, Hu P, Lin X, Han W, Zhu L, Tan X, et al. PTBP1 Induces ADAR1 P110 Isoform Expression Through IRES–Like Dependent Translation Control and Influences Cell Proliferation in Gliomas. Cell Mol Life Sci: CMLS (2015) 72:4383–97. doi: 10.1007/s00018-015-1938-7
111. Pastor F, Kolonias D, Giangrande PH, Gilboa E. Induction of Tumour Immunity by Targeted Inhibition of Nonsense–Mediated mRNA Decay. Nature (2010) 465:227–30. doi: 10.1038/nature08999
112. Li F, Yi Y, Miao Y, Long W, Long T, Chen S, et al. N(6)–Methyladenosine Modulates Nonsense–Mediated mRNA Decay in Human Glioblastoma. Cancer Res (2019) 79:5785–98. doi: 10.1158/0008-5472.CAN-18-2868
113. Garrido G, Schrand B, Rabasa A, Levay A, D’Eramo F, Berezhnoy A, et al. Tumor–Targeted Silencing of the Peptide Transporter TAP Induces Potent Antitumor Immunity. Nat Commun (2019) 10:3773. doi: 10.1038/s41467-019-11728-2
114. Kortylewski M, Yu H. Role of Stat3 in Suppressing Anti–Tumor Immunity. Curr Opin Immunol (2008) 20:228–33. doi: 10.1016/j.coi.2008.03.010
115. Ferguson SD, Srinivasan VM, Heimberger AB. The Role of STAT3 in Tumor–Mediated Immune Suppression. J Neuro–Oncol (2015) 123:385–94. doi: 10.1007/s11060-015-1731-3
116. Wei J, Wu A, Kong LY, Wang Y, Fuller G, Fokt I, et al. Hypoxia Potentiates Glioma–Mediated Immunosuppression. PloS One (2011) 6:e16195. doi: 10.1371/journal.pone.0016195
117. Wei J, Barr J, Kong LY, Wang Y, Wu A, Sharma AK, et al. Glioblastoma Cancer–Initiating Cells Inhibit T–Cell Proliferation and Effector Responses by the Signal Transducers and Activators of Transcription 3 Pathway. Mol Cancer Ther (2010) 9:67–78. doi: 10.1158/1535-7163.MCT-09-0734
118. Kortylewski M, Swiderski P, Herrmann A, Wang L, Kowolik C, Kujawski M, et al. In Vivo Delivery of siRNA to Immune Cells by Conjugation to a TLR9 Agonist Enhances Antitumor Immune Responses. Nat Biotechnol (2009) 27:925–32. doi: 10.1038/nbt.1564
119. Esposito CL, Nuzzo S, Ibba ML, Ricci–Vitiani L, Pallini R, Condorelli G, et al. Combined Targeting of Glioblastoma Stem–Like Cells by Neutralizing RNA–Bio–Drugs for STAT3. Cancers (Basel) (2020) 12:1434. doi: 10.3390/cancers12061434
120. Wei J, Chen P, Gupta P, Ott M, Zamler D, Kassab C, et al. Immune Biology of Glioma–Associated Macrophages and Microglia: Functional and Therapeutic Implications. Neuro–oncology (2020) 22:180–94. doi: 10.1093/neuonc/noz212
121. Sangaletti S, Tripodo C, Sandri S, Torselli I, Vitali C, Ratti C, et al. Osteopontin Shapes Immunosuppression in the Metastatic Niche. Cancer Res (2014) 74:4706–19. doi: 10.1158/0008-5472.CAN-13-3334
122. Zhou W, Ke SQ, Huang Z, Flavahan W, Fang X, Paul J, et al. Periostin Secreted by Glioblastoma Stem Cells Recruits M2 Tumour–Associated Macrophages and Promotes Malignant Growth. Nat Cell Biol (2015) 17:170–82. doi: 10.1038/ncb3090
123. Chang AL, Miska J, Wainwright DA, Dey M, Rivetta CV, Yu D, et al. CCL2 Produced by the Glioma Microenvironment Is Essential for the Recruitment of Regulatory T Cells and Myeloid–Derived Suppressor Cells. Cancer Res (2016) 76:5671–82. doi: 10.1158/0008-5472.CAN-16-0144
124. Zhang X, Chen L, Dang WQ, Cao MF, Xiao JF, Lv SQ, et al. CCL8 Secreted by Tumor–Associated Macrophages Promotes Invasion and Stemness of Glioblastoma Cells via ERK1/2 Signaling. Lab Invest (2020) 100:619–29. doi: 10.1038/s41374-019-0345-3
125. De Boeck A, Ahn BY, D’Mello C, Lun X, Menon SV, Alshehri MM, et al. Glioma–Derived IL–33 Orchestrates an Inflammatory Brain Tumor Microenvironment That Accelerates Glioma Progression. Nat Commun (2020) 11:4997. doi: 10.1038/s41467-020-18569-4
126. Soldevilla MM, Villanueva H, Casares N, Lasarte JJ, Bendandi M, Inoges S, et al. MRP1–CD28 Bi–Specific Oligonucleotide Aptamers: Target Costimulation to Drug–Resistant Melanoma Cancer Stem Cells. Oncotarget (2016) 7:23182–96. doi: 10.18632/oncotarget.8095
127. McNamara JO, Kolonias D, Pastor F, Mittler RS, Chen L, Giangrande PH, et al. Multivalent 4–1BB Binding Aptamers Costimulate CD8+ T Cells and Inhibit Tumor Growth in Mice. J Clin Invest (2008) 118:376–86. doi: 10.1172/JCI33365
128. Pastor F, Kolonias D, McNamara JO 2nd, Gilboa E. Targeting 4–1BB Costimulation to Disseminated Tumor Lesions With Bi–Specific Oligonucleotide Aptamers. Mol Ther (2011) 19:1878–86. doi: 10.1038/mt.2011.145
129. Schrand B, Berezhnoy A, Brenneman R, Williams A, Levay A, Kong LY, et al. Targeting 4–1BB Costimulation to the Tumor Stroma With Bispecific Aptamer Conjugates Enhances the Therapeutic Index of Tumor Immunotherapy. Cancer Immunol Res (2014) 2:867–77. doi: 10.1158/2326-6066.CIR-14-0007
130. Yang Y, Sun X, Xu J, Cui C, Safari Yazd H, Pan X, et al. Circular Bispecific Aptamer–Mediated Artificial Intercellular Recognition for Targeted T Cell Immunotherapy. ACS Nano (2020) 14:9562–71. doi: 10.1021/acsnano.9b09884
131. Zheng J, Zhao S, Yu X, Huang S, Liu HY. Simultaneous Targeting of CD44 and EpCAM With a Bispecific Aptamer Effectively Inhibits Intraperitoneal Ovarian Cancer Growth. Theranostics (2017) 7:1373–88. doi: 10.7150/thno.17826
132. Hu Z, He J, Gong W, Zhou N, Zhou S, Lai Z, et al. TLS11a Aptamer/CD3 Antibody Anti–Tumor System for Liver Cancer. J Biomed Nanotechnol (2018) 14:1645–53. doi: 10.1166/jbn.2018.2619
133. Li Z, Hu Y, An Y, Duan J, Li X, Yang XD. Novel Bispecific Aptamer Enhances Immune Cytotoxicity Against MUC1–Positive Tumor Cells by MUC1–CD16 Dual Targeting. Molecules (2019) 24:478. doi: 10.3390/molecules24030478
134. Boltz A, Piater B, Toleikis L, Guenther R, Kolmar H, Hock B. Bi–Specific Aptamers Mediating Tumor Cell Lysis. J Biol Chem (2011) 286:21896–905. doi: 10.1074/jbc.M111.238261
135. Soldevilla MM, Villanueva H, Meraviglia–Crivelli D, Menon AP, Ruiz M, Cebollero J, et al. ICOS Costimulation at the Tumor Site in Combination With CTLA–4 Blockade Therapy Elicits Strong Tumor Immunity. Mol Ther (2019) 27:1878–91. doi: 10.1016/j.ymthe.2019.07.013
136. Lee H, Kim TH, Park D, Jang M, Chung JJ, Kim SH, et al. Combinatorial Inhibition of Cell Surface Receptors Using Dual Aptamer–Functionalized Nanoconstructs for Cancer Treatment. Pharmaceutics (2020) 12:689. doi: 10.3390/pharmaceutics12070689
137. Godlewski J, Ferrer–Luna R, Rooj AK, Mineo M, Ricklefs F, Takeda YS, et al. MicroRNA Signatures and Molecular Subtypes of Glioblastoma: The Role of Extracellular Transfer. Stem Cell Rep (2017) 8:1497–505. doi: 10.1016/j.stemcr.2017.04.024
138. Fareh M, Almairac F, Turchi L, Burel–Vandenbos F, Paquis P, Fontaine D, et al. Cell–Based Therapy Using miR–302–367 Expressing Cells Represses Glioblastoma Growth. Cell Death Dis (2017) 8:e2713. doi: 10.1038/cddis.2017.117
139. Alvarez–Erviti L, Seow Y, Yin H, Betts C, Lakhal S, Wood MJ. Delivery of siRNA to the Mouse Brain by Systemic Injection of Targeted Exosomes. Nat Biotechnol (2011) 29:341–5. doi: 10.1038/nbt.1807
140. Deng SZ, Lai MF, Li YP, Xu CH, Zhang HR, Kuang JG. Human Marrow Stromal Cells Secrete microRNA–375–Containing Exosomes to Regulate Glioma Progression. Cancer Gene Ther (2020) 27:203–15. doi: 10.1038/s41417-019-0079-9
141. De Toro J, Herschlik L, Waldner C, Mongini C. Emerging Roles of Exosomes in Normal and Pathological Conditions: New Insights for Diagnosis and Therapeutic Applications. Front Immunol (2015) 6:203. doi: 10.3389/fimmu.2015.00203
142. Ha D, Yang N, Nadithe V. Exosomes as Therapeutic Drug Carriers and Delivery Vehicles Across Biological Membranes: Current Perspectives and Future Challenges. Acta Pharm Sin B (2016) 6:287–96. doi: 10.1016/j.apsb.2016.02.001
143. Steg AD, Katre AA, Goodman B, Han HD, Nick AM, Stone RL, et al. Targeting the Notch Ligand JAGGED1 in Both Tumor Cells and Stroma in Ovarian Cancer. Clin Cancer Res (2011) 17:5674–85. doi: 10.1158/1078-0432.CCR-11-0432
144. Noh K, Mangala LS, Han HD, Zhang N, Pradeep S, Wu SY, et al. Differential Effects of EGFL6 on Tumor Versus Wound Angiogenesis. Cell Rep (2017) 21:2785–95. doi: 10.1016/j.celrep.2017.11.020
145. Lee TJ, Haque F, Shu D, Yoo JY, Li H, Yokel RA, et al. RNA Nanoparticle as a Vector for Targeted siRNA Delivery Into Glioblastoma Mouse Model. Oncotarget (2015) 6:14766–76. doi: 10.18632/oncotarget.3632
146. Zatsepin TS, Kotelevtsev YV, Koteliansky V. Lipid Nanoparticles for Targeted siRNA Delivery – Going From Bench to Bedside. Int J Nanomed (2016) 11:3077–86. doi: 10.2147/IJN.S106625
147. Sun X, Chen Y, Zhao H, Qiao G, Liu M, Zhang C, et al. Dual–Modified Cationic Liposomes Loaded With Paclitaxel and Survivin siRNA for Targeted Imaging and Therapy of Cancer Stem Cells in Brain Glioma. Drug Delivery (2018) 25:1718–27. doi: 10.1080/10717544.2018.1494225
148. Alshaer W, Hillaireau H, Vergnaud J, Mura S, Deloménie C, Sauvage F, et al. Aptamer–Guided siRNA–Loaded Nanomedicines for Systemic Gene Silencing in CD–44 Expressing Murine Triple–Negative Breast Cancer Model. J Controlled Rel (2018) 271:98–106. doi: 10.1016/j.jconrel.2017.12.022
149. Wang C, Wang Z, Chen C, Fu X, Wang J, Fei X, et al. A Low MW Inhibitor of CD44 Dimerization for the Treatment of Glioblastoma. Br J Pharmacol (2020) 177:3009–23. doi: 10.1111/bph.15030
150. Lächelt U, Wagner E. Nucleic Acid Therapeutics Using Polyplexes: A Journey of 50 Years (and Beyond). Chem Rev (2015) 115:11043–78. doi: 10.1021/cr5006793
151. Adams C, Israel LL, Ostrovsky S, Taylor A, Poptani H, Lellouche JP, et al. Development of Multifunctional Magnetic Nanoparticles for Genetic Engineering and Tracking of Neural Stem Cells. Adv Healthc Mater (2016) 5:841–9. doi: 10.1002/adhm.201500885
Keywords: noncoding RNAs, tumor microenvironment, microRNA, siRNA, aptamer, antisense oligonucleotide, glioblastoma
Citation: Wei J, Gilboa E, Calin GA and Heimberger AB (2021) Immune Modulatory Short Noncoding RNAs Targeting the Glioblastoma Microenvironment. Front. Oncol. 11:682129. doi: 10.3389/fonc.2021.682129
Received: 17 March 2021; Accepted: 11 August 2021;
Published: 31 August 2021.
Edited by:
Payal Watchmaker, University of California, San Francisco, United StatesCopyright © 2021 Wei, Gilboa, Calin and Heimberger. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Amy B. Heimberger, YW15LmhlaW1iZXJnZXJAbm9ydGh3ZXN0ZXJuLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.