 Pin Zhao
Pin Zhao Samiullah Malik
Samiullah Malik Shaojun Xing
Shaojun Xing- 1Guandong Key Laboratory for Biomedical Measurements and Ultrasound Imaging, School of Biomedical Engineering, Shenzhen University Health Science Center, Shenzhen, China
- 2Department of Pathogen Biology, Shenzhen University Health Science Center, Shenzhen, China
Hepatocellular carcinoma (HCC), is the third leading cause of cancer-related deaths, which is largely caused by virus infection. About 80% of the virus-infected people develop a chronic infection that eventually leads to liver cirrhosis and hepatocellular carcinoma (HCC). With approximately 71 million HCV chronic infected patients worldwide, they still have a high risk of HCC in the near future. However, the mechanisms of carcinogenesis in chronic HCV infection have not been still fully understood, which involve a complex epigenetic regulation and cellular signaling pathways. Here, we summarize 18 specific gene targets and different signaling pathways involved in recent findings. With these epigenetic alterations requiring histone modifications and DNA hyper or hypo-methylation of these specific genes, the dysregulation of gene expression is also associated with different signaling pathways for the HCV life cycle and HCC. These findings provide a novel insight into a correlation between HCV infection and HCC tumorigenesis, as well as potentially preventable approaches. Hepatitis C virus (HCV) infection largely causes hepatocellular carcinoma (HCC) worldwide with 3 to 4 million newly infected cases diagnosed each year. It is urgent to explore its underlying molecular mechanisms for therapeutic treatment and biomarker discovery. However, the mechanisms of carcinogenesis in chronic HCV infection have not been still fully understood, which involve a complex epigenetic regulation and cellular signaling pathways. Here, we summarize 18 specific gene targets and different signaling pathways involved in recent findings. With these epigenetic alterations requiring histone modifications and DNA hyper or hypo-methylation of these specific genes, the dysregulation of gene expression is also associated with different signaling pathways for the HCV life cycle and HCC. These findings provide a novel insight into a correlation between HCV infection and HCC tumorigenesis, as well as potentially preventable approaches.
Introduction
Hepatocellular carcinoma (HCC), is the third leading cause of cancer-related deaths with about 1 million deaths annually worldwide (1, 2). Around 70% to 90% of all patients with chronic liver diseases will develop hepatocellular carcinoma (3). By summing up the main risk factors worldwide, researchers have discovered that chronic hepatitis B virus (HBV) infection is the main reason in eastern Asia and sub-Saharan Africa, whereas in North America, Europe, and Japan, hepatitis C virus (HCV) infection and alcohol abuse are the main factors (2). Here we try to discuss the relationship between HCV infection and hepatocellular carcinoma in an epigenetic aspect.
Hepatitis C virus (HCV) is a single-stranded, positive-sense RNA with approximately 9,600 nucleotide bases. The RNA genome is initiated for translation and transcription in host cells (4). The studies on replication system of HCV life cycle are crucial for our understanding of acute and chronic HCV infection. There are several steps of HCV life cycle, including attachment, endocytosis, fusion, HCV RNA translation, proteolytic processing, viral RNA replication, assembly, maturation, and release (5). It begins with viral attachment to two host receptors, low-density lipoprotein receptor (LDLR) and heparin sulfate proteoglycans (HSPGs), which triggering HCV outer membrane protein E1/E2 heterodimer to bind to CD81 and scavenger receptor B1 (SRB1) (6–8). It is reported that initial attachment not only ensures a tight junction between HCV and hepatocytes, but also possibly leads to HCV particle degradation (9). HCV particle is then folded into an internal endosome by hepatocyte cell membrane and clathrin, as a result of interaction between CD81 and Claudin1 (CLDN1) (10, 11). Along with viral entry into the cell, clathrin is dissociated into the cytosol and then HCV RNA is also released into the cytosol through the acidic pH-dependent endosome fusion and endocytosis (12). Subsequently, HCV RNA, combined with ribosome in rough endoplasmic reticulum, functions as a template for HCV polyprotein translation. A single polyprotein precursor with approximately 3000 amino acids is translated and further processed into 10 mature viral proteins, including structural and nonstructural glycoproteins (E1, E2, p7, NS2, NS3, NS4A, NS4B, NS5A, and NS5B). RNA-dependent RNA polymerase, NA5B, generates a negative-sense RNA intermediate with the template of HCV RNA for the synthesis of numerous HCV RNA. HCV assembly, maturations, and release are complex. During assembly, HCV core proteins form a protective shell with HCV RNA. This immature HCV particle recruits a luminal lipid droplet (LuLD) to create a high-density HCV precursor. The pre-very-low-density lipoprotein (pre-VLDLs) is formed in rough endoplasmic reticulum (13). In the Golgi, the orderly combination of pre-VLDLs, large triacylglycerol (TG)-rich lipid droplets, and high-density HCV precursors is required for the cellular secretory machinery to transport these multivesicular bodies to the cell surface (14, 15).
Since HCV was discovered, it has been reported to be tightly associated with HCC. For example, HCV replication has been detected IN HCC tissue (16, 17). Overexpression of HCV core protein causes HCC in transgenic mice (18). And HCV infection in HepG2 (human hepatoblastoma) cells promotes cell growth and tumor formation in vivo (19). However, HCV-induced HCC is a long-term process that progresses more than 20 years and involves a serial of steps, including chronic HCV infection, chronic hepatic inflammation, liver fibrosis, steatosis, cirrhosis, irreversible genetic or epigenetic alterations, and progression of the malignant carcinogenic cells (20). Different from HBV-induced HCC by integrating its DNA into the host genome and leading to potential directly tumorigenesis, the carcinogenic potential of HCV is considered to indirectly link to HCV infection, inflammation, and genetic or epigenetic alterations. Some studies showed that HCV infection induced oxidative stress by viral proteins. Oxidative stress is essential for inflammatory progression and promote immunological tolerance in hepatitis. The genotype 3 of HCV is reported to more associated with steatosis and fibrosis progression in HCV patients (21).
As a member of the Flaviviridae family, hepatitis C virus (HCV) is a major risk factor of liver cirrhosis and hepatocellular carcinoma (HCC). With approximately 71 million HCV chronic infected patients worldwide, they still have a high risk of HCC in the near future, although direct-acting antivirals (DAA) powerfully eliminate most chronic HCV infection in patients (22). However, some multiple epidemiological studies demonstrate that after DAA treatment, sustained virologic responses (SVR) disturbs normal interferon regulation, which is helpful for HCC tumor development and metastasis (23–25). Moreover, liver transplantation and the transmission of undiagnosed chronic HCV infection will also exceptionally increase the risk of HCV infection. Some vaccines are designed to replace these direct-acting antivirals (DAA) by interrupting HCV transmission or stimulating the production of neutralizing antibodies (nAbs) (26). However, there is not yet an effective vaccine to prevent HCV infection. A number of challenges to cure HCV infection have aroused people to uncover more molecular mechanisms of the immune response to dysfunction.
One of the remarkable molecular alterations in HCV-infected patients is the dysregulation of some specific genes and signaling pathways, which resulted from dynamic epigenetic changes in the genome. Epigenetic regulation is specifically dictated by DNA methylation, post-translational histone modifications, chromatin remodeling, and noncoding RNA-mediated gene silencing. Previous results showed that HCV infection induces genome-wide epigenetic changes in active and repressed chromatin, where altered histone modifications and DNA methylation of the host gene decide their mRNA levels and expression. And then these changes influence the downstream signaling pathways associated with the HCV life cycle and HCC (27). Besides, infection with HCV generally leads to chromosomal instability by condensing chromatin and forming multi-centrosomes (28). In this review, we will focus on epigenetic changes induced by HCV-infected HCC tumorigenesis, including DNA methylation, histone modifications, non-coding RNAs, and related signaling pathways.
Methylation Profiles in HCV-Infected HCC
DNA methylation-related alterations, including DNA hypermethylation, hypomethylation, and imprinting loss, tightly associated with various human diseases, like cancers. Specifically, DNA hypermethylation at the promoter regions of some tumor suppressor genes represses the expression of these genes, which immediately contributes to the carcinogenic processes of HCV-infected HCC. And hypermethylation of promoter CpG island is the main mechanism of these genes’ inactivation. Studies have shown that the methylation disorders for the inactivation of tumor suppressor genes and activation of oncogenic genes are the one common reason for the occurrence of liver cancer. However, our understanding of liver-specific genes remains quite limited. In summary, 18 genes, including tumor suppressors and oncogenic genes, are methylated with different levels (29–35) (Figure 1A). RNA-seq analysis has shown that promoter hypermethylation of tumor-suppressive genes contributes to low protein and hypomethylation of oncogenic genes leads to relatively high expression (Figure 1B). To investigate the properties of the gene regulatory network in HCV-induced HCC, we mapped these 18 genes by STING network. It showed that there were 14 genes involved in and SFRP1 displayed most connected nodes and edges (Figure 1C). Also, these genes are involved in different cellular processes (Figure 1D). It is shown that cell apoptotic and development processes are the most influenced in HCC cells with the greatest number of enriched genes, and cell differentiation is the second, which suggests that HCV infection induces HCC mainly through dysregulation of cell apoptosis and development.
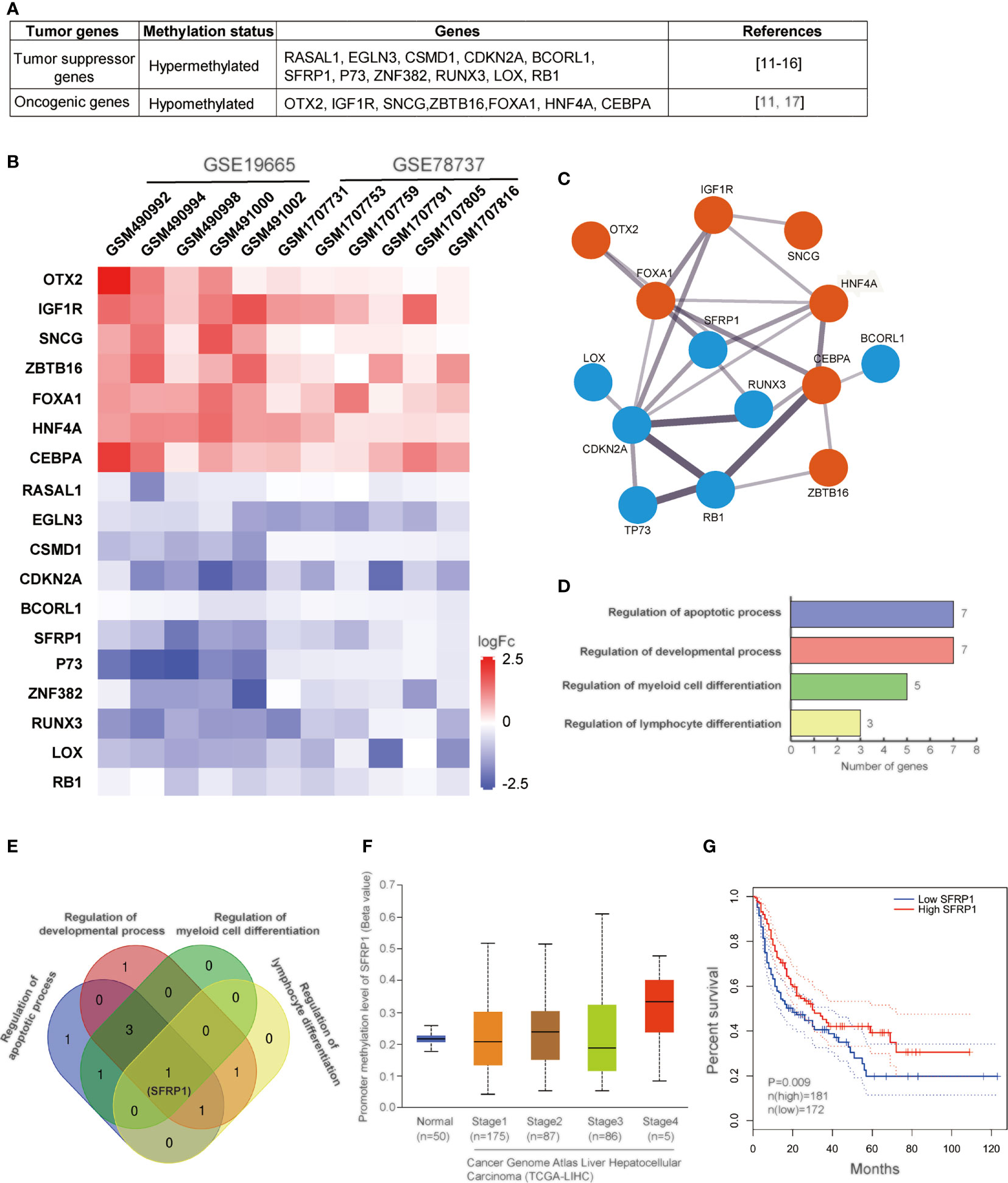
Figure 1 Methylation analysis of genes in HCV-induced HCC. (A) Table of 11 tumor suppressor genes and 7 oncogenic genes regulated in HCV-induced HCC. (B) Heatmap of the correlation between the expression levels of 18 genes and HCV-infected HCC samples among TCGA data sets. The output thresholds are FDR < 0.05, P < 0.05 and |log2 FC| > 1. (C) Functional interaction network of 14 genes. Protein interactions are presented by STING online tool (https://stingnetwork.com). Blue dots indicate down-regulated, red, up-regulated. Line thickness indicates the strength of interactions. (D) The biological processes with 18 genes are analyzed. There are four different processes involving in corresponding genes. Gene Ontology analysis is conducted using online website PANTHER Tools (http://pantherdb.org/data/). (E) Venn results of 18 genes which are regulated in the four biological processes. It is showed that only SFRP1 is commonly regulated in all biological processes. (F) The DNA methylation levels of SFRP1 in clinicopathological stages of HCC in the TCGA data set. (G) Kaplan–Meier curves showing that patients with different expression levels of SFRP1 had different overall survival.
Except for tumor suppressor, p53, a number of tumor suppressor genes have also been reported to be hypermethylated in previous researches. There are 11 tumor suppressor genes involved in HCV-infected HCC development, including RASAL1, EGLN3, CSMD1, CDKN2A, BCORL1, SFRP1, ZNF382, RUNX3, LOX, RB1, P73. In general, hypermethylation of these genes transcriptionally represses protein expression, thus all of these tumor suppressor genes are downregulated in HCV-related HCC cells.
RAS protein activator like 1 (RASAL1) is a member of the GTPase-activating proteins (GAPs) family. By transforming the GTPase activity of Ras proteins into the inactive GDP-bound form of Ras, RASAL1 inhibits Ras/MAPK signaling. The hypermethylation of RASAL1 leads to low protein expression, which stimulates cell proliferation and differentiation through Ras/MAPK signaling in HCV-induced tumorigenesis. RASAL1 has been consistently reported to be downregulated in HCC cells (36).
Egl-9 family hypoxia inducible factor 3 (EGLN3) is one of the hypoxia-related prognostic genes and correlated with a poor prognosis in HCC (37). The family of prolyl hydroxylases (PHDs/EGLNs) is reported to inhibit tumor growth through degradation of hypoxia-inducible factor (HIF) (38). Therefore, HCV infection may downregulate EGLN3 expression, which stimulates HCC development.
CUB and Sushi multiple domains 1 (CSMD1) is a membrane-bound complement inhibitor, of which lncRNA, lncCSMD1, is associated with tumor progression of HCC cohorts (39). The expression of lncCSMD1 elevates MYC protein in the nucleus of HCC cells, resulting in cell proliferation and invasion. The activation of the MYC signaling pathway promotes tumor growth and metastasis of HCC patients. In addition, miR-10b binds to the 3′-untranslated region (3’-UTR) of CSMD1 for downregulating CSMD1 expression, whereas miR-10b is highly expressed in HCC tissues compared to normal tissues (40).
CDKN2A, also known as cyclin-dependent kinase inhibitor 2A by binding to cyclin-dependent kinases (CDKs), acts as a tumor suppressor by blocking the cell cycle from G1 to S phase (41). Besides P53, CDKN2A is also mostly inactivated in human cancers. It is first discovered that DNA hypermethylation occurs at the CDKN2A promoter region in the plasma and serum of liver cancer patients (42). And subsequently, CDKN2A promoter methylation is reported to increase HCC risk in tissues (43). Meta-analysis has validated that promoter methylation of CDKN2A links to an enhancive HCC risk and likely to be a triage marker for HCC (44). Deletion or knockdown of CDKN2A in HCV-induced HCC tissues is also common, as well as mutations of TP53, AXIN1, and CDKN2A (45).
BCL6 corepressor like 1 (BCORL1) is abnormally expressed in human HCC (46). It is reported that BCORL1 prominently promotes facilitated epithelial-mesenchymal transition (EMT) of HCC by negatively regulating E-cadherin expression. BCORL1 protein is a transcriptional corepressor and can interact with several histone deacetylases to repress the transcription of E-cadherin. It is suggested to be an independent prognostic marker for human hepatocellular carcinoma.
Zinc-finger protein 382 (ZNF382), as a member of the zinc-finger protein family, has been evidenced to be downregulated in several types of cancers. In hepatitis B virus (HBV)-related HCC cells, ZNF382 is commonly downregulated by promoter hypermethylation (47). Tumor suppressor ZNF382 mainly impairs of Wnt/β-catenin pathway and activates p53 signaling to induce cell apoptosis in HCC cells.
RUNX family transcription factor 3 (RUNX3) is reported to act as a tumor suppressor in breast cancer and lung cancer. It is known to transcriptionally regulate genes associated with the development and differentiation of cells within the immune and nervous system. In HCC cells, promoter hypermethylation of RUNX3 is reported to occur at an early stage of HCC development (48). Knockdown of RUNX3 stimulates expression of a multidrug resistance-associated protein (MRP) and results in chemotherapy resistance in HCC (49). Consistently, RUNX3 restoration downregulates cancer stem cell (CSC) signaling in hepatocellular carcinoma through Jagged1-Notch signaling (50).
Lysyl oxidase (LOX) is a copper‐dependent amine oxidase for cross‐linking of collagen and elastin molecules in the extracellular matrix. In HCC cells, LOX induces epithelial–mesenchymal transition (51). And the expression of LOX is reported to be regulated by HIF‐1α or hypoxia. However, it is still unclear about the connection between the expression of LOX and early recurrence and poor survival in patients with HCC.
RB transcriptional corepressor 1 (RB1) is a key regulator of multiple cellular processes, such as stability of chromatin structure, DNA replication, and cell cycle. As a negative regulator of the cell cycle, RB1 functions to control cells from dividing too fast. Only when it is phosphorylated by CDK3/cyclinC, RB1 promotes G0/G1 transition in the cell cycle. RB1 exerts its tumor suppression by interacting with E2F1, subsequently repressing its transcription activity (52, 53). Therefore, RB1 is frequently reported to be downregulated in many types of human cancers. It is evident that RB1 genes are silenced in HCC, but different from other cancers, RB1 is rare to be mutated in HCC cells (54). Moreover, low expression of RB1 in HCC cells mainly contributes to promoter methylation (55).
Tumor protein P73 is a homology of p53 protein with a similar DNA binding domain and frequency of mutation in cancers. In contrast to p53, p73 is poorly understood. It is reported that p73 expression is significantly related to HCC prognosis and could be an indicator of HCC patients (56). However, this study concludes that P73 is upregulated in HCC cells, which is controversial to other research results. It has been reported that the promoter methylation level of P73 is significantly higher in HCC than in normal tissues (57). So, it is still necessary to clarify the expression level and methylation level of P73 in HCC patients.
One of the important genes we mentioned above is SFRP1. Secreted frizzled-related protein 1 (SFRP1) functions as a regulator of the Wnt signaling pathway by interacting with Wnts. By inhibition of Wnt signaling, SFRP1 expression increases, and thus, it is classified as a tumor suppressor. In various human cancers, SFRP1 is transcriptionally silenced via DNA methylation (58). The high expression level of SFRP1 has a meaningful impact on survival rates of HCC patients, even though the underline mechanisms are still unknown. HCV infection promotes epigenetic inactivation of SFRP1, which is helpful for hepatocellular carcinoma. It has been evident that promoter methylation of SFRP1 upregulated in all stages of HCC and low SFRP1 expression decreases HCC patient’s survival rate (Figures 1E–G). Similar to another solid tumor, the development of HCC is thought to require at least three dysregulations of cellular processes (proliferation, cell cycle, apoptosis) (59). Epigenetic repression of SFRP1 may cause dysregulation of cell proliferation, cell cycle, migration, and differentiation, which result in cancer cell formation and poor prognosis, and drug-treated resistance. Hence, SFRP1 protein expression provides the possibility for a new therapy approach of HCV-induced HCC.
As for oncogenic genes, it mainly includes ZBTB16, OTX2, IGF1R, SNCG, FOXA1, HNF4A, CEBPA. The high expression of Zinc Finger and BTB Domain Containing 16 (ZBTB16) and orthodenticle homeobox 2 (OTX2) have also been found. However, their functions in HCC remain to be further studied. Type 1 receptor (IGF1R) is reported to interact with insulin-like growth factor 1 and 2 (IGF1 and IGF2) for increasing fibrosis and impair liver functions from the start of preneoplastic alterations up to the developed hepatocellular carcinoma (HCC) stage (60). The neuronal protein, synuclein-gamma (SNCG), is highly expressed in advanced hepatocellular carcinomas, and SNCG gene activation via demethylation in tumor tissue of HCC is not limited to HBV and HCV infection (35). Collectively, both IGF1R and SNCG is regarded as a potential indicator that demethylation of their CpG islands is an early sign of genetic abnormality in liver cirrhosis preceding hepatocarcinogenesis.
Forkhead box A1 (FOXA1) is a transcription factor and involved in embryonic development, metabolism, and epithelial lineage differentiation (61). It is reported that the expression of FOXA1 is relatively higher in both HCC tissues and human hepatic cell lines than non-tumor tissue and cells. It is concluded that FOXA1 promotes cell proliferation and inhibits apoptosis (62), whereas FOXA1 is also found to suppress hepatocellular carcinoma cell viability and motility by transcriptional inactivation of phosphoinositide-3-kinase regulatory subunit 1 (PIK3R1) in PI3K/Akt signaling (63). Therefore, the functions of FOXA1 as an oncogenic gene or tumor suppressor are still controversial. The mechanism of FOXA1 in the development of HCC is necessary to be further explored.
Hepatocyte nuclear factor 4a (HNF4a) is a member of liver-enriched transcriptional factors and regulates hepatic gluconeogenic program and lipid metabolism by binding to specific genomic regions of particular genes (64). It is reported that HNF4a expression is associated with aggressive HCC (65). Recent evidence has explained that two isoforms of HNF4a are determined by two separated promoters, P1 and P2. P1-HNF4a is distributed in normal adult liver, whereas P2 products are in fetal liver and liver cancer (66). The reduced nuclear P1-HNF4a contributes to proliferation and inflammation with HCC risk (67). In contrast, P2-HNF4a is induced in HCC (68). P2-HNF4a prevents P1-HNF4α re-localization from the nucleus to cytoplasm for tumor growth by downregulating the circadian clock gene ARNTL in healthy hepatocytes. However, to date, the role of HNF4a in HCC progression is still to be elucidated.
CCAAT/enhancer-binding protein alpha (CEBPA) is also a transcriptional factor, which contains a basic leucine zipper (bZIP) domain and recognizes the CCAAT motif in the promoter of target genes. CEBPA is reported to inhibit cell proliferation, cell motility, and metastasis. And thus, CEBPA is known as a putative tumor suppressor, and it is downregulated in solid tumors (69). In the liver, it also functions in liver homeostasis and hematopoietic myeloid cell lineage. By activating CEBPA in the HCC tumor microenvironment with a first-in-class small activating RNA oligonucleotide drug, MTL-CEBPA, white cell count significantly increases, and HCC progression delays in combination with Sorafenib treatment (70). Functional alterations of CEBPA have also been investigated in HCC cell lines. It is upregulated in a subset of HCC (71). In HCC tissues, CEBPA expression epigenetically promotes cell proliferation, which suggests a novel oncogenic mechanism in HCC.
HCV-infected HCC has been reported to associate with some repetitive elements (REs) in non-coding regions of the genome, mainly including long interspersed nuclear element-1 (LINE1) and Alu element (Alu). It is reported that DNA methylation of this locus-specific REs is down-regulated in HCV-induced HCC, although the molecular mechanisms in liver cancer development are still unclear (72). In addition, gene silencing induced by DNA methylation is governed by DNA methyltransferases (DNMTs). The expression level of DNMTs is also influenced by HCV infection. It has been reported that different HCV genotypes regulate both mRNA and protein expression of DNMTs. For example, mRNA levels of DNMT1, 3a, 3b are upregulated in genotype 1b and 3a, and DNMT 3b mRNA is also increased in genotype 2a (73).
Histone Modifications in HCV-Infected HCC
Histone modifications have been intensively studied in various cancer models, especially histone methylation, acetylation, phosphorylation, sumoylation, and ubiquitination. During HCC development, histone methylation, and acetylation are widely investigated. It has been reported that putative vasohibin 2 (VASH2) abnormally overexpresses to promote HCC proliferation and inhibits apoptosis, which resulted from the lower H3K27 trimethylation, and higher high H3K4 trimethylation and H3 acetylation at VASH2 promoters (74).
For the roles of histone methylation on special HCV-induced HCC, it has been reported that overexpression of KDM5B/JARID1B, a member of JmjC histone demethylase, could enhance HCC cell proliferation through the regulation of its downstream genes, E2F1 and E2F2 (75). Another histone lysine-specific demethylase (LSD1) is also upregulated, and its expression was linked with HCV infection in HCC. With the Kaplan-Meier method, LSD1 protein is shown to decrease rates of overall survival and disease-free survival and regarded as an independent prognostic factor in HCC patients. LSD1 knockout cells display a higher level of H3K4me1/2 and H3K9me1/2 than normal cells (76). Histone lysine or arginine methylase, including G9a, EZH2, SUV39H1, and SUV39H2, tightly associate with the progression of HCC prognosis. G9a, a histone methyltransferase for lysine 9 of histone 3 (H3K9), mainly functions in gene inactivation, which is essential for early embryogenesis (77, 78). A synthetic triazole analog of guanosine for anti-HCV therapy, ribavirin (RBV) exerts its function by downregulating the mRNA levels of several preactivated interferon (IFN)-stimulated genes (ISGs) in HCC patients. By recruiting more G9a, these selected ISGs are modified with histone H3 lysine9 dimethylation/trimethylation (H3K9me2/H3K9me3) for transcription repression. Defect of G9a impairs ISG downregulation and inhibits RBV to activate IFN for anti-HCV action (79). EZH2 is a histone methyltransferase for lysine 27 of histone 3, of which gene belongs to polycomb. EZH2 plays a crucial role in gene silencing, chromatin remodeling through the interaction with other transcription factors (80). The overexpression of EZH2 is regarded as a promising biomarker for HCC patients in China (81). To patients with HCV infection to HCC, there are not only DNA methylation of targeted enhancers, enriched for the binding sites of the transcription factors, FOXA1, FOXA2, and HNF4A, and decreased histone H3 lysine 27 trimethylation (H3K27me3), caused by the EZH2 inhibitor GSK343, but also negatively associated with increased CG dinucleotide density and polycomb-mediated repression on genes involved in stem cell development (34). SET domain-containing histone lysine methyltransferase 1 (SUV39H1) is also upregulated in HCC patients from Hong Kong, but not patients from the Japanese population (82, 83). However, compared with SUV39H1, SUV39H2 is more correlated with HCV infection (84).
Histone acetylation, regulated dynamically by histone acetyltransferases (HATs) and histone deacetylases (HDACs), also widely occurs in the development of HCV-infected HCC, such as histone H3 acetylated on lysine 9 (H3K9Ac), histone 3 acetylated on lysine 27 (H3K27Ac), H2A acetylated on lysine 5 (H2AK5ac), and H3 acetylated on lysine 14 (H3K14Ac) (85, 86). Knocking out histone deacetylase 3 (HDAC3) immediately leads to hyperacetylated H3K9, along with lower trimethylation of H3K9 (H3K9me3) in mice, which disturbs double-strand break (DSB) repair and promotes HCC tumorigenesis (87). H3K27Ac, mainly localized at central euchromatin regions in cancer cells, activates global cell type-specific gene expression. HCC tissues also show higher H3K27Ac levels than the normal liver no matter it is caused by HCV infection or HBV infection (88). HCC-related histone acetylation usually involves multiple biological processes. For example, iron overload is a risk factor of HCV to HCC progression in the liver, which is normally regulated by a small peptide, human hepcidin, produced by our body. With HCV infection, the expression level of hepcidin is decreased by histone deacetylase (HDAC) activation on histone acetylation, not DNA methylation (89). The role of hepcidin against HCV replication with signal transducer and activator of transcription 3 (STAT3) signaling activation provides a potential treatment for anti-HCV defense. Furthermore, histone deacetylase (HDAC) has been reported to be positively associated with HCV and HCC. The inhibitor of HDAC, therefore, has been considered for clinical HCC treatment because people found that HDAC3 inhibitor suppressed HCV replication by increasing liver-expressed antimicrobial peptide 1(LEAP1) and decreasing apolipoprotein A1 (ApoA1). The antiviral effect of HDAC3 inhibitor may be explained to alter histone acetylation of these gene promoter regions with the transcription factors, including hypoxia-inducible factor 1α (HIF1α) and STAT3 (90).
Measurement of dynamic changes on histone modifications in specific HCV-induced HCC remains limited for characterizing clinical features and treatments of HCC. With the development of detection methods, including immunohistochemistry, Western blot, and ChIP-sequencing, more specific alterations on histone status and DNA–protein modifications will be traced for potential epigenetic therapies in HCV-specific HCC.
Regulation of Noncoding RNAs (ncRNAs) in HCV-Infected HCC
High throughput transcriptome studies have discovered abundant noncoding RNAs in eukaryotes. These novel RNAs seem to involve in a larger scope of cellular functions beyond our understanding. Noncoding RNAs (ncRNAs) play a critical role in the regulation of various cancers, including HCC (91). In these 18 selected genes, several genes are tightly associated with ncRNAs. For example, the promoter region of SFRP1 is reported to be methylated by non-coding RNA HOTTIP-recruited DNMT3B and promotes inflammation in rheumatoid arthritis (92). And lncRNA-maternally expressed gene 3 (lncRNA-MEG3) upregulates IGF1R expression in hypoxia-induced proliferation of pulmonary artery smooth muscle cells (PASMCs) proliferation (93). FOXA1 expression is activated by long noncoding RNA (lncRNA) SBF2-AS1 and repressed by lncRNA PVT1 (94, 95). lncRNA CEBPA-AS1 promotes tumorigenesis via CEBPA/Bcl2 in oral squamous cell carcinoma (96). However, the mechanisms by which ncRNAs regulate HCV-induced HCC need to be further explored. Two kinds of ncRNAs, including microRNAs (miRNAs) and long noncoding RNAs (lncRNAs), have been most studied in recent years.
MicroRNAs (miRNAs) are small non-coding RNA with 20-23 nucleotides in length and originated from 60 to 70 nucleotides miRNA precursor (pre-miRNA), which play a crucial regulatory role in various cellular functions by targeting mRNAs for mRNA degradation and translational repression (97–99). Aberrant miRNAs profiles have been displayed in human cancers, including HCC. In HCV infection HCC cells, miRNAs exert an antiviral effect on HCV replication and infection through virus–host interaction (100). More than 1,000 miRNAs have been identified to date. Among them, miR-122 is the one of most abundant miRNAs in the liver and tightly associated with HCV replication and HCC (101, 102). It is reported that miR-122 binds 5′-UTR of the HCV genome and promotes the replication and translation of HCV RNA (103). Moreover, the upregulation of miRNA-122 in human HCC cells results from the transcription activation of the DR1 and DR2 motifs at its promoter by binding peroxisome proliferator-activated receptor-gamma (PPARγ) and retinoid X receptor alpha (RXRα) complex (104). However, hepatitis B virus X protein (HBX), not HCV protein, binds PPARγ and suppresses miR-122 gene transcription. Viral infectivity was enhanced by miR-122 but significantly reduced by miR-200c. miR-200c is reported to target the 3′UTR of occludin (OCLN) and reduce OCLN mRNA level, and therefore, prevents HCV entry to cells (105). HCV infection also decreases miR-124 level via Dnmt1, resulting in cell migration and invasion in intrahepatic cholangiocarcinoma (ICC) cells (106). In addition, miR-182 is dysregulated in HCV-infected NK cells, which influences HCV replication (107). Also, miRNA-484, 524, 615, and 628 are also discovered in HCV-mediated HCC patients and are potential important biomarkers for diagnosis of fibrosis and cirrhosis (108).
LncRNAs are non-coding RNAs that are more than 200 nucleotides in length, which participate in various biological processes and associate with human disease. Current evidence points to lncRNAs affect the diverse cellular functions of the immune response, including innate and adaptive immunity (109, 110). IFN is an antiviral factor to prevent viral entry, replication, transcription, translation, packaging, and release. Recent studies have shown that several lncRNAs positively or negatively regulate the IFN response (111). For example, lncRNA 8 (Lnc-ITM2C-1/LOC151484) has been identified to be increased for HCV replication in HCV-infected Huh-7.5 hepatoma cells (112). It positively regulates HCV infection by indirectly promoting the expression of G protein-coupled receptor 55 (GPR55) and repressing the expression of several interferon-stimulated genes (ISGs), including ISG15, Mx1, and IFITM1 during the IFN response. In contrast, lncRNAs are regulated by interferon-alpha (IFNα) in Huh7.5.1 cells, of which gRNA significantly inhibits HCV infection through its spatial domain. And also, IFI6 is negatively regulated by lncRNAs (113). As for HCV-related HCC, there are only two lncRNAs reported, including HIF and PAR5 (114). These lncRNAs are significantly dysregulated in HCV-related HCC tissues. Other seven lncRNAs, including LINC01419, BC014579, AK021443, RP11-401P9.4, RP11-304 L19.5, CTB-167B5.2, and AF070632, are differently expressed and could be potential biomarkers for the early detection of HCC (115).
Signaling Pathways in HCV-Induced HCC
The molecular mechanisms of HCV-induced HCC are complex. Recent evidence has shown that HCV RNA-encoded proteins can modulate several signaling pathways, including the Wnt signaling pathway, Ras/MAPK signaling pathway, p53 signaling pathway, JAK-STAT pathway, and PI3K-AKT pathway (Figure 2). These pathways are significantly linked with cell cycle regulation, proliferation, and apoptosis. The dysregulation of signaling pathways by HCV proteins is obvious in the development of HCC.
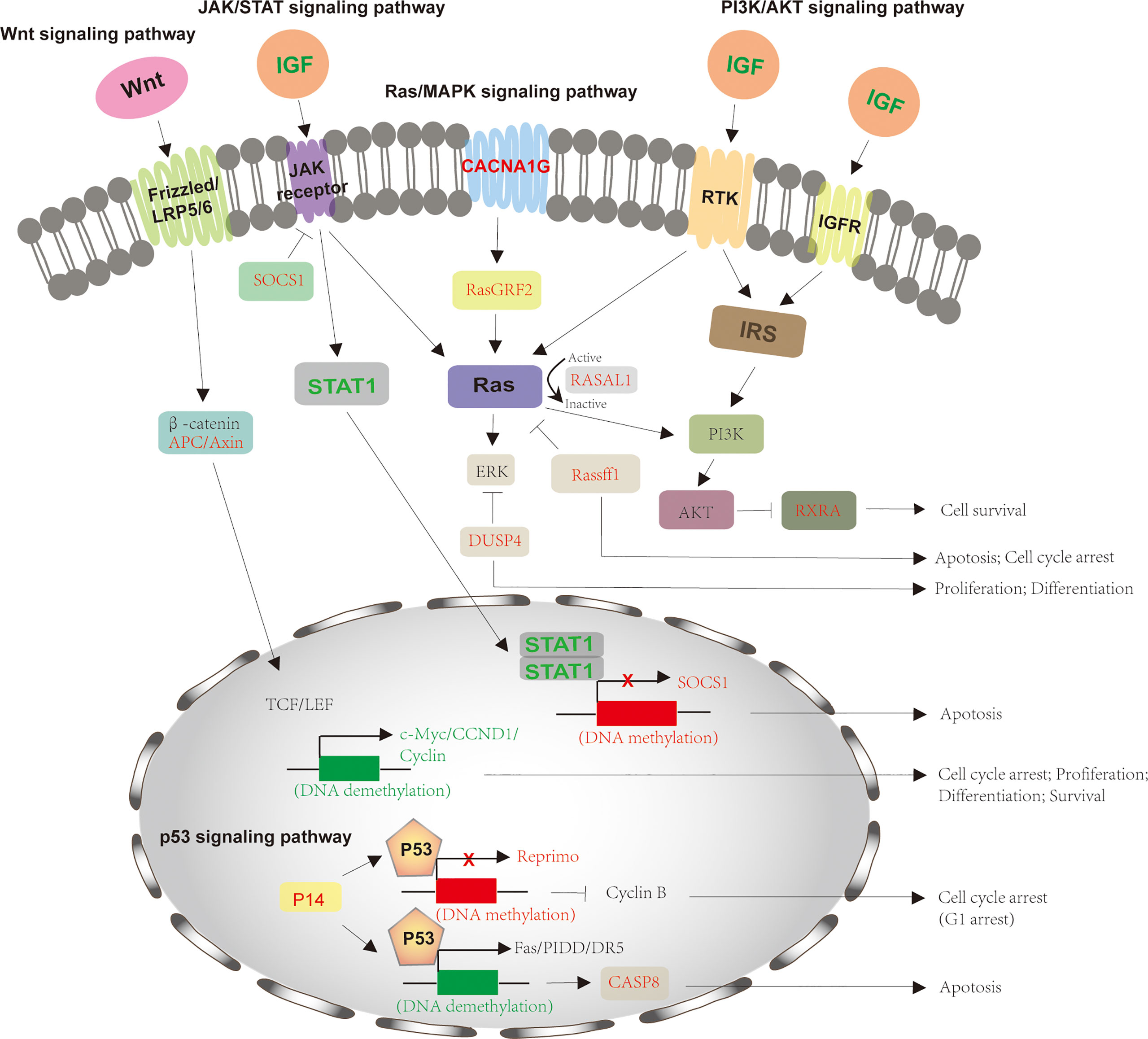
Figure 2 Signaling pathways involved in HCV-induced HCC. The process of HCV-induced HCC involves in several signaling pathways, including Wnt signaling pathway, Ras/MAPK signaling pathway, p53 signaling pathway, JAK-STAT pathway, and PI3K-AKT pathway. For Wnt signaling pathway, HCV infection downregulates APC and Axin2 expression via hypermethylation, which activates LEF/TCF-dependent downstream genes, such as c-Myc, CCND1, and Cyclin and thus promotes cell proliferation and differentiation. The HCV-mediated activation of JAK-STAT pathway by decreasing SOCS1 and increasing IGF, STAT1 to forms a negative feedback and inhibits cell apoptosis. The downregulation of DUSP4, RASAL1, and RasGRF2 by HCV infection also stimulates Ras/MAPK signaling pathway for cell growth. IGF also activates PI3K-AKT pathway by reducing RXRA expression. p53 signaling pathway is also modulated by HCV infection through downregulation of P14, Reprimo and CASP8, preventing from cell cycle arrest and apoptosis. The crosstalk between different signaling pathways happens and cooperates for cell growth and tumorigenesis. The proteins with normal expression are black; the downregulated proteins, resulted from these gene promoters’ DNA methylation, are red and upregulated proteins for gene promoters’ DNA demethylation are green. APC, adenomatous polyposis coli; DUSP1, dual‐specificity phosphatase 1; JAK/STAT, Janus kinase-signal transducer and activator of transcription; IGF, insulin-like growth factor; CCND1, cyclin D1; SOCS1, suppressor of cytokine signaling 1; RASAL1, Ras protein activator like 1; RasGRF2, Ras protein-specific guanine nucleotide-releasing factor 2; CASP8, caspase 8; MAPK, mitogen-activated protein kinase.
Wnt Signaling Pathway
The conserved Wnt signaling pathway regulates numerous cellular processes, including cell polarity, motility, migration, and neural patterning during embryonic development (116). More than 50% of HCC patients display an alteration in the Wnt signaling pathway, including protein expression and mutation (117). And around 30% of HCC occurs mutation of β-catenin for its accumulation (118). Wnt is a secreted, hydrophobic cysteine-rich lipid-modified glycoprotein (119). It is bound by cell surface receptor (Frizzled receptor) and co-receptor (low-density lipoprotein–related protein 5 or 6 [LRP5/6]) and activated Wnt to prevent the β-catenin degradation complex from degradation (120). This complex is composed of casein kinase I, glycogen synthase kinase 3β (GSK3β), adenomatous polyposis coli (APC), Diversin, and Axin, which phosphorylate β-catenin and subsequently induce β-catenin degradation by the proteasome (121). The downregulation of APC or Axin2 by HCV infection, to some extent, leads to the accumulation of de-phospho-β-catenin, thereby more β-catenins translocate to the nucleus for the aberrant activation of LEF/TCF target genes, such as c-Myc, CCND1, and Cyclin. Furthermore, β-catenin accumulation also stimulates the malignant transformation of hepatocytes to HCC cells by overexpressing Deltanp73 for ant-apoptosis and chemoresistance of cells (122, 123). Deltanp73 is a repressor of tumor suppressor p53 and its human homology tap73 (124). In addition, when HCV causes chronic liver injuries, including liver fibrosis, epigenetic downregulation of peroxisome proliferator-activated receptor γ (PPARγ) is the main reason for myofibroblastic transdifferentiation (MTD) during liver fibrogenesis, which is proved to be associated with the Wnt signaling pathway. Inhibition of the Wnt signaling pathway restores PPARγ expression and hepatic stellate cells (HSCs) differentiation for liver regeneration (125). These studies suggest that the Wnt signaling pathway notably regulates HCV-infected HCC.
Ras/MAPK Signaling Pathway
The aberrant activation of the Ras/MAPK pathway has been evident in the occurrence and development of HCC (126, 127). This pathway is activated by various extracellular growth factors-mediated cell surface receptors, either receptor tyrosine kinase (RTK) or G‐protein‐coupled receptor (GPCR). Ras, a small GTPase, plays a key role in this pathway. Once activated by these growth factors, GTP-Ras recruits Raf1 to phosphorylate MAP2Ks and ERK 1/2, activating two key transcription factors of the AP‐1 family, c‐Jun and c‐Fos for expression of downstream genes involved in cell cycle and differentiation, through the translocation of ERK into the nucleus (128, 129). In this pathway, RASSF1, a member of the Ras association domain family (RASSF), is a Ras inhibitor, of which the expression is reduced by the DNA methylation of its gene’s promoter in the condition of HCV infection (130). The member of GTPase‐activating proteins (GAPs), RASAL1, promotes the switch of GTP to GDP, causing Ras from GTP-activated form to GDP-inactivated form (131). However, it is downregulated by DNA methylation during the development of HCV-induced HCC. And dual‐specificity phosphatase 1(DUSP1) is a specific ERK regulator (132). It is also downregulated by HCV infection. For the HCV replicon system, activation of the MEK1/2 kinase activity of Ras/MAPK signaling pathway inhibits the anti-HCV effect of IFN-gamma, which is partly because of the direct or indirect regulation of NS5A protein phosphorylation (133). The Ras/MAPK signaling pathway has a key role in HCV infection and the development of HCC.
PI3K/AKT Signaling Pathway
PI3KAkt/PKB plays a major role in cancer development and progression by inhibiting apoptosis and stimulating cell proliferation. This pathway is activated by multiple growth factors (IGF and EGF) and cytokines (134). Once activated, phosphoinositol-3-kinase (PI3K) then activates Akt, a serine-threonine kinase, through a lipid messenger phosphoinositol triphosphate 3 (PIP3). By phosphorylating multiple downstream substrates from cytoplasmic proteins, Akt regulates various cellular activities, such as cell proliferation, cell division, apoptosis, and RNA processing. It has been reported that PI3K/Akt signaling is activated by HCV, which promotes HCV entry and replication (135, 136). Akt phosphorylation occurs when PI3K activates SREBP, a downstream effector of this signaling pathway, resulting in HCV translocation by HCV NS5A. NS5A can protect cells from apoptosis, which is one underlying mechanism that has been uncovered. Specifically, the tumor suppressor gene, PTEN, is a negative regulator of the PI3K/Akt pathway by dephosphorylating PIP3 and inactivating Akt. Downregulating phosphatase and tensin homolog (PTEN) by NS5A enhances PI3K/Akt pathway and accumulates more Akt for HCV-infected HCC pathogenesis (137). In addition, myeloid-derived suppressor cells (MDSCs) function to maintain HCV persistent infection, and MDSC-like monocytes are induced by HCV through PI3K/Akt signaling pathway (138). Taken together, the PI3K/AKT signaling pathway strongly associates with the life cycle of HCV, and its inhibition could be a promising HCC treatment strategy.
JAK/STAT Signaling Pathway
Janus kinase-signal transducer and activator of transcription (JAK/STAT) regulates multiple cellular functions, such as differentiation, proliferation, and apoptosis, by transmitting extracellular signals to the nucleus for the transcriptional activation or repression of genes. It has been reported that this signaling pathway plays a crucial role in HCV-induced HCC. Especially, when stimulated, HCV-infected cells with interleukin 6 (IL6), the phosphorylation of JAK1/2 and STAT3, and STAT3-mediated transcription are inhibited by HCV core-binding JAKs, whereas IFN-gamma stimulation promotes JAK1/2 phosphorylation and STAT3-mediated transcription (139). The mechanistic analysis shows that HCV prevents the effect of IFN stimulation on JAK/STAT signaling pathway by degrading components in this pathway (140). Furthermore, the downregulation of STAT promotes virus replication and prevents anti-viral IFN-stimulated genes (ISG) expression, including PKR, OAS2, MxB, and ISG15 (141).
The expression level of signaling-related protein STAT1 is significantly down-regulated in the HCC tumor tissues, of which the underlying mechanisms are complex (142). On one hand, although STAT1 is essential for HCV replication, it is obviously decreased in HCV-infected cells as a result of proteasome-dependent degradation by HCV core/E1/E2, and NS3-4A proteins (143). On the other hand, STAT1 inhibits HCC growth by promoting p53-related cell cycling and apoptotic cell death in the STAT1 signaling pathway (142). Also, a recent study uncovers that STAT1 overexpression induces G0/G1 cell cycle arrest via downregulated expressions of cyclin A, cyclin D1, cyclin E, and CDK2 protein and promotes apoptosis by decreasing the expression of NF-κB p65 and Hes-1 and increasing the expression of p53 and Fbxw7 (144).
In addition, activation of JAK-STAT signaling and IFN-stimulated genes (ISG) expression is also regulated by a novel innate immune regulator, stromal antigen 2 (encoded by STAG2) (145). STAG2, a subunit of nuclear cohesion complex, plays an important role in genomic DNA stability, activation of cGAS-STING-IRF3 signaling, and recognition of cytoplasmic microchromatin. The deficiency of STAG2 in cells inhibits virus replication via the activation of JAK-STAT signaling.
P53 Signaling Pathway
P53 tumor suppressor is a nuclear transcription factor that regulates several genes involved in apoptosis, cell cycle arrest, and growth. It is activated by genotoxic or cellular stresses like oxidative stress, DNA damage, and nutrient deprivation. In normal tissues, p53 restricts tumor growth and malignant transformation, not only by autonomously triggering cell cycle arrest or apoptosis but also by non-autonomously releasing senescence-related secretory phenotype in the tumor microenvironment (146). However, the P53 signaling pathway is one of the main apoptosis pathways. As a tumor repressor, p53 negatively regulates the progression of various cancers in some ways, one of which is called the MDM2-p53 feedback pathway (147). The alterations of the murine double minute 2 (MDM2) -p53 feedback pathway are common in HCC (148–150). MDM2 is an E3 ubiquitin ligase. Specifically, P53 binds the promoter region of MDM2 and activates MDM2 transcription, whereas the expressed MDM2 blocks P53 transcription through the interaction of the P53 transactivation domain and promotes P53 proteasome-dependent degradation. Mutations of p53 in HCC mainly occur in the DNA-binding domain of p53, which weakens the affinity of p53 to its target genes and reduces MDM2 expression. Mutations of P53 occur at an early stage of tumor development and fail to induce p53-mediated apoptosis or sentence, which disturbs cancer therapy. Therefore, mutations of the P53 gene lead to patients with poorer survival rates for reduced expression of normal p53 protein.
Recent evidence has shown that p53 is overexpressed in the early stage of HCV-induced HCC, especially at HCV-infected damaged liver (151). HCV core-protein is reported to enhance p53 transcriptional activity and repress p21(waf1) expression. By acetylating lys273 and lys382 and phosphorylating ser15, HCV core-protein augments the ability of p53 to bind promoter regions of downstream genes (152). P21 inhibits cyclin-dependent kinase (CDK), and the HCV-mediated downregulation of p21 promotes cell cycle progression and cell growth. However, it is also reported that HCV nonstructural protein 5A (NS5A), transcriptionally represses p53 transactivation by directly interacting with p53 (153). Also, a coactivator of p53, hTAFII32, also binds to HCV NS5A, which incorporately inhibits p53-mediated transcriptional activation during HCV infection and may promote hepatocarcinogenesis. When p53 is knocked down in Huh7 cells, HCV RNA replication and viral protein significantly increase, suggesting that the p53 signaling pathway negatively regulates HCV infection and subsequently tumor development (154).
Conclusions
HCV infection is a major etiological factor for liver diseases. About 80% of the infected people develop a chronic infection, which eventually leads to a more severe form of liver disease, including liver cirrhosis and hepatocellular carcinoma (HCC). Over 90% of primary liver cancer belongs to HCC, and it may progress through dysregulation of a complex epigenetic network (155). The alterations in transcriptional regulation, along with downstream signaling pathways, might play a critical role in HCV-induced HCC. In this review, we have summarized 18 genes (7 genes with hypomethylation and 11 genes with hypermethylation) that are commonly regulated for HCV-mediated hepatocarcinogenesis. Perhaps, there are a wide variety of genes, including these 18 genes, which are transcriptionally activated or repressed through site-specific DNA methylation at genes’ promoter regions. Complementing our upstream regulations is the activation or blocking of tumor-associated signaling pathways, indicating that HCV infection plays a crucial role in the development of HCC.
HCV infection induces the alterations of genomic methylation levels in the promoter regions of cellular genes, which transcriptionally influence protein expression. These dysregulations of proteins directly or indirectly promote tumor growth through various signaling pathways. In detail, the inactivation of cellular genes by DNA hypermethylation, especially tumor suppress genes promotes a chronic HCV infection to liver cirrhosis, even HCC. And demethylation of signaling-related genes and oncogenic genes is also an important cause of hepatic carcinogenesis resulting from HCV infection. For application, DNA methylation of cancer-related genes might be useful as a prognostic marker. However, detection of candidate gene methylation was started with HCV-infected HCC patients, and further study is necessary to explore the functional involvement in the development of hepatic carcinogenesis.
Aberrant regulation of histone modifications is also linked to hepatocellular carcinogenesis. On one hand, histone enzymes, including various acetyltransferases and demethylase, are directly influenced by HCV infection and tumorigenesis. On another hand, the dynamics of histone modifications on different targeted genes, especially histone acetylation for gene activation and methylation for gene silencing, happens in the process of HCV infection and HCC development. Further studies on histone modifications are helpful for exploring clinical biomarkers or potential treatment on hepatic carcinogenesis. Moreover, small RNAs are believed to regulate more than 60% of whole human genes (156). Accumulated evidence suggests that small RNAs are tightly involved in cancer. The aberrant expressions of microRNAs (miRNAs) and small non-coding RNAs influence the levels of various protein expressions, which could play key roles in cell proliferation and differentiation and HCC patients’ survival rate in the condition of HCV infection.
As mentioned above, no one pathway acts alone but signaling cross-talking between diverse pathways exists in liver cancer. The interplay between pathways becomes complicated and influents positively or negatively. For example, the upstream protein Ras is involved in various pathways, such as MEK/ERK, PI3K/AKT, and RalEGF/Ral pathways. And in turn, activating Raf and Akt gene simultaneously Ras/MEK and PI3K/Akt pathways (48). The receptors of the JAK/STAT signaling pathway also stimulates the Ras/MEK pathway via Ras protein, as well as the receptors of the PI3K/Akt pathway. So, the alterations of DNA methylation, histone modifications, and ncRNAs, epigenetically impact downstream signaling pathways when HCV infection occurs. Taken together, epigenetic regulation of HCV-induced HCC is complex and necessary to be deeply studied. The clinical application of its epigenetic mechanisms is, therefore, being further evaluated.
Taken together, with our understanding of the molecular mechanism of HCV-indued HCC via a complex epigenetic network, different strategies that targeted these 18 proteins and five signaling pathways should be potentially investigated. In consideration that HCV-induced HCC transcriptionally lead to the activation of oncogenic genes and repression of tumor suppressor genes, a combination therapy may be necessary for targeting different proteins. Also, it is also a great strategy to identify some chemical drugs, which inhibit simultaneously more than two major signaling pathways. Besides, targeted drug delivery systems (DDS) can also be helpful to deliver these therapeutic drugs to specific tumor sites for HCV-induced HCC treatment (157).
Author Contributions
PZ prepared the original draft. SM discussed and contributed to the manuscript writing. SX supervised the project and contributed to the final version of the manuscript. All authors have made a direct and intellectual contribution to the work. All authors contributed to the article and approved the submitted version.
Funding
This work has been supported by the Shenzhen Science and Technology Program (KQTD20190929172538530) and the National Natural Science Foundation (81971492).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Glossary

References
1. Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2015. CA: Cancer J Clin (2015) 65:5–29. doi: 10.3322/caac.21254
2. Petruzziello A. Epidemiology of Hepatitis B Virus (HBV) and Hepatitis C Virus (HCV) Related Hepatocellular Carcinoma. Open Virol J (2018) 12:26–32. doi: 10.2174/1874357901812010026
3. Sherman M. Hepatocellular Carcinoma: Epidemiology, Surveillance, and Diagnosis. Semin Liver Dis (2010) 30:003–16. doi: 10.1055/s-0030-1247128
4. Alazard-Dany N, Denolly S, Boson B, Cosset F-L. Overview of HCV Life Cycle With a Special Focus on Current and Possible Future Antiviral Targets. Viruses (2019) 11:30. doi: 10.3390/v11010030
5. D’souza S, Lau KC, Coffin CS, Patel TR. Molecular Mechanisms of Viral Hepatitis Induced Hepatocellular Carcinoma. World J Gastroenterol (2020) 26:5759–83. doi: 10.3748/wjg.v26.i38.5759
6. Albecka A, Belouzard S, De Beeck AO, Descamps V, Goueslain L, Bertrand-Michel J, et al. Role of Low-Density Lipoprotein Receptor in the Hepatitis C Virus Life Cycle. Hepatology (2012) 55:998–1007. doi: 10.1002/hep.25501
7. Wack A, Soldaini E, Tseng C-TK, Nuti S, Klimpel GR, Abrignani S. Binding of the Hepatitis C Virus Envelope Protein E2 to CD81 Provides a Co-Stimulatory Signal for Human T Cells. Eur J Immunol (2001) 31:166–75. doi: 10.1002/1521-4141(200101)31:1<166::aid-immu166>3.0.co;2-l
8. Scarselli E, Ansuini H, Cerino R, Roccasecca RM, Acali S, Filocamo G, et al. The Human Scavenger Receptor Class B Type I Is a Novel Candidate Receptor for the Hepatitis C Virus. EMBO J (2002) 21:5017–25. doi: 10.1093/emboj/cdf529
9. Evans MJ, Von Hahn T, Tscherne DM, Syder AJ, Panis M, Wölk B, et al. Claudin-1 Is a Hepatitis C Virus Co-Receptor Required for a Late Step in Entry. Nature (2007) 446:801–5. doi: 10.1038/nature05654
10. Tscherne DM, Jones CT, Evans MJ, Lindenbach BD, Mckeating JA, Rice CM. Time- and Temperature-Dependent Activation of Hepatitis C Virus for low-pH-Triggered Entry. J Virol (2006) 80:1734–41. doi: 10.1128/JVI.80.4.1734-1741.2006
11. Blanchard E, Belouzard S, Goueslain L, Wakita T, Dubuisson J, Wychowski C, et al. Hepatitis C Virus Entry Depends on Clathrin-Mediated Endocytosis. J Virol (2006) 80:6964–72. doi: 10.1128/JVI.00024-06
12. Huang H, Sun F, Owen DM, Li W, Chen Y, Gale M, et al. Hepatitis C Virus Production by Human Hepatocytes Dependent on Assembly and Secretion of Very Low-Density Lipoproteins. Proc Natl Acad Sci (2007) 104:5848. doi: 10.1073/pnas.0700760104
13. Coller KE, Heaton NS, Berger KL, Cooper JD, Saunders JL, Randall G. Molecular Determinants and Dynamics of Hepatitis C Virus Secretion. PloS Pathog (2012) 8:e1002466. doi: 10.1371/journal.ppat.1002466
14. Tamai K, Shiina M, Tanaka N, Nakano T, Yamamoto A, Kondo Y, et al. Regulation of Hepatitis C Virus Secretion by the Hrs-Dependent Exosomal Pathway. Virology (2012) 422:377–85. doi: 10.1016/j.virol.2011.11.009
15. Syed GH, Khan M, Yang S, Siddiqui A. Hepatitis C Virus Lipoviroparticles Assemble in the Endoplasmic Reticulum (ER) and Bud Off From the ER to the Golgi Compartment in COPII Vesicles. J Virol (2017) 91:e00499–00417. doi: 10.1128/JVI.00499-17
16. Horiike N, Nonaka T, Kumamoto I, Kajino K, Onji M, Ohta Y. Hepatitis C Virus Plus- and Minus- Strand RNA in Hepatocellular Carcinoma and Adjoining Nontumorous Liver. J Med Virol (1993) 41:312–5. doi: 10.1002/jmv.1890410410
17. Gerber MA, Shieh YS, Shim KS, Thung SN, Demetris AJ, Schwartz M, et al. Detection of Replicative Hepatitis C Virus Sequences in Hepatocellular Carcinoma. Am J Pathol (1992) 141:1271–7.
18. Moriya K, Fujie H, Shintani Y, Yotsuyanagi H, Tsutsumi T, Ishibashi K, et al. The Core Protein of Hepatitis C Virus Induces Hepatocellular Carcinoma in Transgenic Mice. Nat Med (1998) 4:1065–7. doi: 10.1038/2053
19. Sun BS, Pan J, Clayton MM, Liu J, Yan X, Matskevich AA, et al. Hepatitis C Virus Replication in Stably Transfected HepG2 Cells Promotes Hepatocellular Growth and Tumorigenesis. J Cell Physiol (2004) 201:447–58. doi: 10.1002/jcp.20083
20. Goto K, Suarez AAR, Wrensch F, Baumert TF, Lupberger J. Hepatitis C Virus and Hepatocellular Carcinoma: When the Host Loses Its Grip. Int J Mol Sci (2020) 21:3057. doi: 10.3390/ijms21093057
21. Westin J, Nordlinder H, Lagging M, Norkrans G, Wejstål R. Steatosis Accelerates Fibrosis Development Over Time in Hepatitis C Virus Genotype 3 Infected Patients. J Hepatol (2002) 37:837–42. doi: 10.1016/s0168-8278(02)00299-4
22. The Polaris Observatory Hcv Collaborators. Global Prevalence and Genotype Distribution of Hepatitis C Virus Infection in 2015: A Modelling Study. Lancet Gastroenterol Hepatol (2017) 2:161–76. doi: 10.1016/S2468-1253(16)30181-9
23. Gao X, Zhan M, Wang L, Ding Y, Niu J. Timing of DAA Initiation After Curative Treatment and Its Relationship With the Recurrence of HCV-Related HCC. J Hepatocell Carcinoma (2020) 7:347–60. doi: 10.2147/JHC.S279657
24. Dash S, Aydin Y, Widmer KE, Nayak L. Hepatocellular Carcinoma Mechanisms Associated With Chronic HCV Infection and the Impact of Direct-Acting Antiviral Treatment. J Hepatocell Carcinoma (2020) 7:45–76. doi: 10.2147/JHC.S221187
25. Kanwal F, Kramer J, Asch SM, Chayanupatkul M, Cao Y, El-Serag HB. Risk of Hepatocellular Cancer in HCV Patients Treated With Direct-Acting Antiviral Agents. Gastroenterology (2017) 153:996–1005.e1001. doi: 10.1053/j.gastro.2017.06.012
26. Pawlotsky J-M, Feld JJ, Zeuzem S, Hoofnagle JH. From non-A, non-B Hepatitis to Hepatitis C Virus Cure. J Hepatol (2015) 62:S87–99. doi: 10.1016/j.jhep.2015.02.006
27. Perez S, Kaspi A, Domovitz T, Davidovich A, Lavi-Itzkovitz A, Meirson T, et al. Hepatitis C Virus Leaves an Epigenetic Signature Post Cure of Infection by Direct-Acting Antivirals. PloS Genet (2019) 15:e1008181–e1008181. doi: 10.1371/journal.pgen.1008181
28. Machida K, Mcnamara G, Cheng KTH, Huang J, Wang C-H, Comai L, et al. Hepatitis C Virus Inhibits DNA Damage Repair Through Reactive Oxygen and Nitrogen Species and by Interfering With the ATM-NBS1/Mre11/Rad50 DNA Repair Pathway in Monocytes and Hepatocytes. J Immunol (2010) 185:6985–98. doi: 10.4049/jimmunol.1000618
29. Lim JS, Park S-H, Jang KL. Hepatitis C Virus Core Protein Overcomes Stress-Induced Premature Senescence by Down-Regulating P16 Expression via DNA Methylation. Cancer Lett (2012) 321:154–61. doi: 10.1016/j.canlet.2012.01.044
30. Abd El-Rahman Nabawy Zekri AA-M N, El-Din El-Rouby MN, Shousha HI, Barakat AB, El-Desouky ED, Zayed NA, et al. Disease Progression From Chronic Hepatitis C to Cirrhosis and Hepatocellular Carcinoma Is Associated With Increasing DNA Promoter Methylation. Asian Pac J Cancer Prev (2013) 14:6721–6. doi: 10.7314/apjcp.2013.14.11.6721
31. Sun G, Zhang C, Feng M, Liu W, Xie H, Qin Q, et al. Methylation Analysis of P16, SLIT2, SCARA5, and Runx3 Genes in Hepatocellular Carcinoma. Med (Baltimore) (2017) 96:e8279–9. doi: 10.1097/MD.0000000000008279
32. Quan H, Zhou F, Nie D, Chen Q, Cai X, Shan X, et al. Hepatitis C Virus Core Protein Epigenetically Silences SFRP1 and Enhances HCC Aggressiveness by Inducing Epithelial–Mesenchymal Transition. Oncogene (2014) 33:2826–35. doi: 10.1038/onc.2013.225
33. Sun S, Li Y, Han S, Jia H, Li X, Li X. A Comprehensive Genome-Wide Profiling Comparison Between HBV and HCV Infected Hepatocellular Carcinoma. BMC Med Genomics (2019) 12:147–7. doi: 10.1186/s12920-019-0580-x
34. Wijetunga NA, Pascual M, Tozour J, Delahaye F, Alani M, Adeyeye M, et al. A Pre-Neoplastic Epigenetic Field Defect in HCV-Infected Liver at Transcription Factor Binding Sites and Polycomb Targets. Oncogene (2017) 36:2030–44. doi: 10.1038/onc.2016.340
35. Zhao W, Liu H, Liu W, Wu Y, Chen W, Jiang B, et al. Abnormal Activation of the Synuclein-Gamma Gene in Hepatocellular Carcinomas by Epigenetic Alteration. Int J Oncol (2006) 28:1081–8. doi: 10.3892/ijo.28.5.1081
36. Calvisi DF, Ladu S, Conner EA, Seo D, Hsieh J-T, Factor VM, et al. Inactivation of Ras GTPase-Activating Proteins Promotes Unrestrained Activity of Wild-Type Ras in Human Liver Cancer. J Hepatol (2011) 54:311–9. doi: 10.1016/j.jhep.2010.06.036
37. Jiang L, Liu Q-L, Liang Q-L, Zhang H-J, Ou W-T, Yuan G-L. Association of PHD3 and HIF2α Gene Expression With Clinicopathological Characteristics in Human Hepatocellular Carcinoma. Oncol Lett (2018) 15:545–51. doi: 10.3892/ol.2017.7302
38. Erez N, Milyavsky M, Eilam R, Shats I, Goldfinger N, Rotter V. Expression of Prolyl-Hydroxylase-1 (PHD1/EGLN2) Suppresses Hypoxia Inducible Factor-1α Activation and Inhibits Tumor Growth. Cancer Res (2003) 63:8777.
39. Liu J, Xu R, Mai S-J, Ma Y-S, Zhang M-Y, Cao P-S, et al. LncRNA CSMD1-1 Promotes the Progression of Hepatocellular Carcinoma by Activating MYC Signaling. Theranostics (2020) 10:7527–44. doi: 10.7150/thno.45989
40. Zhu Q, Gong L, Wang J, Tu Q, Yao L, Zhang J-R, et al. miR-10b Exerts Oncogenic Activity in Human Hepatocellular Carcinoma Cells by Targeting Expression of CUB and Sushi Multiple Domains 1 (CSMD1). BMC Cancer (2016) 16:806–6. doi: 10.1186/s12885-016-2801-4
41. Rayess H, Wang MB, Srivatsan ES. Cellular Senescence and Tumor Suppressor Gene P16. Int J Cancer (2012) 130:1715–25. doi: 10.1002/ijc.27316
42. Wong IHN, Lo YMD, Zhang J, Liew C-T, Ng MHL, Wong N, et al. Detection of Aberrant P16 Methylation in the Plasma and Serum of Liver Cancer Patients. Cancer Res (1999) 59:71.
43. Zang J-J, Xie F, Xu J-F, Qin Y-Y, Shen R-X, Yang J-M, et al. P16 Gene Hypermethylation and Hepatocellular Carcinoma: A Systematic Review and Meta-Analysis. World J Gastroenterol (2011) 17:3043–8. doi: 10.3748/wjg.v17.i25.3043
44. Shelton JG, Chang JT, Fau-Lee F, Lee RA, Fau-Franklin J, Franklin LS, et al. B-Raf and Insulin Synergistically Prevent Apoptosis and Induce Cell Cycle Progression in Hematopoietic Cells. Cell Cycle (2004) 3:189–96. doi: 10.4161/cc.3.2.624
45. Forbes SA, Tang G, Bindal N, Bamford S, Dawson E, Cole C, et al. COSMIC (the Catalogue of Somatic Mutations in Cancer): A Resource to Investigate Acquired Mutations in Human Cancer. Nucleic Acids Res (2010) 38:D652–7. doi: 10.1093/nar/gkp995
46. Yin G, Liu Z, Wang Y, Dou C, Li C, Yang W, et al. BCORL1 Is an Independent Prognostic Marker and Contributes to Cell Migration and Invasion in Human Hepatocellular Carcinoma. BMC Cancer (2016) 16:103–3. doi: 10.1186/s12885-016-2154-z
47. Dang S, Zhou J, Chen Y, Chen P, Ji M, Shi B, et al. Dynamic Expression of ZNF382 and Its Tumor-Suppressor Role in Hepatitis B Virus-Related Hepatocellular Carcinogenesis. Oncogene (2019) 38:4804–19. doi: 10.1038/s41388-019-0759-9
48. Park WS, Cho YG, Kim CJ, Song JH, Lee YS, Kim SY, et al. Hypermethylation of the RUNX3 Gene in Hepatocellular Carcinoma. Cell Cycle (2005) 37:276–81. doi: 10.1038/emm.2005.37
49. Kataoka J, Shiraha H, Horiguchi S, Sawahara H, Uchida D, Nagahara T, et al. Loss of Runt-Related Transcription Factor 3 Induces Resistance to 5-Fluorouracil and Cisplatin in Hepatocellular Carcinoma. Oncol Rep (2016) 35:2576–82. doi: 10.3892/or.2016.4681
50. Nishina S-I, Shiraha H, Nakanishi Y, Tanaka S, Matsubara M, Takaoka N, et al. Restored Expression of the Tumor Suppressor Gene RUNX3 Reduces Cancer Stem Cells in Hepatocellular Carcinoma by Suppressing Jagged1-Notch Signaling. Oncogene Rep (2011) 26:523–31. doi: 10.3892/or.2011.1336
51. Umezaki N, Nakagawa S, Yamashita Y-I, Kitano Y, Arima K, Miyata T, et al. Lysyl Oxidase Induces Epithelial-Mesenchymal Transition and Predicts Intrahepatic Metastasis of Hepatocellular Carcinoma. Cancer Sci (2019) 110:2033–43. doi: 10.1111/cas.14010
52. Kouzarides T. Transcriptional Control by the Retinoblastoma Protein. Semin Cancer Biol (1995) 6:91–8. doi: 10.1006/scbi.1995.0012
53. Riley DJ, Lee WH, Lee WH. The Retinoblastoma Protein: More Than a Tumor Suppressor. Annu Rev Cell Biol (1994) 10:1–29. doi: 10.1146/annurev.cb.10.110194.000245
54. Anwar SL, Krech T, Hasemeier B, Schipper E, Schweitzer N, Vogel A, et al. Deregulation of RB1 Expression by Loss of Imprinting in Human Hepatocellular Carcinoma. J Pathol (2014) 233:392–401. doi: 10.1002/path.4376
55. Edamoto Y, Hara A, Biernat W, Terracciano L, Cathomas G, Riehle H-M, et al. Alterations of RB1, P53 and Wnt Pathways in Hepatocellular Carcinomas Associated With Hepatitis C, Hepatitis B and Alcoholic Liver Cirrhosis. Int J Cancer (2003) 106:334–41. doi: 10.1002/ijc.11254
56. Tannapfel A, Wasner M, Krause K, Geissler F, Katalinic A, Hauss J, et al. Expression of P73 and Its Relation to Histopathology and Prognosis in Hepatocellular Carcinoma. J Natl Cancer Inst (1999) 91:1154–8. doi: 10.1093/jnci/91.13.1154
57. Liu WJ, Wang J-P, Li J-Q, Zhang C-Q, Zheng LZ, Yuan Y-F, et al. Correlations of CpG Island Methylator Phenotype and OPCML Gene Methylation to Carcinogenesis of Hepatocellular Carcinoma. Ai Zheng (2006) 25:696–700.
58. Baharudin R, Tieng FYF, Lee L-H, Ab Mutalib NS. Epigenetics of SFRP1: The Dual Roles in Human Cancers. Cancers (Basel) (2020) 12:445. doi: 10.3390/cancers12020445
59. Whittaker S, Marais R, Zhu AX. The Role of Signaling Pathways in the Development and Treatment of Hepatocellular Carcinoma. Oncogene (2010) 29:4989–5005. doi: 10.1038/onc.2010.236
60. Adamek A, Kasprzak A. Insulin-Like Growth Factor (IGF) System in Liver Diseases. Int J Mol Sci (2018) 19:1308. doi: 10.3390/ijms19051308
61. Song B, Park S-H, Zhao JC, Fong K-W, Li S, Lee Y, et al. Targeting FOXA1-Mediated Repression of TGF-β Signaling Suppresses Castration-Resistant Prostate Cancer Progression. J Clin Invest (2019) 129:569–82. doi: 10.1172/JCI122367
62. Yuan Z, Ye M, Qie J, Ye T. FOXA1 Promotes Cell Proliferation and Suppresses Apoptosis in HCC by Directly Regulating miR-212-3p/FOXA1/AGR2 Signaling Pathway. Onco Targets Ther (2020) 13:5231–40. doi: 10.2147/OTT.S252890
63. He S, Zhang J, Zhang W, Chen F, Luo R. FOXA1 Inhibits Hepatocellular Carcinoma Progression by Suppressing PIK3R1 Expression in Male Patients. J Exp Clin Cancer Res (2017) 36:175–5. doi: 10.1186/s13046-017-0646-6
64. Chandra V, Huang P, Potluri N, Wu D, Kim Y, Rastinejad F. Multidomain Integration in the Structure of the HNF-4α Nuclear Receptor Complex. Nature (2013) 495:394–8. doi: 10.1038/nature11966
65. Ning B-F, Ding J, Liu J, Yin C, Xu W-P, Cong W-M, et al. Hepatocyte Nuclear Factor 4α-Nuclear Factor-κb Feedback Circuit Modulates Liver Cancer Progression. Hepatology (2014) 60:1607–19. doi: 10.1002/hep.27177
66. Guo S, Lu H. Novel Mechanisms of Regulation of the Expression and Transcriptional Activity of Hepatocyte Nuclear Factor 4α. J Cell Biochem (2019) 120:519–32. doi: 10.1002/jcb.27407
67. Fekry B, Ribas-Latre A, Baumgartner C, Mohamed AMT, Kolonin MG, Sladek FM, et al. Hnf4α-Deficient Fatty Liver Provides a Permissive Environment for Sex-Independent Hepatocellular Carcinoma. Cancer Res (2019) 79:5860–73. doi: 10.1158/0008-5472.CAN-19-1277
68. Fekry B, Ribas-Latre A, Baumgartner C, Deans JR, Kwok C, Patel P, et al. Incompatibility of the Circadian Protein BMAL1 and HNF4α in Hepatocellular Carcinoma. Nat Commun (2018) 9:4349–9. doi: 10.1038/s41467-018-06648-6
69. Lourenço AR, Coffer PJ. A Tumor Suppressor Role for C/Ebpα in Solid Tumors: More Than Fat and Blood. Oncogene (2017) 36:5221–30. doi: 10.1038/onc.2017.151
70. Sarker D, Plummer R, Meyer T, Sodergren MH, Basu B, Chee CE, et al. MTL-CEBPA, a Small Activating RNA Therapeutic Upregulating C/EBP-α, in Patients With Advanced Liver Cancer: A First-in-Human, Multicenter, Open-Label, Phase I Trial. Clin Cancer Res (2020) 26:3936. doi: 10.1158/1078-0432.CCR-20-0414
71. Lu G, Leung CH, Yan B, Tan CM, Low SY, Aung MO, et al. C/EBPalpha Is Up-Regulated in a Subset of Hepatocellular Carcinomas and Plays a Role in Cell Growth and Proliferation. Gastroenterology (2010) 139:632–43. doi: 10.1053/j.gastro.2010.03.051
72. Zheng Y, Hlady RA, Joyce BT, Robertson KD, He C, Nannini DR, et al. DNA Methylation of Individual Repetitive Elements in Hepatitis C Virus Infection-Induced Hepatocellular Carcinoma. Clin Epigenet (2019) 11:145–5. doi: 10.1186/s13148-019-0733-y
73. Siddiqui NN, Ul Haq A, Ali Siddiqui O, Khan R. DNA Methyltransferase 1, 3a, and 3b Expression in Hepatitis C Associated Human Hepatocellular Carcinoma and Their Clinicopathological Association. Tumor Biol (2016) 37:10487–97. doi: 10.1007/s13277-016-4941-1
74. Xue X, Gao W, Sun B, Xu Y, Han B, Wang F, et al. Vasohibin 2 Is Transcriptionally Activated and Promotes Angiogenesis in Hepatocellular Carcinoma. Oncogene (2013) 32:1724–34. doi: 10.1038/onc.2012.177
75. Shigekawa Y, Hayami S, Ueno M, Miyamoto A, Suzaki N, Kawai M, et al. Overexpression of KDM5B/JARID1B Is Associated With Poor Prognosis in Hepatocellular Carcinoma. Oncotarget (2018) 9:34320–35. doi: 10.18632/oncotarget.26144
76. Kim S, Bolatkan A, Kaneko S, Ikawa N, Asada K, Komatsu M, et al. Deregulation of the Histone Lysine-Specific Demethylase 1 Is Involved in Human Hepatocellular Carcinoma. Biomolecules (2019) 9:810. doi: 10.3390/biom9120810
77. Tachibana M, Sugimoto K, Nozaki M, Ueda J, Ohta T, Ohki M, et al. G9a Histone Methyltransferase Plays a Dominant Role in Euchromatic Histone H3 Lysine 9 Methylation and Is Essential for Early Embryogenesis. Genes Dev (2002) 16:1779–91. doi: 10.1101/gad.989402
78. Rice JC, Briggs SD, Ueberheide B, Barber CM, Shabanowitz J, Hunt DF, et al. Histone Methyltransferases Direct Different Degrees of Methylation to Define Distinct Chromatin Domains. Mol Cell (2003) 12:1591–8. doi: 10.1016/s1097-2765(03)00479-9
79. Testoni B, Durantel D, Lebossé F, Fresquet J, Helle F, Negro F, et al. Ribavirin Restores Ifnα Responsiveness in HCV-Infected Livers by Epigenetic Remodelling at Interferon Stimulated Genes. Gut (2016) 65:672. doi: 10.1136/gutjnl-2014-309011
80. Simon JA, Tamkun JW. Programming Off and on States in Chromatin: Mechanisms of Polycomb and Trithorax Group Complexes. Curr Opin Genet Dev (2002) 12:210–8. doi: 10.1016/s0959-437x(02)00288-5
81. Cai M-Y, Tong Z-T, Zheng F, Liao Y-J, Wang Y, Rao H-L, et al. EZH2 Protein: A Promising Immunomarker for the Detection of Hepatocellular Carcinomas in Liver Needle Biopsies. Gut (2011) 60:967. doi: 10.1136/gut.2010.231993
82. Fan DN-Y, Tsang FH-C, Tam AH-K, Au SL-K, Wong CC-L, Wei L, et al. Histone Lysine Methyltransferase, Suppressor of Variegation 3-9 Homolog 1, Promotes Hepatocellular Carcinoma Progression and Is Negatively Regulated by microRNA-125b. Hepatology (2013) 57:637–47. doi: 10.1002/hep.26083
83. Kondo Y, Shen L, Suzuki S, Kurokawa T, Masuko K, Tanaka Y, et al. Alterations of DNA Methylation and Histone Modifications Contribute to Gene Silencing in Hepatocellular Carcinomas. Hepatol Res (2007) 37:974–83. doi: 10.1111/j.1872-034X.2007.00141.x
84. Hung S-Y, Lin H-H, Yeh K-T, Chang J-G. Histone-Modifying Genes as Biomarkers in Hepatocellular Carcinoma. Int J Clin Exp Pathol (2014) 7:2496–507.
85. Zhou P, Xia J, Zhou Y-J, Wan J, Li L, Bao J, et al. Proportions of Acetyl-Histone-Positive Hepatocytes Indicate the Functional Status and Prognosis of Cirrhotic Patients. World J Gastroenterol (2015) 21:6665–74. doi: 10.3748/wjg.v21.i21.6665
86. Glozak MA, Seto E. Acetylation/deacetylation Modulates the Stability of DNA Replication Licensing Factor Cdt1. J Biol Chem (2009) 284:11446–53. doi: 10.1074/jbc.M809394200
87. Ji H, Zhou Y, Zhuang X, Zhu Y, Wu Z, Lu Y, et al. HDAC3 Deficiency Promotes Liver Cancer Through a Defect in H3K9ac/H3K9me3 Transition. Cancer Res (2019) 79:3676–88. doi: 10.1158/0008-5472.CAN-18-3767
88. Hayashi A, Yamauchi N, Shibahara J, Kimura H, Morikawa T, Ishikawa S, et al. Concurrent Activation of Acetylation and Tri-Methylation of H3K27 in a Subset of Hepatocellular Carcinoma With Aggressive Behavior. PloS One (2014) 9:e91330–0. doi: 10.1371/journal.pone.0091330
89. Liu H, Trinh TL, Dong H, Keith R, Nelson D, Liu C. Iron Regulator Hepcidin Exhibits Antiviral Activity Against Hepatitis C Virus. PloS One (2012) 7:e46631–1. doi: 10.1371/journal.pone.0046631
90. Zhou Y, Wang Q, Yang Q, Tang J, Xu C, Gai D, et al. Histone Deacetylase 3 Inhibitor Suppresses Hepatitis C Virus Replication by Regulating Apo-A1 and LEAP-1 Expression. Virol Sin (2018) 33:418–28. doi: 10.1007/s12250-018-0057-7
91. Unfried JP, Fortes P. LncRNAs in HCV Infection and HCV-Related Liver Disease. Int J Mol Sci (2020) 21:2255. doi: 10.3390/ijms21062255
92. Hu X, Tang J, Hu X, Bao P, Deng W, Wu J, et al. Silencing of Long non-Coding RNA HOTTIP Reduces Inflammation in Rheumatoid Arthritis by Demethylation of SFRP1. Mol Ther Nucleic Acids (2020) 19:468–81. doi: 10.1016/j.omtn.2019.11.015
93. Xing Y, Zheng X, Fu Y, Qi J, Li M, Ma M, et al. Long Noncoding RNA-Maternally Expressed Gene 3 Contributes to Hypoxic Pulmonary Hypertension. Mol Ther (2019) 27:2166–81. doi: 10.1016/j.ymthe.2019.07.022
94. Dai J-H, Huang W-Z, Li C, Deng J, Lin S-J, Luo J. Silencing of Long Noncoding RNA SBF2-AS1 Inhibits Proliferation, Migration and Invasion and Contributes to Apoptosis in Osteosarcoma Cells by Upregulating microRNA-30a to Suppress FOXA1 Expression. Cell Cycle (2019) 18:2727–41. doi: 10.1080/15384101.2019.1656478
95. Liu D-W, Zhang J-H, Liu F-X, Wang X-T, Pan S-K, Jiang D-K, et al. Silencing of Long Noncoding RNA PVT1 Inhibits Podocyte Damage and Apoptosis in Diabetic Nephropathy by Upregulating FOXA1. Exp Mol Med (2019) 51:1–15. doi: 10.1038/s12276-019-0259-6
96. Guo Y, Ma Y, Hu X, Song R, Zhu L, Zhong M. Long non-Coding RNA CEBPA-AS1 Correlates With Poor Prognosis and Promotes Tumorigenesis via CEBPA/Bcl2 in Oral Squamous Cell Carcinoma. Cancer Biol Ther (2018) 19:205–13. doi: 10.1080/15384047.2017.1416276
97. Pasquinelli AE, Hunter S, Bracht J. MicroRNAs: A Developing Story. Curr Opin Genet Dev (2005) 15:200–5. doi: 10.1016/j.gde.2005.01.002
98. Bartel DP. MicroRNAs: Genomics, Biogenesis, Mechanism, and Function. Cell (2004) 116:281–97. doi: 10.1016/s0092-8674(04)00045-5
100. Pedersen IM, Cheng G, Wieland S, Volinia S, Croce CM, Chisari FV, et al. Interferon Modulation of Cellular microRNAs as an Antiviral Mechanism. Nature (2007) 449:919–22. doi: 10.1038/nature06205
101. Jopling CL, Yi M, Lancaster AM, Lemon SM, Sarnow P. Modulation of Hepatitis C Virus RNA Abundance by a Liver-Specific microRNA. Science (2005) 309:1577. doi: 10.1126/science.1113329
102. Lagos-Quintana M, Rauhut R, Yalcin A, Meyer J, Lendeckel W, Tuschl T. Identification of Tissue-Specific microRNAs From Mouse. Curr Biol (2002) 12:735–9. doi: 10.1016/s0960-9822(02)00809-6
103. Henke JI, Goergen D, Zheng J, Song Y, Schüttler CG, Fehr C, et al. microRNA-122 Stimulates Translation of Hepatitis C Virus RNA. EMBO J (2008) 27:3300–10. doi: 10.1038/emboj.2008.244
104. Song K, Han C, Zhang J, Lu D, Dash S, Feitelson M, et al. Epigenetic Regulation of MicroRNA-122 by Peroxisome Proliferator Activated Receptor-Gamma and Hepatitis B Virus X Protein in Hepatocellular Carcinoma Cells. Hepatology (2013) 58:1681–92. doi: 10.1002/hep.26514
105. Elhelw DS, Riad SE, Shawer H, El-Ekiaby N, Salah A, Zekri A, et al. Ectopic Delivery of miR-200c Diminishes Hepatitis C Virus Infectivity Through Transcriptional and Translational Repression of Occludin. Arch Virol (2017) 162:3283–91. doi: 10.1007/s00705-017-3449-3
106. Zeng B, Li Z, Chen R, Guo N, Zhou J, Zhou Q, et al. Epigenetic Regulation of miR-124 by Hepatitis C Virus Core Protein Promotes Migration and Invasion of Intrahepatic Cholangiocarcinoma Cells by Targeting SMYD3. FEBS Lett (2012) 586:3271–8. doi: 10.1016/j.febslet.2012.06.049
107. El Sobky SA, El-Ekiaby NM, Mekky RY, Elemam NM, Mohey Eldin MA, El-Sayed M, et al. Contradicting Roles of miR-182 in Both NK Cells and Their Host Target Hepatocytes in HCV. Immunol Lett (2016) 169:52–60. doi: 10.1016/j.imlet.2015.10.013
108. El Maraghy SA, Adel O, Zayed N, Yosry A, El-Nahaas SM, Gibriel AA. Circulatory miRNA-484, 524, 615 and 628 Expression Profiling in HCV Mediated HCC Among Egyptian Patients; Implications for Diagnosis and Staging of Hepatic Cirrhosis and Fibrosis. J Adv Res (2019) 22:57–66. doi: 10.1016/j.jare.2019.12.002
109. Imamura K, Akimitsu N. Long Non-Coding RNAs Involved in Immune Responses. Front Immunol (2014) 5:573. doi: 10.3389/fimmu.2014.00573
110. Fitzgerald KA, Caffrey DR. Long Noncoding RNAs in Innate and Adaptive Immunity. Curr Opin Immunol (2014) 26:140–6. doi: 10.1016/j.coi.2013.12.001
111. Valadkhan S, Fortes P. Regulation of the Interferon Response by incRNAs in HCV Infection. Front Microbiol (2018) 9:181. doi: 10.3389/fmicb.2018.00181
112. Hu P, Wilhelm J, Gerresheim GK, Shalamova LA, Niepmann M. Lnc-ITM2C-1 and GPR55 Are Proviral Host Factors for Hepatitis C Virus. Viruses (2019) 11:549. doi: 10.3390/v11060549
113. Liu X, Duan X, Holmes JA, Li W, Lee SH, Tu Z, et al. A Long Noncoding RNA Regulates Hepatitis C Virus Infection Through Interferon Alpha-Inducible Protein 6. Hepatology (2019) 69:1004–19. doi: 10.1002/hep.30266
114. Zhang Q, Matsuura K, Kleiner DE, Zamboni F, Alter HJ, Farci P. Analysis of Long Noncoding RNA Expression in Hepatocellular Carcinoma of Different Viral Etiology. J Transl Med (2016) 14:328–8. doi: 10.1186/s12967-016-1085-4
115. Zhang H, Zhu C, Zhao Y, Li M, Wu L, Yang X, et al. Long Non-Coding RNA Expression Profiles of Hepatitis C Virus-Related Dysplasia and Hepatocellular Carcinoma. Oncotarget (2015) 6:43770–8. doi: 10.18632/oncotarget.6087
116. Komiya Y, Habas R. Wnt Signal Transduction Pathways. Organogenesis (2008) 4:68–75. doi: 10.4161/org.4.2.5851
117. Ozturk M. Genetic Aspects of Hepatocellular Carcinogenesis. Semin Liver Dis (1999) 19:235–42. doi: 10.1055/s-2007-1007113
118. Zucman-Rossi J, Benhamouche S, Godard C, Boyault S, Grimber G, Balabaud C, et al. Differential Effects of Inactivated Axin1 and Activated β-Catenin Mutations in Human Hepatocellular Carcinomas. Oncogene (2007) 26:774–80. doi: 10.1038/sj.onc.1209824
119. Kikuchi A, Yamamoto H, Sato A, Matsumoto S. New Insights Into the Mechanism of Wnt Signaling Pathway Activation. Int Rev Cell Mol Biol (2011) 291:21–71. doi: 10.1016/B978-0-12-386035-4.00002-1
120. Pal Monga S. β-Catenin Signaling and Roles in Liver Homeostasis, Injury, and Tumorigenesis. Gastroenterology (2015) 148:1294–310. doi: 10.1053/j.gastro.2015.02.056
121. Morin PJ, Sparks AB, Korinek V, Barker N, Clevers H, Vogelstein B, et al. Activation of β-Catenin-Tcf Signaling in Colon Cancer by Mutations in β-Catenin or APC. Science (1997) 275:1787. doi: 10.1126/science.275.5307.1787
122. Müller M, Schilling T, Sayan AE, Kairat A, Lorenz K, Schulze-Bergkamen H, et al. Tap73/Δnp73 Influences Apoptotic Response, Chemosensitivity and Prognosis in Hepatocellular Carcinoma. Cell Death Ddifferent (2005) 12:1564–77. doi: 10.1038/sj.cdd.4401774
123. Levrero M. Viral Hepatitis and Liver Cancer: The Case of Hepatitis C. Oncogene (2006) 25:3834–47. doi: 10.1038/sj.onc.1209562
124. Zaika AI, Slade N, Erster SH, Sansome C, Joseph TW, Pearl M, et al. DeltaNp73, a Dominant-Negative Inhibitor of Wild-Type P53 and TAp73, Is Up-Regulated in Human Tumors. J Exp Med (2002) 196:765–80. doi: 10.1084/jem.20020179
125. Miao C-G, Yang Y-Y, He X, Huang C, Huang Y, Zhang L, et al. Wnt Signaling in Liver Fibrosis: Progress, Challenges and Potential Directions. Biochimie (2013) 95:2326–35. doi: 10.1016/j.biochi.2013.09.003
126. Li L, Zhao G-D, Shi Z, Qi L-L, Zhou L-Y, Fu Z-X. Ras/Raf/MEK/ERK Signaling Pathway and Its Role in the Occurrence and Development of HCC. Oncol Lett (2016) 12:3045–50. doi: 10.3892/ol.2016.5110
127. Delire B, Stärkel P. The Ras/MAPK Pathway and Hepatocarcinoma: Pathogenesis and Therapeutic Implications. Eur J Clin Invest (2015) 45:609–23. doi: 10.1111/eci.12441
128. Ito Y, Sasaki Y, Horimoto M, Wada S, Tanaka Y, Kasahara A, et al. Activation of Mitogen-Activated Protein Kinases/Extracellular Signal-Regulated Kinases in Human Hepatocellular Carcinoma. Hepatology (1998) 27:951–8. doi: 10.1002/hep.510270409
129. Dhillon AS, Hagan S, Rath O, Kolch W. MAP Kinase Signalling Pathways in Cancer. Oncogene (2007) 26:3279–90. doi: 10.1038/sj.onc.1210421
130. Volodko N, Gordon M, Salla M, Abu Ghazaleh H, Baksh S. RASSF Tumor Suppressor Gene Family: Biological Functions and Regulation. FEBS Lett (2014) 588:2671–84. doi: 10.1016/j.febslet.2014.02.041
131. Jin H, Wang X, Ying J, Wong AHY, Cui Y, Srivastava G, et al. Epigenetic Silencing of a Ca(2+)-Regulated Ras GTPase-Activating Protein RASAL Defines a New Mechanism of Ras Activation in Human Cancers. Proc Natl Acad Sci USA (2007) 104:12353–8. doi: 10.1073/pnas.0700153104
132. Calvisi DF, Pinna F, Meloni F, Ladu S, Pellegrino R, Sini M, et al. Dual-Specificity Phosphatase 1 Ubiquitination in Extracellular Signal-Regulated Kinase–Mediated Control of Growth in Human Hepatocellular Carcinoma. Cancer Res (2008) 68:4192. doi: 10.1158/0008-5472.CAN-07-6157
133. Huang Y, Chen XC, Konduri M, Fomina N, Lu J, Jin L, et al. Mechanistic Link Between the Anti-HCV Effect of Interferon Gamma and Control of Viral Replication by a Ras-MAPK Signaling Cascade. Hepatology (2006) 43:81–90. doi: 10.1002/hep.21011
134. Avila MA, Berasain C, Sangro B, Prieto J. New Therapies for Hepatocellular Carcinoma. Oncogene (2006) 25:3866–84. doi: 10.1038/sj.onc.1209550
135. Liu Z, Tian Y, Machida K, Lai MMC, Luo G, Foung SKH, et al. Transient Activation of the PI3K-AKT Pathway by Hepatitis C Virus to Enhance Viral Entry. J Biol Chem (2012) 287:41922–30. doi: 10.1074/jbc.M112.414789
136. Shi Q, Hoffman B, Liu Q. PI3K-Akt Signaling Pathway Upregulates Hepatitis C Virus RNA Translation Through the Activation of SREBPs. Virology (2016) 490:99–108. doi: 10.1016/j.virol.2016.01.012
137. Cheng D, Zhang L, Yang G, Zhao L, Peng F, Tian Y, et al. Hepatitis C Virus NS5A Drives a PTEN-PI3K/Akt Feedback Loop to Support Cell Survival. Liver Int (2015) 35:1682–91. doi: 10.1111/liv.12733
138. Zhai N, Li H, Song H, Yang Y, Cui A, Li T, et al. Hepatitis C Virus Induces MDSCs-Like Monocytes Through TLR2/PI3K/AKT/STAT3 Signaling. PloS One (2017) 12:e0170516–e0170516. doi: 10.1371/journal.pone.0170516
139. Hosui A, Ohkawa K, Ishida H, Sato A, Nakanishi F, Ueda K, et al. Hepatitis C Virus Core Protein Differently Regulates the JAK-STAT Signaling Pathway Under Interleukin-6 and Interferon-γ Stimuli. J Biol Chem (2003) 278:28562–71. doi: 10.1074/jbc.M210485200
140. Stevenson NJ, Bourke NM, Ryan EJ, Binder M, Fanning L, Johnston JA, et al. Hepatitis C Virus Targets the Interferon-α JAK/STAT Pathway by Promoting Proteasomal Degradation in Immune Cells and Hepatocytes. FEBS Lett (2013) 587:1571–8. doi: 10.1016/j.febslet.2013.03.041
141. Mahony R, Gargan S, Roberts KL, Bourke N, Keating SE, Bowie AG, et al. A Novel Anti-Viral Role for STAT3 in IFN-α Signalling Responses. Cell Mol Life Sci (2017) 74:1755–64. doi: 10.1007/s00018-016-2435-3
142. Chen G, Wang H, Xie S, Ma J, Wang G. STAT1 Negatively Regulates Hepatocellular Carcinoma Cell Proliferation. Oncol Rep (2013) 29:2303–10. doi: 10.3892/or.2013.2398
143. Lin W, Choe WH, Hiasa Y, Kamegaya Y, Blackard JT, Schmidt EV, et al. Hepatitis C Virus Expression Suppresses Interferon Signaling by Degrading STAT1. Gastroenterology (2005) 128:1034–41. doi: 10.1053/j.gastro.2005.02.006
144. Chen J, Wang H, Wang J, Huang S, Zhang W. STAT1 Inhibits Human Hepatocellular Carcinoma Cell Growth Through Induction of P53 and Fbxw7. Cancer Cell Int (2015) 15:111–1. doi: 10.1186/s12935-015-0253-6
145. Ding S, Diep J, Feng N, Ren L, Li B, Ooi YS, et al. STAG2 Deficiency Induces Interferon Responses via cGAS-STING Pathway and Restricts Virus Infection. Nat Commun (2018) 9:1485–5. doi: 10.1038/s41467-018-03782-z
146. Lujambio A, Akkari L, Simon J, Grace D, Tschaharganeh DF, Bolden JE, et al. Non-Cell-Autonomous Tumor Suppression by P53. Cell (2013) 153:449–60. doi: 10.1016/j.cell.2013.03.020
147. Brooks CL, Gu W. Dynamics in the P53-Mdm2 Ubiquitination Pathway. Cell Cycle (2004) 7:895–9. doi: 10.4161/cc.3.7.997
148. Hussain SP, Schwank J, Staib F, Wang XW, Harris CC. TP53 Mutations and Hepatocellular Carcinoma: Insights Into the Etiology and Pathogenesis of Liver Cancer. Oncogene (2007) 26:2166–76. doi: 10.1038/sj.onc.1210279
149. Nose H, Imazeki-Ohto M, Ohto-Omata M, Omata M. P53 Gene Mutations and 17p Allelic Deletions in Hepatocellular Carcinoma From Japan. Cancer (1993) 72:355–60. doi: 10.1002/1097-0142(19930715)72:2<355::aid-cncr2820720208>3.0.co;2-w
150. Nishida N, Kudo M. Recent Advancements in Comprehensive Genetic Analyses for Human Hepatocellular Carcinoma. Oncology (2013) 84:93–7. doi: 10.1159/000345897
151. Loguercio C, Cuomo A, Tuccillo C, Gazzerro P, Cioffi M, Molinari AM, et al. Liver P53 Expression in Patients With HCV-Related Chronic Hepatitis. J Viral Hepatitis (2003) 10:266–70. doi: 10.1046/j.1365-2893.2003.00432.x
152. Kao C-F, Chen S-Y, Chen J-Y, Lee Y-HW. Modulation of P53 Transcription Regulatory Activity and Post-Translational Modification by Hepatitis C Virus Core Protein. Oncogene (2004) 23:2472–83. doi: 10.1038/sj.onc.1207368
153. Lan K-H, Sheu M-L, Hwang S-J, Yen S-H, Chen S-Y, Wu J-C, et al. HCV NS5A Interacts With P53 and Inhibits P53-Mediated Apoptosis. Oncogene (2002) 21:4801–11. doi: 10.1038/sj.onc.1205589
154. Dharel N, Kato N, Muroyama R, Taniguchi H, Otsuka M, Wang Y, et al. Potential Contribution of Tumor Suppressor P53 in the Host Defense Against Hepatitis C Virus. Hepatology (2008) 47:1136–49. doi: 10.1002/hep.22176
155. Hsu IC, Metcalf RA, Sun T, Welsh JA, Wang NJ, Harris CC. Mutational Hot Spot in the P53 Gene in Human Hepatocellular Carcinomas. Nature (1991) 350:427–8. doi: 10.1038/350427a0
156. Schickel R, Boyerinas B, Park SM, Peter ME. MicroRNAs: Key Players in the Immune System, Differentiation, Tumorigenesis and Cell Death. Oncogene (2008) 27:5959–74. doi: 10.1038/onc.2008.274
Keywords: hepatitis C virus, hepatocellular carcinoma, DNA methylation, histone modifications, signaling pathways
Citation: Zhao P, Malik S and Xing S (2021) Epigenetic Mechanisms Involved in HCV-Induced Hepatocellular Carcinoma (HCC). Front. Oncol. 11:677926. doi: 10.3389/fonc.2021.677926
Received: 14 April 2021; Accepted: 28 June 2021;
Published: 15 July 2021.
Edited by:
Gong Zhang, Jinan University, ChinaCopyright © 2021 Zhao, Malik and Xing. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shaojun Xing, c2hhb2p1bi14aW5nQHN6dS5lZHUuY24=