 Ying-Cheng Chiang
Ying-Cheng Chiang Po-Han Lin
Po-Han Lin Wen-Fang Cheng
Wen-Fang Cheng
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 14 October 2021
Sec. Gynecological Oncology
Volume 11 - 2021 | https://doi.org/10.3389/fonc.2021.675972
This article is part of the Research Topic Harnessing the DNA damage response in gynecologic malignancies View all 6 articles
Epithelial ovarian cancer (EOC) patients are generally diagnosed at an advanced stage, usually relapse after initial treatments, which include debulking surgery and adjuvant platinum-based chemotherapy, and eventually have poor 5-year survival of less than 50%. In recent years, promising survival benefits from maintenance therapy with poly(ADP-ribose) polymerase (PARP) inhibitor (PARPi) has changed the management of EOC in newly diagnosed and recurrent disease. Identification of BRCA mutations and/or homologous recombination deficiency (HRD) is critical for selecting patients for PARPi treatment. However, the currently available HRD assays are not perfect predictors of the clinical response to PARPis in EOC patients. In this review, we introduce the concept of synthetic lethality, the rationale of using PARPi when HRD is present in tumor cells, the clinical trials of PARPi incorporating the HRD assays for EOC, the current HRD assays, and other HRD assays in development.
Epithelial ovarian cancer (EOC) is a major cause of death in women worldwide (1–5). Due to a lack of specific symptoms and biological markers for early diagnosis, most ovarian cancer patients are diagnosed at an advanced stage in which the disease has spread beyond the pelvis, with an associated 5-year survival of less than 50% (6). The primary standard treatment, debulking surgery, and adjuvant chemotherapy with a platinum and paclitaxel regimen, can achieve good initial response rates, but the majority of ovarian cancer patients eventually relapse (7). Based on the evidence to date, antiangiogenic agents and poly-adenosine diphosphate ribose polymerase (PARP) inhibitors (PARPis) are the most promising targeted therapies for EOC in the past decade (8, 9). Maintenance therapy with PARPis has rewritten the management of EOC in newly diagnosed and recurrent disease (10–15). In the era of precision medicine, it is important to select the appropriate patients to benefit from the targeted therapy. Evaluating homologous recombination deficiency (HRD) in tumor cells as a potential predictor of the response to a PARPi is an important clinical issue. Identification of BRCA mutations and/or HRD status in clinical specimens is critical to selecting EOC patients for PARPi treatment, which has been evaluated in several clinical trials (10–15). The US Food and Drug Administration (FDA) has also approved companion diagnostic tests for PARPi use based on these trials. Cost-effective analysis has shown that PARPi therapy should preferably be reserved for HRD-positive EOC patients until the cost is significantly reduced (16, 17). The measurement of HRD is important for the appropriate use of PARPis in EOC patients, and understanding the various HRD assays will aid clinical practice (18). The HRD and HRD assay terminology should be intrepreted with caution as it might appear to be somewhat confusing (Table 1). In general, HRD is different from HRD test positive. HRD test positive means that the tumors or patients have deficiency of homologous recombination repair pathway, including those with germline and somatic BRCA mutations. HRD test negative actually means that the tumors or patients have intact homologous recombination repair pathway, indicating homologous recombination repair (HRR) proficient. However, the currently available HRD assays are not perfect predictors of the PARPi response because, in previous trials, HRD-positive and HRD-negative patients defined by the current assays both benefited from PARPi. In this review, we introduce the concept of synthetic lethality, the rationale of using PARPis when HRD is present in tumor cells, the clinical trials of PARPis incorporating the HRD assays for EOC, the current HRD assays, and other HRD assays in development (Figure 1).

Table 1 Terminology of homologous recombination.
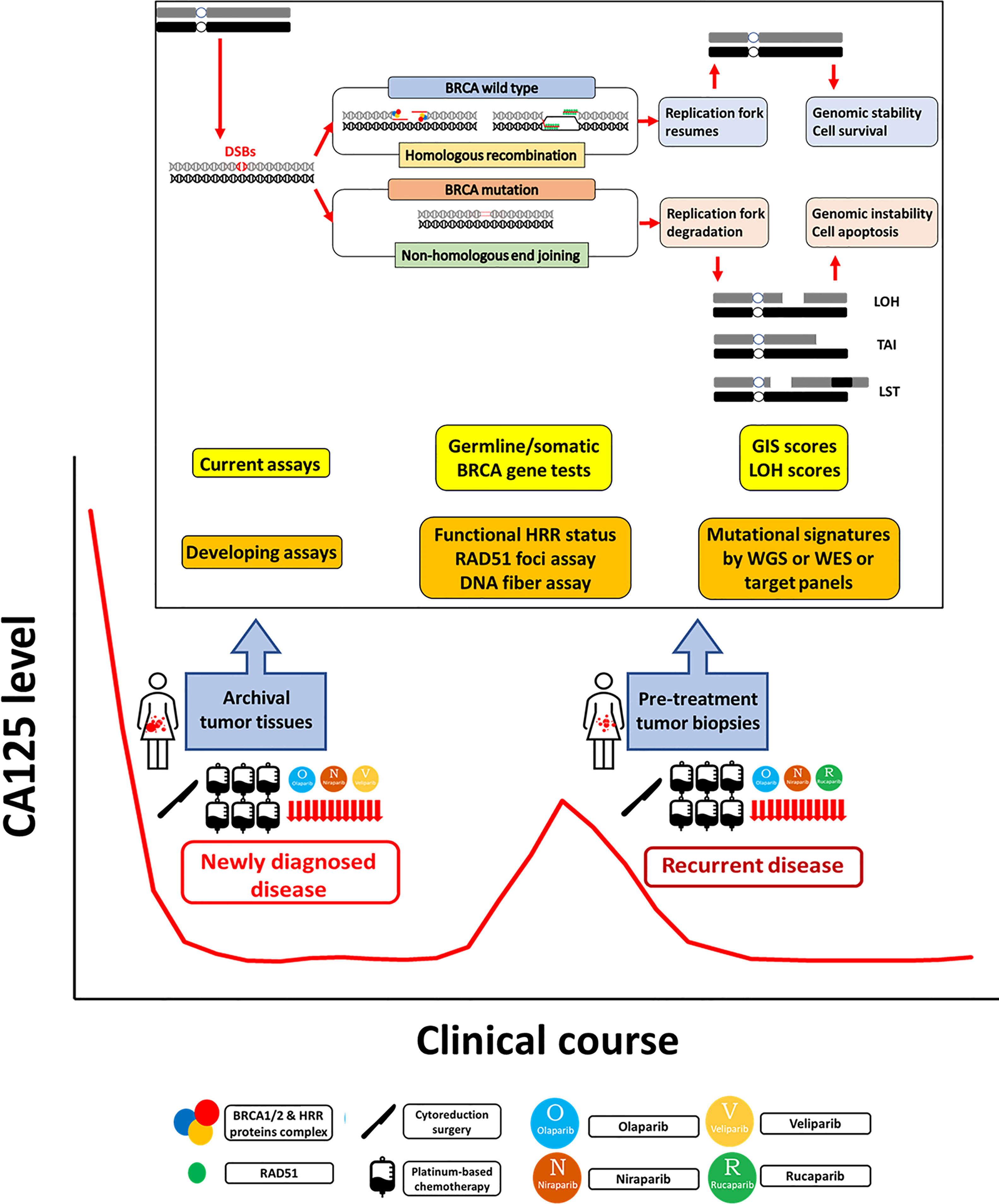
Figure 1 Homologous recombination deficiency assays in epithelial ovarian cancer. Homologous recombination repair (HRR) is an important pathway for repairing DNA double-strand breaks (DSBs). In HRR, DSBs are detected and bound by BRCA1/2 and other HRR protein complexes to localize RAD51 to the exposed single-strand DNA, which then invades the DNA sequence on a homologous sister chromatid to synthesize the new DNA strand. Homologous recombination deficiency (HRD) occurs when HRR is impaired, especially by BRCA mutation. DSBs are repaired by other error-prone repair pathways, including nonhomologous end joining (NHEJ), which may cause point mutations, small insertions or deletions, and even large-scale chromosomal rearrangements in the repaired DNA strands. In the S phase of the cell cycle, the replication forks stop when encountering DNA damage to allow DNA repair before replication continues. After the damage is repaired, the stalled fork resumes replication. If the damage cannot be repaired, the stalled replication fork undergoes fork collapse, comprising fork degradation to cell apoptosis. In general, HRD assays have three main categories: germline or somatic mutations of genes in the HRR pathway, genomic scars or mutational signatures representing the patterns of genomic instability, and checking the functional status of HRR. Germline and somatic BRCA tests should be performed in all patients with newly diagnosed epithelial ovarian cancer (EOC). The current HRD assays based on SNP-based microarray technologies measure loss of heterozygosity (LOH), telomeric allelic imbalance (TAI), and large-scale state transitions (LSTs). The genomic instability score (GIS) combines the information derived from the LOH, TAI, and LST to represent the degree of genomic instability. Functional assays of HRR status include RAD51 foci assays and DNA fiber assays. The RAD51 subnuclear foci is generally present after DNA damage in normal cells, but HRD cells cannot form RAD51 foci. The DNA fiber assay was developed to evaluate the dynamics of the replication fork. Mutational signatures describe distinct patterns of nucleotide transitions with the surrounding nucleotide context from next-generation sequencing (NGS) data for human tumors. The developing comprehensive genomic scar assays include whole genome sequencing (WGS), whole exome sequencing (WES), and target gene panels consisting of variable sizes of selected cancer-susceptible genes. PARPi maintenance therapy benefits newly diagnosed advanced stage and platinum-sensitive recurrent EOC patients. The current genomic scar-based HRD assays can identify additional wild-type BRCA patients who may benefit from PARPi therapy. However, some issues need to be resolved. For tissue retrieval, multistep sampling to obtain archival and pretreatment tumor tissues over the clinical course has the potential to guide the therapy. For precision medicine, it is ideal to develop a comprehensive model to integrate clinical platinum sensitivity, genomic scar/mutational signatures, and functional tests to provide both past evidence of HRD and the current functional ability of HRR.
DNA damage in cells may result from exogenous or endogenous sources, such as oxidative damage, radiation, ultraviolet light, cytotoxic materials, and replication errors, among others (19). Accumulation of unrepaired DNA damage is harmful to cells, leading to genomic instability and, eventually, apoptosis. Several DNA damage response (DDR) pathways are present in cells to fix single-strand breaks (SSBs) or double-strand breaks (DSBs) in the damaged DNA. Dysregulation of the DDR during the cell cycle is associated with carcinogenesis (20). HRR is an important DDR pathway for the repair of DNA DSBs that generally acts in the S and G2 phase of the cell cycle. HRR is an error-proof repair mechanism in which the original sequence at the DSB site is restored by homologous recombination. In HRR, DSBs are detected and bound by the MRE11-RAD50-NSB1 (MRN) complex, which in turn recruits ATM and BRCA1. A small part of the DNA sequence at the DSB site is removed to expose the single-stranded DNA. With localization of BRCA1, BRCA2, and PALB2 at the exposed DNA, the DNA recombinase RAD51 binds to the single-stranded DNA and invades the DNA sequence on a homologous sister chromatid, which is used as a template for synthesizing the new DNA strand, effectively preserving the original genetic information (21, 22). Homologous recombination deficiency (HRD) occurs when HRR is impaired and DSBs are repaired by another, error-prone repair pathway, such as nonhomologous end joining (NHEJ), microhomology-mediated end joining, or single-strand annealing, which may cause point mutations, small insertions or deletions, and even large-scale chromosomal rearrangements in the repaired DNA strands (23–25).
PARP plays multiple roles in repairing both SSBs and DSBs, such as binding to DNA breaks, recruiting repair proteins, and resolving collapsed replication forks (26–30). The small molecule inhibitors of PARP (i.e., PARPis) trap PARP at the sites of SSBs, stalling the replication fork. When the stalling replication fork is encountered by the DNA replication machinery, it creates a DSB. The PARPi-induced DSB undergoes HRR in normal cells. However, in cells with HRD, PARPi-induced DSBs are repaired by the error-prone pathways, leading to genomic instability, apoptosis, and cell death, the so-called synthetic lethality (26–31). The development of PARPis for clinical management is based on the increasing sensitivity of BRCA-mutated cancer cells to PARPis (32, 33). Cancers with HRD are also more sensitive to platinum drugs and topoisomerase inhibitors (26, 34). Although platinum sensitivity has been considered a marker of PARPi sensitivity, the correlation is unsatisfactory (35). In contrast, homologous recombination-proficient (HRP) cells are often resistant to PARPis or platinum drugs.
DDR pathways are a potential target for cancer therapy based on the concept of synthetic lethality, with the aim of specifically killing cancer cells dependent on a compensatory DNA repair pathway for survival (20). In recent decades, several phases II and III clinical trials have demonstrated the survival benefits of PARPi use in the treatment of EOC, especially high-grade serous type, in recurrent and newly diagnosed disease. In these trials, BRCA gene mutation tests and/or HRD assays were investigated using a primary study design or retrospective exploratory analysis to determine the predictive value of these assays in stratifying EOC patients that benefit from PARPi therapy. Table 2 summarizes several important trials of PARPis in the setting of recurrent and newly diagnosed disease.
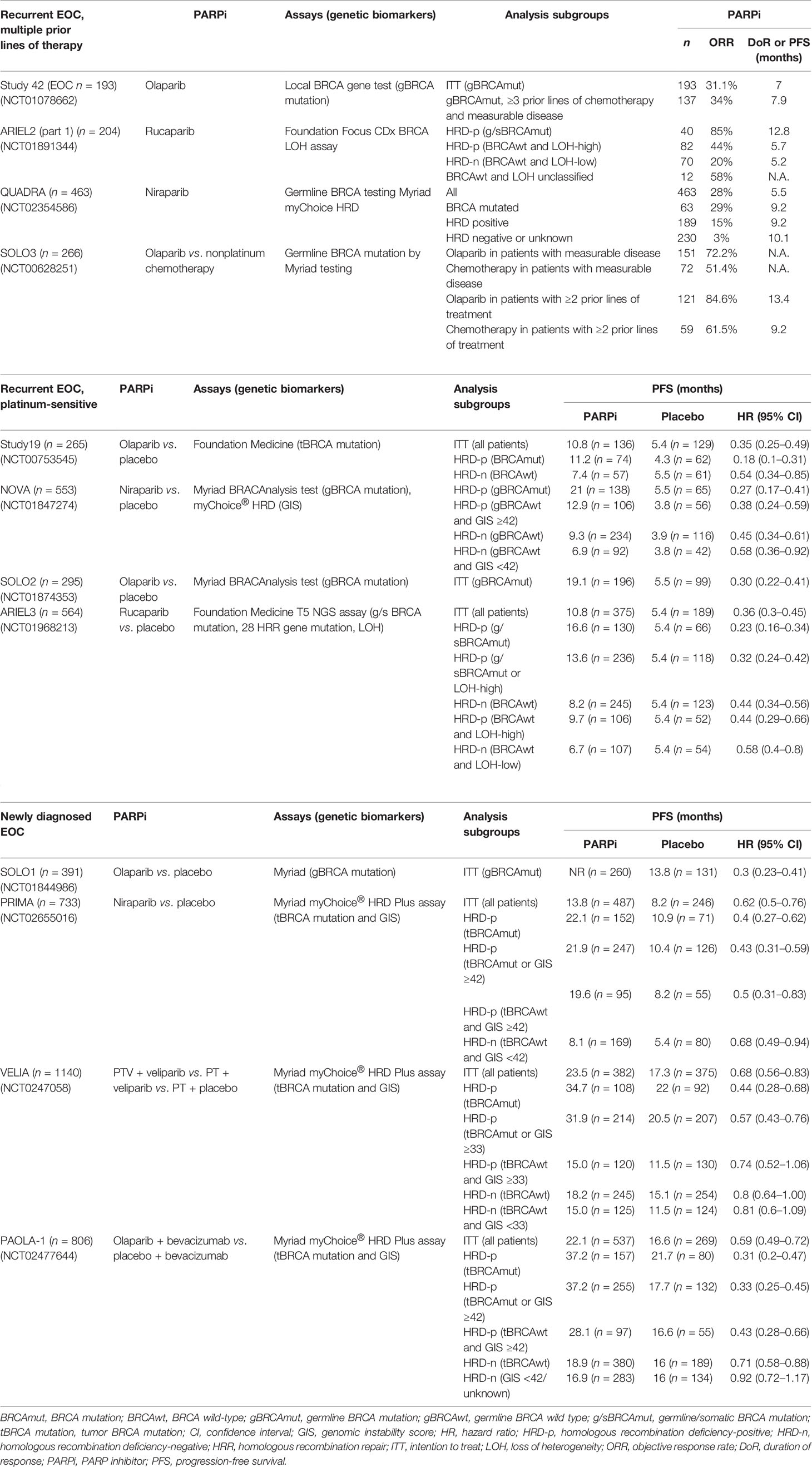
Table 2 Clinical trials of PARPi in recurrent or newly diagnosed epithelial ovarian cancer (EOC).
In the phase II study 42 trial (36, 37), the objective response rate (ORR) to olaparib in patients with germline BRCA1/2 (gBRCA1/2)-mutated advanced ovarian cancer who had received ≥3 prior lines of chemotherapy was 34% (95% CI, 26–42) and median duration of the response (DoR) was 7.9 months (95% CI, 5.6–9.6). The median DoR for platinum-sensitive and platinum-resistant disease was 8.2 months (95% CI, 5.6–13.5) and 8.0 months (95% CI, 4.8–14.8), respectively. These findings suggest that olaparib monotherapy has antitumor activity in patients with gBRCA1/2-mutated advanced ovarian cancer following more than three prior lines of chemotherapy.
In the phase II ARIEL2 (Part 1) trial (38), the status of homologous recombination was determined by the Foundation Focus CDx BRCA loss of heterozygosity (LOH) assay. HRD was defined as more than 14% genomic LOH based on the TCGA dataset. The median progression-free survival (PFS) after rucaparib treatment was 12.8 months in the BRCA-mutated subgroup, 5.7 months in the LOH-high subgroup, and 5.2 months in the LOH-low subgroup. PFS was significantly longer in the BRCA-mutated (HR = 0.27; 95% CI, 0.16–0.44; p < 0.0001) and LOH-high (HR = 0.62; 95% CI, 0.42–0.90; p = 0.011) subgroups compared with the LOH-low subgroup. Thus, ARIEL2 (Part 1) concluded that rucaparib provides significant PFS benefits in platinum-sensitive relapsed EOC patients with BRCA-mutated or BRCA wild-type LOH-high. The findings suggest that tumor LOH can be used to identify platinum-sensitive EOC patients with BRCA wild-type who might benefit from rucaparib. The LOH threshold was adjusted to ≥16% in a post-hoc analysis to improve the selection of patients who will benefit from rucaparib (39).
In the phase II QUADRA trial (40), a clinical benefit of niraparib was observed in heavily pretreated EOC patients (median of four previous lines of therapy), including platinum-sensitive, platinum-resistant, and platinum-refractory patients. In BRCA-mutated patients, niraparib was more active in patients who were platinum sensitive to last line platinum-based chemotherapy (39%) compared with platinum-resistant (33%) and platinum-refractory (19%) patients. In the BRCA wild-type HRD-positive cohort, 20% of platinum-sensitive patients achieved a response, but only 2.4% of platinum-resistant and refractory patients had a response similar to the HRD-negative or unknown cohort (3%) (40–42).
In the phase III SOLO3 trial (43), monotherapy olaparib resulted in significant and clinically relevant improvements in ORR (72.2% vs. 51.4%) and PFS (13.4 vs. 9.2 months) compared with nonplatinum chemotherapy in patients with gBRCA-mutated platinum-sensitive recurrent EOC patients with at least two prior lines of platinum-based chemotherapy.
In the phase II study 19 trial (44), the median PFS was significantly longer in the olaparib group than in the placebo group among the BRCA-mutated cohort (11.2 vs. 4.3 months; HR = 0.18; 95% CI, 0.10–0.31; p < 0.0001) and BRCA wild-type cohort (7.4 vs. 5.5 months; HR = 0.54; 95% CI, 0.34–0.85; p = 0.0075). Study 19 concluded that platinum-sensitive recurrent serous ovarian cancer patients with a BRCA mutation receive the greatest benefits from olaparib maintenance therapy.
In the phase III NOVA trial (13), the status of homologous recombination was determined by the BRCA mutation or myChoice assay with a genomic instability score (GIS) ≥42. The median PFS was significantly longer in the niraparib group than in the placebo group among the gBRCA-mutated cohort (21.0 vs. 5.5 months; HR = 0.27; 95% CI, 0.17–0.41; p < 0.001), the non-gBRCA-mutated HRD-positive cohort (12.9 vs. 3.8 months; HR = 0.38; 95% CI, 0.24–0.59; p < 0.001), and the non-gBRCA-mutated cohort (9.3 vs. 3.9 months; HR = 0.45; 95% CI, 0.34–0.61; p < 0.001). NOVA concluded that the median PFS was significantly longer among the platinum-sensitive recurrent EOC patients receiving niraparib maintenance therapy regardless of the gBRCA mutation or HRD status.
In the phase III SOLO2 trial, the median PFS was significantly longer in the olaparib group than in the placebo group among the BRCA-mutated cohort (19.1 vs. 5.5 months; HR = 0.30; 95% CI, 0.22–0.41; p < 0.0001) (15). The median overall survival was significantly longer in the olaparib group than in the placebo group among the BRCA-mutated cohort (51.7 vs. 38.8 months; HR = 0.74; 95% CI, 0.54–1.00; p = 0.054) (45). SOLO2 concluded that olaparib maintenance therapy provides significant PFS benefits and a median overall survival benefit of 12.9 months compared with placebo in platinum-sensitive recurrent EOC patients with BRCA mutation.
In the phase III ARIEL 3 trial (14), the status of homologous recombination was determined by the Foundation Focus CDx BRCA LOH assay with the threshold ≥16% genomic LOH. The median PFS was significantly longer in the rucaparib group than in the placebo group among the BRCA-mutated cohort (16.6 vs. 5.4 months; HR = 0.23; 95% CI, 0.16–0.34; p < 0.0001), the HRD cohort (13.6 vs. 5.4 months; HR = 0.32; 95% CI, 0.24–0.42; p < 0.0001), and the intention-to-treat population (10.8 vs. 5.4 months; HR = 0.36; 95% CI, 0.30–0.45; p < 0.0001). The benefit was greatest in BRCA-mutated patients, followed by the HRD-positive (defined as BRCA mutated or LOH-high), HRD-positive (defined as BRCA wild-type and LOH-high), and HRD-negative (defined as BRCA wild-type and LOH-low) cohorts. ARIEL3 concluded that maintenance rucaparib significantly improves PFS in platinum-sensitive relapsed EOC patients following a complete or partial response to second-line or later platinum-based chemotherapy.
In the SOLO1 trial (12), newly diagnosed advanced stage EOC patients with germline or somatic BRCA mutations receiving olaparib maintenance therapy had a significant improvement in PFS than the placebo group (median not reached vs. 13.8 months; HR = 0.30; 95% CI, 0.23–0.41; p < 0.001). In a 5-year follow-up, the median PFS was significantly longer in patients who received olaparib than those who received placebo (56.0 vs. 13.8 months; HR = 0.33; 95% CI, 0.25–0.43) (46).
In the PRIMA trial (11), the status of homologous recombination was determined by the BRCA mutation or myChoice assay with GIS ≥42. The median PFS was significantly longer in the niraparib group than in the placebo group among the HRD cohort (21.9 vs. 10.4 months; HR = 0.43; 95% CI, 0.31–0.59; p < 0.001) and the intention-to-treat population (13.8 vs. 8.2 months; HR = 0.62; 95% CI, 0.50–0.76; p < 0.001). Analyses of GIS in the BRCA wild-type cohort was a preplanned exploratory analysis. The results showed a benefit in all BRCA wild-type patients regardless of the GIS, though it was higher in the GIS-high subgroup compared with the GIS-low subgroup (HR = 0.5; 95% CI, 0.31–0.83 vs. HR = 0.68; 95% CI, 0.49–0.94). PRIMA concluded that niraparib maintenance therapy significantly prolonged the PFS in newly diagnosed advanced stage EOC patients who responded to platinum after primary treatment regardless of homologous recombination status (11).
In the VELIA trial (10), the status of homologous recombination was determined by the BRCA mutation or myChoice assay with GIS ≥33. The median PFS was significantly longer in the veliparib-throughout group than in the control group among the BRCA-mutated cohort (34.7 vs. 22.0 months; HR = 0.44; 95% CI, 0.28–0.68; p < 0.001), the HRD-positive cohort (31.9 vs. 20.5 months; HR = 0.57; 95% CI, 0.43–0.76; p < 0.001), and the overall population (23.5 vs. 17.3 months; HR = 0.68; 95% CI, 0.56–0.83; p < 0.001). VELIA concluded that a regimen of carboplatin, paclitaxel, and veliparib induction therapy followed by veliparib maintenance therapy leads to significantly longer PFS than carboplatin plus paclitaxel induction therapy alone. The PFS benefit was less pronounced in VELIA than in the other frontline studies, possibly because the HRD cohort contained more false positives, with a higher percentage of patients classified as having HRD (55%) despite a lower percentage of BRCA-mutated patients (26%).
In the PAOLA-1 trial (47), the status of homologous recombination was determined by tumor BRCA mutations or myChoice assay with GIS ≥42. The median PFS was significantly longer in the olaparib plus bevacizumab group than in the placebo plus bevacizumab group among the overall population (22.1 vs. 16.6 months; HR = 0.59; 95% CI, 0.49–0.72; p < 0.001), the BRCA-mutated cohort (37.2 vs. 21.7 months; HR = 0.31; 95% CI, 0.20–0.47), the BRCA wild-type cohort (18.9 vs. 16.0 months; HR = 0.71; 95% CI, 0.58–0.88), the BRCA-mutated HRD-positive cohort (37.2 vs. 17.7 months; HR = 0.33; 95% CI, 0.25–0.45), and the BRCA wild-type HRD-positive cohort (28.1 vs. 16.6 months; HR = 0.43; 95% CI, 0.28–0.66). PAOLA-1 concluded that maintenance olaparib provides a significant PFS benefit in advanced EOC patients receiving first-line standard chemotherapy with bevacizumab, especially in patients with HRD-positive tumors, including those without a BRCA mutation. In the HRP cohort, the addition of olaparib to bevacizumab did not improve the PFS (16.6 vs. 16.2 months; HR = 0.43; 95% CI, 0.75–1.35). The findings indicate that the GIS has the potential to identify HRD-negative EOC patients who do not derive a benefit from olaparib in combination with bevacizumab (15).
Based on these trials, PARPis were approved by the US FDA and European Medicines Agency (EMA) in the management of EOC patients: (1) salvage monotherapy in BRCA mutant or HRD-positive disease after multiple prior lines of therapy, (2) second-line maintenance therapy in recurrent platinum-sensitive disease regardless of BRCA mutation or HRD status, and (3) first-line maintenance therapy for newly diagnosed advanced stage platinum-sensitive disease with/without BRCA mutation or HRD-positive status (Table 3). The homologous recombination status of EOC patients is critical to achieving survival benefits from PARPi treatment. Therefore, how to define the homologous recombination status of EOC patients using HRD assays is an important issue for clinicians.
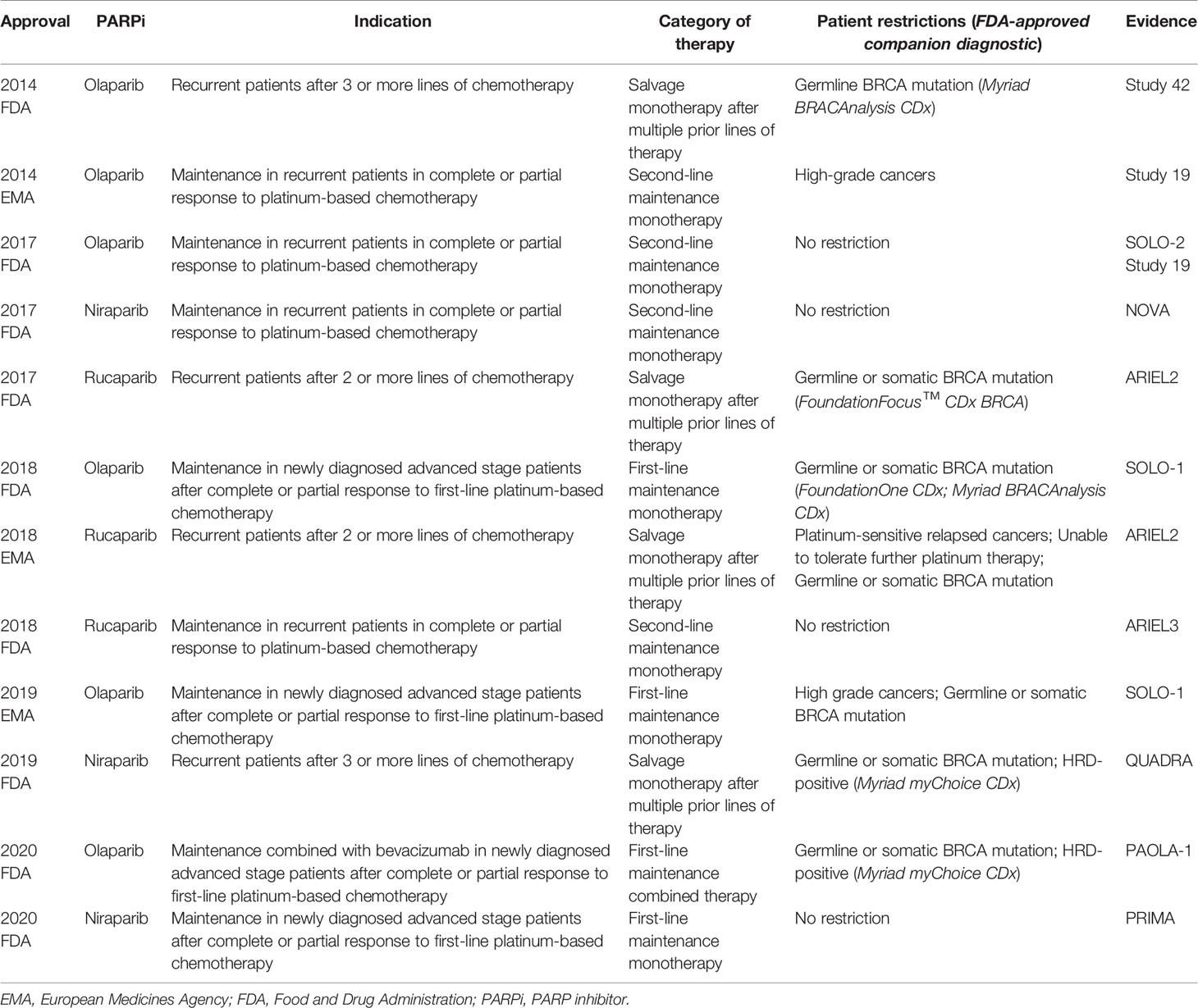
Table 3 FDA and EMA approval of PARP inhibitors in epithelial ovarian cancer.
In general, HRD assays are of three main categories: germline or somatic mutations in genes in the HRR pathway, genomic scars or mutational signatures representing patterns of genomic instability, and checking the function of RAD51 localization to sites of DNA damage (18, 21, 34, 48–50).
BRCA1/2 mutation tests are well known in the clinical management of EOC patients. The gBRCA tests with genetic counseling should be performed near the time of diagnosis for all patients with newly diagnosed EOC (51–53). Both germline and tumor BRCA tests can identify EOC patients who would benefit from PARPi maintenance therapy. Notably, a somatic test cannot substitute for a germline test because 5% of gBRCA-mutated patients test negative for tumor BRCA. Approximately 11%–18% of patients have a gBRCA mutation. The gBRCA test not only informs the patient but can also identify family members at risk of possible associated malignancies, which will be helpful for early detection or prevention (51, 52, 54). In patients with negative gBRCA tests, somatic BRCA tests can identify another 6%–7% of patients with somatic BRCA mutations (34, 48, 55–57).
The majority of clinical trials have demonstrated that advanced stage, mainly high-grade serous type, EOC patients with gBRCA or somatic BRCA mutations derive the greatest benefit from PARPi maintenance therapy in primary first-line management or in platinum-sensitive recurrent disease (10–15, 44, 47). The tumor BRCA status, including germline and somatic BRCA mutations, was generally used as a response predictor in these trials (14, 44, 47). A few studies presented data on PARPi treatments only for EOC patients with somatic BRCA mutations. The PFS benefit of PARPi compared with placebo in EOC patients with somatic BRCA mutations and platinum-sensitive recurrent disease was similar to the benefit in those with gBRCA mutations in study 19 (olaparib, HR = 0.23 vs. 0.17, respectively) (58), NOVA (niraparib, HR = 0.27 vs. 0.27, respectively) (13), and ARIEL2 Part 1 (rucaparib, response rate 74% vs. 85%, respectively) (38). For newly diagnosed advanced disease, a similar trend was noted for patients with somatic and gBRCA mutations (veliparib, HR = 0.35 vs. 0.5, respectively) in the VELIA trial (10). However, patients with BRCA wild-type had a smaller, but still significant, benefit from PARPi use in primary first-line management or in platinum-sensitive recurrent disease (13–15). This indicated a poor negative predictive value of the BRCA mutation status as a predictor of the PARPi response.
In addition to BRCA1/2 genes, non-BRCA genes involved in the HRR pathway include ATM, BRIP1, NBN, PALB2, RAD51B, RAD51C, and RAD51D (34, 56, 59–62). Patients with germline or somatic mutations in non-BRCA HRR genes also derive a survival benefit from DNA repair inhibitors (34, 63–66). However, the prevalence of non-BRCA HRR gene mutations is quite low, making it difficult to determine the association of individual genes with clinical outcomes, and all non-BRCA HRR gene mutations are usually pooled together for interpretation in these studies. For example, in study 19, 21 platinum-sensitive recurrent patients with non-BRCA somatic mutations, including BRIP1, CDK12, RAD54L, and RAD51B, derived a similar PFS benefit as those with BRCA mutations (HR = 0.21 and 0.18, respectively) (67). In ARIEL2, non-BRCA HRR gene mutations were noted in 20 patients (10%), including ATM, BRIP1, CHEK2, FANCA, FANCI, FANCM, NBN, RAD51B, RAD51C, and RAD54L. The sensitivity of non-BRCA HR gene mutations in discriminating the rucaparib response was only 11%. Furthermore, how many non-BRCA HRR genes should be included in the testing list is still unclear. A major challenge for HRR gene testing is the annotation of variants of uncertain significance (VUS) (68–70). In the broader gene panel tests, the functional and clinical impacts of most individual mutations in the genomic loci have not been well characterized. In many cancers, the somatic VUS are more numerous and diverse than germline VUS (71). There is a difference in the reporting rate of BRCA VUS (3%–50%), the protocols for detection, and the strategies for management between individual laboratories (72). Currently, the evidence is not sufficient to determine which individual or panel of non-BRCA HRR genes could be used to predict the PARPi response.
Defects in the HRR pathway generate genetic variations, chromosome structural abnormalities, and genomic instability. These so-called genomic scars or mutational signatures are permanent even if the function of the HRR pathway is restored, which represents a record of DNA damage repair through different pathways in response to harmful exposure in cells (26). Genetic variations generally consist of single nucleotide variants, small insertions and deletions (indels), copy number variations (CNVs), and large chromosomal rearrangements. Single nucleotide variants (SNVs) are base substitutions that may lead to deleterious missense or nonsense mutations. Indels less than 1,000 kbp in length may cause frameshift mutations. CNVs with DNA insertions or deletions more than 1 kb in size may lead to increased expression of oncogenes or decreased expression of tumor suppressor genes. Large chromosomal rearrangements, such as translocations, are a common cause of LOH. The current methods of detecting “genomic scars” use SNP-based microarray technologies to measure somatic CNV, including LOH, telomeric allelic imbalance (TAI), and large-scale state transitions (LSTs) (73). LOH indicates permanent loss of an allele copy in DNA of more than 15 Mb, which renders the tumor cell homozygous at that locus (74). TAI refers to different allele copy numbers of more than 11 Mb in the subtelomere but not crossing the centromere (75, 76). An LST indicates chromosomal breaks between adjacent genomic regions of more than 10 Mb as a result of translocations, copy gains, and copy losses (77). The degree of HRD highly correlates with the LOH, TAI, or number of LSTs in the chromosomes. The GIS combines the information derived from LOH, TAI, and LSTs to represent the degree of genomic instability (76).
The myChoice CDx (Myriad Genetics) and Foundation Focus CDx BRCA LOH (Foundation Medicine) are currently the most common commercial assays (78, 79). The myChoice CDx (Myriad Genetics) includes a tumor BRCA mutation test and GIS. The Foundation Focus CDx BRCA LOH (Foundation Medicine) includes a tumor BRCA mutation test and genomic LOH. Both assays use next-generation sequencing (NGS) platforms to identify SNVs, indels, and large rearrangements in genes of tumor tissue (42, 78), and the BRCA gene mutation results are concordant in these two assays (67). In the myChoice CDx assay, GIS ≥42 is regarded as “HRD positive” (78). Briefly, tumor tissue is sequenced with a panel of single-nucleotide polymorphisms (SNPs) to generate allele-specific copy number profiles to calculate the sum of LOH, TAI, and LST, resulting in a continuous score from 0 to 100, the GIS. A score of 42 is the 5th percentile in a cohort of ovarian and breast cancers in which HRD was defined as biallelic loss of BRCA1/2 (78). In the Foundation Focus CDx assay, the percent genomic LOH, calculated as the fraction of genome regions with LOH determined by sequencing SNPs in tumor DNA, is used to measure HRD status. The LOH cutoff value is determined independent of tumor BRCA status, and 14% genomic LOH was considered to indicate HRD-positive cells in the ARIEL2 trial (38). In the subsequent ARIEL3, this was adjusted to 16% genomic LOH as the threshold (14). However, the compatibility between HRD defined by the GIS and percent genomic LOH needs to be determined.
FDA has approved companion diagnostic HRD assays for salvage monotherapy in recurrent disease after multiple prior lines of therapy and in first-line maintenance therapy. In the first-line maintenance setting, ESMO suggests that germline and somatic BRCA mutation tests are routinely recommended to identify EOC patients who should receive PARPi therapy. A validated HRD assay is reasonable to stratify BRCA wild-type EOC patients who would benefit from PARPi therapy (21) although PARPis were approved as maintenance therapy for all platinum-sensitive recurrent EOC patients regardless of BRCA mutation or HRD status. The PFS benefits of PARPi maintenance therapy decrease incrementally from BRCA-mutated to HRD-positive to HRD-negative cohorts according to the current HRD assays. It may be helpful for the platinum-sensitive relapsed EOC patients to choose bevacizumab or PARPi as maintenance therapy. ESMO suggests that BRCA mutation tests and validated genomic scar-based HRD assays are reasonable for predicting the benefits of PARPi use in platinum-sensitive recurrent disease (21). An unmet need is determining whether the HRD assays can consistently identify a subgroup of patients who do not benefit from PARPi therapy. The HRD-negative (BRCA wild-type and GIS <42) subgroup constituted approximately 50% of all participants in these trials (10, 11, 47). All of these trials showed a benefit in the intention-to-treat population, which is not sufficient to determine whether the HRD-negative EOC patients benefitted from maintenance PARPi therapy. None of these trials were prospectively designed to stratify the patients into subgroups by HRD assay, and the clinical benefit of PARPi maintenance therapy in HRD-related cohorts was obtained from exploratory analyses.
The cost-effective analysis in platinum-sensitive recurrent EOC showed that maintenance PARPi therapy is the preferred strategy over a treat-all strategy for patients with BRCA mutations or in HRD-positive patients (16). In the model, mean costs and progression-free quality-adjusted life years were $827 and 3.4 months for observation, $46,157 and 5.7 months for a BRCA-only strategy, $109,368 and 8.5 months for a gBRCA and HRD-positive strategy, and $169,127 and 8.8 months for a treat-all strategy. Another cost-effective analysis of “PARPi-for-all” compared with an HRD assay-directed strategy using the models established from the PRIMA, VELIA, and PAOLA-1 trials showed that front-line PARPi maintenance therapy should be reserved for HRD-positive EOC patients until the cost of PARPi is significantly reduced (17). In the analysis, the mean cost per patient for the “PARPi-for-all” strategy was $166,269, $286,715, and $366,506 for the PRIMA, VELIA, and PAOLA-1 trials, respectively. For the HRD assay-directed strategy, the mean cost per patient was $98,188, $167,334, and $260,671 for the PRIMA, VELIA, and PAOLA-1 trials, respectively. However, to the best of our knowledge, a cost-effective analysis between HRD assays is currently lacking for EOC.
The current HRD assays have some limitations (Table 4). Multiple causes of discordance between clinical PARPi responses and HRD assay results are possible, including timing, quality and quantity of samples, tumor heterogeneity, reversion mutations, and non-HRD-related PARPi resistance mechanisms. The assays define HRD based on genomic scars, but these assays are unable to represent dynamic changes in the homologous recombination function of the cells. For example, in ARIEL2 (38), pretreatment biopsies and archival tumor biopsies (median time lapse: 2.7 years) were both used for genomic LOH assays in 117 patients. Approximately 34% of patients with LOH-low in the initial archival tissue biopsy turned out to be LOH-high in the pretreatment biopsy, and 30% of these patients had partial responses to rucaparib, which is similar to the LOH-high cohort. Fresh tumor samples may differ from archival tissue samples due to tumor heterogeneity and clonal evolution, and assays using fresh samples more accurately reflect the current status of the tumor (80, 81).

Table 4 Limitations of the current HRD assays.
A BRCA or HRR mutation may form a genomic scar detected by HRD assays, but the tumor could restore the HRR function through reversion or secondary mutations, causing resistance to chemotherapy or PARPis over time (82–84). Reversion or secondary somatic mutations have been identified in BRCA1/2, RAD51C, RAD51D, and PALB2 in platinum-resistant EOC tumors (85–87). BRCA reversion mutations have been identified in 10%–30% of tumors after platinum exposure, which could lead to platinum or PARP resistance (85, 88). The HRD assays are unable to account for reversion mutations because the original gene mutations have resulted in a permanent, irreversible genomic scar. In platinum-resistant EOC patients, the current HRD assays may be inadequate for predicting PARPi responses, as in the QUADRA trial. However, the reversion mutations may be found only in fresh tissue samples and are difficult to detected in germline or archival samples. For tumor suppressor genes, retention of one wild-type allele reduces the effect of a somatic or gBRCA1/2 mutation (89); however, current HRD assays are unable to report the status of both alleles. In addition, there are other mechanisms of treatment resistance that cannot be detected by HRD assays, including upregulation of membrane drug transporters, modulation of DSB end resection, protected stalled replication forks, and stabilization of the BRCA1 C-terminal domains (90–94). Intratumor heterogeneity, clonal adaptation, and cancer evolution are challenges for precision medicine (95). The current HRD assays were developed to stratify EOC patients by PARPi benefit and it is uncertain that these assays could be utilized for other inhibitors targeting other genes or molecules involved in the DNA repair pathway. The genomic scar-based HRD assays cannot provide a real-time dynamic evaluation of the current status of the tumor, which may limit the predictive ability, especially when the testing samples are obtained from archival tissue.
Several HRD assays are in development and are summarized in Table 5. Tumor NGS assays by targeted gene panel are clinically available mutation tests with panels consisting of variable sizes of select cancer-susceptible genes to identify SNVs, small indels, CNVs, and large chromosomal rearrangements by NGS techniques (96). Larger panels provide more clinically useful information but increase the VUS rates (97). However, mutations or variants identified on tumor NGS assays may not correlate with the clinical response (98). Tumor NGS assays using whole genome sequencing (WGS) or whole exome sequencing (WES) are also in development to assess the tumor HRD status. WGS identifies the whole genome, whereas WES identifies all protein coding regions of tumor DNA, which comprise 1%–2% of the genome but approximately 85% of all disease-causing mutations (99, 100). The raw data generated by WGS is, on average, 30 times larger than the raw data generated by WES; thus, WGS is more time consuming and expensive (101, 102).
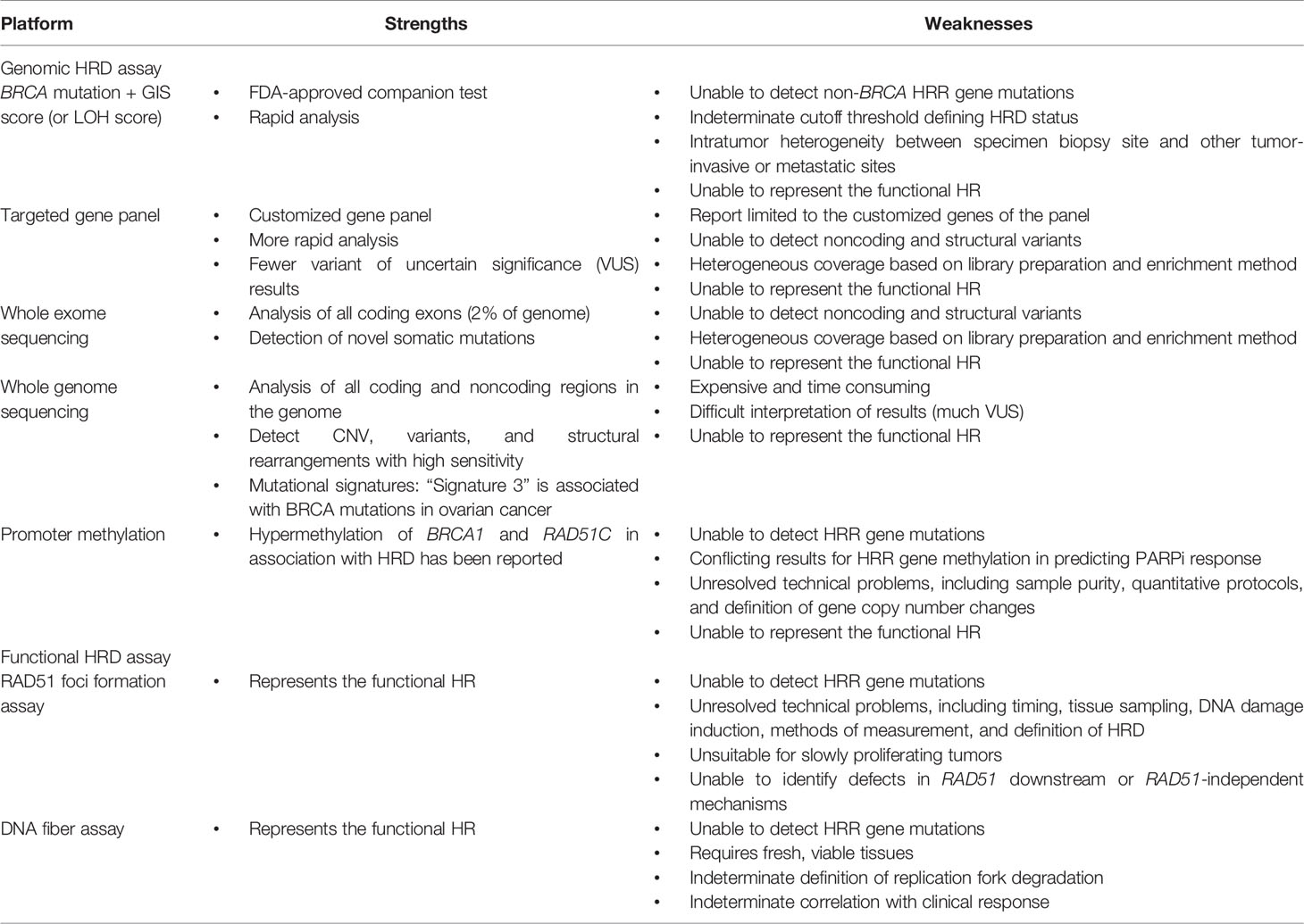
Table 5 Main strengths and weaknesses of the HRD assays.
Mutational signatures describe distinct patterns of nucleotide transitions with the context of the surrounding nucleotides from the WGS data of human tumors (71). Different DNA damage processes, such as aging, UV light, radiation, cytotoxic drugs, and DNA repair pathway defects, have been correlated with different patterns of mutations, which can generate characteristic mutational signatures using computational methodologies (103, 104). “Signature 3” is associated with BRCA1/2 mutations and BRCA1 promoter methylation in breast, ovarian, pancreatic, and stomach cancers (71, 105, 106). This is a pattern of single-base substitutions in which mutations are distributed among the six possible types (e.g., C > A, C > G, C > T, T > A, T > C, T > G) and the surrounding sequence contexts (bases on either side of the mutated base). Signature 3 has been proposed as a biomarker of HRD, but some challenges make it unsuitable for guiding PARPi therapy in EOC (50, 107). The majority of high-grade serous EOCs contain some contribution from signature 3, and it probably lacks specificity as a HRD biomarker. In addition, signature 3 remains a static representation of HRD on the genome rather than the functional state. HRDetect is a WGS-based assay based on aggregating six HRD-associated signatures into a single score, including microhomology-mediated deletions, base substitution signature 3, rearrangement signature 3, rearrangement signature 5, HRD index, and base substitution signature 8 (105). HRDetect has been shown to predict BRCA deficiency with a sensitivity of 98.7% in 560 breast cancers (including the training cohort), 86% in a validation breast cancer cohort (n = 80), and nearly 100% in validation ovarian cancer (n = 73) and pancreatic cancer (n = 96) cohorts. However, the ability of HRDetect to predict the PARPi response in EOC has not been confirmed (108–110). The major limitations of HRDetect include requiring fresh frozen tissue with >50% tumor cells, higher expense, and longer time to carry out the assay (111). Another limitation is that it represents the historical existence of a genomic scar but not the current HRR status.
Promoter methylation results in decreased gene transcription, and an association of gene promoter methylation in BRCA1 and RAD51C with HRD had been reported. However, clinical studies of HRR gene methylation have reported conflicting results, and its role in predicting PARPi responses in EOC patients is controversial (38, 56, 112–115). For example, BRCA1 promoter hypermethylation was detected in 13% of tumors and RAD51C promoter hypermethylation in 2% of tumors in ARIEL2. Genomic LOH (test sensitivity 78%) performed better than non-BRCA HRR gene mutations (sensitivity 11%, p < 0.001) or promotor hypermethylation (sensitivity 48%, p < 0.02) in predicting a response to rucaparib. Some technical problems still need to be resolved, including sample purity, quantitative methylation assays, and gene copy number changes (112, 113, 116–118)
In order to define HRD in real time, the tests should theoretically check an important downstream factor integrating multiple upstream HRR pathways to provide a dynamic evaluation of the actual HRD status (119). RAD51 encodes a recombinase that plays an important role in HRR and replication fork processing (119, 120). In HRR of DSBs, RAD51 units are localized to the site of the DNA break with the help of numerous mediator protein complexes, including BRCA1 and BRCA2, to facilitate DNA strand invasion into the sister chromatid. The RAD51 subnuclear foci is generally present after DNA damage in normal cells, but cells with HRD cannot form RAD51 foci. Therefore, measurement of RAD51 subnuclear foci is a functional assay for detecting HRD in tumor samples (121, 122).
Studies have demonstrated that the absence or decrease of subnuclear RAD51 foci correlates with BRCA mutations, the response to chemotherapy or PARPi treatment, and clinical prognosis (123–129). Immunofluorescence has been used for evaluation of RAD51 foci formation in live cells derived from tumors, ascites, or patient-derived xenograft models in which DNA damage was induced by irradiation, platinum, or PARPis (126, 128, 130, 131). Mukhopadhyay et al. reported the first functional assay from live cells/tumor tissue from a prospective series of consecutive EOC patients. The functional assay for HRD using RAD51 foci formation correctly predicted that approximately 50% of EOC are HRD ahead of the TCGA publication in 2011 (130); the series was subsequently expanded by the same group to show its validity in a various live cell storage and sample transport conditions (132). The homologous recombination Repair CAPacity (RECAP) assay developed by Meijer et al. measures RAD51 foci in proliferating cells from fresh breast cancer tissue after ex vivo radiation. They found a strong correlation between RECAP-defined HRD and BRCA mutations. Thus, the RECAP assay could identify HRP tumors that developed BRCA reversion mutations in patients with gBRCA mutations (133). Immunohistochemistry of RAD51 foci was demonstrated in formalin-fixed paraffin-embedded (FFPE) samples of breast cancer, and positive RAD51 foci correlated with resistance to platinum or PARPi (110, 124, 134). Waks et al. found that the positive RAD51 foci were also associated with BRCA reversion mutations, through which the HRR pathway was restored and correlated with platinum resistance (134).
Hill et al. developed another method of checking RAD51 expression and replication fork stability in organoid cultures (124). The organoids derived from ascites or EOC tumors were checked for the numbers of cells with RAD51 foci after irradiation. They also stained γH2AX and geminin to mark DNA damage and the S/G2 phase of the cell cycle. The HRD score was generally calculated as the number of RAD51-positive cells over the number of cells in S/G2 (marked by geminin or cyclin A2). Samples were defined as HR-low (<10%–20% RAD51-positive cells), HR-intermediate (10%–20% to 35%–50% RAD51-positive cells), or HR-proficient (>35%–50% RAD51-positive cells). However, the correlation of the clinical response with the organoid RAD51 foci by immunohistochemistry needs to be determined (135).
The DNA fiber assay was developed to evaluate the dynamics of the replication fork (136–139). The chromosome unzips the intertwined DNA strands to make copies during S phase of the cell cycle, forming the replication fork. The replication fork stops when encountering DNA damage to allow DNA repair before replication continues. Stalled replication forks require numerous proteins, such as BRCA1, BRCA2, and RAD51, to protect from degradation by nucleases. After damage is repaired, the stalled fork resumes its progress. If the damage cannot be repaired to resume DNA synthesis, the stalled replication fork undergoes degradation, with the fork collapse leading to cell apoptosis (140–143). Extensive replication fork degradation is associated with chemosensitivity in BRCA-mutated tumors (91, 119). Briefly, the DNA fiber assay evaluates the dynamics of replication forks by incorporating DNA with two labeled thymidine analogs, iododeoxyuridine (IdU) and chlorodeoxyuridine (CldU), which can be visualized by an immunofluorescence-based approach (139, 144, 145). Degradation of the stalled forks leads to shortening of the thymidine-labeled tract (124, 144, 145), and it is commonly detected in the absence of key factors, such as BRCA1 or BRCA2 (91, 146–148). A preclinical study showed that DNA fiber assays correlate better with platinum sensitivity than PARPi sensitivity (124). Further studies are needed to verify the feasibility of DNA fiber assays in predicting PARPi sensitivity.
The challenges with the RAD51 foci assay include exam timing (pre- or posttreatment), tissue resources (primary tumor, ascites, or FFPE), methods of inducing DNA damage (radiation, platinum, or PARPi), methods of detection (immunofluorescence or immunohistochemistry), measurement of RAD51 foci (numbers or percentages), intratumor or interobserver variability, and the definition of HRD in correlation with the clinical response (125, 126, 149). In addition, RAD51 foci assays are not suitable for slowly proliferating tumors and are unable to identify defects in the HRR pathway downstream of RAD51 or RAD51-independent mechanisms (150–155). The challenges of DNA fiber assays include requiring fresh viable tissues, the definition of replication fork degradation, and the correlation with clinical responses (135). The RAD51 foci assay and DNA fiber assay are both potential functional HRD assays, but when the commercial assays will be available is unclear.
PARPi maintenance therapy has made great progress in the management of EOC, with the successful translation of the concept of “synthetic lethality” into clinical practice. PARPis are expensive, and the cost-effective analysis showed that PARPi therapy for BRCA-mutated or HRD-positive EOC patients is a preferable strategy over a treat-all strategy. Although FDA-approved companion HRD assays are available for PARPi use, an important unmet problem is that the current HRD assays are unable to consistently identify a subgroup of patients that does not benefit from PARPis, which will lead to increasing medical expenses and possible resistance to PARPi therapy. There are still some issues that need to be resolved, including the quality of tissue samples, intratumor heterogeneity, the cutoff threshold for the definition of HRD, functional HRD status, reversion or secondary mutations of HRR genes, discordance with the clinical response, and cancer evolution. Ongoing development of new comprehensive HRD assays, such as WES/WGS-based assays or functional RAD51 foci/DNA fiber assays, will improve our ability to select appropriate EOC patients who benefit from PARPi. The comprehensive genomic scar assays based on WGS or WES could provide the HRR gene mutations, mutational signatures, and reversion mutations simultaneously. For tissue retrieval, multistep sampling to obtain archival and pretreatment tumor tissues has the potential to disclose the cancer evolutionary changes over the clinical course to guide the therapy. For precision medicine, it is necessary to develop a comprehensive model integrating the clinical factors, genomic mutational signatures, and functional tests to provide both past evidence of HRD and the current functional ability of HRR. Prospectively designed head-to-head comparison between the various HRD assays incorporating into clinical trials of PARPi monotherapy or in combination with traditional chemotherapy, antiangiogenetic agents, checkpoint inhibitors, or other DNA repair inhibitors is important to establish the optimal clinical implications of HRD assays.
Conception and study design: Y-CC and W-FC. Writing and revision of the manuscript: Y-CC, P-HL, and W-FC. Study supervision: W-FC. All authors contributed to the article and approved the submitted version.
This work was supported by research grants from National Taiwan University Hospital (NTUH. 105-N02, NTUH. UN105-059, and NTUH. 108-S4230).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The authors thank the 7th Core Laboratory Facility of the Department of Medical Research of National Taiwan University Hospital and A1 Laboratory of the National Taiwan University Hospital for supporting this work.
1. Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2020. CA Cancer J Clin (2020) 70(1):7–30. doi: 10.3322/caac.21590
2. Torre LA, Trabert B, DeSantis CE, Miller KD, Samimi G, Runowicz CD, et al. Ovarian Cancer Statistics, 2018. CA Cancer J Clin (2018) 68(4):284–96. doi: 10.3322/caac.21456
3. Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2019. CA Cancer J Clin (2019) 69(1):7–34. doi: 10.3322/caac.21551
4. Allemani C, Matsuda T, Di Carlo V, Harewood R, Matz M, Niksic M, et al. Global Surveillance of Trends in Cancer Survival 2000-14 (CONCORD-3): Analysis of Individual Records for 37 513 025 Patients Diagnosed With One of 18 Cancers From 322 Population-Based Registries in 71 Countries. Lancet (2018) 391(10125):1023–75. doi: 10.1016/S0140-6736(17)33326-3
5. Chiang YC, Chen CA, Chiang CJ, Hsu TH, Lin MC, You SL, et al. Trends in Incidence and Survival Outcome of Epithelial Ovarian Cancer: 30-Year National Population-Based Registry in Taiwan. J Gynecol Oncol (2013) 24(4):342–51. doi: 10.3802/jgo.2013.24.4.342
7. Hiss D. Optimizing Molecular-Targeted Therapies in Ovarian Cancer: The Renewed Surge of Interest in Ovarian Cancer Biomarkers and Cell Signaling Pathways. J Oncol (2012) 2012:737981. doi: 10.1155/2012/737981
8. Basu P, Mukhopadhyay A, Konishi I. Targeted Therapy for Gynecologic Cancers: Toward the Era of Precision Medicine. Int J Gynecol Obstet (2018) 143:131–6. doi: 10.1002/ijgo.12620
9. Berek JS, Kehoe ST, Kumar L, Friedlander M. Cancer of the Ovary, Fallopian Tube, and Peritoneum. Int J Gynecol Obstet (2018) 143:59–78. doi: 10.1002/ijgo.12614
10. Coleman RL, Fleming GF, Brady MF, Swisher EM, Steffensen KD, Friedlander M, et al. Veliparib With First-Line Chemotherapy and as Maintenance Therapy in Ovarian Cancer. N Engl J Med (2019) 381(25):2403–15. doi: 10.1056/NEJMoa1909707
11. González-Martín A, Pothuri B, Vergote I, DePont Christensen R, Graybill W, Mirza MR, et al. Niraparib in Patients With Newly Diagnosed Advanced Ovarian Cancer. N Engl J Med (2019) 381(25):2391–402. doi: 10.1056/NEJMoa1910962
12. Moore K, Colombo N, Scambia G, Kim BG, Oaknin A, Friedlander M, et al. Maintenance Olaparib in Patients With Newly Diagnosed Advanced Ovarian Cancer. N Engl J Med (2018) 379(26):2495–505. doi: 10.1056/NEJMoa1810858
13. Mirza MR, Monk BJ, Herrstedt J, Oza AM, Mahner S, Redondo A, et al. Niraparib Maintenance Therapy in Platinum-Sensitive, Recurrent Ovarian Cancer. N Engl J Med (2016) 375(22):2154–64. doi: 10.1056/NEJMoa1611310
14. Coleman RL, Oza AM, Lorusso D, Aghajanian C, Oaknin A, Dean A, et al. Rucaparib Maintenance Treatment for Recurrent Ovarian Carcinoma After Response to Platinum Therapy (ARIEL3): A Randomised, Double-Blind, Placebo-Controlled, Phase 3 Trial. Lancet (2017) 390(10106):1949–61. doi: 10.1016/s0140-6736(17)32440-6
15. Pujade-Lauraine E, Ledermann JA, Selle F, Gebski V, Penson RT, Oza AM, et al. Olaparib Tablets as Maintenance Therapy in Patients With Platinum-Sensitive, Relapsed Ovarian Cancer and a BRCA1/2 Mutation (SOLO2/ENGOT-Ov21): A Double-Blind, Randomised, Placebo-Controlled, Phase 3 Trial. Lancet Oncol (2017) 18(9):1274–84. doi: 10.1016/s1470-2045(17)30469-2
16. Dottino JA, Moss HA, Lu KH, Secord AA, Havrilesky LJ. U.S. Food and Drug Administration-Approved Poly (ADP-Ribose) Polymerase Inhibitor Maintenance Therapy for Recurrent Ovarian Cancer: A Cost-Effectiveness Analysis. Obstet Gynecol (2019) 133(4):795–802. doi: 10.1097/aog.0000000000003171
17. Gonzalez R, Havrilesky LJ, Myers ER, Secord AA, Dottino JA, Berchuck A, et al. Cost-Effectiveness Analysis Comparing “PARP Inhibitors-for-All” to the Biomarker-Directed Use of PARP Inhibitor Maintenance Therapy for Newly Diagnosed Advanced Stage Ovarian Cancer. Gynecol Oncol (2020) 159(2):483–90. doi: 10.1016/j.ygyno.2020.08.003
18. Hoppe MM, Sundar R, Tan DSP, Jeyasekharan AD. Biomarkers for Homologous Recombination Deficiency in Cancer. JNCI: J Natl Cancer Institute (2018) 110(7):704–13. doi: 10.1093/jnci/djy085
19. Vilenchik MM, Knudson AG. Endogenous DNA Double-Strand Breaks: Production, Fidelity of Repair, and Induction of Cancer. Proc Natl Acad Sci USA (2003) 100(22):12871–6. doi: 10.1073/pnas.2135498100
20. Curtin NJ. DNA Repair Dysregulation From Cancer Driver to Therapeutic Target. Nat Rev Cancer (2012) 12(12):801–17. doi: 10.1038/nrc3399
21. Miller RE, Leary A, Scott CL, Serra V, Lord CJ, Bowtell D, et al. ESMO Recommendations on Predictive Biomarker Testing for Homologous Recombination Deficiency and PARP Inhibitor Benefit in Ovarian Cancer. Ann Oncol (2020) 31(12):1606–22. doi: 10.1016/j.annonc.2020.08.2102
22. Whelan DR, Lee WTC, Yin Y, Ofri DM, Bermudez-Hernandez K, Keegan S, et al. Spatiotemporal Dynamics of Homologous Recombination Repair at Single Collapsed Replication Forks. Nat Commun (2018) 9(1):3882. doi: 10.1038/s41467-018-06435-3
23. Chang HHY, Pannunzio NR, Adachi N, Lieber MR. Non-Homologous DNA End Joining and Alternative Pathways to Double-Strand Break Repair. Nat Rev Mol Cell Biol (2017) 18(8):495–506. doi: 10.1038/nrm.2017.48
24. Hastings PJ, Lupski JR, Rosenberg SM, Ira G. Mechanisms of Change in Gene Copy Number. Nat Rev Genet (2009) 10(8):551–64. doi: 10.1038/nrg2593
25. Ottaviani D, LeCain M, Sheer D. The Role of Microhomology in Genomic Structural Variation. Trends Genetics: TIG (2014) 30(3):85–94. doi: 10.1016/j.tig.2014.01.001
26. Lord CJ, Ashworth A. The DNA Damage Response and Cancer Therapy. Nature (2012) 481(7381):287–94. doi: 10.1038/nature10760
27. Konstantinopoulos PA, Ceccaldi R, Shapiro GI, D’Andrea AD. Homologous Recombination Deficiency: Exploiting the Fundamental Vulnerability of Ovarian Cancer. Cancer Discovery (2015) 5(11):1137–54. doi: 10.1158/2159-8290.Cd-15-0714
28. Pommier Y, O’Connor MJ, de Bono J. Laying a Trap to Kill Cancer Cells: PARP Inhibitors and Their Mechanisms of Action. Sci Trans Med (2016) 8(362):362ps17. doi: 10.1126/scitranslmed.aaf9246
29. Pilié PG, Tang C, Mills GB, Yap TA. State-Of-the-Art Strategies for Targeting the DNA Damage Response in Cancer. Nat Rev Clin Oncol (2019) 16(2):81–104. doi: 10.1038/s41571-018-0114-z
30. Ray Chaudhuri A, Nussenzweig A. The Multifaceted Roles of PARP1 in DNA Repair and Chromatin Remodelling. Nat Rev Mol Cell Biol (2017) 18(10):610–21. doi: 10.1038/nrm.2017.53
31. George A, Kaye S, Banerjee S. Delivering Widespread BRCA Testing and PARP Inhibition to Patients With Ovarian Cancer. Nat Rev Clin Oncol (2017) 14(5):284–96. doi: 10.1038/nrclinonc.2016.191
32. Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, et al. Specific Killing of BRCA2-Deficient Tumours With Inhibitors of Poly(ADP-Ribose) Polymerase. Nature (2005) 434(7035):913–7. doi: 10.1038/nature03443
33. Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, et al. Targeting the DNA Repair Defect in BRCA Mutant Cells as a Therapeutic Strategy. Nature (2005) 434(7035):917–21. doi: 10.1038/nature03445
34. Pennington KP, Walsh T, Harrell MI, Lee MK, Pennil CC, Rendi MH, et al. Germline and Somatic Mutations in Homologous Recombination Genes Predict Platinum Response and Survival in Ovarian, Fallopian Tube, and Peritoneal Carcinomas. Clin Cancer Res (2014) 20(3):764–75. doi: 10.1158/1078-0432.ccr-13-2287
35. Gelmon KA, Tischkowitz M, Mackay H, Swenerton K, Robidoux A, Tonkin K, et al. Olaparib in Patients With Recurrent High-Grade Serous or Poorly Differentiated Ovarian Carcinoma or Triple-Negative Breast Cancer: A Phase 2, Multicentre, Open-Label, non-Randomised Study. Lancet Oncol (2011) 12(9):852–61. doi: 10.1016/s1470-2045(11)70214-5
36. Kaufman B, Shapira-Frommer R, Schmutzler RK, Audeh MW, Friedlander M, Balmaña J, et al. Olaparib Monotherapy in Patients With Advanced Cancer and a Germline BRCA1/2 Mutation. J Clin Oncol (2015) 33(3):244–50. doi: 10.1200/jco.2014.56.2728
37. Domchek SM, Aghajanian C, Shapira-Frommer R, Schmutzler RK, Audeh MW, Friedlander M, et al. Efficacy and Safety of Olaparib Monotherapy in Germline BRCA1/2 Mutation Carriers With Advanced Ovarian Cancer and Three or More Lines of Prior Therapy. Gynecol Oncol (2016) 140(2):199–203. doi: 10.1016/j.ygyno.2015.12.020
38. Swisher EM, Lin KK, Oza AM, Scott CL, Giordano H, Sun J, et al. Rucaparib in Relapsed, Platinum-Sensitive High-Grade Ovarian Carcinoma (ARIEL2 Part 1): An International, Multicentre, Open-Label, Phase 2 Trial. Lancet Oncol (2017) 18(1):75–87. doi: 10.1016/s1470-2045(16)30559-9
39. Coleman RL, Swisher EM, Oza AM, Scott CL, Giordano H, Lin KK, et al. Refinement of Prespecified Cutoff for Genomic Loss of Heterozygosity (LOH) in ARIEL2 Part 1: A Phase II Study of Rucaparib in Patients (Pts) With High Grade Ovarian Carcinoma (HGOC). J Clin Oncol (2016) 34: (15_suppl):5540. doi: 10.1200/JCO.2016.34.15_suppl.5540
40. Moore KN, Secord AA, Geller MA, Miller DS, Cloven N, Fleming GF, et al. Niraparib Monotherapy for Late-Line Treatment of Ovarian Cancer (QUADRA): A Multicentre, Open-Label, Single-Arm, Phase 2 Trial. Lancet Oncol (2019) 20(5):636–48. doi: 10.1016/s1470-2045(19)30029-4
41. Moore KN, Secord AA, Geller MA, Miller DS, Cloven NG, Fleming GF, et al. QUADRA: A Phase 2, Open-Label, Single-Arm Study to Evaluate Niraparib in Patients (Pts) With Relapsed Ovarian Cancer (ROC) Who Have Received ≥3 Prior Chemotherapy Regimens. J Clin Oncol (2018) 36: (15_suppl):5514. doi: 10.1200/JCO.2018.36.15_suppl.5514
42. Haunschild CE, Tewari KS. The Current Landscape of Molecular Profiling in the Treatment of Epithelial Ovarian Cancer. Gynecol Oncol (2020) 160(1):333–45. doi: 10.1016/j.ygyno.2020.09.043
43. Penson RT, Valencia RV, Cibula D, Colombo N, Leath CA 3rd, Bidziński M, et al. Olaparib Versus Nonplatinum Chemotherapy in Patients With Platinum-Sensitive Relapsed Ovarian Cancer and a Germline BRCA1/2 Mutation (SOLO3): A Randomized Phase III Trial. J Clin Oncol (2020) 38(11):1164–74. doi: 10.1200/jco.19.02745
44. Ledermann J, Harter P, Gourley C, Friedlander M, Vergote I, Rustin G, et al. Olaparib Maintenance Therapy in Patients With Platinum-Sensitive Relapsed Serous Ovarian Cancer: A Preplanned Retrospective Analysis of Outcomes by BRCA Status in a Randomised Phase 2 Trial. Lancet Oncol (2014) 15(8):852–61. doi: 10.1016/s1470-2045(14)70228-1
45. Poveda A, Floquet A, Ledermann JA, Asher R, Penson RT, Oza AM, et al. Olaparib Tablets as Maintenance Therapy in Patients With Platinum-Sensitive Relapsed Ovarian Cancer and a BRCA1/2 Mutation (SOLO2/ENGOT-Ov21): A Final Analysis of a Double-Blind, Randomised, Placebo-Controlled, Phase 3 Trial. Lancet Oncol (2021) 22(5):620–31. doi: 10.1016/S1470-2045(21)00073-5
46. Banerjee S, Moore KN, Colombo N, Scambia G, Kim BG, Oaknin A, et al. 811mo Maintenance Olaparib for Patients (Pts) With Newly Diagnosed, Advanced Ovarian Cancer (OC) and a BRCA Mutation (BRCAm): 5-Year (Y) Follow-Up (F/U) From SOLO1. Ann Oncol (2020) 31:S613. doi: 10.1016/j.annonc.2020.08.950
47. Ray-Coquard I, Pautier P, Pignata S, Pérol D, González-Martín A, Berger R, et al. Olaparib Plus Bevacizumab as First-Line Maintenance in Ovarian Cancer. New Engl J Med (2019) 381(25):2416–28. doi: 10.1056/NEJMoa1911361
48. Walsh T, Casadei S, Lee MK, Pennil CC, Nord AS, Thornton AM, et al. Mutations in 12 Genes for Inherited Ovarian, Fallopian Tube, and Peritoneal Carcinoma Identified by Massively Parallel Sequencing. Proc Natl Acad Sci USA (2011) 108(44):18032–7. doi: 10.1073/pnas.1115052108
49. Wagle N, Berger MF, Davis MJ, Blumenstiel B, Defelice M, Pochanard P, et al. High-Throughput Detection of Actionable Genomic Alterations in Clinical Tumor Samples by Targeted, Massively Parallel Sequencing. Cancer Discovery (2012) 2(1):82–93. doi: 10.1158/2159-8290.Cd-11-0184
50. Stover EH, Fuh K, Konstantinopoulos PA, Matulonis UA, Liu JF. Clinical Assays for Assessment of Homologous Recombination DNA Repair Deficiency. Gynecol Oncol (2020) 159(3):887–98. doi: 10.1016/j.ygyno.2020.09.029
51. Konstantinopoulos PA, Norquist B, Lacchetti C, Armstrong D, Grisham RN, Goodfellow PJ, et al. Germline and Somatic Tumor Testing in Epithelial Ovarian Cancer: ASCO Guideline. J Clin Oncol (2020) 38(11):1222–45. doi: 10.1200/jco.19.02960
52. Lancaster JM, Powell CB, Chen LM, Richardson DL. Society of Gynecologic Oncology Statement on Risk Assessment for Inherited Gynecologic Cancer Predispositions. Gynecol Oncol (2015) 136(1):3–7. doi: 10.1016/j.ygyno.2014.09.009
53. Daly MB, Pilarski R, Yurgelun MB, Berry MP, Buys SS, Dickson P, et al. NCCN Guidelines Insights: Genetic/Familial High-Risk Assessment: Breast, Ovarian, and Pancreatic, Version 1.2020. J Natl Compr Canc Netw (2020) 18(4):380–91. doi: 10.6004/jnccn.2020.0017
54. Vergote I, Banerjee S, Gerdes AM, van Asperen C, Marth C, Vaz F, et al. Current Perspectives on Recommendations for BRCA Genetic Testing in Ovarian Cancer Patients. Eur J Cancer (2016) 69:127–34. doi: 10.1016/j.ejca.2016.10.006
55. Hennessy BT, Timms KM, Carey MS, Gutin A, Meyer LA, Flake DD 2nd, et al. Somatic Mutations in BRCA1 and BRCA2 Could Expand the Number of Patients That Benefit From Poly (ADP Ribose) Polymerase Inhibitors in Ovarian Cancer. J Clin Oncol (2010) 28(22):3570–6. doi: 10.1200/jco.2009.27.2997
56. Bell D, Berchuck A, Birrer M, Chien J, Cramer DW, Dao F, et al. Integrated Genomic Analyses of Ovarian Carcinoma. Nature (2011) 474(7353):609–15. doi: 10.1038/nature10166
57. Alsop K, Fereday S, Meldrum C, deFazio A, Emmanuel C, George J, et al. BRCA Mutation Frequency and Patterns of Treatment Response in BRCA Mutation-Positive Women With Ovarian Cancer: A Report From the Australian Ovarian Cancer Study Group. J Clin Oncol (2012) 30(21):2654–63. doi: 10.1200/jco.2011.39.8545
58. Dougherty BA, Lai Z, Hodgson DR, Orr MCM, Hawryluk M, Sun J, et al. Biological and Clinical Evidence for Somatic Mutations in BRCA1 and BRCA2 as Predictive Markers for Olaparib Response in High-Grade Serous Ovarian Cancers in the Maintenance Setting. Oncotarget (2017) 8(27):43653–61. doi: 10.18632/oncotarget.17613
59. Daly MB, Pilarski R, Axilbund JE, Berry M, Buys SS, Crawford B, et al. Genetic/Familial High-Risk Assessment: Breast and Ovarian, Version 2.2015. J Natl Compr Canc Netw (2016) 14(2):153–62. doi: 10.6004/jnccn.2016.0018
60. Norquist BM, Harrell MI, Brady MF, Walsh T, Lee MK, Gulsuner S, et al. Inherited Mutations in Women With Ovarian Carcinoma. JAMA Oncol (2016) 2(4):482–90. doi: 10.1001/jamaoncol.2015.5495
61. de Bono J, Mateo J, Fizazi K, Saad F, Shore N, Sandhu S, et al. Olaparib for Metastatic Castration-Resistant Prostate Cancer. N Engl J Med (2020) 382(22):2091–102. doi: 10.1056/NEJMoa1911440
62. Mateo J, Porta N, Bianchini D, McGovern U, Elliott T, Jones R, et al. Olaparib in Patients With Metastatic Castration-Resistant Prostate Cancer With DNA Repair Gene Aberrations (TOPARP-B): A Multicentre, Open-Label, Randomised, Phase 2 Trial. Lancet Oncol (2020) 21(1):162–74. doi: 10.1016/s1470-2045(19)30684-9
63. Norquist BM, Brady MF, Harrell MI, Walsh T, Lee MK, Gulsuner S, et al. Mutations in Homologous Recombination Genes and Outcomes in Ovarian Carcinoma Patients in GOG 218: An NRG Oncology/Gynecologic Oncology Group Study. Clin Cancer Res (2018) 24(4):777–83. doi: 10.1158/1078-0432.Ccr-17-1327
64. Loveday C, Turnbull C, Ramsay E, Hughes D, Ruark E, Frankum JR, et al. Germline Mutations in RAD51D Confer Susceptibility to Ovarian Cancer. Nat Genet (2011) 43(9):879–82. doi: 10.1038/ng.893
65. Bajrami I, Frankum JR, Konde A, Miller RE, Rehman FL, Brough R, et al. Genome-Wide Profiling of Genetic Synthetic Lethality Identifies CDK12 as a Novel Determinant of PARP1/2 Inhibitor Sensitivity. Cancer Res (2014) 74(1):287–97. doi: 10.1158/0008-5472.Can-13-2541
66. McCabe N, Turner NC, Lord CJ, Kluzek K, Bialkowska A, Swift S, et al. Deficiency in the Repair of DNA Damage by Homologous Recombination and Sensitivity to Poly(ADP-Ribose) Polymerase Inhibition. Cancer Res (2006) 66(16):8109–15. doi: 10.1158/0008-5472.Can-06-0140
67. Hodgson DR, Dougherty BA, Lai Z, Fielding A, Grinsted L, Spencer S, et al. Candidate Biomarkers of PARP Inhibitor Sensitivity in Ovarian Cancer Beyond the BRCA Genes. Br J Cancer (2018) 119(11):1401–9. doi: 10.1038/s41416-018-0274-8
68. Bonnet C, Krieger S, Vezain M, Rousselin A, Tournier I, Martins A, et al. Screening BRCA1 and BRCA2 Unclassified Variants for Splicing Mutations Using Reverse Transcription PCR on Patient RNA and an Ex Vivo Assay Based on a Splicing Reporter Minigene. J Med Genet (2008) 45(7):438–46. doi: 10.1136/jmg.2007.056895
69. Cartegni L, Chew SL, Krainer AR. Listening to Silence and Understanding Nonsense: Exonic Mutations That Affect Splicing. Nat Rev Genet (2002) 3(4):285–98. doi: 10.1038/nrg775
70. Li H, LaDuca H, Pesaran T, Chao EC, Dolinsky JS, Parsons M, et al. Classification of Variants of Uncertain Significance in BRCA1 and BRCA2 Using Personal and Family History of Cancer From Individuals in a Large Hereditary Cancer Multigene Panel Testing Cohort. Genet Med (2020) 22(4):701–8. doi: 10.1038/s41436-019-0729-1
71. Alexandrov LB, Kim J, Haradhvala NJ, Huang MN, Tian Ng AW, Wu Y, et al. The Repertoire of Mutational Signatures in Human Cancer. Nature (2020) 578(7793):94–101. doi: 10.1038/s41586-020-1943-3
72. Toland AE, Forman A, Couch FJ, Culver JO, Eccles DM, Foulkes WD, et al. Clinical Testing of BRCA1 and BRCA2: A Worldwide Snapshot of Technological Practices. NPJ Genom Med (2018) 3:7. doi: 10.1038/s41525-018-0046-7
73. Watkins JA, Irshad S, Grigoriadis A, Tutt AN. Genomic Scars as Biomarkers of Homologous Recombination Deficiency and Drug Response in Breast and Ovarian Cancers. Breast Cancer Res (2014) 16(3):211. doi: 10.1186/bcr3670
74. Abkevich V, Timms KM, Hennessy BT, Potter J, Carey MS, Meyer LA, et al. Patterns of Genomic Loss of Heterozygosity Predict Homologous Recombination Repair Defects in Epithelial Ovarian Cancer. Br J Cancer (2012) 107(10):1776–82. doi: 10.1038/bjc.2012.451
75. Birkbak NJ, Wang ZC, Kim JY, Eklund AC, Li Q, Tian R, et al. Telomeric Allelic Imbalance Indicates Defective DNA Repair and Sensitivity to DNA-Damaging Agents. Cancer Discovery (2012) 2(4):366–75. doi: 10.1158/2159-8290.Cd-11-0206
76. Timms KM, Abkevich V, Hughes E, Neff C, Reid J, Morris B, et al. Association of BRCA1/2 Defects With Genomic Scores Predictive of DNA Damage Repair Deficiency Among Breast Cancer Subtypes. Breast Cancer Res (2014) 16(6):475. doi: 10.1186/s13058-014-0475-x
77. Popova T, Manié E, Rieunier G, Caux-Moncoutier V, Tirapo C, Dubois T, et al. Ploidy and Large-Scale Genomic Instability Consistently Identify Basal-Like Breast Carcinomas With BRCA1/2 Inactivation. Cancer Res (2012) 72(21):5454–62. doi: 10.1158/0008-5472.Can-12-1470
78. Telli ML, Timms KM, Reid J, Hennessy B, Mills GB, Jensen KC, et al. Homologous Recombination Deficiency (HRD) Score Predicts Response to Platinum-Containing Neoadjuvant Chemotherapy in Patients With Triple-Negative Breast Cancer. Clin Cancer Res (2016) 22(15):3764–73. doi: 10.1158/1078-0432.Ccr-15-2477
79. Frampton GM, Fichtenholtz A, Otto GA, Wang K, Downing SR, He J, et al. Development and Validation of a Clinical Cancer Genomic Profiling Test Based on Massively Parallel DNA Sequencing. Nat Biotechnol (2013) 31(11):1023–31. doi: 10.1038/nbt.2696
80. Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, et al. Intratumor Heterogeneity and Branched Evolution Revealed by Multiregion Sequencing. N Engl J Med (2012) 366(10):883–92. doi: 10.1056/NEJMoa1113205
81. Talmadge JE. Clonal Selection of Metastasis Within the Life History of a Tumor. Cancer Res (2007) 67(24):11471–5. doi: 10.1158/0008-5472.Can-07-2496
82. Edwards SL, Brough R, Lord CJ, Natrajan R, Vatcheva R, Levine DA, et al. Resistance to Therapy Caused by Intragenic Deletion in BRCA2. Nature (2008) 451(7182):1111–5. doi: 10.1038/nature06548
83. Kondrashova O, Nguyen M, Shield-Artin K, Tinker AV, Teng NNH, Harrell MI, et al. Secondary Somatic Mutations Restoring RAD51C and RAD51D Associated With Acquired Resistance to the PARP Inhibitor Rucaparib in High-Grade Ovarian Carcinoma. Cancer Discovery (2017) 7(9):984–98. doi: 10.1158/2159-8290.Cd-17-0419
84. Sakai W, Swisher EM, Karlan BY, Agarwal MK, Higgins J, Friedman C, et al. Secondary Mutations as a Mechanism of Cisplatin Resistance in BRCA2-Mutated Cancers. Nature (2008) 451(7182):1116–20. doi: 10.1038/nature06633
85. Norquist B, Wurz KA, Pennil CC, Garcia R, Gross J, Sakai W, et al. Secondary Somatic Mutations Restoring BRCA1/2 Predict Chemotherapy Resistance in Hereditary Ovarian Carcinomas. J Clin Oncol (2011) 29(22):3008–15. doi: 10.1200/jco.2010.34.2980
86. Swisher EM, Sakai W, Karlan BY, Wurz K, Urban N, Taniguchi T. Secondary BRCA1 Mutations in BRCA1-Mutated Ovarian Carcinomas With Platinum Resistance. Cancer Res (2008) 68(8):2581–6. doi: 10.1158/0008-5472.Can-08-0088
87. Noordermeer SM, van Attikum H. PARP Inhibitor Resistance: A Tug-Of-War in BRCA-Mutated Cells. Trends Cell Biol (2019) 29(10):820–34. doi: 10.1016/j.tcb.2019.07.008
88. Lin KK, Harrell MI, Oza AM, Oaknin A, Ray-Coquard I, Tinker AV, et al. BRCA Reversion Mutations in Circulating Tumor DNA Predict Primary and Acquired Resistance to the PARP Inhibitor Rucaparib in High-Grade Ovarian Carcinoma. Cancer Discovery (2019) 9(2):210–9. doi: 10.1158/2159-8290.Cd-18-0715
89. Ledermann JA, Oza AM, Lorusso D, Aghajanian C, Oaknin A, Dean A, et al. Rucaparib for Patients With Platinum-Sensitive, Recurrent Ovarian Carcinoma (ARIEL3): Post-Progression Outcomes and Updated Safety Results From a Randomised, Placebo-Controlled, Phase 3 Trial. Lancet Oncol (2020) 21(5):710–22. doi: 10.1016/s1470-2045(20)30061-9
90. Burger H, Loos WJ, Eechoute K, Verweij J, Mathijssen RH, Wiemer EA. Drug Transporters of Platinum-Based Anticancer Agents and Their Clinical Significance. Drug Resist Updates: Rev Commentaries Antimicrobial Anticancer Chemother (2011) 14(1):22–34. doi: 10.1016/j.drup.2010.12.002
91. Ray Chaudhuri A, Callen E, Ding X, Gogola E, Duarte AA, Lee JE, et al. Replication Fork Stability Confers Chemoresistance in BRCA-Deficient Cells. Nature (2016) 535(7612):382–7. doi: 10.1038/nature18325
92. Jaspers JE, Kersbergen A, Boon U, Sol W, van Deemter L, Zander SA, et al. Loss of 53BP1 Causes PARP Inhibitor Resistance in Brca1-Mutated Mouse Mammary Tumors. Cancer Discovery (2013) 3(1):68–81. doi: 10.1158/2159-8290.Cd-12-0049
93. Rottenberg S, Jaspers JE, Kersbergen A, van der Burg E, Nygren AO, Zander SA, et al. High Sensitivity of BRCA1-Deficient Mammary Tumors to the PARP Inhibitor AZD2281 Alone and in Combination With Platinum Drugs. Proc Natl Acad Sci USA (2008) 105(44):17079–84. doi: 10.1073/pnas.0806092105
94. Xu G, Chapman JR, Brandsma I, Yuan J, Mistrik M, Bouwman P, et al. REV7 Counteracts DNA Double-Strand Break Resection and Affects PARP Inhibition. Nature (2015) 521(7553):541–4. doi: 10.1038/nature14328
95. Lipinski KA, Barber LJ, Davies MN, Ashenden M, Sottoriva A, Gerlinger M. Cancer Evolution and the Limits of Predictability in Precision Cancer Medicine. Trends Cancer (2016) 2(1):49–63. doi: 10.1016/j.trecan.2015.11.003
96. Khan SS, Chen AP, Takebe N. Impact of NCI-MATCH: A Nationwide Oncology Precision Medicine Trial. Expert Rev Precis Med Drug Dev (2019) 4(4):251–8. doi: 10.1080/23808993.2019.1623023
97. Kurian AW, Ward KC, Howlader N, Deapen D, Hamilton AS, Mariotto A, et al. Genetic Testing and Results in a Population-Based Cohort of Breast Cancer Patients and Ovarian Cancer Patients. J Clin Oncol (2019) 37(15):1305–15. doi: 10.1200/jco.18.01854
98. Prasad V. Why the US Centers for Medicare and Medicaid Services (CMS) Should Have Required a Randomized Trial of Foundation Medicine (F1CDx) Before Paying for it. Ann Oncol (2018) 29(2):298–300. doi: 10.1093/annonc/mdx786
99. Meienberg J, Zerjavic K, Keller I, Okoniewski M, Patrignani A, Ludin K, et al. New Insights Into the Performance of Human Whole-Exome Capture Platforms. Nucleic Acids Res (2015) 43(11):e76. doi: 10.1093/nar/gkv216
100. Barbitoff YA, Polev DE, Glotov AS, Serebryakova EA, Shcherbakova IV, Kiselev AM, et al. Systematic Dissection of Biases in Whole-Exome and Whole-Genome Sequencing Reveals Major Determinants of Coding Sequence Coverage. Sci Rep (2020) 10(1):2057. doi: 10.1038/s41598-020-59026-y
101. Clark MJ, Chen R, Lam HY, Karczewski KJ, Chen R, Euskirchen G, et al. Performance Comparison of Exome DNA Sequencing Technologies. Nat Biotechnol (2011) 29(10):908–14. doi: 10.1038/nbt.1975
102. Seaby EG, Pengelly RJ, Ennis S. Exome Sequencing Explained: A Practical Guide to its Clinical Application. Brief Funct Genomics (2016) 15(5):374–84. doi: 10.1093/bfgp/elv054
103. Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, et al. Signatures of Mutational Processes in Human Cancer. Nature (2013) 500(7463):415–21. doi: 10.1038/nature12477
104. Funnell T, Zhang AW, Grewal D, McKinney S, Bashashati A, Wang YK, et al. Integrated Structural Variation and Point Mutation Signatures in Cancer Genomes Using Correlated Topic Models. PloS Comput Biol (2019) 15(2):e1006799. doi: 10.1371/journal.pcbi.1006799
105. Davies H, Glodzik D, Morganella S, Yates LR, Staaf J, Zou X, et al. HRDetect Is a Predictor of BRCA1 and BRCA2 Deficiency Based on Mutational Signatures. Nat Med (2017) 23(4):517–25. doi: 10.1038/nm.4292
106. Polak P, Kim J, Braunstein LZ, Karlic R, Haradhavala NJ, Tiao G, et al. A Mutational Signature Reveals Alterations Underlying Deficient Homologous Recombination Repair in Breast Cancer. Nat Genet (2017) 49(10):1476–86. doi: 10.1038/ng.3934
107. Gulhan DC, Lee JJ, Melloni GEM, Cortés-Ciriano I, Park PJ. Detecting the Mutational Signature of Homologous Recombination Deficiency in Clinical Samples. Nat Genet (2019) 51(5):912–9. doi: 10.1038/s41588-019-0390-2
108. Zhao EY, Shen Y, Pleasance E, Kasaian K, Leelakumari S, Jones M, et al. Homologous Recombination Deficiency and Platinum-Based Therapy Outcomes in Advanced Breast Cancer. Clin Cancer Res (2017) 23(24):7521–30. doi: 10.1158/1078-0432.Ccr-17-1941
109. Staaf J, Glodzik D, Bosch A, Vallon-Christersson J, Reuterswärd C, Häkkinen J, et al. Whole-Genome Sequencing of Triple-Negative Breast Cancers in a Population-Based Clinical Study. Nat Med (2019) 25(10):1526–33. doi: 10.1038/s41591-019-0582-4
110. Chopra N, Tovey H, Pearson A, Cutts R, Toms C, Proszek P, et al. Homologous Recombination DNA Repair Deficiency and PARP Inhibition Activity in Primary Triple Negative Breast Cancer. Nat Commun (2020) 11(1):2662. doi: 10.1038/s41467-020-16142-7
111. Robbe P, Popitsch N, Knight SJL, Antoniou P, Becq J, He M, et al. Clinical Whole-Genome Sequencing From Routine Formalin-Fixed, Paraffin-Embedded Specimens: Pilot Study for the 100,000 Genomes Project. Genet Med (2018) 20(10):1196–205. doi: 10.1038/gim.2017.241
112. Bernards SS, Pennington KP, Harrell MI, Agnew KJ, Garcia RL, Norquist BM, et al. Clinical Characteristics and Outcomes of Patients With BRCA1 or RAD51C Methylated Versus Mutated Ovarian Carcinoma. Gynecol Oncol (2018) 148(2):281–5. doi: 10.1016/j.ygyno.2017.12.004
113. Ruscito I, Dimitrova D, Vasconcelos I, Gellhaus K, Schwachula T, Bellati F, et al. BRCA1 Gene Promoter Methylation Status in High-Grade Serous Ovarian Cancer Patients–a Study of the Tumour Bank Ovarian Cancer (TOC) and Ovarian Cancer Diagnosis Consortium (OVCAD). Eur J Cancer (2014) 50(12):2090–8. doi: 10.1016/j.ejca.2014.05.001
114. Swisher EM, Gonzalez RM, Taniguchi T, Garcia RL, Walsh T, Goff BA, et al. Methylation and Protein Expression of DNA Repair Genes: Association With Chemotherapy Exposure and Survival in Sporadic Ovarian and Peritoneal Carcinomas. Mol Cancer (2009) 8:48. doi: 10.1186/1476-4598-8-48
115. Lheureux S, Lai Z, Dougherty BA, Runswick S, Hodgson DR, Timms KM, et al. Long-Term Responders on Olaparib Maintenance in High-Grade Serous Ovarian Cancer: Clinical and Molecular Characterization. Clin Cancer Res (2017) 23(15):4086–94. doi: 10.1158/1078-0432.Ccr-16-2615
116. Chiang JW, Karlan BY, Cass L, Baldwin RL. BRCA1 Promoter Methylation Predicts Adverse Ovarian Cancer Prognosis. Gynecol Oncol (2006) 101(3):403–10. doi: 10.1016/j.ygyno.2005.10.034
117. Stefansson OA, Villanueva A, Vidal A, Martí L, Esteller M. BRCA1 Epigenetic Inactivation Predicts Sensitivity to Platinum-Based Chemotherapy in Breast and Ovarian Cancer. Epigenet: Off J DNA Methylation Soc (2012) 7(11):1225–9. doi: 10.4161/epi.22561
118. Cunningham JM, Cicek MS, Larson NB, Davila J, Wang C, Larson MC, et al. Clinical Characteristics of Ovarian Cancer Classified by BRCA1, BRCA2, and RAD51C Status. Sci Rep (2014) 4:4026. doi: 10.1038/srep04026
119. Byrum AK, Vindigni A, Mosammaparast N. Defining and Modulating ‘Brcaness’. Trends Cell Biol (2019) 29(9):740–51. doi: 10.1016/j.tcb.2019.06.005
120. Mason JM, Chan YL, Weichselbaum RW, Bishop DK. Non-Enzymatic Roles of Human RAD51 at Stalled Replication Forks. Nat Commun (2019) 10(1):4410. doi: 10.1038/s41467-019-12297-0
121. Ceccaldi R, Rondinelli B, D’Andrea AD. Repair Pathway Choices and Consequences at the Double-Strand Break. Trends Cell Biol (2016) 26(1):52–64. doi: 10.1016/j.tcb.2015.07.009
122. Wilson AJ, Stubbs M, Liu P, Ruggeri B, Khabele D. The BET Inhibitor INCB054329 Reduces Homologous Recombination Efficiency and Augments PARP Inhibitor Activity in Ovarian Cancer. Gynecol Oncol (2018) 149(3):575–84. doi: 10.1016/j.ygyno.2018.03.049
123. Mukhopadhyay A, Plummer ER, Elattar A, Soohoo S, Uzir B, Quinn JE, et al. Clinicopathological Features of Homologous Recombination-Deficient Epithelial Ovarian Cancers: Sensitivity to PARP Inhibitors, Platinum, and Survival. Cancer Res (2012) 72(22):5675–82. doi: 10.1158/0008-5472.Can-12-0324
124. Hill SJ, Decker B, Roberts EA, Horowitz NS, Muto MG, Worley MJ Jr., et al. Prediction of DNA Repair Inhibitor Response in Short-Term Patient-Derived Ovarian Cancer Organoids. Cancer Discovery (2018) 8(11):1404–21. doi: 10.1158/2159-8290.Cd-18-0474
125. Graeser M, McCarthy A, Lord CJ, Savage K, Hills M, Salter J, et al. A Marker of Homologous Recombination Predicts Pathologic Complete Response to Neoadjuvant Chemotherapy in Primary Breast Cancer. Clin Cancer Res (2010) 16(24):6159–68. doi: 10.1158/1078-0432.Ccr-10-1027
126. Meijer TG, Verkaik NS, Sieuwerts AM, van Riet J, Naipal KAT, van Deurzen CHM, et al. Functional Ex Vivo Assay Reveals Homologous Recombination Deficiency in Breast Cancer Beyond BRCA Gene Defects. Clin Cancer Res (2018) 24(24):6277–87. doi: 10.1158/1078-0432.Ccr-18-0063
127. Cruz C, Castroviejo-Bermejo M, Gutiérrez-Enríquez S, Llop-Guevara A, Ibrahim YH, Gris-Oliver A, et al. RAD51 Foci as a Functional Biomarker of Homologous Recombination Repair and PARP Inhibitor Resistance in Germline BRCA-Mutated Breast Cancer. Ann Oncol (2018) 29(5):1203–10. doi: 10.1093/annonc/mdy099
128. Tumiati M, Hietanen S, Hynninen J, Pietilä E, Färkkilä A, Kaipio K, et al. A Functional Homologous Recombination Assay Predicts Primary Chemotherapy Response and Long-Term Survival in Ovarian Cancer Patients. Clin Cancer Res (2018) 24(18):4482–93. doi: 10.1158/1078-0432.Ccr-17-3770
129. Shah MM, Dobbin ZC, Nowsheen S, Wielgos M, Katre AA, Alvarez RD, et al. An Ex Vivo Assay of XRT-Induced Rad51 Foci Formation Predicts Response to PARP-Inhibition in Ovarian Cancer. Gynecol Oncol (2014) 134(2):331–7. doi: 10.1016/j.ygyno.2014.05.009
130. Mukhopadhyay A, Elattar A, Cerbinskaite A, Wilkinson SJ, Drew Y, Kyle S, et al. Development of a Functional Assay for Homologous Recombination Status in Primary Cultures of Epithelial Ovarian Tumor and Correlation With Sensitivity to Poly(ADP-Ribose) Polymerase Inhibitors. Clin Cancer Res (2010) 16(8):2344–51. doi: 10.1158/1078-0432.Ccr-09-2758
131. Castroviejo-Bermejo M, Cruz C, Llop-Guevara A, Gutiérrez-Enríquez S, Ducy M, Ibrahim YH, et al. A RAD51 Assay Feasible in Routine Tumor Samples Calls PARP Inhibitor Response Beyond BRCA Mutation. EMBO Mol Med (2018) 10(12):e9172. doi: 10.15252/emmm.201809172
132. O’Donnell RL, McCormick A, Mukhopadhyay A, Woodhouse LC, Moat M, Grundy A, et al. The Use of Ovarian Cancer Cells From Patients Undergoing Surgery to Generate Primary Cultures Capable of Undergoing Functional Analysis. PloS One (2014) 9(6):e90604. doi: 10.1371/journal.pone.0090604
133. Torquato S, Pallavajjala A, Goldstein A, Toro PV, Silberstein JL, Lee J, et al. Genetic Alterations Detected in Cell-Free DNA Are Associated With Enzalutamide and Abiraterone Resistance in Castration-Resistant Prostate Cancer. JCO Precis Oncol (2019) 3:PO.18.00227. doi: 10.1200/po.18.00227
134. Waks AG, Cohen O, Kochupurakkal B, Kim D, Dunn CE, Buendia Buendia J, et al. Reversion and non-Reversion Mechanisms of Resistance to PARP Inhibitor or Platinum Chemotherapy in BRCA1/2-Mutant Metastatic Breast Cancer. Ann Oncol (2020) 31(5):590–8. doi: 10.1016/j.annonc.2020.02.008
135. Fuh K, Mullen M, Blachut B, Stover E, Konstantinopoulos P, Liu J, et al. Homologous Recombination Deficiency Real-Time Clinical Assays, Ready or Not? Gynecol Oncol (2020) 159(3):877–86. doi: 10.1016/j.ygyno.2020.08.035
136. Bensimon A, Simon A, Chiffaudel A, Croquette V, Heslot F, Bensimon D. Alignment and Sensitive Detection of DNA by a Moving Interface. Science (1994) 265(5181):2096–8. doi: 10.1126/science.7522347
137. Michalet X, Ekong R, Fougerousse F, Rousseaux S, Schurra C, Hornigold N, et al. Dynamic Molecular Combing: Stretching the Whole Human Genome for High-Resolution Studies. Science (1997) 277(5331):1518–23. doi: 10.1126/science.277.5331.1518
138. Parra I, Windle B. High Resolution Visual Mapping of Stretched DNA by Fluorescent Hybridization. Nat Genet (1993) 5(1):17–21. doi: 10.1038/ng0993-17
139. Técher H, Koundrioukoff S, Azar D, Wilhelm T, Carignon S, Brison O, et al. Replication Dynamics: Biases and Robustness of DNA Fiber Analysis. J Mol Biol (2013) 425(23):4845–55. doi: 10.1016/j.jmb.2013.03.040
140. Ait Saada A, Lambert SAE, Carr AM. Preserving Replication Fork Integrity and Competence via the Homologous Recombination Pathway. DNA Repair (2018) 71:135–47. doi: 10.1016/j.dnarep.2018.08.017
141. Yao NY, O’Donnell M. Replisome Structure and Conformational Dynamics Underlie Fork Progression Past Obstacles. Curr Opin Cell Biol (2009) 21(3):336–43. doi: 10.1016/j.ceb.2009.02.008
142. Berti M, Vindigni A. Replication Stress: Getting Back on Track. Nat Struct Mol Biol (2016) 23(2):103–9. doi: 10.1038/nsmb.3163
143. Zeman MK, Cimprich KA. Causes and Consequences of Replication Stress. Nat Cell Biol (2014) 16(1):2–9. doi: 10.1038/ncb2897
144. Quinet A, Tirman S, Jackson J, Šviković S, Lemaçon D, Carvajal-Maldonado D, et al. PRIMPOL-Mediated Adaptive Response Suppresses Replication Fork Reversal in BRCA-Deficient Cells. Mol Cell (2020) 77(3):461–74.e9. doi: 10.1016/j.molcel.2019.10.008
145. Quinet A, Carvajal-Maldonado D, Lemacon D, Vindigni A. DNA Fiber Analysis: Mind the Gap! Methods Enzymol (2017) 591:55–82. doi: 10.1016/bs.mie.2017.03.019
146. Schlacher K, Christ N, Siaud N, Egashira A, Wu H, Jasin M. Double-Strand Break Repair-Independent Role for BRCA2 in Blocking Stalled Replication Fork Degradation by MRE11. Cell (2011) 145(4):529–42. doi: 10.1016/j.cell.2011.03.041
147. Ying S, Hamdy FC, Helleday T. Mre11-Dependent Degradation of Stalled DNA Replication Forks Is Prevented by BRCA2 and PARP1. Cancer Res (2012) 72(11):2814–21. doi: 10.1158/0008-5472.Can-11-3417
148. Schlacher K, Wu H, Jasin M. A Distinct Replication Fork Protection Pathway Connects Fanconi Anemia Tumor Suppressors to RAD51-Brca1/2. Cancer Cell (2012) 22(1):106–16. doi: 10.1016/j.ccr.2012.05.015
149. Naipal KA, Verkaik NS, Ameziane N, van Deurzen CH, Ter Brugge P, Meijers M, et al. Functional Ex Vivo Assay to Select Homologous Recombination-Deficient Breast Tumors for PARP Inhibitor Treatment. Clin Cancer Res (2014) 20(18):4816–26. doi: 10.1158/1078-0432.Ccr-14-0571
150. Kawamoto T, Araki K, Sonoda E, Yamashita YM, Harada K, Kikuchi K, et al. Dual Roles for DNA Polymerase Eta in Homologous DNA Recombination and Translesion DNA Synthesis. Mol Cell (2005) 20(5):793–9. doi: 10.1016/j.molcel.2005.10.016
151. Wiese C, Dray E, Groesser T, San Filippo J, Shi I, Collins DW, et al. Promotion of Homologous Recombination and Genomic Stability by RAD51AP1 via RAD51 Recombinase Enhancement. Mol Cell (2007) 28(3):482–90. doi: 10.1016/j.molcel.2007.08.027
152. Balmus G, Pilger D, Coates J, Demir M, Sczaniecka-Clift M, Barros AC, et al. ATM Orchestrates the DNA-Damage Response to Counter Toxic non-Homologous End-Joining at Broken Replication Forks. Nat Commun (2019) 10(1):87. doi: 10.1038/s41467-018-07729-2
153. Gogola E, Duarte AA, de Ruiter JR, Wiegant WW, Schmid JA, de Bruijn R, et al. Selective Loss of PARG Restores PARylation and Counteracts PARP Inhibitor-Mediated Synthetic Lethality. Cancer Cell (2018) 33(6):1078–93.e12. doi: 10.1016/j.ccell.2018.05.008
154. Guillemette S, Serra RW, Peng M, Hayes JA, Konstantinopoulos PA, Green MR, et al. Resistance to Therapy in BRCA2 Mutant Cells Due to Loss of the Nucleosome Remodeling Factor CHD4. Genes Dev (2015) 29(5):489–94. doi: 10.1101/gad.256214.114
Keywords: homologous recombination deficiency, epithelial ovarian cancer, PARP inhibitor, genomic scar, RAD51 foci formation, mutational signatures
Citation: Chiang Y-C, Lin P-H and Cheng W-F (2021) Homologous Recombination Deficiency Assays in Epithelial Ovarian Cancer: Current Status and Future Direction. Front. Oncol. 11:675972. doi: 10.3389/fonc.2021.675972
Received: 04 March 2021; Accepted: 17 September 2021;
Published: 14 October 2021.
Edited by:
David S. P. Tan, National University of Singapore, SingaporeReviewed by:
Sarah Taylor, University of Pittsburgh, United StatesCopyright © 2021 Chiang, Lin and Cheng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wen-Fang Cheng, d2VuZmFuZ2NoZW5nQHlhaG9vLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.