 Pedro S. de Carvalho
Pedro S. de Carvalho Fabio E. Leal
Fabio E. Leal Marcelo A. Soares
Marcelo A. Soares- 1Programa de Oncovirologia, Instituto Nacional do Câncer, Rio de Janeiro, Brazil
- 2Departamento de Genética, Universidade Federal do Rio de Janeiro, Rio de Janeiro, Brazil
Non-Hodgkin lymphoma is the most common malignancy affecting people living with HIV (PLWH). Among its several subtypes, diffuse large B-cell lymphoma (DLBCL) is an important manifestation within the HIV-infected compartment of the population. Since HIV is able to modulate B cells and promote lymphomagenesis through direct and indirect mechanisms, HIV-related DLBCL has specific characteristics. In this review, we address the clinical and molecular properties of DLBCL disease in the context of HIV infection, as well as the mechanisms by which HIV is able to modulate B lymphocytes and induce their transformation into lymphoma.
Introduction
Historically, lymphomas have been assigned as either Hodgkin or non-Hodgkin types according to histological features [for example, the Reed-Sternberg cells (1, 2)], clinical presentation and response to therapies (3, 4). Non-Hodgkin lymphomas (NHLs) are a large spectrum of diseases that arise from lymphocytes at different stages of development, affecting virtually any organ (5). Also, NHLs are more frequent than Hodgkin lymphomas (6). According to the latest Globocan estimates, NHLs show incidence and mortality of 5.8 and 2.6 per 100.000 inhabitants worldwide, respectively (6). Currently, the World Health Organization (WHO) recognizes more than 60 distinct entities as members of the NHL group, excluding lymphoproliferative disorders and non-malignant manifestations (7). NHLs may arise from B lymphocytes, T lymphocytes or NK cells (7). Generally, NHLs subtypes from B cells are more common and represent about 85% of all cases (8, 9). Although data from Globocan represent collective incidence of NHLs, the distribution of subtypes is not homogenous and varies according to the study population (10). Among B cell-derived NHLs, the diffuse large B cell lymphoma (DLBCL) is consistently reported as the most common manifestation worldwide, followed by follicular lymphoma (FL, especially in Central and South Americas) and chronic lymphocytic leukemia (CLL, especially in southeast Europe) (10, 11). Individuals with Caucasian ancestry were associated with increased incidence of DLBCL, FL and CLL when compared with subjects with Hispanic, Asian or African ancestries (12). Few epidemiological data address specifically DLBCL and, in general, NHLs are reported as a group. However, a study performed with the US population from 1975 to 2017 showed DLBCL incidence estimates of 5.6 per 100.000 individuals per year (13). Also, between 45-60% of all NHL cases reported in Central and South America were DLBCL (14). A study performed with 27,796 NHLs diagnosed between 2000 and 2016 in Sweden reported DLBCL as the most common subtype, accounting for 35% of all cases (15). Those results indicate the relevance of DLBCL as a frequent NHL manifestation. The epidemiological data described above refers to the general population, however, it is known that specific subpopulations, such as people living with HIV (PLWH), harbor even greater burden of NHLs and DLBCL.
Among the risk factors linked with NHL development, immunosuppression is one of the well-documented. For example, solid organ transplant recipients had six-fold higher risk of developing NHL than the general population (16). In this scenario, HIV infection and lymphomagenesis were also extensively associated due to the immunosuppression induced by HIV infection (8). Indeed, before the advent of highly active antiretroviral therapy (HAART), HIV-infected patients had increased incidence of cancers associated with infection by other oncogenic viruses, such as Kaposi’s sarcoma (HHV8 - human herpes virus 8), cervical cancer (HPV - human papillomavirus) and NHL (EBV – Epstein-Barr virus) (17). Those cancers, including the group of NHLs, were subsequently named AIDS defining cancers (ADCs), given their relationship with HIV-associated immunosuppression, while all the other tumors observed among PLWH were called non-AIDS defining cancers (NADCs) (17, 18). Specifically, PLWH had a 113-fold higher risk of developing NHLs than uninfected counterparts (18). Accordingly, it was observed that the increase in NHL cases during the 1980s in USA was due to, in large part, the increase in AIDS-associated NHL (19). Nevertheless, after the advent of HAART, the epidemiology of cancers in PLWH suffered significant changes. With access to HAART, restoration of immunocompetence and aging of PLWH, the incidence of ADC started to decline while the incidence of NADC increased (20). According to a study performed with an European database, NHL incidence was 462.6 per 100.000 HIV-infected people that did not receive treatment, while the incidence was 205.1 per 100.000 HAART-treated patients (21). However it is clear that, despite the decreasing number of NHL cases in PLWH after the introduction of HAART, their estimate is not yet comparable to that of the general population and the incidence remains higher in HIV-infected people (20). Currently, even in the post-HAART era, NHLs are still reported as the most common neoplasia in PLWH (22). Among the numerous subtypes of NHLs, DLBCL and Burkitt lymphoma are the most frequent manifestations in PLWH (23, 24). In fact, about 60-70% of all NHLs from HIV-infected subjects are reported as DLBCL (24–26), indicating its relevance in this specific subcompartment of the population.
Regarding clinical presentation, NHLs may affect lymph nodes (nodal manifestation) or other tissues outside lymph nodes (extranodal manifestation), among which gastrointestinal tract and central nervous system are relatively common sites (27). NHL patients may present lymphadenomegalia and/or B symptoms (≥ 10% weight loss during the last six months, night sweating and fever ≥ 38°C) (28). NHL staging is based on the Ann Arbor system, which defines four stages according to the disease dissemination (29, 30), while the International Prognostic Index (IPI) is used for the prognosis of NHL patients (31).
In the current review, we address one of the most relevant subtypes of NHLs, DLBCL. We cover its molecular characteristics and its specific features when manifested in PLWH.
The Molecular Basis of DLBCL
The DLBCL is not a homogenous disease and can be further stratified into subtypes according to gene expression signatures (Figure 1). The first report that identified DLBCL subgroups based on molecular properties was performed by Alizadeh and coworkers (2000). Using microarray assays, the authors investigated the expression pattern of genes related with B cell development and lymphomagenesis, which allowed the clusterization of DLBCL samples into two main subgroups called GCB (germinal center B-like) and ABC (activated B-like) (32). The GCB subtype was characterized by elevated expression of genes associated with germinal center (GC) phenotype, such as BCL-6, BCL-7A, LMO2 and others, while the ABC subtype was characterized by low or undetectable levels of GC markers accompanied by high expression of genes related to plasmacytic differentiation, a post-GC phenotype (32, 33). Interestingly, even though the ABC subtype initiates the transition towards the pro-plasmacytic program, as observed by the high levels of IFR4 and other markers (34), ABC cells are incapable to conclude the plasmacytic differentiation because they fail to stimulate transcription factors ultimately required for the acquisition of plasma cell phenotype (35), such as Blimp-1 (36, 37). In fact, Blimp-1 mutations that inhibit its expression or destabilize the protein were described exclusively in ABC cases (38), emphasizing its incapacity to follow the plasmacytic program completely. Therefore, ABC subtype represents an abnormal stage of B cell ontogenesis in which the differentiation is arrested in between the end of germinal center reaction and the commitment with plasma cell generation (33). The distinction between GCB and ABC is relevant since it adds prognostic value and, classically, ABC patients exhibit worse survival estimates than GCB subjects (32).
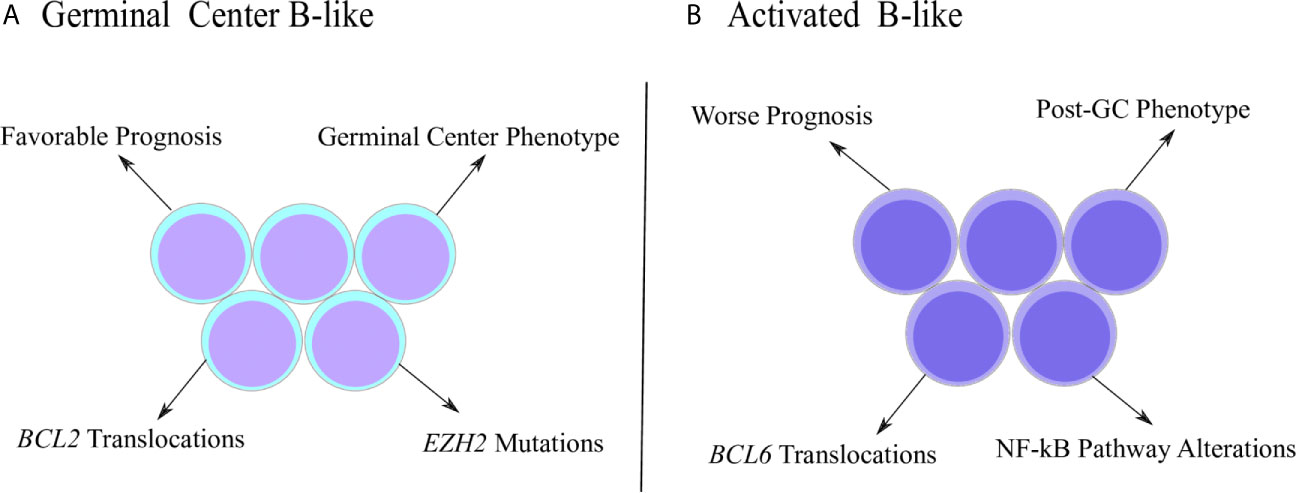
Figure 1 Distinctive characteristics of DLBCL subtypes. (A) Germinal center B-like (GCB) subtype was originally characterized by a gene expression pattern that resembled the germinal center (GC) phenotype. GCB cases are associated with better prognosis, as well as molecular properties, such as increased frequency of BCL2 translocations and EZH2 mutations. (B) Activated B-like (ABC) subtype was described with a gene expression pattern associated with the post-GC phenotype. ABC subjects show worse prognosis and specific molecular properties, such as higher frequency of BCL6 translocations and alterations involving the NF-kB pathway.
After the first description of DLBCL subtypes, new methodologies for subtype identification were published. Wright and coworkers (2003) identified a list of 27 genes with predictive value to distinguish between ABC, GCB and a third group called unclassified, since it did not fit into either of the two categories established (39). In agreement with the original report (32), the subtypes described according the list of 27 genes also had prognostic value and the overall survival estimates were 31%, 59% and 47% for ABC, GCB and unclassified cases respectively (39). On the other hand, the Hans algorithm proposes subtype identification based on immunohistochemical assessment of three key proteins: BCL-6, IRF4 and CD10. According to this method, the authors were able to distinguish between GCB (CD10+, IRF4- and/or BCL6+) with overall survival of 76% and non-GCB cases (CD10- and BCL6- or CD10-, BCL6+ and IFR4+) that showed only 34% overall survival estimates (40). Another technology applied in DLBCL subtype identification is the Lymph2Cx panel, which interrogates the expression levels of 20 key genes using digital gene expression analysis (34). It is a method especially relevant for formalin-fixed paraffin-embedded (FFPE) samples and also aggregates prognostic values like the abovementioned methodologies (34). Interestingly, even though DLBCL subtype identification had its prognostic relevance confirmed in several reports, its predictive value was not observed among HIV-infected patients with the disease (41), indicating that the lymphoma has unique properties in the HIV infection context that must be addressed apart.
Molecular events that happen during B lymphocyte development are also a source of genetic alterations found in DLBCL (42). Besides somatic hypermutation and class switch recombination (43), nucleotide substitutions catalyzed by AID (activation-induced deaminase) may also produce somatic mutations and large-scale alterations, such as chromosomal translocations (33). Break sites in genes commonly translocated in DLBCL are often AID-targeted regions (44–46). Moreover, DLBCL samples exhibited proto-oncogene mutations at sites recognized by AID (47). Therefore, deregulation and errors resulting from B lymphocyte ontogenesis may also promote DLBCL development.
Chromosomal translocations consists on the exchange of DNA segments between distinct chromosomes and are among the most common DLBCL genetic alterations (48). The pattern generally observed is the transfer of a protoncogene segment to a site downstream of an immunoglobulin locus, usually immunoglobulin heavy-chain (IgH) (49). This mechanism positions protoncogenes under the regulatory control of IgH loci and not their original regulatory regions, affecting gene expression (33, 46, 49). BCL2, BCL6 and MYC are common translocation targets in DLBCL (50). BCL2 translocation is described as t(14;18) because it is the transfer of a chromosome 18 segment containing BLC2 to an IgH downstream site on chromosome 14 (51). This translocation is not observed in ABC subtypes, but is especially frequent among GCB cases, affecting 35-40% of cases (52, 53). Also, evidence suggests that t(14;18) is acquired early in ontogenesis during VDJ recombination breaks (54). BCL2 translocation was associated with increased protein expression, whose antiapoptotic effect favors the survival of transformed cells (52, 53, 55). In ABC subtype, increased expression of BCL2 was also reported, but was associated with chromosomal duplications and not t(14;18) (55). Moving forward, BCL6 translocations were described either affecting IgH locus on chromosome 14 or non-Ig loci. The t(3;14) event occurs between IgH and a fragment of chromosome 3 containing BCL6 (56). Even though this translocation affects only 10% of GCB cases, 25% of ABC samples comprise that event (57). This may also be another mechanism by which the ABC subtype is unable to completely assume the plasmacytic program, since t(3;14) positions BCL-6 under the command of IgH regulatory regions and prevents its complete inhibition after the end of the germinal center reaction (58). ABC samples carrying t(3;14) expressed higher levels of BCL6 than non-carriers of the same subtype (57). However, in analysis without subtype stratification, BCL6 expression was not significantly affected by t(3;14) (57, 59). On the other hand, BCL6 translocation with non-Ig partners (such as histone H4) were associated with increased gene expression (60).
MYC translocation towards the IgH locus was reported as t(8;14) (61). It is a less common event than BCL2 and BCL6 translocations, affecting about 10% of DLBCL cases (62, 63). Among t(8;14) carriers, the majority belongs to GCB subtype (64). MYC is a transcription factor related with cell cycle and survival, and therefore its increased expression after juxtaposition to IgH locus also contributes to carcinogenesis (65). BCL2, BCL6 and MYC translocations are not mutually exclusive and may occur together in the same patient, forming the double-hit and even triple-hit lymphomas (66, 67). A study with 155 DLBCL subjects showed that 2.3% were double-hit for MYC/BCL2, 2.3% were double-hit for MYC/BCL6 and 0.8% (one patient) was triple-hit and carried the three translocations (68). In general, carriers of multiple hits (two or three translocations) are associated with worse survival (55, 64, 69).
Besides chromosomal translocations, some point mutations in DLBCL were also reported. Nucleotide substitutions, deletions and duplications were described in several genes, such as BCL6, MYC, PAX5, PIM1, RhoH and others (47). A large genomic analysis performed with 1,001 DLBCL cases described a list with the 60 most frequently mutated genes (70). Among them, they described mutations in MYC, PAX5, BCL2, CARD11, CDKN2A and other targets, indicating the genetic heterogeneity found in this cancer. However, the authors also observed some common patterns. For example, BCL2, with known antiapoptotic properties (71) and CARD11, a gene associated with lymphocyte activation (72) suffered missense mutations or copy number gains, while genes involved with cell cycle inhibition, such as CDKN2A (73) suffered nonsense mutations or copy number losses (70). Overall, mutations found in DLBCL favored pathways associated with survival or cell cycle progression and blocked pathways with the opposite effect (74). Accordingly, CARD11 was pointed as an important oncogene for DLBCL development in other reports (75, 76).
The distribution of some mutations also varies according to the DLBCL subtypes (77). Specifically, in GCB cases lesions that allow the persistence of the germinal center transcriptional program are observed, preventing the transformed cells to move forward to the following stages of normal B cell development, a process called “locking in” (33). One of the main components responsible for keeping the germinal center phenotype in GCB cases is the EZH2 enzyme, a known repressor of transcription factors needed for plasma cell differentiation, such as Blimp1 and IRF4 (78). The inhibition occurs with the triple methylation of lysine 27 residue on histone H3 located in regulatory regions of those genes (78). Thereby, the enzyme suppresses the pro-plasmacytic program and confines the transformed cell within a GC phenotype (58). Gain of function mutations in EZH2 are found in 20% of GCB cases and are not found in ABC subtype (79), a pattern already described in the literature (70, 74, 80). Those are missense mutations in which a tyrosine residue from the catalytic site of the enzyme is replaced by another amino acid, affecting directly its affinity for the substrate, which results in hypermethylation of the target regions and leads to an inhibition even more pronounced than by wild-type EZH2 (81). On the other hand, ABC subtype is characterized by alterations in genes of the NF-kB pathway (70, 80). NF-kB is a transcription factor important for B cell development, mediating proliferation, survival and apoptotic pathways (82). The NF-kB pathway is constitutively activated in ABC subtype (83). In vitro administration of NF-kB pharmacological inhibitors was toxic exclusively in ABC cell lines and did not have an effect in GCB models (83). Some genetic lesions are responsible for NF-kB constitutive activation. TNFAIP3, a NF-kB repressor, suffers deletions or loss of function mutations in 30% of ABC cases (84). In parallel, MYD88, an activator of NF-kB, showed gain of function mutations in 29% of ABC samples (85). Alterations in those genes are rare or not reported for GCB (70, 84, 85).
Regarding DLBCL treatment, the main option is chemotherapy. Initially, a combination of drugs called CHOP (cyclophosphamide, hydroxydaunorubicin, vincristine/oncovin and prednisolone) was used (86–88). The advent of an anti-CD20 monoclonal antibody (rituximab) with antitumoral activity (89) led to its addition to chemotherapeutic regimens, creating the R-CHOP treatment. R-CHOP-treated patients showed significantly greater survival estimates when compared to CHOP-treated subjects (90–93). For example, progression-free survival estimates were 66% and 45% for R-CHOP and CHOP groups, respectively (90). Currently, R-CHOP is the principal treatment option for DLBCL, but 30% of relapsed cases are still observed (90). More recent studies are searching for treatment strategies according to DLBCL subtypes, especially for ABC given its worse survival performance. Since NF-kB pathway function is crucial in ABC subjects, a study raised the hypothesis that bortezomib, an NF-kB inhibitor, could exert selective efficacy on this subtype (86). However, the combination of bortezomib and R-CHOP did not improve survival estimates when compared to R-CHOP regimen alone (94, 95), indicating that new subtype-specific targets must be studied. Accordingly, the combination of R-CHOP and ibrutinib, a BTK inhibitor, resulted in better survival estimates in ABC patients that were younger than 60 years when compared to R-CHOP alone (96). Although statistical significance was not observed, the R-CHOP and lenalidomide combination showed a trend towards an improved survival among ABC patients with worse IPI scores (≥ 3) (97).
HIV-Induced Modifications in B Cells
Although the general molecular mechanisms involved with DLBCL pathogenesis are well documented and summarized above, the specificities of DLBCL in an HIV-infected context are yet unclear. Despite T-CD4 lymphocyte depletion being one of the hallmarks of HIV infection, viral-induced effects are also reported for the B cell compartment (Figure 2). Knowing that B lymphocytes are the progenitor cells of DLBCL, we will first approach how HIV is able to exert its influence over B lymphocytes either by direct or indirect mechanisms throughout its life cycle in this section. As suggested by Moir & Fauci, most of the B cell alterations are due to unbalanced distribution of subpopulations, that is, while certain subgroups of B cells suffer a shrinkage under the influence of HIV, others will undergo enrichment (98) and even participate in lymphomagenesis.
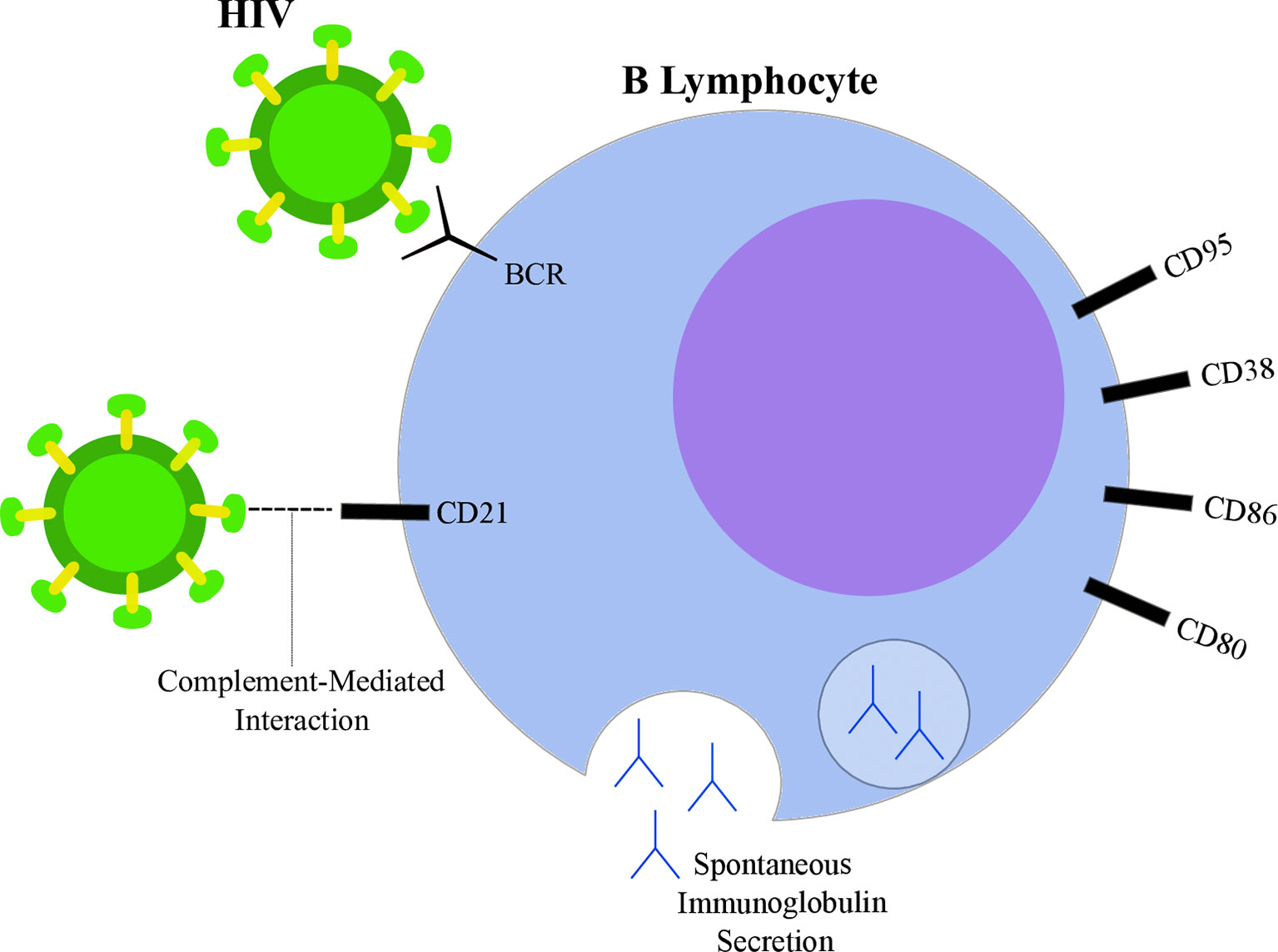
Figure 2 Highlights of HIV-induced B lymphocyte alterations. HIV particle (green) is able to bind B lymphocyte (blue) through direct interactions with surface immunoglobulins (B-cell receptor, BCR) and through CD21 interactions mediated by complement proteins. Spontaneous immunoglobulin secretion leading to hyperglobulinemia is observed in the context of HIV infection, as well as alterations in surface markers, such as increased expression of CD80, CD86, CD38 and CD95.
Studies suggest that HIV is able to directly interact with surface molecules on B lymphocytes. Despite not expressing CD4, interaction between the viral glycoprotein gp120 and membrane immunoglobulins from the variable heavy chain 3 (VH3) family has been reported (99). Moreover, gp120 was shown to interact with C-type lectin receptors on B lymphocytes (100, 101). CD21 is another known binding site of HIV in B cells through interactions mediated by complement proteins (102). However, whether this interaction is able to induce direct effects on lymphocytes is still being discussed (98). On the other hand, a known participation of CD21-dependent interactions contributes to the maintenance of a extracellular reservoir of surface-bound viral particles potentially transmittable to T-CD4 cells (103, 104). Furthermore, HIV may also influence B cells through indirect mechanisms by altering the cytokine secretion pattern of different cell types. Indeed, several cytokines were found overexpressed in plasma samples from PLWH when compared to uninfected controls (105). For example, HIV increased T-CD4 lymphocyte spontaneous IL6 secretion (106), IL10 and TNFα production by monocytes exposed to viral glycoproteins (107) and IFNα secretion by plasmacytoid dendritic cells (108). Those cytokines contribute to B cell activation, differentiation (109, 110) and may also guide the modifications observed upon HIV infection.
A common feature observed within the B cell compartment during HIV infection is the acquisition of a hyperactivation phenotype, characterized by a plethora of physiological alterations. One of the first studies to suggest chronic B cell activation in PLWH was performed in 1983 by Lane and coworkers. The authors indicate that, although the peripheral B cells extracted from AIDS donors were less responsive to antigenic stimulation, those cells exhibited spontaneous secretion of IgG, IgA and IgM at levels 10 times higher than HIV-uninfected controls (111). In agreement, the increased serum levels of immunoglobulin molecules, a state defined as hyperglobulinemia, was also associated with HIV infection (112–114). Analyses regarding the cellular source of immunoglobulin secretion were suggestive of a polyclonal B cell activation in PLWH (115). Taken together, these data suggest a shift in the B cell compartment towards a pro-plasmacytic pathway, since increased antibody secretion was reported under the influence of HIV infection (98, 116). This pattern was also observed when the peripheral B cells from PLWH were shown to be enriched in a population with low levels of CD21. This subgroup (CD21low) was associated with diminished proliferative capacity after antigenic stimuli and with plasmacytoid morphological features (116). Indeed, both CD21 downregulation and decreased proliferative response are related to cell priming into the plasmacytic differentiation program (117). The favoring of plasmacytoid pathways upon HIV infection was reported by others (118, 119), emphasizing the viral effect on reorganizing the pool of B cells towards a less responsive and terminally differentiated route. Interestingly, while CD21low cells showed enhanced antibody secretion in PLWH (116), they were not able to spontaneously secrete immunoglobulin in uninfected individuals (117). Another aspect of HIV-induced B cell hyperactivation is the modification of surface markers, such as the increased expression of activation-related molecules (120). Nevertheless, although the baseline levels of CD86 and CD80 were higher among HIV subjects, their B cells were not capable of upregulating the expression of those markers upon receiving proper stimulation (121). Similarly, CD38 levels were higher in B cells from HIV-infected donors, but those cells were also more susceptible to apoptosis since the levels of the proapoptotic CD95 molecule were increased (122). Those results are in agreement with, and help to delineate the paradoxical nature of the hyperactivation phenotype: while B cells show evidence of activation at resting state, they are poorly responsive to adequate stimuli, which contributes to impaired humoral responses.
Additional HIV-induced effects on the B cell compartment were reported in the literature, such as effects on memory B cells and immune exhaustion, a state defined by diminished proliferative response and effector functions upon antigenic stimulation (123). As described above, the loss of CD21 expression is a valuable marker of HIV disease in B cells. The CD21low subpopulation forms a heterogeneous cluster that reveals multiple B cell modifications under influence of HIV (124), including evidence of immune exhaustion. Although CD21low cells were initially associated with plasmacytoid features, confirming the hyperactivated phenotype, not all of them belong to the pro-plasmacytoid compartment (98). A fraction of CD21low cells characterized by low levels of CD27 and high expression of FCLR4 has been observed in HIV+ patients (125). The expression of those markers also described a specific subpopulation of memory B cells found originally in tonsils (126). Therefore, those CD21LOW CD27- FCRL4+ cells were subsequently named as tissue-like memory cells and were shown to be enriched in PLWH (127). Interestingly, evidence of premature exhaustion was found in tissue-like memory cells, demonstrating their poor responsiveness to appropriate BCR stimulation when compared to conventional memory B cells (127, 128). Those results indicate the expansion of an abnormal B cell subpopulation in PLWH, suggesting the enrichment of an exhausted memory B cell phenotype and impaired humoral responses.
In this scenario, an interesting topic is whether highly active antiretroviral treatment (HAART) is able to reverse HIV-induced B cell modifications. Although it is well established that the therapy is capable of minimizing B lymphocyte alterations, the extension of that activity is suggested to be partial and not valid for all the observed HIV-induced effects. Hyperactivation and hyperglobulinemia are examples of alterations completely resolved by HAART (129). When compared to untreated patients, immunoglobulin serum levels were significantly decreased after HAART administration and correlated with HIV viral load reduction (129, 130). However, effects regarding the reorganization of B cell compartments are incompletely reversed by HAART (131). As demonstrated by Moir and collaborators, some B cell subpopulations from HAART-treated PLWH reached percentages comparable to those found in uninfected donors, however the increase in resting memory cells was slow and incomplete, revealing the persistence of memory B cell deficiency (131). The partial restoration of that compartment was confirmed by others, indicating that HAART-treated subjects still have lower levels of memory B cells than uninfected counterparts (132). In agreement with those findings, the CD21low subset enrichment, one of the main B cell alterations upon HIV acquisition, was not completely abolished after HAART introduction, and CD21low counts were still significantly higher in HAART-treated patients than in uninfected controls, demonstrating a partial restoration of B cell subsets (133). In contrast, a report by Tanko and coworkers showed that, after HAART treatment of HIV subjects, the percentages of immature transitional, naive and memory B cell groups were equivalent to non-HIV individuals, except for plasmablasts, which continued to be enriched in PLWH even after HAART (134). Altogether, these results suggest that even after HAART, residual modifications in B cell subpopulations persist in PLWH.
HIV-Induced Lymphomagenesis
Classically, HIV has been associated with lymphomagenesis (and carcinogenesis in general) because of its immunosuppressive activities. PLWH, while immunosuppressed, loose immunological vigilance over other pathogens and become permissive to oncogenic viral infection (135). Regarding lymphoma development, EBV (Epstein-Barr Virus) co-infection has a meaningful role (136, 137). EBV is a gamma-herpes virus able to infect B lymphocytes through interactions with CD21 (138). EBV infection is worldwide disseminated and progresses as an asymptomatic condition throughout life in the majority of carriers (139, 140). The activity of T lymphocytes is crucial to control EBV infection, assuring its asymptomatic status (141). However, HIV-induced immunosuppression favors EBV oncogenic activities, worsening the risk of lymphoproliferative diseases and lymphomas (142). The exact mechanisms by which EBV is able to induce oncogenesis are beyond the scope of this review and are addressed elsewhere (143–145). Some examples of oncogenic activities are found in EBV proteins produced during its life cycle. LMP1 (Latent Membrane Protein 1) is able to transform B cells and to stimulate lymphomagenesis (146). Its mechanism of action consists on the mimicry of CD40 physiological signaling, stimulating lymphocyte proliferation (147) and antiapoptotic pathways in an NFKB- (148) and Akt- (149) dependent manner. Another example is EBNA2 (EBV-encoded Nuclear Antigen 2), which also exhibits pro-survival activity and favors B cell lymphoma development (150). On the scope of reduced immunological surveillance, the HIV-infected context, especially in intravenous drug users, is associated with higher rates of HCV infection (151). HCV positivity was associated with lymphomagenesis (152). NHL patients with HIV showed even greater frequency of HCV infection than PLWH that did not exhibit cancer (153). HCV infection was also associated with worse overall survival and increased the risk of NHL development (153), suggesting another tumorigenic mechanism in HIV-infected subjects. Even though immunosuppression is one of the main oncogenic mechanisms of HIV infection, direct effects induced by the virus are also being reported as important contributors to lymphomagenesis.
As discussed in the previous section, HIV exerts influence over the host’s B cell population. In this scenario, effects on B lymphocytes contributing to lymphomagenesis have been reported. The B cell hyperactivation under HIV influence (111, 116, 133) is also associated with immunoglobulin (Ig) class switching (154, 155), one of the main sources of genetic variability in B cell lymphomas (33, 44, 46). Therefore, it may contribute to the acquisition of genetic lesions related to lymphomagenesis, such as chromosomal translocations (45, 156). Activation-induced deaminase (AID) is an enzyme whose activity is linked with the double-strand breaks needed for translocation events (43, 45). The interaction between the HIV gp120 glycoprotein and C-type lectin receptors on B lymphocytes is able to upregulate AID expression and, consequently, to trigger class switching events (100, 101). An additional triggering mechanism was observed where HIV particles with envelopes carrying CD40L host molecules stimulate B lymphocytes to, similarly, upregulate AID and proceed through Ig class switch (157). Indeed, previous reports had already observed high expression of immunological markers associated with Ig class switch in PLWH. Those studies showed that, when compared to HIV subjects that did not progress to lymphoma, high serum levels of IL6, IL10 and IgE were observed specifically among the ones that developed cancer until three years after enrolment (158, 159), reassuring the relevance of this pathway for HIV-associated lymphomas. Indeed, both IL6 and IL10 are cytokines associated with Ig class switch induction (109). Therefore, such favouring of AID expression and Ig class switch upon HIV infection may contribute to the acquisition of genetic lesions potentially able to drive lymphoma.
An additional mechanism recently proposed for HIV-induced carcinogenesis is the release of pro-tumoral exosomes. Exosomes derived from infected cells were enriched in HIV transactivator response element (TAR) RNA and associated with antiapoptotic properties (160). Chen and coworkers showed that exosomes from T cells or from PLWH plasma samples were able to directly induce cancer cell proliferation and oncogene expression through the EGFR/TLR3 axis followed by ERK1/2 phosphorylation (161). Interestingly, those effects were not observed when B lymphoma cell lines were used, apparently due to the lack of EGFR expression (161). However, exosome-derived microRNAs (miRNAs) were proposed as relevant biomarkers for Hodgkin lymphoma diagnosis in HIV-infected donors (162) and, therefore, it remains to be elucidated whether or not exosomes may have a role in lymphomagenesis or if their effect is cancer-specific.
Besides the contribution of HIV infection to class switching events, more recent reports have demonstrated direct participation of viral proteins during lymphoma development (163). For example, viral p17 matrix variants were able to stimulate proliferative and antiapoptotic pathways in B lymphocytes, facilitating their clonal expansion (164). The p17 variants with proliferative effect pass through a conformational change and expose an amino acid sequence originally enclosured in wild-type p17 three-dimensional structure (165). Even though the identity of the receptors involved in p17 signaling is not fully understood, interactions between p17 and CXCR1 or CXCR2 were demonstrated and associated with angiogenesis (166), another pro-tumoral effect. Besides p17, oncogenic activities were also reported for other viral proteins. Kundu and coworkers showed that Tat (transactivator of transcription) expression in mice favored B cell lymphoma generation in approximately 30% of animals (167). Accordingly, a study pointed that Tat was able to activate DNA repair proteins among B cells cultivated together with HIV (168), as well as to induce angiogenesis (169). More evidence indicates that Vpr (viral protein R) and Vpu (viral protein U) also participate in HIV-induced lymphomagenesis. For example, Vpr induced DNA double-strand breaks in infected cells (170), while Vpu was important for lymphoma adhesion in endothelial cells (171). Altogether, given the relevance of HIV in inducing specific lymphoma promoting pathways, we will approach the characteristics of HIV-related DLBCL in the next sections.
Molecular Characteristics of HIV-Related DLBCL
The HIV-related DLBCL has molecular properties underpinning its specific clinical features described above. In this context, a report sought to investigate whether well-established prognostic genes would be equally informative for the HIV-related DLBCL. The authors reported that, although relevant for immunocompetent hosts, the expression of BLC2, Blimp-1 or FOXP1 did not correlate with patient outcome in PLWH. Interestingly, neither DLBCL subtype (ABC and GCB) was informative of survival in the HIV+ cohort (172), a pattern later confirmed by others (41, 173), suggesting that HIV-associated DLBCL is a particular disease and has its own molecular and pathological properties. In this section, we will address molecular differences reported in the literature when comparing HIV-DLBCL and IC (Immunocompetent)-DLBCL such as gene expression, miRNA levels and chromosomal organization, since the predictors applicable to DLBCL in the general population are not completely valid for PLWH (Table 1).
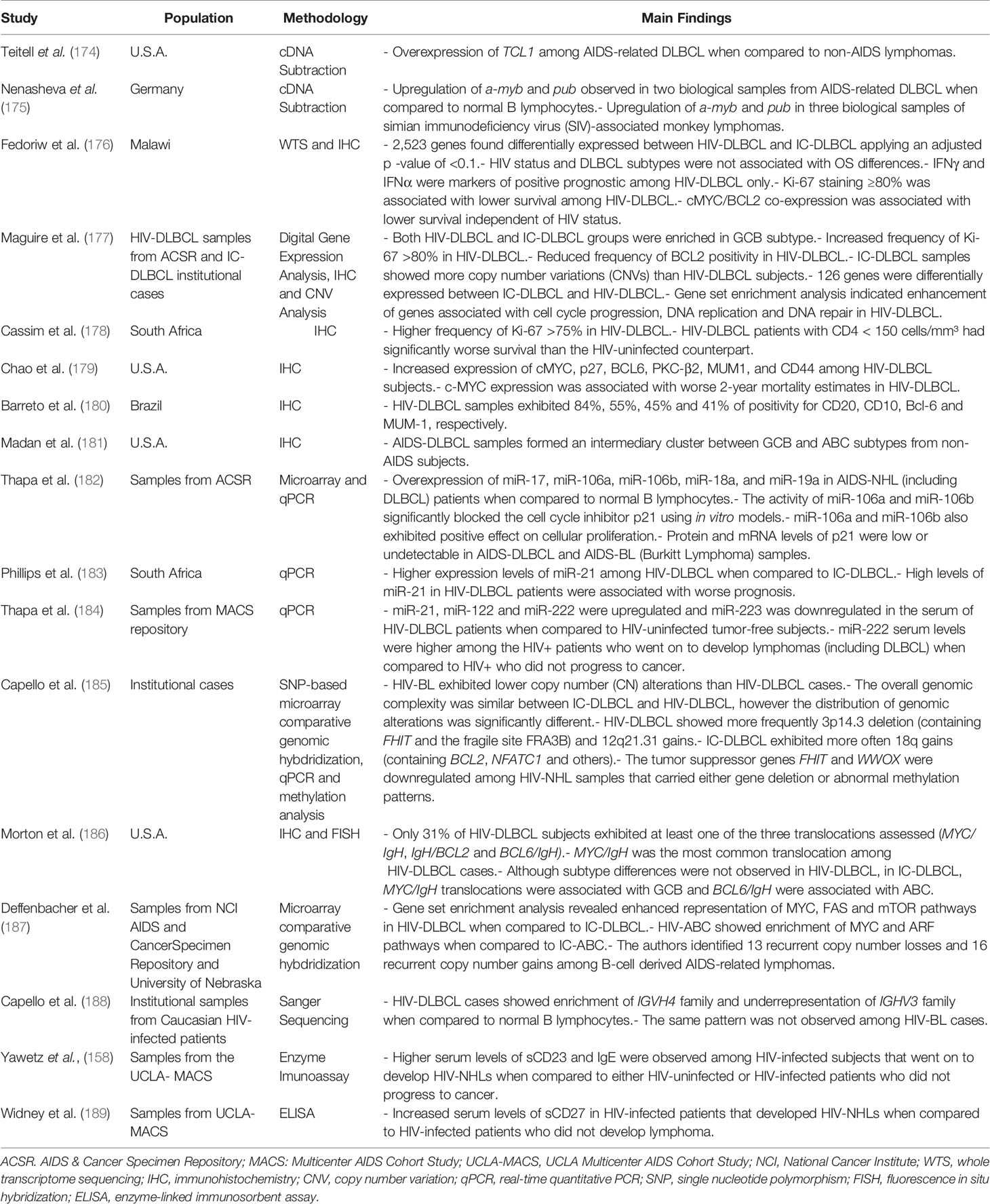
Table 1 Highlights of reports that explored the molecular characteristics of HIV-DLBCL.
One of the first reports to address gene expression signatures in HIV-DLBCL was performed by Teitell and coworkers (1999). Using cDNA subtraction techniques, they described overexpression of TCL1 (T-cell leukemia 1) oncogene in AIDS-related DLBCL samples when compared to non-AIDS tumors (174), which was later confirmed by the same group (190). Moreover, upregulation of a-myb and pub genes was also described in HIV-DLBCL cases, despite the limited number of human biological samples (n = 2) available in the study (175). More recent data compared the transcriptional profiles between 22 HIV-DLBCL and 14 IC-DLBCL from Malawi (Africa) using the whole transcriptome sequencing technology (176). The analysis revealed HIV status as a major contributor to differences observed in the expression levels of 2,523 genes. In fact, HIV-DLBCL samples were enriched in pathways related to hypoxia and cell metabolism when compared to the immunocompetent counterparts. Likewise, higher expression of IFNγ and IFNα were associated with better outcomes only in HIV-DLBCL patients (176). Another study found 126 differentially expressed genes when comparing the expression profiles between GCB subtypes from either PLWH or HIV-uninfected donors. The HIV-related GCB showed upregulation in genes associated with cell cycle progression, downregulation of cell cycle inhibitors and enhanced expression of DNA repair genes (177), indicating greater proliferative capacity. In fact, Ki67 staining, a proliferation marker, was reported as a valuable prognostic marker in HIV-DLBCL (172, 176). Consistently, stronger Ki67 staining was observed in HIV-DLBCL samples when compared to immunocompetent donors (177, 178). Immunohistochemistry studies have also provided valuable insights about HIV-DLBCL. The protein levels of cMYC, BCL6, PKC-β2, MUM1 and CD44 were significantly increased in HIV-DLBCL patients, while p27 levels were reduced (179). Among all those differentially expressed markers, cMYC positivity was associated with inferior survival in HIV-infected subjects (179), indicating new possible predictors for this population. cMYC and BCL2 simultaneous overexpression (double expressor lymphomas) comprehends a relevant inferior prognostic predictor in non-infected donors (191, 192). Some studies showed that the frequency of double expressors is similar between HIV-DLBCL and immunocompetent patients (193). However, its prognostic relevance was not yet addressed in the HIV-infected context, even though cMYC (179) and BCL2 (194) expression were described separately as prognostic factors for HIV-DLBCL. Similarly, another immunohistochemistry study detected high frequency (around 40%) of HIV-DLBCL samples positive for BCL6 and MUM1 (180).
Since DLBCL molecular subtypes are also determined by gene expression profiling (32), some reports investigated which one of them (ABC or GCB) were predominant in HIV-DLBCL. Those investigations, however, resulted in conflicting data. Some reports consistently show enrichment of GCB phenotype in HIV-DLBCL samples (172, 176, 177, 179, 180). On the other hand, enhanced expression of ABC markers was also demonstrated for those cancers (186, 187, 195). A third approach has indicated that HIV-DLBCL possess intermediate features between ABC and GCB, co-expressing markers of both subtypes simultaneously (181, 195, 196), which may explain discrepancies between reports. Indeed, a clusterization analysis performed by Madan and collaborators revealed that AIDS-related DLBCL samples formed an intermediary group between GCB and ABC clusters originated from non-AIDS donors (181). Taken together, the heterogeneous results on the cellular origin of DLBCL in the HIV+ context indicate that more studies are necessary to elucidate whether there is a subtype prevalence among HIV-DLBCL subjects or if the frequencies vary according to the study population.
Differential miRNA expression was also associated with HIV-DLBCL phenotype. When compared to non-neoplastic B cells from healthy donors, overexpression of miR-17, miR-106a, miR-106b, miR-18a and miR-19a was detected in lymphomas arising in the HIV+ context, including DLBCL. Among those, miR-106a and miR-106b significantly blocked the p21 cell cycle inhibitor and, consequently, enhanced cellular proliferation (182), which corroborates with the increased proliferative behavior already described in HIV-DLBCL. Also, miR-21 expression was higher in HIV-DLBCL than in IC-DLBCL and those patients considered “miR-21 high” exhibited poorer survival (183). Another report demonstrated elevated serum levels of miR-222 in PLWH prior to DLBCL development, suggesting its measurement as a valuable marker for the identification of HIV+ subjects at risk of developing lymphoma before diagnosis (184).
Besides miRNA expression signatures, different chromosomal alterations were reported comparing HIV-DLBCL and IC-DLBCL. A study showed that, although the overall number of genomic alterations was similar between groups, the distribution of certain lesions was significantly associated with HIV status (185). The most common alterations in HIV-DLBCL were deletions in 3p14.3 and gains of 12q21.31, while gains of 18q (a region containing BCL2) were the most frequent rearrangements in IC-DLBCL. The authors also showed that the alterations had functional impact. The chromosomal deletions enriched in HIV+ samples were associated with reduced expression of known tumor suppressor genes, such as WWOX, FHIT, DCC and PARK2 (185), another example of a carcinogenic pathway exclusively detected in HIV-DLBCL. Morton and coworkers (2014) also showed that, among the three most common translocation targets in DLBCL, translocation of MYC was the most frequent in HIV-infected individuals, while translocations of BCL2 or BCL6 were rare (186). In agreement with both previous reports, enrichment of MYC targets, as well as losses affecting WWOX and FHIT were observed in HIV-DLBCL by Deffenbacher and colleagues (2010) (187). Altogether, multiple data show various molecular mechanisms altered specifically in PLWH and support the assumption that HIV-DLBCL is a particular disease with a considerable amount of differences from its manifestation in immunocompetent hosts.
Lastly, few studies have also indicated deregulation of immunological pathways in HIV-related lymphomas (155). For example, the pattern of immunoglobulin gene rearrangement products observed in HIV-DLBCL is particular and differs from that of HIV-uninfected individuals (189). Capello and coworkers (2008) showed an enrichment of IGHV4 family (especially the IGHV4-34 gene) and an underrepresentation of IGHV3 family (in particular, the IGHV3-23 gene) among HIV-DLBCL patients (188). Moreover, serum levels of the soluble forms of CD23 and CD27 were increased in AIDS patients who went on to develop lymphomas, including DLBCL, when compared to HIV+ or even to AIDS subjects who did not progress to cancer (158, 189). Although there is a lack of data addressing immune-related gene expression signatures specifically in HIV-DLBCL, those seminal reports provided valuable insights about immune pathways possibly altered. Interestingly, in immunocompetent subjects, sCD27 was not associated with increased risk of DLBCL (197), corroborating its relevance and biomarker potential in HIV-DLBCL only.
HIV-Related DLBCL: Particular Clinical Features and Treatment Outcomes
The HIV+ population not only harbors greater incidence estimates of non-Hodgkin lymphomas (24–26), but also DLBCL is clinically distinct between PLWH and the general HIV-uninfected population. Some reports comparing clinical variables between HIV+ and HIV- people affected by DLBCL or NHLs in general have indicated differences regarding age at diagnosis, tumor staging, frequency of symptoms and frequency of extranodal site involvement (Figure 3). A report from Spanish patients with DLBCL described that HIV+ individuals were significantly younger at the time of diagnosis compared to the HIV-uninfected counterparts (median 44 years vs. 62 years, respectively). Also, HIV+ patients with DLBCL exhibited more frequently B-symptoms, later clinical staging (III-IV) and worse ECOG score (≥ 2) than HIV- subjects (198). The younger age at diagnosis appears as a common feature of lymphomas from PLWH and was also noticed in a report studying HIV-related NHL cases in Italy (199). In agreement with those previous reports, data from USA and Puerto Rico confirmed the earlier age presentation, higher frequency of B-symptoms and more advanced clinical staging (III-IV) in HIV+ patients with either DLBCL or NHLs (24, 200). Altogether, the results consistently show particular clinical features of DLBCL in PLWH from different populations worldwide. Additionally, other clinical specificities were reported in the literature. Increased risk of DLBCL occurring at a extranodal site was observed in PLWH when compared to HIV-uninfected patients (25, 200, 201), although this difference was not observed in the work by Baptista and collaborators (198) possibly due to differences in study population and sample size. Among extranodal sites, gastrointestinal tract was significantly more common in HIV+ patients (25, 201, 202) and an increase in central nervous system involvement was also reported (24). It is noteworthy that, in general, the reports did not observe statistical differences between HIV+ and HIV- subjects regarding the international prognostic index (IPI) (25, 198, 203). However, an exception is the work by Spina and collaborators which found worse IPI scores (≥ 2), significantly more frequent in PLWH (201). Taken together, data from different reports indicate that, in fact, the DLBCL (and the NHL as a whole) from PLWH has a particular clinical presentation suggestive of more aggressive features at diagnosis.
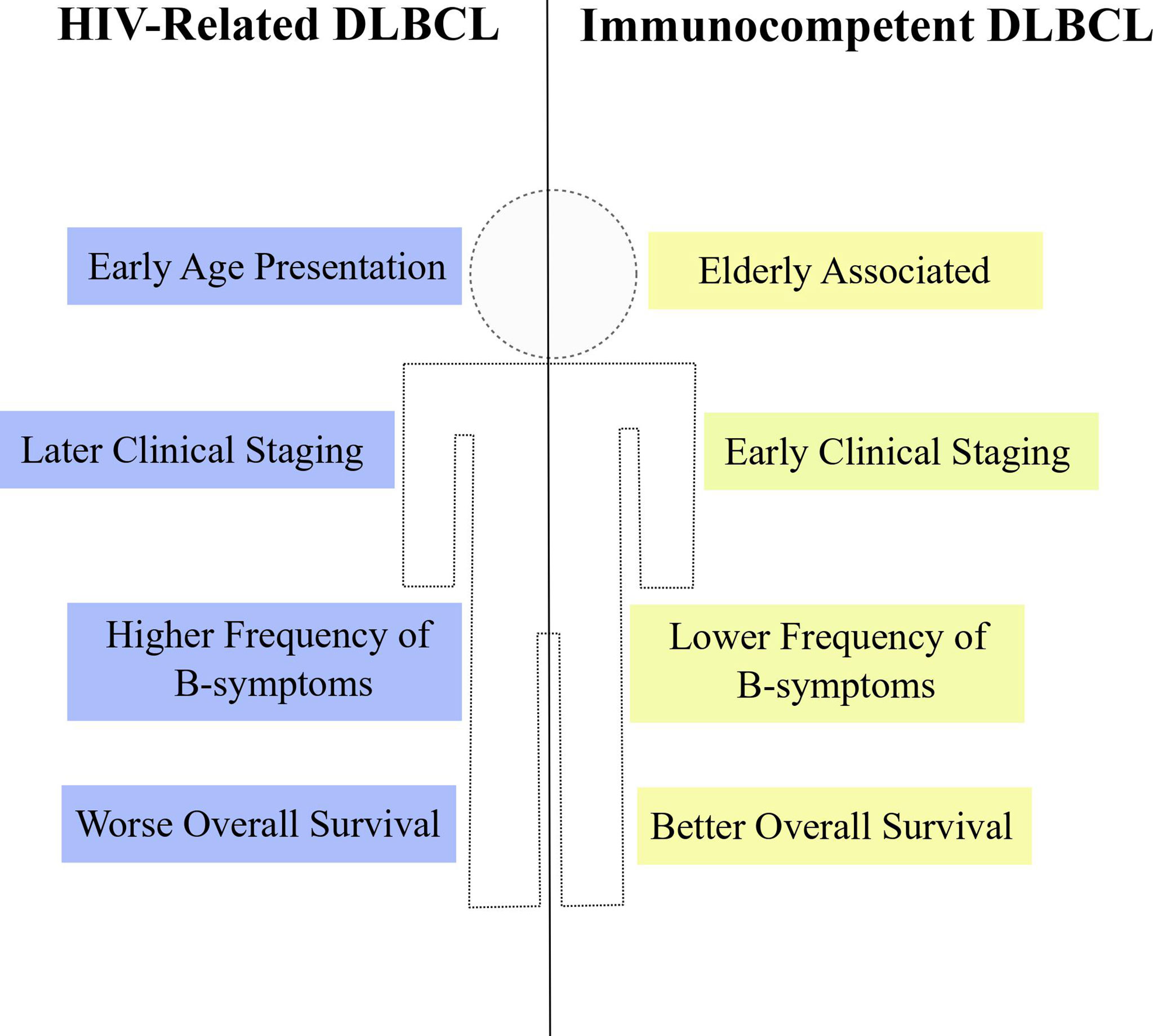
Figure 3 Clinical properties of HIV-DLBCL. HIV-related DLBCL (blue) is commonly associated with particular clinical features when compared to immunocompetent DLBCL (yellow), such as early age at diagnosis, later clinical staging, higher frequency of B-symptoms and worse overall survival estimates.
Besides analyzing clinical features, some reports have also compared survival estimates between HIV+ and HIV- patients with DLBCL or NHL. The overall survival (OS) of HIV+ subjects with DLBCL was lower than in the HIV-uninfected counterparts even in multivariate models adjusted for type of treatment (24, 198–200). However, differences in the survival estimates according to the HIV status are not clear when analyzing specifically the death by lymphoma instead of death by any cause (overall survival). Indeed, the disease-free survival (DFS) did not differ between HIV-DLBCL and immunocompetent DLBCL (IC-DLBCL) treated with R-CHOP (198). Also, the 2-year lymphoma specific mortality remained unchanged between NHL patients with or without HIV when the former group had CD4 counts of at least 200 cells/μL and no history of other AIDS-defining illnesses, although the ones with less than 200 cells/μL and/or AIDS-defining illness performed worse when compared to non-HIV subjects (24). An analysis with HIV+ patients with different types of cancers showed that there were no differences in cancer-specific mortality for DLBCL subjects stratified according to the HIV status and, in fact, 64.8% of all deaths from PLWH with DLBCL were attributable to HIV-related complications and not to the lymphoma itself (204). Nevertheless, in contrast with those previous reports, Coutinho and collaborators reported unexpected better estimates of both OS and DFS in HIV+ individuals with DLBCL (25). Taken together, the results indicate that the HIV-DLBCL is accompanied by worse OS and equal estimates of lymphoma-specific death, suggesting that, independently of HIV, the response to lymphoma treatment could be similar in both groups. In agreement with that, a systematic review showed that the R-CHOP regimen is a lymphoma treatment associated with improvement in OS and progression-free survival (PFS) in HIV-infected patients. The authors also demonstrate that the use of R-CHOP together with HAART did not affect survival or response to treatment (26), emphasizing the effectiveness of this chemotherapeutic regimen in PLWH. Moreover, the administration of R-CHOP significantly improved the survival and reduced the frequency of death due to lymphoma in HIV-DLBCL patients under HAART treatment (205), which may explain the unchanged estimates of lymphoma-specific mortality seen in previous studies.
In contrast with the data described above (26, 205), the first studies to address the response to chemotherapy in HIV-related DLBCL cases raised the concern of treatment-related toxicity being increased in this group. For example, the work by Kaplan and collaborators indicated that the addition of rituximab to the CHOP drug combination (the R-CHOP regimen) was associated with greater incidence of adverse effects in PLWH (206). The addition of rituximab to the CDE (cyclophosphamide, doxorubicin and etoposide) regimen was also suggestive of increased frequency of adverse events in HIV-related NHL subjects (207). Nevertheless, after these first publications, the feasibility of adding rituximab in different drug combinations for lymphoma treatment in PLWH has been reassessed by several clinical trials. Indeed, different reports showed the effectiveness and safety of rituximab-containing chemotherapeutic regimens for the HIV+ population (26, 208–211) and, interestingly, even in severely immunosuppressed HIV-infected patients (with CD4 counts less than 100 cells/μL), rituximab was associated with improved survival without increasing the rate of adverse effects. A possible difference between reports, as suggested by Dunleavy & Wilson (212), was that HIV+ patients with very low CD4 counts (less than 50 cells/mm3) were the most affected by the treatment-related adverse effects reported primarily by Kaplan and collaborators, while other reports excluded patients with clinical signs of advanced HIV disease (209). In fact, HAART improved the survival in patients with HIV-related B-cell lymphomas (205, 213), as well as low CD4 counts were associated with worse OS in HIV-DLBCL (214). Therefore, despite having more aggressive clinical features at diagnosis, HIV-infected patients are consistently being reported as equally eligible for chemotherapeutic regimens as well as their HIV-uninfected parallels, especially in the context of the HAART era and with comparable CD4 levels.
Concluding Remarks
HIV-DLBCL is a particular illness with specific characteristics. As shown previously by diverse data, DLBCL from PLWH is accompanied by specific clinical features, such as early age at diagnosis, higher frequency of B symptoms and extranodal involvement, as well as later tumor staging. Interestingly, unique molecular properties are also observed in HIV-DLBCL subjects, including gene expression signatures, chromosomal rearrangements and miRNAs altered levels. All in all, HIV-DLBCL-related properties may occur due to viral modulation of B cell compartments and direct influence during lymphomagenesis. Even though HIV-DLBCL patients consistently exhibit equal estimates of survival when compared to IC-DLBCL subjects (198, 204), PLWH are commonly excluded from clinical trials. In fact, regarding lymphoma-associated clinical trials, the estimates of PLWH exclusion are around 70% (215, 216). Given the intrinsic relationship between lymphoma and HIV, the inclusion of HIV-DLBCL in clinical trial protocols may benefit and improve the understanding of the disease in this particularly susceptible population.
Author Contributions
PC: wrote the manuscript. PC, FL, and MS: reviewed and edited the manuscript. All authors contributed to the article and approved the submitted version.
Funding
MS was supported by grants by the Rio de Janeiro State Science Foundation (FAPERJ) # E-26/202.894/2017 and by the Brazilian Research Council (CNPq) # 305765/2015-9. PC is recipient of a Master’s scholarship by the Brazilian National Cancer Institute (INCA).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Küppers R, Hansmann ML. The Hodgkin and Reed/Sternberg Cell. Int J Biochem Cell Biol (2005) 37:511–7. doi: 10.1016/j.biocel.2003.10.025
2. Ehrlich PF. “The Hodgkin and Reed/Sternberg Cell”. In: Coran AG, Caldamone A, Adzick AS, Krummel TM, Laberge J-M, Shamberger R editors. Elsevier: Pediatric Surgery (2012). p. 517–27. doi: 10.1016/B978-0-323-07255-7.00038-6
3. Lakhtakia R, Burney I. A Historical Tale of Two Lymphomas: Part Ii: Non-Hodgkin Lymphoma. Sultan Qaboos Univ Med J (2015) 15:e317–21. doi: 10.18295/squmj.2015.15.03.003
4. Aisenberg AC. Historical Review of Lymphomas. Br J Haematol (2000) 109:466–76. doi: 10.1046/j.1365-2141.2000.01988.x
5. Armitage JO, Gascoyne RD, Lunning MA, Cavalli F. Non-Hodgkin Lymphoma. Lancet (2017) 390:298–310. doi: 10.1016/S0140-6736(16)32407-2
6. World Health Organization (WHO). Non-Hodgkin lymphoma Globocan The Global Cancer Observatory (2020). Available at: https://gco.iarc.fr/today/data/factsheets/cancers/34-Non-hodgkin-lymphoma-fact-sheet.pdf [Accessed April 22, 2021].
7. Swerdlow SH, Campo E, Pileri SA, Harris NL, Stein H, Siebert R, et al. The 2016 Revision of the World Health Organization Classification of Lymphoid Neoplasms. Blood (2016) 127:2375–90. doi: 10.1182/blood-2016-01-643569
8. Shankland KR, Armitage JO, Hancock BW. Non-Hodgkin Lymphoma. Lancet (2012) 380:848–57. doi: 10.1016/S0140-6736(12)60605-9
9. Laurent C, Do C, Gourraud PA, De Paiva GR, Valmary S, Brousset P. Prevalence of Common non-Hodgkin Lymphomas and Subtypes of Hodgkin Lymphoma by Nodal Site of Involvement. Med (United States) (2015) 94:e987. doi: 10.1097/MD.0000000000000987
10. Anderson JR, Armitage JO, Weisenburger DD. Epidemiology of the non-Hodgkin’s Lymphomas: Distributions of the Major Subtypes Differ by Geographic Locations. Ann Oncol (1998) 9:717–20. doi: 10.1023/A:1008265532487
11. Perry AM, Diebold J, Nathwani BN, Maclennan KA, Müller-Hermelink HK, Bast M, et al. Non-Hodgkin Lymphoma in the Developing World: Review of 4539 Cases From the International non-Hodgkin Lymphoma Classification Project. Haematologica (2016) 101:1244–50. doi: 10.3324/haematol.2016.148809
12. Li Y, Wang Y, Wang Z, Yi D, Ma S. Racial Differences in Three Major NHL Subtypes: Descriptive Epidemiology. Cancer Epidemiol (2015) 39:8–13. doi: 10.1016/j.canep.2014.12.001
13. National Cancer Institute. Seer Cancer Stat Facts: Diffuse Large B-Cell Lymphoma . Available at: https://seer.cancer.gov/statfacts/html/dlbcl.html.
14. Diumenjo MC, Abriata G, Forman D, Sierra MS. The Burden of non-Hodgkin Lymphoma in Central and South America. Cancer Epidemiol (2016) 44:S168–77. doi: 10.1016/j.canep.2016.05.008
15. Ekberg S E, Smedby K, Glimelius I, Nilsson-Ehle H, Goldkuhl C, Lewerin C, et al. Trends in the Prevalence, Incidence and Survival of non-Hodgkin Lymphoma Subtypes During the 21st Century – a Swedish Lymphoma Register Study. Br J Haematol (2020) 189:1083–92. doi: 10.1111/bjh.16489
16. Clarke CA, Morton LM, Lynch C, Pfeiffer RM, Hall EC, Gibson TM, et al. Risk of Lymphoma Subtypes After Solid Organ Transplantation in the United States. Br J Cancer (2013) 109:280–8. doi: 10.1038/bjc.2013.294
17. Dal Maso L, Serraino D, Franceschi S. Epidemiology of AIDS-related Tumours in Developed and Developing Countries. Eur J Cancer (2001) 37:1188–201. doi: 10.1016/S0959-8049(01)00120-4
18. Franceschi S, Vecchia C, Maso LD, Serraino D, Rezza G. Spectrum of AIDS-associated Malignant Disorders. Lancet (1998) 352:906–7. doi: 10.1016/S0140-6736(05)60047-5
19. Coté TR, Biggar RJ, Rosenberg PS, Devesa SS, Percy C, Yellin FJ, et al. Non-Hodgkin’s Lymphoma Among People With Aids: Incidence, Presentation and Public Health Burden. Int J Cancer (1997) 73:645–50. doi: 10.1002/(SICI)1097-0215(19971127)73:5<645::AID-IJC6>3.0.CO;2-X
20. Robbins HA, Shiels MS, Pfeiffer RM, Engels EA. Epidemiologic Contributions to Recent Cancer Trends Among HIV-infected People in the United States. AIDS (2014) 28:881–90. doi: 10.1097/QAD.0000000000000163
21. Bohlius J, Schmidlin K, Costagliola D, Fätkenheuer G, May M, Caro-Murillo AM, et al. Incidence and Risk Factors of HIV-related non- Hodgkin’s Lymphoma in the Era of Combination Antiretroviral Therapy: A European Multicohort Study. Antivir Ther (2009) 14:1065–74. doi: 10.3851/IMP1462
22. Robbins HA, Pfeiffer RM, Shiels MS, Li J, Hall HI, Engels EA. Excess Cancers Among HIV-infected People in the United States. J Natl Cancer Inst (2015) 107:1–8. doi: 10.1093/jnci/dju503
23. Brunnberg U, Hentrich M, Hoffmann C, Wolf T, Hübel K. Hiv-Associated Malignant Lymphoma. Oncol Res Treat (2017) 40:82–7. doi: 10.1159/000456036
24. Chao C, Xu L, Abrams D, Leyden W, Horberg M, Towner W, et al. Survival of non-Hodgkin Lymphoma Patients With and Without Hiv Infection in the Era of Combined Antiretroviral Therapy. AIDS (2010) 24:1765–70. doi: 10.1097/QAD.0b013e32833a0961
25. Coutinho R, Pria AD, Gandhi S, Bailey K, Fields P, Cwynarski K, et al. Hiv Status Does Not Impair the Outcome of Patients Diagnosed With Diffuse Large B-Cell Lymphoma Treated With R-CHOP in the cART Era. AIDS (2014) 28:689–97. doi: 10.1097/QAD.0000000000000133
26. Barta SK, Xue X, Wang D, Tamari R, Lee JY, Mounier N, et al. Treatment Factors Affecting Outcomes in HIV-associated non-Hodgkin Lymphomas: A Pooled Analysis of 1546 Patients. Blood (2013) 122:3251–62. doi: 10.1182/blood-2013-04-498964
27. Padhi S, Paul TR, Challa S, Prayaga AK, Rajappa S, Raghunadharao D, et al. Primary Extra Nodal non Hodgkin Lymphoma: A 5 Year Retrospective Analysis. Asian Pac J Cancer Prev (2012) 13:4889–95. doi: 10.7314/APJCP.2012.13.10.4889
28. Ninkovic S, Lambert J. Non-Hodgkin Lymphoma. Med (United Kingdom) (2017) 45:297–304. doi: 10.1016/j.mpmed.2017.02.008
29. Carbone PP, Kaplan HS, Musshoff K, Smithers DW, Tubiana M. Report of the Committee on Hodgkin’s Disease Staging Classification. Cancer Res (1971) 31:1860–1.
30. Armitage JO. Staging Non-Hodgkin Lymphoma. CA Cancer J Clin (2005) 55:368–76. doi: 10.3322/canjclin.55.6.368
31. International Non-Hodgkin’s Lymphoma Prognostic Factors Project. A Predictive Model for Aggressive Non-Hodgkin’s Lymphoma. N Engl J Med (1993) 329:987–94. doi: 10.1056/NEJM199309303291402
32. Alizadeh AA, Elsen MB, Davis RE, Ma CL, Lossos IS, Rosenwald A, et al. Distinct Types of Diffuse Large B-cell Lymphoma Identified by Gene Expression Profiling. Nature (2000) 403:503–11. doi: 10.1038/35000501
33. Blombery PA, Wall M, Seymour JF. The Molecular Pathogenesis of B-cell non-Hodgkin Lymphoma. Eur J Haematol (2015) 95:280–93. doi: 10.1111/ejh.12589
34. Scott DW, Wright GW, Williams PM, Lih C-J, Walsh W, Jaffe ES, et al. Determining Cell-of-Origin Subtypes of Diffuse Large B-cell Lymphoma Using Gene Expression in Formalin-Fixed Paraffin-Embedded Tissue. Blood (2014) 123:1214–7. doi: 10.1182/blood-2013-11-536433
35. Lenz G. Insights Into the Molecular Pathogenesis of Activated B-Cell-Like Diffuse Large B-cell Lymphoma and its Therapeutic Implications. Cancers (Basel) (2015) 7:811–22. doi: 10.3390/cancers7020812
36. Kallies A, Hasbold J, Tarlinton DM, Dietrich W, Corcoran LM, Hodgkin PD, et al. Plasma Cell Ontogeny Defined by Quantitative Changes in Blimp-1 Expression. J Exp Med (2004) 200:967–77. doi: 10.1084/jem.20040973
37. Shaffer AL, Lin K-I, Kuo TC, Yu X, Hurt EM, Rosenwald A, et al. Blimp-1 Orchestrates Plasma Cell Differentiation by Extinguishing the Mature B Cell Gene Expression Program. Immunity (2002) 17:51–62. doi: 10.1016/S1074-7613(02)00335-7
38. Mandelbaum J, Bhagat G, Tang H, Mo T, Brahmachary M, Shen Q, et al. Blimp1 Is a Tumor Suppressor Gene Frequently Disrupted in Activated B Cell-Like Diffuse Large B Cell Lymphoma. Cancer Cell (2010) 18:568–79. doi: 10.1016/j.ccr.2010.10.030
39. Wright G, Tan B, Rosenwald A, Hurt EH, Wiestner A, Staudt LM. A Gene Expression-Based Method to Diagnose Clinically Distinct Subgroups of Diffuse Large B Cell Lymphoma. Proc Natl Acad Sci USA (2003) 100:9991–6. doi: 10.1073/pnas.1732008100
40. Hans CP, Weisenburger DD, Greiner TC, Gascoyne RD, Delabie J, Ott G, et al. Confirmation of the Molecular Classification of Diffuse Large B-cell Lymphoma by Immunohistochemistry Using a Tissue Microarray. Blood (2004) 103:275–82. doi: 10.1182/blood-2003-05-1545
41. Nandikolla AG, Wang Y, Shah UA, Derman O, Kornblum N, Mantzaris I, et al. Cell of Origin by Immunohistochemistry is Not Prognostic of Overall Survival in HIV Associated DLBCL- Analysis of Clinical Pathological Markers in HIV Dlbcl. Blood (2016) 128:1867–7. doi: 10.1182/blood.V128.22.1867.1867
42. Shaffer AL, Rosenwald A, Staudt LM. Lymphoid Malignancies: The Dark Side of B-cell Differentiation. Nat Rev Immunol (2002) 2:920–32. doi: 10.1038/nri953
43. Di Noia JM, Neuberger MS. Molecular Mechanisms of Antibody Somatic Hypermutation. Annu Rev Biochem (2007) 76:1–22. doi: 10.1146/annurev.biochem.76.061705.090740
44. Burmeister T, Molkentin M, Schwartz S, Gökbuget N, Hoelzer D, Thiel E, et al. Erroneous Class Switching and False Vdj Recombination: Molecular Dissection of T(8;14)/MYC-IGH Translocations Inburkitt-Type Lymphoblastic Leukemia/B-Cell Lymphoma. Mol Oncol (2013) 7:850–8. doi: 10.1016/j.molonc.2013.04.006
45. Lu Z, Tsai AG, Akasaka T, Ohno H, Jiang Y, Melnick AM, et al. Bcl6 Breaks Occur At Different AID Sequence Motifs in Ig–BCL6 and non-Ig–BCL6 Rearrangements. Blood (2013) 121:4551–4. doi: 10.1182/blood-2012-10-464958
46. Akasaka H, Akasaka T, Kurata M, Ueda C, Shimizu A, Uchiyama T, et al. Molecular Anatomy of BCL6 Translocations Revealed by Long-Distance Polymerase Chain Reaction-Based Assays. Cancer Res (2000) 60:2335–41.
47. Pasqualucci L, Neumeister P, Goossens T, Nanjangud G, Chaganti RSK, Küppers R, et al. Hypermutation of Multiple Proto-Oncogenes in B-cell Diffuse Large-Cell Lymphomas. Nature (2001) 412:341–6. doi: 10.1038/35085588
48. Vega F, Medeiros LJ. Chromosomal Translocations Involved in non-Hodgkin Lymphomas. Arch Pathol Lab Med (2003) 127:1148–60. doi: 10.1043/1543-2165(2003)127<1148:CTIINL>2.0.CO;2
49. Willis TG, Dyer MJS. The Role of Immunoglobulin Translocations in the Pathogenesis of B-cell Malignancies. Blood (2000) 96:808–22. doi: 10.1182/blood.v96.3.808.015k30_808_822
50. Miles RR, Shah RK, Frazer JK. Molecular Genetics of Childhood, Adolescent and Young Adult non-Hodgkin Lymphoma. Br J Haematol (2016) 173:582–96. doi: 10.1111/bjh.14011
51. Pegoraro L, Palumbo A, Erikson J, Falda M, Giovanazzo B, Emanuel BS, et al. A 14;18 and an 8;14 Chromosome Translocation in a Cell Line Derived From an Acute B-Cell Leukemia. Proc Natl Acad Sci (1984) 81:7166–70. doi: 10.1073/pnas.81.22.7166
52. Iqbal J, Sanger WG, Horsman DE, Rosenwald A, Pickering DL, Dave B, et al. Bcl2 Translocation Defines a Unique Tumor Subset Within the Germinal Center B-Cell-Like Diffuse Large B-Cell Lymphoma. Am J Pathol (2004) 165:159–66. doi: 10.1016/S0002-9440(10)63284-1
53. Huang JZ, Sanger WG, Greiner TC, Staudt LM, Weisenburger DD, Pickering DL, et al. The T(14;18) Defines a Unique Subset of Diffuse Large B-cell Lymphoma With a Germinal Center B-cell Gene Expression Profile. Blood (2002) 99:2285–90. doi: 10.1182/blood.V99.7.2285
54. Bakhshi A, Jensen JP, Goldman P, Wright JJ, McBride OW, Epstein AL, et al. Cloning the Chromosomal Breakpoint of T(14;18) Human Lymphomas: Clustering Around Jh on Chromosome 14 and Near a Transcriptional Unit on 18. Cell (1985) 41:899–906. doi: 10.1016/S0092-8674(85)80070-2
55. Visco C, Tzankov A, Xu-Monette ZY, Miranda RN, Tai YC, Li Y, et al. Patients With Diffuse Large B-Cell Lymphoma of Germinal Center Origin With BCL2 Translocations Have Poor Outcome, Irrespective of MYC Status: A Report From an International Dlbcl Rituximab-CHOP Consortium Program Study. Haematologica (2013) 98:255–63. doi: 10.3324/haematol.2012.066209
56. Baron BW, Nucifora G, McCabe N, Espinosa R, Le Beau MM, McKeithan TW. Identification of the Gene Associated With the Recurring Chromosomal Translocations T(3;14)(q27;q32) and T(3;22)(q27;q11) in B-cell Lymphomas. Proc Natl Acad Sci (1993) 90:5262–6. doi: 10.1073/pnas.90.11.5262
57. Iqbal J, Greiner TC, Patel K, Dave BJ, Smith L, Ji J, et al. Distinctive Patterns of BCL6 Molecular Alterations and Their Functional Consequences in Different Subgroups of Diffuse Large B-cell Lymphoma. Leukemia (2007) 21:2332–43. doi: 10.1038/sj.leu.2404856
58. Testoni M, Zucca E, Young KH, Bertoni F. Genetic Lesions in Diffuse Large B-cell Lymphomas. Ann Oncol (2015) 26:1069–80. doi: 10.1093/annonc/mdv019
59. Skinnider BF, Horsman DE, Dupuis B, Gascoyne RD. Bcl-6 and Bcl-2 Protein Expression in Diffuse Large B-cell Lymphoma and Follicular Lymphoma: Correlation With 3q27 and 18q21 Chromosomal Abnormalities. Hum Pathol (1999) 30:803–8. doi: 10.1016/S0046-8177(99)90141-7
60. Ohno H. Pathogenetic Role of BCL6 Translocation in B-cell non-Hodgkin’s Lymphoma. Histol Histopathol (2004) 19:637–50. doi: 10.14670/HH-19.637
61. Boxer LM, Dang CV. Translocations Involving C-Myc and C-Myc Function. Oncogene (2001) 20:5595–610. doi: 10.1038/sj.onc.1204595
62. Kramer MHH, Hermans J, Wijburg E, Philippo K, Geelen E, van Krieken JHJM, et al. Clinical Relevance of BCL2, BCL6, and MYC Rearrangements in Diffuse Large B-Cell Lymphoma. Blood (1998) 92:3152–62. doi: 10.1182/blood.V92.9.3152.421a07_3152_3162
63. Niitsu N, Okamoto M, Miura I, Hirano M. Clinical Features and Prognosis of De Novo Diffuse Large B-cell Lymphoma With T(14;18) and 8q24/c-MYC Translocations. Leukemia (2009) 23:777–83. doi: 10.1038/leu.2008.344
64. Copie-Bergman C, Cuillière-Dartigues P, Baia M, Briere J, Delarue R, Canioni D, et al. Myc-IG Rearrangements are Negative Predictors of Survival in DLBCL Patients Treated With Immunochemotherapy: A Gela/Lysa Study. Blood (2015) 126:2466–74. doi: 10.1182/blood-2015-05-647602
65. Klapproth K, Wirth T. Advances in the Understanding of MYC-induced Lymphomagenesis. Br J Haematol (2010) 149:484–97. doi: 10.1111/j.1365-2141.2010.08159.x
66. Rosenthal A, Younes A. High Grade B-Cell Lymphoma With Rearrangements of MYC and BCL2 and/or BCL6: Double Hit and Triple Hit Lymphomas and Double Expressing Lymphoma. Blood Rev (2017) 31:37–42. doi: 10.1016/j.blre.2016.09.004
67. Dunleavy K. Aggressive B Cell Lymphoma: Optimal Therapy for MYC-positive, Double-Hit, and Triple-Hit Dlbcl. Curr Treat Options Oncol (2015) 16:58. doi: 10.1007/s11864-015-0374-0
68. Peroja P, Pedersen M, Mantere T, Nørgaard P, Peltonen J, Haapasaari KM, et al. Mutation of TP53, Translocation Analysis and Immunohistochemical Expression of MYC, BCL-2 and BCL-6 in Patients With DLBCL Treated With R-Chop. Sci Rep (2018) 8:1–9. doi: 10.1038/s41598-018-33230-3
69. Campo E. MYC in DLBCL: Partners Matter. Blood (2015) 126:2439–40. doi: 10.1182/blood-2015-10-671362
70. Reddy A, Zhang J, Davis NS, Moffitt AB, Love CL, Waldrop A, et al. Genetic and Functional Drivers of Diffuse Large B Cell Lymphoma. Cell (2017) 171:481–94.e15. doi: 10.1016/j.cell.2017.09.027
71. Tsujimoto Y. Role of Bcl-2 Family Proteins in Apoptosis: Apoptosomes or Mitochondria? Genes Cells (1998) 3:697–707. doi: 10.1046/j.1365-2443.1998.00223.x
72. Rawlings DJ, Sommer K, Moreno-García ME. The CARMA1 Signalosome Links the Signalling Machinery of Adaptive and Innate Immunity in Lymphocytes. Nat Rev Immunol (2006) 6:799–812. doi: 10.1038/nri1944
73. Foulkes WD, Flanders TY, Pollock PM, Hayward NK. The CDKN2A (P16) Gene and Human Cancer. Mol Med (1997) 3:5–20. doi: 10.1007/BF03401664
74. Mungall K, Morin RD, Pleasance E, Mungall AJ, Goya R, Huff RD, et al. Mutational and Structural Analysis of Diffuse Large B-cell Lymphoma Using Whole-Genome Sequencing. Blood (2013) 122:1256–65. doi: 10.1182/blood-2013-02-483727
75. Lenz G, Davis RE, Ngo VN, Lam L, George TC, Wright GW, et al. Oncogenic CARD11 Mutations in Human Diffuse Large B Cell Lymphoma. Science (2008) 319:1676–9. doi: 10.1126/science.1153629
76. Bu R, Bavi P, Abubaker J, Jehan Z, Al-Haqawi W, Ajarim D, et al. Role of Nuclear Factor-κb Regulators TNFAIP3 and CARD11 in Middle Eastern Diffuse Large B-cell Lymphoma. Leuk Lymphoma (2012) 53:1971–7. doi: 10.3109/10428194.2012.668286
77. Lenz G, Wright GW, Emre NCT, Kohlhammer H, Dave SS, Davis RE, et al. Molecular Subtypes of Diffuse Large B-cell Lymphoma Arise by Distinct Genetic Pathways. Proc Natl Acad Sci USA (2008) 105:13520–5. doi: 10.1073/pnas.0804295105
78. Caganova M, Carrisi C, Varano G, Mainoldi F, Zanardi F, Germain P, et al. Germinal Center Dysregulation by Histone Methyltransferase EZH2 Promotes Lymphomagenesis. J Clin Invest (2013) 123:5009–22. doi: 10.1172/JCI70626
79. Morin RD, Johnson NA, Severson TM, Mungall AJ, An J, Goya R, et al. Somatic Mutations Altering EZH2 (Tyr641) in Follicular and Diffuse Large B-cell Lymphomas of Germinal-Center Origin. Nat Genet (2010) 42:181–5. doi: 10.1038/ng.518
80. Arthur SE, Jiang A, Grande BM, Alcaide M, Cojocaru R, Rushton CK, et al. Genome-Wide Discovery of Somatic Regulatory Variants in Diffuse Large B-cell Lymphoma. Nat Commun (2018) 9:4001. doi: 10.1038/s41467-018-06354-3
81. Yap DB, Chu J, Berg T, Schapira M, Cheng SWG, Moradian A, et al. Somatic Mutations At EZH2 Y641 Act Dominantly Through a Mechanism of Selectively Altered PRC2 Catalytic Activity, to Increase H3k27 Trimethylation. Blood (2011) 117:2451–9. doi: 10.1182/blood-2010-11-321208
82. Pohl T. Rel/NF-κb Transcription Factors: Key Mediators of B-cell Activation. Immunol Rev (2000) 176:134–40. doi: 10.1034/j.1600-065X.2000.00615.x
83. Davis RE, Brown KD, Siebenlist U, Staudt LM. Constitutive Nuclear Factor κb Activity is Required for Survival of Activated B Cell–Like Diffuse Large B Cell Lymphoma Cells. J Exp Med (2001) 194:1861–74. doi: 10.1084/jem.194.12.1861
84. Compagno M, Lim WK, Grunn A, Nandula SV, Brahmachary M, Shen Q, et al. Mutations of Multiple Genes Cause Deregulation of NF-κb in Diffuse Large B-cell Lymphoma. Nature (2009) 459:717–21. doi: 10.1038/nature07968
85. Ngo VN, Young RM, Schmitz R, Jhavar S, Xiao W, Lim K, et al. Oncogenically Active Myd88 Mutations in Human Lymphoma. Nature (2011) 470:115–9. doi: 10.1038/nature09671
86. Kwak J-Y. Treatment of Diffuse Large B Cell Lymphoma. Korean J Intern Med (2012) 27:369. doi: 10.3904/kjim.2012.27.4.369
87. Fisher RI, Gaynor ER, Dahlberg S, Oken MM, Grogan TM, Mize EM, et al. Comparison of a Standard Regimen (Chop) With Three Intensive Chemotherapy Regimens for Advanced non-Hodgkin’s Lymphoma. N Engl J Med (1993) 328:1002–6. doi: 10.1056/NEJM199304083281404
88. Itoh K, Ohtsu T, Wakita H, Igarashi T, Ishizawa K, Onozawa Y, et al. Dose-Escalation Study of CHOP With or Without Prophylactic G-CSF in Aggressive non-Hodgkin’s Lymphoma. Ann Oncol (2000) 11:1241–8. doi: 10.1023/A:1008361513544
89. Cartron G, Watier H, Golay J, Solal-Celigny P. From the Bench to the Bedside: Ways to Improve Rituximab Efficacy. Blood (2004) 104:2635–42. doi: 10.1182/blood-2004-03-1110
90. Feugier P, Van Hoof A, Sebban C, Solal-Celigny P, Bouabdallah R, Fermé C, et al. Long-Term Results of the R-CHOP Study in the Treatment of Elderly Patients With Diffuse Large B-Cell Lymphoma: A Study by the Groupe D’etude Des Lymphomes De L’adulte. J Clin Oncol (2005) 23:4117–26. doi: 10.1200/JCO.2005.09.131
91. Tariq A, Aziz MT, Mehmood Y, Asghar SA, Khurshid A. Clinical Response to CHOP vs. R-CHOP in Adult Patients With Diffuse Large B-Cell Lymphomas. Asian Pac J Cancer Prev (2018) 19:1181–4. doi: 10.22034/APJCP.2018.19.5.1181
92. Coiffier B, Thieblemont C, Van Den Neste E, Lepeu G, Plantier I, Castaigne S, et al. Long-Term Outcome of Patients in the LNH-98.5 Trial, the First Randomized Study Comparing rituximab-CHOP to Standard Chop Chemotherapy in DLBCL Patients: A Study by the Groupe D’etudes Des Lymphomes De L’adulte. Blood (2010) 116:2040–5. doi: 10.1182/blood-2010-03-276246
93. Coiffier B, Lepage E, Brière J, Herbrecht R, Tilly H, Bouabdallah R, et al. Chop Chemotherapy Plus Rituximab Compared With Chop Alone in Elderly Patients With Diffuse Large-B-Cell Lymphoma. N Engl J Med (2002) 346:235–42. doi: 10.1056/NEJMoa011795
94. Davies A, Cummin TE, Barrans S, Maishman T, Mamot C, Novak U, et al. Gene-Expression Profiling of Bortezomib Added to Standard Chemoimmunotherapy for Diffuse Large B-cell Lymphoma (Remodl-B): An Open-Label, Randomised, Phase 3 Trial. Lancet Oncol (2019) 20:649–62. doi: 10.1016/S1470-2045(18)30935-5
95. Leonard JP, Kolibaba KS, Reeves JA, Tulpule A, Flinn IW, Kolevska T, et al. Randomized Phase Ii Study of R-CHOP With or Without Bortezomib in Previously Untreated Patients With non-Germinal Center B-Cell-Like Diffuse Large B-cell Lymphoma. J Clin Oncol (2017) 35:3538–46. doi: 10.1200/JCO.2017.73.2784
96. Younes A, Sehn LH, Johnson P, Zinzani PL, Hong X, Zhu J, et al. Randomized Phase Iii Trial of Ibrutinib and Rituximab Plus Cyclophosphamide, Doxorubicin, Vincristine, and Prednisone in Non–Germinal Center B-Cell Diffuse Large B-Cell Lymphoma. J Clin Oncol (2019) 37:1285–95. doi: 10.1200/JCO.18.02403
97. Nowakowski GS, Chiappella A, Gascoyne RD, Scott DW, Zhang Q, Jurczak W, et al. Robust: A Phase III Study of Lenalidomide Plus R-CHOP Versus Placebo Plus R-CHOP in Previously Untreated Patients With ABC-Type Diffuse Large B-Cell Lymphoma. J Clin Oncol (2021) 39:1317–28. doi: 10.1200/JCO.20.01366
98. Moir S, Fauci AS. B Cells in HIV Infection and Disease. Nat Rev Immunol (2009) 9:235–45. doi: 10.1038/nri2524
99. Berberian L, Goodglick L, Kipps TJ, Braun J. Immunoglobulin VH3 Gene Products: Natural Ligands for HIV Gp120. Science (1993) 261:1588–91. doi: 10.1126/science.7690497
100. He B, Qiao X, Klasse PJ, Chiu A, Chadburn A, Knowles DM, et al. Hiv-1 Envelope Triggers Polyclonal Ig Class Switch Recombination Through a CD40-Independent Mechanism Involving BAFF and C-Type Lectin Receptors. J Immunol (2006) 176:3931–41. doi: 10.4049/jimmunol.176.7.3931
101. Cagigi A, Du L, Dang LVP, Grutzmeier S, Atlas A, Chiodi F, et al. Cd27- B-cells Produce Class Switched and Somatically Hyper-Mutated Antibodies During Chronic Hiv-1 Infection. PloS One (2009) 4:1–9. doi: 10.1371/journal.pone.0005427
102. Moir S, Malaspina A, Li Y, Chun TW, Lowe T, Adelsberger J, et al. B Cells of HIV-1-infected Patients Bind Virions Through CD21-Complement Interactions and Transmit Infectious Virus to Activated T Cells. J Exp Med (2000) 192:637–45. doi: 10.1084/jem.192.5.637
103. Malaspina A, Moir S, Nickle DC, Donoghue ET, Ogwaro KM, Ehler LA, et al. Human Immunodeficiency Virus Type 1 Bound to B Cells: Relationship to Virus Replicating in CD4+ T Cells and Circulating in Plasma. J Virol (2002) 76:8855–63. doi: 10.1128/jvi.76.17.8855-8863.2002
104. Ho J, Moir S, Kulik L, Malaspina A, Donoghue ET, Miller NJ, et al. Role for CD21 in the Establishment of an Extracellular Hiv Reservoir in Lymphoid Tissues. J Immunol (2007) 178:6968–74. doi: 10.4049/jimmunol.178.11.6968
105. Bordoni V, Sacchi A, Casetti R, Cimini E, Tartaglia E, Pinnetti C, et al. Impact of ART on Dynamics of Growth Factors and Cytokines in Primary Hiv Infection. Cytokine (2020) 125:154839. doi: 10.1016/j.cyto.2019.154839
106. Weimer R, Zipperle S, Daniel V, Zimmermann R, Schimpf K, Opelz G. Hiv-Induced IL-6/IL-10 Dysregulation of CD4 Cells is Associated With Defective B Cell Help and Autoantibody Formation Against Cd4 Cells. Clin Exp Immunol (1998) 111:20–9. doi: 10.1046/j.1365-2249.1998.00429.x
107. Planès R, Serrero M, Leghmari K, BenMohamed L, Bahraoui E. Hiv-1 Envelope Glycoproteins Induce the Production of TNF-α and IL-10 in Human Monocytes by Activating Calcium Pathway. Sci Rep (2018) 8:1–15. doi: 10.1038/s41598-018-35478-1
108. Beignon A-S. Endocytosis of HIV-1 Activates Plasmacytoid Dendritic Cells Via Toll-like Receptor- Viral Rna Interactions. J Clin Invest (2005) 115:3265–75. doi: 10.1172/JCI26032
109. Urashima M, Chauhan D, Hatziyanni M, Ogata A, Hollenbaugh D, Aruffo A, et al. Cd40 Ligand Triggers Interleukin-6 Mediated B Cell Differentiation. Leuk Res (1996) 20:507–15. doi: 10.1016/0145-2126(95)00098-4
110. MacChia D, Almerigogna F, Parronchi P, Ravina A, Maggi E, Romagnani S. Membrane Tumour Necrosis Factor-α is Involved in the Polyclonal B-Cell Activation Induced by HIV-infected Human T Cells. Nature (1993) 363:464–6. doi: 10.1038/363464a0
111. Lane HC, Masur H, Edgar LC, Whalen G, Rook AH, Fauci AS. Abnormalities of B-Cell Activation and Immunoregulation in Patients With the Acquired Immunodeficiency Syndrome. N Engl J Med (1983) 309:453–8. doi: 10.1056/NEJM198308253090803
112. Lugada ES, Mermin J, Asjo B, Kaharuza F, Downing R, Langeland N, et al. Immunoglobulin Levels Amongst Persons With and Without Human Immunodeficiency Virus Type 1 Infection in Uganda and Norway. Scand J Immunol (2004) 59:203–8. doi: 10.1111/j.0300-9475.2004.01376.x
113. Onifade AA, Ojerinde AO, Emeka GJ, Agbedana, Oluogun WA. The Role of IgG, Iga and IgM as Immunological Markers of HIV/AIDS Progression. J AIDS HIV Res (2017) 9:152–8. doi: 10.5897/JAHR2017.0421
114. Hønge BL, Petersen MS, Jespersen S, Medina C, Té DDS, Kjerulff B, et al. T-Cell and B-cell Perturbations are Similar in ART-naive HIV-1 and HIV-1/2 Dually Infected Patients. Aids (2019) 33:1143–53. doi: 10.1097/QAD.0000000000002185
115. Shirai A, Cosentino M, Leitman-Klinman SF, Klinman DM. Human Immunodeficiency Virus Infection Induces Both Polyclonal and Virus-Specific B Cell Activation. J Clin Invest (1992) 89:561–6. doi: 10.1172/JCI115621
116. Moir S, Malaspina A, Ogwaro KM, Donoghue ET, Hallahan CW, Ehler LA, et al. Hiv-1 Induces Phenotypic and Functional Perturbations of B Cells in Chronically Infected Individuals. Proc Natl Acad Sci USA (2001) 98:10362–7. doi: 10.1073/pnas.181347898
117. Lau D, Lan LY-L, Andrews SF, Henry C, Rojas KT, Neu KE, et al. Low CD21 Expression Defines a Population of Recent Germinal Center Graduates Primed for Plasma Cell Differentiation. Sci Immunol (2017) 2:eaai8153. doi: 10.1126/sciimmunol.aai8153
118. Nagase H, Agematsu K, Kitano K, Takamoto M, Okubo Y, Komiyama A, et al. Mechanism of Hypergammaglobulinemia by HIV Infection: Circulating Memory B-cell Reduction With Plasmacytosis. Clin Immunol (2001) 100:250–9. doi: 10.1006/clim.2001.5054
119. Conge AM, Tarte K, Reynes J, Segondy M, Gerfaux J, Zembala M, et al. Impairment of B-lymphocyte Differentiation Induced by Dual Triggering of the B-cell Antigen Receptor and CD40 in Advanced Hiv-1-Disease. Aids (1998) 12:1437–49. doi: 10.1097/00002030-199812000-00005
120. Martínez-Maza O, Crabb E, Mitsuyasu RT, Fahey JL, Giorgi JV. Infection With the Human Immunodeficiency Virus (HIV) is Associated With an In Vivo Increase in B Lymphocyte Activation and Immaturity. J Immunol (1987) 138:3720–4.
121. Malaspina A, Moir S, Kottilil S, Hallahan CW, Ehler LA, Liu S, et al. Deleterious Effect of HIV-1 Plasma Viremia on B Cell Costimulatory Function. J Immunol (2003) 170:5965–72. doi: 10.4049/jimmunol.170.12.5965
122. Yan J, Zhang S, Sun J, Xu J, Zhang X. Irreversible Phenotypic Perturbation and Functional Impairment of B Cells During Hiv-1 Infection. Front Med (2017) 11:536–47. doi: 10.1007/s11684-017-0592-x
123. Khaitan A, Unutmaz D. Revisiting Immune Exhaustion During Hiv Infection. Curr HIV/AIDS Rep (2011) 8:4–11. doi: 10.1007/s11904-010-0066-0
124. Thorarinsdottir K, Camponeschi A, Gjertsson I, Mårtensson IL. Cd21-/Low B Cells: A Snapshot of a Unique B Cell Subset in Health and Disease. Scand J Immunol (2015) 82:254–61. doi: 10.1111/sji.12339
125. Sciaranghella G, Tong N, Mahan AE, Suscovich TJ, Alter G. Decoupling Activation and Exhaustion of B Cells in Spontaneous Controllers of HIV Infection. AIDS (2013) 27:175–80. doi: 10.1097/QAD.0b013e32835bd1f0
126. Ehrhardt GRA, Hsu JT, Gartland L, Leu CM, Zhang S, Davis RS, et al. Expression of the Immunoregulatory Molecule FcRH4 Defines a Distinctive Tissue-Based Population of Memory B Cells. J Exp Med (2005) 202:783–91. doi: 10.1084/jem.20050879
127. Moir S, Ho J, Malaspina A, Wang W, DiPoto AC, O’Shea MA, et al. Evidence for HIV-Associated B Cell Exhaustion in a Dysfunctional Memory B Cell Compartment in HIV-infected Viremic Individuals. J Exp Med (2008) 205:1797–805. doi: 10.1084/jem.20072683
128. Liechti T, Kadelka C, Braun DL, Kuster H, Boni J, Robbiani M, et al. Widespread B Cell Perturbations in HIV-1 Infection Afflict Naive and Marginal Zone B Cells. J Exp Med (2019) 216:2071–90. doi: 10.1084/jem.20181124
129. Jacobson MA, Khayam-Bashi H, Martin JN, Black D, Ng V. Effect of Long-Term Highly Active Antiretroviral Therapy in Restoring Hiv-Induced Abnormal B-Lymphocyte Function. JAIDS J Acquir Immune Defic Syndr (2002) 31:472–7. doi: 10.1097/00126334-200212150-00003
130. Morris L, Binley JM, Clas BA, Bonhoeffer S, Astill TP, Kost R, et al. Hiv-1 Antigen–specific and –Nonspecific B Cell Responses are Sensitive to Combination Antiretroviral Therapy. J Exp Med (1998) 188:233–45. doi: 10.1084/jem.188.2.233
131. Moir S, Malaspina A, Ho J, Wang W, DiPoto AC, O’Shea MA, et al. Normalization of B Cell Counts and Subpopulations After Antiretroviral Therapy in Chronic Hiv Disease. J Infect Dis (2008) 197:572–9. doi: 10.1086/526789
132. Chong Y, Ikematsu H, Kikuchi K, Yamamoto M, Murata M, Nishimura M, et al. Selective CD27+ (Memory) B Cell Reduction and Characteristic B Cell Alteration in Drug-Naive and HAART-Treated Hiv Type 1-Infected Patients. AIDS Res Hum Retroviruses (2004) 20:219–26. doi: 10.1089/088922204773004941
133. Abudulai LN, Fernandez S, Corscadden K, Hunter M, Kirkham LAS, Post JJ, et al. Chronic HIV-1 Infection Induces B-Cell Dysfunction That is Incompletely Resolved by Long-Term Antiretroviral Therapy. J Acquir Immune Defic Syndr (2016) 71:381–9. doi: 10.1097/QAI.0000000000000869
134. Tanko RF, Soares AP, Müller TL, Garrett NJ, Samsunder N, Abdool Karim Q, et al. Effect of Antiretroviral Therapy on the Memory and Activation Profiles of B Cells in HIV-Infected African Women. J Immunol (2017) 198:1220–8. doi: 10.4049/jimmunol.1601560
135. Linke-Serinsöz E, Fend F, Quintanilla-Martinez L. Human Immunodeficiency Virus (HIV) and Epstein-Barr Virus (Ebv) Related Lymphomas, Pathology View Point. Semin Diagn Pathol (2017) 34:352–63. doi: 10.1053/j.semdp.2017.04.003
136. Van Baarle D, Hovenkamp E, Callan MFC, Wolthers KC, Kostense S, Tan LC, et al. Dysfunctional Epstein-Barr Virus (Ebv)-Specific Cd8+ T Lymphocytes and Increased Ebv Load in HIV-1 Infected Individuals Progressing to AIDS-related non-Hodgkin Lymphoma. Blood (2001) 98:146–55. doi: 10.1182/blood.V98.1.146
137. Green M, Michaels MG, Katz BZ, Burroughs M, Gerber D, Shneider BL, et al. Cmv-IVIG for Prevention of Epstein Barr Virus Disease and Posttransplant Lymphoproliferative Disease in Pediatric Liver Transplant Recipients. Am J Transplant (2006) 6:1906–12. doi: 10.1111/j.1600-6143.2006.01394.x
138. Fingeroth JD, Weis JJ, Tedder TF, Strominger JL, Biro PA, Fearon DT. Epstein-Barr Virus Receptor of Human B Lymphocytes is the C3d Receptor Cr2. Proc Natl Acad Sci USA (1984) 81:4510–4. doi: 10.1073/pnas.81.14.4510
139. Young LS, Rickinson AB. Epstein-Barr Virus: 40 Years on. Nat Rev Cancer (2004) 4:757–68. doi: 10.1038/nrc1452
140. Luftig MA, Stanfield BA. Recent Advances in Understanding Epstein-Barr Virus. F1000Research (2017) 6:1–10. doi: 10.12688/f1000research.10591.1
141. Chijioke O, Marcenaro E, Moretta A, Capaul R, Münz C. Role of the 2B4 Receptor in CD8+ T-Cell-Dependent Immune Control of Epstein-Barr Virus Infection in Mice With Reconstituted Human Immune System Components. J Infect Dis (2015) 212:803–7. doi: 10.1093/infdis/jiv114
142. Ambinder RF. Epstein-Barr Virus Associated Lymphoproliferations in the AIDS Setting. Eur J Cancer (2001) 37:1209–16. doi: 10.1016/S0959-8049(01)00123-X
143. Carbone A, Cesarman E, Spina M, Gloghini A, Schulz TF. Hiv-Associated Lymphomas and Gamma-Herpesviruses. Blood (2009) 113:1213–24. doi: 10.1182/blood-2008-09-180315
144. Grywalska E, Rolinski J. Epstein-Barr Virus–Associated Lymphomas. Semin Oncol (2015) 42:291–303. doi: 10.1053/j.seminoncol.2014.12.030
145. Shannon-Lowe C, Rickinson AB, Bell AI. Epstein–Barr Virus-Associated Lymphomas. Philos Trans R Soc B Biol Sci (2017) 372:20160271. doi: 10.1098/rstb.2016.0271
146. Ma S-D, Tsai M-H, Romero-Masters JC, Ranheim EA, Huebner SM, Bristol JA, et al. Latent Membrane Protein 1 (LMP1) and LMP2A Collaborate To Promote Epstein-Barr Virus-Induced B Cell Lymphomas in a Cord Blood-Humanized Mouse Model But are Not Essential. J Virol (2017) 91:1–17. doi: 10.1128/jvi.01928-16
147. Uchida J, Yasui T, Takaoka-Shichijo Y, Muraoka M, Kulwichit W, Raab-Traub N, et al. Mimicry of CD40 Signals by Epstein-Barr Virus LMP1 in B Lymphocyte Responses. Science (1999) 286:300–3. doi: 10.1126/science.286.5438.300
148. D’Souza BN, Edelstein LC, Pegman PM, Smith SM, Loughran ST, Clarke A, et al. Nuclear Factor κb-Dependent Activation of the Antiapoptotic Bfl-1 Gene by the Epstein-Barr Virus Latent Membrane Protein 1 and Activated Cd40 Receptor. J Virol (2004) 78:1800–16. doi: 10.1128/jvi.78.4.1800-1816.2004
149. Yun S, Kim YS, Hur DY. LMP1 and 2A Induce the Expression of Nrf2 Through Akt Signaling Pathway in Epstein-Barr Virus–Transformed B Cells. Transl Oncol (2019) 12:775–83. doi: 10.1016/j.tranon.2019.02.009
150. Kim JH, Kim WS, Hong JY, Ryu KJ, Kim SJ, Park C. Epstein-Barr Virus Ebna2 Directs Doxorubicin Resistance of B Cell Lymphoma Through CCL3 and CCL4-mediated Activation of NF-κb and Btk. Oncotarget (2017) 8:5361–70. doi: 10.18632/oncotarget.14243
151. Platt L, Easterbrook P, Gower E, McDonald B, Sabin K, McGowan C, et al. Prevalence and Burden of HCV Co-Infection in People Living With Hiv: A Global Systematic Review and Meta-Analysis. Lancet Infect Dis (2016) 16:797–808. doi: 10.1016/S1473-3099(15)00485-5
152. Canioni D, Michot J-M, Rabiega P, Molina TJ, Charlotte F, Lazure T, et al. In Situ Hepatitis C Ns3 Protein Detection Is Associated With High Grade Features in Hepatitis C-Associated B-Cell non-Hodgkin Lymphomas. PloS One (2016) 11:e0156384. doi: 10.1371/journal.pone.0156384
153. Besson C, Noel N, Lancar R, Prevot S, Algarte-Genin M, Rosenthal E, et al. Hepatitis C Virus or Hepatitis B Virus Coinfection and Lymphoma Risk in People Living With Hiv. Aids (2020) 34:599–608. doi: 10.1097/QAD.0000000000002461
154. Epeldegui M, Vendrame E, Martínez-Maza O. Hiv-Associated Immune Dysfunction and Viral Infection: Role in the Pathogenesis of AIDS-related Lymphoma. Immunol Res (2010) 48:72–83. doi: 10.1007/s12026-010-8168-8
155. Martínez-Maza O, Widney D, Van Der Meijden M, Knox R, Echeverri A, Breen EC, et al. Immune Dysfunction and the Pathogenesis of AIDS-associated non-Hodgkin’s Lymphoma. Mem Inst Oswaldo Cruz (1998) 93:373–81. doi: 10.1590/S0074-02761998000300019
156. Chiarle R, Zhang Y, Frock RL, Lewis SM, Molinie B, Ho YJ, et al. Genome-Wide Translocation Sequencing Reveals Mechanisms of Chromosome Breaks and Rearrangements in B Cells. Cell (2011) 147:107–19. doi: 10.1016/j.cell.2011.07.049
157. Epeldegui M, Thapa DR, de la Cruz J, Kitchen S, Zack JA, Martínez-Maza O. Cd40 Ligand (Cd154) Incorporated Into HIV Virions Induces Activation-Induced Cytidine Deaminase (Aid) Expression in Human B Lymphocytes. PloS One (2010) 5:e11448. doi: 10.1371/journal.pone.0011448
158. Yawetz S, Cumberland W, van der Meyden M, Martinez-Maza O. Elevated Serum Levels of Soluble CD23 (Scd23) Precede the Appearance Ofacquired Immunodeficiency Syndrome–Associated non-Hodgkin’s Lymphoma. Blood (1995) 85:1843–9. doi: 10.1182/blood.V85.7.1843.bloodjournal8571843
159. Vendrame E, Hussain SK, Breen EC, Magpantay LI, Widney DP, Jacobson LP, et al. Serum Levels of Cytokines and Biomarkers for Inflammation and Immune Activation, and HIV-Associated non-Hodgkin B-Cell Lymphoma Risk. Cancer Epidemiol Biomarkers Prev (2014) 23:343–9. doi: 10.1158/1055-9965.EPI-13-0714
160. Narayanan A, Iordanskiy S, Das R, Van Duyne R, Santos S, Jaworski E, et al. Exosomes Derived From HIV-1-infected Cells Contain Trans-Activation Response Element Rna. J Biol Chem (2013) 288:20014–33. doi: 10.1074/jbc.M112.438895
161. Chen L, Feng Z, Yue H, Bazdar D, Mbonye U, Zender C, et al. Exosomes Derived From HIV-1-infected Cells Promote Growth and Progression of Cancer Via HIV Tar Rna. Nat Commun (2018) 9:4585. doi: 10.1038/s41467-018-07006-2
162. Hernández-Walias FJ, Vázquez E, Pacheco Y, Rodríguez-Fernández JM, Pérez-Elías MJ, Dronda F, et al. Risk, Diagnostic and Predictor Factors for Classical Hodgkin Lymphoma in HIV-1-Infected Individuals: Role of Plasma Exosome-Derived miR-20a and Mir-21. J Clin Med (2020) 9:760. doi: 10.3390/jcm9030760
163. Dolcetti R, Gloghini A, Caruso A, Carbone A. A Lymphomagenic Role for HIV Beyond Immune Suppression? Blood (2016) 127:1403–9. doi: 10.1182/blood-2015-11-681411
164. Giagulli C, D’Ursi P, He W, Zorzan S, Caccuri F, Varney K, et al. A Single Amino Acid Substitution Confers B-Cell Clonogenic Activity to the HIV-1 Matrix Protein P17. Sci Rep (2017) 7:1–13. doi: 10.1038/s41598-017-06848-y
165. He W, Mazzuca P, Yuan W, Varney K, Bugatti A, Cagnotto A, et al. Identification of Amino Acid Residues Critical for the B Cell Growth-Promoting Activity of HIV-1 Matrix Protein p17 Variants. Biochim Biophys Acta Gen Subj (2019) 1863:13–24. doi: 10.1016/j.bbagen.2018.09.016
166. Caccuri F, Giagulli C, Bugatti A, Benetti A, Alessandri G, Ribatti D, et al. Hiv-1 Matrix Protein P17 Promotes Angiogenesis Via Chemokine Receptors CXCR1 and CXCR2. Proc Natl Acad Sci USA (2012) 109:14580–5. doi: 10.1073/pnas.1206605109
167. Kundu RK, Sangiorgi F, Wu L-Y, Pattengale PK, Hinton DR, Gill PS, et al. Expression of the Human Immunodeficiency Virus-Tat Gene in Lymphoid Tissues of Transgenic Mice Is Associated With B-Cell Lymphoma. Blood (1999) 94:275–82. doi: 10.1182/blood.V94.1.275.413a30_275_282
168. Srivastava DK, Tendler CL, Milani D, English MA, Licht JD, Wilson SH. The HIV-1 Transactivator Protein Tat is a Potent Inducer of the Human Dna Repair Enzyme β-Polymerase. Aids (2001) 15:433–40. doi: 10.1097/00002030-200103090-00001
169. Mitola S, Soldi R, Zanon I, Barra L, Gutierrez MI, Berkhout B, et al. Identification of Specific Molecular Structures of Human Immunodeficiency Virus Type 1 Tat Relevant for Its Biological Effects on Vascular Endothelial Cells. J Virol (2000) 74:344–53. doi: 10.1128/jvi.74.1.344-353.2000
170. Tachiwana H, Shimura M, Nakai-Murakami C, Tokunaga K, Takizawa Y, Sata T, et al. Hiv-1 Vpr Induces DNA Double-Strand Breaks. Cancer Res (2006) 66:627–31. doi: 10.1158/0008-5472.CAN-05-3144
171. Henderson WW, Ruhl R, Lewis P, Bentley M, Nelson JA, Moses AV. Human Immunodeficiency Virus (Hiv) Type 1 Vpu Induces the Expression of CD40 in Endothelial Cells and Regulates Hiv-Induced Adhesion of B-Lymphoma Cells. J Virol (2004) 78:4408–20. doi: 10.1128/jvi.78.9.4408-4420.2004
172. Chadburn A, Chiu A, Lee JY, Chen X, Hyjek E, Banham AH, et al. Immunophenotypic Analysis of AIDS-Related Diffuse Large B-Cell Lymphoma and Clinical Implications in Patients From AIDS Malignancies Consortium Clinical Trials 010 and 034. J Clin Oncol (2009) 27:5039–48. doi: 10.1200/JCO.2008.20.5450
173. Rusconi C, Re A, Bandiera L, Crucitti L, Spina M, Gini G, et al. Cell-of-Origin Identification and Prognostic Correlation in HIV-Associated Diffuse Large B-Cell Lymphomas: Results of an Italian Multicentric Study. Blood (2018) 132:5294–4. doi: 10.1182/blood-2018-99-116437
174. Teitell M, Damore MA, Sulur GG, Turner DE, Stern MH, Said JW, et al. Tcl1 Oncogene Expression in AIDS-related Lymphomas and Lymphoid Tissues. Proc Natl Acad Sci USA (1999) 96:9809–14. doi: 10.1073/pnas.96.17.9809
175. Nenasheva VV, Nikolaev AI, Martynenko AV, Kaplanskaya IB, Bodemer W, Hunsmann G, et al. Differential Gene Expression in HIV/SIV-associated and Spontaneous Lymphomas. Int J Med Sci (2005) 2:122–8. doi: 10.7150/ijms.2.122
176. Fedoriw Y, Selitsky S, Montgomery ND, Kendall SM, Richards KL, Du W, et al. Identifying Transcriptional Profiles and Evaluating Prognostic Biomarkers of HIV-associated Diffuse Large B-Cell Lymphoma From Malawi. Mod Pathol (2020) 2:122–8. doi: 10.1038/s41379-020-0506-3
177. Maguire A, Chen X, Wisner L, Malasi S, Ramsower C, Kendrick S, et al. Enhanced DNA Repair and Genomic Stability Identify a Novel HIV-Related Diffuse Large B-cell Lymphoma Signature. Int J Cancer (2019) 145:3078–88. doi: 10.1002/ijc.32381
178. Cassim S, Antel K, Chetty DR, Oosthuizen J, Opie J, Mohamed Z, et al. Diffuse Large B-Cell Lymphoma in a South African Cohort With a High Hiv Prevalence: An Analysis by Cell-of-Origin, Epstein–Barr Virus Infection and Survival. Pathology (2020) 145:3078–88. doi: 10.1016/j.pathol.2020.02.007
179. Chao C, Silverberg MJ, Xu L, Chen L-H, Castor B, Martínez-Maza O, et al. Said J. A Comparative Study of Molecular Characteristics of Diffuse Large B-cell Lymphoma From Patients With and Without Human Immunodeficiency Virus Infection. Clin Cancer Res (2015) 21:1429–37. doi: 10.1158/1078-0432.CCR-14-2083
180. Barreto L, Azambuja D, De Morais JC. Expression of Immunohistochemical Markers in Patients With AIDS-related Lymphoma. Braz J Infect Dis (2012) 16:74–7. doi: 10.1016/S1413-8670(12)70278-7
181. Madan R, Gormley R, Dulau A, Xu D, Walsh D, Ramesh KH, et al. AIDS and non-AIDS Diffuse Large B-cell Lymphomas Express Different Antigen Profiles. Mod Pathol (2006) 19:438–46. doi: 10.1038/modpathol.3800493
182. Thapa DR, Li X, Jamieson BD, Martínez-Maza O. Overexpression of Micrornas From the Mir-17-92 Paralog Clusters in AIDS-related non-Hodgkin’s Lymphomas. PloS One (2011) 6:21–3. doi: 10.1371/journal.pone.0020781
183. Mcgrath E PP. MicroRNA 21 Expression Levels in HIV Negative and HIV Positive Diffuse Large B Cell Lymphoma. Hered Genet (2015) 04:2–5. doi: 10.4172/2161-1041.1000143
184. Thapa DR, Hussain SK, Tran W-C, D'souza G, Bream JH, Achenback CJ, et al. Serum MicroRNAs in HIV-Infected Individuals as Pre-Diagnosis Biomarkers for AIDS-NHL. JAIDS J Acquir Immune Defic Syndr (2014) 66:229–37. doi: 10.1097/QAI.0000000000000146
185. Capello D, Scandurra M, Poretti G, Rancoita PMV, Mian M, Gloghini A, et al. Genome Wide DNA-Profiling of HIV-related B-Cell Lymphomas. Br J Haematol (2010) 148:245–55. doi: 10.1111/j.1365-2141.2009.07943.x
186. Morton LM, Kim CJ, Weiss LM, Bhatia K, Cockburn M, Hawes D, et al. Molecular Characteristics of Diffuse Large B-cell Lymphoma in Human Immunodeficiency Virus-Infected and -Uninfected Patients in the Pre-Highly Active Antiretroviral Therapy and Pre-Rituximab Era. Leuk Lymphoma (2014) 55:551–7. doi: 10.3109/10428194.2013.813499
187. Deffenbacher KE, Iqbal J, Liu Z, Fu K, Chan WC. Recurrent Chromosomal Alterations in Molecularly Classified AIDS-related Lymphomas: An Integrated Analysis of DNA Copy Number and Gene Expression. J Acquir Immune Defic Syndr (2010) 54:18–26. doi: 10.1097/QAI.0b013e3181d3d9eb
188. Capello D, Martini M, Gloghini A, Cerri M, Rasi S, Deambrogi C, et al. Molecular Analysis of Immunoglobulin Variable Genes in Human Immunodeficiency Virus-Related non-Hodgkin’s Lymphoma Reveals Implications for Disease Pathogenesis and Histogenesis. Haematologica (2008) 93:1178–85. doi: 10.3324/haematol.12705
189. Widney D, Gundapp G, Said JW, Van Der Meijden M, Bonavida B, Demidem A, et al. Aberrant Expression of CD27 and Soluble Cd27 (sCD27) in HIV Infection and in AIDS-associated Lymphoma. Clin Immunol (1999) 93:114–23. doi: 10.1006/clim.1999.4782
190. Patrone L, Henson SE, Teodorovic J, Malone CS, French SW, Wall R, et al. Gene Expression Patterns in AIDS Versus non-AIDS-Related Diffuse Large B-cell Lymphoma. Exp Mol Pathol (2003) 74:129–39. doi: 10.1016/S0014-4800(03)00007-8
191. Ting C-Y, Chang K-M, Kuan J-W, Sathar J, Chew L-P, Wong O-LJ, et al. Clinical Significance of BCL2 , C- MYC , and BCL6 Genetic Abnormalities, Epstein-Barr Virus Infection, CD5 Protein Expression, Germinal Center B Cell/non-Germinal Center B-Cell Subtypes, Co-Expression of MYC/BCL2 Proteins and Co-expression of MYC/BCL2/BCL. Int J Med Sci (2019) 16:556–66. doi: 10.7150/ijms.27610
192. Hu S, Xu-Monette ZY, Tzankov A, Green T, Wu L, Balasubramanyam A, et al. Myc/Bcl2 Protein Coexpression Contributes to the Inferior Survival of Activated B-Cell Subtype of Diffuse Large B-cell Lymphoma and Demonstrates High-Risk Gene Expression Signatures: A Report From the International Dlbcl Rituximab-Chop Consortium Program. Blood (2013) 121:4021–31. doi: 10.1182/blood-2012-10-460063
193. Shponka V, Reveles CY, Alam S, Jaramillo M, Maguire A, Rimsza LM, et al. Frequent Expression of Activation-Induced Cytidine Deaminase in Diffuse Large B-cell Lymphoma Tissues From Persons Living With Hiv. AIDS (2020) 34:2025–35. doi: 10.1097/QAD.0000000000002653
194. Philippe L, Lancar R, Laurent C, Algarte-Genin M, Chassagne-Clément C, Fabiani B, et al. In Situ Bcl2 Expression is an Independent Prognostic Factor in HIV-associated DLBCL, a LYMPHOVIR Cohort Study. Br J Haematol (2020) 188:413–23. doi: 10.1111/bjh.16176
195. Carbone A, Gloghini A, Larocca LM, Capello D, Pierconti F, Canzonieri V, et al. Expression Profile of MUM1/IRF4, Bcl-6, and CD138/syndecan-1 Defines Novel Histogenetic Subsets of Human Immunodeficiency Virus–Related Lymphomas. Blood (2001) 97:744–51. doi: 10.1182/blood.V97.3.744
196. Hoffmann C, Tiemann M, Schrader C, Janssen D, Wolf E, Vierbuchen M, et al. Aids-Related B-cell Lymphoma (Arl): Correlation of Prognosis With Differentiation Profiles Assessed by Immunophenotyping. Blood (2005) 106:1762–9. doi: 10.1182/blood-2004-12-4631
197. Hosnijeh FS, Portengen L, Späth F, Bergdahl IA, Melin B, Mattiello A, et al. Soluble B-cell Activation Marker of sCD27 and sCD30 and Future Risk of B-cell Lymphomas: A Nested Case-Control Study and Meta-Analyses. Int J Cancer (2016) 138:2357–67. doi: 10.1002/ijc.29969
198. Baptista MJ, Garcia O, Morgades M, Gonzalez-Barca E, Miralles P, Lopez-Guillermo A, et al. Hiv-Infection Impact on Clinical-Biological Features and Outcome of Diffuse Large B-cell Lymphoma Treated With R-CHOP in the Combination Antiretroviral Therapy Era. Aids (2015) 29:811–8. doi: 10.1097/QAD.0000000000000624
199. Cingolani A, Lepri AC, Teofili L, Galli L, Mazzotta V, Baldin GM, et al. Survival and Predictors of Death in People With HIV-associated Lymphoma Compared to Those With a Diagnosis of Lymphoma in General Population. PloS One (2017) 12:1–15. doi: 10.1371/journal.pone.0186549
200. Han X, Jemal A, Hulland E, Simard EP, Nastoupil L, Ward E, et al. Hiv Infection and Survival of Lymphoma Patients in the Era of Highly Active Antiretroviral Therapy. Cancer Epidemiol Biomarkers Prev (2017) 26:303–11. doi: 10.1158/1055-9965.EPI-16-0595
201. Spina M, Carbone A, Vaccher E, Gloghini A, Talamini R, Cinelli R, et al. Outcome in Patients With Non-Hodgkin Lymphoma and With or Without Human Immunodeficiency Virus Infection. Clin Infect Dis (2004) 38:142–4. doi: 10.1086/380129
202. Vaccher E, Tirelli U, Spina M, Talamini R, Errante D, Simonelli C, et al. Age and Serum Lactate Dehydrogenase Level are Independent Prognostic Factors in Human Immunodeficiency Virus-Related non-Hodgkin’s Lymphomas: A Single-Institute Study of 96 Patients. J Clin Oncol (1996) 14:2217–23. doi: 10.1200/JCO.1996.14.8.2217
203. Navarro J-T, Lloveras N, Ribera J-M, Oriol A, Mate J-L, Feliu E. The Prognosis of HIV-infected Patients With Diffuse Large B-Cell Lymphoma Treated With Chemotherapy and Highly Active Antiretroviral Therapy is Similar to That of HIV-negative Patients Receiving Chemotherapy. Haematologica (2005) 90:704–6.
204. Coghill AE, Shiels MS, Suneja G, Engels EA. Elevated Cancer-Specific Mortality Among Hiv-Infected Patients in the United States. J Clin Oncol (2015) 33:2376–83. doi: 10.1200/JCO.2014.59.5967
205. Ezzat H, Filipenko D, Vickars L, Galbraith P, Li C, Murphy K, et al. Improved Survival in HIV-Associated Diffuse Large B-Cell Lymphoma With the Addition of Rituximab to Chemotherapy in Patients Receiving Highly Active Antiretroviral Therapy. HIV Clin Trials (2007) 8:132–44. doi: 10.1310/hct0803-132
206. Kaplan LD. Rituximab Does Not Improve Clinical Outcome in a Randomized Phase 3 Trial of CHOP With or Without Rituximab in Patients With HIV-associated non-Hodgkin Lymphoma: Aids-Malignancies Consortium Trial 010. Blood (2005) 106:1538–43. doi: 10.1182/blood-2005-04-1437
207. Spina M, Jaeger U, Sparano JA, Talamini R, Simonelli C, Michieli M, et al. Rituximab Plus Infusional Cyclophosphamide, Doxorubicin, and Etoposide in HIV-associated non-Hodgkin Lymphoma: Pooled Results From 3 Phase 2 Trials. Blood (2005) 105:1891–7. doi: 10.1182/blood-2004-08-3300
208. Ribera J-M, Oriol A, Morgades M, González-Barca E, Miralles P, López-Guillermo A, et al. Safety and Efficacy of Cyclophosphamide, Adriamycin, Vincristine, Prednisone and Rituximab in Patients With Human Immunodeficiency Virus-Associated Diffuse Large B-Cell Lymphoma: Results of a Phase Ii Trial. Br J Haematol (2008) 140:411–9. doi: 10.1111/j.1365-2141.2007.06943.x
209. Boué F, Gabarre J, Gisselbrecht C, Reynes J, Cheret A, Bonnet F, et al. Phase II Trial of CHOP Plus Rituximab in Patients With HIV-Associated non-Hodgkin’s Lymphoma. J Clin Oncol (2006) 24:4123–8. doi: 10.1200/JCO.2005.05.4684
210. Miralles P, Berenguer J, Ribera JM, Rubio R, Mahillo B, Téllez MJ, et al. Prognosis of AIDS-related Systemic non-Hodgkin Lymphoma Treated With Chemotherapy and Highly Active Antiretroviral Therapy Depends Exclusively on Tumor-Related Factors. J Acquir Immune Defic Syndr (2007) 44:167–73. doi: 10.1097/QAI.0b013e31802bb5d0
211. Sparano JA, Lee JY, Kaplan LD, Levine AM, Ramos JC, Ambinder RF, et al. Rituximab Plus Concurrent Infusional Epoch Chemotherapy is Highly Effective in HIV-associated B-Cell non-Hodgkin Lymphoma. Blood (2010) 115:3008–16. doi: 10.1182/blood-2009-08-231613
212. Dunleavy K, Wilson WH. How I Treat HIV-associated Lymphoma. Blood (2012) 119:3245–55. doi: 10.1182/blood-2011-08-373738
213. Thirlwell C, Sarker D, Stebbing J, Bower M. Acquired Immunodeficiency Syndrome - Related Lymphoma in the Era of Highly Active Antiretroviral Therapy. Clin Lymphoma (2003) 4:86–92. doi: 10.3816/CLM.2003.n.016
214. Dunleavy K, Little RF, Pittaluga S, Grant N, Wayne AS, Carrasquillo JA, et al. The Role of Tumor Histogenesis, FDG-PET, and Short-Course Epoch With Dose-Dense Rituximab (Sc-EPOCH-RR) in HIV-associated Diffuse Large B-cell Lymphoma. Blood (2010) 115:3017–24. doi: 10.1182/blood-2009-11-253039
215. Venturelli S, Pria AD, Stegmann K, Smith P, Bower M. The Exclusion of People Living With Hiv (Plwh) From Clinical Trials in Lymphoma. Br J Cancer (2015) 113:861–3. doi: 10.1038/bjc.2015.301
Keywords: NHL, DLBCL, HIV, ABC, GCB, PLWH
Citation: de Carvalho PS, Leal FE and Soares MA (2021) Clinical and Molecular Properties of Human Immunodeficiency Virus-Related Diffuse Large B-Cell Lymphoma. Front. Oncol. 11:675353. doi: 10.3389/fonc.2021.675353
Received: 03 March 2021; Accepted: 14 April 2021;
Published: 29 April 2021.
Edited by:
Robert Ohgami, University of California, San Francisco, United StatesReviewed by:
Michele Merli, University of Insubria, ItalyMichael Bayerl, Penn State Milton S. Hershey Medical Center, United States
Copyright © 2021 de Carvalho, Leal and Soares. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marcelo A. Soares, bWFzb2FyZXNAaW5jYS5nb3YuYnI=