 Yosr Hamdi1,2*
Yosr Hamdi1,2* Najah Mighri1†
Najah Mighri1† Maroua Boujemaa1†
Maroua Boujemaa1† Nesrine Mejri1,3
Nesrine Mejri1,3 Sonia Ben Nasr1,4
Sonia Ben Nasr1,4 Mariem Ben Rekaya1,5
Mariem Ben Rekaya1,5 Olfa Messaoud1
Olfa Messaoud1 Hanen Bouaziz1,6
Hanen Bouaziz1,6 Yosra Berrazega3Haifa Rachdi3
Yosra Berrazega3Haifa Rachdi3 Olfa Jaidane6Nouha Daoud3Aref Zribi4
Olfa Jaidane6Nouha Daoud3Aref Zribi4 Jihene Ayari4
Jihene Ayari4 Houda El Benna1,3Soumaya Labidi1,3Jamel Ben Hassouna6Abderazzek Haddaoui4Khaled Rahal6Farouk Benna7
Houda El Benna1,3Soumaya Labidi1,3Jamel Ben Hassouna6Abderazzek Haddaoui4Khaled Rahal6Farouk Benna7 Ridha Mrad8Slim Ben Ahmed9
Ridha Mrad8Slim Ben Ahmed9 Hamouda Boussen1,3Samir Boubaker1,2
Hamouda Boussen1,3Samir Boubaker1,2 Sonia Abdelhak1
Sonia Abdelhak1- 1Laboratory of Biomedical Genomics and Oncogenetics, LR20IPT05, Institut Pasteur de Tunis, University of Tunis El Manar, Tunis, Tunisia
- 2Laboratory of Human and Experimental Pathology, Institut Pasteur de Tunis, Tunis, Tunisia
- 3Medical Oncology Department, Abderrahman Mami Hospital, Faculty of Medicine Tunis, University Tunis El Manar, Tunis, Tunisia
- 4Department of Medical Oncology, Military Hospital of Tunis, Tunis, Tunisia
- 5UR17ES15, Oncotheranostic Biomarkers, Faculty of Medicine of Tunis, University Tunis El Manar, Tunis, Tunisia
- 6Surgical Oncology Department, Salah Azaiez Institute of Cancer, Tunis, Tunisia
- 7Department of Radiation Oncology, University of Tunis, Tunis, Tunisia
- 8Department of Human Genetics, Charles Nicolle Hospital, Tunis, Tunisia
- 9Faculty of Medicine of Sousse Department of Medical Oncology Farhat Hached University Hospital University of Sousse, Sousse, Tunisia
Background: Breast cancer is the world’s most common cancer among women. It is becoming an increasingly urgent problem in low- and middle-income countries (LMICs) where a large fraction of women is diagnosed with advanced-stage disease and have no access to treatment or basic palliative care. About 5-10% of all breast cancers can be attributed to hereditary genetic components and up to 25% of familial cases are due to mutations in BRCA1/2 genes. Since their discovery in 1994 and 1995, as few as 18 mutations have been identified in BRCA genes in the Tunisian population. The aim of this study is to identify additional BRCA mutations, to estimate their contribution to the hereditary breast and ovarian cancers in Tunisia and to investigate the clinicopathological signatures associated with BRCA mutations.
Methods: A total of 354 patients diagnosed with breast and ovarian cancers, including 5 male breast cancer cases, have been investigated for BRCA1/2 mutations using traditional and/or next generation sequencing technologies. Clinicopathological signatures associated with BRCA mutations have also been investigated.
Results: In the current study, 16 distinct mutations were detected: 10 in BRCA1 and 6 in BRCA2, of which 11 are described for the first time in Tunisia including 3 variations that have not been reported previously in public databases namely BRCA1_c.915T>A; BRCA2_c.-227-?_7805+? and BRCA2_c.249delG. Early age at onset, family history of ovarian cancer and high tumor grade were significantly associated with BRCA status. BRCA1 carriers were more likely to be triple negative breast cancer compared to BRCA2 carriers. A relatively high frequency of contralateral breast cancer and ovarian cancer occurrence was observed among BRCA carriers and was more frequent in patients carrying BRCA1 mutations.
Conclusion: Our study provides new insights into breast and ovarian cancer genetic landscape in the under-represented North African populations. The prevalence assessment of novel and recurrent BRCA1/2 pathogenic mutations will enhance the use of personalized treatment and precise screening strategies by both affected and unaffected North African cancer cases.
Introduction
Breast cancer is the most common malignancy among women worldwide (1). Incidence and mortality rates of breast cancer differ between populations (1). In Tunisia, it remains the most common cancer among females and represents the first leading cause of cancer mortality among women. The mean age at diagnosis of Tunisian breast cancer cases is around 50 years old, a decade younger than Western countries (2, 3).
BRCA1 and BRCA2 are the most prominent breast cancer susceptibility genes that convey high risk of breast and ovarian cancers (4). Since their discovery, a wide range of mutational spectrum have been described for both genes. So far, more than 1800 distinct BRCA1 and 2000 BRCA2 mutations have been reported in the Breast Cancer Information Core (BIC) database. These mutations explain around 20-30% of breast cancer genetic component and seem to be associated with different other cancers such as prostate, pancreatic, endometrial and melanoma (5). The identification of novel BRCA1/2 mutations has important clinical implications. Indeed, unaffected BRCA mutation carriers have various preventive options including extensive and regular surveillance, chemoprevention, and risk-reducing surgery (6–8), while, affected cases carrying BRCA mutations could benefit from personalized therapeutic options such as platinum-based chemotherapy and poly (ADP-ribose) polymerase (PARP) inhibitors (9, 10). However, a full BRCA1 and BRCA2 gene screening remains a labor and time-consuming challenge due to the large gene size, diverse mutations, or variants of unknown significance (VUS) and complexity of large genomic rearrangements (LRs) and copy number variations (CNVs) requiring special technical approaches. Recent advances in high throughput sequencing technologies including Target panels and whole exome sequencing (WES) allowed rapid, sensitive, and cost-effective screening of the large BRCA genes. In addition, the decreased cost of genotyping and sequencing offered affordable targeted testing options.
Sequencing thousands of cancer samples showed that the frequency of germline mutations in BRCA genes varies widely among populations. Some mutations are shared between different populations and others are ethnic specific (11, 12). Indeed, in certain countries and ethnic communities, the BRCA mutation spectrum is limited to a few founder mutations (13, 14). This is mainly observed in geographically, culturally, or religiously isolated populations and in countries with high rates of consanguinity and endogamy that undergo rapid expansion from a limited number of ancestors. Consequently, some alleles become more frequent which explain the high frequency of some founder mutations in these populations. The founder effect may, therefore, influence mutation prevalence and gene penetrance. Since cancer risk is a function of mutation prevalence and penetrance that seems to vary by ethnicity, investigating the prevalence, the frequency, and the penetrance of novel BRCA mutation in different populations will bring new insights on cancer risk and etiology.
In Tunisia, previous studies on BRCA genes have focused only on breast cancer patients. In some studies, the genetic investigation concerned all the coding regions of BRCA1 and/or BRCA2 genes and in others only hotspot exons have been investigated. These reports have revealed a total of only 18 distinct mutations of which 12 are localized within BRCA1 gene including 2 large rearrangements encompassing exons 5 and 20. Among the identified mutations c.211dupA, c.5266dupC in BRCA1 and c.1310_1313delAAGA in BRCA2 were the most recurrent mutations encountered among the hereditary breast cancer cases (11, 15–23). Despite these efforts, the mutational spectrum of BRCA1/2 genes is still not well established. The main goal of the present study was to identify additional novel BRCA mutations and to investigate the contribution of these mutations to the missing heredity of breast and ovarian cancers. We also aimed to compare breast cancer clinicopathological characteristics in BRCA+ vs BRCA- Tunisian breast cancer cases.
Materials And Methods
Patients
A total of 354 breast and ovarian cancer patients (335 breast cancer patients and 19 ovarian cancer patients) were included in this study referred from different medical oncology departments in Tunisia including those of Abderrahman Mami Hospital, Military Hospital of Tunis, Salah Azaiez Institute of Cancer and Farhat Hachad University Hospital University of Sousse. Written informed consents were obtained from all participants. The study has been conducted according to the Declaration of Helsinki Principles and ethical approval was obtained from the biomedical ethics committee of Institut Pasteur de Tunis (2017/16/E/Hôpital A-M). Clinico-pathological characteristics and follow-up data were collected from patients’ medical records. Probands were selected based on the following selection criteria (1): Presence of at least 3 related first or second-degree breast cancer cases at any age (2), Young cancer patients aged less than 35 years (3), Presence of at least two cases of breast or ovarian cancer, regardless of age, and at least one case of pancreatic cancer or prostate cancer in a related first- or second-degree patient (4), one case with triple negative breast cancer (TNBC) at an age ≤40 years (5), one breast cancer and one ovarian cancer cases diagnosed at first or second degree relatives at any age. A study flowchart is illustrated in Figure 1.
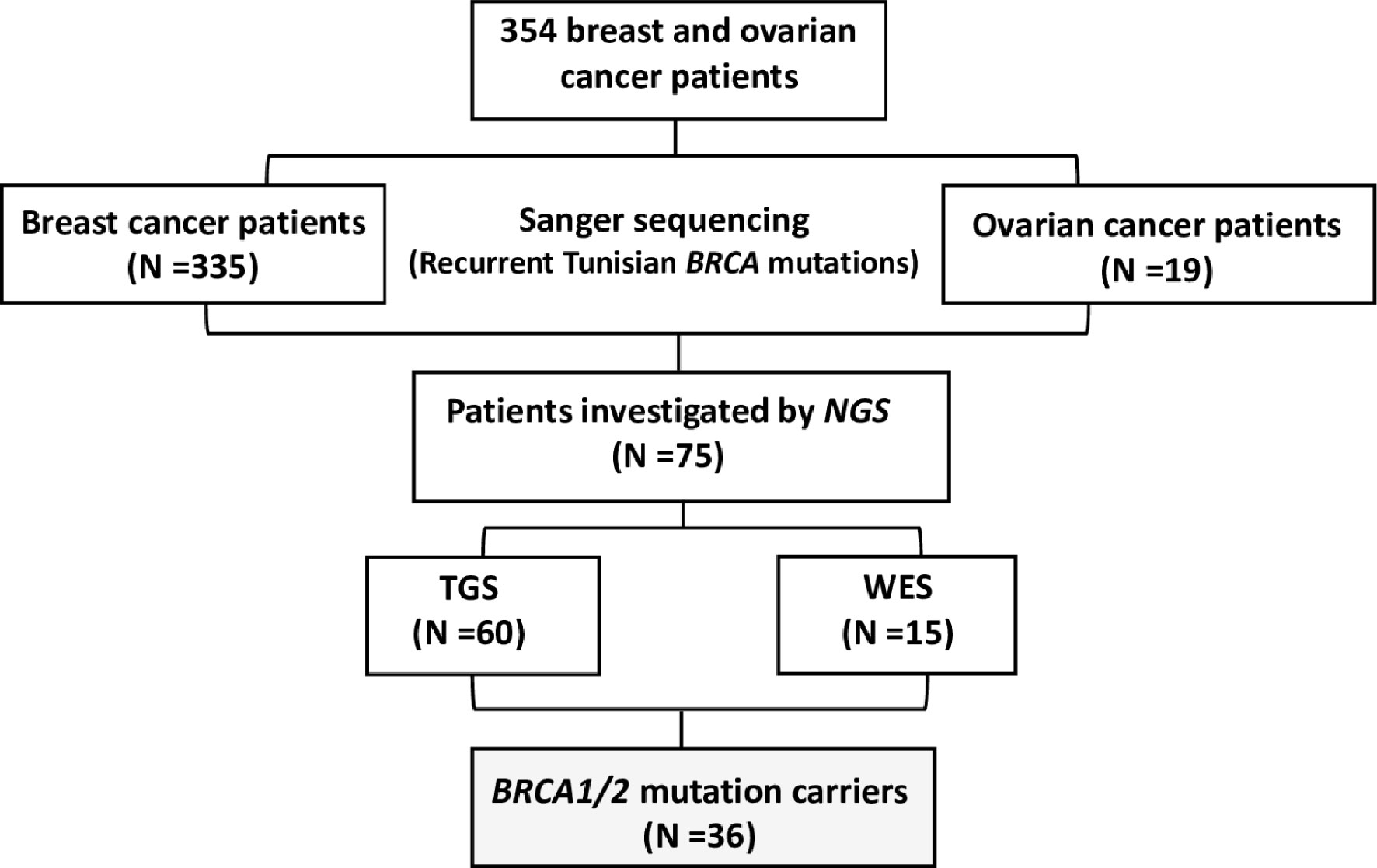
Figure 1 Study flowchart.
DNA Isolation
Total genomic DNA was isolated from peripheral blood using DNeasy blood DNA extraction Kit (Qiagen) according to the manufacturer’s instructions. DNA purity and concentration were measured using a NanoDrop™ spectrophotometer.
Screening for Recurrent Mutations in BRCA1 and BRCA2 Genes Using Sanger Sequencing
Before performing Next Generation Sequencing (NGS) analysis, the studied cohort was screened for at least one of the recurrent BRCA1/2 mutations previously reported in the Tunisian population, namely exon5-c.211dupA (rs397508938), exon11-c.798_799delTT (rs80357724), exon11-c.2551delG (rs397508977), exon11-c.3331_3334delCAAG (rs80357701) and exon20-c.5266dupC (rs80357906) of the BRCA1 gene and exon10-c.1310_1313 delAAGA (rs80359277), exon16-c.7654dupA (rs879255463) in BRCA2 gene respectively. The reference sequences used were NM_007294.3 for BRCA1 and NM_000059.3 for BRCA2.
PCR reactions were performed on genomic DNA (gDNA), following standard protocols. Sanger sequencing has been performed using an automated sequencer (ABI 3500; Applied Biosystems, Foster City, CA) and a cycle sequencing reaction kit (Bigdye Terminator v3.1 kit, Applied Biosystems). The data were analyzed using BioEdit software version 7.2.5.
Sanger sequencing technique was then used to validate the identified mutations resulting from NGS.
NGS was performed on 75 breast and ovarian cancer cases. Targeted BRCA1/2 sequencing and whole exome sequencing were performed on 60 and 15 patients respectively.
Targeted Gene Sequencing
Targeted gene sequencing was performed on BRCA1/2 for 60 breast and ovarian cancer patients with strong family history. All targeted coding exons and exon–intron boundaries of BRCA1/2 genes were amplified with 253 pooled primer pairs. After the targeted amplification and construction of a library through QIAGEN Library Kit v2.0, the libraries were pooled prior to emulsion PCR and bead enrichment steps that were carried out using an automated protocol on the GeneRead QIAcube (QIAGEN, Hilden, Germany) using the GeneRead Clonal Amp Q Kit (QIAGEN, Hilden, Germany), according to the manufacturer’s protocol. Following bead enrichment, the pooled libraries were sequenced using the GeneReader platform (QIAGEN, Hilden, Germany).
Whole Exome Sequencing
WES was performed for 15 breast cancer Tunisian patients. Samples were prepared according to Agilent’s SureSelect Protocol Version 1.2 and enrichment was carried out according to Agilent SureSelect protocols. Enriched samples were sequenced on the Illumina HiSeq2000 platform using TruSeq v3 chemistry with paired-end (2 × 100). Exome DNA sequences were mapped to their location in the build of the human genome (hg19/b37) using the Burrows–Wheeler Aligner (BWA) package. The subsequent SAM files were converted to BAM files using Samtools. Duplicate reads were removed using Picard. GATK was then used to recalibrate the base quality scores as well as for SNP and short INDEL calling. Annotation and prioritization of potential disease-causing variants were performed using VarAFT (Variant Annotation and Filtering Tool) (http://varaf t.eu). To annotate variants, VarAFT uses ANNOVAR, a command line tool. INDELs and SNPs annotated were filtered according to several criteria (1): considering breast cancer as autosomal dominant disease and removing variants that were found in a homozygous state (2), variants identified as intronic, intergenic, and non-coding or synonymous were discarded (3), assuming that causal variants are rare, we removed all variants with an allele frequency > 1% either in ExAC (24), 1000 genomes (25) or ESP6500 (http://evs.gs.washington.edu/EVS/) (4), Using different in silico prediction tools, the functional impact of all identified variants has been assessed. Based on this assessment, Benign and tolerated variants were removed. Finally, significant candidate variants were obtained after filtering against their phenotypic relevance.
Clinico-Pathological Features of BRCA1 and BRCA2 Carriers
Clinical and pathological features of BRCA+ vs BRCA- patients as well as BRCA1 vs BRCA2 carriers were compared and evaluated. Statistical analysis was performed using SPSS software (version 23). Quantitative variables with normal distribution were analyzed by Student’s t test. Comparison of qualitative data was performed using Chi-square test. Fisher’s exact test was used for the study of small sample size. Correlation is considered statistically significant between two variables if the P value is less than or equal to 0.05.
Results
Epidemiological and Clinico-Pathological Features of Investigated Breast and Ovarian Cancer Patients
A family history of breast and ovarian cancer was present in 35.24% and 11.14% of patients respectively. In addition, 2.68% of patients presented both breast and ovarian cancers. Consanguineous families represent 35.31% of the studied patients. Mean age at menarche was 12.81 years. Mean age at first pregnancy was 26.62 years. Oral contraception was reported by 47.31% of patients, 25.99% of patients have never breastfed and 31.85% were premenopausal.
The mean age at diagnosis of breast cancer was 43.10 years and 31.94% of patients were ≤35 years. Among investigated patients1.49% were male breast cancer (MBC) cases. Inflammatory breast cancer (IBC) (T4d) was seen in 8.65% of patients. Invasive ductal carcinoma (IDC) was the most frequent (90.04%) while infiltrating lobular carcinoma (ILC) was observed in only 3.94% of cases. Scarff-Bloom-Richardson (SBR) grade III was the most common (47.71%). Mean Tumor size was 33.62mm. Patients with positive lymph node disease represented 53.37% of our cohort, 88.67% of patients had Ki-67>14%. Luminal B tumors were the most common (56.88%) followed by triple negative breast cancer (TNBC) (23.85%), Her2+ (11.93%) and luminal A (7.34%). Distant metastases were observed in 26.34% of patients.
For ovarian cancer cases, the mean age at diagnosis was 52.62 years and the majority with serous ovarian carcinoma.
Genetic Analysis
Genetic analysis results showed that 36 out of 354 tested breast and ovarian cancer patients were BRCA1/2 mutation carriers (31 breast cancer cases and 5 ovarian cancer patients), including 21 patients with BRCA1 mutation and 15 patients carrying BRCA2 mutation. A total of 16 mutations have been identified including 11 short indels, 4 single nucleotide variations (3 nonsense & 1 splicing) and 1 large rearrangement.
Identified BRCA1/2 Pathogenic Mutations in Breast Cancer Cases
Within the studied breast cancer cohort, 13 pathogenic mutations have been identified: 8 in BRCA1 and 5 in BRCA2 genes. Among the identified mutations, 9 are described for the first time in Tunisian population (6 in BRCA1 and 3 in BRCA2) (Table 1).
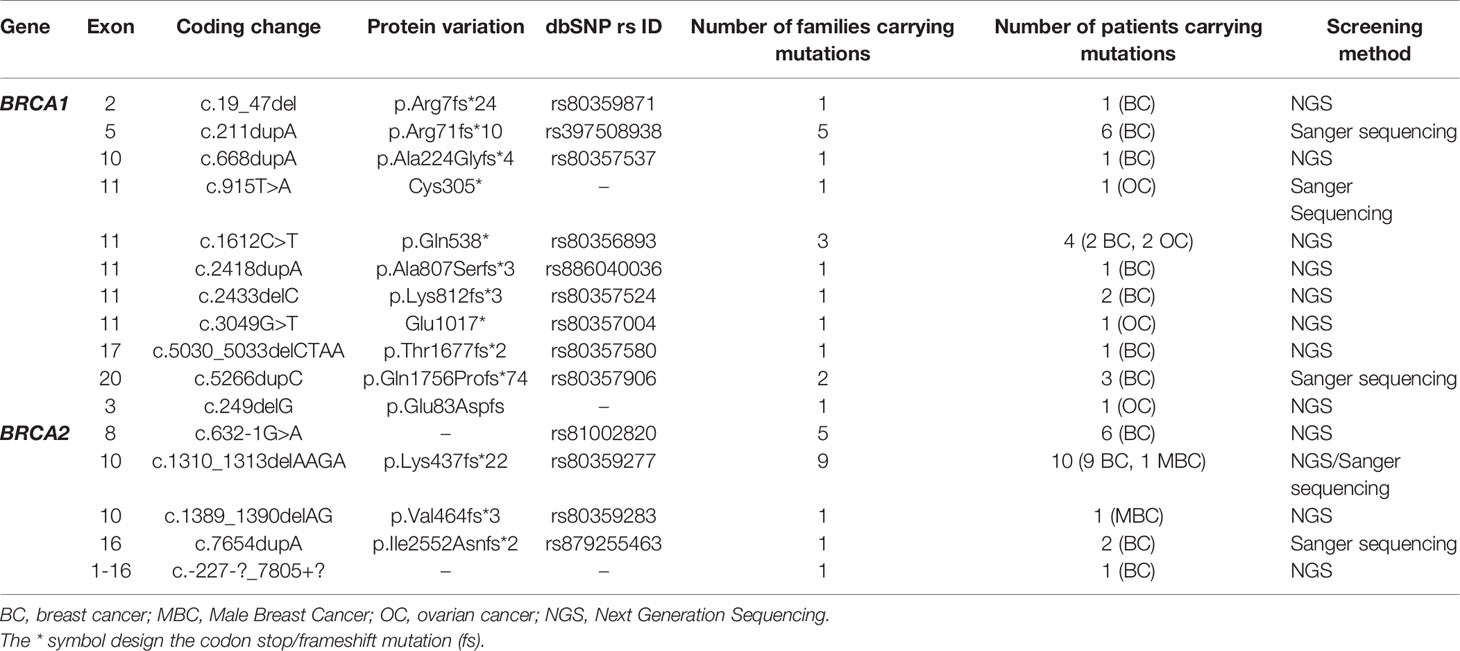
Table 1 Mutations in the BRCA1/2 genes identified in breast cancer and ovarian cancer patients by Sanger and next generation sequencing technologies.
Considering the BRCA1 gene, 6 patients belonging to 5 unrelated families were carriers of the recurrent c.211dupA mutation. Three patients belonging to 2 unrelated families were positive for c.5266dupC mutation. The missense c.1612C>T mutation has been identified in 2 related patients. c.19_47del, c.668dupA, c.2418dupA and c.5030_5033delCTAA mutations have been identified each in one patient. c.2433delC has been identified among 2 related patients. Except c.211dupA and c.5266dupC mutations, all remaining BRCA1 mutations are reported for the first time in the Tunisian population.
In the BRCA2 gene, 3 frameshift mutations as well as 1 splicing and 1 large rearrangement mutation were detected. Our results revealed 6 patients belonging to 5 unrelated families that are double heterozygous for BRCA2 gene. Indeed, these families were carrying two mutations classified as pathogenic in the ClinVar database namely c.632-1G>A and c.1310_1313delAAGA. Four additional patients carrying only the c.1310_1313delAAGA mutation have been identified including one male breast cancer. The c.7654dupA mutation was identified in 2 related patients with a strong family history of hereditary breast and ovarian cancer. The c.1389_1390delAG mutation has been identified in 1 additional male breast cancer case. Similarly, a large rearrangement mutation of BRCA2 gene (Del exons 1-16) has been identified in one patient. Among the identified BRCA2 mutations, BRCA2-Del exons 1-16 mutation is novel and was not described in public databases. c.632-1G>A and c.1389_1390delAG are described for the first time in the Tunisian population.
BRCA1/2 Pathogenic Mutations Identified in Ovarian Cancer Cases
A total of 19 ovarian cancer patients were screened for BRCA pathogenic mutations using Sanger and/or NGS. Four distinct deleterious mutations were identified: 3 mutations in BRCA1 gene (c.915T>A, c.1612C>T and c.3049G>T) and one BRCA2 mutation (c.249delG).
BRCA1-c.915T>A and BRCA2-c.249delG mutations are novel and not described in public databases. The other identified mutations are described for the first time in the Tunisian population. c.1612C>T mutation was identified among 2 patients. This same mutation was also identified in 2 related breast cancer patients. Screening for additional carriers of the identified mutations, based on their geographic origin, was performed using Sanger sequencing. Consequently, the geographic origin of the identified BRCA1/2 mutations has been clearly established (Figure 2). We have also illustrated the distribution of BRCA mutations identified in hereditary breast and ovarian cases on BRCA1 and BRCA2 genes (Figure 3). Breast cancer cluster regions (BCCRs) and ovarian cancer cluster regions (OCCRs) were assigned in Figure 3 according to the study of Rebbeck et al., 2015 (26). Among BRCA mutations identified in breast cancer patients, BRCA1_c.211dupA, BRCA1_c.5266dupC, BRCA2_c.-227-?_7805+?, BRCA2_1310_1313delAAGA, BRCA2_c.1389_1390delAG and BRCA2_c.7654dupA occurred in BCCRs. Considering BRCA mutations identified in ovarian cancer patients BRCA1_c.1612C>T and BRCA1_3049G>T arose in OCCRs.
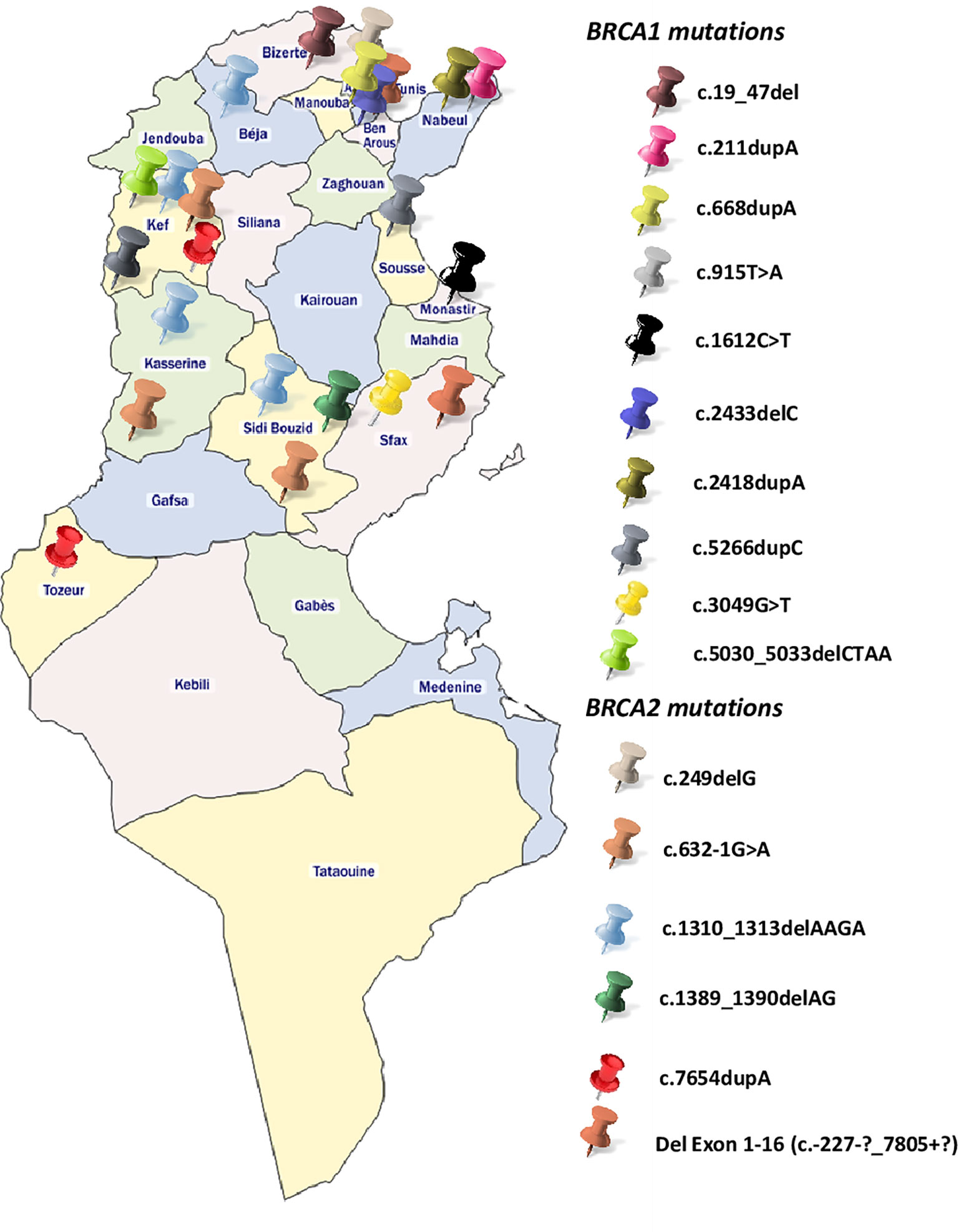
Figure 2 Geographical distribution of the identified BRCA1 and BRCA2 mutations.
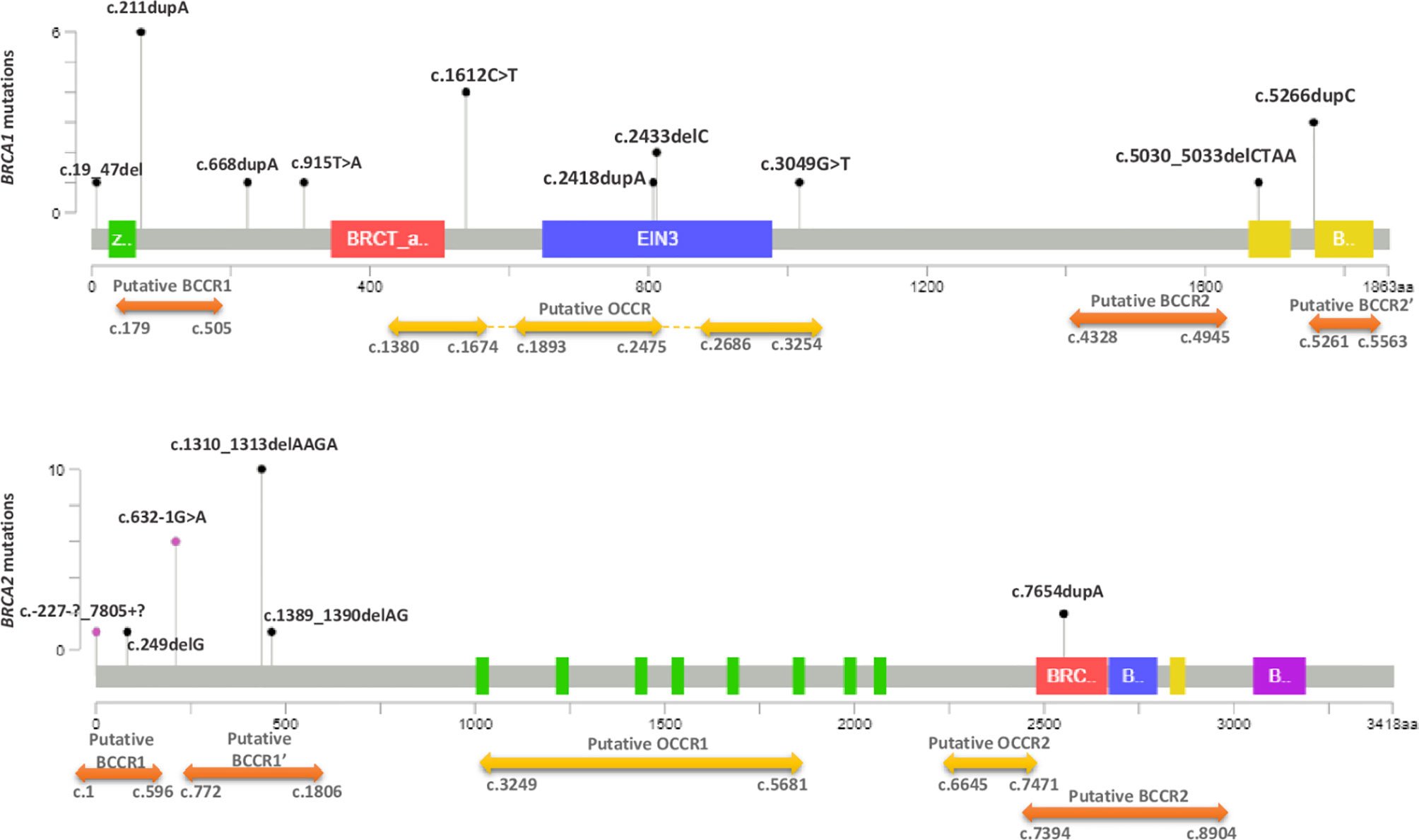
Figure 3 Distribution of BRCA1 and BRCA2 mutations identified in hereditary breast and ovarian cancer cases. The Length of mutation indicator reflects the number of observed carriers. The diagrams linearly represent BRCA1/2 protein domains (x-axis). BRCA1 domains: Zinc/Ring finger (green); BRCT_assoc: serine-rich domain associated with BRCT (red); Ethylene insensitive 3 (blue); BRCA1 C terminus domain (yellow). BRCA2 domains: BRC repeats (green); BRCA-2_helical (red); oligonucleotide/oligosaccharide-binding, domain 1 (blue); tower domain (yellow) and oligonucleotide/oligosaccharide-binding, domain 3 (purple). Black mutation indicators depict truncating mutations and purple indicators represent the other types of mutations (splicing, LR). Cluster regions (breast cancer cluster regions (BCCRs) (orange) and ovarian cancer cluster regions (OCCRs) (yellow) were assigned according to the study of Rebbeck et al. (26).
Polymorphisms and Variant of Unknown Significance Identified in BRCA1 and BRCA2 Genes
In addition to the pathogenic mutations that have been identified in BRCA genes, several SNPs and variants of unknown significance (VUS) have been observed (Supplementary Table 1). Among the 101 identified variants, 45.54% were coding, 50.49% were intronic and 3.96% were localized within regulatory regions. The majority of variations were classified as benign or likely benign in the ClinVar database (91.08%) and five intronic variations were not reported. One patient carried a VUS rs397507308 in BRCA2 and 3 other patients carried 2 intronic variations (rs276174878 and rs276174816) that have conflicting interpretations of pathogenicity. Another patient diagnosed with early onset bilateral breast cancer had an in-frame variant with conflicting interpretations of pathogenicity rs80358343 (c.5017_5019delCAC) in the BRCA1 gene.
Clinico-Pathological Features of BRCA Carriers Among Breast Cancer Cohort
Clinico-pathological characteristics of breast cancer cases carrying BRCA1 and BRCA2 mutations are described in Tables 2, 3 respectively. We investigated these clinico-pathological features in BRCA1/2 mutation carriers vs BRCA negative patients (Table 4) and between BRCA1 and BRCA2 mutation carriers (Table 5), as well.
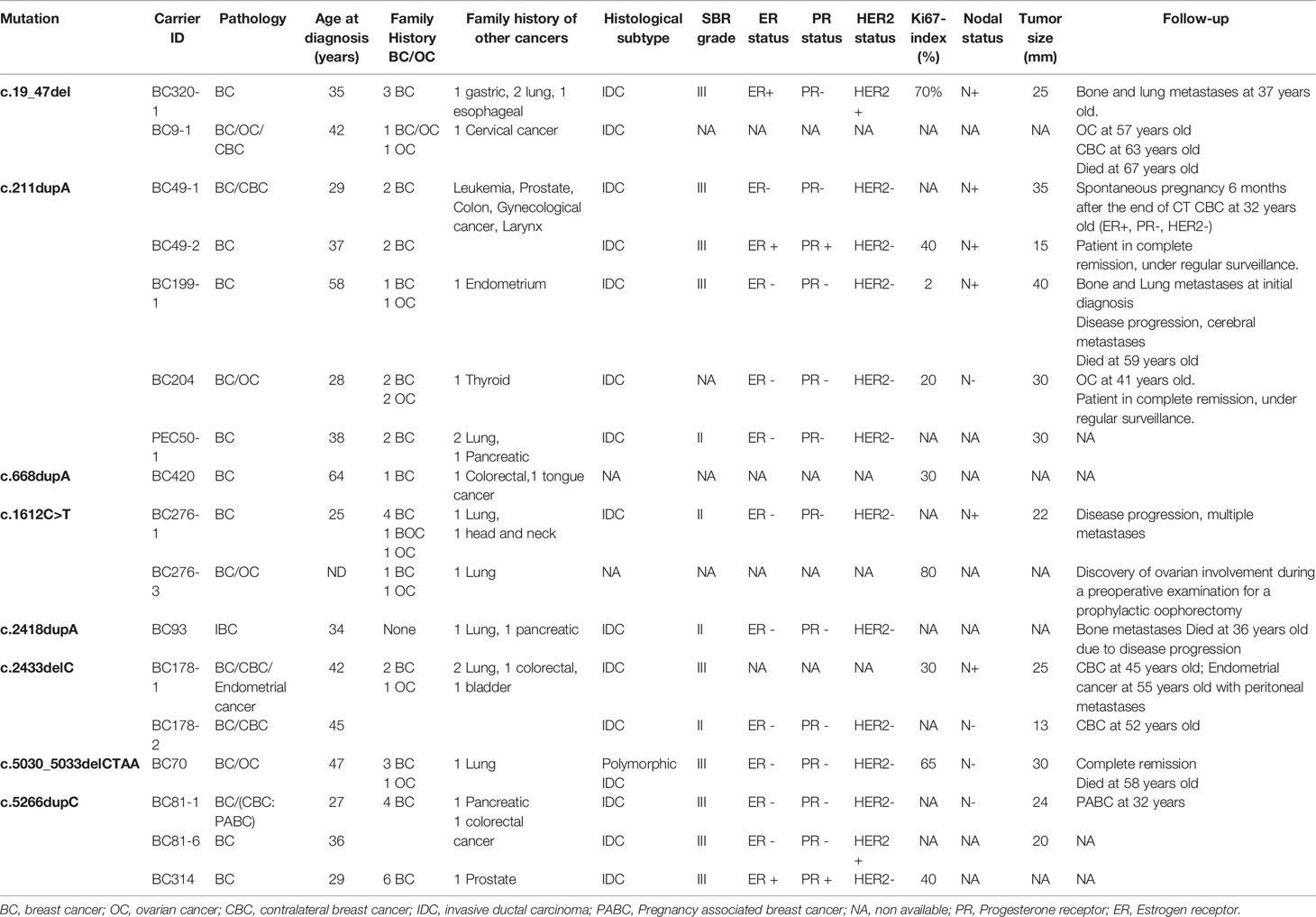
Table 2 Clinicopathological features of BRCA1 carriers.
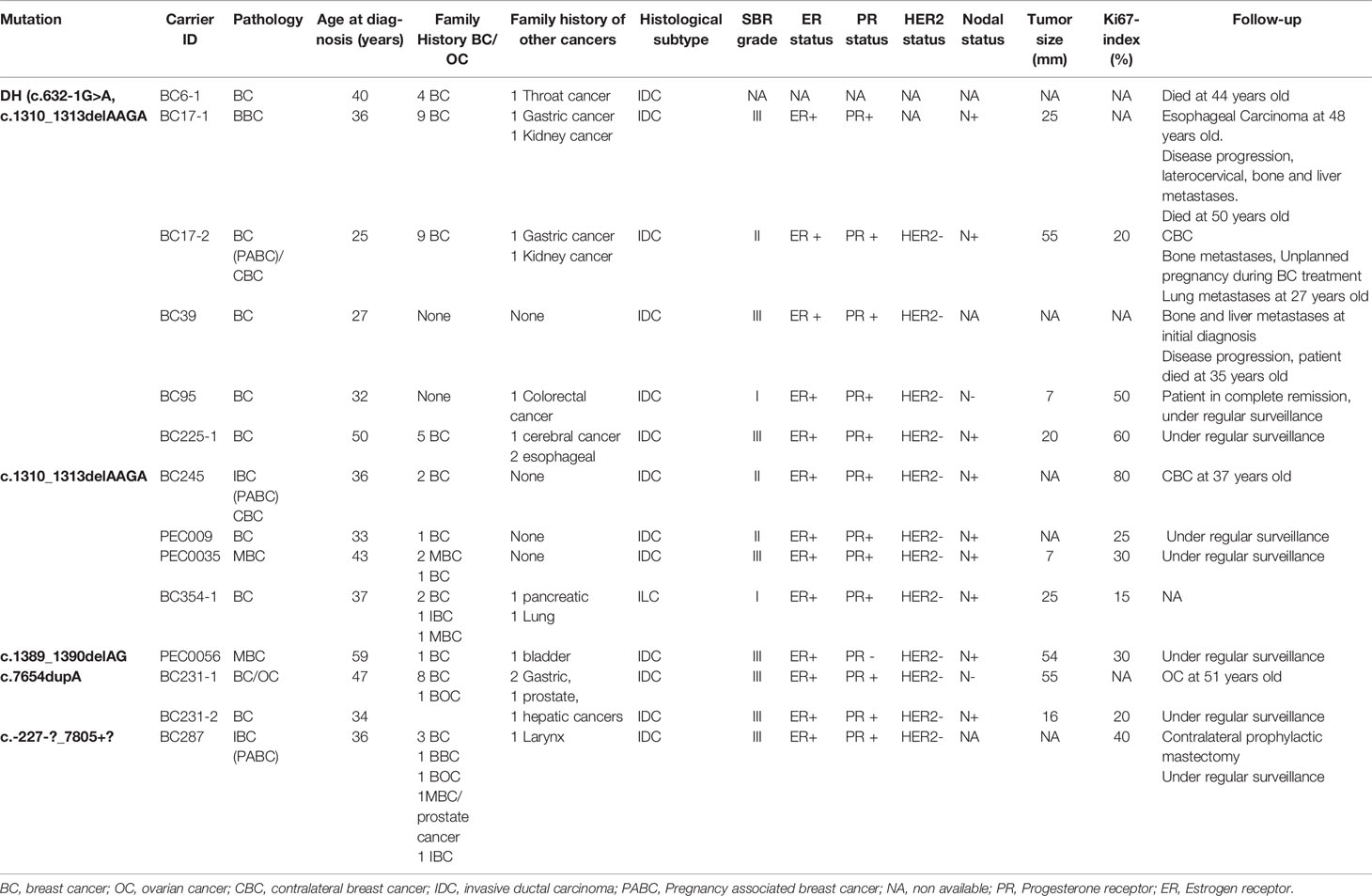
Table 3 Clinico-pathological features of BRCA2 carriers.
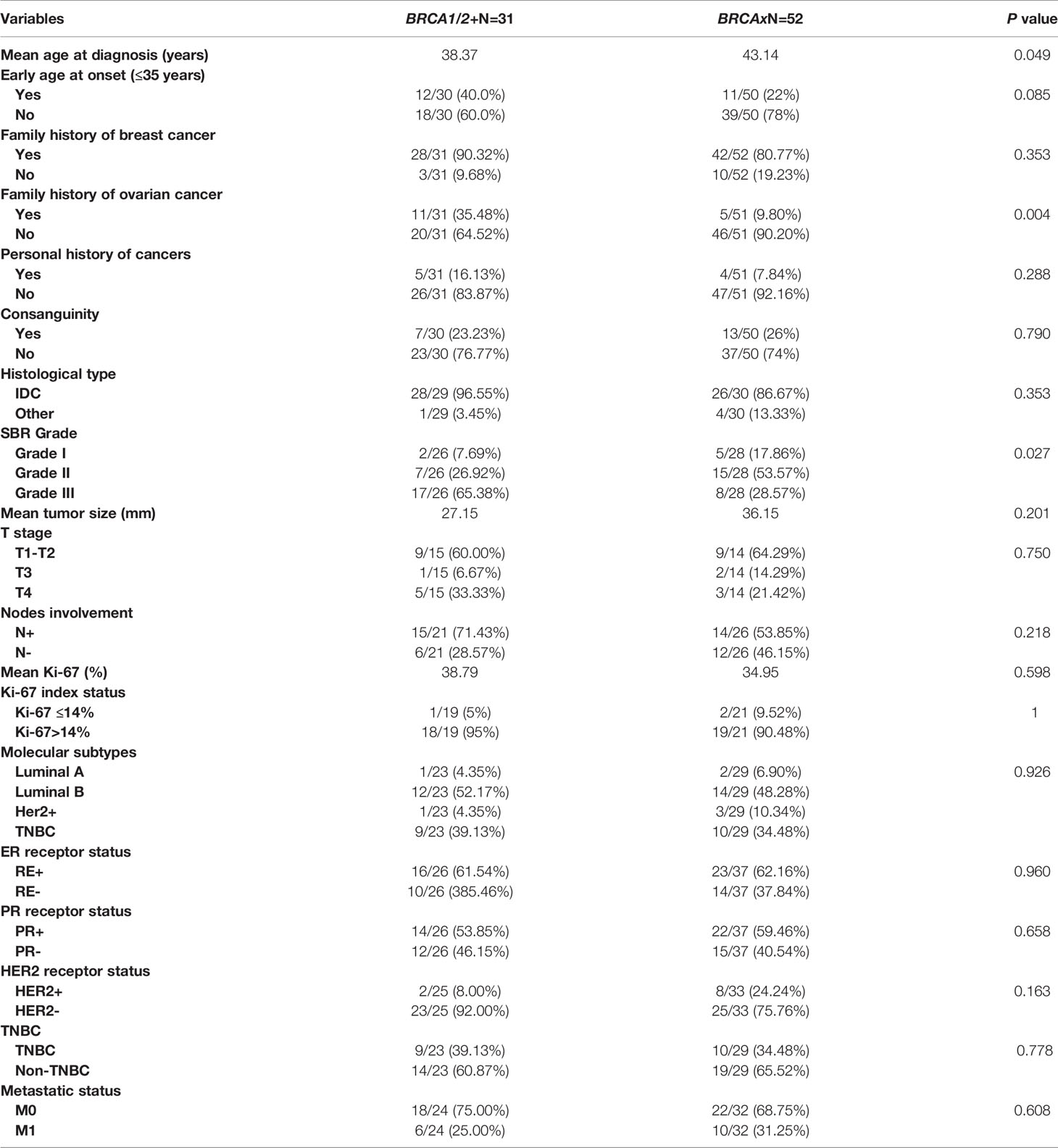
Table 4 Epidemiological and clinico-pathological characteristics of patients carrying or not BRCA1/2 mutations.
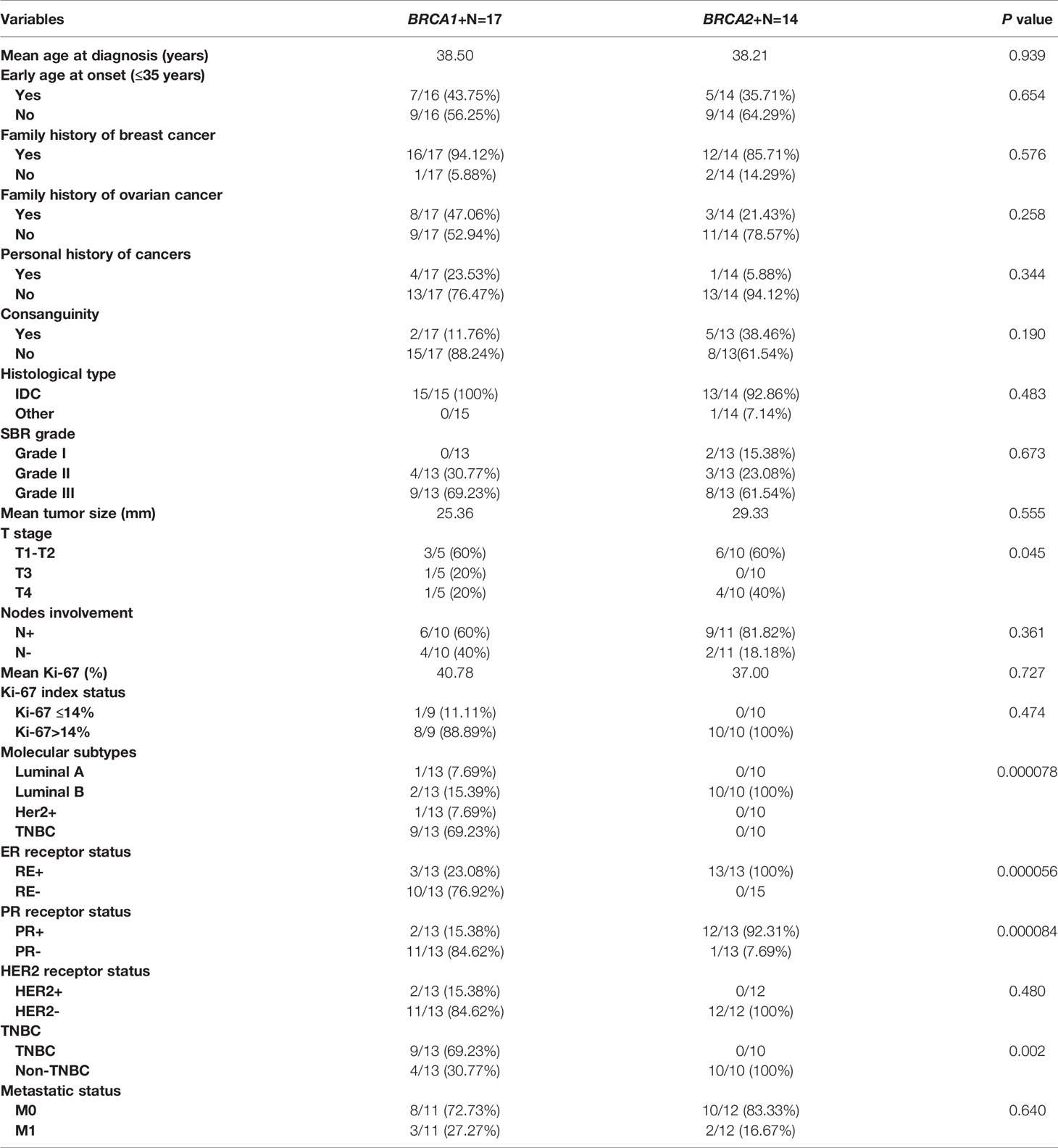
Table 5 Epidemiological and clinico-pathological characteristics of patients carrying BRCA1 and BRCA2 mutations.
BRCA+ vs BRCA-
Family history of ovarian cancer was significantly associated with BRCA positive status (p=0.004). Regarding the mean age at diagnosis BRCA carriers seem to be younger than BRCA- patients (38.37 vs 43.14) (p= 0.049). However, no significant difference has been observed between both groups regarding family history of breast cancer, personal history of cancer and consanguinity. Similarly, no significant differences have been observed between the 2 groups in histological subtype, nodal involvement, tumor stage, hormonal receptors status, HER2 status, molecular subtypes, Ki-67 index, and metastases (Table 4). Nevertheless, SBR grade III was found in 65.38% of patients with BRCA1/2 mutations against a frequency of 28.57% among non-carriers, this difference appears to be statistically significant (p= 0.027).
BRCA1 vs BRCA2
Association between clinico-pathological features and BRCA status (BRCA1+, BRCA2+ and BRCAx) was shown in Figures 4, 5. Our results showed that there were no significant differences between BRCA1 and BRCA2 mutated groups regarding the mean age at diagnosis, the family history of personal cancers, of breast cancer and ovarian cancer (Table 5).
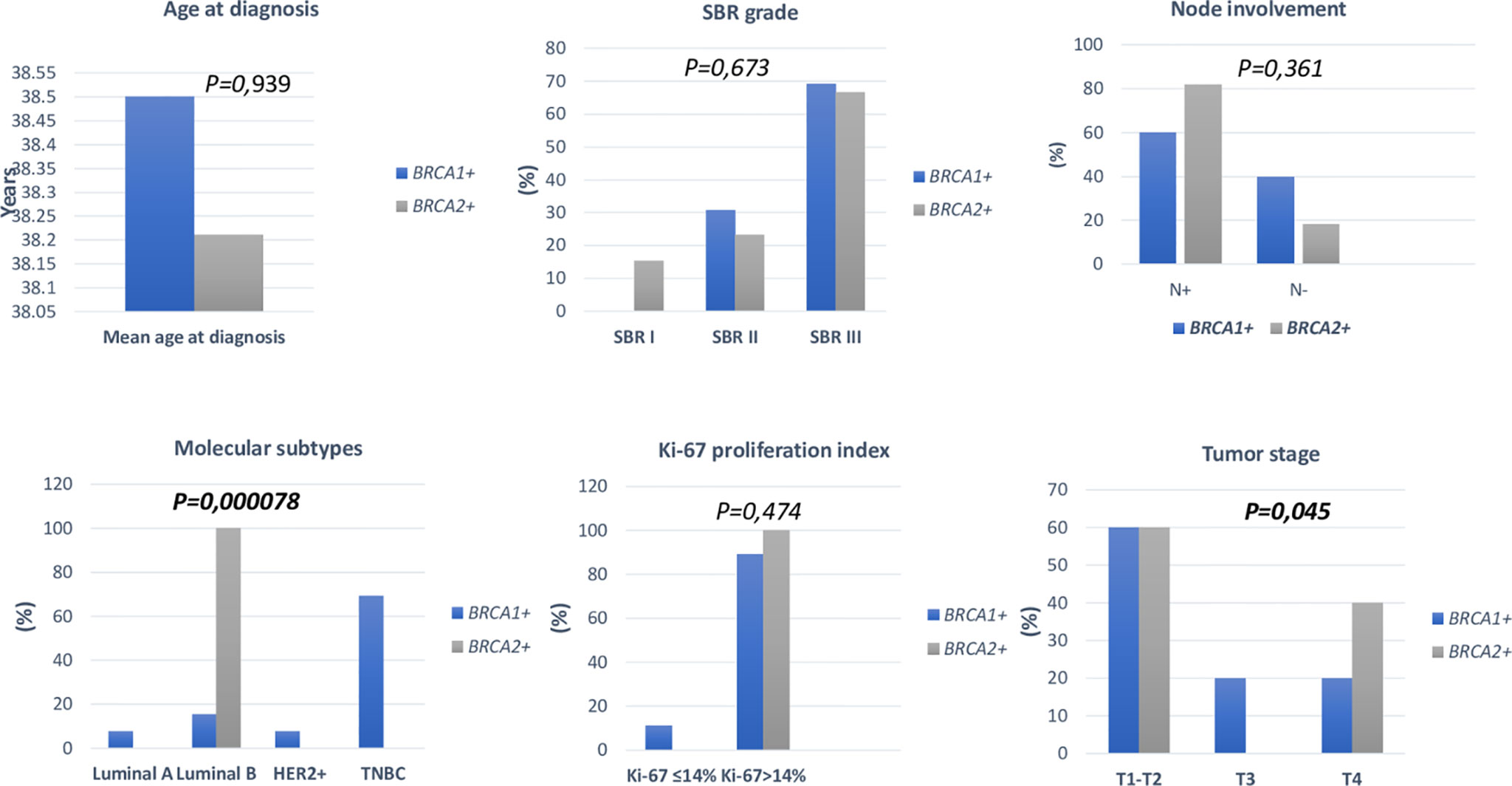
Figure 4 Distribution of clinico-pathological features of breast cancer in BRCA1+ and BRCA2+ patients.
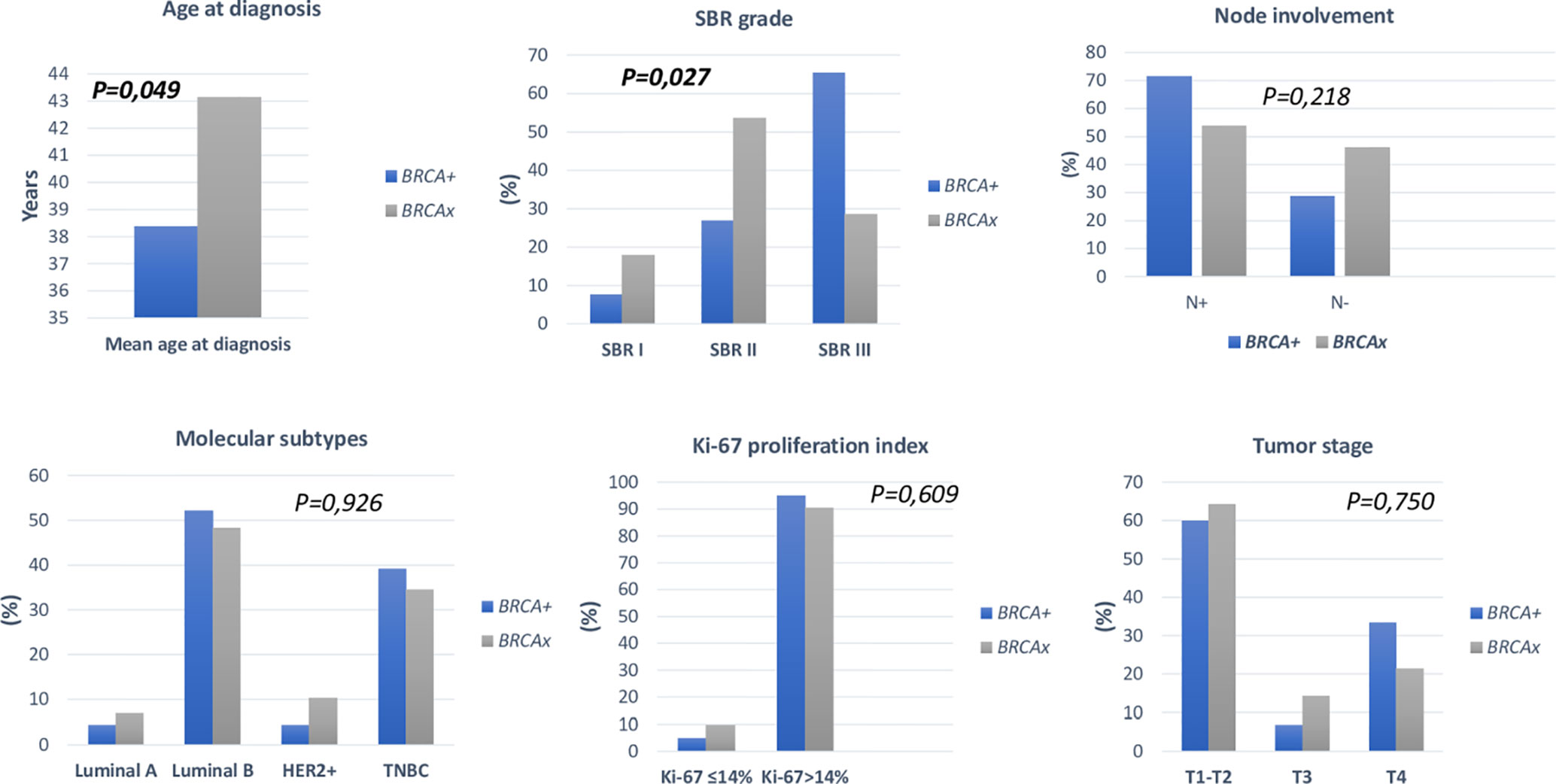
Figure 5 Distribution of clinico-pathological features of breast cancer in BRCA+ and BRCAx patients.
Pathology showed that the infiltrating ductal carcinoma was the most common histological type in both groups (100% and 92.86%). HER2 status, lymph node involvement, SBR grade, tumor size, Ki-67 index and metastatic status showed no statistically significant difference between both studied groups. However, BRCA1 carriers were more likely to have triple negative breast cancer (p=0.002) and BRCA2 carriers were more likely to have luminal B breast cancer tumors (p=0.000078). In addition, positive estrogen receptor (ER) status and positive progesterone receptor (PR) status studied separately were both associated with BRCA2 mutated tumors (p=0.000056 and p=0.000084), respectively.
Follow Up of BRCA1 and BRCA2 Carriers
Among BRCA carriers, contralateral breast cancer and ovarian cancer co-occurrence were observed respectively in 22.58% and 16.12% of cases. One patient diagnosed with early onset breast cancer has undergone a contralateral prophylactic mastectomy and is currently under regular surveillance. Both contralateral breast cancer and ovarian cancer occurrence were more frequent in BRCA1 than BRCA2 carriers. Also, 22.58% of the carriers have developed distant metastases and 5 cases died due to disease progression.
Discussion
Detection of mutations in hereditary breast and ovarian cancer related BRCA1 and BRCA2 genes is an effective method of cancer prevention, early detection, and treatment. Mutations in the highly penetrant BRCA genes explain around a quarter of these cases (27). The frequency of germline mutations identified on both genes varies depending on the geographic and ethnic distributions. In some populations, a wide spectrum of different mutations is present, whereas in other groups specific recurrent BRCA mutations have been reported that may be due to the founder mutation effect (28–32).
Our previous studies, investigating breast cancer loci and Nucleotide Excision Repair pathway, have shown that the Tunisian population is an admixed and intermediate population between Sub-Saharan Africans and Europeans (33, 34). This genetic diversity reflects the inter-ethnic variability in the frequency distribution of the studied polymorphisms. Indeed, allele frequencies of several variants were found to be statistically different between Tunisian and other populations including rs2046210 and rs941764 that site in breast cancer susceptibility loci (33). These findings are in favor of the genetic heterogeneity to breast cancer predisposition in the Tunisian population. So far, only 18 deleterious BRCA mutations have been reported. In the current study, 16 BRCA mutations, including 11 novel variations, have been identified in a cohort of 354 Tunisian breast and ovarian cancer patients. For breast cancer cases, high fractions of young patients (31.94%), cases with family history of breast cancer (35.24%), Triple negative breast cancer (24.31%) and high tumor grade (47.41%) have been observed. As reported in previous studies, the high fractions of early onset, triple negative cases and also the presence of family history of breast cancer may be associated with germline BRCA mutations (35, 36). Indeed, it is now well documented that breast cancer patients in North Africa are almost 10 years younger than patients from western countries (37). In Tunisia, around 11% of breast cancer cases are under 35 years old (38). In fact, at a young age, the human organism usually functions as well as it ever will. However, interactions between some genetic and environmental factors (GxE) may cause a physiological decline of some organism systems leading to early disease presentation. Therefore, the influence of specific genetic background, differences in variant penetrance and frequency between populations along with environmental factors may explain this early onset of the disease. Large cohorts of young breast cancer patients should be studied to elucidate these GxE factors.
For ovarian cancer cases, the mean age at diagnosis was 52.62 years and the majority presented with serous ovarian carcinoma. Previous studies have shown that among all patients diagnosed with serous ovarian carcinoma, which is the most common subtype, over 15% will have germline BRCA mutations (39).
Among the 16 distinct deleterious mutations that have been observed c.19_47del, c.668dupA, c.915T>A, c.1612C>T, c.2418dupA, c.2433delC, c.3049G>T and c.5030_5033delCTAA in BRCA1 and c.-227-?_7805+? (Del exons 1-16), c.249delG, c.632-1G>A, c.1389_1390delAG in BRCA2, are reported for the first time in the Tunisian population. We have also identified an inframe deletion reported to have a conflicting interpretation of pathogenicity effect in early onset bilateral breast cancer patient BRCA1_c.5017_5019delCAC. This variation has been described in multiple breast and ovarian cancer cases, with some families showing incomplete co-segregation of the variation (40–42).
Among BRCA mutations identified in breast cancer patients BRCA1_c.211dupA, BRCA1_c.5266dupC,BRCA2_c.-227-?_7805+?,BRCA2_1310_1313delAAGA, BRCA2_c.1389_1390delAG and BRCA2_c.7654dupA occurred in BCCRs that are considered to be associated with an increased likelihood of breast cancer compared to ovarian cancer. Considering BRCA mutations identified in ovarian cancer patients BRCA1_c.1612C>T and BRCA1_3049G>T arose in OCCRs. Other mutations, namely c.19_47del, c.668dupA, c.915T>A, c.2418dupA, c.2433delC, c.5030_5033delCTAA in BRCA1 and c.249delG, c.632-1G>A in BRCA2 do not overlap with previously reported breast or ovarian cancer cluster regions. This could be explained by ethnic differences in BRCA mutation spectrum or it may indicate shared cluster regions for both breast and ovarian cancer.
In the BRCA1 gene, the c.19_47del mutation was identified in one breast cancer patient. This mutation was previously described only in the Algerian population (43). The c.2433delC mutation was described in Korean breast and ovarian patients (44, 45), and in Mexican patients (46, 47). The pathogenic c.1612C>T mutation was identified in 4 breast and ovarian cancer patients. This mutation has been identified in Brazilian population (48), in ovarian cancer patients from Israeli population (49) and in Macedonian population (50). We also detected the c.668dupA mutation in one patient. This latter has not been reported in previous studies neither in Tunisia nor in other populations. Nevertheless, it is already listed and classified as pathogenic in ClinVar and predicted to result in the substitution of Alanine to Glycine (p.Ala224Glyfs) which leads to BRCA1 protein truncation. Another new mutation was identified in BRCA1 gene, c.2418dupA, that was reported by our group for the first time in the Tunisian population and was not reported previously in other populations (51). c.3049G>T has been identified in one ovarian cancer patient. This mutation has been reported in Thai patients with non-mucinous epithelial ovarian cancer (52). The c.5030_5033delCTAA mutation was identified among one patient with breast and ovarian cancers and it is reported in Brazilian population (48). The c.915T>A mutation is novel and not described in public databases.
In addition to the identification of rare and novel BRCA1 mutations, other mutations seem to be recurrent and/or were described in previous Tunisian reports. The c.211dupA mutation was shared by 6 patients belonging to the same geographical origin. This mutation has so far been reported only in hereditary breast/ovarian cancer families of Tunisian origin, particularly in the North-East region, suggesting a founder effect. In order to unravel the genetic specificities of this mutation and to trace its origin a haplotype analysis has been conducted by our group on the North Eastern region (51). Results have determined the founder haplotype segregating with this mutation and have revealed that it arose in the period of colonization approximately 130 years ago.
The c.5266dupC mutation has been identified among two families. This mutation was previously described in 8 Tunisian breast cancer families (11, 16, 17, 20). It was originally described as an Ashkenazi founder mutation. Haplotype analysis has shown that this mutation arose approximately 1800 years ago in Northern Europe (53). Then, it has been reported in several other populations such as, Italian, Russian Slovenian and Greek (54).
Interestingly for BRCA2 gene, 6 breast cancer patients were double heterozygous carrying the two deleterious mutations c.632-1G>A and c.1310_1313delAAGA, and 4 other unrelated patients carried only the c.1310_1313delAAGA mutation including one male breast cancer (MBC). c.632-1G>A mutation appears to be rare in other populations since it was only reported in one patient with prostate cancer in the UK (55). However, c.1310_1313delAAGA seems to be a founder mutation in Maghrebin countries (16, 17, 56, 57). It has been also identified in patients with Lebanese (58), European (59–62), African (63), Asian (64) and Latino ancestry (65) as well as in Caribbean cohorts (66, 67). These results show the genetic heterogeneity of breast and ovarian cancers in Tunisian patients and the admixed origins of BRCA mutations in Tunisia.
In addition, 5 male breast cancer cases were investigated among which 2 carried BRCA2 mutations (c.1310_1313delAAGA and c.1389_1390delAG). Male breast cancer is a rare disease accounting for less than 1% of all breast cancer cases and it was previously shown that nearly 90% of MBC arising in BRCA mutation carriers are found to harbor a BRCA2 mutation (68). Unfortunately, being a man with “a women’s disease” makes MBC a disease surrounded by social taboo and lack of awareness especially in underdeveloped countries. Indeed, the treatment of MBC has been extrapolated from the knowledge of female breast cancer, despite the multiple differences in the pathogenesis, biology and genetics of these two disease entities. These evidence make MBC a gender issue that requires more attention from the scientific community.
The introduction of the c.1310_1313delAAGA mutation, that have been encountered in diverse populations, in the Tunisian population could be explained by the immigration of Andalusians in Tunisia which has been intensified after the fall of Granada in 1492 and lasted for two centuries before the total expulsion of all Andalusian Moriscos from the Iberian Peninsula in 1610. The diverse geographical distribution of this mutation may further suggest independent origins as shown for the 4184del4 BRCA1 mutation reported to have at least three independent origins in the study of Neuhausen et al. (69). The c.7654dupA BRCA2 gene mutation which was identified in a unique family with a strong family history of breast and ovarian cancer is reported previously and exclusively in Algerian population (70) and could be therefore specific to North African countries.
Through this report and despite the identification of novel mutations in Tunisian population, it is clear that the genetic susceptibility to breast cancer is explained in a vast majority of cases by recurrent mutations. Indeed, more than 44.44% of carriers harbor BRCA1-c.211dupA or BRCA2-1310_1313deAAGA mutations which highlights the importance of screening these mutations in the treatment workflow of cases with early onset or strong family history of breast cancer. In fact, identifying germline BRCA1 and BRCA2 pathogenic mutations is a crucial component in the medical management of affected patients. Regular surveillance and/or prophylactic mastectomy of the second breast or prophylactic salpingo oophorectomies, which have been shown to reduce the risk of developing cancer, are recommended to these carriers. Moreover, relatives who test positive for a germline BRCA pathogenic mutation may take appropriate action to prevent cancer or have cancer diagnosed as early as possible for better treatment options (59).
In addition, mutations in the BRCA genes and their associations with clinico-pathological features were reported in several studies (71–74). However, in Tunisia this aspect was not previously investigated. This point was raised in the present study and our results showed that patients with BRCA1 and BRCA2 mutations were similar with regard to several epidemiological and clinico-pathological parameters. Nevertheless, BRCA1 carriers were more likely to be triple negative breast cancer compared to BRCA2 carriers (p=0.002) and BRCA2 carriers were more likely to be luminal B breast cancer tumors (p=0.000078). Consistent with our findings, various previous studies reported that there is a much higher rate of TNBC among BRCA1 mutation carriers (75, 76) and BRCA2-related breast cancer is often luminal (77). Additionally, positive ER was significantly associated with BRCA2+ tumors (p=0.000056). PR status was significantly different between BRCA1 and BRCA2 mutation carriers; BRCA2 carriers are more likely to develop progesterone receptor (PR) positive tumors and PR-negative breast cancer are associated with BRCA1 mutation carriers (p=0.000084). It was reported that the ER positivity was predominantly seen in BRCA2 mutation carriers, which is consistent with our findings (71, 78). Furthermore, a previous report has found that BRCA2-associated cancers are mainly PR positive (79). Other studies have raised some pathological differences between BRCA1/2 mutation carriers and BRCAx patients. In our study, BRCA carriers seem to be younger than BRCA-negative patients (p=0.049). Furthermore, patients with a positive family history of ovarian cancer are more likely to be BRCA positive (p=0.004). We also observed a significant predominance of SBR grade III tumors among BRCA1/2 mutations carriers (p= 0.027). These findings are in line with previous literature (35, 80, 81).
Furthermore, we have assessed disease outcomes in BRCA carriers, and we have observed a relatively high proportion of contralateral breast cancer and ovarian cancer occurrence that were more frequently observed in BRCA1 carriers. Previous reports have demonstrated that women carrying a pathogenic mutation in the BRCA1 or BRCA2 genes have an increased risk of developing a second primary cancer in the contralateral breast. The cumulative risk 20 years after breast cancer diagnosis was estimated to be 40% for BRCA1 carriers and about 26% for BRCA2 carriers (82). In accordance with our findings, it was shown also that the occurrence of both breast and ovarian cancer in a woman is associated with a high likelihood of a germline BRCA1 mutation (83).
Besides the BRCA genetic mutations that have been identified in our study, mutations on other high to moderate breast cancer genes such as TP53, ATM, BLM and CHEK2 have been also identified for the first time in North African populations (data not shown). All these findings reflect the genetic heterogeneity of cancer predisposition in Tunisia and highlights the importance of the use of NGS to identify clinically actionable genetic variants that have a crucial role in disease management. Therefore, technological advances in terms of array and DNA sequencing technologies made the route towards the examination of genetic risk largely clear. However, practical challenges related to marked population-specific differences still exist. In this context, Manolio and colleagues conveniently classified LRRK2 as a high penetrant gene associated with Parkinson disease (84) with G2019S mutation being the main cause of Parkinson familial cases. Recently the international LRRK2 consortium reported a worldwide frequency of 1% of LRRK2 G2019S, 30–40% in Arab patients from North Africa and 10–30% in Ashkenazi Jews, but is very rare in Asians (85, 86). As a variant´s frequency has a direct impact on its penetrance, this example shows the ethnic-dependent penetrance of some important variants involved in complex diseases and the role of consanguinity and endogamy in shaping the genetic susceptibility to these diseases. Therefore, the same reflection can be applied on high and low penetrant breast cancer variants in order to review their penetrance in underrepresented populations such as North Africans. A disproportionate distribution of the identified mutations is observed between the Northern and Southern regions of Tunisia (Figure 2), with the vast majority found in the North. This can be explained by a selection bias because most of the recruited participants come from Northern governorates, but it can also be explained by the very high consanguinity rates in the South that reaches 98% in some cities and that may have an impact on BRCA mutations frequency and prevalence.
Additional limitations of our study have been observed. Indeed, to our best knowledge, this work represents the largest BRCA1/2 study in Africa. However, we believe that the sample size is still small and larger cohorts are needed to trace a clear and complete BRCA1/2 mutational spectrum in Tunisia. In addition, because of the limited resources dedicated to this work, we were not able to perform a complete sequencing of both genes for the whole cohort. Therefore, the frequency and prevalence of the identified mutations need to be assessed in larger studies. Clearly, the prevalence assessment of BRCA1 and BRCA2 mutations also rely on the quality of both cohort selection criteria and mutation ascertainment methods. The identification of novel BRCA mutations and the assessment of their penetrance in a specific population will help to implement more affordable and cost effective targeted genetic testing strategies.
Finally, up until now, most data on BRCA1/2 mutations associated with high risk for hereditary breast and ovarian cancer do not cover the North African populations. Accordingly, the novel mutations identified in this study will help to improve knowledge on the genetic component of hereditary breast and ovarian cancer in the North African region and will lead to a better clinical management of cancer patients. In addition, we are aiming to share genetic and phenotypic data with larger multi-ethnic Consortia of BRCA1/2 mutation carriers such as the Consortium of Investigators of Modifiers of BRCA1/2 (CIMBA) (87). This will make our findings more broadly useful and will give us a global overview of the similarities and differences that the Tunisian population has compared to other ethnicities.
Conclusion
In conclusion we have identified 16 distinct BRCA mutations in breast and ovarian cancer patients including 11 novel mutations in the Tunisian population. The recognition of the BRCA mutational spectrum and its geographical distribution in Tunisia is of keen interest for the scientific and medical communities as it helps to develop precise risk assessment tools, accurate genetic testing, cost-effective approaches for prevention and early detection of the disease as well as personalized treatments of BRCA related cancers for both affected and unaffected cancer cases.
Data Availability Statement
The minimal dataset that would be necessary to interpret, replicate and build upon the findings reported in this study are included in this article and in its supplementary files. All identified mutations with their related details have been shared in the public database ClinVar under the following link “https://www.ncbi.nlm.nih.gov/clinvar/submitters/507986/”. Any additional datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Ethics Statement
The studies involving human participants were reviewed and approved by The Biomedical Ethics Committee of Institut Pasteur de Tunis (2017/16/E/Hôpital A-M). The patients/participants provided their written informed consent to participate in this study. Written informed consent was obtained from participants for the publication of any potentially identifiable images or data included in this article.
Author Contributions
YH prepared the study concept and design, supervised the study, did data analysis, data interpretation, drafted, and critically revised the manuscript. NMi and MB did the experiments, participated in participant recruitment, and participated in drafting and reviewing the manuscript. NMe contributed to clinical data analysis and reviewed the manuscript. SN contributed to participants recruitment and reviewed the manuscript. MR and OM contributed to data collection and reviewed the manuscript. HanB, YB, HR, OJ, ND, AZ, JA, HEB, SL and JBH contributed to the clinical investigation and recruitment of patients. AH, KR, FB, RM, SBA and HamB critically revised the clinicopathological section and the whole manuscript. SB and SA contributed to the study concept, design and supervision and critically revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Tunisian Ministry of Higher Education and Scientific Research (LR16IPT05) and the Tunisian Ministry of Public Health (PEC-4-TUN). MB and MBR are recipient of a MOBIDOC fellowship funded by the EU through the EMORI and PASRI programs managed by the ANPR.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
This manuscript is dedicated to the memory of Prof. Farouk Benna, an imminent radiotherapist who died from Covid19 when ensuring his medical activity. We would like to thank Dr. Zied Zidi, Dr Ghazi Jerbi, Dr Samir Khalfallah, Dr Monia Hechiche, Dr Achraf Chaari, Dr Olfa gharbi, Dr Monia Hechiche, Dr Sami Bellil, Dr Ichraf Jbir, Dr Olfa Daldoul Ben Fraj, Dr Bassem Allani/Dr David Khayat, Dr Tarek Bouzid, Dr El Fatmi Rym/Dr Mokrani Amina, Dr Samia Chatti Dey, Dr Hatem Chaaba, Dr Med Ali Ayadi, Dr Lotfi Kochbati, Dr Fethi Messoudi and Dr Chadha Elward Abdelhedi for their contribution in this work. We are also grateful to all participants and their family members for their participation in the study.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2021.674965/full#supplementary-material
Abbreviations
BCCRs, Breast Cancer Cluster Regions; ER, Estrogen receptor; gDNA, Genomic DNA; GxE, Genetic and Environmental factors; IDC, Invasive Ductal Carcinoma; LMICs, Low- and middle-income countries; OCCRs, Ovarian cancer cluster regions; PR, Progesterone receptor; SBR, Scarff-Bloom-Richardson; TNBC, Triple Negative Breast Cancer; VUS, Variants of Unknown Significance; WES, Whole Exome Sequencing.
References
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global Cancer Statistics 2018: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA: Cancer J Clin (2018) 68(6):394–424. doi: 10.3322/caac.21492
2. Chalabi N, Bernard-Gallon DJ, Bignon YJ, Breast Med C, Kwiatkowski F, Agier M, et al. Comparative Clinical and Transcriptomal Profiles of Breast Cancer Between French and South Mediterranean Patients Show Minor But Significative Biological Differences. Cancer Genomics Proteomics (2008) 5(5):253–61.
3. Boussen H, Bouzaiene H, Ben Hassouna J, Dhiab T, Khomsi F, Benna F, et al. Inflammatory Breast Cancer in Tunisia: Epidemiological and Clinical Trends. Cancer (2010) 116(11 Suppl):2730–5. doi: 10.1002/cncr.25175
4. Antoniou A, Pharoah PD, Narod S, Risch HA, Eyfjord JE, Hopper JL, et al. Average Risks of Breast and Ovarian Cancer Associated With BRCA1 or BRCA2 Mutations Detected in Case Series Unselected for Family History: A Combined Analysis of 22 Studies. Am J Hum Genet (2003) 72(5):1117–30. doi: 10.1086/375033
5. Mersch J, Jackson MA, Park M, Nebgen D, Peterson SK, Singletary C, et al. Cancers Associated With BRCA 1 and BRCA 2 Mutations Other Than Breast and Ovarian. Cancer (2015) 121(2):269–75. doi: 10.1002/cncr.29041
6. Domchek SM, Friebel TM, Singer CF, Evans DG, Lynch HT, Isaacs C, et al. Association of Risk-Reducing Surgery in BRCA1 or BRCA2 Mutation Carriers With Cancer Risk and Mortality. JAMA (2010) 304(9):967–75. doi: 10.1001/jama.2010.1237
7. Rebbeck TR, Lynch HT, Neuhausen SL, Narod SA, Van't Veer L, Garber JE, et al. Prophylactic Oophorectomy in Carriers of BRCA1 or BRCA2 Mutations. N Engl J Med (2002) 346(21):1616–22. doi: 10.1056/NEJMoa012158
8. Menkiszak J, Chudecka-Glaz A, Gronwald J, Cymbaluk-Ploska A, Celewicz A, Swiniarska M, et al. Prophylactic Salpingo-Oophorectomy in BRCA1 Mutation Carriers and Postoperative Incidence of Peritoneal and Breast Cancers. J Ovarian Res (2016) 9:11. doi: 10.1186/s13048-016-0220-4
9. Turner NC, Tutt AN. Platinum Chemotherapy for BRCA1-Related Breast Cancer: Do We Need More Evidence? Breast Cancer Res (2012) 14(6):115. doi: 10.1186/bcr3332
10. Mateo J, Lord CJ, Serra V, Tutt A, Balmana J, Castroviejo-Bermejo M, et al. A Decade of Clinical Development of PARP Inhibitors in Perspective. Ann Oncol (2019) 30(9):1437–47. doi: 10.1093/annonc/mdz192
11. Mahfoudh W, Bouaouina N, Ahmed SB, Gabbouj S, Shan J, Mathew R, et al. Hereditary Breast Cancer in Middle Eastern and North African (MENA) Populations: Identification of Novel, Recurrent and Founder BRCA1 Mutations in the Tunisian Population. Mol Biol Rep (2012) 39(2):1037–46. doi: 10.1007/s11033-011-0829-8
12. Kadouri L, Hubert A, Rotenberg Y, Hamburger T, Sagi M, Nechushtan C, et al. Cancer Risks in Carriers of the BRCA1/2 Ashkenazi Founder Mutations. J Med Genet (2007) 44(7):467–71. doi: 10.1136/jmg.2006.048173
13. Fackenthal JD, Olopade OI. Breast Cancer Risk Associated With BRCA1 and BRCA2 in Diverse Populations. Nat Rev Cancer (2007) 7(12):937–48. doi: 10.1038/nrc2054
14. Ramus SJ, Gayther SA. The Contribution of BRCA1 and BRCA2 to Ovarian Cancer. Mol Oncol (2009) 3(2):138–50. doi: 10.1016/j.molonc.2009.02.001
15. Mestiri S, Monastiri K, Ben SA, Bouaouina N, Presneau N, Bignon Y, et al. Mutational Analysis of Breast/Ovarian Cancer Hereditary Predisposition Gene BRCA1 in Tunisian Women. Arch l'Institut Pasteur Tunis (2000) 77(1-4):11–5.
16. Troudi W, Uhrhammer N, Sibille C, Dahan C, Mahfoudh W, Bouchlaka Souissi C, et al. Contribution of the BRCA1 and BRCA2 Mutations to Breast Cancer in Tunisia. J Hum Genet (2007) 52(11):915–20. doi: 10.1007/s10038-007-0195-5
17. Fourati A, Louchez MM, Fournier J, Gamoudi A, Rahal K, El May MV, et al. Screening for Common Mutations in BRCA1 and BRCA2 Genes: Interest in Genetic Testing of Tunisian Families With Breast and/or Ovarian Cancer. Bull Cancer (2014) 101(11):E36–40. doi: 10.1684/bdc.2014.2049
18. Msolly A. BRCA1 and BRCA2 Mutations Are They Related to Breast Cancer in a Sample of Tunisian Population? Cancer Ther Oncol Int J (2015) 1(1):1–5. doi: 10.19080/CTOIJ.2015.01.555551
19. Riahi A, Chabouni-Bouhamed H, Kharrat M. Prevalence of BRCA1 and BRCA2 Large Genomic Rearrangements in Tunisian High Risk Breast/Ovarian Cancer Families: Implications for Genetic Testing. Cancer Genet (2017) 210:22–7. doi: 10.1016/j.cancergen.2016.11.002
20. Riahi A, Kharrat M, Ghourabi ME, Khomsi F, Gamoudi A, Lariani I, et al. Mutation Spectrum and Prevalence of BRCA1 and BRCA2 Genes in Patients With Familial and Early-Onset Breast/Ovarian Cancer From Tunisia. Clin Genet (2015) 87(2):155–60. doi: 10.1111/cge.12337
21. Hadiji-Abbes N, Trifa F, Choura M, Khabir A, Sellami-Boudawara T, Frikha M, et al. A Novel BRCA2 in Frame Deletion in a Tunisian Woman With Early Onset Sporadic Breast Cancer. Pathol Biol (Paris) (2015) 63(4-5):185–9. doi: 10.1016/j.patbio.2015.07.009
22. Mahfoudh W, Bettaieb I, Ghedira R, Snoussi K, Bouzid N, Klayech Z, et al. Contribution of BRCA1 5382insc Mutation in Triple Negative Breast Cancer in Tunisia. J Trans Med (2019) 17(1):123. doi: 10.1186/s12967-019-1873-8
23. Ben Kridis-Rejeb W, Ben Ayed-Guerfali D, Ammous-Boukhris N, Ayadi W, Kifagi C, Charfi S, et al. Identification of Novel Candidate Genes by Exome Sequencing in Tunisian Familial Male Breast Cancer Patients. Mol Biol Rep (2020) 47(9):6507–16. doi: 10.1007/s11033-020-05703-0
24. Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, et al. Analysis of Protein-Coding Genetic Variation in 60,706 Humans. Nature (2016) 536(7616):285–91. doi: 10.1038/nature19057
25. Genomes Project C, Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, et al. A Global Reference for Human Genetic Variation. Nature (2015) 526(7571):68–74. doi: 10.1038/nature15393
26. Rebbeck TR, Mitra N, Wan F, Sinilnikova OM, Healey S, McGuffog L, et al. Association of Type and Location of BRCA1 and BRCA2 Mutations With Risk of Breast and Ovarian Cancer. JAMA (2015) 313(13):1347–61. doi: 10.1001/jama.2014.5985
27. Melchor L, Benitez J. The Complex Genetic Landscape of Familial Breast Cancer. Hum Genet (2013) 132(8):845–63. doi: 10.1007/s00439-013-1299-y
28. Laraqui A, Uhrhammer N, Rhaffouli HE, Sekhsokh Y, Lahlou-Amine I, Bajjou T, et al. BRCA Genetic Screening in Middle Eastern and North African: Mutational Spectrum and Founder BRCA1 Mutation (C.798_799deltt) in North African. Dis Markers (2015) 2015:194293. doi: 10.1155/2015/194293
29. Laitman Y, Friebel TM, Yannoukakos D, Fostira F, Konstantopoulou I, Figlioli G, et al. The Spectrum of BRCA1 and BRCA2 Pathogenic Sequence Variants in Middle Eastern, North African, and South European Countries. Hum Mutat (2019) 40(11):e1–e23. doi: 10.1002/humu.23842
30. Behl S, Hamel N, de Ladurantaye M, Lepage S, Lapointe R, Mes-Masson AM, et al. Founder BRCA1/BRCA2/PALB2 Pathogenic Variants in French-Canadian Breast Cancer Cases and Controls. Sci Rep (2020) 10(1):6491. doi: 10.1038/s41598-020-63100-w
31. Oosthuizen J, Kotze MJ, van der Merwe N, Myburgh EJ, Bester P, van der Merwe NC. Globally Rare BRCA2 Variants With Founder Haplotypes in the South African Population: Implications for Point-Of-Care Testing Based on a Single-Institution BRCA1/2 Next-Generation Sequencing Study. Front Oncol (2020) 10:619469. doi: 10.3389/fonc.2020.619469
32. Tennen RI, Laskey SB, Koelsch BL, McIntyre MH, Tung JY. Identifying Ashkenazi Jewish BRCA1/2 Founder Variants in Individuals Who Do Not Self-Report Jewish Ancestry. Sci Rep (2020) 10(1):7669. doi: 10.1038/s41598-020-63466-x
33. Hamdi Y, Ben Rekaya M, Jingxuan S, Nagara M, Messaoud O, Benammar Elgaaied A, et al. A Genome Wide SNP Genotyping Study in the Tunisian Population: Specific Reporting on a Subset of Common Breast Cancer Risk Loci. BMC Cancer (2018) 18(1):1295. doi: 10.1186/s12885-018-5133-8
34. Hamdi Y, Jerbi M, Romdhane L, Ben Rekaya M, El Benna H, Chouchane L, et al. Genetic Diversity and Functional Effect of Common Polymorphisms in Genes Involved in the First Heterodimeric Complex of the Nucleotide Excision Repair Pathway. DNA Repair (Amst) (2020) 86:102770. doi: 10.1016/j.dnarep.2019.102770
35. Fang M, Zhu L, Li H, Li X, Wu Y, Wu K, et al. Characterization of Mutations in BRCA1/2 and the Relationship With Clinic-Pathological Features of Breast Cancer in a Hereditarily High-Risk Sample of Chinese Population. Oncol Lett (2018) 15(3):3068–74. doi: 10.3892/ol.2017.7717
36. Godet I, Gilkes DM. BRCA1 and BRCA2 Mutations and Treatment Strategies for Breast Cancer. Integr Cancer Sci Ther (2017) 4(1):10. doi: 10.15761/ICST.1000228
37. Chouchane L, Boussen H, Sastry KSR. Breast Cancer in Arab Populations: Molecular Characteristics and Disease Management Implications. Lancet Oncol (2013) 14(10):e417–e24. doi: 10.1016/S1470-2045(13)70165-7
38. Zehani S, Maalej M, Hsairi M, Hechiche M, Romdhane B, Boussen H, et al. Breast Cancer in Tunisia: Epidemiologic Characteristics and Trends in Incidence. La Tunisie Medicale (2009) 87(7):417–25.
39. Neff RT, Senter L, Salani R. BRCA Mutation in Ovarian Cancer: Testing, Implications and Treatment Considerations. Ther Adv Med Oncol (2017) 9(8):519–31. doi: 10.1177/1758834017714993
40. Meindl A, German Consortium for Hereditary B, Ovarian C. Comprehensive Analysis of 989 Patients With Breast or Ovarian Cancer Provides BRCA1 and BRCA2 Mutation Profiles and Frequencies for the German Population. Int J Cancer (2002) 97(4):472–80. doi: 10.1002/ijc.1626
41. Lim MC, Kang S, Seo SS, Kong SY, Lee BY, Lee SK, et al. BRCA1 and BRCA2 Germline Mutations in Korean Ovarian Cancer Patients. J Cancer Res Clin Oncol (2009) 135(11):1593–9. doi: 10.1007/s00432-009-0607-3
42. Zuntini R, Cortesi L, Calistri D, Pippucci T, Martelli PL, Casadio R, et al. BRCA1 P. His1673del Is a Pathogenic Mutation Associated With a Predominant Ovarian Cancer Phenotype. Oncotarget (2017) 8(14):22640. doi: 10.18632/oncotarget.15151
43. Cherbal F, Bakour R, Adane S, Boualga K. BRCA1 and BRCA2 Germline Mutation Spectrum in Hereditary Breast/Ovarian Cancer Families From Maghrebian Countries. Breast Dis (2012) 34(1):1–8. doi: 10.3233/BD-130348
44. Ahn SH, Son BH, Yoon KS, Noh DY, Han W, Kim SW, et al. BRCA1 and BRCA2 Germline Mutations in Korean Breast Cancer Patients at High Risk of Carrying Mutations. Cancer Lett (2007) 245(1-2):90–5. doi: 10.1016/j.canlet.2005.12.031
45. Kim H, Cho DY, Choi DH, Choi SY, Shin I, Park W, et al. Characteristics and Spectrum of BRCA1 and BRCA2 Mutations in 3,922 Korean Patients With Breast and Ovarian Cancer. Breast Cancer Res Treat (2012) 134(3):1315–26. doi: 10.1007/s10549-012-2159-5
46. Weitzel JN, Clague J, Martir-Negron A, Ogaz R, Herzog J, Ricker C, et al. Prevalence and Type of BRCA Mutations in Hispanics Undergoing Genetic Cancer Risk Assessment in the Southwestern United States: A Report From the Clinical Cancer Genetics Community Research Network. J Clin Oncol (2013) 31(2):210–6. doi: 10.1200/JCO.2011.41.0027
47. Torres-Mejia G, Royer R, Llacuachaqui M, Akbari MR, Giuliano AR, Martinez-Matsushita L, et al. Recurrent BRCA1 and BRCA2 Mutations in Mexican Women With Breast Cancer. Cancer Epidemiol Biomarkers Prev (2015) 24(3):498–505. doi: 10.1158/1055-9965.EPI-13-0980
48. Palmero EI, Carraro DM, Alemar B, Moreira MAM, Ribeiro-Dos-Santos A, Abe-Sandes K, et al. The Germline Mutational Landscape of BRCA1 and BRCA2 in Brazil. Sci Rep (2018) 8(1):9188. doi: 10.1038/s41598-018-27315-2
49. Barnes-Kedar I, Bernstein-Molho R, Ginzach N, Hartmajer S, Shapira T, Magal N, et al. The Yield of Full BRCA1/2 Genotyping in Israeli High-Risk Breast/Ovarian Cancer Patients Who Do Not Carry the Predominant Mutations. Breast Cancer Res Treat (2018) 172(1):151–7. doi: 10.1007/s10549-018-4887-7
50. Jakimovska M, Maleva Kostovska I, Popovska-Jankovic K, Kubelka-Sabit K, Karadjozov M, Stojanovska L, et al. BRCA1 and BRCA2 Germline Variants in Breast Cancer Patients From the Republic of Macedonia. Breast Cancer Res Treat (2018) 168(3):745–53. doi: 10.1007/s10549-017-4642-5
51. Mighri N, Hamdi Y, Boujemaa M, Othman H, Ben Nasr S, El Benna H, et al. Identification of Novel BRCA1 and RAD50 Mutations Associated With Breast Cancer Predisposition in Tunisian Patients. Front Genet (2020) 11:552971. doi: 10.3389/fgene.2020.552971
52. Manchana T, Phoolcharoen N, Tantbirojn P. BRCA Mutation in High Grade Epithelial Ovarian Cancers. Gynecol Oncol Rep (2019) 29:102–5. doi: 10.1016/j.gore.2019.07.007
53. Hamel N, Feng BJ, Foretova L, Stoppa-Lyonnet D, Narod SA, Imyanitov E, et al. On the Origin and Diffusion of BRCA1 C.5266dupc (5382insc) in European Populations. Eur J Hum Genet (2011) 19(3):300–6. doi: 10.1038/ejhg.2010.203
54. Janavicius R. Founder BRCA1/2 Mutations in the Europe: Implications for Hereditary Breast-Ovarian Cancer Prevention and Control. EPMA J (2010) 1(3):397–412. doi: 10.1007/s13167-010-0037-y
55. Edwards SM, Evans DGR, Hope Q, Norman A, Barbachano Y, Bullock S, et al. Prostate Cancer in BRCA2 Germline Mutation Carriers is Associated With Poorer Prognosis. Br J Cancer (2010) 103(6):918–24. doi: 10.1038/sj.bjc.6605822
56. Laarabi FZ, Ratbi I, Elalaoui SC, Mezzouar L, Doubaj Y, Bouguenouch L, et al. High Frequency of the Recurrent C.1310_1313delaaga BRCA2 Mutation in the North-East of Morocco and Implication for Hereditary Breast-Ovarian Cancer Prevention and Control. BMC Res Notes (2017) 10(1):188. doi: 10.1186/s13104-017-2511-2
57. Cherbal F, Bakour R, Adane S, Boualga K, Benais-Pont G, Maillet P. BRCA1 and BRCA2 Germline Mutations Screening in Algerian Breast/Ovarian Cancer Families. Dis Markers (2010) 28(6):377–84. doi: 10.1155/2010/585278
58. El Saghir NS, Zgheib NK, Assi HA, Khoury KE, Bidet Y, Jaber SM, et al. BRCA1 and BRCA2 Mutations in Ethnic Lebanese Arab Women With High Hereditary Risk Breast Cancer. Oncologist (2015) 20(4):357–64. doi: 10.1634/theoncologist.2014-0364
59. Caputo S, Benboudjema L, Sinilnikova O, Rouleau E, Beroud C, Lidereau R, et al. Description and Analysis of Genetic Variants in French Hereditary Breast and Ovarian Cancer Families Recorded in the UMD-BRCA1/BRCA2 Databases. Nucleic Acids Res (2012) 40(Database issue):D992–1002. doi: 10.1093/nar/gkr1160
60. Vos JR, Teixeira N, van der Kolk DM, Mourits MJ, Rookus MA, van Leeuwen FE, et al. Variation in Mutation Spectrum Partly Explains Regional Differences in the Breast Cancer Risk of Female BRCA Mutation Carriers in the Netherlands. Cancer Epidemiol Biomarkers Prev (2014) 23(11):2482–91. doi: 10.1158/1055-9965.EPI-13-1279
61. Meisel C, Sadowski CE, Kohlstedt D, Keller K, Staritz F, Grubling N, et al. Spectrum of Genetic Variants of BRCA1 and BRCA2 in a German Single Center Study. Arch Gynecol Obstet (2017) 295(5):1227–38. doi: 10.1007/s00404-017-4330-z
62. Thomassen M, Hansen TV, Borg A, Lianee HT, Wikman F, Pedersen IS, et al. BRCA1 and BRCA2 Mutations in Danish Families With Hereditary Breast and/or Ovarian Cancer. Acta Oncol (2008) 47(4):772–7. doi: 10.1080/02841860802004974
63. Zhang J, Fackenthal JD, Zheng Y, Huo D, Hou N, Niu Q, et al. Recurrent BRCA1 and BRCA2 Mutations in Breast Cancer Patients of African Ancestry. Breast Cancer Res Treat (2012) 134(2):889–94. doi: 10.1007/s10549-012-2136-z
64. Jang JH, Lee JE, Kwon MJ, Ki CS, Kim JW, Nam SJ, et al. Spectra of BRCA1 and BRCA2 Mutations in Korean Patients With Breast Cancer: The Importance of Whole-Gene Sequencing. J Hum Genet (2012) 57(3):212–5. doi: 10.1038/jhg.2011.139
65. Cruz-Correa M, Perez-Mayoral J, Dutil J, Echenique M, Mosquera R, Rivera-Roman K, et al. Hereditary Cancer Syndromes in Latino Populations: Genetic Characterization and Surveillance Guidelines. Hered Cancer Clin Pract (2017) 15:3. doi: 10.1186/s13053-017-0063-z
66. Akbari MR, Donenberg T, Lunn J, Curling D, Turnquest T, Krill-Jackson E, et al. The Spectrum of BRCA1 and BRCA2 Mutations in Breast Cancer Patients in the Bahamas. Clin Genet (2014) 85(1):64–7. doi: 10.1111/cge.12132
67. Donenberg T, Ahmed H, Royer R, Zhang S, Narod SA, George S, et al. A Survey of BRCA1, BRCA2, and PALB2 Mutations in Women With Breast Cancer in Trinidad and Tobago. Breast Cancer Res Treat (2016) 159(1):131–8. doi: 10.1007/s10549-016-3870-4
68. Silvestri V, Barrowdale D, Mulligan AM, Neuhausen SL, Fox S, Karlan BY, et al. Male Breast Cancer in BRCA1 and BRCA2 Mutation Carriers: Pathology Data From the Consortium of Investigators of Modifiers of BRCA1/2. Breast Cancer Res (2016) 18(1):15. doi: 10.1186/s13058-016-0671-y
69. Neuhausen SL, Mazoyer S, Friedman L, Stratton M, Offit K, Caligo A, et al. Haplotype and Phenotype Analysis of Six Recurrent BRCA1 Mutations in 61 Families: Results of an International Study. Am J Hum Genet (1996) 58(2):271.
70. Henouda S, Bensalem A, Reggad R, Serrar N, Rouabah L, Pujol P. Contribution of BRCA1 and BRCA2 Germline Mutations to Early Algerian Breast Cancer. Dis Markers (2016) 2016:7869095. doi: 10.1155/2016/7869095
71. Veronesi A, de Giacomi C, Magri MD, Lombardi D, Zanetti M, Scuderi C, et al. Familial Breast Cancer: Characteristics and Outcome of BRCA 1-2 Positive and Negative Cases. BMC Cancer (2005) 5:70. doi: 10.1186/1471-2407-5-70
72. Atchley DP, Albarracin CT, Lopez A, Valero V, Amos CI, Gonzalez-Angulo AM, et al. Clinical and Pathologic Characteristics of Patients With BRCA-Positive and BRCA-Negative Breast Cancer. J Clin Oncol (2008) 26(26):4282–8. doi: 10.1200/JCO.2008.16.6231
73. Jouhadi H, Tazzite A, Azeddoug H, Naim A, Nadifi S, Benider A. Clinical and Pathological Features of BRCA1/2 Tumors in a Sample of High-Risk Moroccan Breast Cancer Patients. BMC Res Notes (2016) 9:248. doi: 10.1186/s13104-016-2057-8
74. Atci MM, Geredeli C, Ay S, Sakin A, Erturk B, Secmeler S, et al. Clinical and Pathological Characteristics of Patients With High-Risk Breast Cancer Based on BRCA Mutation Profiles: A Retrospective Study. Eur J Breast Health (2021) 17(2):123–7. doi: 10.4274/ejbh.galenos.2020.6346
75. Lee E, McKean-Cowdin R, Ma H, Spicer DV, Van Den Berg D, Bernstein L, et al. Characteristics of Triple-Negative Breast Cancer in Patients With a BRCA1 Mutation: Results From a Population-Based Study of Young Women. J Clin Oncol (2011) 29(33):4373–80. doi: 10.1200/JCO.2010.33.6446
76. Muendlein A, Rohde BH, Gasser K, Haid A, Rauch S, Kinz E, et al. Evaluation of BRCA1/2 Mutational Status Among German and Austrian Women With Triple-Negative Breast Cancer. J Cancer Res Clin Oncol (2015) 141(11):2005–12. doi: 10.1007/s00432-015-1986-2
77. Stefansson OA, Jonasson JG, Olafsdottir K, Bjarnason H, Th Johannsson O, Bodvarsdottir SK, et al. Genomic and Phenotypic Analysis of BRCA2 Mutated Breast Cancers Reveals Co-Occurring Changes Linked to Progression. Breast Cancer Res (2011) 13(5):R95. doi: 10.1186/bcr3020
78. Lakhani SR, Reis-Filho JS, Fulford L, Penault-Llorca F, van der Vijver M, Parry S, et al. Prediction of BRCA1 Status in Patients With Breast Cancer Using Estrogen Receptor and Basal Phenotype. Clin Cancer Res (2005) 11(14):5175–80. doi: 10.1158/1078-0432.CCR-04-2424
79. Keeney MG, Couch FJ, Visscher DW, Lindor NM. Non-BRCA Familial Breast Cancer: Review of Reported Pathology and Molecular Findings. Pathology (2017) 49(4):363–70. doi: 10.1016/j.pathol.2017.03.002
80. Zhang S, Royer R, Li S, McLaughlin JR, Rosen B, Risch HA, et al. Frequencies of BRCA1 and BRCA2 Mutations Among 1,342 Unselected Patients With Invasive Ovarian Cancer. Gynecol Oncol (2011) 121(2):353–7. doi: 10.1016/j.ygyno.2011.01.020
81. Lang GT, Shi JX, Hu X, Zhang CH, Shan L, Song CG, et al. The Spectrum of BRCA Mutations and Characteristics of BRCA-Associated Breast Cancers in China: Screening of 2,991 Patients and 1,043 Controls by Next-Generation Sequencing. Int J Cancer (2017) 141(1):129–42. doi: 10.1002/ijc.30692
82. Kuchenbaecker KB, Hopper JL, Barnes DR, Phillips K-A, Mooij TM, Roos-Blom M-J, et al. Risks of Breast, Ovarian, and Contralateral Breast Cancer for BRCA1 and BRCA2 Mutation Carriers. Jama (2017) 317(23):2402–16. doi: 10.1001/jama.2017.7112
83. Einbeigi Z, Bergman A, Meis-Kindblom JM, Flodin A, Bjursell C, Martinsson T, et al. Occurrence of Both Breast and Ovarian Cancer in a Woman Is a Marker for the BRCA Gene Mutations: A Population-Based Study From Western Sweden. Familial Cancer (2007) 6(1):35–41. doi: 10.1007/s10689-006-9101-0
84. Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, Hunter DJ, et al. Finding the Missing Heritability of Complex Diseases. Nature (2009) 461(7265):747–53. doi: 10.1038/nature08494
85. Lesage S, Belarbi S, Troiano A, Condroyer C, Hecham N, Pollak P, et al. Is the Common LRRK2 G2019S Mutation Related to Dyskinesias in North African Parkinson Disease? Neurology (2008) 71(19):1550–2. doi: 10.1212/01.wnl.0000338460.89796.06
86. Hulihan MM, Ishihara-Paul L, Kachergus J, Warren L, Amouri R, Elango R, et al. LRRK2 Gly2019Ser Penetrance in Arab-Berber Patients From Tunisia: A Case-Control Genetic Study. Lancet Neurol (2008) 7(7):591–4. doi: 10.1016/S1474-4422(08)70116-9
87. Chenevix-Trench G, Milne RL, Antoniou AC, Couch FJ, Easton DF, Goldgar DE, et al. An International Initiative to Identify Genetic Modifiers of Cancer Risk in BRCA1 and BRCA2 Mutation Carriers: The Consortium of Investigators of Modifiers of BRCA1 and BRCA2 (CIMBA). Breast Cancer Res (2007) 9(2):104. doi: 10.1186/bcr1670
Keywords: BRCA cancers, genetic testing, novel BRCA mutations, clinicopathological signatures, precision medicine
Citation: Hamdi Y, Mighri N, Boujemaa M, Mejri N, Ben Nasr S, Ben Rekaya M, Messaoud O, Bouaziz H, Berrazega Y, Rachdi H, Jaidane O, Daoud N, Zribi A, Ayari J, El Benna H, Labidi S, Ben Hassouna J, Haddaoui A, Rahal K, Benna F, Mrad R, Ben Ahmed S, Boussen H, Boubaker S and Abdelhak S (2021) Identification of Eleven Novel BRCA Mutations in Tunisia: Impact on the Clinical Management of BRCA Related Cancers. Front. Oncol. 11:674965. doi: 10.3389/fonc.2021.674965
Received: 02 March 2021; Accepted: 27 July 2021;
Published: 20 August 2021.
Edited by:
Zodwa Dlamini, University of Pretoria, South AfricaReviewed by:
Yan Du, Fudan University, ChinaUmamaheswaran Gurusamy, University of California, San Francisco, United States
Copyright © 2021 Hamdi, Mighri, Boujemaa, Mejri, Ben Nasr, Ben Rekaya, Messaoud, Bouaziz, Berrazega, Rachdi, Jaidane, Daoud, Zribi, Ayari, El Benna, Labidi, Ben Hassouna, Haddaoui, Rahal, Benna, Mrad, Ben Ahmed, Boussen, Boubaker and Abdelhak. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yosr Hamdi, eW9zci5oYW1kaUBwYXN0ZXVyLnRu
†These authors have contributed equally to this work