
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol., 27 April 2021
Sec. Cancer Immunity and Immunotherapy
Volume 11 - 2021 | https://doi.org/10.3389/fonc.2021.672747
This article is part of the Research TopicUnderstanding the Interplay Between the Tumor Immune Microenvironment and Genetic Alterations in Thoracic MalignanciesView all 13 articles
 Nicola Principe1,2
Nicola Principe1,2 Joel Kidman1,2
Joel Kidman1,2 Richard A. Lake1,2
Richard A. Lake1,2 Willem Joost Lesterhuis2,3
Willem Joost Lesterhuis2,3 Anna K. Nowak1,4
Anna K. Nowak1,4 Alison M. McDonnell3
Alison M. McDonnell3 Jonathan Chee1,2*
Jonathan Chee1,2*The success of immunotherapy that targets inhibitory T cell receptors for the treatment of multiple cancers has seen the anti-tumor immune response re-emerge as a promising biomarker of response to therapy. Longitudinal characterization of T cells in the tumor microenvironment (TME) helps us understand how to promote effective anti-tumor immunity. However, serial analyses at the tumor site are rarely feasible in clinical practice. Malignant pleural effusions (MPE) associated with thoracic cancers are an abnormal accumulation of fluid in the pleural space that is routinely drained for patient symptom control. This fluid contains tumor cells and immune cells, including lymphocytes, macrophages and dendritic cells, providing a window into the local tumor microenvironment. Recurrent MPE is common, and provides an opportunity for longitudinal analysis of the tumor site in a clinical setting. Here, we review the phenotype of MPE-derived T cells, comparing them to tumor and blood T cells. We discuss the benefits and limitations of their use as potential dynamic biomarkers of response to therapy.
A malignant pleural effusion (MPE) is an abnormal accumulation of fluid in the pleural space associated with advanced stage disease and poor clinical outcomes (1, 2). These effusions are present at diagnosis in over 90% of patients with mesothelioma (3) and 40% of patients with advanced lung cancer (1), and are a common feature of metastatic disease to the lung in patients with breast cancer, lymphoma, ovarian and stomach cancers (2, 4). MPEs are an exudative fluid, resulting from increased vascular permeability, inflammation and plasma leakage caused in part by tumor cells blocking the outflow of fluid from the pleural space (5). This build up of fluid leads to symptoms of various severity including breathlessness, chest pain and cough (5), with current therapy consisting largely of palliative measures designed to drain or eliminate the pleural space to prevent accumulation of fluid (6–9). With the exception of pleurectomy, recurrent MPE can occur throughout disease progression.
As MPE is adjacent to both primary and metastatic lung tumor tissue, it is a unique peri-tumoral environment populated with tumor cells, cytokines, growth factors, enzymes and immune cells (10, 11). MPEs are routinely drained, providing an attractive option to longitudinally study the tumor microenvironment (TME) in thoracic cancers such as mesothelioma, where a major hurdle is the inability to collect serial tumor biopsies. Our review focuses on the adaptive immune cells in MPEs, and how they could inform responses to cancer immunotherapies.
Immune checkpoint blockade (ICB) targeting T cell inhibitory receptors: cytotoxic T lymphocyte associated protein-4 (CTLA-4) and programmed cell death protein-1/ligand-1 (PD-1/PD-L1) have revolutionized cancer treatment. Single or dual agent ICB provides an durable survival benefit in patients with mesothelioma and non-small cell (NSCLC) lung cancer patients (12–15). Four ICB therapies: pembrolizumab, nivolumab, atezolizumab and durvulamab that target the PD-1/PD-L1 pathway are approved first and second-line treatments for patients with advanced NSCLC (16). Combination of ipilimumab (anti-CTLA-4) and nivolumab (anti-PD-1) provides a survival benefit over chemotherapy in malignant pleural mesothelioma (14). Platinum-based chemotherapies may synergize with ICB, with single arm studies showing that combination chemo-immunotherapy reduces tumor burden and shows promising progression-free and overall survival outcomes for mesothelioma (17–19). In addition, complete tumor regression has been observed in some NSCLC (20–22) and SCLC (23–25) patients treated with chemo-immunotherapy. Atezolizumab and durvalumab are also approved in combination with platinum-based chemotherapy for advanced SCLC patients (26). However, these best-case responses are only observed in a minority of patients. ICB is also expensive and can cause severe immune-related toxicities, highlighting the need to develop biomarkers that can accurately predict patient outcomes and inform clinical decisions (27). To date, several predictive biomarkers have been associated with ICB outcomes in some cancers, including intratumoral expression of the inhibitory receptor PD-L1 (28), the tumor mutational landscape (29), immune gene signatures within the TME (30, 31), and the presence of tumor infiltrating lymphocytes (TILs) and their expression of PD-1/PD-L1 (32–35). However, there is no common biomarker that can accurately predict ICB outcomes across different thoracic cancers, and there is a need to develop more nuanced biomarkers of response.
As ICB primarily acts through T cells, in-depth characterization of T cell subsets within the TME, and how they correlate with ICB outcomes, has been extensively investigated. CD8+ T cell subsets characterized by expression of activation/memory associated markers and their T cell receptor (TCR) usage have been linked with outcome to ICB (34, 36–39), highlighting the potential utility of T cell subsets to inform ICB responses. As T cells are also enriched in MPEs, they could offer insight into anti-tumor responses if they accurately reflect TIL phenotype, frequency and function. Longitudinal analysis of MPEs could reveal dynamic changes in the TME without the need for serial biopsies, and aid development of a biomarker of response.
Below, we review studies that have characterized matched MPE, TME and peripheral blood derived T cells, focusing on whether MPE T cells are similar in phenotype, function and specificity to their tumor counterparts. We also review changes in T cells derived from MPEs of patients undergoing ICB, and whether these changes were associated with treatment outcomes. Lastly, we discuss the unique opportunities and challenges a longitudinal study of MPE T cells brings, in terms of improving our understanding of therapeutic mechanisms, and developing a biomarker of response to ICB.
MPEs contain multiple cell types including tumor cells, pleural mesothelial cells, and innate and adaptive immune cells (40). Innate immune cells in the MPE include monocytes, macrophages, neutrophils, mast cells, dendritic cells and natural killer cells (41). These cells release cytokines, growth factors and chemokines including monocyte chemotactic protein (MCP-1), vascular endothelial growth factor (VEGF), IL-8, IL-6, IL-1β, interferon gamma (IFN-γ), tumor necrosis factor alpha (TNFα), and transforming growth factor beta (TGFβ) (40, 42–48). These cytokines can be proinflammatory and in some cases protumorigenic, promoting angiogenesis, vascular permeability and protecting cancer cells from apoptosis. MPE have increased lactate dehydrogenase, and a lower pH than non-malignant pleural fluid, suggestive of an immunosuppressive environment (11, 41, 49). The characterization of MPE proteins, cytokine milieu, innate cells, tumor cells, and their relation to overall survival have been extensively reviewed elsewhere (50–52), so this review will focus on T cells.
The proportion of total T cells in the MPE is greater than in matched peripheral blood samples from both mesothelioma and lung cancer patients (53, 54). CD4+ T cells are the predominant T cell subset in MPE both prior to and after chemotherapy (41, 54–57). Of these CD4+ T cells, an increased proportion of regulatory T cells (CD4+CD25+) are recruited in MPEs by chemokines and pro-inflammatory cytokines, compared to matched peripheral blood (58–61). Despite this abundance of CD4+ T cells, several studies have shown that the CD4+/CD8+ T cell ratio in MPEs is similar to matched peripheral blood samples in patients with mesothelioma (54, 62). For lung cancer patients, CD8+ T cell frequencies were greater in the peripheral blood compared to the MPE for one study (53) but were similar between both compartments in others (41, 63), suggesting CD8+ T cell infiltration in the pleural space may be more cancer or chemotherapy specific.
Several recent studies have examined the effect of chemotherapy on the immune milieu of matched MPE and tumor samples. At baseline, MPEs contain a lower frequency of CD4+ and CD4+ regulatory (Foxp3+) T cells compared to matched NSCLC tumor tissue, whereas CD8+ T cells were increased in the MPE compared to tumor samples (63). After chemotherapy, matched MPEs and tumor tissue from patients with mesothelioma displayed similar proportions of CD3+ T cells, CD4+ helper (CD25-) and CD4+ regulatory (CD25+CD127lo) T cells post-chemotherapy (64), but similarly, CD8+ T cells were greater in the MPE than matched tumor tissue (10, 64). Increased pre- and post-chemotherapy frequencies of CD4+ T cells in MPE and tumors were associated with complete response and improved survival in chemotherapy treated mesothelioma patients (56, 65, 66). Post-chemotherapy regulatory T cell frequencies in tumors negatively associated with survival, but this association was not observed in matched MPE samples (64). Comparison of T cell proportions between tumor and MPE in these studies are limited by small sample sizes, and whether proportions of CD4+ and CD8+ T cells are similar in matched tumor and MPE samples are unknown.
MPEs are typically enriched with CD4+ T cells, particularly regulatory CD4+ T cells. CD4+/CD8+ T cell ratios in MPEs vary between patients, likely because of patient heterogeneity such as prior treatment, disease stage and amount of fluid drained. The surface phenotype, effector function, and differentiation status of MPE T cells offers further insight into the immune status of the MPE.
T cells upregulate inhibitory checkpoint receptors in the presence of chronic tumor antigen exposure. Checkpoint receptor signaling inhibits T cell function, and immunosuppressive TMEs exploit these signaling pathways to curtail an effective anti-tumor response. Although CTLA-4 and PD-1 are the most common targets in ICB therapy, other inhibitory checkpoint receptors are expressed on TILs including TIM-3, LAG-3, TIGIT and PD-L1. Increased frequencies of CD8+PD-1+ T cells in tumors post anti-PD-1 treatment have been associated with complete and partial responses in NSCLC (34). Hence, the expression of checkpoint receptors on MPE T cells is of great interest because these T cells could be potential targets for ICB, and predictors of response.
The expression of inhibitory receptors on CD4+ and CD8+ T cells in MPE have been reported in multiple studies for mesothelioma and lung cancer. While this varies between patients, ~30% of CD4+ and ~40% of CD8+ T cells express PD-1 in the MPE (56, 62, 63) and these frequencies are greater than in matched peripheral blood T cells (62, 63, 67–70). Inhibitory receptors TIM-3, LAG-3, CTLA-4 and PD-L1 are also expressed on MPE CD4+ and CD8+ T cells at greater proportions than matched peripheral blood samples in mesothelioma and lung cancer patients (56, 62, 63, 67, 69). In addition, regulatory T cells constitutively express the inhibitory receptor TIGIT (71), and display increased expression of CTLA-4 and PD-1 in the MPE compared to peripheral blood (58, 63).
In comparison to tumor tissue, the frequencies of CD8+ T cells expressing PD-1 and TIM-3 are greater than the MPE prior to treatment (63). However, they are similar in frequency between the two compartments post-chemotherapy (64). For CD4+ helper (Foxp3-) T cells and regulatory (Foxp3+) T cells, the expression of PD-1 and TIM-3 are similar between matched MPE and tumor tissue both pre- and post-chemotherapy (63, 64). In addition, the proportion of CD4+LAG-3+ and CD8+LAG-3+ T cells in MPE after chemotherapy are similar to tumor tissue in one study (64), but not another (56). Co-expression of inhibitory receptors on T cells, in particular PD-1 and TIM-3, indicates further T cell dysfunction which has been reported to be unfavorable for ICB efficacy (37). To date, there is only one report of co-expression of these receptors, which found that the majority of CD4+PD-1+ and CD8+PD-1+ MPE T cells prior to treatment did not co-express LAG-3 or TIM-3. Less than 2% of CD8+ T cells were PD-1+TIM-3+ and less than 6% of CD4+ T cells were PD-1+LAG-3+ (68), suggesting that most of the CD8+ T cells in the MPE could be amenable to anti-PD-1 therapy.
MPE T cells are similar to TILs in that they both express increased inhibitory checkpoint receptors compared to blood T cells. Although the expression of inhibitory receptors TIM-3, PD-1 or LAG-3 on MPE T cells did not associate with improved survival post chemotherapy (64), inhibitory receptor co-expression on MPE T cells, and their correlation to ICB therapy outcomes are still of interest.
A hallmark of antigen-specific T cell responses is their ability to differentiate into memory T cells after activation, and mount a rapid response upon re-exposure to their cognate antigen. Memory CD8+ T cells are loosely classified into effector memory (TEM: CD45RO+/CD62L-, CD45RA-CCR7-), central memory (TCM: CD45RO+CD62L+, CD45RA-CCR7+) and resident memory (TRM: CD45RO+CD103+) subsets based on surface expression of differentiation markers and tissue localization. TEM and TCMs are generally found circulating in the peripheral blood and lymphatics, whilst TRMs are non-circulatory and tissue tropic. Understanding CD8+ memory T cell differentiation status is crucial because inhibitory checkpoint receptors are highly expressed on memory CD8+ T cells in tumors (72–74) and are potential cellular targets of ICB. ICB also drives changes in CD8+ memory T cell differentiation (75). Importantly, tumor infiltration of memory T cell subsets and their gene signatures correlated with ICB response and overall survival in melanoma and lung cancer patients (36, 73). MPE-derived CD4+ and CD8+ T cells in mesothelioma and lung cancer exhibit a memory phenotype prior to treatment. MPEs have increased frequencies of TCM and TEM cells for both cancer types compared to peripheral blood (53, 57). MPEs also have greater frequencies of TCM but reduced TEM compared to non-malignant pleural fluid (54).
Memory T cell subsets found in MPE are phenotypically similar to subsets found in mesothelioma tumors but there are limited studies on matched samples. Both MPE and tumors have a greater frequency of TEM cells than the circulation (76), suggesting that the proportion of TEM cells in the MPE may reflect the TME. Recent studies have also shone a spotlight on the role of TRMs in tumor immunosurveillance. Increased pre-treatment frequencies of TRMs in tumors associate with improved survival (72, 74), and increase pre- or post-treatment frequencies associate with response to anti-PD-1 therapy in lung cancer patients (73). CD8+ TRMs prior to chemotherapy have been reported in MPE of lung cancer patients, but in lesser proportions compared to matched tumor samples (77). Similar to their tumor counterparts, memory T cell subsets in the MPE could offer a predictor of therapeutic response.
CD8+ T cells proliferate and produce effector molecules such as cytotoxic granules (granzyme B, perforin) and proinflammatory cytokines (IFNγ) to mediate tumor cell killing. The ability of T cells to produce effector molecules ex vivo is a measure of T cell effector function. Understanding the effector status of T cells is important as ICB induces activation and proliferation of circulating and intratumoral T cells which correlates with response (78, 79). While MPE CD8+ T cells can produce IFNγ, granzyme B and perforin ex vivo, the frequency of MPE T cells that secrete these molecules is reduced compared to T cells from matched peripheral blood samples (67, 70). Specifically, blood derived effector (CD45RA+CD27-) T cells have increased perforin secretion than these T cell subsets from the MPE (53). However, these reports have used non-specific stimuli ex vivo to measure effector molecule production from MPE and peripheral blood T cells. Antigen-specific assays are required to understand if impairment is restricted to tumor antigen-specific T cells only.
There are limited comparisons of matched MPE-derived T cell and TIL effector function. TILs and matched MPE T cells from advanced NSCLC patients were hypofunctional, with decreased frequency of CD8+IFNγ+ T cells than tumors from patients with early stage NSCLC (80). Impaired effector function of MPE T cells could be due to an immunosuppressive environment characterized by high levels of TGFβ, tumor associated macrophages and myeloid derived suppressor cells (42, 55, 64, 67).
Effector CD4+ T cells can differentiate into helper T cell (Th) subtypes which have been identified in MPE from people with lung cancer and mesothelioma. The CD4+ helper T cell subtypes include Th1, Th2, Th17, Th9 and Th22 which are each identified by unique transcriptional signatures, and production of different cytokines (81, 82). ICB induces expansion of effector Th1 and Th17 cells in the TME, therefore it is important to determine if Th subtypes in the MPE is associated with ICB outcomes (83–85).
Th1 cells are pro-inflammatory, secreting IFNγ to stimulate effector CD8+ T cell differentiation. Approximately 45% of CD4+ MPE T cells produce IFNγ indicating a predominant Th1 phenotype in the MPE which is greater in frequency than matched peripheral blood samples (53). In comparison to Th1, Th2 promotes humoral immunity by producing cytokines IL-4, IL-5 and IL-10. The balance of these two subsets in the MPE remains controversial. Some reports suggest the MPE favors the Th2 over the Th1 pathway in comparison to pleural fluid from tuberculosis patients (86, 87). However, IL-4 was detected in the MPE in some studies (86–88) but was below 1% or undetected in others (53, 55, 89). In addition, IL-4 was detected at low levels and IFNγ was undetected in both paired MPE and mesothelioma tumor supernatant in another study (55).
The role of Th17 cells in the TME also remains controversial. The production of IL-17 has been reported to stimulate recruitment of dendritic cells, NK cells and CD8+ T cells into the TME (90), but also can promote tumor growth through IL-17R signaling (91, 92). Frequencies of Th17 cells are greater in the MPE than peripheral blood and exhibit an TEM phenotype (CD45RO+CD45RA) (93). The proportion of Th17 cells negatively correlated with regulatory T cells in the MPE, suggesting that regulatory T cells inhibited generation and differentiation of Th17 cells in the pleural space (61). For tumor tissue, one study found IL-17 in mesothelioma tumor supernatant but not in matched MPE (55).
In comparison, Th9 and Th22 cells suppress anti-tumor immunity. Both Th9 and Th22 cell proportions in the MPE are greater than the peripheral blood and also express an TEM phenotype (CD45RO+CD45RA) in both compartments (94, 95). Th9 cells produce IL-9 which has been identified to promote tumor angiogenesis (96). Th9 cell frequencies correlate to regulatory T cell frequencies in the MPE, and higher Th9 cells in MPEs associated with poor survival in lung cancer patients (95). There are no reports of Th9 cells in matched MPE and TME. One study suggests that Th9 cells may infiltrate into the MPE from the circulation as CCR7 expression was decreased on Th9 cells in the MPE compared to matched blood (97). Th22 cells produce IL-22 which has been identified to promote migration and proliferation of cancer cells and resist apoptosis and chemotherapy (98). In NSCLC patients, IL-22 was greater in matched tumor tissue than MPE (99), and IL-22 expression in MPE promoted cancer cell migration (94), and protected cancer cells from apoptosis by chemotherapies (99).
Taken together, it is evident that multiple CD4+ T helper subtypes are present in the MPE, but varying frequencies of different subtypes have been reported. Further analysis is required to understand if any of these cell types in the MPE associate with ICB efficacy.
Importantly, tumor antigen-specific T cells can be found in the MPE. Co-culture of MPE-derived lymphocytes with tumor cells or known tumor antigens from lung cancer patients resulted in IFNγ production (41, 69) and CD137 expression (68), suggesting tumor reactivity (100). In addition, tumor reactive MPE-derived CD8+ T cells displayed a memory phenotype with checkpoint expression (PD-1+TIM-3-) (68). However, most studies have included a T cell expansion step prior to assessing tumor reactivity, so the actual proportion of MPE T cells specific for tumor-antigens is unclear.
In addition to screening for reactivity, T cell receptor (TCR) analyses are used to study antigen-specific T cell responses. Individual TCRα/β chains are highly variable across complementarity determining regions (CDR), the regions crucial for antigen-specificity. Antigen-specific clonal expansion can be estimated by quantifying the distribution of TCR variable genes, or CDR sequences. ICB induces a peripheral expansion of TCR clonotypes which correlates to clinical benefit in lung cancer (101, 102). In lung tumors, the TCR repertoire clonality and the number of expanded TCR clones was greater in ICB responders compared to non-responders post-treatment (38).
TCR analyses of matched MPE and blood from lung cancer patients revealed over expression of particular TCRβ variable genes in the MPE compared to matched blood samples (103), suggesting that MPE T cells had undergone clonal expansion. The presence of shared, highly expanded TCR clonotypes in MPE and tumors would greatly support the notion that T cells in both compartments are similar. High throughput TCR sequencing is used in this area, as shared TCRβs have been found in ascites and tumors in other studies (104). We previously reported that CD4+PD-1+ MPE T cells consist of distinct, clonally expanded TCRβs from CD4+PD-1+ T cells in matched peripheral blood (62). TCR analyses of matched TILs and MPE T cells in thoracic cancers are currently limited, and would greatly inform the similarities in antigen-specificity between the compartments.
The expression of inhibitory checkpoint receptors, enrichment of CD4+ regulatory T cells, presence of CD8+ memory T cells and impaired cytotoxic, effector T cell function in MPEs suggest that they exist in an immunosuppressed environment. They are more similar to TILs than peripheral blood T cells (Figure 1). However, there are reported differences in the CD4+/CD8+ ratios, co-expression patterns of checkpoint receptors, and CD4+ Th subtypes between TILs and MPE T cells. The similarities in antigen-specificity, or TCR usage of T cells between the two compartments are also unknown. Characterizing the phenotypes of T cell clones at both sites would help researchers understand how the MPE or TME shapes the development of these cells. Next, we review how therapies could shape the phenotype of MPE T cells, because such changes could inform biomarker development.
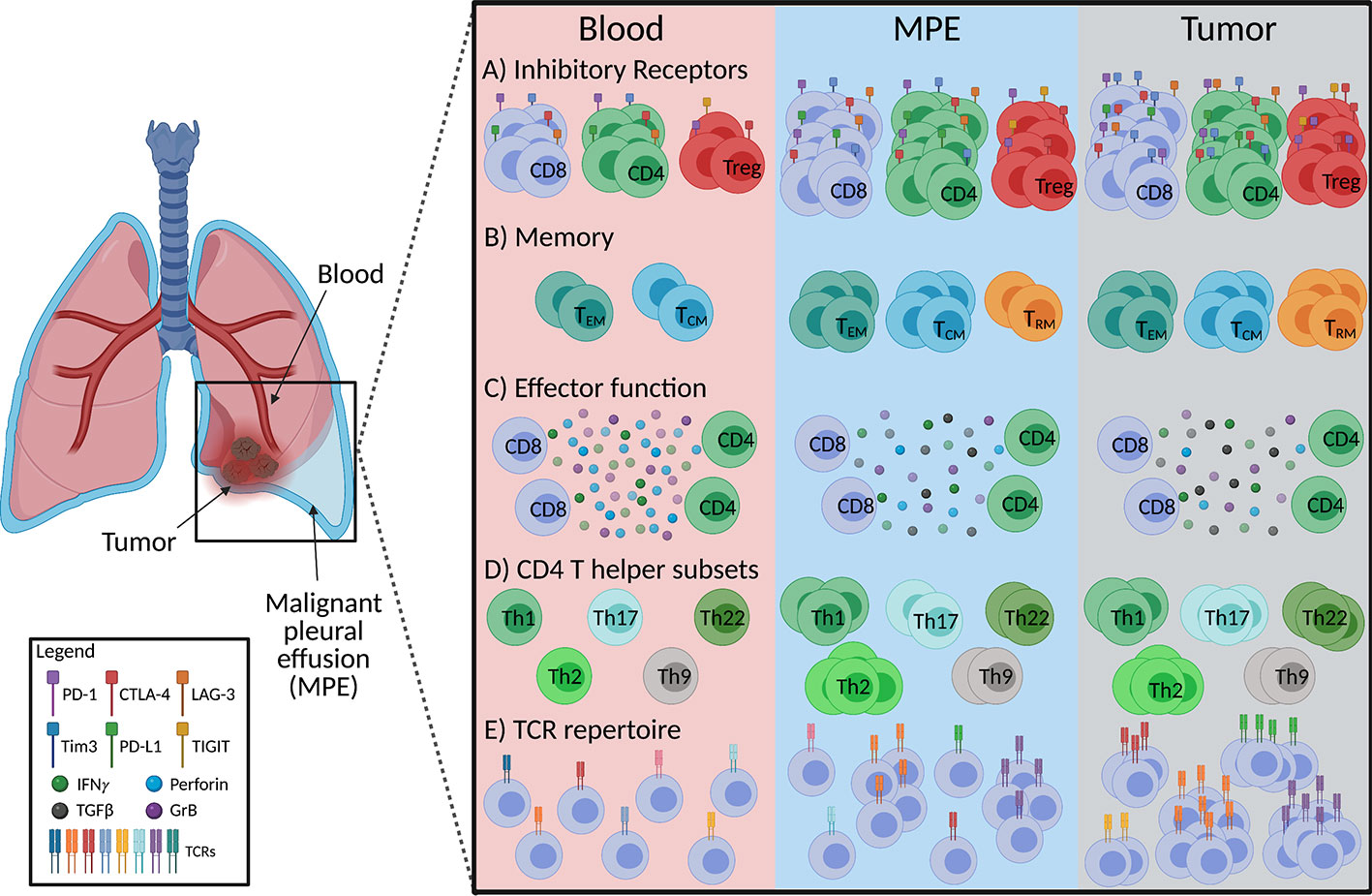
Figure 1 Schematic diagram summarizing characteristics of MPE-derived T cells in comparison to tumor and peripheral blood in mesothelioma and lung cancer. (A) Frequencies of CD8+, CD4+ and CD4+ regulatory (Treg) T cells expressing inhibitory receptors in the MPE are similar to tumor infiltrating T cells, however co-expression of inhibitory receptors on T cells is greater in tumors than MPE. (B) MPE and tumor contain greater proportions of effector memory (TEM) and central memory (TCM) T cells than the circulation with tissue resident memory T cell (TRM) frequencies the greatest at the tumor site. (C) Production of T cell effector cytokines (IFNγ, granzyme B; GrB, perforin) are similar between the MPE and tumor infiltrating T cells but lower than those in peripheral blood. (D) MPE are likely enriched with a Th2 phenotype along with a greater proportion of Th1, Th2, Th17, Th9, Th22 than circulating T cells, while tumors display a greater frequency of Th17 and Th22 than MPE. Th9 between tumor and MPE is undefined. (E) MPE contains tumor reactive T cells, implying a more clonal T cell receptor (TCR) repertoire than the peripheral blood. Figure created with BioRender.com.
Serial analyses of MPE-derived T cells in patients undergoing ICB are rare, but a study of MPE that developed after ICB has been reported. Ikematsu and colleagues characterized T cells in MPE samples drained from lung cancer patients after ICB. There were greater frequencies of CD4+TIM-3+, CD4+TIGIT+ and CD8+PD-L1+ MPE T cells from ICB treated patients compared to MPEs from chemotherapy treated patients (105). However, there were no differences in frequencies of CD8+ and CD4+ MPE T cells expressing PD-1, TIM-3, TIGIT, PD-L1 or IFNγ between responders and non-responders to anti-PD-1 therapy (105, 106). Interestingly, post treatment frequencies of Th17 (CD4+IL-17+) and CD4+LAG-3+ T cells in the MPE negatively associated with clinical outcome to anti-PD-1 ICB (105). Two NSCLC patients who became resistant to anti-PD-L1 therapy and developed recurrent MPE had increased frequencies of effector memory (CCR7-CD45RA-) CD8+ T cells and TIM-3 or CTLA-4 expressing CD8+ and CD4+ T cells cells in the MPE post-treatment, but this was compared to untreated rather than responding patients (107). Together this suggests inhibitory receptor expression increases on MPE T cells in anti-PD-1 and anti-PD-L1 treated patients. There are no studies which have analyzed serial samples of MPE in ICB treated patients.
MPE T cells have been studied in serial samples from patients treated with chemotherapy. Longitudinal analysis of MPE in a mesothelioma patient identified that the percentage of CD3+ T cells decreased in the MPE following 4 cycles of cisplatin-pemetrexed based chemotherapy, producing a partial response (10). When on-treatment changes were examined, the first dose of methotrexate chemotherapy reduced total CD3+ T cells in the MPE but these frequencies returned back to baseline levels after the second dose of methotrexate in NSCLC patients (108). Frequencies of MPE CD4+ T cells increased, regulatory T cells decreased and CD8+ T cells were unchanged following methotrexate. In terms of T cell function, it increased frequencies of MPE-derived IFNγ+ and IL-2+ T cells (108). This suggests that the MPE environment is dynamic, and changes in MPE T cells can be shaped by therapies, similar to T cells in other compartments (65, 66, 109–111).
There are very few studies of serial analyses of MPE-derived T cell phenotype, function and antigen-specificity, and how they associate with ICB outcomes. However, those studies inform us of potential T cell phenotypes from TILs and MPEs that are of interest because they could associate with ICB responses (Table 1). Serial analysis of MPE T cells in patients undergoing ICB therapies would be greatly informative.
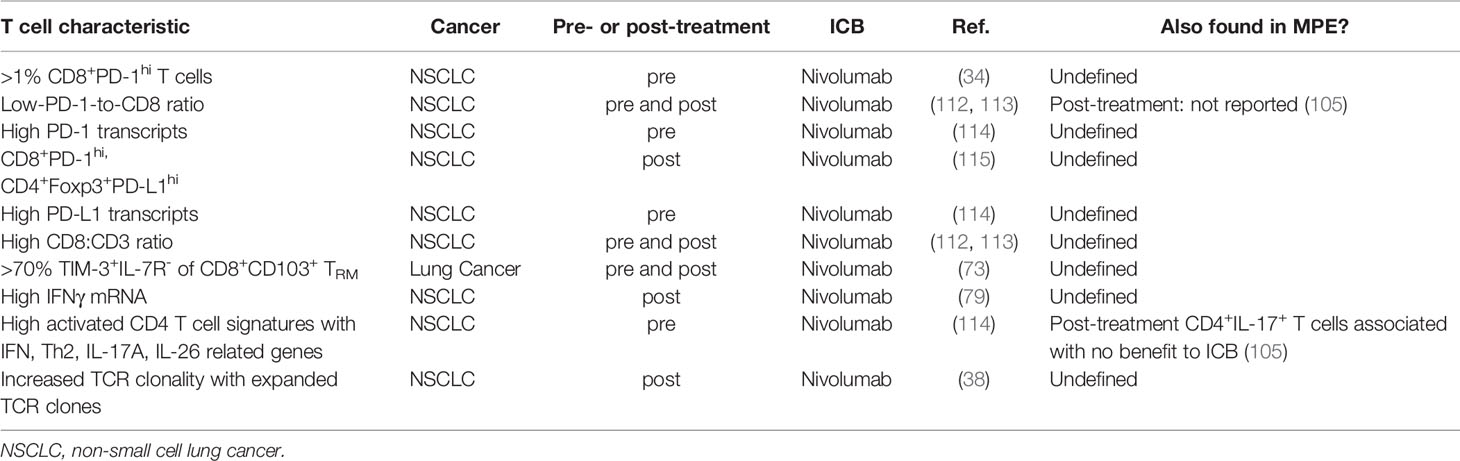
Table 1 Intratumoral T cell characteristics that associate with clinical benefit to ICB in lung cancer patients.
The potential of MPE-derived T cells as a biomarker for therapy responses is attractive for several reasons (Table 2). Firstly, MPE-derived T cells may be more closely related to TILs than circulating T cells. The presence of memory CD8+ T cells that express inhibitory receptors and CD4+ regulatory T cells in MPEs suggest that T cell responses are suppressed, similar to the TME. Furthermore, the MPE environment also consists of tumor cells, MDSCs and immunosuppressive cytokines that may shape T cell phenotype in a similar manner to the TME. Secondly, because pleural fluid is often serially drained, a dynamic biomarker could be developed. We previously argued that not all determinants of ICB response can be found prior to treatment, and changes in TME or blood that occur early on treatment could offer a more accurate, dynamic biomarker of response. Indeed, changes in T cell repertoire phenotype, diversity, and immune gene signatures early during ICB treatment correlate with ICB responses in murine and clinical studies (36, 75, 116–119). While most studies of tumor and blood suggest that changes in CD8+ T cells correlate with ICB outcomes, other T cell populations in the MPE, such as CD4+ helper T cells, could also be predictive of ICB outcomes. Regular drainage of MPEs provides a unique opportunity to study these dynamic changes. Although this review focuses only on T cells, how MPE-derived T cell frequencies and phenotypes change in relation to other components in the fluid, such as tumor cell, MDSC numbers, and suppressive cytokine levels is informative for biomarker development.

Table 2 Benefits and limitations for using the MPE to develop T cell biomarkers for ICB therapy response.
MPE-derived T cells exhibit memory phenotypes indicative of chronic antigen-specific activation. The antigen-specificity of MPE TEM and TRM cells, and how they change with therapy are of great interest. It is promising that tumor-reactive T cells can be expanded from the MPE, but the overlap in antigen-specificities between TILs and MPE T cells, if any, are unknown. TCR sequencing offers a complementary method to study the extent of clonal overlap between TILs, blood and MPE-derived T cell populations, and to track the changes in antigen-specific T cells without prior knowledge of any tumor antigens. The extent in which the MPE environment drives T cell differentiation is unclear. We speculate that MPE consist of T cells that have migrated from the local tumor and blood. However, to what extent the MPE environment changes T cell phenotype is unclear. It is possible that further activation and differentiation of T cells in the MPE drives the distinct phenotypes of MPE-derived T cells (Figure 2). Single cell technology is a powerful tool to comprehensively study the interactions of different cells in the MPE, and will greatly help our understanding in this area. The transcriptome and TCRαβ usage of individual T cells can be determined, allowing researchers to match phenotypes to individual T cell clones. These TCRs of interest can be subsequently screened for tumor reactivity. Single cell technology is now used in numerous studies of TMEs, and can be applied to MPE samples.
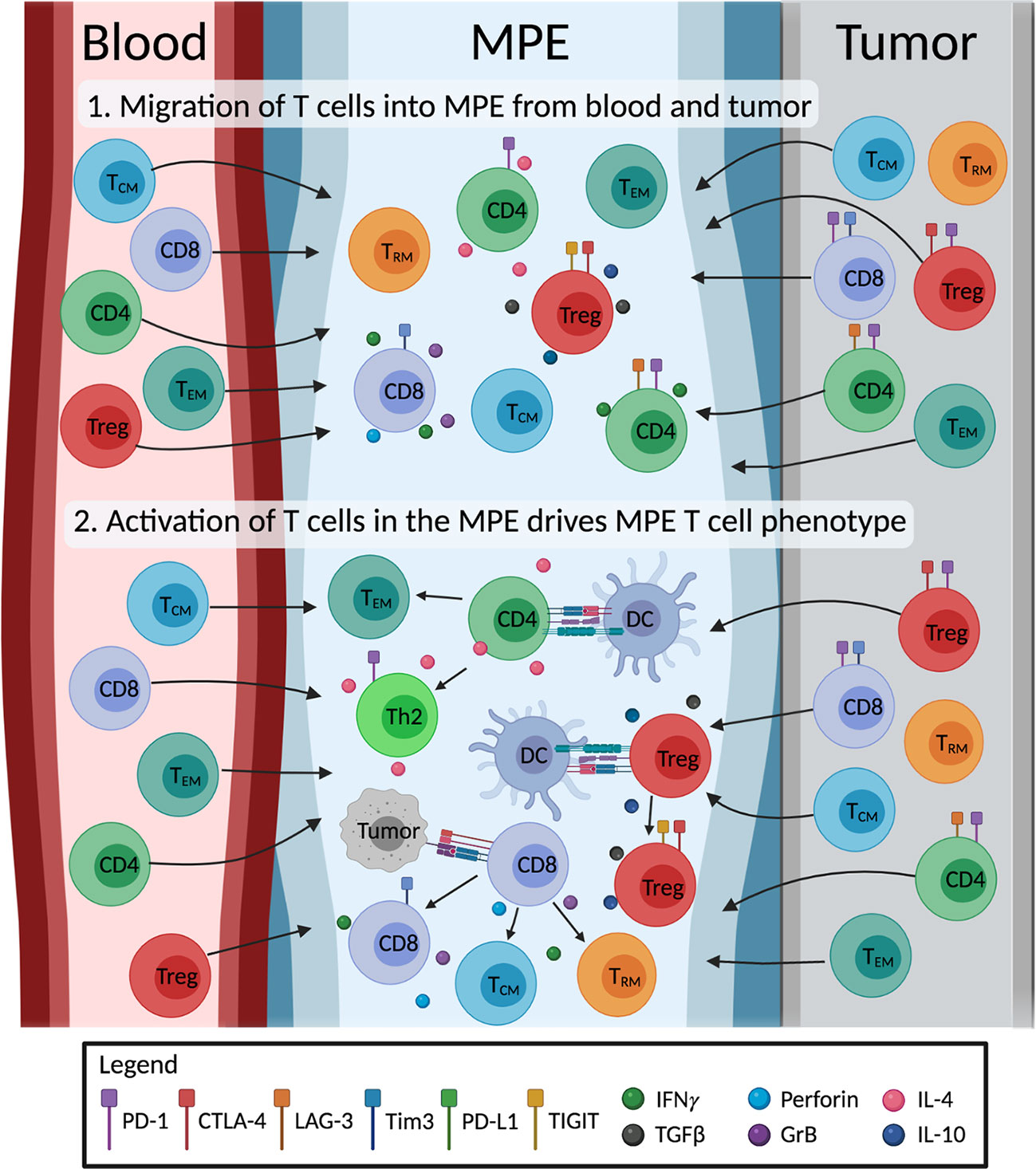
Figure 2 Illustration of the proposed origin and development of MPE T cells. The impact of the MPE environment on T cell differentiation is unclear. We hypothesize that 1) MPE acts as a sink, containing a mix of T cells originally from the blood and the tumor site. 2) MPE environment including cytokines and other cells (e.g. tumor cells, dendritic cells; DC) drive changes in phenotype of MPE T cells. MPE T cells differentiate into effector subtypes, producing immunostimulatory (IFNγ, perforin, granzyme B; GrB, IL-4) or immunosuppressive (TGF-β, IL-10) cytokines; exhausted T cells expressing inhibitory receptors; and differentiate into memory T cells (i.e. effector (TEM), central (TCM) and tissue resident (TRM) memory T cells). Figure created with BioRender.com.
However, there are some limitations with studying MPEs (Table 2). The volume of fluid drained, and cellularity of MPE samples varies between patients (62, 120, 121). In some instances, MPE cell numbers are too few for meaningful downstream analysis, especially for rare T cell subsets. Even though longitudinal analysis can be performed with MPE samples, the number of drainage events vary between patients and the timing of them cannot be predicted. Furthermore, differences in MPE immunophenotype measured over time are not always attributed to tumor or treatment. External factors such as infections and lung inflammation could alter the T cell phenotype and are confounding factors that have to be accounted for (122–124). Lastly, management regimens to treat MPEs including talc pleurodesis and VATS pleurodesis and pleurectomy, which is a palliative therapeutic option for malignant pleural mesothelioma patients, aim to obliterate the pleural space and prevent MPE recurrence. This then eliminates the opportunity to serially sample the MPE for biomarkers of response to therapy.
Although the cellular components of MPE have been studied extensively, recent developments in cancer immunotherapy and the need for biomarkers of response have led researchers to focus on MPE T cells. These cells share phenotypic features with TILs, but further study is required to elucidate if MPE T cells are truly reflective of their tumor counterparts. We think that dynamic analyses of MPE T cells in relation to ICB outcomes will lead to a robust and clinically useful ICB response biomarker.
NP wrote the article and made the figures. JK, WL, RL, AN, AM, and JC critically revised the manuscript. All authors contributed to the article and approved the submitted version.
NP was supported by Cancer Council WA and UWA Richard Walter Gibbon Medical Research scholarships. JK was supported by icare Dust Diseases Board. WL was supported by a Simon Lee Fellowship, an NHMRC Fellowship, and a Cancer Council WA fellowship. AM was supported by grants and fellowship from Cancer Council WA, Raine Medical Research Foundation and icare Dust Diseases Board. JC was supported by grants and fellowship from the Raine Medical Research Foundation, Cancer Council WA, WA Department of Health, and icare Dust Diseases Board. The National Centre for Asbestos Related Diseases receives funding through the National Health and Medical Research Council Centres of Research Excellence scheme APP1197652.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Porcel JM, Gasol A, Bielsa S, Civit C, Light RW, Salud A. Clinical Features and Survival of Lung Cancer Patients With Pleural Effusions. Respirology (2015) 20(4):654–9. doi: 10.1111/resp.12496
2. Zamboni MM, da Silva CT Jr, Baretta R, Cunha ET, Cardoso GP. Important Prognostic Factors for Survival in Patients With Malignant Pleural Effusion. BMC Pulm Med (2015) 15:29. doi: 10.1186/s12890-015-0025-z
3. Cheah HM, Lansley SM, Varano Della Vergiliana JF, Tan AL, Thomas R, Leong SL, et al. Malignant Pleural Fluid From Mesothelioma has Potent Biological Activities. Respirology (2017) 22(1):192–9. doi: 10.1111/resp.12874
4. Khaleeq G, Musani AI. Emerging Paradigms in the Management of Malignant Pleural Effusions. Respir Med (2008) 102(7):939–48. doi: 10.1016/j.rmed.2008.01.022
5. Lee YC, Light RW. Management of Malignant Pleural Effusions. Respirology (2004) 9(2):148–56. doi: 10.1111/j.1440-1843.2004.00566.x
6. Porcel JM, Lui MM, Lerner AD, Davies HE, Feller-Kopman D, Lee YC. Comparing Approaches to the Management of Malignant Pleural Effusions. Expert Rev Respir Med (2017) 11(4):273–84. doi: 10.1080/17476348.2017.1300532
7. Davies HE, Mishra EK, Kahan BC, Wrightson JM, Stanton AE, Guhan A, et al. Effect of an Indwelling Pleural Catheter vs Chest Tube and Talc Pleurodesis for Relieving Dyspnea in Patients With Malignant Pleural Effusion: The TIME2 Randomized Controlled Trial. JAMA (2012) 307(22):2383–9. doi: 10.1001/jama.2012.5535
8. Thomas R, Fysh ETH, Smith NA, Lee P, Kwan BCH, Yap E, et al. Effect of an Indwelling Pleural Catheter vs Talc Pleurodesis on Hospitalization Days in Patients With Malignant Pleural Effusion: The AMPLE Randomized Clinical Trial. JAMA (2017) 318(19):1903–12. doi: 10.1001/jama.2017.17426
9. Muruganandan S, Azzopardi M, Thomas R, Fitzgerald DB, Kuok YJ, Cheah HM, et al. The Pleural Effusion And Symptom Evaluation (Please) Study of Breathlessness in Patients With a Symptomatic Pleural Effusion. Eur Respir J (2020) 55(5). doi: 10.1183/13993003.00980-2019
10. Lievense LA, Bezemer K, Cornelissen R, Kaijen-Lambers ME, Hegmans JP, Aerts JG. Precision Immunotherapy; Dynamics in the Cellular Profile of Pleural Effusions in Malignant Mesothelioma Patients. Lung Cancer (2017) 107:36–40. doi: 10.1016/j.lungcan.2016.04.015
11. Thomas R, Cheah HM, Creaney J, Turlach BA, Lee YC. Longitudinal Measurement of Pleural Fluid Biochemistry and Cytokines in Malignant Pleural Effusions. Chest (2016) 149(6):1494–500. doi: 10.1016/j.chest.2016.01.001
12. Peters S, Gettinger S, Johnson ML, Janne PA, Garassino MC, Christoph D, et al. Phase II Trial of Atezolizumab as First-Line or Subsequent Therapy for Patients With Programmed Death-Ligand 1-Selected Advanced non-Small-Cell Lung Cancer (Birch). J Clin Oncol (2017) 35(24):2781–9. doi: 10.1200/JCO.2016.71.9476
13. Hellmann MD, Paz-Ares L, Bernabe Caro R, Zurawski B, Kim S-W, Carcereny Costa E, et al. Nivolumab Plus Ipilimumab in Advanced non–Small-Cell Lung Cancer. New Engl J Med (2019) 381(21):2020–31. doi: 10.1056/NEJMoa1910231
14. Baas P, Scherpereel A, Nowak AK, Fujimoto N, Peters S, Tsao AS, et al. First-Line Nivolumab Plus Ipilimumab in Unresectable Malignant Pleural Mesothelioma (CheckMate 743): A Multicentre, Randomised, Open-Label, Phase 3 Trial. Lancet (2021) 397(10272):375–86. doi: 10.1016/S0140-6736(20)32714-8
15. Gandhi L, Rodriguez-Abreu D, Gadgeel S, Esteban E, Felip E, De Angelis F, et al. Pembrolizumab Plus Chemotherapy in Metastatic non-Small-Cell Lung Cancer. N Engl J Med (2018) 378(22):2078–92. doi: 10.1056/NEJMoa1801005
16. Brahmer JR, Govindan R, Anders RA, Antonia SJ, Sagorsky S, Davies MJ, et al. The Society for Immunotherapy of Cancer Consensus Statement on Immunotherapy for the Treatment of non-Small Cell Lung Cancer (Nsclc). J Immunother Cancer (2018) 6(1):75. doi: 10.1186/s40425-018-0382-2
17. Nowak AK, Lesterhuis WJ, Kok PS, Brown C, Hughes BG, Karikios DJ, et al. Durvalumab With First-Line Chemotherapy in Previously Untreated Malignant Pleural Mesothelioma (Dream): A Multicentre, Single-Arm, Phase 2 Trial With a Safety Run-in. Lancet Oncol (2020) 21(9):1213–23. doi: 10.1016/S1470-2045(20)30462-9
18. Alley EW, Lopez J, Santoro A, Morosky A, Saraf S, Piperdi B, et al. Clinical Safety and Activity of Pembrolizumab in Patients With Malignant Pleural Mesothelioma (Keynote-028): Preliminary Results From a non-Randomised, Open-Label, Phase 1b Trial. Lancet Oncol (2017) 18(5):623–30. doi: 10.1016/S1470-2045(17)30169-9
19. Scherpereel A, Mazieres J, Greillier L, Lantuejoul S, Do P, Bylicki O, et al. Nivolumab or Nivolumab Plus Ipilimumab in Patients With Relapsed Malignant Pleural Mesothelioma (Ifct-1501 Maps2): A Multicentre, Open-Label, Randomised, non-Comparative, Phase 2 Trial. Lancet Oncol (2019) 20(2):239–53. doi: 10.1016/S1470-2045(18)30765-4
20. Paz-Ares L, Ciuleanu TE, Cobo M, Schenker M, Zurawski B, Menezes J, et al. First-Line Nivolumab Plus Ipilimumab Combined With Two Cycles of Chemotherapy in Patients With non-Small-Cell Lung Cancer (CheckMate 9la): An International, Randomised, Open-Label, Phase 3 Trial. Lancet Oncol (2021) 22(2):198-211. doi: 10.1016/S1470-2045(20)30641-0
21. Faivre-Finn C, Vicente D, Kurata T, Planchard D, Paz-Ares L, Vansteenkiste JF, et al. Brief Report: Four-Year Survival With Durvalumab After Chemoradiotherapy in Stage Iii NSCLC - an Update From the PACIFIC Trial. J Thorac Oncol (2021) S1556-0864(21):00022-8. doi: 10.1016/j.jtho.2020.12.015
22. Garassino MC, Cho BC, Kim JH, Mazieres J, Vansteenkiste J, Lena H, et al. Durvalumab as Third-Line or Later Treatment for Advanced non-Small-Cell Lung Cancer (Atlantic): An Open-Label, Single-Arm, Phase 2 Study. Lancet Oncol (2018) 19(4):521–36. doi: 10.1016/S1470-2045(18)30144-X
23. Paz-Ares L, Dvorkin M, Chen Y, Reinmuth N, Hotta K, Trukhin D, et al. Durvalumab Plus Platinum–Etoposide Versus Platinum–Etoposide in First-Line Treatment of Extensive-Stage Small-Cell Lung Cancer (Caspian): A Randomised, Controlled, Open-Label, Phase 3 Trial. Lancet (2019) 394(10212):1929–39. doi: 10.1016/S0140-6736(19)32222-6
24. Reck M, Bondarenko I, Luft A, Serwatowski P, Barlesi F, Chacko R, et al. Ipilimumab in Combination With Paclitaxel and Carboplatin as First-Line Therapy in Extensive-Disease-Small-Cell Lung Cancer: Results From a Randomized, Double-Blind, Multicenter Phase 2 Trial. Ann Oncol (2013) 24(1):75–83. doi: 10.1093/annonc/mds213
25. Ready NE, Ott PA, Hellmann MD, Zugazagoitia J, Hann CL, de Braud F, et al. Nivolumab Monotherapy and Nivolumab Plus Ipilimumab in Recurrent Small Cell Lung Cancer: Results From the CheckMate 032 Randomized Cohort. J Thorac Oncol (2020) 15(3):426–35. doi: 10.1016/j.jtho.2019.10.004
26. Mathieu L, Shah S, Pai-Scherf L, Larkins E, Vallejo J, Li X, et al. Fda Approval Summary: Atezolizumab and Durvalumab in Combination With Platinum-Based Chemotherapy in Extensive Stage Small Cell Lung Cancer. Oncologist (2021). doi: 10.1002/onco.13752
27. Lesterhuis WJ, Bosco A, Millward MJ, Small M, Nowak AK, Lake RA. Dynamic Versus Static Biomarkers in Cancer Immune Checkpoint Blockade: Unravelling Complexity. Nat Rev Drug Discovery (2017) 16(4):264–72. doi: 10.1038/nrd.2016.233
28. Herbst RS, Soria JC, Kowanetz M, Fine GD, Hamid O, Gordon MS, et al. Predictive Correlates of Response to the Anti-PD-L1 Antibody MPDL3280A in Cancer Patients. Nature (2014) 515(7528):563–7. doi: 10.1038/nature14011
29. Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, et al. Cancer Immunology. Mutational Landscape Determines Sensitivity to PD-1 Blockade in non-Small Cell Lung Cancer. Science (2015) 348(6230):124–8. doi: 10.1126/science.aaa1348
30. Ayers M, Lunceford J, Nebozhyn M, Murphy E, Loboda A, Kaufman DR, et al. Ifn-Gamma-Related Mrna Profile Predicts Clinical Response to PD-1 Blockade. J Clin Invest (2017) 127(8):2930–40. doi: 10.1172/JCI91190
31. Lee HS, Jang HJ, Choi JM, Zhang J, de Rosen VL, Wheeler TM, et al. Comprehensive Immunoproteogenomic Analyses of Malignant Pleural Mesothelioma. JCI Insight (2018) 3(7):e98575. doi: 10.1172/jci.insight.98575
32. Yu Y, Zeng D, Ou Q, Liu S, Li A, Chen Y, et al. Association of Survival and Immune-Related Biomarkers With Immunotherapy in Patients With Non-Small Cell Lung Cancer: A Meta-Analysis and Individual Patient-Level Analysis. JAMA Netw Open (2019) 2(7):e196879. doi: 10.1001/jamanetworkopen.2019.6879
33. Althammer S, Tan TH, Spitzmuller A, Rognoni L, Wiestler T, Herz T, et al. Automated Image Analysis of NSCLC Biopsies to Predict Response to anti-PD-L1 Therapy. J Immunother Cancer (2019) 7(1):121. doi: 10.1186/s40425-019-0589-x
34. Thommen DS, Koelzer VH, Herzig P, Roller A, Trefny M, Dimeloe S, et al. A Transcriptionally and Functionally Distinct PD-1(+) Cd8(+) T Cell Pool With Predictive Potential in non-Small-Cell Lung Cancer Treated With PD-1 Blockade. Nat Med (2018) 24(7):994–1004. doi: 10.1038/s41591-018-0057-z
35. Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, et al. Pd-1 Blockade Induces Responses by Inhibiting Adaptive Immune Resistance. Nature (2014) 515(7528):568–71. doi: 10.1038/nature13954
36. Sade-Feldman M, Yizhak K, Bjorgaard SL, Ray JP, de Boer CG, Jenkins RW, et al. Defining T Cell States Associated With Response to Checkpoint Immunotherapy in Melanoma. Cell (2018) 175(4):998–1013. doi: 10.1016/j.cell.2018.10.038
37. Miller BC, Sen DR, Al Abosy R, Bi K, Virkud YV, LaFleur MW, et al. Subsets of Exhausted Cd8(+) T Cells Differentially Mediate Tumor Control and Respond to Checkpoint Blockade. Nat Immunol (2019) 20(3):326–36. doi: 10.1038/s41590-019-0312-6
38. Zhang J, Ji Z, Caushi JX, El Asmar M, Anagnostou V, Cottrell TR, et al. Compartmental Analysis of T-cell Clonal Dynamics as a Function of Pathologic Response to Neoadjuvant Pd-1 Blockade in Resectable non-Small Cell Lung Cancer. Clin Cancer Res (2020) 26(6):1327–37. doi: 10.1158/1078-0432.CCR-19-2931
39. Cha E, Klinger M, Hou Y, Cummings C, Ribas A, Faham M, et al. Improved Survival With T Cell Clonotype Stability After Anti-CTLA-4 Treatment in Cancer Patients. Sci Transl Med (2014) 6(238):238ra70. doi: 10.1126/scitranslmed.3008211
40. Batra H, Antony VB. Pleural Mesothelial Cells in Pleural and Lung Diseases. J Thorac Dis (2015) 7(6):964–80. doi: 10.3978/j.issn.2072-1439.2015.02.19
41. Dhupar R, Okusanya OT, Eisenberg SH, Monaco SE, Ruffin AT, Liu D, et al. Characteristics of Malignant Pleural Effusion Resident Cd8(+) T Cells From a Heterogeneous Collection of Tumors. Int J Mol Sci (2020) 21(17):6178. doi: 10.3390/ijms21176178
42. Kaczmarek M, Sikora J. Macrophages in Malignant Pleural Effusions - Alternatively Activated Tumor Associated Macrophages. Contemp Oncol (Pozn) (2012) 16(4):279–84. doi: 10.5114/wo.2012.30054
43. Saraya T, Ohkuma K, Watanabe T, Mikura S, Kobayashi F, Aso J, et al. Diagnostic Value of Vascular Endothelial Growth Factor, Transforming Growth Factor-beta, Interleukin-8, and the Ratio of Lactate Dehydrogenase to Adenosine Deaminase in Pleural Effusion. Lung (2018) 196(2):249–54. doi: 10.1007/s00408-018-0090-1
44. Lieser EA, Croghan GA, Nevala WK, Bradshaw MJ, Markovic SN, Mansfield AS. Up-Regulation of Pro-Angiogenic Factors and Establishment of Tolerance in Malignant Pleural Effusions. Lung Cancer (2013) 82(1):63–8. doi: 10.1016/j.lungcan.2013.07.007
45. Hirayama N, Tabata C, Tabata R, Maeda R, Yasumitsu A, Yamada S, et al. Pleural Effusion Vegf Levels as a Prognostic Factor of Malignant Pleural Mesothelioma. Respir Med (2011) 105(1):137–42. doi: 10.1016/j.rmed.2010.10.010
46. Yeh HH, Lai WW, Chen HH, Liu HS, Su WC. Autocrine IL-6-induced Stat3 Activation Contributes to the Pathogenesis of Lung Adenocarcinoma and Malignant Pleural Effusion. Oncogene (2006) 25(31):4300–9. doi: 10.1038/sj.onc.1209464
47. Yokoyama A, Maruyama M, Ito M, Kohno N, Hiwada K, Yano S. Interleukin 6 Activity in Pleural Effusion. Its Diagnostic Value and Thrombopoietic Activity. Chest (1992) 102(4):1055–9. doi: 10.1378/chest.102.4.1055
48. Giannou AD, Marazioti A, Spella M, Kanellakis NI, Apostolopoulou H, Psallidas I, et al. Mast Cells Mediate Malignant Pleural Effusion Formation. J Clin Invest (2015) 125(6):2317–34. doi: 10.1172/JCI79840
49. Nieto JC, Zamora C, Porcel JM, Mulet M, Pajares V, Munoz-Fernandez AM, et al. Migrated T Lymphocytes Into Malignant Pleural Effusions: An Indicator of Good Prognosis in Lung Adenocarcinoma Patients. Sci Rep (2019) 9(1):2996. doi: 10.1038/s41598-018-35840-3
50. Murthy P, Ekeke CN, Russell KL, Butler SC, Wang Y, Luketich JD, et al. Making Cold Malignant Pleural Effusions Hot: Driving Novel Immunotherapies. Oncoimmunology (2019) 8(4):e1554969. doi: 10.1080/2162402X.2018.1554969
51. Stathopoulos GT, Kalomenidis I. Malignant Pleural Effusion: Tumor-Host Interactions Unleashed. Am J Respir Crit Care Med (2012) 186(6):487–92. doi: 10.1164/rccm.201203-0465PP
52. Kassis J, Klominek J, Kohn EC. Tumor Microenvironment: What can Effusions Teach Us? Diagn Cytopathol (2005) 33(5):316–9. doi: 10.1002/dc.20280
53. Prado-Garcia H, Aguilar-Cazares D, Flores-Vergara H, Mandoki JJ, Lopez-Gonzalez JS. Effector, Memory and Naive Cd8+ T Cells in Peripheral Blood and Pleural Effusion From Lung Adenocarcinoma Patients. Lung Cancer (2005) 47(3):361–71. doi: 10.1016/j.lungcan.2004.07.046
54. Scherpereel A, Grigoriu BD, Noppen M, Gey T, Chahine B, Baldacci S, et al. Defect in Recruiting Effector Memory Cd8+ T-Cells in Malignant Pleural Effusions Compared to Normal Pleural Fluid. BMC Cancer (2013) 13:324. doi: 10.1186/1471-2407-13-324
55. Lievense LA, Cornelissen R, Bezemer K, Kaijen-Lambers ME, Hegmans JP, Aerts JG. Pleural Effusion of Patients With Malignant Mesothelioma Induces Macrophage-Mediated T Cell Suppression. J Thorac Oncol (2016) 11(10):1755–64. doi: 10.1016/j.jtho.2016.06.021
56. Marcq E, Waele J, Audenaerde JV, Lion E, Santermans E, Hens N, et al. Abundant Expression of TIM-3, Lag-3, PD-1 and PD-L1 as Immunotherapy Checkpoint Targets in Effusions of Mesothelioma Patients. Oncotarget (2017) 8(52):89722–35. doi: 10.18632/oncotarget.21113
57. Atanackovic D, Block A, de Weerth A, Faltz C, Hossfeld DK, Hegewisch-Becker S. Characterization of Effusion-Infiltrating T Cells: Benign Versus Malignant Effusions. Clin Cancer Res (2004) 10(8):2600–8. doi: 10.1158/1078-0432.CCR-03-0239
58. Chen YQ, Shi HZ, Qin XJ, Mo WN, Liang XD, Huang ZX, et al. Cd4+Cd25+ Regulatory T Lymphocytes in Malignant Pleural Effusion. Am J Respir Crit Care Med (2005) 172(11):1434–9. doi: 10.1164/rccm.200504-588OC
59. Qin XJ, Shi HZ, Deng JM, Liang QL, Jiang J, Ye ZJ. Ccl22 Recruits CD4-Positive CD25-Positive Regulatory T Cells Into Malignant Pleural Effusion. Clin Cancer Res (2009) 15(7):2231–7. doi: 10.1158/1078-0432.CCR-08-2641
60. Lv M, Xu Y, Tang R, Ren J, Shen S, Chen Y, et al. Mir141-CXCL1-CXCR2 Signaling-Induced Treg Recruitment Regulates Metastases and Survival of non-Small Cell Lung Cancer. Mol Cancer Ther (2014) 13(12):3152–62. doi: 10.1158/1535-7163.MCT-14-0448
61. Ye ZJ, Zhou Q, Zhang JC, Li X, Wu C, Qin SM, et al. Cd39+ Regulatory T Cells Suppress Generation and Differentiation of Th17 Cells in Human Malignant Pleural Effusion Via a LAP-dependent Mechanism. Respir Res (2011) 12:77. doi: 10.1186/1465-9921-12-77
62. Chee J, Watson MW, Chopra A, Nguyen B, Cook AM, Creaney J, et al. Tumour Associated Lymphocytes in the Pleural Effusions of Patients With Mesothelioma Express High Levels of Inhibitory Receptors. BMC Res Notes (2018) 11(1):864. doi: 10.1186/s13104-018-3953-x
63. Kim HR, Park HJ, Son J, Lee JG, Chung KY, Cho NH, et al. Tumor Microenvironment Dictates Regulatory T Cell Phenotype: Upregulated Immune Checkpoints Reinforce Suppressive Function. J Immunother Cancer (2019) 7(1):339. doi: 10.1186/s40425-019-0785-8
64. Salaroglio IC, Kopecka J, Napoli F, Pradotto M, Maletta F, Costardi L, et al. Potential Diagnostic and Prognostic Role of Microenvironment in Malignant Pleural Mesothelioma. J Thorac Oncol (2019) 14(8):1458–71. doi: 10.1016/j.jtho.2019.03.029
65. Pasello G, Zago G, Lunardi F, Urso L, Kern I, Vlacic G, et al. Malignant Pleural Mesothelioma Immune Microenvironment and Checkpoint Expression: Correlation With Clinical-Pathological Features and Intratumor Heterogeneity Over Time. Ann Oncol (2018) 29(5):1258–65. doi: 10.1093/annonc/mdy086
66. Parra ER, Villalobos P, Behrens C, Jiang M, Pataer A, Swisher SG, et al. Effect of Neoadjuvant Chemotherapy on the Immune Microenvironment in non-Small Cell Lung Carcinomas as Determined by Multiplex Immunofluorescence and Image Analysis Approaches. J Immunother Cancer (2018) 6(1):48. doi: 10.1186/s40425-018-0368-0
67. Li L, Yang L, Wang L, Wang F, Zhang Z, Li J, et al. Impaired T Cell Function in Malignant Pleural Effusion is Caused by TGF-beta Derived Predominantly From Macrophages. Int J Cancer (2016) 139(10):2261–9. doi: 10.1002/ijc.30289
68. Prado-Garcia H, Romero-Garcia S, Puerto-Aquino A, Rumbo-Nava U. The PD-L1/PD-1 Pathway Promotes Dysfunction, But Not “Exhaustion”, in Tumor-Responding T Cells From Pleural Effusions in Lung Cancer Patients. Cancer Immunol Immunother (2017) 66(6):765–76. doi: 10.1007/s00262-017-1979-x
69. Khanna S, Thomas A, Abate-Daga D, Zhang J, Morrow B, Steinberg SM, et al. Malignant Mesothelioma Effusions Are Infiltrated by CD3(+) T Cells Highly Expressing PD-L1 and the PD-L1(+) Tumor Cells Within These Effusions Are Susceptible to ADCC by the Anti-PD-L1 Antibody Avelumab. J Thorac Oncol (2016) 11(11):1993–2005. doi: 10.1016/j.jtho.2016.07.033
70. Hu CY, Zhang YH, Wang T, Chen L, Gong ZH, Wan YS, et al. Interleukin-2 Reverses Cd8(+) T Cell Exhaustion in Clinical Malignant Pleural Effusion of Lung Cancer. Clin Exp Immunol (2016) 186(1):106–14. doi: 10.1111/cei.12845
71. Gu FF, Wu JJ, Liu YY, Hu Y, Liang JY, Zhang K, et al. Human Inflammatory Dendritic Cells in Malignant Pleural Effusions Induce Th1 Cell Differentiation. Cancer Immunol Immunother (2020) 69(5):779–88. doi: 10.1007/s00262-020-02510-1
72. Djenidi F, Adam J, Goubar A, Durgeau A, Meurice G, de Montpreville V, et al. Cd8+Cd103+ Tumor-Infiltrating Lymphocytes are Tumor-Specific Tissue-Resident Memory T Cells and a Prognostic Factor for Survival in Lung Cancer Patients. J Immunol (2015) 194(7):3475–86. doi: 10.4049/jimmunol.1402711
73. Clarke J, Panwar B, Madrigal A, Singh D, Gujar R, Wood O, et al. Single-Cell Transcriptomic Analysis of Tissue-Resident Memory T Cells in Human Lung Cancer. J Exp Med (2019) 216(9):2128–49. doi: 10.1084/jem.20190249
74. Ganesan AP, Clarke J, Wood O, Garrido-Martin EM, Chee SJ, Mellows T, et al. Tissue-Resident Memory Features are Linked to the Magnitude of Cytotoxic T Cell Responses in Human Lung Cancer. Nat Immunol (2017) 18(8):940–50. doi: 10.1038/ni.3775
75. Kurtulus S, Madi A, Escobar G, Klapholz M, Nyman J, Christian E, et al. Checkpoint Blockade Immunotherapy Induces Dynamic Changes in PD-1(-)CD8(+) Tumor-Infiltrating T Cells. Immunity (2019) 50(1):181–94. doi: 10.1016/j.immuni.2018.11.014
76. Klampatsa A, O’Brien SM, Thompson JC, Rao AS, Stadanlick JE, Martinez MC, et al. Phenotypic and Functional Analysis of Malignant Mesothelioma Tumor-Infiltrating Lymphocytes. Oncoimmunology (2019) 8(9):e1638211. doi: 10.1080/2162402X.2019.1638211
77. Qu QX, Zhu XY, Du WW, Wang HB, Shen Y, Zhu YB, et al. 4-1bb Agonism Combined With Pd-L1 Blockade Increases the Number of Tissue-Resident Cd8+ T Cells and Facilitates Tumor Abrogation. Front Immunol (2020) 11:577. doi: 10.3389/fimmu.2020.00577
78. Mankor JM, Disselhorst MJ, Poncin M, Baas P, Aerts J, Vroman H. Efficacy of Nivolumab and Ipilimumab in Patients With Malignant Pleural Mesothelioma is Related to a Subtype of Effector Memory Cytotoxic T Cells: Translational Evidence From Two Clinical Trials. EBioMedicine (2020) 62:103040. doi: 10.1016/j.ebiom.2020.103040
79. Karachaliou N, Gonzalez-Cao M, Crespo G, Drozdowskyj A, Aldeguer E, Gimenez-Capitan A, et al. Interferon Gamma, an Important Marker of Response to Immune Checkpoint Blockade in Non-Small Cell Lung Cancer and Melanoma Patients. Ther Adv Med Oncol (2018) 10:1758834017749748. doi: 10.1177/1758834017749748
80. O’Brien SM, Klampatsa A, Thompson JC, Martinez MC, Hwang WT, Rao AS, et al. Function of Human Tumor-Infiltrating Lymphocytes in Early-Stage non-Small Cell Lung Cancer. Cancer Immunol Res (2019) 7(6):896–909. doi: 10.1158/2326-6066.CIR-18-0713
81. Yi FS, Zhai K, Shi HZ. Helper T Cells in Malignant Pleural Effusion. Cancer Lett (2021) 500:21–8. doi: 10.1016/j.canlet.2020.12.016
82. Chraa D, Naim A, Olive D, Badou A. T Lymphocyte Subsets in Cancer Immunity: Friends or Foes. J Leukoc Biol (2019) 105(2):243–55. doi: 10.1002/JLB.MR0318-097R
83. Wei SC, Levine JH, Cogdill AP, Zhao Y, Anang NAS, Andrews MC, et al. Distinct Cellular Mechanisms Underlie Anti-CTLA-4 and Anti-PD-1 Checkpoint Blockade. Cell (2017) 170(6):1120–33. doi: 10.1016/j.cell.2017.07.024
84. Liakou CI, Kamat A, Tang DN, Chen H, Sun J, Troncoso P, et al. Ctla-4 Blockade Increases IFNgamma-producing Cd4+Icoshi Cells to Shift the Ratio of Effector to Regulatory T Cells in Cancer Patients. Proc Natl Acad Sci U S A (2008) 105(39):14987–92. doi: 10.1073/pnas.0806075105
85. Jiao S, Subudhi SK, Aparicio A, Ge Z, Guan B, Miura Y, et al. Differences in Tumor Microenvironment Dictate T Helper Lineage Polarization and Response to Immune Checkpoint Therapy. Cell (2019) 179(5):1177–90 e13. doi: 10.1016/j.cell.2019.10.029
86. Okamoto M, Hasegawa Y, Hara T, Hashimoto N, Imaizumi K, Shimokata K, et al. T-Helper Type 1/T-Helper Type 2 Balance in Malignant Pleural Effusions Compared to Tuberculous Pleural Effusions. Chest (2005) 128(6):4030–5. doi: 10.1378/chest.128.6.4030
87. Oshikawa K, Yanagisawa K, Ohno S, Tominaga S, Sugiyama Y. Expression of ST2 in Helper T Lymphocytes of Malignant Pleural Effusions. Am J Respir Crit Care Med (2002) 165(7):1005–9. doi: 10.1164/ajrccm.165.7.2105109
88. Heymann D, L’Her E, Nguyen J-M, Raher S, Canfrère I, Coupey L, et al. Leukaemia Inhibitory Factor (Lif) Production in Pleural Effusions: Comparison With Production of IL-4, Il-8, IL-10 and Macrophage-Colony Stimulating Factor (M-Csf). Cytokine (1996) 8(5):410–6. doi: 10.1006/cyto.1996.0056
89. DeLong P, Carroll RG, Henry AC, Tanaka T, Ahmad S, Leibowitz MS, et al. Regulatory T Cells and Cytokines in Malignant Pleural Effusions Secondary to Mesothelioma and Carcinoma. Cancer Biol Ther (2005) 4(3):342–6. doi: 10.4161/cbt.4.3.1644
90. Zou W, Restifo NP. T(H)17 Cells in Tumour Immunity and Immunotherapy. Nat Rev Immunol (2010) 10(4):248–56. doi: 10.1038/nri2742
91. Bailey SR, Nelson MH, Himes RA, Li Z, Mehrotra S, Paulos CM. Th17 Cells in Cancer: The Ultimate Identity Crisis. Front Immunol (2014) 5:276. doi: 10.3389/fimmu.2014.00276
92. Chang SH. T Helper 17 (Th17) Cells and interleukin-17 (Il-17) in Cancer. Arch Pharm Res (2019) 42(7):549–59. doi: 10.1007/s12272-019-01146-9
93. Ye Z-J, Zhou Q, Gu Y-Y, Qin S-M, Ma W-L, Xin J-B, et al. Generation and Differentiation of IL-17–producing Cd4+ T Cells in Malignant Pleural Effusion. J Immunol (2010) 185(10):6348–54. doi: 10.4049/jimmunol.1001728
94. Ye ZJ, Zhou Q, Yin W, Yuan ML, Yang WB, Xiang F, et al. Interleukin 22-Producing Cd4+ T Cells in Malignant Pleural Effusion. Cancer Lett (2012) 326(1):23–32. doi: 10.1016/j.canlet.2012.07.013
95. Ye ZJ, Zhou Q, Yin W, Yuan ML, Yang WB, Xiong XZ, et al. Differentiation and Immune Regulation of IL-9-producing Cd4+ T Cells in Malignant Pleural Effusion. Am J Respir Crit Care Med (2012) 186(11):1168–79. doi: 10.1164/rccm.201207-1307OC
96. He J, Wang L, Zhang C, Shen W, Zhang Y, Liu T, et al. Interleukin-9 Promotes Tumorigenesis Through Augmenting Angiogenesis in non-Small Cell Lung Cancer. Int Immunopharmacol (2019) 75:105766. doi: 10.1016/j.intimp.2019.105766
97. Bu XN, Zhou Q, Zhang JC, Ye ZJ, Tong ZH, Shi HZ. Recruitment and Phenotypic Characteristics of Interleukin 9-Producing Cd4+ T Cells in Malignant Pleural Effusion. Lung (2013) 191(4):385–9. doi: 10.1007/s00408-013-9474-4
98. Hernandez P, Gronke K, Diefenbach A. A Catch-22: Interleukin-22 and Cancer. Eur J Immunol (2018) 48(1):15–31. doi: 10.1002/eji.201747183
99. Zhang W, Chen Y, Wei H, Zheng C, Sun R, Zhang J, et al. Antiapoptotic Activity of Autocrine interleukin-22 and Therapeutic Effects of Interleukin-22-Small Interfering RNA on Human Lung Cancer Xenografts. Clin Cancer Res (2008) 14(20):6432–9. doi: 10.1158/1078-0432.CCR-07-4401
100. Sneddon S, Rive CM, Ma S, Dick IM, Allcock RJN, Brown SD, et al. Identification of a CD8+ T-Cell Response to a Predicted Neoantigen in Malignant Mesothelioma. Oncoimmunology (2020) 9(1):1684713. doi: 10.1080/2162402X.2019.1684713
101. Han J, Duan J, Bai H, Wang Y, Wan R, Wang X, et al. Tcr Repertoire Diversity of Peripheral Pd-1+Cd8+ T Cells Predicts Clinical Outcomes After Immunotherapy in Patients With Non–Small Cell Lung Cancer. Cancer Immunol Res (2020) 8(1):146–54. doi: 10.1158/2326-6066.CIR-19-0398
102. Formenti SC, Rudqvist NP, Golden E, Cooper B, Wennerberg E, Lhuillier C, et al. Radiotherapy Induces Responses of Lung Cancer to CTLA-4 Blockade. Nat Med (2018) 24(12):1845–51. doi: 10.1038/s41591-018-0232-2
103. Duncan SR, Elias DJ, Roglic M, Pekny KW. Theofilopoulos AN. T-Cell Receptor Biases and Clonal Proliferations in Blood and Pleural Effusions of Patients With Lung Cancer. Hum Immunol (1997) 53(1):39–48. doi: 10.1016/S0198-8859(96)00296-0
104. Jang M, Yew PY, Hasegawa K, Ikeda Y, Fujiwara K, Fleming GF, et al. Characterization of T Cell Repertoire of Blood, Tumor, and Ascites in Ovarian Cancer Patients Using Next Generation Sequencing. Oncoimmunology (2015) 4(11):e1030561. doi: 10.1080/2162402X.2015.1030561
105. Ikematsu Y, Tanaka K, Yanagihara T, Liu R, Inoue H, Yoneshima Y, et al. Immune Checkpoint Protein and Cytokine Expression by T Lymphocytes in Pleural Effusion of Cancer Patients Receiving anti-PD-1 Therapy. Lung Cancer (2019) 138:58–64. doi: 10.1016/j.lungcan.2019.10.011
106. Tseng YH, Ho HL, Lai CR, Luo YH, Tseng YC, Whang-Peng J, et al. Pd-L1 Expression of Tumor Cells, Macrophages, and Immune Cells in Non-Small Cell Lung Cancer Patients With Malignant Pleural Effusion. J Thorac Oncol (2018) 13(3):447–53. doi: 10.1016/j.jtho.2017.10.034
107. Koyama S, Akbay EA, Li YY, Herter-Sprie GS, Buczkowski KA, Richards WG, et al. Adaptive Resistance to Therapeutic Pd-1 Blockade is Associated With Upregulation of Alternative Immune Checkpoints. Nat Commun (2016) 7:10501. doi: 10.1038/ncomms10501
108. Guo M, Wu F, Hu G, Chen L, Xu J, Xu P, et al. Autologous Tumor Cell-Derived Microparticle-Based Targeted Chemotherapy in Lung Cancer Patients With Malignant Pleural Effusion. Sci Transl Med (2019) 11(474):eaat5690. doi: 10.1126/scitranslmed.aat5690
109. Zhang L, Dermawan K, Jin M, Liu R, Zheng H, Xu L, et al. Differential Impairment of Regulatory T Cells Rather Than Effector T Cells by Paclitaxel-Based Chemotherapy. Clin Immunol (2008) 129(2):219–29. doi: 10.1016/j.clim.2008.07.013
110. McCoy MJ, Lake RA, van der Most RG, Dick IM, Nowak AK. Post-Chemotherapy T-cell Recovery is a Marker of Improved Survival in Patients With Advanced Thoracic Malignancies. Br J Cancer (2012) 107(7):1107–15. doi: 10.1038/bjc.2012.362
111. Li JY, Duan XF, Wang LP, Xu YJ, Huang L, Zhang TF, et al. Selective Depletion of Regulatory T Cell Subsets by Docetaxel Treatment in Patients With Nonsmall Cell Lung Cancer. J Immunol Res (2014) 2014:286170. doi: 10.1155/2014/286170
112. Mazzaschi G, Facchinetti F, Missale G, Canetti D, Madeddu D, Zecca A, et al. The Circulating Pool of Functionally Competent NK and CD8+ Cells Predicts the Outcome of Anti-PD1 Treatment in Advanced Nsclc. Lung Cancer (2019) 127:153–63. doi: 10.1016/j.lungcan.2018.11.038
113. Mazzaschi G, Madeddu D, Falco A, Bocchialini G, Goldoni M, Sogni F, et al. Low PD-1 Expression in Cytotoxic Cd8(+) Tumor-Infiltrating Lymphocytes Confers an Immune-Privileged Tissue Microenvironment in NSCLC With a Prognostic and Predictive Value. Clin Cancer Res (2018) 24(2):407–19. doi: 10.1158/1078-0432.CCR-17-2156
114. Prat A, Navarro A, Pare L, Reguart N, Galvan P, Pascual T, et al. Immune-Related Gene Expression Profiling After PD-1 Blockade in Non-Small Cell Lung Carcinoma, Head and Neck Squamous Cell Carcinoma, and Melanoma. Cancer Res (2017) 77(13):3540–50. doi: 10.1158/0008-5472.CAN-16-3556
115. Wu SP, Liao RQ, Tu HY, Wang WJ, Dong ZY, Huang SM, et al. Stromal PD-L1-Positive Regulatory T Cells and PD-1-Positive Cd8-Positive T Cells Define the Response of Different Subsets of Non-Small Cell Lung Cancer to PD-1/PD-L1 Blockade Immunotherapy. J Thorac Oncol (2018) 13(4):521–32. doi: 10.1016/j.jtho.2017.11.132
116. Huang AC, Postow MA, Orlowski RJ, Mick R, Bengsch B, Manne S, et al. T-Cell Invigoration to Tumour Burden Ratio Associated With anti-PD-1 Response. Nature (2017) 545(7652):60–5. doi: 10.1038/nature22079
117. Kim K, Park S, Park SY, Kim G, Park SM, Cho JW, et al. Single-Cell Transcriptome Analysis Reveals TOX as a Promoting Factor for T Cell Exhaustion and a Predictor for Anti-PD-1 Responses in Human Cancer. Genome Med (2020) 12(1):22. doi: 10.1186/s13073-020-00722-9
118. Julia EP, Mando P, Rizzo MM, Cueto GR, Tsou F, Luca R, et al. Peripheral Changes in Immune Cell Populations and Soluble Mediators After Anti-PD-1 Therapy in non-Small Cell Lung Cancer and Renal Cell Carcinoma Patients. Cancer Immunol Immunother (2019) 68(10):1585–96. doi: 10.1007/s00262-019-02391-z
119. Siddiqui I, Schaeuble K, Chennupati V, Fuertes Marraco SA, Calderon-Copete S, Pais Ferreira D, et al. Intratumoral Tcf1(+)Pd-1(+)Cd8(+) T Cells With Stem-Like Properties Promote Tumor Control in Response to Vaccination and Checkpoint Blockade Immunotherapy. Immunity (2019) 50(1):195–211.e10. doi: 10.1016/j.immuni.2018.12.021
120. Abouzgheib W, Bartter T, Dagher H, Pratter M, Klump W. A Prospective Study of the Volume of Pleural Fluid Required for Accurate Diagnosis of Malignant Pleural Effusion. Chest (2009) 135(4):999–1001. doi: 10.1378/chest.08-2002
121. Carter J, Miller JA, Feller-Kopman D, Ettinger D, Sidransky D, Maleki Z. Molecular Profiling of Malignant Pleural Effusion in Metastatic non-Small-Cell Lung Carcinoma. The Effect of Preanalytical Factors. Ann Am Thorac Soc (2017) 14(7):1169–76. doi: 10.1513/AnnalsATS.201609-709OC
122. Biton J, Ouakrim H, Dechartres A, Alifano M, Mansuet-Lupo A, Si H, et al. Impaired Tumor-Infiltrating T Cells in Patients With Chronic Obstructive Pulmonary Disease Impact Lung Cancer Response to PD-1 Blockade. Am J Respir Crit Care Med (2018) 198(7):928–40. doi: 10.1164/rccm.201706-1110OC
123. Qama D, Choi WI, Kwon KY. Immune Responses in the Lungs of Patients With Tuberculous Pleural Effusion Without Pulmonary Tuberculosis. BMC Immunol (2012) 13:45. doi: 10.1186/1471-2172-13-45
124. Mateu-Jimenez M, Curull V, Pijuan L, Sanchez-Font A, Rivera-Ramos H, Rodriguez-Fuster A, et al. Systemic and Tumor Th1 and Th2 Inflammatory Profile and Macrophages in Lung Cancer: Influence of Underlying Chronic Respiratory Disease. J Thorac Oncol (2017) 12(2):235–48. doi: 10.1016/j.jtho.2016.09.137
Keywords: malignant pleural effusions (MPE), T cells, immune checkpoint therapy, checkpoint receptors, memory T cells
Citation: Principe N, Kidman J, Lake RA, Lesterhuis WJ, Nowak AK, McDonnell AM and Chee J (2021) Malignant Pleural Effusions—A Window Into Local Anti-Tumor T Cell Immunity? Front. Oncol. 11:672747. doi: 10.3389/fonc.2021.672747
Received: 26 February 2021; Accepted: 07 April 2021;
Published: 27 April 2021.
Edited by:
Emanuela Felley-Bosco, University of Zurich, SwitzerlandReviewed by:
Evelien Smits, University of Antwerp, BelgiumCopyright © 2021 Principe, Kidman, Lake, Lesterhuis, Nowak, McDonnell and Chee. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jonathan Chee, am9uYXRoYW4uY2hlZUB1d2EuZWR1LmF1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.