 Jan P. Bogen1,2*
Jan P. Bogen1,2* Julius Grzeschik
Julius Grzeschik Harald Kolmar
Harald Kolmar
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 27 May 2021
Sec. Genitourinary Oncology
Volume 11 - 2021 | https://doi.org/10.3389/fonc.2021.672262
Bladder cancer is a frequent malignancy and has a clinical need for new therapeutic approaches. Antibody and protein technologies came a long way in recent years and new engineering approaches were applied to generate innovative therapeutic entities with novel mechanisms of action. Furthermore, mRNA-based pharmaceuticals recently reached the market and CAR-T cells and viral-based gene therapy remain a major focus of biomedical research. This review focuses on the engineering of biologics, particularly therapeutic antibodies and their application in preclinical development and clinical trials, as well as approved monoclonal antibodies for the treatment of bladder cancer. Besides, newly emerging entities in the realm of bladder cancer like mRNA, gene therapy or cell-based therapeutics are discussed and evaluated. As many discussed molecules exhibit unique mechanisms of action based on innovative protein engineering, they reflect the next generation of cancer drugs. This review will shed light on the engineering strategies applied to develop these next generation treatments and provides deeper insights into their preclinical profiles, clinical stages, and ongoing trials. Furthermore, the distribution and expression of the targeted antigens and the intended mechanisms of action are elucidated.
The genitourinary system encompasses reproductive organs and the urinary system. The latter comprises the kidneys, which are being connected to the bladder in the lower pelvis via the ureters. The bladder is a hollow organ, which releases upon muscle contraction urine via the urethra. Bladder cancer (BC) is the second most common genitourinary malignant disease and the 10th most common cancer, causing nearly 570.000 new cases and 210.000 deaths worldwide each year (1, 2). It typically affects older adults, and peak incidence occurs in the seventh and eighth decades of life. Males are four times more likely to develop bladder cancers. Hence BC is the sixth most common cancer in males and the ninth leading cause of cancer-related deaths (2).
Bladder cancer is categorized by cell type and by stage of tissue invasion. Besides urothelial carcinoma/transitional cell carcinoma (UC/TCC), which is by far the most common type of bladder cancer (90-95%), other malignancies as squamous cell carcinoma (2-5%), adenocarcinoma (0.5–2%), and small-cell carcinoma (<1%) all describe tumors of the bladder (3). Even though UC/TCC predominantly occurs in the bladder, the ureters and the urethra can be affected as well.
In general, UCs are classified as either non-muscle-invasive bladder cancer (NMIBC), which accounts for approximately 75% of cases, or muscle-invasive bladder cancer (MIBC), which is found in approximately 25% of patients (4, 5). Both are further staged using the Tumor, Node, Metastasis (TNM) classification. This describes how far a tumor has grown (T), whether the cancer has spread to nearby lymph nodes (N) and whether it has started to metastasize (M). Ta describes a non-invasive papillary carcinoma confined to the mucosa growing from the inner lining (urothelium) into the lumen of the bladder (Figure 1). Tumors growing flat on the urothelium, not invading the connective tissue, and not extending into the lumen are classified as carcinoma in situ (CIS), known as Cis/Tis. Papillary tumors invading the lamina propria and the connective tissue are classified as T1. Ta, Tis, and T1 are classified as NMIBC, while stages ≥ T2 are classified as MIBC. In T2a or T2b, the cancer has invaded the inner or the outer detrusor muscle, and at T3, the tumor has invaded beyond the muscle layer and the surrounding fat tissue. At T4, the tumor has begun to grow through the bladder wall into the pelvic or abdominal wall, invading adjacent organs and/or started to spread to nearby lymph nodes or even to more distant organs (metastasis, M1) (6).
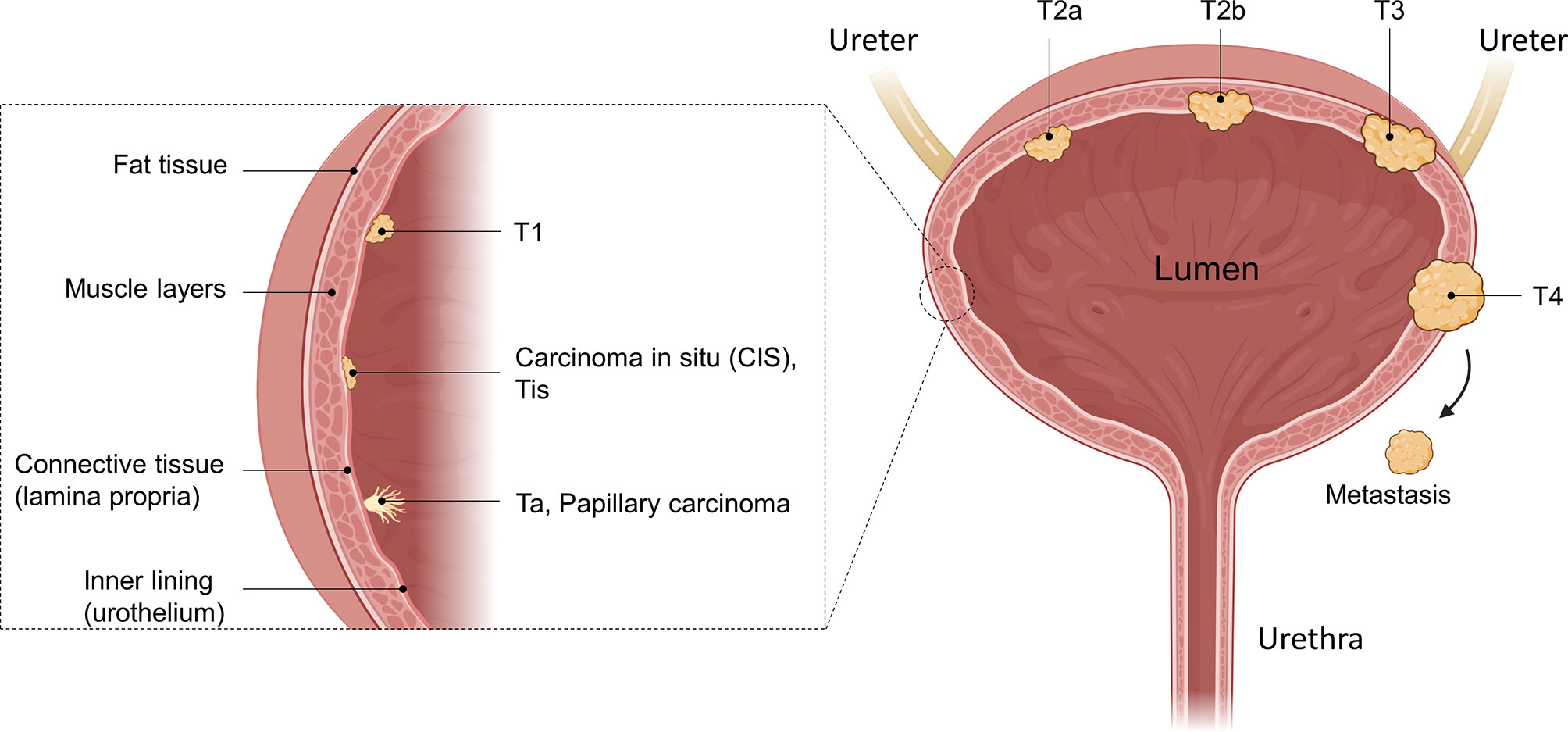
Figure 1 Anatomical illustration of different bladder cancer stages. The bladder as a whole, as well as a zoomed-in illustration of the bladder wall is shown. Different tumors in different tumor stages are illustrated as well as their invasion into the muscle tissue. While Ta, Tis, and T1 are defined as non-muscle-invasive, muscle-invasive bladder cancer (MIBC) disease begins with T2. After penetrating the muscle and reaching the fatty tissue, the T3 stage is reached, where T4 is characterized as growth of the tumor into other organs or metastasizing. Created with BioRender.com.
This staging system was created by the American Joint Committee on Cancer (AJCC) and the International Union Against Cancer (UICC). Histological grading follows the 2004 World Health Organization (WHO) grading system for flat and papillary lesions (4). The system was an upgrade to the previous system from 1973, which uses grades 1, 2, and 3 and may still be used. In 2016 the WHO further improved the classification system to the most recent version (7). Today tumors can be classified as low grade or high grade. While the previously defined grade 2 can be classified as low or high grade, CIS belongs to high grade.
Today, the diagnosis of bladder cancer is performed more and more utilizing molecular analysis (8, 9). Genetic profiling allows for the subdivision of tumors into the basal and luminal subtype, which has a distinct impact on the success of chemotherapeutic treatment (10–12). Following diagnosis and tumor staging, NMIBC tumors are stratified by risk of recurrence and/or progression: While low-grade tumors consist of slowly growing, more differentiated cells, high-grade tumors are poorly differentiated and tend to spread faster. Furthermore, low-grade tumors have a lower risk of recurrence, while high-grade tumors have a higher risk of progression and metastasizing (6). Physicians have multiple options for diagnosing of UCs. However, cystoscopy is considered the key diagnostic tool for bladder cancer. To evaluate the cell type and stage of tissue invasion, a Transurethral Resection of the Bladder (TURB) is performed to confirm the diagnosis and stage the patient correctly.
All papillary tumors are resected completely during the TURB, which in some cases can be considered a cure. CIS cannot be resected completely during the TURB and thus need additional instillation therapy to reduce tumor burden, recurrence, and/or progression (4). For high and intermediate risk NMIBC patients, there is evidence of significantly reducing recurrence rate by additional instillation therapy to the bladder of either a chemotherapeutic agent, like mitomycin c, or Bacillus Calmette Guérin (BCG). Single instillation (SI) of mitomycin c (postoperative), a potent chemotherapeutic agent which crosslinks DNA, has been shown to reduce the 5-year recurrence rate by 14% (59%-45%) (13). For most NMIBC patients, intravesical immunotherapy utilizing BCG is the standard of care (14, 15). BCG is a non-pathogenic bacillus derived from Mycobacterium bovis and represents the only available vaccine for tuberculosis (16). Most of the time, BCG treatment starts after TURB, aiming at preventing tumor recurrence (17).
Although failure after BCG instillation therapy is observed in 30% to 40% of cases (18, 19), patients who experience early failure are defined as having BCG unresponsive NMIBC disease (20, 21). These patients are very unlikely to benefit from further BCG therapy and are recommended radical cystectomy, which describes the complete removal of the bladder. The primary treatment for patients with MIBC depends on the M (metastasis) status. For localized MIBC (M0), the primary and standard treatment is the complete removal of the bladder and creation of a urinary diversion (radical cystectomy) +/- neoadjuvant therapy. In very select cases, a bladder-sparing approach can be adopted. Controversies exist regarding age and type of diversion (5). Non-eligible patients can be offered radiotherapy. 10-15% of patients with muscle-invasive disease are already metastatic at diagnosis (22). As first-line treatment in advanced stages, platinum-based chemotherapeutics, like cisplatin, are utilized, which interfere with DNA replication in fast-growing cells (23). Furthermore, intravenous application of the nucleoside-analog gemcitabine, structurally related to cytidine, and instillation of mitomycin c are chemotherapeutic treatment options (24). Even though bladder cancer is relatively chemosensitive, reoccurrence after first-line treatment with the chemotherapeutics gemcitabine plus cisplatin results in poor prognosis (25). Resistance mechanisms include the elevated drug efflux as well as reduced influx. Furthermore, increased repair of the DNA leads to drug resistance. Multiple enzymes and repair mechanisms, including ERCC1, PARP, Nrf2, CTR1, are involved in the development of drug resistance (26). A detailed review on resistance of bladder cancer against conventional therapies can be found elsewhere (26, 27). If treatment fails, it might become necessary to perform cystectomy, where the bladder is partly or completely removed (28). As this is a radical measure with a significant impact on the quality of life for respective patients, new treatment options are necessary, allowing for a more precise treatment and circumventing side effects that are observed in classical chemotherapies.
Immunotherapy approaches revolutionized cancer therapy in the last decades. It is based on the concept of activating the patients’ immune system to eradicate the malignant cells. Even though a successful biotherapy applied for approximately 40 years, the anti-tumor mechanisms associated with BCG are not fully understood (29). Nevertheless, BCG-mediated effects are presumably based on the involvement of CD4+ and CD8+ lymphocytes, natural killer (NK) cells, and granulocytes, as well as a variety of cytokines (29). As BCG-failure is observed in 30% to 40% of NMIBC patients (18, 19), there is a need for new immunotherapy-based drugs. Different engineering strategies and antibody formats were investigated in (pre-)clinical settings over the last decades to employ new mechanism of actions. On the one hand, novel approaches based on antibody engineering like monoclonal antibodies (mAbs), antibody-drug-conjugates (ADCs), and bispecific molecules have been explored. On the other hand, gene therapeutics, mRNA- and cell-based therapies are emerging as promising tools to treat urologic cancers. Here we review those engineering strategies, illuminate the mechanism of action, and discuss clinical trials (Table 1).
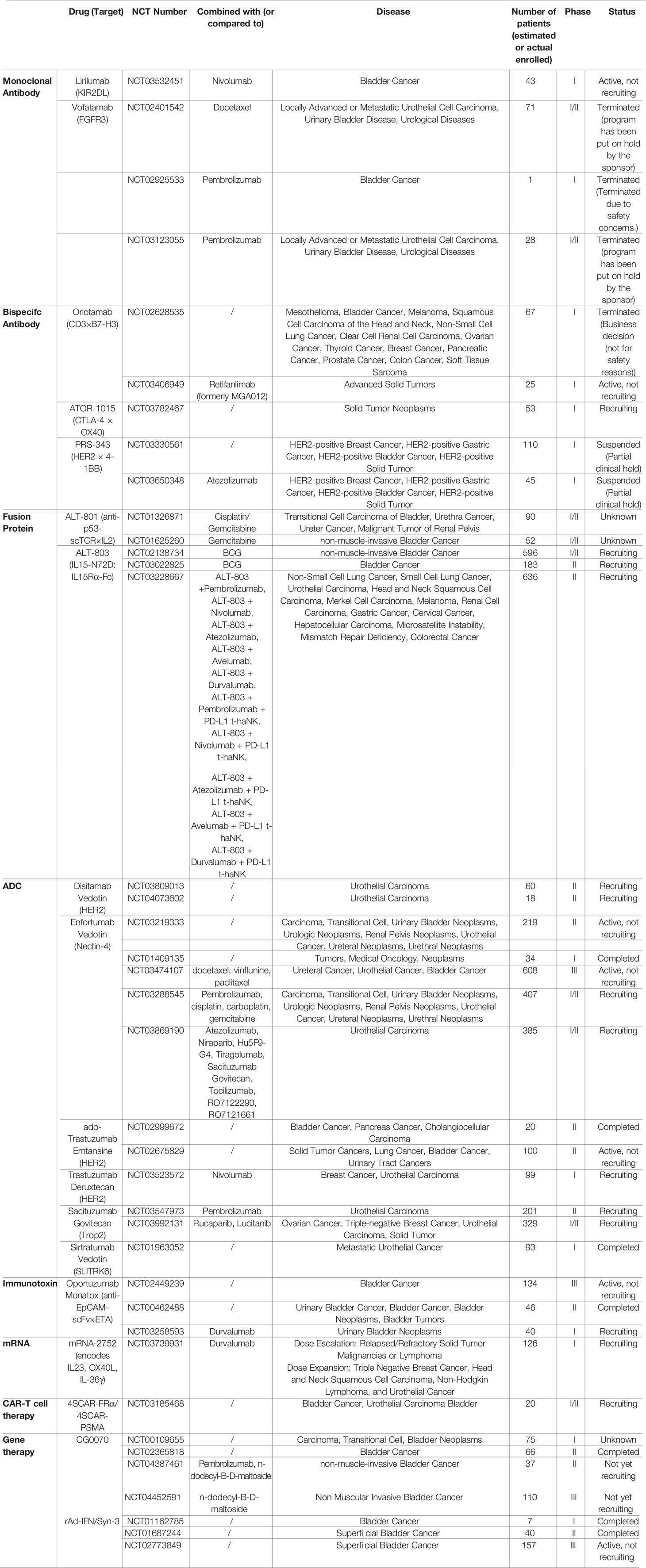
Table 1 Overview of clinical trials targeting bladder cancer with engineered biologics.
With over 90 mAbs approved for a diverse set of diseases like cancer, infectious and autoimmune diseases, monoclonal antibodies are essential in modern medical therapy options (30). While some rely on Fc-mediated effector functions like antibody-dependent cellular cytotoxicity (ADCC) and complement-dependent cytotoxicity (CDC), others achieve clinical benefit through blocking the interaction between the tumor cell and the immune system, like checkpoint inhibitors.
Over the last years, immune checkpoint inhibition showed a massive impact on cancer therapies. Particularly antibodies blocking the interaction of the programmed cell death protein 1 (PD-1) and its ligand (PD-L1) showed impressive outcomes in clinical applications (31), resulting in the approval of six anti-PD-(L)1 antibodies (Pembrolizumab, Nivolumab, Cemiplimab, Durvalumab, Avelumab, Atezolizumab). Today, except Cemiplimab, all these anti-PD-(L)1 antibodies are in clinical use for UCs (32, 33).
Studies investigating the expression of PD-1 and PD-L1 in UC samples showed an elevated expression level of PD-(L)1 in high-risk tumors compared to low-risk tumors (34). Immunohistochemistry (IHC) studies on surgically resected urothelial cancer specimens found PD-L1 expression in 21.1% of cases (35). In patients failing BCG-treatment, PD-L1 expression correlated with higher tumor grades. While 7% of pTa tumors expressed PD-L1, 30% of pT3/4 and 45% of CIS tumors exhibited PD-L1 expression. The frequent observation that BCG treatment strongly induces PD-L1 expression might be a reason for the lost effectiveness of BCG treatment over time (36). Furthermore, PD-L1 expression was associated with tumor infiltration of mononuclear cells (36). CD4+ and CD8+ tumor-infiltrating lymphocytes (TILs) were analyzed by flow cytometry for the expression of PD-1. In most patients, PD-1 expression on TILs was highly upregulated compared to peripheral blood lymphocytes (35). While in one study no correlation in PD-(L)1 expression on overall survival was observed (34), another study found that PD-L1 is a significant prognostic factor for postoperative recurrence and survival (35).
After receiving breakthrough therapy designation, priority review status, and accelerated approval, the anti-PD-L1 mAb Atezolizumab (Tecentriq) was the first approved checkpoint inhibitor for MIBC in 2016 (37). In the following year, the Food and Drug Administration (FDA) granted approval for the PD-1 specific mAbs Nivolumab (Opdivo) (37) and Pembrolizumab (Keytruda) (38) as well as for the PD-L1 targeting antibodies Avelumab (Bavencio) (37, 38) and Durvalumab (Imfinzi) (38, 39) for locally advanced or metastatic UC showing tumor progression during or after adjuvant therapy utilizing platinum-containing chemotherapy or neoadjuvant treatment. Today, PD-1/PD-L1 specific antibodies became the standard of care as a second-line treatment for bladder cancer patients.
At the beginning of 2020, Pembrolizumab received approval for the treatment of BCG-unresponsive NMIBC patients unwilling or unfit for cystectomy with CIS with or without papillary tumors after being granted priority review. Today, Pembrolizumab is the only antibody approved for the treatment of NMIBC.
While Atezolizumab, Avelumab, and Durvalumab are IgG1 antibodies, Nivolumab and Pembrolizumab belong to the IgG4 isotype (37, 38). Given the number of approved PD-1/PD-L1 specific mAbs for UCs and their impact in other oncologic areas, checkpoint inhibitors have become an essential tool for the treatment of bladder cancer. Their mechanism of action is depicted in Figure 2A.
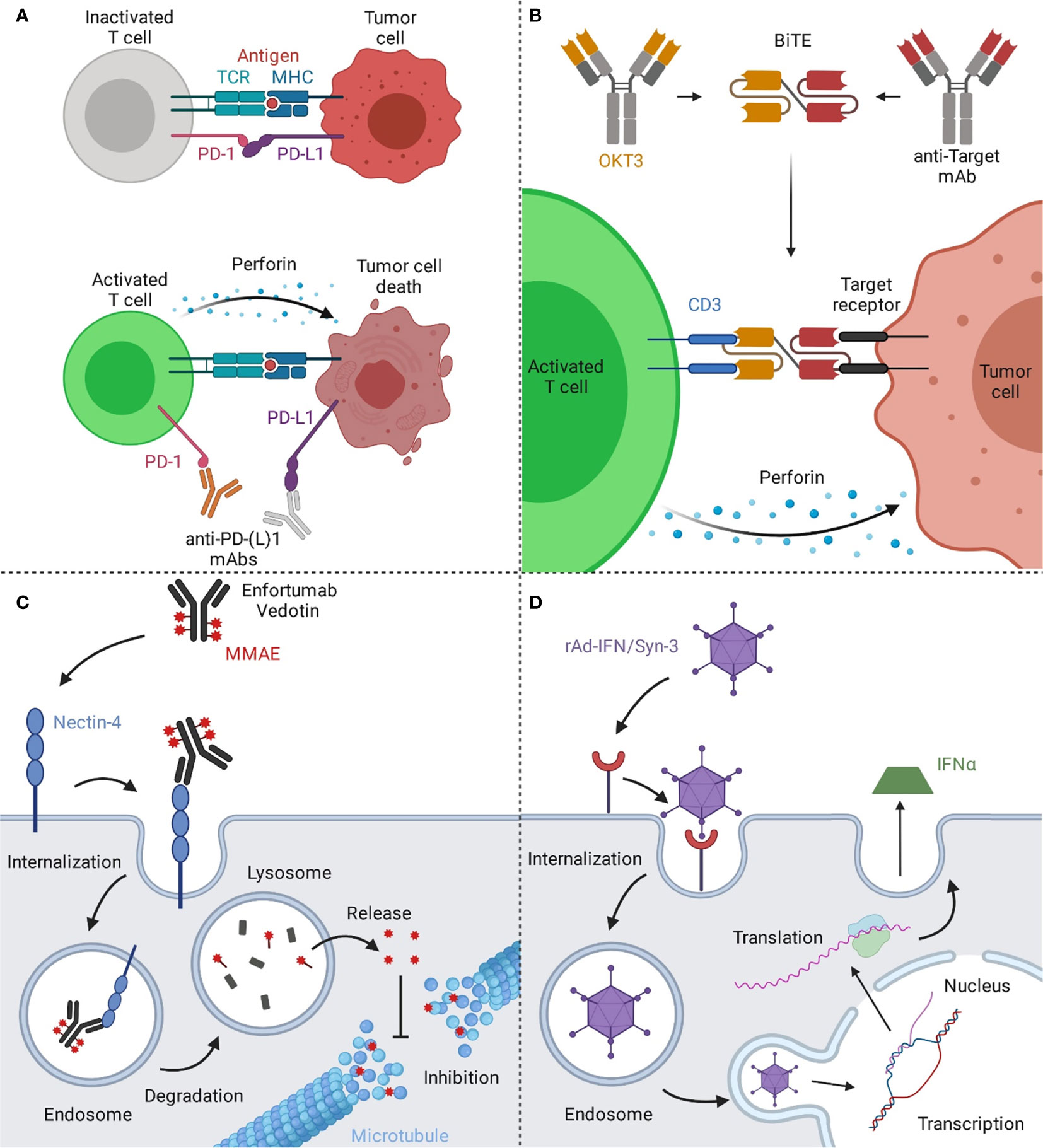
Figure 2 Mechanisms of action of immune and viral therapies for bladder cancer. (A) T cells can recognize cancer cells via the interaction of their T cell receptor (TCR) with the major histocompatibility complex (MHC) on the target cell, resulting in cytotoxic activity eventually leading to cell death of the malignant cells. However, a prominent escape mechanism of cancer is the upregulation of checkpoint inhibitors like PD-L1, inhibiting cancer-specific T cells from attacking the malignant cells. By blocking the interaction of PD-1 on T cells with its ligand PD-L1 on the tumor cell, T cells are reactivated and can effectively target the tumor. (B) Bispecific T cell engagers (BiTEs) consist of two binding arms, simultaneously binding to a tumor-associated antigen on the tumor cell and CD3, a part of the TCR complex, on T cells, generating a synthetic immunological synapse. This results in T cell activation, eventually leading to the induction of a cytotoxic effect against the targeted tumor cell. (C) Upon binding of Enfortumab Vedotin to nectin-4, the antibody-drug-conjugate (ADC) gets subsequently internalized into the endosome. Next, the complex is transported to the lysosome, where the ADC is degraded. The valine-citrulline linker is cleaved by cathepsin B resulting in the release of monomethyl auristatin E (MMAE) and the subsequent inhibition of the microtubule, eventually leading to cell death. (D) The rAd-IFN/Syn-3 adenovirus is internalized into cells of the bladder after prior instillation. The genetic information of the virus is translocated into the nucleus and upon transduction, these cells produce and secrete IFNα, resulting in an anti-tumor response by activating nearby immune cells. Created with BioRender.com.
Human natural killer (NK) cell activity is modulated by killer cell immunoglobulin-like receptors (KIR) (40). KIRs are classified as activating and inhibitory (41) and recognize predominantly HLA class I as their ligand (42). Blockage of the inhibitory KIR2DL1 has been shown to promote ADCC in tumor models (43).
In 2009, Romagné et al. immunized transgenic mice (44) with BW5417 thymoma cells stably transfected with KIR2DL1, with subsequent booster immunizations utilizing soluble KIR2DL3 (45). Screening of the resulting antibodies resulted in the isolation of the IgG4 antibody IPH2101 (1-7F9), which effectively antagonized KIR signaling by blocking the interaction of HLA class I to inhibitory KIR2DLs. IPH2101 (1-7F9) showed enhanced NK-cell mediated cytotoxicity in vitro and in vivo (45). An antibody variant with stabilized hinge region termed IPH2102 (Lirilumab) is currently in a phase I study aimed at evaluating its effect in combination with Nivolumab in bladder cancer patients (NCT03532451).
In 2018, Chen and coworkers generated murine monoclonal antibodies by immunizing mice with the bladder cancer cell line EJ. Subsequently, hybridomas were generated, and resulting antibodies were verified for binding to the tumor cells, resulting in the isolation of KMP1. By affinity chromatography and mass spectrometry, CD44 was identified as the antigen of KMP1. IHC studies with patient-derived biopsies showed that patients exhibiting the epitope recognized by KMP1 had a worse prognosis compared to those with a weak KMP1-staining. Furthermore, KMP1 mediated inhibition of proliferation, migration, and adhesion in EJ cells in vitro and inhibited tumor growth of EJ-derived tumors in xenografts (46).
CD44 emerged as a promising bladder cancer target as its expression correlates with a higher aggressiveness of the tumor defined by a higher invasion ability compared to CD44 negative cells in vitro. Interestingly, it was shown that Interleukin-6 (IL-6) signaling facilitates a favorable microenvironment for CD44 expression (47). Furthermore, CD44 overexpressing tumors exhibited a lower complete response rate and lower survival. Interestingly, in vivo experiments showed that CD44+ tumors were more resistant to irradiation, both for immunocompromised and immunocompetent hosts (48).
Vofatamab is a clinical-stage mAb targeting the fibroblast growth factor receptor 3 (FGFR3). FGFR3 signaling has been linked to development, differentiation, growth, and survival (49). In patients suffering from bladder cancer, 13 different activating missense mutations have been identified, with three alternations, R248C, S249C, and Y375C, accounting for over 85% of all observed mutations (50). The cysteine residues generated by the mutations presumably lead to ligand-independent receptor dimerization and subsequent activation (51). Furthermore, FGFR3-fusion proteins were observed, where the complete FGFR3 sequence except the final exon was C-terminally fused with the sequences of TACC3 (52, 53) or BAIAP2L1 (54), resulting in a phenotype associated with higher grades (50, 55).
One study on FGFR3 in the context of NMIBC found that 32% of T1 tumors exhibit FGFR3 mutation variants, and 96% of those are overexpressed. In tumors exhibiting a wild-type FGFR3 gene (68%), overexpression was found in 47% of cases. Overall, 63% of the study population exhibited overexpression of FGFR3 (56). Consistently, another study showed that 85% of tumors exhibiting mutations within FGFR3 showed overexpression while being low-grade. Furthermore, it seems that higher FGFR3 expression is elevated in more differentiated tumors (57). This is in line with the finding that higher FGFR3 expression was found in pTa stage tumors compared to pT1 tumors and is therefore associated with lower-grade (57, 58) as well as a favorable prognosis. In contrast, wtFGFR3 expression did not show any influence on clinical parameters (56). Only 42% of tumors exhibiting wtFGFR3 showed overexpression, where 66% were high-grade, and 68% invasive (58).
In 2009, Qing and coworkers isolated an anti-FGFR3 monoclonal antibody, termed R3mab via phage display, which blocked the ligand binding as well as receptor dimerization (59). Furthermore, R3mab was able to bind not only wtFGFR3 but also the most common FGFR3 mutants and inhibited proliferation as well as FGFR3 signaling in bladder cancer cells, exhibiting wild type and mutated variants of FGFR3. In subsequent in vivo studies utilizing xenografts of either WT or mutated FGFR3 cells, R3mab revealed remarkable anti-tumor effects. In 2015 R3mab, now termed as Vofatamab, was tested in a phase I/II dose-escalation study alone or in combination with docetaxel for metastatic urothelial cell carcinoma (NCT02401542). 20% of enrolled patients experienced serious adverse events when treated with Vofatamab as a monotherapy and over 50% when treated in combination with docetaxel, resulting in the termination of the study. Furthermore, a phase I study in 2016 combined with Pembrolizumab (NCT02925533) was conducted, but terminated as well due to safety concerns. A phase I/II study in 2017 for locally advanced or metastatic urothelial cell carcinoma (NCT03123055) was terminated since over 46% of patients experienced serious adverse events.
Over recent years, bispecific antibodies (bsAb) have gained interest of pharmaceutical and academic research. The dual targeting binding properties can mediate mechanisms of action, being impossible to accomplish with their monospecific counterparts or a combination of multiple standard IgGs (60). To this day, only three bispecific antibodies were approved. One is the by now withdrawn Catumaxomab for the treatment of ascites, the second one is Blinatumomab, which is approved for the treatment of acute lymphatic leukemia (ALL), and Emicizumab, approved for hemophilia A treatment, is the third. However, a variety of bsAbs is in (pre-)clinical development for the treatment of different indications, including bladder cancer, exhibiting a diverse spectrum of antibody formats.
T-cell engagers are bispecific molecules that bridge cytotoxic T cells to tumor cells leading to the generation of a synthetic immunologic synapse. This results in cytotoxic activity on tumor cells mediated by T-cells. Commonly, CD3-specific antibodies are utilized as T cell binding moieties (61). The mechanism of action of T cell engagers is depicted in Figure 2B. The molecular structure T cell engagers, as well as of all other discussed molecules, is illustrated in Figure 3.
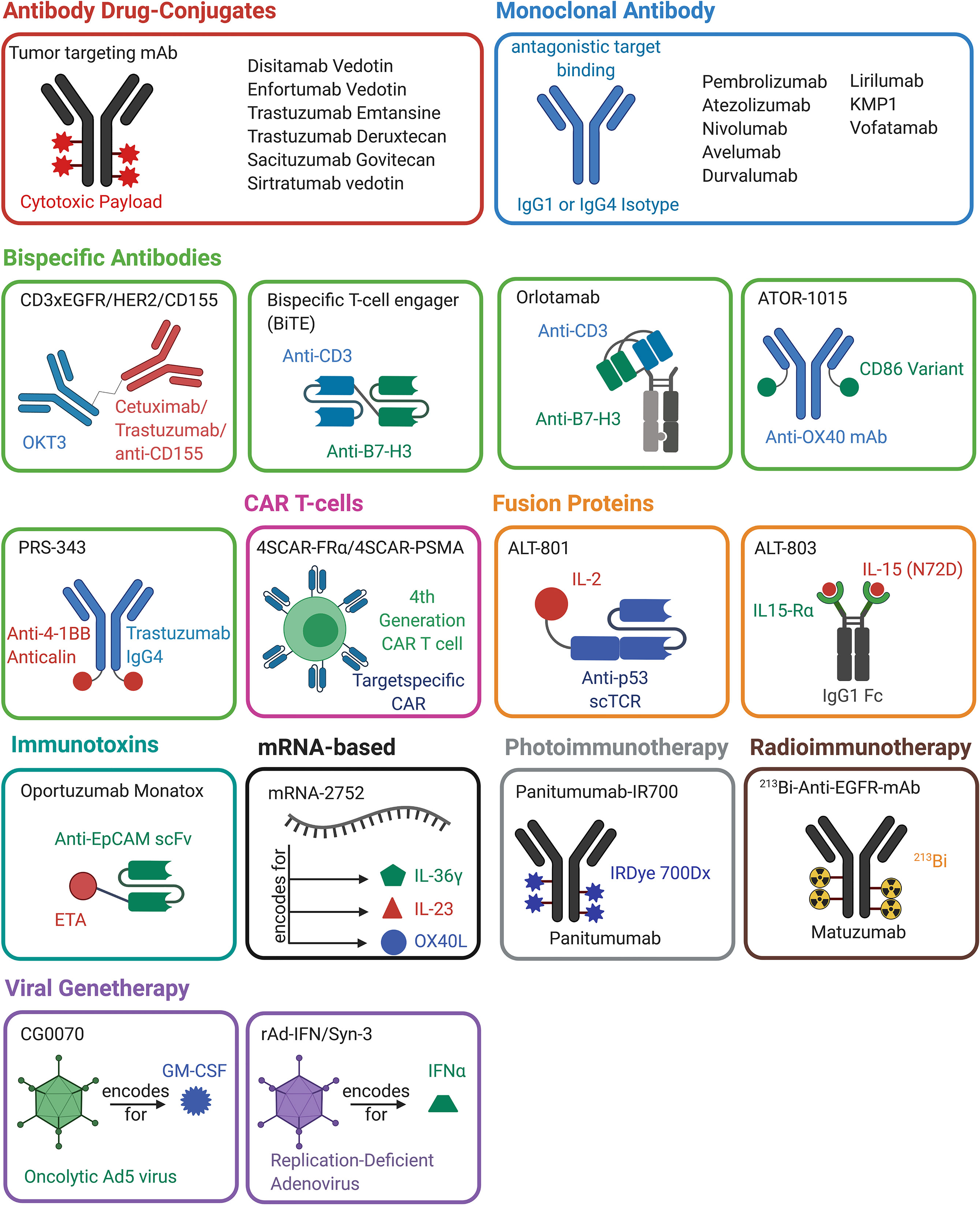
Figure 3 Overview of antibody-, protein-, mRNA-, cell- and viral-based drugs that are approved or are in (pre-)clinical development for the treatment of bladder cancer. The molecules belonging to one drug class are marked in the same color. A schematic representation of the molecular architecture is given. Created with BioRender.com.
In 2018 Juan Ma and coworkers generated CD3×EGFR and CD3×HER2 bispecific antibodies by reacting the anti-epidermal growth factor receptor (EGFR) mAb Cetuximab or the anti-HER2 mAb Trastuzumab, respectively, with sulfo-succinimidyl 4-(N-maleimidomethyl)cyclohexane-1-carboxylate (sulfo-SMCC), followed by conjugation with OKT3 after activation with Traut’s reagent (62). The mouse-derived OKT3 mAb recognizes the CD3ϵ domain in the T cell receptor (TCR) complex and is commonly used for the construction of T cell engagers, as e.g., Blinatumomab (63). By analyzing human-derived active T cells, it could be demonstrated that the bispecific constructs exhibited a stronger cytotoxic activity against bladder cancer cells and mediated secretion of the activation markers Interferon gamma (IFN-γ), tumor necrosis factor alpha (TNF-α), and IL-2 (62). The following year, the same group utilized this system to generate a CD3×CD155 bispecific antibody (64).
CD155, also known as nectin-like protein 5, belongs to the Ig superfamily and is overexpressed in multiple malignancies, including lung adenocarcinoma, pancreatic, colon cancer as well as in muscle-invasive bladder cancer (65–70).
Peripheral blood mononuclear cells (PBMC), isolated from healthy donors and bladder cancer patients, were activated and subsequently treated with the CD3×CD155 bsAb, resulting in cytotoxic effects against bladder cancer cell lines (64). A major drawback in chemical antibody conjugation strategy lies in the fact that it is not site-specific, and therefore it yields a heterogenous population of bispecific molecules.
Several alternative routes exist to overcome this problem. A frequently used format is based on the genetic fusion of two single chain Fv (scFv) fragments that consist of an antibody variable domain of the heavy and the light chain connected by a linker sequence, each addressing different targets resulting in a bispecific T cell engager (BiTE)-like architecture (Figure 3) (71). In frame of development of bispecific T cell engagers for the treatment of bladder cancer, Li and coworkers generated a tandem BiTE-based CD3×B7-H3 bispecific antibody (72, 73).
B7-H3 is naturally expressed by antigen-presenting cells (APC) and is involved in inhibition of T cells (74–76). However, overexpression can also be found on several cancer cells (77–83), where B7-H3 plays a role in cell migration and invasion (84). Interestingly, B7-H3 exhibits a high expression level in bladder cancer cells. In vitro cytotoxicity studies of this CD3×B7-H3 BiTE, as well as in vivo experiments, revealed a notable cell-killing activity of bladder cancer cells, which was further improved by combination with trametinib, a small molecule MEK inhibitor (72).
The only T cell engaging molecule that was in clinical testing for bladder cancer was Orlotamab/MGD009 (NCT02628535), a humanized Dual Affinity Re-Targeting (DART) protein which comprises a CD3 and a B7-H3 binding moiety (85). DART molecules exhibit a diabody-like architecture with the VH domain of one binder linked to the VL of the second binder and the VH of the second binding moiety attached to the VL of the first. Furthermore, disulfides are utilized to stabilize the construct (60, 86). The DART of MGD009 is linked to an Fc fragment, resulting in a monovalent antibody-like molecule (Figure 3). The Fc part is mutated to reduce effector functions while allowing recyclization via the neonatal Fc receptor to mediate extended in vivo half-life (85). The first clinical testing of Orlotamab/MGD009 against bladder cancer, among others, was terminated due to business decisions and not for safety issues (85) (NCT02628535). However, a phase I study is currently ongoing for the treatment of advanced solid tumors (87) (NCT03406949).
Immune checkpoint inhibitors such as the anti-CTLA-4 mAb Ipilimumab can have significant effects in cancer patients. Kvarnhammar and coworkers developed a bispecific CTLA-4×OX40 bsAb (ATOR-1015) (88). To this end, OX40 binding Fab domains were isolated from an scFv phage display library. For CTLA-4 binding, the V-like domain of the CTLA-4 ligand CD86 was engineered in five residues to achieve a 100-fold increased affinity towards CTLA-4. This engineered CD86 domain was C-terminally fused to the kappa light chain of the anti-OX40 IgG1 molecule (88) (Figure 3). The resulting bispecific antibody bound CTLA-4 with 3.0 nM affinity and blocked the interaction with CD80 and CD86.
The co-stimulatory molecule OX40 (CD134) belongs to the next generation of immune therapeutic targets. CTLA-4 and OX40 are upregulated in tumor-infiltrating, but not peripheral, regulatory T cells (Tregs). ATOR-1015 binds domain 2 on OX40 with an affinity of 1.6 nM and blocks interaction with OX40L and furthermore induced T cell activation in vitro by blocking CTLA-4. By crosslinking OX40 upon bind to either CTLA-4 or to FcγRIIIa (CD16a), ATOR-1015 leads to NK-cell or T cell-mediated depletion of OX40-overexpressing Tregs. Those Tregs can negatively regulate the immune response (89–92), and their depletion can enhance anti-tumor activity (93–95). By targeting both receptors in a bispecific manner, the activity of the bsAb is not only directed to the tumor side, eventually introducing anti-tumor responses, but also may result in a more favorable safety profile and better efficacy compared to standard anti-CTLA4 mAbs (88, 96).
In vivo experiments utilizing the MB49 bladder cancer model showed a reduction in tumor growth and prolonged survival. Furthermore, ATOR-1015 enhanced the effect of anti-PD1 treatment in in vivo models. As ATOR-1015 shows promising results in preclinical cancer models, including colon, pancreas, and bladder cancer, it is currently in phase I clinical trials (NCT03782467).
PRS-343 is a bispecific fusion protein targeting HER2 and 4-1BB (CD137), which is a costimulatory receptor on T cells. Despite 4-1BB being a therapeutic target of major interest, prior clinical studies utilizing the monospecific 4-1BB-binding IgG4 antibody Urelumab were stopped due to hepatoxicity (97). The bispecific antibody PRS-343 has the potential to overcome these side-effects by its tumor-specific mechanism of action. Upon accumulation in HER2-positive tumors and clustering of HER2, 4-1BB is clustered on nearby T cells, resulting in T cell activation and a cytotoxic response against the tumor.
The PRS-343 architecture is based on a Trastuzumab variant and a 4-1BB specific anticalin (98) (Figure 3). Anticalins are engineered variants of tear lipocalin and neutrophil -gelatinase–associated lipocalin (NGAL), where multiple loops are randomized by concerted mutagenesis (98–100). Via phage display, a 4-1BB-binding anticalin, termed J10, was isolated from an anticalin library. This molecule exhibits a 2 nM affinity, shows cross-specific binding to 4-1BB from cynomolgus monkeys, and does not block ligand binding (98). Trastuzumab VH and VL domains were subcloned into an IgG4 format to silence Fc-mediated effector functions. Furthermore, the S228P mutation was implemented, suppressing the naturally occurring Fab-arm exchange found in IgG4 molecules (101). Fab-arm exchange describes the naturally occurring and dynamic dissociation of an IgG4 homo-tetramer into two half-molecules consisting of one heavy and one light chain. This is followed by the reassociation with a second half antibody, leading to an antibody with two different Fab arms (102). This mechanism would lead to antibodies with reduced specificity and therapeutic efficacy and is therefore avoided in PRS-343 (103). Additionally, the F234A and the L235A mutation, which reduce the binding to Fcγ-receptors and further silence effector function, were implemented in PRS-343 (104, 105). Since the distance between effector and target cells is crucial for T cell activation and cytotoxicity (105), the anticalin moiety was N- and C-terminally fused to the heavy and light chains of the engineered Trastuzumab variant utilizing a (G4S)3 linker. While all variants exhibited similar biophysical properties, the C-terminal heavy chain fusion showed the strongest T cell activation.
In vivo experiments demonstrated recruitment of TILs, while lymphocytes in peripheral blood remained unaffected (98). Due to its favorable properties, phase I dose-escalation studies were conducted in patients with HER2-positive cancers, including bladder cancer, either as a monotherapy (NCT03330561) or in combination with Atezolizumab (NCT03650348). Both studies were suspended, but very recently, the clinical hold was lifted by the FDA.
Like bsAbs, fusion proteins can mediate mechanisms of action that are impossible to achieve with a mixture of the parental, monofunctional fusion partners. For example, Etanercept or Belatacept are antibody-derived Fc fusion proteins where the Fab fragment of the antibody is replaced by either the extracellular domain of tumor necrosis factor receptor or CTLA4, respectively. While those molecules are approved for the treatment of rheumatoid arthritis or organ transplant rejection for years (106–108), no fusion proteins are approved for bladder cancer yet, but some have entered clinical development.
In 1995, Theobald and coworkers used transgenic mice to generate cytotoxic T lymphocytes (CTL) targeting the human p53 protein (109). From the population of CTLs the clone no. 5, specific to the p53 peptide 264–272, was isolated by limiting dilution (110). To generate a soluble T-cell receptor, Belmont et al. amplified the Vα and Vβ/Cβ regions by RT-PCR. Subsequently, the C-terminal end of the Vα was fused to the N-terminal end of the Vβ utilizing a (G4S)4 linker. The Cβ region directly fused to Vβ was truncated to lack the final cysteine as well as the transmembrane and cytoplasmic regions. Via a peptide linker (encoding for VNAKTTAPSVYPLAPV), human IL-2 was linked to the single-chain TCR (scTCR) (Figure 3). The construct was produced in mammalian cell culture and the resulting fusion protein bound to p53-peptide-loaded MHC on T2 cells, as well as to the IL-2 receptor. Furthermore, the scTCR-IL-2 construct exhibited similar biologic effects in vitro as recombinant IL-2. It was shown that this construct could bring p53-peptide-loaded MHC-bearing cells in proximity with cells exhibiting the IL-2R. Pharmacokinetic studies showed a half-life in mice of 1.6 to 3h, which is elevated compared to free IL-2, as it has a half-life of 5 min. In vivo experiments using xenografts showed beneficial outcomes of the scTCR-IL-2 construct in comparison to IL-2 alone (111). To reduce the immunogenicity of the construct, the murine Cβ was exchanged to its human counterpart while retaining a promising profile in in vivo experiments (112). In 2011 and 2012, phase I/II studies of NMIBC and bladder cancer were initiated, but the current status is unknown (NCT01326871, NCT01625260).
However, this engineering approach’s versatility was demonstrated as Zhu and coworkers substituted the IL-2 moiety with an IL-15 variant (N72D), which exhibits an improved binding towards the IL-15Rβ, resulting in an scTCR fusion construct exhibiting a super agonistic mechanism of action for the IL-15 pathway (113).
Based on the IL-15 N72D mutein, Han and coworkers constructed in 2011 a fusion protein of the sushi domain of IL-15Rα (aa 1-66) and a C-terminal human IgG1 Fc part. Subsequently, plasmids encoding for IL-15 N72D and IL-15Rα-hFc were co-transfected in Chinese hamster ovary (CHO) cells. IL-15 is poorly produced in mammalian cell culture due to its fast degradation. However, in complex with IL-15Rα, the cytokine is shielded from degradation in the endoplasmic reticulum, resulting in high expression levels of the IL-15 N72D:IL-15Rα-hFc complex. Utilizing ion-exchange chromatography methods allowed for the specific purification of IL-15Rα-hFc molecules fully occupied by two IL-15 N72D variants (114) (Figure 3).
IL-15 stimulates the proliferation of NK cells and T cells and induces the generation of CD8+ cytotoxic lymphocytes. Naturally, both IL-15 and IL-15Rα are expressed on dendritic cells and interact with NK cells and T cells in trans, which express cell-bound IL-2/IL-15Rβ and the common gamma chain (γc), resulting in heterotrimeric receptor formed inside the immunologic synapse (115). ALT-803 can bind to IL-2/-15Rβ and γc on T cells and NK cells and induce the IL-15 pathway leading to stimulation of cell proliferation.
Pharmacokinetic experiments demonstrated that the complex exhibited an in vivo half-life of 18 to 25 h, which is significantly enhanced compared to wtIL-15 (0.64 h). In vivo, the complex mediated proliferation of NK cells and CD8+ T cells in mice (114). As this super agonistic construct can activate NK cells, it was in vitro demonstrated that the ALT-803 is able to modulate ADCC activity of a cancer-specific antibody. Furthermore, it led to the upregulation of activating NK cell receptors, antiapoptotic factors, and factors involved in the NK cytotoxicity while simultaneously downregulating NK cell inhibiting factors (116). Furuya and coworkers investigated the effect of ALT-803 in an orthotopic mouse model, demonstrating that subcutaneous administration of ALT-803 is non-inferior to intravesical treatment of BCG. While subcutaneous administered ALT-803 activated CTLs, NK cells and NKT cells, in combination with intravesical BCG additional immunomodulating cytokines in the serum were detected (117). Additional mouse models demonstrated that ALT-803 increased PD-L1 expression in tumors in vivo and elucidated the safety and efficacy of combination treatment with PD-L1-specific checkpoint blockage, paving the way for clinical combination treatment (118). Currently, ALT-803 is in phase I/II and phase II studies investigating its effect on patients suffering from NMIBC or bladder cancer, respectively, as monotherapy or in combination with BCG (NCT02138734, NCT03022825). Furthermore, a currently recruiting phase II study will investigate the efficacy of ALT-803 in combination with the checkpoint inhibitors Pembrolizumab, Nivolumab, Atezolizumab, Avelumab or Durvalumab. One cohort, in addition to the combination of ALT-803 with one of the checkpoint inhibitors, will be weekly dosed with PD-L1 t-haNK cells (NCT03228667). These cells are derived from NK-92 cells, exhibit the high-affinity CD16 receptor, produce an IL-2 variant, which is retained in the endoplasmic reticulum, and carry an additional chimeric antigen receptor addressing PD-L1 (119).
Antibody-drug-conjugates (ADC) comprise a cancer-targeting monoclonal antibody, which is conjugated to a cytotoxic agent (Figure 3). The specificity of the mAb ensures a selective targeting of tumor cells. Upon antigen-binding, ADCs are internalized by receptor-mediated endocytosis and trafficked to the endosome and ultimately to the lysosome. Depending on the linker utilized, acidic pH or lysosomal proteases mediate linker cleavage, resulting in the release of the toxin and eventually cell death (120, 121). The linkers, the toxins, and the conjugation sites of all clinical-stage ADCs for bladder cancer are illustrated in Figure 4. While coupling efficiency and heterogeneity of the conjugate are common hurdles (122), nine ADCs are approved for several malignancies, including solid tumors such as breast cancer and hematologic malignancies (123–127). Currently, 16 ADCs are in phase II and III studies, with many more in phase I studies and preclinical development (121). With the recent approval of Enfortumab Vedotin (Padcev) and the clinical evaluation of multiple other ADCs, the focus is shifting to urothelial malignancies (128).
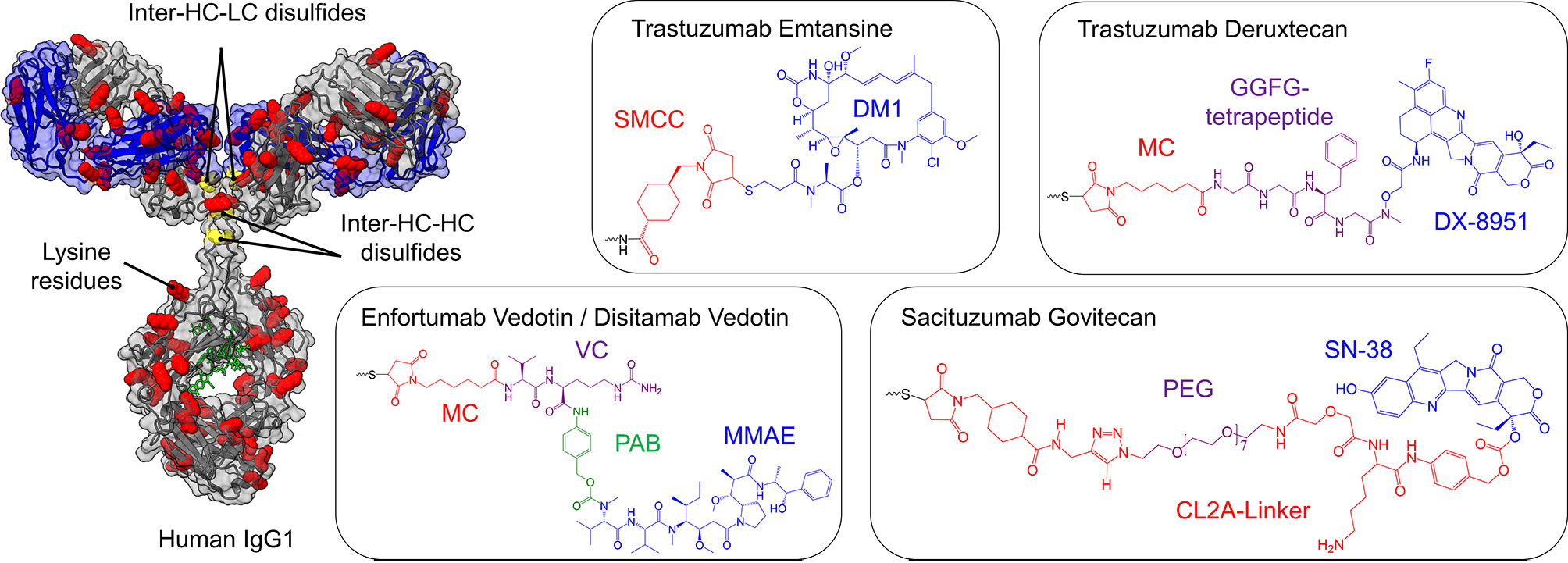
Figure 4 Cytotoxic payloads and conjugation strategies of approved and clinical-stage antibody-drug-conjugates (ADCs) for the treatment of bladder cancer. On the left, a human IgG1 antibody is shown, where cysteine (yellow) and lysine residues (red) are highlighted, the glycosylation is marked in green, while the light chains are depicted in blue. The heavy chain is illustrated in grey. Depending on the conjugation strategy, either cysteine residues or lysine residues are modified with the toxins illustrated on the right side. Except for ado-Trastuzumab Emtansine, all clinical-stage ADCs for bladder cancer are based on the partial reduction of inter-chain disulfide bonds (yellow), resulting in a maximal drug to antibody ratio (DAR) of 8, while simultaneously reducing the number of potential ADC variants. In ado-Trastuzumab Emtansine conjugation is achieved by coupling to lysine residues (red). The respective linkers and toxins of the payloads are color-coded and named separately. SMCC, succinimidyl 4-(N-maleimidomethyl)cyclohexane-1-carboxylate; DM1, maytansine; MC, maleimidocaproyl; PAB, p-aminobenzyl; MMAE, monomethyl auristatin E; PEG, polyethylene glycol.
Two marketed ADCs are based on the monoclonal antibody Trastuzumab targeting the human epidermal growth factor receptor 2 (HER2), a well-studied tumor antigen overexpressed in a variety of malignant diseases, including breast cancer (129, 130) and lung cancer (131, 132). Furthermore, HER2 emerged as a promising therapeutic target for bladder cancer:
Fluorescence in situ hybridization (FISH) and IHC studies of tissue arrays derived from high- and low-grade UCs, as well as papillary urothelial neoplasms, revealed that up to 9% of high-grade UCs exhibit a HER2 gene amplification, which can be associated with higher recurrence and a worse prognosis. Additionally, a subset of high-grade NMIBC patients exhibits a HER2 gene amplification, which can be correlated with aggressive tumor growth (133). Consistently, a silver-enhanced in situ hybridization (SISH) analysis of NMIBC patients showed that 4.2% of the tumors exhibited stronger HER2 staining. All HER2-positive NMIBC samples belonged to the high-grade subset, and HER2-expression is an independent predictor of tumor prognosis (134). Today, multiple anti-HER2-ADCs are in clinical development for the treatment of bladder cancer (128).
In order to generate a novel HER2-specific antibody, Yao and coworkers immunized mice with the extracellular domain (ECD) of HER2, extracted the spleen, and generated hybridomas (135). The clone exhibiting the best binding activity was sequenced and subsequently humanized by CDR grafting. The resulting mAb variant was termed Hertuzumab and showed a higher affinity compared to gold-standard Trastuzumab. By partial reduction of intermolecular disulfide bonds and a subsequent conjugation utilizing a thiol-reactive maleimido-valine-citrulline-dipeptide-linker, monomethyl auristatin E (MMAE) was conjugated, resulting in the ADC RC48 (135). MMAE inhibits tubulin polymerization, resulting in phase arrest and apoptosis (136). The linker is effectively cleaved by human cathepsin B upon internalization in HER2 positive cells but stable in human plasma (137). In vivo studies using xenografts demonstrated a significant and durable regression, underlining the high potency of this ADC (135).
In 2015, a dose-escalating phase I study was conducted in HER2-overexpressing cancer patients, showing a manageable safety profile while exhibiting low liver toxicity (137, 138) (NCT02881190). Currently, RC48, renamed as Disitamab Vedotin, is in phase II studies for locally advanced or metastatic urothelial cancer in HER2 overexpressing (NCT03809013) or HER2-negative patients (NCT04073602) and recently gained breakthrough therapy status.
For the treatment of PD-1/PD-L1 unresponsive patients suffering from locally advanced or metastatic UC, the FDA approved Enfortumab Vedotin in 2018 (139). This was based on a clinical trial including 125 participants (NCT03219333), where 44% of enrolled patients had tumor shrinkage or growth arrest, including 12% showing a complete response, meaning the tumor disappeared entirely (140). This ADC consists of a nectin-4 targeting antibody, conjugated to a microtubule inhibitor and was granted breakthrough therapy designation, priority review status, and accelerated approval.
The cell adhesion molecule (CAM) nectin-4 belongs to the family of nectin-like proteins (Necl), consisting of nectin-1, -2, -3, -4, and -5, which trans-interact with one another (141–143). Besides breast (144), lung (145), and ovarian cancer (146), it is expressed in malignancies of the bladder (147), where it is involved in metastasis and proliferation (145, 148–150).
The parental human IgG1 antibody (AGS-22M6) was generated by utilizing the XenoMouse technology, where transgenic mice are genetically engineered with a humanized humoral immune system (151). Immunization was performed with the extracellular domain (ECD) of human nectin-4, followed by the generation of over 50 hybridomas (152). Besides binding to human nectin-4, AGS-22M6 showed cross-specificity to its monkey and rat orthologues. Using flow cytometry, it was demonstrated that AGS-22M6 bound its human target with a KD value of 10 pM, which was also confirmed for its MMAE-conjugated counterpart. By deletion mutagenesis of nectin-4, it was shown that the epitope of AGS-22M6 is located on the V-domain, enabling inhibition of the heterodimerization with nectin-1 (152). For recombinant expression, VH and VL genes were extracted from the hybridoma and subcloned into expression vectors for production in CHO cells. For ADC generation, the microtubule-disrupting toxin MMAE was conjugated utilizing the cleavable linker maleimidocaproylvaline-citrulline-p- aminobenzyloxycarbonyl (136), which was conjugated by partial reduction of interchain disulfide bond (153, 154), resulting in a drug to antibody ratio (DAR) of approximately 4 (152). In preclinical xenograft models, the ADC showed significant growth inhibition in breast, pancreatic, lung, and bladder cancer models (152), paving the way for its phase I study, which started in 2011 (NCT01409135).
Currently, Enfortumab Vedotin is in further clinical trials investigating its effect on bladder cancer in comparison to chemotherapy (NCT03474107), in combination with checkpoint inhibitors and chemotherapy (NCT03288545), or combined with other immunotherapies after failure of platinum-containing chemotherapy (NCT03869190).
In 2008, Phillips and coworkers conjugated the HER2-specific antibody Trastuzumab to a maytansine derivative (DM1) via the non-reducible thioether linker SMCC (155, 156). Trastuzumab exhibits 88 lysine residues that could potentially be utilized for conjugation. While the DAR is approximately 3.5, at least 70 lysine residues are partially conjugated, resulting in a heterogeneous product (157). The resulting ADC, termed T-DM1, was effective in Trastuzumab-sensitive and -insensitive cancer models in vitro as well as in vivo. T-DM1 is internalized upon binding to HER2, resulting in accumulation in the endosome (155, 158). As the linker utilized is non-reducible, DM1 release occurs by antibody degradation in the lysosome (156, 159). After lysosomal escape, DM1 inhibits microtubule assembly, resulting in cell death (155, 160). T-DM1, further termed as ado-Trastuzumab Emtansine, was approved in 2013 for HER2 positive breast cancer. Recently, a phase II study investigating the tumor response in HER2 expressing tumors, including bladder cancer, was completed (NCT02999672). In cohort 1, 13 patients with locally advanced or metastatic urothelial bladder carcinoma were dosed with 3.6 mg/kg qw, after the safety of Trastuzumab Emtansine was assessed by an independent data management committee after dosing the first six patients with 2.4 mg/kg qw. Partial response (defined as a 30% decrease in the sum of the diameters of the target lesions taking as a reference the baseline sum diameter according to the RECIST 1.1 criteria) was observed in 38.5% of patients. No patients achieved complete response. Median OS (95%CI) was 7.03 months (3.75 to NE). A second study on HER2 amplified or mutated cancers, including bladder cancer, is currently ongoing (NCT02675829).
DS-8201a is another HER2-targeting ADC. It is based on Trastuzumab, which was conjugated with DX-8951 (DXd), a topoisomerase I inhibitor via a GGFG peptide linker, using maleimide chemistry, after partial reduction of interchain disulfides. This tetrapeptide linker is cleaved by lysosomal proteases cathepsins B and L in HER2-positive tumor cells upon receptor-mediated endocytosis (161). The coupling results in a high DAR of 7.7, enabling anti-tumor effects even in HER2 low-expressing models not being feasible with ADCs exhibiting lower DAR (including T-DM1/ado-Trastuzumab Emtansine) (161, 162).
This higher DAR might help to overcome drug resistance, which is observed in T-DM1 treated patients. Even though the drug resistance in the context of T-DM1 is not fully understood, one of the main reasons might be the downregulation of HER2, impaired internalization, enhanced recycling of HER2, and lysosomal degradation (155, 163). However, due to its different mechanism of action and efficacy in low HER2-expressing cancers, it is hypothesized that DS-8201a might be effective against T-DM1 resistant tumors (161). Currently, DS8201a, renamed as Trastuzumab Deruxtecan, is under clinical investigation for advanced breast and urothelial cancer in combination with Nivolumab (NCT03523572) (164). Trastuzumab Deruxtecan is approved for treatment of patients suffering from unresectable or metastatic HER2-positive breast cancer.
In 1990, Stein and coworkers utilized the hybridoma technology to generate the antibody RS7-3G11 by immunizing mice with crude membrane preparations of squamous cell carcinoma of the lung (165). It was observed that the resulting mAb RS7-3G11 is specific to multiple malignancies, including breast, colon, renal, and prostate cancers (166). In 1995, Basu and coworkers identified Trop2 as the target of RS7-3G11 (167).
Trop2, also known as human trophoblastic cell surface antigen 2, is an oncogene involved in tumorigenesis and tumor invasion (168–170). With a low expression in healthy tissue and being upregulated in several cancers, it emerged as a promising target (171, 172).
It was observed that RS7 facilitates fast internalization and was evaluated for in vivo efficacy (173). Upon humanization by CDR-grafting, the now termed hRS7 was subjected to radio-labeled in vivo studies (174). Additionally, ADCC effects in cervical cancers were observed utilizing hRS7 (174). Later on, hRS7 was conjugated with the topoisomerase inhibitor SN-38, resulting in the ADC IMMU-132 or Sacituzumab Govitecan (175, 176). Conjugation was performed by mild reduction of interchain disulfides followed by formation of a thioether bond, resulting in DARs of less than 4 (176). Subsequent phase I studies showed acceptable toxicity and promising results (177). Currently, Sacituzumab Govitecan is in clinical phase II studies for metastatic UC (NCT03547973) and phase I/II studies in combination with chemotherapy for multiple carcinomas, including UC (NCT03992131). In early 2020, Sacituzumab Govitecan was approved under the name Trodelvy for the treatment of triple-negative breast cancer.
Utilizing tumor microarrays, SLIT and NTRK-like protein 6 (SLITRK6) were found to be expressed in 88% of bladder cancer specimens, of which 67% showed strong to moderate expression. Furthermore, transitional cell carcinoma exhibited SLITRK6 expression in 90% of cases and metastatic bladder cancer in 100% of cases (178).
In order to generate an anti-SLITRK6 ADC, the Xenomouse technology was employed by performing immunization with SLITRK6 expressing cells or recombinant SLITRK6 protein, resulting in hybridomas clones. The clone AGS-15C was identified for its binding to SLITRK6 and subcloned for recombinant expression (178). Subsequently, ASG-15C was conjugated to MMAE via a valine-citrulline dipeptide linker, resulting in the ADC ASG-15ME. The ADC caused complete growth inhibition in xenograft lung cancer models. Interestingly, approximately 5% of tumor cells were negative for SLITRK6 expression, underlining the effect this ADC has on heterogenic cancers, which are typically found in patients (178). AGS-15ME, also termed Sirtratumab Vedotin, was tested in a phase I clinical study on metastatic urothelial cancer (NCT01963052).
With the recent approval of Enfortumab Vedotin and the large number of additional toxin-coupled mAbs in clinical testing for bladder cancer, ADCs have the chance to gain a profound impact in the treatment of bladder cancer. The mechanism of action of the approved Enfortumab Vedotin is depicted in Figure 2C.
The photoimmunotherapeutic approach is based on an ADC-like molecule that carries an inactive cytotoxic agent to the tumor cell by conjugation to a monoclonal antibody (Figure 3). By irradiation with light of the appropriate wavelength, the cytotoxic agent is activated and is able to mediate effective tumor-killing (179).
The photoimmuno-conjugate Panitumumab-IR700 is based on the EGFR-specific mAb Panitumumab. RT-PCR analysis of TURB bladder washings of stage Ta, T1, and Tis NMIBC patients revealed that EGFR expression is 1.7-fold elevated in low-risk patients and up to 3-fold higher in high-risk patients (180). Other studies found by IHC that 15.1% of UC patients had low and 26.2% had high EGFR expression (181), where one study found EGFR overexpression in 74% of UC patients (182). No association of EGFR expressing UCs and mutations in the tyrosine kinase domain are known to this date (182). However, EGFR expression was correlated with prognostic factors like grade, deep muscle invasion, and recurrence (180, 181). Other IHC studies of tissue microarrays revealed that 70% of samples exhibited a relative staining intensity of 1 or higher staining. Of those samples, 16% had staining intensities of 3, and 31% had staining intensities of 4 stainings. A higher rate of positive stainings was observed in squamous tumors (94%) in comparison with nonsquamous tumors (69%). Interestingly, no correlation between EGFR staining intensity and T stage was observed (183). However, Kassouf et al. found that tumors simultaneously showing a high EGFR and low HER4 expression display a more invasive phenotype in conjunction with a short recurrence time (184). In consistence, a study investigating the expression of all HER-family members in bladder cancer found that while EGFR and HER2 expression rates alone do not correlate with survival, higher EGFR or HER2 expression combined with low HER3 and HER4 expression result in a worse prognosis (185).
In 2017 Railkar et al. conjugated the EGFR-specific and FDA-approved mAb Panitumumab via NHS ester chemistry with IRDye 700Dx, generating Panitumumab-IR700, intending to create a targeted phototherapy, termed photoimmunotherapy (PIT) for bladder cancer (183). IR700 is a hydrophilic agent with low cytotoxicity. Its cytotoxic nature is activated by the association to the cell membrane and subsequent activation using near-infrared radiation (NIR), resulting in the generation of cytotoxic oxygen species (186). Since the EGFR-specific antibody mediates membrane proximity, only EGFR-positive cells are targeted by this approach. In vitro, Panitumumab-IR700 showed cytotoxic effects on EGFR-positive bladder cancer cells in a dose-dependent manner (183). Additionally, the effects are dependent on the expression level of EGFR and the applied NIR. As EGFR expression increases, lower NIR is needed to achieve similar IC50 values. In xenograft models, the bladder cancer cell line UMUC-5 was utilized to induce tumors that were subsequently treated with Panitumumab-IR700. NIR treated tumors showed regression, which was not observed in non-NIR-treated control groups. Furthermore, xenografts generated with cells exhibiting a lower EGFR-expression profile, showed no significant difference between NIR-treated and NIR-untreated animals, underlining the importance of antigen expression level on the tumor (183).
Since anti-tumor effects were limited by the surface expression of the antigen, Siddiqui and coworkers generated a combinatorial approach, consisting of Panitumumab-IR700 and Trastuzumab-IR700 (187). In vitro and in vivo experiments utilizing the EGFR/HER2 double-positive bladder cancer cell line SW-780 showed more potent effects upon combinatorial treatment in comparison to mono treatments (187). Even though there is no clinical study ongoing utilizing Panitumumab-IR700, the IR700 conjugated anti-EGFR PIT Cetuximab Salotarocan is currently under investigation in phase I/II studies for EGFR expressing advanced solid tumors in combination with anti-PD1 treatment, as well as phase III studies for head and neck cancer (NCT03769506). Under the name Acalux, Cetuximab Salotarocan was approved for the treatment of head and neck cancer in Japan in 2020.
The radioimmune approach combines the advantages of radiation therapy with the precision of immunotherapy by bringing a radioactive isotope in proximity to the tumor, mediated by the specific targeting of a mAb (188).
In 2009, Pfost and coworkers generated a conjugate of the anti-EGFR antibody Matuzumab and SCN-CHX-A”-diethylenetriaminepentaacetic acid (DTPA), resulting in a mAb-chelator conjugate (189, 190). Subsequently, the anti-EGFR-mAb was incubated with 213Bi, leading to chelation of the alpha-emitter by the conjugated mAb (Figure 3). The labeling yield of 213Bi to the mAb after 7 minutes was up to 97% and a specific activity of 0.35-1.4 MBq per µg antibody could be determined. Unbound 213Bi was separated by size exclusion chromatography. For in vivo experiments, mice were intravesical treated with luciferase transfected bladder carcinoma cells. Subsequently, mice were intravesical treated with 213Bi-Anti-EGFR-mAb, unlabeled mAb or the chemotherapeutic mitomycin c, respectively. Reaction to the treatment was analyzed utilizing bioluminescence. 213Bi-Anti-EGFR-mAb showed significantly higher anti-tumor activity compared to unlabeled mAb and did not show nephrotoxicity in contrast to mitomycin c (189).
In a subsequent study in 2015, the same group showed that application of even higher radiation doses did not lead to toxic side effects after instillation of 213Bi-Anti-EGFR-mAb in bladder cancer-bearing xenografts. Fractionated applications in advanced stage tumors were investigated, based on the idea, that an alpha-emitter is most potent when the diameter of the tumor does not exceed the emission range of the radioisotope. By applying multiple treatments, the tumor can be eradicated step-wise by eradicating the outer layer of the tumor in each treatment. It was demonstrated that various applications show a stronger potency in vivo (191). Still, radioimmunotherapeutic approaches for bladder cancer have not yet entered the clinic.
Immunotoxins comprise the high toxicity and specificity of ADCs while bypassing the conjugation and heterogenicity problem by genetically fusing a targeting moiety (like an antibody) to a protein-based toxin (192).
Oportuzumab Monatox is an immunotoxin, binding specifically to the epithelial cell adhesion molecule (EpCAM), which is a transmembrane glycoprotein, frequently overexpressed in different cancers (193), inter alia malignancies of the colon, intestine, breast, lung, and prostate (194).
Analysis of microarrays of high-grade and advanced stage bladder cancer tissues for EpCAM expression by IHC showed positive staining results in 54% of samples (195). EpCAM expression was linked to advanced stage, higher-grade tumors, and worse survival outcomes (195). This finding could be confirmed by another study performing enzyme-linked immunosorbent assay (ELISA) measurements utilizing the urine of bladder tumor patients (196). Nevertheless, EpCAM expression was no independent prognostic factor. It is hypothesized that EpCAM is involved in tumor progression, making it a potential immunotherapeutic target (195). The soluble variant of EpCAM, found in the urine, is generated by cleavage between A243 and G244 (196).
In 1997 Zimmermann and coworkers chemically conjugated a truncated Pseudomonas aeruginosa exotoxin A (ETA) variant (252–613) via thioether bonds to the EpCAM-specific, hybridoma derived mAb MOC31 (197). The lack of the ETA domain I, which mediates cell-binding, leads to an immunotoxin, which is only targeting EpCAM positive cells and showed potent cytotoxic effects in vitro and in vivo. Small and medium-sized tumors were either nearly completely eradicated or stopped tumor growth but failed to do so in larger tumors due to insufficient tumor penetration and limited dose-escalation due to unspecific toxicity (197). In the same year, Krebber and coworkers utilized the phage display technology to isolate the coding sequence of the hybridoma MOC31 (198). Even though its affinity was measured to be 3 nM, it failed to enrich at the tumor site in SW2 lung cancer xenografts (199). As it was hypothesized that this is due to its insufficient thermal stability, the antibody was humanized by grafting its CDR loops onto that Trastuzumab scFv 4D5 (199).
Furthermore, eight framework residues were transferred to generate a more stable antibody that exhibited parental specificity and affinity, resulting in enrichment in lung tumor xenografts (199). Paolo and coworkers further engineered the molecule by fusing it genetically C-terminal to a truncated Pseudomonas aeruginosa exotoxin A (ETA252-608) (200). In wild type ETA, amino acids 609-613, which was still used in the thiol conjugated immunotoxin (197), are encoding for the endoplasmic reticulum (ER) retention sequence REDLK. This sequence was altered to its mammalian counterpart, KDEL, which increases cytotoxicity (200, 201). The linker exhibited a furin cleavage site that allowed the release of the toxin in the endosome upon binding of the scFv to EpCAM and subsequent internalization (Figure 3). Two terminal His-Tags facilitate the purification of the immunotoxin after production in E.coli. As this construct showed potent and selective cytotoxicity in vitro, in vivo experiments were conducted, resulting in favorable anti-tumor effects (200).
After phase I/II studies in head and neck cancers, where the molecule resulted in a 88% response rate with 25% complete response, the renamed molecule (Oportuzumab Monatox) was subjected to local treatment in bladder cancer in a phase III study by intravesical administration twice weekly for six weeks, followed by six weeks with one administration per week (NCT02449239) (202). Enrolled patients were selected to suffer from NMIBC, especially CIS and/or exhibited high-grade Ta or T1 papillary disease and have not responded to previous BCG treatment. Additionally, a co-therapy phase I study is recruiting, combining Oportuzumab Monatox with the checkpoint inhibitor antibody Durvalumab (NCT03258593).
With the current SARS-CoV2 pandemic ongoing and the swift development of vaccine candidates, mRNA-based options are currently in the focus of the scientific community. Encoding for one or multiple proteins, the mRNA-based therapy enables the expression of the therapeutic entity within the patient, surpassing development issues of single proteins (203). Very recently, the first mRNA-based vaccines received emergency authorization to fight the SARS-CoV2 pandemic, potentially paving the way for mRNA-based therapeutics in the field of oncology.
Formulated as a liquid nanoparticle encapsulated mRNA, mRNA-2752 encodes OX40L, IL-23, and IL-36γ, combining two different approaches: While IL-23 and IL-36γ are intended to act as adjuvants and to mediate a pro-inflammatory response within the tumor microenvironment (TME), the OX40 ligand activates T cells in order to generate an anti-tumor response (Figure 3). This, in turn, leads to the T cell antigen-dependent proliferation of CD4+ and CD8+ T cells and increases simultaneously their effector functions and the survival of memory T cells (204). In an ongoing phase I study, patients suffering from urothelial cancer and other malignancies are intratumorally injected with mRNA-2752 as monotherapy or in combination with Durvalumab (NCT03739931).
In recent years, transgenic T cells, exhibiting a chimeric antigen receptor (CAR), gained tremendous interest for cancer therapy. The concept dates back to the 1980s when an scFvs was fused to TCR in order to redirect the T cell specificity in a non-major histocompatibility complex (MHC)-restricted manner (205). CARs consist of an antibody-fragment, usually an scFv, that is genetically fused with CD3ζ (1st generation) or additionally with the activation domain of CD28 or 4-1BB (2nd generation). In 3rd generation CARs, all three activation domains are implemented. 2nd generation CAR T cells additionally equipped with transgene expression of cytokines or costimulatory ligand domains are referred to as 4th generation (206). Upon antigen recognition, cytotoxic effects mediate target cell killing (207). Today, two CAR-T cell therapies are approved (Kymriah and Yescarta). While both target CD19, Kymriah is approved for refractory or relapse (R/R) B cell precursor acute lymphoblastic leukemia and Yescarta for R/R large B cell lymphoma (208, 209).
Currently, two CAR-T cell approaches (Figure 3) targeting the folate receptor alpha (FRα) and the prostate-specific membrane antigen (PSMA) are in phase I/II studies for bladder cancer (NCT03185468). PSMA is significantly overexpressed in UCs and exhibits a stronger expression in NMIBC compared to MIBC, where its expression is correlated with recurrence and progression (210). FRα overexpression is often found in cancers of epithelial origin such as breast, ovary lung, or kidney cancer (211–213). In bladder cancer, FRα is significantly higher expressed in low-grade tumor tissue compared to high-grade tumors (214).
In these 4th generation CAR-T cells, the targeting scFv is coupled to the costimulating intracellular domains of CD28, 4-1BB, CD27, and CD3ζ. Furthermore, as a “safety switch”, a suicide expression cassette was implemented consisting of the inducible caspase 9 (iCasp9) fused to FKBP12-F36V (215, 216). The latter can bind to the small molecule AP1903, resulting in homodimerization and activation of iCasp9. This way, activated CAR-T cells can be depleted by apoptosis in case of safety concerns (217). As CAR-T cell therapy showed poor performance in solid tumors (218), the result of this ongoing trial will illuminate the possibility of CAR-T therapy in the context of urethral cancers.
Gene therapy describes the delivery of genetic information to a host cell to introduce a therapeutic effect (219, 220). Besides genetic diseases, gene therapy has a strong focus on cancer therapy (221). As a promising tool for gene delivery, viral vectors were engineered to allow for a favorable safety and deliverability profile (222). As the bladder is a closed compartment, instillation of viral vectors can lead to a local treatment, which is not possible to achieve with systemic application of various drugs.
CG0070 is a cancer-selective adenovirus encoding for human GM-CSF (Figure 3). In an Ad5 backbone, the endogenous E1a promoter was replaced by the human E2F-1 promotor, which has a 5’ SV40 polyadenylation signal and promotes tumor-selective gene expression. Furthermore, the adenovirus exhibits the entire E3 region except for the gp19kD gene, which was replaced with the human GM-CSF gene under the control of the E3 promoter. The rest of the vector backbone, encoding for e.g., packaging signals, E2, E4 late protein regions, as well as inverted terminal repeat (ITR) sequences, were identical to the wild-type Ad5 (223).
As an oncolytic virus, CG0070 replicates selectively in malignant cells, resulting in the production of additional adenoviruses. Defects of the retinoblastoma (Rb) pathway are a shared trait of multiple cancers, including bladder cancer. The E2F-1 promoter, which controls the essential E1a gene, is selectively active in cells exhibiting an Rb pathway defect, resulting in its oncolytic characteristic. Furthermore, the production of the anti-tumoral GM-CSF is controlled by the E3 promoter, which in turn is dependent on the expression of the E1a gene product, resulting in E2F-1 dependent, and therefore tumor-selective, GM-CSF expression (223).
In vitro experiments confirmed the selectivity to Rb defective cells in terms of E1a and GM-CSF mRNA levels as well as Rb status dependent viral replication and cytotoxicity. Furthermore, in vivo experiments confirmed the anti-tumoral activity of CG0070 in bladder cancer xenografts, which showed synergistic effects in combination with docetaxel. However, these effects are due to the oncolytic effects of CG-0070 since human GM-CSF is not active in mice (223, 224). A first in-human study confirmed a favorable safety profile since no maximum tolerated dose was reached. The most common adverse event was grade 1-2 bladder toxicity.
Additionally, high doses of GM-CFS were observed in the urine of patients. CG0070 treatment resulted in a complete response rate of 48.6% for a median duration of 10.4 months (NCT00109655) (225). Intravesical administration of CG-0070 was established in a subsequent phase II study after prior bladder rise with saline solution and 0.1% dodecylmaltoside (DDM) to enhance transduction efficiently. The interim report described a complete response in 47% of all patients after six months and 50% in CIS patients while exhibiting an acceptable toxicity profile (NCT02365818) (226). Enrolled patients failed intravesical BCG treatment 12 to 24 months prior to CG0070 treatment, thus patients are not BCG unresponsive as per FDA definition. Currently, a phase II study of CG0070 in combination with checkpoint inhibitor Pembrolizumab is ongoing (NCT04387461), as well as a phase III study of CG0070 as a monotherapy (NCT04452591). Both studies focus on the treatment of NMIBC.
In 2001 Iqbal Ahmed and coworkers generated a recombinant replication-deficient adenovirus carrying the genetic information for human interferon-α2b under the control of a CMV promoter (Figure 3) (227). This constructed rAdIFNα2b viral vector revealed significant anti-tumor activity and suppressed primary and metastatic tumor growth in vivo (227). These findings reflect previous findings, which demonstrated that adenoviral-based interferon-assisted immunotherapy yields promising results (228, 229).
A major obstacle for viral gene therapy in the context of bladder cancer was the glycosaminoglycan layer (GAG), which shields the surface of urothelial cells from viral infection and hindered effective transduction (230, 231). To overcome this problem, Connor and coworkers identified polyamides that enable efficient viral transduction in vivo (232). Further bioanalytical characterization revealed that one impurity of the initial heterogenic detergent was the bioactive compound (231, 232). Subsequent chemical modification led to the generation of Syn-3, which mediated an enhanced in vivo transduction efficacy in the bladder while exhibiting a favorable safety profile (231, 233).
In 2004 Benedict and coworkers combined the adenoviral IFN approach with Syn3 and performed intravesical instillation in mice, resulting in high urinary IFN levels for at least seven days, while intravesical instillation of rIFN protein alone was detectable for only 12h. Regression of GFP-labeled tumor cells was detected after 1h treatment to rAd-IFN/Syn-3, while no cytotoxic effect on normal tissue was observed. Even cell lines previously unresponsive to rIFNα treatment showed cytotoxic effects in vitro and in vivo (234). Intravesical instillation of IFN alone or the rAd-IFN without Syn3 showed no impact on tumor burden.
A phase I study evaluating the safety and efficacy showed no dose-limiting toxicities (NCT01162785). Except for patients receiving the lowest dose, effective gene transfer was found in all patients with detectable IFNα levels. 43% of the enrolled patients experienced a complete response at three months, where two remained disease-free at month 29 and 39, respectively (235). The subsequent phase II study enrolling high-grade BCG refractory or relapsed NMIBC patients showed that 35% of patients achieved 12 month without high-grade recurrence (NCT01687244) (236). Very recently, the primary efficacy analysis of a phase III study in BCG-unresponsive NMIBC patients was published (NCT02773849), where 53.4% of enrolled CIS patients had a complete response at 3 months and 45.5% of those maintained this outcome at 12 months (237). The mechanism of action of Nadofaragene Firadenovec is depicted in Figure 2D.
A variety of engineered antibodies and proteins are in preclinical development and clinical trials for the treatment of BC. Among them, checkpoint inhibitors were the first approved biologics for bladder cancer, but the list is likely to be extended. In particular, ADCs are promising entities to have a major impact on the treatment of bladder cancer in the future (128). By combining the specificity of an antibody with the strong cytotoxic effect of its payload, ADCs circumvent systemic exposure of the toxin and specifically target the tumor. Engineering of the linkers to be cleaved by endoplasmic enzymes or pH-dependent release of the payload further contributes to the efficacy and safety of this drug class. Enfortumab Vedotin was recently approved for BC, but the list of approved ADCs will most probably expand in the near future. While Trastuzumab Deruxtecan and Sirtratumab Vedotin are currently in phase I studies, phase II studies are ongoing for Disitamab Vedotin, ado-Trastuzumab Emtansine, and Sacituzumab Govitecan investigating efficacy in bladder cancer patients (Table 1).
Bispecific antibodies and fusion proteins are also next generation biologics and are under investigation for bladder cancer. Even though exhibiting interesting molecular designs, most bispecifics are in early stages of clinical development (Table 1). While preclinical assessment of T cell engagers like Orlotamab, Treg depleting bsAb like ATOR-1015 or super agonistic cytokine traps like ALT-803 exhibit promising profiles, only ongoing and future trials will show whether these classes of molecules will have an impact in uro-oncology.
However, ADCs, bispecifics, and most fusion proteins are applied systemically and despite the specific tumor targeting mediated by the binding moiety, systemic side effects and resulting dose-limitations may accrue. Furthermore, molecule heterogeneity remains a challenge for ADC development. The immunotoxin Oportuzumab Monatox circumvent both problems by genetic fusion of the bioactive drug ETA to the scFv and local administration to the bladder via instillation. As clinical trials utilizing this molecule resulted in encouraging results (Table 1), a rolling biologics license application (BLA) submission is currently under review by the FDA, which might lead to the FDA approval of Oportuzumab Monatox.
The instillation of Oportuzumab Monatox results in a local treatment of the tumor but also in a shorter half-life. This limitation is circumvented by gene therapeutic approaches, leading to a prolonged local treatment. CG0070 and rAd-IFN/Syn-3 are adenovirus-based vectors targeting the bladder tissue and lead to the production of anti-tumor cytokines upon transduction. While CG0070 does this in a non-tumor-selective manner, resulting in an oncolytic mechanism of action, rAd-IFN/Syn-3 transduces cells in a non-tumor selectivemanner, leading to sustained high IFNα levels and a subsequent convenient quarterly treatment schedule with an excellent safety profile, where only 1.9% of the patients discontinued treatment. A phase III study for CG0070 is still ongoing, while the results of the phase III study of rAd-IFN/Syn-3 recently showed very promising outcomes, paving the way for a potential future approval (237). rAd-IFN/Syn-3 was granted priority review, fast track and breakthrough therapy designations by the FDA with the BLA currently under investigation.
Of note, the clinical outcomes of CG0070 and rAd-IFN/Syn-3 are not directly comparable as the patient populations are not identical. The phase II study of CG0070 enrolled patients described as BCG-unresponsive but allowed for patients with disease recurrence up to 24 months to be included and did not solely request patients to have received adequate prior BCG treatment according to the BCG Unresponsive criteria (NCT02365818). By now, BCG unresponsive patients must show recurrence within 12 months for CIS, or 6 months for Ta/T1, or 3 months for T1 to be defined as BCG-unresponsive and depicts a population very unlikely to respond to further BCG treatment (20, 21). The patient in the GC0070 phase II study most likely had a lower risk of recurrence/progression compared to the population of rAd-IFN/Syn-3 trial (NCT01687244) and might have shown a positive response to other instillation treatments, including rechallenge with BCG compared to a true BCG-unresponsive population. Additionally, the dosing scheme of rAd-IFN/Syn-3 is less time consuming and more convenient compared to CG0070. Therefore, it is tempting to speculate that rAd-IFN/Syn-3 has a better stand in the direct comparison and might be the favorable treatment of choice. In respect to all these information, Kulkarni very recently elaborated the potential of rAd-IFN/Syn-3 to become the “New Gold Standard” for BCG-unresponsive bladder cancer patients (238).
Due to the ongoing SARS-CoV2 pandemic, mRNA-based medicine is in the spotlight. Due to its intratumoral application, mRNA-2752 is locally active in the tumor of the patient, where the expression of immunoregulatory proteins might lead to a strong anti-tumor effect. Furthermore, mRNA-based therapies are more straightforward to develop compared to virus-based therapeutics. Even though mRNA-based vaccines have recently gained emergency use authorization, the clinical studies of mRNA-based therapeutics for cancer treatment are still in early testing.
Next-generation molecules like ADCs, immunotoxins, and gene therapeutic approaches will most probably change the way bladder cancer is treated. Conventional antibodies, on the other hand, except checkpoint inhibitors, will probably not have a major impact on UC treatment. Vofatamab, an anti-FGFR3 antibody, was in three phase I and phase I/II studies for bladder cancer, all of which were terminated, even though FGFR3 seems to be an interesting target, especially for NMIBC.
For the development of future therapeutics, a special focus will be set on the route of administration. As the bladder is relatively uncomplicatedy to reach from the outside and local treatments will result in less systemic side effects, future molecules will focus on application by instillation. Oportuzumab Monatox demonstrates that this is a feasible route to administer biologics. In line with current events, mRNA- and viral-based therapeutics will play a tremendous role in the coming years and will most probably have a major impact on pharmaceutical research, leading to new molecules with novel mechanisms of action.
Taken together, there is a broad pipeline of molecules exhibiting different design strategies and various mechanisms of action. From stimulating the immune system within the TME, recruiting of effector cells to directly or indirectly kill cancerous cells, several therapeutic approaches are under clinical testing for bladder cancer, giving hope to patients in need. Future approval of ADCs, immunotoxins, and gene therapeutic approaches will have a strong impact on bladder cancer treatment. These additional options are expected to result in beneficial clinical outcomes and less severe side effects compared to present-day treatments.
JB, BH, and HK conceived this review. JB wrote the first draft of the manuscript. JB, JJ, BH, and HK wrote sections of the manuscript. JG and AB gave scientific advice. All authors contributed to the article and approved the submitted version.
This work was supported by the Ferring Darmstadt Labs at Technical University of Darmstadt and by the department of GPRD at Ferring Holding S.A., Saint-Prex. The funder bodies were not involved in the study design, collection, analysis, interpretation of data, the writing of this article or the decision to submit it for publication.
JG, JJ, AB, and BH were employed by the company Ferring Pharmaceuticals. JB is employed by TU Darmstadt in frame of a collaboration project with Ferring Pharmaceuticals.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We would like to thank the department of GPRD at Ferring Holding S.A., Saint-Prex, for funding. Figures were created with BioRender.com.
1. Kaufman DS, Shipley WU, Feldman AS. Bladder Cancer. Lancet (2009) 374:239–49. doi: 10.1016/S0140-6736(09)60491-8
2. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin (2021) 71:209–49. doi: 10.3322/caac.21660
3. Martin JW, Carballido EM, Ahmed A, Farhan B, Dutta R, Smith C, et al. Squamous Cell Carcinoma of the Urinary Bladder: Systematic Review of Clinical Characteristics and Therapeutic Approaches. Arab J Urol (2016) 14:183–91. doi: 10.1016/j.aju.2016.07.001
4. Babjuk M, Burger M, Compérat EM, Gontero P, Mostafid AH, Palou J, et al. European Association of Urology Guidelines on Non-muscle-invasive Bladder Cancer (TaT1 and Carcinoma in Situ) - 2019 Update. Eur Urol (2019) 76:639–57. doi: 10.1016/j.eururo.2019.08.016
5. Witjes JA, Compérat E, Cowan NC, De Santis M, Gakis G, Lebret T, et al. EAU Guidelines on Muscle-Invasive and Metastatic Bladder Cancer: Summary of the 2013 Guidelines. Eur Urol (2014) 65:778–92. doi: 10.1016/j.eururo.2013.11.046
6. Edge S, Byrd DR, Compton CC, Fritz AG, Greene F, Trotti A. Ajcc Cancer Staging Handbook. New York: Springer-Verlag New York (2010).
7. Compérat EM, Burger M, Gontero P, Mostafid AH, Palou J, Rouprêt M, et al. Grading of Urothelial Carcinoma and The New “World Health Organisation Classification of Tumours of the Urinary System and Male Genital Organs 2016”. Eur Urol focus (2019) 5:457–66. doi: 10.1016/j.euf.2018.01.003
8. Urquidi V, Rosser CJ, Goodison S. Molecular Diagnostic Trends in Urological Cancer: Biomarkers for non-Invasive Diagnosis. Curr Med Chem (2012) 19:3653–63. doi: 10.2174/092986712801661103
9. Schmitz-Dräger BJ, Droller M, Lokeshwar VB, Lotan Y, Hudson MA, van Rhijn BW, et al. Molecular Markers for Bladder Cancer Screening, Early Diagnosis, and Surveillance: The WHO/ICUD Consensus. Urol Int (2015) 94:1–24. doi: 10.1159/000369357
10. Guo CC, Bondaruk J, Yao H, Wang Z, Zhang L, Lee S, et al. Assessment of Luminal and Basal Phenotypes in Bladder Cancer. Sci Rep (2020) 10:9743. doi: 10.1038/s41598-020-66747-7
11. Dadhania V, Zhang M, Zhang L, Bondaruk J, Majewski T, Siefker-Radtke A, et al. Meta-Analysis of the Luminal and Basal Subtypes of Bladder Cancer and the Identification of Signature Immunohistochemical Markers for Clinical Use. EBioMedicine (2016) 12:105–17. doi: 10.1016/j.ebiom.2016.08.036
12. Trilla-Fuertes L, Gámez-Pozo A, Prado-Vázquez G, Zapater-Moros A, Díaz-Almirón M, Arevalillo JM, et al. Biological Molecular Layer Classification of Muscle-Invasive Bladder Cancer Opens New Treatment Opportunities. BMC Cancer (2019) 19:636. doi: 10.1186/s12885-019-5858-z
13. Sylvester RJ, Oosterlinck W, Holmang S, Sydes MR, Birtle A, Gudjonsson S, et al. Systematic Review and Individual Patient Data Meta-Analysis of Randomized Trials Comparing a Single Immediate Instillation of Chemotherapy After Transurethral Resection With Transurethral Resection Alone in Patients With Stage Pta-Pt1 Urothelial Carcinoma of the Bladder: Which Patients Benefit From the Instillation? Eur Urol (2016) 69:231–44. doi: 10.1016/j.eururo.2015.05.050
14. Hall MC, Chang SS, Dalbagni G, Pruthi RS, Seigne JD, Skinner EC, et al. Guideline for the Management of Nonmuscle Invasive Bladder Cancer (Stages Ta, T1, and Tis): 2007 Update. J Urol (2007) 178:2314–30. doi: 10.1016/j.juro.2007.09.003
15. Babjuk M, Oosterlinck W, Sylvester R, Kaasinen E, Böhle A, Palou-Redorta J, et al. EAU Guidelines on non-Muscle-Invasive Urothelial Carcinoma of the Bladder, the 2011 Update. Eur Urol (2011) 59:997–1008. doi: 10.1016/j.eururo.2011.03.017
16. Guallar-Garrido S, Julián E. Bacillus Calmette-Guérin (Bcg) Therapy for Bladder Cancer: An Update. Immunotargets Ther (2020) 9:1–11. doi: 10.2147/ITT.S202006
17. Babjuk M, Böhle A, Burger M, Capoun O, Cohen D, Compérat EM, et al. Eau Guidelines on Non–Muscle-invasive Urothelial Carcinoma of the Bladder: Update 2016. Eur Urol (2017) 71:447–61. doi: 10.1016/j.eururo.2016.05.041
18. Zlotta AR, Fleshner NE, Jewett MA. The Management of BCG Failure in non-Muscle-Invasive Bladder Cancer: An Update. Can Urol Assoc J J Assoc Des Urol du Canada (2009) 3:S199–205. doi: 10.5489/cuaj.1196
19. Witjes JA. Management of BCG Failures in Superficial Bladder Cancer: A Review. Eur Urol (2006) 49:790–7. doi: 10.1016/j.eururo.2006.01.017
20. Kamat AM, Sylvester RJ, Böhle A, Palou J, Lamm DL, Brausi M, et al. Definitions, End Points, and Clinical Trial Designs for Non-Muscle-Invasive Bladder Cancer: Recommendations From the International Bladder Cancer Group. J Clin Oncol Off J Am Soc Clin Oncol (2016) 34:1935–44. doi: 10.1200/jco.2015.64.4070
21. Lerner SP, Dinney C, Kamat A, Bivalacqua TJ, Nielsen M, O’Donnell M, et al. Clarification of Bladder Cancer Disease States Following Treatment of Patients With Intravesical Bcg. Bladder Cancer (2015) 1:29–30. doi: 10.3233/blc-159002
22. Rosenberg JE, Carroll PR, Small EJ. Update on Chemotherapy for Advanced Bladder Cancer. J Urol (2005) 174:14–20. doi: 10.1097/01.ju.0000162039.38023.5f
23. Dasari S, Tchounwou PB. Cisplatin in Cancer Therapy: Molecular Mechanisms of Action. Eur J Pharmacol (2014) 740:364–78. doi: 10.1016/j.ejphar.2014.07.025
24. Porten SP, Leapman MS, Greene KL. Intravesical Chemotherapy in non-Muscle-Invasive Bladder Cancer. Indian J Urol (2015) 31:297–303. doi: 10.4103/0970-1591.166446
25. Yafi FA, North S, Kassouf W. First- and Second-Line Therapy for Metastatic Urothelial Carcinoma of the Bladder. Curr Oncol (2011) 18:e25–34. doi: 10.3747/co.v18i1.695
26. Massari F, Santoni M, Ciccarese C, Brunelli M, Conti A, Santini D, et al. Emerging Concepts on Drug Resistance in Bladder Cancer: Implications for Future Strategies. Crit Rev Oncol Hematol (2015) 96:81–90. doi: 10.1016/j.critrevonc.2015.05.005
27. Zangouei AS, Rahimi HR, Mojarrad M, Moghbeli M. Non Coding RNAs as the Critical Factors in Chemo Resistance of Bladder Tumor Cells. Diagn Pathol (2020) 15:136. doi: 10.1186/s13000-020-01054-3
28. Patel VG, Oh WK, Galsky MD. Treatment of Muscle-Invasive and Advanced Bladder Cancer in 2020. CA Cancer J Clin (2020) 70:404–23. doi: 10.3322/caac.21631
29. Redelman-Sidi G, Glickman MS, Bochner BH. The Mechanism of Action of BCG Therapy for Bladder Cancer–a Current Perspective. Nat Rev Urol (2014) 11:153–62. doi: 10.1038/nrurol.2014.15
30. Kaplon H, Muralidharan M, Schneider Z, Reichert JM. Antibodies to Watch in 2020. mAbs (2020) 12:1703531. doi: 10.1080/19420862.2019.1703531
31. Han Y, Liu D, Li L. Pd-1/Pd-L1 Pathway: Current Researches in Cancer. Am J Cancer Res (2020) 10:727–42.
32. Gopalakrishnan D, Koshkin VS, Ornstein MC, Papatsoris A, Grivas P. Immune Checkpoint Inhibitors in Urothelial Cancer: Recent Updates and Future Outlook. Ther Clin Risk Manag (2018) 14:1019–40. doi: 10.2147/tcrm.S158753
33. Bellmunt J, Powles T, Vogelzang NJ. A Review on the Evolution of PD-1/PD-L1 Immunotherapy for Bladder Cancer: The Future is Now. Cancer Treat Rev (2017) 54:58–67. doi: 10.1016/j.ctrv.2017.01.007
34. Kawahara T, Ishiguro Y, Ohtake S, Kato I, Ito Y, Ito H, et al. PD-1 and PD-L1 are More Highly Expressed in High-Grade Bladder Cancer Than in Low-Grade Cases: PD-L1 Might Function as a Mediator of Stage Progression in Bladder Cancer. BMC Urol (2018) 18:97. doi: 10.1186/s12894-018-0414-8
35. Nakanishi J, Wada Y, Matsumoto K, Azuma M, Kikuchi K, Ueda S. Overexpression of B7-H1 (Pd-L1) Significantly Associates With Tumor Grade and Postoperative Prognosis in Human Urothelial Cancers. Cancer Immunol Immunother CII (2007) 56:1173–82. doi: 10.1007/s00262-006-0266-z
36. Inman BA, Sebo TJ, Frigola X, Dong H, Bergstralh EJ, Frank I, et al. Pd-L1 (B7-H1) Expression by Urothelial Carcinoma of the Bladder and BCG-induced Granulomata: Associations With Localized Stage Progression. Cancer (2007) 109:1499–505. doi: 10.1002/cncr.22588
37. Farina MS, Lundgren KT, Bellmunt J. Immunotherapy in Urothelial Cancer: Recent Results and Future Perspectives. Drugs (2017) 77:1077–89. doi: 10.1007/s40265-017-0748-7
38. Wołącewicz M, Hrynkiewicz R, Grywalska E, Suchojad T, Leksowski T, Roliński J, et al. Immunotherapy in Bladder Cancer: Current Methods and Future Perspectives. Cancers (2020) 12. doi: 10.3390/cancers12051181
39. Raja R, Kuziora M, Brohawn PZ, Higgs BW, Gupta A, Dennis PA, et al. Early Reduction in Ctdna Predicts Survival in Patients With Lung and Bladder Cancer Treated With Durvalumab. Clin Cancer Res Off J Am Assoc Cancer Res (2018) 24:6212–22. doi: 10.1158/1078-0432.Ccr-18-0386
40. Campbell KS, Purdy AK. Structure/Function of Human Killer Cell Immunoglobulin-Like Receptors: Lessons From Polymorphisms, Evolution, Crystal Structures and Mutations. Immunology (2011) 132:315–25. doi: 10.1111/j.1365-2567.2010.03398.x
41. Pende D, Falco M, Vitale M, Cantoni C, Vitale C, Munari E, et al. Killer Ig-Like Receptors (Kirs): Their Role in NK Cell Modulation and Developments Leading to Their Clinical Exploitation. Front Immunol (2019) 10:1179. doi: 10.3389/fimmu.2019.01179
42. Béziat V, Hilton HG, Norman PJ, Traherne JA. Deciphering the Killer-Cell Immunoglobulin-Like Receptor System At Super-Resolution for Natural Killer and T-cell Biology. Immunology (2017) 150:248–64. doi: 10.1111/imm.12684
43. Binyamin L, Alpaugh RK, Hughes TL, Lutz CT, Campbell KS, Weiner LM. Blocking NK Cell Inhibitory Self-Recognition Promotes Antibody-Dependent Cellular Cytotoxicity in a Model of Anti-Lymphoma Therapy. J Immunol (Baltimore Md 1950) (2008) 180:6392–401. doi: 10.4049/jimmunol.180.9.6392
44. Ishida I, Tomizuka K, Yoshida H, Tahara T, Takahashi N, Ohguma A, et al. Production of Human Monoclonal and Polyclonal Antibodies in TransChromo Animals. Cloning Stem Cells (2002) 4:91–102. doi: 10.1089/153623002753632084
45. Romagné F, André P, Spee P, Zahn S, Anfossi N, Gauthier L, et al. Preclinical Characterization of 1-7F9, a Novel Human anti-KIR Receptor Therapeutic Antibody That Augments Natural Killer-Mediated Killing of Tumor Cells. Blood (2009) 114:2667–77. doi: 10.1182/blood-2009-02-206532
46. Chen Y, Wang H, Zuo Y, Li N, Ding M, Li C. A Novel Monoclonal Antibody KMP1 has Potential Antitumor Activity of Bladder Cancer by Blocking CD44 In Vivo and In Vitro. Cancer Med (2018) 7:2064–77. doi: 10.1002/cam4.1446
47. Wu CT, Lin WY, Chen WC, Chen MF. Predictive Value of CD44 in Muscle-Invasive Bladder Cancer and Its Relationship With IL-6 Signaling. Ann Surg Oncol (2018) 25:3518–26. doi: 10.1245/s10434-018-6706-0
48. Wu CT, Lin WY, Chang YH, Chen WC, Chen MF. Impact of CD44 Expression on Radiation Response for Bladder Cancer. J Cancer (2017) 8:1137–44. doi: 10.7150/jca.18297
49. Helsten T, Schwaederle M, Kurzrock R. Fibroblast Growth Factor Receptor Signaling in Hereditary and Neoplastic Disease: Biologic and Clinical Implications. Cancer Metastasis Rev (2015) 34:479–96. doi: 10.1007/s10555-015-9579-8
50. di Martino E, Tomlinson DC, Williams SV, Knowles MA. A Place for Precision Medicine in Bladder Cancer: Targeting the Fgfrs. Future Oncol (London England) (2016) 12:2243–63. doi: 10.2217/fon-2016-0042
51. d’Avis PY, Robertson SC, Meyer AN, Bardwell WM, Webster MK, Donoghue DJ. Constitutive Activation of Fibroblast Growth Factor Receptor 3 by Mutations Responsible for the Lethal Skeletal Dysplasia Thanatophoric Dysplasia Type I. Cell Growth Differ Mol Biol J Am Assoc Cancer Res (1998) 9:71–8.
52. Guo G, Sun X, Chen C, Wu S, Huang P, Li Z, et al. Whole-Genome and Whole-Exome Sequencing of Bladder Cancer Identifies Frequent Alterations in Genes Involved in Sister Chromatid Cohesion and Segregation. Nat Genet (2013) 45:1459–63. doi: 10.1038/ng.2798
53. Costa R, Carneiro BA, Taxter T, Tavora FA, Kalyan A, Pai SA, et al. Fgfr3-TACC3 Fusion in Solid Tumors: Mini Review. Oncotarget (2016) 7:55924–38. doi: 10.18632/oncotarget.10482
54. Nakanishi Y, Akiyama N, Tsukaguchi T, Fujii T, Satoh Y, Ishii N, et al. Mechanism of Oncogenic Signal Activation by the Novel Fusion Kinase Fgfr3-Baiap2l1. Mol Cancer Ther (2015) 14:704–12. doi: 10.1158/1535-7163.Mct-14-0927-t
55. Sfakianos JP, Cha EK, Iyer G, Scott SN, Zabor EC, Shah RH, et al. Genomic Characterization of Upper Tract Urothelial Carcinoma. Eur Urol (2015) 68:970–7. doi: 10.1016/j.eururo.2015.07.039
56. Neuzillet Y, van Rhijn BW, Prigoda NL, Bapat B, Liu L, Bostrom PJ, et al. FGFR3 Mutations, But Not FGFR3 Expression and FGFR3 Copy-Number Variations, are Associated With Favourable non-Muscle Invasive Bladder Cancer. Virchows Arch Int J Pathol (2014) 465:207–13. doi: 10.1007/s00428-014-1596-4
57. Mhawech-Fauceglia P, Cheney RT, Fischer G, Beck A, Herrmann FR. FGFR3 and p53 Protein Expressions in Patients With pTa and pT1 Urothelial Bladder Cancer. Eur J Surg Oncol J Eur Soc Surg Oncol Br Assoc Surg Oncol (2006) 32:231–7. doi: 10.1016/j.ejso.2005.11.018
58. Tomlinson DC, Baldo O, Harnden P, Knowles MA. FGFR3 Protein Expression and its Relationship to Mutation Status and Prognostic Variables in Bladder Cancer. J Pathol (2007) 213:91–8. doi: 10.1002/path.2207
59. Qing J, Du X, Chen Y, Chan P, Li H, Wu P, et al. Antibody-Based Targeting of FGFR3 in Bladder Carcinoma and T(4;14)-Positive Multiple Myeloma in Mice. J Clin Invest (2009) 119:1216–29. doi: 10.1172/jci38017
60. Kontermann RE, Brinkmann U. Bispecific Antibodies. Drug Discovery Today (2015) 20:838–47. doi: 10.1016/j.drudis.2015.02.008
61. Vafa O, Trinklein ND. Perspective: Designing T-Cell Engagers With Better Therapeutic Windows. Front Oncol (2020) 10. doi: 10.3389/fonc.2020.00446
62. Ma J, Ge J, Xue X, Xiu W, Ma P, Sun X, et al. Targeting Bladder Cancer Using Activated T cells Armed With Bispecific Antibodies. Oncol Rep (2018) 39:1245–52. doi: 10.3892/or.2018.6211
63. Newman MJ, Benani DJ. A Review of Blinatumomab, a Novel Immunotherapy. J Oncol Pharm Pract Off Publ Int Soc Oncol Pharm Pract (2016) 22:639–45. doi: 10.1177/1078155215618770
64. Ma W, Ma J, Lei T, Zhao M, Zhang M. Targeting Immunotherapy for Bladder Cancer by Using anti-CD3 × CD155 Bispecific Antibody. J Cancer (2019) 10:5153–61. doi: 10.7150/jca.29937
65. Mendelsohn CL, Wimmer E, Racaniello VR. Cellular Receptor for Poliovirus: Molecular Cloning, Nucleotide Sequence, and Expression of a New Member of the Immunoglobulin Superfamily. Cell (1989) 56:855–65. doi: 10.1016/0092-8674(89)90690-9
66. Nakai R, Maniwa Y, Tanaka Y, Nishio W, Yoshimura M, Okita Y, et al. Overexpression of Necl-5 Correlates With Unfavorable Prognosis in Patients With Lung Adenocarcinoma. Cancer Sci (2010) 101:1326–30. doi: 10.1111/j.1349-7006.2010.01530.x
67. Bevelacqua V, Bevelacqua Y, Candido S, Skarmoutsou E, Amoroso A, Guarneri C, et al. Nectin Like-5 Overexpression Correlates With the Malignant Phenotype in Cutaneous Melanoma. Oncotarget (2012) 3:882–92. doi: 10.18632/oncotarget.594
68. Nishiwada S, Sho M, Yasuda S, Shimada K, Yamato I, Akahori T, et al. Clinical Significance of CD155 Expression in Human Pancreatic Cancer. Anticancer Res (2015) 35:2287–97.
69. Masson D, Jarry A, Baury B, Blanchardie P, Laboisse C, Lustenberger P, et al. Overexpression of the CD155 Gene in Human Colorectal Carcinoma. Gut (2001) 49:236–40. doi: 10.1136/gut.49.2.236
70. Zhang J, Zhu Y, Wang Q, Kong Y, Sheng H, Guo J, et al. Poliovirus Receptor CD155 is Up-Regulated in Muscle-Invasive Bladder Cancer and Predicts Poor Prognosis. Urol Oncol: Semin Orig Investig (2020) 38:41.e11–8. doi: 10.1016/j.urolonc.2019.07.006
71. Krishnamurthy A, Jimeno A. Bispecific Antibodies for Cancer Therapy: A Review. Pharmacol Ther (2018) 185:122–34. doi: 10.1016/j.pharmthera.2017.12.002
72. Li H, Huang C, Zhang Z, Feng Y, Wang Z, Tang X, et al. Mek Inhibitor Augments Antitumor Activity of B7-H3-Redirected Bispecific Antibody. Front Oncol (2020) 10:1527. doi: 10.3389/fonc.2020.01527
73. Zheng M, Yu L, Hu J, Zhang Z, Wang H, Lu D, et al. Efficacy of B7-H3-Redirected BiTE and CAR-T Immunotherapies Against Extranodal Nasal Natural Killer/T Cell Lymphoma. Trans Oncol (2020) 13:100770. doi: 10.1016/j.tranon.2020.100770
74. Pardoll DM. The Blockade of Immune Checkpoints in Cancer Immunotherapy. Nat Rev Cancer (2012) 12:252–64. doi: 10.1038/nrc3239
75. Vigdorovich V, Ramagopal UA, Lázár-Molnár E, Sylvestre E, Lee JS, Hofmeyer KA, et al. Structure and T Cell Inhibition Properties of B7 Family Member, B7-H3. Struct (London Engl 1993) (2013) 21:707–17. doi: 10.1016/j.str.2013.03.003
76. Chen C, Shen Y, Qu QX, Chen XQ, Zhang XG, Huang JA. Induced Expression of B7-H3 on the Lung Cancer Cells and Macrophages Suppresses T-cell Mediating Anti-Tumor Immune Response. Exp Cell Res (2013) 319:96–102. doi: 10.1016/j.yexcr.2012.09.006
77. Mao Y, Chen L, Wang F, Zhu D, Ge X, Hua D, et al. Cancer Cell-Expressed B7-H3 Regulates the Differentiation of Tumor-Associated Macrophages in Human Colorectal Carcinoma. Oncol Lett (2017) 14:6177–83. doi: 10.3892/ol.2017.6935
78. Zhang X, Ji J, Zhang G, Fang C, Jiang F, Ma S, et al. Expression and Significance of B7-H3 and Tie-2 in the Tumor Vasculature of Clear Cell Renal Carcinoma. OncoTargets Ther (2017) 10:5417–24. doi: 10.2147/ott.S147041
79. Yonesaka K, Haratani K, Takamura S, Sakai H, Kato R, Takegawa N, et al. B7-H3 Negatively Modulates CTL-Mediated Cancer Immunity. Clin Cancer Res Off J Am Assoc Cancer Res (2018) 24:2653–64. doi: 10.1158/1078-0432.Ccr-17-2852
80. Varki V, Ioffe OB, Bentzen SM, Heath J, Cellini A, Feliciano J, et al. Pd-L1, B7-H3, and PD-1 Expression in Immunocompetent vs. Immunosuppressed Patients With Cutaneous Squamous Cell Carcinoma. Cancer Immunol Immunother CII (2018) 67:805–14. doi: 10.1007/s00262-018-2138-8
81. Zhao X, Li DC, Zhu XG, Gan WJ, Li Z, Xiong F, et al. B7-H3 Overexpression in Pancreatic Cancer Promotes Tumor Progression. Int J Mol Med (2013) 31:283–91. doi: 10.3892/ijmm.2012.1212
82. Wang F, Wang G, Liu T, Yu G, Zhang G, Luan X. B7-H3 was Highly Expressed in Human Primary Hepatocellular Carcinoma and Promoted Tumor Progression. Cancer Invest (2014) 32:262–71. doi: 10.3109/07357907.2014.909826
83. Cong F, Yu H, Gao X. Expression of CD24 and B7-H3 in Breast Cancer and the Clinical Significance. Oncol Lett (2017) 14:7185–90. doi: 10.3892/ol.2017.7142
84. Li Y, Yang X, Wu Y, Zhao K, Ye Z, Zhu J, et al. B7-H3 Promotes Gastric Cancer Cell Migration and Invasion. Oncotarget (2017) 8:71725–35. doi: 10.18632/oncotarget.17847
85. Tolcher AW, Alley EW, Chichili G, Baughman JE, Moore PA, Bonvini E, et al. Phase 1, First-in-Human, Open Label, Dose Escalation Ctudy of MGD009, a Humanized B7-H3 X CD3 Dual-Affinity Re-Targeting (DART) Protein in Patients With B7-H3-expressing Neoplasms or B7-H3 Expressing Tumor Vasculature. J Clin Oncol (2016) 34:TPS3105–TPS05. doi: 10.1200/JCO.2016.34.15_suppl.TPS3105
86. Moore PA, Zhang W, Rainey GJ, Burke S, Li H, Huang L, et al. Application of Dual Affinity Retargeting Molecules to Achieve Optimal Redirected T-cell Killing of B-cell Lymphoma. Blood (2011) 117:4542–51. doi: 10.1182/blood-2010-09-306449
87. Shankar S, Spira AI, Strauss J, Liu L, La Motte-Mohs R, Wu T, et al. A Phase 1, Open Label, Dose Escalation Study of MGD009, a Humanized B7-H3 X CD3 DART Protein, in Combination With MGA012, an anti-PD-1 Antibody, in Patients With Relapsed or Refractory B7-H3-expressing Tumors. J Clin Oncol (2018) 36:TPS2601–TPS01. doi: 10.1200/JCO.2018.36.15_suppl.TPS2601
88. Kvarnhammar AM, Veitonmäki N, Hägerbrand K, Dahlman A, Smith KE, Fritzell S, et al. The CTLA-4 X OX40 Bispecific Antibody ATOR-1015 Induces Anti-Tumor Effects Through Tumor-Directed Immune Activation. J Immunother Cancer (2019) 7:103. doi: 10.1186/s40425-019-0570-8
89. Kitamura N, Murata S, Ueki T, Mekata E, Reilly RT, Jaffee EM, et al. OX40 Costimulation can Abrogate Foxp3+ Regulatory T Cell-Mediated Suppression of Antitumor Immunity. Int J Cancer (2009) 125:630–38. doi: 10.1002/ijc.24435
90. Mason D, Powrie F. Control of Immune Pathology by Regulatory T Cells. Curr Opin Immunol (1998) 10:649–55. doi: 10.1016/s0952-7915(98)80084-8
91. Curotto de Lafaille MA, Lafaille JJ. CD4(+) Regulatory T Cells in Autoimmunity and Allergy. Curr Opin Immunol (2002) 14:771–8. doi: 10.1016/s0952-7915(02)00408-9
92. Bluestone JA, Abbas AK. Natural Versus Adaptive Regulatory T Cells. Nat Rev Immunol (2003) 3:253–7. doi: 10.1038/nri1032
93. Morse MA, Hobeika AC, Osada T, Serra D, Niedzwiecki D, Lyerly HK, et al. Depletion of Human Regulatory T Cells Specifically Enhances Antigen-Specific Immune Responses to Cancer Vaccines. Blood (2008) 112:610–18. doi: 10.1182/blood-2008-01-135319
94. Hobeika AC, Morse MA, Osada T, Peplinski S, Lyerly HK, Clay TM. Depletion of Human Regulatory T Cells. Methods Mol Biol (Clifton N.J.) (2011) 707:219–31. doi: 10.1007/978-1-61737-979-6_14
95. Onda M, Kobayashi K, Pastan I. Depletion of Regulatory T Cells in Tumors With an anti-CD25 Immunotoxin Induces CD8 T Cell-Mediated Systemic Antitumor Immunity. Proc Natl Acad Sci (2019) 116:4575. doi: 10.1073/pnas.1820388116
96. Perez-Santos M, Anaya-Ruiz M, Herrera-Camacho I, Millán-Pérez Peña L, Rosas-Murrieta NH. Bispecific anti-OX40/CTLA-4 Antibodies for Advanced Solid Tumors: A Patent Evaluation of WO2018202649. Expert Opin Ther Patents (2019) 29:921–24. doi: 10.1080/13543776.2019.1681400
97. Ascierto PA, Simeone E, Sznol M, Fu YX, Melero I. Clinical Experiences With anti-CD137 and anti-PD1 Therapeutic Antibodies. Semin Oncol (2010) 37:508–16. doi: 10.1053/j.seminoncol.2010.09.008
98. Hinner MJ, Aiba RSB, Jaquin TJ, Berger S, Dürr MC, Schlosser C, et al. Tumor-Localized Costimulatory T-Cell Engagement by the 4-1BB/HER2 Bispecific Antibody-Anticalin Fusion Prs-343. Clin Cancer Res Off J Am Assoc Cancer Res (2019) 25:5878–89. doi: 10.1158/1078-0432.Ccr-18-3654
99. Skerra A. Alternative Binding Proteins: Anticalins - Harnessing the Structural Plasticity of the Lipocalin Ligand Pocket to Engineer Novel Binding Activities. FEBS J (2008) 275:2677–83. doi: 10.1111/j.1742-4658.2008.06439.x
100. Rothe C, Skerra A. Anticalin(®) Proteins as Therapeutic Agents in Human Diseases. BioDrugs Clin Immunother Biopharma Gene Ther (2018) 32:233–43. doi: 10.1007/s40259-018-0278-1
101. Lewis KB, Meengs B, Bondensgaard K, Chin L, Hughes SD, Kjaer B, et al. Comparison of the Ability of Wild Type and Stabilized Human IgG(4) to Undergo Fab Arm Exchange With Endogenous IgG(4)in Vitro and In Vivo. Mol Immunol (2009) 46:3488–94. doi: 10.1016/j.molimm.2009.07.009
102. Rispens T, Ooijevaar-de Heer P, Bende O, Aalberse RC. Mechanism of Immunoglobulin G4 Fab-Arm Exchange. J Am Chem Soc (2011) 133:10302–11. doi: 10.1021/ja203638y
103. Silva J-P, Vetterlein O, Jose J, Peters S, Kirby H. The S228P Mutation Prevents In Vivo and In Vitro IgG4 Fab-Arm Exchange as Demonstrated Using a Combination of Novel Quantitative Immunoassays and Physiological Matrix Preparation. J Biol Chem (2015) 290:5462–69. doi: 10.1074/jbc.M114.600973
104. Vafa O, Gilliland GL, Brezski RJ, Strake B, Wilkinson T, Lacy ER, et al. An Engineered Fc Variant of an IgG Eliminates All Immune Effector Functions Via Structural Perturbations. Methods (San Diego Calif) (2014) 65:114–26. doi: 10.1016/j.ymeth.2013.06.035
105. Glaesner W, Vick AM, Millican R, Ellis B, Tschang SH, Tian Y, et al. Engineering and Characterization of the Long-Acting Glucagon-Like Peptide-1 Analogue LY2189265, an Fc Fusion Protein. Diabetes Metabol Res Rev (2010) 26:287–96. doi: 10.1002/dmrr.1080
106. Baldo BA. Chimeric Fusion Proteins Used for Therapy: Indications, Mechanisms, and Safety. Drug Saf (2015) 38:455–79. doi: 10.1007/s40264-015-0285-9
107. Perez CP, Patel N, Mardis CR, Meadows HB, Taber DJ, Pilch NA. Belatacept in Solid Organ Transplant: Review of Current Literature Across Transplant Types. Transplantation (2018) 102:1440–52. doi: 10.1097/tp.0000000000002291
108. Jarvis B, Faulds D. Etanercept: A Review of its Use in Rheumatoid Arthritis. Drugs (1999) 57:945–66. doi: 10.2165/00003495-199957060-00014
109. Theobald M, Biggs J, Dittmer D, Levine AJ, Sherman LA. Targeting p53 as a General Tumor Antigen. Proc Natl Acad Sci USA (1995) 92:11993–7. doi: 10.1073/pnas.92.26.11993
110. Theobald M, Biggs J, Hernández J, Lustgarten J, Labadie C, Sherman LA. Tolerance to p53 by A2.1-restricted Cytotoxic T Lymphocytes. J Exp Med (1997) 185:833–41. doi: 10.1084/jem.185.5.833
111. Card KF, Price-Schiavi SA, Liu B, Thomson E, Nieves E, Belmont H, et al. A Soluble Single-Chain T-cell Receptor IL-2 Fusion Protein Retains MHC-restricted Peptide Specificity and IL-2 Bioactivity. Cancer Immunol Immunother CII (2004) 53:345–57. doi: 10.1007/s00262-003-0450-3
112. Belmont HJ, Price-Schiavi S, Liu B, Card KF, Lee HI, Han KP, et al. Potent Antitumor Activity of a Tumor-Specific Soluble TCR/IL-2 Fusion Protein. Clin Immunol (Orlando Fla.) (2006) 121:29–39. doi: 10.1016/j.clim.2006.05.005
113. Zhu X, Marcus WD, Xu W, Lee HI, Han K, Egan JO, et al. Novel Human interleukin-15 Agonists. J Immunol (Baltimore Md 1950) (2009) 183:3598–607. doi: 10.4049/jimmunol.0901244
114. Han KP, Zhu X, Liu B, Jeng E, Kong L, Yovandich JL, et al. Il-15:IL-15 Receptor Alpha Superagonist Complex: High-Level Co-Expression in Recombinant Mammalian Cells, Purification and Characterization. Cytokine (2011) 56:804–10. doi: 10.1016/j.cyto.2011.09.028
115. Waldmann TA, Dubois S, Miljkovic MD, Conlon KC. Il-15 in the Combination Immunotherapy of Cancer. Front Immunol (2020) 11. doi: 10.3389/fimmu.2020.00868
116. Fantini M, David JM, Wong HC, Annunziata CM, Arlen PM, Tsang KY. An IL-15 Superagonist, ALT-803, Enhances Antibody-Dependent Cell-Mediated Cytotoxicity Elicited by the Monoclonal Antibody NEO-201 Against Human Carcinoma Cells. Cancer Biother Radiopharmaceuticals (2019) 34:147–59. doi: 10.1089/cbr.2018.2628
117. Furuya H, Chan OTM, Pagano I, Zhu C, Kim N, Peres R, et al. Effectiveness of Two Different Dose Administration Regimens of an IL-15 Superagonist Complex (ALT-803) in an Orthotopic Bladder Cancer Mouse Model. J Trans Med (2019) 17:29. doi: 10.1186/s12967-019-1778-6
118. Knudson KM, Hicks KC, Alter S, Schlom J, Gameiro SR. Mechanisms Involved in IL-15 Superagonist Enhancement of anti-PD-L1 Therapy. J Immunother Cancer (2019) 7:82. doi: 10.1186/s40425-019-0551-y
119. Fabian KP, Padget MR, Donahue RN, Solocinski K, Robbins Y, Allen CT, et al. Pd-L1 Targeting High-Affinity NK (t-haNK) Cells Induce Direct Antitumor Effects and Target Suppressive MDSC Populations. J Immunother Cancer (2020) 8. doi: 10.1136/jitc-2019-000450
120. Khongorzul P, Ling CJ, Khan FU, Ihsan AU, Zhang J. Antibody–Drug Conjugates: A Comprehensive Review. Mol Cancer Res (2020) 18:3. doi: 10.1158/1541-7786.MCR-19-0582
121. Zhao P, Zhang Y, Li W, Jeanty C, Xiang G, Dong Y. Recent Advances of Antibody Drug Conjugates for Clinical Applications. Acta Pharm Sin B (2020) 10:1589–600. doi: 10.1016/j.apsb.2020.04.012
122. Nejadmoghaddam MR, Minai-Tehrani A, Ghahremanzadeh R, Mahmoudi M, Dinarvand R, Zarnani AH. Antibody-Drug Conjugates: Possibilities and Challenges. Avicenna J Med Biotechnol (2019) 11:3–23.
123. Petersdorf SH, Kopecky KJ, Slovak M, Willman C, Nevill T, Brandwein J, et al. A Phase 3 Study of Gemtuzumab Ozogamicin During Induction and Postconsolidation Therapy in Younger Patients With Acute Myeloid Leukemia. Blood (2013) 121:4854–60. doi: 10.1182/blood-2013-01-466706
124. Connors JM, Jurczak W, Straus DJ, Ansell SM, Kim WS, Gallamini A, et al. Brentuximab Vedotin With Chemotherapy for Stage III or IV Hodgkin’s Lymphoma. New Engl J Med (2018) 378:331–44. doi: 10.1056/NEJMoa1708984
125. Kantarjian HM, DeAngelo DJ, Stelljes M, Martinelli G, Liedtke M, Stock W, et al. Inotuzumab Ozogamicin Versus Standard Therapy for Acute Lymphoblastic Leukemia. New Engl J Med (2016) 375:740–53. doi: 10.1056/NEJMoa1509277
126. Modi S, Saura C, Yamashita T, Park YH, Kim S-B, Tamura K, et al. Trastuzumab Deruxtecan in Previously Treated HER2-Positive Breast Cancer. New Engl J Med (2019) 382:610–21. doi: 10.1056/NEJMoa1914510
127. Verma S, Miles D, Gianni L, Krop IE, Welslau M, Baselga J, et al. Trastuzumab Emtansine for HER2-Positive Advanced Breast Cancer. New Engl J Med (2012) 367:1783–91. doi: 10.1056/NEJMoa1209124
128. Lattanzi M, Rosenberg JE. The Emerging Role of Antibody-Drug Conjugates in Urothelial Carcinoma. Expert Rev Anticancer Ther (2020) 20:551–61. doi: 10.1080/14737140.2020.1782201
129. Mitri Z, Constantine T, O’Regan R. The HER2 Receptor in Breast Cancer: Pathophysiology, Clinical Use, and New Advances in Therapy. Chemother Res Pract (2012) 2012:743193. doi: 10.1155/2012/743193
130. Hsu JL, Hung MC. The Role of HER2, EGFR, and Other Receptor Tyrosine Kinases in Breast Cancer. Cancer Metastasis Rev (2016) 35:575–88. doi: 10.1007/s10555-016-9649-6
131. Pillai RN, Behera M, Berry LD, Rossi MR, Kris MG, Johnson BE, et al. HER2 Mutations in Lung Adenocarcinomas: A Report From the Lung Cancer Mutation Consortium. Cancer (2017) 123:4099–105. doi: 10.1002/cncr.30869
132. Li BT, Ross DS, Aisner DL, Chaft JE, Hsu M, Kako SL, et al. Her2 Amplification and HER2 Mutation Are Distinct Molecular Targets in Lung Cancers. J Thorac Oncol Off Publ Int Assoc Study Lung Cancer (2016) 11:414–9. doi: 10.1016/j.jtho.2015.10.025
133. Chen PC, Yu HJ, Chang YH, Pan CC. Her2 Amplification Distinguishes a Subset of non-Muscle-Invasive Bladder Cancers With a High Risk of Progression. J Clin Pathol (2013) 66:113–9. doi: 10.1136/jclinpath-2012-200944
134. Lim SD, Cho YM, Choi GS, Park HK, Paick SH, Kim WY, et al. Clinical Significance of Substaging and HER2 Expression in Papillary Nonmuscle Invasive Urothelial Cancers of the Urinary Bladder. J Korean Med Sci (2015) 30:1068–77. doi: 10.3346/jkms.2015.30.8.1068
135. Yao X, Jiang J, Wang X, Huang C, Li D, Xie K, et al. A Novel Humanized anti-HER2 Antibody Conjugated With MMAE Exerts Potent Anti-Tumor Activity. Breast Cancer Res Treat (2015) 153:123–33. doi: 10.1007/s10549-015-3503-3
136. Doronina SO, Toki BE, Torgov MY, Mendelsohn BA, Cerveny CG, Chace DF, et al. Development of Potent Monoclonal Antibody Auristatin Conjugates for Cancer Therapy. Nat Biotechnol (2003) 21:778–84. doi: 10.1038/nbt832
137. Rinnerthaler G, Gampenrieder SP, Greil R. Her2 Directed Antibody-Drug-Conjugates Beyond T-DM1 in Breast Cancer. Int J Mol Sci (2019) 20. doi: 10.3390/ijms20051115
138. Gong J, Shen L, Wang W, Fang J. Safety, Pharmacokinetics and Efficacy of RC48-ADC in a Phase I Study in Patients With HER2-overexpression Advanced Solid Cancer. J Clin Oncol (2018) 36:e16059–e59. doi: 10.1200/JCO.2018.36.15_suppl.e16059
139. Chang E, Weinstock C, Zhang L, Charlab R, Dorff SE, Gong Y, et al. Fda Approval Summary: Enfortumab Vedotin for Locally Advanced or Metastatic Urothelial Carcinoma. Clin Cancer Res Off J Am Assoc Cancer Res (2020). doi: 10.1158/1078-0432.Ccr-20-2275
140. Rosenberg JE, O’Donnell PH, Balar AV, McGregor BA, Heath EI, Yu EY, et al. Pivotal Trial of Enfortumab Vedotin in Urothelial Carcinoma After Platinum and Anti-Programmed Death 1/Programmed Death Ligand 1 Therapy. J Clin Oncol Off J Am Soc Clin Oncol (2019) 37:2592–600. doi: 10.1200/jco.19.01140
141. Takai Y, Nakanishi H. Nectin and Afadin: Novel Organizers of Intercellular Junctions. J Cell Sci (2003) 116:17. doi: 10.1242/jcs.00167
142. Takai Y, Ikeda W, Ogita H, Rikitake Y. The Immunoglobulin-Like Cell Adhesion Molecule Nectin and Its Associated Protein Afadin. Annu Rev Cell Dev Biol (2008) 24:309–42. doi: 10.1146/annurev.cellbio.24.110707.175339
143. Takai Y, Miyoshi J, Ikeda W, Ogita H. Nectins and Nectin-Like Molecules: Roles in Contact Inhibition of Cell Movement and Proliferation. Nat Rev Mol Cell Biol (2008) 9:603–15. doi: 10.1038/nrm2457
144. Fabre-Lafay S, Monville F, Garrido-Urbani S, Berruyer-Pouyet C, Ginestier C, Reymond N, et al. Nectin-4 is a New Histological and Serological Tumor Associated Marker for Breast Cancer. BMC Cancer (2007) 7:73–3. doi: 10.1186/1471-2407-7-73
145. Takano A, Ishikawa N, Nishino R, Masuda K, Yasui W, Inai K, et al. Identification of Nectin-4 Oncoprotein as a Diagnostic and Therapeutic Target for Lung Cancer. Cancer Res (2009) 69:6694. doi: 10.1158/0008-5472.CAN-09-0016
146. Derycke MS, Pambuccian SE, Gilks CB, Kalloger SE, Ghidouche A, Lopez M, et al. Nectin 4 Overexpression in Ovarian Cancer Tissues and Serum: Potential Role as a Serum Biomarker. Am J Clin Pathol (2010) 134:835–45. doi: 10.1309/AJCPGXK0FR4MHIHB
147. Hoffman-Censits JH, Choi W, Lombardo K, Hahn NM, McConkey DJ, McGuire B, et al. Expression of Nectin-4 in Bladder Cancer With Variant Histology. J Clin Oncol (2020) 38:546–46. doi: 10.1200/JCO.2020.38.6_suppl.546
148. Zhang Y, Liu S, Wang L, Wu Y, Hao J, Wang Z, et al. A Novel PI3K/AKT Signaling Axis Mediates Nectin-4-induced Gallbladder Cancer Cell Proliferation, Metastasis and Tumor Growth. Cancer Lett (2016) 375:179–89. doi: 10.1016/j.canlet.2016.02.049
149. Pavlova NN, Pallasch C, Elia AE, Braun CJ, Westbrook TF, Hemann M, et al. A Role for PVRL4-driven Cell-Cell Interactions in Tumorigenesis. eLife (2013) 2:e00358. doi: 10.7554/eLife.00358
150. Siddharth S, Goutam K, Das S, Nayak A, Nayak D, Sethy C, et al. Nectin-4 is a Breast Cancer Stem Cell Marker That Induces WNT/β-Catenin Signaling Via Pi3k/Akt Axis. Int J Biochem Cell Biol (2017) 89:85–94. doi: 10.1016/j.biocel.2017.06.007
151. Green LL. Antibody Engineering Via Genetic Engineering of the Mouse: XenoMouse Strains are a Vehicle for the Facile Generation of Therapeutic Human Monoclonal Antibodies. J Immunol Methods (1999) 231:11–23. doi: 10.1016/s0022-1759(99)00137-4
152. Challita-Eid PM, Satpayev D, Yang P, An Z, Morrison K, Shostak Y, et al. Enfortumab Vedotin Antibody-Drug Conjugate Targeting Nectin-4 is a Highly Potent Therapeutic Agent in Multiple Preclinical Cancer Models. Cancer Res (2016) 76:3003–13. doi: 10.1158/0008-5472.Can-15-1313
153. Sussman D, Smith LM, Anderson ME, Duniho S, Hunter JH, Kostner H, et al. Sgn-LIV1A: A Novel Antibody-Drug Conjugate Targeting LIV-1 for the Treatment of Metastatic Breast Cancer. Mol Cancer Ther (2014) 13:2991–3000. doi: 10.1158/1535-7163.Mct-13-0896
154. Lyon RP, Meyer DL, Setter JR, Senter PD. Conjugation of Anticancer Drugs Through Endogenous Monoclonal Antibody Cysteine Residues. Methods Enzymol (2012) 502:123–38. doi: 10.1016/b978-0-12-416039-2.00006-9
155. Barok M, Joensuu H, Isola J. Trastuzumab Emtansine: Mechanisms of Action and Drug Resistance. Breast Cancer Res BCR (2014) 16:209. doi: 10.1186/bcr3621
156. Lewis Phillips GD, Li G, Dugger DL, Crocker LM, Parsons KL, Mai E, et al. Targeting HER2-positive Breast Cancer With trastuzumab-DM1, an Antibody-Cytotoxic Drug Conjugate. Cancer Res (2008) 68:9280–90. doi: 10.1158/0008-5472.Can-08-1776
157. Chen L, Wang L, Shion H, Yu C, Yu YQ, Zhu L, et al. In-Depth Structural Characterization of Kadcyla® (Ado-Trastuzumab Emtansine) and its Biosimilar Candidate. mAbs (2016) 8:1210–23. doi: 10.1080/19420862.2016.1204502
158. Ritchie M, Tchistiakova L, Scott N. Implications of Receptor-Mediated Endocytosis and Intracellular Trafficking Dynamics in the Development of Antibody Drug Conjugates. mAbs (2013) 5:13–21. doi: 10.4161/mabs.22854
159. Erickson HK, Park PU, Widdison WC, Kovtun YV, Garrett LM, Hoffman K, et al. Antibody-Maytansinoid Conjugates are Activated in Targeted Cancer Cells by Lysosomal Degradation and Linker-Dependent Intracellular Processing. Cancer Res (2006) 66:4426–33. doi: 10.1158/0008-5472.Can-05-4489
160. Chari RV. Targeted Cancer Therapy: Conferring Specificity to Cytotoxic Drugs. Accounts Chem Res (2008) 41:98–107. doi: 10.1021/ar700108g
161. Ogitani Y, Aida T, Hagihara K, Yamaguchi J, Ishii C, Harada N, et al. DS-8201a, A Novel HER2-Targeting ADC With a Novel Dna Topoisomerase I Inhibitor, Demonstrates a Promising Antitumor Efficacy With Differentiation From T-DM1. Clin Cancer Res Off J Am Assoc Cancer Res (2016) 22:5097–108. doi: 10.1158/1078-0432.Ccr-15-2822
162. Andrikopoulou A, Zografos E, Liontos M, Koutsoukos K, Dimopoulos M-A, Zagouri F. Trastuzumab Deruxtecan (DS-8201a): The Latest Research and Advances in Breast Cancer. Clin Breast Cancer (2020). doi: 10.1016/j.clbc.2020.08.006
163. Li G, Guo J, Shen B-Q, Yadav DB, Sliwkowski MX, Crocker LM, et al. Mechanisms of Acquired Resistance to Trastuzumab Emtansine in Breast Cancer Cells. Mol Cancer Ther (2018) 17:1441. doi: 10.1158/1535-7163.MCT-17-0296
164. Koshkin VS, O’Donnell P, Yu EY, Grivas P. Systematic Review: Targeting HER2 in Bladder Cancer. Bladder Cancer (2019) 5:1–12. doi: 10.3233/BLC-180196
165. Stein R, Chen S, Sharkey RM, Goldenberg DM. Murine Monoclonal Antibodies Raised Against Human non-Small Cell Carcinoma of the Lung: Specificity and Tumor Targeting. Cancer Res (1990) 50:1330–6.
166. Stein R, Basu A, Chen S, Shih LB, Goldenberg DM. Specificity and Properties of MAb Rs7-3G11 and the Antigen Defined by This Pancarcinoma Monoclonal Antibody. Int J Cancer (1993) 55:938–46. doi: 10.1002/ijc.2910550611
167. Basu A, Goldenberg DM, Stein R. The Epithelial/Carcinoma Antigen EGP-1, Recognized by Monoclonal Antibody RS7-3G11, is Phosphorylated on Serine 303. Int J Cancer (1995) 62:472–9. doi: 10.1002/ijc.2910620419
168. Cubas R, Li M, Chen C, Yao Q. Trop2: A Possible Therapeutic Target for Late Stage Epithelial Carcinomas. Biochim Biophys Acta (2009) 1796:309–14. doi: 10.1016/j.bbcan.2009.08.001
169. Shvartsur A, Bonavida B. Trop2 and its Overexpression in Cancers: Regulation and Clinical/Therapeutic Implications. Genes Cancer (2015) 6:84–105. doi: 10.18632/genesandcancer.40
170. Bignotti E, Zanotti L, Calza S, Falchetti M, Lonardi S, Ravaggi A, et al. Trop-2 Protein Overexpression is an Independent Marker for Predicting Disease Recurrence in Endometrioid Endometrial Carcinoma. BMC Clin Pathol (2012) 12:22–2. doi: 10.1186/1472-6890-12-22
171. Stepan LP, Trueblood ES, Hale K, Babcook J, Borges L, Sutherland CL. Expression of Trop2 Cell Surface Glycoprotein in Normal and Tumor Tissues: Potential Implications as a Cancer Therapeutic Target. J Histochem Cytochem Off J Histochem Soc (2011) 59:701–10. doi: 10.1369/0022155411410430
172. Chen X, Pang B, Liang Y, Xu S-C, Xin T, Fan H-T, et al. Overexpression of EpCAM and Trop2 in Pituitary Adenomas. Int J Clin Exp Pathol (2014) 7(11):7907–14.
173. Shih LB, Xuan H, Aninipot R, Stein R, Goldenberg DM. In Vitro and In Vivo Reactivity of an Internalizing Antibody, RS7, With Human Breast Cancer. Cancer Res (1995) 55:5857s–63s.
174. Govindan SV, Stein R, Qu Z, Chen S, Andrews P, Ma H, et al. Preclinical Therapy of Breast Cancer With a Radioiodinated Humanized Anti-EGP-1 Monoclonal Antibody: Advantage of a Residualizing Iodine Radiolabel. Breast Cancer Res Treat (2004) 84:173–82. doi: 10.1023/B:BREA.0000018417.02580.ef
175. Vlachostergios PJ, Jakubowski CD, Niaz MJ, Lee A, Thomas C, Hackett AL, et al. Antibody-Drug Conjugates in Bladder Cancer. Bladder Cancer (2018) 4:247–59. doi: 10.3233/blc-180169
176. Goldenberg DM, Sharkey RM. Sacituzumab Govitecan, a Novel, Third-Generation, Antibody-Drug Conjugate (ADC) for Cancer Therapy. Expert Opin Biol Ther (2020) 20:871–85. doi: 10.1080/14712598.2020.1757067
177. Starodub AN, Ocean AJ, Shah MA, Guarino MJ, Picozzi VJ Jr., Vahdat LT, et al. First-in-Human Trial of a Novel Anti-Trop-2 Antibody-SN-38 Conjugate, Sacituzumab Govitecan, for the Treatment of Diverse Metastatic Solid Tumors. Clin Cancer Res Off J Am Assoc Cancer Res (2015) 21:3870–8. doi: 10.1158/1078-0432.Ccr-14-3321
178. Morrison K, Challita-Eid PM, Raitano A, An Z, Yang P, Abad JD, et al. Development of ASG-15ME, a Novel Antibody-Drug Conjugate Targeting SLITRK6, a New Urothelial Cancer Biomarker. Mol Cancer Ther (2016) 15:1301–10. doi: 10.1158/1535-7163.Mct-15-0570
179. Kobayashi H, Choyke PL. Near-Infrared Photoimmunotherapy of Cancer. Accounts Chem Res (2019) 52:2332–39. doi: 10.1021/acs.accounts.9b00273
180. Di Maida F, Mari A, Scalici Gesolfo C, Cangemi A, Allegro R, Sforza S, et al. Epidermal Growth Factor Receptor (Egfr) Cell Expression During Adjuvant Treatment After Transurethral Resection for Non-Muscle-Invasive Bladder Cancer: A New Potential Tool to Identify Patients At Higher Risk of Disease Progression. Clin Genitourin Cancer (2019) 17:e751–e58. doi: 10.1016/j.clgc.2019.04.008
181. Hashmi AA, Hussain ZF, Irfan M, Khan EY, Faridi N, Naqvi H, et al. Prognostic Significance of Epidermal Growth Factor Receptor (EGFR) Over Expression in Urothelial Carcinoma of Urinary Bladder. BMC Urol (2018) 18:59. doi: 10.1186/s12894-018-0373-0
182. Chaux A, Cohen JS, Schultz L, Albadine R, Jadallah S, Murphy KM, et al. High Epidermal Growth Factor Receptor Immunohistochemical Expression in Urothelial Carcinoma of the Bladder is Not Associated With EGFR Mutations in Exons 19 and 21: A Study Using Formalin-Fixed, Paraffin-Embedded Archival Tissues. Hum Pathol (2012) 43:1590–5. doi: 10.1016/j.humpath.2011.11.016
183. Railkar R, Krane LS, Li QQ, Sanford T, Siddiqui MR, Haines D, et al. Epidermal Growth Factor Receptor (EGFR)-Targeted Photoimmunotherapy (PIT) for the Treatment of EGFR-expressing Bladder Cancer. Mol Cancer Ther (2017) 16:2201–14. doi: 10.1158/1535-7163.Mct-16-0924
184. Kassouf W, Black PC, Tuziak T, Bondaruk J, Lee S, Brown GA, et al. Distinctive Expression Pattern of ErbB Family Receptors Signifies an Aggressive Variant of Bladder Cancer. J Urol (2008) 179:353–8. doi: 10.1016/j.juro.2007.08.087
185. Memon AA, Sorensen BS, Meldgaard P, Fokdal L, Thykjaer T, Nexo E. The Relation Between Survival and Expression of HER1 and HER2 Depends on the Expression of HER3 and HER4: A Study in Bladder Cancer Patients. Br J Cancer (2006) 94:1703–9. doi: 10.1038/sj.bjc.6603154
186. Kishimoto S, Bernardo M, Saito K, Koyasu S, Mitchell JB, Choyke PL, et al. Evaluation of Oxygen Dependence on In Vitro and In Vivo Cytotoxicity of Photoimmunotherapy Using IR-700-antibody Conjugates. Free Radical Biol Med (2015) 85:24–32. doi: 10.1016/j.freeradbiomed.2015.03.038
187. Siddiqui MR, Railkar R, Sanford T, Crooks DR, Eckhaus MA, Haines D, et al. Targeting Epidermal Growth Factor Receptor (EGFR) and Human Epidermal Growth Factor Receptor 2 (Her2) Expressing Bladder Cancer Using Combination Photoimmunotherapy (Pit). Sci Rep (2019) 9:2084. doi: 10.1038/s41598-019-38575-x
188. Larson SM, Carrasquillo JA, Cheung N-KV, Press OW. Radioimmunotherapy of Human Tumours. Nat Rev Cancer (2015) 15:347–60. doi: 10.1038/nrc3925
189. Pfost B, Seidl C, Autenrieth M, Saur D, Bruchertseifer F, Morgenstern A, et al. Intravesical Alpha-Radioimmunotherapy With 213Bi-anti-EGFR-mAb Defeats Human Bladder Carcinoma in Xenografted Nude Mice. J Nucl Med Off Publication Soc Nucl Med (2009) 50:1700–8. doi: 10.2967/jnumed.109.065961
190. Mirzadeh S, Brechbiel MW, Atcher RW, Gansow OA. Radiometal Labeling of Immunoproteins: Covalent Linkage of 2-(4-Isothiocyanatobenzyl)Diethylenetriaminepentaacetic Acid Ligands to Immunoglobulin. Bioconjug Chem (1990) 1:59–65. doi: 10.1021/bc00001a007
191. Fazel J, Rötzer S, Seidl C, Feuerecker B, Autenrieth M, Weirich G, et al. Fractionated Intravesical Radioimmunotherapy With (213)Bi-anti-EGFR-MAb is Effective Without Toxic Side-Effects in a Nude Mouse Model of Advanced Human Bladder Carcinoma. Cancer Biol Ther (2015) 16:1526–34. doi: 10.1080/15384047.2015.1071735
192. Akbari B, Farajnia S, Ahdi Khosroshahi S, Safari F, Yousefi M, Dariushnejad H, et al. Immunotoxins in Cancer Therapy: Review and Update. Int Rev Immunol (2017) 36:207–19. doi: 10.1080/08830185.2017.1284211
193. Went PT, Lugli A, Meier S, Bundi M, Mirlacher M, Sauter G, et al. Frequent EpCam Protein Expression in Human Carcinomas. Hum Pathol (2004) 35:122–8. doi: 10.1016/j.humpath.2003.08.026
194. Keller L, Werner S, Pantel K. Biology and Clinical Relevance of Epcam. Cell Stress (2019) 3:165–80. doi: 10.15698/cst2019.06.188
195. Brunner A, Prelog M, Verdorfer I, Tzankov A, Mikuz G, Ensinger C. EpCAM is Predominantly Expressed in High Grade and Advanced Stage Urothelial Carcinoma of the Bladder. J Clin Pathol (2008) 61:307–10. doi: 10.1136/jcp.2007.049460
196. Bryan RT, Shimwell NJ, Wei W, Devall AJ, Pirrie SJ, James ND, et al. Urinary EpCAM in Urothelial Bladder Cancer Patients: Characterisation and Evaluation of Biomarker Potential. Br J Cancer (2014) 110:679–85. doi: 10.1038/bjc.2013.744
197. Zimmermann S, Wels W, Froesch BA, Gerstmayer B, Stahel RA, Zangemeister-Wittke U. A Novel Immunotoxin Recognising the Epithelial Glycoprotein-2 has Potent Antitumoural Activity on Chemotherapy-Resistant Lung Cancer. Cancer Immunol Immunother (1997) 44:1–9. doi: 10.1007/s002620050348
198. Krebber A, Bornhauser S, Burmester J, Honegger A, Willuda J, Bosshard HR, et al. Reliable Cloning of Functional Antibody Variable Domains From Hybridomas and Spleen Cell Repertoires Employing a Reengineered Phage Display System. J Immunol Methods (1997) 201:35–55. doi: 10.1016/S0022-1759(96)00208-6
199. Willuda J, Honegger A, Waibel R, Schubiger PA, Stahel R, Zangemeister-Wittke U, et al. High Thermal Stability Is Essential for Tumor Targeting of Antibody Fragments. Cancer Res (1999) 59:5758.
200. Di Paolo C, Willuda J, Kubetzko S, Lauffer I, Tschudi D, Waibel R, et al. A Recombinant Immunotoxin Derived From a Humanized Epithelial Cell Adhesion Molecule-Specific Single-Chain Antibody Fragment has Potent and Selective Antitumor Activity. Clin Cancer Res Off J Am Assoc Cancer Res (2003) 9:2837–48.
201. Seetharam S, Chaudhary VK, FitzGerald D, Pastan I. Increased Cytotoxic Activity of Pseudomonas Exotoxin and Two Chimeric Toxins Ending in KDEL. J Biol Chem (1991) 266:17376–81. doi: 10.1016/S0021-9258(19)47383-3
202. Baeuerle PA, Gires O. EpCAM (CD326) Finding its Role in Cancer. Br J Cancer (2007) 96:417–23. doi: 10.1038/sj.bjc.6603494
203. Schlake T, Thess A, Fotin-Mleczek M, Kallen KJ. Developing mRNA-vaccine Technologies. RNA Biol (2012) 9:1319–30. doi: 10.4161/rna.22269
204. Bauer T, Patel M, Jimeno A, Wang D, McDermott J, Zacharek S, et al. Abstract CT210: A Phase I, Open-Label, Multicenter, Dose Escalation Study of mRNA-2752, a Lipid Nanoparticle Encapsulating mRNAs Encoding Human OX40L, Il-23, and IL-36γ, for Intratumoral Injection Alone and in Combination With Immune Checkpoint Blockade. Cancer Res (2019) 79:CT210. doi: 10.1158/1538-7445.AM2019-CT210
205. Gross G, Waks T, Eshhar Z. Expression of immunoglobulin-T-cell Receptor Chimeric Molecules as Functional Receptors With Antibody-Type Specificity. Proc Natl Acad Sci USA (1989) 86:10024–28. doi: 10.1073/pnas.86.24.10024
206. Subklewe M, von Bergwelt-Baildon M, Humpe A. Chimeric Antigen Receptor T Cells: A Race to Revolutionize Cancer Therapy. Transfus Med Hemother Offizielles Organ Dtsch Ges fur Transfusionsmed und Immunhamatol (2019) 46:15–24. doi: 10.1159/000496870
207. Feins S, Kong W, Williams EF, Milone MC, Fraietta JA. An Introduction to Chimeric Antigen Receptor (CAR) T-cell Immunotherapy for Human Cancer. Am J Hematol (2019) 94:S3–s9. doi: 10.1002/ajh.25418
208. Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, et al. Chimeric Antigen Receptor-Modified T Cells for Acute Lymphoid Leukemia. New Engl J Med (2013) 368:1509–18. doi: 10.1056/NEJMoa1215134
209. Zheng P-P, Kros JM, Li J. Approved CAR T Cell Therapies: Ice Bucket Challenges on Glaring Safety Risks and Long-Term Impacts. Drug Discov Today (2018) 23:1175–82. doi: 10.1016/j.drudis.2018.02.012
210. Schreiber H, Hänze J, Nimphius W, Verburg FA, Luster M, Hofmann R, et al. Prostate Specific Membrane Antigen (PSMA) in Urothelial Cell Carcinoma (UCC) is Associated With Tumor Grading and Staging. J Cancer Res Clin Oncol (2020) 146:305–13. doi: 10.1007/s00432-019-03113-9
211. Cheung A, Bax HJ, Josephs DH, Ilieva KM, Pellizzari G, Opzoomer J, et al. Targeting Folate Receptor Alpha for Cancer Treatment. Oncotarget (2016) 7:52553–74. doi: 10.18632/oncotarget.9651
212. Parker N, Turk MJ, Westrick E, Lewis JD, Low PS, Leamon CP. Folate Receptor Expression in Carcinomas and Normal Tissues Determined by a Quantitative Radioligand Binding Assay. Anal Biochem (2005) 338:284–93. doi: 10.1016/j.ab.2004.12.026
213. Lu Y, Low PS. Immunotherapy of Folate Receptor-Expressing Tumors: Review of Recent Advances and Future Prospects. J Controlled Release Off J Controlled Release Soc (2003) 91:17–29. doi: 10.1016/s0168-3659(03)00215-3
214. Yu D-S, Yan H-Y, Wu C-L. Folate Receptor Expression in Bladder Cancer and its Correlation With Tumor Behaviors and Clinical Outcome. J Cancer Res Pract (2017) 4:130–33. doi: 10.1016/j.jcrpr.2017.05.001
215. NCI Autologous Fra-4SCAR-Expressing T Cells 4SCAR-Fra . National Cancer Institute. Available at: https://www.cancer.gov/publications/dictionaries/cancer-drug/def/autologous-fra-4scar-expressing-t-cells-4scar-fra (Accessed 11.02.2021).
216. NCI Autologous PSMA-4SCAR-Expressing T Cells 4SCAR-PSMA . National Cancer Institute. Available at: https://www.cancer.gov/publications/dictionaries/cancer-drug/def/autologous-psma-4scar-expressing-t-cells-4scar-psma (Accessed 11.02.2021).
217. Li J, Li W, Huang K, Zhang Y, Kupfer G, Zhao Q. Chimeric Antigen Receptor T Cell (CAR-T) Immunotherapy for Solid Tumors: Lessons Learned and Strategies for Moving Forward. J Hematol Oncol (2018) 11:22. doi: 10.1186/s13045-018-0568-6
218. Ma S, Li X, Wang X, Cheng L, Li Z, Zhang C, et al. Current Progress in CAR-T Cell Therapy for Solid Tumors. Int J Biol Sci (2019) 15:2548–60. doi: 10.7150/ijbs.34213
219. Kaji EH, Leiden JM. Gene and Stem Cell Therapies. JAMA (2001) 285:545–50. doi: 10.1001/jama.285.5.545
220. Mulligan RC. The Basic Science of Gene Therapy. Sci (New York N.Y.) (1993) 260:926–32. doi: 10.1126/science.8493530
221. Walther W, Schlag PM. Current Status of Gene Therapy for Cancer. Curr Opin Oncol (2013) 25:659–64. doi: 10.1097/cco.0000000000000004
222. Lundstrom K. Viral Vectors in Gene Therapy. Dis (Basel Switzerland) (2018) 6. doi: 10.3390/diseases6020042
223. Ramesh N, Ge Y, Ennist DL, Zhu M, Mina M, Ganesh S, et al. CG0070, a Conditionally Replicating Granulocyte Macrophage Colony-Stimulating Factor–Armed Oncolytic Adenovirus for the Treatment of Bladder Cancer. Clin Cancer Res Off J Am Assoc Cancer Res (2006) 12:305–13. doi: 10.1158/1078-0432.Ccr-05-1059
224. Broudy VC, Kaushansky K, Harlan JM, Adamson JW. Interleukin 1 Stimulates Human Endothelial Cells to Produce Granulocyte-Macrophage Colony-Stimulating Factor and Granulocyte Colony-Stimulating Factor. J Immunol (Baltimore Md 1950) (1987) 139:464–8.
225. Burke JM, Lamm DL, Meng MV, Nemunaitis JJ, Stephenson JJ, Arseneau JC, et al. A First in Human Phase 1 Study of CG0070, a GM-CSF Expressing Oncolytic Adenovirus, for the Treatment of Nonmuscle Invasive Bladder Cancer. J Urol (2012) 188:2391–7. doi: 10.1016/j.juro.2012.07.097
226. Packiam VT, Lamm DL, Barocas DA, Trainer A, Fand B, Davis RL 3rd, et al. An Open Label, Single-Arm, Phase II Multicenter Study of the Safety and Efficacy of CG0070 Oncolytic Vector Regimen in Patients With BCG-unresponsive non-Muscle-Invasive Bladder Cancer: Interim Results. Urol Oncol (2018) 36:440–47. doi: 10.1016/j.urolonc.2017.07.005
227. Iqbal Ahmed CM, Johnson DE, Demers GW, Engler H, Howe JA, Wills KN, et al. Interferon α2b Gene Delivery Using Adenoviral Vector Causes Inhibition of Tumor Growth in Xenograft Models From a Variety of Cancers. Cancer Gene Ther (2001) 8:788–95. doi: 10.1038/sj.cgt.7700364
228. Zhang JF, Hu C, Geng Y, Selm J, Klein SB, Orazi A, et al. Treatment of a Human Breast Cancer Xenograft With an Adenovirus Vector Containing an Interferon Gene Results in Rapid Regression Due to Viral Oncolysis and Gene Therapy. Proc Natl Acad Sci USA (1996) 93:4513–8. doi: 10.1073/pnas.93.9.4513
229. Ahmed CM, Sugarman BJ, Johnson DE, Bookstein RE, Saha DP, Nagabhushan TL, et al. In Vivo Tumor Suppression by Adenovirus-Mediated Interferon alpha2b Gene Delivery. Hum Gene Ther (1999) 10:77–84. doi: 10.1089/10430349950019200
230. Klingler CH. Glycosaminoglycans: How Much do We Know About Their Role in the Bladder? Urologia (2016) 83 Suppl 1:11–4. doi: 10.5301/uro.5000184
231. Duplisea JJ, Mokkapati S, Plote D, Schluns KS, McConkey DJ, Yla-Herttuala S, et al. The Development of Interferon-Based Gene Therapy for BCG Unresponsive Bladder Cancer: From Bench to Bedside. World J Urol (2019) 37:2041–49. doi: 10.1007/s00345-018-2553-7
232. Connor RJ, Engler H, Machemer T, Philopena JM, Horn MT, Sutjipto S, et al. Identification of Polyamides That Enhance Adenovirus-Mediated Gene Expression in the Urothelium. Gene Ther (2001) 8:41–8. doi: 10.1038/sj.gt.3301348
233. Yamashita M, Rosser CJ, Zhou J-H, Zhang X-Q, Connor RJ, Engler H, et al. Syn3 Provides High Levels of Intravesical Adenoviral-Mediated Gene Transfer for Gene Therapy of Genetically Altered Urothelium and Superficial Bladder Cancer. Cancer Gene Ther (2002) 9:687–91. doi: 10.1038/sj.cgt.7700488
234. Benedict WF, Tao Z, Kim C-S, Zhang X, Zhou J-H, Adam L, et al. Intravesical Ad-Ifnα Causes Marked Regression of Human Bladder Cancer Growing Orthotopically in Nude Mice and Overcomes Resistance to IFN-α Protein. Mol Ther (2004) 10:525–32. doi: 10.1016/j.ymthe.2004.05.027
235. Dinney Colin PN, Fisher Mark B, Navai N, O’Donnell Michael A, Cutler D, Abraham A, et al. Phase I Trial of Intravesical Recombinant Adenovirus Mediated Interferon-α2b Formulated in Syn3 for Bacillus Calmette-Guérin Failures in Nonmuscle Invasive Bladder Cancer. J Urol (2013) 190:850–56. doi: 10.1016/j.juro.2013.03.030
236. Shore ND, Boorjian SA, Canter DJ, Ogan K, Karsh LI, Downs TM, et al. Intravesical Rad-Ifnα/Syn3 for Patients With High-Grade, Bacillus Calmette-Guerin-Refractory or Relapsed non-Muscle-Invasive Bladder Cancer: A Phase II Randomized Study. J Clin Oncol Off J Am Soc Clin Oncol (2017) 35:3410–16. doi: 10.1200/jco.2017.72.3064
237. Boorjian SA, Alemozaffar M, Konety BR, Shore ND, Gomella LG, Kamat AM, et al. Intravesical Nadofaragene Firadenovec Gene Therapy for BCG-unresponsive non-Muscle-Invasive Bladder Cancer: A Single-Arm, Open-Label, Repeat-Dose Clinical Trial. Lancet Oncol (2020) 22:107–17. doi: 10.1016/S1470-2045(20)30540-4
Keywords: antibody engineering, protein engineering, bladder cancer, urothelial carcinoma, ADC, immunotherapy, immuno-oncology, recombinant viral vector vaccines
Citation: Bogen JP, Grzeschik J, Jakobsen J, Bähre A, Hock B and Kolmar H (2021) Treating Bladder Cancer: Engineering of Current and Next Generation Antibody-, Fusion Protein-, mRNA-, Cell- and Viral-Based Therapeutics. Front. Oncol. 11:672262. doi: 10.3389/fonc.2021.672262
Received: 25 February 2021; Accepted: 11 May 2021;
Published: 27 May 2021.
Edited by:
Ja Hyeon Ku, Seoul National University, South KoreaReviewed by:
Massimo Fantini, Precision Biologics, Inc., United StatesCopyright © 2021 Bogen, Grzeschik, Jakobsen, Bähre, Hock and Kolmar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Harald Kolmar, SGFyYWxkLktvbG1hckBUVS1EYXJtc3RhZHQuZGU=; Jan P. Bogen, Qm9nZW5AQmlvY2hlbWllLVRVRC5kZQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.