
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Oncol., 12 October 2021
Sec. Gastrointestinal Cancers
Volume 11 - 2021 | https://doi.org/10.3389/fonc.2021.668466
 Luigi Tornillo1,2*†
Luigi Tornillo1,2*† Frank Serge Lehmann3†Andrea Garofoli1,4Viola Paradiso1
Frank Serge Lehmann3†Andrea Garofoli1,4Viola Paradiso1 Charlotte K. Y. Ng5,6
Charlotte K. Y. Ng5,6 Salvatore Piscuoglio1,4*
Salvatore Piscuoglio1,4*Serrated lesions of the colorectum are the precursors of 15–30% of colorectal cancers (CRCs). These lesions have a peculiar morphological appearance, and they are more difficult to detect than conventional adenomatous polyps. In this study, we sought to define the genomic landscape of these lesions using high-depth targeted sequencing. Eight sessile serrated lesions without dysplasia (SSL), three sessile serrated lesions with dysplasia (SSL/D), two traditional serrated adenomas (TSA), and three tubular adenomas (TA) were retrieved from the files of the Institute of Pathology of the University Hospital Basel and from the GILAB AG, Allschwil, Switzerland. Samples were microdissected together with the matched normal counterpart, and DNA was extracted for library preparation. Library preparation was performed using the Oncomine Comprehensive Assay targeting 161 common cancer driver genes. Somatic genetic alterations were defined using state-of-the-art bioinformatic analysis. Most SSLs, as well as all SSL/Ds and TSAs, showed the classical BRAF p.V600E mutation. The BRAF-mutant TSAs showed additional alterations in CTNNB1, NF1, TP53, NRAS, PIK3CA, while TA showed a consistently different profile, with mutations in ARID1A (two cases), SMAD4, CDK12, ERBB3, and KRAS. In conclusion, our results provide evidence that SSL/D and TSA are similar in somatic mutations with the BRAF hotspot somatic mutation as a major driver of the disease. On the other hand, TAs show a different constellation of somatic mutations such as ARID1A loss of function.
Colorectal cancer (CRC) is one of the most frequent tumors worldwide (1). The development of CRC represents a classical example of carcinogenesis, with the adenoma-carcinoma sequence being a well-established model for several epithelial tumors (2). Multiple (epi)genetic alterations in the epithelial cells of the intestine lead to the development first of an adenoma, that in a minority of cases may transform into CRC (2). In the last 20 years, however, the landscape of colorectal carcinogenesis has been partially modified by the explosion of molecular biology techniques. CRC is nowadays seen as a heterogeneous disease (3, 4). While two-thirds of CRCs arise through the “classical” chromosomal instability pathway, 15–30% probably arise through the “serrated neoplasia pathway” (5–7). The molecular events underlying the development of serrated neoplasia have been partially unraveled. In general, CRC can be roughly classified according to the following criteria (1): APC-status (mutated or wild-type) (2); microsatellite status (stable or unstable, MSS or MSI) (3); KRAS status (mutated or wild-type) (4); BRAF status (mutated or wild-type) (5); methylation status (CpG Islands methylation phenotype high or low, CIMP-H or CIMP-L) (6, 8, 9). The most interesting and important topic is that the molecular and morphological heterogeneity of CRC corresponds to clinical heterogeneity (e.g., localization, prognosis, response to therapy). This has led to dramatic changes in the surveillance, prevention, and treatment of CRC (3).
The morphological features of the serrated pathway are the so-called serrated polyps. Currently, the WHO classification recognizes three major categories of serrated lesions, namely, the hyperplastic polyp (HP), the sessile serrated lesion (SSL, with or without dysplasia), and the traditional serrated adenoma (TSA), based mainly on the work of Torlakovic and Snover (9, 10). This classification is based on the distinct histological and cytological features such as morphology and the “serrated” (i.e., resembling “sawtooth”) architecture of colon crypts, and the position and the extent of the proliferative zone in the crypts (7, 11, 12). While the preneoplastic potential of HPs is still debated, “true” preneoplastic lesions are SSL and TSA. Because SSLss have a higher incidence, they can be viewed as the most important precursors of malignancy in the serrated pathway (12).
The morphology of the serrated pathway seems to match, at least partially, specific driver molecular alterations. The most frequent is the V600E mutation in the BRAF proto-oncogene (13, 14). Additional epigenetic changes as observed in the CIMP-H phenotype, namely, hypermethylation of the MLH1 gene, may lead to microsatellite instability (7, 13, 14) and subsequent development of CRC. However, no complete correspondence exists between the CIMP-H phenotype and the serrated pathway (5). Additionally, the study by the Cancer Genome Atlas (TCGA) has identified six pathways that are altered in CRC (WNT, TGF-beta, RTK/RAS, PI3K, TP53, MS) (15), and more recently a molecular classification distinguishing four subgroups has been suggested (16).
In this study, we analyzed a series of histologically well-characterized precursor lesions of CRC by NGS to characterize the genetic landscape of serrated lesions and to compare it with tubular adenomas.
Eight sessile serrated lesions (SSL), three sessile serrated lesions with dysplasia (SSL/D), and two traditional serrated adenomas (TSA) were retrieved from the files of the Institute of Pathology of the University Hospital Basel, Switzerland, and from the GILAB AG, Allschwil, Switzerland, between January 1 and June 30, 2016. For all selected samples, a matched normal mucosa was available and was used as germline control. The histological classification for the samples is summarized in Table 1. Three tubular adenomas (TA) were used as control. All the slides were reviewed by an experienced pathologist (LT) to confirm the histological diagnosis, using the current WHO classification (9). The study has been approved by the Institutional Review Board of the Institute of Pathology, University Hospital Basel, and the Ethics Committee of Nordwest/Central Switzerland (EKNZ).
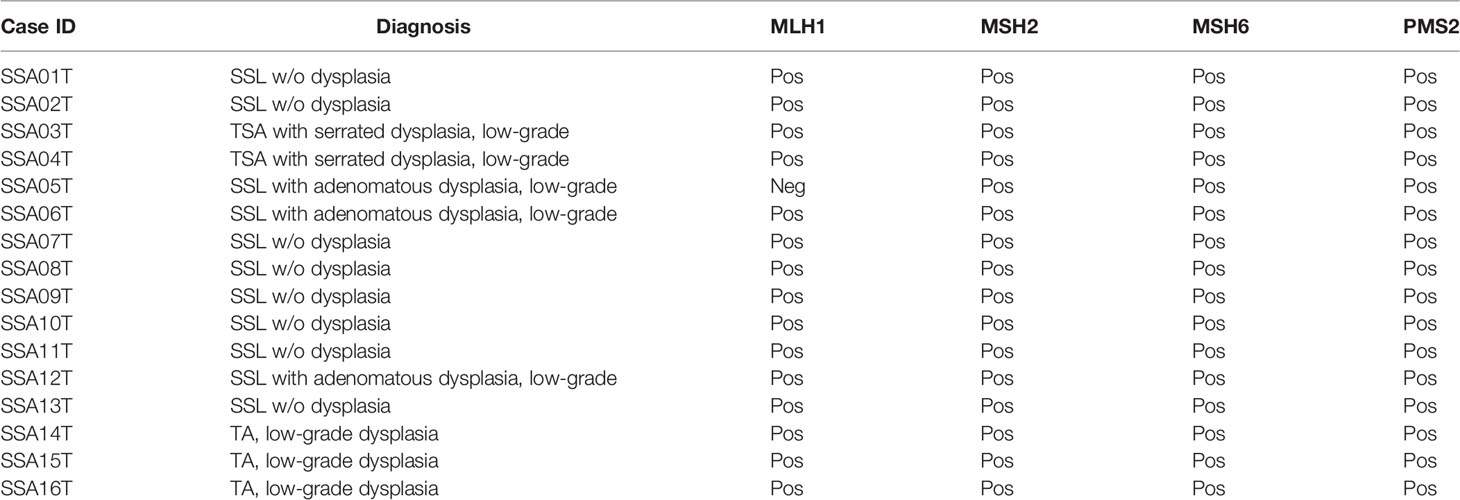
Table 1 Histologic and immunohistochemical characterization (mismatch repair proteins) of the samples included in the study.
Eight-μm-thick sections from representative formalin-fixed paraffin-embedded (FFPE) histological blocks for each lesion and matched normal mucosa were stained with Nuclear Fast Red in RNase-free conditions and subjected to microdissection with a sterile needle under a stereomicroscope (Olympus) to ensure a percentage of tumor cells >90%, as described previously (17). DNA was extracted from the microdissected tissues using the DNeasy Blood and Tissue Kit (Qiagen), according to the manufacturer’s guidelines, and quantified using the Qubit Fluorometer assay (Life Technologies). All samples yielded DNA of sufficient quantity and quality for sequencing analysis.
Library preparation was performed using the Oncomine Comprehensive Assay v3 (Thermo Fisher Scientific) using 20 ng DNA according to the manufacturer’s guidelines. This multiplex-PCR based technique targets 161 common cancer driver genes. Quantification and quality control were performed with the Ion Library TaqMan Quantitation Kit (Thermo Fisher Scientific). Pooled libraries diluted to 30 pM each were loaded on Ion 530 Chip (Thermo Fisher Scientific) and processed in Ion Chef Instrument (Thermo Fisher Scientific). Sequencing was performed on the Ion S5 system (Thermo Fisher Scientific).
Somatic mutation calling was performed using PipeIT (18), which performs the initial variant calling step by Torrent Variant Caller (TVC, v5.0.3, Thermo Fisher Scientific) using low stringency variant calling parameters. PipeIT whitelists hotspot variants (19, 20) then filters out variants covered by fewer than 10 reads in either the tumor or the matched normal sample, supported by fewer than eight reads or for which the tumor variant allele frequency (VAF) was <10 times that of the matched normal VAF. Whitelisted hotspot variants that did not pass the above read count and/or VAF filters were manually reviewed. All BRAF V600E mutations were confirmed to be somatic by Sanger sequencing as previously described (21).
Immunohistochemical staining was performed as previously described (22, 23). Briefly, 8 μm-thick sections were cut, dehydrated, and processed on a Bond III (Leica Biosystems Wetzlar, Germany). The ready-to-use (RTU) primary antibodies were from Novocastra (Leica Biosystems, Wetzlar, Germany), optimized for use on BOND III. The staining was performed with the following antibodies:
MLH1 (clone ES 05): Epitope retrieval (ER) 1, pH 7.5/8, 30’ 100°C; incubation time: 30’
MSH2 (clone 79H11): ER 2, pH9, 20’ 100°C; incubation time: 30’
MSH6 (clone EP 49): ER2, pH9, 20’ 100°C; incubation time: 30’
PMS2 (clone EP 51); ER2, pH9, 10’ 100°C; incubation time: 30’
We performed targeted sequencing of eight sessile serrated lesions without dysplasia (SSL), three sessile serrated lesions with dysplasia (SSL/D), two traditional serrated adenomas (TSA, Figure 1), as well as three tubular adenomas (TA) as control. Sequencing was performed at a mean depth of 670× (range from 382× to 942×; Supplementary Table 1). On average, SSLs harbored 1.75 mutations (range 1–4, n=8), SSL/Ds 2 mutations (range 2–2, n=3), TSAs 55 mutations (range 5–5, n=2), and TAs 3 mutations (range 2–4, n=3) (Figure 2). Annotation of the somatic mutations reveals that all but two samples harbored hotspot alteration in cancer-related genes.

Figure 1 Histological micrographs (A, C, E, G) and immunohistochemical stain results for MLH1 (B, D, F, H). (A, B) SSL without dysplasia (SSA08T); (C, D) SSL with dysplasia (ID SSA05T); (E, F) TSA (SSA04T); (G, H) TA (SSA15T). Note negativity for MLH1 in SSA05T (D).
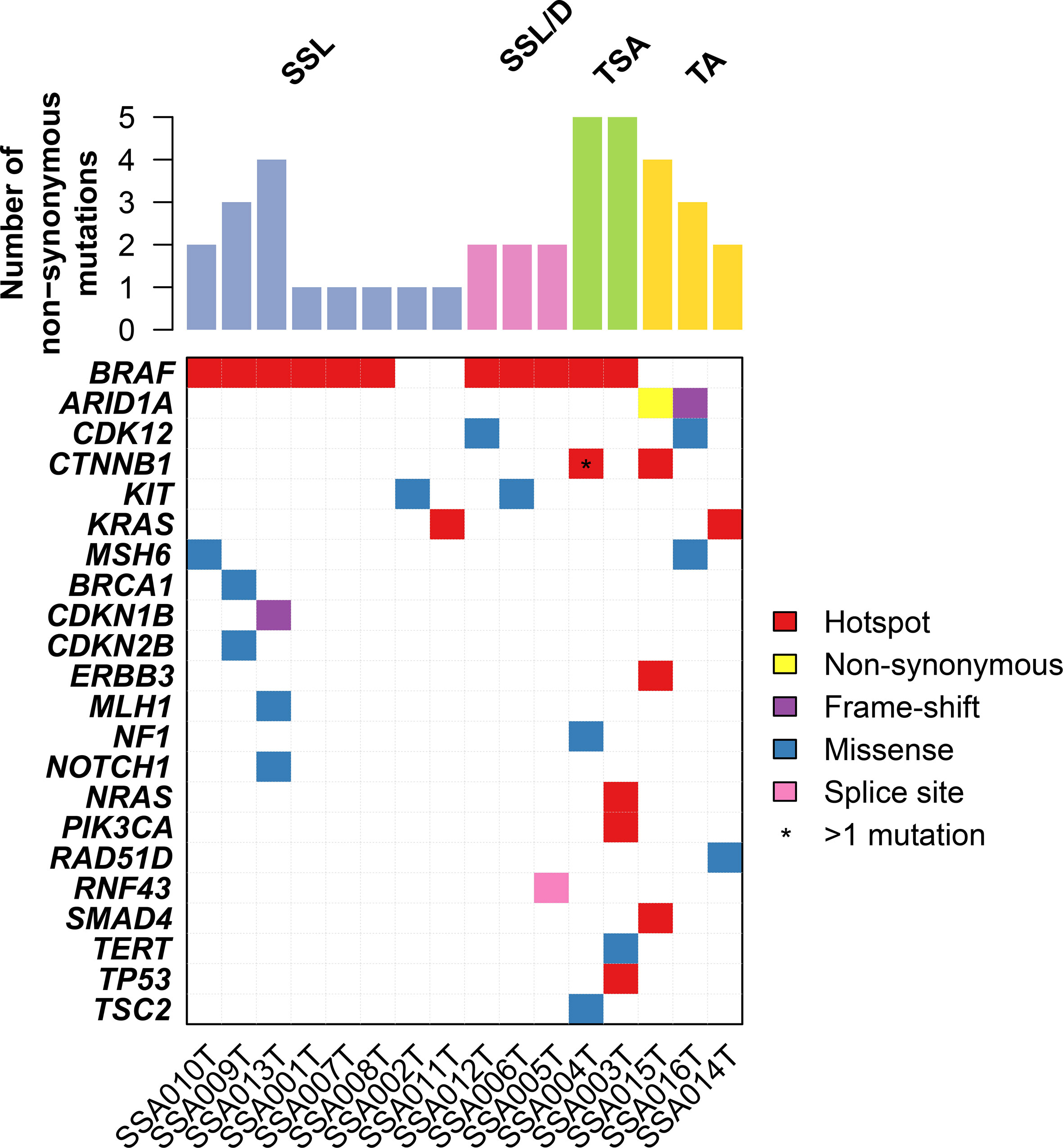
Figure 2 Repertoire of somatic genetic alterations in the serrated lesion of the colorectum. Heatmap indicating somatic genetic alterations identified in samples subjected to sequencing. Somatic alterations are color-coded according to the legend. Hotspot mutations are non-synonymous mutations that are in hotspot residues (see Materials and Methods).
All SSL/D and TSAs showed the classical BRAF p.V600E mutation, together with 75% (6/8) of SSLs. Of note, one of the two BRAF-wildtype SSLs harbored a hotspot KRAS Q61K mutation (Figure 2 and Supplementary Table 2). Additionally, the BRAF-mutant TSAs showed additional alterations in CTNNB1, NF1, TP53, NRAS, PIK3CA.
We further identified alterations involving MLH1 and MSH6, part of the “mismatch repair” machinery, in two SSLs. However, these mutations were missense rather than truncating, and their variant allele fractions were ~5%, suggesting they may not result in mismatch repair deficiency. Indeed, immunohistochemistry of MLH1, MSH2, MSH6, and PMS2 showed that all SSLs were positive, indicating mismatch repair proficiency (Figure 1 and Table 1). On the other hand, we identified one SSL/D (SSA005T) that was MLH1-negative.
On the other hand, TAs showed a consistently different profile and were all wild-type for BRAF (p = 0.02, Fisher’s exact test compared to the serrated lesions). By contrast, TAs harbored mutations in ARID1A (two cases), SMAD4, CDK12, ERBB3, and KRAS. The complete list of mutations is shown in Supplementary Table 2.
Taken together, our results provide evidence that SSL/Ds and TSAs are similar in somatic mutations having the BRAF hotspot somatic mutations as a major driver of the disease. On the other hand, TAs show a different constellation of somatic mutations such as ARID1A loss of function.
CRC is the third neoplasia for incidence and the fourth cause of death for neoplasia worldwide (1). Although CRC represents the classical model of development of epithelial cancer through the so-called adenoma-carcinoma sequence (2), it is now clear that there are other genetic events underlying its origin and growth. In particular, the clinical and biological relevance of serrated lesions of the colon has been pointed out in the last 25 years (5, 6). Serrated lesions of the colon are very well defined from the pathological point of view (12), and patients with serrated lesions are followed up similarly to “classical” tubular (TA), tubulovillous (TVA), and villous adenomas (VA) of the colon.
In general, in our series, only TSA showed a higher frequency of mutations, while SSL, TA, and TVA had fewer mutations/cases. This is in agreement with a recent study, in which no difference was found between SSL and conventional adenomas regarding the frequency of mutations (24). Although we only have two TSAs in our series, the presence of five mutations/case in comparison to ≈1.75–2 in SSL or TA may suggest that genomic instability could underlie the morphology of TSA.
We found the classical mutation BRAF p.V600E in most serrated lesions in our series, in particular in all SSLs with dysplasia and TSAs. SSLs with dysplasia are considered the “true” premalignant lesion to CRC in the so-called “serrated pathway” and progress relatively quickly, whereas TSAs are more similar to “conventional” adenomas regarding the development of cancer (25, 26). BRAF is one of the key players in the development of CRC through serrated adenomas (27, 28). BRAF is a molecular switch in the RAS–RAF–MEK–ERK pathway, which is crucial in the control of proliferation and differentiation (28). In our study, we found KRAS mutations in 1/4 conventional adenomas and in 1/12 serrated polyps (SSL without dysplasia, BRAF-wildtype), which is in keeping with the results of several previous studies, where the frequency of KRAS mutations in both conventional and serrated adenomas was between 8 and 10% (29–32). BRAF mutation has been suggested to be an early phenomenon in the genesis of SSL, whereas the genetic landscape of TSA seems to be more heterogeneous (4). In a recent study using whole-exome sequencing, BRAF V600E was found to be the only consistent mutation in serrated polyps (33). In our study, two SSLs without dysplasia had no BRAF mutation. One case had a p.Q61K mutation in KRAS, and the other had a p.A507P substitution in KIT. The former is a rare mutation that has been described in non-small-cell lung cancer and in colon cancer, with oncogenic potential. The latter has been described in GISTs as oncogenic.
Other mutations found in SSLs affected BRCA1, a very important tumor suppressor gene, involved in the genesis and development of many different tumors (34), CDKN1B and CDKN2B (also known as p27KIP1 and p15INK4B), two important tumor suppressor genes involved in the regulation of cell cycle. We also found two SSLs with mutations affecting proteins of the DNA-repair machinery (MLH1 and MSH6). Immunohistochemistry showed that all SSLs were mismatch repair proficient, thus these MLH1 and MSH6 mutations are not likely to have affected the mismatch repair pathway, and it is also unlikely that these SSL polyps were driven by MLH1 methylation. Similarly, NOTCH1 has been shown to act as an oncogene in CRC (35), so it is likely that the missense mutation we found is simply a bystander phenomenon.
SSL/D had a monotonous genomic landscape. They showed the classical BRAF V600E mutation, as described in the literature (5, 33, 36). They also showed mutations in CDK12 and in RNF43. CDK12 is a recently characterized cyclin-dependent kinase (CDK) that has been claimed to be involved in the genesis of prostate and ovarian cancers (37). RNF43 belongs to a superfamily of thrombospondin type 1 repeat-containing proteins (R-spondins) (38). R-spondins control the activity of WNT signaling also in adult stem cells (38). Interestingly, missense mutations in RNF43 have been found consistently in microsatellite unstable CRCs (39) and in some hereditary serrated polyposes (40). It is however a well-known factor in the development of gastric adenocarcinoma (41) and pancreatic intraductal papillary mucinous neoplasm (42). We also identified one SSL/D with loss of MLH1 expression without MLH1 somatic mutations. One could speculate here that the dysplasia, in this case, was driven by MLH1 methylation.
TSA showed in contrast more mutations (five/case). BRAF p.V600E was found in both cases. Other mutations were found in CTNNB1 (beta-catenin), NF1, TSC2, NRAS, PIK3CA, TERT, TP53. The role of beta-catenin in Wnt signaling and colon carcinogenesis is very well known (15). It has been shown that TSC2, through its interaction with mTOR, may increase the activity of Wnt signaling (43). Moreover, there is a cross-talk between PIK3CA and Wnt signaling, probably again mediated by TSC2 (43). NF1 changes have not been described previously in serrated lesions. Interestingly, it was claimed to be a target of mutational changes in MSI-CRCs (44). TERT is a well-known factor involved in the progression of colorectal carcinogenesis and higher activity (45). p.Q61K mutation in NRAS is a well-known activating mutation, with oncogenic potential. p.C275Y missense mutation in TP53 has been found in colorectal cancer (15), and mutations in TP53 are associated with the development of dysplasia in TSAs (46).
TAs showed three mutations/case, but they were more variable. One case showed the classical KRAS p.G12C. Other involved genes were ARID1A, CDK12, CTNNB1, KIT, MSH6, ERBB3, RAD51D. Truncating mutations of ARID1A, a chromatin remodeler, have been related to the development of endometrioid carcinoma of the ovary (47) and of early CRC (48). ERBB3 belongs to the HER receptor tyrosine kinase (RTK) family and has been found to be mutated in 11% of CRCs (49). RAD51D, involved in DNA repair, has been found to be rarely mutated in ovarian, breast, and colon cancer (50). Of note, our sequencing panel does not include APC, which is an important cancer gene in the conventional pathway. A previous study found that APC mutations are rare in serrated lesions, which would suggest APC mutations may be another distinguishing molecular feature that distinguishes the conventional and the serrated pathways (51).
The main limitation of our work is that it is a small retrospective series. The use of a targeted sequencing panel also limits our ability to characterize the global genomic features of these lesions. The study of the methylation profiles of these samples could also provide additional insights into the pathogenesis of the serrated pathway. Further study in larger cohorts will be required to confirm our results.
In summary, we have studied by NGS a small series of colorectal serrated lesions (SSL, SSL/D, and TSA) and have compared their genomic profiles with conventional TAs. Serrated lesions have a simpler genomic profile in comparison with TAs. BRAF p.V600E is the most frequent genomic alteration in serrated lesions.
Sequencing data are be available on the European Genome-Phenome Archive database under the accession number EGAS00001005648.
The studies involving human participants were reviewed and approved by the Ethics Committee of Nordwest/Central Switzerland (EKNZ). The patients/participants provided their written informed consent to participate in this study.
LT and SP conceived and supervised the study. LT performed the histologic review. AG performed the bioinformatics analyses under the supervision of CN. VP extracted the DNA and performed the sequencing reactions. LT, FL, LMT, CN, and SP analyzed the data and critically discussed the results. LT, CN, and SP wrote the manuscript. All authors contributed to the article and approved the submitted version.
The study was supported by grants from Oncosuisse KFS-4988-02-2020-R to SP. SP was also supported by the Theron Foundation, Vaduz (LI) and from the Surgery Department of the University Hospital Basel. The funding bodies had no role in study design; in the collection, analysis, and interpretation of data; in the writing of the report; and in the decision to submit the article for publication.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2021.668466/full#supplementary-material
Supplementary Table 1 | Sequencing statistics.
Supplementary Table 2 | Somatic single nucleotide variants (SNVs) and insertion/deletions (indels) identified in our cohort.
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global Cancer Statistics 2018: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin (2018) 68:394–424. doi: 10.3322/caac.21492
2. Kinzler KW, Vogelstein B. Lessons From Hereditary Colorectal Cancer. Cell (1996) 87:159–70. doi: 10.1016/s0092-8674(00)81333-1
3. Colussi D, Brandi G, Bazzoli F, Ricciardiello L. Molecular Pathways Involved in Colorectal Cancer: Implications for Disease Behavior and Prevention. Int J Mol Sci (2013) 14:16365–85. doi: 10.3390/ijms140816365
4. IJspeert JEG, Vermeulen L, Meijer GA, Dekker E. Serrated Neoplasia—Role in Colorectal Carcinogenesis and Clinical Implications. Nat Rev Gastroenterol Hepatol (2015) 12:401–9. doi: 10.1038/nrgastro.2015.73
5. Bettington M, Walker N, Clouston A, Brown I, Leggett B, Whitehall V. The Serrated Pathway to Colorectal Carcinoma: Current Concepts and Challenges. Histopathology (2013) 62:367–86. doi: 10.1111/his.12055
6. Jass JR. Classification of Colorectal Cancer Based on Correlation of Clinical, Morphological and Molecular Features. Histopathology (2007) 50:113–30. doi: 10.1111/j.1365-2559.2006.02549.x
7. Snover DC. Update on the Serrated Pathway to Colorectal Carcinoma. Hum Pathol (2011) 42:1–10. doi: 10.1016/j.humpath.2010.06.002
8. O’Brien MJ, Zhao Q, Yang S. Colorectal Serrated Pathway Cancers and Precursors. Histopathology (2015) 66:49–65. doi: 10.1111/his.12564
9. Who Classification of Tumours Editorial Board. Digestive System Tumours. Lyon, IARC: Who Press (2019).
10. Torlakovic E, Skovlund E, Snover DC, Torlakovic G, Nesland JM. Morphologic Reappraisal of Serrated Colorectal Polyps. Am J Surg Pathol (2003) 27:65–81. doi: 10.1097/00000478-200301000-00008
11. Ensari A, Bilezikçi B, Carneiro F, Doğusoy GB, Driessen A, Dursun A, et al. Serrated Polyps of the Colon: How Reproducible Is Their Classification? Virchows Arch (2012) 461:495–504. doi: 10.1007/s00428-012-1319-7
12. Rex DK, Ahnen DJ, Baron JA, Batts KP, Burke CA, Burt RW, et al. Serrated Lesions of the Colorectum: Review and Recommendations From an Expert Panel. Am J Gastroenterol (2012) 107:1315–29. doi: 10.1038/ajg.2012.161
13. Clancy C, Burke JP, Kalady MF, Coffey JC. BRAF Mutation Is Associated With Distinct Clinicopathological Characteristics in Colorectal Cancer: A Systematic Review and Meta-Analysis. Colorectal Dis (2013) 15:e711–8. doi: 10.1111/codi.12427
14. Kambara T, Simms LA, Whitehall VLJ, Spring KJ, Wynter CVA, Walsh MD, et al. BRAF Mutation Is Associated With DNA Methylation in Serrated Polyps and Cancers of the Colorectum. Gut (2004) 53:1137–44. doi: 10.1136/gut.2003.037671
15. Network TCGA. The Cancer Genome Atlas Network. Comprehensive Molecular Characterization of Human Colon and Rectal Cancer. Nature (2012) 487:330–7. doi: 10.1038/nature11252
16. Guinney J, Dienstmann R, Wang X, de Reyniès A, Schlicker A, Soneson C, et al. The Consensus Molecular Subtypes of Colorectal Cancer. Nat Med (2015) 21:1350–6. doi: 10.1038/nm.3967
17. Piscuoglio S, Ng CKY, Murray MP, Guerini-Rocco E, Martelotto LG, Geyer FC, et al. The Genomic Landscape of Male Breast Cancers. Clin Cancer Res (2016) 22:4045–56. doi: 10.1158/1078-0432.ccr-15-2840
18. Garofoli A, Paradiso V, Montazeri H, Jermann PM, Roma G, Tornillo L, et al. PipeIT: A Singularity Container for Molecular Diagnostic Somatic Variant Calling on the Ion Torrent Next-Generation Sequencing Platform. J Mol Diagn (2019) 21:884–94. doi: 10.1016/j.jmoldx.2019.05.001
19. Chang MT, Asthana S, Gao SP, Lee BH, Chapman JS, Kandoth C, et al. Identifying Recurrent Mutations in Cancer Reveals Widespread Lineage Diversity and Mutational Specificity. Nat Biotechnol (2016) 34:155–63. doi: 10.1038/nbt.3391
20. Gao J, Chang MT, Johnsen HC, Gao SP, Sylvester BE, Sumer SO, et al. 3D Clusters of Somatic Mutations in Cancer Reveal Numerous Rare Mutations as Functional Targets. Genome Med (2017) 9:4. doi: 10.1186/s13073-016-0393-x
21. Paradiso V, Garofoli A, Tosti N, Lanzafame M, Perrina V, Quagliata L, et al. Diagnostic Targeted Sequencing Panel for Hepatocellular Carcinoma Genomic Screening. J Mol Diagn (2018) 20:836–48. doi: 10.1016/j.jmoldx.2018.07.003
22. Coto-Llerena M, Tosti N, Taha-Mehlitz S, Kancherla V, Paradiso V, Gallon J, et al. Transcriptional Enhancer Factor Domain Family Member 4 Exerts an Oncogenic Role in Hepatocellular Carcinoma by Hippo-Independent Regulation of Heat Shock Protein 70 Family Members. Hepatol Commun (2021) 5:661–74. doi: 10.1002/hep4.1656
23. Taha-Mehlitz S, Bianco G, Coto-Llerena M, Kancherla V, Bantug GR, Gallon J, et al. Adenylosuccinate Lyase Is Oncogenic in Colorectal Cancer by Causing Mitochondrial Dysfunction and Independent Activation of NRF2 and mTOR-MYC-Axis. Theranostics (2021) 11:4011–29. doi: 10.7150/thno.50051
24. Lin S-H, Raju GS, Huff C, Ye Y, Gu J, Chen J-S, et al. The Somatic Mutation Landscape of Premalignant Colorectal Adenoma. Gut (2018) 67:1299–305. doi: 10.1136/gutjnl-2016-313573
25. Lazarus R, Junttila OE, Karttunen TJ, Mäkinen MJ. The Risk of Metachronous Neoplasia in Patients With Serrated Adenoma. Am J Clin Pathol (2005) 123:349–59. doi: 10.1309/vbagv3br96n2eqtr
26. Lu F-I, van Niekerk DW, Owen D, Tha SPL, Turbin DA, Webber DL. Longitudinal Outcome Study of Sessile Serrated Adenomas of the Colorectum: An Increased Risk for Subsequent Right-Sided Colorectal Carcinoma. Am J Surg Pathol (2010) 34:927–34. doi: 10.1097/PAS.0b013e3181e4f256
27. Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, et al. Mutations of the BRAF Gene in Human Cancer. Nature (2002) 417:949–54. doi: 10.1038/nature00766
28. Kolch W. Meaningful Relationships: The Regulation of the Ras/Raf/MEK/ERK Pathway by Protein Interactions. Biochem J (2000) 351 Pt 2:289–305. doi: 10.1042/bj3510289
29. Rajagopalan H, Bardelli A, Lengauer C, Kinzler KW, Vogelstein B, Velculescu VE. Tumorigenesis: RAF/RAS Oncogenes and Mismatch-Repair Status. Nature (2002) 418:934. doi: 10.1038/418934a
30. Fernando WC, Miranda MS, Worthley DL, Togashi K, Watters DJ, Leggett BA, et al. The CIMP Phenotype in BRAF Mutant Serrated Polyps From a Prospective Colonoscopy Patient Cohort. Gastroenterol Res Pract (2014) 2014:374926. doi: 10.1155/2014/374926
31. Kakar S, Deng G, Cun L, Sahai V, Kim YS. CpG Island Methylation Is Frequently Present in Tubulovillous and Villous Adenomas and Correlates With Size, Site, and Villous Component. Hum Pathol (2008) 39:30–6. doi: 10.1016/j.humpath.2007.06.002
32. O’Brien MJ, Yang S, Mack C, Xu H, Huang CS, Mulcahy E, et al. Comparison of Microsatellite Instability, CpG Island Methylation Phenotype, BRAF and KRAS Status in Serrated Polyps and Traditional Adenomas Indicates Separate Pathways to Distinct Colorectal Carcinoma End Points. Am J Surg Pathol (2006) 30:1491–501. doi: 10.1097/01.pas.0000213313.36306.85
33. Dehghanizadeh S, Khoddami V, Mosbruger TL, Hammoud SS, Edes K, Berry TS, et al. Active BRAF-V600E Is the Key Player in Generation of a Sessile Serrated Polyp-Specific DNA Methylation Profile. PloS One (2018) 13:e0192499. doi: 10.1371/journal.pone.0192499
34. Roy R, Chun J, Powell SN. BRCA1 and BRCA2: Different Roles in a Common Pathway of Genome Protection. Nat Rev Cancer (2012) 12:68–78. doi: 10.1038/nrc3181
35. Wang Z, Li Y, Sarkar FH. Notch Signaling Proteins: Legitimate Targets for Cancer Therapy. Curr Protein Pept Sci (2010) 11:398–408. doi: 10.2174/138920310791824039
36. Voorham QJM, Carvalho B, Spiertz AJ, Claes B, Mongera S, van Grieken NCT, et al. Comprehensive Mutation Analysis in Colorectal Flat Adenomas. PloS One (2012) 7:e41963. doi: 10.1371/journal.pone.0041963
37. Greenleaf AL. Human CDK12 and CDK13, Multi-Tasking CTD Kinases for the New Millenium. Transcription (2019) 10:91–110. doi: 10.1080/21541264.2018.1535211
38. Raslan AA, Yoon JK. R-Spondins: Multi-Mode WNT Signaling Regulators in Adult Stem Cells. Int J Biochem Cell Biol (2019) 106:26–34. doi: 10.1016/j.biocel.2018.11.005
39. Dienstmann R, Tabernero J. Spectrum of Gene Mutations in Colorectal Cancer: Implications for Treatment. Cancer J (2016) 22:149–55. doi: 10.1097/PPO.0000000000000191
40. Valle L, de Voer RM, Goldberg Y, Sjursen W, Försti A, Ruiz-Ponte C, et al. Update on Genetic Predisposition to Colorectal Cancer and Polyposis. Mol Aspects Med (2019) 69:10–26. doi: 10.1016/j.mam.2019.03.001
41. Min B-H, Hwang J, Kim NK, Park G, Kang SY, Ahn S, et al. Dysregulated Wnt Signalling and Recurrent Mutations of the Tumour Suppressor RNF43 in Early Gastric Carcinogenesis. J Pathol (2016) 240:304–14. doi: 10.1002/path.4777
42. Sakamoto H, Kuboki Y, Hatori T, Yamamoto M, Sugiyama M, Shibata N, et al. Clinicopathological Significance of Somatic RNF43 Mutation and Aberrant Expression of Ring Finger Protein 43 in Intraductal Papillary Mucinous Neoplasms of the Pancreas. Mod Pathol (2015) 28:261–7. doi: 10.1038/modpathol.2014.98
43. Jin T, George Fantus I, Sun J. Wnt and Beyond Wnt: Multiple Mechanisms Control the Transcriptional Property of Beta-Catenin. Cell Signal (2008) 20:1697–704. doi: 10.1016/j.cellsig.2008.04.014
44. Wang Q, Montmain G, Ruano E, Upadhyaya M, Dudley S, Liskay MR, et al. Neurofibromatosis Type 1 Gene as a Mutational Target in a Mismatch Repair-Deficient Cell Type. Hum Genet (2003) 112:117–23. doi: 10.1007/s00439-002-0858-4
45. Pellatt AJ, Wolff RK, Herrick J, Lundgreen A, Slattery ML. Tert’s Role in Colorectal Carcinogenesis. Mol Carcinog (2013) 52:507–13. doi: 10.1002/mc.21885
46. Bettington ML, Chetty R. Traditional Serrated Adenoma: An Update. Hum Pathol (2015) 46:933–8. doi: 10.1016/j.humpath.2015.04.002
47. Samartzis EP, Noske A, Dedes KJ, Fink D, Imesch P. ARID1A Mutations and PI3K/AKT Pathway Alterations in Endometriosis and Endometriosis-Associated Ovarian Carcinomas. Int J Mol Sci (2013) 14:18824–49. doi: 10.3390/ijms140918824
48. Kishida Y, Oishi T, Sugino T, Shiomi A, Urakami K, Kusuhara M, et al. Associations Between Loss of ARID1A Expression and Clinicopathologic and Genetic Variables in T1 Early Colorectal Cancer. Am J Clin Pathol (2019) 152:463–70. doi: 10.1093/ajcp/aqz062
49. Jaiswal BS, Kljavin NM, Stawiski EW, Chan E, Parikh C, Durinck S, et al. Oncogenic ERBB3 Mutations in Human Cancers. Cancer Cell (2014) 25:543–4. doi: 10.1016/j.ccr.2014.03.030
50. Pelttari LM, Kiiski J, Nurminen R, Kallioniemi A, Schleutker J, Gylfe A, et al. A Finnish Founder Mutation in RAD51D: Analysis in Breast, Ovarian, Prostate, and Colorectal Cancer. J Med Genet (2012) 49:429–32. doi: 10.1136/jmedgenet-2012-100852
Keywords: colorectal cancer, serrated lesions, DNA sequencing, BRAF, tubulovillous adenoma
Citation: Tornillo L, Lehmann FS, Garofoli A, Paradiso V, Ng CKY and Piscuoglio S (2021) The Genomic Landscape of Serrated Lesion of the Colorectum: Similarities and Differences With Tubular and Tubulovillous Adenomas. Front. Oncol. 11:668466. doi: 10.3389/fonc.2021.668466
Received: 16 February 2021; Accepted: 15 September 2021;
Published: 12 October 2021.
Edited by:
David Gibbons, St. Vincent’s University Hospital, IrelandReviewed by:
Mark Bettington, The University of Queensland, AustraliaCopyright © 2021 Tornillo, Lehmann, Garofoli, Paradiso, Ng and Piscuoglio. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Salvatore Piscuoglio, cy5waXNjdW9nbGlvQHVuaWJhcy5jaA==; Luigi Tornillo, bHVpZ2kudG9ybmlsbG9AdW5pYmFzLmNo
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.