 Oscar G. W. Wong
Oscar G. W. Wong Jing Li
Jing Li Annie N. Y. Cheung
Annie N. Y. Cheung- Department of Pathology, The University of Hong Kong, Hong Kong, China
Ovarian clear cell carcinoma (OCCC) is one of the major types of ovarian cancer and is of higher relative prevalence in Asians. It also shows higher possibility of resistance to cisplatin-based chemotherapy leading to poor prognosis. This may be attributed to the relative lack of mutations and aberrations in homologous recombination-associated genes, which are crucial in DNA damage response (DDR), such as BRCA1, BRCA2, p53, RAD51, and genes in the Fanconi anemia pathway. On the other hand, OCCC is characterized by a number of genetic defects rendering it vulnerable to DDR-targeting therapy, which is emerging as a potent treatment strategy for various cancer types. Mutations of ARID1A, PIK3CA, PTEN, and catenin beta 1 (CTNNB1), as well as overexpression of transcription factor hepatocyte nuclear factor-1β (HNF-1β), and microsatellite instability are common in OCCC. Of particular note is the loss-of-function mutations in ARID1A, which is found in approximately 50% of OCCC. ARID1A is crucial for processing of DNA double-strand break (DSB) and for sustaining DNA damage signaling, rendering ARID1A-deficient cells prone to impaired DNA damage checkpoint regulation and hence sensitive to poly ADP ribose polymerase (PARP) inhibitors. However, while preclinical studies have demonstrated the possibility to exploit DDR deficiency in OCCC for therapeutic purpose, progress in clinical application is lagging. In this review, we will recapitulate the preclinical studies supporting the potential of DDR targeting in OCCC treatment, with emphasis on the role of ARID1A in DDR. Companion diagnostic tests (CDx) for predicting susceptibility to PARP inhibitors are rapidly being developed for solid tumors including ovarian cancers and may readily be applicable on OCCC. The potential of various available DDR-targeting drugs for treating OCCC by drawing analogies with other solid tumors sharing similar genetic characteristics with OCCC will also be discussed.
Introduction
Ovarian carcinoma is the most lethal gynecologic malignancy in many countries, with an estimated incidence of 290,000 cases and a mortality of 180,000 cases per year worldwide (1). Histologically, ovarian carcinomas are classified into several different subtypes, including the four major types: serous, endometrioid, clear cell, and mucinous (2). Each subtype exhibits distinct genetic alterations and clinical and prognostic characteristics. Ovarian clear cell carcinoma (OCCC) accounts for approximately 5~27% of all ovarian carcinomas (3–5). It is characterized by high recurrence rate and generally displays worse prognosis owing to frequent de novo resistance to chemotherapeutic agents in advanced stages (6, 7). A number of targeted therapy strategies, including anti-angiogenesis, multitargeted tyrosine kinase inhibition, and PIK3CA/AKT/mTOR pathway inhibition as well as immunotherapy were evaluated in clinical trials, but only modest efficacies or non-histotype selective effects were observed (8). Thus, discovering effective targeted therapies for OCCC remains a challenging goal.
Components of the DNA damage response (DDR) are rich sources of molecular targets for cancer therapy (9) (Figure 1). DDR is a collection of interdependent signaling pathways, and machineries evolved to cope with DNA damage, which happens constantly in every cell (12). Activation of DDR can manifest into cell-cycle arrest, regulation of DNA replication, and the repair or bypass of DNA damage, ultimately alleviating the devastating effect of replicating damaged DNA. In the event of unsuccessful or suboptimal repair of DNA leading to unsustainable genomic instability, DDR can signify senescence or elimination of the affected cells by programmed cell death (13–16). DNA can be damaged in multiple ways, and thus there are multiple interrelated DDR pathways (reviewed below). Intriguingly, DDR activation is an early event during tumorigenesis (14, 15), whilst defects in one or more DDR pathways are a hallmark of cancer, leading to genomic instability and a greater dependency on the remaining pathways for survival (17). While some DDR-targeting agents, mainly of the Poly (ADP-ribose) polymerase (PARP) inhibitors (PARPi) class, have been approved for management of ovarian cancers, relatively little was known about the utility of other DDR-targeting agents in an OCCC-specific context. Given the distinct genetics features of OCCC, we argue that the disease should be vulnerable to many more DDR-targeting agents. The rationale and agents for targeting DDR have been excellently reviewed previously (10, 17–20). Here in this article, DDR and its role in cancer therapy will be briefly reviewed, then we will recapitulate the preclinical studies pointing to the potential of DDR targeting in OCCC treatment, with emphasis on the role of ARID1A in DDR.
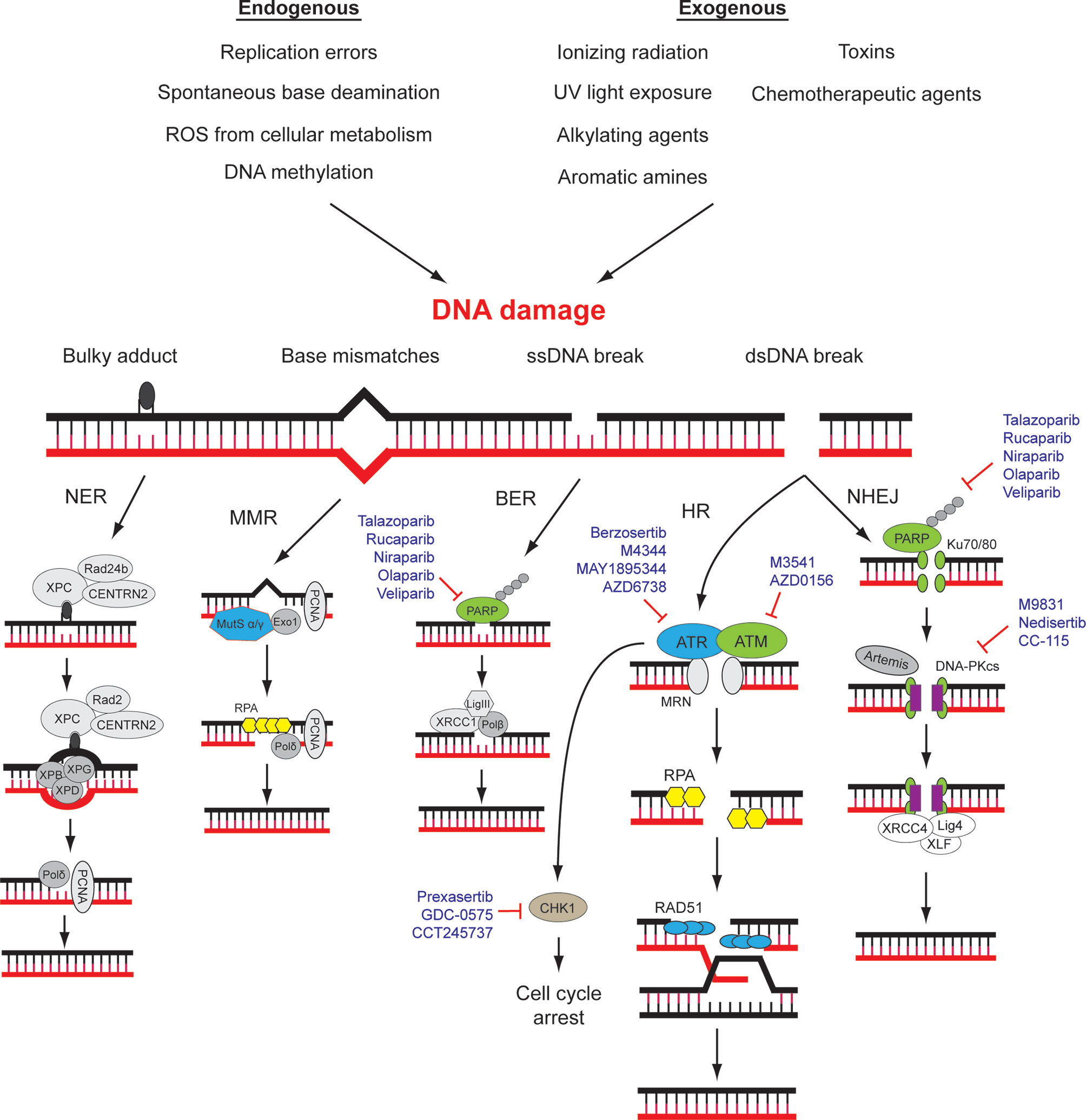
Figure 1 DNA damage repair (DDR) pathways and related targets for therapy. DNA damage may be caused by various environmental and endogenous stresses. Different types of DNA damage are repaired by specific signaling and repair pathways. A number of drugs that target different components of DDR pathways are approved for use or in clinical trials. NER, nucleotide excision repair; MMR, mismatch repair; BER, base excision repair; HR, homologous recombination; NHEJ, Non-homologous end-joining. Modified from (10, 11).
DNA Damage Repair Pathways and Cancer
The genomic DNA of eukaryotes is highly folded, organized, and wrapped around histones in the form of chromatin in the nucleus of mammalian cells (21). Chromatin fibers possess different degrees of compression and multiple types of chemical modifications, ensuring the accurate gene expression and the integrity of genomic structure (21, 22). DDR plays a pivotal role in the maintenance of human genomic integrity and stability through repairing and reducing DNA damage resulting from a wide variety of endogenous and exogenous threats, including ultraviolet light, ionizing radiation, topoisomerase agents, reactive oxygen species, and error-prone (non-proofreading) DNA polymerases (12, 23–26). DNA damage generates aberrant nucleotides or nucleotide fragments in the DNA strands. Five distinct DNA repair pathways have been identified in mammalian cells, including base excision repair (BER), mismatch repair (MMR), and nucleotide excision repair (NER) for single-strand break, homologous recombination (HR) and non-homologous end joining (NHEJ) for double-strand break (12, 27) (Figure 1). Deficiencies in DNA damage repair system may cause increased risks of generating mutations and chromosomal instability, which may ultimately lead to tumorigenesis (28). The molecular details of DDR pathways have been extensively studied, and readers are referred to a number of excellent reviews (10, 17, 27). Here, the key players of HR and NHEJ will be briefly introduced since their mechanisms and interactions are underlying determinants of cancer cell sensitivity towards some current DDR-targeting drugs, namely, PARP inhibitors.
Non-Homologous End Joining and Homologous Recombination
DNA double-strand break (DSB) is the most severe type of DNA damages occurring within eukaryotic cells, mainly caused by the irradiation and chemotherapeutic agents (29). Irreparable DSB may lead to cell death, while its aberrant repair may result in changes of heredity materials such as chromosome deletion and translocation (30). DSB repair is generally performed through two main mechanisms: non-homologous end joining (NHEJ) and homologous recombination (HR).
In the NHEJ pathway, Ku70/80 heterodimer is responsible for recognizing and binding the ends of the DSB, acting as a scaffold to recruit other NHEJ components to the DSB for the DSB ends-processing, including DNA-dependent protein kinase catalytic subunit (DNA-PKcs), X-ray repair cross complementing 4 (XRCC4), LIG4, XRCC4-like factors (XLF), and Aprataxin and PNK-like factors (APLF) (29, 31). Once the sites of DSB are detected and secured, several NHEJ repairing proteins, including Artemis, Polynucleotide kinase/phosphatase, APLF, and Werner syndrome protein, are recruited to remove non-ligatable end groups and excise the naked strands. Then, the gap is filled in by Family X polymerase μ/λ and LIG4 (29).
The triggering of the HR pathway initiates with the formation of MRN complex (MRE11-RAD50-NBS1) for recognizing and binding to the DSB sites (32). Subsequently, several HR components, including ataxia-telangiectasia mutated kinase (ATM), CtIP, and BRCA1, are recruited to initiate a series of HR events including phosphorylation of MRE11, MDC1, and H2AX (33). The initial nucleotides excision is performed by the MRN complex through its endonuclease activity in the presence of CtlP. Then, under the promotion of BRCA1, nucleases Exonuclease 1 and DNA2 thoroughly cut the 5’ end and generate a RPA-coated single stranded DNA (ssDNA) at the 3’ end (34). Next, the BRCA1-PALB2-BRCA2 complex assembles the recombinase RAD51 onto the ssDNA to form presynaptic filaments. RAD52 also interacts with RPA and promotes RAD51 to displace the RPA wrapped on ssDNA (32, 35). Subsequently, the filament invades the homologous sequence, initiating the remaining re-synthesis of damaged DNA strands using the sister chromatid as a template for error-free repair.
NHEJ can be activated in different cell types and at various cell cycle stages but mainly in G0 and G1 phases, while HR pathway occurs mainly in the S and G2 phases (34). NHEJ pathway can quickly repair up to 85% of DNA double-strand break damage generated by irradiation. However, NHEJ is an extremely error-prone repairing mechanism since it reconnects broken DNA strands together through end processing without a DNA template and some DNA fragments might be lost during this sort of end processing (29). In contrast, HR pathway may be the most important repairing pathway as it utilizes undisturbed sister chromatid as template DNA to restore the original DNA sequence in a high-fidelity manner (35).
Synthetic Lethality of HR-Deficient Phenotype
A cancer cell with deficiency in a DDR pathway is dependent on another DDR pathway for survival, potentiating single-agent activity of an inhibitor of that dependent pathway—an approach that has been described as synthetic lethality. Since the deficiency is specific to cancer cells, such approach is expected to spare normal cells and provide superior specificity in comparison to conventional chemotherapies (36, 37). Up to now, the most successful example of therapeutic agents realizing the synthetic lethality principle is the pharmaceutical inhibition of poly (ADP-ribose) polymerase 1 (PARP1) (37). PARP1 is a key enzyme for the detection of single-strand DNA breaks (SSBs). Inhibition of PARP1 is synthetic lethal with mutations in the genes encoding the HR proteins BRCA1, BRCA2, as well as partner and localizer of BRCA2 (PALB2). Mutations of these genes are found in small percentage of sporadic and significant proportion of hereditary high-grade serous ovarian cancer (38). A number of PARP inhibitors (PARPis) such as olaparib, rucaparib, and niraparib have been approved for the management of ovarian cancers in various settings (39–45) (Figure 1). Efforts are also underway to expand the experience acquired from the use of PARP1 inhibitors into a broader range of defects in HR (46).
Predictors of Response to PARP Inhibitors
Tremendous effort has been put on extending the utility of PARPis in treating tumors without BRCA1/2 mutations, as HR deficiency may be resulted from defects in HR genes other than BRCA1/2, or through epigenetics mechanisms such as promoter methylation of BRCA1/2 (47). Thus, various approaches have been explored to identify biomarkers for PARPi sensitivity such as sequencing all known HR genes, multiplex-ligation-dependent probe amplification (MLPA) (48), promoter hypermethylation assays (47, 49), gene expression signatures (50–52), candidate single genes expression, e.g., SLFN11 (53), gross chromosomal structural changes (54–56), and direct assay for HR functioning and PARPi sensitivity such as RAD51 foci detection and dose-response test on patient-derived tumor organoids (57–60). Nowadays, identification of patients who will most likely benefit from PARPi treatment is achieved through the use of next-generation sequencing (NGS) of targeted gene panels or genomic signatures (genomic scars). International consensus is still lacking regarding the optimal approach to define HR deficiency (60).
Currently, there are five commercially available FDA-approved CDx tests for evaluating PARPi eligibility: BRACAnalysis CDx and MyChoice CDx from Myriad Genetic Laboratories, Inc., as well as FoundationOne CDx, FoundationFocus CDxBRCA, and FoundationOne Liquid CDx from Foundation Medicine (60, 61). NGS-based CDx tests follow similar workflows: DNA is extracted from tissues and sequenced through NGS platforms, and then raw reads are processed through proprietary bioinformatics pipelines for variants calling and medical interpretation. For instance, according to the ACGM classification, variants can be classified as: pathogenic (class 5), likely pathogenic (class 4), variant of uncertain significance (class 3), likely benign (class 2), and benign (class 1) (62). The five CDx mentioned can all detect pathogenic or likely pathogenic variants of BRCA1/2. Among them, MyChoice CDx and FoundationOne CDx assays are prospectively validated assays for evaluation of HR deficiency status (61). The HR deficiency status can be assessed by checking the percentage of genomic regions with loss of heterozygosity (LOH) using single nucleotide polymorphism (SNP) sequencing or by calculation of the Genomic Instability Score (GIS) via coalescing three parameters: LOH, large scale transitions (LSTs) and telomeric allelic imbalance (TAI). Other assays such as the FDA-approved MSK-IMPACT, which provides integrated mutation profiling of actionable cancer targets developed by Memorial Sloan Kettering’s Department of Pathology, also cover detection of somatic variants in DDR genes and allow evaluation of experimental HR deficiency scores (63, 64). Recently, HRDetect, a new computational method dedicated to measuring HR deficiency based on whole-genome sequencing (WGS) data, is gaining momentum as a predictor of PARPi response. The HRDetect algorithm extracts a tumor’s mutational signatures and identify those specific for HR deficiency, such as multiple small duplications throughout the genome or numerous large tandem duplications (65). HRDetect successfully identified HR-deficient triple negative breast cancer (TNBC) and predicted their rucaparib sensitivities in a translational clinical trial RIO (EudraCT 2014-003319-12) (66). The algorithm is easily accessible through the reference web-based tool Signal (67).
The benefits of these CDx tests and related technical platforms were supported by clinical trials. In the SOLO1 trial (NCT01844986), patients with newly diagnosed advanced ovarian cancers were tested for BRCA1/2 mutation by BRACAnalysis (Myriad) or BRCA1/2 genetic testing assay (BGI) before randomization into olaparib and placebo groups as maintenance therapy. The risk of disease progression or death was 70% lower with olaparib than with placebo (68). NGS platform from Foundation Medicine was adopted to evaluate BRCA mutation status and LOH in patients with platinum sensitive recurrent ovarian high-grade serous carcinomas (44). The findings that patients with BRCA mutant or BRCA wild-type and LOH high cancers treated with rucaparib showed longer progression-free survival than patients with BRCA wild-type LOH low cancers support the efficacy of evaluating LOH in cancers to identify BRCA wild-type patients who are likely to benefit from rucaparib therapy. LIGHT (NCT02983799) is a phase 2 multicenter study to assess the efficacy and safety of olaparib in patients with BRCA mutation and HR deficiency status evaluated by BRACAnalysis CDx and myChoice CDx tests. Four cohorts were identified: subjects with germline BRCA mutations (gBRCAm), somatic BRCA mutations (sBRCAm), or potential aberrations in homologous recombination deficiency (HRD) (HRD-positive), as well as in subjects without identifiable HRD (HRD-negative). Consistent with the maintenance therapy setting, similar efficacy of olaparib was observed among gBRCAm and sBRCAm patients. More importantly, for non-BRCAm patients, a longer median progress-free survival and higher objective response rate were observed in the HRD positive cohort compared with the HRD-negative cohort (69). Recently, short-term patient-derived ovarian cancer organoids have been proposed to serve as testing platform of real-time HR deficiency and PARPi sensitivity, capable of supplementing sequencing-based approaches (70). Most of these investigations were performed in high-grade serous ovarian cancer. There is a relative lack of studies regarding biomarkers and clinical trials involving OCCC in the literature. It will be worthwhile to investigate the general HR deficiency status of OCCC in relation to PARPi and other DRR-targeting therapies’ sensitivity.
Clear Cell Carcinoma of the Ovary
Epidemiology, Morphology, Immunophenotype, and Genetic Features of OCCC
The incidence of OCCC shows wide variation among different geographic populations. OCCC contributes to less than 12% in Western countries where HGSC is the most common ovarian cancer subtype. In contrast, up to 20–27% of ovarian cancers in Asian countries are OCCC (5, 71, 72). OCCC generally shows morphologic and molecular biological characteristics distinct from other subtypes of epithelial ovarian cancers. Differences in pathogenesis and genetic profiles between OCCC and other major histological subtypes of ovarian cancers are summarized in Table 1.
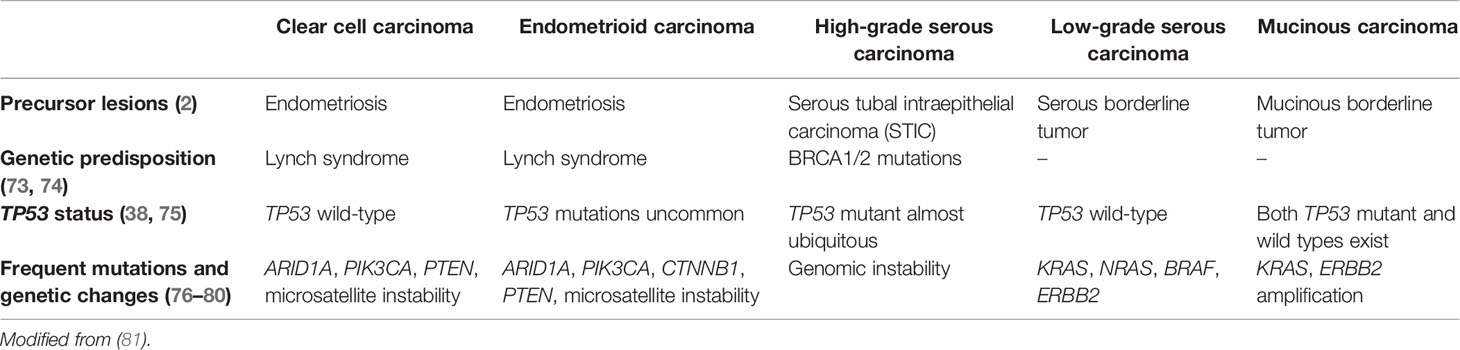
Table 1 Distinct features of OCC as compared with other major histotypes of ovarian cancer.
Morphologically, OCCCs are mainly composed of tumor cells with clear cytoplasm forming tubule-cystic, papillary, and solid architectures. Tumor cells with eosinophilic cytoplasm are often present. Hobnail cells may be found lining the tubules and cysts (82) (Figure 2A). OCCC usually shows strong positive immunoreactivity for cytokeratin 7, PAX8, HNF-1β, and Napsin A but are often negative for estrogen receptor (ER), progesterone receptor (PR), and Wilms tumor protein 1 (WT1) (83–86) (Figures 2B–D). Assessment of the morphology and immunoprofile is important to differentiate OCCC from other histological subtypes of ovarian carcinomas, particularly high-grade serous carcinoma and endometrioid carcinoma.
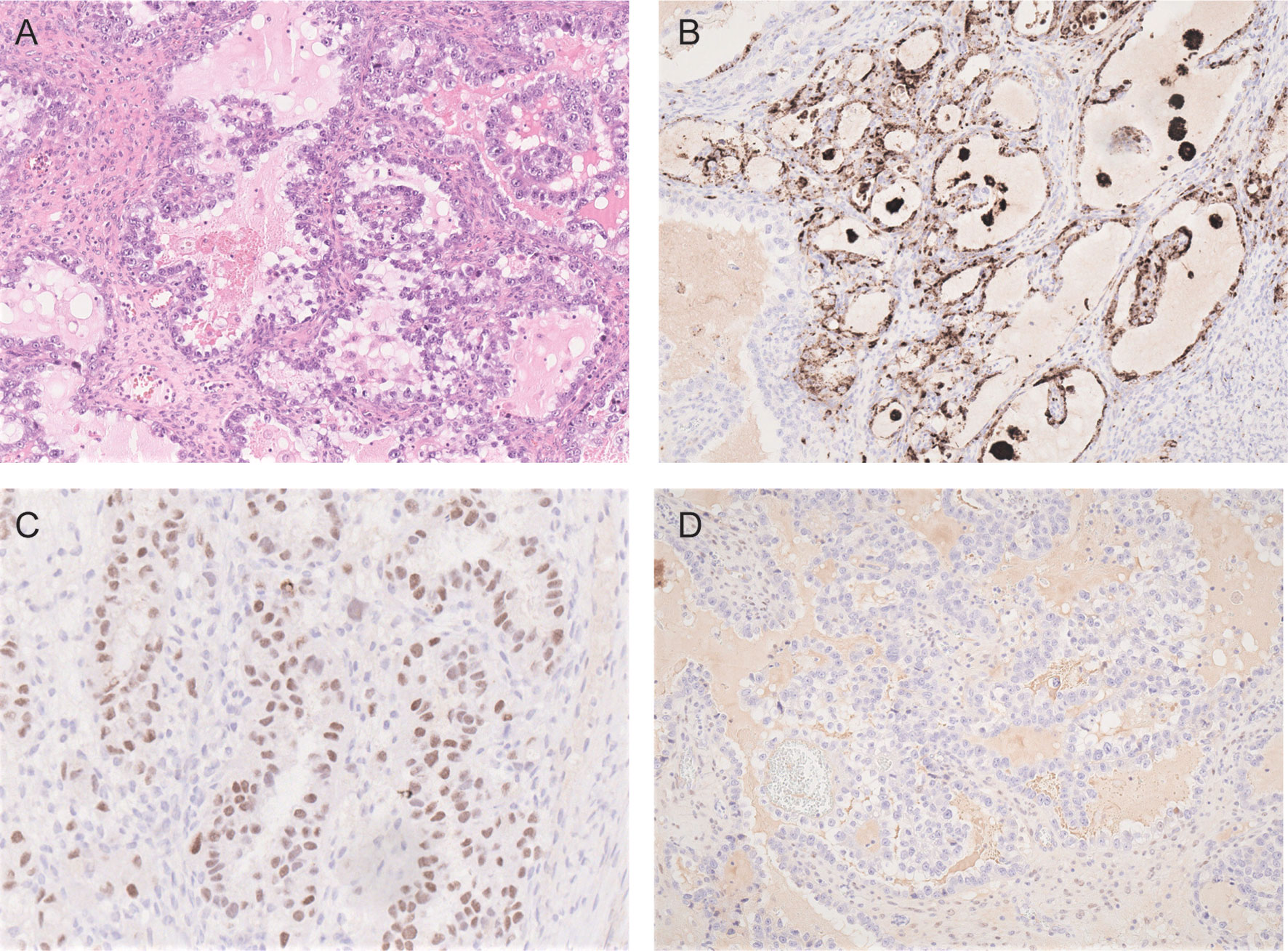
Figure 2 Photomicrographs of OCCC showing (A) tubulocystic pattern with tubules lined by hobnail, clear and eosinophilic cells (H&E). The carcinoma cells are immunoreactive for napsin A (B) and HNF-1β (C) but negative for estrogen receptor (D).
Genetically, loss of heterozygosity at chromosome 3p25 region is observed in nearly half of primary and metastatic OCCC (87). The high incidence of distinct genetic alterations, including mutations in ARID1A (46–66.7%) (76, 77, 88) and PIK3CA (32–50%) (88–90), as well as loss of PTEN activity (28–38%) (91, 92), are recognized to be key driving events in the pathogenesis of OCCC. In comparison with high-grade serous carcinoma, OCCC seldom exhibits mutations in TP53 and BRAF genes (90, 93). In addition, upregulated expression of hepatocyte nuclear factor-1β (HNF-1β) is commonly reported in ovarian clear cell tumors, including borderline tumors and carcinomas, suggesting a critical role of HNF-1β in the tumorigenesis of OCCC (72, 94, 95). OCCC also shows a high expression level of vascular endothelial growth factor (VEGF) that is commonly associated with poor clinical outcome (96). Compared to other subtypes, OCCC is more often diagnosed at an early stage, and the prognosis is good when no metastasis occurs (97). However, OCCCs in advanced stages or recurrent status usually show poor prognosis and are commonly resistant to most of existing chemotherapeutic treatments (6). Hence, exploring novel predictive biomarker and efficient therapeutic strategy is badly needed for improving the survival of OCCC patients.
DDR Status in OCCC
A significant proportion of OCCC shows HR deficiency and hence should be susceptible to PARP inhibitor therapy. Using targeted capture and deep sequencing analysis for germline and somatic loss-of-function mutations in 30 genes, including BRCA1, BRCA2, and 11 other genes in the HR pathway in 390 ovarian carcinomas, Pennington et al. documented that 26% of OCCC in their cohort harbored HR-related mutations (98). Similarly, about one-third of a Japanese cohort of OCCC was reported to have germline and somatic mutations in HR genes. The percentage is lower than that of high-grade serous carcinoma in which 44% present such mutations. Notably, the lists of mutated HR genes in high-grade serous carcinoma and CCC exhibited significant differences, with ATM being the most frequently mutated HR gene in OCCC (99). Moreover, as much as half of the OCCC exhibited the BRCAness phenotype (mutation in 18 different HR genes or having BRCA1 hypermethylation or loss of BRCA1 protein expression) in a Danish ovarian cancer cohort comprising 20 cases of OCCC (100). Genetic abnormalities of OCCC have therefore been suggested to be synthetic lethal with PARP inhibitors (5).
Some OCCCs are suggested to have defective DNA mismatch repair (MMR), and Lynch syndrome confers susceptibility to OCCC (101, 102). Indeed, endometrioid and clear-cell carcinomas are the most common tumors in the ovary to be associated with Lynch syndrome (103–105). By assessing microsatellite markers, Cai et al. concluded in an earlier study that about 21% of their OCCC samples exhibited at least some levels of microsatellite instability (MSI). A strong correlation existed between alterations in the expression of hMLH1 and hMSH2, two major DNA MMR genes, and the high level of MSI (MSI-H) in these tumors (106). Bennett et al. evaluated the expression MMR proteins in 109 unselected OCCC samples with known clinical characteristics. In their cohort, 6 (5.5%) of the tumors exhibited loss of MMR proteins (107). Howitt et al. reported that MSI-H or loss of MMR protein expression can be detected in 10% of OCCC and that these tumors with MSI-H status are associated with increased tumor-infiltrating lymphocytes and increased PD-L1 expression, thus should be susceptible to immune checkpoint therapy (108). However, PD-L1 expression was found to be more prevalent among OCCCs when compared with endometrial CCC irrespective of their MSI status (109). In another study, exome sequencing of 48 pairs of OCCC tumor/non-tumor samples identified 3 (6.25%) cases showing somatic hypermutated phenotype with ≥12-fold somatic mutations than the other 45 cases. These hypermutated OCCC cases have 8.1-fold higher frameshift indels/non-frameshift indels ratio indicating faulty MMR mechanisms consistent with MSI, leading to elevated single nucleotide indel insertion or deletion (88).
Compared with HR and MMR, less is known about the NER or NHEJ capability of OCCC. The mRNA levels of endonuclease ERCC1 and helicase XPB (ERCC3), key players in the NER pathway, tend to be higher in OCCC as compared to other types of ovarian cancers (110). Elevated ERCC1 is also suggested to contribute to chemoresistance in ovarian cancer (108), although the number of OCCC in the study was very small.
ARID1A Is a DDR Component and Promising Target of Synthetic Lethality Therapy
ARID1A is a subunit of the SWI/SNF chromatin-remodeling complex encompassing multiple proteins including also ARID1B, SMARCA4, and SMARCB1 that have tumor-suppressor functions (111). By altering chromatin structure and presumably modulating the accessibility of DNA, SWI/SNF complexes assume important roles in controlling DNA replication, transcription, and repair (112). Consistent with this view, SWI/SNF complexes or some of their components were found to be recruited to damaged DNA early in the repair process (113–115). Of note, ARID1A, which is frequently mutated in OCCC (38), and its homolog ARID1B participate in cellular resistance to various types of DNA damage, including DSB (Figure 3).
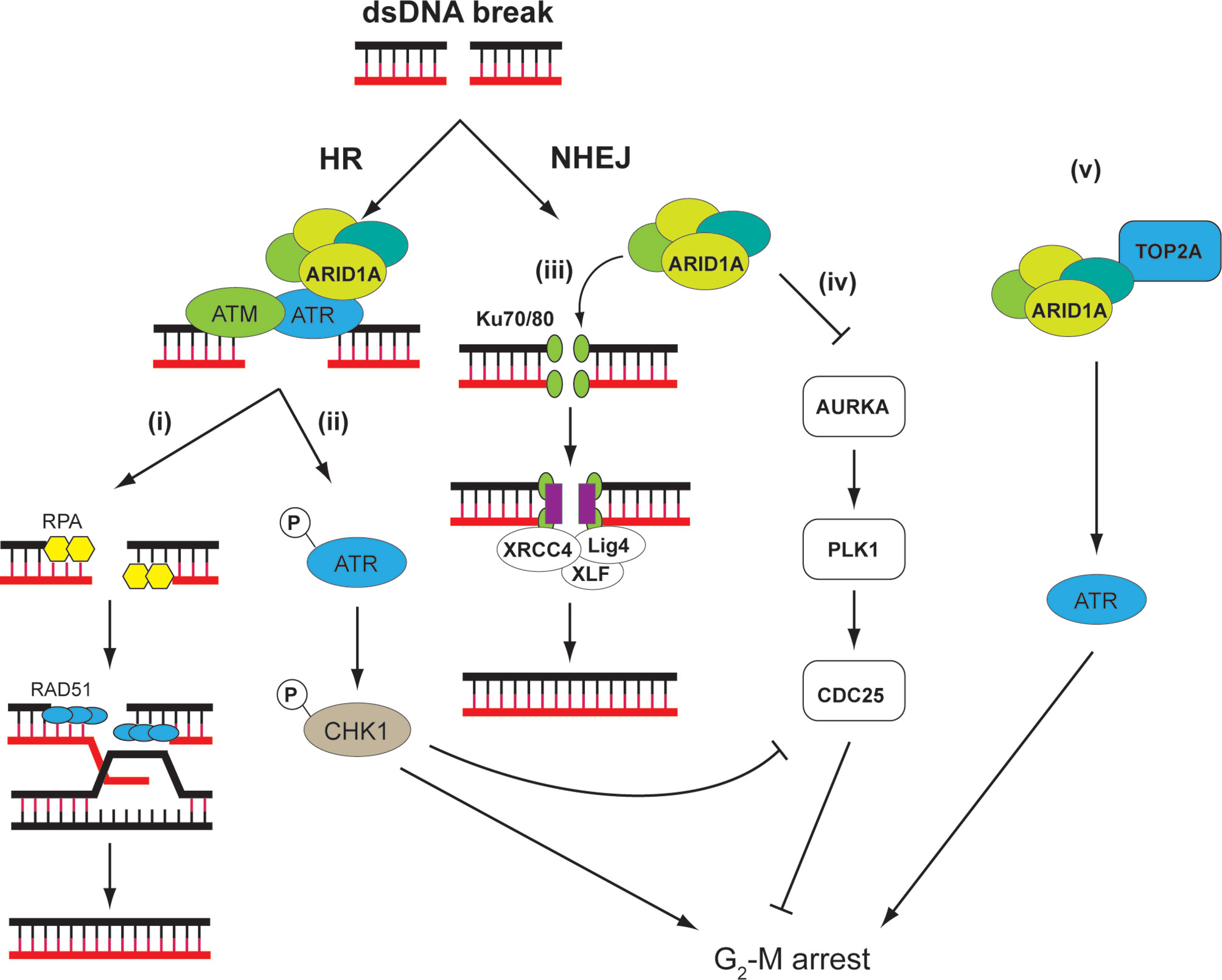
Figure 3 ARID1A is involved in DDR through different mechanisms. ARID1A is implicated on both HR and NHEJ. On DNA double-strand breaks (DSB), ATR directly interacts and recruits ARID1A containing SWI/SNF complex, which serves to (i) reduce nucleosome density at the DSB and facilitate DNA end resection to create single-strand for RPA deposition. The RPA-covered single-strand DNA is necessary for RAD51-mediated homology search and strand invasion (116). (ii) In addition, ARID1A is necessary for ATR activation, which, through CHK1, signals G2-M arrest (117). (iii) In NHEJ, SWI/SNF complex is necessary for recruitment of Ku70/80 and XRCC4 at DSB. Thus, loss of ARID1A leads to reduced NHEJ activity (118). (iv) ARID1A transcriptionally represses the mitosis driver kinase AURKA. In cells with ARID1A loss, AURKA-PLK1-CDC25C pathway is upregulated. In addition, CDC25C activity is negatively regulated by ARID1A–ATR–CHK pathway under DNA damage conditions, thereby strictly controlling the CDC25 activity in ARID1A WT cells. CDC25C activity is hence de-repressed in ARID1A mutated cells, leading to weakened G2-M checkpoint (119). (v) In addition, SWI/SNF complex may recruit TOP2A to chromatin, which, through ATR and other mechanisms, may signal S and G2-M arrest. These factors might render tumor cells sensitive to small-molecule ATR inhibitors as these agents impair the ability of cells to mount adequate DDRs while at the same time accelerating mitotic entry (120).
ARID1A and ARID1B are two mutually exclusive subunits in the SWI/SNF complex. Knocking down ARID1B in ARID1A-deficient colorectal cancer cells sensitized the cells to DNA damage caused by irradiation (121). It is therefore proposed that ARID1B depletion can work with radiotherapy to enhance response of patients with ARID1A-deficient malignancies. ARID1A and ARID1B accumulate at DNA damage site, and suppression of their expression reduces NHEJ activity. Several other components of the SWI/SNF complex including SNF5, BAF60a, BAF60c, BAF155, and BAF170 exhibit a similar NHEJ attenuated phenotype (118).
ARID1A mutation is prevalent in cancers, and synthetic lethal therapies associated with these mutations are being actively explored (122). For instance, it has been reported that ARID1A-deficient HEC151 uterine endometrial cancer and TOV21G OCCC cell lines acquire high sensitivity to PARP inhibition after exposure to exogenously induced DNA breaks such as ionizing radiation (123). A higher level of DSBs and cell death, demonstrated by immunoreactivity for phosphor-H2AX and cleaved caspase-3 respectively, was found in ARID1A-deficient cancers after combined radiation and PARP inhibitor therapy.
Moreover, in endometrial atypical hyperplasia, which demonstrates heterogeneous and patchy loss of ARID1A, higher immunoreactivity for phospho-H2AX, a marker for DSBs, was found in ARID1A-negative tumor foci than in areas with intact ARID1A expression. Immortalized endometrial epithelial cells with ARID1A knockdown also had lowered NHEJ activity and consequently higher susceptibility towards irradiation and PARP inhibitor olaparib (123). Indeed, the ARID1A-deficient CCC cell line TOV21G exhibited sensitivity towards olaparib, whereas the ARID1A wild-type CCC line RMG1 was apparently refractory to the drug (123). NUCOLL43, a novel OCCC cell line derived from an ARID1A-positive tumor, also exhibited intact DNA HR capacity (124).
ATR, a serine/threonine-specific protein kinase also known as ataxia telangiectasia and Rad3-related protein, is pivotal in detecting DNA damage and activating DNA damage checkpoint response (125, 126). Oncogene activation can impose replication stress on tumor cells, which may rely on ATR checkpoint function for survival. This suggested a rationale for applying ATR inhibitors as anticancer drugs (127). Potent and specific ATR inhibitors have been discovered including EPT-46464, AZ20, VE-821, and VX-970, some of which have entered clinical trials (120).
In the colorectal carcinoma cell line HCT 116, ARID1A is recruited to DSBs by its interaction with ATR where it sustains ATR activation and facilitates the generation of RPA coated single-strand DNA. Loss of ARID1A leads to impaired checkpoint activation and repair of DNA DSBs, which sensitizes cells to DSB-inducing agents such as radiation and PARP inhibitors (117).
Screening of genetic profile synthetic lethal with the ATR inhibitor VE-821 also identified ARID1A suppression as contributing factor to cytotoxicity of VE-821. ARID1A deficiency results in topoisomerase 2A and cell cycle defects, which cause an increased reliance on ATR checkpoint activity. Such mechanistic impact sensitizes ARID1A deficient triple-negative breast cancer cells to clinical inhibitors of ATR, both in vitro and in vivo (120). Indeed, a recent phase I clinical trial of an ATR inhibitor VX-970 on various advanced solid malignancies including cancers of the ovary, breast, colon, and stomach as monotherapy or in combination with carboplatin showed that the drug was well tolerated and showed satisfactory response (128). Mechanistically, significant inhibition of phosphorylation of CHK1, an ATR downstream substrate, was demonstrated.
Similarly, in germ cell tumors, ARID1A deficiency produced by CRISPR/Cas9 gene editing or pharmacological inhibition considerably sensitized tumor cells towards ATR inhibition (129). Mechanistically, ARID1A was postulated to control gene transcription, DDR, and epigenetic profile through putative downstream effectors DNA methyltransferase 1-associated protein DMAP1 and DNA Polymerase POLE. These data suggested ATR inhibition to be a promising approach to work with ARID1A mutations and warrant further investigation in OCCC.
Aurora kinases are serine/threonine kinases controlling multiple functions in mitosis including chromatid segregation. These kinases are related to the G1 DNA damage checkpoint via the p53 and p73 pathways. Aurora kinase A (AURKA) or serine/threonine protein kinase 6, triggers G2/M transition by phosphorylating a number of substrates including Polo-like kinase-1 (PLK1), ultimately leading to the nuclear localization of cell division cycle 25C (CDC25C). AURKA overexpression is common in many malignancies including leukemia as well as cancers of the ovary, lung, pancreas, liver, and colon (119). Hence, AURKA inhibitors are actively being investigated as cancer therapeutics (130). Inhibition of AURKA may contribute to the G2 DNA damage checkpoint through AURKA’s effect on PLK1 and CDC25B activation (131). Intriguingly, we have earlier reported high expression of PLK1 in OCCC, and PLK1 was shown to regulate apoptosis and autophagy of OCCC cells in relation to chemosensitivity (132).
ARID1A can attenuate AURKA transcription via epigenetic mechanism. On the other hand, ARID1A has a synthetic lethal interaction with AURKA in colorectal cancer cells such that AURKA inhibition can selectively impede the growth of ARID1A-deficient colorectal cancer cells. HCT116 with ARID1A knockout was most sensitive to AURAKA inhibitors in a screen of epigenetic drug library. Interestingly, knockout cells were more sensitive to AURKA inhibitors than PARP inhibitors. The ARID1A knockout genetic background aggravated the abnormal chromosome arrangement and segregation of AURKA inhibition and subsequently more apoptosis (119). In cells lacking ARID1A, AURKA upregulation leads to persistent activation of CDC25C (119). Hyperactivation of AURKA/PLK1/CDC25 axis was therefore observed in ARID1A knockout cells as ARID1A was found to be a transcription repressor of AURKA (119).
OCCC is the malignancy with highest proportion of ARID1A mutation (~50%). Given the fact that ARID1A interacts closely with key players in several DDR pathways, we hypothesize that most OCCC will be responsive to DDR-targeting drugs synthetic lethal to ARID1A mutations.
PI3K/AKT Pathway Alterations in OCCC May Enhance DDR Therapy
Compared with other histologic subtypes, OCCC exhibits higher prevalence of PIK3CA mutations and PTEN deletion (20–46% and 20%, respectively, compared with 2.3–3.7% and 7%, respectively, in high-grade serous ovarian carcinoma) (133). However, using PI3K pathway inhibitors as monotherapies for ovarian cancer was only met with limited success (8). Hence a new paradigm for utilizing PI3K inhibitors in ovarian cancer management is to explore combination strategies to improve the efficacy of PI3K pathway blockades (134). Since the PI3K/Akt/mTOR signaling pathway exerts multiple layers of regulation on the repair of DNA DSBs and SSBs, it has been suggested that DDR proteins may represent attractive targets of synthetic lethality with PI3K inhibitors (134). Indeed, it has been shown that PI3K inhibitors and CHK1 inhibitors combination treatments exhibit remarkably higher cytotoxicity in high-grade serous ovarian carcinoma cells compared with each individual drug alone, with evidence of increased DNA damage, chromosomal breaks, and mitotic catastrophe (135).
While targeting of PI3K/Akt/mTOR in in vitro models of OCCC has shown promising effectiveness (136–140), little is known about the impact of the targeting agents on the DDR capacity of OCCC cells. Since hyperactivation of PI3K3A and AKT via gain-of-function mutations is common in human cancers, perhaps insight could be drawn from investigations in other malignancies. In cervical cancer, PIK3CA mutation and increased expression of pAKT have been found to be related with resistance to radiotherapy (141, 142). Activated AKT can stimulate DNA repair such as DSB repair after radiotherapy (143). Common hotspot mutation of PIK3CA, PIK3CA-E545K mutation has also been found to enhance DNA repair in cervical cancer cells as demonstrated by fewer pH2A.X foci and more highly activated Chk1/Chk2, regulators of DDR in mutated cells (144). On the other hand, PI3K inhibitor (LY294002) can, through DSB repair, sensitize cervical cancer cells to radiation in vivo and in vitro (145–147).
The PI3K/AKT and Wnt/β-catenin pathways interact in the impact on DDR. PIK3CA-E545K is reported to confer resistance to ionizing radiation in cervical cancer cells by inducing overexpression and nuclear accumulation of β-catenin (144). Inhibiting β-catenin enhances radiosensitivity of cervical cancer cells in vitro and in vivo particularly in PIK3CA-E545K mutated cells. The Wnt/β-catenin inhibitor XAV-939 boosted radiation-induced DNA damage but attenuated DDR (144).
The activity of these pathways may also be related to the MMR status of the malignancies. The MMR-deficient colorectal carcinoma was found to be more related to PI3K, DDR, and WNT pathway aberrations, a profile different from MMR proficient cancer cells. Among pivotal genes in the WNT pathway, mutual exclusivity between mutations of CTNNB1 and APC or RNF43 was also demonstrated. It is interesting since CTNNB1 gene encoded β-catenin, the major effector of the WNT signal pathway, is negatively controlled by APC and RNF43. Moreover, MLH1-methylated MMR-deficient carcinomas have less CTNNB1 mutations than MLH1-unmethylated MMR-deficient cancers (148).
The PI3K/AKT and Wnt/β-catenin pathways also play a role in synergistic lethality in cancer therapy. In gastric cancer, synergistic lethality for combination therapy with paclitaxel and PI3K p110α-specific inhibitor alpelisib was found to be stronger in PIK3CA-mutant cells related to enhanced DDR and apoptosis, as demonstrated by γ-H2ax and caspase 3/7 assays, respectively (149). Indeed, Juvekar et al. have demonstrated that PI3K3CA inhibition of breast cancer cells by alpelisib can produce raised nucleotide depletion-mediated DNA damage and thus death of cancer cells (150). PI3K3CA inhibition was also found to be more effective in inducing DNA damage than inhibition of AKT.
Since aberration of these pathways are common in OCCC, exploration of DDR-targeted therapy in relation to the PI3K/AKT and Wnt/β-catenin pathways in OCCC may trigger novel treatment approaches
HNF-1β and DDR
Overexpression of HNF-1β is a clinically useful marker for discriminating OCCC from other subtypes of ovarian cancer (151). As a transcription factor HNF-1β assume several functional roles during the carcinogenesis of OCCC, such as metabolic reprogramming of the cancer cells (152, 153), causing tumor-associated thrombosis (154), and conferring carboplatin resistance (155). More importantly, HNF1β appears to be integral to the transformation of endometriosis lesions, serving as the master regulator of an antioxidant detoxification system responsible for resisting the oxidative microenvironment caused by hemolysis during the development of endometriosis (156). In terms of DDR, HNF-1β takes part in the Chk1 regulated cell cycle checkpoint in endometriotic cells, contributing to their transformation (157). Similarly, HNF-1β activates Chk1-mediated cell checkpoint in the presence of DNA damaging agents such as bleomycin and thereby enhancing cell survival by maximizing DNA repair efficiency (158). CHK1 inhibitor can abolish such activity and hence sensitize cells to the chemotherapy (159). These studies suggested HNF-1β is involved in the DDR of OCCC and that DDR targeting may be effect in HNF-1β overexpressing tumors.
Further Evidence of DDR-Targeting Drugs’ Efficacies in OCCC
The utilities of DDR-targeting drugs as cytotoxic agents of OCCC cells have been evaluated in several preclinical models. By assessing RAD51 foci formation in the presence of ionizing radiation-induced DNA DSBs, Wilkerson et al. categorized a panel of 12 OCCC cell lines as HR-competent and HR-deficient. The HR-deficient cells were also demonstrated to exhibit increased susceptibility towards cisplatin and the PARP inhibitor BMN-673 when compared with the HR competent cancer cells (160). Moreover, loss of PTEN function was found to be related to HR DNA repair deficiency in a portion of OCCC cells. Indeed, it is known that PTEN plays a crucial role in DDR in several cancers such as colorectal cancers, and PTEN polymorphism was associated with response to neoadjuvant chemoradiotherapy (161).
Predictors of Response to PARP Inhibitors and Immunotherapy for OCCC
According to the recent guideline from the American Society of Clinical Oncology (ASCO) (162), all women with ovarian cancers irrespective of histological subtypes should have germline genetic tests of ovarian cancer susceptibility genes as a multigene panel that should at least include BRCA1, BRCA2, RAD51C, RAD51D, BRIP1, MLH1, MSH2, MSH6, PMS2, and PALB2. Somatic genetic testing on tumor tissue should be performed if germline pathogenic or likely pathogenic variants cannot be found. It is considered that such genetic testing should not be restricted to high-grade serous carcinoma (98, 163) since women having clear cell, endometrioid, low-grade serous, or carcinosarcoma subtypes of ovarian cancer were also found to still carry a significant risk of harboring germline BRCA mutation near to that of high-grade serous carcinoma (98).
MMR deficiency is found in about 10–12% of unselected ovarian cancers although it is more common in non-serous histological subtypes (101, 104, 164). It is reported that 11.5% of clear cell carcinoma exhibit MMR deficiency. Microsatellite instability is also demonstrated in a subset of OCCC, rendering them immunogenic (108). Hence, routine testing of MMR deficiency in OCCC is also recommended for consideration of treatment with pembrolizumab in the setting of recurrent disease.
Conclusion
OCCC is characterized by chemoresistance and worse prognosis particularly cases with extraovarian dissemination demanding novel therapeutic approaches, especially targeted therapy. Like other solid tumors, OCCC exhibits frequent DDR defects that can be exploited to specifically kill cancer cells, under the principle of synthetic lethality. In fact, one may expect that DDR targeting can be exceptionally effective given the distinct genetic characteristics of the malignancy. While PARP inhibitors have proven utilities in high-grade serous ovarian carcinomas, the efficacies of other DDR-targeting approaches in OCCC await exploration, particularly by comprehensive clinical trials. Biomarkers for predicting sensitivity to PARP inhibitors and other DDR-targeting strategies as well as identifying synergistic combinations may readily be applicable in OCCC and worth exploring.
Author Contributions
OW and AC conceived and wrote the manuscript. JL wrote the manuscript. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global Cancer Statistics 2018: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA: Cancer J Clin (2018) 68:394–424. doi: 10.3322/caac.21492
2. McCluggage WG, Lax SF, Longacre TA, Malpica A, Soslow RA. Tumours of the Ovary. In: WHO Classification of Female Genital Tumours. International Agency of Research on Cancer (2020).
3. Okamoto A, Glasspool RM, Mabuchi S, Matsumura N, Nomura H, Itamochi H, et al. Gynecologic Cancer Intergroup (GCIG) Consensus Review for Clear Cell Carcinoma of the Ovary. Int J Gynecol Cancer (2014) 24:S20–25. doi: 10.1097/igc.0000000000000289
4. Matsuzaki S, Yoshino K, Ueda Y, Matsuzaki S, Kakuda M, Okazawa A, et al. Potential Targets for Ovarian Clear Cell Carcinoma: A Review of Updates and Future Perspectives. Cancer Cell Int (2015) 15:117. doi: 10.1186/s12935-015-0267-0
5. Machida H, Matsuo K, Yamagami W, Ebina Y, Kobayashi Y, Tabata T, et al. Trends and Characteristics of Epithelial Ovarian Cancer in Japan Between 2002 and 2015: A JSGO-JSOG Joint Study. Gynecol Oncol (2019) 153:589–96. doi: 10.1016/j.ygyno.2019.03.243
6. Sugiyama T, Kamura T, Kigawa J, Terakawa N, Kikuchi Y, Kita T, et al. Clinical Characteristics of Clear Cell Carcinoma of the Ovary: A Distinct Histologic Type With Poor Prognosis and Resistance to Platinum-Based Chemotherapy. Cancer (2000) 88:2584–9. doi: 10.1002/1097-0142(20000601)88:11<2584::AID-CNCR22>3.0.CO;2-5
7. Takano M, Tsuda H, Sugiyama T. Clear Cell Carcinoma of the Ovary: Is There a Role of Histology-Specific Treatment? J Exp Clin Cancer Res (2012) 31:53. doi: 10.1186/1756-9966-31-53
8. Ogasawara A, Sato S, Hasegawa K. Current and Future Strategies for Treatment of Ovarian Clear Cell Carcinoma. J Obstet Gynaecol Res (2020) 46:1678–89. doi: 10.1111/jog.14350
9. Pearl LH, Schierz AC, Ward SE, Al-Lazikani B, Pearl FM. Therapeutic Opportunities Within the DNA Damage Response. Nat Rev Cancer (2015) 15:166–80. doi: 10.1038/nrc3891
10. Pilie PG, Tang C, Mills GB, Yap TA. State-of-the-Art Strategies for Targeting the DNA Damage Response in Cancer. Nat Rev Clin Oncol (2019) 16:81–104. doi: 10.1038/s41571-018-0114-z
11. Chatterjee N, Walker GC. Mechanisms of DNA Damage, Repair, and Mutagenesis. Environ Mol Mutagen (2017) 58:235–63. doi: 10.1002/em.22087
12. Jackson SP, Bartek J. The DNA-Damage Response in Human Biology and Disease. Nature (2009) 461:1071–8. doi: 10.1038/nature08467
13. d'Adda di Fagagna F, Reaper PM, Clay-Farrace L, Fiegler H, Carr P, Von Zglinicki T, et al. A DNA Damage Checkpoint Response in Telomere-Initiated Senescence. Nature (2003) 426:194–8. doi: 10.1038/nature02118
14. Bartkova J, Horejsi Z, Koed K, Kramer A, Tort F, Zieger K, et al. DNA Damage Response as a Candidate Anti-Cancer Barrier in Early Human Tumorigenesis. Nature (2005) 434:864–70. doi: 10.1038/nature03482
15. Gorgoulis VG, Vassiliou LV, Karakaidos P, Zacharatos P, Kotsinas A, Liloglou T, et al. Activation of the DNA Damage Checkpoint and Genomic Instability in Human Precancerous Lesions. Nature (2005) 434:907–13. doi: 10.1038/nature03485
16. Bartkova J, Rezaei N, Liontos M, Karakaidos P, Kletsas D, Issaeva N, et al. Oncogene-Induced Senescence is Part of the Tumorigenesis Barrier Imposed by DNA Damage Checkpoints. Nature (2006) 444:633–7. doi: 10.1038/nature05268
17. O'Connor MJ. Targeting the DNA Damage Response in Cancer. Mol Cell (2015) 60:547–60. doi: 10.1016/j.molcel.2015.10.040
18. Darzynkiewicz Z, Traganos F, Wlodkowic D. Impaired DNA Damage Response–an Achilles' Heel Sensitizing Cancer to Chemotherapy and Radiotherapy. Eur J Pharmacol (2009) 625:143–50. doi: 10.1016/j.ejphar.2009.05.032
19. Jeggo PA, Pearl LH, Carr AM. DNA Repair, Genome Stability and Cancer: A Historical Perspective. Nat Rev Cancer (2016) 16:35–42. doi: 10.1038/nrc.2015.4
20. Klinakis A, Karagiannis D, Rampias T. Targeting DNA Repair in Cancer: Current State and Novel Approaches. Cell Mol Life Sci (2020) 77:677–703. doi: 10.1007/s00018-019-03299-8
21. Oberdoerffer P, Sinclair DA. The Role of Nuclear Architecture in Genomic Instability and Ageing. Nat Rev Mol Cell Biol (2007) 8:692–702. doi: 10.1038/nrm2238
22. Klose RJ, Zhang Y. Regulation of Histone Methylation by Demethylimination and Demethylation. Nat Rev Mol Cell Biol (2007) 8:307–18. doi: 10.1038/nrm2143
23. Henle ES, Linn S. Formation, Prevention, and Repair of DNA Damage by Iron/Hydrogen Peroxide. J Biol Chem (1997) 272:19095–8. doi: 10.1074/jbc.272.31.19095
24. Loeb LA, Monnat RJ Jr. DNA Polymerases and Human Disease. Nature Reviews. Genetics (2008) 9:594–604. doi: 10.1038/nrg2345
25. Desouky O, Ding N, Zhou G. Targeted and Non-Targeted Effects of Ionizing Radiation. J Radiat Res Appl Sci (2015) 8:247–54. doi: 10.1016/j.jrras.2015.03.003
26. Rastogi RP, Kumar A, Tyagi MB, Sinha RP. Molecular Mechanisms of Ultraviolet Radiation-Induced DNA Damage and Repair. J Nucleic Acids (2010) 2010:592980–0. doi: 10.4061/2010/592980
27. Giglia-Mari G, Zotter A, Vermeulen W. DNA Damage Response. Cold Spring Harbor Perspect Biol (2011) 3:a000745. doi: 10.1101/cshperspect.a000745
28. Hanahan D, Robert WA. Hallmarks of Cancer: The Next Generation. Cell (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
29. Mahaney BL, Meek K, Lees-Miller SP. Repair of Ionizing Radiation-Induced DNA Double-Strand Breaks by non-Homologous End-Joining. Biochem J (2009) 417:639–50. doi: 10.1042/BJ20080413
30. Qiu Z, Zhang Z, Roschke A, Varga T, Aplan PD. Generation of Gross Chromosomal Rearrangements by a Single Engineered DNA Double Strand Break. Sci Rep (2017) 7:43156. doi: 10.1038/srep43156
31. Pannunzio NR, Watanabe G, Lieber MR. Nonhomologous DNA End-Joining for Repair of DNA Double-Strand Breaks. J Biol Chem (2018) 293:10512–23. doi: 10.1074/jbc.TM117.000374
32. Vignard J, Mirey G, Salles B. Ionizing-Radiation Induced DNA Double-Strand Breaks: A Direct and Indirect Lighting Up. Radiotherapy Oncol J Eur Soc Ther Radiol Oncol (2013) 108:362–9. doi: 10.1016/j.radonc.2013.06.013
33. Borsos BN, Majoros H, Pankotai T. Ubiquitylation-Mediated Fine-Tuning of DNA Double-Strand Break Repair. Cancers (Basel) (2020) 12:1617. doi: 10.3390/cancers12061617
34. Krajewska M, Fehrmann RSN, de Vries EGE, van Vugt MATM. Regulators of Homologous Recombination Repair as Novel Targets for Cancer Treatment. Front Genet (2015) 6:96. doi: 10.3389/fgene.2015.00096
35. San Filippo J, Sung P, Klein H. Mechanism of Eukaryotic Homologous Recombination. Annu Rev Biochem (2008) 77:229–57. doi: 10.1146/annurev.biochem.77.061306.125255
36. Curtin NJ. DNA Repair Dysregulation From Cancer Driver to Therapeutic Target. Nat Rev Cancer (2012) 12:801–17. doi: 10.1038/nrc3399
37. Lord CJ, Ashworth A. The DNA Damage Response and Cancer Therapy. Nature (2012) 481:287–94. doi: 10.1038/nature10760
38. Cancer Genome Atlas Research, N. Integrated Genomic Analyses of Ovarian Carcinoma. Nature (2011) 474:609–15. doi: 10.1038/nature10166
39. Ledermann J, Harter P, Gourley C, Friedlander M, Vergote I, Rustin G, et al. Olaparib Maintenance Therapy in Platinum-Sensitive Relapsed Ovarian Cancer. N Engl J Med (2012) 366:1382–92. doi: 10.1056/NEJMoa1105535
40. Ledermann J, Harter P, Gourley C, Friedlander M, Vergote I, Rustin G, et al. Olaparib Maintenance Therapy in Patients With Platinum-Sensitive Relapsed Serous Ovarian Cancer: A Preplanned Retrospective Analysis of Outcomes by BRCA Status in a Randomised Phase 2 Trial. Lancet Oncol (2014) 15:852–61. doi: 10.1016/S1470-2045(14)70228-1
41. Pujade-Lauraine E, Ledermann JA, Selle F, Gebski V, Penson RT, Oza AM, et al. Olaparib Tablets as Maintenance Therapy in Patients With Platinum-Sensitive, Relapsed Ovarian Cancer and a BRCA1/2 Mutation (SOLO2/ENGOT-Ov21): A Double-Blind, Randomised, Placebo-Controlled, Phase 3 Trial. Lancet Oncol (2017) 18:1274–84. doi: 10.1016/S1470-2045(17)30469-2
42. Kaufman B, Shapira-Frommer R, Schmutzler RK, Audeh MW, Friedlander M, Balmana J, et al. Olaparib Monotherapy in Patients With Advanced Cancer and a Germline BRCA1/2 Mutation. J Clin Oncol (2015) 33:244–50. doi: 10.1200/JCO.2014.56.2728
43. Coleman RL, Oza AM, Lorusso D, Aghajanian C, Oaknin A, Dean A, et al. Rucaparib Maintenance Treatment for Recurrent Ovarian Carcinoma After Response to Platinum Therapy (ARIEL3): A Randomised, Double-Blind, Placebo-Controlled, Phase 3 Trial. Lancet (2017) 390:1949–61. doi: 10.1016/S0140-6736(17)32440-6
44. Swisher EM, Lin KK, Oza AM, Scott CL, Giordano H, Sun J, et al. Rucaparib in Relapsed, Platinum-Sensitive High-Grade Ovarian Carcinoma (ARIEL2 Part 1): An International, Multicentre, Open-Label, Phase 2 Trial. Lancet Oncol (2017) 18:75–87. doi: 10.1016/S1470-2045(16)30559-9
45. Mirza MR, Monk BJ, Herrstedt J, Oza AM, Mahner S, Redondo A, et al. Niraparib Maintenance Therapy in Platinum-Sensitive, Recurrent Ovarian Cancer. N Engl J Med (2016) 375:2154–64. doi: 10.1056/NEJMoa1611310
46. Lord CJ, Tutt AN, Ashworth A. Synthetic Lethality and Cancer Therapy: Lessons Learned From the Development of PARP Inhibitors. Annu Rev Med (2015) 66:455–70. doi: 10.1146/annurev-med-050913-022545
47. Kondrashova O, Topp M, Nesic K, Lieschke E, Ho GY, Harrell MI, et al. Methylation of All BRCA1 Copies Predicts Response to the PARP Inhibitor Rucaparib in Ovarian Carcinoma. Nat Commun (2018) 9:3970. doi: 10.1038/s41467-018-05564-z
48. Lips EH, Laddach N, Savola SP, Vollebergh MA, Oonk AM, Imholz AL, et al. Quantitative Copy Number Analysis by Multiplex Ligation-Dependent Probe Amplification (MLPA) of BRCA1-Associated Breast Cancer Regions Identifies Brcaness. Breast Cancer Res (2011) 13:R107. doi: 10.1186/bcr3049
49. Ruscito I, Dimitrova D, Vasconcelos I, Gellhaus K, Schwachula T, Bellati F, et al. BRCA1 Gene Promoter Methylation Status in High-Grade Serous Ovarian Cancer Patients–a Study of the Tumour Bank Ovarian Cancer (TOC) and Ovarian Cancer Diagnosis Consortium (OVCAD). Eur J Cancer (2014) 50:2090–8. doi: 10.1016/j.ejca.2014.05.001
50. Jazaeri AA, Yee CJ, Sotiriou C, Brantley KR, Boyd J, Liu ET. Gene Expression Profiles of BRCA1-Linked, BRCA2-Linked, and Sporadic Ovarian Cancers. J Natl Cancer Inst (2002) 94:990–1000. doi: 10.1093/jnci/94.13.990
51. Larsen MJ, Kruse TA, Tan Q, Laenkholm AV, Bak M, Lykkesfeldt AE, et al. Classifications Within Molecular Subtypes Enables Identification of BRCA1/BRCA2 Mutation Carriers by RNA Tumor Profiling. PloS One (2013) 8:e64268. doi: 10.1371/journal.pone.0064268
52. Peng G, Chun-Jen Lin C, Mo W, Dai H, Park YY, Kim SM, et al. Genome-Wide Transcriptome Profiling of Homologous Recombination DNA Repair. Nat Commun (2014) 5:3361. doi: 10.1038/ncomms4361
53. Lok BH, Gardner EE, Schneeberger VE, Ni A, Desmeules P, Rekhtman N, et al. PARP Inhibitor Activity Correlates With SLFN11 Expression and Demonstrates Synergy With Temozolomide in Small Cell Lung Cancer. Clin Cancer Res (2017) 23:523–35. doi: 10.1158/1078-0432.CCR-16-1040
54. Joosse SA, van Beers EH, Tielen IH, Horlings H, Peterse JL, Hoogerbrugge N, et al. Prediction of BRCA1-Association in Hereditary Non-BRCA1/2 Breast Carcinomas With Array-CGH. Breast Cancer Res Treat (2009) 116:479–89. doi: 10.1007/s10549-008-0117-z
55. Vollebergh MA, Lips EH, Nederlof PM, Wessels LFA, Schmidt MK, van Beers EH, et al. An Acgh Classifier Derived From BRCA1-Mutated Breast Cancer and Benefit of High-Dose Platinum-Based Chemotherapy in HER2-Negative Breast Cancer Patients. Ann Oncol (2011) 22:1561–70. doi: 10.1093/annonc/mdq624
56. Watkins JA, Irshad S, Grigoriadis A, Tutt AN. Genomic Scars as Biomarkers of Homologous Recombination Deficiency and Drug Response in Breast and Ovarian Cancers. Breast Cancer Res (2014) 16:211. doi: 10.1186/bcr3670
57. Mukhopadhyay A, Elattar A, Cerbinskaite A, Wilkinson SJ, Drew Y, Kyle S, et al. Development of a Functional Assay for Homologous Recombination Status in Primary Cultures of Epithelial Ovarian Tumor and Correlation With Sensitivity to Poly(ADP-Ribose) Polymerase Inhibitors. Clin Cancer Res (2010) 16:2344–51. doi: 10.1158/1078-0432.CCR-09-2758
58. Lee JM, Gordon N, Trepel JB, Lee MJ, Yu M, Kohn EC. Development of a Multiparameter Flow Cytometric Assay as a Potential Biomarker for Homologous Recombination Deficiency in Women With High-Grade Serous Ovarian Cancer. J Transl Med (2015) 13:239. doi: 10.1186/s12967-015-0604-z
59. Cruz C, Castroviejo-Bermejo M, Gutierrez-Enriquez S, Llop-Guevara A, Ibrahim YH, Gris-Oliver A, et al. RAD51 Foci as a Functional Biomarker of Homologous Recombination Repair and PARP Inhibitor Resistance in Germline BRCA-Mutated Breast Cancer. Ann Oncol (2018) 29:1203–10. doi: 10.1093/annonc/mdy099
60. Morice PM, Coquan E, Weiswald LB, Lambert B, Vaur D, Poulain L. Identifying Patients Eligible for PARP Inhibitor Treatment: From NGS-Based Tests to 3D Functional Assays. Br J Cancer (2021) 125:7–14. doi: 10.1038/s41416-021-01295-z
61. Ngoi NYL, Tan DSP. The Role of Homologous Recombination Deficiency Testing in Ovarian Cancer and its Clinical Implications: Do We Need It? ESMO Open (2021) 6:100144. doi: 10.1016/j.esmoop.2021.100144
62. Plon SE, Eccles DM, Easton D, Foulkes WD, Genuardi M, Greenblatt MS, et al. Sequence Variant Classification and Reporting: Recommendations for Improving the Interpretation of Cancer Susceptibility Genetic Test Results. Hum Mutat (2008) 29:1282–91. doi: 10.1002/humu.20880
63. Rosenbaum E, Jonsson P, Seier K, Qin LX, Chi P, Dickson M, et al. Clinical Outcome of Leiomyosarcomas With Somatic Alteration in Homologous Recombination Pathway Genes. JCO Precis Oncol (2020) 4:1350–60. doi: 10.1200/PO.20.00122
64. Cheng DT, Mitchell TN, Zehir A, Shah RH, Benayed R, Syed A, et al. Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): A Hybridization Capture-Based Next-Generation Sequencing Clinical Assay for Solid Tumor Molecular Oncology. J Mol Diagn (2015) 17:251–64. doi: 10.1016/j.jmoldx.2014.12.006
65. Davies H, Glodzik D, Morganella S, Yates LR, Staaf J, Zou X, et al. Hrdetect is a Predictor of BRCA1 and BRCA2 Deficiency Based on Mutational Signatures. Nat Med (2017) 23:517–25. doi: 10.1038/nm.4292
66. Chopra N, Tovey H, Pearson A, Cutts R, Toms C, Proszek P, et al. Homologous Recombination DNA Repair Deficiency and PARP Inhibition Activity in Primary Triple Negative Breast Cancer. Nat Commun (2020) 11:2662. doi: 10.1038/s41467-020-16142-7
67. Degasperi A, Amarante TD, Czarnecki J, Shooter S, Zou X, Glodzik D, et al. A Practical Framework and Online Tool for Mutational Signature Analyses Show Inter-Tissue Variation and Driver Dependencies. Nat Cancer (2020) 1:249–63. doi: 10.1038/s43018-020-0027-5
68. Moore K, Colombo N, Scambia G, Kim BG, Oaknin A, Friedlander M, et al. Maintenance Olaparib in Patients With Newly Diagnosed Advanced Ovarian Cancer. N Engl J Med (2018) 379:2495–505. doi: 10.1056/NEJMoa1810858
69. Cadoo KA, Simpkins F, Mathews CA, Kabil N, Bennett J, Aghajanian C. Olaparib Treatment in Patients (Pts) With Platinum-Sensitive Relapsed (PSR) Ovarian Cancer (OC) by BRCA Mutation (Brcam) and Homologous Recombination Deficiency (HRD) Status: Phase II LIGHT Study. J Clin Oncol (2020) 38:6013–3. doi: 10.1200/JCO.2020.38.15_suppl.6013
70. Hill SJ, Decker B, Roberts EA, Horowitz NS, Muto MG, Worley MJ Jr, et al. Prediction of DNA Repair Inhibitor Response in Short-Term Patient-Derived Ovarian Cancer Organoids. Cancer Discov (2018) 8:1404–21. doi: 10.1158/2159-8290.CD-18-0474
71. del Carmen MG, Birrer M, Schorge JO. Clear Cell Carcinoma of the Ovary: A Review of the Literature. Gynecol Oncol (2012) 126:481–90. doi: 10.1016/j.ygyno.2012.04.021
72. Kwok LM. Clear Cell Carcinoma of the Ovary : Epidemiology and Biomarker HNF-1beta. Hong Kong: University of Hong Kong Libraries (2015).
73. Leskela S, Romero I, Cristobal E, Perez-Mies B, Rosa-Rosa JM, Gutierrez-Pecharroman A, et al. Mismatch Repair Deficiency in Ovarian Carcinoma: Frequency, Causes, and Consequences. Am J Surg Pathol (2020) 44:649–56. doi: 10.1097/PAS.0000000000001432
74. Risch HA, McLaughlin JR, Cole DE, Rosen B, Bradley L, Kwan E, et al. Prevalence and Penetrance of Germline BRCA1 and BRCA2 Mutations in a Population Series of 649 Women With Ovarian Cancer. Am J Hum Genet (2001) 68:700–10. doi: 10.1086/318787
75. Kobel M, Kalloger SE, Boyd N, McKinney S, Mehl E, Palmer C, et al. Ovarian Carcinoma Subtypes are Different Diseases: Implications for Biomarker Studies. PloS Med (2008) 5:e232. doi: 10.1371/journal.pmed.0050232
76. Jones S, Wang T-L, Shih I-M, Mao T-L, Nakayama K, Roden R, et al. Frequent Mutations of Chromatin Remodeling Gene ARID1A in Ovarian Clear Cell Carcinoma. Science (2010) 330:228–31. doi: 10.1126/science.1196333
77. Wiegand KC, Shah SP, Al-Agha OM, Zhao Y, Tse K, Zeng T, et al. ARID1A Mutations in Endometriosis-Associated Ovarian Carcinomas. N Engl J Med (2010) 363:1532–43. doi: 10.1056/NEJMoa1008433
78. Anglesio MS, Kommoss S, Tolcher MC, Clarke B, Galletta L, Porter H, et al. Molecular Characterization of Mucinous Ovarian Tumours Supports a Stratified Treatment Approach With HER2 Targeting in 19% of Carcinomas. J Pathol (2013) 229:111–20. doi: 10.1002/path.4088
79. Anglesio MS, Arnold JM, George J, Tinker AV, Tothill R, Waddell N, et al. Mutation of ERBB2 Provides a Novel Alternative Mechanism for the Ubiquitous Activation of RAS-MAPK in Ovarian Serous Low Malignant Potential Tumors. Mol Cancer Res (2008) 6:1678–90. doi: 10.1158/1541-7786.MCR-08-0193
80. McBride DJ, Etemadmoghadam D, Cooke SL, Alsop K, George J, Butler A, et al. Tandem Duplication of Chromosomal Segments is Common in Ovarian and Breast Cancer Genomes. J Pathol (2012) 227:446–55. doi: 10.1002/path.4042
81. Ovary - Epithelial Carcinoma . Available at: http://www.bccancer.bc.ca/health-professionals/clinical-resources/cancer-management-guidelines/gynecology/ovary-epithelial-carcinoma#.
82. Walker RA. World Health Organization Classification of Tumours. Pathol Genet Tumours Breast Female Genital Organs (2005) 46:229–9. doi: 10.1111/j.1365-2559.2004.02026.x
83. Kobel M, Kalloger SE, Carrick J, Huntsman D, Asad H, Oliva E, et al. A Limited Panel of Immunomarkers can Reliably Distinguish Between Clear Cell and High-Grade Serous Carcinoma of the Ovary. Am J Surg Pathol (2009) 33:14–21. doi: 10.1097/PAS.0b013e3181788546
84. Kobel M, Rahimi K, Rambau PF, Naugler C, Le Page C, Meunier L, et al. An Immunohistochemical Algorithm for Ovarian Carcinoma Typing. Int J Gynecol Pathol (2016) 35:430–41. doi: 10.1097/PGP.0000000000000274
85. Lim D, Ip PP, Cheung AN, Kiyokawa T, Oliva E. Immunohistochemical Comparison of Ovarian and Uterine Endometrioid Carcinoma, Endometrioid Carcinoma With Clear Cell Change, and Clear Cell Carcinoma. Am J Surg Pathol (2015) 39:1061–9. doi: 10.1097/PAS.0000000000000436
86. Kobel M, Luo L, Grevers X, Lee S, Brooks-Wilson A, Gilks CB, et al. Ovarian Carcinoma Histotype: Strengths and Limitations of Integrating Morphology With Immunohistochemical Predictions. Int J Gynecol Pathol (2019) 38:353–62. doi: 10.1097/PGP.0000000000000530
87. Simsir A, Palacios D, Linehan WM, Merino MJ, Abati A. Detection of Loss of Heterozygosity at Chromosome 3p25–26 in Primary and Metastatic Ovarian Clear-Cell Carcinoma: Utilization of Microdissection and Polymerase Chain Reaction in Archival Tissues. Diagn Cytopathol (2001) 24:328–32. doi: 10.1002/dc.1070
88. Shibuya Y, Tokunaga H, Saito S, Shimokawa K, Katsuoka F, Bin L, et al. Identification of Somatic Genetic Alterations in Ovarian Clear Cell Carcinoma With Next Generation Sequencing. Genes Chromosomes Cancer (2018) 57:51–60. doi: 10.1002/gcc.22507
89. Takenaka M, Saito M, Iwakawa R, Yanaihara N, Saito M, Kato M, et al. Profiling of Actionable Gene Alterations in Ovarian Cancer by Targeted Deep Sequencing. Int J Oncol (2015) 46:2389–98. doi: 10.3892/ijo.2015.2951
90. Zannoni GF, Improta G, Pettinato A, Brunelli C, Troncone G, Scambia G, et al. Molecular Status of PI3KCA, KRAS and BRAF in Ovarian Clear Cell Carcinoma: An Analysis of 63 Patients. J Clin Pathol (2016) 69:1088–92. doi: 10.1136/jclinpath-2016-203776
91. Hashiguchi Y, Tsuda H, Inoue T, Berkowitz RS, Mok SC. PTEN Expression in Clear Cell Adenocarcinoma of the Ovary. Gynecol Oncol (2006) 101:71–5. doi: 10.1016/j.ygyno.2005.09.047
92. Ho CM, Lin MC, Huang SH, Huang CJ, Lai HC, Chien TY, et al. PTEN Promoter Methylation and LOH of 10q22-23 Locus in PTEN Expression of Ovarian Clear Cell Adenocarcinomas. Gynecol Oncol (2009) 112:307–13. doi: 10.1016/j.ygyno.2008.09.040
93. Vang R, Whitaker BP, Farhood AI, Silva EG, Ro JY, Deavers MT. Immunohistochemical Analysis of Clear Cell Carcinoma of the Gynecologic Tract. Int J Gynecol Pathol (2001) 20:252–9. doi: 10.1097/00004347-200107000-00008
94. DeLair D, Han G, Irving JA, Leung S, Ewanowich CA, Longacre TA, et al. Hnf-1β in Ovarian Carcinomas With Serous and Clear Cell Change. Int J gynecological Pathol Off J Int Soc Gynecological Pathologists (2013) 32:541–6. doi: 10.1097/PGP.0b013e318273fd07
95. Huang W, Cheng X, Ji J, Zhang J, Li Q. The Application Value of HNF-1β Transcription Factor in the Diagnosis of Ovarian Clear Cell Carcinoma. Int J Gynecol Pathol (2016) 35:66–71. doi: 10.1097/pgp.0000000000000213
96. Mabuchi S, Kawase C, Altomare DA, Morishige K, Hayashi M, Sawada K, et al. Vascular Endothelial Growth Factor is a Promising Therapeutic Target for the Treatment of Clear Cell Carcinoma of the Ovary. Mol Cancer Ther (2010) 9:2411–22. doi: 10.1158/1535-7163.MCT-10-0169
97. Liu H, Xu Y, Ji J, Dong R, Qiu H, Dai X. Prognosis of Ovarian Clear Cell Cancer Compared With Other Epithelial Cancer Types: A Population−Based Analysis. Oncol Lett (2020) 19:1947–57. doi: 10.3892/ol.2020.11252
98. Pennington KP, Walsh T, Harrell MI, Lee MK, Pennil CC, Rendi MH, et al. Germline and Somatic Mutations in Homologous Recombination Genes Predict Platinum Response and Survival in Ovarian, Fallopian Tube, and Peritoneal Carcinomas. Clin Cancer Res (2014) 20:764–75. doi: 10.1158/1078-0432.CCR-13-2287
99. Sugino K, Tamura R, Nakaoka H, Yachida N, Yamaguchi M, Mori Y, et al. Germline and Somatic Mutations of Homologous Recombination-Associated Genes in Japanese Ovarian Cancer Patients. Sci Rep (2019) 9:17808. doi: 10.1038/s41598-019-54116-y
100. Hjortkjaer M, Malik Aagaard Jorgensen M, Waldstrom M, Ornskov D, Sogaard-Andersen E, Jakobsen A, et al. The Clinical Importance of Brcaness in a Population-Based Cohort of Danish Epithelial Ovarian Cancer. Int J Gynecol Cancer (2019) 29:166–73. doi: 10.1136/ijgc-2018-000017
101. Jensen KC, Mariappan MR, Putcha GV, Husain A, Chun N, Ford JM, et al. Microsatellite Instability and Mismatch Repair Protein Defects in Ovarian Epithelial Neoplasms in Patients 50 Years of Age and Younger. Am J Surg Pathol (2008) 32:1029–37. doi: 10.1097/PAS.0b013e31816380c4
102. Chui MH, Ryan P, Radigan J, Ferguson SE, Pollett A, Aronson M, et al. The Histomorphology of Lynch Syndrome-Associated Ovarian Carcinomas: Toward a Subtype-Specific Screening Strategy. Am J Surg Pathol (2014) 38:1173–81. doi: 10.1097/PAS.0000000000000298
103. Ketabi Z, Bartuma K, Bernstein I, Malander S, Gronberg H, Bjorck E, et al. Ovarian Cancer Linked to Lynch Syndrome Typically Presents as Early-Onset, non-Serous Epithelial Tumors. Gynecol Oncol (2011) 121:462–5. doi: 10.1016/j.ygyno.2011.02.010
104. Pal T, Permuth-Wey J, Kumar A, Sellers TA. Systematic Review and Meta-Analysis of Ovarian Cancers: Estimation of Microsatellite-High Frequency and Characterization of Mismatch Repair Deficient Tumor Histology. Clin Cancer Res (2008) 14:6847–54. doi: 10.1158/1078-0432.CCR-08-1387
105. Rosen DG, Cai KQ, Luthra R, Liu J. Immunohistochemical Staining of Hmlh1 and Hmsh2 Reflects Microsatellite Instability Status in Ovarian Carcinoma. Mod Pathol (2006) 19:1414–20. doi: 10.1038/modpathol.3800672
106. Cai KQ, Albarracin C, Rosen D, Zhong R, Zheng W, Luthra R, et al. Microsatellite Instability and Alteration of the Expression of Hmlh1 and Hmsh2 in Ovarian Clear Cell Carcinoma. Hum Pathol (2004) 35:552–9. doi: 10.1016/j.humpath.2003.12.009
107. Bennett JA, Morales-Oyarvide V, Campbell S, Longacre TA, Oliva E. Mismatch Repair Protein Expression in Clear Cell Carcinoma of the Ovary: Incidence and Morphologic Associations in 109 Cases. Am J Surg Pathol (2016) 40:656–63. doi: 10.1097/PAS.0000000000000602
108. Howitt BE, Strickland KC, Sholl LM, Rodig S, Ritterhouse LL, Chowdhury D, et al. Clear Cell Ovarian Cancers With Microsatellite Instability: A Unique Subset of Ovarian Cancers With Increased Tumor-Infiltrating Lymphocytes and PD-1/PD-L1 Expression. Oncoimmunology (2017) 6:e1277308. doi: 10.1080/2162402X.2016.1277308
109. Willis BC, Sloan EA, Atkins KA, Stoler MH, Mills AM. Mismatch Repair Status and PD-L1 Expression in Clear Cell Carcinomas of the Ovary and Endometrium. Mod Pathol (2017) 30:1622–32. doi: 10.1038/modpathol.2017.67
110. Reed E, Yu JJ, Davies A, Gannon J, Armentrout SL. Clear Cell Tumors Have Higher Mrna Levels of ERCC1 and XPB Than Other Histological Types of Epithelial Ovarian Cancer. Clin Cancer Res (2003) 9:5299–305.
111. Kadoch C, Hargreaves DC, Hodges C, Elias L, Ho L, Ranish J, et al. Proteomic and Bioinformatic Analysis of Mammalian SWI/SNF Complexes Identifies Extensive Roles in Human Malignancy. Nat Genet (2013) 45:592–601. doi: 10.1038/ng.2628
112. Hohmann AF, Vakoc CR. A Rationale to Target the SWI/SNF Complex for Cancer Therapy. Trends Genet (2014) 30:356–63. doi: 10.1016/j.tig.2014.05.001
113. Chai B, Huang J, Cairns BR, Laurent BC. Distinct Roles for the RSC and Swi/Snf ATP-Dependent Chromatin Remodelers in DNA Double-Strand Break Repair. Genes Dev (2005) 19:1656–61. doi: 10.1101/gad.1273105
114. Aydin OZ, Marteijn JA, Ribeiro-Silva C, Rodriguez Lopez A, Wijgers N, Smeenk G, et al. Human ISWI Complexes are Targeted by SMARCA5 Atpase and SLIDE Domains to Help Resolve Lesion-Stalled Transcription. Nucleic Acids Res (2014) 42:8473–85. doi: 10.1093/nar/gku565
115. Densham RM, Garvin AJ, Stone HR, Strachan J, Baldock RA, Daza-Martin M, et al. Human BRCA1-BARD1 Ubiquitin Ligase Activity Counteracts Chromatin Barriers to DNA Resection. Nat Struct Mol Biol (2016) 23:647–55. doi: 10.1038/nsmb.3236
116. Hays E, Nettleton E, Carter C, Morales M, Vo L, Passo M, et al. The SWI/SNF Atpase BRG1 Stimulates DNA End Resection and Homologous Recombination by Reducing Nucleosome Density at DNA Double Strand Breaks and by Promoting the Recruitment of the Ctip Nuclease. Cell Cycle (2020) 19:3096–114. doi: 10.1080/15384101.2020.1831256
117. Shen J, Peng Y, Wei L, Zhang W, Yang L, Lan L, et al. ARID1A Deficiency Impairs the DNA Damage Checkpoint and Sensitizes Cells to PARP Inhibitors. Cancer Discov (2015) 5:752–67. doi: 10.1158/2159-8290.CD-14-0849
118. Watanabe R, Ui A, Kanno S, Ogiwara H, Nagase T, Kohno T, et al. SWI/SNF Factors Required for Cellular Resistance to DNA Damage Include ARID1A and ARID1B and Show Interdependent Protein Stability. Cancer Res (2014) 74:2465–75. doi: 10.1158/0008-5472.CAN-13-3608
119. Wu C, Lyu J, Yang EJ, Liu Y, Zhang B, Shim JS. Targeting AURKA-CDC25C Axis to Induce Synthetic Lethality in ARID1A-Deficient Colorectal Cancer Cells. Nat Commun (2018) 9:3212. doi: 10.1038/s41467-018-05694-4
120. Williamson CT, Miller R, Pemberton HN, Jones SE, Campbell J, Konde A, et al. ATR Inhibitors as a Synthetic Lethal Therapy for Tumours Deficient in ARID1A. Nat Commun (2016) 7:13837. doi: 10.1038/ncomms13837
121. Niedermaier B, Sak A, Zernickel E, Xu S, Groneberg M, Stuschke M. Targeting ARID1A-Mutant Colorectal Cancer: Depletion of ARID1B Increases Radiosensitivity and Modulates DNA Damage Response. Sci Rep (2019) 9:18207. doi: 10.1038/s41598-019-54757-z
122. Bitler BG, Fatkhutdinov N, Zhang R. Potential Therapeutic Targets in ARID1A-Mutated Cancers. Expert Opin Ther Targets (2015) 19:1419–22. doi: 10.1517/14728222.2015.1062879
123. Park Y, Chui MH, Suryo Rahmanto Y, Yu ZC, Shamanna RA, Bellani MA, et al. Loss of ARID1A in Tumor Cells Renders Selective Vulnerability to Combined Ionizing Radiation and PARP Inhibitor Therapy. Clin Cancer Res (2019) 25:5584–94. doi: 10.1158/1078-0432.CCR-18-4222
124. Franklin M, Gentles L, Matheson E, Bown N, Cross P, Ralte A, et al. Characterization and Drug Sensitivity of a Novel Human Ovarian Clear Cell Carcinoma Cell Line Genomically and Phenotypically Similar to the Original Tumor. Cancer Med (2018) 7:4744–54. doi: 10.1002/cam4.1724
125. Sancar A, Lindsey-Boltz LA, Unsal-Kacmaz K, Linn S. Molecular Mechanisms of Mammalian DNA Repair and the DNA Damage Checkpoints. Annu Rev Biochem (2004) 73:39–85. doi: 10.1146/annurev.biochem.73.011303.073723
126. Williams RM, Zhang X. Roles of ATM and ATR in DNA Double Strand Breaks and Replication Stress. Prog Biophys Mol Biol (2020) 161:27–38. doi: 10.1016/j.pbiomolbio.2020.11.005
127. Karnitz LM, Zou L. Molecular Pathways: Targeting ATR in Cancer Therapy. Clin Cancer Res (2015) 21:4780–5. doi: 10.1158/1078-0432.CCR-15-0479
128. Yap TA, O'Carrigan B, Penney MS, Lim JS, Brown JS, de Miguel Luken MJ, et al. Phase I Trial of First-in-Class ATR Inhibitor M6620 (VX-970) as Monotherapy or in Combination With Carboplatin in Patients With Advanced Solid Tumors. J Clin Oncol (2020) 38:3195–204. doi: 10.1200/JCO.19.02404
129. Kurz L, Miklyaeva A, Skowron MA, Overbeck N, Poschmann G, Becker T, et al. ARID1A Regulates Transcription and the Epigenetic Landscape via POLE and DMAP1 While ARID1A Deficiency or Pharmacological Inhibition Sensitizes Germ Cell Tumor Cells to ATR Inhibition. Cancers (Basel) (2020) 12:905. doi: 10.3390/cancers12040905
130. D'Assoro AB, Haddad T, Galanis E. Aurora-a Kinase as a Promising Therapeutic Target in Cancer. Front Oncol (2015) 5:295. doi: 10.3389/fonc.2015.00295
131. Ma HT, Poon RYC. Aurora Kinases and DNA Damage Response. Mutat Res (2020) 821:111716. doi: 10.1016/j.mrfmmm.2020.111716
132. Chan KK, Wong OG, Wong ES, Chan KK, Ip PP, Tse KY, et al. Impact of Iaspp on Chemoresistance Through PLK1 and Autophagy in Ovarian Clear Cell Carcinoma. Int J Cancer (2018) 143:1456–69. doi: 10.1002/ijc.31535
133. Ediriweera MK, Tennekoon KH, Samarakoon SR. Role of the PI3K/AKT/Mtor Signaling Pathway in Ovarian Cancer: Biological and Therapeutic Significance. Semin Cancer Biol (2019) 59:147–60. doi: 10.1016/j.semcancer.2019.05.012
134. Huang TT, Lampert EJ, Coots C, Lee JM. Targeting the PI3K Pathway and DNA Damage Response as a Therapeutic Strategy in Ovarian Cancer. Cancer Treat Rev (2020) 86:102021. doi: 10.1016/j.ctrv.2020.102021
135. Huang TT, Brill E, Nair JR, Zhang X, Wilson KM, Chen L, et al. Targeting the PI3K/Mtor Pathway Augments CHK1 Inhibitor-Induced Replication Stress and Antitumor Activity in High-Grade Serous Ovarian Cancer. Cancer Res (2020) 80:5380–92. doi: 10.1158/0008-5472.CAN-20-1439
136. Oishi T, Itamochi H, Kudoh A, Nonaka M, Kato M, Nishimura M, et al. The PI3K/Mtor Dual Inhibitor NVP-BEZ235 Reduces the Growth of Ovarian Clear Cell Carcinoma. Oncol Rep (2014) 32:553–8. doi: 10.3892/or.2014.3268
137. Caumanns JJ, Berns K, Wisman GBA, Fehrmann RSN, Tomar T, Klip H, et al. Integrative Kinome Profiling Identifies Mtorc1/2 Inhibition as Treatment Strategy in Ovarian Clear Cell Carcinoma. Clin Cancer Res (2018) 24:3928–40. doi: 10.1158/1078-0432.CCR-17-3060
138. Caumanns JJ, van Wijngaarden A, Kol A, Meersma GJ, Jalving M, Bernards R, et al. Low-Dose Triple Drug Combination Targeting the PI3K/AKT/Mtor Pathway and the MAPK Pathway is an Effective Approach in Ovarian Clear Cell Carcinoma. Cancer Lett (2019) 461:102–11. doi: 10.1016/j.canlet.2019.07.004
139. Makii C, Ikeda Y, Oda K, Uehara Y, Nishijima A, Koso T, et al. Anti-Tumor Activity of Dual Inhibition of Phosphatidylinositol 3-Kinase and MDM2 Against Clear Cell Ovarian Carcinoma. Gynecol Oncol (2019) 155:331–9. doi: 10.1016/j.ygyno.2019.08.028
140. Shigeta S, Lui GYL, Shaw R, Moser R, Gurley KE, Durenberger G, et al. Targeting BET Proteins BRD2 and BRD3 in Combination With PI3K-AKT Inhibition as a Therapeutic Strategy for Ovarian Clear Cell Carcinoma. Mol Cancer Ther (2021) 20:691–703. doi: 10.1158/1535-7163.MCT-20-0809
141. McIntyre JB, Wu JS, Craighead PS, Phan T, Kobel M, Lees-Miller SP, et al. PIK3CA Mutational Status and Overall Survival in Patients With Cervical Cancer Treated With Radical Chemoradiotherapy. Gynecol Oncol (2013) 128:409–14. doi: 10.1016/j.ygyno.2012.12.019
142. Kim TJ, Lee JW, Song SY, Choi JJ, Choi CH, Kim BG, et al. Increased Expression of Pakt is Associated With Radiation Resistance in Cervical Cancer. Br J Cancer (2006) 94:1678–82. doi: 10.1038/sj.bjc.6603180
143. Iida M, Harari PM, Wheeler DL, Toulany M. Targeting AKT/PKB to Improve Treatment Outcomes for Solid Tumors. Mutat Res (2020) 819-820:111690. doi: 10.1016/j.mrfmmm.2020.111690
144. Jiang W, Wu Y, He T, Zhu H, Ke G, Xiang L, et al. Targeting of Beta-Catenin Reverses Radioresistance of Cervical Cancer With the PIK3CA-E545K Mutation. Mol Cancer Ther (2020) 19:337–47. doi: 10.1158/1535-7163.MCT-19-0309
145. Lee CM, Fuhrman CB, Planelles V, Peltier MR, Gaffney DK, Soisson AP, et al. Phosphatidylinositol 3-Kinase Inhibition by LY294002 Radiosensitizes Human Cervical Cancer Cell Lines. Clin Cancer Res (2006) 12:250–6. doi: 10.1158/1078-0432.CCR-05-1084
146. Fuhrman CB, Kilgore J, LaCoursiere YD, Lee CM, Milash BA, Soisson AP, et al. Radiosensitization of Cervical Cancer Cells via Double-Strand DNA Break Repair Inhibition. Gynecol Oncol (2008) 110:93–8. doi: 10.1016/j.ygyno.2007.08.073
147. Liu Y, Cui B, Qiao Y, Zhang Y, Tian Y, Jiang J, et al. Phosphoinositide-3-Kinase Inhibition Enhances Radiosensitization of Cervical Cancer In Vivo. Int J Gynecol Cancer (2011) 21:100–5. doi: 10.1097/IGC.0b013e3182021bfd
148. Wang J, Li R, He Y, Yi Y, Wu H, Liang Z. Next-Generation Sequencing Reveals Heterogeneous Genetic Alterations in Key Signaling Pathways of Mismatch Repair Deficient Colorectal Carcinomas. Mod Pathol (2020) 33:2591–601. doi: 10.1038/s41379-020-0612-2
149. Kim KJ, Kim JW, Sung JH, Suh KJ, Lee JY, Kim SH, et al. PI3K-Targeting Strategy Using Alpelisib to Enhance the Antitumor Effect of Paclitaxel in Human Gastric Cancer. Sci Rep (2020) 10:12308. doi: 10.1038/s41598-020-68998-w
150. Juvekar A, Hu H, Yadegarynia S, Lyssiotis CA, Ullas S, Lien EC, et al. Phosphoinositide 3-Kinase Inhibitors Induce DNA Damage Through Nucleoside Depletion. Proc Natl Acad Sci USA (2016) 113:E4338–4347. doi: 10.1073/pnas.1522223113
151. Kato N, Toukairin M, Asanuma I, Motoyama T. Immunocytochemistry for Hepatocyte Nuclear Factor-1beta (HNF-1beta): A Marker for Ovarian Clear Cell Carcinoma. Diagn Cytopathol (2007) 35:193–7. doi: 10.1002/dc.20623
152. Mandai M, Amano Y, Yamaguchi K, Matsumura N, Baba T, Konishi I. Ovarian Clear Cell Carcinoma Meets Metabolism; HNF-1beta Confers Survival Benefits Through the Warburg Effect and ROS Reduction. Oncotarget (2015) 6:30704–14. doi: 10.18632/oncotarget.5228
153. Okamoto T, Mandai M, Matsumura N, Yamaguchi K, Kondoh H, Amano Y, et al. Hepatocyte Nuclear Factor-1beta (HNF-1beta) Promotes Glucose Uptake and Glycolytic Activity in Ovarian Clear Cell Carcinoma. Mol Carcinog (2015) 54:35–49. doi: 10.1002/mc.22072
154. Cuff J, Salari K, Clarke N, Esheba GE, Forster AD, Huang S, et al. Integrative Bioinformatics Links HNF1B With Clear Cell Carcinoma and Tumor-Associated Thrombosis. PloS One (2013) 8:e74562. doi: 10.1371/journal.pone.0074562
155. Lopes-Coelho F, Gouveia-Fernandes S, Goncalves LG, Nunes C, Faustino I, Silva F, et al. HNF1beta Drives Glutathione (GSH) Synthesis Underlying Intrinsic Carboplatin Resistance of Ovarian Clear Cell Carcinoma (OCCC). Tumour Biol (2016) 37:4813–29. doi: 10.1007/s13277-015-4290-5
156. Kajihara H, Yamada Y, Kanayama S, Furukawa N, Noguchi T, Haruta S, et al. Clear Cell Carcinoma of the Ovary: Potential Pathogenic Mechanisms (Review). Oncol Rep (2010) 23:1193–203. doi: 10.3892/or_00000750
157. Shigetomi H, Higashiura Y, Kajihara H, Kobayashi H. A Potential Link of Oxidative Stress and Cell Cycle Regulation for Development of Endometriosis. Gynecol Endocrinol (2012) 28:897–902. doi: 10.3109/09513590.2012.683071
158. Ito F, Yoshimoto C, Yamada Y, Sudo T, Kobayashi H. The HNF-1beta-USP28-Claspin Pathway Upregulates DNA Damage-Induced Chk1 Activation in Ovarian Clear Cell Carcinoma. Oncotarget (2018) 9:17512–22. doi: 10.18632/oncotarget.24776
159. Shigetomi H, Sudo T, Shimada K, Uekuri C, Tsuji Y, Kanayama S, et al. Inhibition of Cell Death and Induction of G2 Arrest Accumulation in Human Ovarian Clear Cells by HNF-1beta Transcription Factor: Chemosensitivity Is Regulated by Checkpoint Kinase CHK1. Int J Gynecol Cancer (2014) 24:838–43. doi: 10.1097/IGC.0000000000000136
160. Wilkerson PM, Dedes KJ, Samartzis EP, Dedes I, Lambros MB, Natrajan R, et al. Preclinical Evaluation of the PARP Inhibitor BMN-673 for the Treatment of Ovarian Clear Cell Cancer. Oncotarget (2017) 8:6057–66. doi: 10.18632/oncotarget.14011
161. De Mattia E, Roncato R, Palazzari E, Toffoli G, Cecchin E. Germline and Somatic Pharmacogenomics to Refine Rectal Cancer Patients Selection for Neo-Adjuvant Chemoradiotherapy. Front Pharmacol (2020) 11:897. doi: 10.3389/fphar.2020.00897
162. Konstantinopoulos PA, Norquist B, Lacchetti C, Armstrong D, Grisham RN, Goodfellow PJ, et al. Germline and Somatic Tumor Testing in Epithelial Ovarian Cancer: ASCO Guideline. J Clin Oncol (2020) 38:1222–45. doi: 10.1200/JCO.19.02960
163. Norquist BM, Harrell MI, Brady MF, Walsh T, Lee MK, Gulsuner S, et al. Inherited Mutations in Women With Ovarian Carcinoma. JAMA Oncol (2016) 2:482–90. doi: 10.1001/jamaoncol.2015.5495
Keywords: ovarian cancer (OC), ovarian clear cell carcinoma, DNA damage response (DDR), targeted therapy, ARID1A
Citation: Wong OGW, Li J and Cheung ANY (2021) Targeting DNA Damage Response Pathway in Ovarian Clear Cell Carcinoma. Front. Oncol. 11:666815. doi: 10.3389/fonc.2021.666815
Received: 11 February 2021; Accepted: 08 September 2021;
Published: 19 October 2021.
Edited by:
Sung-Jong Lee, The Catholic University of Korea, South KoreaReviewed by:
Francesco Pepe, University of Naples Federico II, ItalyKonstantinos Venetis, University of Milan, Italy
Copyright © 2021 Wong, Li and Cheung. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Annie N. Y. Cheung, YW55Y2hldW5AcGF0aG9sb2d5LmhrdS5oaw==