 Kai Zimmer
Kai Zimmer Florian Kocher
Florian Kocher Alberto Puccini2
Alberto Puccini2- 1Department of Hematology and Oncology, Comprehensive Cancer Center Innsbruck, Medical University of Innsbruck, Innsbruck, Austria
- 2Medical Oncology Unit 1, Ospedale Policlinico San Martino Istituto di ricovero e cura a carattere scientifico (IRCCS), University of Genoa, Genoa, Italy
Mutated germline alleles in the DNA damage repair (DDR) genes “breast cancer gene 1” (BRCA1) and BRCA2 have originally been identified as major susceptibility genes in breast and ovarian cancers. With the establishment and approval of more cost-effective gene sequencing methods, germline and somatic BRCA mutations have been detected in several cancers. Since the approval of poly (ADP)-ribose polymerase inhibitors (PARPi) for BRCA-mutated cancers, BRCA mutations gained rising therapeutic implications. The impact and significance of BRCA mutations have been evaluated extensively in the last decades. Moreover, other genes involved in the DDR pathway, such as ATM, ATR, or CHK1, have emerged as potential new treatment targets, as inhibitors of these proteins are currently under clinical investigation. This review gives a concise overview on the emerging clinical implications of mutations in the DDR genes in gastrointestinal cancers with a focus on BRCA mutations.
Introduction
The breast and ovarian cancer susceptibility genes “breast cancer gene” (BRCA)1 and BRCA2 have been thoroughly investigated in the last decades. It is estimated, that one out of 400 to 800 individuals (0.125%–0.25%) in the USA may harbor a germline loss-of-function mutation in BRCA1 or BRCA2 (gBRCA-mut) (1, 2). This is associated with an approximately 60% lifetime risk for breast cancer and a 15% to 40% lifetime risk for ovarian cancer development (3–5). Of note, the rate of gBRCA-mut is higher in the Ashkenazi Jewish population, in which 2.5% harbor a pathogenic mutation (6). In this specific population, two founder mutations in BRCA1 and one founder mutation in BRCA2 have been identified (7).
For BRCA-mutated breast and ovarian cancer, treatment with Poly-(ADP) ribose polymerase inhibitors (PARPi) has been established as a standard of care (8, 9). Besides breast and ovarian cancer, PARPi have been recently evaluated for the treatment of BRCA-mut pancreatic cancer implicating a possible further use in other BRCA-mut gastrointestinal (GI) cancers. The frequency of BRCA1 and BRCA2 mutations in GI cancers varies between tumor entities (see Table 1). However, as BRCA1 and BRCA2 play pivotal roles in the cellular response to DNA damage, targeting DNA damage repair (DDR) mechanisms gained focus in drug development. In addition, as oncologists, it is paramount to be aware of the crucial implications related to the broadening population of patients who are going to be tested for and counseled about germline BRCA mutations, since the recognition of hereditary syndromes is important in order to set up dedicated follow-up to reduce the incidence of second tumors among survivors and to reduce mortality among their relatives (22). As treatment options in advanced GI cancers are limited and are associated with a poor overall survival (OS), the quest towards new therapeutics is ongoing, and drugs targeting BRCA and other gene members within the DDR complex are heavily investigated. In this article, we aimed to give a concise overview on the pathophysiology of the DDR machinery, current and upcoming treatment options for GI cancers with BRCA1/2 mutations, and on other DDR genes that are currently under clinical investigation.
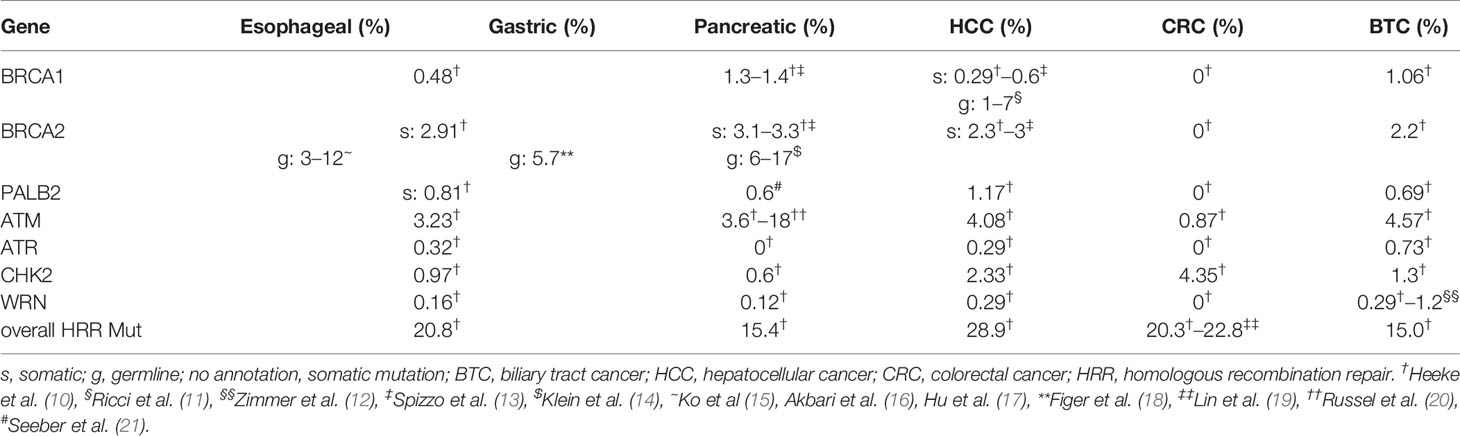
Table 1 Mutation frequencies of BRCA1, BRCA2, and of other frequently mutated DDR genes.
BRCA Genes Are Key Players in the DNA Damage Repair Complex
DDR mechanisms maintain genomic integrity and stability by restoring DNA damage arising from intracellular and extracellular stressors. Those stressors can lead to base alterations or single- (SSBs) or double-strand breaks (DSBs) (23). If left unrepaired, strand breakages can result in the breakdown of chromosomes and, subsequently, in the loss of genes. Mainly, DNA DSBs cannot only arise from stalled or broken DNA replication forks but can also be caused by ionizing radiation, reactive oxygen species (ROS), and physical or mechanical stress (23).
Two mechanisms have been discovered which counteract serious DNA damage. Predominantly, cells utilize a mechanism termed “nonhomologous end joining” (NHEJ), in which the broken DNA strands are brought into proximity and the ends are joined by DNA ligation (24). This mechanism is error prone, as processing of the DNA ends by NHEJ leading to the loss of nucleotides and causing rearrangements, which might lead to an altered DNA sequence at the site of breakage. However, if a sister chromatid is available as a template for the repair machinery during or shortly after DNA replication in the S- and G2-phase of the cell cycle (25), NHEJ is avoided and a mechanism termed “homologous recombination repair” (HRR) is preferably used. BRCA1 and BRCA2 are important players in the molecular machinery of HRR, that accurately repair DSBs and prevents the delay or arrest of the cell cycle, apoptosis, or the passing on of damaged DNA (26). In brief, HRR repairs DSBs via exchange of DNA strands between a pair of homologous duplex DNA sequences, whereas one strand acts as a template to restore the lost or damaged information on the other strand (Figure 1).
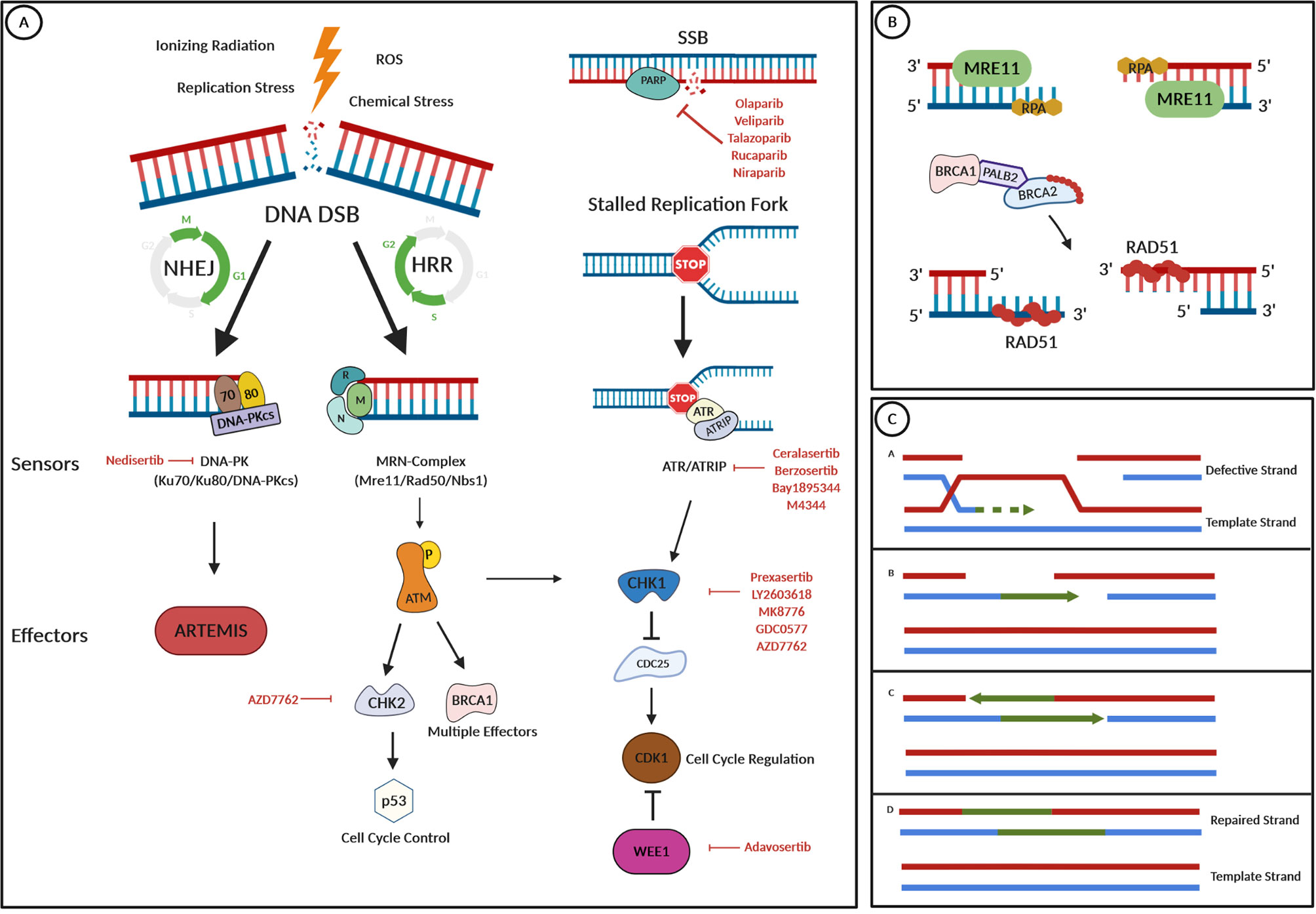
Figure 1 DNA damage repair pathways and drug targets. (A) Several internal and external stressors such as ionizing radiation, replication stress, ROS, or chemical stress can lead to DNA double-strand breaks (DNA DSBs). Depending on cell cycle progression and the availability of a template strand, either the nonhomologous end joining (NHEJ) or the homologous recombination repair (HRR) pathway are the mechanism of choice for DNA repair. In the NHEJ pathway, DNA damage is recognized by the “DNA-dependent protein kinase” (DNA-PK), a nuclear serine/threonine protein kinase complex, composed of a large catalytic subunit named DNA-PKcs and a heterodimer of Ku proteins (Ku70/80). Then, the nuclease Artemis is activated and cleaves 5′- and 3′-DNA overhangs. In HRR, which only occurs in the S and the G2 phase of the cell cycle, a protein complex termed MRN (which is composed of MRE11, Rad50, and Nbs1) recognizes DNA DSBs. After its activation by phosphorylation, the ATM kinase works as a master effector protein for HRR by activating several downstream effectors such as the CHK1 or the CHK2 kinase or the BRCA1 protein, having effects on cell cycle progression via p53 control or CDK1 inhibition. Stalled replication forks lead to an arrest of DNA replication and such replication stress could lead to DNA DSBs. To prevent this and to stop cell cycle progression, the ATR protein kinase is recruited and binds to its partner protein ATRIP. Cell cycle regulation and repair mechanisms are then controlled via CHK1 activation. Repair of DNA single-strand breaks (SSBs) is initiated by PARP enzymes. (B) In HRR, the MRE protein acts as a nuclease and resects the broken DNA ends, which results in the overhanging of single-stranded 3′-ends. Protection from further resection or modification of ssDNA is reached by the binding of the RPA protein. RAD51 is essential for HRR, as it facilitates DNA unwinding, stretching, and invasion into the template strand. To prevent its early polymerization on DNA, RAD51 proteins are kept inactive by binding to BRCA2, which is recruited to the broken DNA in a complex with BRCA1 and the linker protein PALB2 after detection of DNA damage. The RAD51-DNA filament is then capable of invading the template strand. (C) The RAD51-DNA filament invades the template strand and searches for extended homologous regions, which are then stabilized by base pairing. RAD51 disassembles and leaves a heteroduplex of the defective DNA strand and the template strand. The invading strand is extended by a DNA polymerase, and after elongation, the newly synthesized strand segment is displaced and finally ligated with its original strand endings. This figure was created using the BioRender.com online tool.
In the HRR pathway, BRCA1 acts upstream of BRCA2 and has multiple distinct functions, which are mediated by its different genetic domains (27), emphasizing the pivotal role of BRCA1 as a caretaker of the genome, as it directly and indirectly interacts with DNA damage sensors, DDR effectors, tumor suppressors, and cell cycle regulators (28, 29). “partner and localizer of BRCA2” (PALB2) binds directly to both, BRCA1 and BRCA2, and thereby provides a physical link between the two proteins (27, 30, 31).
Lack of a functional BRCA1 or BRCA2 allele leads to a deficiency in DNA DSB repair by HRR. In such a constellation, the cell utilizes more error-prone mechanisms such as NHEJ or single-strand annealing, which in turn, leads to increased genomic instability (32). In BRCA1-deficient cells, chromosomal aberrations, such as triradial or quadriradial chromosomes, or rearrangements, like translocations, have frequently been observed (33). In BRCA2-deficient cells, aneuploidy has been observed due to deregulation of centrosomes and the mitotic spindles (34). An important step in carcinogenesis of BRCA-mutated cells seems to be the loss of heterozygosity (LOH), as this can be observed in most gBRCA-mut cancers (35). However, there are data suggesting that loss of the wild-type allele may not be a prerequisite of BRCA-associated tumorigenesis (27). Besides LOH, promotor methylation of the BRCA genes seems to be an important mechanism for loss of function (LOF) and was more frequently observed in sporadic than in gBRCA-mut cancers (36). A BRCA1 methylation signature has also been associated with a better response to cytotoxic treatment in a small cohort of breast cancer patients (37).
Platinum-Based Regimens Are Considered Efficient in DDR Aberrant Cancers
Cytotoxic chemotherapeutic agents, such as platinum agents, lead to interstrand crosslinks, resulting in distortion of the DNA double helix, which inhibits the transcription, as RNA polymerases stall at the platinum crosslinks. If left unresolved, the cell evokes a programmed cell death pathway (38). However, in the situation of a hypofunctional DDR, e.g., through a LOF or a hypermethylation of the BRCA genes, these lesions are left unrepaired to a greater extend and the viability of cells is reduced dramatically (39). This observation has already been transferred to clinical settings and has been implemented in the treatment of gynecological malignancies. As such, in BRCA-mut breast cancer, the use of platinum-based therapeutic regimens, showed promising results in triple negative breast cancer (40–44), and the first randomized phase III trial (TNT trial) led to a longer progression-free survival (PFS) in patients with BRCA mutations compared with BRCA-wt patients. However, no effect on OS has been observed (45). Some studies have also reported improved PFS, OS, and response rates (RR) in ovarian cancer patients with BRCA mutations undergoing platinum-containing regimens (46–54).
PARP Inhibitors Generate Synthetic Lethality in HRR Defect Cancers
The development of PARPi added further treatment options for BRCA-mut cancers by making use of the concept of synthetic lethality. Synthetic lethality between two genes occurs where individual loss of either gene is compatible with life, but simultaneous loss of both genes results in cell death (55). Endogenous processes (i.e., oxidation) lead to thousands of mutated bases every day, which are mainly repaired by a mechanism called base excision repair (BER). BER generates single-nucleotide gaps (i.e., SSBs), which has to be filled by a DNA polymerase and then be sealed by a DNA ligase (56). The PARP1 protein recruits those DNA repair enzymes to the SSB sites, by binding to these sites of breakage and ADP-ribosylates itself (this has been termed “PARylation”). The resulting poly-ADP tails serves as docking sites for repair enzymes needed to fix the SSBs (57). When PARP1 is blocked, SSBs persist, which generates DSBs when a replication fork passes through (58). In BRCA-deficient cells, those DBSs cannot be repaired during replication, leading to cell death by accumulation of DSBs and the resulting genomic catastrophe (55).
However, this classical model (“SSB replication run-off model”) of our understanding of the mechanism of action of PARPi is still incomplete and has been challenged by the “PARP1 trapping model” and the “replication restart model,” which are reviewed elsewhere (55, 59).
Clinical evidence for the efficacy of PARPi in gBRCA-mut patients has first been observed in breast and ovarian cancer and was later also observed in metastasized castration-resistant prostate cancer and pancreatic cancer (60). To date, the PARPi olaparib, rucaparib, talazoparib, veliparib, and niraparib gained FDA approval in different settings.
Although its promising activity, more than 40% of BRCA-deficient patients fail to respond to PARPi and acquired resistance commonly occurs (60). Four categories of resistance mechanisms have been identified so far (61): cellular availability of the inhibitor, activity and abundance of PAR chains, reactivation of HRR, and replication fork protection. Of these, especially the restoration of HRR genes through genetic reversion mutations in BRCA1 or via synthetic viable loss of 53BP1 has been clinically proven (61–63). Also, resistance to platinum-based chemotherapies strongly predicts PARPi resistance (64).
BRCA Mutations in Pancreatic Cancer
With an estimated 5-year OS below 5% in the metastasized setting, new therapeutic approaches are urgently needed for patients with pancreatic cancer (65).
A family history of pancreatic cancer is reported in about 5%–10% of newly diagnosed cases (66–68) with germline mutations in BRCA2 contributing to approximately 6%–17% of these cases (14, 66, 69) (Table 1). Also, truncating mutations in PALB2 were identified to occur in 1%–3% of familial pancreatic cancer cases (70–72). In a large cohort comprising 2,818 sporadic pancreatic cancers, our group identified gene mutations in BRCA1, BRCA2, or PALB2 in 1.3%, 3.1%, and 0.6%, respectively. Interestingly, none of the patients with a BRCA1 or BRCA2 mutations had a concomitant mutation in PALB2 and vice versa. BRCA-mut and PALB2-mut statuses were also associated with a distinct genomic profile, potentially predictive for response to checkpoint-inhibitor therapy (21). Other estimates of BRCA gene mutation prevalence in sporadic PDAC ranged from 5.5% to 21.6% (73). Before the discovery of PARPi, several case reports and small clinical trials suggested a benefit for DNA-crosslinking chemotherapy, e.g., with platinum, in BRCA-mut pancreatic cancer (74–77). In 2014, Golan et al. retrospectively analyzed clinical data of 71 patients with BRCA1/2-mut pancreatic cancer and observed a significantly longer OS for individuals with an advanced disease when treated with a platinum therapy compared with nonplatinum treatment (22 vs. 9 months) (66).
In the last decade, several clinical trials investigated PARPi in BRCA-mut pancreatic cancer. In a phase II trial including 23 patients with metastatic pancreatic cancer, the PARPi olaparib reached a RR of 21.7%, a stable disease (SD) ≥8 weeks in 35%, and a median PFS and OS of 4.6 and 9.8 months, respectively (78) (Table 2). In the first randomized, placebo-controlled phase III trial evaluating the efficacy of olaparib as a maintenance treatment in patients with gBRCA1/2-mut and metastatic pancreatic cancer (POLO trial), a significant prolongation of PFS was observed. For the 92 patients treated in the interventional arm, median PFS reached 7.4 months compared with 3.8 months in the placebo arm (HR, 0.53). However, an interim overall survival analysis failed to show a survival benefit (86). Interestingly, olaparib maintenance therapy was associated with improved quality of life in the POLO trial (90). As such, olaparib has been approved by the FDA as a first-line maintenance treatment in gBRCA-mut metastatic pancreatic cancer. Promising upcoming clinical trials are currently evaluating the efficacy of olaparib in patients with somatic mutations in BRCA and other HRR genes (NCT02677038) and a combinational approach with the PD-1 immune checkpoint-inhibitor pembrolizumab (NCT04548752).
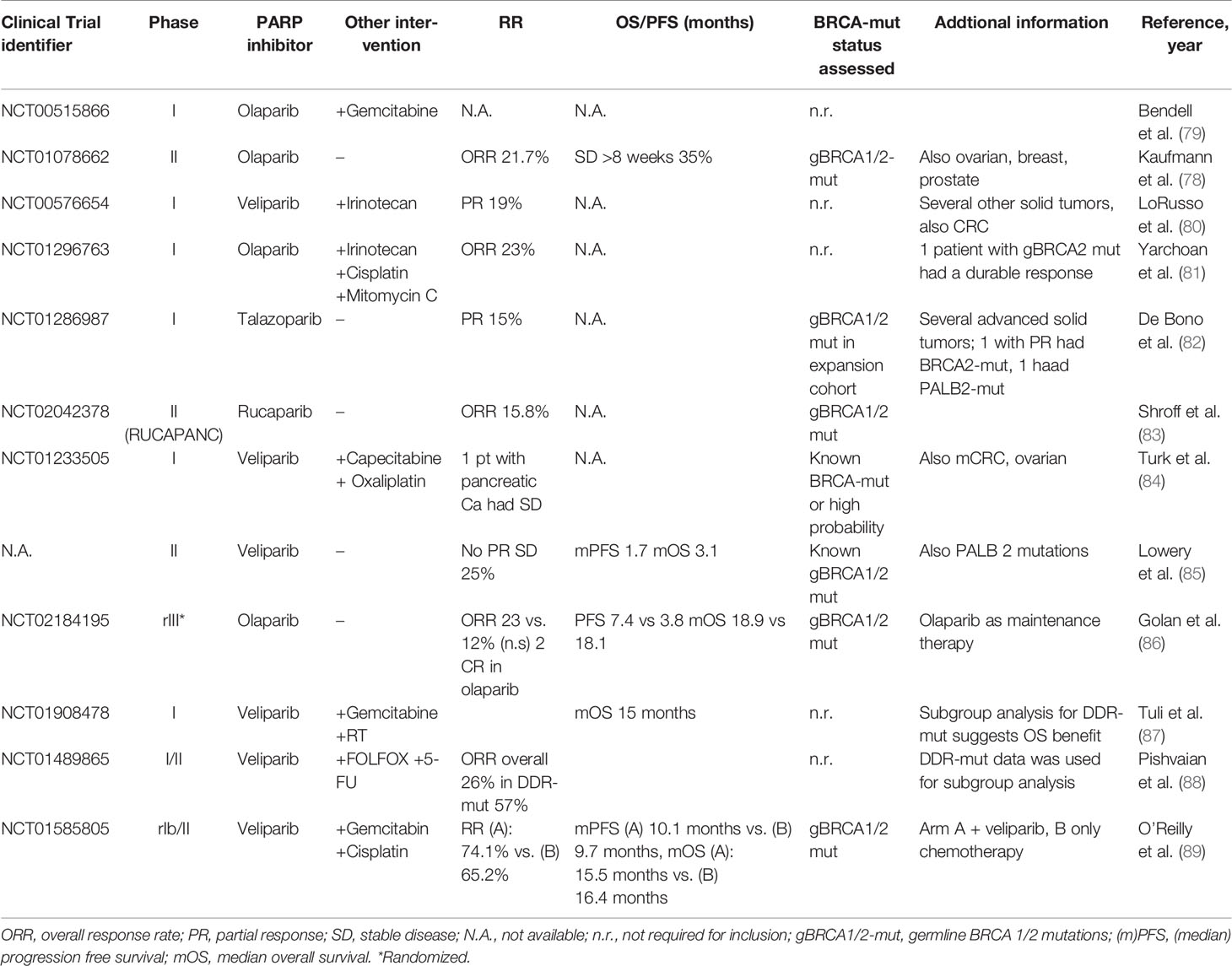
Table 2 Published clinical trials using PARP inhibitors in pancreatic cancer.
In the phase II RUCAPANC trial enrolling 19 BRCA1/2-mut pancreatic cancer patients (with three of those harboring a somatic BRCA mutation), rucaparib led to response in 15.8% (n = 3, 2 PR, 1 CR) of patients (83). Therefore, rucaparib for BRCA-mut pancreatic cancer is currently evaluated in further ongoing clinical trials. A phase Ib/II trial evaluates a combinational approach of rucaparib with irinotecan, fluorouracil, and leucovorin in different GI tumors (NCT03337087). The LODESTAR trial (NCT04171700) is a phase II multicenter open-label study of rucaparib as treatment for solid tumors, associated with deleterious mutations in several HRR genes, and another trial investigates rucaparib as a maintenance treatment for patients that have not progressed on platinum-based therapy (NCT03140670).
In 16 patients with known germline BRCA1/2 mutation receiving the PARPi veliparib in a phase II setting, no partial or complete responses (PR and CR, respectively) was observed. However, four patients had a SD ≥4 months (85).
In 58 patients within a single-arm, open-label phase I/II study, investigating a combination of veliparib with 5-fluorouracil and oxaliplatin, the ORR was 26% (PR: n = 11, CR: n = 4), PFS was 4.0 months and OS was 7.8 months. One patient showed an exceptional response, remaining in CR for nearly four years. Interestingly, in the subgroup of patients with prior platinum treatment, the ORR was only 7% compared with 32% in those who were platinum naive. This finding suggests that mechanisms of acquired resistance to platinum overlap the mechanisms of resistance to PARPi (88).
In an open-label, randomized, multicenter, two-arm phase II trial, 50 patients with a germline BRCA1/2 or PALB2 mutation were randomized to cisplatin and gemcitabine with or without the addition of veliparib. The addition of veliparib resulted in a higher disease control rate (100% vs. 78%), but no prolongation in PFS or OS was observed. The authors concluded that cisplatin and gemcitabine may be a new standard approach in gBRCA/PALB2-mut pancreatic cancer (89, 91).
Other promising clinical trials in BRCA-mut pancreatic cancer include a phase II trial investigating the combination of the PARPi iraparib and the PD-1 Inhibitor dostarolimab (NCT04493060), a phase I trial investigating the platinum salt BTP-114 in several BRCA-mut or other DDR genes mutated solid tumors (NCT02950064) or a phase II trial evaluating the wee1-inhibitor adavosertib (the MATCH Screening trial, NCT02465060) (Supplementary Tables S1 and S2).
Besides the prominent clinical role of BRCA mutations, loss of ATM is an even more common event in pancreatic cancer. This is implicated in the early stages and the progression of PDACs (20) and can be found in about 4% to 18% of cases. To target this alteration, ATR inhibitors are considered effective and are under investigation in currently recruiting clinical trials (Supplementary Table S3). A retrospective clinical characterization of 22 ATM-mutated PDACs showed a prognostic value of these alterations compared with control patients with superior 5-year OS rates of 38.3% vs. 6.6% suggesting a possible role as a biomarker (92).
BRCA Mutations in Other GI Cancers
Esophageal Cancer
Considering the poor prognosis of esophageal cancer patients, there is an urgent need towards novel therapeutic agents. However, trials evaluating novel treatment have been disappointing so far. In an approach to define molecular subgroups of esophageal cancer, Secrier et al. performed whole-genome sequencing in 129 esophageal cancer samples and established three subtypes with genetic implications for potential stratified and targeted therapeutic approaches. Approximately one in three tumors was classified as “C>A/T dominant” due to a C>A/T mutational pattern. About one-fifth was classified as “DDR impaired”, presenting with defects in HRR and chromosome segregation pathways. Fifty percent of the samples was classified “mutagenic,” as they presented with a dominant T>G mutational pattern and the highest mutational burden and the highest neoantigen load (93). However, in the DDR-impaired subgroup, BRCA1/2 mutations were rare, and the authors speculated, that a pathway-level disruption of HRR contributes to a BRCA-like mutational signature rather than mutations of BRCA genes. Large epidemiological studies investigating the incidence of BRCA mutations in esophageal cancer and estimating the risk of esophageal cancer in gBRCA-mut carriers are limited. However, studies from regions with very high incidence rates in esophageal cancer, found BRCA2 mutations in about 3% to 12% in these selected populations (15–17). The frequency of somatic mutations in BRCA1/2 has been estimated for gastroesophageal cancers with 0.48% for BRCA1 and 2.91% for BRCA2 (10) (Table 1).
To the best of our knowledge, no results of large clinical trials investigating the role of HRR targeting agents, such as PARPi, in esophageal cancer are available so far. However, patients with esophageal cancer and with deleterious mutations in HRR genes are eligible for the LODESTAR trial (NCT04171700) in which treatment with rucaparib will be evaluated. Ongoing clinical trials will also investigate niraparib (NCT03840967), a combination of olaparib and anti-VEGFR antibody ramucirumab (NCT03008278), olaparib alone (SOLAR trial, NCT03829345), a combination of rucaparib and ramucirumab with or without the anti-PD1 antibody nivolumab (NCT03995017), or a combination of olaparib with paclitaxel and the anti-PD1 inhibitor pembrolizumab (NCT04592211). Of note, screening for HRR prior to inclusion is not a prerequisite for all of these trials (Supplementary Table S2).
A study performed in 144 patients with advanced or metastatic esophageal cancer receiving cisplatin- or docetaxel-based treatment, found, that a low BRCA1 mRNA expression correlated with an increase in RR and median OS in patients treated with cisplatin, but was associated with decreased RR and OS in patients treated with docetaxel (94). This suggests a role of BRCA1 mRNA expression levels as a predictive and prognostic marker in esophageal cancer.
Gastric Cancer
Considering the low rate of gBRCA-mut carriers with gastric cancer and very limited data on somatic BRCA mutations in gastric cancer (Table 1), only small clinical trials have been conducted. Clinical data of BRCA-mut gastric cancer patients is therefore also limited, but it is suggested, that this trait might be predictive for treatment with DNA damaging agents (95).
In a subgroup analysis of the MEDIOLA basket trial evaluating the combination of olaparib and the anti-PDL1 antibody durvalumab, an ORR of 10% in gastric cancer patients was reported (96). Of note, in this trial, patients with metastatic gastric cancer were not selected by gBRCA-mut status, and no subgroup analysis has been presented.
In 2013, Chen et al. analyzed BRCA1 protein expression by immunohistochemistry (IHC) of 637 gastric cancer samples to evaluate relationships between BRCA1 expression, already established prognostic factors, platinum-based adjuvant chemotherapy, and survival. Positive BRCA1 staining was observed in 34% of patients. Interestingly, BRCA1 positivity was associated with a significant prolongation of survival and BRCA1-negative patients seemed to benefit from platinum-based adjuvant chemotherapy (97). In 318 patients with stage II/III sporadic gastric cancer cases, approximately half of the patients were identified as BRCA1 negative by IHC. BRCA1 negativity seemed to be associated with a reduced disease-free survival but predicted response to adjuvant chemotherapy (98). In a large cohort of 367 patients with sporadic gastric cancer BRCA1 and BRCA2 mRNA levels were investigated by IHC, ISH and RT-qPCR. In this study no association with clinical-pathological biomarkers and survival with BRCA status was observed (99).
Biliary Tract Cancer
Biliary tract cancer (BTC) comprises gallbladder cancer (GBC), intrahepatic (IHCC), and extrahepatic cholangiocellular carcinoma (EHC).
In a molecular profiling approach, 28.9% of BTC tumors harbored pathogenic mutations in several HR-DDR genes (10). Another study found germline or somatic mutations in DDR genes in 63.5% of patients with BTC. Moreover, a significantly prolonged PFS and OS were observed in patients harboring DDR alterations and who received first-line platinum containing chemotherapy (100).
Recently, our group reported results from a NGS study of 1,292 patients with BTC and found BRCA2 mutations in 3% and BRCA1 mutations in 0.6%. BRCA2 mutations were significantly more frequent than BRCA1 mutations in GBC and IHCC (4.0% vs. 0.3% and 3.7% vs 0.4%). No difference was observed in EHC (2.6% BRCA2 vs. 2.1% BRCA1). Also, a significant association of BRCA-mut carriers with MSI-H/dMMR and higher TMB was observed, suggesting a potential rationale for the combination of PARPi and immune checkpoint inhibitors (13).
Clinical trials in BRCA-mut BTC are limited. A small retrospective case series with 18 cases of BRCA associated EHCs, of which 13 received platinum-based therapy and four received a PARPi, the authors reported a median OS of 40.3 months for patients with stage I/II BTC, and an OS of 25 months for stage III/IV BTC (101).
To date, no clinical trial on targeting mutations in BRCA or other DDR genes in BTC has been presented, but several promising trials are ongoing. For example, a phase II trial evaluates olaparib in BTC with mutations in 16 DDR genes (NCT04042831), whereas another trial compares the combination of olaparib and the ATR inhibitor AZD6738 with the combination of AZD6738 and durvalumab (NCT04298021). The PARPi rucaparib will be evaluated in a phase I/II trial in combination with irinotecan, fluorouracil and leucovorin (NCT03337087), and in another trial in combination with nivolumab (NCT03639935).
Hepatocellular Carcinoma
Literature involving BRCA mutations in hepatocellular carcinoma (HCC) is limited. The BCLC reported a fourfold increase of RR in HCC patients with BRCA mutations compared with BRCA wt patients (68, 102).
In a cohort of 214 patients with HCC, DDR gene alterations were observed in 22.8%, of which 85% harbored a somatic mutation (19). Another study analyzing data from a large industrial molecular profiling company was unable to identify any somatic BRCA1/2 or PALB2 mutation in HCC patients, but the overall frequency of mutations in 25 HRR associated genes was estimated with 20%, with ARID1A contributing to nearly half of these cases (10).
Despite promising preclinical evidence (103), no larger clinical trial targeting BRCA-mut in HCC is currently under way.
Colorectal Cancer
The risk for colorectal cancer (CRC) in BRCA carriers seems elevated in women below the age of 50 and also for anal carcinoma (104), making screening measures an important tool in this cohort. In a small case series, BRCA-mutated CRCs were also found to be more often of mucinous histology (105), which is a feature also associated with other defects in DNA repair genes such as the mismatch repair genes suggesting a distinct tumor biology and underlining the need for further investigation.
However, to date, no larger clinical trials targeting somatic BRCA-mut in CRC patients have been published. In several small clinical trials investigating PARPi in CRC, BRCA status has not been assessed as an eligibility criterion and only one upcoming trial (LODESTAR, NCT04171700) will select CRC patients according to BRCA-mut status.
In a small study in CRC patients, monotherapy with PARPi resulted in an ORR of 0% (106). However, used in combination with chemotherapy or radiotherapy, ORR rose up to 57% in a study combining veliparib with FOLFIRI with or without bevacizumab (107). No stratification according to BRCA-mut status was performed.
Beyond BRCA and PARP Inhibitors in GI Cancers
In the currently largest approach to identify DDR gene alterations in GI cancers, 17,486 patients with GI cancer were screened and at least one alteration in a subset of 10 DDR genes was identified in 17.1%, with gastric cancer being the most frequently DDR mutated tumor (27.1% had at least one DDR gene mutated), providing a rationale for clinical trials (108) in this specific subset.
Besides PARPi, emerging DDR associated targets in GI cancers include ATM, ATR, CHK1/2, WRN, or WEE1. ATM plays a pivotal role as a master regulator of DNA damage recognition and repair (Figure 1) and it is among the most commonly aberrant genes in sporadic cancers, especially in hematologic malignancies (109). Heterozygosity for a germline ATM-mut can be observed with a frequency of approximately 1% in the population and increases cancer risk by two- to threefold (110, 111). Frequency of somatic mutations in GI cancers ranges from 0.87% in HCC to 4.5% in CRC (111), making it a considerable druggable target. To date, the ATM inhibitor AZD0156 is the first-in-class agent to be investigated in a phase I clinical trial (AToM trial, NCT02588105) in patients with advanced malignancies including gastric cancer as a monotherapy or in combination with olaparib or with irinotecan/FOLFIRI. Preclinical studies suggested a potentiated effect of olaparib when used in combination with AZD0156 (112). A randomized, double-blind phase II trial investigating the combination of olaparib and paclitaxel stratified patients by ATM protein levels and found a longer OS for the addition of olaparib regardless of the ATM expression levels (113) (Supplementary Table S3). In this trial, signals for a longer OS in the population with low ATM expression led to further analysis in the phase III GOLD trial. Unfortunately, the GOLD trial did not meet its primary endpoint of a significant improvement in OS with the addition of olaparib, neither in the overall population (8.8 vs. 6.9 months) nor in the ATM-negative population (12.0 vs. 10.0 months) (114). Biomarker subgroup analysis (i.e., for BRCA) of the GOLD-trial cohort, was unable to identify any significant association with clinical outcomes (115).
Preclinical data support the hypothesis that loss of ATM leads to increased sensitivity to ATR inhibition (116). Also, preclinical data suggest a dependency on ATR signaling in PARPi-resistant BRCA-mut cells for replication fork stabilization (117–119), providing a rationale to combine ATR inhibitors (ATRi) with PARPi to overcome and prevent secondary resistance mechanisms. Currently, ATRi (AZD6738, VX-970/M6620, M4344, BAY1895344) are investigated in several phase I and phase II trials involving GI cancers (Supplementary Table S1). Combinational approaches include crosslinking agents, PARPi or PD-1/PD-L1 inhibitors. One trial investigates the combination of AZD6738 in combination with olaparib in patients with IDH-mutant cholangiocellular carcinoma (NCT03878095). Another phase II trial investigates the combination of VX-970 and irinotecan in gastric or gastroesophageal junction cancers harboringTP53 mutations. To the best of our knowledge, DDR mutational status (such as ATM) is only assessed in two clinical trials investigating ATRi (NCT04095273, NCT04266912) prior to inclusion.
The combination of AZD6738 (ceralasertib) and paclitaxelin heavy pretreated cancer patients yielded clinical efficacy with an ORR of 25% according to a phase I trial (120). Another phase I trial (NCT02264678), including pancreatic and gastric cancers patients the combination of ceralasertib with olaparib or durvalumab, showed promising preliminary signals of antitumor activity (121). Encouraging safety was found for M6620 (VX-970) as monotherapy or in combination with carboplatin in patients with advanced solid tumors. Of note, one patient with metastatic colorectal cancer, harboring a loss in ATM and an ARID1A mutation, achieved a CR and had an ongoing PFS of 29 months (122). M6620 in combination with gemcitabine showed promising results in preliminary data coming from an phase I trial (123).
The CHK1 kinase has been recognized as one of the key components of cell cycle checkpoint response after DNA damage and is essential for HRR (124). Interestingly, in a study using data from a large industrial molecular profiling provider, no somatic CHK1 mutation was observed in GI cancers, but a prevalence of somatic CHK2 mutations between 0.6% in pancreatic cancer and 4.35% in HCC was observed (10). To date, several CHK1/2 inhibitors are under investigation for clinical use. In a phase I trial including patients with advanced solid tumors monotherapy with the CHK 1/2 inhibitor prexasertib (LY2606368) resulted in a PR rate of 4.4% activity was observed (PR 4.4% comparably, the CHK1 inhibitor GDC-0575 in combination with gemcitabine showed only modest clinical efficacy in a phase 1 trial including 102 patients with several solid tumors with only four patients achieving PR (3.9%) (125).
The WEE1 nuclear kinase is a key regulator of cell-cycle progression (126) and is phosphorylated by CHK1 upon DNA damage. Subsequently, WEE1 leads to the inactivation of the “cyclin-dependent kinase 1” (CDK1)-cyclin B complex, resulting in cell cycle arrest in the G2 phase (Figure 1) (126). Overexpression of WEE1 has been reported in several cancers (126). In pancreatic cancer, preclinical models suggest a combinational approach of a WEE1 inhibitor with radio- and chemotherapy, and a phase I/II trial treated 34 patients with locally advanced PDAC with the WEE1 inhibitor adavosertib (AZD1775) in combination with gemcitabine and radiation therapy. The study found a median PFS of 9.4 months, a median OS of 21.4 months and was well tolerated (127).
In the MATCH screening trial (NCT02465060), patients with a BRCA1 or BRCA2-mut will receive adavosertib. Adavosertib also sensitizes TP53-deficient tumor cells to DNA damage and has already shown antitumor activity in a phase I trial involving 202 patients with refractory solid tumors (128). In gastric (129) and colon cancer (130), WEE1 seems to be overexpressed and is therefore a suggested target in these entities.
Preclinical data support combinational approaches of PARPi with ATRi or WEE1i to overcome resistance mechanisms and sensitize tumors to radiotherapy (RT) (119, 131). A phase I clinical trial investigates such an approach in esophageal cancer by adding adavosertib to RT (NCT04460937). The phase I STAR trial (NCT04197713) investigates a combination of adavosertib with olaparib in patients with germline or somatic mutations in several HRR genes. However, the combination of WEE1i and PARPi is associated with high rates of toxicity, but toxicity might be reduced by a sequential dosing approach (132). Also, the combination of an ATRi and a WEE1i produced synthetic lethality in a preclinical model and supports further clinical investigation (133).
For the NHEJ machinery, DNA-PK is an important mediator and is also investigated as a druggable target. An inhibitor of this kinase, nedisertib, is combined with RT in localized pancreatic cancer (NCT04172532), and in combination with the anti-PD-L1 inhibitor avelumab and RT, among others, in BTC (NCT04068194).
Emerging in the field of DDR targets is the Werner syndrome helicase (WRN) that has been associated with a distinct molecular landscape in CRC and was associated with higher TMB, higher prevalence of MSI-H/dMMR and PD-L1 expression, supporting further trials investigating the use of immune checkpoint inhibitors. Also, WRN inhibitors are under preclinical investigation, but those results have not been transferred to clinical trials, so far (12).
Rationales for other combinational treatments have been reviewed extensively elsewhere (119, 134, 135).
Discussion and Future Perspectives
Here, we presented a review on the clinical perspectives of targeting DDR pathways in GI cancers with a focus on BRCA.
While the role of gBRCA mutations for the lifetime risk of several cancers has been evaluated extensively over the last two decades, the understanding and epidemiology of somatic BRCA mutations and other DDR genes in carcinogenesis, and further, specific treatment of such cancers, is still in an early phase. The risk of developing a specific GI tumor in individuals with a gBRCA mutation varies between entities and between certain BRCA mutations (i.e., BRCA1 and BRCA2). For gBRCA2-mut carriers, the risk of developing pancreatic, esophageal, gastric, and BTC is higher than in gBRCA1-mut carriers. For HCC, risks seem comparable, and for CRC, evidence is unclear if gBRCA1-mut carriers have a higher lifetime risk for CRC compared with gBRCA2-mut carriers (68, 102). The highest estimated lifetime risk for gBRCA2-mut carriers was reported in BTC (fivefold) (68),. This tissue tropism raises fundamental questions on the biology of the respective tissues both in health and carcinogenesis, suggesting different vulnerabilities to intra- and extracellular stressors or environmental factors.
Intensive follow-up and screening for breast cancer has been established in gBRCA-mut carriers, but the value of screening for other cancers, such as GI tumors apart of CRC, is controversial. In pancreatic cancer, for patients with an ATM, BRCA2 or PALB2 mutation, guidelines recommend, that yearly screening should start at age 50 or 10 years younger than the youngest relative with pancreatic cancer, preferably by MRI/MRCP and/or EUS (136). Such imaging procedures would also allow to screen for BTCs. However, consensus was not reached for screening in BRCA1-mut carriers. In CRC, recommendations are not that clear, but clinicians should offer screening at age 40 in BRCA-mut carriers with a first-degree relative with CRC or advanced adenoma (137). In gastric cancer, guidelines recommend a prophylactic total gastrectomy between age 20 and 30 for individuals carrying pathogenic variants in the E-Cadherin (CHD1) gene, which is associated with a very high lifetime risk for gastric cancer (138). However, no consensus recommendations on screening for gastric cancer in gBRCA-mut carriers have been established, which is also true for gastroesophageal cancer (139). Nevertheless, obtaining a detailed family history should be part of every first counseling in newly diagnosed cancers as it could have important implications for the patient and his or her relatives.
While clinical trials targeting the HRR system provide limited evidence for advantage of the use of PARPi in pancreatic cancer (86), such evidence is lacking for other GI cancers. Also, a BRCA-mut status has not been approved as a predictive biomarker, such as MSI for pembrolizumab, which would, i.e., allow a site-agnostic use of PARPi. Some authors suggest that BRCA1/2 mutations are not suitable as site-agnostic biomarkers for PARPi therapy as dependency on BRCA-mut for tumorigenesis differs between tumor lineages, BRCA1 and BRCA2 are merely a small part of the complex phenotype of “BRCAness,” and resistance mechanisms, which restore BRCA function, make BRCA mutations not an ideal site-agnostic marker. However, an accurate estimate of HRR deficiency may be a better predictive biomarker for upcoming clinical trials (140, 141).
In the future, an increasing number of clinical trials will not only focus on BRCA mutations, but on a group of genes defining BRCAness. The concept of BRCAness describes HRR defects in the absence of gBRCA1/2 mutations, but includes somatic BRCA mutations and mutations in several other genes such as PALB2, ATM, ATR, CHEK1/2, ARID1A, RAD51, NBS, etc. (33). Interestingly, a BRCAness genotype seems to predispose cellular sensitivity to PARPi and platinum therapy (33), having critical implication for upcoming clinical trials by providing a rationale to screen for a BRCAness phenotype, rather than screening solely for BRCA1 or BRCA2 mutations. However, exact and consensual definitions of BRCAness have still to be determined (11).
Also, other prognostic biomarkers for PARPi are heavily investigated. HRR-deficient (HRD) tumors seem to prioritize more error-prone DNA repair pathways, such as NHEJ, resulting in the accumulation of small-insertion deletions and LOH events (142). The assessment of HRD comprises different approaches, as a uniform definition has not been made until today. This makes it challenging to compare different trials using different approaches and lead the path for clinicians. In general, HRD testing relies on either the direct detection of genomic perturbations associated with genomic instability (such as mutational or methylation testing of BRCA or other HRR pathway genes), on the use of surrogates of genomic instability by detecting a genomic scar or on functional assays. Mainly used as indirect measures are the LOH determined by SNP-Sequencing, telomeric allelic imbalance (TAI), and large-scale state transitions (LST) for which each HRD score has been developed (143–145). Currently, for clinical routine use, only two prospectively validated and commercially available tests for assessment of HRD status are available (146): the myChoice CDx (Myriad Genetics) calculates a score based on all three genomic instability features and also includes BRCA1/2 mutations. In contrast, the FoundationOne CDx (Foundation Medicine) only calculates the percentage of genomic LOH. However, the clinical use of those tests is limited to ovarian cancer patients as a companian diagnostic for treatment with PARP-inhibitors and therefore no consensus recommendation for the use in other settings are available at the moment. Also, the value can be limited, as scars in the DNA (e.g., through DNA damaging chemotherapy) might be misinterpreted as HRR deficiency (62). To the best of our knowledge, no general consensus on the nomenclature of the genomic phenotype of BRCAness or “HRDness” has been established so far. Distinct definitions to achieve coherence in publications and clinical trials are desirable.
There is a certain rationale for combined treatment with PARPi and immune checkpoint inhibitors coming from several preclinical studies (147). It is suggested that DDR defects lead to a higher neoantigen load and TMB (12, 108). Moreover, PARPi lead to a more inflamed tumor microenvironment by activating the cGAS-STING pathway (147–149) and by increasing immune cell infiltration. PARPi were also found to upregulate PD-L1 (147, 150), which strongly suggests the combination with anti-PD-1/PD-L1 inhibitors. A phase 1a/b trial investigating the PARPi pamiparib and the anti-PD1 antibody tislelizumab in patients with advanced solid tumors, found the combination to be well tolerated and the ORR was 20% (151). In pancreatic cancer, combination with niraparib and dostarolimab (NCT04493060) and olaparib and pembrolizumab (NCT04548752) are currently under investigation. For other GI cancers, several PARPi/ICPi combinations (Supplementary Table S3) are currently evaluated.
Loss of the DNA damage sensor ATM has been associated with a survival benefit in platinum-treated CRC patients (62, 152), and preclinical studies suggest treatment of ATM-deficient tumors with a PARPi and ATRi combination (153–156). Such an approach could be applied to gastric cancers, in which ATM mutations are quite common, while BRCA mutations are rare (135, 157). Of note, the combination of a PARPi with chemotherapy (GOLD trial) did not result in longer OS in an ATM-low subpopulation but addition of an ATR-inhibitor-improved efficacy. In the setting of BTC, a phase I trial investigates the combination of olaparib and the ATR-inhibitor AZD6728 with or without the addition of durvalumab (NCT04298021). However, in this trial, DDR mutational status is not assessed before treatment.
Looking in the near future, results of several clinical trials, of which only some are mentioned above, are awaited to clear the view on the value of DDR targeting drugs in GI cancers. To date, limited and early clinical data are not sufficient to make a prognosis on the success of this approach and only an empirical and therefore long-lasting approach to the best combinational treatment will lead the path.
Conclusion
Frequency of germline and somatic BRCA-mut and other HRR genes in GI cancers varies extensively between sites, and besides BRCA-mut PDAC, clinical implications of such findings must be determined. Soon, various clinical trials will add further evidence for the use of HRR targeting agents in GI cancers. Also, investigating the BRCAness phenotype rather than BRCA1 or BRCA2 alone, is gaining more attention and is supported by evidence of promising combinational treatments. Oncologists must not forget the crucial implications related to finding gBRCA mutations both for the patients and their relatives.
Author Contributions
AS, FK, and KZ contributed to manuscript conception. KZ wrote the first draft of the manuscript. AS, FK and AP wrote sections of the manuscript. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2021.662055/full#supplementary-material
References
1. Ford D, Easton DF, Peto J. Estimates of the Gene Frequency of BRCA1 and Its Contribution to Breast and Ovarian Cancer Incidence. Am J Hum Genet (1995) 57(6):1457–62.
2. Whittemore AS, Gong G, John EM, McGuire V, Li FP, Ostrow KL, et al. Prevalence of BRCA1 Mutation Carriers Among U.S. Non-Hispanic Whites. Cancer Epidemiol Biomarkers Prev (2004) 13(12):2078–83.
3. Antoniou A, Pharoah PD, Narod S, Risch HA, Eyfjord JE, Hopper JL, et al. Average Risks of Breast and Ovarian Cancer Associated With BRCA1 or BRCA2 Mutations Detected in Case Series Unselected for Family History: A Combined Analysis of 22 Studies. Am J Hum Genet (2003) 72(5):1117–30. doi: 10.1086/375033
4. Kuchenbaecker KB, Hopper JL, Barnes DR, Phillips KA, Mooij TM, Roos-Blom MJ, et al. Risks of Breast, Ovarian, and Contralateral Breast Cancer for BRCA1 and BRCA2 Mutation Carriers. JAMA (2017) 317(23):2402–16. doi: 10.1001/jama.2017.7112
5. Chen S, Parmigiani G. Meta-Analysis of BRCA1 and BRCA2 Penetrance. J Clin Oncol (2007) 25(11):1329–33. doi: 10.1200/JCO.2006.09.1066
6. Rubinstein WS. Hereditary Breast Cancer in Jews. Fam Cancer (2004) 3(3-4):249–57. doi: 10.1007/s10689-004-9550-2
7. Levy-Lahad E, Catane R, Eisenberg S, Kaufman B, Hornreich G, Lishinsky E, et al. Founder BRCA1 and BRCA2 Mutations in Ashkenazi Jews in Israel: Frequency and Differential Penetrance in Ovarian Cancer and in Breast-Ovarian Cancer Families. Am J Hum Genet (1997) 60(5):1059–67.
8. Robson M, Im SA, Senkus E, Xu B, Domchek SM, Masuda N, et al. Olaparib for Metastatic Breast Cancer in Patients With a Germline BRCA Mutation. N Engl J Med (2017) 377(6):523–33. doi: 10.1056/NEJMoa1706450
9. Moore K, Colombo N, Scambia G, Kim BG, Oaknin A, Friedlander M, et al. Maintenance Olaparib in Patients With Newly Diagnosed Advanced Ovarian Cancer. N Engl J Med (2018) 379(26):2495–505. doi: 10.1056/NEJMoa1810858
10. Heeke AL, Pishvaian MJ, Lynce F, Xiu J, Brody JR, Chen WJ, et al. Prevalence of Homologous Recombination-Related Gene Mutations Across Multiple Cancer Types. JCO Precis Oncol (2018) 2018:PO.17.00286. doi: 10.1200/PO.17.00286
11. Ricci AD, Rizzo A, Bonucci C, Tober N, Palloni A, Mollica V, et al. PARP Inhibitors in Biliary Tract Cancer: A New Kid on the Block? Medicines (2020) 7(9):54. doi: 10.3390/medicines7090054
12. Zimmer K, Puccini A, Xiu J, Baca Y, Spizzo G, Lenz HJ, et al. -Mutated Colorectal Cancer Is Characterized by a Distinct Genetic Phenotype. Cancers (2020) 12(5):1319. doi: 10.3390/cancers12051319
13. Spizzo G, Puccini A, Xiu J, Goldberg RM, Grothey A, Shields AF, et al. Molecular Profile of BRCA-Mutated Biliary Tract Cancers. ESMO Open (2020) 5(3):e000682. doi: 10.1136/esmoopen-2020-000682
14. Klein AP. Genetic Susceptibility to Pancreatic Cancer. Mol Carcinog (2012) 51(1):14–24. doi: 10.1002/mc.20855
15. Ko JM, Ning L, Zhao XK, Chai AWY, Lei LC, Choi SSA, et al. BRCA2 Loss-of-Function Germline Mutations Are Associated With Esophageal Squamous Cell Carcinoma Risk in Chinese. Int J Cancer (2020) 146(4):1042–51. doi: 10.1002/ijc.32619
16. Akbari MR, Malekzadeh R, Nasrollahzadeh D, Amanian D, Islami F, Li S, et al. Germline BRCA2 Mutations and the Risk of Esophageal Squamous Cell Carcinoma. Oncogene (2008) 27(9):1290–6. doi: 10.1038/sj.onc.1210739
17. Hu N, Wang C, Han XY, He LJ, Tang ZZ, Giffen C, et al. Evaluation of BRCA2 in the Genetic Susceptibility of Familial Esophageal Cancer. Oncogene (2004) 23(3):852–8. doi: 10.1038/sj.onc.1207150
18. Figer A, Irmin L, Geva R, Flex D, Sulkes J, Sulkes A, et al. The Rate of the 6174delt Founder Jewish Mutation in BRCA2 in Patients With Non-Colonic Gastrointestinal Tract Tumours in Israel. Br J Cancer (2001) 84(4):478–81. doi: 10.1054/bjoc.2000.1605
19. Lin J, Shi J, Guo H, Yang X, Jiang Y, Long J, et al. Alterations in DNA Damage Repair Genes in Primary Liver Cancer. Clin Cancer Res (2019) 25(15):4701–11. doi: 10.1158/1078-0432.CCR-19-0127
20. Russell R, Perkhofer L, Liebau S, Lin Q, Lechel A, Feld FM, et al. Loss of ATM Accelerates Pancreatic Cancer Formation and Epithelial-Mesenchymal Transition. Nat Commun (2015) 6:7677. doi: 10.1038/ncomms8677
21. Seeber A, Zimmer K, Kocher F, Puccini A, Xiu J, Nabhan C, et al. Molecular Characteristics of BRCA1/2 and PALB2 Mutations in Pancreatic Ductal Adenocarcinoma. ESMO Open (2020) 5(6):e000942. doi: 10.1136/esmoopen-2020-000942
22. Puccini A, Bregni G, Sciallero S. Maintenance Olaparib for Metastatic Pancreatic Cancer. N Engl J Med (2019) 381(15):1491–2. doi: 10.1056/NEJMc1911185
23. Lieber MR. The Mechanism of Double-Strand DNA Break Repair by the Nonhomologous DNA End-Joining Pathway. Annu Rev Biochem (2010) 79:181–211. doi: 10.1146/annurev.biochem.052308.093131
24. Chang HHY, Pannunzio NR, Adachi N, Lieber MR. Non-Homologous DNA End Joining and Alternative Pathways to Double-Strand Break Repair. Nat Rev Mol Cell Biol (2017) 18(8):495–506. doi: 10.1038/nrm.2017.48
25. Venkitaraman AR. Cancer Susceptibility and the Functions of BRCA1 and BRCA2. Cell (2002) 108(2):171–82. doi: 10.1016/s0092-8674(02)00615-3
26. Her J, Bunting SF. How Cells Ensure Correct Repair of DNA Double-Strand Breaks. J Biol Chem (2018) 293(27):10502–11. doi: 10.1074/jbc.TM118.000371
27. Roy R, Chun J, Powell SN. BRCA1 and BRCA2: Different Roles in a Common Pathway of Genome Protection. Nat Rev Cancer (2011) 12(1):68–78. doi: 10.1038/nrc3181
28. Deng CX, Brodie SG. Roles of BRCA1 and its Interacting Proteins. Bioessays (2000) 22(8):728–37. doi: 10.1002/1521-1878(200008)22:8<728::AID-BIES6>3.0.CO;2-B
29. Huen MS, Sy SM, Chen J. BRCA1 and its Toolbox for the Maintenance of Genome Integrity. Nat Rev Mol Cell Biol (2010) 11(2):138–48. doi: 10.1038/nrm2831
30. Zhang F, Fan Q, Ren K, Andreassen PR. PALB2 Functionally Connects the Breast Cancer Susceptibility Proteins BRCA1 and BRCA2. Mol Cancer Res (2009) 7(7):1110–8. doi: 10.1158/1541-7786.MCR-09-0123
31. Zhang F, Ma J, Wu J, Ye L, Cai H, Xia B, et al. PALB2 Links BRCA1 and BRCA2 in the DNA-Damage Response. Curr Biol (2009) 19(6):524–9. doi: 10.1016/j.cub.2009.02.018
32. Turner N, Tutt A, Ashworth A. Hallmarks of 'Brcaness' in Sporadic Cancers. Nat Rev Cancer (2004) 4(10):814–9. doi: 10.1038/nrc1457
33. Lord CJ, Ashworth A. BRCAness Revisited. Nat Rev Cancer (2016) 16(2):110–20. doi: 10.1038/nrc.2015.21
34. Karaayvaz-Yildirim M, Silberman RE, Langenbucher A, Saladi SV, Ross KN, Zarcaro E, et al. Aneuploidy and a Deregulated DNA Damage Response Suggest Haploinsufficiency in Breast Tissues of. Sci Adv (2020) 6(5):eaay2611. doi: 10.1126/sciadv.aay2611
35. Maxwell KN, Wubbenhorst B, Wenz BM, De Sloover D, Pluta J, Emery L, et al. BRCA Locus-Specific Loss of Heterozygosity in Germline BRCA1 and BRCA2 Carriers. Nat Commun (2017) 8(1):319. doi: 10.1038/s41467-017-00388-9
36. Vos S, Moelans CB, van Diest PJ. BRCA Promoter Methylation in Sporadic Versus BRCA Germline Mutation-Related Breast Cancers. Breast Cancer Res (2017) 19(1):64. doi: 10.1186/s13058-017-0856-z
37. Stefansson OA, Hilmarsdottir H, Olafsdottir K, Tryggvadottir L, Sverrisdottir A, Johannsson OT, et al. Promoter Methylation Status in 1031 Primary Breast Cancers Predicts Favorable Outcomes Following Chemotherapy. JNCI Cancer Spectr (2020) 4(2):pkz100. doi: 10.1093/jncics/pkz100
38. Johnstone TC, Park GY, Lippard SJ. Understanding and Improving Platinum Anticancer Drugs–Phenanthriplatin. Anticancer Res (2014) 34(1):471–6.
39. Kennedy RD, Quinn JE, Mullan PB, Johnston PG, Harkin DP. The Role of BRCA1 in the Cellular Response to Chemotherapy. J Natl Cancer Inst (2004) 96(22):1659–68. doi: 10.1093/jnci/djh312
40. Byrski T, Gronwald J, Huzarski T, Grzybowska E, Budryk M, Stawicka M, et al. Pathologic Complete Response Rates in Young Women With BRCA1-Positive Breast Cancers After Neoadjuvant Chemotherapy. J Clin Oncol (2010) 28(3):375–9. doi: 10.1200/JCO.2008.20.7019
41. Byrski T, Dent R, Blecharz P, Foszczynska-Kloda M, Gronwald J, Huzarski T, et al. Results of a Phase II Open-Label, Non-Randomized Trial of Cisplatin Chemotherapy in Patients With BRCA1-Positive Metastatic Breast Cancer. Breast Cancer Res (2012) 14(4):R110. doi: 10.1186/bcr3231
42. Tutt AN, Lord CJ, McCabe N, Farmer H, Turner N, Martin NM, et al. Exploiting the DNA Repair Defect in BRCA Mutant Cells in the Design of New Therapeutic Strategies for Cancer. Cold Spring Harb Symp Quant Biol (2005) 70:139–48. doi: 10.1101/sqb.2005.70.012
43. Silver DP, Richardson AL, Eklund AC, Wang ZC, Szallasi Z, Li Q, et al. Efficacy of Neoadjuvant Cisplatin in Triple-Negative Breast Cancer. J Clin Oncol (2010) 28(7):1145–53. doi: 10.1200/JCO.2009.22.4725
44. Evers B, Helleday T, Jonkers J. Targeting Homologous Recombination Repair Defects in Cancer. Trends Pharmacol Sci (2010) 31(8):372–80. doi: 10.1016/j.tips.2010.06.001
45. Tutt A, Tovey H, Cheang MCU, Kernaghan S, Kilburn L, Gazinska P, et al. Carboplatin in BRCA1/2-Mutated and Triple-Negative Breast Cancer BRCAness Subgroups: The TNT Trial. Nat Med (2018) 24(5):628–37. doi: 10.1038/s41591-018-0009-7
46. Alsop K, Fereday S, Meldrum C, deFazio A, Emmanuel C, George J, et al. BRCA Mutation Frequency and Patterns of Treatment Response in BRCA Mutation-Positive Women With Ovarian Cancer: A Report From the Australian Ovarian Cancer Study Group. J Clin Oncol (2012) 30(21):2654–63. doi: 10.1200/JCO.2011.39.8545
47. Bolton KL, Chenevix-Trench G, Goh C, Sadetzki S, Ramus SJ, Karlan BY, et al. Association Between BRCA1 and BRCA2 Mutations and Survival in Women With Invasive Epithelial Ovarian Cancer. JAMA (2012) 307(4):382–90. doi: 10.1001/jama.2012.20
48. Lesnock JL, Darcy KM, Tian C, Deloia JA, Thrall MM, Zahn C, et al. BRCA1 Expression and Improved Survival in Ovarian Cancer Patients Treated With Intraperitoneal Cisplatin and Paclitaxel: A Gynecologic Oncology Group Study. Br J Cancer (2013) 108(6):1231–7. doi: 10.1038/bjc.2013.70
49. Naumann RW, Morris JC, Tait DL, Higgins RV, Crane EK, Drury LK, et al. Patients With BRCA Mutations Have Superior Outcomes After Intraperitoneal Chemotherapy in Optimally Resected High Grade Ovarian Cancer. Gynecol Oncol (2018) 151(3):477–80. doi: 10.1016/j.ygyno.2018.10.003
50. Pal T, Permuth-Wey J, Kapoor R, Cantor A, Sutphen R. Improved Survival in BRCA2 Carriers With Ovarian Cancer. Fam Cancer (2007) 6(1):113–9. doi: 10.1007/s10689-006-9112-x
51. Sun C, Li N, Ding D, Weng D, Meng L, Chen G, et al. The Role of BRCA Status on the Prognosis of Patients With Epithelial Ovarian Cancer: A Systematic Review of the Literature With a Meta-Analysis. PloS One (2014) 9(5):e95285. doi: 10.1371/journal.pone.0095285
52. Tan DS, Kaye SB. Chemotherapy for Patients With BRCA1 and BRCA2-Mutated Ovarian Cancer: Same or Different? Am Soc Clin Oncol Educ Book (2015), 114–21. doi: 10.14694/EdBook_AM.2015.35.114
53. Tan DS, Rothermundt C, Thomas K, Bancroft E, Eeles R, Shanley S, et al. "BRCAness" Syndrome in Ovarian Cancer: A Case-Control Study Describing the Clinical Features and Outcome of Patients With Epithelial Ovarian Cancer Associated With BRCA1 and BRCA2 Mutations. J Clin Oncol (2008) 26(34):5530–6. doi: 10.1200/JCO.2008.16.1703
54. Vencken PMLH, Kriege M, Hoogwerf D, Beugelink S, van der Burg MEL, Hooning MJ, et al. Chemosensitivity and Outcome of BRCA1- and BRCA2-Associated Ovarian Cancer Patients After First-Line Chemotherapy Compared With Sporadic Ovarian Cancer Patients. Ann Oncol (2011) 22(6):1346–52. doi: 10.1093/annonc/mdq628
55. Helleday T. The Underlying Mechanism for the PARP and BRCA Synthetic Lethality: Clearing Up the Misunderstandings. Mol Oncol (2011) 5(4):387–93. doi: 10.1016/j.molonc.2011.07.001
56. Wallace SS. Base Excision Repair: A Critical Player in Many Games. DNA Repair (2014) 19:14–26. doi: 10.1016/j.dnarep.2014.03.030
57. Morales J, Li L, Fattah FJ, Dong Y, Bey EA, Patel M, et al. Review of Poly (ADP-Ribose) Polymerase (PARP) Mechanisms of Action and Rationale for Targeting in Cancer and Other Diseases. Crit Rev Eukaryot Gene Expr (2014) 24(1):15–28. doi: 10.1615/critreveukaryotgeneexpr.2013006875
58. Zheng F, Zhang Y, Chen S, Weng X, Rao Y, Fang H. Mechanism and Current Progress of Poly ADP-Ribose Polymerase (PARP) Inhibitors in the Treatment of Ovarian Cancer. BioMed Pharmacother (2020) 123:109661. doi: 10.1016/j.biopha.2019.109661
59. Ström CE, Johansson F, Uhlén M, Szigyarto CA, Erixon K, Helleday T. Poly (ADP-Ribose) Polymerase (PARP) Is Not Involved in Base Excision Repair But PARP Inhibition Traps a Single-Strand Intermediate. Nucleic Acids Res (2011) 39(8):3166–75. doi: 10.1093/nar/gkq1241
60. Rose M, Burgess JT, O’Byrne K, Richard DJ, Bolderson E. PARP Inhibitors: Clinical Relevance, Mechanisms of Action and Tumor Resistance. Front Cell Dev Biol (2020) 8(879):564601. doi: 10.3389/fcell.2020.564601
61. Noordermeer SM, van Attikum H. PARP Inhibitor Resistance: A Tug-Of-War in BRCA-Mutated Cells. Trends Cell Biol (2019) 29(10):820–34. doi: 10.1016/j.tcb.2019.07.008
62. Mateo J, Lord CJ, Serra V, Tutt A, Balmaña J, Castroviejo-Bermejo M, et al. A Decade of Clinical Development of PARP Inhibitors in Perspective. Ann Oncol (2019) 30(9):1437–47. doi: 10.1093/annonc/mdz192
63. Mihailidou C, Karamouzis MV, Schizas D, Papavassiliou AG. Co-Targeting C-Met and DNA Double-Strand Breaks (DSBs): Therapeutic Strategies in BRCA-Mutated Gastric Carcinomas. Biochimie (2017) 142:135–43. doi: 10.1016/j.biochi.2017.09.001
64. Fong PC, Yap TA, Boss DS, Carden CP, Mergui-Roelvink M, Gourley C, et al. Poly(ADP)-Ribose Polymerase Inhibition: Frequent Durable Responses in BRCA Carrier Ovarian Cancer Correlating With Platinum-Free Interval. J Clin Oncol (2010) 28(15):2512–9. doi: 10.1200/JCO.2009.26.9589
65. Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2018. CA Cancer J Clin (2018) 68(1):7–30. doi: 10.3322/caac.21442
66. Golan T, Kanji ZS, Epelbaum R, Devaud N, Dagan E, Holter S, et al. Overall Survival and Clinical Characteristics of Pancreatic Cancer in BRCA Mutation Carriers. Br J Cancer (2014) 111(6):1132–8. doi: 10.1038/bjc.2014.418
67. Golan T, Sella T, O'Reilly EM, Katz MH, Epelbaum R, Kelsen DP, et al. Overall Survival and Clinical Characteristics of BRCA Mutation Carriers With Stage I/II Pancreatic Cancer. Br J Cancer (2017) 116(6):697–702. doi: 10.1038/bjc.2017.19
68. Consortium BCL. Cancer Risks in BRCA2 Mutation Carriers. J Natl Cancer Inst (1999) 91(15):1310–6. doi: 10.1093/jnci/91.15.1310
69. Murphy KM, Brune KA, Griffin C, Sollenberger JE, Petersen GM, Bansal R, et al. Evaluation of Candidate Genes MAP2K4, MADH4, ACVR1B, and BRCA2 in Familial Pancreatic Cancer: Deleterious BRCA2 Mutations in 17%. Cancer Res (2002) 62(13):3789–93.
70. Jones S, Hruban RH, Kamiyama M, Borges M, Zhang X, Parsons DW, et al. Exomic Sequencing Identifies PALB2 as a Pancreatic Cancer Susceptibility Gene. Sci (N Y NY) (2009) 324(5924):217. doi: 10.1126/science.1171202
71. Slater EP, Langer P, Niemczyk E, Strauch K, Butler J, Habbe N, et al. PALB2 Mutations in European Familial Pancreatic Cancer Families. Clin Genet (2010) 78(5):490–4. doi: 10.1111/j.1399-0004.2010.01425.x
72. Tischkowitz MD, Sabbaghian N, Hamel N, Borgida A, Rosner C, Taherian N, et al. Analysis of the Gene Coding for the BRCA2-Interacting Protein PALB2 in Familial and Sporadic Pancreatic Cancer. Gastroenterology (2009) 137(3):1183–6. doi: 10.1053/j.gastro.2009.06.055
73. Luo G, Lu Y, Jin K, Cheng H, Guo M, Liu Z, et al. Pancreatic Cancer: BRCA Mutation and Personalized Treatment. Expert Rev Anticancer Ther (2015) 15(10):1223–31. doi: 10.1586/14737140.2015.1086271
74. Chalasani P, Kurtin S, Dragovich T. Response to a Third-Line Mitomycin C (MMC)-Based Chemotherapy in a Patient With Metastatic Pancreatic Adenocarcinoma Carrying Germline BRCA2 Mutation. JOP (2008) 9(3):305–8.
75. James E, Waldron-Lynch MG, Saif MW. Prolonged Survival in a Patient With BRCA2 Associated Metastatic Pancreatic Cancer After Exposure to Camptothecin: A Case Report and Review of Literature. Anticancer Drugs (2009) 20(7):634–8. doi: 10.1097/CAD.0b013e32832b511e
76. Fogelman D, Sugar EA, Oliver G, Shah N, Klein A, Alewine C, et al. Family History as a Marker of Platinum Sensitivity in Pancreatic Adenocarcinoma. Cancer Chemother Pharmacol (2015) 76(3):489–98. doi: 10.1007/s00280-015-2788-6
77. Sonnenblick A, Kadouri L, Appelbaum L, Peretz T, Sagi M, Goldberg Y, et al. Complete Remission, in BRCA2 Mutation Carrier With Metastatic Pancreatic Adenocarcinoma, Treated With Cisplatin Based Therapy. Cancer Biol Ther (2011) 12(3):165–8. doi: 10.4161/cbt.12.3.16292
78. Kaufman B, Shapira-Frommer R, Schmutzler RK, Audeh MW, Friedlander M, Balmaña J, et al. Olaparib Monotherapy in Patients With Advanced Cancer and a Germline BRCA1/2 Mutation. J Clin Oncol (2015) 33(3):244–50. doi: 10.1200/JCO.2014.56.2728
79. Bendell J, O'Reilly EM, Middleton MR, Chau I, Hochster H, Fielding A, et al. Phase I Study of Olaparib Plus Gemcitabine in Patients With Advanced Solid Tumours and Comparison With Gemcitabine Alone in Patients With Locally Advanced/Metastatic Pancreatic Cancer. Ann Oncol (2015) 26(4):804–11. doi: 10.1093/annonc/mdu581
80. LoRusso PM, Li J, Burger A, Heilbrun LK, Sausville EA, Boerner SA, et al. Phase I Safety, Pharmacokinetic, and Pharmacodynamic Study of the Poly(ADP-Ribose) Polymerase (PARP) Inhibitor Veliparib (ABT-888) in Combination With Irinotecan in Patients With Advanced Solid Tumors. Clin Cancer Res (2016) 22(13):3227–37. doi: 10.1158/1078-0432.Ccr-15-0652
81. Yarchoan M, Hopkins A, Jaffee EM. Tumor Mutational Burden and Response Rate to PD-1 Inhibition. N Engl J Med (2017) 377(25):2500–1. doi: 10.1056/NEJMc1713444
82. de Bono J, Ramanathan RK, Mina L, Chugh R, Glaspy J, Rafii S, et al. Dose-Escalation, Two-Part Trial of the PARP Inhibitor Talazoparib in Patients With Advanced Germline BRCA1/2 Mutations and Selected Sporadic Cancers. Cancer Discovery (2017) 7(6):620–9. doi: 10.1158/2159-8290.Cd-16-1250
83. Shroff RT, Hendifar A, McWilliams RR, Geva R, Epelbaum R, Rolfe L, et al. Rucaparib Monotherapy in Patients With Pancreatic Cancer and a Known Deleterious. JCO Precis Oncol (2018) 2018:PO.17.00316. doi: 10.1200/PO.17.00316
84. Turk AA, Deming DA, Lubner SJ, Mulkerin D, LoConte NK, Tevaarwerk A, et al. A Phase I Study of Veliparib (Vel) in Combination With Oxaliplatin (Ox) and Capecitabine (Cap) in Advanced Solid Tumors. J Clin Conol (2018) 36(4_suppl):314. doi: 10.1200/JCO.2018.36.4_suppl.314
85. Lowery MA, Kelsen DP, Capanu M, Smith SC, Lee JW, Stadler ZK, et al. Phase II Trial of Veliparib in Patients With Previously Treated BRCA-Mutated Pancreas Ductal Adenocarcinoma. Eur J Cancer (2018) 89:19–26. doi: 10.1016/j.ejca.2017.11.004
86. Golan T, Hammel P, Reni M, Van Cutsem E, Macarulla T, Hall MJ, et al. Maintenance Olaparib for Germline BRCA-Mutated Metastatic Pancreatic Cancer. N Engl J Med (2019) 381(4):317–27. doi: 10.1056/NEJMoa1903387
87. Tuli R, Shiao SL, Nissen N, Tighiouart M, Kim S, Osipov A, et al. A Phase 1 Study of Veliparib, a PARP-1/2 Inhibitor, With Gemcitabine and Radiotherapy in Locally Advanced Pancreatic Cancer. EBioMedicine (2019) 40:375–81. doi: 10.1016/j.ebiom.2018.12.060
88. Pishvaian MJ, Wang H, He AR, Hwang JJ, Smaglo BG, Kim SS, et al. A Phase I/II Study of Veliparib (ABT-888) in Combination With 5-Fluorouracil and Oxaliplatin in Patients With Metastatic Pancreatic Cancer. Clin Cancer Res (2020) 26(19):5092–101. doi: 10.1158/1078-0432.CCR-20-1301
89. O'Reilly EM, Lee JW, Zalupski M, Capanu M, Park J, Golan T, et al. Randomized, Multicenter, Phase II Trial of Gemcitabine and Cisplatin With or Without Veliparib in Patients With Pancreas Adenocarcinoma and a Germline. J Clin Oncol (2020) 38(13):1378–88. doi: 10.1200/JCO.19.02931
90. Yoo HK, Kindler HL, McCutcheon S, McGuinness D, Patel N, Hettle R, et al. POLO: Quality-Adjusted (QA) Progression-Free Survival (PFS) and Patient (Pt)-Centered Outcomes With Maintenance Olaparib in Pts With Metastatic Pancreatic Cancer (Mpac). J Clin Oncol (2020) 38(15_suppl):4626. doi: 10.1200/JCO.2020.38.15_suppl.4626
91. O'Reilly EM, Lee JW, Lowery MA, Capanu M, Stadler ZK, Moore MJ, et al. Phase 1 Trial Evaluating Cisplatin, Gemcitabine, and Veliparib in 2 Patient Cohorts: Germline BRCA Mutation Carriers and Wild-Type BRCA Pancreatic Ductal Adenocarcinoma. Cancer (2018) 124(7):1374–82. doi: 10.1002/cncr.31218
92. Hannan Z, Yu S, Domchek S, Mamtani R, Reiss KA. Clinical Characteristics of Patients With Pancreatic Cancer and Pathogenic ATM Mutations. JNCI Cancer Spectr (2021) 5(2):pkaa121. doi: 10.1093/jncics/pkaa121
93. Secrier M, Li X, de Silva N, Eldridge MD, Contino G, Bornschein J, et al. Mutational Signatures in Esophageal Adenocarcinoma Define Etiologically Distinct Subgroups With Therapeutic Relevance. Nat Genet (2016) 48(10):1131–41. doi: 10.1038/ng.3659
94. Gao Y, Zhu J, Zhang X, Wu Q, Jiang S, Liu Y, et al. BRCA1 mRNA Expression as a Predictive and Prognostic Marker in Advanced Esophageal Squamous Cell Carcinoma Treated With Cisplatin- or Docetaxel-Based Chemotherapy/Chemoradiotherapy. PloS One (2013) 8(1):e52589. doi: 10.1371/journal.pone.0052589
95. Halpern N, Grinshpun A, Boursi B, Golan T, Margalit O, Aderka D, et al. Clinical Characteristics and Prognosis of Gastric Cancer Patients With BRCA1/2 Germline Mutations: Report of Ten Cases and a Literature Review. Onco Targets Ther (2020) 13:11637–44. doi: 10.2147/OTT.S276814
96. Bang Y-J, Kaufman B, Geva R, Stemmer SM, Hong S-H, Lee J-S, et al. An Open-Label, Phase II Basket Study of Olaparib and Durvalumab (MEDIOLA): Results in Patients With Relapsed Gastric Cancer. J Clin Oncol (2019) 37(4_suppl):140. doi: 10.1200/JCO.2019.37.4_suppl.140
97. Chen W, Wang J, Li X, Li J, Zhou L, Qiu T, et al. Prognostic Significance of BRCA1 Expression in Gastric Cancer. Med Oncol (2013) 30(1):423. doi: 10.1007/s12032-012-0423-5
98. Kim JW, Cho HJ, Kim M, Lee KH, Kim MA, Han SW, et al. Differing Effects of Adjuvant Chemotherapy According to BRCA1 Nuclear Expression in Gastric Cancer. Cancer Chemother Pharmacol (2013) 71(6):1435–43. doi: 10.1007/s00280-013-2141-x
99. Kim HS, Hwang IG, Min HY, Bang YJ, Kim WH. Clinical Significance of BRCA1 and BRCA2 mRNA and Protein Expression in Patients With Sporadic Gastric Cancer. Oncol Lett (2019) 17(5):4383–92. doi: 10.3892/ol.2019.10132
100. Chae H, Kim D, Yoo C, Kim KP, Jeong JH, Chang HM, et al. Therapeutic Relevance of Targeted Sequencing in Management of Patients With Advanced Biliary Tract Cancer: DNA Damage Repair Gene Mutations as a Predictive Biomarker. Eur J Cancer (2019) 120:31–9. doi: 10.1016/j.ejca.2019.07.022
101. Golan T, Raitses-Gurevich M, Kelley RK, Bocobo AG, Borgida A, Shroff RT, et al. Overall Survival and Clinical Characteristics of BRCA-Associated Cholangiocarcinoma: A Multicenter Retrospective Study. Oncologist (2017) 22(7):804–10. doi: 10.1634/theoncologist.2016-0415
102. Thompson D, Easton DF, Consortium BCL. Cancer Incidence in BRCA1 Mutation Carriers. J Natl Cancer Inst (2002) 94(18):1358–65. doi: 10.1093/jnci/94.18.1358
103. Guillot C, Hall J, Herceg Z, Merle P, Chemin I. Update on Hepatocellular Carcinoma Breakthroughs: Poly(ADP-Ribose) Polymerase Inhibitors as a Promising Therapeutic Strategy. Clin Res Hepatol Gastroenterol (2014) 38(2):137–42. doi: 10.1016/j.clinre.2013.07.006
104. Phelan CM, Iqbal J, Lynch HT, Lubinski J, Gronwald J, Moller P, et al. Incidence of Colorectal Cancer in BRCA1 and BRCA2 Mutation Carriers: Results From a Follow-Up Study. Br J Cancer (2014) 110(2):530–4. doi: 10.1038/bjc.2013.741
105. Grinshpun A, Halpern N, Granit RZ, Hubert A, Hamburger T, Laitman Y, et al. Phenotypic Characteristics of Colorectal Cancer in BRCA1/2 Mutation Carriers. Eur J Hum Genet (2018) 26(3):382–6. doi: 10.1038/s41431-017-0067-1
106. Leichman L, Groshen S, O'Neil BH, Messersmith W, Berlin J, Chan E, et al. Phase II Study of Olaparib (AZD-2281) After Standard Systemic Therapies for Disseminated Colorectal Cancer. Oncologist (2016) 21(2):172–7. doi: 10.1634/theoncologist.2015-0319
107. Gorbunova V, Beck JT, Hofheinz RD, Garcia-Alfonso P, Nechaeva M, Cubillo Gracian A, et al. A Phase 2 Randomised Study of Veliparib Plus FOLFIRI±bevacizumab Versus Placebo Plus FOLFIRI±bevacizumab in Metastatic Colorectal Cancer. Br J Cancer (2019) 120(2):183–9. doi: 10.1038/s41416-018-0343-z
108. Parikh AR, He Y, Hong TS, Corcoran RB, Clark JW, Ryan DP, et al. Analysis of DNA Damage Response Gene Alterations and Tumor Mutational Burden Across 17,486 Tubular Gastrointestinal Carcinomas: Implications for Therapy. Oncologist (2019) 24(10):1340–7. doi: 10.1634/theoncologist.2019-0034
109. Takagi M. DNA Damage Response and Hematological Malignancy. Int J Hematol (2017) 106(3):345–56. doi: 10.1007/s12185-017-2226-0
110. Swift M, Morrell D, Massey RB, Chase CL. Incidence of Cancer in 161 Families Affected by Ataxia-Telangiectasia. N Engl J Med (1991) 325(26):1831–6. doi: 10.1056/NEJM199112263252602
111. Choi M, Kipps T, Kurzrock R. ATM Mutations in Cancer: Therapeutic Implications. Mol Cancer Ther (2016) 15(8):1781–91. doi: 10.1158/1535-7163.MCT-15-0945
112. Riches LC, Trinidad AG, Hughes G, Jones GN, Hughes AM, Thomason AG, et al. Pharmacology of the ATM Inhibitor AZD0156: Potentiation of Irradiation and Olaparib Responses Preclinically. Mol Cancer Ther (2020) 19(1):13–25. doi: 10.1158/1535-7163.MCT-18-1394
113. Bang YJ, Im SA, Lee KW, Cho JY, Song EK, Lee KH, et al. Randomized, Double-Blind Phase II Trial With Prospective Classification by ATM Protein Level to Evaluate the Efficacy and Tolerability of Olaparib Plus Paclitaxel in Patients With Recurrent or Metastatic Gastric Cancer. J Clin Oncol (2015) 33(33):3858–65. doi: 10.1200/JCO.2014.60.0320
114. Bang YJ, Xu RH, Chin K, Lee KW, Park SH, Rha SY, et al. Olaparib in Combination With Paclitaxel in Patients With Advanced Gastric Cancer Who Have Progressed Following First-Line Therapy (GOLD): A Double-Blind, Randomised, Placebo-Controlled, Phase 3 Trial. Lancet Oncol (2017) 18(12):1637–51. doi: 10.1016/S1470-2045(17)30682-4
115. Liu YZ, Hodgson D, Locker G, Lai Z, Balcerzak D, Sharpe A, et al. Olaparib Plus Paclitaxel Sensitivity in Biomarker Subgroups of Gastric Cancer. Ann Oncol (2018) 29:viii25–viii6. doi: 10.1093/annonc/mdy269.081
116. Menezes DL, Holt J, Tang Y, Feng J, Barsanti P, Pan Y, et al. A Synthetic Lethal Screen Reveals Enhanced Sensitivity to ATR Inhibitor Treatment in Mantle Cell Lymphoma With ATM Loss-Of-Function. Mol Cancer Res (2015) 13(1):120–9. doi: 10.1158/1541-7786.Mcr-14-0240
117. Yazinski SA, Comaills V, Buisson R, Genois MM, Nguyen HD, Ho CK, et al. ATR Inhibition Disrupts Rewired Homologous Recombination and Fork Protection Pathways in PARP Inhibitor-Resistant BRCA-Deficient Cancer Cells. Genes Dev (2017) 31(3):318–32. doi: 10.1101/gad.290957.116
118. Kim H, George E, Ragland R, Rafail S, Zhang R, Krepler C, et al. Targeting the ATR/CHK1 Axis With PARP Inhibition Results in Tumor Regression in. Clin Cancer Res (2017) 23(12):3097–108. doi: 10.1158/1078-0432.CCR-16-2273
119. Pilié PG, Gay CM, Byers LA, O'Connor MJ, Yap TA. PARP Inhibitors: Extending Benefit Beyond BRCA-Mutant Cancers. Clin Cancer Res (2019) 25(13):3759–71. doi: 10.1158/1078-0432.CCR-18-0968
120. Lee J, Kim ST, Smith S, Mortimer PG, Loembé B, Hong J, et al. Results From a Phase I, Open-Label Study of Ceralasertib (AZD6738), a Novel DNA Damage Repair Agent, in Combination With Weekly Paclitaxel in Refractory Cancer (NCT02630199). J Clin Oncol (2020) 38(15_suppl):3503–. doi: 10.1200/JCO.2020.38.15_suppl.3503
121. Krebs MG, Lopez J, El-Khoueiry A, Bang Y-J, Postel-Vinay S, Abida W, et al. Abstract CT026: Phase I Study of AZD6738, an Inhibitor of Ataxia Telangiectasia Rad3-Related (ATR), in Combination With Olaparib or Durvalumab in Patients (Pts) With Advanced Solid Cancers. Cancer Res (2018) 78(13 Supplement):CT026–CT. doi: 10.1158/1538-7445.Am2018-ct026
122. Yap TA, O'Carrigan B, Penney MS, Lim JS, Brown JS, de Miguel Luken MJ, et al. Phase I Trial of First-In-Class ATR Inhibitor M6620 (VX-970) as Monotherapy or in Combination With Carboplatin in Patients With Advanced Solid Tumors. J Clin Oncol (2020) 38(27):3195–204. doi: 10.1200/JCO.19.02404
123. Plummer ER, Dean EJ, Evans TRJ, Greystoke A, Herbschleb K, Ranson M, et al. Phase I Trial of First-in-Class ATR Inhibitor VX-970 in Combination With Gemcitabine (Gem) in Advanced Solid Tumors (NCT02157792). J Clin Oncol (2016) 34(15_suppl):2513–. doi: 10.1200/JCO.2016.34.15_suppl.2513
124. Zhang Y, Hunter T. Roles of Chk1 in Cell Biology and Cancer Therapy. Int J Cancer (2014) 134(5):1013–23. doi: 10.1002/ijc.28226
125. Italiano A, Infante JR, Shapiro GI, Moore KN, LoRusso PM, Hamilton E, et al. Phase I Study of the Checkpoint Kinase 1 Inhibitor GDC-0575 in Combination With Gemcitabine in Patients With Refractory Solid Tumors. Ann Oncol (2018) 29(5):1304–11. doi: 10.1093/annonc/mdy076
126. Matheson CJ, Backos DS, Reigan P. Targeting WEE1 Kinase in Cancer. Trends Pharmacol Sci (2016) 37(10):872–81. doi: 10.1016/j.tips.2016.06.006
127. Cuneo KC, Morgan MA, Sahai V, Schipper MJ, Parsels LA, Parsels JD, et al. Dose Escalation Trial of the Wee1 Inhibitor Adavosertib (AZD1775) in Combination With Gemcitabine and Radiation for Patients With Locally Advanced Pancreatic Cancer. J Clin Oncol (2019) 37(29):2643–50. doi: 10.1200/JCO.19.00730
128. Leijen S, van Geel RM, Pavlick AC, Tibes R, Rosen L, Razak AR, et al. Phase I Study Evaluating WEE1 Inhibitor AZD1775 As Monotherapy and in Combination With Gemcitabine, Cisplatin, or Carboplatin in Patients With Advanced Solid Tumors. J Clin Oncol (2016) 34(36):4371–80. doi: 10.1200/JCO.2016.67.5991
129. Kim HY, Cho Y, Kang H, Yim YS, Kim SJ, Song J, et al. Targeting the WEE1 Kinase as a Molecular Targeted Therapy for Gastric Cancer. Oncotarget (2016) 7(31):49902–16. doi: 10.18632/oncotarget.10231
130. Ge XC, Wu F, Li WT, Zhu XJ, Liu JW, Wang BL. Upregulation of WEE1 is a Potential Prognostic Biomarker for Patients With Colorectal Cancer. Oncol Lett (2017) 13(6):4341–8. doi: 10.3892/ol.2017.5984
131. Parsels LA, Parsels JD, Tanska DM, Maybaum J, Lawrence TS, Morgan MA. The Contribution of DNA Replication Stress Marked by High-Intensity, Pan-Nuclear γh2ax Staining to Chemosensitization by CHK1 and WEE1 Inhibitors. Cell Cycle (2018) 17(9):1076–86. doi: 10.1080/15384101.2018.1475827
132. Fang Y, McGrail DJ, Sun C, Labrie M, Chen X, Zhang D, et al. Sequential Therapy With PARP and WEE1 Inhibitors Minimizes Toxicity While Maintaining Efficacy. Cancer Cell (2019) 35(6):851–67.e7. doi: 10.1016/j.ccell.2019.05.001
133. Bukhari AB, Lewis CW, Pearce JJ, Luong D, Chan GK, Gamper AM. Inhibiting Wee1 and ATR Kinases Produces Tumor-Selective Synthetic Lethality and Suppresses Metastasis. J Clin Invest (2019) 129(3):1329–44. doi: 10.1172/JCI122622
134. Ashworth A, Lord CJ. Synthetic Lethal Therapies for Cancer: What's Next After PARP Inhibitors? Nat Rev Clin Oncol (2018) 15(9):564–76. doi: 10.1038/s41571-018-0055-6
135. Pilié PG, Tang C, Mills GB, Yap TA. State-Of-the-Art Strategies for Targeting the DNA Damage Response in Cancer. Nat Rev Clin Oncol (2019) 16(2):81–104. doi: 10.1038/s41571-018-0114-z
136. Goggins M, Overbeek KA, Brand R, Syngal S, Del Chiaro M, Bartsch DK, et al. Management of Patients With Increased Risk for Familial Pancreatic Cancer: Updated Recommendations From the International Cancer of the Pancreas Screening (CAPS) Consortium. Gut (2020) 69(1):7–17. doi: 10.1136/gutjnl-2019-319352
137. Kupfer SS, Gupta S, Weitzel JN, Samadder J. AGA Clinical Practice Update on Colorectal and Pancreatic Cancer Risk and Screening in BRCA1 and BRCA2 Carriers: Commentary. Gastroenterology (2020) 159(2):760–4. doi: 10.1053/j.gastro.2020.03.086
138. van der Post RS, Vogelaar IP, Carneiro F, Guilford P, Huntsman D, Hoogerbrugge N, et al. Hereditary Diffuse Gastric Cancer: Updated Clinical Guidelines With an Emphasis on Germline CDH1 Mutation Carriers. J Med Genet (2015) 52(6):361–74. doi: 10.1136/jmedgenet-2015-103094
139. Oliveira SF IC, Pereira P, Fragoso S, Santos S, Rodrigues P, Parreira J, et al. 4628 - Gastric Cancer Screening in BRCA 2 Gene Mutation Carriers: Should it be Recommended? Ann Oncol (2019) 30:v253–324. doi: 10.1093/annonc/mdz247
140. Curtin NJ, Drew Y, Sharma-Saha S. Why BRCA Mutations Are Not Tumour-Agnostic Biomarkers for PARP Inhibitor Therapy. Nat Rev Clin Oncol (2019) 16(12):725–6. doi: 10.1038/s41571-019-0285-2
141. Johnson N, Johnson SF, Yao W, Li YC, Choi YE, Bernhardy AJ, et al. Stabilization of Mutant BRCA1 Protein Confers PARP Inhibitor and Platinum Resistance. Proc Natl Acad Sci USA (2013) 110(42):17041–6. doi: 10.1073/pnas.1305170110
142. Davies H, Glodzik D, Morganella S, Yates LR, Staaf J, Zou X, et al. HRDetect Is a Predictor of BRCA1 and BRCA2 Deficiency Based on Mutational Signatures. Nat Med (2017) 23(4):517–25. doi: 10.1038/nm.4292
143. Watkins JA, Irshad S, Grigoriadis A, Tutt AN. Genomic Scars as Biomarkers of Homologous Recombination Deficiency and Drug Response in Breast and Ovarian Cancers. Breast Cancer Res (2014) 16(3):211. doi: 10.1186/bcr3670
144. Abkevich V, Timms KM, Hennessy BT, Potter J, Carey MS, Meyer LA, et al. Patterns of Genomic Loss of Heterozygosity Predict Homologous Recombination Repair Defects in Epithelial Ovarian Cancer. Br J Cancer (2012) 107(10):1776–82. doi: 10.1038/bjc.2012.451
145. Popova T, Manié E, Rieunier G, Caux-Moncoutier V, Tirapo C, Dubois T, et al. Ploidy and Large-Scale Genomic Instability Consistently Identify Basal-Like Breast Carcinomas With BRCA1/2 Inactivation. Cancer Res (2012) 72(21):5454–62. doi: 10.1158/0008-5472.CAN-12-1470
146. Ngoi NYL, Tan DSP. The Role of Homologous Recombination Deficiency Testing in Ovarian Cancer and Its Clinical Implications: Do We Need it? ESMO Open (2021) 6(3):100144. doi: 10.1016/j.esmoop.2021.100144
147. Stewart RA, Pilié PG, Yap TA. Development of PARP and Immune-Checkpoint Inhibitor Combinations. Cancer Res (2018) 78(24):6717–25. doi: 10.1158/0008-5472.CAN-18-2652
148. Shen J, Zhao W, Ju Z, Wang L, Peng Y, Labrie M, et al. PARPi Triggers the STING-Dependent Immune Response and Enhances the Therapeutic Efficacy of Immune Checkpoint Blockade Independent of BRCAness. Cancer Res (2019) 79(2):311–9. doi: 10.1158/0008-5472.CAN-18-1003
149. Huang J, Wang L, Cong Z, Amoozgar Z, Kiner E, Xing D, et al. The PARP1 Inhibitor BMN 673 Exhibits Immunoregulatory Effects in a Brca1(-/-) Murine Model of Ovarian Cancer. Biochem Biophys Res Commun (2015) 463(4):551–6. doi: 10.1016/j.bbrc.2015.05.083
150. Jiao S, Xia W, Yamaguchi H, Wei Y, Chen MK, Hsu JM, et al. PARP Inhibitor Upregulates PD-L1 Expression and Enhances Cancer-Associated Immunosuppression. Clin Cancer Res (2017) 23(14):3711–20. doi: 10.1158/1078-0432.CCR-16-3215
151. Friedlander M, Meniawy T, Markman B, Mileshkin L, Harnett P, Millward M, et al. Pamiparib in Combination With Tislelizumab in Patients With Advanced Solid Tumours: Results From the Dose-Escalation Stage of a Multicentre, Open-Label, Phase 1a/B Trial. Lancet Oncol (2019) 20(9):1306–15. doi: 10.1016/S1470-2045(19)30396-1
152. Sundar R, Miranda S, Rodrigues DN, Chénard-Poirier M, Dolling D, Clarke M, et al. Ataxia Telangiectasia Mutated Protein Loss and Benefit From Oxaliplatin-Based Chemotherapy in Colorectal Cancer. Clin Colorectal Cancer (2018) 17(4):280–4. doi: 10.1016/j.clcc.2018.05.011
153. Jette NR, Radhamani S, Arthur G, Ye R, Goutam S, Bolyos A, et al. Combined Poly-ADP Ribose Polymerase and Ataxia-Telangiectasia Mutated/Rad3-Related Inhibition Targets Ataxia-Telangiectasia Mutated-Deficient Lung Cancer Cells. Br J Cancer (2019) 121(7):600–10. doi: 10.1038/s41416-019-0565-8
154. Jette NR, Kumar M, Radhamani S, Arthur G, Goutam S, Yip S, et al. ATM-Deficient Cancers Provide New Opportunities for Precision Oncology. Cancers (2020) 12(3):687. doi: 10.3390/cancers12030687
155. Lloyd RL, Wijnhoven PWG, Ramos-Montoya A, Wilson Z, Illuzzi G, Falenta K, et al. Combined PARP and ATR Inhibition Potentiates Genome Instability and Cell Death in ATM-Deficient Cancer Cells. Oncogene (2020) 39(25):4869–83. doi: 10.1038/s41388-020-1328-y
156. Bradbury A, Hall S, Curtin N, Drew Y. Targeting ATR as Cancer Therapy: A New Era for Synthetic Lethality and Synergistic Combinations? . Pharmacol Ther (2020) 207:107450. doi: 10.1016/j.pharmthera.2019.107450
Keywords: BRCA, DNA damage repair, PARP inhibitors (PARPi), precision oncology, molecular profiling, gastrointenstinal cancer
Citation: Zimmer K, Kocher F, Puccini A and Seeber A (2021) Targeting BRCA and DNA Damage Repair Genes in GI Cancers: Pathophysiology and Clinical Perspectives. Front. Oncol. 11:662055. doi: 10.3389/fonc.2021.662055
Received: 31 January 2021; Accepted: 15 September 2021;
Published: 11 October 2021.
Edited by:
Holger Rumpold, Ordensklinikum Linz, AustriaReviewed by:
Einat Shacham Shmueli, Sheba Medical Center, IsraelYu Sunakawa, St. Marianna University School of Medicine, Japan
Copyright © 2021 Zimmer, Kocher, Puccini and Seeber. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Andreas Seeber, YW5kcmVhcy5zZWViZXJAdGlyb2wta2xpbmlrZW4uYXQ=