 Xin Chen
Xin Chen Jing Xu2†
Jing Xu2†
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Oncol. , 27 April 2021
Sec. Cancer Genetics
Volume 11 - 2021 | https://doi.org/10.3389/fonc.2021.656675
This article is part of the Research Topic Non-coding RNAs in Breast Cancer View all 11 articles
Single-cell RNA sequencing is a powerful tool to explore the heterogeneity of breast cancer. The identification of the cell subtype that responds to estrogen has profound significance in breast cancer research and treatment. The transcriptional regulation of estrogen is an intricate network involving crosstalk between protein-coding and non-coding RNAs, which is still largely unknown, particularly at the single cell level. Therefore, we proposed a novel strategy to specify cell subtypes based on a cell-specific ceRNA network (CCN). The CCN was constructed by integrating a cell-specific RNA-RNA co-expression network (RCN) with an existing ceRNA network. The cell-specific RCN was built based on single cell expression profiles with predefined reference cells. Heterogeneous cell subtypes were inferred by enriching RNAs in CCN to the estrogen response hallmark. Edge biomarkers were identified in the early estrogen response subtype. Topological analysis revealed that NEAT1 was a hub lncRNA for the early response subtype, and its ceRNAs could predict patient survival. Another hub lncRNA, DLEU2, could potentially be involved in GPCR signaling, based on CCN. The CCN method that we proposed here facilitates the inference of cell subtypes from a network perspective and explores the function of hub lncRNAs, which are promising targets for RNA-based therapeutics.
The incidence of breast cancer has increased at a rate of 0.3% per year from 2012 to 2016 in the United States, largely because of the rising rates of local stage and hormone receptor-positive disease (1). As estrogen plays a predominant role in breast cancer, understanding the mechanisms of estrogen regulation holds profound significance in breast cancer research and treatment. The transcriptional regulation of estrogen receptor (ER) is an intricate network of signaling and functional processes that is still largely unknown at the single cell level. Recently, the intra-cell line heterogeneity of breast cancer has been comprehensively characterized through single-cell RNA sequencing (scRNA-seq), revealing transcriptomic subpopulations within cell lines (2). Therefore, investigating the heterogeneity of estrogen regulation at the single cell level could shed more light on estrogen mechanisms and potential breast cancer therapeutics.
As an active metabolite of estrogen, 17β-estradiol (E2) is essential for both normal breast cells and malignant breast cancer cells. Zhu et al. performed scRNA-seq on estrogen receptor alpha positive breast cancer cells stimulated by E2. Their research revealed a dynamic transcriptional network in which estrogen signaling promotes breast cancer cell survival and growth by mediating a metabolic switch (3). They also provided valuable data resources to explore the heterogeneous response of cells from the same cell line upon estrogen stimulation.
Dai et al. proposed a probability theory-based method to construct a cell-specific network for individual cells (4), which innovatively characterized each cell from the perspective of a ‘stable’ gene network rather than ‘unstable’ gene expression. This prompted us to propose a novel strategy to characterize single cells from the perspective of networks. The RNAs interact in a complicated manner within cells. For example, RNA functions as microRNA (miRNA) sponges by competitively binding to the same miRNA, reducing the repression or degradation effect of the miRNA on the target genes. These RNAs are competing endogenous RNAs (ceRNAs). Evidence from studies indicates that long non-coding RNAs (lncRNAs) act as ceRNAs to compete with miRNAs with mRNAs. For example, NEAT1 was reported to serve as a ceRNA of ZEB1, which competes with miR-448 in breast cancer (5). PTEN, a well-known tumor suppressor, regulates MALAT1 expression by potentially sponging oncogenic miRNAs, including miR-17 and miR-20a in breast cancer (6). Therefore, it is adequate for the characterization of single cells from the viewpoint of the ceRNA network. Wang et al. applied the cell-specific network method developed by Dai et al. and integrated public ceRNA regulations to build a database named LnCeCell, which comprised the predicted lncRNA-associated ceRNA networks at single-cell resolution (7). In this study, we aimed to investigate cell heterogeneity upon estrogen stimulation, from the perspective of the ceRNA network.
Liu et al. developed a sample-specific network (SSN) method to construct a personalized network for individual patients, based on the expression profile of these patients (8). Inspired by the SSN method, we designed a novel strategy to construct a cell-specific ceRNA network (CCN) by integrating a cell-specific RNA–RNA co-expression network (RCN) with an existing ceRNA network. The cell-specific RCN was first constructed from single cell expression profiles with the aid of predefined reference cells, provided by the SSN method. After incorporating public ceRNA networks into the RCN, the CCN was obtained. To dissect the heterogeneity of the cell response to estrogen, RNAs in CCN were enriched with estrogen response hallmarks. The edge biomarkers for the early estrogen response subtype were also identified in the CCN; NEAT1 had high average degree among the early response cells, and ceRNA survival analysis indicated that NEAT1 and its ceRNAs could predict patient survival. Moreover, we inferred the function of another hub lncRNA, DLEU2, which might be involved in GPCR signaling, based on both Gene Ontology (GO) and REACTOME pathways. In summary, we established a novel method to construct a CCN and provide single-cell network-related insights into estrogen regulation in breast cancer.
We downloaded the scRNA-seq data from Gene Expression Omnibus (GEO) (accession number: GSE107858). Following the filtering process described in the paper (3), we performed further analysis on 84 MCF-7 cells. RNAs with fragments per kilo base of transcript per million reads mapped (FPKM) >1 in at least 25% (84 × 0.25 = 21) of the cells were used for further analysis. The filtering parameter is referred to the paper (9). The dataset GSE107863 for T47D was an independent validation cohort to support the findings obtained using MCF-7 cells.
The ceRNA network was downloaded (10) (http://starbase.sysu.edu.cn/) using the Web API. The ceRNAs for all mRNAs, lncRNAs, and pseudogenes were downloaded using default parameters. The ceRNA network contained 308,266 ceRNA pairs composed of 18,942 RNAs. The complete information is presented in Table S1.
We obtained the known cancer-related genes from the Cancer Gene Census (CGC) database (http://cancer.sanger.ac.uk/cosmic/census), which contains 576 genes (11). Other 876 cancer-related genes were also downloaded from the Genetic Association Database (GAD) database (http://geneticassociationdb.nih.gov/).
Functional gene sets “HALLMARK_ESTROGEN_RESPONSE_EARLY” and “HALLMARK_ESTROGEN_RESPONSE_LATE” were downloaded and extracted from the hallmark gene sets of MSigDB (https://www.gsea-msigdb.org/gsea/msigdb/, v7.2). REACTOME pathways and biological processes information of GO was also downloaded from MSigDB.
We downloaded the transcript annotation from Ensembl and obtained 215,307 annotations. The transcript ID, transcript type, and HUGO Gene Nomenclature Committee (HGCN) symbols were downloaded from Ensembl. Further, the annotations whose transcript type belonged to “lincRNA” or “antisense” were extracted as the lncRNAs. We obtained a total of 1,794 lncRNAs. In addition, we also downloaded the lncRNA annotation file lncipedia_5_0_hg19.gtf (full database) from LNCipedia (12). Considering that some lncRNAs had alternative names, we extracted the Ensembl ID, gene_alias and gene_id for each lncRNA. The lncRNA information from either Ensembl or LNCipedia was used to annotate the lncNRAs.
ER is the most important hormone receptor in breast cancer. We also screened the differentially expressed genes (DEGs) between ER-positive and ER-negative patients from public cohorts and denoted as ER_DEGs markers. The raw read counts for breast cancer was downloaded from The Cancer Genome Atlas (TCGA). The R package DESeq2 (13) was used for differential analysis. 22,946 DEGs were identified with adjusted p <0.05. The Z-score scaled expression profile of Molecular Taxonomy of Breast Cancer International Consortium (METABRIC) was also downloaded. T-test was used as the statistical method to calculate the p value of gene expression difference between ER+ vs ER− samples. The p value was then adjusted by fdr method using the R package fdrtool (14). As a result, we obtained 2,951 DEGs with fdr adjusted p <0.05.
The SSN method was developed by Liu et al. to construct a personalized network for individual patients based on their expression profiles (8). Briefly, a reference network can be constructed using the Pearson correlation coefficient (PCC) between molecules based on the expression data of the reference samples. After a new sample is added to the reference samples, the perturbed network can be similarly constructed. Then, the differential network is constructed by the edges with significantly changed correlation between the reference and perturbed networks.
The changed correlation follows a new type of distribution, which is called the “volcano distribution”. The tail areas of this distribution are similar to those of the normal distribution based on the Kolmogorov–Smirnov test with random sampling. Therefore, the statistical hypothesis Z-test was used to evaluate the significance level of the changed correlation because of the central limit theorem (15).
Liu et al. selected 8–17 normal samples as reference samples to construct an SSN. They also ensured that the SSN is robust and stable for the different reference sample sizes. Inspired by the SSN method, 20 MCF-7 cells captured at 0 h were selected as the reference cells in this study (Figure 1A). The reference network was constructed using the PCC. The RNA–RNA correlation was deemed significant with a p-value <0.05. For cells at 3, 6, or 12 h, we added one cell to the reference cells and recalculated the RNA–RNA correlation (Figure 1B). We retained a correlation network, named the perturbed network, containing significant RNA–RNA relationships with a p-value <0.05.
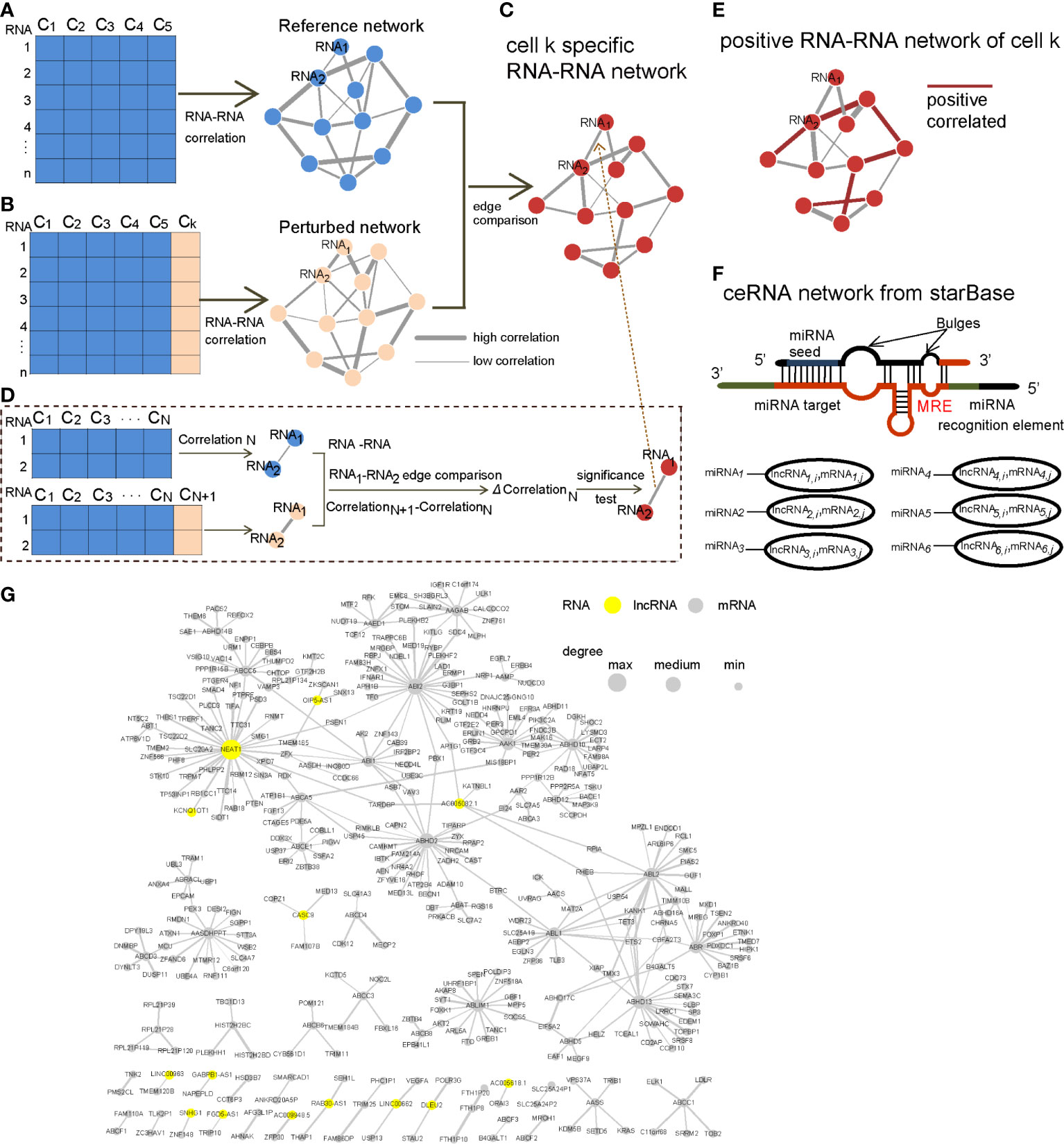
Figure 1 Workflow of constructing the CCN. (A) Reference cells were selected, and the corresponding RCN was constructed and referred to as “Reference network”. (B) One cell k was added to the reference cells, and the corresponding RCN was constructed and referred to as the “Perturbed network”. (C) Edges were compared between the “Reference network” and “Perturbed network” to obtain a cell k-specific RNA–RNA network. (D) One edge composing of two RNAs was compared and tested for its significance level, based on the differential correlation (ΔCorrelationN). (E) Only the positive edges in the cell k-specific RNA–RNA network were candidate edges in the CCN. (F) We downloaded the ceRNA network from starBase. (G) An example of a CCN. The yellow nodes represent lncRNAs, and node size is proportional to its degree in the CCN.
Next, we compared the significant RNA–RNA interactions in the perturbed network and reference network to keep only the edges with significantly changed correlations (Figure 1C). Figure 1D shows how to test the significance of the changed correlation (ΔCorrelationN) between a pair of RNAs. According to the SSN theory proposed by Liu et al. (8), the differential correlation followed a normal distribution, and the significance could be evaluated based on the Z-test.
where N is the number of reference cells.
From the expression perspective, the ceRNAs were positively correlated. Therefore, we considered only the positive and significant differential RNA–RNA interactions as candidate ceRNAs (Figure 1E). The ceRNA network from StarBase was further used to filter the ceRNA network to ensure its biological importance (Figure 1F).
The hypergeometric test was used to evaluate whether the genes in the CCN were significantly enriched in functional gene sets.
where M is the total number of genes in the background network, N is the number of genes in a functional gene set, m is the number of genes in the CCN, and n is the number of CCN genes shared by the functional gene set.
The R package igraph was used to calculate the topological features of RNAs in the CCN (R 4.0.2). The degree of RNA is the number of direct neighbors in the ceRNA network. RNAs with a high degree can be termed as hub RNAs, which play a pivotal role in maintaining the ceRNA–ceRNA relationships within CCN. The betweenness of RNA i can be calculated with the formula
where RNA s and t are RNAs in the CCN different from RNA i, dst represents the number of the shortest paths from s to t, and δst(i) is the number of the shortest paths from s to t that i lies on. For RNA s and t, the ratio is the proportion of the shortest path that RNA i lies on. The sum of the ratios of all RNA pairs is the betweenness centrality of RNA i. The closeness coefficient is the average closeness of RNA i to other RNAs in the network. It is calculated as
where dsi represents the distance between RNA i and other RNAs.
A recently published paper (7) has provided a web tool, “ceRNA survival”, to perform survival analysis for ceRNA composed of lncRNA–miRNA–mRNA, based on The Cancer Genome Atlas (TCGA) datasets. The web tool was used to perform multivariate Cox regression analysis based on miRNA, mRNA, and lncRNA expression, without co-factors.
GSEA (16) was performed using Preranked utility implemented in the standalone version of the GSEA software (v 4.1.0). The RNA sequencing dataset of DLEU2 knockdown was downloaded from the GEO database (https://www.ncbi.nlm.nih.gov/geo/), with accession number GSE162677. We ranked the genes according to the fold change in expression (FPKM.siDLEU2/FPKM.siNC). The fold change was log2 transformed before GSEA.
The CCN was constructed following the workflow shown in Figure 1 (more details in Materials and Methods). Briefly, the edges with significant differential correlation between the reference network (Figure 1A) and perturbed network (Figure 1B) were used to construct a cell-specific RCN (Figure 1C). miRNA targets are negatively regulated by miRNAs. RNAs competitively bind to the same miRNAs as ceRNAs. Chen et al. generated a ceRNA network for each subtype of breast cancer, based on the principle of positive co-expression and shared miRNAs (17). Therefore, the positive RCN appeared to be a candidate ceRNA, based on ceRNA theory (Figure 1E). The ceRNA network from starBase was further used to reduce false-positive ceRNA relations (Figure 1F). As an example, we have demonstrated the ceRNA network for the cell MCF-7 12 h (RHM266) in Figure 1G.
ESR1 plays an important role in breast cancer. Therefore, we first examined its interactors in the cell-specific RCN, which is schematically shown in Figure 1E. We selected cells at 3, 6, and 12 h, in which ESR1 had a degree larger than 25. The known estrogen-regulated genes (ERGs), such as KLF4 (Figures 2A–F) and TSKU (Figures 2A, C), were significantly positively correlated with ESR1. Akaogi et al. reported high expression of KLF4 in ER-α-positive patients. KLF4 was found to bind to the DNA-binding region of ER-α and inhibit the binding of ER-α to estrogen response elements in the promoter regions (18). Known cancer-related genes from CGC, such as MYD88 (Figures 2A, D), DDX10 (Figures 2B, F), MLLT6 (Figure 2D), BCL10 (Figure 2F), and KAT6A (Figure 2F) also interacted with ESR1. The expression of MYD88 could be modulated in a single nucleotide polymorphism (SNP)- and estrogen-dependent fashion (19). Breast cancer-related genes from GAD were also identified as ESR1 interactors, including TP53BP1 (Figure 2B), SRA1 (Figure 2D), and BBS4 (Figure 2E). Low expression of TP53BP1 is associated with increased local recurrence in breast cancer patients treated with conserving surgery and radiotherapy (20). In addition to these protein-coding genes, ESR1 was also co-expressed with non-coding RNAs such as MIR302B (Figure 2C) and MIR4426 (Figure 2E). LncRNAs MIR181A1HG (Figure 2A), ATP1A1OS (Figure 2B), and LINC00094 (Figure 2D) were also shown to cross-talk with ESR1. ER_DEGs frequently interacted with ESR1 within individual cells (Figures 2A–F). These results indicate that the cell-specific RCN reflect the genes’ regulations of breast cancer.
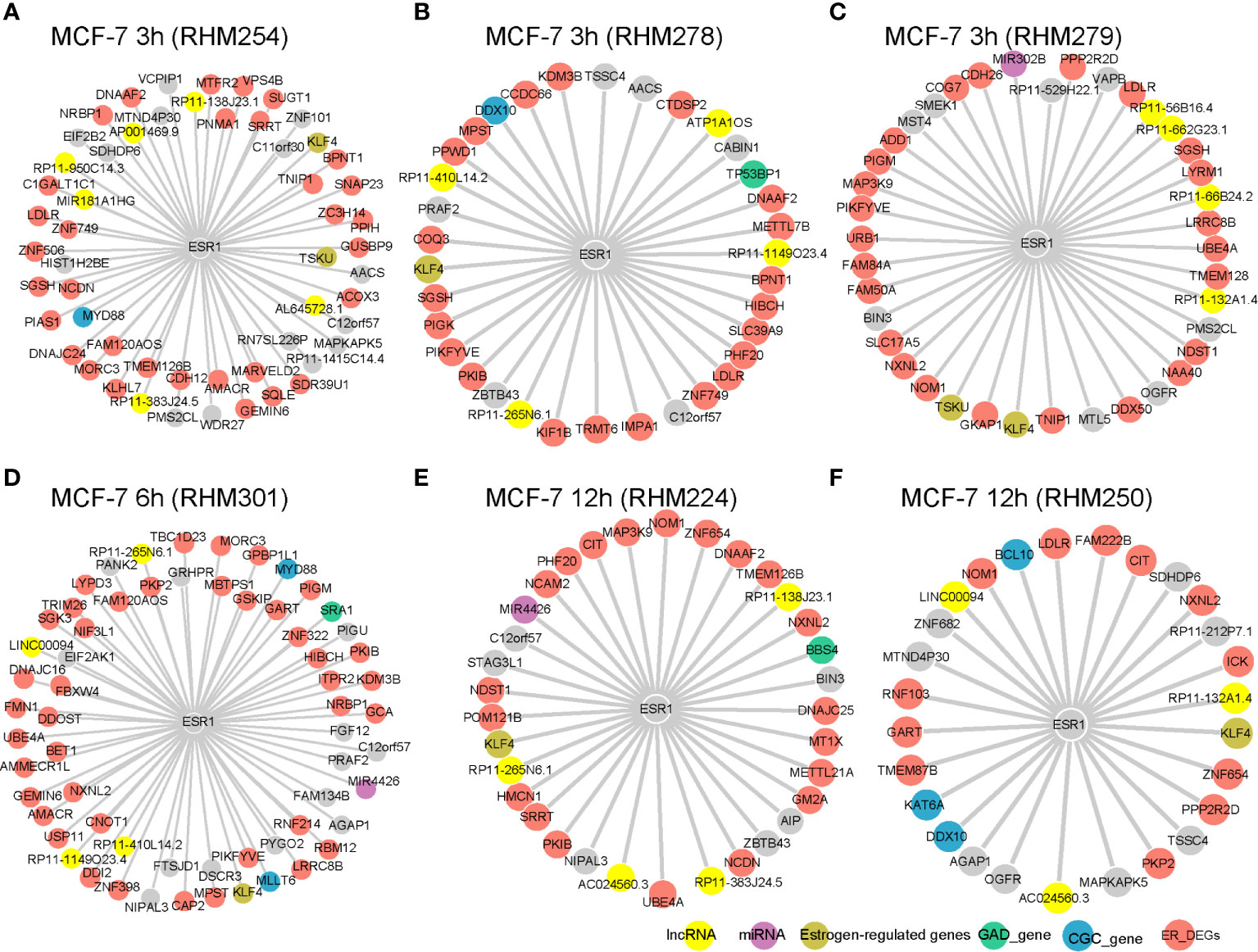
Figure 2 ESR1 interactors in cell-specific RCN. The yellow nodes represent lncRNAs, purple nodes represent miRNAs, green nodes represent GAD genes, blue nodes represent CGC genes, khaki nodes represent estrogen regulated genes, salmon nodes represent DEGs between ER+ vs ER− patients from either TCGA or METABRIC, and gray nodes represent genes with an unknown “biological” label. The RCN is shown for the (A) MCF-7 3 h (RHM254), (B) MCF-7 3 h (RHM278), (C) MCF-7 3 h (RHM279), (D) MCF-7 6 h (RHM301), (E) MCF-7 12 h (RHM224), and (F) MCF-7 12 h (RHM250) cells.
We also constructed a cell-specific RCN based on the expression profiles of the T47D dataset. ESR1 was also co-expressed with known marker genes, including known ERGs, cancer-related genes from CGC, ER-DEGs, and breast cancer-related genes from GAD (Figure S1).
The CCN was then constructed by integrating the RCN with the starBase ceRNA database, which was developed based on CLIP-Seq data. The average numbers of edges and lncRNAs in the CCN are shown in Table 1. The average number of edges decreased after E2 stimulation, from 882 edges (3 h) to 662 edges (12 h). Meanwhile, the average number of lncRNAs decreased from 16 (3 h) to 14 (12 h). In contrast, there were more than 1,600 edges on average in the CCN from the T47D dataset. However, on average, five lncRNAs were involved in the CCN.

Table 1 Average number of edges and lncRNAs in CCN.
Cell type classification assumes high importance in single cell heterogeneity. Therefore, we defined cell subtypes by integrating CCN and the estrogen response hallmark. We retrieved estrogen early response and late response hallmarks from MSigDB. For each CCN, we extracted all the RNAs and performed a hypergeometric test to evaluate the extent of RNA enrichment occurring in these hallmark stages.
We selected one CCN at each time point as an example. The CCN was significantly enriched in the early response hallmark for MCF-7 3 h (RHM223, Figure 3A, p = 0.0002), MCF-7 6 h (RHM300, Figure 3B, p = 6.61E-8), and MCF-7 12 h (RHM265, Figure 3C, p = 3.69E-5). Among all 64 cells (3, 6, and 12 h), 41 were enriched in the estrogen early response hallmark (Figure 3D, p < 0.05). Three out of 64 cells were enriched in the estrogen late response hallmark (Figure S2). Similarly, the cells T47D 3 h (T47D_3 h_2B6), T47D 6 h (T47D_6 h_2H8), and T47D 12 h (T47D_12 h_D8) were enriched in the early response hallmark (Figures S3A–C). In the T47D dataset, seven cells were classified as early response cells (Figure S3D).
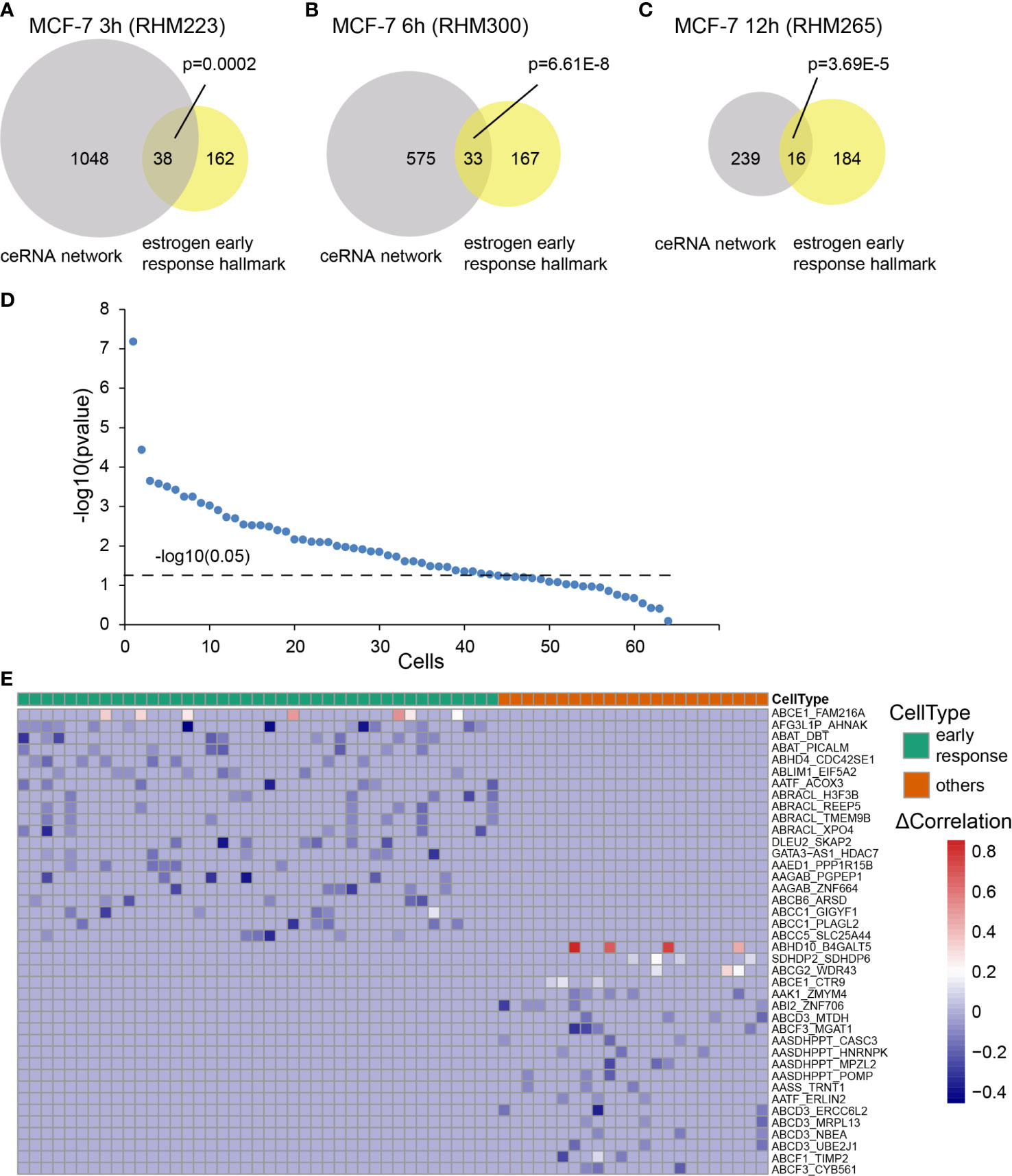
Figure 3 Cell subtypes inferred by the CCN. The RNAs in the CCN were enriched into estrogen early response hallmark, as determined by a hypergeometric test. We showed significant enrichment of RNAs in the CCN of the (A) MCF-7 3 h (RHM223), (B) MCF-7 6 h (RHM300), and (C) MCF-7 12 h (RHM265) cells to the early response hallmarks. (D) The minus log10 transformed p-value calculated by a hypergeometirc test for all cells at 3, 6, and 12 h. The dashed line represents the significance threshold, p = 0.05. (E) The heatmap of differential correlation (ΔCorrelation) in all cells that were classified into two subtypes: early response cells vs. others. Blue represents loss of correlation in the “Perturbed network”, while red refers to the gain of correlation in the “Perturbed network”.
We further classified all 64 cells into two subtypes: early response ones and others. Traditionally, the nodes (RNAs in the network) were screened for biomarker identification. As a complex disease, breast cancer is induced by a set of dysregulated and synergetic genes rather than a single gene. Therefore, network biomarkers are more advantageous for characterizing disease states. Here, we explored the edge markers of both the cell subtypes. We selected the top 20 ceRNA–ceRNA relationships that only appeared in one cell subtype. From the heatmap, we could clearly distinguish the early response subtype from the other subtype (Figure 3E). A similar result is shown in Figure S3E for the T47 dataset.
Topological characterization of the CCN is crucial for identifying the pivotal genes that substantially contribute to gene regulation upon E2 stimulus. For all the CCNs, we analyzed the topological features, including degree, betweenness, and closeness coefficients. We focused on lncRNAs in the CCN with a high degree in the early response subtype. The top five lncRNAs are listed in Table 2. The average degree for all lncRNAs in the early response subtype is in Table S3.

Table 2 LncRNAs with top degree in early response subtype.
The role of NEAT1 in breast cancer has been widely investigated. It is also a hub lncRNA in the CCN of early response cells with an average degree as high as 5.14. Cells with a degree ≥20 for NEAT1were selected. SMAD4 and NF1, the known cancer-related genes in the CGC database, are the common ceRNAs of NEAT1 in all six CCNs (Figures 4A–F). Another CGC gene, PTEN, appears in five of these six ceRNA networks. WWTR1, a CGC gene, is the ceRNA of NEAT1 in cell MCF-7 3 h (RHM254, Figure 4A) and MCF-7 6 h (RHM271, Figure 4B). The GAD genes PRKCA, PRLR, and POLK function as ceRNAs of NEAT1 in cell MCF-7 3 h (RHM254, Figure 4A), MCF-7 6 h (RHM271, Figure 4B), and MCF-7 12 h (RHM265, Figure 4C), respectively. For cell MCF-7 3 h (RHM255, Figure 4D), we identified the ERGs XRCC1 and RAPGEFL1 as ceRNAs of NEAT1. For cell MCF-7 6 h (RHM271), ERGs RAPGEFL1 and SLC7A2 are ceRNAs of NEAT1. TET2, a CGC gene, is the ceRNA of NEAT1 in cell MCF-7 6 h (RHM250, Figure 4E).
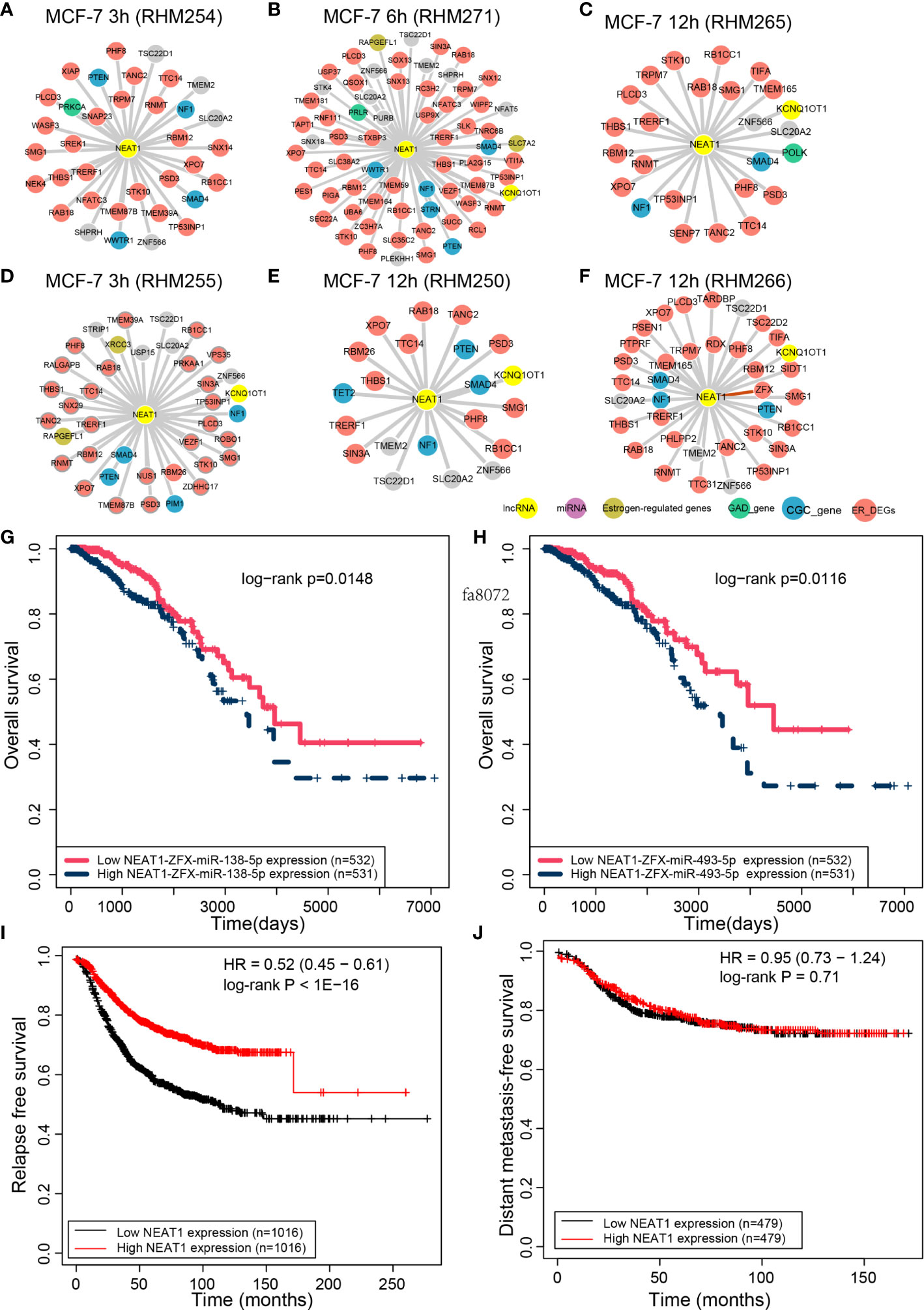
Figure 4 NEAT1 interactors in the CCN. The interactions between NEAT1 and its ceRNAs in the (A) MCF-7 3 h (RHM254), (B) MCF-7 6 h (RHM271), (C) MCF-7 12 h (RHM265), (D) MCF-7 3 h (RHM255), (E) MCF-7 12 h (RHM250), and (F) MCF-7 12 h (RHM266) cells. The survival analysis of NEAT1 and its ceRNA ZFX binding to (G) miR-138-5p and (H) miR-493-5p. The relapse-free (I) and metastasis-free survival analysis (J) performed by Kaplan–Meier Plotter for NEAT1.
Among NEAT1 ceRNAs in the six CCNs, we noted that the ceRNA–ceRNA relationship of NEAT1 and ZFX in MCF-7 12 h (RHM266, Figure 4F) has been recently validated (21). According to a previous study (21), NEAT1 and ZFX competitively bind to miR-138-5p. Next, we performed multivariate Cox regression analysis for the NEAT1–ZFX–miR-138-5p regulation axis using the “ceRNA survival” tool of LnCeCell (7). We divided all breast cancer patients from TCGA into two groups, based on the median expression value of NEAT1–ZFX–miR-138-5p. Patients with high NEAT1–ZFX–miR-138-5p expression had worse overall survival than those with low expression (Figure 4G). Moreover, we curated from starBase that miR-493-5p and miR-513a-5p are significantly shared by NEAT1 and ZFX. Because the expression of miR-513a-5p is not available in the TCGA dataset of breast cancer, we tested the prognostic potential of NEAT1–ZFX–miR-493-5p. As shown in Figure 4H, the NEAT1–ZFX–miR-493-5p axis was also an unfavorable prognostic marker of breast cancer.
As NEAT1 is one of the hubs in CCN, we further used the web tool Kaplan–Meier Plotter (https://kmplot.com/analysis) to perform relapse-free and metastasis-free survival analysis for NEAT1. Three probes from the microarrays were mapped to NEAT1. The mean expression of the probes was used for the survival analysis of NEAT1. High and low NEAT1 expression levels were divided according to the median expression level. As shown in Figure 4I, NEAT1 was a prognostic marker for breast cancer, based on relapse-free survival analysis. However, NEAT1 was not a predictor of metastasis-free survival in breast cancer (Figure 4J).
We also constructed CCNs based on the T47D dataset. NEAT1 is not a hub lncRNA in the CCNs of T47D cells. The top five lncRNAs in the early response cells are shown in Table S2 for the T47D dataset. Topping the list is PITPNA-AS1. But there is no available siLncRNA dataset for PITPNA-AS1. Therefore, we focus on MALAT1, which has the second largest degree. MALAT1 has been widely investigated for its role in breast cancer. Cells with a degree ≥10 for MALAT1 were selected (Figures S4A–C). MALAT1 interacts with known ERGs, cancer-related genes from CGC, ER-DEGs, and breast cancer-related genes from GAD, which is consistent with the results from the MCF-7 dataset. The ceRNA–ceRNA relationships of MALAT1–ZFP36L2, MALAT1–PGRMC2, and MALAT1–PDS5B were shared among the three cells (Figures S4A–C). The ceRNA survival analysis revealed that they were unfavorable prognostic markers for breast cancer (Figures S4D–F). Survival analysis of the hub lncRNA MALAT1 demonstrated that MALAT1 was a prognostic marker of relapse-free survival (Figure S4G) but not metastasis-free survival (Figure S4H).
Function prediction and interpretation of lncRNAs are important factors to dissect their biological mechanisms. Therefore, we tried to infer the function of the hub lncRNA DLEU2, which has not been characterized well in breast cancer.
The silencing or overexpression is a commonly used measure of lncRNA function inference. We searched the GEO database for RNA-sequencing datasets generated by siLncRNA or overexpression of DLEU2. As a result, we found that the dataset for siDLEU2 (GSE162677) matched our criteria.
Differential expression analysis is a commonly used method to explore the function of lncRNAs, especially for in silico experiments. The significantly affected biological functions associated with DLEU2 expression could be theoretically identified based on functional enrichment of the genes affected by DLEU2. However, the dataset GSE162677 is generated from a cervical cell line. Thus, it is not suitable for the functional interpretation of DLEU2 in estrogen regulation in breast cancer.
In MCF-7 cells at 12 h (RHM227), DLEU2 had the highest number of ceRNAs. GSEA was used to enrich RNAs in the CCN. The RNAs in RHM227 were significantly enriched in the up-regulated genes after siDLEU2 (Figure 5A), which indicates that the genes in the CCN may have similar expression changes after siDLEU2 in breast cancer. Therefore, we further predicted DLEU2 function based on the genes in the CCN and used the hypergeometric test to explore the function of DLEU2 by functional enrichment of RNAs in the CCN of RHM227. The functional terms of the GO and REACTOME pathways were downloaded from MSigDB (v7.2). The top 10 most significant biological processes from GO and pathways from REACTOME are shown in Figures 5B, C, respectively. The most significant GO and REACTOME pathway was GPCR signaling. It should be noted that the biological process “ION_TRANSPORT” was also significantly enriched by RNAs in the CCN of RHM227.
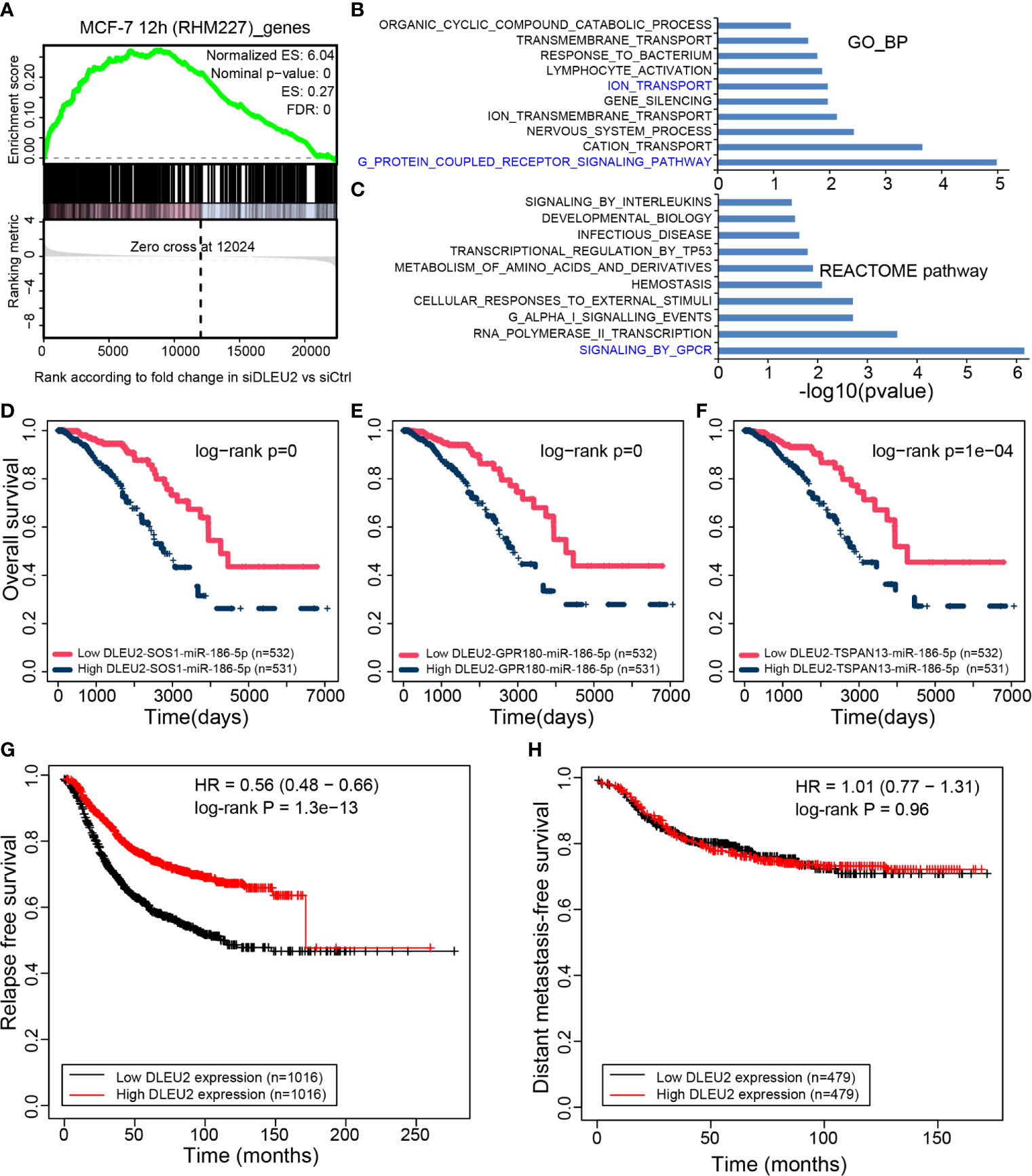
Figure 5 Function inference of DLEU2 via the CCN. (A) GSEA of RNAs in MCF-7 12 h (RHM227) cell to RNAs affected by siDLEU2. The top 10 functional terms from the (B) biological process of GO (GO_BP) and (C) REACTOME pathways enriched by RNAs in the CCN of MCF-7 12 h (RHM227) cell, as determined by the hypergeometric test. The interested terms are colored in blue. DLEU2 and its ceRNAs (D) SOS1, (E) GPR180, and (F) TSPAN13 have prognostic potential for breast cancer. The relapse-free (G) and metastasis-free survival analysis (H) performed by Kaplan–Meier Plotter for DLEU2.
Next, we focused on the ceRNAs of DLEU2 in the CCN of RHM227. SOS1 is involved in GPCR signaling from both GO and REACTOME, while GPR180 participates in GPCR signaling from GO. TSPAN13, a ceRNA of DLEU2, is also a known marker of late estrogen response. It synchronizes with other genes to facilitate ion transport. We retrieved the miRNAs shared by DLEU2, SOS1, GPR180, and TSPAN13. Multivariate Cox regression analysis of ceRNAs demonstrated their prognostic potential in breast cancer (Figures 5D–F). Furthermore, relapse-free and metastasis-free survival analysis using Kaplan–Meier Plotter (https://kmplot.com/analysis) revealed that DLEU2 can predict relapse-free survival (Figure 5G) but not metastasis-free survival (Figure 5H).
To validate the possibility of inferring lncRNA functions via the CCN, we also predicted the function of MALAT1, based on the CCN of T47D cells. The dataset GSE110239 is an mRNA profile generalized by RNA-sequencing for the mammary tumor mouse model PyMT. Mouse genes were mapped to human gene symbols using the R package biomaRt. The genes in the cell T47D_6 h_2C12 was significantly enriched in the up-regulated genes after MALAT1 KO (Figure S5A). Further functional analysis of RNAs in the CCN showed that they were enriched in the pathway of “fatty acid metabolism” (p = 0.015). The “fatty acid metabolism” has also been reported to be enriched by DEGs between BPA exposure and control in mouse liver. Several DEGs were key drivers, such as Apoa2, Akr1c12, and Malat1 (22). This provides additional support for the function inference of lncRNAs via the CCN.
The scRNA-seq technique has become a powerful tool for the elucidation of intra-tumor and intra-cell line heterogeneity in breast cancer (2, 23). Estrogen regulation generally involves not only individual molecules but also molecular networks. Therefore, identifying the CCN upon E2 stimulus is crucial to elucidate the cellular heterogeneity of estrogen regulation at the system level. The CCN is directly constructed based on the single-cell gene expression profile to avoid the bias caused by subjective cluster information. Moreover, Dai et al. demonstrated that gene associations, rather than gene expression, can stably portray biological processes in individual cells (4). We merged ceRNA relations with a cell-specific RCN, which reduced the false-positive RNA–RNA associations.
ESR1 is a pivotal regulator of breast cancer. From the cell-specific RCN, we found that ESR1 interacted with known ERGs, such as KLF4 and TSKU. Other known cancer-related genes from CGC and GAD were also significantly correlated with ESR1. In addition to these protein-coding genes, miRNAs (for example, MIR302B and MIR4426) and lncRNAs (for example, MIR181A1HG, ATP1A1OS, and LINC00094) were predicted to interact with ESR1. MiR-302 (including miR-302b) sensitizes MCF-7 cells to adriamycin and mitoxantrone (24, 25). LINC00094 has been reported as a super-enhancer-associated ce-lncRNA that promotes cell growth in esophageal squamous cell carcinoma (26). It should be noted that GSE107858 only performed polyA RNA sequencing, without miRNA or lncRNA sequencing. The miRNAs or lncRNAs in the expression profile come from the process of mapping reads to the reference genome.
Heterogeneous cell subtypes upon E2 stimulus were inferred by enriching RNAs in CCN to the estrogen response hallmark. The results showed that 68.7% (44/64) of the cells were responsive to E2 stimulation, and 93.2% (41/44) of them were early response cells. We then classified the cells into two subtypes: early response cells and the remaining cells. The correlation differences of the top 20 edges are shown for each subtype in Figure 3E. Among these edge markers, some gene components are not differentially expressed along the time series, which means that they cannot be identified by traditional differential analysis based on gene expression. Regarding the edges of GATA3-AS1 and HDAC7, neither is a differentially expressed gene. However, the edge of GATA3−AS1 and HDAC7 had a significant correlation difference in several early response cells. GATA3–AS1 has been reported to be involved in triple-negative breast cancer progression and immune escape by stabilizing the PD-L1 protein and degrading the GATA3 protein (27). In contrast, the edge of AATF and ERLIN2 showed no correlation difference in early response cells but had a correlation difference in other cells. Although AATF and ERLIN2 are not DEGs, AATF silencing may be utilized to evoke apoptosis and regulate the expression of ERs in MCF-7 cells (28). ERLIN2 has been reported to promote cell survival by regulating endoplasmic reticulum stress in breast cancer. Moreover, its regulation by miR-410 is ER-dependent (29).
LncRNA-associated ceRNAs have been investigated in breast cancer. To explore such key lncRNAs and their ceRNAs, topological features such as degree were utilized to identify lncRNAs that function as hub nodes in the CCN. NEAT1 is the top hub gene observed in the early response subtype, indicating its pivotal role in estrogen regulation. The ceRNAs contain ERGs or cancer-related genes. Intriguingly, NEAT1 and its ceRNAs can also serve as prognostic markers for breast cancer, which further reveals that the constructed CCN has potential clinical applications.
CCN was used to predict lncRNA function. The RNAs in the CCN of one T47D cell were significantly enriched in the up-regulated genes after MALAT1 KO (Figure S5A). Functional enrichment results implied that RNAs in the CCN participated in the pathway of “fatty acid metabolism”, which has also been reported to be associated with BPA exposure, mainly driven by RNAs including MALAT1 (22). This provides evidence for the functional inference of lncRNA via the CCN. We noticed that the hub lncRNA DLEU2 had not been functionally characterized well in breast cancer. Therefore, the public siRNA datasets of DLEU2 were selected to infer the function of DLEU2. The GSEA results (Figure 5A) indicate the feasibility of the functional interpretation of lncRNAs via RNAs in the CCN. Functional terms from GO and REACTOME both demonstrated that DLEU2 is involved in GPCR signaling. In addition, the ceRNAs of DLEU2 can also predict patient survival in breast cancer. These results can facilitate the speculation of the biological functions of hub lncRNAs, which have not been characterized.
The current CCN method had several limitations. We used the gene expression profile in FPKM, which was biased when comparing gene expression among samples. We also did not consider the impact of inter-sample normalization on our results. In this study, we considered only the positive and significantly differential RNA–RNA interactions as candidate ceRNAs, in view of direct miRNA targets. Negative correlations are also important because they might be translated as indirect targets of miRNAs sponged by particular ceRNAs. Therefore, the anti-correlated and significantly differential RNA–RNA interactions were added to the candidate ceRNAs. As a result, the anti-correlation will increase the size of cell-specific RCN (Figure S6 and Table S4) and CCN (Figure S7 and Table S4), but do not consequentially increase the significance of CCN enrichment in estrogen early response hallmarks (Figure S8). Moreover, it does not affect the function inference of lncRNAs via CCN (Figure S7).
To conclude, we proposed a novel strategy for constructing a CCN by integrating reference cell-based cell-specific RCNs and public ceRNA networks. This CCN provides new insights into the inference of cell subtypes by incorporating functional gene set information. Hub lncRNAs in the early response subtype and their ceRNAs could be potential prognostic markers for overall survival and relapse-free survival. This CCN also provides a new perspective to infer the functions of uncharacterized hub lncRNAs, which are potential targets for RNA-based therapeutics.
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
XC and JX conceived the study. YL supervised the project. FZ, CY, and HZ performed the computational analysis. XC and JX drafted the manuscript. WS, TY, YL, and HZ revised the manuscript. All authors contributed to the article and approved the submitted version.
This work was financially supported in part by grants from the National Natural Science Foundation of China [Grant No. 62003094 and 82003615] and Natural Science Foundation of Guangdong Province [Grant No. 2018A0303130080 and 2019A1515011377].
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors are grateful to Haoteng Feng and Xiaoli Wu for their discussions.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2021.656675/full#supplementary-material
ESR1, estrogen receptor alpha; RCN, RNA–RNA co-expression network; ceRNAs, competing endogenous RNAs; CCN, cell-specific ceRNA network; scRNA-seq, single-cell RNA sequencing; lncRNAs, long non-coding RNAs; E2, 17β-estradiol; ERGs, estrogen regulated genes; GO, Gene Ontology; ER, estrogen receptor; SSN, sample-specific networks; CGC, Cancer Gene Census; GAD, Genetic Association Database; METABRIC, Molecular Taxonomy of Breast Cancer International Consortium; TCGA, The Cancer Genome Atlas; DEGs, differentially expressed genes; GEO, Gene Expression Omnibus; FPKM, fragments per kilo base of transcript per million reads mapped.
1. DeSantis CE, Ma J, Gaudet MM, Newman LA, Miller KD, Goding Sauer A, et al. Breast Cancer Statistic. CA Cancer J Clin (2019) 69(6):438–51. doi: 10.3322/caac.21583
2. Chen F, Ding K, Priedigkeit N, Elangovan A, Levine KM, Carleton N, et al. Single-Cell Transcriptomic Heterogeneity in Invasive Ductal and Lobular Breast Cancer Cells. Cancer Res (2020) 81(2):268–81. doi: 10.1158/0008-5472.CAN-20-0696
3. Zhu D, Zhao Z, Cui G, Chang S, Hu L, See YX, et al. Single-Cell Transcriptome Analysis Reveals Estrogen Signaling Coordinately Augments One-Carbon, Polyamine, and Purine Synthesis in Breast Cancer. Cell Rep (2018) 25(8):2285–98.e2284. doi: 10.1016/j.celrep.2018.10.093
4. Dai H, Li L, Zeng T, Chen L. Cell-Specific Network Constructed by Single-Cell RNA Sequencing Data. Nucleic Acids Res (2019) 47(11):e62. doi: 10.1093/nar/gkz172
5. Jiang X, Zhou Y, Sun A J, Xue JL. NEAT1 Contributes to Breast Cancer Progression Through Modulating miR-448 and ZEB1. J Cell Physiol (2018) 233(11):8558–66. doi: 10.1002/jcp.26470
6. Kwok ZH, Roche V, Chew XH, Fadieieva A, Tay Y. A non-Canonical Tumor Suppressive Role for the Long non-Coding RNA MALAT1 in Colon and Breast Cancers. Int J Cancer (2018) 143(3):668–78. doi: 10.1002/ijc.31386
7. Wang P, Guo Q, Hao Y, Liu Q, Gao Y, Zhi H, et al. LnCeCell: A Comprehensive Database of Predicted lncRNA-associated ceRNA Networks At Single-Cell Resolution. Nucleic Acids Res (2020a) 49(D1):D125–33. doi: 10.1093/nar/gkaa1017
8. Liu X, Wang Y, Ji H, Aihara K, Chen L. Personalized Characterization of Diseases Using Sample-Specific Networks. Nucleic Acids Res (2016) 44(22):e164. doi: 10.1093/nar/gkw772
9. Chen X, Hu L, Wang Y, Sun W, Yang C. Single Cell Gene Co-Expression Network Reveals Fech/Crot Signature as a Prognostic Marker. Cells (2019) 8(7):698. doi: 10.3390/cells8070698
10. Li JH, Liu S, Zhou H, Qu L H, Yang JH. starBase v2.0: Decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA Interaction Networks From Large-Scale CLIP-Seq Data. Nucleic Acids Res (2014) 42(Database issue):D92–7. doi: 10.1093/nar/gkt1248
11. Sondka Z, Bamford S, Cole CG, Ward SA, Dunham I, Forbes SA. The COSMIC Cancer Gene Census: Describing Genetic Dysfunction Across All Human Cancers. Nat Rev Cancer (2018) 18(11):696–705. doi: 10.1038/s41568-018-0060-1
12. Volders PJ, Anckaert J, Verheggen K, Nuytens J, Martens L, Mestdagh P, et al. Lncipedia 5: Towards a Reference Set of Human Long non-Coding RNAs. Nucleic Acids Res (2019) 47(D1):D135–9. doi: 10.1093/nar/gky1031
13. Love MI, Huber W, Anders S. Moderated Estimation of Fold Change and Dispersion for RNA-seq Data With Deseq2. Genome Biol (2014) 15(12):550. doi: 10.1186/s13059-014-0550-8
14. Strimmer K. Fdrtool: A Versatile R Package for Estimating Local and Tail Area-Based False Discovery Rates. Bioinformatics (2008) 24(12):1461–2. doi: 10.1093/bioinformatics/btn209
15. Rice JA. Mathematical Statistics and Data Analysis. Cengage Learning. Duxbury: Duxbury Press (2007).
16. Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene Set Enrichment Analysis: A Knowledge-Based Approach for Interpreting Genome-Wide Expression Profiles. Proc Natl Acad Sci U S A (2005) 102(43):15545–50. doi: 10.1073/pnas.0506580102
17. Chen J, Xu J, Li Y, Zhang J, Chen H, Lu J, et al. Competing Endogenous RNA Network Analysis Identifies Critical Genes Among the Different Breast Cancer Subtypes. Oncotarget (2017) 8(6):10171–84. doi: 10.18632/oncotarget.14361
18. Akaogi K, Nakajima Y, Ito I, Kawasaki S, Oie SH, Murayama A, et al. KLF4 Suppresses Estrogen-Dependent Breast Cancer Growth by Inhibiting the Transcriptional Activity of Eralpha. Oncogene (2009) 28(32):2894–902. doi: 10.1038/onc.2009.151
19. Ho MF, Ingle JN, Bongartz T, Kalari KR, Goss PE, Shepherd LE, et al. Tcl1a Single-Nucleotide Polymorphisms and Estrogen-Mediated Toll-Like Receptor-MYD88-Dependent Nuclear Factor-kappaB Activation: Single-Nucleotide Polymorphism- and Selective Estrogen Receptor Modulator-Dependent Modification of Inflammation and Immune Response. Mol Pharmacol (2017) 92(2):175–84. doi: 10.1124/mol.117.108340
20. Neboori HJ, Haffty BG, Wu H, Yang Q, Aly A, Goyal S, et al. Low p53 Binding Protein 1 (53BP1) Expression is Associated With Increased Local Recurrence in Breast Cancer Patients Treated With Breast-Conserving Surgery and Radiotherapy. Int J Radiat Oncol Biol Phys (2012) 83(5):e677–83. doi: 10.1016/j.ijrobp.2012.01.089
21. Yao L, Chen L, Zhou H, Duan F, Wang L, Zhang Y. Long Noncoding Rna NEAT1 Promotes the Progression of Breast Cancer by Regulating miR-138-5p/ZFX Axis. Cancer Biother Radiopharm (2020). doi: 10.1089/cbr.2019.3515
22. Diamante G, Cely I, Zamora Z, Ding J, Blencowe M, Lang J, et al. Systems Toxicogenomics of Prenatal Low-Dose BPA Exposure on Liver Metabolic Pathways, Gut Microbiota, and Metabolic Health in Mice. Environ Int (2021) 146:106260. doi: 10.1016/j.envint.2020.106260
23. Karaayvaz M, Cristea S, Gillespie SM, Patel AP, Mylvaganam R, Luo CC, et al. Unravelling Subclonal Heterogeneity and Aggressive Disease States in TNBC Through Single-Cell RNA-Seq. Nat Commun (2018) 9(1):3588. doi: 10.1038/s41467-018-06052-0
24. Wang Y, Zhao L, Xiao Q, Jiang L, He M, Bai X, et al. miR-302a/b/c/d Cooperatively Inhibit BCRP Expression to Increase Drug Sensitivity in Breast Cancer Cells. Gynecol Oncol (2016) 141(3):592–601. doi: 10.1016/j.ygyno.2015.11.034
25. Zhao L, Wang Y, Jiang L, He M, Bai X, Yu L, et al. MiR-302a/b/c/d Cooperatively Sensitizes Breast Cancer Cells to Adriamycin Via Suppressing P-glycoprotein(P-gp) by Targeting MAP/ERK Kinase Kinase 1 (MEKK1). J Exp Clin Cancer Res (2016) 35:25. doi: 10.1186/s13046-016-0300-8
26. Wang QY, Peng L, Chen Y, Liao LD, Chen JX, Li M, et al. Characterization of Super-Enhancer-Associated Functional lncRNAs Acting as ceRNAs in ESCC. Mol Oncol (2020b) 14(9):2203–30. doi: 10.1002/1878-0261.12726
27. Zhang M, Wang N, Song P, Fu Y, Ren Y, Li Z, et al. Lncrna GATA3-AS1 Facilitates Tumour Progression and Immune Escape in Triple-Negative Breast Cancer Through Destabilization of GATA3 But Stabilization of PD-L1. Cell Prolif (2020) 53(9):e12855. doi: 10.1111/cpr.12855
28. Sharma M. Apoptosis-Antagonizing Transcription Factor (AATF) Gene Silencing: Role in Induction of Apoptosis and Down-Regulation of Estrogen Receptor in Breast Cancer Cells. Biotechnol Lett (2013) 35(10):1561–70. doi: 10.1007/s10529-013-1257-8
Keywords: cell-specific network, ceRNA, estrogen regulation, lncRNA, subtype
Citation: Chen X, Xu J, Zeng F, Yang C, Sun W, Yu T, Zhang H and Li Y (2021) Inferring Cell Subtypes and LncRNA Function by a Cell-Specific CeRNA Network in Breast Cancer. Front. Oncol. 11:656675. doi: 10.3389/fonc.2021.656675
Received: 21 January 2021; Accepted: 30 March 2021;
Published: 27 April 2021.
Edited by:
Naoyuki Kataoka, The University of Tokyo, JapanReviewed by:
Sandra Romero-Córdoba, National Autonomous University of Mexico, MexicoCopyright © 2021 Chen, Xu, Zeng, Yang, Sun, Yu, Zhang and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yan Li, bGl5YW5AbmptdS5lZHUuY24=
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.