Corrigendum: Exome Sequencing in BRCA1-2 Candidate Familias: The Contribution of Other Cancer Susceptibility Genes
 Gabriella Doddato1,2†Floriana Valentino1,2†Annarita Giliberti1,2Filomena Tiziana Papa1,2Rossella Tita3Lucia Pia Bruno1,2Sara Resciniti1,2Chiara Fallerini1,2
Gabriella Doddato1,2†Floriana Valentino1,2†Annarita Giliberti1,2Filomena Tiziana Papa1,2Rossella Tita3Lucia Pia Bruno1,2Sara Resciniti1,2Chiara Fallerini1,2 Elisa Benetti2
Elisa Benetti2 Maria Palmieri1,2Maria Antonietta Mencarelli3Alessandra Fabbiani1,2,3Mirella Bruttini1,3Alfredo Orrico4Margherita Baldassarri1,2Francesca Fava1,2,3Diego Lopergolo1,2,3Caterina Lo Rizzo3Vittoria Lamacchia1,2,3Sara Mannucci1,2,3
Maria Palmieri1,2Maria Antonietta Mencarelli3Alessandra Fabbiani1,2,3Mirella Bruttini1,3Alfredo Orrico4Margherita Baldassarri1,2Francesca Fava1,2,3Diego Lopergolo1,2,3Caterina Lo Rizzo3Vittoria Lamacchia1,2,3Sara Mannucci1,2,3 Anna Maria Pinto3Aurora Currò1,2,3Virginia Mancini5 Oncologic Multidisciplinary Team Azienda Ospedaliera Universitaria Senese Oncologic Multidisciplinary Team Azienda Usl Toscana Sud Est
Anna Maria Pinto3Aurora Currò1,2,3Virginia Mancini5 Oncologic Multidisciplinary Team Azienda Ospedaliera Universitaria Senese Oncologic Multidisciplinary Team Azienda Usl Toscana Sud Est Francesca Mari1,2,3
Francesca Mari1,2,3 Alessandra Renieri1,2,3
Alessandra Renieri1,2,3 Francesca Ariani1,2,3*
Francesca Ariani1,2,3*- 1Medical Genetics, University of Siena, Siena, Italy
- 2Med Biotech Hub and Competence Center, Department of Medical Biotechnologies, University of Siena, Siena, Italy
- 3Genetica Medica, Azienda Ospedaliera Universitaria Senese, Siena, Italy
- 4Molecular Diagnosis and Characterization of Pathogenic Mechanisms of Rare Genetic Diseases, Azienda Ospedaliera Universitaria Senese and Clinical Genetics, ASL Toscana SudEst. Ospedale della Misericordia, Grosseto, Italy
- 5Unit of Pathology, Department of Medical Biotechnology, University of Siena, Siena, Italy
Hereditary Breast and Ovarian Cancer (HBOC) syndrome is a condition in which the risk of breast and ovarian cancer is higher than in the general population. The prevalent pathogenesis is attributable to inactivating variants of the BRCA1-2 highly penetrant genes, however, other cancer susceptibility genes may also be involved. By Exome Sequencing (ES) we analyzed a series of 200 individuals selected for genetic testing in BRCA1-2 genes according to the updated National Comprehensive Cancer Network (NCCN) guidelines. Analysis by MLPA was performed to detect large BRCA1-2 deletions/duplications. Focusing on BRCA1-2 genes, data analysis identified 11 cases with pathogenic variants (4 in BRCA1 and 7 in BRCA1-2) and 12 with uncertain variants (7 in BRCA1 and 5 in BRCA2). Only one case was found with a large BRCA1 deletion. Exome analysis allowed to characterize pathogenic variants in 21 additional genes: 10 genes more traditionally associated to breast and ovarian cancer (ATM, BRIP1, CDH1, PALB2, PTEN, RAD51C, and TP53) (5% diagnostic yield) and 11 in candidate cancer susceptibility genes (DPYD, ERBB3, ERCC2, MUTYH, NQO2, NTHL1, PARK2, RAD54L, and RNASEL). In conclusion, this study allowed a personalized risk assessment and clinical surveillance in an increased number of HBOC families and to broaden the spectrum of causative variants also to candidate “non-canonical” genes.
Introduction
Breast cancer (BC) represents a priority public health problem being the most common cancer in women (1, 2). Families with a history of multiple BC or ovarian cancer (OC) approximately account for 15% of all patients with BC (3). Hereditary breast and ovarian cancer (HBOC) is an adult-onset condition associated with a high risk of breast and ovarian cancer and an increased risk of other cancers such as prostate cancer, pancreatic cancer, and melanoma (4, 5). Since 1994, when BRCA1 (MIM#113705) and BRCA2 (MIM#600185) were identified, pathogenic variants (PVs) in these two genes have been known to be the most important cause of HBOC (6–8). In the majority of cases, the BRCA1 or BRCA2 PVs are inherited from one of the parents accompanied by a cancer susceptibility transmitted as an autosomal dominant trait. On average, the risk of cancer differs between the two genes, as for BRCA1 the assessed average risk of breast and ovarian cancers ranges from 57 to 65% and from 20 to 50%, respectively, and for BRCA2 the risk ranges from 35 to 57% and from 5 to 23%, respectively (9, 10).
The identification of PVs in BRCA1 or BRCA2 genes has important clinical implications, as, when detected, represents important information to guide patients therapeutic and preventive choices. Interventions, such as risk-reducing bilateral mastectomy and salpingo-oophorectomy or annual breast magnetic resonance imaging (MRI) screening are available to subjects who carry BRCA1 or BRCA2 PVs to enable tumor early detection and active risk reduction (11, 12). The presence of BRCA1-2 PVs can also impact cancer treatment decisions, principally with regard to the employment of platinum agents or poly(ADP-ribose) polymerase (PARP) inhibitors (13).
It is now estimated that more than one-half of individuals who meet the National Comprehensive Cancer Network (NCCN) testing criteria for HBOC carry PVs in genes other than BRCA1 or BRCA2. Most of these genes encode proteins sharing the same homologous recombination DNA repair function with BRCA1-2 ATM, BRIP1, CHEK2, NBN, PALB2, RAD50, RAD51C, and RAD51D (14–22). In addition, PVs in genes involved in the overlapping Fanconi anemia (FA) pathway and in the mismatch repair (MMR) pathway have been also found in BC and OC patients (23). Finally, several other genes beyond BRCA1-2, often with a not yet fully cleared clinical significance, have been found to be mutated and involved in HBOC cases by exome sequencing (ES) studies over the last years (24, 25).
The objective of the current study was to analyze by ES a series of 200 individuals who meet the National Comprehensive Cancer Network (NCCN) testing criteria for HBOC. This study allowed to go further beyond BRCA1-2 genes and identify in additional 21 patients with PVs in canonical (ATM, BRIP1, CDH1, PALB2, PTEN, RAD51C, and TP53) and candidate non-canonical (DPYD, ERBB3, ERCC2, MUTYH, NQO2, NTHL1, PARK2, RAD54L, and RNASEL) HBOC genes.
Materials and Methods
Selection of Patients and DNA Samples’ Preparation
Two hundred patients were selected at the Medical Genetics Unit (A.O.U.S, Siena, Italy) between 2019 and 2020 for Exome Sequencing (ES) according to the updated National Comprehensive Cancer Network (NCCN) guidelines (26). The criteria used to access the test are: breast cancer diagnosed <45 years, breast and ovarian cancer, triple negative breast cancer diagnosed <60 years, male breast cancer diagnosed at any age, bilateral breast cancer with the first diagnosed <50 years, epithelial ovarian cancer diagnosed at any age, exocrine pancreatic cancer diagnosed at any age, and personal history of breast cancer and one close blood relative with breast cancer <50 or ovarian cancer or pancreatic cancer.
Genetic counseling was carried out to evaluate each patient’s personal and familial cancer history considering not only breast and ovarian cancers but also other types of cancer. All patients gave their written informed consent to the study that was carried out according to the Helsinki declaration.
Genomic DNA was extracted from EDTA peripheral blood samples using MagCore HF16 (Diatech Lab Line, Jesi, Ancona, Italy) according to the manufacturer’s instructions. DNA quantity was estimated using the Qubit 3.0 Fluorometer (Thermo Fisher Scientific, Waltham, MA, USA).
Exome Sequencing
Sample preparation was performed following the Nextera Flex for Enrichment manufacturer protocol. The workflow uses a bead-based transposome complex to tagment genomic DNA, which is a process that fragments DNA and then tags the DNA with adapter sequences in one step. After saturation with input DNA, the bead-based transposome complex fragments a set number of DNA molecules. This fragmentation provides flexibility to use a wide DNA input range to generate normalized libraries of consistent tight fragment size distribution. Following tagmentation, a limited-cycle PCR adds adapter sequences to the ends of a DNA fragment. A subsequent target enrichment workflow is then applied. Following pooling, the double stranded DNA libraries are denatured and biotinylated TruSight One Expanded Oligonucleotide probes are hybridized to the denatured library fragments. After hybridization, Streptavidin Magnetic Beads (SMB) then capture the targeted library fragments within the regions of interest. The captured and indexed libraries are eluted from beads and further amplified before sequencing. The exome sequencing analysis was performed on the Illumina NovaSeq6000 System (Illumina San Diego, CA, USA) according to the NovaSeq6000 System Guide. Reads were mapped against the hg19 reference genome by using the Burrow-Wheeler aligner BWA (27). Variant calling was obtained using an in-house pipeline which takes advantage of the GATK Best Practices workflow (28).
ES data were filtered using eVai (enGenome) software. Variants’ prioritization was obtained by using increasingly enlarged filters: i) genes (BRCA1 and BRCA2); ii) phenotype (using HPO terms: Breast and ovarian neoplasms); iii) phenotype (using HPO terms: Neoplasms). In order to find PVs we focused our attention on rare variants (minor allele frequency, MAF <0,01). Frameshift, stopgain, and splice site variants were prioritized as pathogenic. Missense variants were predicted to be damaging by CADD-phred prediction tools (score ≥25) and splice site variants by MaxEntScan tool.
The following public databases were used for the interpretation of the variants: ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/), LOVD (https://databases.lovd.nl/shared/genes), the Human Genome Mutation Database (HGMD, http://www.hgmd.cf.ac.uk/ac/index.php), and BRCA Exchange database (https://brcaexchange.org/).
Sanger Sequencing
Selected variants (VUS and PVs) were confirmed by Sanger sequencing. DNAs were sequenced using the PE Big Dye Terminator Cycle Sequencing Kit on an ABI Prism 3130 analyzer (Applied Biosystems). The data were analyzed using the Sequencher version 4.9 software.
MLPA
SALSA MLPA (Multiplex Ligation-dependent Probe Amplification) probe mixes P002-D1 and P090-C1 were used for large deletions/duplications analysis in BRCA1-2, according to the manufacturer’s instructions (MRC-Holland, Amsterdam, Netherlands). MLPA amplicons were run on ABI 3130 Genetic Analyzer (Applied Biosystems, Foster City, USA) while the collected data were analyzed using Coffalyser.net Software (MRC-Holland). Peak heights were normalized and deletions/duplications were defined as recommended by the manufacturer.
RNA Analysis
Total RNA was isolated from PAXgene blood RNA tubes (PreAnalytiX®, Qiagen, Hilden, Germany) (http://www.qiagen.com), with PAXgene Blood RNA Kit (IVD) (PreAnalytiX®), following the manufacturer’s instructions. RNA quality was evaluated by NanoDrop 2000 Spectrophotometer (Thermo Scientific). For each sample, 1 µg of RNA was reverse-transcribed into cDNA using a dedicated Qiagen kit (QuantiTect®Reverse Transcription Kit, Qiagen) according to the manufacturer’s instructions. Amplification was conducted on a Thermal Cycler 2720 (Applied Biosystems) using primers: exons 3-4 Fw (5’-TCACTGGTACTCTTTCTAATCC-3’), exon 7 Rv (3’-TTCAGGAGCAAAGCTGATCTG-5’). Altered transcript pattern was confirmed by comparison with the transcript patterns observed in healthy control. Normal and aberrant bands were excised from the agarose gel, purified using the MinElute Gel Extraction Kit (Qiagen). DNAs were sequenced by Sanger sequencing using the PE Big Dye Terminator Cycle Sequencing Kit on an ABI Prism 3130 analyser (Applied Biosystems). The data were analyzed using the Sequencher version 4.9 software.
Results
In the present study, 200 index patients were selected for analysis of BRCA1-2 genes according to the updated National Comprehensive Cancer Network (NCCN) guidelines. Exome sequencing (ES) followed by a virtual panel focusing on BRCA1 and BRCA2 genes were firstly performed. We obtained a mean coverage depth of 120× for targeted sequenced regions (range, 50–180×). This analysis was flanked by MLPA for the identification of large BRCA1-2 deletions/duplications. Pathogenic variants (PVs) were filtered according to frequency, mutation category, literature, and mutation database data (BRCA exchange, ClinVar database). After BRCA1-2 analysis, PVs in other cancer susceptibility genes were searched and classified according to whether they were traditionally associated (canonical HBOC genes) or not (candidate non-canonical HBOC genes) to breast and ovarian cancer.
Clinical Features of Cancer Patients
The study cohort included a total 200 index subjects (185 females and 15 males; age 34–95 years) with a clinical suspicion of HBOC on the basis of individual and/or family history. The average age of first cancer diagnosis was 55 years (range: 27–87 years) while the average age of second malignancy diagnosis was 61 years (range: 28–94 years). At first diagnosis 150 index patients had breast cancer (BC), 13 ovarian cancer (OC), 4 prostate cancer (PrC), 17 pancreatic cancer (PC), 2 melanoma (M), 8 other types of cancer, and 6 family history of cancer. Considering BC patients, 122 were monolateral and 23 bilateral cases and 5 unknown. Among bilateral BC, 13 patients had metachronous BC and 10 synchronous BC. The range between the first and the second diagnosis was 1–27 years (Table 1). Relating to BC histology, 22 had ductal carcinoma in situ, 78 had invasive ductal carcinoma, 3 had lobular carcinoma in situ, 20 invasive lobular carcinoma, and 27 other types of cancer. As regards the second tumor, 3 had ductal carcinoma in situ, 13 had invasive ductal carcinoma, 3 invasive lobular carcinoma, and 24 other types of cancer. BC therapy was mastectomy in 40 cases, breast conserving therapy in 78 cases, and unknown in 36 cases (Table 2).
TABLE 1

Table 1 Clinical characteristics of HBOC patients.
TABLE 2
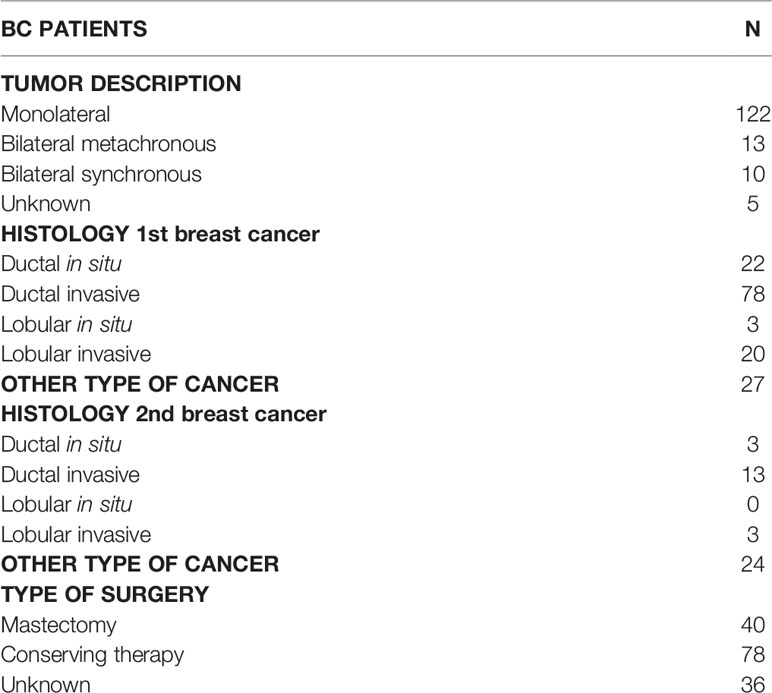
Table 2 Clinical characteristics of Breast Cancer (BC) patients.
BRCA1-2 Variants and Large Deletions/Duplications
BRCA1/2-targeted analysis revealed 11 pathogenic variants (4 in BRCA1 and 7 in BRCA2) and 12 Variants of Uncertain significance (VUS) (7 in BRCA1 and 5 in BRCA2) (Table S1) (Table S2). PVs were two nonsense substitutions, seven frameshift deletions, one splicing, and one missense change (Table S1). Among PVs, nine were already reported in literature or in the BRCA Exchange database (29–35). Seven out of 11 (63%) variants were located in exon 11. One variant (c.4485-1G>T) was intronic and predicted to impact the splicing of mRNA (MaxEntScan). VUS were mostly missense substitutions located in exon 11 (8/12; 66%) (Table S2). Seven VUS were already reported in ClinVar database and five were new (Table S2). MLPA analysis for BRCA1-2 of the same samples revealed only one rearrangement (1/200; 0,5%): a deletion on exon 20 of the BRCA1 gene (rsa 17q21(exon 20)x1) (Figure S1). Clinical features of patients with BRCA1-2 aberrations are reported in Figure S2.
Pathogenic Variants in Canonical HBOC Genes
Beyond BRCA1-2 genes, ES data analysis revealed 10 pathogenic variants in 7 HBOC-related genes (canonical genes): ATM, BRIP1, CDH1, PALB2, PTEN, RAD51C, and TP53. Among these variants, six were nonsense, three were missense, and one splicing variant. Eight variants were already reported in literature or in ClinVar database (Table 3) (36–43).
TABLE 3
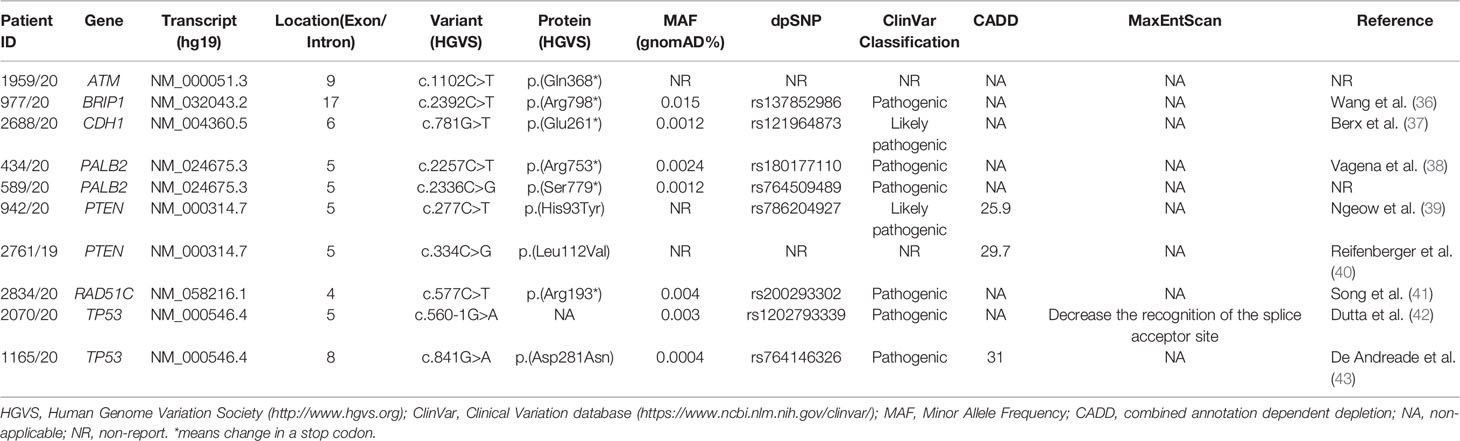
Table 3 Pathogenic variants found in canonical HBOC genes.
With regard to the clinical phenotype, the index patient (III-1) with the PV in the ATM gene (c.1102C>T; p.(Gln368*)) was diagnosed with bilateral BC at 60 years (Figure 1A). Histological analysis indicated multifocal infiltrating ductal carcinoma. As concerns the familial affected individuals, two maternal uncles (II-2 and II-3) resulted to be affected with lung cancer diagnosed at 70 years and 50 years, respectively, a paternal aunt with leukemia (II-7) and a paternal uncle (II-8) with colorectal cancer (Figure 1A). Individual (I-1) had BC at 80 years and the two grandfathers (I-2 and I-4) died of gastric cancer (Figure 1A). The patient (II-1) with the BRIP1 PV (c.2392C>T; p.(Arg798*)) developed colorectal adenocarcinoma at 61 years and ovarian serous carcinoma at 64 years. The mother (I-2) developed gastric cancer at 65 years and her brother (I-1) died of gastric cancer at 65 years. The father (I-3) was diagnosed with lung cancer at 64 years and, among paternal relatives, there are two cases with BC at 46 years (III-1) and at 44 years (II-5), a case with lung cancer (II-4) and another case with gastric cancer at 72 years (I-7) (Figure 1B). The patient with the (c.781G>T; p.(Glu261*)) variant in CDH1 had bilateral lobular BC at 34 years (Figure 1C). The patient (II-5) with PALB2 PV (c.2257C>T; p.(Arg753*)) was affected by prostate cancer at 71 years and by gastric cancer at 72 years. He had one sister (II-4) and one brother (II-3) with lung cancer (both at 70 years), another brother (II-2) with pancreatic cancer (70 years), a nephew (III-1) with liver cancer, and another nephew (III-3) with spinal cord cancer. The father (I-2) died of gastric cancer at 30 years (Figure 1D). The case (III-1) with the (c.2336C>G; p.(Ser7798*)) PV in PALB2 was diagnosed with unilateral infiltrating ductal BC at the age of 41 years (Figure 1E). The father had gastric cancer (II-2), the mother developed BC at 40 years (II-4), and the maternal grandfather prostate cancer at 80 years (I-4) (Figure 1E). Concerning PVs in PTEN, the patient (II-1) harboring the (c.277C>T; p.(His93Tyr)) substitution was diagnosed with ductal invasive BC at 40 years. At 45 years she had uterine adenocarcinoma (Figure 1F). The case (II-1) with the (c.334C>G; p.(Leu112Val)) variant had a personal history of ductal bilateral BC (53 years) (Figure 1G). At the age of 55 years, the patient underwent left partial glossectomy for microinvasive carcinoma of the left lingual edge (Figure 1G). The patient (II-1) with the (c.577C>T; p. (Arg193*)) nonsense variant in RAD51C had BC at 55 years (Figure 1H). Two paternal cousins were affected by BC at the age of 45 (II-3) and 35 (II-5) years, respectively (Figure 1H). The two patients (II-1) with TP53 PVs (c.560-1G>A and c.841G>A; p.(Asp281Asn)) were both diagnosed with ovarian serous carcinoma, one at 79 years (Figure 1I) and the other at 84 years (Figure 1J). The patient with the c.560-1G>A splicing change has two sisters with a diagnosis of BC (at 62, II-3 and at 70 years, II-4) and a sister (II-6) with gastric cancer (69 years) (Figure 1I). The mother (I-2) also had gastric cancer at 63 years and the nephew (III-3) melanoma at 56 years (Figure 1I). As concerns the second patient (c.841G>A; p.(Asp281Asn)), no familiarity for cancer was reported (Figure 1J).
FIGURE 1
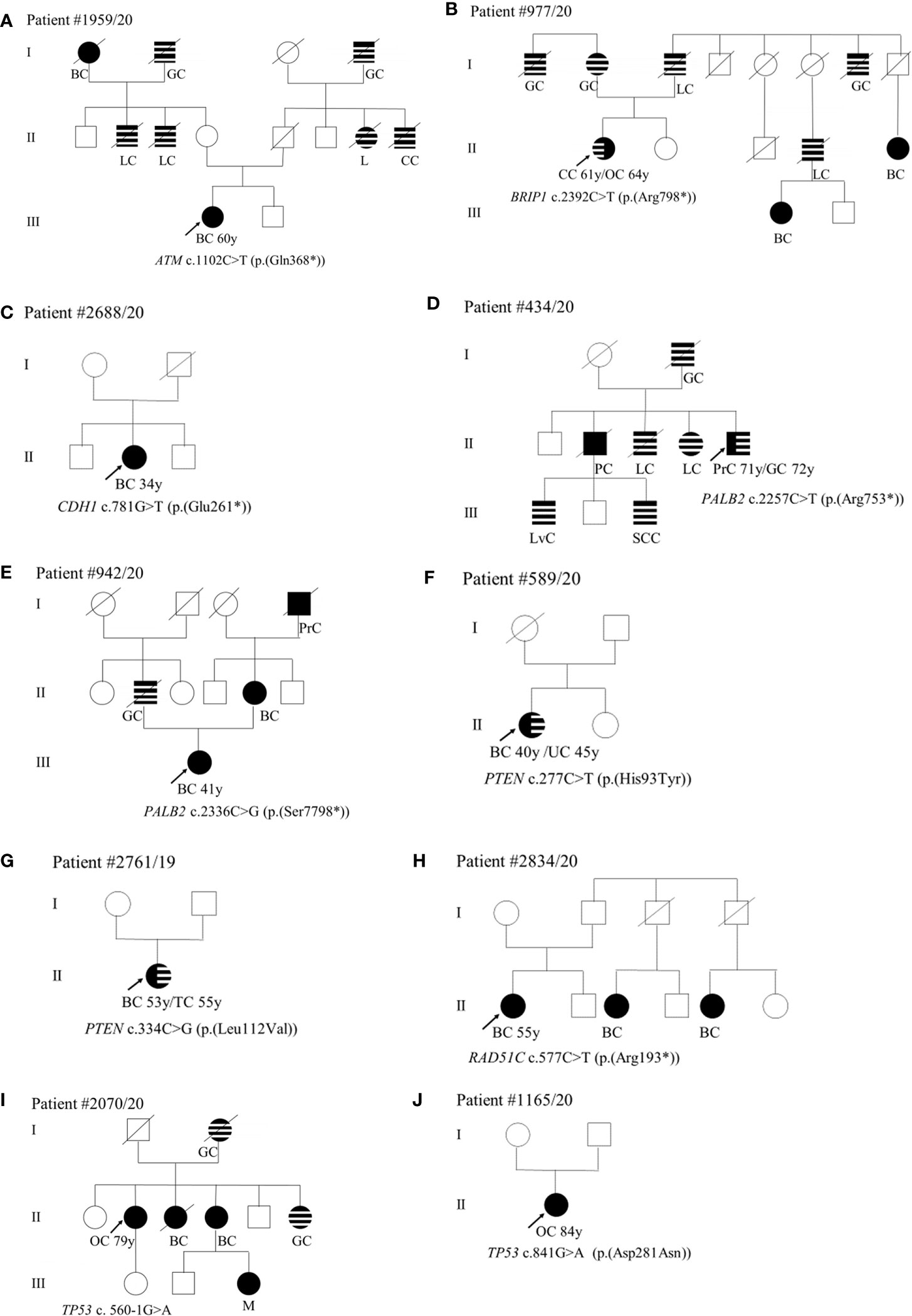
Figure 1 (A–J) Pedigrees of patients carrying pathogenic variants in canonical genes. The gene and their variants are reported below the symbol of each proband. The arrows indicate the proband from each family. The black symbols indicate “HBOC-spectrum phenotypes” while striped symbols indicate “other neoplasms.” AC, abdominal cancer; BC, breast cancer; CC, colorectal cancer; GC, gastric cancer; GIC, gastrointestinal cancer; KC, kidney cancer; L, leukemia; LC, lung cancer; LvC, liver cancer; M, melanoma; OC, ovarian cancer; PC, pancreatic cancer; PrC, prostate cancer, S, sarcoma; SCC, spinal cord cancer; TC, tongue cancer; TyC, Thyroid cancer; UC, uterine cancer; X, not defined.
Pathogenic Variants in Non-Canonical HBOC Genes
Twelve patients had PVs in genes not traditionally associated with HBOC: DPYP, ERBB3, ERCC2, MUTYH, NQO2, NTHL1, PARK2, RAD54L, and RNASEL. Four variants were nonsense, four missense, three splicing, and one frameshift deletion. Nine variants were previously classified as pathogenic or likely pathogenic on the basis of literature and database data (Table 4) (44–49).
TABLE 4
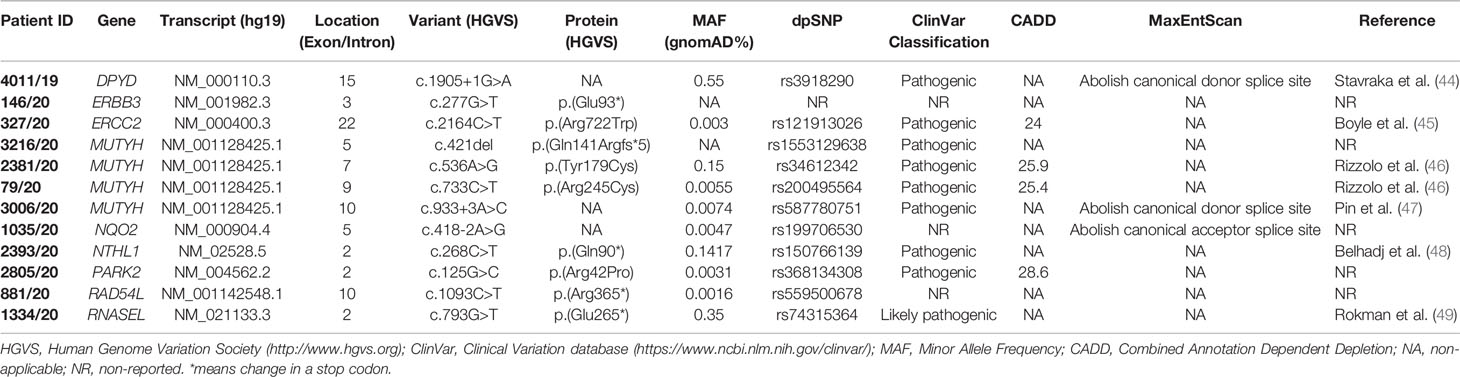
Table 4 Pathogenic variants found in non-canonical HBOC genes.
Comparison of variant frequencies with an internal biobank of 1,022 control samples (age ranging between 34 and 95 years) without any history of tumor and analyzed by ES revealed that variants have frequencies <1% (Table S4).
As concerns phenotype, we found the c.1905+1G>A splice site variant in the DPYP gene in a patient (II-1) diagnosed with unilateral BC at 45 years (Figure 2A). Among paternal relatives, an uncle (I-4) died of gastric cancer at 60 years (Figure 2A). The ERBB3 variant (c.277G>T; p.(Glu93*)) was identified in a patient (III-2) with monolateral infiltrating ductal BC diagnosed at 47 years (Figure 2B). As concerns paternal relatives, there was an uncle (II-2) dead at 72 years of prostate cancer, one cousin of the father (II-5) dead at 50 years of BC, and a grandmother (I-1) with heteroplasia of the uterus (Figure 2B). As regards maternal relatives, there was familiarity for tumors in grandmother siblings (I-6, I-7, I-9) and in cousins of the mother (II-10 and II-11) (Figure 2B). The (c.2164C>T; p.(Arg722Trp)) variant in the ERCC2 gene was found in a patient (III-1) with monolateral infiltrating ductal carcinoma diagnosed at 84 years. The father (II-1) died at 70 years of pancreatic cancer and the paternal grandmother (I-1) died at 60 years of BC (Figure 2C). Among the four patients with MUTYH PVs, the (c.421del; p.(Gln141Argfs*5)) variant was found in a male patient (II-2) who had monolateral ductal infiltrating BC diagnosed at 87 years (Figure 2D). His brother (II-1) had bilateral BC at 71 years and his father (I-1) had colorectal cancer at 55 years (Figure 2D). The patient (II-1) with the (c.536A>G; p.(Tyr179Cys)) variant in MUTYH had angioma of the upper gingival arch at 36 years and monolateral infiltrating BC at 52 years (Figure 2E). Familiarity for gastrointestinal tract neoplasms in the paternal branch (I-2 and I-3) was present. As regards the one with the (c.733C>T; p.(Arg245Cys)) variant had monolateral ductal in situ BC diagnosed at 53 years (Figure 2F). She had a mother (I-7) who died of bilateral BC at the age of 77 years and a strong family history of cancer including gastric (I-1) prostate (I-2), uterus neoplasia (I-4), lung cancer (I-5), and pancreatic cancer (I-9) (Figure 2F). The case (II-1) with the MUTYH c.933+3A>C variant had monolateral ductal in situ BC at 55 years and two previous melanomas, one in the calf at 41 years and one in the back at 52 years. The father (I-6) and two uncles (I-4 and I-7) were diagnosed with lung cancer (Figure 2G). The patient (II-1) with the variant c.418-2A>G in the NQO2 gene had monolateral ductal infiltrating BC at 66 years (Figure 2H). In the family, the father (I-4) had colorectal neoplasm, the mother (I-5) thyroid cancer, one maternal aunt (I-6) and her daughter (II-2) BC, and one cousin (II-3) kidney cancer (Figure 2H). We assessed the pathogenicity of this unclassified variant in NQO2 gene by patient’s mRNA analysis. The analysis showed that this variant abolishes canonical acceptor splice site and results in the formation of two aberrant transcripts: the first skipping of exon 6 p.(Val140_Ala173del)) and the second skipping of exons 5 and 6 p(Phe102_Ala173del)) (data not shown). The (c.268C>T; p.(Gln90*)) stop-gain variant in the NTHL1 gene was identified in a woman (IV-3) with ovarian cancer (62 years) (Figure 2I). In the family, three affected relatives with BC at 44 (III-1), 65 (III-2), and 56 (IV-1) years are present (Figure 2I). The father (III-5) died of lung cancer at 63 years (Figure 2I). The patient (II-3) with the PARK2 variant (c.125G>C; p.(Arg42Pro)) was diagnosed with monolateral lobular infiltrating BC at 75 years (Figure 2J). Previously she was affected by sarcoma of the left leg (61 years) (Figure 2J). In the family, she has a nephew (III-1) who died of BC (46 years) and a maternal cousin (II-1) who died of BC at 50 years (Figure 2J). The case (III-1) with the (c.1093C>T; p.(Arg365*)) variant in RAD54L had a diagnosis of BC at 55 years (Figure 2K). The mother (II-1) had uterus carcinoma at 84 years, a paternal aunt (II-5) and the grandmother (I-1) had juvenile BC (Figure 2K). Finally, the case (II-1) mutated in the RNASEL gene (c.793G>T; p.(Glu265*)) was diagnosed with prostate cancer at 53 years and with pancreatic cancer at 64 years (Figure 2L). His father (I-2) died of prostate cancer at 62 years (Figure 2L).
FIGURE 2
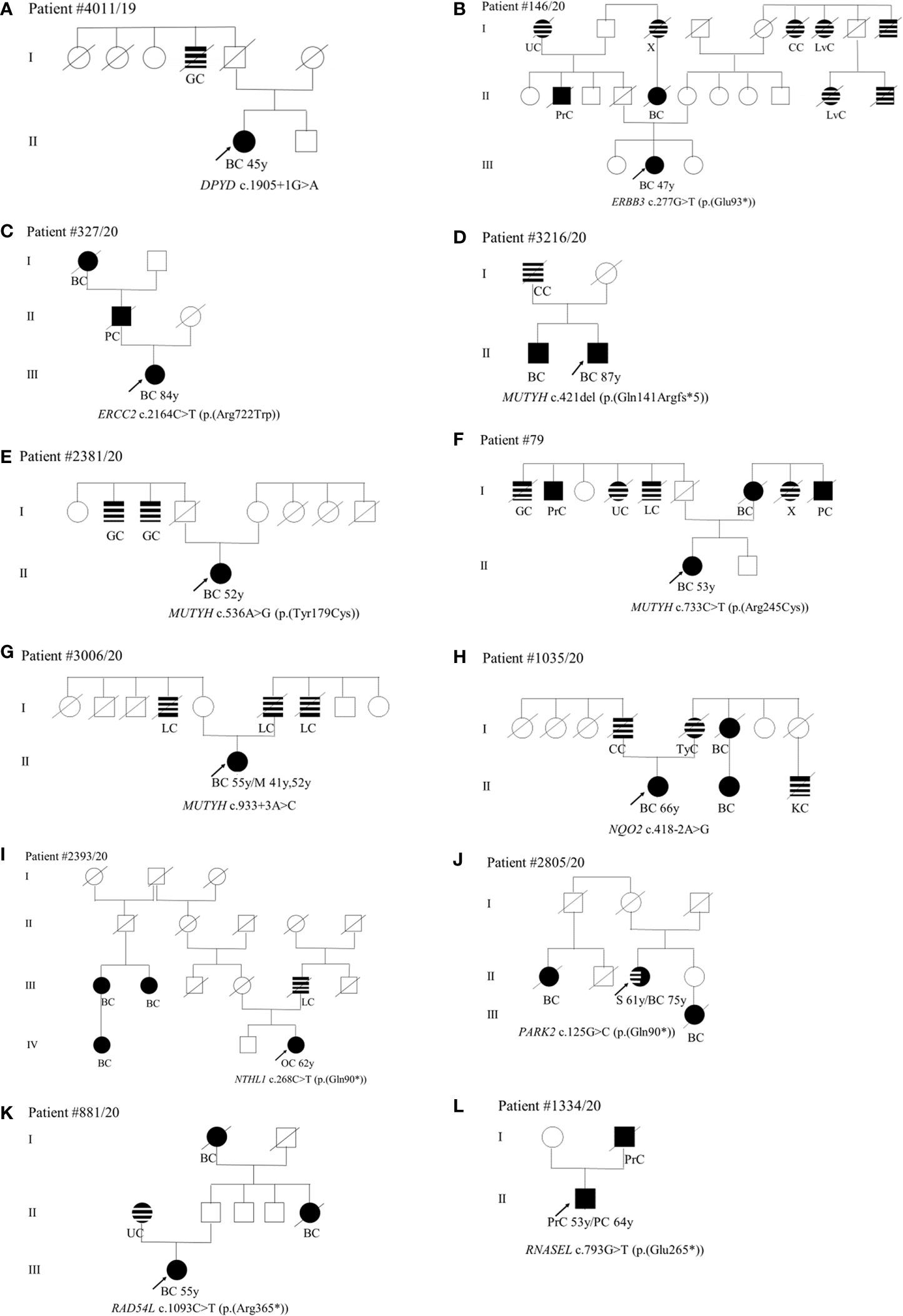
Figure 2 (A–L) Pedigrees of patients carrying pathogenic variants in non-canonical genes. The gene and their variants are reported below the symbol of each proband. The arrows indicate the proband from each family. The black symbols indicate “HBOC-spectrum phenotypes” while striped symbols indicate “other neoplasms.” Abbreviations as in Figure 1.
Uncertain Variants in Cancer Genes Beyond BRCA1-2
In the analysis of ES data, 74 variants were classified as VUS (class 3) on the basis of the IARC recommendations (Table S3). Most VUS were located in ATM and PALB2 genes (Table S3). Fourteen patients carried more than one VUS on different genes. As regards to mutation types, most were missense variants and the remaining were duplications or deletions. Combined Annotation Dependent Depletion (CADD) was considered to predict the effect of the missense variants.
Discussion
Multigene panel genetic tests are increasingly employed for the screening of patients presenting HBOC (14–22). However, multigene panel allows a limited gene analysis while Exome Sequencing (ES) allows the simultaneous assessment of virtually an unlimited number of genes. Previous studies on ES employed for the analysis of HBOC patients demonstrated that it is a powerful tool for the identification of PVs in known HBOC-related genes (canonical) and for the discovery of novel disease factors (non-canonical) (24, 25). However, ES analysis has the limit of not identifying Copy Number Variants (CNVs) and PVs in intronic regulatory elements. Here, we analyzed by ES a cohort of 200 patients with a diagnosis of HBOC according to the updated NCCN guidelines (26). ES data analysis allowed us to identify 11 cases with pathogenic variants (4 in BRCA1 and 7 in BRCA2) (5,5%) and 12 with uncertain variants (7 in BRCA1 and 5 in BRCA2). Only one case was found with a large BRCA1 deletion. The relatively low diagnostic yield in BRCA1-2 genes is probably due to the enlarged updated diagnostic NCCN guidelines. ES analysis allowed to identify PVs in additional 21 individuals: 10 with PVs in genes already associated to breast and ovarian cancer (ATM, BRIP1, CDH1, PALB2, PTEN, RAD51C, and TP53) and 11 in other candidate cancer susceptibility genes (DYPD, ERBB3, ERCC2, MUTYH, NTHL1, NQO2, PARK2, RAD54L, and RNASEL).
Regarding canonical HBOC genes, the genes more frequently mutated were PTEN (2/200; 1%), TP53 (2/200; 1%), and PALB2 (2/200; 1%). It is well known that BC can be included within several other described cancer syndromes, including Li-Fraumeni syndrome and Cowden syndrome. Thus, it is established that women who carry PVs in TP53 and PTEN have an increased risk of BC (50, 51). In accordance, the two patients with PVs in PTEN were diagnosed with BC. The two patients mutated in TP53 were both diagnosed with ovarian serous carcinoma. Loss-of-function mutations in the PALB2 gene confer a predisposition to BC with a prevalence of approximately 1% (52). We found two cases with PALB2 PVs, one with unilateral infiltrating ductal BC and a clear maternal cancer familiarity and one with prostate cancer, a personal history of gastric cancer, and familiarity for other neoplasms. The moderate-penetrance gene ATM, is mutated in 1–2% of BC, while in our study it has been found mutated only once (1/200) in a patient with multifocal infiltrating ductal carcinoma (53). As concerns BRIP1, Cantor et al. firstly reported the gene mutated in early-onset BC patients and subsequently this association was reinforced by Seal and colleagues who reported truncating mutations in BRIP1 as low-penetrance BC susceptibility alleles (54, 55). Our study reported a nonsense BRIP1 variant in a patient with ovarian serous carcinoma. In accordance with literature data reporting the presence of CDH1 PVs in >80% of invasive lobular BC, CDH1 has been found mutated in a patient with lobular BC (56, 57). Since the discovery of RAD51C in 2010 as a gene responsible for hereditary BC and OC, indication of including the gene in routine clinical genetic testing has been controversial, due to the lower prevalence or the absence of mutations found in subsequent studies (58–60). The present study confirms the role of RAD51C in HBOC being found mutated in a patient with BC.
Regarding new candidate non-canonical HBOC genes, the most frequently mutated was MUTYH (4/200; 2%). The MUTYH gene encodes a DNA glycosylase involved in the base excision repair (61). Biallelic MUTYH mutations are involved in the pathogenesis of a form of autosomal recessive colorectal adenomatous polyposis (MUTYH-associated polyposis syndrome, MAP) (62). A study focused on MUTYH-associated extracolonic cancers demonstrated that monoallelic variants are associated with an increased risk of gastric and liver cancers, as well as a slightly increased risk of endometrial and breast cancers (63). Here, we found four BC patients with MUTYH PVs: two with ductal in situ and two with ductal infiltrating carcinoma. They all have a strong family history of cancer. The DPYD gene encodes a protein that is active in the catabolic pathway of 5-fluorouracil and DPYD mutations result in an increased risk of toxicity in cancer patients receiving 5-fluorouracil chemotherapy (64). The rs291593 SNP in DPYD is associated with survival of BC patients (65). In our cohort, DPYP was mutated in a patient affected by unilateral BC. The variant was already demonstrated to affect the transcript resulting in the skipping of exon 14 leading to an inactive DPYD allele (66). ERBB3, encodes for HER3, a member of the EGFR family of receptor tyrosine kinases (67). The role of ERBB3 as somatic driver of tumorigenesis has been well documented, however, there is few data available about the role of germline variants (68). There is only a study reporting the identification of a germline predisposing mutation in a case of erythroleukemia (69). We found an ERBB3 nonsense variant in a patient with monolateral infiltrating ductal BC and a positive cancer history among both paternal and maternal relatives. ERCC2 encodes for an essential component of transcription factor IIH involved in basal cellular transcription and nucleotide excision repair (NER) of DNA lesions (36, 70). Data about its involvement in BC are at present not convincing (71). Here, we report an ERCC2 missense variant in a patient with monolateral infiltrating ductal BC and a positive oncological history. NQO2 encodes a flavoprotein that catalyzes the 2-electron reduction of various quinones, redox dyes, and the vitamin K menadione (72). In 2009, Yu and colleagues proposed NQO2 as a susceptibility gene for breast carcinogenesis (73). Here, we identified a patient with a splice site variant in the NQO2 gene that was never reported. mRNA analysis demonstrated that the variant has splicing impact generating two aberrant transcripts. NTHL1 initiates DNA base excision repair of oxidized ring saturated pyrimidine residues (74). Biallelic mutations in NTHL1 are responsible for familial adenomatous polyposis-3 (FAP3) (48). Subsequently, since NTHL1 mutations can cause a wide variety of cancers in addition to colorectal cancer, the designation “NTHL1 syndrome” was proposed (75). We found a mutation of this gene in a woman affected by ovarian cancer and subsequently by pancreatic cancer, suggesting an HBOC spectrum phenotype. PARK2, encoding a RING domain-containing E3 ubiquitin ligase, was originally identified as a gene responsible for autosomal recessive juvenile Parkinson disease-2 (PARK2, or PDJ) (76). In more recent years, PARK2 has been characterized as a tumor suppressor gene whose loss of heterozygosity (LOH) and loss of expression have been observed in different tumors such as lung, brain, breast, and ovarian cancer (77–79). We found a PARK2 PV in a patient with sarcoma of the left leg and monolateral lobular infiltrating BC. She has a positive BC family history. RAD54L is a gene that was found mutated in primary cancers, including BC (80). In the present study, we identified a BC patient with a truncating variant in RAD54L. RNASEL, encoding the endoribonuclease RNase L, is a key enzyme in the interferon induced antiviral and anti-proliferate pathway (81, 82). Mutations in RNASEL segregate with the disease in prostate cancer families and specific genotypes are associated with an increased risk of prostate cancer (49). In accordance, we found a RNASEL stopgain variant in a patient with a diagnosis of prostate cancer and a father dead at 62 years of prostate cancer.
In conclusion, ES analysis in a cohort of HBOC suspected patients allowed a diagnostic yield of a further −5% in non BRCA1-2 genes. Canonical genes would have been included in a multigene panel for diagnostic purposes while mutations in candidate non-canonical genes would not have emerged if a ES approach had not been carried out, overlapping the boundaries of clinical conditions, often simplistically separated. However, to make a diagnosis other experiments such as segregation analysis should be performed and these genes remains candidates. Variants in DPYD, MUTYH (c.536A>G and c.933+3A>C), NTHL1, and RNASEL genes were also found in controls, suggesting that these variants are probably low–penetrance risk alleles. Finally although it is true that this strategy increases the level of complication of the analysis and the number of VUS per sample, their identification appears to be essential for future definitive classification pooling together the data of different studies.
Supporting Informations
Oncologic Multidisciplinary Team, Azienda Ospedaliera Universitaria Senese, Siena, Italy: Alessandro Neri5,6, Donato Casella6, Andrea Bernini6, Stefania Marsili7, Roberto Petrioli7, Salvatora Tindara Miano7, Alessandra Pascucci7, Ignazio Martellucci7, Monica Crociani8, Marta Vannini8, Federica Fantozzi9, Andrea Stella9, Alessia Carmela Tripodi9, Angelamaria Giusti9, Alfonso Fausto9, Lucia Mantovani9, Francesca Belardi9
Oncologic Multidisciplinary Team, Azienda Usl Toscana Sud Est: Angelo Martignetti10, Amalia Falzetta10, Camilla Casi10, Paolo Petreni10, Beatrice Forzoni11, Enrico Tucci10, Tiziana Baglioni10, Luciana Biscari10, Antonia Borgomastro10, Pamela Torre10, Francesco Di Clemente10, Simona Scali10, Marianna Turrini10, Maria Deligianni10, Sabrina Del Buono11, Simonetta Magnanini11, Alice Baldi11, Tommaso Amato11, Ulpjana Gjondedaj11, Giorgio Bastreghi10, Giovanni Angiolucci11, Carmelo Bengala12, Roberta Di Rocco12
5 Department of Medicine, Surgery and Neuroscience, University of Siena, Siena, Italy
6 Chirurgia Oncologica della mammella, Azienda Ospedaliera Universitaria Senese, Siena, Italy
7 Oncologia Medica, Azienda Ospedaliera Universitaria Senese, Siena, Italy
8 Radioterapia, Azienda Ospedaliera Universitaria Senese, Siena, Italy
9 Senologia, Azienda Ospedaliera Universitaria Senese, Siena, Italy
10 Oncology Department, Azienda Usl Toscana Sud Est, Siena, Italy
11 Oncology Department, Azienda Usl Toscana Sud Est, Arezzo, Italy
12 Division of Medical Oncology, Misericordia Hospital, Azienda USL Toscana Sud Est, Grosseto
Data Availability Statement
NGS data has been deposited in publicly accessible repositories. The data can be found here: https://www.ncbi.nlm.nih.gov/sra/PRJNA727249.
Ethics Statement
Ethical review and approval were not required for the study on human participants in accordance with the local legislation and institutional requirements. The patients/participants provided their written informed consent to participate in this study.
Author Contributions
GD, FV, AR, and FA have made substantial contributions to conceptions and design and have been involved in drafting the paper. AG, FTP, RT, LPB, SR, CF, EB, MP, and MBr have made substantial contributions to acquisition and analysis of the data. MM, AF, AO, MBa, FF, DL, CLR, VL, SM, AP, and AC made clinical evaluation. All authors have given final approval of the version to be published and agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank HBOC patients and their families for their participation to the study.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2021.649435/full#supplementary-material
Supplementary Figure 1 | MLPA results for BRCA1 gene. MLPA results for BRCA1 gene of a patient (#1369/19) with familial BC. MLPA size probes for each exon (top) and genomic position (bottom), are given on the X axis. Calculated probe ratios of test sample normalized to the reference samples is given on the Y axis. Probe ratios are indicated by the dots. Black dots indicate probes within the confidence interval (ratio 0.7 to 1.3 by default). Red dot reveals a heterozygous deletion, probe ratio 0.5, in the test sample.
Supplementary Figure 2 | Pedigrees of patients carrying pathogenic variants in BRCA1-2. Pedigree of patients #3027/20, 352/20, 5632/20, 5647/19, 2734/20, 1568/20, 937/20, 1946/20, 2523/20, 1356/20, 1354/20, and 1369/19. The gene and their variants are reported below the symbol of each proband. The arrows indicate the proband from each family. The black symbols indicate “HBOC-spectrum phenotypes” while striped symbols indicate “other neoplasms.” Abbreviations as in Figure 1.
Supplementary Table 1 | Pathogenic variants found in HBOC genes. HGVS, Human Genome Variation Society (http://www.hgvs.org); ClinVar, Clinical Variation database (https://www.ncbi.nlm.nih.gov/clinvar/); MAF, Minor Allele Frequency; CADD, Combined Annotation Dependent Depletion; NA, non applicable; NR, non reported.
Supplementary Table 2 | VUS variants found in HBOC genes. HGVS, Human Genome Variation Society (http://www.hgvs.org); ClinVar, Clinical Variation database (https://www.ncbi.nlm.nih.gov/clinvar/); MAF, Minor Allele Frequency; CADD, Combined Annotation Dependent Depletion; NA, non applicable; NR, non reported.
Supplementary Table 3 | VUS found in canonical and non-canonical HBOC genes. HGVS, Human Genome Variation Society (http://www.hgvs.org); ClinVar, Clinical Variation database (https://www.ncbi.nlm.nih.gov/clinvar/); MAF, Minor Allele Frequency; CADD, Combined Annotation Dependent Depletion; NA, non applicable; NR, non reported.
Supplementary Table 4 | Comparison of variant frequencies with an internal biobank of 1022 samples (median range 34-95 years) without any history of tumor and analysed by ES. HGVS, Human Genome Variation Society (http://www.hgvs.org).
References
1. Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM. Estimates of Worldwide Burden of Cancer in 2008: GLOBOCAN 2008. Int J Cancer (2010) 127:2893–917. doi: 10.1002/ijc.25516
2. Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2016. CA Cancer J Clin (2016) 69:7–34. doi: 10.3322/caac.21332
3. Ford D, Easton DF, Bishop DT, Narod SA, Goldgar DE. Risks of Cancer in BRCA1-mutation Carriers. Breast Cancer Linkage Consortium. Lancet (London England) (1994) 343:692–5.
4. Moran A, O’Hara C, Khan S, Shack L, Woodward E, Maher ER, et al. Risk of Cancer Other Than Breast or Ovarian in Individuals With BRCA1 and BRCA2 Mutations. Fam Cancer (2012) 11:235–42. doi: 10.1007/s10689-011-9506-2
5. Van Asperen CJ, Brohet RM, Meijers-Heijboer EJ, Hoogerbrugqe N, Verhoef S, Vasen HFA, et al. Cancer Risks in BRCA2 Families: Estimates for Sites Other Than Breast and Ovary. J Med Genet (2005) 42:711–9. doi: 10.1136/jmg.2004.028829
6. Newman B, Austin MA, Lee M, King MC. Inheritance of Human Breast Cancer: Evidence for Autosomal Dominant Transmission in High-Risk Families. Proc Natl Acad Sci USA (1988) 85:3044–8. doi: 10.1073/pnas.85.9.3044
7. Hall JM, Lee MK, Newman B, Morrow JE, Anderson LA, Huey B, et al. Linkage of Early-Onset Familial Breast Cancer to Chromosome 17q21. Science (1990) 250:1684–9. doi: 10.1126/science.2270482
8. Miki Y, Swensen J, Shattuck-Eidens D, Futreal PA, Harshman K, Tavtigian S, et al. A Strong Candidate for the Breast and Ovarian Cancer Susceptibility Gene BRCA1. Science (1994) 266:66–71. doi: 10.1126/science.7545954
9. Chen S, Parmigiani G. Meta-Analysis of BRCA1 and BRCA2 Penetrance. J Clin Oncol (2007) 25:1329–33. doi: 10.1200/JCO.2006.09.1066
10. Kuchenbaecker KB, Hopper JL, Barnes DR, Phillips KA, Mooij TM, Roos-Blom MJ, et al. Risks of Breast, Ovarian, and Contralateral Breast Cancer for BRCA1 and BRCA2 Mutation Carriers. JAMA - J Am Med Assoc (2017) 317:2402–16. doi: 10.1001/jama.2017.7112
11. Domchek SM, Friebel TM, Singer CF, Gareth Evans D, Lynch HT, Isaacs C, et al. Association of Risk-Reducing Surgery in BRCA1 or BRCA2 Mutation Carriers With Cancer Risk and Mortality. JAMA - J Am Med Assoc (2010) 304:967–75. doi: 10.1001/jama.2010.1237
12. Saslow D, Boetes C, Burke W, Harms S, Leach MO, Lehman CD, et al. American Cancer Society Guidelines for Breast Screening With MRI as an Adjunct to Mammography. CA Cancer J Clin (2007) 57:75–89. doi: 10.3322/canjclin.57.2.75
13. Lord CJ, Ashworth A. PARP Inhibitors: Synthetic Lethality in the Clinic. Science (2017) 355:1152–8. doi: 10.1126/science.aam7344
14. Buys SS, Sandbach JF, Gammon A, Patel G, Kidd J, Brown KL, et al. A Study of Over 35,000 Women With Breast Cancer Tested With a 25-Gene Panel of Hereditary Cancer Genes. Cancer (2017) 123:1721–30. doi: 10.1002/cncr.30498
15. Yadav S, Reeves A, Campian S, Paine A, Zakalik D. Outcomes of Retesting BRCA Negative Patients Using Multigene Panels. Fam Cancer (2017) 16:319–28. doi: 10.1007/s10689-016-9956-7
16. Tedaldi G, Tebaldi M, Zampiga V, Danesi R, Arcangeli V, Ravegnani M, et al. Multiple-Gene Panel Analysis in a Case Series of 255 Women With Hereditary Breast and Ovarian Cancer. Oncotarget (2017) 8:47064–75. doi: 10.18632/oncotarget.16791
17. Pinto P, Paulo P, Santos C, Rocha P, Pinto C, Veiga I, et al. Implementation of Next-Generation Sequencing for Molecular Diagnosis of Hereditary Breast and Ovarian Cancer Highlights its Genetic Heterogeneity. Breast Cancer Res Treat (2016) 159:245–56. doi: 10.1007/s10549-016-3948-z
18. Laduca H, Stuenkel AJ, Dolinsky JS, Keiles S, Tandy S, Pesaran T, et al. Utilization of Multigene Panels in Hereditary Cancer Predisposition Testing: Analysis of More Than 2,000 Patients. Genet Med (2014) 6:830–7. doi: 10.1038/gim.2014.40
19. Tung N, Lin NU, Kidd J, Allen BA, Singh N, Wenstrup RJ, et al. Frequency of Germline Mutations in 25 Cancer Susceptibility Genes in a Sequential Series of Patients With Breast Cancer. J Clin Oncol (2016) 34:1460–8. doi: 10.1200/JCO.2015.65.0747
20. Fanale D, Incorvaia L, Filorizzo C, Bono M, Fiorino A, Calò V, et al. Detection of Germline Mutations in a Cohort of 139 Patients With Bilateral Breast Cancer by Multi-Gene Panel Testing: Impact of Pathogenic Variants in Other Genes Beyond Brca1/2. Cancers (Basel) (2020) 12:2415. doi: 10.3390/cancers12092415
21. Kraus C, Hoyer J, Vasileiou G, Wunderle M, Lux MP, Fasching PA, et al. Gene Panel Sequencing in Familial Breast / Ovarian Cancer Genes Others Than BRCA1 / 2. Int J Cancer Gene (2017) 140:95–102.
22. Susswein LR, Marshall ML, Nusbaum R, Vogel Postula KJ, Weissman SM, Yackowski L, et al. Pathogenic and Likely Pathogenic Variant Prevalence Among the First 10,000 Patients Referred for Next-Generation Cancer Panel Testing. Genet Med (2016) 18:823–32. doi: 10.1038/gim.2015.166
23. Nielsen FC, Van Overeem Hansen T, Sørensen CS. Hereditary Breast and Ovarian Cancer: New Genes in Confined Pathways. Nat Rev Cancer (2016) 16:599–612. doi: 10.1038/nrc.2016.72
24. Glentis S, Dimopoulos AC, Rouskas K, Ntritsos G, Evangelou E, Narod SA, et al. Exome Sequencing in BRCA1- and BRCA2-Negative Greek Families Identifies MDM1 and NBEAL1 as Candidate Risk Genes for Hereditary Breast Cancer. Front Genet (2019) 10:1005. doi: 10.3389/fgene.2019.01005
25. Weitzel JN, Neuhausen SL, Adamson A, Tao S, Ricker C, Maoz A, et al. Pathogenic and Likely Pathogenic Variants in PALB2, CHEK2, and Other Known Breast Cancer Susceptibility Genes Among 1054 BRCA-negative Hispanics With Breast Cancer. Cancer (2019) 125:2829–36.
26. Daly MB, Pilarski R, Yurgelun MB, Berry MP, Buys SS, Dickson P, et al. Nccn Guidelines Insights: Genetic/Familial High-Risk Assessment: Breast, Ovarian, and Pancreatic, Version 1.2020. J Natl Compr Cancer Netw (2020) 18:380–91. doi: 10.6004/jnccn.2020.0017
27. Li H, Durbin R. Fast and Accurate Long-Read Alignment With Burrows-Wheeler Transform. Bioinformatics (2010) 26:589–95. doi: 10.1093/bioinformatics/btp698
28. Poplin R, Ruano-Rubio V, DePristo M, Fennell T, Carneiro M, Van der Auwera G, et al. Scaling Accurate Genetic Variant Discovery to Tens of Thousands of Samples. bioRxiv (2017). doi: 10.1101/201178
29. Janavičius R. Founder BRCA1/2 Mutations in the Europe: Implications for Hereditary Breast-Ovarian Cancer Prevention and Control. EPMA J (2010) 1:397–412. doi: 10.1007/s13167-010-0037-y
30. Vendrell JA, Vilquin P, Larrieux M, Van Goethem C, Solassol J. Benchmarking of Amplicon-Based Next-Generation Sequencing Panels Combined With Bioinformatics Solutions for Germline BRCA1 and BRCA2 Alteration Detection. J Mol Diagnostics (2018) 20:754–64. doi: 10.1016/j.jmoldx.2018.06.003
31. Manickam K, Buchanan AH, Schwartz MLB, Hallquist MLG, Williams JL, Rahm AK, et al. Exome Sequencing-Based Screening for BRCA1/2 Expected Pathogenic Variants Among Adult Biobank Participants. JAMA Netw Open (2018) 1:e182140. doi: 10.1001/jamanetworkopen.2018.2140
32. Laitman Y, Friebel TM, Yannoukakos D, Fostira F, Konstantopoulou I, Figlioli G, et al. The Spectrum of BRCA1 and BRCA2 Pathogenic Sequence Variants in Middle Eastern, North African, and South European Countries. Hum Mutat (2019) 40:e1–e23. doi: 10.1002/humu.23842
33. Guo Y, Wang P, Li X, Zhu S, Xu H, Li S, et al. Identifying a BRCA2 c.5722 5723del Mutation in a Han-Chinese Family With Breast Cancer. Biosci Rep (2019) 39:BSR20182471. doi: 10.1042/BSR20182471
34. Parsons MT, Tudini E, Li H, Hahnen E, Wappenschmidt B, Feliubadaló L, et al. Large Scale Multifactorial Likelihood Quantitative Analysis of BRCA1 and BRCA2 Variants: An ENIGMA Resource to Support Clinical Variant Classification. Hum Mutat (2019) 40:1557–78. doi: 10.1002/humu.23818
35. Palmero EI, Carraro DM, Alemar B, Moreira MAM, Ribeiro-Dos-Santos Â, Abe-Sandes K, et al. The Germline Mutational Landscape of BRCA1 and BRCA2 in Brazil. Sci Rep (2018) 8:9188. doi: 10.1038/s41598-018-27315-2
36. Wang J, Li W, Shi Y, Huang Y, Sun T, Tang L, et al. Germline Mutation Landscape of Chinese Patients With Familial Breast/Ovarian Cancer in a Panel of 22 Susceptibility Genes. Cancer Med (2019) 8:2074–84. doi: 10.1002/cam4.2093
37. Berx G, Cleton-Jansen AM, Nollet F, De Leeuw WJF, Van De Vijver MJ, Cornelisse C, et al. E-Cadherin is a Tumour/Invasion Suppressor Gene Mutated in Human Lobular Breast Cancers. EMBO J (1995) 14:6107–15. doi: 10.1002/j.1460-2075.1995.tb00301.x
38. Vagena A, Papamentzelopoulou M, Kalfakakou D, Kollia P, Papadimitriou C, Psyrri A, et al. Palb2 c.2257C>T Truncating Variant is a Greek Founder and is Associated With High Breast Cancer Risk. J Hum Genet (2019) 64:767–73. doi: 10.1038/s10038-019-0612-6
39. Ngeow J, Stanuch K, Mester JL, Barnholtz-Sloan JS, Eng C. Second Malignant Neoplasms in Patients With Cowden Syndrome With Underlying Germline PTEN Mutations. J Clin Oncol (2014) 32:1818–24. doi: 10.1200/JCO.2013.53.6656
40. Reifenberger J, Rauch L, Beckmann MW, Megahed M, Ruzicka T, Reifenberger G. Cowden’s Disease: Clinical and Molecular Genetic Findings in a Patient With a Novel PTEN Germline Mutation. Br J Dermatol (2003) 148:1040–6. doi: 10.1046/j.1365-2133.2003.05322.x
41. Song H, Dicks E, Ramus SJ, Tyrer JP, Intermaggio MP, Hayward J, et al. Contribution of Germline Mutations in the RAD51B, RAD51C, and RAD51D Genes to Ovarian Cancer in the Population. J Clin Oncol (2015) 33:2901–7. doi: 10.1200/JCO.2015.61.2408
42. Dutta S, Pregartner G, Rücker FG, Heitzer E, Zebisch A, Bullinger L, et al. Functional Classification of tp53 Mutations in Acute Myeloid Leukemia. Cancers (Basel) (2020) 12:637. doi: 10.3390/cancers12030637
43. de Andrade KC, Frone MN, Wegman-Ostrosky T, Khincha PP, Kim J, Amadou A, et al. Variable Population Prevalence Estimates of Germline TP53 Variants: A gnomAD-based Analysis. Hum Mutat (2019) 40:97–105. doi: 10.1002/humu.23673
44. Stavraka C, Pouptsis A, Okonta L, DeSouza K, Charlton P, Kapiris M, et al. Clinical Implementation of Pre-Treatment DPYD Genotyping in Capecitabine-Treated Metastatic Breast Cancer Patients. Breast Cancer Res Treat (2019) 175:511–7. doi: 10.1007/s10549-019-05144-9
45. Boyle J, Ueda T, Oh KS, Imoto K, Tamura D, Jagdeo J, et al. Persistence of Repair Proteins At Unrepaired DNA Damage Distinguishes Diseases With ERCC2 (XPD) Mutations: Cancer-prone Xeroderma Pigmentosum vs. non-Cancer-Prone Trichothiodystrophy. Hum Mutat (2008) 29:1194–208. doi: 10.1002/humu.20768
46. Rizzolo P, Silvestri V, Bucalo A, Zelli V, Valentini V, Catucci I, et al. Contribution of MUtYH Variants to Male Breast Cancer Risk: Results From a Multicenter Study in Italy. Front Oncol (2018) 8:583. doi: 10.3389/fonc.2018.00583
47. Pin E, Pastrello C, Tricarico R, Papi L, Quaia M, Fornasarig M, et al. Mutyh c.933+3A>C, Associated With a Severely Impaired Gene Expression, is the First Italian Founder Mutation in MUTYH-Associated Polyposis. Int J Cancer (2013) 132:1060–9. doi: 10.1002/ijc.27761
48. Belhadj S, Quintana I, Mur P, Munoz-Torres PM, Alonso MH, Navarro M, et al. NTHL1 Biallelic Mutations Seldom Cause Colorectal Cancer, Serrated Polyposis or a Multi-Tumor Phenotype, in Absence of Colorectal Adenomas. Sci Rep (2019) 9:9020. doi: 10.1038/s41598-019-45281-1
49. Rökman A, Ikonen T, Seppälä EH, Nupponen N, Autio V, Mononen N, et al. Germline Alterations of the RNASEL Gene, a Candidate HPC1 Gene At 1q25, in Patients and Families With Prostate Cancer. Am J Hum Genet (2002) 70:1299–304. doi: 10.1086/340450.
50. Olivier M, Goldgar DE, Sodha N, Ohgaki H, Kleihues P, Hainaut P, et al. Li-Fraumeni and Related Syndromes: Correlation Between Tumor Type, Family Structure, and TP53 Genotype. Cancer Res (2003) 63:6643–50.
51. Eng C. Will the Real Cowden Syndrome Please Stand Up: Revised Diagnostic Criteria. J Med Genet (2000) 37:828–30. doi: 10.1136/jmg.37.11.828
52. Antoniou AC, Casadei S, Heikkinen T, Barrowdale D, Pylkäs K, Roberts J, et al. Breast-Cancer Risk in Families With Mutations in PALB2. N Engl J Med (2014) 371:497–506. doi: 10.1056/nejmoa1400382
53. Marabelli M, Cheng SC, Parmigiani G. Penetrance of ATM Gene Mutations in Breast Cancer: A Meta-Analysis of Different Measures of Risk. Genet Epidemiol (2016) 40:425–31. doi: 10.1002/gepi.21971
54. Seal S, Thompson D, Renwick A, Elliott A, Kelly P, Barfoot R, et al. Truncating Mutations in the Fanconi Anemia J Gene BRIP1 are Low-Penetrance Breast Cancer Susceptibility Alleles. Nat Genet (2006) 38:1239–41. doi: 10.1038/ng1902
55. Cantor SB, Bell DW, Ganesan S, Kass EM, Drapkin R, Grossman S, et al. BACH1, a Novel Helicase-Like Protein, Interacts Directly With BRCA1 and Contributes to its DNA Repair Function. Cell (2001) 105:149–60. doi: 10.1016/S0092-8674(01)00304-X
56. Ciriello G, Gatza ML, Beck AH, Wilkerson MD, Rhie SK, Pastore A, et al. Comprehensive Molecular Portraits of Invasive Lobular Breast Cancer. Cell (2015) 163:506–19. doi: 10.1016/j.cell.2015.09.033.
57. Desmedt C, Zoppoli G, Gundem G, Pruneri G, Larsimont D, Fornili M, et al. Genomic Characterization of Primary Invasive Lobular Breast Cancer. J Clin Oncol (2016) 34:1872–81. doi: 10.1200/JCO.2015.64.0334
58. Zheng Y, Zhang J, Hope K, Niu Q, Huo D, Olopade OI. Screening RAD51C Nucleotide Alterations in Patients With a Family History of Breast and Ovarian Cancer. Breast Cancer Res Treat (2010) 124:857–61. doi: 10.1007/s10549-010-1095-5
59. Akbari MR, Tonin P, Foulkes WD, Ghadirian P, Tischkowitz M, Narod SA. RAD51C Germline Mutations in Breast and Ovarian Cancer Patients. Breast Cancer Res (2010) 12:404. doi: 10.1186/bcr2619
60. Vuorela M, Pylkäs K, Hartikainen JM, Sundfeldt K, Lindblom A, Von Wachenfeldt Wäppling A, et al. Further Evidence for the Contribution of the RAD51C Gene in Hereditary Breast and Ovarian Cancer Susceptibility. Breast Cancer Res Treat (2011) 130:1003–10. doi: 10.1007/s10549-011-1677-x
61. Venesio T, Balsamo A, D’Agostino VG, Ranzani GN. MUTYH-Associated Polyposis (MAP), the Syndrome Implicating Base Excision Repair in Inherited Predisposition to Colorectal Tumors. Front Oncol (2012) 2:83. doi: 10.3389/fonc.2012.00083
62. Kashfi SMH, Golmohammadi M, Behboudi F, Nazemalhosseini-Mojarad E, Zali MR. MUTYH the Base Excision Repair Gene Family Member Associated With Polyposis Colorectal Cancer. Gastroenterol Hepatol Bed to Bench (2013) 6:S1–S10. doi: 10.22037/ghfbb.v6i0.458
63. Win AK, Reece JC, Dowty JG, Buchanan DD, Clendenning M, Rosty C, et al. Risk of Extracolonic Cancers for People With Biallelic and Monoallelic Mutations in MUTYH. Int J Cancer (2016) 139:1557–63. doi: 10.1002/ijc.30197
64. Van Kuilenburg ABP. Dihydropyrimidine Dehydrogenase and the Efficacy and Toxicity of 5-Fluorouracil. Eur J Cancer (2004) 40:939–50. doi: 10.1016/j.ejca.2003.12.004
65. Pamuła-Piłat J, Tęcza K, Kalinowska-Herok M, Grzybowska E. Genetic 3′UTR Variations and Clinical Factors Significantly Contribute to Survival Prediction and Clinical Response in Breast Cancer Patients. Sci Rep (2020) 10:5736. doi: 10.1038/s41598-020-62662-z
66. Wei X, McLeod HL, McMurrough J, Gonzalez FJ, Fernandez-Salguero P. Molecular Basis of the Human Dihydropyrimidine Dehydrogenase Deficiency and 5-Fluorouracil Toxicity. J Clin Invest (1996) 98:610–5. doi: 10.1172/JCI118830
67. Sithanandam G, Anderson LM. The ERBB3 Receptor in Cancer and Cancer Gene Therapy. Cancer Gene Ther (2008) 15:413–48. doi: 10.1038/cgt.2008.15
68. Kiavue N, Cabel L, Melaabi S, Bataillon G, Callens C, Lerebours F, et al. ERBB3 Mutations in Cancer: Biological Aspects, Prevalence and Therapeutics. Oncogene (2020) 39:487–502. doi: 10.1038/s41388-019-1001-5
69. Braunstein EM, Li R, Sobreira N, Marosy B, Hetrick K, Doheny K, et al. A Germline ERBB3 Variant is a Candidate for Predisposition to Erythroid MDS/Erythroleukemia. Leukemia (2016) 30:2242–5. doi: 10.1038/leu.2016.173
70. Takayama K, Salazar EP, Lehmann A, Stefanini M, Thompson LH, Weber CA. Defects in the DNA Repair and Transcription Gene ERCC2 in the Cancer-prone Disorder Xeroderma Pigmentosum Group D. Cancer Res (1995) 55:5656–63.
71. Rump A, Benet-Pages A, Schubert S, Kuhlmann JD, Janavičius R, Macháčková E, et al. Identification and Functional Testing of ERCC2 Mutations in a Multi-national Cohort of Patients With Familial Breast- and Ovarian Cancer. PloS Genet (2016) 12:e1006248. doi: 10.1371/journal.pgen.1006248
72. Wu K, Knox R, Sun XZ, Joseph P, Jaiswal AK, Zhang D, et al. Catalytic Properties of NAD(P)H:quinone Oxidoreductase-2 (NQO2), a Dihydronicotinamide Riboside Dependent Oxidoreductase. Arch Biochem Biophys (1997) 347:221–8. doi: 10.1006/abbi.1997.0344
73. Yu K, Di GH, Yuan WT, Fan L, Wu J, Hu Z, et al. Functional Polymorphisms, Altered Gene Expression and Genetic Association Link NRH:quinone Oxidoreductase 2 to Breast Cancer With Wild-Type P53. Hum Mol Genet (2009) 18(13):2502–17. doi: 10.1093/hmg/ddp171
74. Ocampo MTA, Chaung W, Marenstein DR, Chan MK, Altamirano A, Basu AK, et al. Targeted Deletion of Mnth1 Reveals a Novel Dna Repair Enzyme Activity. Mol Cell Biol (2002) 22:6111–21. doi: 10.1128/mcb.22.17.6111-6121.2002
75. Grolleman JE, de Voer RM, Elsayed FA, Nielsen M, Weren RDA, Palles C, et al. Mutational Signature Analysis Reveals Nthl1 Deficiency to Cause a Multi-tumor Phenotype. Cancer Cell (2019) 35:256–66. doi: 10.1016/j.ccell.2018.12.011
76. Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, et al. Mutations in the Parkin Gene Cause Autosomal Recessive Juvenile Parkinsonism. Nature (1998) 392:605–8. doi: 10.1038/33416
77. Picchio MC, Martin ES, Cesari R, Calin GA, Yendamuri S, Kuroki T, et al. Alterations of the Tumor Suppressor Gene Parkin in Non-Small Cell Lung Cancer. Clin Cancer Res (2004) 10:2720–4. doi: 10.1158/1078-0432.CCR-03-0086
78. Veeriah S, Taylor BS, Meng S, Fang F, Yilmaz E, Vivanco I, et al. Somatic Mutations of the Parkinson’s Disease-Associated Gene PARK2 in Glioblastoma and Other Human Malignancies. Nat Genet (2010) 42:77–82. doi: 10.1038/ng.491
79. Cesari R, Martin ES, Calin GA, Pentimalli F, Bichi R, McAdams H, et al. Parkin, a Gene Implicated in Autosomal Recessive Juvenile Parkinsonism, is a Candidate Tumor Suppressor Gene on Chromosome 6q25-Q27. Proc Natl Acad Sci USA (2003) 100:5956–61. doi: 10.1073/pnas.0931262100
80. Matsuda M, Miyagawa K, Takahashi M, Fukuda T, Kataoka T, Asahara T, et al. Mutations in the RAD54 Recombination Gene in Primary Cancers. Oncogene (1999) 18:3427–30. doi: 10.1038/sj.onc.1202692
81. Hassel BA, Zhou A, Sotomayor C, Maran A, Silverman RH. A Dominant Negative Mutant of 2-5A-Dependent RNase Suppresses Antiproliferative and Antiviral Effects of Interferon. EMBO J (1993) 12:3297–304. doi: 10.1002/j.1460-2075.1993.tb05999.x
Keywords: BRCA1, BRCA2, cancer susceptibility genes, HBOC, ES (Exome Sequencing)
Citation: Doddato G, Valentino F, Giliberti A, Papa FT, Tita R, Bruno LP, Resciniti S, Fallerini C, Benetti E, Palmieri M, Mencarelli MA, Fabbiani A, Bruttini M, Orrico A, Baldassarri M, Fava F, Lopergolo D, Lo Rizzo C, Lamacchia V, Mannucci S, Pinto AM, Currò A, Mancini V, Oncologic Multidisciplinary Team, Azienda Ospedaliera Universitaria Senese, Oncologic Multidisciplinary Team, Azienda Usl Toscana Sud Est, Mari F, Renieri A and Ariani F (2021) Exome Sequencing in BRCA1-2 Candidate Familias: The Contribution of Other Cancer Susceptibility Genes. Front. Oncol. 11:649435. doi: 10.3389/fonc.2021.649435
Received: 04 January 2021; Accepted: 06 April 2021;
Published: 07 May 2021.
Edited by:
Paola Parrella, Casa Sollievo della Sofferenza (IRCCS), ItalyReviewed by:
Valentina Silvestri, Sapienza University of Rome, ItalyGiovana Tardin Torrezan, A.C.Camargo Cancer Center, Brazil
Copyright © 2021 Doddato, Valentino, Giliberti, Papa, Tita, Bruno, Resciniti, Fallerini, Benetti, Palmieri, Mencarelli, Fabbiani, Bruttini, Orrico, Baldassarri, Fava, Lopergolo, Lo Rizzo, Lamacchia, Mannucci, Pinto, Currò, Mancini, Oncologic Multidisciplinary Team, Azienda Ospedaliera Universitaria Senese, Oncologic Multidisciplinary Team, Azienda Usl Toscana Sud Est, Mari, Renieri and Ariani. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Francesca Ariani, ZnJhbmNlc2NhLmFyaWFuaUB1bmlzaS5pdA==
†These authors have contributed equally to this work