
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Oncol. , 16 June 2021
Sec. Cancer Genetics
Volume 11 - 2021 | https://doi.org/10.3389/fonc.2021.649323
 Junyu Xue1
Junyu Xue1 Shiyong Li2Peijie Shi1
Shiyong Li2Peijie Shi1 Mengke Chen2Shuang Yu1
Mengke Chen2Shuang Yu1 Shubin Hong1Yanbing Li1
Shubin Hong1Yanbing Li1 Rengyun Liu2*
Rengyun Liu2* Haipeng Xiao1*
Haipeng Xiao1*Hotspot mutations in the core promoter region of the telomerase reverse transcriptase (TERT) gene have been well established to associate with aggressive clinical characteristics, radioiodine refractory, tumor recurrence, and mortality in thyroid cancer. Several E-twenty-six (ETS) transcription factors were reported to selectively bound to the mutant TERT promoter and activated TERT expression. In this study we aimed to investigate whether TERT promoter mutations confer sensitivity to ETS inhibitor YK-4-279 in thyroid cancer cells and whether this inhibitor could be served as a potential therapeutic agent for thyroid cancer. In vitro assays showed that YK-4-279 treatment sharply suppressed cell viability, colony formation, migration, and invasion, as well as induced cell cycle arrest and apoptosis in a panel of thyroid cancer cells. The cell viability after YK-4-279 treatment was similar between cell lines harboring mutant and wild-type TERT promoters. Furthermore, YK-4-279 treatment reduced both luciferase activity and mRNA expression of TERT independent of TERT promoter mutation status. Data from RNA-seq further revealed that YK-4-279 significantly affected biological processes including DNA replication and cell cycle. Reduced DNA helicase activity and decreased expression of several helicase genes were observed after YK-4-279 treatment. Moreover, YK-4-279 significantly inhibited tumor growth and induced apoptosis in a xenograft mice model. Thus, ETS inhibitor YK-4-279 suppressed TERT expression and conferred anti-tumor activity in a TERT promoter mutation-independent manner, and it could be a potential agent for the treatment of advanced thyroid cancers.
Thyroid cancer is the most prevalent endocrine cancer worldwide (1). Depending on its histological characteristics, thyroid cancer can be classified as papillary thyroid cancer (PTC), follicular thyroid cancer (FTC), anaplastic thyroid cancer (ATC), and medullary thyroid cancer (MTC). The MTC is derived from parafollicular C cells and accounts for approximately 4% of thyroid cancers (2), while the other three subtypes (PTC, FTC, and ATC) are all developed from follicular thyroid cells and account for the majority of thyroid cancers. Although most of the thyroid cancer cases have a good prognosis after standard clinical treatments, tumor recurrence and/or mortality still occurs in patients with aggressive thyroid cancer.
Genetic alterations drive the progression and contribute to the tumor recurrence and mortality of thyroid cancer (3). Molecular-based management of thyroid cancer is quickly developed in recent years and holds a great advantage to improve the survival of cancer patients with certain genetic alterations (4). RET mutations account for approximately 70% of MTC and associated with aggressiveness of thyroid cancer. Several RET-targeting inhibitors were developed and showed durable anti-tumor efficacy in RET-altered medullary thyroid cancer (5). BRAF V600E mutation, which constitutively activates MEK phosphorylation and MAPK/ERK signaling pathway, is the most prevalent genetic alteration in follicular-derived thyroid cancer, particularly in PTC (6, 7). Several inhibitors targeting BRAF mutation, such as vemurafenib and dabrafenib, have been discovered and show a dramatic response in advanced thyroid cancer patients harboring BRAF V600E mutation (8–10).
Hotspot mutations in the core promoter region of the telomerase reverse telomerase (TERT) gene are considered as the most frequent non-coding mutations in follicular-derived thyroid cancer (11). Numerous studies have demonstrated that TERT promoter mutations, especially when accompanied with BRAF V600E or RAS mutations, showed strong correlation to aggressive clinical characteristics, radioiodine refractory and poor clinical outcomes in patients with thyroid cancer (12–19). Several ETS factors, including GABPA, ETV1, ETV4, and ETV5 were reported to selectively bind to mutant TERT promoter and activate TERT expression (20–22). These findings raised the possibility that ETS inhibitors might be more sensitive to TERT promoter mutation-driven thyroid cancers. YK-4-279 is a well-described ETS factor inhibitor that has showed anti-tumor activity in Ewing’s sarcoma (23), prostate cancer (24), and neuroblastoma (25). Importantly, recent study showed that BRAF/TERT promoter double-mutated brain tumor cells were sensitive to YK-4-279 treatment, providing a therapeutic opportunity to manage brain tumor patients harboring both BRAF and TERT mutations (26). In this study we tested whether BRAF and/or TERT promoter mutations conferred sensitivity to YK-4-279 in thyroid cancer cells and whether this inhibitor could be served as a therapeutic agent for advanced thyroid cancers.
The human thyroid cancer cell lines, including KTC-1, KHM-5M, Hth7, ACT1, CAL62, WRO, and TTA1 were obtained from the Type Culture Collection of the Chinese Academy of Sciences (Shanghai, China). Human thyroid cancer MDA-T41 was purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA). The origins of human thyroid epithelial cell line Nthy-ori 3-1 and human PTC cell lines, TPC-1 and BCPAP, were as documented (27). BCPAP, KTC-1, KHM-5M, MDA-T41, and WRO were grown at 37°C in RPMI-1640 medium (Invitrogen, CA, USA) supplemented with 10% fetal bovine serum (FBS, #10270-106, Gibco, MD, USA). Nthy-ori3-1, TPC-1, ACT-1, CAL62, Hth7, and TTA1 were grown at 37°C in DMEM medium (Invitrogen, CA, USA) supplemented with 10% FBS.
The ETS inhibitor YK-4-279 (#S7679) was purchased from Selleck Chemicals (Houston, TX), dissolved in Dimethyl sulfoxide (DMSO) with a stock concentration of 10 mM and stored at −80°C. For in vitro study, YK-4-279 was used to treat cells for different concentrations as indicated for different experiments. For in vivo study, YK-4-279 was dissolved in DMSO to generated 15 mg/ml stocking solution and stored in −20°C. Working solution was dissolved as following proportion: 5% 15 mg/ml YK-4-279 solution + 40% PEG300 + 5% Tween 80 + 50% ddH2O. Vehicle was defined as working solution by replacing YK-4-279 with DMSO.
Total RNA was extracted from cultured cells using the TRIzol reagent (#15596-018; Ambion, Life Technologies, Carlsbad, CA) and reverse-transcribed to cDNA using RevertAid First Strand cDNA Synthesis Kit (#K1622; ThermoFisher). Gene expression was analyzed in triplicate using PowerUp™ SYBR™ Green Master Mix (#A25742; Applied Biosystem) on the Applied Biosystems QuantStudio™ 7 Pro Real-Time PCR System. Relative expression of each gene was calculated according to the 2−ΔΔCt method. GAPDH was used as an internal control for normalization. The primers for TERT and GAPDH were used as described previously (20).
The luciferase reporter gene constructs containing the wild-type and mutant TERT promoters (pGL4-TERTp-WT, pGL4-TERTp-C250T and pGL4-TERTp-C228T) were obtained from Addgene (#84924, #84925, and #84926, respectively). For promoter activity assay, cells were seeded in triplicate into a 24-well plate and then transfected with 300 ng pGL4 plasmids containing the TERT promoter using Lipofectamine 3000 (Invitrogen) for 24 h followed by 1 μM of YK-4-279 treatment for 16 h. Rinella luciferase (pRL-TK) plasmid was transfected simultaneously with plasmid mentioned above as the normalizing control. After treatments, cells were lysed, and the luciferase activities were measured using the Dual-Luciferase Reporter Assay System (Promega). Relative luciferase activities were calculated by dividing firefly luciferase values with Rinella luciferase values for each set of reading.
For cell proliferation assay, cells (1,000–2,500 cells per well) were seeded on a 96-well plate, and Cell Counting Kit-8 (CCK-8) assay was carried out at day 5 to evaluate cell proliferation following the manufacturer’s instruction. Briefly, at the end of each culture period, 10 µl of the CCK-8 solution (#K1018, ApexBio) was added to each well. After incubation for 2 h, the absorbance was read at 450 nm. For colony-formation assay, 400 of KHM-5M and Hth7 cells or 1,000 of BCPAP cells were plated in duplicate in six-well plates. YK-4-279 treatment starts at 12 h after cell seeding. Culture media with YK-4-279 were replaced every 3 days. The colonies were photographed, and the total number of colonies ≥100 μm in diameter was counted after 2 weeks of culture.
To examine the effect of YK-4-279 on thyroid cancer migration and invasion, cell migration and invasion assays were performed in triplicates using Transwell® chambers in 24-well plates following Ogasawara’s instruction (28). Transwell chambers with 8-μm pore polycarbonate membrane used for cell migration assay were obtained from Corning (Corning, NY). Chambers coated with Matrigel on the upper surface used for invasion assay were obtained from BD Biosciences (Franklin Lakes, NJ). Briefly, cells (5 × 104 KHM-5M and Hth7 cells or 1.5 × 105 BCPAP cells for migration assay; 1 × 105 KHM-5M and Hth7 cells for invasion assay) were suspended in 250 μl of serum-free medium and placed in the upper chamber, while the lower chamber was loaded with 750 μl of cell culture medium with 10% FBS. Cells were incubated in 37°C with 5% CO2 for 6 h (migration) and 16 h (invasion), respectively. After incubation, the non-invaded cells were removed from the upper surface by a cotton swab. The invaded cells on the lower surface of the membrane were fixed in 4% paraformaldehyde for 15 min, washed three times with PBS, and stained with 0.5% crystal violet. Cells from three microscopic fields were photographed and counted.
For cell cycle analysis, experiments were performed using a commercial cell cycle and apoptosis detection kit (#40301ES60, Yeasen, China) following its instruction. Briefly, cells were collected and washed with PBS, fixed in 70% ethanol for 2 h at 4°C and then incubated in the staining solution with propidium iodide and RNase for 1 h. The apoptosis analysis was determined by the Pacific Blue™ Annexin V (#640918, San Diego, CA). Stained cells were detected by flow cytometry. Cell cycle distributions and the percentage of apoptotic cells were determined with FlowJo software.
The activities of caspases-3 and caspase-7 were detected using the CellEvent™ Caspase 3/7 Green ReadyProbe™ Reagent (#R37111, Invitrogen) as previously reported (29). Briefly, cells were seeded on gelatin-coated slides in 24-well plate and treated with YK-4-279 at different doses (0, 0.3, and 1 µM) for 24 h. Cells were then incubated with 500 ul CellEvent™ Caspase 3/7 ReadyProbe™ Reagent dilution (1 drop/ml) for 30 min at 37°C followed by Hochest 33342 counterstaining for nuclei. Cells were then washed and fixed with 3.7% paraformaldehyde for 15 min and mounted with Anti-Fade Mounting media. Caspase-3/7 activated cells were observed under 200× magnificence with OLYMPUS IX83 Inverted Microscope (Olympus, Japan).
The cDNA libraries were constructed for each RNA sample using the TruSeq Stranded mRNA Library Prep Kit (Illumina, US) according to the manufacturer’s instructions. The mRNA library was prepared from total RNA using the NEBNext Small RNA Library Prep. Before read mapping, clean reads were obtained from the raw reads by removing the adaptor sequences and low-quality reads. The clean reads were then aligned to the human genome (GRCh38, NCBI) using Hisat2. HTseq was used to calculate gene counts, and the RPKM method was used to determine relative gene expression. Differential expression analysis of two conditions/groups (two biological replicates per condition) was performed using the DESeq2 R package (1.16.1). The resulting P-values were adjusted using the Benjamini and Hochberg’s approach for controlling the false discovery rate. Protein-coding genes larger than 1.5-fold alteration (0.587 for log2FC) with P-value <0.05 found by DESeq2 were defined Differential Expression Genes (DEGs) for further investigation in this study.
Gene ontology (GO) analysis was performed to elucidate the biological implications of the differentially expressed genes identified in the experiment. We downloaded the GO annotations from NCBI (http://www.ncbi.nlm.nih.gov/), UniProt (http://www.uniprot.org/), and the Gene Ontology (http://www.geneontology.org/). Fisher’s exact test was applied to identify significantly enriched GO categories.
All animal experiments were approved and performed according to the guidelines of the Institutional Animal Care and Use Committee (IACUC) of Sun Yat-sen University. BALB/c nude mice (Female, 4–6 weeks old) were purchased from Vital River (Beijing, China) and housed in the SPF barrier facilities of Sun Yat-sen University. To ensure the similarity of xenograft tumor size, we followed the protocol of Yu lab described previously (30). Briefly, KHM-5M (1 × 107) cells were injected subcutaneously into the axillary cavity of four-week-old nude mice and harvested when reaching 1 cm3. Necrotic area was dissected meanwhile the remaining compartment was fragmented as 1 mm3 pieces and transplanted into flanks of 6-week-old female nude mice (four mice for each group). When tumor sizes reached around 150 mm3, mice were treated daily with vehicle or 150 mg/kg of YK-4-279 by intraperitoneal injection. Tumor size was measured every day on the skin surface of the animal using a caliper, and tumor volume was calculated by the formula (L + W)3π/6. Mice were sacrificed when xenograft tumors in control group reached 1 cm3, and tumors were surgically removed, photographed, and weighted.
The immunohistochemical staining was performed with primary antibodies on 4 μm-thick formalin-fixed paraffin-embedded tissue specimens as previously described (31). The primary antibodies used were as follows: Anti-Human Ki67 antibody (#ab92742, Abcam), anti-cleaved Caspase-3 antibody (#9664, Cell Signaling Technology). Semi-quantification analysis was performed by calculating percentage of positive staining cell under 200× magnification with the following formula: % of positive cell = counts of positive staining tumor nuclei/counts of all nuclei.
Three independent experiments were performed for all the in vitro assays, and each was done in triplicate. Results are reported as mean ± standard deviation (SD). The significance of differences between two groups was assessed by the Student’s t test. The multiple t-test was used to evaluate the difference of tendency of xenograft tumor growth. Statistical analyses were performed with GraphPad Prism v8.0. All the P values were two-sided and P <0.05 was considered as statistically significant.
To investigate the effect of YK-4-279 on thyroid cancer, cell viability assay was first performed in nine thyroid cancer cell lines with different genetic backgrounds. As shown in Figure 1, treatment with 0.3 µM of YK-4-279 significantly inhibited the cell viability in most of the cell lines, and 3 or 10 µM of YK-4-279 almost abolished cell viabilities in all of the nine cell lines. The IC50 values for BRAF-mutant cell lines and BRAF-wild-type (WT) cell lines were 0.800 and 0.737 μM, respectively (P = 0.867); the IC50 values for TERT promoter mutation harboring cells and TERT-WT cells were 0.717 and 0.861 μM, respectively (P = 0.713). Moreover, there is no significant difference of the IC50 among the cell groups when we divided the cell lines into four groups based on the BRAF and TERT mutation status (Supplementary Figure 1). These data suggested that YK-4-279 suppressed thyroid cancer viability independent of BRAF V600E or TERT promoter mutations.
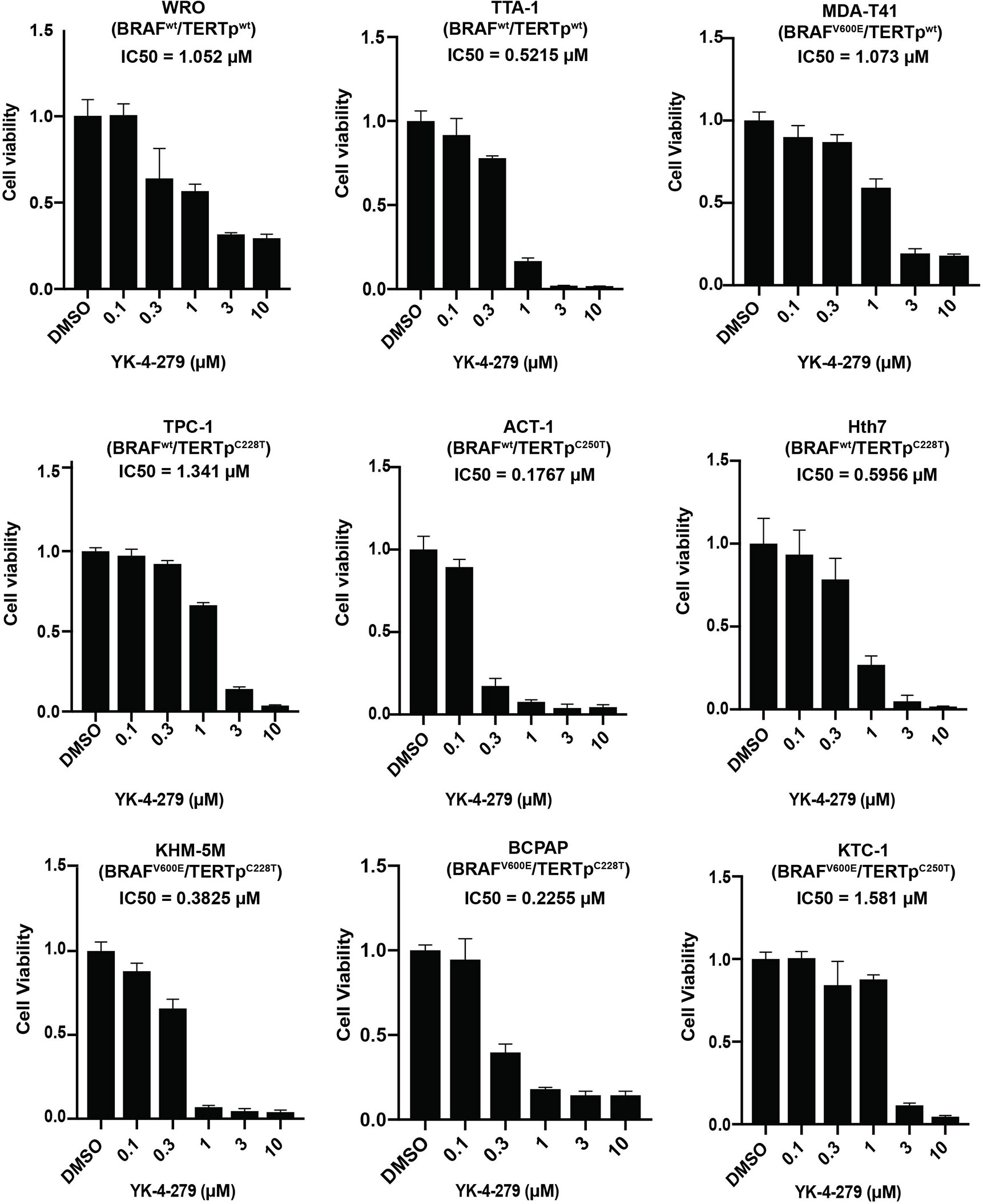
Figure 1 Effects of YK-4-279 on cell viability of thyroid cancer cells. Cells with different genetic backgrounds were treated by YK-4-279 with indicated concentrations for 5 days, followed by the CCK-8 assay. Drug with fresh medium was replenished every other day. Data represent relative cell growth at each indicated concentration of YK-4-279 in comparison with the control (0.1% DMSO). The results were shown as mean ± standard deviation (SD) of three independent experiments.
Since TERT is a master regulator in human cancer and plays vital roles in thyroid cancer, we hypothesized that the effect of YK-4-279 on thyroid cancer viability may be mediated, at least partially, by regulating TERT expression. To address this, we treated cells with YK-4-279 and tested the promoter activity and mRNA expression of TERT in thyroid cells. Luciferase assay showed that YK-4-279 reduced luciferase activities in thyroid cells transfected with wild-type, C228T, and C250T-mutant TERT promoter (Figure 2A). Consistently, treatment of thyroid cancer cells with YK-4-279 for 24 h significantly decreased TERT expression in either TERT promoter-WT or TERT promoter-mutant cell lines in a dose-dependent manner (Figure 2B). These results indicated that YK-4-279 regulated TERT transcription in a TERT promoter mutation-independent manner.
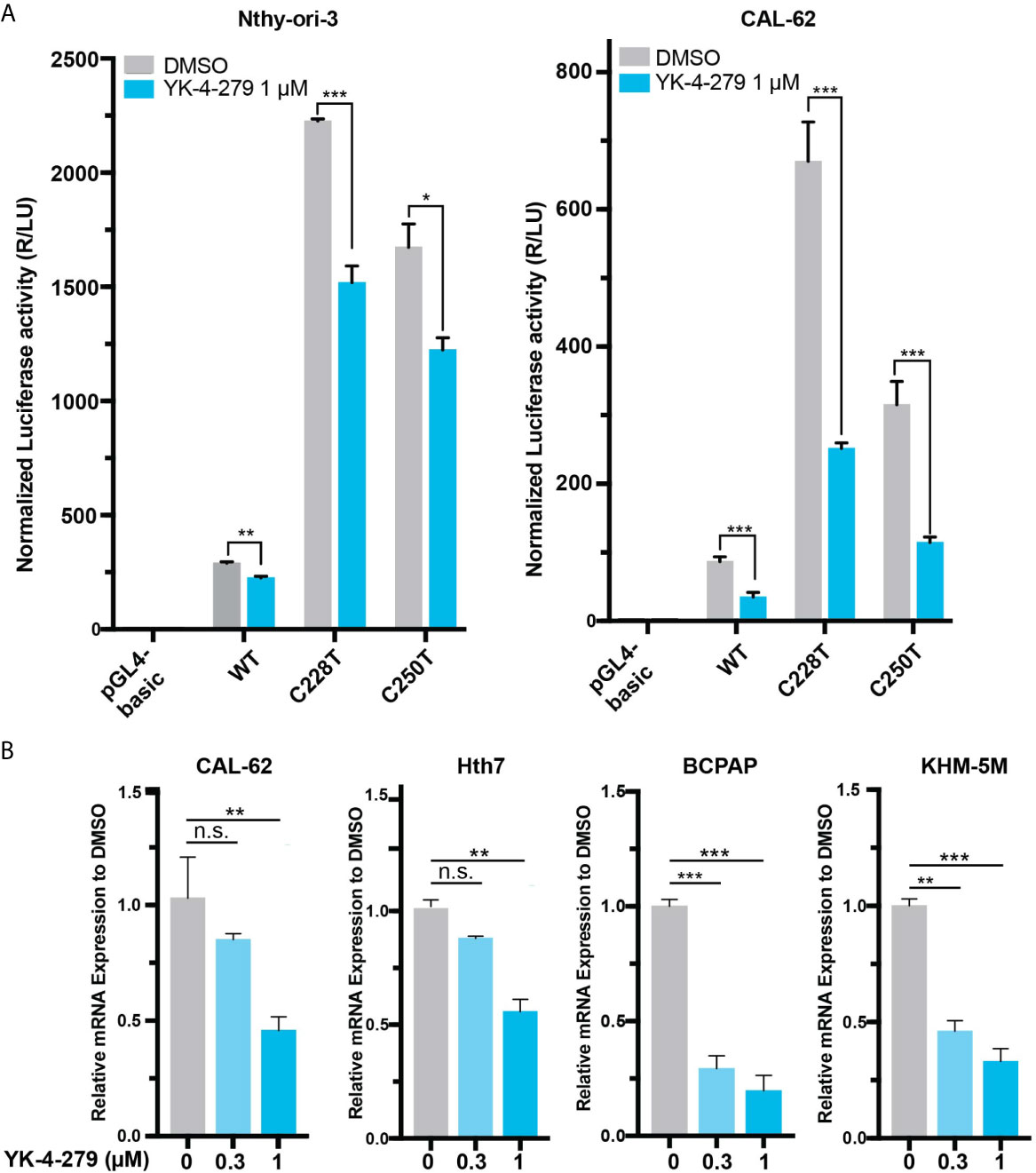
Figure 2 YK-4-249 regulates TERT expression in a TERT promoter mutation-independent manner. (A) YK-4-279 attenuated TERT promoter activities. Luciferase reporter plasmids carrying the wild-type (WT), C228T, or C250T mutant TERT promoters were transient transfected into Nthy-ori-3 and CAL62 cells, and then the cells were treated with 1 μM of YK-4-279 for 16 h, followed by luciferase reporter assay. (B) YK-4-279 treatment inhibited the mRNA expression of TERT. Four cancer cell lines harboring either BRAF V600E and/or TERT promoter mutation were treated with 1 μm of YK-4-279 for 24 h, followed by qRT-PCR for TERT mRNA expression detection. *P < 0.05, **P < 0.01, ***P < 0.001, by two-tailed Student’s t test. n.s., not significant. All the values represent the average ± standard deviation (SD) of triplicate samples from a typical experiment. Similar results were obtained in two additional independent experiments.
To comprehensively investigate the effects of YK-4-279 on aggressive behaviors of thyroid cancer cells, we next used in vitro models to examine the roles of YK-4-279 in oncogenic behaviors of aggressive thyroid cancer cell lines harboring both BRAF V600E and TERT promoter mutations (BCPAP and KHM-5M) or harboring TERT promoter mutation only (Hth7). As shown in Figure 3A, YK-4-279 inhibited anchorage-dependent survival of BCPAP, KHM-5M and Hth7 in plain six-well plates in a dose-dependent manner. Cell cycle is another important parameter for cell proliferation and survival. Previous papers indicated that YK-4-279 impeded cell proliferation via G2/M arrest in Ewing’s sarcoma (32) and neuroblastoma (25). We further investigated the role of YK-4-279 on cell cycle via flow cytometry. After administration of YK-4-279 for 16 h, significantly increased G2/M proportion was observed when treated with 1 μM of YK-4-279 in all cell lines that we tested, showing that YK-4-279 induced G2/M arrest in thyroid cell lines (Figure 3B).
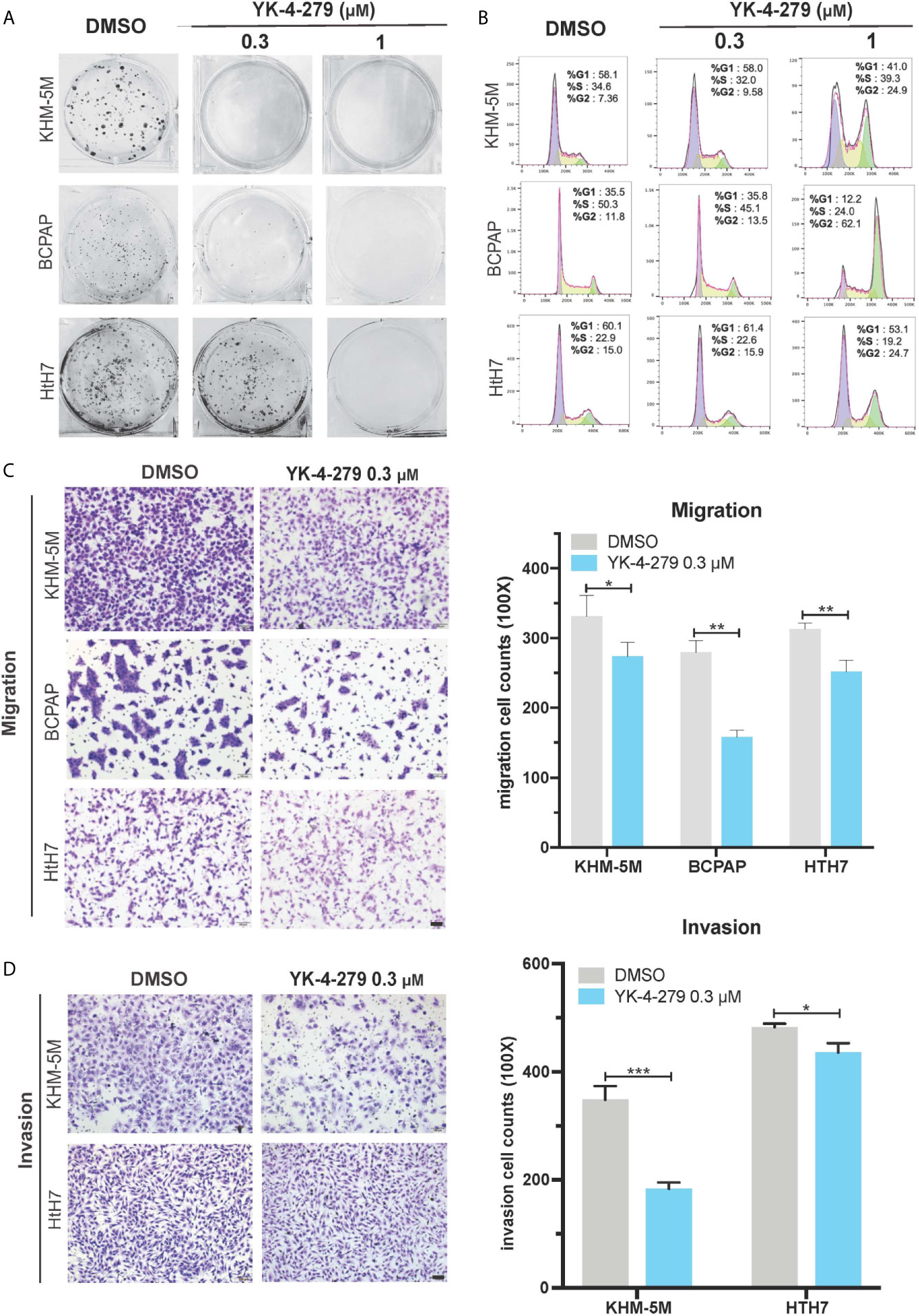
Figure 3 Effects of YK-4-279 on the oncogenic behaviors of thyroid cancer cells. Thyroid cancer cells were treated with DMSO or YK-4-279, followed by performance of monolayer colony formation (A), cell cycle analysis (B), Transwell cell migration (C) and cell invasion assays (D). “DMSO” in (A−D) represented 0.1% of DMSO in fresh culture media. The scale bars in panels (C, D) represent 100 μm. *P < 0.05, **P < 0.01, ***P < 0.001, by two-tailed Student’s t test. All the values represent the average ± standard deviation (SD) of triplicate samples from a typical experiment. Similar results were obtained in two additional independent experiments.
In addition to cell proliferation and survival, migration and invasion are crucial properties of cancer aggressiveness. Since no significant increase of G2/M proportion of cell cycle in thyroid cancer cell lines was observed after 16 h of YK-4-279 treatment at 0.3 μM (Figure 3B), we used this concentration to investigate the effect of cell migration and invasion of thyroid cancer via Boyden chamber with or without Matrigel, respectively. Our data showed that 0.3 μM of YK-4-279 significantly reduced cell migration and invasion in all tested cell lines (Figures 3C, D). Thus, we confirmed that YK-4-279 displayed regressive role on the oncogenic behaviors of thyroid cancer in a dose-dependent manner. Taken together, these data demonstrated that YK-4-279 significantly suppressed aggressive characteristics of thyroid cancer cells in vitro.
After treating thyroid cancer cells with YK-4-279 at different concentrations (0, 0.3, and 1 μM) for 24 h, increased floating apoptotic cells with round-shaped morphology were observed (Figure 4A), indicating that YK-4-279 may induce apoptosis in these cells. To confirm this, we performed PI/Annexin-V double staining assay in YK-4-279 treated cell lines and detected the apoptotic cells by flow cytometry. As a result, we found that the proportions of apoptotic (Annexin V-positive) cells were increased by four to nine times after treatment by 1 μM of YK-4-279 compared to the DMSO groups (Figure 4B, Supplementary Figure 2). Furthermore, we examined activity of caspase-3/7, two major apoptosis-related effector, in treated thyroid cell lines and observed a dose-dependent enhancement of caspase-3/7 activity in all tested cell lines (Figure 4C). These results showed that YK-4-279 induced cell apoptosis in thyroid cancer cells.
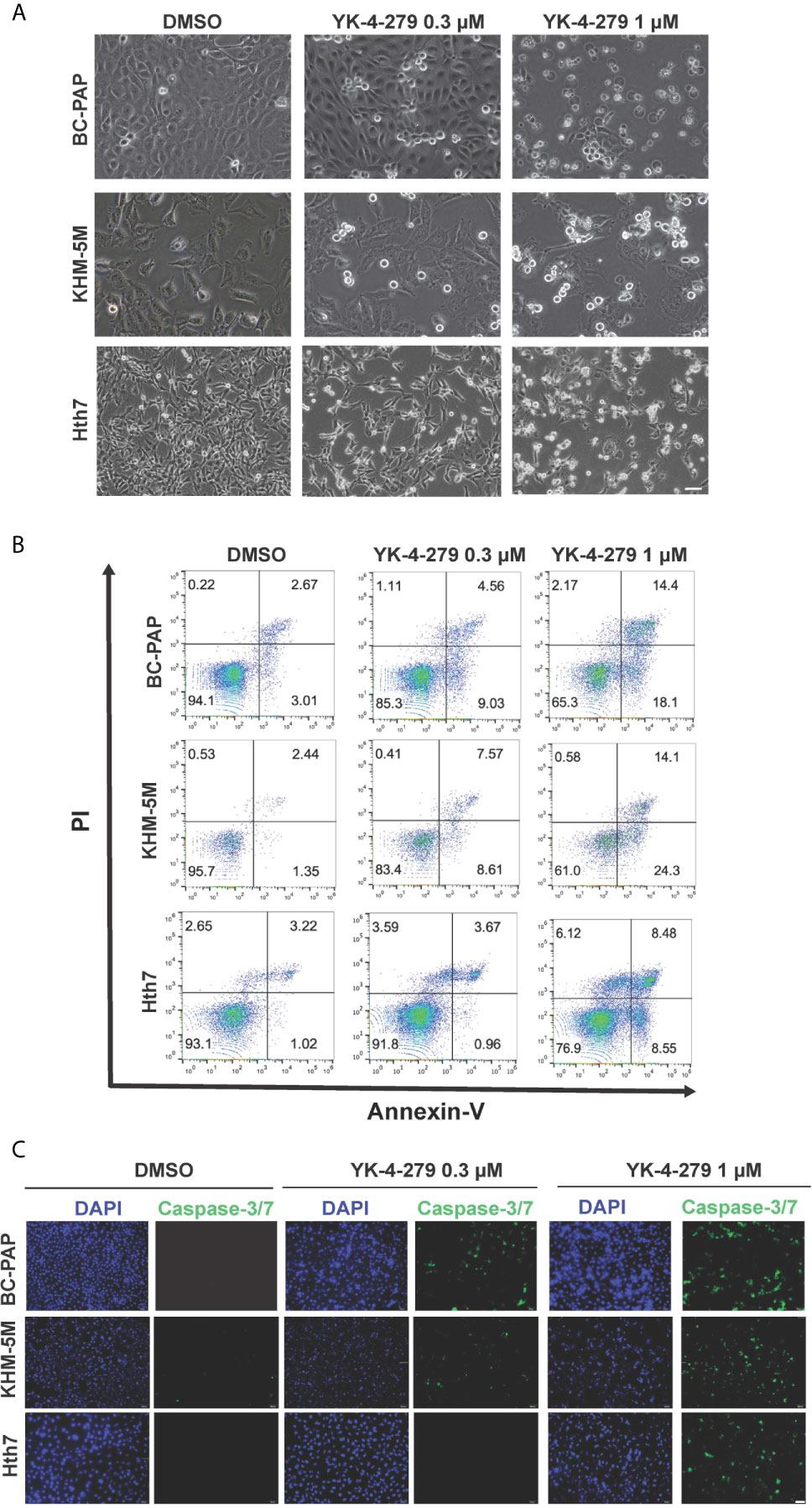
Figure 4 YK-4-279 induces cell apoptosis in thyroid cancer cells. Thyroid cancer cells (KHM-5M, BCPAP, and Hth7) were treated with DMSO or different dosages of YK-4-279 for 24 h. (A) Cell morphological changes (200×) after DMSO or YK-4-279 treatment. (B) Flow cytometry analysis of apoptosis by PI/Annexin-V staining. (C) Examination of caspase-3/7 activity after DMSO or drug treatment. The scale bars in panels (A, C) represent 100 μm. Similar results were obtained in two additional independent experiments.
To systematically explore gene expression profile and pathway affected by YK-4-279, RNA-seq was performed in KHM-5M cells treated with DMSO or YK-4-279 at different dosages (0.3 and 1 μM). Compared with the control group, 390 and 4,377 differentially expressed genes (DEGs) were identified in KHM-5M cells treated with 0.3 and 1 μM of YK-4-279, respectively. Furthermore, 214 DEGs were overlapped in both two treated groups (Figure 5A). The top 20 upregulated and downregulated DEGs were presented in Figure 5B. To interpret the crucial pathways affected by YK-4-279, gene ontology (GO) enrichment analysis based on the DEGs was performed. Results showed that the YK-4-279 treatment was significantly associated with biological processes including DNA replication, nuclear division, chromosome segregation, and meiotic cell cycle (Figure 5C). In addition, molecular function enrichment analysis revealed that catalytic activity, DNA helicase activity, DNA-dependent ATPase activity, and ATP-dependent DNA helicase activity were remarkably correlated with YK-4-279 (Figure 5D). Consistently, a number of DNA helicase genes were significantly downregulated after YK-4-279 treatment (Figure 5E), indicating a potentially pivotal role of DNA helicase activity in mediating the effect of YK-4-279 in thyroid cancer. Moreover, we checked the expression of several genes that were previously reported to be affected by YK-4-279 in Ewing’s sarcoma (32, 33) and found that the expressions of TERT and UBE2C (Ubiquitin Conjugating Enzyme E2-C) were reduced after YK-4-279 treatment in our cell lines (Figure 5F). The expressions of each ETS transcriptional factor were not changed in KHM-5M cells after treatment with YK-4-279 (Supplementary Figure 3).
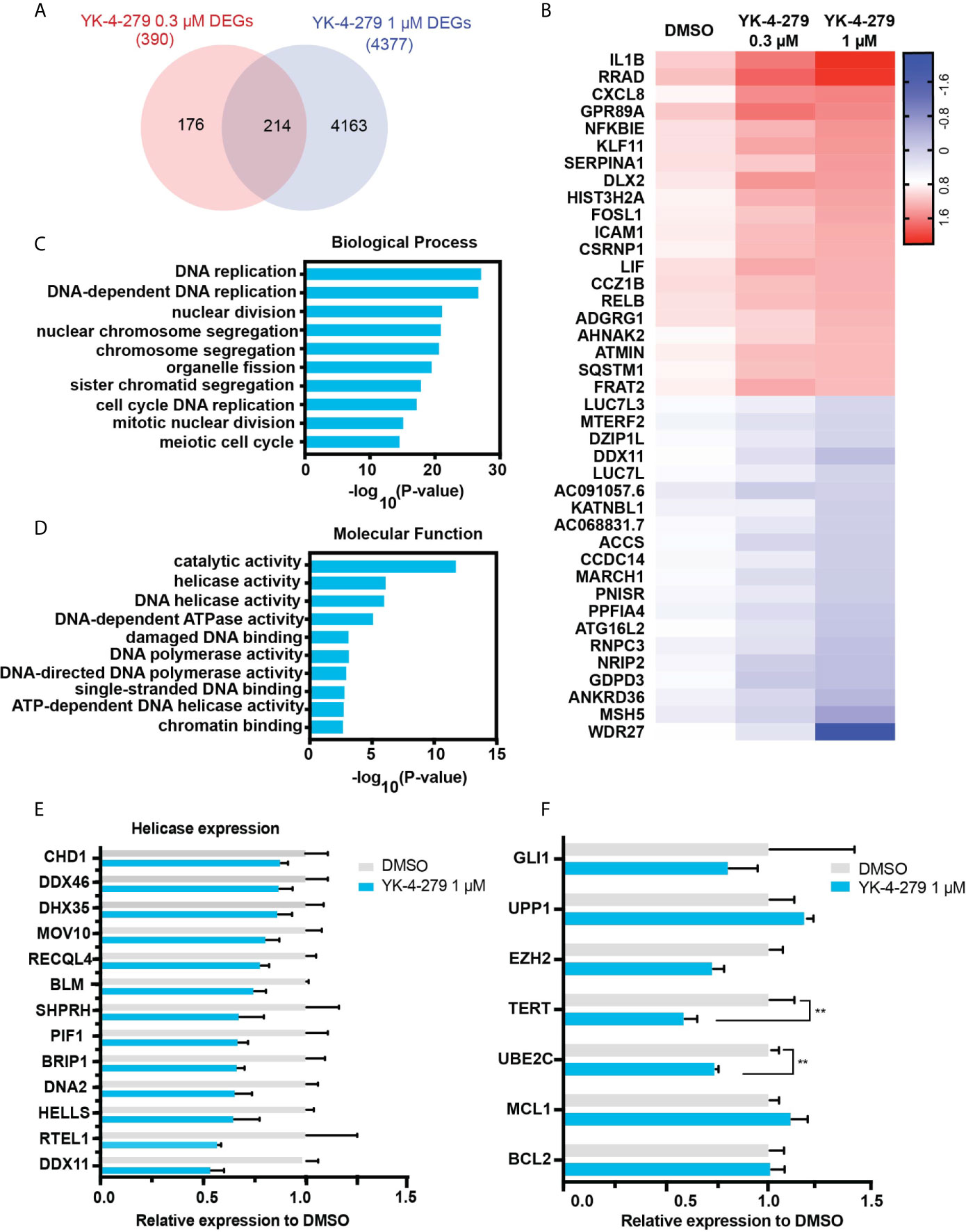
Figure 5 RNA-seq analysis for YK-4-279 treatment. (A) Differentially expressed genes (DEGs) in YK-4-279 treated KHM-5M cells compared with cells treated with DMSO. (B) Representative does-dependent DEGs. Blue referred to downregulated DEGs whereas Red referred to upregulated DEGs. (C, D) Gene ontology enrichment for biological process and molecular function analysis. (E) Significant downregulated DNA helicase in KHM-5M cells treated with 1 μM of YK-4-279 compared to the DMSO group. (F) mRNA expression of selected genes after DMSO or YK-4-279 treatment. ** refers to P < 0.01 by two-tailed Student’s t test.
To determine whether the anti-cancer effects of YK-4-279 observed for thyroid cancer cells was re-capitulated in vivo, a xenograft mouse model was established and treated with YK-4-279 by intraperitoneal injection (Figure 6A). As shown in Figures 6B, C, daily treatment with YK-4-279 at dosage of 150 mg/kg significantly suppressed the growth of xenograft tumors. Consistently, tumor weights in YK-4-279 treatment group were remarkably lower than those in the control group (Figure 6D). Along with the growth suppression, as shown in Figure 6E, immunohistochemistry of xenograft tumors showed that the expression of proliferation marker, Ki-67, was decreased; while the apoptosis marker, cleaved caspase-3, was increased in YK-4-279 treatment group compared to that in controls. Morphological feature of apoptosis was also observed by H&E staining in the drug treated tissue section (Figure 6E). These data showed that YK-4-279 was capable of reducing thyroid tumorigenesis in vivo.
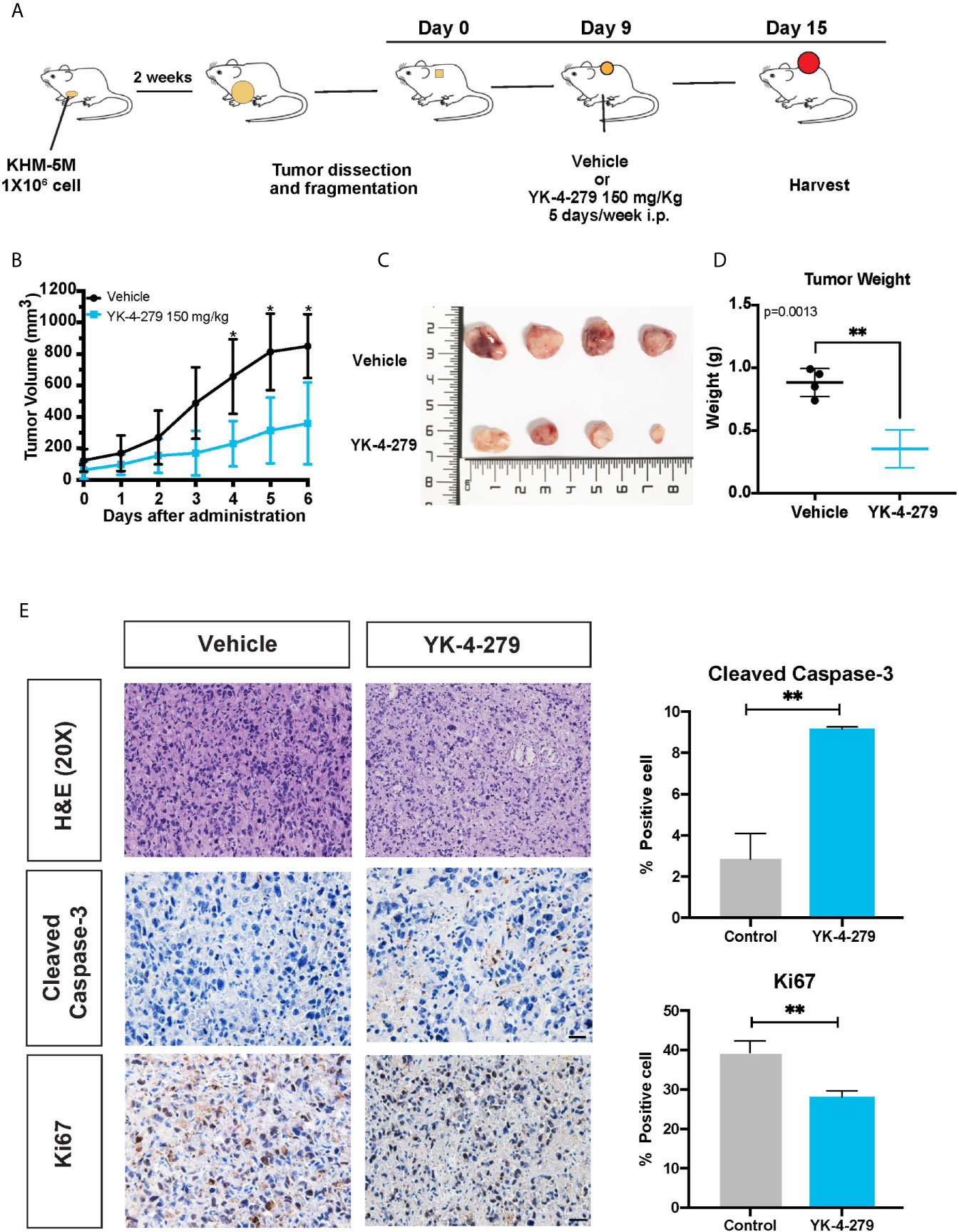
Figure 6 YK-4-279 inhibits in vivo tumor growth. (A) Diagram for the xenograft mice model in this study. (B) The tumor growth curves of xenograft model. (C) Photograph of the xenograft tumors (n = 4). The numbers on ruler refer to centimeter. (D) Weights of xenograft tumors. (E) Representative figures of H&E staining and immunohistochemistry for cleaved caspase-3 and Ki-67. Scale bar, 100 μm. Semi-quantification of immunohistochemistry staining was performed by calculating the percentage of positive staining cell. * refers to P < 0.05, ** refers to P < 0.01, by two-tailed Student’s t test. Triplicated experiments were performed with similar results obtained.
The anti-tumor activity of YK-4-279 was first reported in Ewing’s sarcoma and then confirmed by studies performed in several cancer types, including prostate cancer, neuroblastoma, lymphoma, and glioma (23–26, 34). This is the first study, to the best of our knowledge, to investigate the effect of YK-4-279 on thyroid cancer cells, and our data showed a significant inhibitory effect on the aggressive characteristics of thyroid cancer. Specifically, YK-4-279 treatment sharply suppressed cell growth, survival, migration, and invasion of thyroid cancer cells, and it induced G2/M cell cycle arrest and apoptosis in all the thyroid cancer cell lines we tested. Importantly, our data showed that YK-4-279 robustly suppressed tumor growth in a xenograft thyroid tumor model. These results demonstrated that YK-4-279 efficiently inhibited tumor growth and progression of thyroid cancer and exerted therapeutic potential for aggressive thyroid cancer management.
YK-4-279 was initially reported as a small molecule targeting the interaction of oncogenic protein EWS-FLI1 with RNA helicases DHX9 and DDX5 in Ewing’s sarcoma (23). Recently, several papers revealed that YK-4-279 exerted similar tumor-inhibitory effect in cancer cell lines within overexpression of other ETS-factors. Rahim et al. reported that YK-4-279 specifically reduced tumor growth and metastasis of ETV1-fusion positive prostate cancers by reducing ETV1 transcriptional activity (35). Similarly, exposure to TK-216, the clinical derivative of YK-4-279, reduced the interaction of ETS factors SPIB and SPI1 with these RNA helicases in B-cell lymphomas (34). Furthermore, YK-4-279 showed powerful inhibition of tumor proliferation and mitosis in ETS-FLI1 negative neuroblastoma, suggesting that YK-4-279 might disrupt protein interactions required for mitosis and proliferation (25). These data indicated that novel ETS factor-related mechanism may be involved in YK-4-279 induced solid tumor inhibition.
Several ETS factors, including ETS1, ETS2, ELK1, ELF3, ETV5, and EHF have been shown to be overexpressed during thyroid carcinogenesis, and inhibition of these ETS factors constrained cell proliferation, colony formation, migration, invasion and induced cell cycle arrest and apoptosis (36–42), which was consistent with what we observed after YK-4-279 treatment. Importantly, some classical oncogenes, such as PIK3CA, TWIST1, HER2, and HER3 were identified as the direct targets of ETS factors in thyroid cancer (40–42). Considering that the expression of each ETS factor was not changed after YK-4-279 treatment, the anti-tumor effect of YK-4-279 on thyroid cancer might be implemented by preventing the binding of certain ETS factors to their targets.
To comprehensively identify putative targets and pathway response to YK-4-279 treatment in thyroid cancer, RNA-profiling was performed in the present study. We found that DNA helicase activities were remarkably enriched in GO analysis, while decreased expression of several known DNA helicases was observed after treatment with YK-4-279 in our RNA-seq expression data. These results suggested that DNA helicases are likely to be the key targets and mediators of YK-4-279 treatment in thyroid cancer. We postulated that YK-4-279 may directly impede ETS factor binding to the promoter regions of DNA helicase genes since duplicate ETS motifs were identified as essential transcription regulatory elements in the promoters of multiple DNA helicase-encoding genes (43). However, the essential mechanism of YK-4-279 on DNA helicase regulation required to be further addressed.
Telomerase reverse transcriptase (TERT) is the catalytic component of the telomerase complex, which plays a key role in maintaining telomere length and cell immortality and in controlling cellular activities. Activation of TERT expression promoted cell proliferation without telomere shortening and was linked to cancer hallmark behaviors in several solid cancers (44). Inhibition of TERT expression in thyroid cancer cells significantly reduced cell viability, migration, and invasion in vitro and suppressed tumor growth in vivo (20, 45, 46). In the present study, we found that YK-4-279 treatment reduced TERT expression and the cells treated by YK-4-279 showed similar phenotypes with TERT silencing cells. Therefore, in addition to regulating DNA helicase expression, the anti-tumor activity of YK-4-279 can be explained partially by suppressing the oncogenic effect of TERT in thyroid cancer.
The two hotspot mutations in TERT promoter were frequently observed in patient with PTC, FTC, PDTC, and ATC, but not in MTC (11, 47). The BRAF V600E and the MAPK pathway was reported to activate several ETS transcription factors, the latter then selectively bonded to the mutant promoter of TERT and led to TERT activation in thyroid cancer (20–22). In contrast to the previous findings that BRAF/TERT promoter double-mutated brain tumor cells (26) were hypersensitive against the ETS inhibitor YK-4-279, we found that YK-4-279 reduced the expression of TERT and conferred the anti-tumor activity independent of TERT promoter mutations in thyroid cancer cells. This discrepancy between brain tumors and thyroid cancer can be explained by alternative mechanism of ETS factors on TERT regulation among these cancer types. Takahashi and his colleagues reported that EWSR1–ETS fusion activated hTERT expression in Ewing’s sarcoma by recruiting p300 to TERT promoter in an ETS factor binding site-independent manner (48). In thyroid cancer, phosphorylation and activation of ETS factor ELK1 by BRAF/MAPK pathway were necessary for thyroid-specific FOXE1 recruitment to TERT promoter via direct ELK1–FOXE1 interaction and this recruitment was independent from cis-acting mutations of TERT promoter (49). Thus, it is likely to the case that the interaction between ETS and certain transcription activators, such as FOXE1, play an important role in TERT activation in thyroid cancer cells while YK-4-279 inhibits TERT expression by disrupting ETS factors’ interaction. Further investigation on the mechanism of YK-4-279 on ETS recruitment of co-activators on TERT promoter in thyroid cancer is required.
In conclusion, our results demonstrate that YK-4-279 is powerful in arresting cell cycle and inducing apoptosis, leading to decreased cell growth and progression of thyroid cancer. The anti-tumor activity of YK-4-279 in thyroid cancer is independent of TERT promoter mutations and likely to be explained by inhibiting the expression of TERT and several DNA helicase genes. Although the exact mechanism needs to be further investigated, YK-4-279 is a promising therapeutic agent for the treatment of aggressive thyroid cancers.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: GEO repository and accession GSE171473 https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE171473.
The animal study was reviewed and approved by the Institutional Animal Care and Use Committee (IACUC) of Sun Yat-sen University.
HX and RL designed the study. JX, SL, PS, MC, SY, and SH carried out the experiments. JX, SL, and RL contributed to data analysis. JX prepared the figures and drafted the manuscript under the supervision of RL, YL, and HX. All authors contributed to the article and approved the submitted version.
This study was supported by grants from the National Natural Science Foundation of China (No. 81772850 and No. 82072952), the Guangzhou Science and Technology Project (No. 201803010057) and China Postdoctoral Science Foundation (No. 2020M683125).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2021.649323/full#supplementary-material
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global Cancer Statistics 2018: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin (2018) 68(6):394–424. doi: 10.3322/caac.21492
2. Ceolin L, Duval M, Benini AF, Ferreira CV, Maia AL. Medullary Thyroid Carcinoma Beyond Surgery: Advances, Challenges, and Perspectives. Endocr Relat Cancer (2019) 26(9):R499–518. doi: 10.1530/erc-18-0574
3. Fagin JA, Wells SA Jr. Biologic and Clinical Perspectives on Thyroid Cancer. N Engl J Med (2016) 375(11):1054–67. doi: 10.1056/NEJMra1501993
4. Xing M, Haugen BR, Schlumberger M. Progress in Molecular-Based Management of Differentiated Thyroid Cancer. Lancet (2013) 381(9871):1058–69. doi: 10.1016/s0140-6736(13)60109-9
5. Wirth LJ, Sherman E, Robinson B, Solomon B, Kang H, Lorch J, et al. Efficacy of Selpercatinib in RET-Altered Thyroid Cancers. N Engl J Med (2020) 383(9):825–35. doi: 10.1056/NEJMoa2005651
6. Wan PT, Garnett MJ, Roe SM, Lee S, Niculescu-Duvaz D, Good VM, et al. Mechanism of Activation of the RAF-ERK Signaling Pathway by Oncogenic Mutations of B-RAF. Cell (2004) 116(6):855–67. doi: 10.1016/s0092-8674(04)00215-6
7. Xing M. BRAF Mutation in Papillary Thyroid Cancer: Pathogenic Role, Molecular Bases, and Clinical Implications. Endocr Rev (2007) 28(7):742–62. doi: 10.1210/er.2007-0007
8. Kim KB, Cabanillas ME, Lazar AJ, Williams MD, Sanders DL, Ilagan JL, et al. Clinical Responses to Vemurafenib in Patients With Metastatic Papillary Thyroid Cancer Harboring BRAF(V600E) Mutation. Thyroid (2013) 23(10):1277–83. doi: 10.1089/thy.2013.0057
9. Subbiah V, Kreitman RJ, Wainberg ZA, Cho JY, Schellens JHM, Soria JC, et al. Dabrafenib and Trametinib Treatment in Patients With Locally Advanced or Metastatic BRAF V600-Mutant Anaplastic Thyroid Cancer. J Clin Oncol (2018) 36(1):7–13. doi: 10.1200/jco.2017.73.6785
10. Rosove MH, Peddi PF, Glaspy JA. BRAF V600E Inhibition in Anaplastic Thyroid Cancer. N Engl J Med (2013) 368(7):684–5. doi: 10.1056/NEJMc1215697
11. Liu R, Xing M. TERT Promoter Mutations in Thyroid Cancer. Endocr Relat Cancer (2016) 23(3):R143–55. doi: 10.1530/erc-15-0533
12. Liu R, Bishop J, Zhu G, Zhang T, Ladenson PW, Xing M. Mortality Risk Stratification by Combining BRAF V600E and TERT Promoter Mutations in Papillary Thyroid Cancer: Genetic Duet of BRAF and TERT Promoter Mutations in Thyroid Cancer Mortality. JAMA Oncol (2017) 3(2):202–8. doi: 10.1001/jamaoncol.2016.3288
13. Liu J, Liu R, Shen X, Zhu G, Li B, Xing M. The Genetic Duet of BRAF V600E and TERT Promoter Mutations Robustly Predicts Loss of Radioiodine Avidity in Recurrent Papillary Thyroid Cancer. J Nucl Med (2020) 61(2):177–82. doi: 10.2967/jnumed.119.227652
14. Melo M, da Rocha AG, Vinagre J, Batista R, Peixoto J, Tavares C, et al. TERT Promoter Mutations Are a Major Indicator of Poor Outcome in Differentiated Thyroid Carcinomas. J Clin Endocrinol Metab (2014) 99(5):E754–65. doi: 10.1210/jc.2013-3734
15. Yang X, Li J, Li X, Liang Z, Gao W, Liang J, et al. TERT Promoter Mutation Predicts Radioiodine-Refractory Character in Distant Metastatic Differentiated Thyroid Cancer. J Nucl Med (2017) 58(2):258–65. doi: 10.2967/jnumed.116.180240
16. Xing M, Liu R, Liu X, Murugan AK, Zhu G, Zeiger MA, et al. BRAF V600E and TERT Promoter Mutations Cooperatively Identify the Most Aggressive Papillary Thyroid Cancer With Highest Recurrence. J Clin Oncol (2014) 32(25):2718–26. doi: 10.1200/jco.2014.55.5094
17. Song YS, Lim JA, Choi H, Won JK, Moon JH, Cho SW, et al. Prognostic Effects of TERT Promoter Mutations Are Enhanced by Coexistence With BRAF or RAS Mutations and Strengthen the Risk Prediction by the ATA or TNM Staging System in Differentiated Thyroid Cancer Patients. Cancer (2016) 122(9):1370–9. doi: 10.1002/cncr.29934
18. Shen X, Liu R, Xing M. A Six-Genotype Genetic Prognostic Model for Papillary Thyroid Cancer. Endocr Relat Cancer (2017) 24(1):41–52. doi: 10.1530/erc-16-0402
19. Shi X, Liu R, Qu S, Zhu G, Bishop J, Liu X, et al. Association of TERT Promoter Mutation 1,295,228 C>T With BRAF V600E Mutation, Older Patient Age, and Distant Metastasis in Anaplastic Thyroid Cancer. J Clin Endocrinol Metab (2015) 100(4):E632–7. doi: 10.1210/jc.2014-3606
20. Liu R, Zhang T, Zhu G, Xing M. Regulation of Mutant TERT by BRAF V600E/MAP Kinase Pathway Through FOS/GABP in Human Cancer. Nat Commun (2018) 9(1):579. doi: 10.1038/s41467-018-03033-1
21. Bullock M, Lim G, Zhu Y, Åberg H, Kurdyukov S, Clifton-Bligh R. ETS Factor ETV5 Activates the Mutant Telomerase Reverse Transcriptase Promoter in Thyroid Cancer. Thyroid (2019) 29(11):1623–33. doi: 10.1089/thy.2018.0314
22. Song YS, Yoo SK, Kim HH, Jung G, Oh AR, Cha JY, et al. Interaction of BRAF-induced ETS Factors With Mutant TERT Promoter in Papillary Thyroid Cancer. Endocr Relat Cancer (2019) 26(6):629–41. doi: 10.1530/erc-17-0562
23. Erkizan HV, Kong Y, Merchant M, Schlottmann S, Barber-Rotenberg JS, Yuan L, et al. A Small Molecule Blocking Oncogenic Protein EWS-FLI1 Interaction With RNA Helicase A Inhibits Growth of Ewing’s Sarcoma. Nat Med (2009) 15(7):750–6. doi: 10.1038/nm.1983
24. Rahim S, Beauchamp EM, Kong Y, Brown ML, Toretsky JA, Üren A. YK-4-279 Inhibits ERG and ETV1 Mediated Prostate Cancer Cell Invasion. PloS One (2011) 6(4):e19343. doi: 10.1371/journal.pone.0019343
25. Kollareddy M, Sherrard A, Park JH, Szemes M, Gallacher K, Melegh Z, et al. The Small Molecule Inhibitor YK-4-279 Disrupts Mitotic Progression of Neuroblastoma Cells, Overcomes Drug Resistance and Synergizes With Inhibitors of Mitosis. Cancer Lett (2017) 403:74–85. doi: 10.1016/j.canlet.2017.05.027
26. Gabler L, Lötsch D, Kirchhofer D, van Schoonhoven S, Schmidt HM, Mayr L, et al. TERT Expression Is Susceptible to BRAF and ETS-Factor Inhibition in BRAF(V600E)/TERT Promoter Double-Mutated Glioma. Acta Neuropathol Commun (2019) 7(1):128. doi: 10.1186/s40478-019-0775-6
27. Yu S, Cao S, Hong S, Lin X, Guan H, Chen S, et al. miR-3619-3p Promotes Papillary Thyroid Carcinoma Progression via Wnt/β-Catenin pathway. Ann Transl Med (2019) 7(22):643. doi: 10.21037/atm.2019.10.71
28. Ogasawara M, Matsubara T, Suzuki H. Screening of Natural Compounds for Inhibitory Activity on Colon Cancer Cell Migration. Biol Pharm Bull (2001) 24(6):720–3. doi: 10.1248/bpb.24.720
29. Baek HB, Lombard AP, Libertini SJ, Fernandez-Rubio A, Vinall R, Gandour-Edwards R, et al. XPO1 Inhibition by Selinexor Induces Potent Cytotoxicity Against High Grade Bladder Malignancies. Oncotarget (2018) 9(77):34567–81. doi: 10.18632/oncotarget.26179
30. Wong CC, Kang W, Xu J, Qian Y, Luk STY, Chen H, et al. Prostaglandin E(2) Induces DNA Hypermethylation in Gastric Cancer In Vitro and In Vivo. Theranostics (2019) 9(21):6256–68. doi: 10.7150/thno.35766
31. Zhang Q, Agoston AT, Pham TH, Zhang W, Zhang X, Huo X, et al. Acidic Bile Salts Induce Epithelial to Mesenchymal Transition Via VEGF Signaling in Non-Neoplastic Barrett’s Cells. Gastroenterology (2019) 156(1):130–44.e10. doi: 10.1053/j.gastro.2018.09.046
32. Zöllner SK, Selvanathan SP, Graham GT, Commins RMT, Hong SH, Moseley E, et al. Inhibition of the Oncogenic Fusion Protein EWS-FLI1 Causes G(2)-M Cell Cycle Arrest and Enhanced Vincristine Sensitivity in Ewing’s Sarcoma. Sci Signal (2017) 10(499):eeam8429. doi: 10.1126/scisignal.aam8429
33. Selvanathan SP, Graham GT, Erkizan HV, Dirksen U, Natarajan TG, Dakic A, et al. Oncogenic Fusion Protein EWS-FLI1 Is a Network Hub That Regulates Alternative Splicing. Proc Natl Acad Sci USA (2015) 112(11):E1307–16. doi: 10.1073/pnas.1500536112
34. Spriano F, Chung EYL, Gaudio E, Tarantelli C, Cascione L, Napoli S, et al. The ETS Inhibitors YK-4-279 and TK-216 Are Novel Antilymphoma Agents. Clin Cancer Res (2019) 25(16):5167–76. doi: 10.1158/1078-0432.Ccr-18-2718
35. Rahim S, Minas T, Hong SH, Justvig S, Çelik H, Kont YS, et al. A Small Molecule Inhibitor of ETV1, YK-4-279, Prevents Prostate Cancer Growth and Metastasis in a Mouse Xenograft Model. PloS One (2014) 9(12):e114260. doi: 10.1371/journal.pone.0114260
36. Nakayama T, Ito M, Ohtsuru A, Naito S, Nakashima M, Sekine I. Expression of the Ets-1 Proto-Oncogene in Human Thyroid Tumor. Mod Pathol (1999) 12(1):61–8. doi: 10.1038/modpathol.3880328
37. de Nigris F, Mega T, Berger N, Barone MV, Santoro M, Viglietto G, et al. Induction of ETS-1 and ETS-2 Transcription Factors Is Required for Thyroid Cell Transformation. Cancer Res (2001) 61(5):2267–75.
38. Kong Y, Yin J, Fu Y, Chen Y, Zhou Y, Geng X. Suppression of Elk1 Inhibits Thyroid Cancer Progression by Mediating PTEN Expression. Oncol Rep (2018) 40(3):1769–76. doi: 10.3892/or.2018.6554
39. Chen H, Chen W, Zhang X, Hu L, Tang G, Kong J, et al. E26 Transformation (ETS)−Specific Related Transcription Factor−3 (ELF3) Orchestrates a Positive Feedback Loop That Constitutively Activates the MAPK/Erk Pathway to Drive Thyroid Cancer. Oncol Rep (2019) 41(1):570–8. doi: 10.3892/or.2018.6807
40. Meng D, Li Z, Ma X, Wu L, Fu L, Qin G. ETV5 Overexpression Contributes to Tumor Growth and Progression of Thyroid Cancer Through PIK3CA. Life Sci (2020) 253:117693. doi: 10.1016/j.lfs.2020.117693
41. Puli OR, Danysh BP, McBeath E, Sinha DK, Hoang NM, Powell RT, et al. The Transcription Factor ETV5 Mediates BRAFV600E-Induced Proliferation and TWIST1 Expression in Papillary Thyroid Cancer Cells. Neoplasia (2018) 20(11):1121–34. doi: 10.1016/j.neo.2018.09.003
42. Lv Y, Sui F, Ma J, Ren X, Yang Q, Zhang Y, et al. Increased Expression of EHF Contributes to Thyroid Tumorigenesis Through Transcriptionally Regulating HER2 and HER3. Oncotarget (2016) 7(36):57978–90. doi: 10.18632/oncotarget.11154
43. Uchiumi F, Watanabe T, Tanuma S. Characterization of Various Promoter Regions of the Human DNA Helicase-Encoding Genes and Identification of Duplicated Ets (GGAA) Motifs as an Essential Transcription Regulatory Element. Exp Cell Res (2010) 316(9):1523–34. doi: 10.1016/j.yexcr.2010.03.009
44. Low KC, Tergaonkar V. Telomerase: Central Regulator of All of the Hallmarks of Cancer. Trends Biochem Sci (2013) 38(9):426–34. doi: 10.1016/j.tibs.2013.07.001
45. Teng L, Specht MC, Barden CB, Fahey TJ 3rd. Antisense hTERT Inhibits Thyroid Cancer Cell Growth. J Clin Endocrinol Metab (2003) 88(3):1362–6. doi: 10.1210/jc.2002-021222
46. Maggisano V, Celano M, Lepore SM, Sponziello M, Rosignolo F, Pecce V, et al. Human Telomerase Reverse Transcriptase in Papillary Thyroid Cancer: Gene Expression, Effects of Silencing and Regulation by BET Inhibitors in Thyroid Cancer Cells. Endocrine (2019) 63(3):545–53. doi: 10.1007/s12020-018-01836-2
47. Liu X, Bishop J, Shan Y, Pai S, Liu D, Murugan AK, et al. Highly Prevalent TERT Promoter Mutations in Aggressive Thyroid Cancers. Endocr Relat Cancer (2013) 20(4):603–10. doi: 10.1530/erc-13-0210
48. Takahashi A, Higashino F, Aoyagi M, Yoshida K, Itoh M, Kyo S, et al. EWS/ETS Fusions Activate Telomerase in Ewing’s Tumors. Cancer Res (2003) 63(23):8338–44.
Keywords: YK-4-279, telomerase reverse transcriptase, thyroid cancer, apoptosis, E-twenty-six transcription factor
Citation: Xue J, Li S, Shi P, Chen M, Yu S, Hong S, Li Y, Liu R and Xiao H (2021) The ETS Inhibitor YK-4-279 Suppresses Thyroid Cancer Progression Independent of TERT Promoter Mutations. Front. Oncol. 11:649323. doi: 10.3389/fonc.2021.649323
Received: 04 January 2021; Accepted: 01 March 2021;
Published: 16 June 2021.
Edited by:
Shicheng Guo, University of Wisconsin-Madison, United StatesReviewed by:
Lingli Zhou, Johns Hopkins Medicine, United StatesCopyright © 2021 Xue, Li, Shi, Chen, Yu, Hong, Li, Liu and Xiao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Haipeng Xiao, eGlhb2hwQG1haWwuc3lzdS5lZHUuY24=; Rengyun Liu, bGl1cnk5QG1haWwuc3lzdS5lZHUuY24=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.