 Isaacson B. Adelani
Isaacson B. Adelani Oluwakemi A. Rotimi
Oluwakemi A. Rotimi Emmanuel N. Maduagwu2
Emmanuel N. Maduagwu2 Solomon O. Rotimi
Solomon O. Rotimi- 1Department of Biochemistry, Covenant University, Ota, Nigeria
- 2Department of Biochemistry, Chrisland University, Abeokuta, Nigeria
Hepatocellular carcinoma (HCC) is a unique type of liver cancer instigated by underlying liver diseases. Pre-clinical evidence suggests that HCC progression, like other cancers, could be aided by vitamin D deficiency. Vitamin D is a lipid-soluble hormone usually obtained through sunlight. Vitamin D elucidates its biological responses by binding the vitamin D receptor; thus, promoting skeletal mineralization, and maintain calcium homeostasis. Other reported Vitamin D functions include specific roles in proliferation, angiogenesis, apoptosis, inflammation, and cell differentiation. This review highlighted studies on vitamin D’s functional roles in HCC and discussed the specific therapeutic targets from various in vivo, in vitro and clinical studies over the years. Furthermore, it described recent advancements in vitamin D’s anticancer effects and its metabolizing enzymes’ roles in HCC development. In summary, the review elucidated specific vitamin D-associated target genes that play critical functions in the inhibition of tumorigenesis through inflammation, oxidative stress, invasion, and apoptosis in HCC progression.
Introduction
Hepatocellular carcinoma (HCC) is a unique type of liver cancer instigated by underlying liver diseases. In general, liver cancer constitutes a substantial public health problem that ranks as the sixth most commonly diagnosed cancer and the third most common cause of cancer-related mortality in 2020 (1). Although liver cancer occurs in both genders, the incidence and mortality rates in males are 2 to 3 times higher than in females (1). The loss of the liver’s regenerative ability exacerbates HCC progression, which subsequently potentiates organ failure (2). This loss of regenerative capacity is further compounded by the disruption of various pathways associated with the pathogenesis and progression of HCC, thereby making HCC an outcome of a complex cascade of events (3). Furthermore, the increasing incidence of HCC is mainly associated with viral infections, including hepatitis B (HBV) and C viruses (HCV), as well as other risk factors like non-alcoholic fatty liver disease (4, 5) and mycotoxin exposure (6–9). Aside from these biotic, lifestyle, and environmental factors, pre-clinical evidence suggests that HCC progression, like many other cancers, could be facilitated by vitamin D (VD) deficiency and germline genetic variants in the Vitamin D receptor (VDR) gene, which has been shown to influence the progression of hepatitis to HCC (10, 11). Also, an epidemiological study showed that increased maternal ultraviolet (UV) exposure is associated with a reduced risk of hepatoblastoma in offspring (12). Additionally, evidence from the SEER data showed that the incidence of HCC in the United States is associated with ambient UV exposure (13). Hence, this premise supports the VD - cancer hypothesis and further augments the roles of vitamin D metabolism in hepatocellular carcinogenesis (2).
Vitamin D (VD) is a lipid-soluble hormone usually obtained through the exposure of skin to sunlight. Several factors, including skin pigmentation and a modern lifestyle, could limit VD formation, thus causing VD deficiency (14). In VD synthesis, sunlight UV (B) (280–315 nm) exposure on the skin activates 7-dehydrocholesterol to pre-vitamin D3 and eventually cholecalciferol (VD3) (2). Similarly, UV (B) exposure to ergosterol in plants and fungi produces another form of vitamin D, ergocalciferol (VD2) (15). Asides from the endogenous synthesis of VD, VD3 can also be obtained from diets while VD2 is principally used during vitamin D fortification. Both forms of VD are naturally inactive and are activated via hydroxylation. After synthesis, VD binding protein (DBP) binds VD and transports it to the liver, where hydroxylation at carbon-25 metabolizes VD to 25-hydroxyvitamin D (25(OH)D) through 25-hydroxylase. In this first phase of VD metabolism, hydroxylation occurs predominantly in the hepatic cells although extrahepatic VD hydroxylation reportedly occurs in other tissues with evident 25-hydroxylase activities (16, 17). Most importantly, during this first hydroxylation step, an ubiquitous mitochondrial 25-hydroxylase, CYP27A1 does not hydroxylate VD2 whereas, CYP2R1 usually located in the liver and testes hydroxylates both forms of VD (15). Equally, Zhu et al. (18) reported CYP2R1 as a major but not the only 25-hydroxylase. After the first hydroxylation, the glomerulus filters 25(OH)D transported into the kidney and converts it to a steroid hormone (the active form of VD), 1α, 25 (OH)2D (calcitriol), through 25(OH)D-1α-hydroxylase (19). This metabolic activity in the kidney signifies the second stage of VD hydroxylation. Although 1α-hydroxylation occurs predominantly in the kidney, peripheral tissues including the skin and lymph nodes exhibit extra-renal production of the steroid hormone (20). Finally, in a bid to activate VD’s biological response to regulate gene expression, calcitriol binds VDR (17) in a binding sequence that allows the effective functioning of retinoid X (RXR). RXR belongs to the nuclear receptor family and a member of the steroid/thyroid hormone, primarily functioning as transcription factors (21). RXR also plays essential roles in metabolism and cell differentiation (21). Hence, VD binding enables the VDR – RXR interaction, leading to VD-related functions through gene transcription (22). Thus, VD’s biological action is dependent on VDR, RXR, and the availability of VD (23).
Asides from primary functions, which include promoting skeletal mineralization and maintenance of calcium homeostasis, VD performs pro-apoptotic, pro-differentiation, anti-angiogenetic, anti-proliferative, anti-invasive, and anti-metastatic functions (24). Reports show that VD is an indicator of HCC prognosis and could be vital in predicting HCC patients’ mortality (25). Meanwhile, VD deficiency is fast becoming a global public health challenge (26), and it is continuously associated with an ‘all-cause and cause-specific mortality, despite differences in the VD baseline levels across the world (27, 28). Consequently, there are pieces of evidence showing connections between VD deficiency and HCC progression. For instance, Gaksch et al. (29) meta-analysis proposed an inverse relationship between serum VD (25(OH)D3) level and HCC risk; thereby, suggesting VD’s prospective therapeutic ability in managing HCC. Moreover, increased bioavailability of circulating 25(OH)D3 was also associated with HCC survival as against total or free VD level (30). In contrast, Liu et al. (31) reported that increased 25(OH)D level was associated with an increased risk of HCC incidence. However, they observed that genetic variations related to VD metabolism could influence HCC tumor response, survival, and mortality.
Despite the reported association with HCC development, contrasting reports suggest that baseline VD level could play little or no role in cirrhosis-linked HCC (32). Therefore, this review highlights the in vivo, in vitro, and clinical studies on VD therapeutic targets in HCC. Furthermore, it discussed the significant limitations and possible solutions in using VD as therapeutics.
VD, VDR, and HCC Pathological Conditions
The progression of HCC and pathological conditions like liver cirrhosis are linked to VD deficiency; hence, suggesting that decreased 25(OH)D is associated with poor liver disease prognosis (33). According to Berkan-Kawińska et al. (34) and Yang et al. (35), patients with liver cirrhosis, HBV, and HCV have decreased 25(OH)D levels and could benefit from VD supplementation. VD deficiency has also been linked with infections in patients with HCV-associated liver cirrhosis (36) and the VD deficiency-associated polymorphisms, like rs1993116, rs10741657, rs2282679, rs7944926, and rs12785878, linked with HCV-related HCC (37). This study by Lange et al. (37) also showed that reduced 25(OH)D3 levels in HCV-related HCC patients is associated with genetic variations of CYP2R1, GC, and DHCR7. While the circulating form of VD (25(OH)D3) instigates the hormone’s anti-HCV capacity (38), the active form of VD (1α, 25 (OH)2D3) induces CYP24A1 expression in a VDR-dependent manner. However, VDR expression, repressed by HBV transcript upregulation, affects VD’s binding to the receptor (39). Also, chronic HBV patients are at a higher risk of increased VD deficiency (40).
HCV and HCV-related HCC patients had lower levels of VD and VDR compared to healthy individuals (41). In the same vein, Falleti et al. (42) reported that VDR polymorphisms are associated with the occurrence of HCC in liver cirrhosis patients, specifically in those with alcoholic etiology. In the study, HCC was linked with the b allele of the BsmI A>G (B/b) polymorphism and the T allele of the TaqI T>C (T/t) polymorphism (42). Several studies have reported relationships between VDR polymorphisms and HCC pathological conditions. In a Chinese population hospital-based case-control study, VDR rs2228570 and DBP rs7041 polymorphisms vary between HBV-related HCC patients and healthy individuals thus, suggesting a relationship with increased risk of HBV-related HCC (11). A meta-analysis strengthened these observations, which indicated that VDR rs7975232 and rs2228570 polymorphisms are associated with HCC (43).
Although a non-association of VDR polymorphism and risk of HBV infection in Vietnamese HBV patients was reported by Hoan et al. (44), they suggested that Apal VDR polymorphism (rs7975232) could be associated with clinical outcomes and disease progression. Incidentally, Apa1 VDR polymorphism was shown to be associated with HCC in HCV-cirrhotic patients (45). On the contrary, SNPs of VDR at BsmI, ApaI, and TaqI loci showed no difference between HCC and non-HCC patients, according to Yao et al. (46). However, the authors reported a higher frequency of VDR FokI C > T polymorphism in HCC patients. Also, HCC patients showed a higher prevalence of FokI TT genotype, which is a risk factor for HCC development (46). Interestingly, the FokI TT genotype was also associated with HCC clinicopathology characterized by increased serum alpha-fetoprotein (AFP), advanced tumor stage, cirrhosis, and lymph node metastasis (47). Besides, the Fok1 T allele is linked with a predisposition to reduced VD levels and an increased probability of cancer development in HCV patients (47).
Since there are associations between VD, VDR, and HCC pathological conditions, understanding VD-related mechanisms and therapeutic targets in HCC progression could further substantiate existing evidence and highlight the roles of the hormones in hepatocarcinogenesis.
Therapeutic Effects of Vitamin D in HCC
In Vitro Studies
Over the years, there have been reports of HCC’s resistance to many drugs. An example is resistance to Everolimus, which acts as an mTOR (mechanistic target of rapamycin) inhibitor. mTOR is a serine/threonine-protein kinase found in the PI3K-related kinase (PIKK) family. mTOR’s activation plays critical roles in cell metabolism, proliferation, and HCC progression (48). Hence, inhibiting mTOR is one of the suggested therapeutic targets used to prevent and manage HCC (49). A recent study reported that calcitriol treatment could restore HCC cell sensitivity, thus becoming less resistant to everolimus (50). The reduced cell resistance modulated through the epithelial-mesenchymal transition pathway increased expression of miRNA-375 and decreased expression of target genes, including Metadherin (MTDH), Yes-associated protein-1 (YAP-1), and cellular Myc (c-MYC) (50).
Likewise, Huang et al. (51) investigated calcitriol’s effects on Histone deacetylase 2 (HDAC2) and cell cycle markers to explore the senescence and apoptotic pathway involved in HCC. According to Huang et al. (51), silencing the HDAC2 gene, which is usually highly expressed in HCC tumors, enhances calcitriol’s inhibitory effects. Equally, 1,25(OH)2D3 treatment decreased the expression of HDAC2 with a dose-dependent increased expression of cell cycle marker, cyclin-dependent kinase inhibitor (p21(WAF1/Cip1)) (51). This result suggests that VD could be a potential therapeutic agent in managing HCC via cell cycle modulation. However, VD3 treatment significantly increased Thioredoxin Interacting Protein (TXNIP); thus, enhancing apoptosis while reducing cell proliferation and thioredoxin activities (52). TXNIP is a tumor suppressor gene usually downregulated in HCC; therefore, instigating HCC progression (53). Furthermore, in its hormonal form, VD (1,25(OH)2D3) exhibits anti-proliferative ability and increases the apoptotic ratio in HCC cell lines (54). 1000 nM VD treatment also showed potential cell growth ameliorating ability in HCC cell lines according to the study of Xu et al. (51). Although VD reduced cell viability and proliferation while activating apoptosis, the effects were well enhanced when co-administered with Astemizole (a non-sedating antihistamine). In the same study, VD’s anti-invasive, anti-tumor, and cell migration inhibitory properties were highlighted (55).
Recently, a combination of VD2 analog, Doxercalciferol, and Carnosic acid-enhanced Sorafenib induced HCC cell death through blockage of autophagosomes/lysosomes fusion while also activating autophagy and apoptosis (56). To further elucidate the more apparent HCC related mechanisms, Wang et al. (57) showed that 1,25(OH)2D3 reversed biological alterations of hepatic progenitor cells caused by Aflatoxin B1 (AFB1) in WB-344 cells. Furthermore, VD3 attenuated the activation of Protein kinase B (Akt) while suppressing the expression of cysteine-rich angiogenic inducer 61 (CYR61) and connective tissue growth factor (CTGF), thus indicating anti-tumor effects. Calcitriol, also showed inhibitory roles in HCC by suppressing the hepatocyte growth factor (HGF) and its receptor, c-met (58).
Therefore, it can be deduced from these in vitro studies as summarized in Table 1 that VD acts as an anti-tumor agent in HCC, and it could regulate tumor growth/progression through cell cycle modulation and mTOR inhibition.
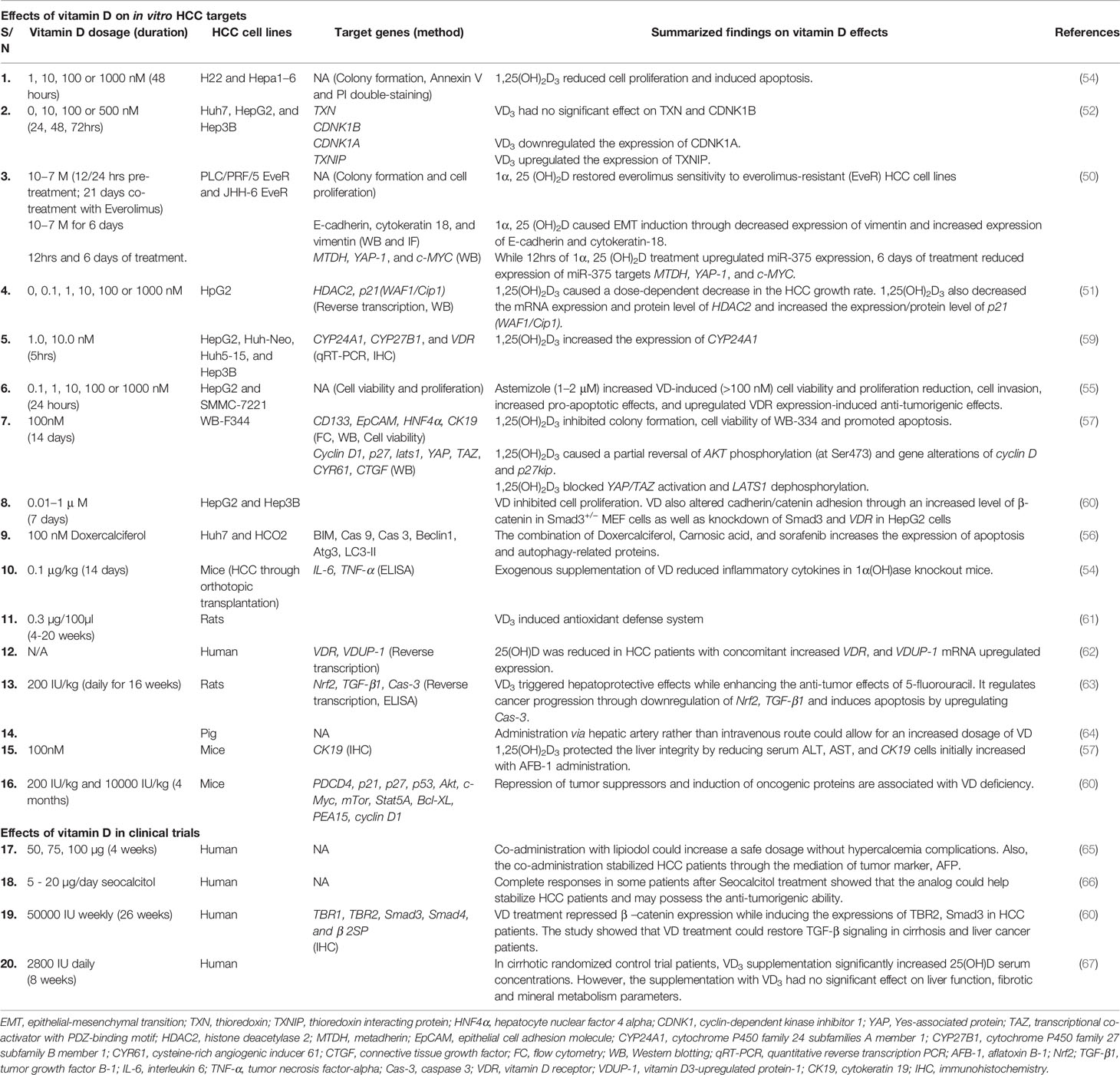
Table 1 Summary of the effects of vitamin D on HCC targets genes.
In Vivo Studies
VD’s anti-inflammatory role in carcinogenesis is now considered an established mechanism of its anti-carcinogenesis property (68). For example, in an activated inflammatory response, dietary VD significantly ameliorated cytokine production observed with diethylnitrosamine (DEN) effects in rats (69). Similarly, a deficient state of 1,25(OH)2D3 triggers inflammatory cytokines production through STAT3 activation (Figure 1) (50). Guo et al. (54) also linked the anti-tumor ability of 1,25(OH)2D3 with the availability of p27kip1 in mice (54). p27kip1 is a cyclin-dependent kinase inhibitor known for its prognostic roles in carcinogenesis. Asides from functioning as a tumor suppressor, p27kip1 promotes apoptosis, regulates tumor drug resistance, protects against inflammatory effects, and enhance cell differentiation as summarized in Table 1 (70). Also, the loss of p27kip1 could negatively affect the anti-tumor ability of 1,25(OH)2D3. The ablation of kidney VD metabolic enzyme, 25(OH)D3-1α-hydroxylase, resulted in tumor formation and increased inflammatory responses in mice (54). However, in the DEN-induced hepatocarcinogenesis mice model, loss of VD3 upregulated protein 1 (VDUP1) promotes carcinogenesis through increased cell proliferation, expression of tumor necrosis factor-α (TNF-α) and nuclear factor-kappa B (NF-kB) activation, thus suggesting VDUP1 as a potential anti-proliferative therapy target (71).
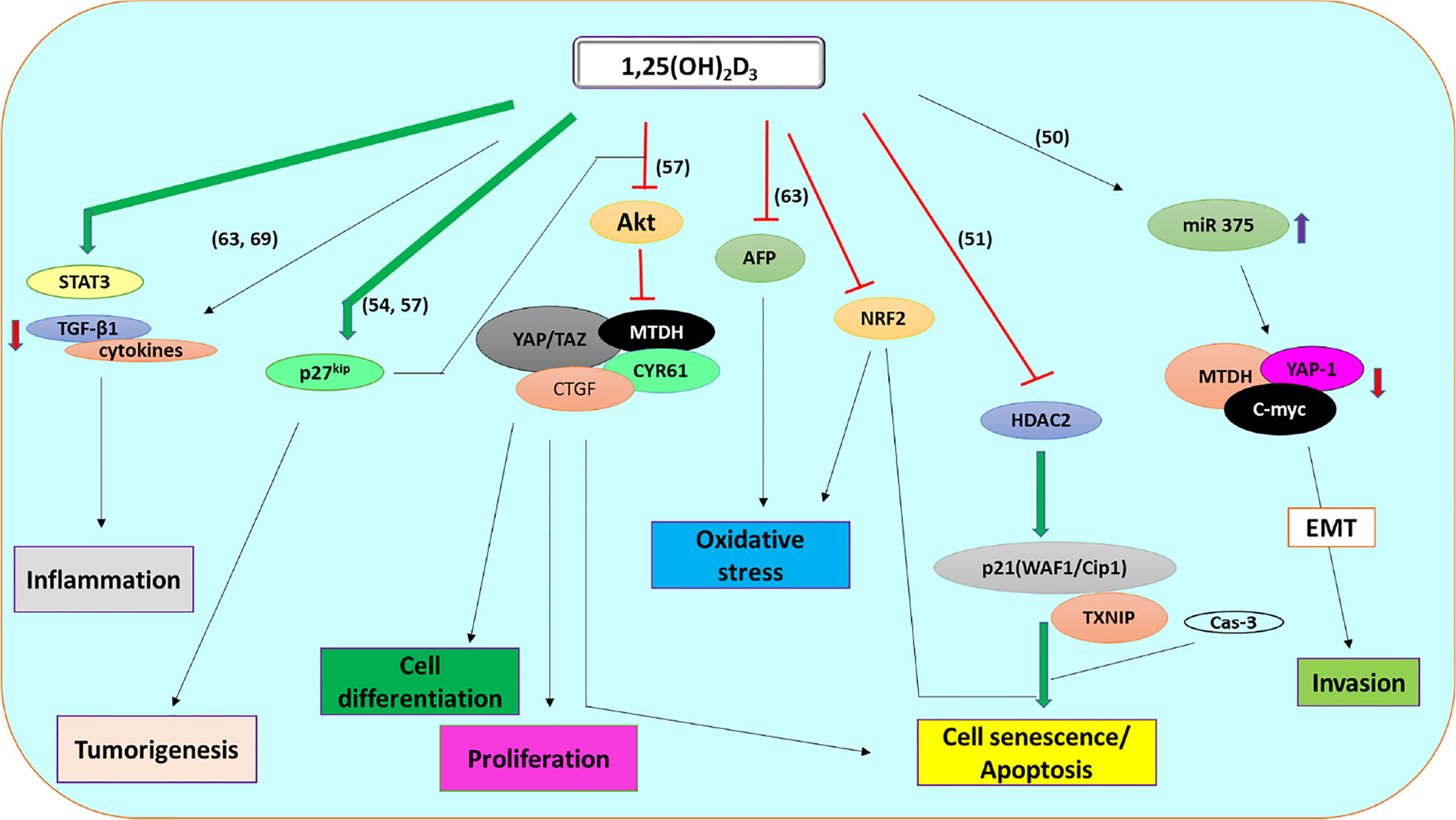
Figure 1 l,25 (OH)2D3 signaling pathway involved in the regulation of HCC through apoptosis, invasion, proliferation, differentiation, tumorigenesis, oxidative stress, and inflammation.
Generally, inflammation induces oxidative stress by activating neutrophils and Kupffer cells, which subsequently triggers cancer progression (72). Oxidative stress is usually associated with the pathogenesis and progression of HCC. However, reports suggest that VD3 could be involved in the attenuation of oxidative stress (61, 69). The physiological advantage of this abuts the vital role of inhibiting oxidative stress in managing hepatocarcinogenesis (73, 74). Besides, VD3 protected against oxidative stress-induced carcinogenesis by reversing different antioxidant enzymes altered in 3’ methyl-4-dimethyl-amino-azobenzene-induced hepatocarcinogenesis (61).
Furthermore, increased serum level and gene expression of the M30 apoptotic marker in HCC patients, amongst others, indicates alteration of the apoptotic pathway in carcinogenesis (62). Thus, the co-regulatory interaction between VD signaling and apoptotic pathway in HCC is imperative in the understanding of VD-related mechanisms (62). Besides, VD3 (cholecalciferol) treatment activates caspase 3 (Cas-3) expression while downregulating protein expression of tumor growth factor (TGF-β) (63). Likewise, co-administration with 5-fluorouracil alleviated the increased liver function enzymes, alpha-fetoprotein (AFP), and nuclear factor erythroid 2-related factor 2 (Nrf2) expression in thioacetamide-induced HCC (63).
Therefore, the in vivo studies showed that VD could regulate HCC progression via activation of apoptosis, reducing oxidative stress and inflammatory effects (Table 1).
Clinical Studies
Despite promising data from in vitro and in vivo studies suggesting VD’s crucial roles in carcinogenesis, established reports and data from clinical studies are still few and far between. These clinical trials included a phase 1 pilot study on VD administration’s effects on serum calcium, hepatic and renal functions by Finlay et al. (75). In the study, HCC patients received up to a 20-fold increased l,25-(OH)2D3 via hepatic arterial infusion without hypercalcemic complications. The study also reported 10 µg/day as a safe dosage with no renal or hepatic complications (75).
However, to eliminate hypercalcemia effects of VD administration, Morris et al. (74) reported in a relatively small pilot study that co-administration of l,25-(OH)2D3 with lipiodol in HCC patients could be an excellent therapeutic measure through stabilization of tumor marker, AFP. From this clinical research, the authors suggested that the use of lipiodol could increase permitted l,25-(OH)2D3 dosage about 50 folds (100 µg) without complications of hypercalcemia. Hence, this positive outcome could have resulted from the intra-arterial hepatic administration route used in the study (65). In addition, Dalhoff et al. (66) administered a starting dose of 10 µg/day seocalcitol (VD analog) and reported that seocalcitol could function as an anti-tumorigenic agent in phase 2 clinical trials. The analog can thus stabilize HCC patients due to its cytostatic rather than cytotoxic capacity (66).
VD may also improve HCC by restoring initially lost tumor growth factor-β (TGF-β) expression in liver tumor (60). In support of this, Chen et al. (63) reported that dysregulated VD-associated genes, including Foxhead box protein O4 (FOXO4) and signal transducer and activator of transcription 1 (STAT1), showed a strong correlation with TGF-β, while VD supplementation reduces cell proliferation.
Furthermore, a selected European population Nested Case-Control Study reported that increased concentration of hormonal VD, l,25-(OH)2D3 decreased the risk of HCC (76). This study informed the idea that l,25-(OH)2D3 treatment could ameliorate HCC development. Likewise, a randomized controlled trial also showed that l,25-(OH)2D3 supplementation of daily 2800 IU resulted in increased serum l,25-(OH)2D3 concentration in cirrhotic patients without significantly altering the mineral metabolism parameters (74).
The Role of Vitamin D Metabolizing Enzymes
Beyond the modulating roles of circulating VD hitherto described, evidence is emerging that these effects are elicited through its metabolizing genes. In this vein, Horvath et al. (59) reported that 1,25(OH)2D3 treatment caused a concurrent dose-dependent mRNA increased expression of CYP24A1 at specific time points in some HCC cell lines. The upregulated expression of CYP24A1 through 1,25(OH)2D3 treatment suggests a positive correlation between the enzyme and VD serum concentration. Chiang et al. (77) also reported that 1,25(OH)2D3 cell line treatment induces upregulation of CYP24A1 expression. Even though 25(OH)D-1α-hydroxylase, CYP27B1 further augmented the upregulation of CYP24A1, as reported by Bikle et al. (78), its transfection also induced cell arrest at the G0/G1 phase through p21/p27; thus, inhibiting tumor cell growth (76). Additionally, single nucleotide polymorphisms of CYP24A1 are associated with an increased risk of HCV infection in some high-risk Chinese population (79). Specifically, rs6013897 (T>A) was significantly associated with an increased risk of HCV infection. In contrast, rs6068816 (C>T), rs3787557 (T>C), rs6022999 (A>G), and rs2248359 (C>T) were associated with increased risk of chronic HCV infection. Consequently, combining VD3 treatment and CYP24A1 inhibitors could annihilate the increased cytoplasmic expression of CYP24A1.
Limitations of the Use of Vitamin D as Therapeutics
VD intoxication, usually characterized by hypercalcemia, is a significant limitation to the therapeutic use of the hormone in alleviating pathological conditions. Consequently, VD analogs have been used in recent years to reduce hypercalcemic effects. For example, a catabolic metabolite of the prodrug, 27 hydroxy BCI-210 (27-OH BCI-210), was reported to inhibit cancer cell growth (80). Although patients take various supplements, including vitamins, to maintain and improve health and prevent disease occurrences (81), there was no observed association between these supplements and HCC patients’ survival (81).
The daily intake of 100,000 IU or more could cause VD toxicity (68), while an increased intake of up to 2000 fold against the prescribed dosage could lead to renal failure (82). It has also been reported that an annual treatment of 500,000 IU VD3 increases fracture risk (83, 84). However, a short-term effect of an accidental overdose of VD3 was minimal; the long-term effect could be detrimental, as van den Ouweland et al. (83) reported. In the study, a single overdose treatment of 2,000,000 IU VD3 caused no short-term clinical toxicity. Therefore, terminating VD and reduced the consumption of calcium and phosphorus helps in managing hypercalcemia. Other interventions integral to controlling hypercalcemia include glucocorticoids, intravenous hydration, diuretics, and calcitonin (85, 86). Equally, to reduce the VD dosage and improve efficacy, combination therapy of VD and its analogs with other chemotherapeutic agents could be explored.
Future Perspectives
It is important to note that VD as an anticancer therapeutic agent could be associated with the administration route. Aside from the hepatic arterial infusion of this lipophilic vitamin, intravenous administration could determine, to some extent, the therapeutic effects and rate of its effectiveness (64). Also, VD supplementation and CYP27B1 gene transfection therapy are other plausible options of exploration in managing and treating HCC (77). Although dosage limitation exists, it will be beneficial to understand the interaction of the VD signaling pathway and carcinogenesis at the genetic level. The genetic interactions could focus on specific targets; thus, alleviating risks that arise with the limitation.
Another line of thought in VD’s therapeutic use could involve understanding the mechanisms in VD’s modulatory roles of the tumor microenvironment (TME). Tumor growth, invasion, and metastasis are generally affected by the interactions between the tumors and their respective microenvironments (87). Understanding these bidirectional interactions between the tumor cells and the environment could open up therapeutic targets and regimes in liver cancer treatment (88–90). Although VD influences angiogenesis, metastasis, and cancer progression in TME, the active form of VD, 1, 25 (OH)2D3 modulates a couple of stroma cells explicitly, suppresses tumor growth, and act as an anti-inflammatory agent within the TME, leading to cancer reduction (91).
Conclusion
Despite positive research findings on VD’s roles in HCC, resulting limitations hinder its progress as a viable therapeutic agent. Although there might be conflicting reports supporting the roles of serum l,25-(OH)2D3 in HCC, there are ample in vitro, in vivo data and some randomized clinical control trials suggesting VD-related mechanism is vital in HCC progression. This research gap could be vital in understanding the mechanisms involved in the VD regulation of HCC. Clinical trials on various combination therapies will also help resolve the research deficiencies recorded in standardizing VD dosage. Therefore, it is strongly recommended that more studies should be carried out on combination therapies of various VD analogs and standard therapeutic agents by targeting crucial genes and pathways involved in VD’s non-classical functions.
Author Contributions
All authors contributed, read, and agreed to the publication of this manuscript. Conceptualization: IA. Supervision: OR, EM, and SR. Roles/writing—original draft: IA. Writing—review and editing: IA, OR, EM, and SR. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors acknowledged the Covenant University Centre for Research, Innovation, and Discovery (CUCRID) for publication support.
References
1. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global Cancer Statistics 2020 : GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin (2021) 0:1–41. doi: 10.3322/caac.21660
2. Elangovan H, Chahal S, Gunton JE. Vitamin D in Liver Disease: Current Evidence and Potential Directions. Biochim Biophys Acta Mol Basis Dis (2017) 1863:907–16. doi: 10.1016/j.bbadis.2017.01.001
3. Moeini A, Cornellà H, Villanueva A. Emerging Signaling Pathways in Hepatocellular Carcinoma. Liver Cancer (2012) 1:83–93. doi: 10.1159/000342405
4. Schlageter M, Terracciano LM, D’Angelo S, Sorrentino P. Histopathology of Hepatocellular Carcinoma. World J Gastroenterol (2014) 20(43):15955–64. doi: 10.3748/wjg.v20.i43.15955
5. Singh AK, Kumar R, Pandey AK. Hepatocellular Carcinoma: Causes, Mechanism of Progression and Biomarkers. Curr Chem Genomics Transl Med (2018) 12:9–26. doi: 10.2174/2213988501812010009
6. Rotimi OA, Rotimi SO, Duru CU, Ebebeinwe OJ, Abiodun AO, Oyeniyi BO, et al. Acute Aflatoxin B1 – Induced Hepatotoxicity Alters Gene Expression and Disrupts Lipid and Lipoprotein Metabolism in Rats. Toxicol Rep (2017) 4:408–14. doi: 10.1016/j.toxrep.2017.07.006
7. McGlynn KA, London WT. Epidemiology and Natural History of Hepatocellular Carcinoma. Best Pract Res Clin Gastroenterol (2005) 19(1):3–23. doi: 10.1016/j.bpg.2004.10.004
8. Omer RE, Kuijsten A, Kadaru AMY, Kok FJ, Idris MO, El Khidir IM, et al. Population-Attributable Risk of Dietary Aflatoxins and Hepatitis B Virus Infection With Respect to Hepatocellular Carcinoma. Nutr Cancer (2004) 48(1):15–21. doi: 10.1207/s15327914nc4801_3
9. Wild CP, Gong YY. Mycotoxins and Human Disease: A Largely Ignored Global Health Issue. Carcinogenesis (2009) 31(1):71–82. doi: 10.1093/carcin/bgp264
10. Hung CH, Chiu YC, Hu TH, Chen CH, Lu SN, Huang CM, et al. Significance of Vitamin D Receptor Gene Polymorphisms for Risk of Hepatocellular Carcinoma in Chronic Hepatitis C. Transl Oncol (2014) 7(4):503–7. doi: 10.1016/j.tranon.2014.05.001
11. Peng Q, Yang S, Lao X, Li R, Chen Z, Wang J, et al. Association of Single Nucleotide Polymorphisms in VDR and DBP Genes With HBV-related Hepatocellular Carcinoma Risk in a Chinese Population. PLoS One (2014) 9(12):e116026. doi: 10.1371/journal.pone.0116026
12. Lombardi C, Heck JE, Cockburn M, Ritz B. Solar UV Radiation and Cancer in Young Children. Cancer Epidemiol Biomarkers Prev (2013) 22(6):1118–28. doi: 10.1158/1055-9965.EPI-12-1316
13. VoPham T, Bertrand KA, Yuan J, Tamimi RM, Hart JE, Laden F. Ambient Ultraviolet Radiation Exposure and Hepatocellular Carcinoma Incidence in the United States. Environ Heal (2017) 16:89. doi: 10.1186/s12940-017-0299-0
14. O’Connor MY, Thoreson CK, Ramsey NLM, Ricks M, Sumner AE. The Uncertain Significance of Low Vitamin D Levels in African Descent Populations: A Review of the Bone and Cardiometabolic Literature. Prog Cardiovasc Dis (2013) 56(3):261–9. doi: 10.1016/j.pcad.2013.10.015
15. Bikle DD. Review Vitamin D Metabolism, Mechanism of Action, and Clinical Applications. Chem Biol (2014) 21(3):319–29. doi: 10.1016/j.chembiol.2013.12.016
16. Mason SS, Kohles SS, Winn SR, Zelick RD. Extrahepatic 25-Hydroxylation of Vitamin D 3 in an Engineered Osteoblast Precursor Cell Line Exploring the Influence on Cellular Proliferation and Matrix Maturation During Bone Development. ISRN BioMed Eng (2013) 2013(956362):1–11. doi: 10.1155/2013/956362
17. Negri M, Gentile A, de Angelis C, Montò T, Patalano R, Colao A, et al. Vitamin D-induced Molecular Mechanisms to Potentiate Cancer Therapy and to Reverse Drug-Resistance in Cancer Cells. Nutrients (2020) 12(1798):1–25. doi: 10.3390/nu12061798
18. Zhu JG, Ochalek JT, Kaufmann M, Jones G, DeLuca HF. CYP2R1 is a Major, But Not Exclusive, Contributor to 25-Hydroxyvitamin D Production In Vivo. Proc Natl Acad Sci (2013) 110(39):15650–5. doi: 10.1073/pnas.1315006110
19. Christakos S, Dhawan P, Verstuyf A, Verlinden L, Carmeliet G. Vitamin D: Metabolism, Molecular Mechanism of Action, and Pleiotropic Effects Vitamin D Analogs. Physiol Rev (2016) 96:365–408. doi: 10.1152/physrev.00014.2015
20. Hewison M, Zehnder D, Bland R, Stewart PM. 1α-Hydroxylase and the Action of Vitamin D. J Mol Endocrinol (2000) 25:141–8. doi: 10.1677/jme.0.0250141
21. Dawson MI, Xia Z. The Retinoid X Receptors and Their Ligands. Biochim Biophys Acta (2012) 1821(1):21–56. doi: 10.1016/j.bbalip.2011.09.014
22. Bettoun DJ, Burris TP, Houck KA, Buck DW, Stayrook KR, Khalifa B, et al. Retinoid X Receptor Is a Nonsilent Major Contributor to Vitamin D Receptor-Mediated Transcriptional Activation. Mol Endocrinol (2003) 17(11):2320–8. doi: 10.1210/me.2003-0148
23. Haussler MR, Whitfield GK, Kaneko I, Haussler CA, Hsieh D, Hsieh JC, et al. Molecular Mechanisms of Vitamin D Action. Calcif Tissue Int (2013) 92(2):77–98. doi: 10.1007/s00223-012-9619-0
24. Jeon SM, Shin EA. Exploring Vitamin D Metabolism and Function in Cancer. Exp Mol Med (2018) 50(20):1–14. doi: 10.1038/s12276-018-0038-9
25. Finkelmeier F, Kronenberger B, Koberle V, Bojunga J, Zeuzem S, Trojan J, et al. Severe 25-Hydroxyvitamin D Deficiency Identifies a Poor Prognosis in Patients With Hepatocellular Carcinoma – a Prospective Cohort Study. Aliment Pharmacol Ther (2014) 39:1204–12. doi: 10.1111/apt.12731
26. Pilz S, Grübler M, Gaksch M, Schwetz V, Trummer C, Hartaigh BÓ, et al. Vitamin D and Mortality. Anticancer Res (2016) 36:1379–87.
27. Schottker B, Jorde R, Peasey A, Thorand B, Jansen EHJM, Groot LD, et al. Vitamin D and Mortality: Meta-Analysis of Individual Participant Data From a Large Consortium of Cohort Studies From Europe and the United States. Br Med J (2014) 348:g3656. doi: 10.1136/bmj.g3656
28. Gaksch M, Jorde R, Grimnes G, Joakimsen R, Schirmer H, Wilsgaard T, et al. Vitamin D and Mortality: Individual Participant Data Meta-Analysis of Standardized 25-Hydroxyvitamin D in 26916 Individuals From a European Consortium. PLoS One (2017) 12(2):e0170791. doi: 10.1371/journal.pone.0170791
29. Zhang Y, Jiang X, Li X, Găman MA, Kord-Varkaneh H, Rahmani J, et al. Serum Vitamin D Levels and Risk of Liver Cancer: A Systematic Review and Dose-Response Meta-Analysis of Cohort Studies. Nutr Cancer (2020) 0:1–9. doi: 10.1080/01635581.2020.1797127
30. Fang AP, Long JA, Zhang YJ, Liu ZY, Li QJ, Zhang DM, et al. Serum Bioavailable, Rather Than Total, 25-Hydroxyvitamin D Levels Are Associated With Hepatocellular Carcinoma Survival. Hepatology (2020) 72(1):169–82. doi: 10.1002/hep.31013
31. Liu H, Jiang X, Qiao Q, Chen L, Matsuda K, Jiang G, et al. Association of Circulating 25-Hydroxyvitamin D and its Related Genetic Variations With Hepatocellular Carcinoma Incidence and Survival. Ann Transl Med (2020) 8(17):1080. doi: 10.21037/atm-20-1637
32. Saumoy M, Ando Y, Jesudian A, Aden B, Jacobson I, Gambarin-Gelwan M. Vitamin D Deficiency is Not Associated With Increased Risk of Hepatocellular Carcinoma in Patients With Cirrhosis. Am J Gastroenterol (2013) 108(Suppl. 1):S108. doi: 10.14309/00000434-201310001-00362
33. Konstantakis C, Tselekouni P, Kalafateli M, Triantos C. Vitamin D Deficiency in Patients With Liver Cirrhosis. Ann Gastroenterol (2016) 29(3):297–306. doi: 10.20524/aog.2016.0037
34. Berkan-Kawińska A, Koślińska-Berkan E, Piekarska A. The Prevalence and Severity of 25-(OH)-Vitamin D Insufficiency in HCV Infected and in HBV Infected Patients: A Prospective Study. Clin Exp Hepatol (2015) 1:5–11. doi: 10.5114/ceh.2015.51373
35. Yang F, Ren H, Gao Y, Zhu Y, Huang W. The Value of Severe Vitamin D Deficiency in Predicting the Mortality Risk of Patients With Liver Cirrhosis: A Meta-Analysis. Clin Res Hepatol Gastroenterol (2019) 43(6):722–9. doi: 10.1016/j.clinre.2019.03.001
36. Buonomo AR, Zappulo E, Scotto R, Pinchera B, Perruolo G, Formisano P, et al. Vitamin D Deficiency is a Risk Factor for Infections in Patients Affected by HCV-related Liver Cirrhosis. Int J Infect Dis (2017) 63:23–9. doi: 10.1016/j.ijid.2017.07.026
37. Lange CM, Miki D, Ochi H, Nischalke HD, Bojunga J, Bibert S, et al. Genetic Analyses Reveal a Role for Vitamin D Insufficiency in HCV-Associated Hepatocellular Carcinoma Development. PLoS One (2013) 8(5):e64053. doi: 10.1371/journal.pone.0064053
38. Ravid A, Rapaport N, Issachar A, Erman A, Bachmetov L, Tur-Kaspa R, et al. 25-Hydroxyvitamin D Inhibits Hepatitis C Virus Production in Hepatocellular Carcinoma Cell Line by a Vitamin D Receptor-Independent Mechanism. Int J Mol Sci (2019) 20:2367. doi: 10.3390/ijms20092367
39. Gotlieb N, Tachlytski I, Lapidot Y, Sultan M, Safran M, Ben-Ari Z. Hepatitis B Virus Downregulates Vitamin D Receptor Levels in Hepatoma Cell Lines, Thereby Preventing Vitamin D-dependent Inhibition of Viral Transcription and Production. Mol Med (2018) 24:53. doi: 10.1186/s10020-018-0055-0
40. Farnik H, Bojunga J, Berger A, Allwinn R, Waidmann O, Kronenberger B, et al. Low Vitamin D Serum Concentration is Associated With High Levels of Hepatitis B Virus Replication in Chronically Infected Patients. Hepatology (2013) 58(4):1270–6. doi: 10.1002/hep.26488
41. Abdel-Mohsen MA, El-Braky AAA, Ghazal AAER, Shamseya MM. Autophagy, Apoptosis, Vitamin D, and Vitamin D Receptor in Hepatocellular Carcinoma Associated With Hepatitis C Virus. Med (United States) (2018) 97(12):1–7. doi: 10.1097/MD.0000000000010172
42. Falleti E, Bitetto D, Fabris C, Cussigh A, Fontanini E, Fornasiere E, et al. Vitamin D Receptor Gene Polymorphisms and Hepatocellular Carcinoma in Alcoholic Cirrhosis. World J Gastroenterol (2010) 16(24):3016–24. doi: 10.3748/wjg.v16.i24.3016
43. Quan Y, Yang J, Qin T, Hu Y. Associations Between Twelve Common Gene Polymorphisms and Susceptibility to Hepatocellular Carcinoma : Evidence From a. World J Surg Oncol (2019) 17:216. doi: 10.1186/s12957-019-1748-8
44. Hoan NX, Khuyen N, Giang DP, Binh MT, Toan NL, Anh DT, et al. Vitamin D Receptor ApaI Polymorphism Associated With Progression of Liver Disease in Vietnamese Patients Chronically Infected With Hepatitis B Virus. BMC Med Genet (2019) 20:201. doi: 10.1186/s12881-019-0903-y
45. Rowida IR, Eshra KA, El-sharaby RM, Eissa RAE, Saied SM, Amer I, et al. Apa1 (Rs7975232) SNP in the Vitamin D Receptor is Linked to Hepatocellular Carcinoma in Hepatitis C Virus Cirrhosis. Br J BioMed Sci (2019) 77(2):53–7. doi: 10.1080/09674845.2019.1680166
46. Yao X, Zeng H, Zhang G, Zhou W, Yan Q, Dai L, et al. The Associated Ion Between the VDR Gene Polymorphisms and Susceptibility to Hepatocellular Carcinoma and the Clinicopathological Features in Subjects Infected With HBV. BioMed Res Int (2013) 2013:953974. doi: 10.1155/2013/953974
47. Cusato J, Boglione L, De Nicolò A, Favata F, Ariaudo A, Mornese Pinna S, et al. Vitamin D Pathway Gene Polymorphisms and Hepatocellular Carcinoma in Chronic Hepatitis C-affected Patients Treated With New Drugs. Cancer Chemother Pharmacol (2018) 81(3):615–20. doi: 10.1007/s00280-018-3520-0
48. Ferrín G, Guerrero M, Amado V, Rodríguez-Perálvarez M, De la Mata M. Activation of mTOR Signaling Pathway in Hepatocellular Carcinoma. Int J Mol Sci (2020) 21(1266):1–16. doi: 10.3390/ijms21041266
49. Matter MS, Decaens T, Andersen JB, Thorgeirsson SS. Targeting the mTOR Pathway in Hepatocellular Carcinoma: Current State and Future Trends. J Hepatol (2014) 60(4):855–65. doi: 10.1016/j.jhep.2013.11.031
50. Provvisiero DP, Negri M, de Angelis C, Di Gennaro G, Patalano R, Simeoli C, et al. Vitamin D Reverts Resistance to the mTOR Inhibitor Everolimus in Hepatocellular Carcinoma Through the Activation of a miR-375/oncogenes Circuit. Sci Rep (2019) 9(11695):1–13. doi: 10.1038/s41598-019-48081-9
51. Huang J, Yang G, Huang Y, Kong W, Zhang S. 1,25(OH)2D3 Inhibits the Progression of Hepatocellular Carcinoma Via Downregulating HDAC2 and Upregulating P21(WAFI/CIP1). Mol Med Rep (2016) 13(2):1373–80. doi: 10.3892/mmr.2015.4676
52. Hamilton JP, Potter JJ, Koganti L, Meltzer SJ, Mezey E. Effects of Vitamin D3 Stimulation of Thioredoxin-Interacting Protein in Hepatocellular Carcinoma. Hepatol Res (2014) 44(13):1357–66. doi: 10.1111/hepr.12302
53. Sheth SS, Bodnar JS, Ghazalpour A, Thipphavong CK, Tsutsumi S, Tward AD, et al. Hepatocellular Carcinoma in Txnip-deficient Mice. Oncogene (2006) 25:3528–36. doi: 10.1038/sj.onc.1209394
54. Guo J, Ma Z, Ma Q, Wu Z, Fan P, Zhou X, et al. 1, 25(Oh)2d3 Inhibits Hepatocellular Carcinoma Development Through Reducing Secretion of Inflammatory Cytokines From Immunocytes. Curr Med Chem (2013) 20(33):4131–41. doi: 10.2174/09298673113209990248
55. Xu J, Wang Y, Zhang Y, Dang S, He S. Astemizole Promotes the Anti-Tumor Effect of Vitamin D Through Inhibiting miR-125a-5p-meidated Regulation of VDR in HCC. BioMed Pharmacother (2018) 107(277):1682–91. doi: 10.1016/j.biopha.2018.08.153
56. Wu Q, Wang X, Pham K, Luna A, Studzinski GP, Liu C. Enhancement of Sorafenib-Mediated Death of Hepatocellular Carcinoma Cells by Carnosic Acid and Vitamin D2 Analog Combination. J Steroid Biochem Mol Biol (2020) 197:105524. doi: 10.1016/j.jsbmb.2019.105524
57. Wang J, Chen Y, Mo PL, Wei YJ, Liu KC, Zhang ZG, et al. 1α,25-Dihydroxyvitamin D3 Inhibits Aflatoxin B1-induced Proliferation and Dedifferentiation of Hepatic Progenitor Cells by Regulating PI3K/Akt and Hippo Pathways. J Steroid Biochem Mol Biol (2018) 183:228–37. doi: 10.1016/j.jsbmb.2018.08.002
58. Wu FS, Zheng SS, Wu LJ, Teng LS, Ma ZM, Zhao WH, et al. Calcitriol Inhibits the Growth of MHCC97 Heptocellular Cell Lines by Down-Modulating C-Met and ERK Expressions. Liver Int (2007) 27(5):700–7. doi: 10.1111/j.1478-3231.2007.01487.x
59. Horvath E, Lakatos P, Balla B, Kósa JP, Tóbiás B, Jozilan H, et al. Marked Increase of CYP24A1 mRNA Level in Hepatocellular Carcinoma Cell Lines Following Vitamin D Administration. Anticancer Res (2012) 32(11):4791–6.
60. Chen J, Katz LH, Muñoz NM, Gu S, Shin JH, Jogunoori WS, et al. Vitamin D Deficiency Promotes Liver Tumor Growth in Transforming Growth Factor-β/Smad3-Deficient Mice Through Wnt and Toll-like Receptor 7 Pathway Modulation. Sci Rep (2016) 6:30217. doi: 10.1038/srep30217
61. Karmakar R, Banik S, Chatterjee M. Inhibitory Effect of Vitamin D3 on 3′methyl-4-dimethyl-amino-azobenzene-induced Rat Hepatocarcinogenesis: A Study on Antioxidant Defense Enzymes. J Exp Ther Oncol (2002) 2(4):193–9. doi: 10.1046/j.1359-4117.2002.01032.x
62. Fingas CD, Altinbas A, Schlattjan M, Beilfuss A, Sowa JP, Sydor S, et al. Expression of Apoptosis- and Vitamin D Pathway-Related Genes in Hepatocellular Carcinoma. Digestion (2013) 87(3):176–81. doi: 10.1159/000348441
63. Ebrahim AR, El-Mesery M, El-Karef A, Eissa LA. Vitamin D Potentiates Anti-Tumor Activity of 5-Fluorouracil Via Modulating Caspase-3 and Tgf-β1 Expression in Hepatocellular Carcinoma-Induced in Rats. Can J Physiol Pharmacol (2018) 96(12):1218–25. doi: 10.1139/cjpp-2018-0445
64. Finlay IG, Stewart GJ, Shirley P, Woolfe S, Pourgholami MH, Morris DL. Hepatic Arterial and Intravenous Administration of 1,25-Dihydroxyvitamin D3 - Evidence of a Clinically Significant Hepatic First-Pass Effect. Cancer Chemother Pharmacol (2001) 48(3):209–14. doi: 10.1007/s002800100333
65. Morris DL, Jourdan JL, Finlay I, Gruenberger T, The MP, Pourgholami MH. Hepatic Intra-Arterial Injection of 1,25-Dihydroxyvitamin D3 in Lipiodol: Pilot Study in Patients With Hepatocellular Carcinoma. Int J Oncol (2002) 21(4):901–6. doi: 10.3892/ijo.21.4.901
66. Dalhoff K, Dancey J, Astrup L, Skovsgaard T, Hamberg KJ, Lofts FJ, et al. A Phase II Study of the Vitamin D Analogue Seocalcitol in Patients With Inoperable Hepatocellular Carcinoma. Br J Cancer (2003) 89:252–7. doi: 10.1038/sj.bjc.6601104
67. Pilz S, Putz-Bankuti C, Gaksch M, Spindelboeck W, Haselberger M, Rainer F, et al. Effects of Vitamin D Supplementation on Serum 25-Hydroxyvitamin D Concentrations in Cirrhotic Patients: A Randomized Controlled Trial. Nutrients (2016) 8:278. doi: 10.3390/nu8050278
68. Liu W, Zhang L, Xu H, Li Y, Hu C, Yang J, et al. The Anti-Inflammatory Effects of Vitamin D in Tumorigenesis. Int J Mol Sci (2018) 19:2736. doi: 10.3390/ijms19092736
69. Adelani IB, Ogadi EO, Onuzulu C, Rotimi OA, Maduagwu EN, Rotimi SO. Dietary Vitamin D Ameliorates Hepatic Oxidative Stress and Inflammatory Effects of Diethylnitrosamine in Rats. Heliyon (2020) 6(9):e04842. doi: 10.1016/j.heliyon.2020.e04842
70. Lloyd RV, Erickson LA, Jin L, Kulig E, Qian X, Cheville JC, et al. P27kip1: A Multifunctional Cyclin-Dependent Kinase Inhibitor With Prognostic Significance in Human Cancers. Am J Pathol (1999) 154(2):313–23. doi: 10.1016/S0002-9440(10)65277-7
71. Kwon H-J, Won Y-S, Suh H-W, Jeon J-H, Shao Y, Yoon S-R, et al. Vitamin D3 Upregulated Protein-1 Suppresses Tnf-α–Induced Nf-κb Activation in Hepatocarcinogenesis. J Immunol (2010) 185(7):3980–9. doi: 10.4049/jimmunol.1000990
72. Fu Y, Chung F. Oxidative Stress and Hepatocarcinogenesis. Hepatoma Res (2018) 2018(4):1–11. doi: 10.20517/2394-5079.2018.29
73. Glauert HP, Calfee-Mason K, Stemm DN, Tharappel JC, Spear BT. Dietary Antioxidants in the Prevention of Hepatocarcinogenesis: A Review. Mol Nutr Food Res (2010) 54(7):875–96. doi: 10.1002/mnfr.200900482
74. Maurya BK, Trigun SK. Fisetin Modulates Antioxidant Enzymes and Inflammatory Factors to Inhibit Aflatoxin-B1 Induced Hepatocellular Carcinoma in Rats. Oxid Med Cell Longev (2016) 2016(1972793):1–9. doi: 10.1155/2016/1972793
75. Finlay IG, Stewart GJ, Ahkter J, Morris DL. A Phase One Study of the Hepatic Arterial Administration of 1,25-Dihydroxyvitamin D3 for Liver Cancers. J Gastroenterol Hepatol (2001) 16(3):333–7. doi: 10.1046/j.1440-1746.2001.02398.x
76. Fedirko V, Duarte-Salles T, Bamia C, Trichopoulou A, Aleksandrova K, Trichopoulos D, et al. Prediagnostic Circulating Vitamin D Levels and Risk of Hepatocellular Carcinoma in European Populations: A Nested Case-Control Study. Hepatology (2014) 60(4):1222–30. doi: 10.1002/hep.27079
77. Chiang K, Yen C, Yeh C, Hsu J, Chen L, Kuo S-F, et al. Hepatocellular Carcinoma Cells Express 25(OH)D-1a-hydroxylase and are Able to Convert 25(OH)D to 1a,25(OH)2D, Leading to the 25(OH)D-Induced Growth Inhibition. J Steroid Biochem Mol Biol (2015) 154:47–52. doi: 10.1016/j.jsbmb.2015.06.008
78. Bikle DD, Patzek S, Wang Y. Bone Reports Physiologic and Pathophysiologic Roles of Extra Renal CYP27b1 : Case Report and Review. Bone Rep (2018) 8:255–67. doi: 10.1016/j.bonr.2018.02.004
79. Fan Hz, Zhang R, Tian T, Zhong Yl, Wu Mp, Xie Cn, et al. CYP24A1 Genetic Variants in the Vitamin D Metabolic Pathway are Involved in the Outcomes of Hepatitis C Virus Infection Among High-Risk Chinese Population. Int J Infect Dis (2019) 84:80–8. doi: 10.1016/j.ijid.2019.04.032
80. Wang Y-R, Wigington DP, Strugnell SA, Knutson JC. Growth Inhibition of Cancer Cells by An Active Metabolite of a Novel Vitamin D Prodrug. Anticancer Res (2005) 25:4333–40.
81. Lee V, Goyal A, Hsu CC, Jacobson JS, Rodriguez RD, Siegel AB. Dietary Supplement Use Among Patients With Hepatocellular Carcinoma. Integr Cancer Ther (2015) 14(1):35–41. doi: 10.1177/1534735414550038
82. Marins TAp, Galvão TdeFG, Korkes F, Malerbi DA ugust C, Ganc AJos., Korn D, et al. Vitamin D Intoxication: Case Report. Einstein (Sao Paulo) (2014) 12(2):242–4. doi: 10.1590/S1679-45082014RC2860
83. van den Ouweland J, Fleuren H, Drabbe M, Vollaard H. Pharmacokinetics and Safety Issues of an Accidental Overdose of 2,000,000 IU of Vitamin D3 in Two Nursing Home Patients: A Case Report. BMC Pharmacol Toxicol (2014) 15(1):1–5. doi: 10.1186/2050-6511-15-57
84. Sanders KM, Stuart AL, Williamson EJ, Simpson JA, Kotowicz MA, Young D, et al. Annual High-Dose Oral Vitamin D and Falls and Fractures in Older Women: A Randomized Controlled Trial. J Am Med Assoc (2010) 303(18):1815–22. doi: 10.1001/jama.2010.594
86. Araki T, Holick MF, Alfonso BD, Charlap E, Romero CM, Rizk D, et al. Vitamin D Intoxication With Severe Hypercalcemia Due to Manufacturing and Labeling Errors of Two Dietary Supplements Made in the United States. J Clin Endocrinol Metab (2011) 96(12):3603–8. doi: 10.1210/jc.2011-1443
87. Baghban R, Roshangar L, Jahanban-Esfahlan R, Seidi K, Ebrahimi-Kalan A, Jaymand M, et al. Tumor Microenvironment Complexity and Therapeutic Implications At a Glance. Cell Commun Signal (2020) 18(59):1–19. doi: 10.1186/s12964-020-0530-4
88. Tsai M-J, Chang W-A, Huang M-S, Kuo P-L. Tumor Microenvironment: A New Treatment Target for Cancer. ISRN Biochem (2014) 2014(351959):1–8. doi: 10.1155/2014/351959
89. Benavente S, Sánchez-García A, Naches S, LLeonart ME, Lorente J. Therapy-Induced Modulation of the Tumor Microenvironment: New Opportunities for Cancer Therapies. Front Oncol (2020) 10:582884. doi: 10.3389/fonc.2020.582884
90. Murciano-Goroff YR, Warner AB, Wolchok JD. The Future of Cancer Immunotherapy: Microenvironment-Targeting Combinations. Cell Res (2020) 30:507–19. doi: 10.1038/s41422-020-0337-2
Keywords: vitamin D, therapeutic, inflammation, apoptosis, differentiation, proliferation, hepatocellular carcinoma
Citation: Adelani IB, Rotimi OA, Maduagwu EN and Rotimi SO (2021) Vitamin D: Possible Therapeutic Roles in Hepatocellular Carcinoma. Front. Oncol. 11:642653. doi: 10.3389/fonc.2021.642653
Received: 17 December 2020; Accepted: 06 April 2021;
Published: 25 May 2021.
Edited by:
Jiang-Jiang Qin, Institute of Cancer and Basic Medicine, Chinese Academy of Sciences (CAS), ChinaReviewed by:
Dandan Lin, The First Affiliated Hospital of Soochow University, ChinaHung-Wen Tsai, National Cheng Kung University, Taiwan
Yan Sun, Sun Yat-Sen University, China
Claudia Pivonello, University of Naples Federico II, Italy
Copyright © 2021 Adelani, Rotimi, Maduagwu and Rotimi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Isaacson B. Adelani, aXNhYWNzb245MDlAZ21haWwuY29t; YmFiYWJvZGUuYWRlbGFuaUBjb3ZlbmFudHVuaXZlcnNpdHkuZWR1Lm5n