 Jerry L. Spivak
Jerry L. Spivak Alison R. Moliterno
Alison R. Moliterno
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Oncol. , 10 March 2021
Sec. Hematologic Malignancies
Volume 11 - 2021 | https://doi.org/10.3389/fonc.2021.641613
This article is part of the Research Topic Novel Treatment Strategies for Myeloproliferative Neoplasms View all 6 articles
The myeloproliferative neoplasms, polycythemia vera, essential thrombocytosis and primary myelofibrosis share driver mutations that either activate the thrombopoietin receptor, MPL, or indirectly activate it through mutations in the gene for JAK2, its cognate tyrosine kinase. Paradoxically, although the myeloproliferative neoplasms are classified as neoplasms because they are clonal hematopoietic stem cell disorders, the mutations affecting MPL or JAK2 are gain-of-function, resulting in increased production of normal erythrocytes, myeloid cells and platelets. Constitutive JAK2 activation provides the basis for the shared clinical features of the myeloproliferative neoplasms. A second molecular abnormality, impaired posttranslational processing of MPL is also shared by these disorders but has not received the recognition it deserves. This abnormality is important because MPL is the only hematopoietic growth factor receptor expressed in hematopoietic stem cells; because MPL is a proto-oncogene; because impaired MPL processing results in chronic elevation of plasma thrombopoietin, and since these diseases involve normal hematopoietic stem cells, they have proven resistant to therapies used in other myeloid neoplasms. We hypothesize that MPL offers a selective therapeutic target in the myeloproliferative neoplasms since impaired MPL processing is unique to the involved stem cells, while MPL is required for hematopoietic stem cell survival and quiescent in their bone marrow niches. In this review, we will discuss myeloproliferative neoplasm hematopoietic stem cell pathophysiology in the context of the behavior of MPL and its ligand thrombopoietin and the ability of thrombopoietin gene deletion to abrogate the disease phenotype in vivo in a JAK2 V617 transgenic mouse model of PV.
The myeloproliferative neoplasms (MPNs), polycythemia vera (PV), essential thrombocytosis (ET), and primary myelofibrosis (PMF), are hematopoietic stem cell (HSC) disorders that express mutations activating JAK2 (1–4), the cognate tyrosine kinase for the type 1 homodimeric hematopoietic growth factor receptors for erythropoietin (EPO) and thrombopoietin (THPO) (5), and the granulocyte-colony stimulating factor receptor as well (6). Although these disorders are genetically distinct (7, 8) (9), have different natural histories (10), different complications and require different therapies (11), they exhibit significant clinical phenotypic mimicry, including overproduction of one or more blood cell types alone or together, extramedullary hematopoiesis (EMH) and transformation to myelofibrosis or to acute leukemia (12). Some of these abnormalities can be directly linked to inappropriate constitutive JAK2 activation, which results in committed hematopoietic progenitor cell (HPC) hypersensitivity to hematopoietic growth factors or complete hematopoietic growth factor-independence, depending on whether the JAK2 mutation is heterozygous or homozygous in the HPC (13). Others cannot, including failure of JAK2 V617F to enhance MPN HSC bone marrow pool size (14), resistance of MPN HSC to tyrosine kinase inhibitors (15) premature release of mutated CD34+ HSC from the bone marrow (16), myelofibrosis or the clinical presentation of PV and PMF as isolated thrombocytosis, suggesting that pathways other than constitutive JAK2 activation are involved.
The MPNs are clonal HSC disorders and the thrombopoietin receptor, MPL, is the only hematopoietic growth factor expressed in these cells. In addition to its role in signal transduction, JAK2 chaperones MPL cell-surface expression and ensures its stability there (17). THPO maintains HSC survival (18) and quiescence within the bone marrow osteoblastic niche (19, 20) and is required for megakaryocytic progenitor cell proliferation (21), but not for megakaryocyte maturation or platelet production (22, 23). MPL or THPO knockout mice appear hematologically normal except for thrombocytopenia but have decreased marrow HSC (24).In contrast, in human congenital amegakaryocytic thrombocytopenia (CAMT), MPL mutations, usually in the MPL distal extracellular cytokine receptor homology domain (CRHD) (25), cause thrombocytopenia, elevated plasma THPO, and severe marrow aplasia (26).
Importantly, the retrovirus MPLV, which caused an acute, fatal PV-like syndrome in mice and in vitro, also immortalized murine HPC, encodes an MPL gene with a truncated extracellular domain (27) (28), indicating that MPL is a proto-oncogene. Murine bone marrow cells expressing ectopic THPO caused a fatal, transplantable myeloproliferative disorder with splenomegaly, osteomyelofibrosis, pancytopenia, and leukemic transformation (29) (30). Conversely, murine marrow cells expressing ectopic EPO (31) or erythroid progenitor cells expressing JAK2 V617F caused erythrocytosis without significant EMH and did not engraft in secondary recipients (32), supporting a primary role for HSC, MPL, and THPO in MPN pathophysiology.
In humans, hereditary or somatic mutations that involve the MPL transmembrane domain or distal CRHD can have an ET or PMF phenotype (2), (33, 34). In addition, MPL bound by mutated CALR is inappropriately transported by this protein chaperone to the cell surface and activated, causing either an ET or PMF phenotype (35–37). Furthermore, germline single nucleotide polymorphisms (SNP) in the MPL distal CRHD that caused a variably penetrant, benign thrombocytosis phenotype with an elevated plasma THPO, occurred in specific ethnic groups (38, 39) and could be recapitulated in the mouse (40, 41). Hereditary THPO mutations causing uncontrolled THPO synthesis are associated with isolated thrombocytosis (42). In one family, however, such a THPO mutation was also associated with leukemic transformation or myelofibrosis (43).
Differing from MPL mutations, JAK2 V617F causes PV, ET, and PMF. However, similarly to hereditary or somatic MPL (2) (38) as well as CALR mutations (35), impaired MPL cell-surface expression is a feature of JAK2 V617F-positive PV, ET, and PMF (44, 45). How impaired expression of the hematopoietic growth factor receptor responsible for HSC survival, expansion and thrombopoiesis could lead to a myeloproliferative state has been a puzzle. In this review, we discuss how the unique dependence of HSC on the MPL–THPO axis together with the unusual pathophysiology of MPL in the MPN, creates a therapeutic target of opportunity to suppress MPN HSC while sparing normal HSC.
Recent developments in HSC biology have reoriented our understanding of both HSC behavior in the bone marrow and their progression from an undifferentiated state to the committed HPC that give rise to erythrocytes, myeloid cells, and platelets. Importantly, HSCs require both the MPL–THPO axis and mature megakaryocytes to remain quiescent in their bone marrow niches (20, 46). The importance of localization of mature megakaryocytes to HSC bone marrow niches is such that a subpopulation of marrow HSC expresses the von Willebrand gene and gives rise directly to self-replicating megakaryocytic HSC (47). Other HSC-generated but lineage-restricted, self-replicating HSCs also give rise directly to megakaryocyte-erythroid and common myeloid repopulating stem cells, in addition to those progressing down the classical HSC pathway of commitment and differentiation (48).
The presence of megakaryocyte-primed HSC at the apex of the HSC hierarchy explains why thrombocytosis is a common presenting manifestation of an MPN. It also explains the mechanisms by which some ET patients only express JAK2 V617F in their platelets (49), and why some JAK2 V617F-positive PV patients only have erythrocytosis and thrombocytosis (50), in addition to those who have a complete panmyelopathy. Additionally, it provides an explanation for the transformation of ET to PV and PMF to PV and vice versa through HSC clonal succession (51).
This behavior must be distinguished from the natural history of MPN driver mutations to produce marrow fibrosis in PV and ET, because such behavior does not necessarily confer the same biologic characteristics as PMF (52). HSC behavior, of course, is not only MPN driver mutation-related, it is also strongly influenced by sex and age (53), and in PV, a proclivity to JAK2 V617F homozygosity by uniparental disomy (54). Most importantly, however, the primacy of the MPL–THPO axis in HSC behavior not only under normal circumstances but in the MPN cannot be ignored from a therapeutic perspective.
The thrombopoietin receptor, MPL, is a member of the type 1 homodimeric cytokine receptor family together with the EPO, prolactin, growth hormone, and granulocyte-colony stimulating factor receptors. Among this receptor family, MPL is unique because it has a reduplicated extracellular CRHD, of which, the distal CRHD is the site of THPO binding. The distal CRHD is also the site of germline SNP causing benign familial thrombocytosis (38, 39), a hot spot for the mutations causing CAMT (25) as well as the site of terminal sialylation during receptor maturation, which is impaired in the MPN (55). Like the other hematopoietic growth factor receptors, MPL is responsible for the metabolism of its cognate ligand, but in contrast to the other hematopoietic growth factors, the production of which is mainly regulated by demand, THPO is constitutively produced in the liver (56), and production of a critical platelet mass is required to maintain a constant platelet count (57).
The observation that MPL cell-surface expression was impaired in JAK2 V617F-positive PV, PMF (44) and ET (45) appeared counterintuitive since MPL is the sole hematopoietic growth factor receptor expressed in HSC, and the MPNs are diseases with autonomous myeloproliferation (58). Additionally, impaired MPL cell-surface expression was universal in the MPN since it also occurred with MPL (59, 60) and CALR (35) mutations, and in familial thrombocytosis due to germline MPL SNP in the MPL distal CRHD as well (38, 39). Mechanisms for impaired MPL cell-surface expression include MPL, CALR or JAK2 mutations, increased MPL turnover, or impaired post-translational processing, and in PMF, reduced GATA1 expression, impaired megakaryocyte differentiation, an associated ribosomal deficiency state and impaired megakaryocyte-specific protein expression (61).
CAMT is due to MPL mutations (25), primarily in the distal CRHD; all three of the above mechanisms are responsible for impaired MPN MPL cell-surface expression (2, 33, 55, 62). Germline SNPs causing impaired MPL cell-surface expression are also located in the distal CRHD and appear to be due to impaired post-translational processing (38, 39). MPL is produced as an incompletely-glycosylated 80 kDa protein, which is fully glycosylated in the Golgi to a 95 kDa mature protein with JAK2 as its obligate chaperone (17). Normally, both incompletely glycosylated and mature MPL proteins are expressed at the cell-surface and both are responsive to THPO-induced signaling (62, 63). While MPN driver mutations and MPL germline SNP result in impaired terminal MPL sialylation in the distal CRHD (55), JAK2 V617F activation adds an additional defect. JAK2 is responsible for enhancing MPL stability and recycling (17, 61), but JAK2 V617F also increases MPL ubiquitination and proteasomal degradation, leading to decreased MPL recycling and half-life, predominantly involving mature MPL (61).
Importantly, since megakaryocytes do not require THPO for megakaryocyte maturation or platelet production, impaired MPL cell-surface expression does not affect these processes (20, 21). It does, however, impair plasma THPO clearance by megakaryocytes and platelets (57). This leads to increased plasma THPO levels in the MPN (64, 65) and a continuous signal for HPC to proliferate, either collectively (MPL S505N, MPL W515 K/L, CALR del/+ and JAK2 V617F), or limited to megakaryopoiesis alone (MPL K39N and MPL P106L), because adequate cell-surface MPL is still expressed in HCP for this purpose (66). Importantly, the MPN phenotype partially emulates that of wild-type mice and humans with unregulated THPO production (42), which is reversible in mice by inhibition of THPO synthesis (30).
Mouse hematopoiesis, of course, differs from human hematopoiesis because in the mouse, the platelet count is approximately three times the human platelet count. The mechanism for this appears to be due to the presence of an asparagine at amino acid residue 39 in the mouse MPL distal CRHD, which is otherwise highly homologous to the human MPL distal CRHD. Importantly, in humans with the germline SNP MPL K39N, where asparagine is substituted for lysine [(38)], MPL expression is impaired and affected individuals’ phenocopy mouse hematopoiesis with thrombocytosis and an elevated plasma THPO, suggesting a human biologic model for MPN pathophysiology.
Because MPL is essential for HSC quiescence and survival in the marrow osteoblastic niche (22, 23, 67) and is also responsible for THPO catabolism (57), we hypothesized that impaired MPL cell-surface expression was essential for MPN phenotypic behavior, causing unregulated HPC proliferation and, eventually, myelofibrosis due to increased plasma THPO (30), depending on the MPN driver mutation allele burden, while also causing HSC loss from the marrow (22).
To test this, we employed a JAK2 V617F transgenic mouse model, which mirrors PV natural history, with erythrocytosis, granulocytosis, thrombocytosis, splenomegaly and eventually anemia and osteomyelofibrosis (68). Unsurprisingly, when crossed with an MPL knockout mouse, there was abolition of the PV histologic phenotype (Figure 1A) and a marked decrease in marrow HSC (Figure 1B), which could only be partially alleviated with expression of one MPL allele. This confirmed an essential role for MPL in this transgenic mouse model of PV (69).
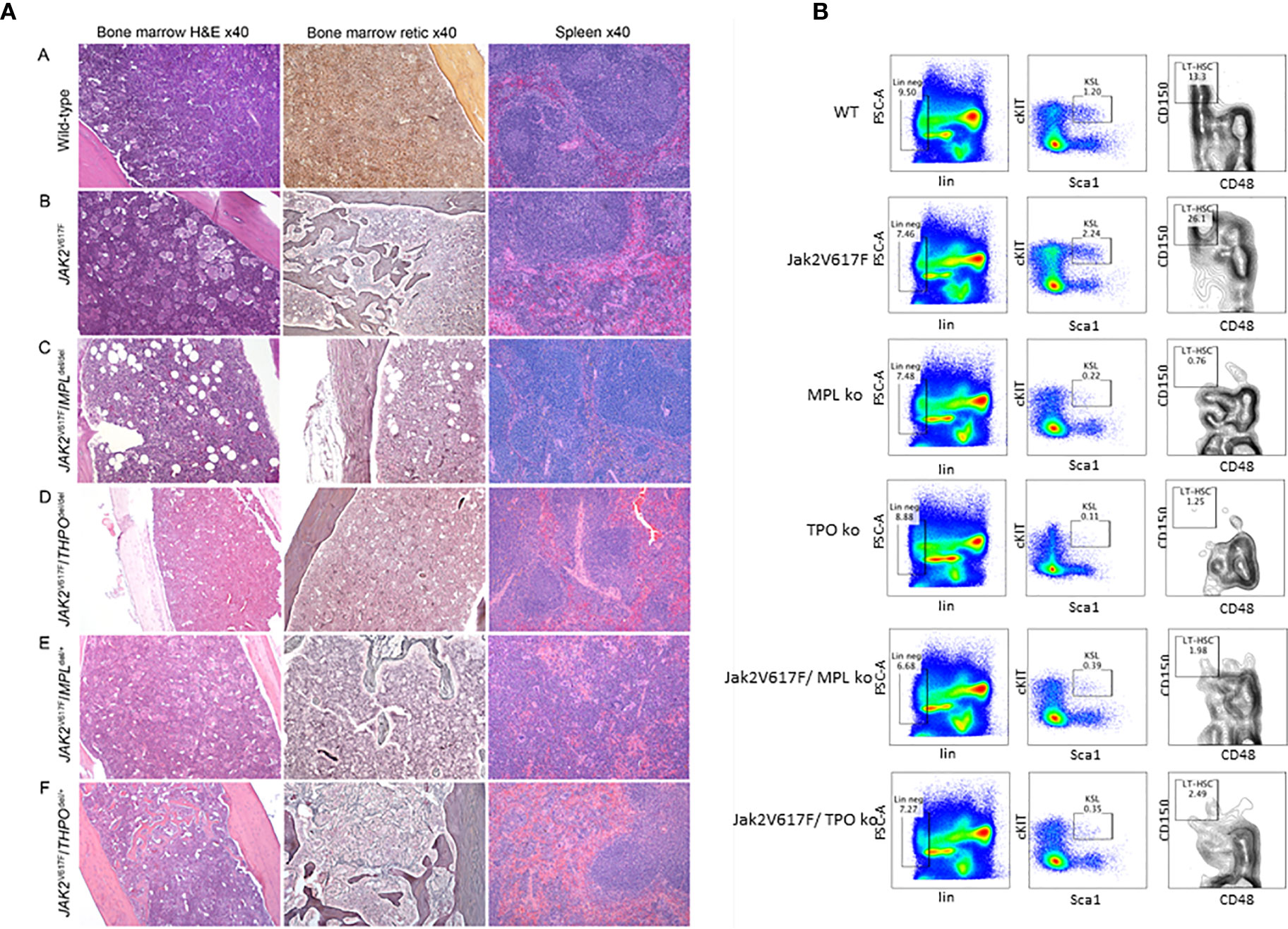
Figure 1 (A) MPL and THPO knockout (del/del) genotypes mitigate marrow and spleen histopathology in a JAK2 V617F transgenic mouse model of PV but this was restored in JAK2 V617F/THPO del/+ mice. Representative marrow and spleen histology at >33 weeks in wild-type mice and JAK2 V617F, JAK2 V617F/MPL del/del JAK2 V617F/THPO del/del, JAK2 V617F/MPL del/+, and JAK2 V617F/THPO del/+ transgenic mice. All images were taken with Zeiss AX10 Imager microscope using a Plano-APO 10×, 0.45 NA lens with a tungsten 3,200 K light source. The imaging medium was digital photomicrography using a bright field microscope and a Pro Res 14 camera with Adobe Photoshop CC acquisition software. Magnification was 40× for all images (69). (B) MPL and THPO knockout genotypes reduce the LT-HSC (CD150+CD48-) population in a JAK2 V617F transgenic mouse model of PV. Flow cytometry of marrow LT-HSC at >16 weeks in wild-type (n = 4), MPL knockout (n = 3), and THPO knockout (n = 3) mice and JAK2 V617 (n = 4), JAK2 V617F/MPL knockout (n = 3) and JAK2 V617F/THPO knockout (n = 3) transgenic mice. The marrow LT-HSC population was 13% of the total LSK population in wild-type mice, 26% of the total LSK population in JAK2 V617F transgenic mice and 0.76 and 1.25% respectively in the MPL knockout and THPO knockout mice and 1.98 and 2.08% respectively of the total LSK population in JAK2 V617F/MPL knockout and JAK2 V617F/THPO knockout transgenic mice. WT vs JAK2 V617F, P <0.029; MPL knockout vs JAK2V 617F, P < 0.022; THPO knockout vs JAK2 V617F, P < 0.021; JAK2 V617F vs JAK2 V617F/MPL knockout, P < 0.023; JAK2 V617F vs JAK2 V617F/THPO knockout, P < 0.034. The number (n) of mice of each genotype studied is in parentheses (69).
PV transgenic mice differed from human PV because the plasma THPO level was reduced, not increased, suggesting that the lower plasma THPO in contrast to wild-type mice was due to increased THPO utilization by the JAK2 V617F-mediated increase in the megakaryocyte and platelet pools, a characteristic also found in mouse models with MPL (59) or CALR mutations (70).
To study the role of THPO in the JAK2 V617F transgenic mouse phenotype, we crossed this mouse with a THPO knockout mouse. Unexpectedly, the PV phenotype was modified; this involved both the reversal of splenomegaly and osteomyelofibrosis, and a reduction in marrow HSC, despite the biallelic expression of functional MPL expressing JAK2 V617F. Restoration of one THPO allele completely restored the PV phenotype, in contrast to the incomplete restoration of the PV phenotype with a single MPL gene (JAK2 V617/MPL del/+). These results indicate that constitutive MPL signaling through JAK2 V617F alone was not sufficient to support the full PV phenotype in this transgenic mouse model.
Our observation that THPO gene deletion abrogated the PV phenotype in a JAK2 V617F transgenic mouse model differs from the observations of Sangkhae et al. (71). Their study employed a JAK2 V617F transgenic mouse model with an ET phenotype (72) and only 16 weeks of observation compared to our study, making their results not comparable with our JAK2 V617F transgenic mouse model, which recapitulated the natural history of PV, and required over 33 weeks of observation for full expression of the disease phenotype. Moreover, although Sangkhae et al. claimed that THPO was not necessary for expression of the ET phenotype in their mouse model, in agreement with our results, in their JAK2 V617F/THPO knockout transgenic mice, thrombocytosis was eliminated and in vitro HPC proliferation, megakaryocyte number and size, and spleen size were also reduced, indicating THPO dependence in their JAK2 V617F ET transgenic mouse model. Importantly, in both transgenic mouse models, abrogation of THPO production did not reduce blood counts below the baseline levels of control mice (68, 70).
Our observation that the MPNs are hematopoietic growth factor-dependent disorders, particularly in the JAK2 V671F heterozygous state is also supported by in vitro studies. For instance, human PV BFU-E heterozygous for JAK2 V617F responded in vitro to erythropoietin like normal BFU-E (13). Similarly, MPN HPC hematopoietic growth factor-responsiveness was observed in vitro with MPL (55) and CALR (4, 37) mutations.
Reduction in marrow HSC in the absence of the MPL or THPO genes in our study also strongly supports our assertion that the MPNs are hematopoietic growth factor-dependent-diseases. Significantly, a small molecule antagonist of MPL preferentially inhibited JAK2 V617F-positive proliferation of PV HSC both in vitro and in vivo compared to normal HSC (73), while in vitro, PV HPC and murine cell lines expressing MPL (55)] or CALR (4, 37) mutations were still THPO-responsive despite the presence of constitutively-activated JAK2.
Furthermore, treatment of wild-type mice with an MPL antagonist antibody allowed non-myeloablative bone marrow transplantation, verifying the requirement for THPO to maintain HSC in their marrow niches (23). Importantly, while the absence of functional MPL causes CAMT in humans, MPL cell-surface expression is impaired, but not absent in MPN HSC and HPC, which, as demonstrated experimentally (59), should give normal HSC and HCP a survival advantage when exposed to a THPO antagonist.
Current models of HSC indicate that they remain quiescent in their bone marrow niches anchored to osteoblasts by a number of adhesive molecules as well as by THPO (Figure 2A). Recent studies, however, suggest that the liver is the primary source of THPO and that osteoblast THPO is not required. In addition, while hepatic THPO production is constitutive, recent studies have identified an important regulatory function of platelet clearance through the Ashwell–Morrell receptor (AMR) (74) (Figure 2A). Hepatic THPO is also markedly increased in inflammatory conditions and is attributed to the direct action of interleukin-6 on hepatic THPO production (75).
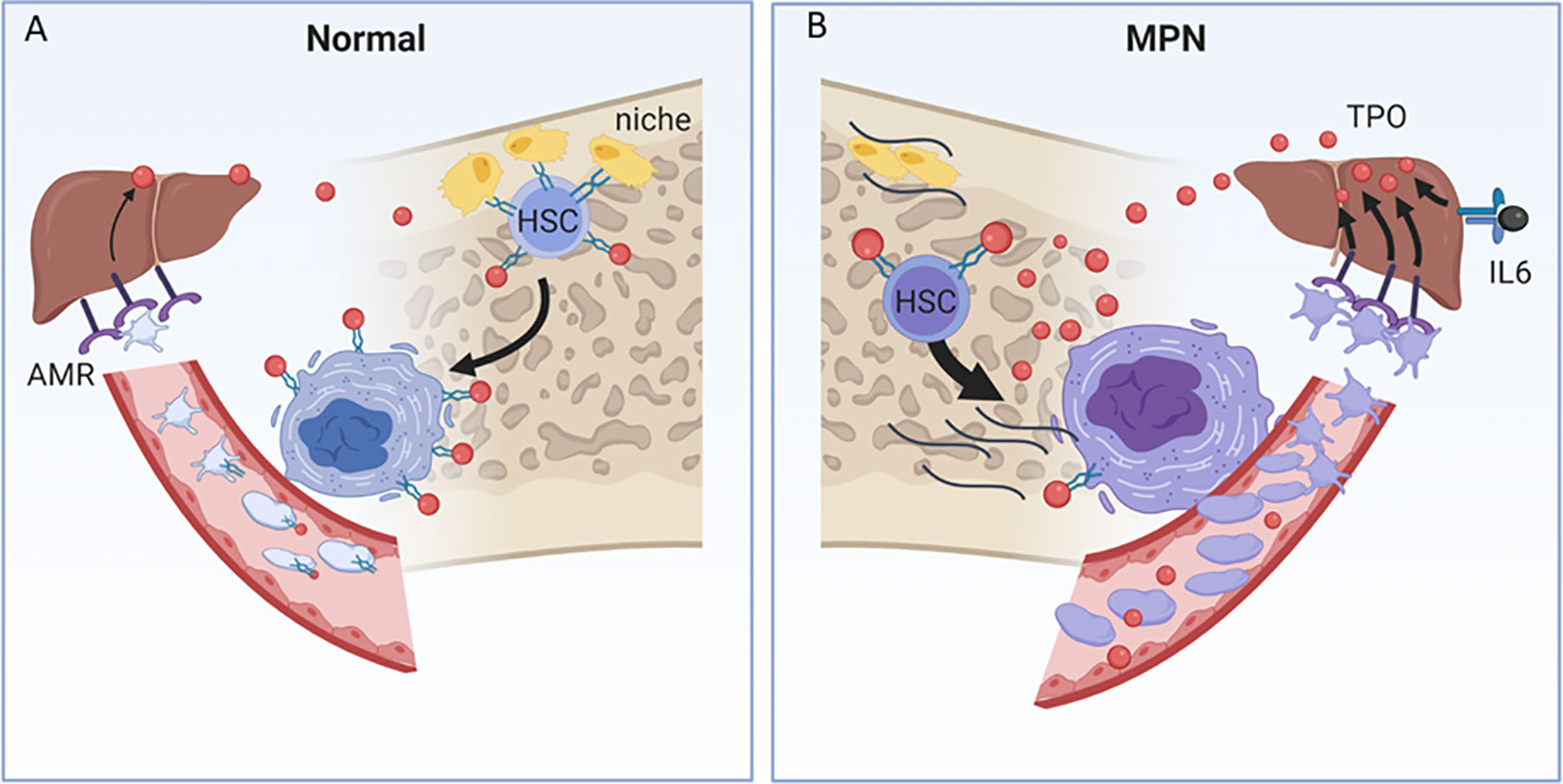
Figure 2 (A) THPO metabolism in normal and MPN contexts. (A) In the endosteal niche, HSCs are tethered to osteoblasts (yellow cells) by adhesive proteins and receptors, including THPO (red ball) and its receptor, MPL (green receptor), and are maintained in a quiescent state. MPL also clears THPO from the circulation by binding and internalizing THPO in megakaryocytes and platelets. Desialyated platelets bind to the Ashwell–Morell receptor (AMR) in the liver, and induce hepatic THPO production, in addition to the constitutive hepatic production of THPO. (B) In the MPN, loss of MPL surface expression and increased THPO/MPL/JAK/STAT signal transduction promote HSC untethering and egress from the endosteal niche to the sinusoidal niche. Mutant, hypersignaling MPN HSCs and megakaryocytes produce inflammatory cytokines, which alter the marrow cellular microenvironment, suppress normal hematopoietic stem cells and promote myelofibrosis (black lines). Loss of MPL cell-surface expression in megakaryocytes and platelets results in the inability to clear plasma THPO, further providing a source of THPO for HSC and megakaryocyte hyperstimulation. MPN platelets are both increased and are more avidly bound to the AMR, which further enhances THPO production. Interleukin-6, produced by the MPN inflammatory milieu, independently enhances hepatic THPO production.
THPO metabolism is markedly altered in the MPN, both due to reduced clearance and enhanced production (Figure 2B). Loss of MPL surface expression in megakaryocytes and platelets, in addition to untethering HSCs from their niche, results in loss of THPO clearance and higher THPO levels that feed back to enhance HSC MPL/JAK/STAT signal transduction in both normal and mutant clonal contexts and drives pathologic myelofibrosis and inflammatory cytokine pathways (44, 76, 77) (Figure 2B). The MPN context is also one of enhanced production of hepatic THPO, both through increased MPN platelet clearance through the AMR, and through IL-6 mediated hepatic stimulation associated with high inflammatory milieu in the MPN (77, 78) (Figure 2B).
Thus, in addition to inhibiting the MPL–THPO interaction at the level of the HSC, antagonizing hepatic THPO production could be a feasible, target-specific, non-myelotoxic therapeutic approach. Gene silencing of hepatic targets can be achieved by using the AMR as entry for modified RNA inference into hepatocytes, and is a safe and effective therapy in human blood diseases (79). Recently, THPO has been effectively silenced in murine models and is effective in lowering THPO levels and platelet counts using this basic technology (80, 81).
Based on our observations, we propose that impaired MPN MPL cell-surface expression leads to inappropriately high plasma THPO because of failure of THPO clearance by MPN platelets and megakaryocytes. Increased plasma THPO enhances activated JAK2 signaling in HPC, while impaired MPL cell-surface expression weakens the ability of HSC to remain in the marrow osteoblastic niche. Eventually, marrow HSC loss due to in situ differentiation or migration from the marrow and sequestration in the spleen, together with continued megakaryocyte stimulation by the elevated plasma THPO, produces a PMF phenotype, regardless of the MPN driver mutation. Viewed from this perspective, the MPNs are in part hematopoietic growth factor-dependent disorders, and targeting the MPL–THPO axis by a THPO antagonist or suppressing hepatic THPO production could be an effective, non-myelotoxic therapeutic strategy.
Both authors planned and wrote the manuscript. All authors contributed to the article and approved the submitted version.
JLS, National Cancer Institute CA108671; ARM, Myeloproliferative Research Foundation Challenge Grant; JS and AM, National Heart, Lung and Blood Institute HL145750. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. James C, Ugo V, Le Couedic JP, Staerk J, Delhommeau F, Lacout C, et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature (2005) 434(7037):1144–8. doi: 10.1038/nature03546
2. Pikman Y, Lee BH, Mercher T, McDowell E, Ebert BL, Gozo M, et al. MPLW515L Is a Novel Somatic Activating Mutation in Myelofibrosis with Myeloid Metaplasia. PloS Med (2006) 3(7):e270. doi: 10.1371/journal.pmed.0030270
3. Nangalia J, Massie CE, Baxter EJ, Nice FL, Gundem G, Wedge DC, et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N Engl J Med (2013) 369(25):2391–405. doi: 10.1056/NEJMoa1312542
4. Klampfl T, Gisslinger H, Harutyunyan AS, Nivarthi H, Rumi E, Milosevic JD, et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N Engl J Med (2013) 369(25):2379–90. doi: 10.1056/NEJMoa1311347
5. Vainchenker W, Dusa A, Constantinescu SN. JAKs in pathology: role of Janus kinases in hematopoietic malignancies and immunodeficiencies. Semin Cell Dev Biol (2008) 19(4):385–93. doi: 10.1016/j.semcdb.2008.07.002
6. Shimoda K, Feng J, Murakami H, Nagata S, Watling D, Rogers NC, et al. Jak1 plays an essential role for receptor phosphorylation and Stat activation in response to granulocyte colony-stimulating factor. Blood (1997) 90(2):597–604. doi: 10.1182/blood.V90.2.597
7. Spivak JL, Considine M, Williams DM, Talbot CC Jr., Rogers O, Moliterno AR, et al. Two clinical phenotypes in polycythemia vera. N Engl J Med (2014) 371(9):808–17. doi: 10.1056/NEJMoa1403141
8. Catani L, Zini R, Sollazzo D, Ottaviani E, Vannucchi AM, Ferrari S, et al. Molecular profile of CD34+ stem/progenitor cells according to JAK2V617F mutation status in essential thrombocythemia. Leukemia (2009) 23(5):997–1000. doi: 10.1038/leu.2008.357
9. Guglielmelli P, Zini R, Bogani C, Salati S, Pancrazzi A, Bianchi E, et al. Molecular profiling of CD34+ cells in idiopathic myelofibrosis identifies a set of disease-associated genes and reveals the clinical significance of Wilms’ tumor gene 1 (WT1). Stem Cells (2007) 25(1):165–73. doi: 10.1634/stemcells.2006-0351
10. Moliterno AR, Williams DM, Rogers O, Isaacs MA, Spivak JL. Phenotypic variability within the JAK2 V617F-positive MPD: Roles of progenitor cell and neutrophil allele burdens. Exp Hematol (2008) 36(11):1480–6. doi: 10.1016/j.exphem.2008.05.006
11. Spivak JL. Myeloproliferative Neoplasms. N Engl J Med (2017) 376(22):2168–81. doi: 10.1056/NEJMra1406186
12. Berk PD, Goldberg JD, Silverstein MN, Weinfeld A, Donovan PB, Ellis JT, et al. Increased incidence of acute leukemia in polycythemia vera associated with chlorambucil therapy. N Engl J Med (1981) 304(8):441–7. doi: 10.1056/NEJM198102193040801
13. Dupont S, Masse A, James C, Teyssandier I, Lecluse Y, Larbret F, et al. The JAK2 617V>F mutation triggers erythropoietin hypersensitivity and terminal erythroid amplification in primary cells from patients with polycythemia vera. Blood (2007) 110(3):1013–21. doi: 10.1182/blood-2006-10-054940
14. Anand S, Stedham F, Beer P, Gudgin E, Ortmann CA, Bench A, et al. Effects of the JAK2 mutation on the hematopoietic stem and progenitor compartment in human myeloproliferative neoplasms. Blood (2011) 118(1):177–81. doi: 10.1182/blood-2010-12-327593
15. Wang X, Ye F, Tripodi J, Hu CS, Qiu J, Najfeld V, et al. JAK2 inhibitors do not affect stem cells present in the spleens of patients with myelofibrosis. Blood (2014) 124(19):2987–95. doi: 10.1182/blood-2014-02-558015
16. Barosi G, Viarengo G, Pecci A, Rosti V, Piaggio G, Marchetti M, et al. Diagnostic and clinical relevance of the number of circulating CD34+ cells in myelofibrosis and myeloid metaplasia. Blood (2001) 98(12):3249–55. doi: 10.1182/blood.V98.12.3249
17. Royer Y, Staerk J, Costuleanu M, Courtoy PJ, Constantinescu SN. Janus kinases affect thrombopoietin receptor cell surface localization and stability. J Biol Chem (2005) 280(29):27251–61. doi: 10.1074/jbc.M501376200
18. Borge OJ, Ramsfjell V, Veiby OP, Murphy MJ Jr., Lok S, Jacobsen SE. Thrombopoietin, but not erythropoietin promotes viability and inhibits apoptosis of multipotent murine hematopoietic progenitor cells in vitro. Blood (1996) 88(8):2859–70. doi: 10.1182/blood.V88.8.2859.bloodjournal8882859
19. Qian H, Buza-Vidas N, Hyland CD, Jensen CT, Antonchuk J, Mansson R, et al. Critical role of thrombopoietin in maintaining adult quiescent hematopoietic stem cells. Cell Stem Cell (2007) 1(6):671–84. doi: 10.1016/j.stem.2007.10.008
20. Yoshihara H, Arai F, Hosokawa K, Hagiwara T, Takubo K, Nakamura Y, et al. Thrombopoietin/MPL signaling regulates hematopoietic stem cell quiescence and interaction with the osteoblastic niche. Cell Stem Cell (2007) 1(6):685–97. doi: 10.1016/j.stem.2007.10.020
21. Goncalves F, Lacout C, Villeval JL, Wendling F, Vainchencker W, Dumenil D. Thrombopoietin does not induce lineage-restricted commitment of Mpl-R expressing pluripotent progenitors but permits their complete erythroid and megakaryocytic differentiation. Blood (1997) 89:3544–53. doi: 10.1182/blood.V89.10.3544.3544_3544_3553
22. de Sauvage FJ, Carver-Moore K, Luoh SM, Ryan A, Dowd M, Eaton DL, et al. Physiological regulation of early and late stages of megakaryocytopoiesis by thrombopoietin. J Exp Med (1996) 183(2):651–6. doi: 10.1084/jem.183.2.651
23. Levin J, Cocault L, Demerens C, Challier C, Pauchard M, Caen J, et al. Thrombocytopenic c-mpl(-/-) mice can produce a normal level of platelets after administration of 5-fluorouracil: the effect of age on the response. Blood (2001) 98(4):1019–27. doi: 10.1182/blood.V98.4.1019
24. Murone M, Carpenter DA, de Sauvage FJ. Hematopoietic deficiencies in c-mpl and TPO knockout mice. Stem Cells (1998) 16(1):1–6. doi: 10.1002/stem.160001
25. Ballmaier M, Germeshausen M. Advances in the understanding of congenital amegakaryocytic thrombocytopenia. Br J Haematol (2009) 146(1):3–16. doi: 10.1111/j.1365-2141.2009.07706.x
26. Geddis AE. Congenital amegakaryocytic thrombocytopenia. Pediatr Blood Cancer (2011) 57(2):199–203. doi: 10.1002/pbc.22927
27. Wendling F, Varlet P, Charon M, Tambourin P. MPLV: a retrovirus complex inducing an acute myeloproliferative leukemic disorder in adult mice. Virology (1986) 149(2):242–6. doi: 10.1016/0042-6822(86)90125-X
28. Souyri M, Vigon I, Penciolelli JF, Heard JM, Tambourin P, Wendling F. A putative truncated cytokine receptor gene transduced by the myeloproliferative leukemia virus immortalizes hematopoietic progenitors. Cell (1990) 63(6):1137–47. doi: 10.1016/0092-8674(90)90410-G
29. Villeval JL, Cohen-Solal K, Tulliez M, Giraudier S, Guichard J, Burstein SA, et al. High thrombopoietin production by hematopoietic cells induces a fatal myeloproliferative syndrome in mice. Blood (1997) 90(11):4369–83. doi: 10.1182/blood.V90.11.4369
30. Yan XQ, Lacey D, Hill D, Chen Y, Fletcher F, Hawley RG, et al. A model of myelofibrosis and osteosclerosis in mice induced by overexpressing thrombopoietin (mpl ligand): reversal of disease by bone marrow transplantation. Blood (1996) 88(2):402–9. doi: 10.1182/blood.V88.2.402.bloodjournal882402
31. Villeval JL, Metcalf D, Johnson GR. Fatal polycythemia induced in mice by dysregulated erythropoietin production by hematopoietic cells. Leukemia (1992) 6(2):107–15. doi: 10.1182/blood.V90.11.4369
32. Mullally A, Lane SW, Ball B, Megerdichian C, Okabe R, Al Shahrour F, et al. Physiological Jak2V617F expression causes a lethal myeloproliferative neoplasm with differential effects on hematopoietic stem and progenitor cells. Cancer Cell (2010) 17(6):584–96. doi: 10.1016/j.ccr.2010.05.015
33. Ding J, Komatsu H, Wakita A, Kato-Uranishi M, Ito M, Satoh A, et al. Familial essential thrombocythemia associated with a dominant-positive activating mutation of the c-MPL gene, which encodes for the receptor for thrombopoietin. Blood (2004) 103(11):4198–200. doi: 10.1182/blood-2003-10-3471
34. Williams DM, Kim AH, Rogers O, Spivak JL, Moliterno AR. Phenotypic variations and new mutations in JAK2 V617F-negative polycythemia vera, erythrocytosis, and idiopathic myelofibrosis. Exp Hematol (2007) 35(11):1641–6. doi: 10.1016/j.exphem.2007.08.010
35. Chachoua I, Pecquet C, El-Khoury M, Nivarthi H, Albu RI, Marty C, et al. Thrombopoietin receptor activation by myeloproliferative neoplasm associated calreticulin mutants. Blood (2016) 127(10):1325–35. doi: 10.1182/blood-2015-11-681932
36. Elf S, Abdelfattah NS, Baral AJ, Beeson D, Rivera JF, Ko A, et al. Defining the requirements for the pathogenic interaction between mutant calreticulin and MPL in MPN. Blood (2018) 131(7):782–6. doi: 10.1182/blood-2017-08-800896
37. Araki M, Yang Y, Masubuchi N, Hironaka Y, Takei H, Morishita S, et al. Activation of the thrombopoietin receptor by mutant calreticulin in CALR-mutant myeloproliferative neoplasms. Blood (2016) 127(10):1307–16. doi: 10.1182/blood-2015-09-671172
38. Moliterno AR, Williams DM, Gutierrez-Alamillo LI, Salvatori R, Ingersoll RG, Spivak JL. Mpl Baltimore: a thrombopoietin receptor polymorphism associated with thrombocytosis. Proc Natl Acad Sci U S A (2004) 101(31):11444–7. doi: 10.1073/pnas.0404241101
39. El Harith E, Roesl C, Ballmaier M, Germeshausen M, Frye-Boukhriss H, von Neuhoff N, et al. Familial thrombocytosis caused by the novel germ-line mutation p.Pro106Leu in the MPL gene. Br J Haematol (2009) 144(2):185–94. doi: 10.1111/j.1365-2141.2008.07430.x
40. Lannutti BJ, Epp A, Roy J, Chen J, Josephson NC. Incomplete restoration of Mpl expression in the mpl-/- mouse produces partial correction of the stem cell-repopulating defect and paradoxical thrombocytosis. Blood (2009) 113(8):1778–85. doi: 10.1182/blood-2007-11-124859
41. Tiedt R, Coers J, Ziegler S, Wiestner A, Hao-Shen H, Bornmann C, et al. Pronounced thrombocytosis in transgenic mice expressing reduced levels of Mpl in platelets and terminally differentiated megakaryocytes. Blood (2009) 113(8):1768–77. doi: 10.1182/blood-2008-03-146084
42. Wiestner A, Schlemper RJ, van der Maas AP, Skoda RC. An activating splice donor mutation in the thrombopoietin gene causes hereditary thrombocythaemia. Nat Genet (1998) 18(1):49–52. doi: 10.1038/ng0198-49
43. Posthuma HL, Skoda RC, Jacob FA, van der Maas AP, Valk PJ, Posthuma EF. Hereditary thrombocytosis not as innocent as thought? Development into acute leukemia and myelofibrosis. Blood (2010) 116(17):3375–6. doi: 10.1182/blood-2010-06-290718
44. Moliterno AR, Hankins WD, Spivak JL. Impaired expression of the thrombopoietin receptor by platelets from patients with polycythemia vera. N Engl J Med (1998) 338(9):572–80. doi: 10.1056/NEJM199802263380903
45. Horikawa Y, Matsumura I, Hashimoto K, Shiraga M, Kosugi S, Tadokoro S, et al. Markedly reduced expression of platelet c-mpl receptor in essential thrombocythemia. Blood (1997) 90(10):4031–8. doi: 10.1182/blood.V90.10.4031
46. Pinho S, Marchand T, Yang E, Wei Q, Nerlov C, Frenette PS. Lineage-Biased Hematopoietic Stem Cells Are Regulated by Distinct Niches. Dev Cell (2018) 44(5):634–41. doi: 10.1016/j.devcel.2018.01.016
47. Sanjuan-Pla A, Macaulay IC, Jensen CT, Woll PS, Luis TC, Mead A, et al. Platelet-biased stem cells reside at the apex of the haematopoietic stem-cell hierarchy. Nature (2013) 502(7470):232–6. doi: 10.1038/nature12495
48. Yamamoto R, Morita Y, Ooehara J, Hamanaka S, Onodera M, Rudolph KL, et al. Clonal analysis unveils self-renewing lineage-restricted progenitors generated directly from hematopoietic stem cells. Cell (2013) 154(5):1112–26. doi: 10.1016/j.cell.2013.08.007
49. Moliterno AR, Williams DM, Rogers O, Spivak JL. Molecular mimicry in the chronic myeloproliferative disorders: reciprocity between quantitative JAK2 V617F and Mpl expression. Blood (2006) 108(12):3913–5. doi: 10.1182/blood-2006-03-008805
51. Spivak JL. How I treat polycythemia vera. Blood (2019) 134(4):341–52. doi: 10.1182/blood.2018834044
52. Passamonti F, Giorgino T, Mora B, Guglielmelli P, Rumi E, Maffioli M, et al. A clinical-molecular prognostic model to predict survival in patients with post polycythemia vera and post essential thrombocythemia myelofibrosis. Leukemia (2017) 31(12):2726–31. doi: 10.1038/leu.2017.169
53. Karantanos T, Chaturvedi S, Braunstein EM, Spivak J, Resar L, Karanika S, et al. Sex determines the presentation and outcomes in MPN and is related to sex-specific differences in the mutational burden. Blood Adv (2020) 4(12):2567–76. doi: 10.1182/bloodadvances.2019001407
54. Kralovics R, Guan Y, Prchal JT. Acquired uniparental disomy of chromosome 9p is a frequent stem cell defect in polycythemia vera. Exp Hematol (2002) 30(3):229–36. doi: 10.1016/S0301-472X(01)00789-5
55. Moliterno AR, Spivak JL. Posttranslational processing of the thrombopoietin receptor is impaired in polycythemia vera. Blood (1999) 94(8):2555–61. doi: 10.1182/blood.V94.8.2555.420k14_2555_2561
56. Decker M, Leslie J, Liu Q, Ding L. Hepatic thrombopoietin is required for bone marrow hematopoietic stem cell maintenance. Science (2018) 360(6384):106–10. doi: 10.1126/science.aap8861
57. Kuter DJ, Rosenberg RD. The reciprocal relationship of thrombopoietin (c-Mpl ligand) to changes in the platelet mass during busulfan-induced thrombocytopenia in the rabbit. Blood (1995) 85(10):2720–30. doi: 10.1182/blood.V85.10.2720.bloodjournal85102720
58. Prchal JF, Axelrad AA. Letter: Bone-marrow responses in polycythemia vera. N Engl J Med (1974) 290(24):1382. doi: 10.1056/NEJM197406132902419
59. Ding J, Komatsu H, Iida S, Yano H, Kusumoto S, Inagaki A, et al. The Asn505 mutation of the c-MPL gene, which causes familial essential thrombocythemia, induces autonomous homodimerization of the c-Mpl protein due to strong amino acid polarity. Blood (2009) 114(15):3325–8. doi: 10.1182/blood-2008-04-149047
60. Marty C, Chaligne R, Lacout C, Constantinescu SN, Vainchenker W, Villeval JL. Ligand-independent thrombopoietin mutant receptor requires cell surface localization for endogenous activity. J Biol Chem (2009) 284(18):11781–91. doi: 10.1074/jbc.M808703200
61. Gilles L, Arslan AD, Marinaccio C, Wen QJ, Arya P, McNulty M, et al. Downregulation of GATA1 drives impaired hematopoiesis in primary myelofibrosis. J Clin Invest (2017) 127(4):1316–20. doi: 10.1172/JCI82905
62. Pecquet C, Diaconu CC, Staerk J, Girardot M, Marty C, Royer Y, et al. Thrombopoietin receptor down-modulation by JAK2 V617F: restoration of receptor levels by inhibitors of pathologic JAK2 signaling and of proteasomes. Blood (2012) 119(20):4625–35. doi: 10.1182/blood-2011-08-372524
63. Cleyrat C, Darehshouri A, Steinkamp MP, Vilaine M, Boassa D, Ellisman MH, et al. Mpl traffics to the cell surface through conventional and unconventional routes. Traffic (2014) 15(9):961–82. doi: 10.1111/tra.12185
64. Wang JC, Chen C, Lou LH, Mora M. Blood thrombopoietin, IL-6 and IL-11 levels in patients with agnogenic myeloid metaplasia. Leukemia (1997) 11(11):1827–32. doi: 10.1038/sj.leu.2400846
65. Wang JC, Chen C, Novetsky AD, Lichter SM, Ahmed F, Friedberg NM. Blood thrombopoietin levels in clonal thrombocytosis and reactive thrombocytosis. Am J Med (1998) 104(5):451–5. doi: 10.1016/S0002-9343(98)00090-4
66. Favale F, Messaoudi K, Varghese LN, Boukour S, Pecquet C, Gryshkova V, et al. An incomplete trafficking defect to the cell-surface leads to paradoxical thrombocytosis for human and murine MPL P106L. Blood (2016) 128(26):3146–58. doi: 10.1182/blood-2016-06-722058
67. Kohlscheen S, Wintterle S, Schwarzer A, Kamp C, Brugman MH, Breuer DC, et al. Inhibition of Thrombopoietin/Mpl Signaling in Adult Hematopoiesis Identifies New Candidates for Hematopoietic Stem Cell Maintenance. PloS One (2015) 10(7):e0131866. doi: 10.1371/journal.pone.0131866
68. Xing S, Wanting TH, Zhao W, Ma J, Wang S, Xu X, et al. Transgenic expression of JAK2V617F causes myeloproliferative disorders in mice. Blood (2008) 111(10):5109–17. doi: 10.1182/blood-2007-05-091579
69. Spivak JL, Merchant A, Williams DM, Rogers O, Zhao W, Duffield A, et al. Thrombopoietin is required for full phenotype expression in a JAK2V617F transgenic mouse model of polycythemia vera. PloS One (2020) 15(6):e0232801. doi: 10.1371/journal.pone.0232801
70. Li J, Prins D, Park HJ, Grinfeld J, Gonzalez-Arias C, Loughran S, et al. Mutant calreticulin knockin mice develop thrombocytosis and myelofibrosis without a stem cell self-renewal advantage. Blood (2018) 131(6):649–61. doi: 10.1182/blood-2017-09-806356
71. Sangkhae V, Etheridge SL, Kaushansky K, Hitchcock IS. The thrombopoietin receptor, MPL, is critical for development of a JAK2V617F-induced myeloproliferative neoplasm. Blood (2014) 124(26):3956–63. doi: 10.1182/blood-2014-07-587238
72. Tiedt R, Hao-Shen H, Sobas MA, Looser R, Dirnhofer S, Schwaller J, et al. Ratio of mutant JAK2-V617F to wild-type Jak2 determines the MPD phenotypes in transgenic mice. Blood (2008) 111(8):3931–40. doi: 10.1182/blood-2007-08-107748
73. Wang X, Haylock D, Hu CS, Kowalczyk W, Jiang T, Qiu J, et al. A thrombopoietin receptor antagonist is capable of depleting myelofibrosis hematopoietic stem and progenitor cells. Blood (2016) 127(26):3398–409. doi: 10.1182/blood-2015-10-674465
74. Grozovsky R, Giannini S, Falet H, Hoffmeister KM. Novel mechanisms of platelet clearance and thrombopoietin regulation. Curr Opin Hematol (2015) 22(5):445–51. doi: 10.1097/MOH.0000000000000170
75. Kaser A, Brandacher G, Steurer W, Kaser S, Offner FA, Zoller H, et al. Interleukin-6 stimulates thrombopoiesis through thrombopoietin: role in inflammatory thrombocytosis. Blood (2001) 98(9):2720–5. doi: 10.1182/blood.V98.9.2720
76. Cerutti A, Custodi P, Duranti M, Noris P, Balduini CL. Thrombopoietin levels in patients with primary and reactive thrombocytosis [see comments]. Br J Haematol (1997) 99(2):281–4. doi: 10.1046/j.1365-2141.1997.3823196.x
77. Di Buduo CA, Giannini S, Abbonante V, Rosti V, Hoffmeister K, Balduini A. Increased beta4GALT1 expression associates with platelet surface galactosylation and thrombopoietin plasma levels in MPNs. Blood (2020). doi: 10.1182/blood.2020007265
78. Hasselbalch HC, Bjorn ME. MPNs as Inflammatory Diseases: The Evidence, Consequences, and Perspectives. Mediators Inflamm (2015) 2015:102476. doi: 10.1155/2015/102476
79. Balwani M, Sardh E, Ventura P, Peiro PA, Rees DC, Stolzel U, et al. Phase 3 Trial of RNAi Therapeutic Givosiran for Acute Intermittent Porphyria. N Engl J Med (2020) 382(24):2289–301. doi: 10.1056/NEJMoa1913147
80. Barrett TJ, Wu BG, Revenko AS, MacLeod AR, Segal LN, Berger JS. Antisense oligonucleotide targeting of thrombopoietin represents a novel platelet depletion method to assess the immunomodulatory role of platelets. J Thromb Haemost (2020) 18(7):1773–82. doi: 10.1111/jth.14808
Keywords: myeloproliferative neoplasms, hematopoietic stem cells, thrombopoietin receptor, thrombopoietin, polycythemia vera, JAK2 V617F, transgenic mice
Citation: Spivak JL and Moliterno AR (2021) The Thrombopoietin Receptor, MPL, Is a Therapeutic Target of Opportunity in the MPN. Front. Oncol. 11:641613. doi: 10.3389/fonc.2021.641613
Received: 14 December 2020; Accepted: 28 January 2021;
Published: 10 March 2021.
Edited by:
Shimin Hu, University of Texas MD Anderson Cancer Center, United StatesReviewed by:
Shannon Elf, University of Chicago, United StatesCopyright © 2021 Spivak and Moliterno. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jerry L. Spivak, amxzcGl2YWtAamhtaS5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.