 Yin Wang
Yin Wang Wen-Jun Weng
Wen-Jun Weng Dun-Hua Zhou
Dun-Hua Zhou Jian-Pei Fang
Jian-Pei Fang Srishti Mishra3
Srishti Mishra3 Li Chai
Li Chai Lu-Hong Xu
Lu-Hong Xu
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Oncol. , 21 April 2021
Sec. Hematologic Malignancies
Volume 11 - 2021 | https://doi.org/10.3389/fonc.2021.632094
This article is part of the Research Topic Novel Diagnostic Tools and Biomarkers in Hematologic Malignancies View all 21 articles
The prognostic impact of Wilms tumor 1 (WT1) mutations remains controversial for patients with acute myeloid leukemia (AML). Here, we aimed to determine the clinical implication of WT1 mutations in a large cohort of pediatric AML. The clinical data of 870 pediatric patients with AML were downloaded from the therapeutically applicable research to generate effective treatment (TARGET) dataset. We analyzed the prevalence, clinical profile, and prognosis of AML patients with WT1 mutations in this cohort. Our results showed that 6.7% of total patients harbored WT1 mutations. These WT1 mutations were closely associated with normal cytogenetics (P<0.001), FMS-like tyrosine kinase 3/internal tandem duplication (FLT3/ITD) mutations (P<0.001), and low complete remission induction rates (P<0.01). Compared to the patients without WT1 mutations, patients with WT1 mutations had a worse 5-year event-free survival (21.7 ± 5.5% vs 48.9 ± 1.8%, P<0.001) and a worse overall survival (41.4 ± 6.6% vs 64.3 ± 1.7%, P<0.001). Moreover, patients with both WT1 and FLT3/ITD mutations had a dismal prognosis. Compared to chemotherapy alone, hematopoietic stem cell transplantation tended to improve the prognoses of WT1-mutated patients. Multivariate analysis demonstrated that WT1 mutations conferred an independent adverse impact on event-free survival (hazard ratio 1.910, P = 0.001) and overall survival (hazard ratio 1.709, P = 0.020). In conclusion, our findings have demonstrated that WT1 mutations are independent poor prognostic factors in pediatric AML.
Acute myeloid leukemia (AML) is a type of blood cancer that originates in the bone marrow from immature white blood cells known as myeloblasts. About 20% of all children with leukemia have AML (1, 2). In the last few years, collaborative studies have revealed a link between the degree of genetic heterogeneity of AML and the clinical outcome, allowing risk stratification before therapy and guiding post-induction treatment (3). The Wilms tumor 1 (WT1) gene, located on chromosome 11p13, encodes a zinc-finger protein that exists in multiple isoforms. It has been implicated in the regulation of cell survival, proliferation and differentiation, and may function both as a tumor suppressor and an oncogene (4, 5). Various mutations across WT1 gene have been reported in solid tumors and AML (6, 7). However, the prognostic impact of WT1 mutations remains controversial for patients with AML (8).
The WT1 mutations have been shown to be independent predictors of worse clinical outcome in some but not all adult AML studies (9–11). Recently, WT1 mutations are proposed to be prognostic markers of risk stratification for adult AML (12). However, the prognostic implications of WT1 mutations have not been clarified in pediatric AML. Moreover, large cohort studies on the clinical significance of WT1 mutations in pediatric AML are scarce. A pediatric study of 298 patients with AML found that WT1 mutations conferred an independent poor prognostic significance (13). However, another study of 842 pediatric AML revealed that the presence of WT1 mutations had no independent prognostic significance in predicting the disease outcome (14). Recently, in a cohort of 353 pediatric patients with AML, Niktoreh et al. (15) have found that WT1 mutations significantly increased the chance of relapse or treatment failure and reduced the probability of 3-year overall survival (OS), but had no significant impact on the 3-year probability of event-free survival (EFS). On the other hand, hematopoietic stem cell transplantation (HSCT) is an important treatment modality for patients with AML. However, the role of HSCT for patients with WT1 mutations remains unknown.
To determine the clinical implication of WT1 mutations, an independent large cohort study of pediatric AML is needed. Therefore, we analyzed the clinical data of 870 pediatric patients with AML from the therapeutically applicable research to generate effective treatment (TARGET) dataset. We found that WT1 mutations are independent poor prognostic factors in pediatric AML in terms of 5-year EFS and OS. Patients with both WT1 and FMS-like tyrosine kinase 3/internal tandem duplication (FLT3/ITD) mutations had a dismal prognosis. Moreover, HSCT might be an effective strategy for patients with WT1 mutations.
The clinical data on patients with AML were downloaded from the TARGET dataset (https://ocg.cancer.gov/programs/target/data-matrix). In total, 870 pediatric patients younger than 18 years old with the information of WT1 mutations were included in our study. The year of diagnosis ranged from 1996 to 2010 while the year of last follow-up ranged from 1997 to 2015. The diagnosis of pediatric AML and risk stratification were defined according to the Children’s Oncology Group (COG) guidelines. Subtype classifications of AML were assigned according to the French–American–British (FAB) classifications. Mutation analyses of WT1, FLT3/ITD, NPM1, and CEBPA were performed as previously described (14, 16–18). Treatment protocols for AML included AAML03P1, AAML0531 and CCG-2961. HSCT was considered for high-risk patients in the first complete remission. Detailed treatments and risk stratification of these studies have been previously described (19).
The data were analyzed with the Statistical Package for the Social Sciences (SPSS®) version, 20.0 (IBM Corporation, Armonk, NY, USA). The χ2 test was used to compare the frequencies of mutations. Fischer’s exact test was used when data were sparse. The nonparametric Mann–Whitney U-test was applied for continuous variables. Complete remission (CR) was defined as bone marrow aspirate with < 5% blasts by morphology. EFS was defined as the time between diagnosis and first event, including induction failure, relapse, or death of any cause. OS was defined as the time between diagnosis and death from any cause. The survival curves were estimated using the Kaplan–Meier method and compared using the log-rank test. Cox proportional hazard models were used to estimate hazard ratios (HR) for multivariate analyses. A two-sided P-value less than 0.05 was considered statistically significant for all statistical analyses.
The patients’ clinical characteristics are shown in Table 1. Overall, among the 870 pediatric patients with AML, 58 patients (6.7%) were identified with WT1 mutations. The white blood cell count (WBC) at diagnosis was significantly higher in WT1-mutated patients (median 56.9×109/L) than in WT1 wild-type patients (median 30.8×109/L; P=0.041). In WT1-mutated group, the FAB subtypes were mainly M1, M2, and M4. A higher proportion of WT1-mutated patients had M4 morphology in comparison with WT1 wild-type patients (41.2% vs 25.9%; P = 0.018). We also evaluated the associations between WT1 mutations and cytogenetic and molecular alterations. In terms of cytogenetics, WT1 mutations were found more frequently in the normal cytogenetics subset (44.2% of WT1-mutated patients had normal cytogenetics compared with 22.3% of those without WT1 mutations; P<0.001). Regarding the molecular alterations, there was a substantial overlap between WT1 mutations and FLT3/ITD, as shown in Table 1, 48.3% of those carrying a WT1 mutation were also FLT3/ITD positive as opposed to 14.7% of patients without WT1 mutations (P<0.001). Moreover, the WT1-mutated patients were classified more frequently as high risk (40.7% vs 12.6%; P<0.001). The treatment protocols for pediatric AML were equally distributed between these two groups (P=0.058). However, there were no significant differences in the median age, the median of FLT3/ITD allelic ratio, NPM1, and CEBPA mutations between the WT1-mutated group and WT1 wild-type group.
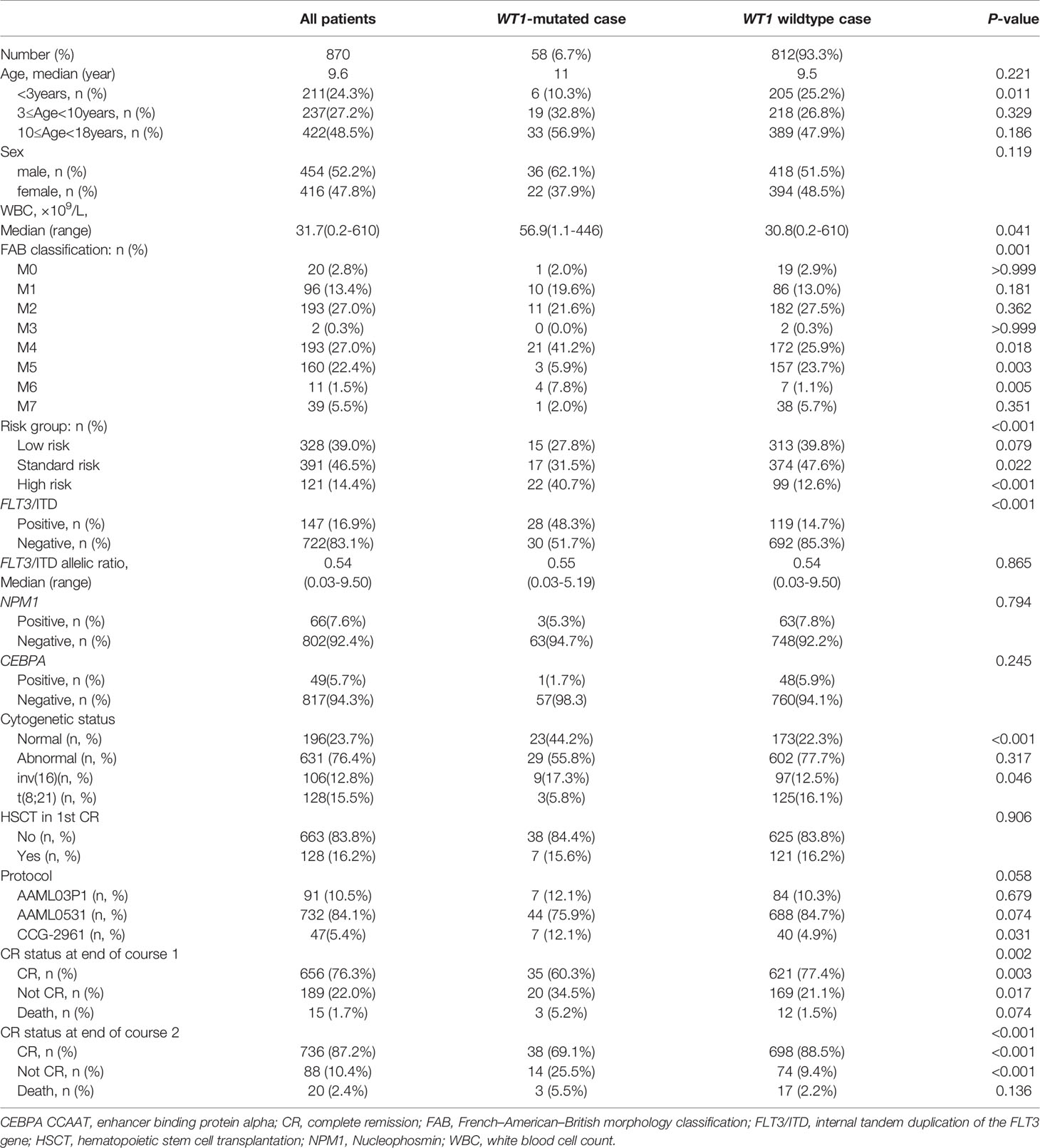
Table 1 Characteristics of pediatric patients with or without WT1 mutations.
The CR rate was determined for all patients after the first and second course of induction therapy. At the end of the first course of therapy, patients with WT1 mutations had a lower rate of CR (60.3%) compared to those without WT1 mutations (77.4%), and the difference was statistically significant (P=0.002). At the end of the second course of therapy, 38(69.1%) of the 55 patients with WT1 mutations achieved a CR compared to 698 (88.5%) of 789 patients without WT1 mutations (P<0.001). Taken together, WT1 mutations were significantly associated with low induction CR rates.
Next, we evaluated the survival data for all the 870 pediatric patients. The median follow-up time for the survivors was 5.6 years. As shown in Figure 1A, WT1-mutated patients had a significantly worse 5-year EFS (21.7 ± 5.5%) compared with WT1 wild-type patients (48.9 ± 1.8%; P<0.001). Moreover, patients with WT1 mutations had a worse 5-year OS (41.4 ± 6.6%) than those without WT1 mutations (64.3 ± 1.7%; P<0.001) (Figure 1B). When analyses were restricted to patients having normal cytogenetics, there were significant differences in the outcome between patients with and without WT1 mutations (Figures 1C, D) (5-year EFS: 15.2 ± 7.8% vs 51.8 ± 3.8%, P<0.001; 5-year OS: 34.4 ± 10.4% vs 66.1± 3.7%, P<0.001). In the subgroup of abnormal cytogenetics (Figures 1E, F), WT1-mutated patients also had a worse survival time compared with WT1 wild-type patients in terms of 5-year EFS (31.0 ± 8.6% vs 48.3 ± 2.1%, P=0.027) and OS (48.0 ± 9.3% vs 64.6± 2.0%, P=0.048).
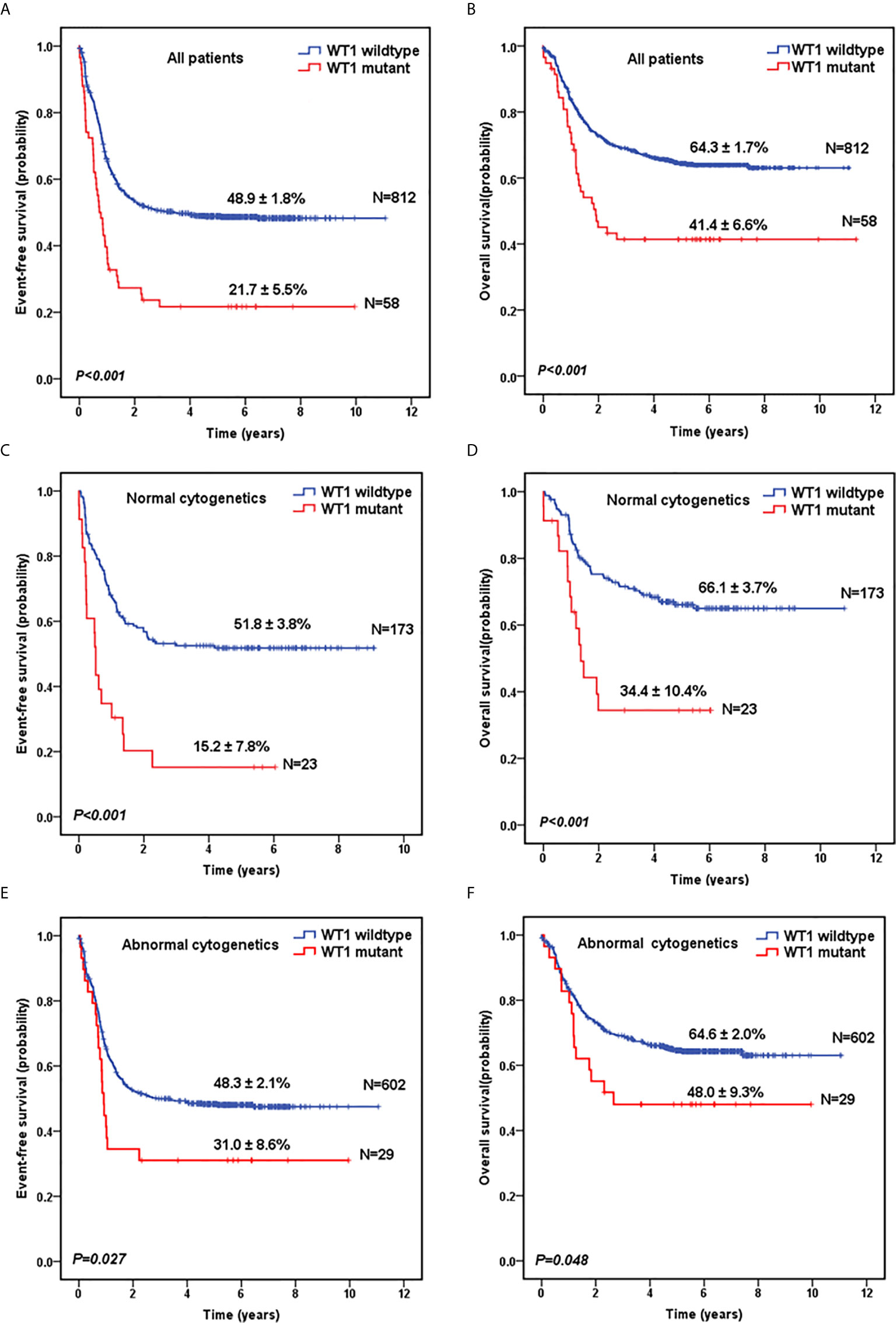
Figure 1 Survival curves of pediatric AML patients with and without WT1 mutations. Probability of EFS (A) and OS (B) for all patients with and without WT1 mutations, respectively. Probability of EFS (C) and OS (D) for cytogenetically normal patients with and without WT1 mutations, respectively. Probability of EFS (E) and OS (F) for cytogenetically abnormal patients with and without WT1 mutations, respectively.
Survival data for patients with FLT3/ITD positive and negative were also explored. As shown in Figure S1A, FLT3/ITD positive was significantly associated with inferior EFS (5-year EFS=33.5± 4.0% vs 49.7± 1.9% for FLT3/ITD-negative; P<0.001). Moreover, the FLT3/ITD positive group had a worse 5-year OS (51.5 ± 4.3%) than the FLT3/ITD-negative group (65.0 ± 1.8%; P=0.003) (Figure S1B).
Given the overlap between WT1 mutations and positive FLT3/ITD status, subset analysis was performed to assess the relative influence of WT1 mutations and FLT3/ITD on the prognosis of children with AML (Figures 2A, B; Table 2). In the FLT3/ITD-positive subgroup, WT1-mutated patients had an extremely dismal prognosis (5-year EFS =12.5 ± 6.5% vs 38.4± 4.5% for WT1 wild-type patients, HR: 2.179 [1.364-3.482], P=0.001; 5-year OS = 27.5± 8.8% vs 57.0 ± 4.7% for WT1 wild-type patients, HR: 2.225[1.305-3.796], P=0.003). When restricted to the FLT3/ITD-negative subgroup, WT1 mutations had an adverse impact on 5-year EFS (HR: 1.861[1.197-2.892], P=0.006) instead of 5-year OS (HR: 1.600[0.933-2.744], P=0.088). Similarly, for the WT1 wild-type patients, FLT3/ITD positive had reduced 5-year EFS (HR: 1.386[1.075-1.788], P=0.012) but not 5-year OS (HR: 1.305[0.961-1.771], P=0.088). However, FLT3/ITD mutations had no significantly negative influence on the outcome of WT1-mutated patients (EFS HR: 1.605[0.886-2.906], P=0.118; OS HR: 1.748[0.870-3.514], P=0.117).
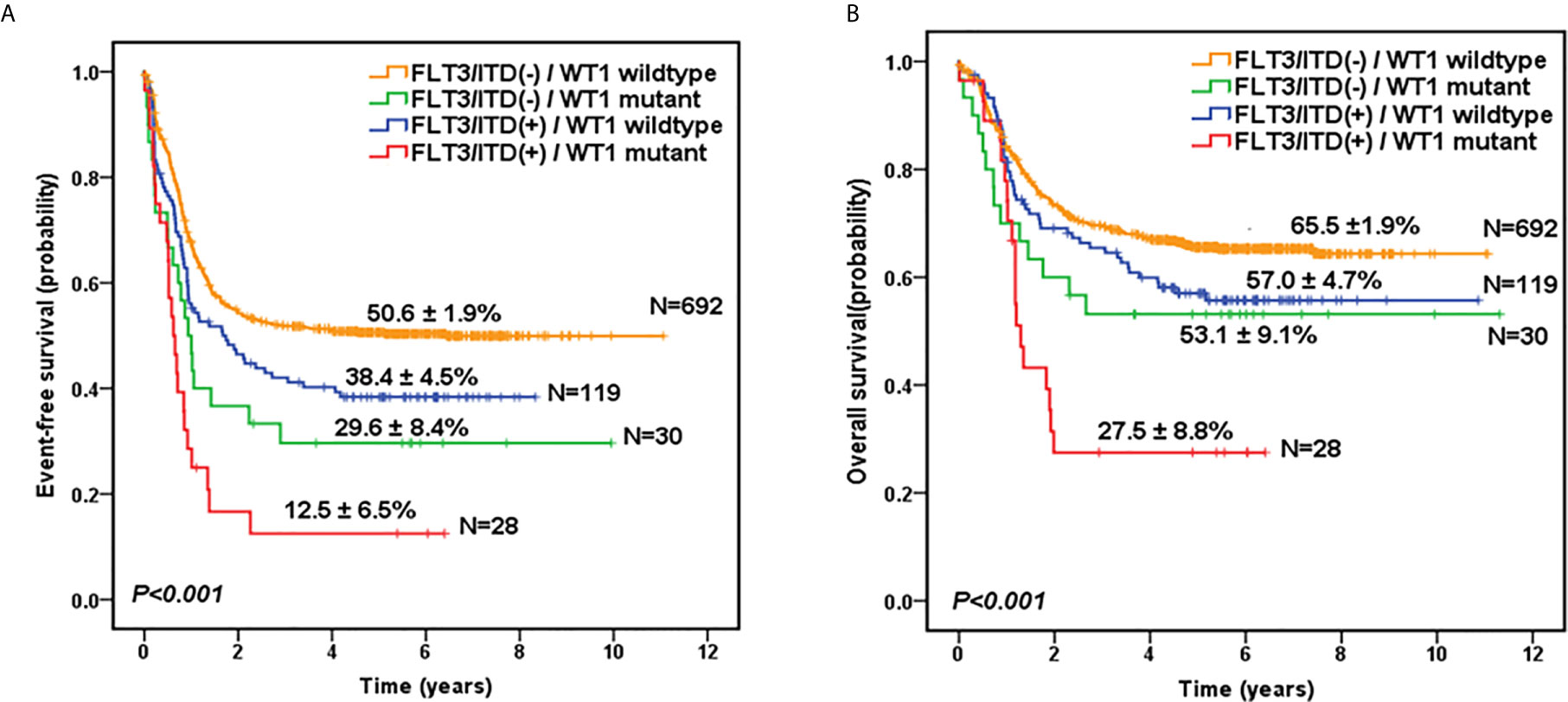
Figure 2 Survival curves of all pediatric AML patients according to the combined WT1 mutations and positive FLT3/ITD status. Probability of EFS (A) and OS (B) for patients according to the combined WT1 mutations and positive FLT3/ITD status, respectively.
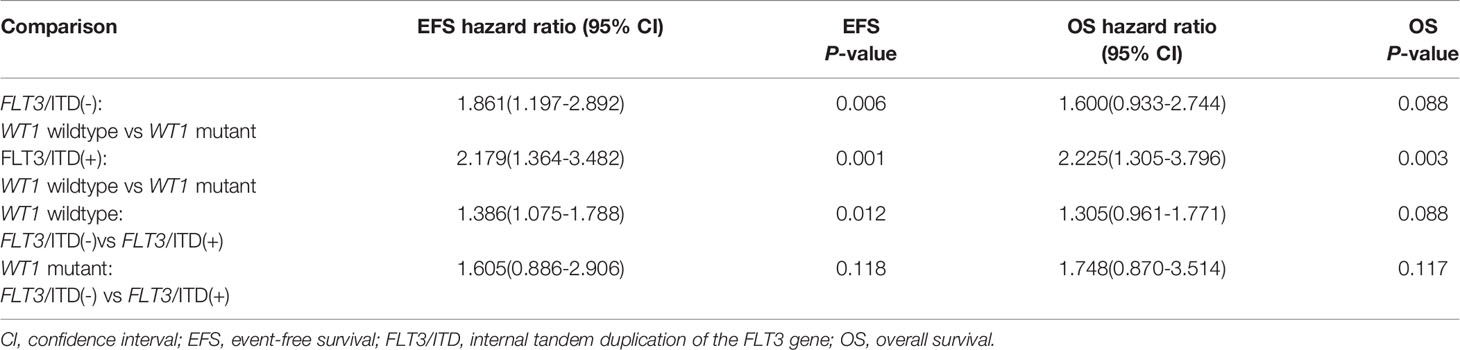
Table 2 Statistical comparison of survival data according to both WT1 and FLT3/ITD status.
Similar results were found in the subgroup of cytogenetically normal AML patients according to the combined WT1 mutations and positive FLT3/ITD status (Figure S2). Of note, the survival curves showed that there were no significant differences between WT1-mutated patients with FLT3/ITD-positive (n=17) and FLT3/ITD negative (n=6), in terms of 5-year EFS (14.1 ± 9.0% vs 16.7 ± 15.2%; P=0.584) and OS (34.5 ± 12.3% vs 33.3 ± 19.2%; P=0.665).
As shown in Table 1, there was no significant difference in the proportion of HSCT in WT1-mutated group and WT1 wild-type group (15.6% vs 16.2%, P=0.906). The survival analysis, after HSCT stratification, showed that for WT1-mutated pediatric AML patients, HSCT conferred a favorable prognostic impact with a trend of better 5-year EFS (42.9 ± 18.7% vs 22.3 ± 7.0% for chemotherapy-only; P=0.316) and OS (57.1 ± 18.7% vs 43.6 ± 8.2% for chemotherapy-only; P=0.483) (Figures 3A, B).
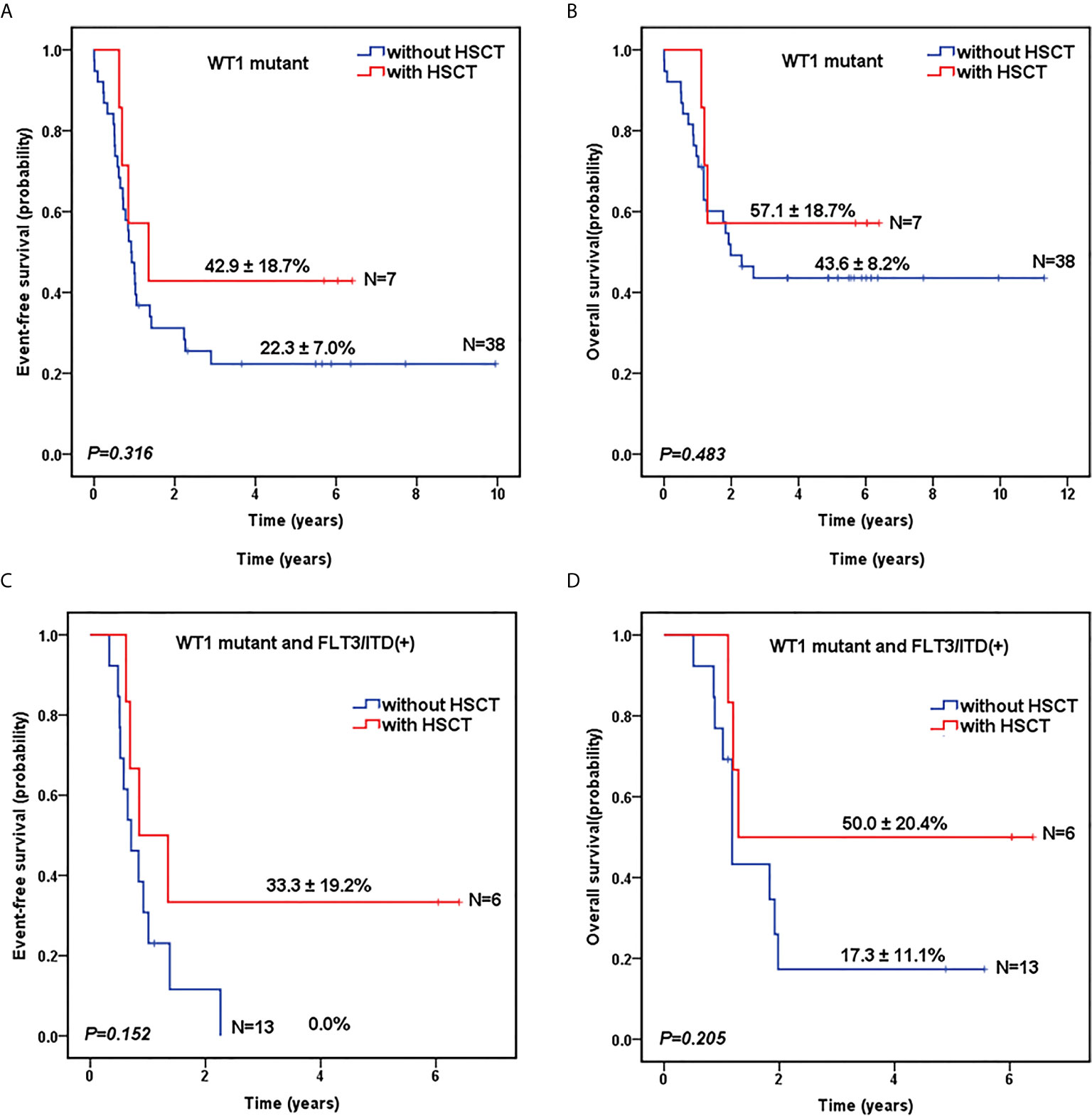
Figure 3 Survival curves of pediatric AML patients according to WT1 mutations and hematopoietic stem cell transplantation (HSCT) status. Probability of EFS (A) and OS (B) for patients with WT1 mutations according to HSCT status, respectively. Probability of EFS (C) and OS (D) for patients with WT1 mutations and FLT3/ITD positive according to HSCT status, respectively.
To further evaluate the role of HSCT in the patients with co-occurring WT1 and FLT3/ITD mutations, we explored the impact of HSCT on those patients. As shown in Figures 3C, D, for AML patients with both WT1 mutations and positive FLT3/ITD, 5-year EFS (33.3 ± 19.2%) and OS (50.0 ± 20.4%) were higher in children with HSCT than those with chemotherapy-only (EFS: 0.0 ± 0.0%, P=0.152; OS: 17.3 ± 11.1%, P=0.205), respectively, although the differences between the two groups were not statistically significant.
Cox regression analyses were then performed to evaluate WT1 mutation status as a predictor of EFS and OS alongside other prognostic factors: age (utilizing 10 years of age as the cutoff value), white blood cell count at diagnosis (utilizing 50×10 9/L as the cutoff value), high risk, standard risk, and HSCT. We identified WT1 mutations as an independent prognostic factor for both EFS and OS in pediatric patients with AML (Table 3). WT1 mutations were significantly associated with inferior EFS (HR: 1.910, 95% CI: 1.297-2.812, P=0.001) and OS (HR: 1.709, 95% CI: 1.090-2.679, P=0.020). Additionally, age (older than 10 years), white blood cell count greater than 50×109/L at first diagnosis, high-risk and standard-risk were significantly related to poor EFS and OS, while HSCT was related to better survival prognosis (HR: 0.431, 95% CI: 0.313-0.593, P<0.001) and OS (HR: 0.594, 95% CI: 0.419-0.843, P=0.004).
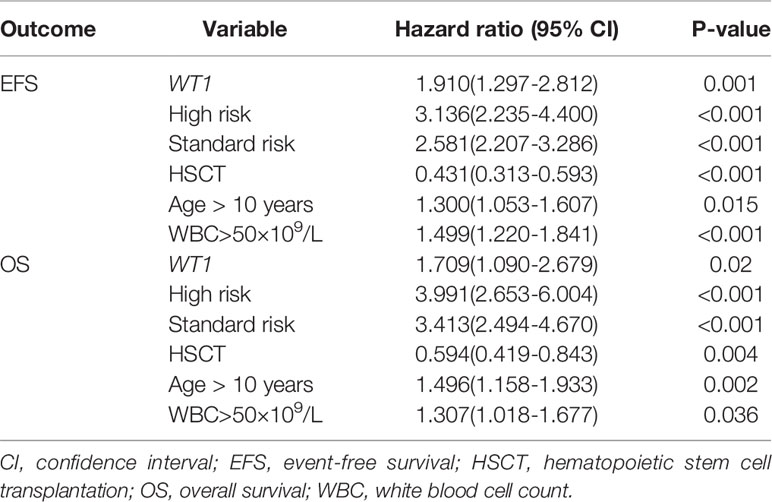
Table 3 Cox regression analysis of WT1 mutations and other prognostic factors.
The TARGET program is a collaborative COG-national cancer institute (NCI) project aiming to comprehensively characterize the mutational, transcriptional, and epigenetic landscapes of a large, well-annotated cohort of pediatric cancer (20). Using this large cohort of subjects, we were able to investigate the clinical implication of WT1 mutations in pediatric AML. Our findings showed that the frequency of WT1 mutations was 6.7% among these 870 pediatric AML patients. This result was similar to the adult AML studies. In a large cohort of adult AML study, the frequency of WT1 mutations among 3157 patients was reported to be 5.5% (21). Next, we found that WT1 mutations were significantly associated with FAB subtypes of M4, with high white blood cell counts at first diagnosis, normal cytogenetics, and FLT3/ITD mutations. However, no association was found between WT1 mutations and CEBPA mutations. These results were different from some of the other studies. For instance, a report by Ho et al. (14) also found that WT1 mutations were related to normal cytogenetics and FLT3/ITD mutations, but they found no correlation between WT1 mutations and white blood cell counts or M4 subtype. A pediatric AML report by Hollink et al. (13) showed that WT1 mutations clustered significantly in the subgroup with normal cytogenetics and were associated with FLT3/ITD and CEBPA mutations.
The prognostic impact of WT1 mutations has not been clarified in pediatric AML. In our study, we found that patients with WT1 mutations had lower CR induction rates, worse EFS and OS rates in comparison to patients without WT1 mutations. Patients with both WT1 and FLT3/ITD mutations had a dismal prognosis. The multivariate analysis showed that WT1 mutations were an independent adverse impact factor. These results are consistent with findings by Hollink et al. (13), though they found the CR induction rates did not differ significantly between patients with WT1-mutated and WT1 wild-type AML. A report from the French study group confirmed that WT1 mutations were an independent prognostic factor for pediatric AML (22). However, a report from the Japanese study group showed that WT1 mutations were related to a poor prognosis in patients with normal cytogenetics, excluding those with FLT3/ITD and those younger than 3 years (23). By contrast, a report from the Nordic Society of Pediatric Hematology and Oncology (NOPHO) revealed that no significant correlation with survival was seen for WT1 mutations (24). Notably, they found that patients with WT1 mutations but negative FLT3/ITD had a superior EFS compared with patients with WT1 wildtype with or without concurrent FLT3/ITD (24). In adult studies, the presence of WT1 mutation has been found to be associated with poor clinical outcomes of AML patients in some but not all studies. In the studies from Cancer and Leukemia Group B (9) and Hou et al. (10), WT1 mutations were correlated with a poor prognosis in AML patients. However, in the study from the German-Austrian Study Group (11), WT1 mutation as a single molecular marker did not seem to impact the patient outcomes. These conflicting results may be due to the differences in sample size, exon of WT1 mutations, and variable treatment protocols across studies. It has been reported that the negative impact of WT1 mutations may be overcome by the use of repetitive cycles of high-dose cytarabine, especially in the subgroup of patients with negative FLT3/ITD genotype (11).
The mechanism of WT1 mutations in leukemogenesis remains elusive. Several different WT1 mutations have been described in AML, which occur primarily in exons 1, 7, and 9. WT1 mutations may result in the loss of DNA binding ability due to loss of the zinc-finger domain or result in loss of expression of the WT1 protein altogether (25–27). WT1 mutations fail to properly direct the ten-eleven translocation-2 to its target sites, either by disruption of the interaction itself or by failing to bind to DNA (28, 29). Recently, Pronier et al. (30) have found that WT1 heterozygous loss enhances stem cell self-renewal, WT1 depletion cooperates with FLT3/ITD mutation to induce fully penetrant AML. Mutational analysis of a large cohort of AML cases revealed that WT1 may play an important role in the epigenetic pathway (31, 32). Given the epigenetic alterations catalogued in WT1 mutant, epigenetic-targeted therapy has been explored as a potential mechanism to deal with this subgroup of leukemia (33). Recently, Sinha et al. (34) have found that mutant WT1 is associated with DNA hypermethylation of polycomb repressor complex 2 targets in AML, and inhibitor of enhancer of zeste homolog 2 (EZH2) may be helpful in this AML subtype.
Alternately, HSCT is one of the most effective treatments for AML. However, it is unknown whether WT1-mutated patients will benefit from HSCT. Our studies showed that compared to chemotherapy alone, HSCT tended to improve the prognoses of WT1-mutated patients, and for patients with both WT1 and FLT3/ITD mutations as well. These results are in agreement with a previous pediatric AML report (14). Recently, Eisfeld et al. (12) have found that co-occurrence of WT1 and NPM1 mutations confers especially poor outcomes in a large cohort of 863 adult AML. They proposed that mutated WT1 co-occurrence with mutated NPM1 would be an adverse marker for risk stratification, indicating patients with both WT1 and NPM1 mutations might be considered for HSCT. However, since NPM1 mutation is relatively rare in children, we could not draw a firm conclusion on this topic due to the small number of patients with both WT1 and NPM1 mutations. Thus, whether WT1 mutation is an indication for HSCT in pediatric AML requires further investigation.
There were several limitations to our study. Firstly, since different WT1 mutations may affect its functions on DNA binding or protein interaction differentially, the details of WT1 mutants can be important to the clinical outcome of AML patients with these mutants. However, the information on the specific mutations of in WT1 is not provided in the TARGET dataset, therefore, we can’t perform further analysis. Secondly, though this is a large pediatric AML cohort study, the sample size is still relatively small in the subgroups of patients with WT1 mutations. We cannot rule out the contribution of FLT3/ITD co-occurrence towards the prognosis. Thirdly, our findings showed that WT1 mutations were associated with poor clinical outcomes, and WT1-mutated patients might benefit from HSCT. These results suggested that WT1 mutations could be used as predictive factors and linked to a specific clinical management plan. However, due to the limitations associated with the TARGET dataset as mentioned above, and the retrospective analysis nature of our study, a large multicentric prospective future study could be of value to further address the prognostic significance of WT1 mutations in AML.
In summary, we analyzed the clinical implication of WT1 mutations in a large pediatric AML cohort. Our findings showed that WT1 mutations are independent poor prognostic factors in pediatric AML. Patients with co-occurring WT1 and FLT3/ITD mutations had a dismal prognosis. Moreover, HSCT might be an effective strategy for patients with WT1 mutations. These results have important implications and might contribute to the refining risk stratification of pediatric AML.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
The studies involving human participants were reviewed and approved by TARGET Publications Committee. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
LC and L-HX participated in project design, data collection, analysis, interpretation and manuscript drafting. YW participated in data interpretation and manuscript drafting. W-JW participated in data collection and analysis. D-HZ and J-PF participated in project design, data interpretation and manuscript drafting. SM participated in manuscript editing. All authors contributed to the article and approved the submitted version.
This work was supported by the Natural Science Foundation of Guangdong Province, China (2018A030313680 to L-HX), Guangdong Basic and Applied Basic Research Foundation (2020A1515010312 to L-HX), Science and Technology Program of Guangzhou City, China (201803010032 to J-PF), Beijing Bethune Charitable Foundation (SCE111DS to J-PF), Xiu Research Fund (to LC).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We would like to thank all study participants and the TARGET group for making these data publicly available.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2021.632094/full#supplementary-material
1. Creutzig U, van den Heuvel-Eibrink MM, Gibson B, Dworzak MN, Adachi S, de Bont E, et al. Diagnosis and management of acute myeloid leukemia in children and adolescents: recommendations from an international expert panel. Blood (2012) 120:3187–205. doi: 10.1182/blood-2012-03-362608
2. de Rooij JD, Zwaan CM, van den Heuvel-Eibrink M. Pediatric AML: From Biology to Clinical Management. J Clin Med (2015) 4:127–49. doi: 10.3390/jcm4010127
3. Conneely SE, Rau RE. The genomics of acute myeloid leukemia in children. Cancer Metastasis Rev (2020) 39:189–209. doi: 10.1007/s10555-020-09846-1
4. Scharnhorst V, van der Eb AJ, Jochemsen AG. WT1 proteins: functions in growth and differentiation. Gene (2001) 273:141–61. doi: 10.1016/s0378-1119(01)00593-5
5. Yang L, Han Y, Suarez Saiz F, Minden MD. A tumor suppressor and oncogene: the WT1 story. Leukemia (2007) 21:868–76. doi: 10.1038/sj.leu.2404624
6. Hohenstein P, Hastie ND. The many facets of the Wilms’ tumour gene, WT1. Hum Mol Genet (2006) 15:R196–201. doi: 10.1093/hmg/ddl196
7. Hastie ND. Wilms’ tumour 1 (WT1) in development, homeostasis and disease. Development (2017) 144:2862–72. doi: 10.1242/dev.153163
8. Owen C, Fitzgibbon J, Paschka P. The clinical relevance of Wilms Tumour 1 (WT1) gene mutations in acute leukaemia. Hematol Oncol (2010) 28:13–9. doi: 10.1002/hon.931
9. Paschka P, Marcucci G, Ruppert AS, Whitman SP, Mrozek K, Maharry K, et al. Wilms’ tumor 1 gene mutations independently predict poor outcome in adults with cytogenetically normal acute myeloid leukemia: a cancer and leukemia group B study. J Clin Oncol (2008) 26:4595–602. doi: 10.1200/JCO.2007.15.2058
10. Hou HA, Huang TC, Lin LI, Liu CY, Chen CY, Chou WC, et al. WT1 mutation in 470 adult patients with acute myeloid leukemia: stability during disease evolution and implication of its incorporation into a survival scoring system. Blood (2010) 115:5222–31. doi: 10.1182/blood-2009-12-259390
11. Gaidzik VI, Schlenk RF, Moschny S, Becker A, Bullinger L, Corbacioglu A, et al. Prognostic impact of WT1 mutations in cytogenetically normal acute myeloid leukemia: a study of the German-Austrian AML Study Group. Blood (2009) 113:4505–11. doi: 10.1182/blood-2008-10-183392
12. Eisfeld AK, Kohlschmidt J, Mims A, Nicolet D, Walker CJ, Blachly JS, et al. Additional gene mutations may refine the 2017 European LeukemiaNet classification in adult patients with de novo acute myeloid leukemia aged <60 years. Leukemia (2020) 34:3215–27. doi: 10.1038/s41375-020-0872-3
13. Hollink IH, van den Heuvel-Eibrink MM, Zimmermann M, Balgobind BV, Arentsen-Peters ST, Alders M, et al. Clinical relevance of Wilms tumor 1 gene mutations in childhood acute myeloid leukemia. Blood (2009) 113:5951–60. doi: 10.1182/blood-2008-09-177949
14. Ho PA, Zeng R, Alonzo TA, Gerbing RB, Miller KL, Pollard JA, et al. Prevalence and prognostic implications of WT1 mutations in pediatric acute myeloid leukemia (AML): a report from the Children’s Oncology Group. Blood (2010) 116:702–10. doi: 10.1182/blood-2010-02-268953
15. Niktoreh N, Walter C, Zimmermann M, von Neuhoff C, von Neuhoff N, Rasche M, et al. Mutated WT1, FLT3-ITD, and NUP98-NSD1 Fusion in Various Combinations Define a Poor Prognostic Group in Pediatric Acute Myeloid Leukemia. J Oncol (2019) 2019:1609128. doi: 10.1155/2019/1609128
16. Meshinchi S, Alonzo TA, Stirewalt DL, Zwaan M, Zimmerman M, Reinhardt D, et al. Clinical implications of FLT3 mutations in pediatric AML. Blood (2006) 108:3654–61. doi: 10.1182/blood-2006-03-009233
17. Brown P, McIntyre E, Rau R, Meshinchi S, Lacayo N, Dahl G, et al. The incidence and clinical significance of nucleophosmin mutations in childhood AML. Blood (2007) 110:979–85. doi: 10.1182/blood-2007-02-076604
18. Ho PA, Alonzo TA, Gerbing RB, Pollard J, Stirewalt DL, Hurwitz C, et al. Prevalence and prognostic implications of CEBPA mutations in pediatric acute myeloid leukemia (AML): a report from the Children’s Oncology Group. Blood (2009) 113:6558–66. doi: 10.1182/blood-2008-10-184747
19. Vujkovic M, Attiyeh EF, Ries RE, Goodman EK, Ding Y, Kavcic M, et al. Genomic architecture and treatment outcome in pediatric acute myeloid leukemia: a Children’s Oncology Group report. Blood (2017) 129:3051–8. doi: 10.1182/blood-2017-03-772384
20. Bolouri H, Farrar JE, Triche T Jr, Ries RE, Lim EL, Alonzo TA, et al. The molecular landscape of pediatric acute myeloid leukemia reveals recurrent structural alterations and age-specific mutational interactions. Nat Med (2018) 24:103–12. doi: 10.1038/nm.4439
21. Krauth MT, Alpermann T, Bacher U, Eder C, Dicker F, Ulke M, et al. WT1 mutations are secondary events in AML, show varying frequencies and impact on prognosis between genetic subgroups. Leukemia (2015) 29:660–7. doi: 10.1038/leu.2014.243
22. Marceau-Renaut A, Duployez N, Ducourneau B, Labopin M, Petit A, Rousseau A, et al. Molecular Profiling Defines Distinct Prognostic Subgroups in Childhood AML: A Report From the French ELAM02 Study Group. Hemasphere (2018) 2:e31. doi: 10.1097/HS9.0000000000000031
23. Sano H, Shimada A, Tabuchi K, Taki T, Murata C, Park MJ, et al. WT1 mutation in pediatric patients with acute myeloid leukemia: a report from the Japanese Childhood AML Cooperative Study Group. Int J Hematol (2013) 98(4):437–45. doi: 10.1007/s12185-013-1409-6
24. Staffas A, Kanduri M, Hovland R, Rosenquist R, Ommen HB, Abrahamsson J, et al. Presence of FLT3-ITD and high BAALC expression are independent prognostic markers in childhood acute myeloid leukemia. Blood (2011) 118:5905–13. doi: 10.1182/blood-2011-05-353185
25. King-Underwood L, Renshaw J, Pritchard-Jones K. Mutations in the Wilms’ tumor gene WT1 in leukemias. Blood (1996) 87:2171–9. doi: 10.1182/blood.V87.6.2171.bloodjournal8762171
26. Chen Z. The possible role and application of WT1 in human leukemia. Int J Hematol (2001) 73:39–46. doi: 10.1007/BF02981901
27. Bielinska E, Matiakowska K, Haus O. Heterogeneity of human WT1 gene. Postepy Hig Med Dosw (Online) (2017) 71:595–601. doi: 10.5604/01.3001.0010.3840
28. Wang Y, Xiao M, Chen X, Chen L, Xu Y, Lv L, et al. WT1 recruits TET2 to regulate its target gene expression and suppress leukemia cell proliferation. Mol Cell (2015) 57:662–73. doi: 10.1016/j.molcel.2014.12.023
29. Rampal R, Figueroa ME. Wilms tumor 1 mutations in the pathogenesis of acute myeloid leukemia. Haematologica (2016) 101:672–9. doi: 10.3324/haematol.2015.141796
30. Pronier E, Bowman RL, Ahn J, Glass J, Kandoth C, Merlinsky TR, et al. Genetic and epigenetic evolution as a contributor to WT1-mutant leukemogenesis. Blood (2018) 132:1265–78. doi: 10.1182/blood-2018-03-837468
31. Patel JP, Gonen M, Figueroa ME, Fernandez H, Sun Z, Racevskis J, et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl J Med (2012) 366:1079–89. doi: 10.1056/NEJMoa1112304
32. Fernandez HF, Sun Z, Yao X, Litzow MR, Luger SM, Paietta EM, et al. Anthracycline dose intensification in acute myeloid leukemia. N Engl J Med (2009) 361:1249–59. doi: 10.1056/NEJMoa0904544
33. Panuzzo C, Signorino E, Calabrese C, Ali MS, Petiti J, Bracco E, et al. Landscape of Tumor Suppressor Mutations in Acute Myeloid Leukemia. J Clin Med (2020) 9(3):802. doi: 10.3390/jcm9030802
Keywords: acute myeloid leukemia, WT1 mutations, pediatric patients, prognostic factors, FLT3/ITD mutations
Citation: Wang Y, Weng W-J, Zhou D-H, Fang J-P, Mishra S, Chai L and Xu L-H (2021) Wilms Tumor 1 Mutations Are Independent Poor Prognostic Factors in Pediatric Acute Myeloid Leukemia. Front. Oncol. 11:632094. doi: 10.3389/fonc.2021.632094
Received: 22 November 2020; Accepted: 29 March 2021;
Published: 21 April 2021.
Edited by:
Alejandro Gru, University of Virginia Health System, United StatesReviewed by:
Deepshi Thakral, All India Institute of Medical Sciences, IndiaCopyright © 2021 Wang, Weng, Zhou, Fang, Mishra, Chai and Xu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Li Chai, bGNoYWlAYndoLmhhcnZhcmQuZWR1; Lu-Hong Xu, eHVsaG9uZ0BtYWlsLnN5c3UuZWR1LmNu
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.