
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 01 April 2021
Sec. Cancer Immunity and Immunotherapy
Volume 11 - 2021 | https://doi.org/10.3389/fonc.2021.627549
This article is part of the Research Topic Understanding the Immuno-Oncological Mechanism of Cancer Using Systems Immunology Approaches View all 43 articles
 Pooria Safarzadeh Kozani1†
Pooria Safarzadeh Kozani1† Pouya Safarzadeh Kozani2,3†
Pouya Safarzadeh Kozani2,3† Fatemeh Rahbarizadeh1,4*
Fatemeh Rahbarizadeh1,4* Shahryar Khoshtinat Nikkhoi5
Shahryar Khoshtinat Nikkhoi5Chimeric antigen receptor (CAR) T cell therapy has offered cancer patients a new alternative therapeutic choice in recent years. This novel type of therapy holds tremendous promise for the treatment of various hematologic malignancies including B-cell acute lymphoblastic leukemia (B-ALL) and lymphoma. However, CAR T cell therapy has experienced its ups and downs in terms of toxicities and efficacy shortcomings. Adverse events such as cytokine release syndrome (CRS), neurotoxicity, graft rejection, on-target off-tumor toxicities, and tumor relapse have tied the rescuing hands of CAR T cell therapies. Moreover, in the case of solid tumor treatment, CAR T cell therapies have not yielded encouraging results mainly due to challenges such as the formidable network of the tumor microenvironments (TME) that operates in a suppressive fashion resulting in CAR T cell dysfunction. In this review, we tend to shine a light on emerging strategies and solutions for addressing the mentioned barriers. These solutions might dramatically help shorten the gap between a successful clinical outcome and the hope for it.
Chimeric antigen receptor (CAR) T cells are genetically engineered T cells that possess the ability to specifically recognize and target tumor cells with significant discrimination from healthy tissues. Unlike the conventional cancer treatment methods such as surgery, radiotherapy, and chemotherapy, CAR T cells target tumor-specific antigens (TSAs) or tumor-associated antigens (TAAs) expressed on the surface of their target tumor cells with the delicate specificity granted to them by their antibody fragment-equipped targeting domain in a non-major histocompatibility complex (MHC) manner (1, 2).
In detail, CARs are made of an extracellular domain comprised of a targeting domain and a hinge, a transmembrane (TM) domain, and an intracellular domain composed of one or more co-stimulatory domains and an activation domain. The targeting domain of CARs are commonly composed of a single-chain variable fragment (scFv) derived from a monoclonal antibody (mAb) but other targeting domains such as single variable domains of heavy-chain antibodies (VHH, also known as Nanobodies®), ligands, and toxins have also been used, even though less commonly (2–4). Researchers have demonstrated the potential of VHH-based CAR T cells against solid tumors and in targeting their tumor microenvironments (TME) (3, 5). Moreover, in an interesting twist, Wang and colleagues used chlorotoxin as their CAR targeting domain because of its potential binding capacity to antigens associated with glioblastoma and they reported acceptable tumor elimination alongside undetectable off-target effects towards healthy cells, even in vivo (4). The authors also concluded that the reactivity of the chlorotoxin-equipped CAR T cells with their target cells is dependent on the expression of matrix metalloproteinase 2 (4).
So far, CD8, CD28, IgG1, and IgG4 have been used as hinges connecting CAR targeting domains to the TM domain. The TM domain of CARs is derived from molecules such as CD3ζ, CD8α, CD4, CD28, and the inducible T cell co-stimulator (ICOS) (6). Moreover, co-stimulatory domains are important components of CARs since they can contribute to different properties of CAR T cells (7–9). To this date, CD28, CD137 (4-1BB), CD134 (OX40), ICOS, CD27, MYD88-CD40, and KIR2DS2 have been used as co-stimulatory domains in various studies (10–12).
The activation domain used in the structure of CARs has the critical role of T cell activation upon target antigen encountering and it is mostly derived from the CD3ζ part of the T cell receptor (TCR) CD3 complex (13). However, other activation domains that have been used include FcϵRIγ, the ζ-chain of TCR-associated protein kinase 70 kDa (ZAP70), and DAP12 (13–17). The early CAR T cells (termed “first-generation CARs”) did not have any co-stimulatory domain and did not show promising antitumor efficacy mainly due to the lack of adequate persistence and activation (18). For addressing these caveats, researchers added co-stimulatory domains to the intracellular domain of CARs generating “second-generation” and “third-generation” CARs which have one and two co-stimulatory domains, respectively (13). These CAR T cells showed enhanced persistence, activation, and effector function in clinical trials in comparison with first-generation CARs (19, 20). Researchers have even stepped further by adding an inducer domain of a specific cytokine such as interleukin (IL)-2 to the intracellular domain of second-generation CARs only to generate T cells redirected for universal cytokine-mediated killing (TRUCKs) or armored CARs (21). These CAR T cells can deliver transgenic products or payloads to the targeted tumor tissues leading to the enhancement of antitumor activity and efficacy of CAR T cell therapy (21). Additionally, according to Kershaw et al., the activation and proliferation of T cells might be more favorable if accompanied by a third cytokine engagement signal besides the other two primary activation and co-stimulation signals (22). In this regard, Kagoya1 et al. redecorated second-generation CARs, originally harboring the CD3ζ activation domain and CD28 co-stimulation domain, by adding a truncated cytoplasmic domain from the IL-2 receptor β (IL-2Rβ) that harbors a STAT3-binding site (23). The in vitro investigations of these researchers revealed that the activation of the JAK kinase, STAT3, and STAT5 pathways are dependent on the engagement of the CAR with its target antigen (23). Moreover, these novel CAR T cells were capable of establishing more outstanding persistence and tumoricidal activity in preclinical models of hematologic and solid tumors compared with their conventional counterparts (23). Figure 1 represents a detailed description of the various components of CARs.
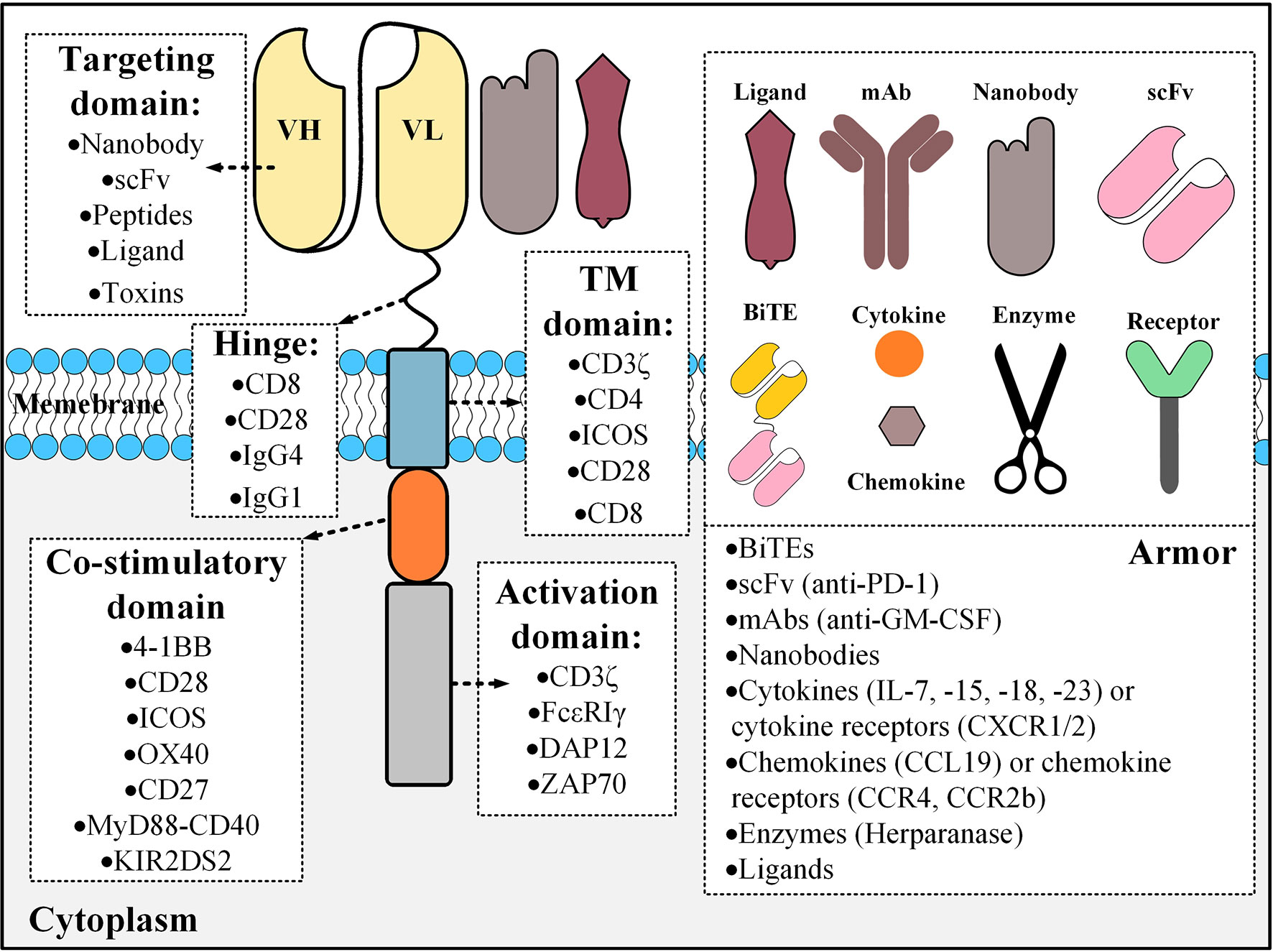
Figure 1 The building blocks of a CAR molecule and examples of different components that could be used for its construction. CAR constructs can also be engineered for the expression of an “armor” molecule that can operate in different aspects including the modifications of the tumor microenvironments, enhancing the homing of CAR T cells to the tumor site, or having immunomodulatory effects resulting in the augmentation of CAR T cell tumoricidal efficacy. Different types of armors are shown in the right panel with examples in parentheses. BiTE, bispecific T cell engager; TM, transmembrane; mAb, monoclonal antibody; scFv, single-chain variable fragment; ICOS, the inducible T cell co-stimulator; ZAP70, the ζ-chain of TCR-associated protein kinase 70 kDa.
CAR T cell therapy has induced remarkable clinical outcomes, especially in the treatment of hematologic malignancies, which has led to the US Food and Drug Administration (FDA) approval of four CD19-targeting CAR T cell products, named Kymriah™ (Tisagenlecleucel), Yescarta™ (Axicabtagene ciloleucel), Tecartus™ (Brexucabtagene autoleucel), and Breyanzi™ (Lisocabtagene maraleucel) (24–27). So far, patients with conventional treatment-resistant relapsed/refractory (R/R) B-cell Acute Lymphoblastic Leukemia (B-ALL), Diffuse Large B Cell Lymphoma (DLBCL), and Mantle cell lymphoma (MCL) can only profit from the therapeutic benefits of the mentioned products (24–27).
However, from the early days of CAR T cell therapy, it has been accompanied by various types of side effects, which can range from manageable mild to irreversible life-threatening toxicities. Such toxicities created various safety concerns that have limited the broader application of CAR T cells in various oncological indications. Therefore, understanding their nature and developing strategies for their mitigation are subjects of paramount importance. Neurologic toxicity and Cytokine Release Syndrome (CRS) (characterized by the overproduction of immune-regulatory cytokines and factors) alongside immune responses mediated by the recipient’s immune system against the infused CAR T cells (recognized as foreign invaders) have been rather frequent in the related clinical settings (28, 29). In the case of CD19-based CAR T cell therapies, the long-term persistence of CAR T cells that fail to discriminate between malignant and healthy B cells leads to the elimination of the latter, long after treatment completion (28, 30). This phenomenon is known as B cell aplasia and it renders the respective patients susceptible to infections caused by numerous bacteria (28, 30). Additionally, due to the poor availability of TSAs, a high proportion of CAR T therapies target TAAs that are also expressed by normal cells at physiological levels (29). Since such CAR T cells fail to discriminate between normal and malignant cells, there have been cases of serious adverse events against healthy tissues (which are known as on-target off-tumor toxicities) (29). Furthermore, disease relapse has also been observed both in the cases of blood-based malignancies and solid tumors (31). This occurrence is a result of target antigen loss or extreme antigen downregulation which is undertaken by tumor cells as a potential mechanism for immune evasion (31). Ultimately, the harsh immunosuppressive features of the TME can also impinge on CAR T cell functionality, in the context of solid tumors (32). In this review, we tend to present a detailed description of the mentioned caveats and then discuss intelligent strategies aimed at removing them. Furthermore, we brief how such elaborate strategies might enable a safer and more efficient CAR T cell therapy.
CRS is the most common side effect of CAR T cell therapy that is usually observed several days following the adoptive transfer (28, 33). CRS is commonly characterized by elevated levels of IL-1, IL-2, IL-6, IL-8, IL-10, interferon-γ (INF-γ), granulocyte-macrophage colony-stimulating factor (GM-CSF), and tumor necrosis factor α (TNF-α) in a patient’s serum (28, 33). Patients experiencing CRS usually manifest hypotension, fever, pulmonary edema, and hypoxia (28). However, depending on the grade and severity of CRS, other symptoms could also be observed in the affected patients including tachycardia, myalgias, diarrhea, nausea, acute kidney injury, anemia, arrhythmias, and hyperbilirubinemia (28). Such manifestations could be resultant from CRS-related damages to various vital organs of the patients which require meticulous medical care (28). The starting point of this so-called storm is the activation of CAR T cells following their engagement with their target antigen (28, 33). This activation leads to the production and secretion of inflammatory cytokines by CAR T cells (28, 33). In response to these cytokines, other innate immune cells, such as macrophages, begin to release inflammatory cytokines such as IL-1 and IL-6, thus creating a loop of inflammation (28, 33). To take the situation under control, the mentioned self-intensifying loop needs to be disrupted.
In contrast with CRS, the rate of neurotoxicity in CAR T therapies has been rather inconsistent in different reports (28, 34). Patients with neurologic toxicity often experience seizures, hallucinations, delirium, brain edema, headache, or tremor (28, 34). To this date, the exact mechanism that gives rise to CAR T cell-induced neurotoxicity remains a mystery (28). However, investigators have reported the presence of CD19-redirected CAR T cells in the patients’ cerebrospinal fluid alongside elevated levels of pro-inflammatory cytokines (28, 34). Moreover, a high concentration of such cytokines might activate the endothelium of the brain vessels and the blood-brain barrier (BBB) resulting in their permeabilization and consequent cerebral edema (35). Based on a recent report by Parker and colleagues, in the case of CD19-based CAR T cell therapies, the observed neurotoxicity could be attributed to the targeting of CD19-expressing brain mural cells by CAR T cells (36). Since mural cells provide vital support for the BBB, their elimination might facilitate CAR T cell infiltration into the brain (hence a high possibility of cerebral edema emergence) (36). In this section, we briefly discuss strategies that could be beneficial in the management of CAR T cell-mediated CRS and neurologic toxicity.
GM-CSF is a macrophage- and monocyte-activating cytokine known to be an important factor in mediating CRS (28, 29). GM-CSF can be neutralized using mAbs, such as lenzilumab, which can result in a significant reduction of myeloid and T cell infiltration in the central nervous system (CNS) (37). This reduction has been helpful in the mitigation of neuroinflammation (NI) and the prevention of CAR T cell-mediated CRS in preclinical models (37). Additionally, not only this method does not interfere with CAR T cell functionality, but it also elevates their tumoricidal efficacy by reducing the risk of CAR T cell-mediated CRS and NI (37). In detail, GM-CSF neutralization inhibits the secretion of CRS-causing cytokines such as IL-6 and reduces the production of other CRS-mediating pro-inflammatory factors including IL-8 and monocyte chemoattractant protein 1 (MCP-1), which act as immune cell trafficking mediators (38, 39). In addition to antibody-mediated neutralization, genetic engineering methods can also be used for the manipulation of CAR T cells that are less likely to mediate CRS and neurotoxicity (37, 40). Recently, studies have shown that knocking out the GM-CSF gene in CAR T cells using transcription activator-like effector nucleases (TALEN) or CRISPR/Cas9 can significantly reduce the production and secretion of GM-CSF, which can consequently abrogate the macrophage-dependent secretion of CRS-associated biomarkers such as MCP-1, IL-6, and IL-8 (37, 40). Also, this approach has reduced the levels of key CRS-mediators and enhanced the antitumor activity of CAR T cells in preclinical models (37). Above all, CAR T cells can also be genetically engineered to secrete GM-CSF neutralizing antibodies which can further mitigate the risk of CRS and neurotoxicity.
Studies have shown that monocyte- and macrophage-released IL-1 and IL-6 are associated with CAR T cell-mediated CRS and immune effector cell-associated neurotoxicity syndrome (ICANS) (41, 42). Preclinical data indicate that monocytes are a more responsible source of IL-1 and IL-6 during CRS occurrence (42). It has been demonstrated that CRS can be prevented by methods such as monocyte ablation or IL-6 receptor blockade using tocilizumab (42). However, it has been reported that tocilizumab does not prevent delayed lethal neurotoxicity in preclinical mouse models (42). In this case, Anakinra, which is an immunosuppressive drug and an IL-1 receptor antagonist, has shown promising results after administration into preclinical mouse CRS models by protecting them from both lethal neurotoxicity and CRS (42). Of note, anakinra can be as effective as tocilizumab in rescuing preclinical mouse models from lethal CRS (42). Furthermore, other studies have reported that IL-6 receptor blockade may not be completely sufficient in controlling severe CRS and it might be necessary to use high-dose corticosteroids for this aim (43, 44). Other studies have engineered CAR T cells to secrete IL-1 receptor antagonists which have induced promising effects in preventing or reducing CRS and neurotoxicity in preclinical mouse models (41). In conclusion, IL-1 and IL-6 are both key players in the development and progression of post-CAR T cell infusion CRS and neurotoxicity (42). Targeting strategies against IL-1 can be an applicable approach for the prevention and mitigation of both CAR T cell-induced CRS and neurologic toxicities.
Recently, it has been found that high levels of circulating catecholamines can mediate various types of immune-dysregulation, including CRS, through a self-augmenting loop in macrophages (45). Catecholamines have effective roles in the release of cytokines induced by T cell-activating therapeutic agents (45). It has been found that inhibition of catecholamine synthesis can result in a significant reduction in the level of cytokine release both in vitro and in vivo (45). Also, it has been demonstrated that atrial natriuretic peptide (ANP) can reduce the levels of circulating catecholamines without interfering with the tumoricidal activity of CAR T cells (45, 46). Furthermore, myeloid-secreted catecholamines are also known as critical mediators of CRS (45). Researchers have indicated that myeloid-specific ablation of tyrosine hydroxylase (an essential enzyme involved in the synthesis of catecholamines) using metyrosine can protect mouse models of lymphoma with stimulated macrophages from lethal complications of CRS after CD19-based CAR T cell therapy (45). Such studies indicate that catecholamines are key modulators of cytokine release, and not only blocking their synthesis pathway does not lead to side effects or CAR T cell functionality impairment but it also might reduce the incidence of CRS development and progression (45, 46). Such tactics also suggest that the modification of cellular pathways involved in CRS progression might reduce the risk of this life-threatening toxicity (45).
From the emerging days of CAR T cell therapy, the pros and cons of the cell sources, from which CAR T cells are generated, have been under investigation. Using autologous T cells (derived from the patients themselves) for producing CAR T cells is not always feasible because of the patients’ disease burden or the particular treatment course they are under. On the other hand, allogeneic T cells (obtained from healthy donors) are not completely limitation-free as they might be rejected by the recipients’ immune system (28, 29). This unfavorable event is mostly mediated by the recipient’s T cells and natural killer (NK) cells, as these cells recognize the allogeneic CAR T cells as invading foreign cells that should be eradicated from the host’s body (47–51).
One of the most recent strategies for addressing the issue of allogeneic CAR T cell rejection exploits the 4-1BB cell surface receptor present on the recipients’ T and NK cells (51). The expression of this receptor is upregulated in activated T cells and NK cells (51). This strategy uses an engineered receptor named Alloimmune Defense Receptor (ADR), which is made of a 4-1BB-recognizing domain derived from 4-1BB ligand (4-1BBL), an intracellular CD3ζ domain, a spacer, and a transmembrane domain (51). The ADR is designed to be expressed on the surface of CAR T cells (51). In detail, the ADR recognizes the upregulated 4-1BB molecule on the surface of activated alloreactive T cells and NK cells which leads to the activation of the ADR-expressing CAR T cells and results in the elimination of the recipients’ alloreactive immune cells (51). Moreover, ADR expression does not impinge on the effector function of CAR T cells, therefore, this approach can give allogeneic CAR T cells a new weapon that they can use against immune cells trying to interfere with their fight against tumors (51).
However, since ADR-expressing CAR T cells upregulate 4-1BB expression (following activation), they might become subjects of fratricide (self-cytotoxicity) (52). Mo et al. also reported limited fratricide both in preclinical models and in vitro (however, transient) (52). An interesting mechanism could be attributed to this limited fratricide based on similar situations already observed in other studies (52). Ruella and colleagues reported that accidental transduction of leukemic B cells (during the manufacturing process of CD19-redirected CAR T cells) might give rise to the resistance of such leukemic cells to CD19-based CAR T cell therapy (52). The underlying mechanism for this resistance is that the transduced leukemic B cells start to express the CD19-specific CAR molecule which eventually manages to engage with their CD19 antigen (52). This self-reactivity leads to the masking of the CD19 epitope recognized by the CD19-redirected CAR T cells, therefore, the CAR-expressing leukemic B cells evade the antitumor reactions of the mentioned CAR T therapy (52). This scenario might somehow be possible in the case of activated ADR-positive CAR T cells as it helps them become fratricide-resistant (51). In detail, following activation, ADR-positive CAR T cells start to upregulate 4-1BB on their surface which consequently manages to engage with their ADR (51). This self-engagement masks the 4-1BB molecule, therefore, it can no longer be recognized by other ADR-positive CAR T cells (the emergence of fratricide-resistant ADR-positive CAR T cells) (51). Furthermore, this self-engagement might also provide the mentioned fratricide-resistant CAR T cells with amplified proliferation and persistence signals (51, 53, 54). Figure 2 represents a detailed description of ADR-expressing CAR T cells.
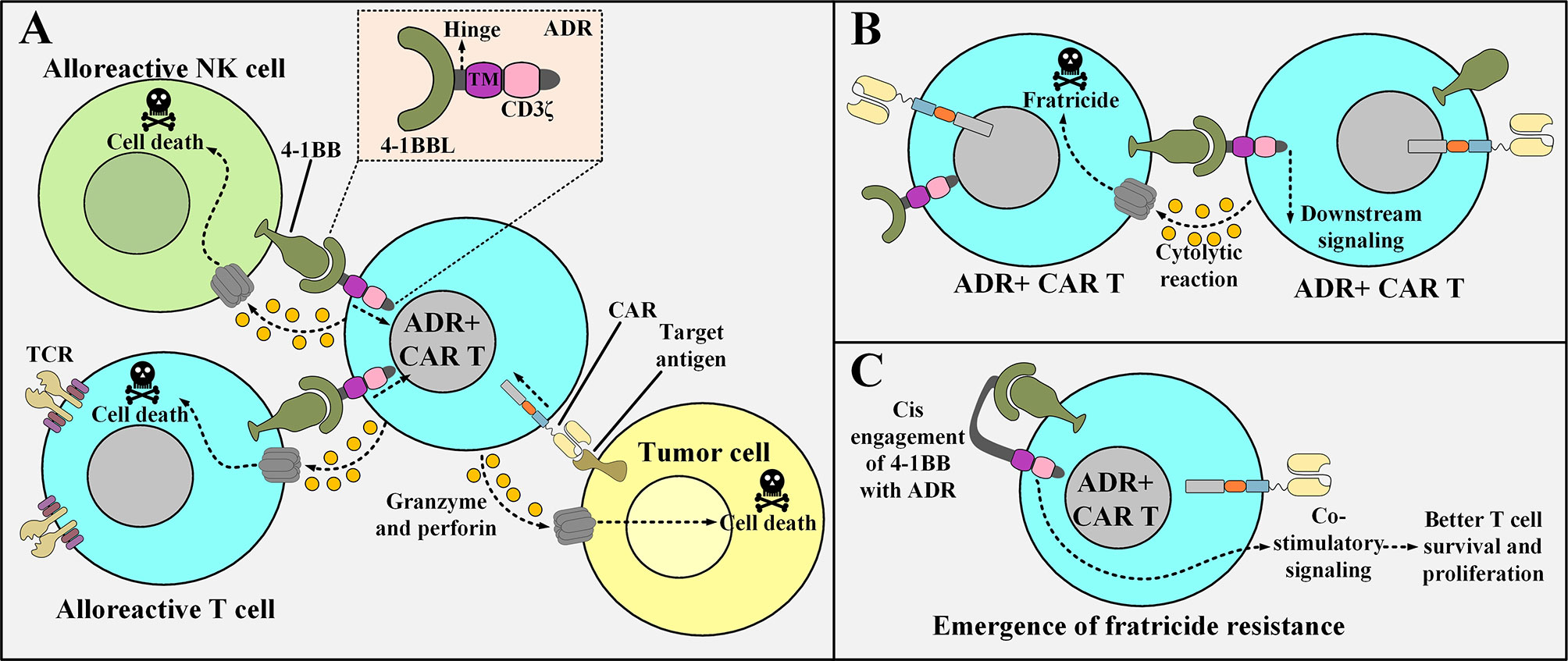
Figure 2 The underlying of mechanism of action of ADR-expressing CAR T cells, the issue of their fratricide, and the possible mechanism of action for the emergence of fratricide-resistant ADR-positive CAR T cells. (A) ADR-expressing CAR T cells enforce cytolytic reactions only against alloreactive T cells and NK cells (that are activated with their 4-1BB upregulated) and manage to spare resting T cells and NK cell (that are non-alloreactive). On the other hand, they can also enforce tumoricidal responses against their target tumor cells. (B) Following their activation, ADR-positive CAR T cells upregulate 4-1BB. This makes them susceptible to fratricide (self-cytotoxicity). (C) A proposed underlying mechanism for the emergence of ADR-expressing CAR T cells that are resistant to fratricide. The surface expressed ADR reacts with the 4-1BB molecule that is upregulated after the activation of the ADR-positive CAR T cells. This phenomenon renders 4-1BB hidden from being recognized by other ADR-expressing CAR T cells (hence the ADR-expressing CAR T cell represented in the figure becomes fratricide-resistant). TM, transmembrane domain; 4-1BBL, a fragment derived from the 4-1BB ligand; ADR, alloimmune defense receptor; NK, natural killer; CAR, chimeric antigen receptor; TCR, T cell receptor.
CD47 is a transmembrane protein responsible for mediating a “do not eat me” signal in numerous types of malignant cells (55). The signal regulatory protein-α (SIRPα) is recognized as the receptor for CD47 on various immune cells including macrophages (55). Once tumor cells that express the CD47 antigen on their surface encounter macrophages, CD47 binds to SIRPα which leads to the transmission of the “do not eat me” signal and consequent abrogation of phagocytosis by macrophages (55). Therefore, using this mechanism, malignant cells can easily evade immune system-mediated eradication (55). This mechanism can be applied when using allogeneic CAR T cells to avert macrophage-assisted CAR T cell rejection and subsequent clearance. In this regard, CAR T cells can be engineered to express CD47 on their surface as a means of evading phagocytosis by macrophages.
Investigators have also exploited genetic engineering methods for diminishing the level of alloreactivity when using allogeneic CAR T cells. The TRAC gene is one of the foremost targets in this regard whose knock-out using diverse genetic manipulation tactics, such as TALEN, zinc-finger nuclease (ZFN), and CRISPR-Cas9, has been established to be effective in the ablation of both TCR α and β chains and alleviating alloreactivity (56–60). Furthermore, other researchers have highlighted the use of allogeneic CAR T cells with TCR and CD52 knock-outs and they have demonstrated that these cells act as satisfactory universal CAR T cell candidates since they do not cause alloreactivity and are able to mediate molecular remission in patients with R/R B-ALL (57). It is worth mentioning that the knock-out of CD52 renders these CAR T cells resistant to the depletion impacts of the anti-CD52 antibody alemtuzumab (57). Additionally, CRISPR-Cas9-mediated CAR transgene knock-in in the TRAC gene locus has also been investigated by other researchers as they have suggested that this method can also be as efficient as the other mentioned methods for disrupting the endogenous TCR expression in allogeneic CAR T cells (61). Also, CRISPR-Cas9 and ZFN both have been utilized for the ablation of HLA expression to diminish the level of alloreactivity when using allogeneic CAR T cells as well (62, 63).
TAAs targeted by CAR T cells are usually expressed by healthy tissues as well, even though at lower rates. However, despite this limited target antigen expression, CAR T cells still manage to recognize these normal cells and initiate cytolytic reactions against them. This phenomenon results in the elimination of those healthy cells (known as “on-target off-tumor” toxicities), thus causing life-threatening side effects such as multi-organ failure for the respective patients. B cell aplasia, as characterized by the elimination and absence of B cells, is a renowned off-tumor incident after CD19- or CD22-based CAR T cell therapy mediating hypogammaglobulinemia in the recipients that subjects them to various types of infectious diseases (30, 64). In detail, CD19 and CD22 are both expressed on normal B cells as well as the malignant ones (64). Therefore, CAR T cells targeting either of these antigens happen to eradicate normal B cells as well (64). In this regard, B cell aplasia is considered as a parameter for the assessment of CD19 and CD22 CAR T cell therapy efficacy and persistence as well as their success rate (64). It can also act as a marker for the possibility of disease relapse (64). To tackle these limitations, scientists have engineered smart CAR constructs with tumor-selectivity mechanisms capable of precise discrimination between malignant and healthy cells. In this section, we will briefly discuss some of these strategies alongside highlighting their advantages and disadvantages.
TME exhibit an upregulated expression profile for multiple classes of tumor-associated proteases such as plasmin (65, 66), matrix metalloproteases (67), cathepsins (68–70), and legumain (71, 72) which could be exploited to engineer smart CAR platforms. A conditionally active CAR construct whose antigen-recognition domain is composed of a probody constitutes the novel strategy of “masked CARs” which increase the applicability of CAR T cells in the treatment of cancers that lack definitive TAAs (73). In detail, a probody is an antibody with its antigen recognition site covered by a masking peptide recombinantly linked to it by a protease-sensitive linker that is susceptible to proteolytic cleavage only by TME proteases (73, 74). Conceptually, the protease-sensitive linker is cleaved in the presence of tumor-associated proteases leading to the subsequent disengagement of the masking peptide and unveiling of the antigen-binding site of the targeting domain (73). This occurrence opens the gate for the downstream tumoricidal responses of the effector cells (Figure 3A) (73). Probodies, as compared to conventional mAbs, have shown a tremendously increased safety index due to their prolonged pharmacokinetic half-life which enables them to reach higher exposure rates while dosed at the same level as that of conventional mAbs (74). This expanded safety zone might be translatable in the field of masked CAR T cell therapy in a way that higher infusion dosages can impose more effective therapeutic impacts without crossing the red line of safety (73). Furthermore, the use of universal linkers sensitive to several proteases secreted by different types of TME makes the masked CAR platform a reliable candidate for targeting multiple tumor types in a concurrent manner (73). Despite the quiescence of masked CAR T cells in the circulation until their trafficking into the TME, there are still some off-tumor toxicities delivered to healthy tissues that secret the proteases to which the linker peptide is sensitive (73). However, such occurrences do not yet defame the generosity and expanded safety profile of this CAR platform (73).
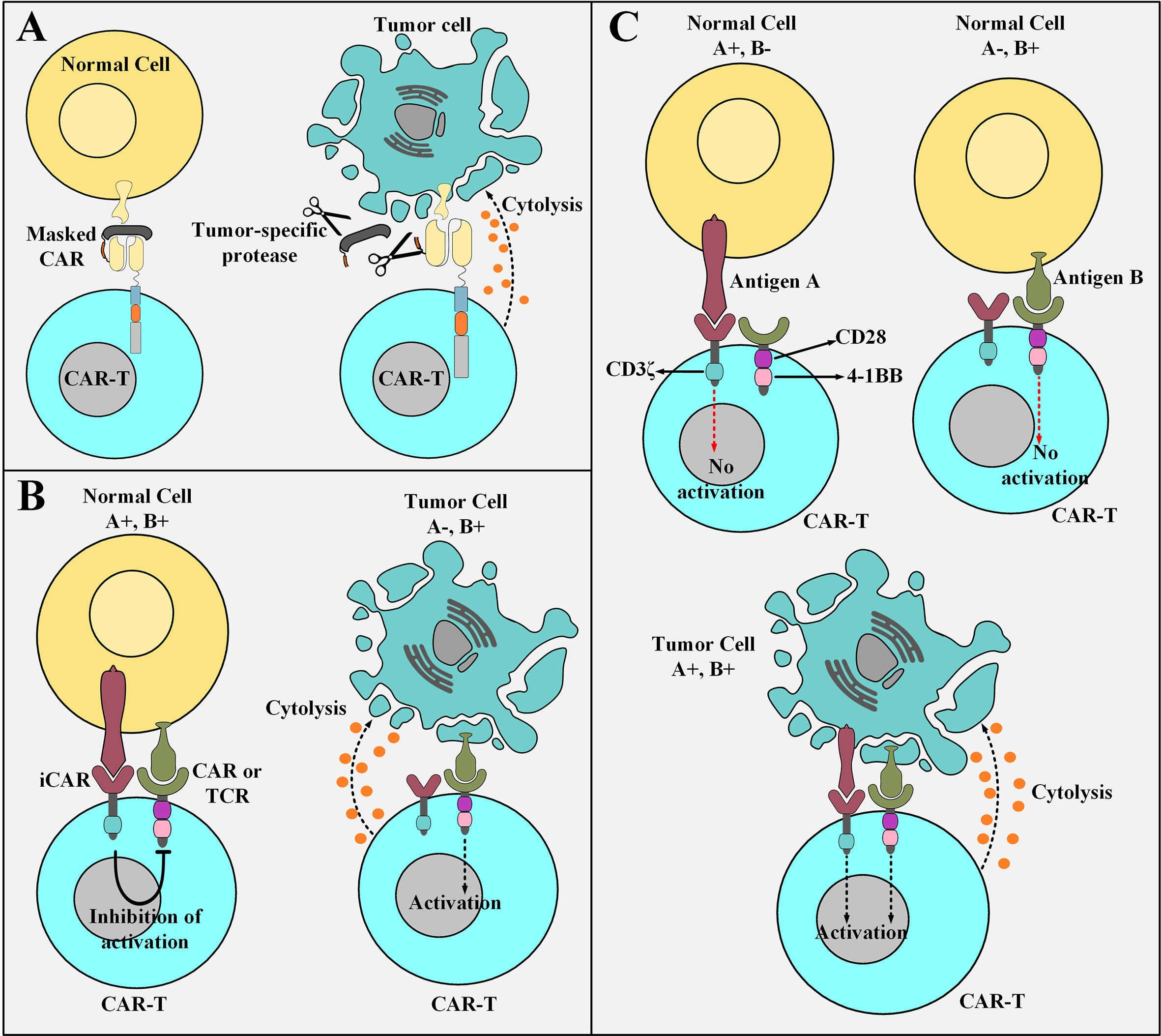
Figure 3 Elaborate strategies for tackling the on-target off-tumor toxicity of CAR T cells. (A) Masked CAR T cells and their mechanism of action. Masked CAR T cells are unable to cytotoxically attack healthy cells because their targeting domains are veiled. In the tumor microenvironment, the masking peptide is dissociated due to the presence of tumor-specific proteases. This phenomenon results in the recognition of the tumor cells by masked CAR T cells leading to the cytolysis of the tumor cells. (B) iCAR T cells and their mechanism of action. iCAR T cells are cytotoxically inert while encountering healthy cells simultaneously expressing their inhibitory and activatory antigens. Upon encountering tumor cells that only express the activatory antigen and are deficient in the expression of the inhibitory antigen, iCAR T cells are activated to carry out their tumoricidal effects. (C) The logic-gated CAR platform and its mechanism of action. Logic-gated CAR T cells require the presence of two antigens for their activation and triggering of cytotoxic effects. This strategy increases their safety index while encountering healthy tissues expressing one of the antigens. CAR-T, chimeric antigen receptor T cell; iCAR, inhibitory CAR; TCR, T cell receptor.
Another strategy to minimize the deleterious damages of “on-target off-tumor toxicities” or bystander healthy tissue damages is the use of an antigen-specific iCAR. This platform provides a self-regulating dynamic safety switch to circumvent the consequences of unwanted T cell responses and divert them away from the undesired tissues (75). The general concept is to have a surface antigen recognition domain fused to the signaling domains of endogenous immunoinhibitory receptors such as CTLA-4 or PD-1 to reversibly restrict T cell cytokine secretion, cytotoxicity, and proliferation despite concurrent engagement of an activating receptor (which can be a CAR or simply just an engineered TCR) (75). The iCAR platform allows T cells to discriminate between healthy and cancerous cells in an antigen selective manner (Figure 3B) (75). The transgenic expression of the iCAR construct does not impinge on the basic functionality of the T cells in the absence of its specific inhibitory antigen (75). Moreover, other T cell-restricted inhibitory receptors such as BTLA, 2B4, and LAG-3 or their combination in a single second-generation iCAR or as an iCAR with multiple combined cytoplasmic domains can also be used to regulate the cytotoxic functionality of CAR T cells (75–77).
Despite being experimentally evident that iCAR T cells can still sustain their tumoricidal functionality even after exposure to inhibitory antigens, the possibility of a proportion of iCAR T cells becoming anergized over repeated exposure to an inhibitory antigen is not completely ruled out (75, 78). Moreover, since this elaborate regulatory approach is antigen-specific, it requires tissue-specific target antigens that are expressed by healthy tissues but are absent from or down-regulated by tumor cells (75). Human leukocyte antigen (HLA) can be a suitable antigen of such characteristics since it is expressed in all cell types but substantially downregulated by tumor cells to confer them the ability to escape T cell-mediated immune system responses (79).
A trans-signaling CAR strategy in which T cell activation signal and co-stimulatory signal are physically dissociated from each other in two antigen-specificity different CARs has been developed to equip CAR T cells with a “double or nothing” strategy (80–84). These CAR T cells (known as logic-gated CAR T cells) can only attack tumor cells that simultaneously express both of the antigens recognized by the antigen recognition domains of the two different CARs (hence they manage to spare healthy cell expressing only one of the antigens) (80–84). Conceptually, T cells are genetically modified to express two CARs; one CAR that only harbors the CD3ζ signaling domain and recognizes an antigen of interest with low affinity and a chimeric co-stimulatory receptor (CCR) that recognizes a different antigen of interest with high affinity (80–84). Moreover, CCR engagement with antigen provides the co-stimulatory signaling cascades necessary for T cell activation and potent cytotoxicity (80–84). Genetically modified T cells expressing these two constructs are not potently activated while they encounter normal cells, which only express one of the two antigens, due to insufficient activation signals (Figure 3C) (80–84). However, several issues question the practicality of this proposed strategy. Limitations such as identification of two tumor antigens which are only expressed by a given type of cancer with nonoverlapping expression in normal tissues (81). Additionally, another limitation relates to the difficulty of designing a CAR with a narrow optimum affinity range or a CAR that is practically applicable to almost a broad spectrum of patients (80). In vitro findings have demonstrated weak cytokine secretion by trans-signaling CAR T cells against cells expressing only one TAA and pronounced cytokine secretion upon encountering tumor cells co-expressing both antigens (81). These findings suggest that the dual-specificity trans-signaling CAR platform might potentiate the therapeutic efficacy of CAR T cells against target cancer cells while diminishing their cross-reactivity with normal tissues (81).
Recently, researchers have used γδ T cells, which are a subset of T cells that harbor TCRs with γδ subunits, instead of the more common αβ subunits (85, 86). γδ T cells are about 1-10% of circulating T cells but they act as important components of the immune system (87). Vγ9Vδ2 T cells, which are a subset of γδ T cells, have an intrinsic tumor-distinguishing ability since they can recognize the phosphoantigens that are non-peptidic tumor antigens and are typical features of metabolism-dysregulated tumor cells (88). Researchers have investigated a novel tactic using Vγ9Vδ2 T cells as the backbone for generating unique “co-stimulatory domain-only CARs” (89). Dissimilar from the conventional αβ T cells (used as the primary source for the production of CAR T cells), γδ T cells recognize their target antigens with no dependence on MHC class I or II (90). The Vγ9Vδ2 TCR is the most prevalent γδ TCR expressed by γδ T cells (90). These TCRs recognize phosphoantigens such as isopentenyl pyrophosphate (IPP) over-produced in cancer cells and not in healthy ones (90). γδ T cells differentiate between cancerous and normal cells by identifying these antigens as a “danger alarm” (90). Studies have demonstrated that GD2-targeting co-stimulation-only CAR T cells generated from T cells with Vγ9Vδ2 TCRs are functional and show robust cytolytic responses only against GD2-positive neuroblastoma cells, but not against GD2-positive normal cells, in vitro (89). This fact highlights the role of the endogenous Vγ9Vδ2 TCR since the CD3ζ signal is only provided by the tumor cells that interact with the endogenous Vγ9Vδ2 TCR (Figure 4A) (89).
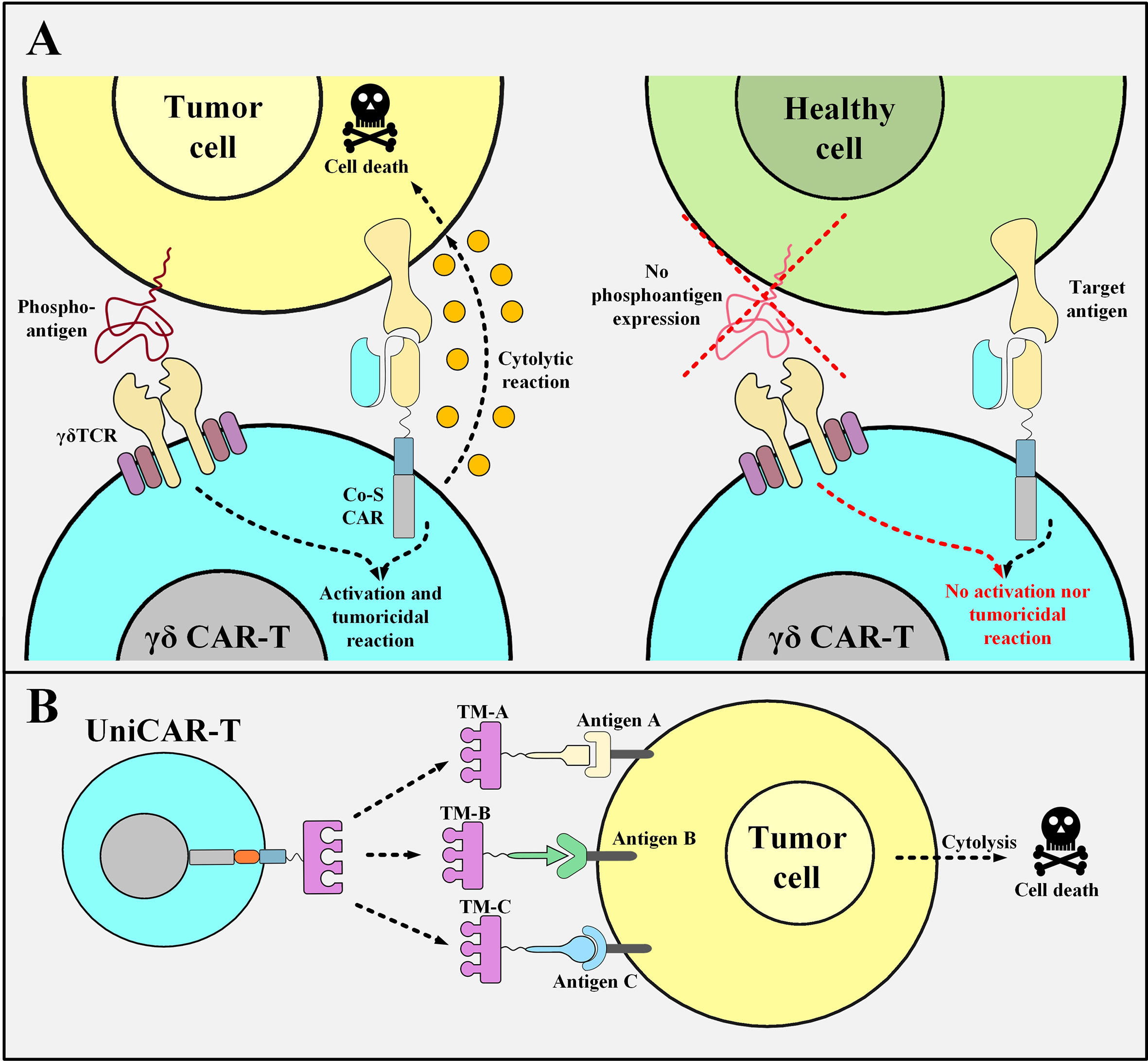
Figure 4 Co-stimulation-only γδ CAR T Cells and UniCAR T cells and their mechanism of action. (A) The mechanism of action of γδ CAR T cells with “co-stimulation-only” CARs and “Vγ9Vδ2 TCRs”. The tumoricidal activity of γδ CAR T cells will be in action when the CD3ζ activation and co-stimulation signals are provided. The activation signal is mediated through the Vγ9Vδ2 TCR of these CAR T cells only when they encounter the phosphoantigens expressed by the metabolically dysregulated tumor cells. Furthermore, the co-stimulation signal is provided once the co-stimulation-only CAR molecules recognize their specific TAA. Since healthy cells do not express such phosphoantigens, γδ CAR T cells will not be able to mediate cytotoxic reactions against them. (B) UniCAR T cells. UniCAR T cells can be activated and redirected towards different tumor cells based on the presence of different TMs. The clearance of the TM from the circulation simply results in the quiescence of the UniCAR T cells. Co-S CAR, co-stimulation-only CAR; TM, target module; UniCAR-T, universal chimeric antigen receptor T cell.
Moreover, other studies have also presented similar promising results demonstrating that CAR T cells generated using Vδ2 T cells (termed γδ CAR T cells) can migrate towards tumor cells and perform antigen cross-presentation (91). These findings propose that γδ CAR T cells can enter the tumor site and eliminate tumor cells alongside uptaking the target antigens which lead to stimulatory antigen presentation to tumor-infiltrating lymphocytes (TILs) with αβ TCRs (91). It has been proposed that tumors such as melanoma can be the right battlefield for these fighting cells since they harbor high tumor antigen frequency and large numbers of tumor-reactive lymphocytes and TILs (91). This fact may be considered as an advantage of γδ CAR T cells over conventional CAR T cells which could be quite worthwhile in the treatment of solid tumors (91). Moreover, various studies have also indicated that graft-versus-host disease (GVHD) is mediated by alloreactive T cells harboring the αβ TCR, while T cells with the γδ TCR do not mediate alloreactivity, therefore they are not capable of GVHD induction (92). Besides, γδT cells also can orchestrate various compelling antileukemic and anti-infectious effects (92). However, the functionality of γδ CAR T cells in comparison with conventional CAR T cells, their in-detail characterization, and their large-scale manufacturing protocols are yet to be explored. In a nutshell, it can still be concluded that γδT CAR T cells might show promise for prospective clinical evaluations in solid tumors since they present exclusive useful properties over conventional CAR T cells.
Another elaborate strategy to diminish the risk of off-tumor side effects entails the use of a modular CAR platform known as UniCAR. This strategy makes it possible to reversibly turn off the CAR system as fast as possible in case of unwanted CAR T cell-mediated side effects (93). UniCARs are designed as two different components that form an immune complex together to be guided towards the desired target cells. Conceptually, UniCARs consist of the UniCAR effector T cells and engineered recombinant target modules which direct them to the surface of the appropriate target cells (Figure 4B) (93). The specificity of the target module dictates the UniCAR T cells exactly which target cells they should attack and their rapid elimination from the circulation sufficiently vouches for their safety index as it switches UniCAR T cells “On” and “Off” (93). Since the antibody domain of the UniCAR T cells is directed against a unique epitope on the target module, they can establish an immune complex in their presence (93). This would guide UniCAR T cells towards their target cells (93). On the other hand, in the absence of a target module, UniCAR T cells automatically turn off which makes their controlling much more feasible than that of conventional CAR T cells (93–95). To minimize the considerable risks of CRS during a UniCAR T cell therapy, the administration of a rapidly eliminated target module should start at low doses and then be adjusted and increased based on the emergence of unexpected side effects (93). Once the desired target cells are eliminated or any life-threatening adverse events happen, the termination of targeting module administration will simply cause the UniCAR T cells to be turned off (93). Additionally, the potential benefits of the UniCAR platform in the cases of disease relapse are discussed in an upcoming section.
So far, various attempts have been made for controlling the activity of CAR T cells after their infusion into patients. This topic deserves special attention since it can contribute to the control and prevention of the previously mentioned CAR T cell-mediated toxicities which can sometimes be life-threatening. In this section, we briefly summarize strategies aimed at controlling the expression of CARs on the surface of the engineered T cells as well as some of the most potent strategies developed for overall control over CAR T cells after their administration.
It has been demonstrated that the tyrosine kinase inhibitor Dasatinib, an FDA-approved treatment for Philadelphia chromosome-positive chronic myeloid leukemia (CML) and ALL, inhibits LCK and thereby prevents the phosphorylation of CD3ζ and ZAP70 (96). Mestermann and colleagues have exploited dasatinib to increase the safety index of CAR T cells (96). The mentioned mechanism can mediate the disruption of the downstream signaling cascade in CARs harboring either CD28-CD3ζ or 4-1BB-CD3ζ activation modules (96). Moreover, dasatinib can induce a quickly occurring (3 hours) hybernation in CD8- and CD4-positive CAR T cells which can continue for several days without imposing any negative effects on T cell viability (96). Moreover, different dosing schemes of dasatinib can be used for partial or complete inhibition of CAR T cell activity (96). It has been shown that the administration of dasatinib shortly after CAR T cell infusion in preclinical CRS mouse models protects them from CRS that could otherwise be lethal in models not receiving dasatinib (96). The main advantage of this method is that upon the discontinuation of dasatinib administration, its inhibitory effect rapidly and completely reverses, therefore, the previously affected CAR T cells can continue their normal signaling pathway and antitumor activity (96). The favorable pharmacodynamics of dasatinib is another advantage of this approach allowing for multiple-time utilization of this drug for sequential CAR T cell activity turning “off” and “on”. Conclusively, dasatinib administration in CAR T cell-receiving preclinical models and in vitro assays pauses cytolytic activity, cytokine production, and expansion of CAR T cells and it can be applied as a pharmacologic on/off switch for CAR T cells (96). Aside from these, this approach might suffer from several limitations. One is that the inhibitory impact of dasatinib over CAR T cells that have already been activated is less pronounced (96). Furthermore, since dasatinib exerts its inhibitory effect through TCR signaling, the endogenous T cells will also be affected in terms of their effector function (96, 97). Another limitation of this approach affects patients with aggressive tumors whose malignant cells tend to proliferate at a high speed (96). In such patients, over the course of toxicities, while dasatinib is administered to control CAR T cell functionality, the CAR T cell-mediated tumoricidal reactions are halted temporarily which leaves more room for tumor progression (96).
STOP CAR is a recently developed CAR platform made of a recognition (R) chain, responsible for antigen binding, and a signaling (S) chain, responsible for T cell activation (98). The endodomains of these two distinct chains have a computationally designed protein pair that helps them dimerize into a functional heterodimer without the need for a dimerizing agent (98). This heterodimer is a chemically disruptable heterodimer (CDH) and it can be exclusively disrupted and dissociated into two monomers by the administration of small molecules such as A1331852 and A1155463 (which are Bcl-XL inhibitors) (98). The availability of disruptive small molecules that have valid clinical applications, prolonged half-life, and significant tolerance in humans are the principles for the design of such CDHs (98). The basic aim of the STOP CAR platform is to utilize globular domains from modular proteins that do not disturb the synapse-proximal T cell signaling (98). In detail, the CDH is made of human apolipoprotein E4 (apoE4), which is located on the R chain, and Bcl-XL, which is located on the S chain (98). apoE4 and Bcl-XL are human-originated proteins with very few numbers of amino acid substitutions (98). Therefore, they might not lead to transgene immune rejection in the recipients receiving these CAR T cells (99, 100). This strategy enables us to decrease the activity of the infused CAR T cells temporarily instead of shutting it down permanently (which happens in the case of suicide switches, as discussed in the upcoming subsections) (98). When there are no disruptive small molecules administered, apoE4 and Bcl-XL are paired together allowing CAR T cell activation upon the recognition of the antigen of interest (98). On the other hand, in the presence of the disruptive small molecules, apoE4 and Bcl-XL are dissociated from each other and maintain their monomeric form which does not allow CAR T cell activation upon antigen recognition (Figure 5A) (98). STOP CAR T cells have been tested against two different antigens (PSMA and CD19), and it has been found that their efficacy is equal to their respective conventional second-generation CAR T cells (98). Taken together, STOP CARs can be utilized for controlling the post-infusion effector function of CAR T cells in a very safe, cost-effective, reversible, and efficient manner (98). However, when such protein engineering techniques are applied for the development of a CAR molecule, there is a slight possibility for the emergence of immunogenic epitopes (98). In this regard, elaborate computational approaches can be used for the depletion of such T cell epitopes (98, 101).
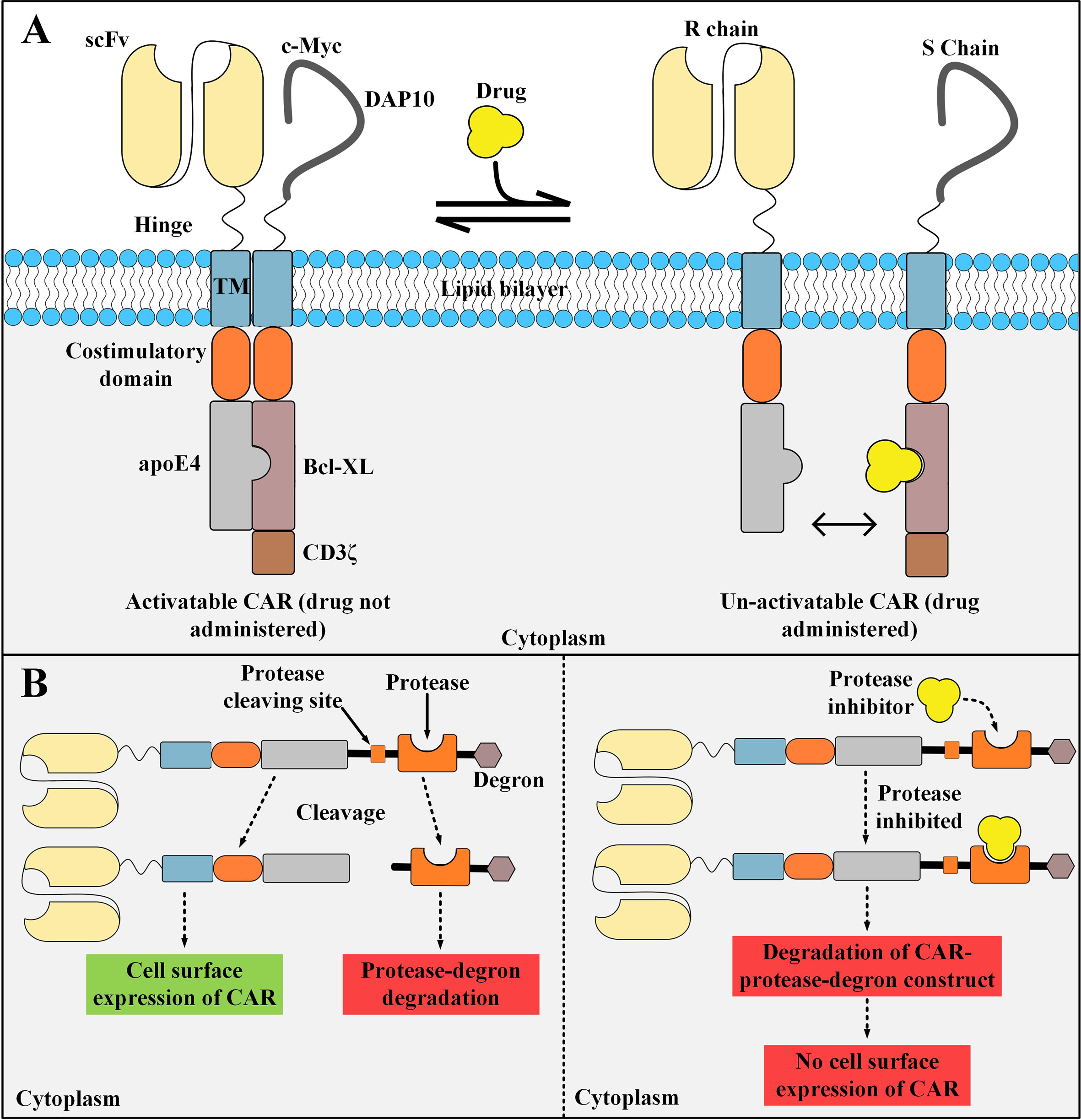
Figure 5 STOP CAR and SWIFF-CARs structures and action mechanisms. (A) The STOP-CAR platform. The S chain is made of a c-myc and DAP10 (which are used for enhancing the stability and expression of the S chain), a hinge, a transmembrane domain, a co-stimulatory domain, Bcl-XL, and CD3ζ. On the other hand, the R chain is made of an scFv, a hinge, a transmembrane domain, a co-stimulatory domain, and human apolipoprotein E4. When the disruptive drug is not administered, the R and S chain can bind to each other and the CAR can be activated upon target antigen engagement. After the disruptive drug administration, it binds to its binding site on the Bcl-XL domain located on the S chain, therefore, it renders the R and S chain unable to pair and the CAR un-activatable. (B) A simplified illustration of the SWIFF-CAR platform. In the absence of the protease inhibitor (left panel), the protease retains its proteolytic activity and binds to its cleaving site leading to the dissociation of the CAR molecule from the rest of the construct and its translocation to the cell surface resulting in normal CAR-mediated antitumor activity. In the presence of the protease inhibitor (right panel), the inhibitor binds to its binding site located on the protease and mediates its inhibition, therefore, the protease cannot bind to its cleaving site which results in the degradation of the CAR molecule and the rest of the construct in the T cell proteolytic pathways (right panel). apoE4, human apolipoprotein E4; scFv, single-chain variable fragment; CAR, chimeric antigen receptor. R chain, recognition chain; S chain, signaling chain.
The activity of CAR T cells can be controlled through mechanisms that can regulate the expression of CAR molecules on the surface of T cells (as an on- and off-switch). Recently, Juillerat et al. have generated a CAR T cell activity-controlling platform termed switch-off CARs (SWIFF-CARs) which entails using the protease-based small molecule-assisted shutoff (SMASh) (102). In this platform, the SWIFF-CAR construct is made of the CAR molecule followed by a protease cleaving site, a protease (the HCV NS3 protease), and a degradation moiety named “degron” (102). In the absence of the cell-permeable protease inhibitor Asunaprevir, the protease cleaves its target site leading to dissociation of the CAR from the protease and degron (102). This change will result in the translocation of the CAR molecule to the cell surface allowing its normal activity and signaling cascade (a state called “ON”) (102). On the other hand, in the presence of asunaprevir, it binds to its binding site on the protease and inhibits its cleaving activity (102). Therefore, the CAR molecule will not dissociate from the protease cleaving site, protease, and degron which will lead to the proteolytic degradation of the CAR molecule (a state called “OFF”) (Figure 5B) (102). This study shows that it might be feasible to directly incorporate an off-switch into the CAR construct which enables reversible control of the CAR surface expression (102). However, this tactic is just considered as an in vitro-tested prototype of a seemingly applicable idea for now. In-depth in vivo preclinical investigations may better highlight the applicability of CAR T cells equipped with this switch in terms of in vivo expansion and tumoricidal activity as well as the ability to discriminate the healthy cells from the malignant ones.
Selective and permanent ablation of CAR T cells in emergencies including the occurrence of GVHD or on-target off-tumor toxicities has been the subject of numerous investigations over the past years. The need for safety switches capable of irreversible elimination of CAR T cells during the mentioned adverse events and the implementation of such strategies have been recognized as an efficient way for addressing these challenges. One of these safety switches is based on suicide gene technologies which function through different mechanisms such as metabolic pathways, agent dimerization as well as targeting via therapeutic mAbs. These switches are discussed in detail throughout the upcoming section. It is worth mentioning that biological quiescence alongside favorable bioavailability and biodistribution profiles are all among the desired characteristics of an ideal suicide switch activation agent (103).
Suicide switches can be based on converting a non-toxic compound into a toxic one which eventually acts to kill the suicide switch-harboring cell. Herpes simplex virus thymidine kinase (HSV-TK), unlike mammalian cell thymidine kinase, shows an incredibly high affinity to ganciclovir (GCV), which is a nucleoside analog (104, 105). GCV is phosphorylated by HSV-TK to GCV-monophosphate (MP) and which is eventually converted to GCV-trisphosphate (TP). DNA polymerase incorporates GCV-TP into the leading strand of DNA which results in GCV-induced chain termination (106, 107). The HSV-TK/GCV suicide switch is also capable of triggering death-inducing signaling cascades through the formation of Fas-associated death domain protein (FADD) and the activation of caspases through ligand-independent CD95 aggregation (108). Despite the gradual effectiveness and potential immunogenicity risks of the HSV-TK switch (due to its viral origin), its benefit-to-risk ratio might still be clinically favorable (Figure 6A) (104, 105). Another example of this type of suicide-inducing switch involves cytosine deaminase (CD), which is a pyrimidine salvage enzyme (109). Mechanistically, 5-fluorouracil (5-FU) is the product of the deamination of the antifungal medication 5-fluorocytosine (5-FC) by CD, and therefore it plays the role of the highly cytotoxic compound capable of cell death induction (109). In this regard, equipping CAR T cells with the genes encoding enzymes such as HSV-TK or CD enables irreversible elimination of the infused CAR T cells in times of encountering adverse complications. Furthermore, type 1 HSV-TK gene has also been known as a positron emission tomography (PET) reporter gene that can be leveraged for providing insights into CAR T cell trafficking into tumor sites (110).
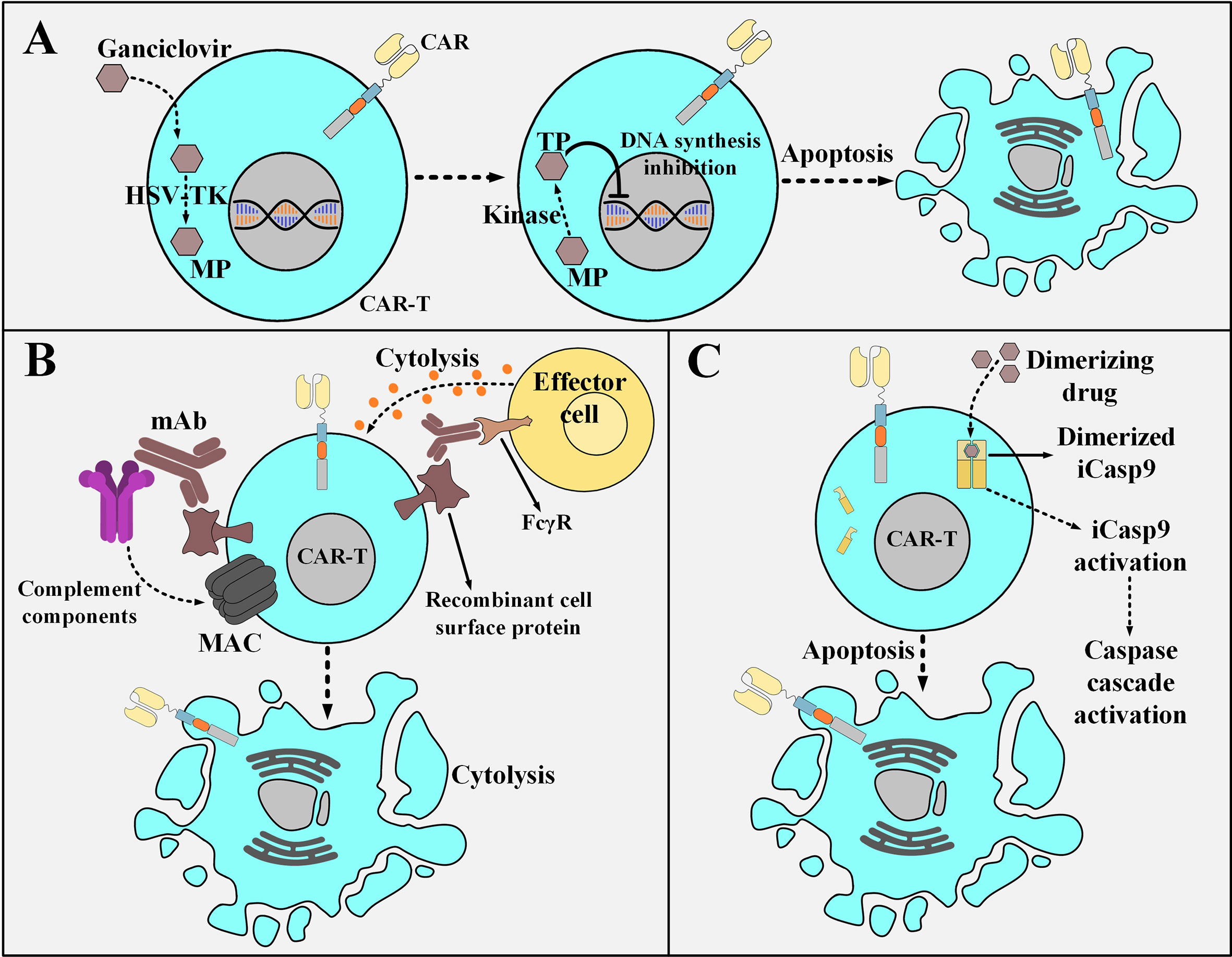
Figure 6 Various strategies for the post-infusion controlling of CAR T cells. (A) The HSV-TK safety switch and its mechanism of action. Upon the administration of ganciclovir, it is catalyzed to MP by HSV-TK. MP will eventually be phosphorylated to TP. Subsequently, TP disrupts DNA synthesis leading to the selective ablation of CAR T cells. (B) An example a mAb-based safety switch and its mechanism of action. Co-expression of a desired recombinant cell surface protein enables selective elimination of CAR T cells expressing that molecule through the administration of pharmaceutical-grade mAbs which are specific for it. This elimination is mediated by the engagement of immune effector cells or complement fixation. (C) The iCasp9 suicide switch and its mechanism of action. The administration of the dimerizing drug results in the dimerization of the iCasp molecule. The dimerized iCasp is now activated and can trigger downstream apoptotic cascades that result in the death of the CAR T cells harboring this switch. HSV-TK, Herpes simplex virus thymidine kinase; MP, ganciclovir-monophosphate; TP, ganciclovir-trisphosphate; MAC, membrane attack complex; mAb, monoclonal antibody; FcγR, Fcγ receptor; iCasp9, inducible caspase 9.
Another suicide switch strategy for the selective in vivo ablation of CAR T cells is their genetic engineering to coordinately express a CAR and a recombinant cell surface protein (111–113). This recombinant protein should retain a conformationally intact binding epitope recognized by a given pharmaceutical-grade mAb such as cetuximab (EGFR-specific mAb) or rituximab (CD20-specific mAb) (111–113). This approach renders the mentioned CAR T cells susceptible to antibody-dependent cell-mediated cytotoxicity (ADCC) or complement-dependent cytotoxicity (CDC) upon exposure to that reagent without altering their cytotoxic functionality (Figure 6B) (111–113). The suicide system based on the use of cetuximab can be considered a more compatible option for application in patients with hematologic malignancies (including ALL) since it would not ablate B cells and it is also a more considerate option for those leukemia patients who are seeking adoptive T cell therapy but are already under rituximab treatment. Moreover, there has not been any truncated form of the tetraspan transmembrane protein of CD20 that can conformationally retain its capacity for rituximab binding (112, 114–116). It is also encouraging to mention that there have been no signs of immunogenicity mounted against the EGFRt molecule (a truncated form of the epidermal growth factor receptor) of the EGFRt-positive CAR T cells in preclinical mouse models (113). These findings support the hypothesis that the administration of cetuximab might result in the recovery of the B cell compartment in patients who have undergone CD19-based CAR T cell therapy and experienced prolonged persistence of CD19-redirected CAR T cells, B cell aplasia, and complete tumor regression (113). However, there are still some concerns about the clinical applicability of such mAb-based switches since the administration of mAbs into patients might result in serious damages to healthy tissues that express the native form of the recombinant protein (103). Even though such concerns might limit the broader development of safety switches based on this platform, their clinical evaluation completed in the upcoming years will elucidate these matters.
Another example of an inducible safety switch is based on the recombinant fusion of a modified FKBP12 (human FK506-binding protein) to the human caspase 9 or the membrane-anchored intracellular domain of Fas (117, 118). This approach enables at-will dimerization in the presence of a biologically inert dimerizing agent (such as AP1903) (117, 118). Conceptually, the modified FKBP12 binds to the synthetic dimerizing drug with high affinity allowing for the dimerization and subsequent activation of the inducible caspase 9 (iCasp9) or the Fas-based suicide switch (117, 118). This results in a caspase cascade that leads to the apoptosis of the cells expressing these constructs (Figure 6C) (117, 118). Furthermore, studies have reported the elimination of >90% of the iCasp9-equipped T cells within 30 minutes following a single-dose administration of the dimerizing drug in patients with GVHD (118). Moreover, this rapid elimination correlated with the resolution of GVHD without recurrence (118). Furthermore, the iCasp9 switch has been known to possess several advantages over other safety switches such as its low immunogenicity profile (due to the human origin of the iCasp9 suicide gene) and the utilization of a biologically inert small molecule for its activation (instead of antiviral agents such as ganciclovir) (118). These advantages make this safety switch a more suitable option for application in the field of cellular therapy. Moreover, the rapid cell death mediation of this switch through the engagement of the endogenous apoptotic pathway in the cell (occurring minutes after the administration of the dimerizing drug) is much faster than other safety switches that require interference with DNA synthesis for cell death induction (119–124)
No single antigen might be considered a universal one because of the antigenic heterogeneity profile within a single tumor and among different patients. Antigen loss, antigen downregulation, or the emergence of alternatively spliced antigens (that no longer can be targeted by CAR T cells due to the loss of the recognized epitope) are all among elaborate antigen-dependent strategies performed by tumor cells to escape immune recognition (125–127). Such phenomena consequently limit the tumoricidal efficacy of targeted immunotherapy resulting in poor clinical responses (125–127). Simultaneous multispecific targeting is one of the proposed strategies aimed at offsetting tumor antigen escape variants which might provide enhanced durability of immunotherapy-mediated remission. CAR T cells whose CAR constructs are equipped with bispecific targeting domains in a tandem manner (Tandem CAR or TanCAR) or T cells co-expressing two different chimeric receptors specific for two distinct TAAs might have superiorities compared with conventional CAR T cells. These genetically manipulated T cells are endowed with the ability to cytotoxically target tumor cells expressing either antigen or both antigens simultaneously. Such CAR T cells have exhibited accentuated antitumor activity in vitro and in animal models of human tumors such as glioblastoma and B cell malignancies (128–134). According to a recent report from a clinical trial (NCT03185494) evaluating the tumoricidal efficacy of bispecific CD19/CD22-redirected CAR T cells in adult R/R B-ALL patients, all of the 6 patients (100%) experienced MRD-negative CR without the onset of neurotoxicity (135).
Furthermore, in cases of disease relapse, UniCAR T cells can also be beneficial since they can initiate cytotoxic reactions against the evading tumor cells upon the introduction of targeting modules that target a new tumor antigen (rather than the ones alternatively spliced or with expression loss or downregulation) (93). This capability demonstrates why this CAR platform is universally applicable towards different target antigens of interest without the need for redesigning a new CAR construct.
The major differences in the nature of normal and cancerous tissues can be exploited for developing smart TME-responsive or -dodging therapeutic approaches. The poor level of nutrition availability, low extracellular pH (acidosis), and low oxygenation level (hypoxia) are among various TME-specific characteristics (136, 137). The hypoxic microenvironment is characterized by oxygenation levels often below 1-2% (136, 137). Moreover, the immunosuppressive hypoxia-A2-adenosinergic axis is a very interesting characteristic of many treatment-resistant tumors (138). Discovering the key roles of the upstream factors in this pathway has led to the development of unique counterstrategies for inhibiting the hypoxia/hypoxia-inducible factor-1α (HIF-1α) axis (139, 140). Preclinical investigations targeting the A2A adenosine receptor (A2AR) and the adenosine-generating ectoenzyme CD73 have led to significant therapeutic efficacy (139–142). In detail, the hypoxic environment of the TME stabilizes HIF-1α leading to an elevation in the expression level of adenosine-generating ectoenzymes including CD39 and CD73 (143). The increments in the level of CD39 and CD73 elevate the level of adenosine (143). Further on, adenosine binds to A2AR on T cells which leads to the elevation in the level of cAMP (143). This elevation activates the cAMP-dependent protein kinase A (PKA), through the binding of cAMP to PKA (143). The downstream signaling cascade resulting from PKA activation in T cells leads to the blockage of TCR signaling and the expression of immunosuppression-responsible genes through the cAMP Response Element (CRE) (143). HIF-1α additionally controls the expression of factors responsible for tumorigenicity and immunosuppression through the Hypoxia Response Element (HRE) in a straightforward manner (143). Therefore, the CRE and HRE downstream signaling cascades decrease the level of INF-γ, IL-12, and IL-2 secretion and elevate the level of the TGF-β signaling pathway and IL-10, PD-1, CTLA-4, COX-2, and T-regulatory (Treg) expression (143). Conclusively, the hypoxic nature of the TME mediates an elevated level of anergy and exhaustion and a reduced level of cytokine production and secretion in T cells and CAR T cells (143). Researchers have shown that supplemental oxygenation and utilizing oxygenation agents can reverse hypoxia in the TME (143–145). They have suggested that this method can avert the stabilization of HIF-1α and impair the hypoxia-adenosinergic immunosuppressive axis (143–145). They have demonstrated that this method can reprogram the nature of the TME from “immunosuppressive” to “immunopermissive” (143–145). Moreover, they have underlined the clinical application of systemic oxygenation and oxygenation agents in conjunction with the A2AR blockade to further tackle the TME immunosuppressive nature (143). This strategy disrupts the upstream and downstream (hypoxia-HIF-1α and adenosine-A2AR, respectively) cascades of the immunosuppressive hypoxia-adenosinergic signaling axis and can maximize the therapeutic benefits of A2AR antagonists alongside elevating the susceptibility of tumors to cancer treatments (Figure 7) (143). Furthermore, other researchers have exploited the hypoxia of TME and have designed smart self-decision-making CAR T cells (146). They have fused an oxygen-sensitive subdomain of HIF-1α to a CAR scaffold and generated CAR T cells that are responsive to a hypoxic environment (146). This strategy has been developed to restrict the expression of CAR to only those CAR T cells residing in the hypoxic TME (rather than the ones in the non-hypoxic environment of non-malignant tissues) (146). Therefore, these CAR T cells can reduce the off-tumor effects of conventional CAR T cells (146).
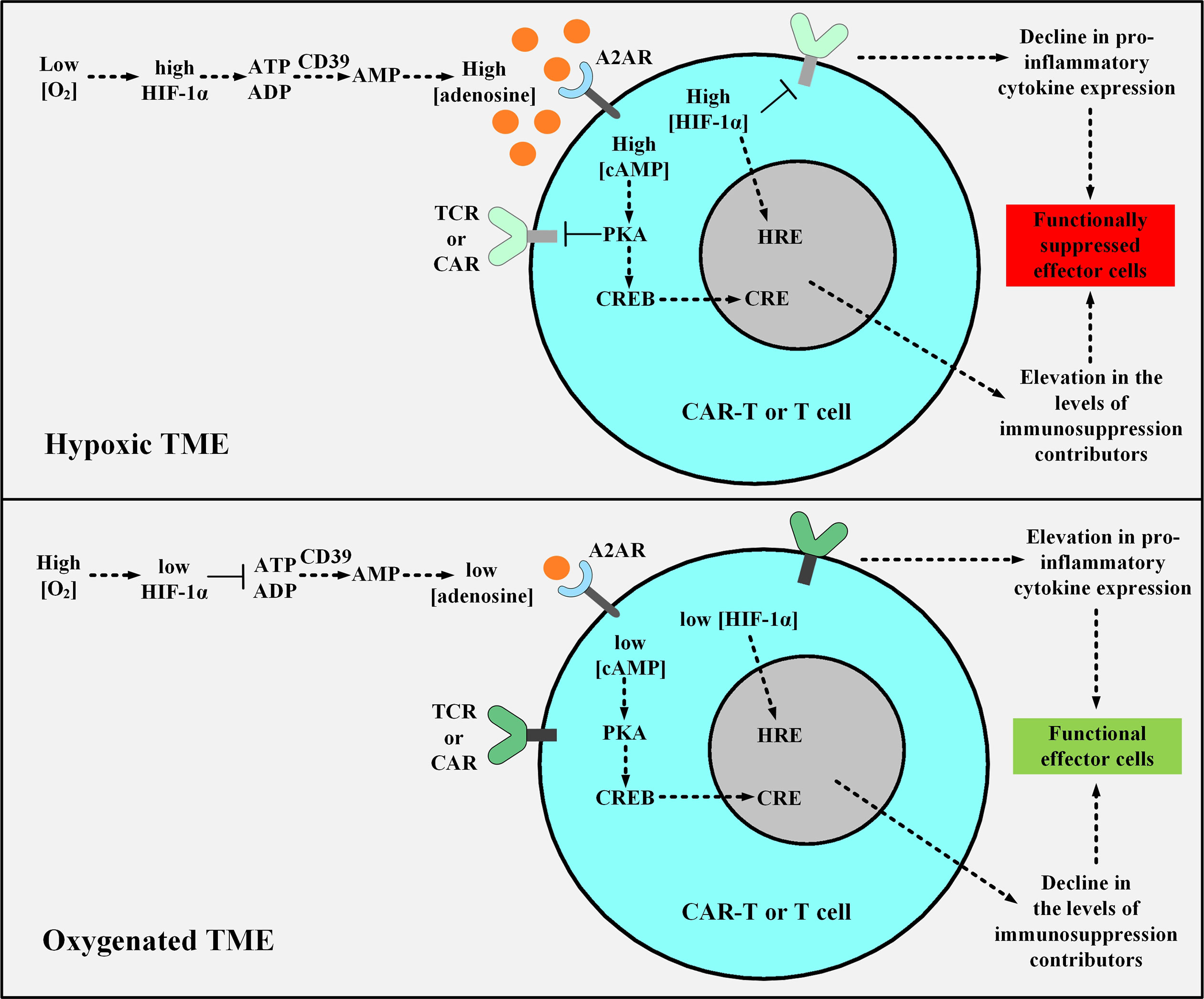
Figure 7 The effects of hypoxic or oxygenated TME on the fate, functionality, and antitumor activity of T cell or CAR T cells. The upper panel shows the hypoxic TME. In this condition, the low level of O2 leads to the stabilization of HIF-1α which elevates the expression level of CD39 and CD73 leading to the excessive production of adenosine. Adenosine binds to A2AR causing an elevation in the level of cAMP, and cAMP activates PKA. Further on, PKA inhibits TCR and CAR signaling and upregulates the expression of immunosuppresion contributors via CRE. Moreover, HIF-1α elevates the expression level of immunosuppressive genes through HRE. The activated CRE and HRE signaling cascades together reduce the level of INF-γ, IL-12, and IL-2 expression and upregulate TGF-β, IL-10, PD-1, CTLA-4, COX-2, and Treg expression. In the lower panel, supplemental oxygenation or using oxygenation agents elevates the level of O2, therefore, the downstream signaling pathway cannot proceed as it would in the case of a hypoxic microenvironment. TME, tumor microenvironment; HIF-1α, Hypoxia-inducible factor-1α; A2AR, A2A adenosine receptor; PKA, protein kinase A; CRE, cAMP Response Element; CREB, cAMP Response Element-Binding Protein; HRE, Hypoxia Response Element; TCR, T cell receptor; CAR, chimeric antigen receptor.
The effector function and differentiation state of T cells are highly impacted by their cellular metabolism condition (147). Moreover, the components of CARs expressed in transduced T cells have impacts on their nutritional intake and metabolic state (147). The metabolism-functionality relationship found in T cells can be used as a tool for defining their fate, activity, and effector function (147). For instance, studies have shown that the presence of 4-1BB co-stimulatory domain in the construct of CARs persuades the T cells to develop central memory phenotype and have an enhanced oxidative breakdown of fatty acids alongside improving their expansion capacity and persistence (147). On the other hand, the CD28 co-stimulatory domain improves glycolysis and makes CAR T cells develop effector memory phenotype (147). Moreover, supplementation such as supplementing with L-arginine can balance the elevated arginine metabolism in activated T cells alongside improving tumoricidal functionality and inducing central memory phenotype development (148).
Having a detailed gene expression profile of the genes mostly involved in cellular metabolism can help us achieve the goal of metabolic reprogramming of T cells by modifying the expression level of metabolic genes. This topic has been at the center of T cell reprogramming investigations since it has recently shown encouraging results. In this regard, it has been found that leukemic cells inhibit the Akt/mTORC1 signaling of T cells triggering their impaired functionality (149). Leukemic cells also mediate the downregulation of the glucose transporter Glut1 leading to a decreased level of glucose uptake in T cells (149). Overexpressing the Akt pathway or the Glut1 transporter is the proposed strategy to tackle this caveat caused by leukemic cells (149). This strategy can somewhat bring back the functionality of the T cells to the level before the negative impacts were imposed on them by the tumor cells (149). Moreover, PPAR-gamma co-activator 1a, also known as PGC1a, is a transcription factor co-activator affecting various cellular metabolic pathways. This metabolic regulator is downregulated in T cells infiltrating tumor sites (150). Researchers have found that the overexpression of PGC1a in T cells rebuilds their effector functionality as well as their metabolic and mitochondrial activity (150). Additionally, genetically suppressing Acetyl-CoA acetyltransferase (ACAT1), which is a cholesterol esterification enzyme converting excess cholesterol to cholesterol esters, in T cells leads to an elevation in the cholesterol concentration of the T cell plasma membrane (151). As a consequence, this phenomenon will effectively improve T cell signaling, thus mediating a better effector function and antitumor activity (151).
Furthermore, studies have used the characteristics and behavior of tumor cells and tissues to reprogram the metabolism of T cells (152, 153). In this regard, it has been discovered that necrotic tumor cells release potassium (K+) in the TME which leads to the excessive accumulation of this ion (152). This phenomenon elevates the intracellular concentration of K+ in the tumor-infiltrating T cells more than the normal level leading to a limitation in their nutrient uptake (152). Moreover, this accumulation in T cells downregulates their Protein Kinase B (Akt)/mammalian target of rapamycin (mTOR) signaling and interferes with T cell activation signaling (152). Researchers have shown that overexpressing K+ channels can act to reduce the elevated intracellular K+ levels, promote Akt/mTOR activity, and bring back the diminished effector function of T cells (152). Moreover, researchers have found a solution for the K+ accumulation-mediated limited nutrient uptake of T cells in the TME (153). They have found that ex vivo culturing and activation of T cells in a K+-elevated condition, which resembles the restricted nutrient uptake state in the K+-accumulated TME, prepares the T cells for the mentioned condition through their metabolic reprogramming (153). This preparation of T cells keeps their stemness and improves their antitumor cytolytic properties (153). Taken together, the abovementioned metabolic reprogramming strategies either improve T cell and CAR T cell responses, activity, and effector function in the TME or they avert the negative effects of particular TME-specific modifications performed by the tumor cells on the infiltrating T cells.
30 years after the first genetic manipulation of T cells for generating CAR T cells, today, they can be known as the lifeblood of immunotherapeutics. In 2018, the American Society of Clinical Oncology (ASCO) named CAR T cell therapy “Advance of the Year” which further highlights the key role of this fighting soldier in the cancer treatment revolution. However, CAR T cell therapy toxicities and limitations appear as stones thrown at its fragile success. Therefore, clinical and basic science research efforts are highly required for addressing these ongoing/unsolved caveats. The herein discussed strategies might pave the way for less toxic and more effectual CAR T cell therapies with more favorable clinical outcomes since such toxicities are success-limiting factors themselves. However, as discussed throughout this review, each of these strategies might have its advantages and disadvantages over another which will define their applicability depending on the need. Moreover, the majority of these strategies are still in the laboratory or preclinical development which highlights the fact that they might require further optimization for translational purposes. Also, some of these strategies are currently under clinical evaluation and their clinical potential and efficacy are to be determined in the upcoming years. Furthermore, the fact that CAR T cell therapy has remarkably achieved four FDA approvals in the case of hematologic malignancies indicates that this type of immunotherapy might soon be a popular choice for the treatment of a wide spectrum of oncological indications (and even immunological indications). This should encourage scientists to optimize the already introduced strategies or to design and develop novel ones to address the remaining hindrances. In a nutshell, these strategies might be applied in a synergistic fashion to orchestrate a safer CAR T cell therapy whilst maximizing its tumoricidal efficacy in a way that it is just good news for patients with difficult-to-treat malignancies.
PooS: conceptualization, investigation, writing—original draft, writing—review and editing, validation, supervision. PouS: conceptualization, investigation, writing—original draft, writing—review and editing, validation, supervision. FR: writing—review and editing, validation, supervision. SK: writing—review and editing, validation. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. June CH, O’Connor RS, Kawalekar OU, Ghassemi S, Milone MC. CAR T cell immunotherapy for human cancer. Science (2018) 359(6382):1361–5. doi: 10.1126/science.aar6711
2. Hashem Boroojerdi M, Rahbarizadeh F, Safarzadeh Kozani P, Kamali E, Safarzadeh Kozani P. Strategies for having a more effective and less toxic CAR T-cell therapy for acute lymphoblastic leukemia. Med Oncol (2020) 37(11):100. doi: 10.1007/s12032-020-01416-3
3. Xie YJ, Dougan M, Jailkhani N, Ingram J, Fang T, Kummer L, et al. Nanobody-based CAR T cells that target the tumor microenvironment inhibit the growth of solid tumors in immunocompetent mice. Proc Natl Acad Sci U.S.A. (2019) 116(16):7624–31. doi: 10.1073/pnas.1817147116
4. Wang D, Starr R, Chang WC, Aguilar B, Alizadeh D, Wright SL, et al. Chlorotoxin-directed CAR T cells for specific and effective targeting of glioblastoma. Sci Transl Med (2020) 12(533):eaaw2672. doi: 10.1126/scitranslmed.aaw2672
5. Rajabzadeh A, Rahbarizadeh F, Ahmadvand D, Kabir Salmani M, Hamidieh AA. A VHH-Based Anti-MUC1 Chimeric Antigen Receptor for Specific Retargeting of Human Primary T Cells to MUC1-Positive Cancer Cells. Cell J (2021) 22(4):502–13. doi: 10.22074/cellj.2021.6917
6. Stoiber S, Cadilha BL, Benmebarek MR, Lesch S, Endres S, Kobold S. Limitations in the Design of Chimeric Antigen Receptors for Cancer Therapy. Cells (2019) 8(5):472. doi: 10.3390/cells8050472
7. Drent E, Poels R, Ruiter R, van de Donk N, Zweegman S, Yuan H, et al. Combined CD28 and 4-1BB Costimulation Potentiates Affinity-tuned Chimeric Antigen Receptor-engineered T Cells. Clin Cancer Res (2019) 25(13):4014–25. doi: 10.1158/1078-0432.CCR-18-2559
8. Sommer C, Cheng HY, Nguyen D, Dettling D, Yeung YA, Sutton J, et al. Allogeneic FLT3 CAR T Cells with an Off-Switch Exhibit Potent Activity against AML and Can Be Depleted to Expedite Bone Marrow Recovery. Mol Ther (2020) 28(10):2237–51. doi: 10.1016/j.ymthe.2020.06.022
9. Chapman NM, Gottschalk S, Chi H. Preventing Ubiquitination Improves CAR T Cell Therapy via ‘CAR Merry-Go-Around’. Immunity (2020) 53(2):243–5. doi: 10.1016/j.immuni.2020.07.023
10. Collinson-Pautz MR, Chang WC, Lu A, Khalil M, Crisostomo JW, Lin PY, et al. Constitutively active MyD88/CD40 costimulation enhances expansion and efficacy of chimeric antigen receptor T cells targeting hematological malignancies. Leukemia (2019) 33(9):2195–207. doi: 10.1038/s41375-019-0417-9
11. Wang E, Wang LC, Tsai CY, Bhoj V, Gershenson Z, Moon E, et al. Generation of Potent T-cell Immunotherapy for Cancer Using DAP12-Based, Multichain, Chimeric Immunoreceptors. Cancer Immunol Res (2015) 3(7):815–26. doi: 10.1158/2326-6066.CIR-15-0054
12. Guedan S, Madar A, Casado-Medrano V, Shaw C, Wing A, Liu F, et al. Single residue in CD28-costimulated CAR-T cells limits long-term persistence and antitumor durability. J Clin Invest (2020) 130(6):3087–97. doi: 10.1172/JCI133215
13. Tokarew N, Ogonek J, Endres S, von Bergwelt-Baildon M, Kobold S. Teaching an old dog new tricks: next-generation CAR T cells. Br J Cancer (2019) 120(1):26–37. doi: 10.1038/s41416-018-0325-1
14. Caratelli S, Sconocchia T, Arriga R, Coppola A, Lanzilli G, Lauro D, et al. FCγ Chimeric Receptor-Engineered T Cells: Methodology, Advantages, Limitations, and Clinical Relevance. Front Immunol (2017) 8:457. doi: 10.3389/fimmu.2017.00457
15. Karlsson H, Svensson E, Gigg C, Jarvius M, Olsson-Strömberg U, Savoldo B, et al. Evaluation of Intracellular Signaling Downstream Chimeric Antigen Receptors. PloS One (2015) 10(12):e0144787. doi: 10.1371/journal.pone.0144787
16. Ng YY, Tay JC, Li Z, Wang J, Zhu J, Wang S. T Cells Expressing NKG2D CAR with a DAP12 Signaling Domain Stimulate Lower Cytokine Production While Being Effective in Tumor Eradication. Mol Ther (2020) 29:75–85. doi: 10.1016/j.ymthe.2020.08.016
17. Chen J, Moore A, Ringshausen I. ZAP-70 Shapes the Immune Microenvironment in B Cell Malignancies. Front Oncol (2020) 10:595832. doi: 10.3389/fonc.2020.595832
18. Duell J, Lurati S, Dittrich M, Bedke T, Pule M, Einsele H, et al. First Generation Chimeric Antigen Receptor Display Functional Defects In Key Signal Pathways Upon Antigen Stimulation. Blood (2010) 116(21):2088–8. doi: 10.1182/blood.V116.21.2088.2088
19. van der Stegen SJ, Hamieh M, Sadelain M. The pharmacology of second-generation chimeric antigen receptors. Nat Rev Drug Discovery (2015) 14(7):499–509. doi: 10.1038/nrd4597
20. Zhong XS, Matsushita M, Plotkin J, Riviere I, Sadelain M. Chimeric antigen receptors combining 4-1BB and CD28 signaling domains augment PI3kinase/AKT/Bcl-XL activation and CD8+ T cell-mediated tumor eradication. Mol Ther (2010) 18(2):413–20. doi: 10.1038/mt.2009.210
21. Chmielewski M, Abken H. TRUCKs: the fourth generation of CARs. Expert Opin Biol Ther (2015) 15(8):1145–54. doi: 10.1517/14712598.2015.1046430
22. Kershaw MH, Westwood JA, Darcy PK. Gene-engineered T cells for cancer therapy. Nat Rev Cancer (2013) 13(8):525–41. doi: 10.1038/nrc3565
23. Kagoya Y, Tanaka S, Guo T, Anczurowski M, Wang CH, Saso K, et al. A novel chimeric antigen receptor containing a JAK-STAT signaling domain mediates superior antitumor effects. Nat Med (2018) 24(3):352–9. doi: 10.1038/nm.4478
24. Prasad V. Tisagenlecleucel—the first approved CAR-T-cell therapy: implications for payers and policy makers. Nat Rev Clin Oncol (2018) 15(1):11–2. doi: 10.1038/nrclinonc.2017.156
25. Bouchkouj N, Kasamon YL, de Claro RA, George B, Lin X, Lee S, et al. FDA approval summary: Axicabtagene ciloleucel for relapsed or refractory large B-cell lymphoma. Clin Cancer Res (2019) 25(6):1702–8. doi: 10.1158/1078-0432.CCR-18-2743
26. Voelker R. CAR-T Therapy Is Approved for Mantle Cell Lymphoma. Jama (2020) 324(9):832. doi: 10.1001/jama.2020.15456
27. Mullard A. FDA approves fourth CAR-T cell therapy. Nat Rev Drug Discovery (2021) 20:166. doi: 10.1038/d41573-021-00031-9
28. Brudno JN, Kochenderfer JN. Toxicities of chimeric antigen receptor T cells: recognition and management. Blood (2016) 127(26):3321–30. doi: 10.1182/blood-2016-04-703751
29. Brudno JN, Kochenderfer JN. Recent advances in CAR T-cell toxicity: Mechanisms, manifestations and management. Blood Rev (2019) 34:45–55. doi: 10.1016/j.blre.2018.11.002
30. Hill JA, Giralt S, Torgerson TR, Lazarus HM. CAR-T - and a side order of IgG, to go? - Immunoglobulin replacement in patients receiving CAR-T cell therapy. Blood Rev (2019) 38:100596. doi: 10.1016/j.blre.2019.100596
31. Shah NN, Fry TJ. Mechanisms of resistance to CAR T cell therapy. Nat Rev Clin Oncol (2019) 16(6):372–85. doi: 10.1038/s41571-019-0184-6
32. Scarfò I, Maus MV. Current approaches to increase CAR T cell potency in solid tumors: targeting the tumor microenvironment. J Immunother Cancer (2017) 5:28. doi: 10.1186/s40425-017-0230-9
33. Larson RC, Maus MV. Recent advances and discoveries in the mechanisms and functions of CAR T cells. Nat Rev Cancer (2021) 21:145–61. doi: 10.1038/s41568-020-00323-z
34. Kochenderfer JN, Dudley ME, Kassim SH, Somerville RP, Carpenter RO, Stetler-Stevenson M, et al. Chemotherapy-refractory diffuse large B-cell lymphoma and indolent B-cell malignancies can be effectively treated with autologous T cells expressing an anti-CD19 chimeric antigen receptor. J Clin Oncol (2015) 33(6):540–9. doi: 10.1200/jco.2014.56.2025
35. Gust J, Hay KA, Hanafi LA, Li D, Myerson D, Gonzalez-Cuyar LF, et al. Endothelial Activation and Blood-Brain Barrier Disruption in Neurotoxicity after Adoptive Immunotherapy with CD19 CAR-T Cells. Cancer Discovery (2017) 7(12):1404–19. doi: 10.1158/2159-8290.CD-17-0698
36. Parker KR, Migliorini D, Perkey E, Yost KE, Bhaduri A, Bagga P, et al. Single-Cell Analyses Identify Brain Mural Cells Expressing CD19 as Potential Off-Tumor Targets for CAR-T Immunotherapies. Cell (2020) 183(1):126–42.e17. doi: 10.1016/j.cell.2020.08.022
37. Sterner RM, Sakemura R, Cox MJ, Yang N, Khadka RH, Forsman CL, et al. GM-CSF inhibition reduces cytokine release syndrome and neuroinflammation but enhances CAR-T cell function in xenografts. Blood (2019) 133(7):697–709. doi: 10.1182/blood-2018-10-881722
38. Russo RC, Garcia CC, Teixeira MM, Amaral FA. The CXCL8/IL-8 chemokine family and its receptors in inflammatory diseases. Expert Rev Clin Immunol (2014) 10(5):593–619. doi: 10.1586/1744666X.2014.894886
39. Deshmane SL, Kremlev S, Amini S, Sawaya BE. Monocyte chemoattractant protein-1 (MCP-1): an overview. J Interferon Cytokine Res (2009) 29(6):313–26. doi: 10.1089/jir.2008.0027
40. Sachdeva M, Duchateau P, Depil S, Poirot L, Valton J. Granulocyte-macrophage colony-stimulating factor inactivation in CAR T-cells prevents monocyte-dependent release of key cytokine release syndrome mediators. J Biol Chem (2019) 294(14):5430–7. doi: 10.1074/jbc.AC119.007558
41. Giavridis T, van der Stegen SJC, Eyquem J, Hamieh M, Piersigilli A, Sadelain M. CAR T cell-induced cytokine release syndrome is mediated by macrophages and abated by IL-1 blockade. Nat Med (2018) 24(6):731–8. doi: 10.1038/s41591-018-0041-7
42. Norelli M, Camisa B, Barbiera G, Falcone L, Purevdorj A, Genua M, et al. Monocyte-derived IL-1 and IL-6 are differentially required for cytokine-release syndrome and neurotoxicity due to CAR T cells. Nat Med (2018) 24(6):739–48. doi: 10.1038/s41591-018-0036-4
43. Park JH, Rivière I, Gonen M, Wang X, Sénéchal B, Curran KJ, et al. Long-Term Follow-up of CD19 CAR Therapy in Acute Lymphoblastic Leukemia. N Engl J Med (2018) 378(5):449–59. doi: 10.1056/NEJMoa1709919
44. Neelapu SS, Tummala S, Kebriaei P, Wierda W, Gutierrez C, Locke FL, et al. Chimeric antigen receptor T-cell therapy - assessment and management of toxicities. Nat Rev Clin Oncol (2018) 15(1):47–62. doi: 10.1038/nrclinonc.2017.148
45. Staedtke V, Bai RY, Kim K, Darvas M, Davila ML, Riggins GJ, et al. Disruption of a self-amplifying catecholamine loop reduces cytokine release syndrome. Nature (2018) 564(7735):273–7. doi: 10.1038/s41586-018-0774-y
46. Vollmar AM. The role of atrial natriuretic peptide in the immune system. Peptides (2005) 26(6):1086–94. doi: 10.1016/j.peptides.2004.08.034
47. Elliott TJ, Eisen HN. Allorecognition of purified major histocompatibility complex glycoproteins by cytotoxic T lymphocytes. Proc Natl Acad Sci U S A (1988) 85(8):2728–32. doi: 10.1073/pnas.85.8.2728
48. Ciccone E, Pende D, Viale O, Di Donato C, Tripodi G, Orengo AM, et al. Evidence of a natural killer (NK) cell repertoire for (allo) antigen recognition: definition of five distinct NK-determined allospecificities in humans. J Exp Med (1992) 175(3):709–18. doi: 10.1084/jem.175.3.709
49. Ruggeri L, Capanni M, Urbani E, Perruccio K, Shlomchik WD, Tosti A, et al. Effectiveness of donor natural killer cell alloreactivity in mismatched hematopoietic transplants. Science (2002) 295(5562):2097–100. doi: 10.1126/science.1068440
50. Colonna M, Brooks EG, Falco M, Ferrara GB, Strominger JL. Generation of allospecific natural killer cells by stimulation across a polymorphism of HLA-C. Science (1993) 260(5111):1121–4. doi: 10.1126/science.8493555
51. Mo F, Watanabe N, McKenna MK, Hicks MJ, Srinivasan M, Gomes-Silva D, et al. Engineered off-the-shelf therapeutic T cells resist host immune rejection. Nat Biotechnol (2020) 39:56–63 . doi: 10.1038/s41587-020-0601-5
52. Ruella M, Xu J, Barrett DM, Fraietta JA, Reich TJ, Ambrose DE, et al. Induction of resistance to chimeric antigen receptor T cell therapy by transduction of a single leukemic B cell. Nat Med (2018) 24(10):1499–503. doi: 10.1038/s41591-018-0201-9.
53. Menk AV, Scharping NE, Rivadeneira DB, Calderon MJ, Watson MJ, Dunstane D, et al. 4-1BB costimulation induces T cell mitochondrial function and biogenesis enabling cancer immunotherapeutic responses. J Exp Med (2018) 215(4):1091–100. doi: 10.1084/jem.20171068
54. Stephan MT, Ponomarev V, Brentjens RJ, Chang AH, Dobrenkov KV, Heller G, et al. T cell-encoded CD80 and 4-1BBL induce auto- and transcostimulation, resulting in potent tumor rejection. Nat Med (2007) 13(12):1440–9. doi: 10.1038/nm1676
55. Veillette A, Chen J. SIRPα-CD47 Immune Checkpoint Blockade in Anticancer Therapy. Trends Immunol (2018) 39(3):173–84. doi: 10.1016/j.it.2017.12.005
56. Torikai H, Reik A, Liu P-Q, Zhou Y, Zhang L, Maiti S, et al. A foundation for universal T-cell based immunotherapy: T cells engineered to express a CD19-specific chimeric-antigen-receptor and eliminate expression of endogenous TCR. Blood (2012) 119(24):5697–705. doi: 10.1182/blood-2012-01-405365
57. Qasim W, Zhan H, Samarasinghe S, Adams S, Amrolia P, Stafford S, et al. Molecular remission of infant B-ALL after infusion of universal TALEN gene-edited CAR T cells. Sci Transl Med (2017) 9(374):eaaj2013. doi: 10.1126/scitranslmed.aaj2013
58. Valton J, Guyot V, Marechal A, Filhol JM, Juillerat A, Duclert A, et al. A Multidrug-resistant Engineered CAR T Cell for Allogeneic Combination Immunotherapy. Mol Ther (2015) 23(9):1507–18. doi: 10.1038/mt.2015.104
59. Georgiadis C, Preece R, Nickolay L, Etuk A, Petrova A, Ladon D, et al. Long Terminal Repeat CRISPR-CAR-Coupled “Universal” T Cells Mediate Potent Anti-leukemic Effects. Mol Ther (2018) 26(5):1215–27. doi: 10.1016/j.ymthe.2018.02.025
60. Eyquem J, Mansilla-Soto J, Giavridis T, van der Stegen SJ, Hamieh M, Cunanan KM, et al. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature (2017) 543(7643):113–7. doi: 10.1038/nature21405
61. MacLeod DT, Antony J, Martin AJ, Moser RJ, Hekele A, Wetzel KJ, et al. Integration of a CD19 CAR into the TCR Alpha Chain Locus Streamlines Production of Allogeneic Gene-Edited CAR T Cells. Mol Ther (2017) 25(4):949–61. doi: 10.1016/j.ymthe.2017.02.005
62. Ren J, Zhang X, Liu X, Fang C, Jiang S, June CH, et al. A versatile system for rapid multiplex genome-edited CAR T cell generation. Oncotarget (2017) 8(10):17002–11. doi: 10.18632/oncotarget.15218
63. Torikai H, Reik A, Soldner F, Warren EH, Yuen C, Zhou Y, et al. Toward eliminating HLA class I expression to generate universal cells from allogeneic donors. Blood (2013) 122(8):1341–9. doi: 10.1182/blood-2013-03-478255
64. Maude SL, Teachey DT, Porter DL, Grupp SA. CD19-targeted chimeric antigen receptor T-cell therapy for acute lymphoblastic leukemia. Blood (2015) 125(26):4017–23. doi: 10.1182/blood-2014-12-580068
65. Barthel BL, Rudnicki DL, Kirby TP, Colvin SM, Burkhart DJ, Koch TH. Synthesis and biological characterization of protease-activated prodrugs of doxazolidine. J medicinal Chem (2012) 55(14):6595–607. doi: 10.1021/jm300714p
66. Chakravarty PK, Carl PL, Weber MJ, Katzenellenbogen JA. Plasmin-activated prodrugs for cancer chemotherapy. 1. Synthesis and biological activity of peptidylacivicin and peptidylphenylenediamine mustard. J medicinal Chem (1983) 26(5):633–8. doi: 10.1021/jm00359a003
67. Albright CF, Graciani N, Han W, Yue E, Stein R, Lai Z, et al. Matrix metalloproteinase–activated doxorubicin prodrugs inhibit HT1080 xenograft growth better than doxorubicin with less toxicity. Mol Cancer Ther (2005) 4(5):751–60. doi: 10.1158/1535-7163.MCT-05-0006
68. Baurain R, Masquelier M, Deprez-De Campeneere D, Trouet A. Amino acid and dipeptide derivatives of daunorubicin. 2. Cellular pharmacology and antitumor activity of L1210 leukemic cells in vitro and in vivo. J medicinal Chem (1980) 23(11):1171–4. doi: 10.1021/jm00185a004
69. Ueki N, Lee S, Sampson NS, Hayman MJ. Selective cancer targeting with prodrugs activated by histone deacetylases and a tumour-associated protease. Nat Commun (2013) 4(1):1–8. doi: 10.1038/ncomms3735
70. Zhong Y-J, Shao L-H, Li Y. Cathepsin B-cleavable doxorubicin prodrugs for targeted cancer therapy. Int J Oncol (2013) 42(2):373–83. doi: 10.3892/ijo.2012.1754
71. Liu C, Sun C, Huang H, Janda K, Edgington T. Overexpression of legumain in tumors is significant for invasion/metastasis and a candidate enzymatic target for prodrug therapy. Cancer Res (2003) 63(11):2957–64.
72. Stern L, Perry R, Ofek P, Many A, Shabat D, Satchi-Fainaro R. A novel antitumor prodrug platform designed to be cleaved by the endoprotease legumain. Bioconjugate Chem (2009) 20(3):500–10. doi: 10.1021/bc800448u
73. Han X, Bryson PD, Zhao Y, Cinay GE, Li S, Guo Y, et al. Masked chimeric antigen receptor for tumor-specific activation. Mol Ther (2017) 25(1):274–84. doi: 10.1016/j.ymthe.2016.10.011
74. Desnoyers LR, Vasiljeva O, Richardson JH, Yang A, Menendez EE, Liang TW, et al. Tumor-specific activation of an EGFR-targeting probody enhances therapeutic index. Sci Trans Med (2013) 5(207):207ra144–207ra144. doi: 10.1126/scitranslmed.3006682
75. Fedorov VD, Themeli M, Sadelain M. PD-1–and CTLA-4–based inhibitory chimeric antigen receptors (iCARs) divert off-target immunotherapy responses. Sci Trans Med (2013) 5(215):215ra172–215ra172. doi: 10.1126/scitranslmed.3006597
76. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer (2012) 12(4):252–64. doi: 10.1038/nrc3239
77. Sharma P, Wagner K, Wolchok JD, Allison JP. Novel cancer immunotherapy agents with survival benefit: recent successes and next steps. Nat Rev Cancer (2011) 11(11):805–12. doi: 10.1038/nrc3153
78. Amarnath S, Mangus CW, Wang JC, Wei F, He A, Kapoor V, et al. The PDL1-PD1 axis converts human TH1 cells into regulatory T cells. Sci Transl Med (2011) 3(111):111ra120. doi: 10.1126/scitranslmed.3003130
79. Campoli M, Ferrone S. HLA antigen changes in malignant cells: epigenetic mechanisms and biologic significance. Oncogene (2008) 27(45):5869–85. doi: 10.1038/onc.2008.273
80. Hanada K, Restifo NP. Double or nothing on cancer immunotherapy. Nat Biotechnol (2013) 31(1):33–4. doi: 10.1038/nbt.2471
81. Lanitis E, Poussin M, Klattenhoff AW, Song D, Sandaltzopoulos R, June CH, et al. Chimeric antigen receptor T Cells with dissociated signaling domains exhibit focused antitumor activity with reduced potential for toxicity in vivo. Cancer Immunol Res (2013) 1(1):43–53. doi: 10.1158/2326-6066.CIR-13-0008
82. Blankenstein T. Receptor combinations hone T-cell therapy. Nat Biotechnol (2016) 34(4):389–91. doi: 10.1038/nbt.3539
83. Kloss CC, Condomines M, Cartellieri M, Bachmann M, Sadelain M. Combinatorial antigen recognition with balanced signaling promotes selective tumor eradication by engineered T cells. Nat Biotechnol (2013) 31(1):71–5. doi: 10.1038/nbt.2459
84. Wilkie S, van Schalkwyk MC, Hobbs S, Davies DM, van der Stegen SJ, Pereira AC, et al. Dual targeting of ErbB2 and MUC1 in breast cancer using chimeric antigen receptors engineered to provide complementary signaling. J Clin Immunol (2012) 32(5):1059–70. doi: 10.1007/s10875-012-9689-9
85. Braza MS, Klein B. Anti-tumour immunotherapy with Vγ9Vδ2 T lymphocytes: from the bench to the bedside. Br J Haematol (2013) 160(2):123–32. doi: 10.1111/bjh.12090
86. Fisher JP, Flutter B, Wesemann F, Frosch J, Rossig C, Gustafsson K, et al. Effective combination treatment of GD2-expressing neuroblastoma and Ewing’s sarcoma using anti-GD2 ch14.18/CHO antibody with Vγ9Vδ2+ γδT cells. Oncoimmunology (2016) 5(1):e1025194. doi: 10.1080/2162402X.2015.1025194
87. Fisher JP, Yan M, Heuijerjans J, Carter L, Abolhassani A, Frosch J, et al. Neuroblastoma killing properties of Vδ2 and Vδ2-negative γδT cells following expansion by artificial antigen-presenting cells. Clin Cancer Res (2014) 20(22):5720–32. doi: 10.1158/1078-0432.CCR-13-3464
88. Fisher J, Anderson J. Engineering Approaches in Human Gamma Delta T Cells for Cancer Immunotherapy. Front Immunol (2018) 9:1409. doi: 10.3389/fimmu.2018.01409
89. Fisher J, Abramowski P, Wisidagamage Don ND, Flutter B, Capsomidis A, Cheung GW, et al. Avoidance of On-Target Off-Tumor Activation Using a Co-stimulation-Only Chimeric Antigen Receptor. Mol Ther (2017) 25(5):1234–47. doi: 10.1016/j.ymthe.2017.03.002
90. Morita CT, Jin C, Sarikonda G, Wang H. Nonpeptide antigens, presentation mechanisms, and immunological memory of human Vgamma2Vdelta2 T cells: discriminating friend from foe through the recognition of prenyl pyrophosphate antigens. Immunol Rev (2007) 215:59–76. doi: 10.1111/j.1600-065X.2006.00479.x
91. Capsomidis A, Benthall G, Van Acker HH, Fisher J, Kramer AM, Abeln Z, et al. Chimeric Antigen Receptor-Engineered Human Gamma Delta T Cells: Enhanced Cytotoxicity with Retention of Cross Presentation. Mol Ther (2018) 26(2):354–65. doi: 10.1016/j.ymthe.2017.12.001
92. Handgretinger R, Schilbach K. The potential role of γδ T cells after allogeneic HCT for leukemia. Blood (2018) 131(10):1063–72. doi: 10.1182/blood-2017-08-752162
93. Albert S, Arndt C, Feldmann A, Bergmann R, Bachmann D, Koristka S, et al. A novel nanobody-based target module for retargeting of T lymphocytes to EGFR-expressing cancer cells via the modular UniCAR platform. Oncoimmunology (2017) 6(4):e1287246. doi: 10.1080/2162402X.2017.1287246
94. Cao Y, Rodgers DT, Du J, Ahmad I, Hampton EN, Ma JS, et al. Design of switchable chimeric antigen receptor T cells targeting breast cancer. Angewandte Chemie Int Edition (2016) 55(26):7520–4. doi: 10.1002/anie.201601902
95. Chang ZL, Lorenzini MH, Chen X, Tran U, Bangayan NJ, Chen YY. Rewiring T-cell responses to soluble factors with chimeric antigen receptors. Nat Chem Biol (2018) 14(3):317. doi: 10.1038/nchembio.2565
96. Mestermann K, Giavridis T, Weber J, Rydzek J, Frenz S, Nerreter T, et al. The tyrosine kinase inhibitor dasatinib acts as a pharmacologic on/off switch for CAR T cells. Sci Transl Med (2019) 11(499):eaau5907. doi: 10.1126/scitranslmed.aau5907
97. Blake S, Hughes TP, Mayrhofer G, Lyons AB. The Src/ABL kinase inhibitor dasatinib (BMS-354825) inhibits function of normal human T-lymphocytes in vitro. Clin Immunol (2008) 127(3):330–9. doi: 10.1016/j.clim.2008.02.006
98. Giordano-Attianese G, Gainza P, Gray-Gaillard E, Cribioli E, Shui S, Kim S, et al. A computationally designed chimeric antigen receptor provides a small-molecule safety switch for T-cell therapy. Nat Biotechnol (2020) 38(4):426–32. doi: 10.1038/s41587-019-0403-9
99. Berger C, Flowers ME, Warren EH, Riddell SR. Analysis of transgene-specific immune responses that limit the in vivo persistence of adoptively transferred HSV-TK-modified donor T cells after allogeneic hematopoietic cell transplantation. Blood (2006) 107(6):2294–302. doi: 10.1182/blood-2005-08-3503
100. Jensen MC, Popplewell L, Cooper LJ, DiGiusto D, Kalos M, Ostberg JR, et al. Antitransgene rejection responses contribute to attenuated persistence of adoptively transferred CD20/CD19-specific chimeric antigen receptor redirected T cells in humans. Biol Blood Marrow Transplant (2010) 16(9):1245–56. doi: 10.1016/j.bbmt.2010.03.014
101. Zhao H, Verma D, Li W, Choi Y, Ndong C, Fiering SN, et al. Depletion of T cell epitopes in lysostaphin mitigates anti-drug antibody response and enhances antibacterial efficacy in vivo. Chem Biol (2015) 22(5):629–39. doi: 10.1016/j.chembiol.2015.04.017
102. Juillerat A, Tkach D, Busser BW, Temburni S, Valton J, Duclert A, et al. Modulation of chimeric antigen receptor surface expression by a small molecule switch. BMC Biotechnol (2019) 19(1):44. doi: 10.1186/s12896-019-0537-3
103. Jones BS, Lamb LS, Goldman F, Di Stasi A. Improving the safety of cell therapy products by suicide gene transfer. Front Pharmacol (2014) 5:254. doi: 10.3389/fphar.2014.00254
104. Traversari C, Marktel S, Magnani Z, Mangia P, Russo V, Ciceri F, et al. The potential immunogenicity of the TK suicide gene does not prevent full clinical benefit associated with the use of TK-transduced donor lymphocytes in HSCT for hematologic malignancies. Blood J Am Soc Hematol (2007) 109(11):4708–15. doi: 10.1182/blood-2006-04-015230
105. Marin V, Cribioli E, Philip B, Tettamanti S, Pizzitola I, Biondi A, et al. Comparison of different suicide-gene strategies for the safety improvement of genetically manipulated T cells. Hum Gene Ther Methods (2012) 23(6):376–86. doi: 10.1089/hgtb.2012.050
106. Moolten FL. Tumor chemosensitivity conferred by inserted herpes thymidine kinase genes: paradigm for a prospective cancer control strategy. Cancer Res (1986) 46(10):5276–81.
107. Elion GB, Furman PA, Fyfe JA, De Miranda P, Beauchamp L, Schaeffer HJ. Selectivity of action of an antiherpetic agent, 9-(2-hydroxyethoxymethyl) guanine. Proc Natl Acad Sci (1977) 74(12):5716–20. doi: 10.1073/pnas.74.12.5716
108. Beltinger C, Fulda S, Kammertoens T, Meyer E, Uckert W, Debatin K-M. Herpes simplex virus thymidine kinase/ganciclovir-induced apoptosis involves ligand-independent death receptor aggregation and activation of caspases. Proc Natl Acad Sci (1999) 96(15):8699–704. doi: 10.1073/pnas.96.15.8699
109. Tiraby M, Cazaux C, Baron M, Drocourt D, Reynes J-P, Tiraby G. Concomitant expression of E. coli cytosine deaminase and uracil phosphoribosyltransferase improves the cytotoxicity of 5-fluorocytosine. FEMS Microbiol Lett (1998) 167(1):41–9. doi: 10.1111/j.1574-6968.1998.tb13205.x
110. Murty S, Labanieh L, Murty T, Gowrishankar G, Haywood T, Alam IS, et al. PET reporter gene imaging and ganciclovir-mediated ablation of chimeric antigen receptor T-cells in solid tumors. Cancer Res (2020) 80:4731–40. doi: 10.1158/0008-5472.CAN-19-3579
111. Wang X, Chang W-C, Wong CW, Colcher D, Sherman M, Ostberg JR, et al. A transgene-encoded cell surface polypeptide for selection, in vivo tracking, and ablation of engineered cells. Blood J Am Soc Hematol (2011) 118(5):1255–63. doi: 10.1182/blood-2011-02-337360
112. Serafini M, Manganini M, Borleri G, Bonamino M, Imberti L, Biondi A, et al. Characterization of CD20-transduced T lymphocytes as an alternative suicide gene therapy approach for the treatment of graft-versus-host disease. Hum Gene Ther (2004) 15(1):63–76. doi: 10.1089/10430340460732463
113. Paszkiewicz PJ, Fräßle SP, Srivastava S, Sommermeyer D, Hudecek M, Drexler I, et al. Targeted antibody-mediated depletion of murine CD19 CAR T cells permanently reverses B cell aplasia. J Clin Invest (2016) 126(11):4262–72. doi: 10.1172/JCI84813
114. Griffioen M, van Egmond EH, Kester MG, Willemze R, Falkenburg JF, Heemskerk MH. Retroviral transfer of human CD20 as a suicide gene for adoptive T-cell therapy. haematologica (2009) 94(9):1316–20. doi: 10.3324/haematol.2008.001677
115. van Meerten T, Claessen M-J, Hagenbeek A, Ebeling SB. The CD20/α CD20 ‘suicide’system: novel vectors with improved safety and expression profiles and efficient elimination of CD20-transgenic T cells. Gene Ther (2006) 13(9):789–97. doi: 10.1038/sj.gt.3302705
116. Vogler I, Newrzela S, Hartmann S, Schneider N, Von Laer D, Koehl U, et al. An improved bicistronic CD20/tCD34 vector for efficient purification and in vivo depletion of gene-modified T cells for adoptive immunotherapy. Mol Ther (2010) 18(7):1330–8. doi: 10.1038/mt.2010.83
117. Thomis DC, Marktel S, Bonini C, Traversari C, Gilman M, Bordignon C, et al. A Fas-based suicide switch in human T cells for the treatment of graft-versus-host disease. Blood J Am Soc Hematol (2001) 97(5):1249–57. doi: 10.1182/blood.V97.5.1249
118. Di Stasi A, Tey S-K, Dotti G, Fujita Y, Kennedy-Nasser A, Martinez C, et al. Inducible apoptosis as a safety switch for adoptive cell therapy. N Engl J Med (2011) 365:1673–83. doi: 10.1056/NEJMoa1106152
119. Bonini C, Ferrari G, Verzeletti S, Servida P, Zappone E, Ruggieri L, et al. HSV-TK gene transfer into donor lymphocytes for control of allogeneic graft-versus-leukemia. Science (1997) 276(5319):1719–24. doi: 10.1126/science.276.5319.1719
120. Tiberghien P, Reynolds CW, Keller J, Spence S, Deschaseaux M, Certoux J-M, et al. Ganciclovir treatment of herpes simplex thymidine kinase-transduced primary T lymphocytes: an approach for specific in vivo donor T-cell depletion after bone marrow transplantation? Blood (1994) 84:1333–41. doi: 10.1182/blood.V84.4.1333.bloodjournal8441333
121. Ciceri F, Bonini C, Stanghellini MTL, Bondanza A, Traversari C, Salomoni M, et al. Infusion of suicide-gene-engineered donor lymphocytes after family haploidentical haemopoietic stem-cell transplantation for leukaemia (the TK007 trial): a non-randomised phase I–II study. Lancet Oncol (2009) 10(5):489–500. doi: 10.1016/s1470-2045(09)70074-9.
122. Tiberghien P, Ferrand C, Lioure B, Milpied N, Angonin R, Deconinck E, et al. Administration of herpes simplex–thymidine kinase–expressing donor T cells with a T-cell–depleted allogeneic marrow graft. Blood J Am Soc Hematol (2001) 97(1):63–72. doi: 10.1182/blood.V97.1.63
123. Spencer DM, Wandless TJ, Schreiber SL, Crabtree GR. Controlling signal transduction with synthetic ligands. Science (1993) 262(5136):1019–24. doi: 10.1126/science.7694365
124. Clackson T, Yang W, Rozamus LW, Hatada M, Amara JF, Rollins CT, et al. Redesigning an FKBP–ligand interface to generate chemical dimerizers with novel specificity. Proc Natl Acad Sci (1998) 95(18):10437–42. doi: 10.1073/pnas.95.18.10437
125. Dunn GP, Old LJ, Schreiber RD. The three Es of cancer immunoediting. Annu Rev Immunol (2004) 22:329–60. doi: 10.1146/annurev.immunol.22.012703.104803
126. Nagaraj S, Gupta K, Pisarev V, Kinarsky L, Sherman S, Kang L, et al. Altered recognition of antigen is a mechanism of CD8+ T cell tolerance in cancer. Nat Med (2007) 13(7):828–35. doi: 10.1038/nm1609
127. Leen AM, Rooney CM, Foster AE. Improving T cell therapy for cancer. Annu Rev Immunol (2007) 25:243–65. doi: 10.1146/annurev.immunol.25.022106.141527
128. De Munter S, Ingels J, Goetgeluk G, Bonte S, Pille M, Weening K, et al. Nanobody based dual specific CARs. Int J Mol Sci (2018) 19(2):403. doi: 10.3390/ijms19020403
129. Fry TJ, Shah NN, Orentas RJ, Stetler-Stevenson M, Yuan CM, Ramakrishna S, et al. CD22-targeted CAR T cells induce remission in B-ALL that is naive or resistant to CD19-targeted CAR immunotherapy. Nat Med (2018) 24(1):20. doi: 10.1038/nm.4441
130. Grada Z, Hegde M, Byrd T, Shaffer DR, Ghazi A, Brawley VS, et al. TanCAR: a novel bispecific chimeric antigen receptor for cancer immunotherapy. Mol Therapy Nucleic Acids (2013) 2:e105. doi: 10.1038/mtna.2013.32
131. Hegde M, Corder A, Chow KK, Mukherjee M, Ashoori A, Kew Y, et al. Combinational targeting offsets antigen escape and enhances effector functions of adoptively transferred T cells in glioblastoma. Mol Ther (2013) 21(11):2087–101. doi: 10.1038/mt.2013.185
132. Hegde M, Mukherjee M, Grada Z, Pignata A, Landi D, Navai SA, et al. Tandem CAR T cells targeting HER2 and IL13Rα2 mitigate tumor antigen escape. J Clin Invest (2016) 126(8):3036–52. doi: 10.1172/JCI83416
133. Martyniszyn A, Krahl A-C, Andre MC, Hombach AA, Abken H. CD20-CD19 bispecific CAR T cells for the treatment of B-cell malignancies. Hum Gene Ther (2017) 28(12):1147–57. doi: 10.1089/hum.2017.126
134. Zah E, Lin M-Y, Silva-Benedict A, Jensen MC, Chen YY. T cells expressing CD19/CD20 bispecific chimeric antigen receptors prevent antigen escape by malignant B cells. Cancer Immunol Res (2016) 4(6):498–508. doi: 10.1158/2326-6066.CIR-15-0231
135. Dai H, Wu Z, Jia H, Tong C, Guo Y, Ti D, et al. Bispecific CAR-T cells targeting both CD19 and CD22 for therapy of adults with relapsed or refractory B cell acute lymphoblastic leukemia. J Hematol Oncol (2020) 13(1):30. doi: 10.1186/s13045-020-00856-8
136. Brown JM, Wilson WR. Exploiting tumour hypoxia in cancer treatment. Nat Rev Cancer (2004) 4(6):437–47. doi: 10.1038/nrc1367
137. Vaupel P, Mayer A. Hypoxia in cancer: significance and impact on clinical outcome. Cancer Metastasis Rev (2007) 26(2):225–39. doi: 10.1007/s10555-007-9055-1
138. Sitkovsky MV. Lessons from the A2A Adenosine Receptor Antagonist-Enabled Tumor Regression and Survival in Patients with Treatment-Refractory Renal Cell Cancer. Cancer Discovery (2020) 10(1):16–9. doi: 10.1158/2159-8290.CD-19-1280
139. Ohta A, Sitkovsky M. Role of G-protein-coupled adenosine receptors in downregulation of inflammation and protection from tissue damage. Nature (2001) 414(6866):916–20. doi: 10.1038/414916a
140. Ohta A, Gorelik E, Prasad SJ, Ronchese F, Lukashev D, Wong MK, et al. A2A adenosine receptor protects tumors from antitumor T cells. Proc Natl Acad Sci USA (2006) 103(35):13132–7. doi: 10.1073/pnas.0605251103
141. Allard B, Pommey S, Smyth MJ, Stagg J. Targeting CD73 enhances the antitumor activity of anti-PD-1 and anti-CTLA-4 mAbs. Clin Cancer Res (2013) 19(20):5626–35. doi: 10.1158/1078-0432.CCR-13-0545
142. Young A, Ngiow SF, Barkauskas DS, Sult E, Hay C, Blake SJ, et al. Co-inhibition of CD73 and A2AR Adenosine Signaling Improves Anti-tumor Immune Responses. Cancer Cell (2016) 30(3):391–403. doi: 10.1016/j.ccell.2016.06.025
143. Halpin-Veszeleiova K, Hatfield SM. Oxygenation and A2AR blockade to eliminate hypoxia/HIF-1α-adenosinergic immunosuppressive axis and improve cancer immunotherapy. Curr Opin Pharmacol (2020) 53:84–90. doi: 10.1016/j.coph.2020.07.005
144. Hatfield SM, Kjaergaard J, Lukashev D, Belikoff B, Schreiber TH, Sethumadhavan S, et al. Systemic oxygenation weakens the hypoxia and hypoxia inducible factor 1α-dependent and extracellular adenosine-mediated tumor protection. J Mol Med (Berl) (2014) 92(12):1283–92. doi: 10.1007/s00109-014-1189-3
145. Hatfield SM, Kjaergaard J, Lukashev D, Schreiber TH, Belikoff B, Abbott R, et al. Immunological mechanisms of the antitumor effects of supplemental oxygenation. Sci Transl Med (2015) 7(277):277ra30. doi: 10.1126/scitranslmed.aaa1260
146. Juillerat A, Marechal A, Filhol JM, Valogne Y, Valton J, Duclert A, et al. An oxygen sensitive self-decision making engineered CAR T-cell. Sci Rep (2017) 7:39833. doi: 10.1038/srep39833
147. Kawalekar OU, O'Connor RS, Fraietta JA, Guo L, McGettigan SE, Posey AD Jr, et al. Distinct Signaling of Coreceptors Regulates Specific Metabolism Pathways and Impacts Memory Development in CAR T Cells. Immunity (2016) 44(2):380–90. doi: 10.1016/j.immuni.2016.01.021
148. Geiger R, Rieckmann JC, Wolf T, Basso C, Feng Y, Fuhrer T, et al. L-Arginine Modulates T Cell Metabolism and Enhances Survival and Anti-tumor Activity. Cell (2016) 167(3):829–42.e13. doi: 10.1016/j.cell.2016.09.031
149. Siska PJ, van der Windt GJ, Kishton RJ, Cohen S, Eisner W, MacIver NJ, et al. Suppression of Glut1 and Glucose Metabolism by Decreased Akt/mTORC1 Signaling Drives T Cell Impairment in B Cell Leukemia. J Immunol (2016) 197(6):2532–40. doi: 10.4049/jimmunol.1502464
150. Scharping NE, Menk AV, Moreci RS, Whetstone RD, Dadey RE, Watkins SC, et al. The Tumor Microenvironment Represses T Cell Mitochondrial Biogenesis to Drive Intratumoral T Cell Metabolic Insufficiency and Dysfunction. Immunity (2016) 45(2):374–88. doi: 10.1016/j.immuni.2016.07.009
151. Yang W, Bai Y, Xiong Y, Zhang J, Chen S, Zheng X, et al. Potentiating the antitumour response of CD8(+) T cells by modulating cholesterol metabolism. Nature (2016) 531(7596):651–5. doi: 10.1038/nature17412
152. Eil R, Vodnala SK, Clever D, Klebanoff CA, Sukumar M, Pan JH, et al. Ionic immune suppression within the tumour microenvironment limits T cell effector function. Nature (2016) 537(7621):539–43. doi: 10.1038/nature19364
Keywords: chimeric antigen receptor, immunotherapy, tumor microenvironment, toxicities, adoptive cell therapy, solid tumors
Citation: Safarzadeh Kozani P, Safarzadeh Kozani P, Rahbarizadeh F and Khoshtinat Nikkhoi S (2021) Strategies for Dodging the Obstacles in CAR T Cell Therapy. Front. Oncol. 11:627549. doi: 10.3389/fonc.2021.627549
Received: 09 November 2020; Accepted: 08 March 2021;
Published: 01 April 2021.
Edited by:
Daniel Olive, Aix Marseille Université, FranceReviewed by:
Manel Juan, Hospital Clínic de Barcelona, SpainCopyright © 2021 Safarzadeh Kozani, Safarzadeh Kozani, Rahbarizadeh and Khoshtinat Nikkhoi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fatemeh Rahbarizadeh, cmFoYmFyaWZAbW9kYXJlcy5hYy5pcg==
†These authors share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.