 Lin Dong
Lin Dong Shuangmei Zou1
Shuangmei Zou1 Haizhen Lu
Haizhen Lu
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Oncol., 14 May 2021
Sec. Molecular and Cellular Oncology
Volume 11 - 2021 | https://doi.org/10.3389/fonc.2021.627460
This article is part of the Research TopicMolecular Mechanisms and Targeted Therapies for Colorectal CancerView all 15 articles
Background: A large proportion of patients with Lynch syndrome (LS) have MSH2 abnormalities, but genotype-phenotype studies of MSH2 mutations in LS are still lacking. The aim of this study was to comprehensively analyze the clinicopathological characteristics and molecular basis of colorectal cancer (CRC) in patients with uncommon MSH2 cytoplasmic expression.
Methods: We retrospectively reviewed 4195 consecutive cases of CRC patients diagnosed between January 2015 and December 2017 at the Cancer Hospital Chinese Academy of Medical Sciences. Of the 4195 patients with CRC, 69 were indicated to have abnormal MSH2 expression through tumor immunohistochemical staining. Genetic tests, such as next-generation sequencing, large genomic rearrangement (LGR) analysis, microsatellite instability status analysis and genomic breakpoint analysis, were performed. Clinicopathological and molecular characteristics and clinical immunotherapy response were analyzed.
Results: Forty-five of 69 patients were identified to have LS with pathogenic germline mutations in MSH2 and/or EPCAM. Of these LS patients, 26.7% were confirmed to harbor large genomic rearrangements (LGRs). Of note, three tumors from two unrelated family pedigrees exhibited a rare cytoplasmic MSH2 staining pattern that was found in LS patients with EPCAM/MSH2 deletions. RNA analysis showed that two novel mRNA fusions of EPCAM and MSH2 resulted in the predicted protein fusion with MSH2 cytoplasmic localization. Analyses of genomic breakpoints indicated that two novel deletions of EPCAM and MSH2 originated from Alu repeat-mediated recombination events. Our study also provides clinical evidence for the beneficial effect of the PD-1 inhibitor pembrolizumab for CRC patients that exhibit cytoplasmic MSH2 staining.
Conclusion: Our study demonstrates that the rare cytoplasmic MSH2 staining pattern should be fully recognized by pathologists and geneticists. Given the specific genotype-phenotype correlation in LS screening, we advocate that all CRC patients with cytoplasmic MSH2 staining in histology should be screened for LGRs of EPCAM and MSH2.
Lynch syndrome (LS), an autosomal dominant hereditary disorder, is the most common colorectal cancer (CRC) predisposition syndrome, accounting for 1%–3% of all newly diagnosed CRCs (1). LS is caused by pathogenic germline mutations in one of several dMMR genes (MLH1, MSH2, MSH6 and PMS2) and deletions in EPCAM (2–4). Deficient DNA mismatch repair (dMMR) is defined as a lack of immunohistochemically detectable MMR protein expression in tumors and microsatellite instability (MSI), and it is the diagnostic hallmark of LS (5). Concurrent loss of MSH2 and MSH6 proteins, which can be revealed by a universal reflex testing program using immunohistochemistry (IHC), is a common dMMR expression pattern that generally indicates the presence of a germline MSH2 mutation (6, 7). In addition, deletions of the 3’ end of EPCAM are thought to lead to tissue-specific epigenetic silencing of MSH2 through aberrant promoter methylation (2). EPCAM deletions account for approximately 20% of cases in which MSH2 and/or MSH6 are lost but there is no detectable MSH2 germline mutation (8–10). These unique cases cannot be distinguished from those in which MSH2 mutations are revealed by IHC analysis of MMR proteins (11). Multiplex ligation-dependent probe amplification (MLPA) analysis, which is used to detect large genomic rearrangements (LGRs), is a complementary diagnostic tool in comprehensive genetic testing strategies for LS (12, 13).
IHC analysis of MMR proteins is a cost-effective initial screening method for LS (14, 15). A previous study suggested that the protein expression pattern of MSH2 and MSH6 proteins can be categorized into three types: intact staining of both proteins, loss of both proteins, and isolated loss of MSH6 (16). Mutations in MSH2 are generally thought to result in the loss of IHC-detectable MSH2 and MSH6. Some challenging cases present with loss of MSH2 and with patchy loss of MSH6, as reported by Dr. Pearlman (17). Cytoplasmic staining is commonly interpreted as having no known significance, with previous literature citing questionable IHC staining quality (14). Dr. Sekine delineated a cryptic nonfunctional in-frame EPCAM-MSH2 fusion protein resulting from a genomic rearrangement between EPCAM intron 5 and MSH2 intron 2 in one LS patient with aberrant cytoplasmic MSH2 localization in colon cancer (18).
Mismatch repair status has been widely used as a positive predictive marker for clinical benefit of immune checkpoint blockade approved by US Food and Drug Administration in metastatic CRCs with dMMR or MSI-high (19). Immunotherapy treatment becomes a new and promising therapeutic option for advanced CRC patients. The importance of accurate interpretation of MMR protein IHC has been paid more attention by clinicians and pathologist (20, 21).
Unfortunately, no other studies are available that might shed light on whether this observation is simply an artifact or a valid finding in some MSH2-related LS cases. Due to this uncertainty, it is necessary to systematically assess rare cytoplasmic MSH2 abnormalities to avoid missing potential LS probands and to stratify CRC patients for immunotherapy. In this study, we investigated clinicopathological characteristics and performed molecular characterizations of MSH2 abnormalities in a large cohort of 4195 CRC patients with a particular focus on elucidating the association of cytoplasmic MSH2 staining with genotype in real-world LS patients.
Among 4195 eligible patients from the Colorectal Cancer Initiative Screening Program for Lynch Syndrome (CRISPLS) in the Cancer Hospital of the Chinese Academy of Medical Sciences between January 2015 and December 2017, we identified a cohort of 69 patients with loss of the MSH2 and/or MSH6 proteins who had been screened by IHC staining for tumor MMR proteins. Detailed information on the CRISPLS cohort was previously reported (22). Clinicopathological characteristics and information about cancer personal/family history were collected for patients from the CRISPLS cohort who had undergone surgical resection and for whom a sufficient DNA sample was available. The study was approved by the Ethics Committee of NCC/CICAMS (NCC1790). Individual informed consent was waived because of the retrospective nature of the study. Patients were informed if they were identified as having LS.
IHC analyses of MMR proteins, including MLH1, PMS2, MSH2, MSH6 and BRAF V600E, were routinely performed in CRC patients. One representative block of formalin-fixed, paraffin-embedded tumor tissue was selected per patient. Monoclonal antibodies against MLH1 (clone ES05), PMS2 (clone EPR3947), MSH2 (clone FE11), MSH6 (clone EP49) (Beijing Zhongshan Golden Bridge Biotechnology, China), and BRAF V600E (VE1) (Ventana Medical Systems, AZ, USA) were used. Briefly, after deparaffinization, rehydration and antigen-retrieval, 4-μm-thick sections were stained in a Ventana Benchmark IHC automated slide stainer and visualized using the OptiView DAB IHC detection kit (Ventana Medical Systems). The absence of nuclear staining in tumor cells or very faint nuclear staining in focal tumor cells was defined as loss of protein expression (abnormal staining). Stromal/lymphoid cells and nearby normal glandular epithelium of the bowel served as positive internal controls.
Microsatellite instability (MSI) testing was performed on tumor and normal DNA using a fluorescence PCR-based assay (MSI-Reader MSI Analysis System; MICROREAD, Beijing, China) in which six mononucleotide repeat markers (NR-21, NR-24, NR-27, BAT-25, BAT-26 and MONO-27) and two pentanucleotide repeat loci (Penta-C and Penta-D) were amplified to confirm the identity of paired tumor and benign tissues. The PCR products were run on an Applied Biosystems 3500 Genetic Analyzer and analyzed using GeneMapper v5.0 software (Applied Biosystems, CA, USA). Tumors with shifts in two or more markers were classified as unstable MSI-high (23, 24).
Formalin-fixed, paraffin-embedded tumors and adjacent normal tissue were collected from the cohort. Genomic DNA was extracted using a TGuide Genomic DNA One-Step Kit and a TGuide Automated Nucleic Acid Preparation Instrument (TIANGEN BIOTECH, Beijing, China) according to the manufacturer’s instructions as previously described (22).
Next-generation sequencing technology was performed with the Agilent SureSelect-XT Low Input Target Enrichment kit (Agilent Technologies, CA, USA) for germline mutation testing of MMR genes from genomic DNA extracted from normal FFPE samples according to the manufacturer’s instructions. Molecular barcoded DNA libraries were hybridized with a commercial ClearSeq Inherited Disease multigene panel that covered total exons and intron boundaries within at least ±20 bases of the EPCAM, MLH1, PMS2, MSH2 and MSH6 genes (Agilent Technologies). A detailed protocol for variant annotation and classification was described previously (22, 25). Diagnosis of LS is dependent on identifying the pathogenic germline variants of MMR genes. Interpretations of germline variants are classified according to the database of the International Society of Gastrointestinal Hereditary Tumors (InSiGHT) and guideline of the American College of Medical Genetics and Genomics (ACMG). The carriers with likely pathogenic or pathogenic variants are defined as LS patients.
Large genomic rearrangements (LGRs) in MSH2 and EPCAM genes among MSH2-deficient patients with no germline mutations identified by next-generation sequencing were assessed by MLPA using the SALSA MLPA P003 MLH1/MSH2 kit (including the 3’ end of EPCAM) and P072 MSH6 kit (including the EPCAM/MSH2 region) (MRC-Holland, Amsterdam, The Netherlands). Fragment analysis of amplified genomic DNA extracted from normal FFPE samples was performed on an ABI3500 capillary sequencer (Applied Biosystems). The MLPA data were quantitatively analyzed using Coffalyser.Net software (www.mlpa.com).
Total RNA was extracted from the peripheral blood leukocytes of patients using TRIzol reagent (Agilent Technologies). cDNA was synthesized using a PrimeScript II 1st Strand cDNA Synthesis Kit (Takara, Japan) and analyzed for EPCAM-MSH2 fusion transcripts. Polymerase chain reaction (PCR) products were loaded directly on 2% agarose gels and visualized under UV illumination. Selected PCR products were sequenced on an ABI 3500xl capillary DNA analyzer (Applied Biosystems, CA, USA). Details of the PCR primers used are provided in Table S1.
A series of long-range PCR experiments designed to span the putative deletion region were performed to characterize the exact breakpoints in the EPCAM and MSH2 genes using a TaKaRa LA PCR Kit (Takara, Japan) according to the manufacturer’s protocol. The PCR products were analyzed by electrophoresis on ethidium bromide-stained 1% agarose gels and then subjected to UV detection. The expected fragment was purified and sequenced on an ABI 3500xl capillary DNA analyzer (Applied Biosystems). Details of the PCR primers are provided in Table S2.
A univariate analysis of categorical variables was performed by cross tabulation using a chi-square test to compute p-values. An unpaired t test was used for continuous variables. Statistical descriptions or analyses were conducted using SPSS (Version 22; SPSS Inc., Chicago, IL, USA) or Prism (Version 7; San Diego, CA, USA) software. All tests were 2-tailed, and p-values < 0.05 were considered statistically significant.
We retrospectively enrolled a consecutive cohort of 4195 CRC patients. Among these patients, 345 were eligible, exhibiting dMMR, and 69 exhibited abnormal MSH2 protein expression (Figure S1). The frequency of MSH2 deficiency (dMSH2) among the dMMR group was 20% (69 of 345). The demographic and clinical characteristics of patients with MSH2-deficient CRC are summarized in Table 1. Briefly, the mean age was 50 years at diagnosis of CRC (standard deviation, 12.7), 94.2% was aged 70 years or younger, 65.2% was male, 47.8% occurred in the proximal colon, 46.4% was at tumor stage II, adenocarcinoma was most common histological type (91.3%). The differences in age of onset, personal history of cancer and family history of LS-related cancers between LS and dMSH2 were statistically significant.
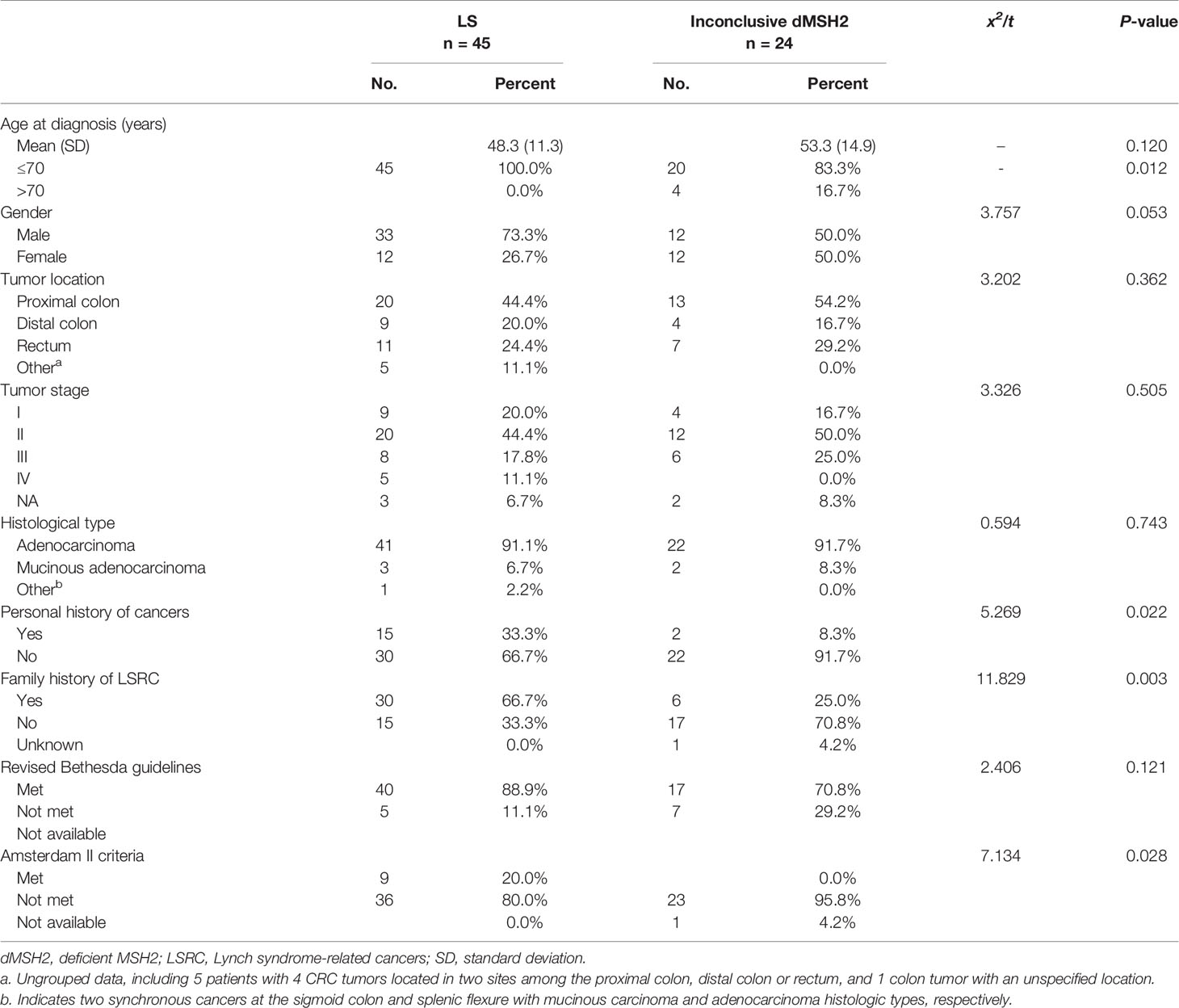
Table 1 Clinicopathological characteristics of patients with MSH2-deficient CRC.
To estimate the frequency and specificity of MSH2 germline mutations among patients with CRC in the real world, we systematically analyzed a consecutive CRC patient cohort that had been universally screened for LS in our previous study (22). Germline analyses were performed on samples from 69 patients with MSH2-deficient CRC. The frequencies of MSH2/EPCAM mutations among different categories of CRC subgroups are presented in detail in Table S3. Forty-five patients (1.1%) were identified as having LS with pathogenic germline mutations in MSH2 and/or EPCAM. Of these, 12 (26.7%) were confirmed to carry LGRs in MSH2/EPCAM by MLPA, including six probands with MSH2 genomic deletions, four with MSH2-EPCAM deletions (two cases from a family pedigree), and two with EPCAM deletions. The clinicopathological and molecular findings for these 12 patients with LGRs are presented in Table 2. All cases were identified as microsatellite instability-high (MSI-H) by PCR-MSI. Notably, all 12 of these patients were also ascertained to have a strong cancer family history. Patients with MSH2/EPCAM LGRs exhibited an earlier age of CRC onset (mean: 43.8 years) than those with LS with MSH2 SNV/indel (mean: 49.9 years).
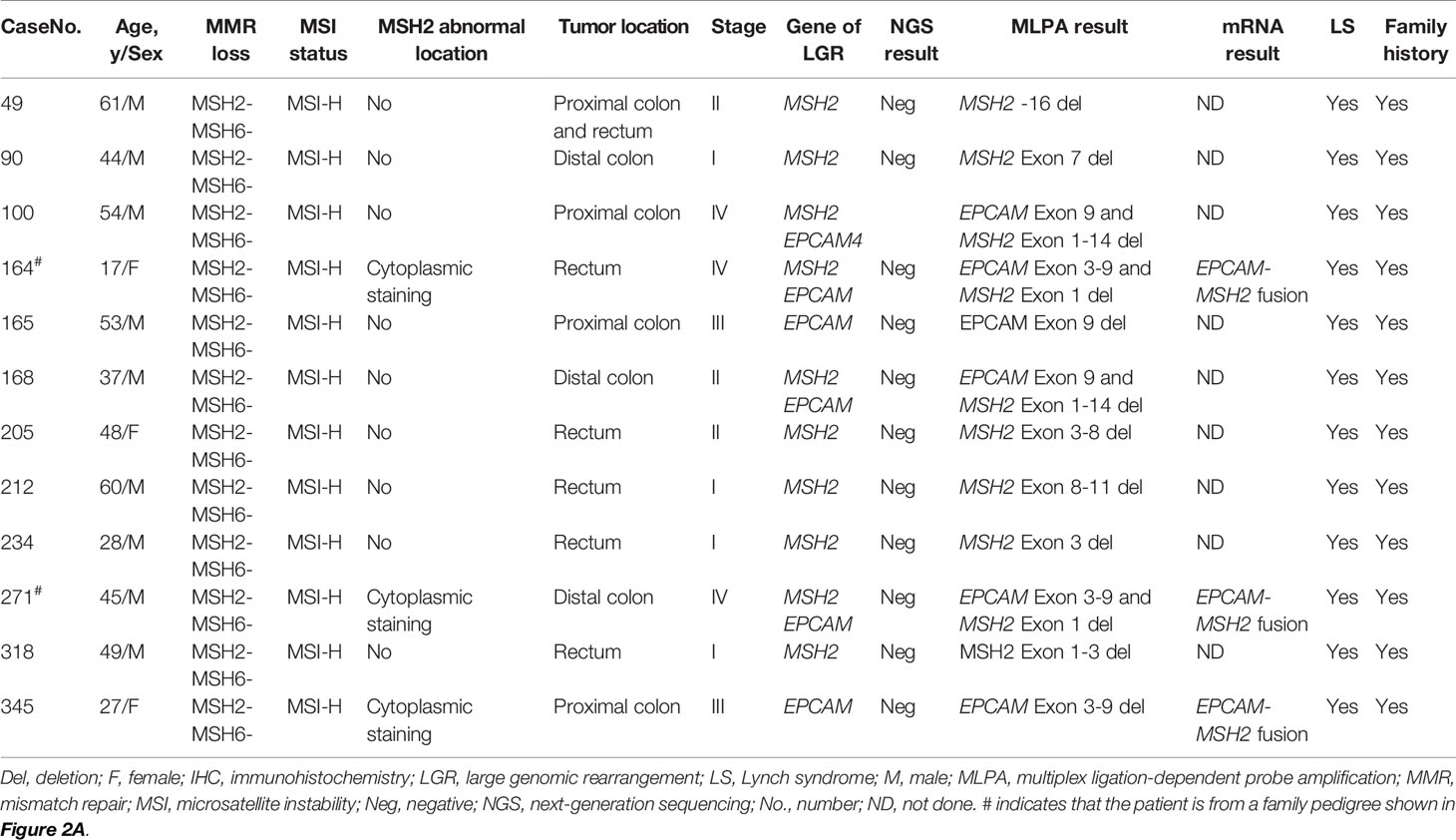
Table 2 Clinical, pathological and molecular findings in CRC tumors with MSH2 LGR-associated LS.
MSH2 abnormalities usually manifest as the absence of nuclear staining in tumor cells. Among the 69 patients with an MSH2 abnormality, we noted that three (4.3%) exhibited rare cytoplasmic MSH2 localization in tumor cells but showed patchy expression of the MSH6 protein that was somewhat weaker in tumor cells than in internal control cells (patient 164 and patient 271 from one family pedigree and patient 345) (Figures 1 and S2). They presented a classical family history of LS cancer (Figure 2). PCR-MSI tests indicated a status of MSI-high in the tumors of these patients (Figure S2). MLPA tests identified combined deletions of MSH2 and EPCAM in all three patients. Two patients had a heterozygous large genomic deletion in EPCAM (exons 3–9) and MSH2 (exon 1). One patient harbored heterozygous deletion of exons 3–9 of EPCAM (Figure S4).
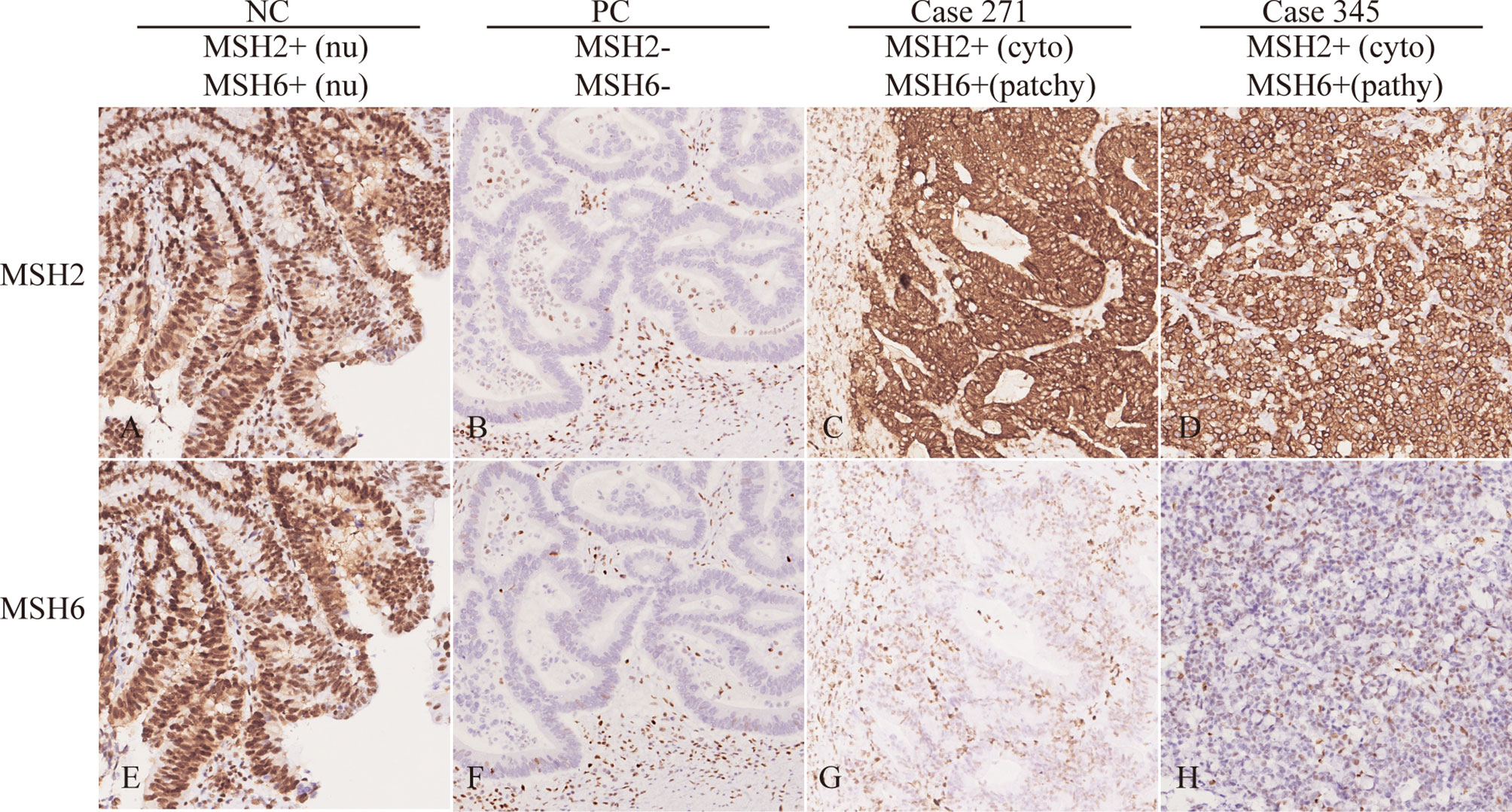
Figure 1 Rare MSH2 cytoplasmic staining in CRC. IHC of MSH2 and MSH6 in selected patients showing protein expression in tumor cells. The NC (negative control with proficient MMR) showed nuclear staining of MSH2 and MSH6. The PC (positive control with deficient MMR) showed loss of nuclear staining of MSH2 and MSH6. However, patient 271 and 345 showed cytoplasmic MSH2 and patchy/weak MSH6 nuclear staining in tumor cells. Nu, nuclear; cyto, cytoplasmic.
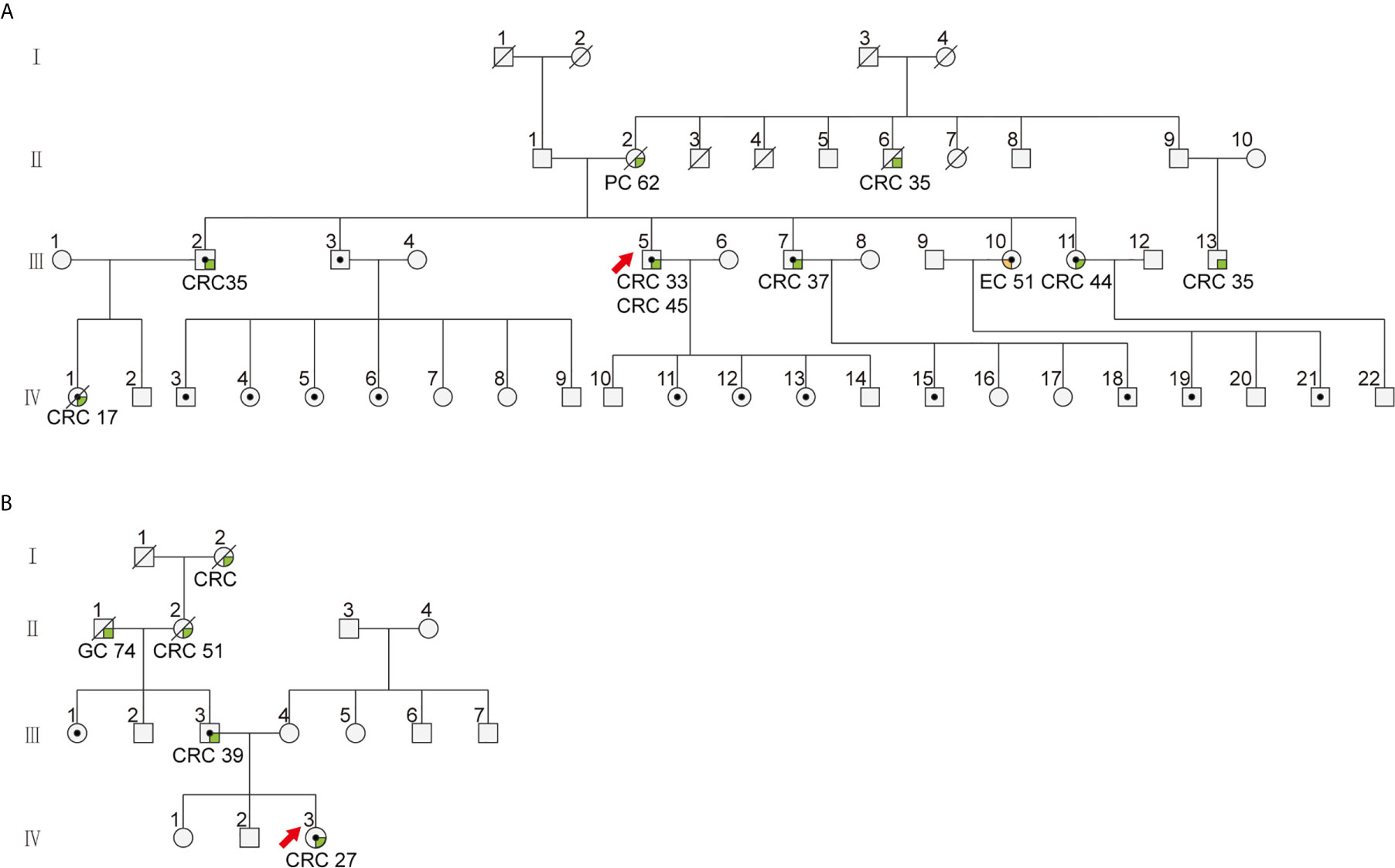
Figure 2 The family pedigree of patients with cytoplasmic staining of MSH2 in colorectal cancer. The family pedigrees of (A) Patient 271 (III5) and 164 (IV1) and (B) patient 345 (IV3) investigated in this study. The proband is indicated by the red arrow. The black dots indicate mutation carriers confirmed by genetic testing. The cancer types and age of onset are listed beneath each affected family member.
To elucidate the molecular characteristics of cytoplasmic localization of MSH2 in tumors, we performed mapping analysis of gene fusions and gene breakpoints for two individual patients. Sanger sequencing of cDNA from the blood lymphocytes of the patients suggested that a fusion transcript of EPCAM and MSH2 was the source of aberrant MSH2 localization in tumors. Sequencing revealed a common fusion of exon 1 of EPCAM and exon 2 of MSH2 in patient 271 and patient 345. On the basis of this fusion transcript analysis, we performed a series of long-range PCR experiments on the region of interest between intron 1 of EPCAM and intron 1 of MSH2 to identify the precise breakpoint in the MSH2/EPCAM LGRs (Figures S6 and S7). Ultimately, we mapped the breakpoint to intron 1 of EPCAM (hg38, chr2:47372970)/MSH2 upstream [hg38, chr2:47400492]) in patient 271 and to intron 1 of EPCAM (hg38, chr2:47370462)/intron 1 of MSH2 [hg38, chr2:47404894]) in patient 345. Both were novel mutations that were not reported in the International Society for Gastrointestinal Hereditary Tumours (InSiGHT) variant database (http://insight-database.org). Repeated Alu elements are implicated in the etiology of genomic rearrangement for many inherited cancers (26). Our analysis using RepeatMasker (http://www.repeatmasker.org) revealed that these breakpoints lay within Alu elements that share high sequence identities. A schematic diagram is shown in Figure 3.
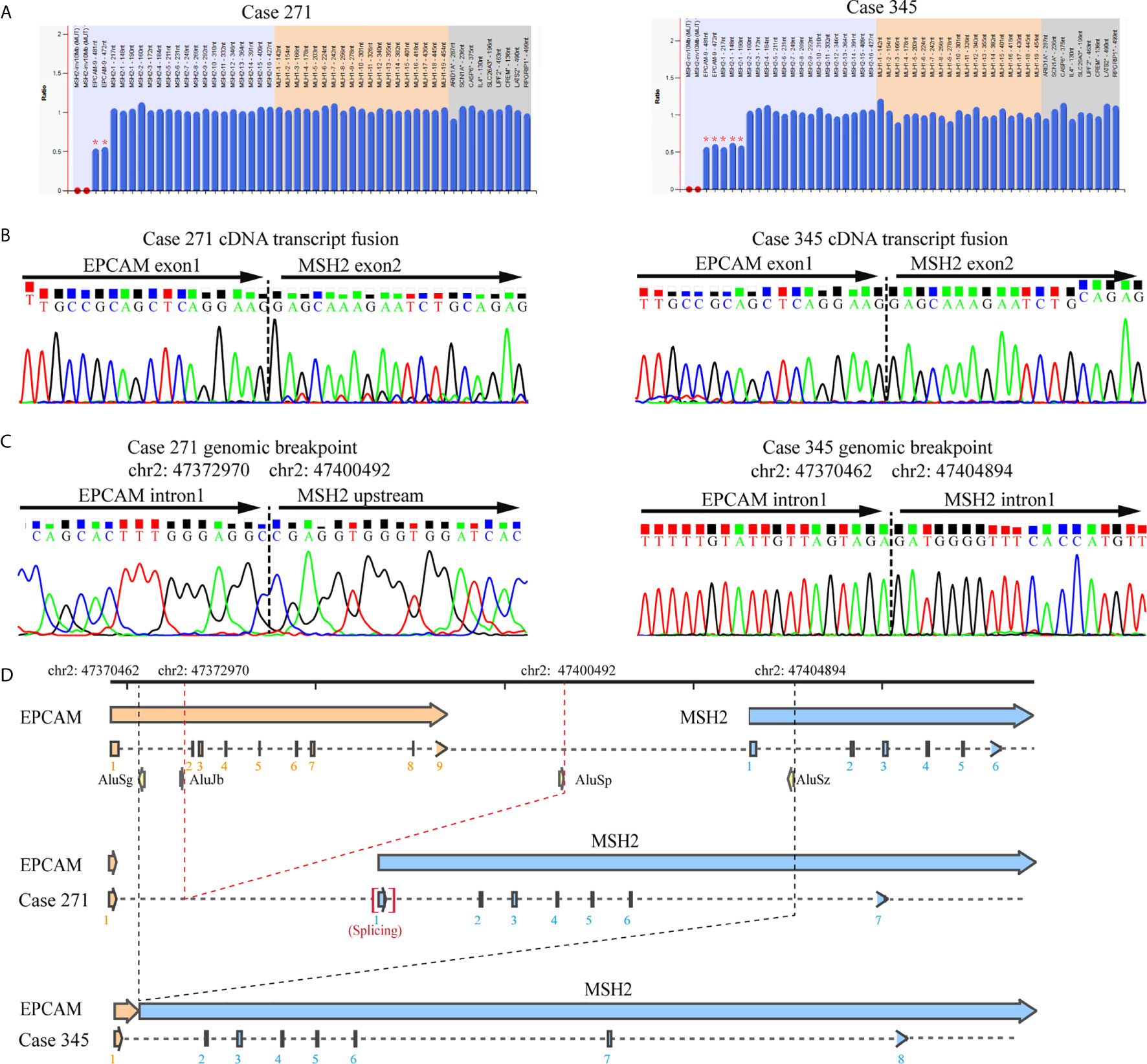
Figure 3 Identification of the germline EPCAM-MSH2 fusion in patients with suspected LS and cytoplasmic MSH2 staining. (A) MLPA testing identified the presence of a large genomic deletion in the MSH2 and EPCAM genes in patients 271 and 345. (B) Sanger sequencing of EPCAM and MSH2 transcripts amplified by the E1M23-1F and E1M23-2R primer pairs in the peripheral blood cDNA of the proband revealed a fusion between exon 1 of EPCAM and exon 2 of MSH2. (C) Sequencing analysis of long-range PCR products from genomic DNA defined the exact breakpoint, as indicated by the genomic locus. (D) Schematic representation showing the genomic deletions mediated by recombination between Alu elements.
Among patients with tumors showing abnormal MSH2 expression in our cohort, a 45-year-old male (patient 271) was diagnosed with retroperitoneal lymph node metastasis of stage IV colon cancer with rare MSH2 cytoplasmic localization. This patient underwent dissection of distal colon cancer at the age of 33. A cancer family history survey for this patient showed that nine members in three consecutive generations suffered from LS-related cancers (LSRC), including CRC in II6, III2, III5, III7, III11, III13, IV1; pancreatic cancer in II6; and endometrial cancer in III10 (Figure 2A). Patient IV1 (case 164) was retrospectively analyzed and identified as having the same MMR pattern as proband III5 (case 271). PCR-MSI confirmed that both patients showing a rare MSH2 cytoplasmic localization were MSI-high, which should be considered an uncommon dMMR pattern. Genetic testing identified this pedigree as LS harboring a novel pathogenic genomic deletion of EPCAM and MSH2 genes. Patient 271 was treated with the PD-1 inhibitor pembrolizumab in combination with capecitabine every 3 weeks for 19 cycles. The patient achieved a clinical partial response (PR) with a 56% reduction in the short axis diameter of the enlarged retroperitoneal lymph node, as revealed by computed tomography (CT) scans after a 19-month course of treatment (Figure 4).
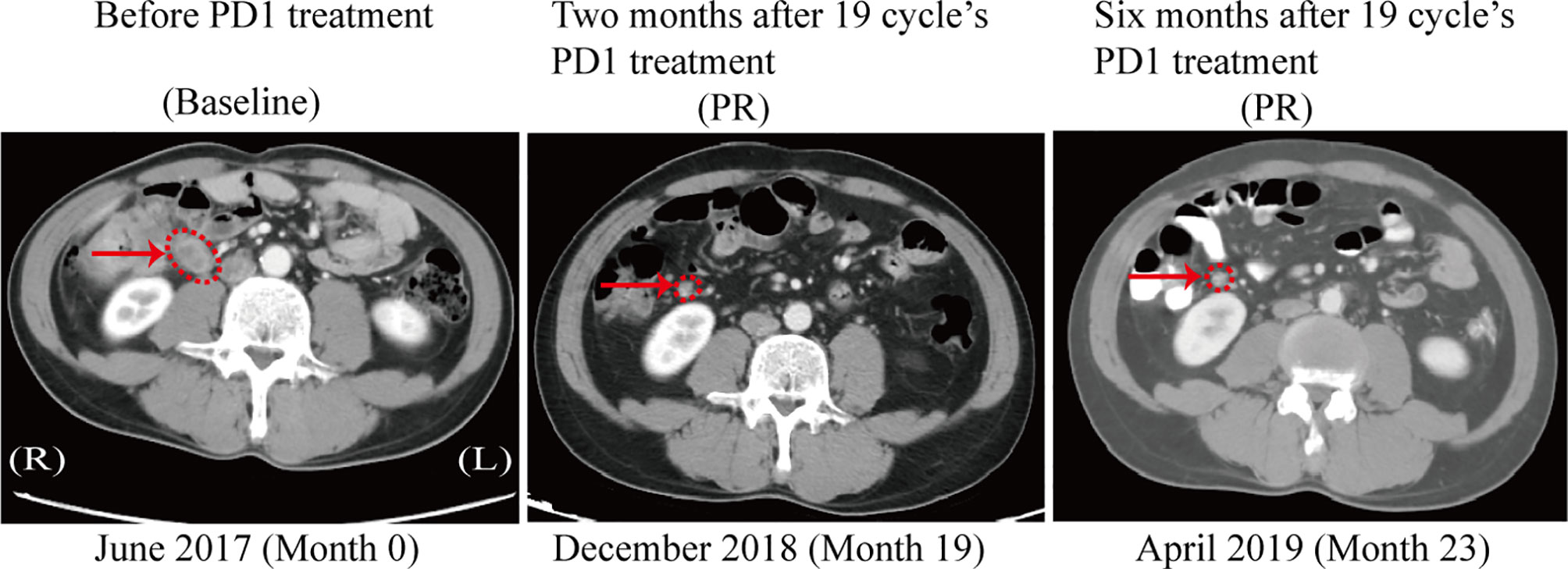
Figure 4 CT scan after PD1 treatment in the patient with MSH2 cytoplasmic staining. A computed tomography (CT) scan revealed the clinical benefits of the PD-1 inhibitor pembrolizumab in the patient with cytoplasmic staining of MSH2. In June 2017, a baseline CT scan showed an enlarged retroperitoneal lymph node (1.8x2.0 cm) with a diagnosis of metastatic colon cancer (arrows). No disease progression was confirmed in this patient 6 months after completion of 19 cycles of immunotherapy, as demonstrated by a stable lymph node size of 0.8x0.8 cm between December 2018 and April 2019 (arrows).
In recent years, there has been increasing demand for immunotherapy and LS screening among CRC patients displaying MSI-high or dMMR (27–30). These developments highlight the importance of closely integrating pathologic diagnosis, clinical counseling, and molecular testing for precision medicine approaches to CRC therapy. The purpose of further emphasizing molecular pathology in CRC is to personalize patient treatment and screening of suspected LS patients. In our previous study, we found that a significant proportion of LS patients have LGRs in MMR genes in China, a finding that has often been missed by previous next-generation sequencing (22, 31). In the current study, we systematically investigated the diverse mutation patterns of MSH2 and the clinicopathological characteristics of patients with MSH2 abnormalities in our cohort, especially LGRs, and the corresponding specific genotype-phenotype associations. We found that 12 of 45 (26.7%) LS patients were carriers of MSH2 and/or EPCAM LGRs. Three of 12 (25%) probands with MSH2/EPCAM LGRs harbored a rare MSH2 chimeric fusion protein that was detectable in the cytoplasm of tumor cells by IHC. We also provided clinical evidence that a CRC patient harboring cytoplasmic MSH2 fusion proteins was responsive to treatment with immune checkpoint inhibitors.
MMR IHC profiles help discriminate which genes may be deficient in MMR function. Previous studies have indicated that genomic deletion of MSH2 is a frequent causal event among LS patients (13, 32). One breakthrough study demonstrated that germline deletion of EPCAM also leads to inactivation of MSH2 in families with LS (2). Diverse mutation patterns, especially large genomic deletions/duplications in the MSH2 and/or EPCAM genes, increase the complexity of MSH2 IHC interpretation and molecular testing during LS screening (12, 18, 33–36). dMMR has emerged as a major predictive biomarker for the efficacy of immune checkpoint inhibitors in CRC (19). However, a post hoc analysis of clinical trials found that misinterpretation of IHC for MMR proteins was responsible for primary resistance to immune checkpoint inhibitors (21). One rare case of a patient with a MSH2/EPCAM LGR was reported to show distinct cytoplasmic localization of MSH2 (18). The study also revealed that there was an indication of EPCAM-MSH2 protein fusion rather than artificial nonspecific staining. The cases in this study and others demonstrate that there are still some challenges and pitfalls that pathologists need to avoid. However, because these atypical situations have not been well documented in the literature, they have not been effectively translated into clinical practice guidelines and are easily misinterpreted by pathologists (37–39). In the current study, characterization of three probands with abnormal MSH2 localization by IHC revealed detectable and specific cytoplasmic staining of MSH2 and loss of nuclear MSH2 staining with patchy MSH6 expression in the nucleus. Complementary PCR-MSI tests confirmed an MSI-high pattern suggestive of dMMR in tumors with cytoplasmic MSH2 staining. All three patients were identified as having LS with combined deletion of MSH2 and EPCAM, indicating an interestingly distinct phenotype-genotype association in LS screening. Together, the findings of our study based on a large consecutive CRC cohort demonstrate the pathologic characteristics of this novel MSH2 staining pattern and its possible association with germline mutations in MMR genes. Cases in which cytoplasmic MSH2 staining is combined with patchy or weak MSH6 staining in tumor cells are highly suspected to be LS.
IHC staining of MMR proteins should always be interpreted with caution. When interpreting staining as abnormal, pathologists should consider the localization, proportion, intensity and internal control of staining in tumor cells (40). Tumors carrying an MSH2 germline mutation generally show a complete loss of MSH2 and MSH6 proteins, whereas other uncommon staining patterns, such as MSH2 loss and patchy MSH6 nuclear staining, as well as retained staining of MSH2 and MSH6, have drawn the attention of pathologists and genetic counselors during IHC analysis of MMR proteins (17, 41). The findings of this study highlight another uncommon staining pattern: MSH2 cytoplasmic staining and patchy MSH6 nuclear staining, rather than the absence of staining, in tumor cells. IHC staining of adjacent normal colorectal tissues showed possible MSH2 cytoplasmic expression and nuclear staining. This indicates that a “double hit” in MSH2 causes inactivation of the normal allele, resulting in its absence in the nuclei of tumor cells. This study expands the pathologic interpretations of MSH2 IHC staining during LS prescreening and immunotherapy testing. The three patients with MSH2 abnormalities were all identified as having LS. The correlation between MSH2 cytoplasmic staining and large genomic deletions in MSH2/EPCAM is significant. Therefore, the dMMR pattern of cytoplasmic MSH2 staining and patchy/weak MSH6 nuclear staining should be helpful diagnostically in cases where LS is highly suspected by incorporating LGR analysis of MSH2/EPCAM. In addition, on the basis of these LS cases with ambiguous patchy/weak MSH6 staining, we also advocate for the use of four MMR proteins in IHC screens instead of the two-protein (MSH6 and PMS2) staining method (17, 42, 43).
EPCAM is an epithelial cell adhesion molecule located on the cell surface. The protein consists of a signal peptide, extracellular domain (N-terminal), transmembrane domain and cytoplasmic domain (C-terminal). Patients with cytoplasmic staining have a common feature of MSH2 C-terminal fusion with an EPCAM N-terminal fragment of 25 amino acids, including a complete signal peptide (44). The signal peptide is required for cytoplasmic membrane localization of the EPCAM protein. It is possible that the EPCAM signal peptide guides the cytoplasmic membrane translocation of EPCAM-MSH2 fusion proteins in this study. MSH2 and MSH6 in humans form an hMutSα heterodimer and are subsequently imported to the nucleus in a manner dependent on MSH6 nuclear localization sequences (45, 46). The EPCAM-MSH2 fusion proteins may interfere with binding and dimerization with MSH6. This defect in protein interaction in turn results in MSH6 protein instability without heterodimerization with MSH2, as indicated by weak or absent MSH6 staining in the nucleus (17).
In this study, 26.7% of LS CRC patients with MSH2 abnormalities carried pathogenic mutations in MSH2/EPCAM LGRs, consistent with previous studies (8, 11, 13). EPCAM deletions are generally considered to result in promoter hypermethylation and epigenetic silencing of the neighboring MSH2 gene through transcriptional read-through (2). Our study indicates that nonfunctional chimeric proteins derived from fusion transcripts of MSH2/EPCAM LGRs were underestimated in LS screening. This was not accidental. Three out of 12 patients with MSH2/EPCAM LGRs exhibited a similar phenotype of cytoplasmic staining of MSH2. Although the three identified patients from two families harbored two different genomic aberrations, they were found to have the same EPCAM-MSH2 transcript that involves the fusion of exon 1 of EPCAM and exon 2 of MSH2. A subsequent detailed analysis of breakpoint junctions indicated that the molecular mechanism underlying the novel rearrangements was intrachromosomal recombination mediated by Alu-Alu elements. They harbored different breakpoints in EPCAM-MSH2 than those reported by Sekine et al. (18). Alu repeats are a family of short interspersed nuclear elements (SINEs) that are prevalent in the human genome. It has been shown that some genes, such as MSH2 and EPCAM, are more prone to LGRs because of the presence of abundant homologous Alu elements (10, 47, 48). Therefore, molecular characterizations of uncommon cases from emerging clinical practice data definitely increase our understanding of LS etiology and contribute to refinements in corresponding genetic diagnostic approaches.
From a clinical standpoint, MMR-deficient CRC cases respond poorly to fluorouracil-based chemotherapeutics and are highly sensitive to immune checkpoint inhibitors (19, 30, 49–52). Misdiagnosis of dMMR and MSI status are the primary factors underlying resistance to immunotherapy among CRC patients (21). A lack of familiarity with the nuances of MMR IHC can lead to interpretive errors (15). Our findings suggest that CRCs with cytoplasmic localization of MSH2 should be considered dMMR and MSI-high. One case also demonstrated that anti-PD-1 immunotherapy shows a durable clinical benefit. Although this is only one example, we believe that, given their dMMR status, such patients should expect good efficacy with immune checkpoint inhibitor therapy. We suggest that PCR-MSI should be endorsed as a complementary test for abnormal patients that show equivocal immunostaining patterns to improve personalized targeted therapy.
In conclusion, our study demonstrates that the rare cytoplasmic MSH2 staining pattern in LS patients should be fully recognized by pathologists and geneticists. Given the specific genotype-phenotype correlation in LS screening, we advocate that all CRC patients with cytoplasmic MSH2 staining in histology should be screened for LGRs of EPCAM and MSH2 in clinical practice.
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding authors.
The studies involving human participants were reviewed and approved by Cancer Hospital of the Chinese Academy of Medical Sciences. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
JC and JY designed and supervised the overall project. LD, SZ, and YZ compiled and analyzed the data and performed statistical analyses. LD, HL, and LG interpreted the data and drafted the manuscript. JY critically revised the manuscript for intellectual content. All authors contributed to the article and approved the submitted version.
This work was supported by grants from the Special Fund of the Chinese Central Government for Basic Scientific Research in Commonweal Research Institutes (2016ZX310024 and 2016ZX310176), Beijing Hope Run Special Fund of the Cancer Foundation of China (LC2017B14), the Non-Profit Central Research Institute Fund of Chinese Academy of Medical Sciences (2019PT310026) and the National Key Research and Development Program (2017YFC1311005). The funders had no role in the study design, data acquisition, analysis, interpretation, writing or submission of the manuscript.
Author YZ was employed by company Beijing Microread Genetics.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2021.627460/full#supplementary-material
1. Moreira L, Balaguer F, Lindor N, de la Chapelle A, Hampel H, Aaltonen LA, et al. Identification of Lynch Syndrome Among Patients With Colorectal Cancer. JAMA (2012) 308:1555–65. doi: 10.1001/jama.2012.13088
2. Ligtenberg MJ, Kuiper RP, Chan TL, Goossens M, Hebeda KM, Voorendt M, et al. Heritable Somatic Methylation and Inactivation of MSH2 in Families With Lynch Syndrome Due to Deletion of the 3’ Exons of TACSTD1. Nat Genet (2009) 41:112–7. doi: 10.1038/ng.283
3. Li GM. Mechanisms and Functions of DNA Mismatch Repair. Cell Res (2008) 18:85–98. doi: 10.1038/cr.2007.115
4. Hampel H, Frankel WL, Martin E, Arnold M, Khanduja K, Kuebler P, et al. Screening for the Lynch Syndrome (Hereditary Nonpolyposis Colorectal Cancer). N Engl J Med (2005) 352:1851–60. doi: 10.1056/NEJMoa043146
5. Palter VN, Baker NA, Rabeneck L, Tinmouth J, Gagliardi AR, Kennedy ED, et al. A Framework to Build Capacity for a Reflex-Testing Program for Lynch Syndrome. Genet Med (2019) 21:1381–9. doi: 10.1038/s41436-018-0342-8
6. Pai RK, Pai RK. A Practical Approach to the Evaluation of Gastrointestinal Tract Carcinomas for Lynch Syndrome. Am J Surg Pathol (2016) 40:e17–34. doi: 10.1097/PAS.0000000000000620
7. Chen W, Swanson BJ, Frankel WL. Molecular Genetics of Microsatellite-Unstable Colorectal Cancer for Pathologists. Diagn Pathol (2017) 12:24. doi: 10.1186/s13000-017-0613-8
8. Rumilla K, Schowalter KV, Lindor NM, Thomas BC, Mensink KA, Gallinger S, et al. Frequency of Deletions of EPCAM (TACSTD1) in MSH2-Associated Lynch Syndrome Cases. J Mol Diagn (2011) 13:93–9. doi: 10.1016/j.jmoldx.2010.11.011
9. Cini G, Quaia M, Canzonieri V, Fornasarig M, Maestro R, Morabito A, et al. Toward a Better Definition of EPCAM Deletions in Lynch Syndrome: Report of New Variants in Italy and the Associated Molecular Phenotype. Mol Genet Genomic Med (2019) 7:e587. doi: 10.1002/mgg3.587
10. Perez-Cabornero L, Infante SM, Velasco SE, Lastra AE, Acedo BA, Miner PC, et al. Frequency of Rearrangements in Lynch Syndrome Cases Associated With MSH2: Characterization of a New Deletion Involving Both EPCAM and the 5’ Part of MSH2. Cancer Prev Res (Phila) (2011) 4:1556–62. doi: 10.1158/1940-6207.CAPR-11-0080
11. Kloor M, Voigt AY, Schackert HK, Schirmacher P, von Knebel DM, Blaker H. Analysis of EPCAM Protein Expression in Diagnostics of Lynch Syndrome. J Clin Oncol (2011) 29:223–7. doi: 10.1200/JCO.2010.32.0820
12. Huth C, Kloor M, Voigt AY, Bozukova G, Evers C, Gaspar H, et al. The Molecular Basis of EPCAM Expression Loss in Lynch Syndrome-Associated Tumors. Mod Pathol (2012) 25:911–6. doi: 10.1038/modpathol.2012.30
13. Baudhuin LM, Ferber MJ, Winters JL, Steenblock KJ, Swanson RL, French AJ, et al. Characterization of Hmlh1 and Hmsh2 Gene Dosage Alterations in Lynch Syndrome Patients. Gastroenterology (2005) 129:846–54. doi: 10.1053/j.gastro.2005.06.026
14. Shia J. Immunohistochemistry Versus Microsatellite Instability Testing for Screening Colorectal Cancer Patients At Risk for Hereditary Nonpolyposis Colorectal Cancer Syndrome. Part I. the Utility of Immunohistochemistry. J Mol Diagn (2008) 10:293–300. doi: 10.2353/jmoldx.2008.080031
15. Hissong E, Crowe EP, Yantiss RK, Chen YT. Assessing Colorectal Cancer Mismatch Repair Status in the Modern Era: A Survey of Current Practices and Re-Evaluation of the Role of Microsatellite Instability Testing. Mod Pathol (2018) 31:1756–66. doi: 10.1038/s41379-018-0094-7
16. Kantelinen J, Kansikas M, Candelin S, Hampel H, Smith B, Holm L, et al. Mismatch Repair Analysis of Inherited MSH2 and/or MSH6 Variation Pairs Found in Cancer Patients. Hum Mutat (2012) 33:1294–301. doi: 10.1002/humu.22119
17. Pearlman R, Markow M, Knight D, Chen W, Arnold CA, Pritchard CC, et al. Two-Stain Immunohistochemical Screening for Lynch Syndrome in Colorectal Cancer May Fail to Detect Mismatch Repair Deficiency. Mod Pathol (2018) 31:1891–900. doi: 10.1038/s41379-018-0058-y
18. Sekine S, Ogawa R, Saito S, Ushiama M, Shida D, Nakajima T, et al. Cytoplasmic MSH2 Immunoreactivity in a Patient With Lynch Syndrome With an EPCAM-MSH2 Fusion. Histopathology (2017) 70:664–9. doi: 10.1111/his.13104
19. Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, et al. PD-1 Blockade in Tumors With Mismatch-Repair Deficiency. N Engl J Med (2015) 372:2509–20. doi: 10.1056/NEJMoa1500596
20. Oliveira AF, Bretes L, Furtado I. Review of PD-1/PD-L1 Inhibitors in Metastatic Dmmr/MSI-H Colorectal Cancer. Front Oncol (2019) 9:396. doi: 10.3389/fonc.2019.00396
21. Cohen R, Hain E, Buhard O, Guilloux A, Bardier A, Kaci R, et al. Association of Primary Resistance to Immune Checkpoint Inhibitors in Metastatic Colorectal Cancer With Misdiagnosis of Microsatellite Instability or Mismatch Repair Deficiency Status. JAMA Oncol (2019) 5:551–5. doi: 10.1001/jamaoncol.2018.4942
22. Dong L, Jin X, Wang W, Ye Q, Li W, Shi S, et al. Distinct Clinical Phenotype and Genetic Testing Strategy for Lynch Syndrome in China Based on a Large Colorectal Cancer Cohort. Int J Cancer (2020) 146:3077–86. doi: 10.1002/ijc.32914
23. Suraweera N, Duval A, Reperant M, Vaury C, Furlan D, Leroy K, et al. Evaluation of Tumor Microsatellite Instability Using Five Quasimonomorphic Mononucleotide Repeats and Pentaplex PCR. Gastroenterology (2002) 123:1804–11. doi: 10.1053/gast.2002.37070
24. Rodriguez-Bigas MA, Boland CR, Hamilton SR, Henson DE, Jass JR, Khan PM, et al. A National Cancer Institute Workshop on Hereditary Nonpolyposis Colorectal Cancer Syndrome: Meeting Highlights and Bethesda Guidelines. J Natl Cancer Inst (1997) 89:1758–62. doi: 10.1093/jnci/89.23.1758
25. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med (2015) 17:405–24. doi: 10.1038/gim.2015.30
26. Elliott B, Richardson C, Jasin M. Chromosomal Translocation Mechanisms At Intronic Alu Elements in Mammalian Cells. Mol Cell (2005) 17:885–94. doi: 10.1016/j.molcel.2005.02.028
27. Nebot-Bral L, Brandao D, Verlingue L, Rouleau E, Caron O, Despras E, et al. Hypermutated Tumours in the Era of Immunotherapy: The Paradigm of Personalised Medicine. Eur J Cancer (2017) 84:290–303. doi: 10.1016/j.ejca.2017.07.026
28. Kloor M, von Knebel DM. The Immune Biology of Microsatellite-Unstable Cancer. Trends Cancer (2016) 2:121–33. doi: 10.1016/j.trecan.2016.02.004
29. Luchini C, Bibeau F, Ligtenberg M, Singh N, Nottegar A, Bosse T, et al. ESMO Recommendations on Microsatellite Instability Testing for Immunotherapy in Cancer, and Its Relationship With PD-1/PD-L1 Expression and Tumour Mutational Burden: A Systematic Review-Based Approach. Ann Oncol (2019) 30:1232–43. doi: 10.1093/annonc/mdz116
30. Overman MJ, McDermott R, Leach JL, Lonardi S, Lenz HJ, Morse MA, et al. Nivolumab in Patients With Metastatic DNA Mismatch Repair-Deficient or Microsatellite Instability-High Colorectal Cancer (Checkmate 142): An Open-Label, Multicentre, Phase 2 Study. Lancet Oncol (2017) 18:1182–91. doi: 10.1016/S1470-2045(17)30422-9
31. Zhang L, Bhaskaran SP, Huang T, Dong H, Chandratre K, Wu X, et al. Variants of DNA Mismatch Repair Genes Derived From 33,998 Chinese Individuals With and Without Cancer Reveal Their Highly Ethnic-Specific Nature. Eur J Cancer (2020) 125:12–21. doi: 10.1016/j.ejca.2019.11.004
32. Wijnen J, van der Klift H, Vasen H, Khan PM, Menko F, Tops C, et al. MSH2 Genomic Deletions are a Frequent Cause of HNPCC. Nat Genet (1998) 20:326–8. doi: 10.1038/3795
33. Kovacs ME, Papp J, Szentirmay Z, Otto S, Olah E. Deletions Removing the Last Exon of TACSTD1 Constitute a Distinct Class of Mutations Predisposing to Lynch Syndrome. Hum Mutat (2009) 30:197–203. doi: 10.1002/humu.20942
34. Liu Q, Hesson LB, Nunez AC, Packham D, Williams R, Ward RL, et al. A Cryptic Paracentric Inversion of MSH2 Exons 2-6 Causes Lynch Syndrome. Carcinogenesis (2016) 37:10–7. doi: 10.1093/carcin/bgv154
35. Kang SY, Park CK, Chang DK, Kim JW, Son HJ, Cho YB, et al. Lynch-Like Syndrome: Characterization and Comparison With EPCAM Deletion Carriers. Int J Cancer (2015) 136:1568–78. doi: 10.1002/ijc.29133
36. Martinez-Bouzas C, Ojembarrena E, Beristain E, Errasti J, Viguera N, Minguez MT. High Proportion of Large Genomic Rearrangements in Hmsh2 in Hereditary Nonpolyposis Colorectal Cancer (HNPCC) Families of the Basque Country. Cancer Lett (2007) 255:295–9. doi: 10.1016/j.canlet.2007.05.004
37. Sepulveda AR, Hamilton SR, Allegra CJ, Grody W, Cushman-Vokoun AM, Funkhouser WK, et al. Molecular Biomarkers for the Evaluation of Colorectal Cancer: Guideline From the American Society for Clinical Pathology, College of American Pathologists, Association for Molecular Pathology, and American Society of Clinical Oncology. Arch Pathol Lab Med (2017) 141:625–57. doi: 10.5858/arpa.2016-0554-CP
38. Gupta S, Provenzale D, Llor X, Halverson AL, Grady W, Chung DC, et al. Nccn Guidelines Insights: Genetic/Familial High-Risk Assessment: Colorectal, Version 2.2019. J Natl Compr Canc Netw (2019) 17:1032–41. doi: 10.6004/jnccn.2019.0044
39. Muller A, Giuffre G, Edmonston TB, Mathiak M, Roggendorf B, Heinmoller E, et al. Challenges and Pitfalls in HNPCC Screening by Microsatellite Analysis and Immunohistochemistry. J Mol Diagn (2004) 6:308–15. doi: 10.1016/S1525-1578(10)60526-0
40. Overbeek LI, Ligtenberg MJ, Willems RW, Hermens RP, Blokx WA, Dubois SV, et al. Interpretation of Immunohistochemistry for Mismatch Repair Proteins is Only Reliable in a Specialized Setting. Am J Surg Pathol (2008) 32:1246–51. doi: 10.1097/PAS.0b013e31816401bb
41. Ollila S, Sarantaus L, Kariola R, Chan P, Hampel H, Holinski-Feder E, et al. Pathogenicity of MSH2 Missense Mutations is Typically Associated With Impaired Repair Capability of the Mutated Protein. Gastroenterology (2006) 131:1408–17. doi: 10.1053/j.gastro.2006.08.044
42. Hall G, Clarkson A, Shi A, Langford E, Leung H, Eckstein RP, et al. Immunohistochemistry for PMS2 and MSH6 Alone Can Replace a Four Antibody Panel for Mismatch Repair Deficiency Screening in Colorectal Adenocarcinoma. Pathology (2010) 42:409–13. doi: 10.3109/00313025.2010.493871
43. Mojtahed A, Schrijver I, Ford JM, Longacre TA, Pai RK. A Two-Antibody Mismatch Repair Protein Immunohistochemistry Screening Approach for Colorectal Carcinomas, Skin Sebaceous Tumors, and Gynecologic Tract Carcinomas. Mod Pathol (2011) 24:1004–14. doi: 10.1038/modpathol.2011.55
44. Schnell U, Cirulli V, Giepmans BN. Epcam: Structure and Function in Health and Disease. Biochim Biophys Acta (2013) 1828:1989–2001. doi: 10.1016/j.bbamem.2013.04.018
45. Guerrette S, Wilson T, Gradia S, Fishel R. Interactions of Human Hmsh2 With Hmsh3 and Hmsh2 With Hmsh6: Examination of Mutations Found in Hereditary Nonpolyposis Colorectal Cancer. Mol Cell Biol (1998) 18:6616–23. doi: 10.1128/MCB.18.11.6616
46. Alani E, Sokolsky T, Studamire B, Miret JJ, Lahue RS. Genetic and Biochemical Analysis of Msh2p-Msh6p: Role of ATP Hydrolysis and Msh2p-Msh6p Subunit Interactions in Mismatch Base Pair Recognition. Mol Cell Biol (1997) 17:2436–47. doi: 10.1128/MCB.17.5.2436
47. Deininger P. Alu Elements: Know the Sines. Genome Biol (2011) 12:236. doi: 10.1186/gb-2011-12-12-236
48. Jiang Y, Zong W, Ju S, Jing R, Cui M. Promising Member of the Short Interspersed Nuclear Elements (Alu Elements): Mechanisms and Clinical Applications in Human Cancers. J Med Genet (2019) 56:639–45. doi: 10.1136/jmedgenet-2018-105761
49. Sargent DJ, Marsoni S, Monges G, Thibodeau SN, Labianca R, Hamilton SR, et al. Defective Mismatch Repair as a Predictive Marker for Lack of Efficacy of Fluorouracil-Based Adjuvant Therapy in Colon Cancer. J Clin Oncol (2010) 28:3219–26. doi: 10.1200/JCO.2009.27.1825
50. Ribic CM, Sargent DJ, Moore MJ, Thibodeau SN, French AJ, Goldberg RM, et al. Tumor Microsatellite-Instability Status as a Predictor of Benefit From Fluorouracil-Based Adjuvant Chemotherapy for Colon Cancer. N Engl J Med (2003) 349:247–57. doi: 10.1056/NEJMoa022289
51. Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK, et al. Mismatch Repair Deficiency Predicts Response of Solid Tumors to PD-1 Blockade. Science (2017) 357:409–13. doi: 10.1126/science.aan6733
Keywords: DNA mismatch repair, Lynch syndrome, colorectal cancer, genetic testing, immunotherapy
Citation: Dong L, Zou S, Jin X, Lu H, Zhang Y, Guo L, Cai J and Ying J (2021) Cytoplasmic MSH2 Related to Genomic Deletions in the MSH2/EPCAM Genes in Colorectal Cancer Patients With Suspected Lynch Syndrome. Front. Oncol. 11:627460. doi: 10.3389/fonc.2021.627460
Received: 27 November 2020; Accepted: 27 April 2021;
Published: 14 May 2021.
Edited by:
Glaucia N M Hajj, International Center for Research, AC Camargo Cancer Center, BrazilReviewed by:
Giovana Tardin Torrezan, A.C.Camargo Cancer Center, BrazilCopyright © 2021 Dong, Zou, Jin, Lu, Zhang, Guo, Cai and Ying. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jianqiang Cai, Y2FpamlhbnFpYW5nMTg4QHNpbmEuY29t; Jianming Ying, am15aW5nQGNpY2Ftcy5hYy5jbg==
†These authors have contributed equally to this work and share senior authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.