 Alejandra Suares
Alejandra Suares María Victoria Medina
María Victoria Medina Omar Coso
Omar Coso
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol., 08 March 2021
Sec. Molecular and Cellular Oncology
Volume 11 - 2021 | https://doi.org/10.3389/fonc.2021.603224
This article is part of the Research TopicNew Roles of Autophagy Pathways in CancerView all 24 articles
Autophagy is a complex degradative process by which eukaryotic cells capture cytoplasmic components for subsequent degradation through lysosomal hydrolases. Although this catabolic process can be triggered by a great variety of stimuli, action in cells varies according to cellular context. Autophagy has been previously linked to disease development modulation, including cancer. Autophagy helps suppress cancer cell advancement in tumor transformation early stages, while promoting proliferation and metastasis in advanced settings. Oncoviruses are a particular type of virus that directly contribute to cell transformation and tumor development. Extensive molecular studies have revealed complex ways in which autophagy can suppress or improve oncovirus fitness while still regulating viral replication and determining host cell fate. This review includes recent advances in autophagic cellular function and emphasizes its antagonistic role in cancer cells.
Living organisms survive and are naturally preserved thanks to the combination of complex systems that coordinate to maintain homeostatic balance (1). The immune and endocrine systems represent good examples, as specialized cells and chemical mediators work together with antibodies and hormones to generate a specific response in the body (2). Individuals constantly face tissue damage due to stressful and environmental signals, as well as normal body deterioration and aging consequences (3). This is why organisms need intracellular signaling mechanisms that allow them to protect themselves from damaged cells, either by killing them or inhibiting their spread (4). In this way, organisms are prevented from preserving defective cell lines with potential mutation or error accumulation that may contribute to disease risk (5).
Macroautophagy (autophagy) is a metabolic process of intracellular component autodegradation, such as proteins and organelles, crucial for maintaining metabolism and cellular homeostasis (6). Normal levels of basal autophagy prevent cells from gradually accumulating proteins and damaged organelles that can become toxic to cells over time (7). Identifying the mechanistic components of this process at the cellular and molecular levels has been of great interest to researchers worldwide since the late 1950s (8). The first scientists to study and coin the name this catabolic mechanism believed that autophagy was just a cytoplasmic “cleaning mechanism” by which cells remove harmful components that accumulate in the cytoplasm (9). This explains the etymology of the term, which comes from the Greek words “phagy,” meaning “eat,” and “auto,” meaning “me.” However, the role of autophagy in cells is now considered to be much broader as well as strongly influenced by the cellular environment. Autophagy modulation is related to human pathophysiology, and its implications affect different medical fields (10). This review summarizes the advances in molecular biology in relation to how this catabolic process helps develop different human diseases, focusing primarily on autophagy’s dual role in health maintenance and tumor progression, with special interest in tumors associated with viral infections.
Up to date, 32 atg (autophagy-related genes), involved in regulating different autophagy stages have been identified in mammals. These genes encode numerous proteins (ATG) that regulate the autophagic machinery (11). Autophagy can be divided mechanistically into different stages: 1) initiation and nucleation (molecule recruitment for isolation membrane extension), 2) phagophore elongation and closure (autophagosome), 3) fusion with lysosomes (autolysosomes), 4) degradation, and 5) cytoplasmic material recycling (12) (Figure 1). Mammalian cells induce the autophagic machinery in response to various cellular stimuli, such as prolonged starvation (13), decreased glucose levels (14), hypoxia (15–17), increased levels of reactive oxygen species (ROS) (18, 19), and ER (endoplasmic reticulum) stress (20, 21), among others (22).

Figure 1 Steps involved in autophagic responses. Autophagy begins with the progressive segregation of cytoplasmic material by double-membrane structures, commonly known as phagophores or isolation membranes. In general, this process is preceded by the inactivation of the PI3K/Akt/mTOR signaling axis. Phagophores are nucleated primarily from the endoplasmic reticulum (ER), but other organelles, such as the Golgi apparatus, plasma membrane, mitochondria, and recycling endosomes have also been shown to participate in this process. The complexes ULK1 and PI3KCIII are involved during phagophore initiation and elongation. Pro-LC3 is free in the cytoplasm and by the action of ATG4 and ATG7, LC3-I is formed. This molecule interacts with the complex ATG16L, ATG3, to later incorporate a phosphatidylethanolamine (PE) molecule into its structure. This results in LC3-II, which binds to autophagosomal membranes and contributes to phagophore elongation as well as closure. Then, these membranous structures seal, and autophagosomes are entirely assembled. Subsequently, autophagosomes fuse with lysosomes to form single membrane structures called autolysosomes, where the degradative process takes place through lysosomal hydrolases. The degradation products of these catabolic reactions reach the cytosol through transporters in the lysosomal membrane and are recycled by bioenergetic circuits.
AMPK protein (AMP-activated kinase) is the main inducer of the autophagic machinery thus reducing intracellular ATP levels. Recent studies have revealed AMPK’s fundamental role in autophagosome maturation and its fusion with lysosomes (23). Similarly, mTOR (mammalian target of rapamycin) activity is regulated by amino acid and glucose levels in mammalian cells (24), mTOR being their main autophagy inhibitor (25). Specifically, mTORC1 (mTOR complex 1) detects the cell’s nutritional status and activates various signaling pathways to regulate cell fate (26). mTORC1 detects and responds to fluctuations of intra and extracellular nutrient levels, mainly amino acids and oxygen, as well as various growth factors (27). A complex dynamic between mTOR and AMPK enables coordinated regulation of signaling pathways in response to cellular environment changes (28).
High-nutrient levels promote mTORC1 inactivation and the induction of anabolic pathways involving protein, lipid, and nucleotide synthesis through S6K (ribosomal protein kinase S6) and 4E-BP1 (initiation factor of the eukaryotic translation 4E - binding protein 1) phosphorylation (29). At the same time, catabolic cellular programs are suppressed as ULK1 is inhibited (Unc-51-like kinase 1, mammalian homolog of atg1), thus leading -in turn- to autophagy inhibition (30). Multiprotein complex ULK1 (ULK1, Beclin-1 (BECN1), and PI3KCIII (phosphatidylinositol-3 kinase class III)) mainly regulates the autophagic mechanism initiation process (31). Once active, the complex is recruited to the isolation membrane, where it contributes to PIP3 (phosphatidylinositol 3-phosphate) formation and Akt activation (32). These cellular events dampen TSC 1/2 (tuberous sclerosis protein 1 and 2)inhibitory effect, a protein heterodimer homologous to RHEB (RAS enriched in brain protein) (33, 34). Akt can also be inhibited by mTORC2, further contributing to autophagy inhibition (35). PTEN (phosphatidylinositol-3,4,5-trisphosphate 3-phosphatase) can hinder PIP3 formation, by activating the PI3K/Akt/mTOR pathway (36).
How the isolation membrane resulting in the phagophore forms still remains unclear. However, this membrane has been reported to derive from the plasma membrane, the endoplasmic reticulum (37), the Golgi apparatus (38), the ER-Golgi intermediate compartment (39), and the mitochondria (40). In these membranes, nucleation occurs (41) (Figure 1). All proteins involved in pagophore elongation, maturation, and closure are recruited through this process (42). The phagophore incorporates and degrades cytoplasmic material during extension and then it closes up, forming double-membrane vesicular structures called autophagosomes (35).
The action of PI3KCIII multiprotein complex (VPS34 (vacuolar proteins sorting 34), BECN1, p150 (ortholog of mammals of VPS15), mAtg14) in the protein recruitment process to the isolation membrane is fundamental (12). At this point, achieving active recycling is essential, a process involving the intervention of the ATG9 protein anchored in the membranes (31). Some Bcl-2 (B-cell lymphoma 2) family members, such as Bcl-2 and Bcl-xL (B-cell lymphoma extra-large), are known inhibitors of programmed cell death but can also inhibit autophagy through their interaction with BECN1 (43–45). Such interaction does not allow BECN1 to interact with VPS34 (46–48).
After nucleation, the ATG16L complex (ATG12, ATG5, ATG16L1) is recruited into the membrane, where it contributes to LC3 (light chain microtubule-associated protein-1 or MAP1LC3B) (49), GATE-16 (Golgi-associated ATPase enhancer of 16 kDa), and GABARAP (aminobutyric protein associated with the γ-acid receptor) lipidation (11, 50). These three groups belong to the ATG8 protein family, highly conserved across the evolutionary scale (51). Several proteins are anchored to the phagophore membrane, which is shed and returned to the cytoplasm before closure. Meanwhile, LC3 remains attached to the autophagosomal membranes throughout the process, making it a useful autophagosome marker (52) (Figure 1).
Several cellular receptors are involved in the selective recognition and recruitment of the cytoplasmic material that is later degraded in autolysosomes. The best-characterized autophagy receptor to date is p62 (Sequestosome 1 or SQSTM1), a molecular adapter with a ubiquitin-binding site and another for LC3 (35). p62 can also promote inflammatory gene expression through NF-kB (nuclear factor kB) regulation, activated when binding to TRAF6 (tumor necrosis factor receptor-associated factor 6) (53). Furthermore, p62 has been shown to activate an antioxidant response by sequestering Keap-1 (Kelch-like ECH-associated protein-1) through an Nrf2-dependent (erythroid-derived nuclear factor 2) mechanism (54) as well as to activate mTORC1 and regulate c-Myc (55).
After formation, autophagosomes relocalize to the perinuclear region through microtubules, where they fuse with lysosomes to form single membrane vesicles called autolysosomes (56), a complex process requiring anchoring factors and about which very little is known so far (57). SNARE proteins (soluble NSF binding protein) take part in the recognition and fusion of these structures. Studies in mice have revealed the importance of a SNARE complex [VTI1B, syntaxin 8, syntaxin 7, and VAMP-8 (vesicle-associated membrane protein 8)] in late fusion with the lysosome (Vadim Atlashkin2003). UVRAG (a gene associated with ultraviolet radiation) can activate GTPase RAB7 to promote fusion (58). Syntaxin 17, located in mature autophagosomes, can also regulate this mechanism (59). Successful binding to lysosomes is necessary for complete autophagy, as lysosomes provide the lytic enzymes needed for the degradation of cytoplasmic components in vesicles (60).
Historically, autophagy was considered to be a mechanism benefitting cell survival, as it recycles damaged and potentially toxic cytoplasmic components to increase vitality in cells subjected to stressful conditions, such as nutrient deprivation (61). Depending on cellular context, autophagy is selective or non-selective (massive autophagy). The former shows high specificity in degradation load selection and delivery, while in the latter, cytoplasmic particles are incorporated randomly (62). Selective autophagy is defined by the cytoplasmic material digested in mitophagy (affects mitochondria) (63), pexophagy (peroxisomes) (64), reticulophagy (endoplasmic reticulum and ribosomes) (65), nucleophagy (nucleus) (66), lipophagy (fat cells) (67), xenophagy (involves pathogens and other non-host entities) (68), and aggrephagy (damaged protein aggregates) (69).
With the advancement of knowledge, autophagy has been found not only to promote cell survival, but also to be induced in dying cells (70). The role of autophagy in death is, even today, a critical controversial point among researchers. While some scientists consider autophagy to been an independent death mechanism (autosis) (71), others argue that activation in dying cells occurs as a failure to rescue the cells from the stressful stimuli leading them to death in the first place (72, 73). In light of the growing number of physiological functions related to the autophagic mechanism, connections with numerous human pathologies have also been strengthened (74).
For cell transformation and tumor development to happen, several basic cellular alterations -referred to as the hallmarks of cancer- must occur (75, 76). Increasing evidence suggests a link between autophagy and cancer (77).
However, establishing the role of autophagy in cancer has proved problematic as it can both contribute to tumor promotion and inhibition, depending on cellular context and disease stage (78). In the early stages of tumor transformation, autophagy can be activated to help cells mitigate mutations and damage their various components. But, once the transformation is complete, tumor cells can make use of the autophagic machinery to meet the high metabolic requirements of these uncontrolled dividing cells (79) (Figure 2).
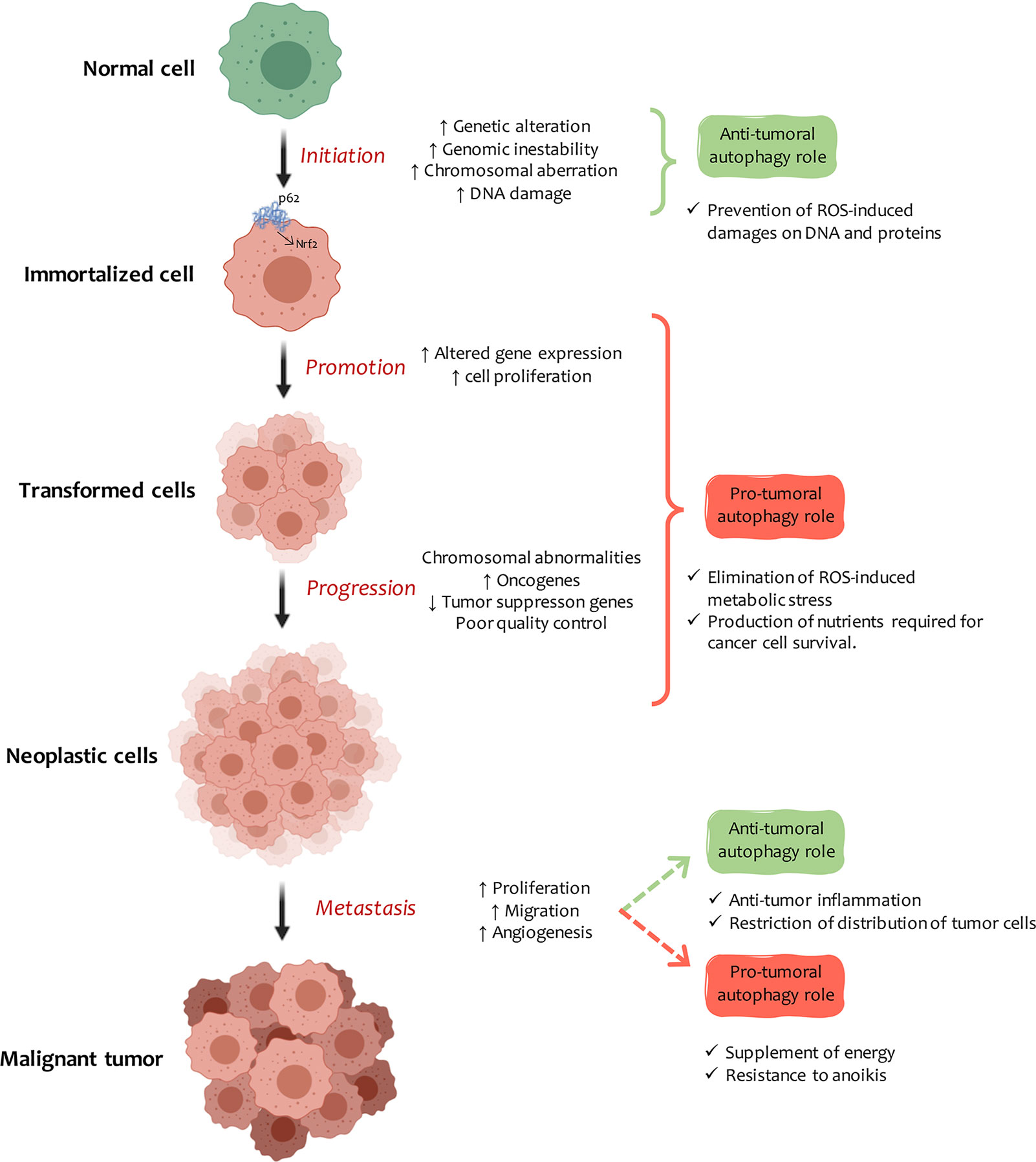
Figure 2 Autophagy and cancer. Autophagy plays a dual role in the development of cancer, the nature of which depends on the tissue, stage, and type of tumor. In carcinogenesis early stage, autophagy induction protects cells from DNA and protein damage due to the indiscriminate increase in ROS and cell-toxic components. Once cells are immortalized, autophagy plays an essential role in promoting tumors. Autophagy induction can be modified, or at least some of the steps involved in the mechanism can be changed. The excessive accumulation of autophagic vesicles leads to p62 molecule accumulation and intracellular signaling activation, which leads to Nrf2 transformation, inflammation, and cell necrosis. In tumor progression, autophagy provides high-energy nutrients to actively dividing transformed cells. When transformed cells metastasize, autophagy can inhibit the process by promoting anti-tumor inflammatory responses or by restricting the expansion of dormant metastatic tumor cells. On the contrary, it supports metastasis by improving cell fitness against a stressful microenvironment (anoikis).
The most relevant findings related to autophagy and its role as a tumor suppressor come from studies on BECN1 (80). Mice with depletion of an allele for this gene showed a higher predisposition to different neoplasm spontaneous development (81). BECN1 has a BH3 domain, so it is not surprising that it can interact with various members of the Bcl-2 protein family and homologous viral proteins (82). Through these interactions, BECN1 can regulate autophagic and programmed cell death (83). Under normal conditions, Bcl-2 inhibits BECN1, whereas under stress conditions, they dissociate. This allows BECN1 to interact with VPS34 and modulate autophagy (84).
BECN1 dysregulation has been associated with the development of several cancers, including 50%–70% of prostate, breast, and ovarian cancers (85). In response to stressful stimuli, BECN1 can interact with Bif-1 (bax1 interaction factor or endophilin B1) through a mechanism involving UVRAG, thus leading to PI3KCIII and VPS34 activation. This regulatory mechanism has been evidenced in various tumor models (86).
Under cellular stress conditions, autophagy induction mitigates oxidative stress by eliminating damaged mitochondria, a crucial source in ROS production (80). When defective autophagy occurs, debris cannot be removed, leading to increased ROS levels and DNA damage with consequent loss of genomic integrity (87, 88). Therefore, autophagy prevents tumor generation by regulating ROS levels (89).
Atg genes expression has been directly linked to this process. Studies in mouse liver with silenced atg7 and atg5 showed autophagy malfunction as contributing to benign hepatic adenoma development (90). These tumors did not progress over time, suggesting that the loss of autophagy may be sufficient for tumorigenesis onset, but not for progression towards advanced stages of the disease (91). On the other hand, mice with deficient in atg4C showed alterations in autophagy and greater predisposition to fibrosarcomas induced by carcinogens (92).
Numerous tumor suppressor proteins promote autophagy (93–95). Tumor suppressor p53 is a usually deregulated protein in many human neoplasms which promotes autophagy when activated by nutrient deprivation or genotoxic stress (91). p53 functional loss is therefore expected to lead to autophagy inhibition (96). However, p53 can act as either an activator or an inhibitor of autophagy depending on its subcellular localization and its action mode (97). Mice with pancreatic oncogenic alleles for k-ras develop precancerous lesions and PDCA (pancreatic ductal adenocarcinoma) over time. Here, p53 expression blocks autophagy, thus inhibiting initial carcinogenesis (98). On the other hand, p53 can contribute to autophagy activation through DRAM1 (DNA damage-regulated autophagy modulator protein 1) (99), atg7, and ulk1 (100) modulation. Another p53 target gene is isg20l1, which promotes autophagy induction and cell death when activated (101).
Numerous studies have revealed an increase in autophagy levels as carcinogenesis progresses (89). In metastasis early stages, cells acquire migratory properties and detach from the tumor to enter the bloodstream and flow through the body to colonize new tissue. At this point, autophagy exerts an anti-tumor role by modulating inflammation and cell shedding, but it also promotes motility and invasion (102). These results suggest that the autophagic machinery is a regulatory mechanism that can inhibit tumor generation in the early stages of the disease and in metastasis (Figure 2).
High metabolism requires a stressful condition to which tumor cells must adapt to proliferate actively in combination with a hypoxic cellular environment (86). Under these conditions, cells can activate autophagy to address various cellular needs and promote oncogenesis (77). Autophagy is activated in the hypoxic regions of tumors to counteract cellular oxygen demand (103). When tumor cells blood supply is insufficient, the autophagic machinery can be activated through an HIF-1-dependent mechanism (hypoxia-inducible factor-1) (15), VEGF (vascular endothelial growth factor) (104), PDGF (platelet-derived growth factor) (105) and oxide synthase (106). Hence, autophagy plays an essential role in promoting tumor cell survival under metabolic stress (107). Furthermore, cell division high rate translates into increased energy and biosynthetic needs, which can be satisfied by rising autophagy levels to obtain ATP and metabolic intermediates (108).
Transcription factor p53 acts as a cellular stress sensor in response to DNA damage and oncogenic stress (94) and often mutates in different types of human cancers (109). Moreover, point mutations in p53 prevent it from inhibiting autophagy in some breast cancer models (96). Consequently, this catabolic signaling pathway is activated to help repair damaged DNA and benefit tumor cells (110).
p62 cell adapter is another crucial molecule in nutrient detection. It can also act as a mitotic transit modulator, an oxidative detoxifying protein inducer, and genomic stability regulator (111). It also contributes to the autophagic mechanism by recruiting proteins and organelles into the autophagosomal compartments for subsequent degradation (112). In liver carcinoma cells, mTORC1 inhibition and ER stress promote p62 accumulation and autophagy induction (113). In colorectal cancer cells, p62 promotes invasion and metastasis by inhibiting apoptosis through a mechanism involving the vitamin D receptor and Nrf2 (114). p62 deletion produces significant autophagy inhibition and affects tumor growth in in vivo and xenograft models (115). Upregulated p62 is commonly found in various tumor models (111).
Approximately 33% of neoplasms developed in mammals present mutations of the ras gene, which indicates the importance of this gene for medical science (116). Recent studies have revealed that autophagy promotes tumor development, invasion, and metastasis in epithelial cells transformed by this oncogene (117). Furthermore, autophagy inhibition in mice with lung tumors induced by k-ras expression was fatal, as specimens died of pneumonia (118). In pancreatic adenocarcinoma models with k-ras mutations, tumorigenicity was associated with increases in IL-1 (interleukin-1), NF-kB, and p62 levels (119).
Many patients manifest metastatic bodies many years after the primary tumor has appeared (120). Autophagy can suppress cell division and motility, thus conserving dormant tumor cells energy. At some point, latent cells can reactivate proliferation and colonize new tissue in response to changes in the tumor microenvironment (121). Anoikis is an apoptotic cell death model triggered by insufficient interaction between the cell and the extracellular matrix, a critical factor for transformed cell invasion and metastasis (122). When cells detach from their matrix to enter the bloodstream, autophagy protects them from anoikis and promotes metastasis (123). On the other hand, autophagy contributes to carcinogenesis by inhibiting apoptosis caused by mitochondrial dysfunction and excessive ROS production (124) (Figure 2).
The tumor microenvironment is the tumor stroma and occupies most of the neoplasm (125). We can therefore hypothesize that as carcinogenesis progresses, the interaction between tumor cells and surrounding stromal cells increases (126). Compared to normal tissues, the tumor microenvironment is characterized by low oxygen levels, high lactate levels, extracellular acidosis, and decreased nutrients (127). It presents great cellular heterogeneity, composed of mesenchymal stem cells, fibroblasts, endothelial cells, immune cells, cytokines, and growth factors (128). Cancer-associated fibroblasts (CAF) are the most studied cells in the tumor microenvironment as they play an active role in tumor promotion (129). These components cooperate to contribute to tumor development (121) (Figure 3, top panel).
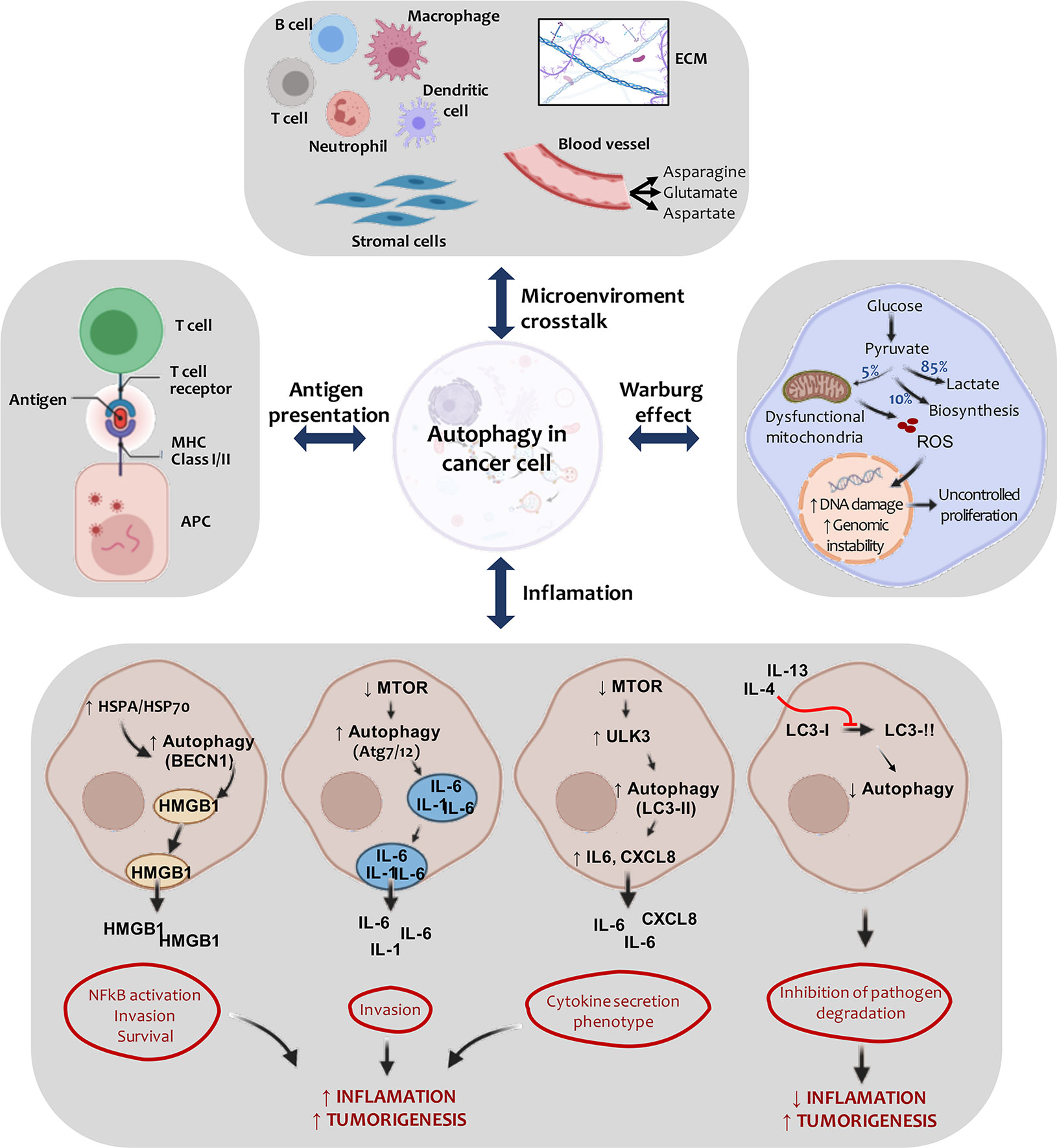
Figure 3 Active crosstalk between autophagy and tumor microenvironment. Carcinogenesis is regulated by autophagy in transformed cells and cells belonging to the tumor microenvironment. Signaling is triggered to the ECM (extracellular matrix) and to stromal cells (such as fibroblasts and pericytes), generating a favorable context for tumor development. As tumor development progresses, cell autophagy activation in the tumor microenvironment serves, in part, to compensate for the inadequate nutritional supply associated with rapidly growing tumors. Here, proper quality control of the mitochondria is necessary to aid glycolysis in tumor cells. In this way, the energy balance (Warburg effect) typically found in malignant cells is maintained. Also, the autophagic process is involved in multiple aspects of lymphocyte development, innate immune signaling and antigen presentation by APCs (antigen presenting cells), processes that are relevant to the disease pathogenesis. HSPA/HSP70 overexpression can induce HMGB1 release in a BECN1-dependent process. This event culminates in the activation of NFκB and promotes tumor proliferation and invasion. Similarly, autophagy induction can contribute to IL-6, IL-1, and IL-8 secretion and promote inflammation. The expression of specific cytokines may inhibit this process, generating a decrease in the inflammatory process.
Autophagy is activated in the tumor microenvironment and in adjacent transformed cells to ensure tissue remodeling, angiogenesis, and interaction with surrounding immune cells (126). Autophagy has been detected in most cells of the tumor microenvironment, but with the ability of performing different actions. For example, when faced with a specific stimulus, the autophagic machinery in fibroblasts promotes tumorigenesis. Simultaneously, in some immune cells, such as cytotoxic T cells, it facilitates immune-response execution against neoplastic cells (130). Under extreme physiological conditions, the stroma activates autophagy to supply energy for adjacent tumor cells (131). In addition, tumor cells can modulate autophagic vesicles’ induction in specific stromal cells (108).
For many years, cancer-related studies focused exclusively on transformed cells, ignoring the tumor environment. At present, the tumor microenvironment is considered to play a fundamental role in tumor development, and its study is essential to form a cohesive idea of what happens within the tumor (132). The extracellular matrix is a fundamental component of the tumor microenvironment. In addition to providing a physical scaffold, it contributes to the secretion of key factors for the tumor’s proper development. Macrophages and fibroblasts associated with cancer are the main cellular models associated with the tumor microenvironment (133).
The bone marrow is the leading site of hematopoiesis and bone formation in most vertebrates as well as the location of inactive and undifferentiated hematopoietic stem cells (HSC) (134). In response to specific stimuli, HSC can differentiate into various blood cells (135). As they have a short life cycle, HSC functional differentiation becomes necessary. During differentiation, cells are exposed to low oxygen levels, a sufficient condition for autophagy induction (136). Recent studies have revealed that autophagy is essential both for self-renewal and for HSC differentiation (137, 138).
After extravasation, monocytes are stimulated by CSF-1 (colony-stimulating factor-1) to induce differentiation. However, this stimulating factor is also associated with autophagy induction through ULK1 activation (139) and PI3K/Akt inhibition (140). CSF-2, another cytokine related to macrophage differentiation, prevents BECN1 and Bcl-2 interaction through a mechanism that includes JNK and triggers autophagy (141). At the same time, G-CSF (granulocyte colony-stimulating factor) contributes to cell survival through apoptosis inhibition and autophagy induction (142).
In recent years, studies related to the role of autophagy and CAFs in the tumor microenvironment have increased (143) and autophagic machinery induction has been associated with the NF-kB pathway and Caveolin-1 (Cav-1) expression in the tumor microenvironment (144, 145). Cav-1 is an essential component of the plasma membrane caveolar, contributing to modulating various signaling pathways. Other scientific sources have revealed a direct relationship between the autophagy induction and BNIP3 (E1B-binding protein 19K/Bcl-2 Nip3) expression in fibroblasts. This induction is accompanied by the loss of Cav-1 expression and the increase in BECN1 and ATG16L (146). Pancreatic stellate cells are a specialized type of fibroblast that can be found in tumor stroma, including PDAC (48, 147). Extensive studies have shown that stellate cells can secrete extracellular matrix molecules and cytokines that contribute to tumor aggressiveness (148). The baseline level of autophagy in PDAC patients is high, and active interaction between the tumor and stromal cell autophagy has been observed (149).
Tumor vasculature is involved in immune cell trafficking and activity. However, it also increases nutrient and oxygen circulation to meet solid tumors’ high energy demands (150). A constant imbalance between pro-, and anti-angiogenic signaling in the tumor microenvironment exists, which contributes to new vessel formation through a VEGF-dependent mechanism (151, 152). Endothelial cells that reside in tumors are exposed to high VEGF levels, nutrient deprivation, and aberrant blood circulation, thus leading to increased autophagy levels (150). In fact, in tumor endothelial cells, autophagy levels are higher than in healthy endothelium (153). Said induction, as mentioned in the previous paragraph, can have anti-angiogenic (154) or pro-angiogenic (155) functions and, therefore, contribute to a different cell fate (156).
Autophagy can induce immune system cells to exert specific responses (157). Recently, autophagy has been shown to influence not only the antigenic profile of antigen donor cells and their ability to release immunogenic signals (158, 159), but also the survival, differentiation, and function of antigen-presenting cells (APC) (160–162).
Innate immunity is the body’s first line of defense against attack by pathogens; it is an active process and it favors the complement system as well as inflammation (163). At the cellular level, the presence of intracellular pathogens is detected by PRRs (pattern recognition receptors) located in the plasma membrane (TLR (Toll-like receptors), 1, 2, 4, 5, and 6), in endosomal membranes (TLR3, TLR7, TLR8, TLR9) or in the cytosol (NOD (Nod-type receptors), RIG-I (gene I-like receptors), RLR (retinoic acid-inducible receptors), and CLR (C-Type lectin-like receptors) (164). PRRs recognize surface antigens of microbes called PAMPs (pathogen-associated molecular patterns), such as lipopolysaccharides of the bacterial cell wall (LPS), flagellin, bacterial, and viral nucleic acids, and finally, some components of the fungal cell walls (165, 166).
After PPRs recognize pathogens, cells can induce autophagy to eliminate them through lysosomal degradation, although this is highly dependent on cellular context and cell type (167). Evidence has shown several TLRs, including TLR1, TLR3, TLR4, TLR5, TLR6, and TLR7, inducing autophagy in humans and mice macrophages (168). The connection between TLR signaling and autophagy is believed to be mediated by adapter proteins TRIF (adapter-inducing interferon-β containing the TIR domain) and Myd88 (Myeloid differentiation primary response 88), which inhibit the interaction between BECN1 and Bcl-2, thus contributing to autophagy induction (169). Furthermore, the link between Myd88 and mTOR has been reported to allow the activation of transcription factors (IRF-5 (interferon regulatory factor 5), IRF-7) that encode for pro-inflammatory cytokine genes and IFN-I (interferon type-I) (170).
Adaptive immunity, on the other hand, produces/makes a more robust and specific response (171), which involves capturing foreign material by APCs (macrophages, B cells, and dendritic cells) to stimulate T lymphocytes and give specific cellular responses (Figure 3, left panel) (172). APCs present antigens to major histocompatibility complex (MHC) molecules through the complex interaction of various cellular factors (173). Autophagy inhibition has been found to reduce MHC-I (MHC-class I) molecules in murine B16 melanoma cells and subsequent cytolysis of tumor cells by CD8+ T cells through cross-presentation (174). CD8+ T cells can respond to exogenous antigens and material undergoing phagocytosis (175). CD4 + T cells recognize antigens from MHC-II molecules (MHC-class II) that are processed in endolysosomal compartments (176). Autophagy may be an essential source of MHC-II antigens derived from intracellular sources through lysosome material supply (177). Like T cells, B cells are regulated by the autophagic machinery. For example, deletion of atg7 or atg5 in the hematopoietic system results in a reduced number of peripheral B cells (178, 179). Dendritic cells are responsible for presenting pathogenic antigens to CD4+ and CD8+ lymphocytes, a process that is favored by autophagy induction (180).
Autophagy can regulate immunity and inflammation in tumor transformation in order to regulate carcinogenesis (181). Cytokine signaling is involved in tumor-associated inflammation and has been linked to promoting tumor-initiating cell self-renewal, tumor growth, angiogenesis, and metastasis (182). Cytokine secretion is variable depending on cancer type, but generally involves IL-1, IL-6, CXCL8/IL-8, IL-10, and interferon-gamma (183). In estrogen-receptor-negative breast tumors, IL-1 expression has been associated with autophagy induction with p62 and LC3 accumulation (184). In liver tissue carcinomas, IL-37 expression regulates autophagy by inhibiting the PI3K/Akt/mTOR axis (185). Some cytokines stimulate autophagy (Th1, TNF-α (tumoral necrosis factor α), IL-2), while others inhibit it (Th2, IL-4, IL-13, IL-10) (121) (Figure 3, bottom panel).
ROS accumulation in tumor development can cause mutations, protein and mitochondrial damage, and increased secretion of inflammatory and antimicrobial agents (186). An increase of intracellular ROS levels is commonly related to inflammatory signaling activation involving NF-kB and regulating the inflammatory response, angiogenesis, and the function of tumor-initiating cells, according to cellular context (187). Furthermore, interaction between NF-kB and the autophagic machinery in order to alter apoptosis and benefit tumor cell survival is common (181). Studies carried out in a murine model of lung adenocarcinoma have revealed that p62 deletion inhibits tumor development through a mechanism that prevents RELA/65 nuclear localization and NF-kB activation (188).
In the 1920s, scientist Otto Heinrich Warburg discovered that tumor cells produce more energy than usual and absorb more glucose than healthy cells through glycolysis regulation, a process known as the “Warburg effect” (189). Numerous scientific reports support the idea that autophagy can generate ATP through a mechanism that involves glycolysis (Figure 3, right panel). Through studies linking these cellular processes, a new paradigm known as the “reverse Warburg effect” has emerged. Stromal cells have been postulated as the key generators of fuel for transformed cells (190). Nowadays, both tumor cells and adjacent stromal cells are believed to contribute to meeting tumor energy needs (191). The importance of autophagy in glycolysis has also been observed in chronic myeloid leukemia and breast cancer cells (192, 193). In mice with K-Ras-driven lung tumors, loss of atg7 leads to defective autophagy, which alters tumor fate, forming benign tumors called oncocytomas (118). These tumor masses with low autophagy levels show defective mitochondria and neutral lipid accumulation (particularly cholesterol esters) due to fatty acid oxidation defects (194).
Mitochondria are central regulators of cell metabolism, which is why they must function correctly. In general, autophagy plays a vital role in the cleaning and quality control of these organelles (195). Both glucose-dependent metabolic pathways and mitochondrial metabolism are essential in tumorigenesis modulation (196). Under hypoxic conditions, pro-apoptotic receptors (BNIP3 and NIX) are activated to induce mitophagy and promote cell survival through HIF-1 regulation (197, 198). In breast cancer cell lines, IGF-I (insulin-like growth factor 1) expression induces BNIP3 expression through a HIF-1-dependent mechanism (199).
Likewise, in glioblastoma cells, PINK1 deletion (the kinase 1 induced by PTEN is a mitochondrial protein of the serine/threonine kinase type) promotes the Warburg effect through ROS and HIF-1 level stabilization. It also reduces PKM2 (pyruvate kinase isoenzyme M2) activity, both regulators of aerobic glycolysis (200). Glycolysis can also be modulated by the interaction between p53 signaling pathways and mitophagy in head and neck squamous carcinoma cell lines (201). Although the molecular mechanism that links these two metabolic regulators has not yet been fully comprehended, Parkin has been found to regulate p53, a ubiquitin ligase that modulates mitochondrial energy metabolism, antioxidant defense, and radiation-induced tumorigenesis (201, 202).
Acetyl-coenzyme A (AcCoA) is a critical metabolic intermediate in autophagy regulation: when its intracellular levels decrease, the autophagic machinery is activated (203). Recent studies in glioblastoma cells have shown that AcCoA increase as a glycolysis product can regulate genes involved in cell migration and adhesion (204). Pancreatic cancers are highly desmoplastic, leading to highly inhospitable environments for cells with high ROS levels, hypoxia, and insufficient nutrient levels (147). Pancreatic stellate cells are a specialized type of fibroblast, commonly found in this type of neoplasm, which contributes to mitochondrial metabolism (205). In the face of stressful conditions, these cells can secrete alanine through an autophagy-dependent mechanism and then be absorbed and used by tumor cells (206). This amino acid fuels the Krebs cycle in PDAC and allows glucose to be used for other anabolic processes such as serine/glycine biosynthesis. Autophagy inhibition in pancreatic stellate cells has recently been shown to decrease tumor growth in transplantation models (149).
HIF-1 and the c-Myc oncogenes coordinated expression regulate cellular glucose transporters, glycolytic enzymes, and mitophagy through choline metabolism (207–209). In B lymphoma cells, c-Myc activates the choline phosphate cytidyltransferase A (PCYT1A) enzyme, inducing mitophagy and preventing cells from dying of necroptosis (210). Arginine is another amino acid that can be dysregulated in cancer cells. Autophagic regulator AMBRA1 (regulator 1 of BECN1 and autophagy) can influence tumor metabolism by regulating c-Myc degradation. When mTOR is inhibited, AMBRA1 is activated. This protein favors the interaction between c-Myc and its phosphatase PP2A (protein phosphatase 2A) to result in the dephosphorylation and degradation of c-Myc, thus reducing cell division rate (211). Cell transformation mediated by c-Myc or RAS-v12 overexpression increases AMPK and FoxO3 expression, which results in increased levels of positive autophagy for BNIP3 and LC3 (212). Finally, the close link between the signaling mechanisms triggered in tumor cells and the adjacent stroma should be taken into consideration; all these events together contribute to tumor metabolism (206, 213).
Autophagy is an essential cellular response element for various types of infections. In general, intracellular pathogens are sequestered and selectively degraded by autophagosomal vesicles (214). However, many pathogens use the host cell’s autophagic machinery to survive and spread (68). Viruses are a good example: once inside the cell, they modulate autophagy to regulate almost all viral life cycle, including insertion and entry of the virus into the host cell, exposure of viral components, and viral protein production (215). Some viruses use autophagosomal membranes as anchors in the replication process, while others inhibit autophagy from avoiding being degraded by lysosomal enzymes in autolysosomes (216). A more detailed examination of the molecular mechanisms modulated during viral infection in relation to the autophagy degradation pathway will be made in the following sections.
Among the various PRRs involved in detecting pathogenic components, TLR receptors located in the plasma membrane and the cytosol stand out (217) (Figure 4A). TLRs activation in endosomes requires PAMPs endocytosis, such as viral RNA, damage-associated molecular patterns (DAMPs), apoptotic cells, or autophagy induction (218). Upon activation, TLRs recruit the Myd88 primary response protein or adapter molecule 1 as an NF-kB activator, contributing to the synthesis of inflammatory cytokines, which in turn trigger IFN production (219) (Figure 4A). NF-kB, IRF-3, and IRF-7 activation can determine the inflammasome assembly and ultimately result in caspase-1 and IL-1β, and IL-18 activation (220). On the other hand, astrocytes infected with three different Zika virus (ZIKV) strains show an increase in inflammatory molecule release (IP-10, IFN-β, NF-kB) and autophagy activation by a mechanism involving TLR3 (221).
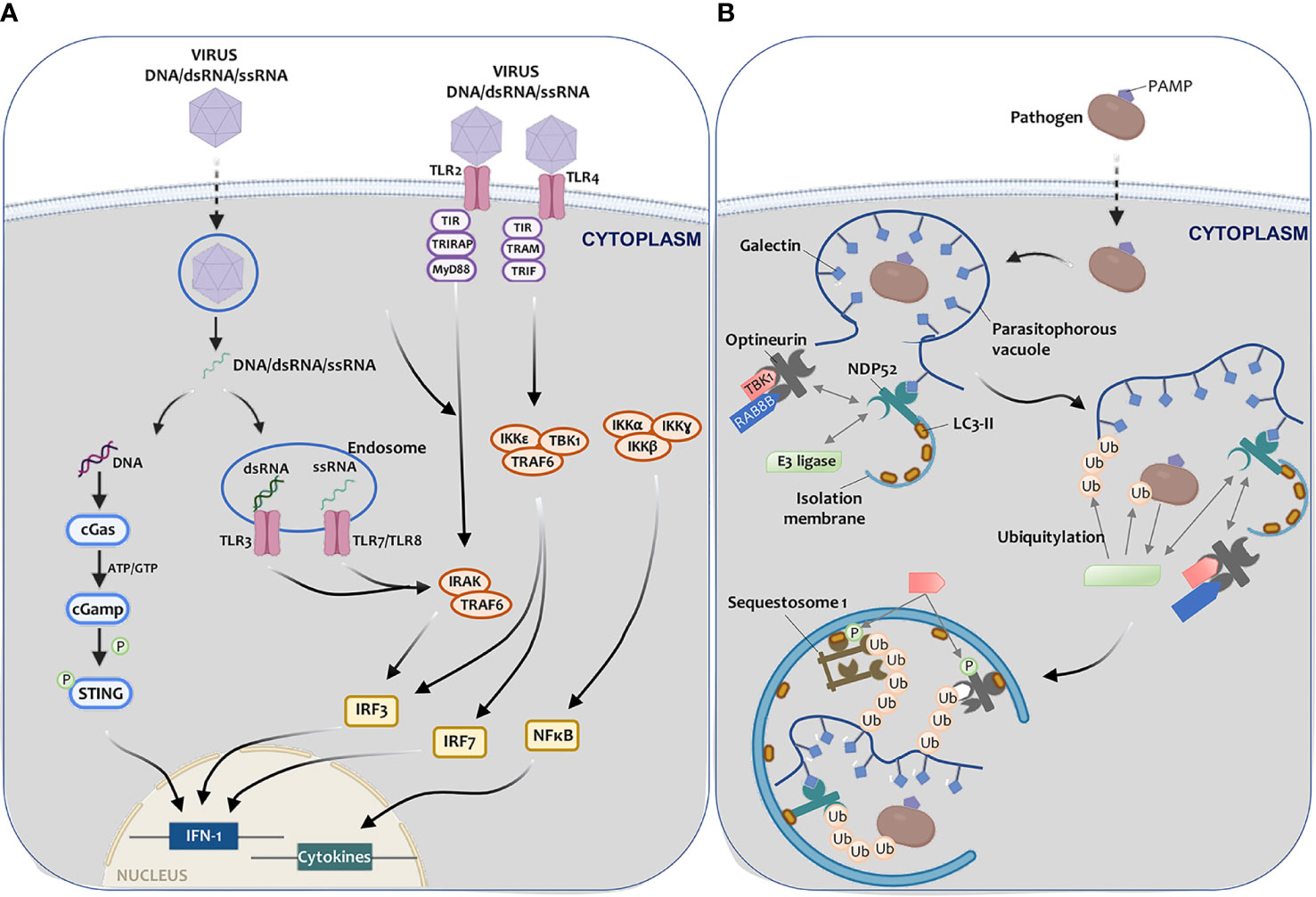
Figure 4 Autophagy-mediated elimination of pathogens. (A) Once a virus enters a cell, (I) viral DNA may be exposed in the cytoplasm, leading to cGAS-STING pathway activation and IFN-I expression, thus triggering the antiviral defense; or (II) viral RNA (endosome) can be recognized by TLR3 (dsRNA) and by TLR7 or TLR8 (ssRNA), triggering IRAK and TRAF6, which leads to IFN-I synthesis. Virus recognition by TLR2 or TLR4 may also trigger the cascade that leads to IFN-I (IRF3, IRF7, or NF-kB) and pro-inflammatory cytokine activation transcription factors. (B) Once pathogens enter the cell, a parasitophorous vacuole with glycosylated molecules starts enclose them. Galectins present in this vacuole bind to NDP52, which interacts directly with E3 ligase LRSAM1 and indirectly with TBK1. TBK1 interacts with optineurin. NDP52 binds to LC3-II, resulting in xenophagy activation. LRSAM1 polymerize ubiquitin at different targets that are yet to be identified. This hypothetical model includes the pathogen and the parasitophorous vesicle as targets. NDP52 recognizes the ubiquitin tags, optineurin, and sequestosome-I. TBK1 phosphorylates optineurin and sequestosome (I) After these steps, the autophagic isolation membrane elongates to capture the pathogen to degrade it.
In mice dendritic cells infected with the human herpes simplex virus-1 (HSV-1), atg5 deletion causes deficiency in the processing and presentation capacity of viral antigens into MHC-II. In this process, TLRs located in endosomal membranes and associated with LC3 have been shown to improve viral antigen processing (222). Moreover, atg5 has been proven necessary for IFN-α production and TLR9 activation through CpG (DNA regions that make up 40% of gene promoters in mammals, with high concentrations of phosphate-linked cytosine and guanine) in HSV-2 infected cells (223).
The second most important PRRs family are the NOD receptors (224), which constitute the cytosolic counterpart of TLRs and include 23 members in humans (225). NOD1 and NOD2 are two receptors that recognize peptidoglycan, a component of the bacterial cell wall; the stimulation of these two receptors forms a transducer complex called the NOD signalosome (226). This signalosome leads to NF-kB activation, which stimulates chemokines and cytokines production, and which in turn initiates the pro-inflammatory response involved in eliminating pathogens (227). Recent studies have revealed an increase in NOD1 and ATG5-dependent autophagy in hepatic ischemia/reperfusion injuries (228). On the other hand, epithelial cell infection with adherent/invasive Escherichia Coli have increased ATG16L1 and LC3 through interaction with NOD2 cytosolic receptor (229).
The CRL receptors family binds to carbohydrates present in pathogens (lectins are proteins that recognize sugars) through a Ca2+ dependent mechanism (230). Type C lectin receptor, Mincle (macrophage-inducible Ca2+ dependent lectin receptor), and TLR4, induce autophagy by activating Myd88 in macrophages (231). RIG-I receptors are characterized by having a C-terminal regulatory domain and a DExD/H helicase domain that contribute to recognizing and unwinding the viral RNA duplex (232). Melanoma differentiation-associated protein 5 (MDA5) is another essential protein in this family. Both proteins detect viral RNA in the cytosol by interacting with mitochondrial signaling through its caspase recruitment domain and regulating autophagy (233).
The last group of receptors includes cytosolic DNA and RNA sensors. An excellent example of these sensors is the cyclic GMP-AMP synthase (cGAS), which plays a fundamental role in recognizing DNA viruses and IFN pathway induction (234). Excessive IFN stimulation can damage the body, so complex regulatory mechanisms have been developed, one of them being autophagy (235). After infection with HSV-1, BECN1 interacts with cGAS, thus altering its nucleotidyltransferase function and triggering the autophagic machinery. When free in the cytosol, cGAS recognizes virus DNA and activates IRF-3 and STING (interferon gene stimulator) to increase IFN production (236) (Figure 4A).
Autophagy can also modulate the adaptive immune response to infection through intracellular pathogens. MHC-I molecules are responsible for presenting pathogenic antigens to CD8+ T lymphocytes in order to trigger different cellular responses, such as endocytosis, vesicle trafficking, and autophagy (237). In the conventional mechanism, the proteasome breaks down these protein antigens into peptide fragments and transports them to the endoplasmic reticulum, where their processing concludes (238) (Figure 4B). Some cells, such as dendritic cells, can present pathogenic antigens to lymphocytes through “cross-presentation” (239). This mechanism can occur through three different signaling pathways (240). In the first case, pathogens are recognized and transported to the proteasome, where small peptide fragments are released and transported to the ER by TAP1 (transporter associated with antigen processing 1) and TAP2 to be presented to HMC-I molecules (240) (Figure 4B). The second pathway is independent of the proteasome, and its lysosomal proteolysis helps facilitate antigen processing (241). Finally, in the last signaling pathway, degradation is proteasomal but independent of TAP (242).
The various death mechanisms involved in dendritic cell cross-presentation have been assessed in comparative studies, which show that atg5 inhibition also inhibited said presentation. This finding has made it possible to associate autophagy with antigens effective presentation to CD8 + T cells (243). Subsequent studies have revealed that autophagy not only influences antigen processing on HMC-I molecules but that is also a prime antigen source for HMC-II molecules, such as CD4 + T cells (244).
Viruses are particles that cannot survive on their own. This is why they have evolved alongside their respective hosts, a process that has given them the ability to use host cell signaling pathways to their advantage (245). To this end, viral particles promote the expression of various viral proteins that mimic host protein structure and function (215). These proteins modulate many cell signaling pathways in favor of viral replication, and autophagy is not exempt from this regulation (246, 247). Viruses with RNA in their genetic material usually contribute to autophagic membrane accumulation, regardless of whether their replication is nuclear or cytosolic (248).
Many viruses that regulate autophagy to facilitate viral survival and replication have been discovered, including poliovirus (249, 250), Coxsackievirus (CVB3) (251, 252), CVB4 (253), Enterovirus 71 (EV71) (254), human rhinovirus (HRV) (255), foot-and-mouth disease virus (FADV) (256), encephalomyocarditis virus (EMCV) (257), dengue virus (DENV) (258, 259), ZIKV (260, 261), mouse hepatitis virus (MHV) (262), Newcastle disease virus (NDV) (263), severe acute respiratory syndrome coronavirus (SARS-CoV) (264), Chikungunya virus (CHIKV) (265), and Japanese encephalitis virus (JEV) (266), among others.
In cells infected with human poliovirus, viral proteins 2BC and 3A promote the formation of autophagic vesicles where viral replication takes place. The virus induces tubular structures in early stages of infection, while forming double-membrane vesicles in advanced settings (267, 268). Recent studies have provided a novel and deeper understanding, by revealing that these viruses can regulate autophagy through a ULK1-independent mechanism (269). During infection with EV71, the ERK inhibition pathway and autophagy impairs viral replication (270).
Measles virus (MeV) belongs to the Paramyxoviridae family and manifests itself mainly in children as high fever, acute respiratory infections and typical papular rashes (271). MeV binds to the host cell through CD46 (CD46 complement regulatory protein), a receptor on the plasma membrane that initiates the autophagic cascade when activated (272). Once active, this receptor binds to the VPS34-BECN1 complex via the GOPC scaffold protein (containing Golgi-associated PDZ and spiral-spiral motif) (273). Virulent MeV samples recognized by CD150 membrane receptors have not shown to have the ability to regulate autophagy in early stages of infection. However, these strains modulate autophagy late in the mechanism to prevent cell death and benefit viral replication (274).
The human immunodeficiency virus (HIV) is a member of the Retroviridae family, affecting more than 30 million people worldwide (275). This human disease progresses towards immune system failure, resulting in infection development or tumor transformation (276), generating alterations in the host’s signaling pathways, and therefore damage accumulation. Critical regulation aspects of the cell cycle are altered, which determine cell transformation and tumor progression, mainly of B cells (277). HIV tat protein, HIV-induced immunosuppression, and a hyperinflammatory state facilitate the oncogenic activity of Kaposi’s sarcoma-associated oncovirus (278). Average survival after HIV infection is estimated to be nine to eleven years without treatment, depending on HIV subtype (279). This retrovirus is transmitted by body fluids and infects CD4+ lymphocytes and macrophages, ensuring prevalence in the host through complex cellular processes (280). In macrophages, autophagy can contribute to HIV degradation or replication. Nef viral protein blocks autophagy initiation by promoting BECN1 binding to Bcl-2 through a PRKN-ligase dependent mechanism (Parkin RBR E3 Ubiquitin Protein Ligase) (281). In studies on CD8+ lymphocytes infected with HIV from patients with lymphoblastic leukemia, the virus was able to inhibit autophagy by reducing ATG8 and BECN1. The opposite result was seen in HIV-infected CD4+ cells (282). Furthermore, an active modulation of ATG1, ATG4D, and ATG5-ATG12 proteins by the virus has been shown. The findings reported so far indicate that HIV can modulate autophagy at different stages to benefit its replication and escape cell degradation (215, 283).
The group of RNA viruses belonging to the Coronaviridae (CoV) family has recently gained relevance. SARS-CoV-2 is the etiological agent of COVID-19, a disease that has had a devastating impact in the past year around the world (284). Before COVID-19, six human CoV pathogens had been identified, two of which are aggressive enough to develop massive infections (285). SARS-CoV infected cells can use the autophagic machinery to degrade viral particles or promote their replication and prevalence. However, more studies are needed to better understand the signaling pathways involved in these viral replication processes (286).
The influenza virus (family Orthomyxoviridae) is another group of viruses implicated in developing human respiratory diseases. Three influenza serotypes have been characterized, only one of them responsible for generating epidemics annually and pandemics at irregular intervals (287). Viral protein M2 is a proton channel that facilitates the acidification of viral particles and allows their decomposition in the host cell nucleus (288). This protein also blocks autophagosome degradation and redirects LC3 to the plasma membrane, generating a cellular redistribution of membranes coupled with this protein. Through this process, these viruses can lead to the formation of filamentous buds, which appear to increase virus stability (289).
CHIKV is transmitted to humans by the bite of some mosquito species and can induce the autophagic machinery through ER stress, increased ROS levels, and reactive nitrogen species (290). Some studies have reported that when the virus is actively replicating, it induces autophagy through the AMPK pathway. However, this has not been observed when the virus is latent (291). Another virus transmitted to humans by mosquitoes is DENV, a Flaviviridae family member that can cause acute or chronic infections (292). These viruses replicate in ER invaginations, so autophagy does not have a structural role in replication. However, cells activate lipophagy to break down cellular triglycerides as well as increase B oxidation and energy production (293). Notably, infections caused by ZIKV, an RNA flavivirus, has generated epidemic outbreaks throughout the world from 2007 to the present (294). A recent study has shown that autophagy can facilitate viral replication through autophagosomal vesicle production or inhibit it in in vivo and in vitro models (295)
Traditionally, cell transformation has been associated with chronic exposure to various carcinogens, such as ionizing radiation and chemical carcinogens, or to genetic predisposition. However, scientific evidence linking viral infections to tumor development has increased (296). Currently, between 15% and 20% of neoplasms are considered to be related to primary viral infections (297). These oncogenic viruses integrate into the host cell genome and utilize host signaling pathways to regulate cell proliferation and differentiation, genomic stability, apoptosis, and immune system recognition (298, 299). Oncoviruses can be classified as direct and indirect carcinogens, although there is some overlap between both. Indirect regulation is related to chronic inflammation modulation contributing to carcinogenesis (300).
Human viral oncogenesis is a complex process in which only a low percentage of individuals develop cancer years after viral infections (301). During this coexistence between the virus and the host, multiple genetic and epigenetic alterations accumulate, contributing to oncogenic pathway dysregulation. In this context, oncoviruses are considered necessary but not sufficient to determine cell transformation (302). Patients with this type of cancer have reduced viral replication, which is required for the cell to actively divide. The virus remains within the cell as a naked nucleic acid in the form of a plasmid or an episome, or it integrates into the cell genome and remains latent (303). Co-evolution of viruses with hosts has shown that the autophagic machinery can be used alongside various proviral and antiviral functions, depending on virus type, cell, and cellular environment (304, 305).
Up to date, eight different oncogenic viruses have been characterized (Table 1) and are described as follows. The human papillomavirus (HPV) and the Merkel cell polyomavirus (MCPyV) are both involved in the development of neoplasms associated with mucosa and skin (306). Hepatitis B virus (HBV) and hepatitis C virus (HVC) are associated with 80% of hepatocellular carcinomas (HCC) (307). The herpes virus associated with the development of Kaposi’s sarcoma (KSHV) and the Epstein-Barr virus (EBV) are mainly viruses associated with endothelial carcinogenesis (308). Cytomegalovirus (HCVM) is another herpes virus that has an onco-modulatory function (309). And human T-cell lymphotropic virus-1 (HTLV-1) is an RNA retrovirus responsible for the development of adult T-cell tumors (ATLL) (310). These viruses can modulate oncogenic responses by regulating autophagy at different points (300, 311) (Figure 5).
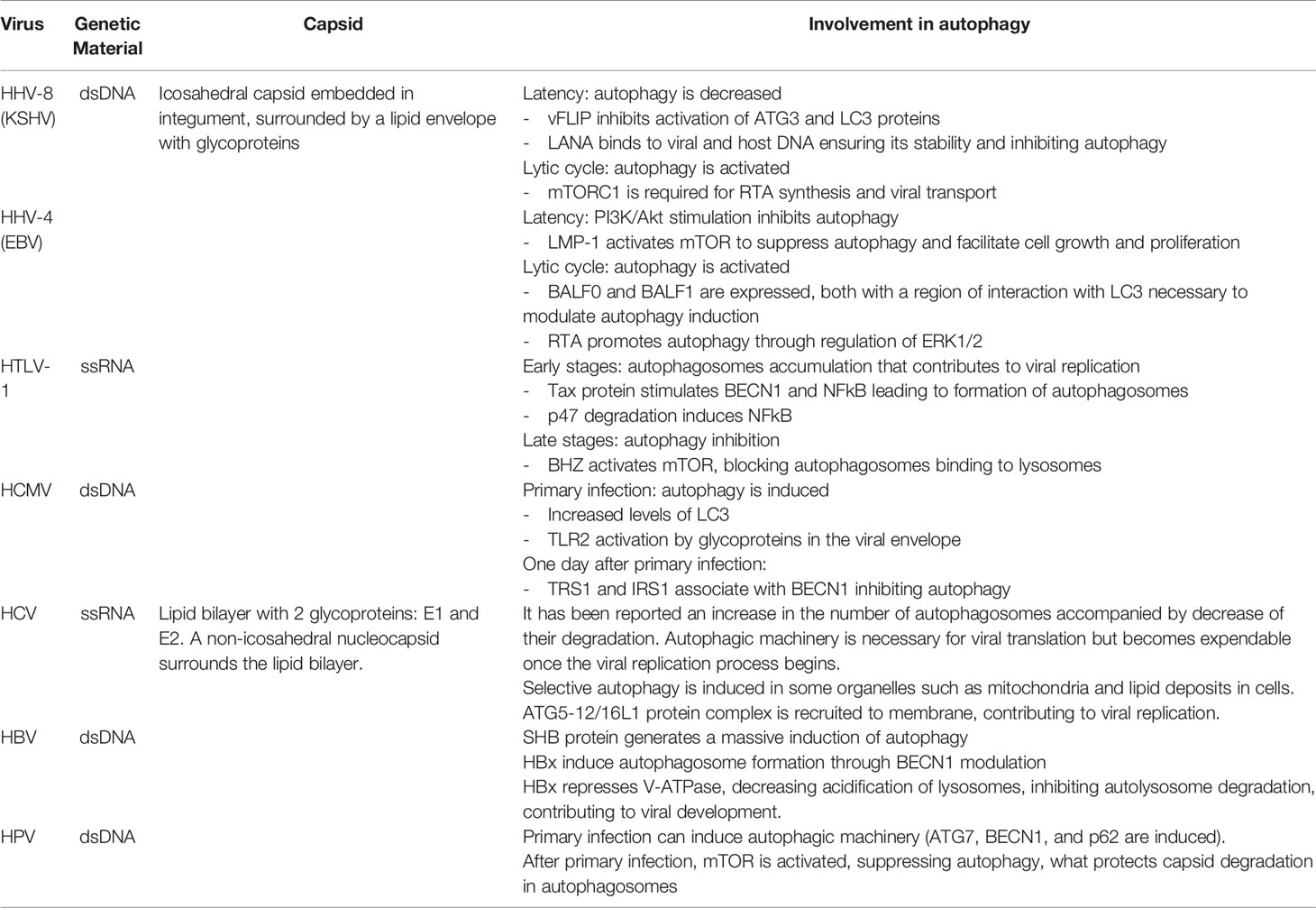
Table 1 Oncogenic viruses and their involvement in autophagy.
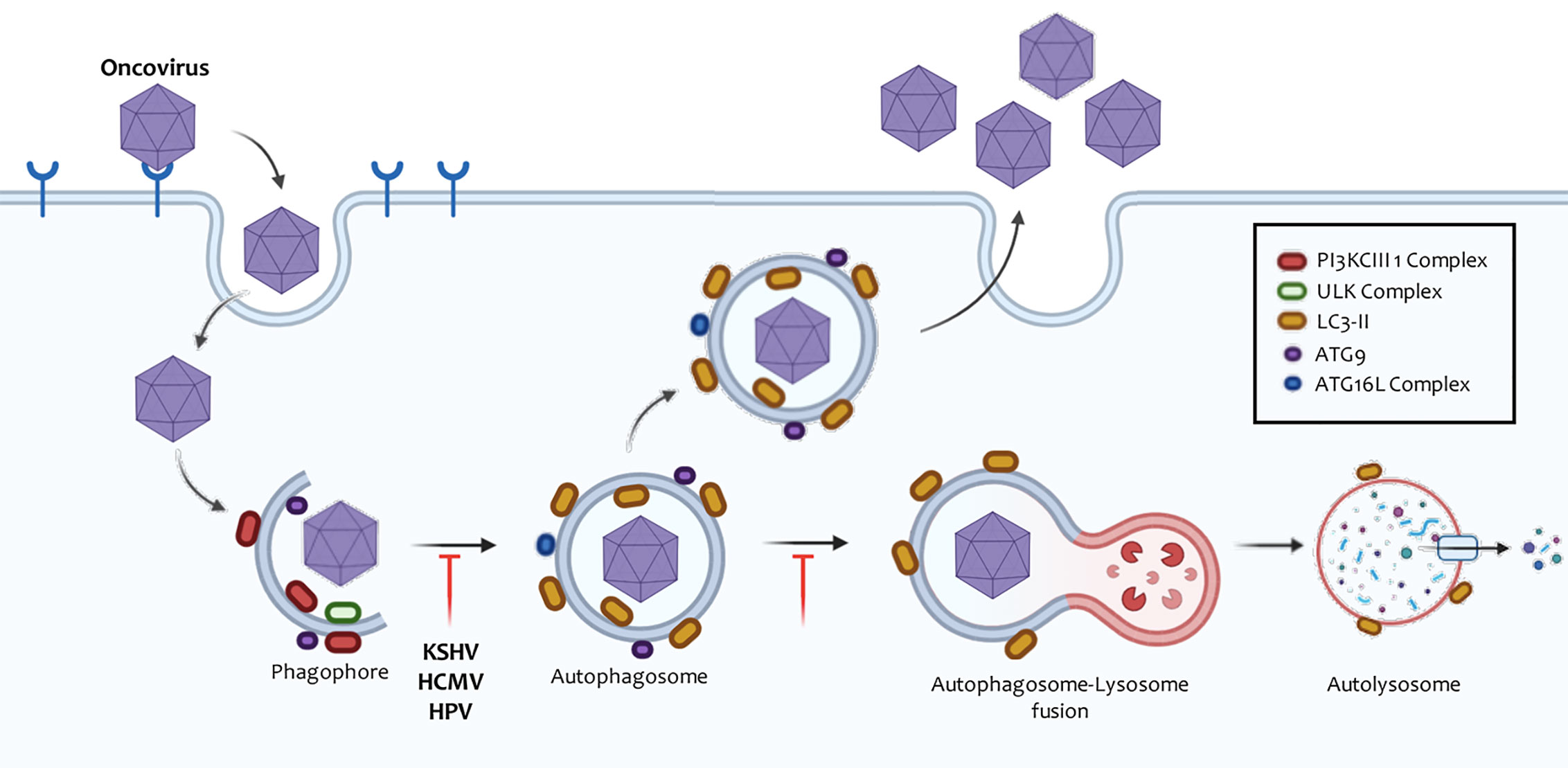
Figure 5 Manipulation of autophagy by oncoviruses. Oncoviruses regulate autophagosome formation or degradation in order to promote their replication cycle. Like many DNA viruses, Herpesviruses prevent their degradation by inhibiting the creation of these vesicles, whereas RNA viruses often induce the accumulation of autophagic membranes independent of their replication in the cytosol or the nucleus. These autophagic membranes can form autolysosomes and finally conclude. In other cases, the autophagic membranes may be used as scaffolds, where the viral replication complexes are positioned or serve as support for the release of the particles. Poliovirus was the first virus where this mechanism has been evidenced.
Herpesviruses are biological structures that have linear double-stranded DNA, ranging from 100 to 200 kb. The viral genome is packaged in an icosahedral capsule embedded in a protein integument and surrounded by a host-derived lipid envelope (312). The viral envelope has glycoproteins that mediate the union with the cytoplasmic membrane of the host for subsequent entry of the virus accompanied by the nucleocapsid and the integument. The capsid travels through microtubules until it reaches the nucleus, coupled with protein complexes that allow nuclear pore opening. It then injects its genetic material into the nucleus (313).
Eight herpesviruses have been identified in humans with a common evolutionary origin, classified according to their genomic sequence and biological characteristics into three subfamilies (alpha, beta, and gamma) (314). Within the gamma-herpesviruses, we can find human herpesviruses 4 (EBV) and 8 (KSHV). These viruses have a high affinity for B lymphocytes, and both establish latent and lytic infections that promote the development of hematological diseases and various types of solid human cancers (315). Surprisingly, the ability of viruses to regulate autophagy can vary according to infection stage (316). HCMV is another herpes virus that primarily infects fibroblasts, but which can also be found in epithelial cells, endothelial cells, hepatocytes, stromal cells, monocytes/macrophages, astrocytes, and neural stem/progenitor cells (309).
KSHV was discovered in 1993 from a tissue sample of a patient with Kaposi’s sarcoma (KS) (317). This virus is generally transmitted by body fluids, although transmission has been observed through the placenta to the fetus in some rare occasions (318, 319). KSHV seroprevalence is estimated to range between 5%–20% worldwide. Yet, only a small number of patients develop secondary diseases associated with the virus, the population mostly at risk being immunosuppressed individuals or individuals with immune system abnormalities, as is the case of AIDS patients (320). KSHV infection is associated with the development of various human pathologies, including Kaposi’s sarcoma, primary effusion lymphoma (PEL), multicentric Castleman’s disease (CMD), and inflammatory cytokine syndromes (KICS) (321). The main reservoir of this virus in its latent form are B lymphocytes, but it can also infect monocytes, fibroblasts, endothelial, epithelial, and dendritic cells through association with receptors on the plasma membrane (322).
After primary infection, the virus remains in the cell as an episome and regulates various host signaling pathways to replicate correctly. The virus produces proteins associated with the viral latency phase which are essential for cell transformation (323). Some of the encoded proteins are complement-fixing proteins (v-CBP), viral interleukin-6 (v-IL-6), viral inflammatory protein type-I (v-MIPI) and type-II (v-MIPII), viral Bcl-2 (v-Bcl-2), viral interferon regulatory factor (v-IRF), viral cyclin (v-Cyclin), latency nuclear antigen (LANA), viral adhesin (v-ADH), receptor-coupled G-protein (v-GCR), thymidylate synthetase, thymidine kinase, ribonucleotide reductase (300, 324)
In various models, autophagy has been found to be a cellular mechanism commonly regulated by viral KSHV proteins (325). After initial expansion at the infection site, the new viral particles spread throughout the body and reach the cells in order to establish a latency phase, especially in the B lymphocytes and endothelial cells of the blood/lymphatic vessels (326). During the latency phase, autophagy and other cellular mechanisms contribute to creating a cellular microenvironment favorable to tumor initiation and progression (321).
The transmembrane glycoprotein K1, encoded by the first KSHV open reading frame, is a signaling protein capable of causing B cell activation (327). V-cyclin and K1 have been found to promote autophagy by stimulating the AMPK pathway (328, 329) (Figure 6). On the other hand, vFLIP restricts the autophagic machinery by inhibiting ATG3 and LC3 proteins (330) (Figure 6). Once viral latency is established, LANA plays a fundamental role in maintaining this phase through NF-kB activation (331). Granato et al. observed viral particles inside autophagic vesicles in the cytoplasm of PEL cells in active replication, thus postulating that autophagy may also be related to viral transport (332). Finally, another protein linked to viral latency is STAT3 (signal transducer and activator of transcription 3), which remains active in a state of viral latency (330). In dendritic cells, KSHV infection induces STAT3 phosphorylation, promoting cell survival and viral latency. Moreover, the release of IL-10, IL-6, and IL-23, cytokines that contribute to keeping STAT3 active, is also induced (333). This allows the viral genome to remain unchanged and the particles to replicate successfully (334).
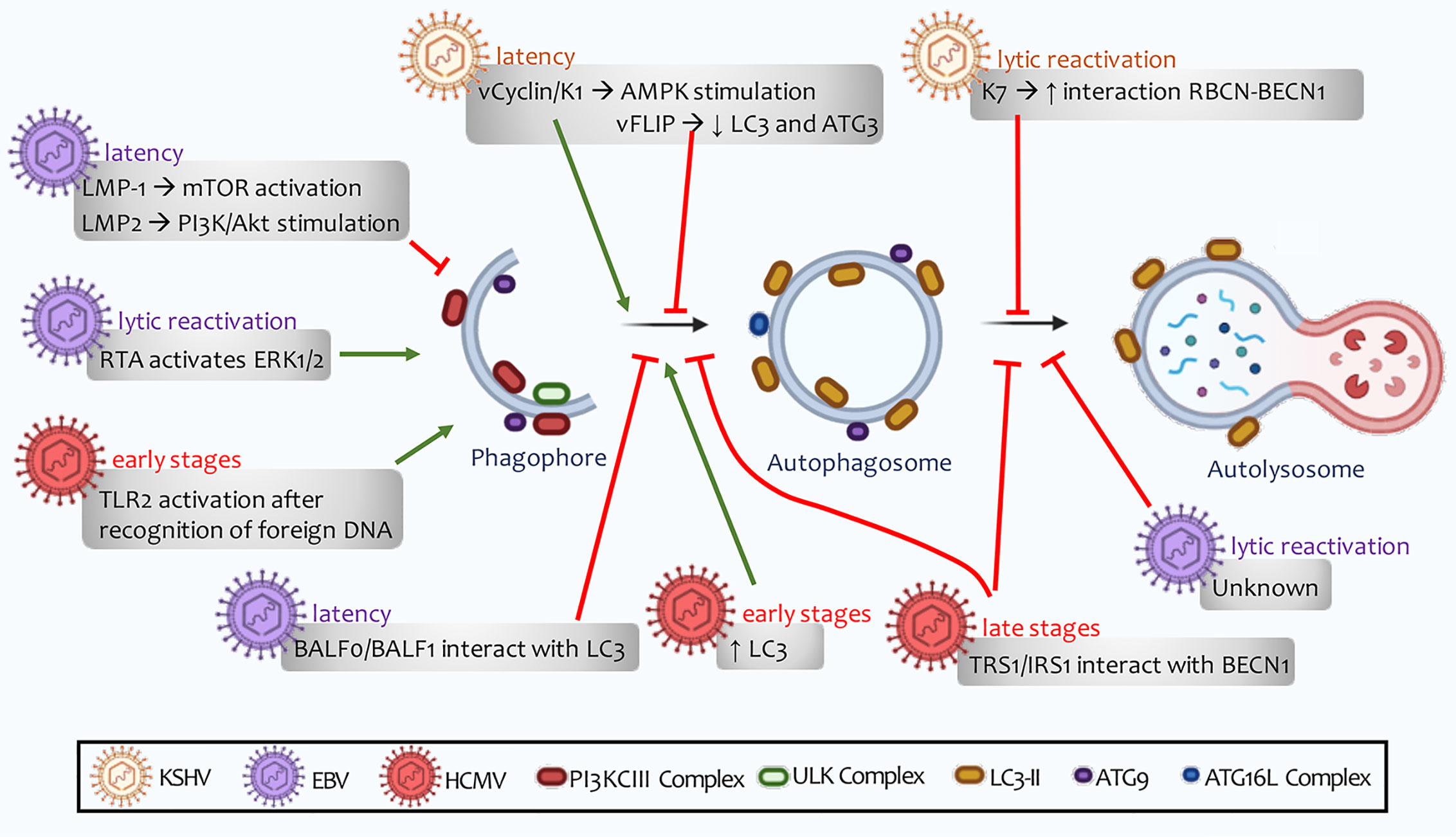
Figure 6 Human Herpesvirus regulation of autophagy. The viral ability to regulate autophagy depends on stage of infection. This regulation can alternate between latent, de novo, or lytic reactivation infections. A distinguishing feature of these lymphotropic viruses is their ability to promote autophagy induction, a process followed by inhibition of autophagosome maturation. This defective autophagy does not allow autolysosomes to form, and therefore to perform successful clearance.
Numerous cellular events can activate lytic KSHV reactivation. Cellular microenvironment can shape the viral epigenome to facilitate latency reactivation (335). ER stress can induce rad21 cleavage, a member of the cohesin complex that generates dramatic changes in the KSHV genome. The loss of DNA loops triggers virus reactivation (336). During this process, cells activate the autophagy machinery with various functionalities (335).
A fundamental component for lytic reactivation is the expression of lytic switch master proteins (RTA), targeting 100 different sites in the KSHV genome and transactivating 34 lytic genes (337). Pringle et al. have reported that mTORC1 is required for lytic replication and RTA synthesis as an activator of cellular transcription. In contrast, this autophagy inhibitor complex has not shown more significant involvement in genomic replication, late gene expression, or in the release of infectious progeny (338). Furthermore, autophagy inhibition has been reported to reduce lytic KSHV reactivation (339).
In contrast to the nuclear LANA function, cytoplasmic isoforms of this viral protein mediate lytic reactivation by antagonizing cellular DNA sensors. These isoforms bind to cGAS, a process that involves STING and NF-kB induction (340). Viral protein K7 expression stimulates RBCN (Rubicon autophagy regulator) interaction with BECN1. These events promote the blocking of autophagosome maturation (333) (Figure 6). KSHV monocyte infection counteract ROS increase induced by macrophage colony-stimulating factor (M-CSF), preventing JNK and Bcl-2’s phosphorylation and inhibiting autophagy. Together with the decrease in TNFα and the increase in the immunosuppressive cytokine IL-10, all these events lead to impaired macrophage survival and differentiation (341). Findings so far allow us to infer that these viruses may induce autophagosomes formation, in which they are transported to the cell surface. Viral proteins inhibit lysosome-to autophagosome-binding and, therefore, autolysosome formation is not degraded by lysosomal hydrolases (330) (Figure 6).
EBV is a very easily transmitted herpesvirus that is mainly contracted in childhood through body fluids. 90% of the world’s population is believed to have been infected at some point of their lives (342, 343). This virus has contributed to the development of various secondary pathologies, such as infectious mononucleosis and some neoplasms of epithelial and lymphocytic origin (344). After entering host cells, viruses amplify and rapidly enter a state of latency. A distinct fact is the presence of three different latency types that can be independently regulated, autophagy playing an essential role in this regulation (345, 346). Each cell presents multiple copies of viral DNA episomes and produces a series of proteins associated with latency, including six nuclear antigens (EBNA 1, 2, 3A, 3B, 3C, and -LP) and three latent membrane proteins (LMP1, 2A and 2B) (347).
LMP2 viral protein has been reported in most neoplasms associated with this virus. This protein stimulates the PI3K/Akt signaling pathway, a process that triggers apoptosis and autophagy inhibition, thus contributing to tumor cell proliferation (348) (Figure 6). This regulation has been seen in gastric carcinomas associated with EBV infection, where the presence of PI3K mutations has been associated with higher tumor occurrence and metastasis (349). Furthermore, LMP2 has been found to contribute to cell proliferation through p27 degradation (350, 351).
Additionally, LMP1 binds to membrane-bound death receptors TRAF and TRADD (tumor necrosis factor receptor type 1-associated death) or activate signaling pathways that include NF-kB, JNK, p38, small GTPases (Cdc42), and the JAK/AP-1/STAT cascades. Moreover, LMP-1 has been reported to activate mTOR to suppress autophagy and facilitate tumor growth and proliferation (352) (Figure 6). In contrast, Hurwitz has shown that LMP-1 can inhibit mTOR by secreting CD63-dependent vesicular proteins, contributing to autophagy induction. This induction is not complete since LMP1 inhibits lysosomes’ binding to autophagic vesicles in order to avoid the viral particles’ degradation by lysosomal hydrolases (353). On the other hand, EBNA3C nuclear antigen activates autophagosome formation through transcriptional induction of several autophagy regulators, including ATG3, ATG5, and ATG7 (354).
During the EBV lytic cycle, autophagy may present bimodal modulation, showing an early stimulation phase in combination with the inhibition of the late phases of the autophagic mechanism (degradation of cytoplasmic material by lysosomal hydrolases) (355). This final regulation favors the acquisition of envelopes and components of the autophagic machinery by newly synthesized virions (356, 357). The viral proteins associated with this virus that regulate autophagy in the EBV lytic cycle have not yet been fully characterized. RTA function, an early expression protein regulating autophagy through an ERK-dependent mechanism, has been recently highlighted (358) (Figure 7).
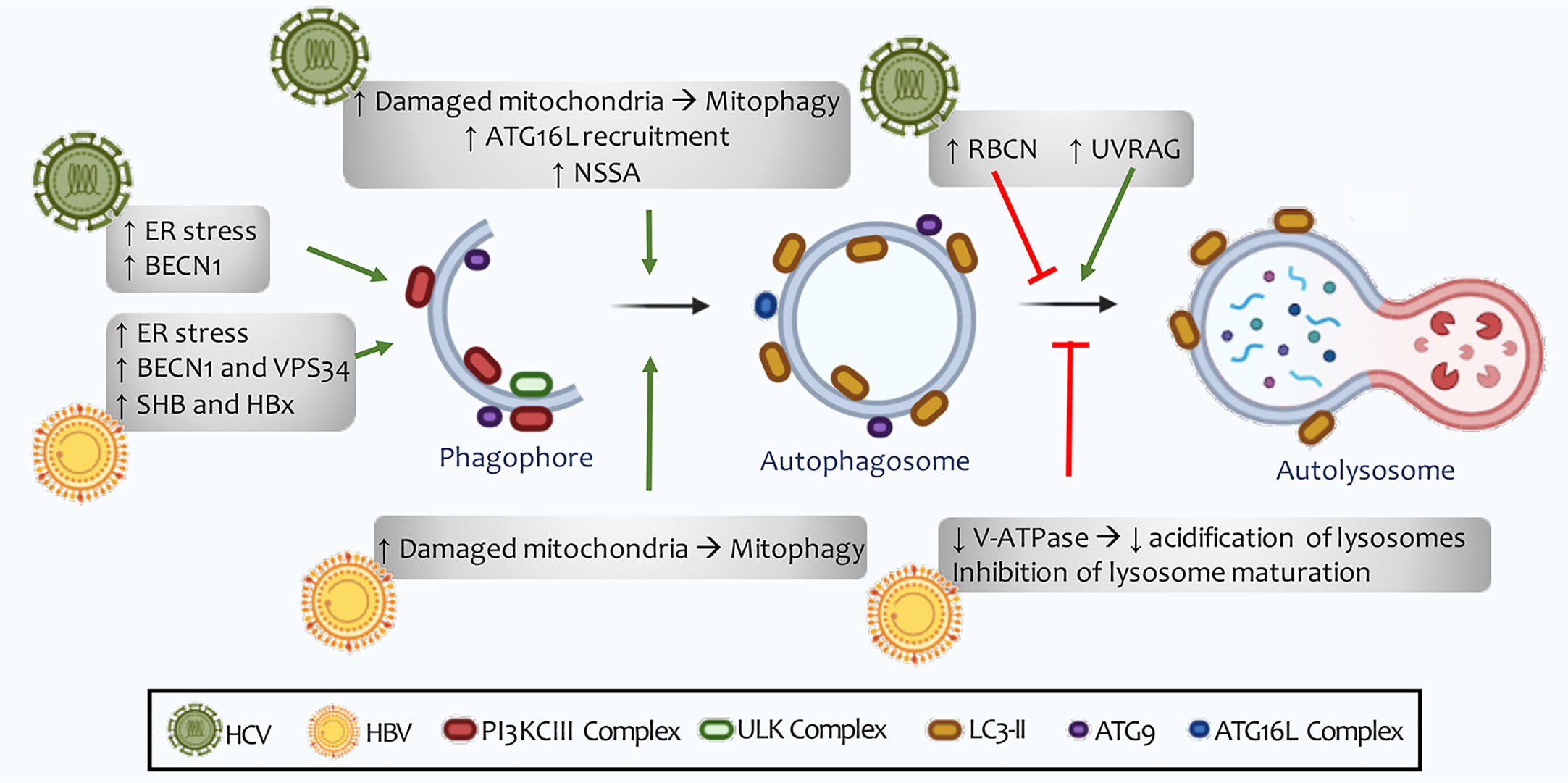
Figure 7 HCV and HBV regulation of autophagy. The autophagic machinery is necessary for incoming viral RNA translation, but it becomes expendable once the viral replication process begins. An increase in the number of autophagosomes does not correlate with an increase in the rate of lysosomal degradation. HCV has been shown to induce the expression of Rubicon, thus inhibiting the maturation of autophagosomes. For its part, HBC reduces the acidification of lysosomes and autolysosomes through the inhibition of V-ATPase (H + type vacuolar enzyme). However, in vitro studies show good fusion of autophagosomes with lysosomes, as well as successful autophagic flow, indicating that these viruses are able to induce selective autophagy in some cellular organelles, such as the mitochondria and lipid deposits in cells.
EBV codes for the expression of two Bcl-2 homologous viral proteins called BHRF1 and BALF0/1 (359, 360). Both viral proteins prevent apoptosis during early B-cell infection but may be dispensable once the latent infection is established (361). BHRF1 anti-apoptotic activity has been studied extensively (362). However, BALF0/1 expression and function remain ambiguous. Two in-frame methionine codons are present near the beginning of the BALF0/1 open reading frame (ORF), suggesting that two proteins with different N-termini may be encoded (363). So far, the BALF1 protein is known to be encoded by the shortest ORF, while the BALFO protein is encoded by the first non-conserved methionine. During the lytic cycle initial phase, BALF0 and BALF1 are expressed, both with a region of interaction with LC3, and modulate autophagy induction (364). So far, we can infer that EBV inhibits autophagy during the latency phase in its initial steps. Simultaneously, during lytic reactivation, the formation of dysfunctional autophagosomes is promoted (Figure 6).
HCMV is a double-stranded DNA herpes virus transmitted through body fluids, not prevalent in any particular age range. This virus has an extensive genome of 236 kilobases, one of the largest viruses that can infect humans (365). Primary infections are generally asymptomatic, although congenital diseases can lead to various severe disabilities or fetal death (309).
HCMV particles have been detected in different cell types, including epithelial cells, connective tissue, hepatocytes, various populations of leukocytes, and vascular endothelial cells (366). HCMV also infects tumor cells and contributes to transformation when affecting healthy tissue by modulating various cellular signaling pathways (309). This virus regulates autophagy in a bimodal manner (367). First, during early stages of infection, it induces autophagic vesicle formation. Later in infection, HMCV blocks autophagy through viral proteins synthesized in the host cell (368). Two proteins involved in autophagy inhibition by association with BECN1 (TRS1 and TRS2) have been identified (Figure 6). Mouna et al. has found that co-expression of viral proteins TRS1 and IRS1 is essential for autophagy inhibition in various cell models (369).
Recent attention has been given to viral components that determine HMCV latency and lytic reactivation, with special focus on the uLb’ gene locus (ul133-138) that restrict viral replication by modulating viral latency and immune evasion through the expression of a considerable number of viral proteins (370). An example of this is UL138: this viral protein is presented to HMC-I to regulate the host’s adaptive immunity in fibroblast, and the autophagic machinery holds this event (371). In contrast, it was reported that autophagy inhibition generates a high response of CD8 + lymphocytes due to the internalization of molecules in MHC-I (372).
Early expression of viral proteins associated with HMCV genes 1 and 2 (IE1 and IE2) is necessary for lytic reactivation of host cell virus and immunomodulation (373). IE2 can interact with itself and UL84 as well as with many specific cell transcription factors to regulate gene expression. This protein plays a critical role in viral DNA synthesis and is also considered to counteract host response (374, 375). IE2 overexpression has been recently found to induce autophagy in HMCV-infected cells (376). These results show that in the early stages of HCMV infection, viral proteins contribute to autophagosomal vesicle formation. At the same time, they inhibit vesicles-to-lysosomes binding in later stages, thereby losing their degradative capacity (Figure 6).
HTLV-1 is a member of the Retroviridae family and is represented by a small single-stranded RNA genome approximately nine kilobases long (377). This retrovirus was first isolated in 1979 in samples from patients with cutaneous manifestations of rapidly growing T-cell lymphoma (ATLL) (378). Furthermore, HTLV-1 is associated with the development of poliomyelitis, HTLV-1 associated myelopathy, infectious dermatitis, arthropathy, and facial nerve palsy (379). Evidence suggests that there are between 5 and 20 million HTLV-1 carriers worldwide, but only 3-5% of them develop secondary pathologies (380, 381).
Immediately after entering the host cell, the viral RNA undergoes reverse transcription, and then binds to the cell’s genome as a provirus. This binding generally occurs in areas close to the binding sites of transcription factors such as STAT1, TP53, and HDAC6 (histone deacetylase 6). Dysregulation generates alterations in cell signaling in the expression of specific genes and autophagy is not exempt from this viral regulation.
Regulatory protein HTLV-1 Tax is an oncoprotein that plays an essential transcriptional role in viral replication and participates in T lymphocytes’ transformation. It can also transactivate or transrepression more than 100 cellular genes by linking and modulating stability and activity (300). Recently, HTLV-1 infection has been reported to induce autophagosomes in cells and inhibit their binding to lysosomes through a tax-dependent mechanism. In this way, the number of non-degrading autophagic vesicles, where viruses can replicate, increases considerably (382). Therefore, Tax viral protein, located in the plasma membrane’s lipid microdomains, binds to the IKK complex to stimulate BECN1 and NF-kB activity (383).
Cell adhesion molecule 1 (CADM1) is a member of the immunoglobulin superfamily and is considered to be an excellent cell surface marker of HTLV-1 infected T cells (384). For CADM1 to be correctly expressed, Tax and NF-kB induction and p47 (a negative NF-kB regulator) degradation are necessary. Autophagy is p47 primary degradation mechanism, and it is active in most ATLL cells infected with HTLV-1 (385).
Another essential viral protein for ATLL development is HBZ (bZIP factor) (303). HBZ inhibits both apoptosis and autophagy and may induce the expression of brain-derived neurotrophic factor (BDNF) and its receptor (386). According to the subcellular location of HBZ, it contributes to tumor progression (nuclear) or favourably contributes to inflammation induction (cytoplasmic) (387). When HBZ is exported from the nucleus to the cytoplasm, it activates mTOR through the DNA damage-inducible protein PPP1R15A (a regulatory subunit of protein phosphatase -1 15A) (386). As in other viral infections, HTLV-1 induces autophagosome formation and inhibits binding to lysosomes to prevent degradation. Consequently, the number of autophagosomal vesicles in the cytosol increases, creating a physical obstacle for developing other cellular processes and a favorable environment for viral particle formation (388).
HCV belongs to the Flaviviridae family, characterized by viral particles with an RNA helix of 9.6 kb in length and wrapped in a lipid bilayer with two anchored glycoproteins (E1 and E2). In general, it presents a non-icosahedral nucleocapsid, although it is possible to find viruses without nucleocapsid in infected patients’ blood (389). Chronic HCV infection can trigger liver fibrosis and cirrhosis, and it is also associated with hepatocellular carcinoma (HCC) and non-Hodgkin lymphomas development (390). This virus can promote carcinogenesis directly through the modulation of specific signaling pathways and indirectly through chronic inflammation (391).
Once inside the host cell, the virus forms a membranous network used during replication (392). Three membranous rearrangements associated with the virus have been identified: cluster vesicles, contiguous vesicles and double-membrane vesicles. In general, cluster vesicles are associated with viral infection early stages. As the infection progresses, the number of double-membrane vesicles also increases (393).
Some viral proteins (CORE, NS2, NS5B, NS3, NS5A) directly benefit carcinogenesis through the induction of proliferation, angiogenesis, apoptosis, immune response, and inhibition of tumor suppressors (303). HCV can induce autophagosome formation and inhibit binding to lysosomes (311) (Figure 7). However, some in vitro studies have revealed that the virus can cause successful autophagic flux. This conflicting result may be explained by HCV probably inducing selective autophagy in some cellular organelles, such as the mitochondria and lipid deposits in cells (394).
Furthermore, the ATG16L complex is recruited into the membranous network, contributing to viral particle replication, and subsequent ATG12 removal suppresses viral RNA replication. Autophagy activation can counteract HCV infection, and the virus has developed different strategies to strengthen its persistence by temporarily regulating the autophagic process (395). The autophagic machinery is necessary for the translation of viral RNA in early stages of infection but becomes dispensable later. Once the replication process is complete, autophagy contributes to releasing viruses to the extracellular space, thus benefiting HCV transmission (300).
HCV infection has been found to induce autophagy through the direct interaction of viral proteins with autophagy effectors. In contrast, HCV has been shown to induce stress autophagy of the endoplasmic reticulum by inducing the three response pathways to misfolded proteins (p-ERK, ATF6 (activating transcription factor 6), IRE1) (396) (Figure 7). Reticulum stress produces calcium release, which disrupts mitochondrial activity and leads to ROS accumulation and damaged mitochondria. This leads to mitophagy activation through NS5A expression. This HCV non-structural protein causes an increase in LC3 levels and reduces p62 in host cells (397) (Figure 7). In patients with chronic HCV infections, mitophagy was observed to be induced due to PINK1 and Parkin’s translocation to the mitochondria outer surface. This mechanism correlates with oxidative phosphorylation dysregulation and mitochondria depletion, contributing to liver injury (398). HCV modulates the autophagic machinery in order to exist in the host cell. Like other viruses, it increases the formation of dysfunctional autophagic vesicles within which it replicates. Furthermore, in tumors associated with this virus chronic infections, high levels of mitophagy were presented (Figure 7).
HBV belongs to the Hepdnaviridae family and is responsible for developing acute and chronic viral hepatitis as well as long-term complications ranging from fibrosis and cirrhosis to cancer. Chronic infection progression is predominant in infected patients during the perinatal and infantile periods (399). Two billion people are estimated to be infected with HBV worldwide, and more than 350 million to be chronic carriers. Only 25% of infected patients develop liver neoplasms (300).
These viruses present circular double-stranded DNA of 3.2 kb in length, which has four open reading frames and has the peculiarity of forming an incomplete chain (400). The end of one strand is associated with viral DNA polymerase (401). Immediately after entering the cell, the virus undergoes reverse transcription within the nucleocapsid. Consequently, linear DNA is formed and secreted as virions or transported to the nucleus, integrating into the host genome to regulate tumor transformation (402).
HBV has a complex replication cycle and needs to encode at least seven viral proteins that regulate different processes in the host cell in order to complete it successfully (403). HBx (hepatitis B virus protein x) and SHB (small surface protein) expression is associated with autophagy regulation in several biological models (404) (Figure 7). SHB protein travels through the cytosol to the endoplasmic reticulum, where stress increases and generates massive autophagy induction. Signaling pathways initiated by stress sensors such as IRE1, p-ERK, and ATF6 help regulate cell homeostasis (405).
As for HBx, it is a multifunctional regulatory protein that may be involved in viral pathogenesis and carcinogenesis (406). Molecular studies have revealed that HBx generates strong autophagy induction by activating DAPK (death-associated protein kinase) and increasing BECN1 (407). However, in later stages of the autophagic mechanism, HBx can repress V-ATPase (vacuolar enzyme type H+), thus reducing lysosome and autolysosome acidification (408) (Figure 7). As they become less acidic, autolysosomes lose their degradative capacity as cytoplasmic debris increases in the cell, creating an inhospitable environment that contributes to carcinogenesis (408). The virus can also interfere with autophagic degradation through RAB7 (Ras-related protein), a small GTPase involved in autophagosome maturation and their fusion with lysosomes (409).
HBV-infected patient biopsies showed that viral persistence is correlated with the expression of mitophagy effectors, Parkin, and PINK (410). Furthermore, this mechanism is believed to be regulated by the AMPK/mTOR/ULK1 axis (411). Liver cells infected with HBV show that miR-155 expression contributes to viral replication and enhances autophagy induction (412). At least four miRNAs have been identified to inhibit viral replication in clinical samples from virus-infected patients, (let-7, miR-433, miR-345, miR-511) (413).
Liver cancer is a global concern due to its high resistance to chemotherapeutic drugs, which has been linked to exosome formation in transformed cells (414). These structures increase drug resistance by inducing chaperone-related signaling pathways and LAMP-2-dependent (type-2 lysosomal membrane protein) autophagy. Patients with liver tumors associated with primary HBV infection show greater tumor volume and greater pathogenicity. This suggests that the virus contributes to generating a more aggressive and resistant HCC phenotype (415).
Therefore, we can infer that HBV generates bimodal autophagy regulation: first by inducing the mechanism and then by inhibiting the formation of mature autolysosomes, thus contributing to viral replication and carcinogenesis (Figure 7).
Papillomaviruses (HPV) and human polyomaviruses (PyV) were initially considered members of the same virus family due to their morphological similarity and genome organization, but have now been classified into separate families: Papillomaviridae and Polyomaviridae, respectively (416), both composed of viruses with a double-stranded circular DNA that encodes various regulatory and structural proteins, some of which have oncogenic properties (417).
Merkel cell polyomavirus (MCPyV) is implicated in developing a very aggressive skin cancer called Merkel carcinoma (418). Most findings about MCPyV oncogenic potential are associated with LT (large antigen T) and sT (small antigen T) expression (419, 420). LT has a J domain (heat shock protein binding domain), a retinoblastoma binding motif (RB, inhibits members of this protein family), and a C-terminus binding to helicase/ATPase domain (required for viral DNA replication) (418). LT’s oncogenic role is mainly associated to its high binding affinity to RB, which promotes the sequestration and inactivation of this tumor suppressor (421).
Regarding the sT antigen, it shares a 78 amino acid N-terminal region (includes the J domain) with LT and has a unique C-terminal domain. This antigen is considered to be the main MCPyV-induced oncogenesis regulator (422). LTs unique expression is sufficient to transform fibroblasts in vitro (423) and in vivo models (424). sT keeps the eukaryotic 4EB-P1 binding protein hyperphosphorylated and inactive, which leads to deregulation of cellular translation events and contributes to cell proliferation and transformation (422). Through these surface antigen expression, viruses modulate the autophagic machinery and contribute to immunosuppression and viral oncogenesis. In MCC tumors associated with MCPyV infection, sT and LT antigen expression suppresses autophagy through miR-375, miR-30a-3p, and miR30a-5p gene modulation. These microRNAs act on atg7, p62, and bcn1 to inhibit autophagic initial stages. These events protect cancer cells from cell death (425).
Human papillomavirus (HPV) is the leading cause of cervical cancer (426). It is also associated with the development of non-melanoma skin cancers, cancer of the head, neck, oropharynx, and the development of various anogenital neoplasms (427). The viral genome is integrated into the host cell’s DNA and encodes the expression of early viral genes (E1 to E7) essential for cell replication, transcription, and transformation. These viruses can also regulate the expression of late genes L1 and L2, which encode viral capsid proteins (428).
Binding and internalizing the virus are processes closely related to manipulating the host cell autophagic machinery. The entry of HPV into cells is associated with autophagy suppression through mTOR activation (429). This event promotes mTORC1, 4EB-P1, and S6K1 (ribosomal protein kinase 1) phosphorylation and activation (430) and ULK1 inactivation. This -in turn- contributes to inhibiting the initial steps of the autophagic mechanism (431).
Once internalized in cells, HPV circulates through an endosomal compartment where capsid proteins are degraded within acidified endosomes, and the viral genome enters the nucleus (432). Here, HPV DNA is amplified and maintained as episomes in the epithelium basal cells by mechanisms mediated by E1 and E2 (433). These early expression proteins are involved in various cellular signaling mechanisms. In particular, E5, E6, and E7 oncoproteins modulate the host cell autophagic machinery. Keratinocytes with HPV-16 infection, followed by E5 expression, produce a decrease in LC3 levels and prevent p62 degradation. E5 interferes mechanically with the transcriptional activation of the autophagic machinery, regulating the expression of bcn1, atg5, lc3, ulk1, atg4a, and atg7 (434).
Unlike E5, viral gene E6/E7 inhibits autolysosome formation by a mechanism involving p53 (435). HPV-16 and HPV-18 infections are associated with the development of squamous cell carcinomas of the head and neck, two neoplasms with a high incidence worldwide (436). In addition to regulating the autophagic machinery, viral proteins E6 and E7 contribute to p53 and p-Rb degradation of. All these events cause the activation of specific transcription factors that modulate cell fate (437). The information obtained so far reveals that autophagy inhibition promotes HPV life cycle and tumor progression.
In recent decades, studying the different cellular functions associated with autophagy has kept specialized scientists alert. This degradative mechanism, used by mammalian cells to maintain cell homeostasis, also directly contributes to modulating the progression of various diseases, such as cancer. Cellular context is essential to determine the functionality of autophagy. In general, cells accumulate damage at disease initial stages, affecting critical points in cell cycle regulation and thus determining cell transformation. Cells can therefore activate autophagy to shrink the damaged organelles and counteract the stressful stimuli to which they are exposed and restore normal state. However, in the context of tumor progression and invasion, the situation may be different. Here, cells present multiple alterations in their signaling pathways, which develops an aggressive cell phenotype, active and uncontrolled division, and high metabolism. Tumor cells can then induce autophagy to generate a fuel supply to maintain tumor cell metabolism.
Furthermore, autophagy can regulate tumor angiogenesis and immunity to benefit disease progression. Before infection, cells can eliminate intracellular pathogens by enzymatic digestion in autophagic vesicles. However, many viruses have developed strategies that allow them to bypass host attack and achieve successful replication and permanence. The number of viruses that modulate the autophagic mechanism for their benefit is increasing. In general, these intracellular organisms regulate the autophagic machinery in a bimodal manner. Upon entering cells, they promote autophagosome formation and inhibit binding to lysosomes, thus losing their degradative capacity. Viruses can use these dysfunctional vesicles to replicate within them, while debris and damage accumulate in cell organelles due to the mechanism’s inefficiency. Other viruses, on the other hand, directly inhibit the autophagic machinery from preventing its degradation. Dysfunctional vesicle accumulation contributes to cell damage accumulation, which benefits cell transformation and tumor development over time. However, more studies are needed to clarify autophagy’s relation to viral infections and tumor development. A thorough understanding of these molecular mechanisms is crucial for developing new antiviral drugs and targeted oncogenic therapies.
AS received an invitation to contribute to the Research topic: New functions of autophagy pathways in cancer. This author designed and wrote the manuscript, MM created the schematic figures and provided critical reading, and OC contributed to the overall design, supervision of the work, and essential reading. All authors contributed to the article and approved the submitted version.
This work was supported by NIH GRANT 1U54CA221208-01 (to OC), UBACYT GRANT 20020150100200BA (to OC), and PICT GRANT 2015-3436 (to OC). The funders had no role in study design, data collection, and analysis, decision to publish or preparation of the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling Editor declared a shared affiliation, though no other collaboration, with the authors.
1. Hegyi G, Vincze G, Szasz A. On the Dynamic Equilibrium in Homeostasis. Open J Biophys (2012) 02(03):60–7. doi: 10.4236/ojbiphy.2012.23009
2. Greives TJ, Dochtermann NA, Stewart EC. Estimating heritable genetic contributions to innate immune and endocrine phenotypic correlations: A need to explore repeatability. Horm Behav (2017) 88:106–11. doi: 10.1016/j.yhbeh.2016.11.015
3. Bektas A, Schurman SH, Sen R, Ferrucci L. Aging, inflammation and the environment. Exp Gerontol (2018) 105(October):10–8. doi: 10.1016/j.exger.2017.12.015
4. Chatterjee N, Walker GC. Mechanisms of DNA damage, repair, and mutagenesis. Environ Mol Mutagen (2017) 58(5):235–63. doi: 10.1002/em.22087
5. Costas MA, Rubio MF. Autofagia, una estrategia de supervivencia celular. Medicina (Buenos Aires) (2017) 77(4):314–20.
6. Feng Y, He D, Yao Z, Klionsky DJ. The machinery of macroautophagy. Cell Res (2014) 24(1):24–41. doi: 10.1038/cr.2013.168
7. White E. The role for autophagy in cancer. J Clin Invest (2015) 125(1):42–6. doi: 10.1172/JCI73941
8. Yu L, Chen Y, Tooze SA. Autophagy pathway: Cellular and molecular mechanisms. Autophagy (2018) 14(2):207–15. doi: 10.1080/15548627.2017.1378838
9. Anding AL, Baehrecke EH. Cleaning House: Selective Autophagy of Organelles. Dev Cell (2017) 41(1):10–22. doi: 10.1016/j.devcel.2017.02.016
10. Noda NN, Inagaki F. Mechanisms of Autophagy. Annu Rev Biophys (2015) 44(1):101–22. doi: 10.1146/annurev-biophys-060414-034248
11. Mizushima N. The ATG conjugation systems in autophagy. Curr Opin Cell Biol (2020) 63:1–10. doi: 10.1016/j.ceb.2019.12.001
12. Pyo J-O, Nah J, Jung Y-K. Molecules and their functions in autophagy. Exp Mol Med (2012) 44(2):73. doi: 10.3858/emm.2012.44.2.029
13. Russell RC, Yuan HX, Guan K-L. Autophagy regulation by nutrient signaling. Cell Res (2014) 24(1):42–57. doi: 10.1038/cr.2013.166
14. Ha J, Guan K-L, Kim J. AMPK and autophagy in glucose/glycogen metabolism. Mol Aspects Med (2015) 46:46–62. doi: 10.1016/j.mam.2015.08.002
15. Gui L, Liu B, LV G. Hypoxia induces autophagy in cardiomyocytes via a hypoxia-inducible factor 1-dependent mechanism. Exp Ther Med (2016) 11(6):2233–9. doi: 10.3892/etm.2016.3190
16. Lai MC, Chang CM, Sun HS. Hypoxia Induces Autophagy through Translational Up-Regulation of Lysosomal Proteins in Human Colon Cancer Cells. Trajkovic V, ed. PloS One (2016) 11(4):e0153627. doi: 10.1371/journal.pone.0153627
17. Chen A, Sceneay J, Gödde N, Kinwel T, Ham S, Thompson EW, et al. Intermittent hypoxia induces a metastatic phenotype in breast cancer. Oncogene (2018) 37(31):4214–25. doi: 10.1038/s41388-018-0259-3
18. Xia G, Zhu T, Li X, Jin Y, Zhou J, Xiao J. ROS−mediated autophagy through the AMPK signaling pathway protects INS−1 cells from human islet amyloid polypeptide−induced cytotoxicity. Mol Med Rep (2018) 18(3):2744–52. doi: 10.3892/mmr.2018.9248
19. Han X, Zhong Z, Kou J, Zheng Y, Liu Z, Jiang Y, et al. ROS Generated by Upconversion Nanoparticle-Mediated Photodynamic Therapy Induces Autophagy Via PI3K/AKT/ mTOR Signaling Pathway in M1 Peritoneal Macrophage. Cell Physiol Biochem (2018) 48(4):1616–27. doi: 10.1159/000492283
20. Yorimitsu T, Klionsky DJ. Endoplasmic Reticulum Stress: A New Pathway to Induce Autophagy. Autophagy (2007) 3(2):160–2. doi: 10.4161/auto.3653
21. Nie T, Yang S, Ma H, Zhang L, Lu F, Tao K, et al. Regulation of ER stress-induced autophagy by GSK3β-TIP60-ULK1 pathway. Cell Death Dis (2016) 7(12):e2563–3. doi: 10.1038/cddis.2016.423
22. Corona Velazquez AF, Jackson WT. So Many Roads: the Multifaceted Regulation of Autophagy Induction. Mol Cell Biol (2018) 38(21):1–14. doi: 10.1128/MCB.00303-18
23. Jang M, Park R, Kim H, Namkoong S, Jo D, Huh YH, et al. AMPK contributes to autophagosome maturation and lysosomal fusion. Sci Rep (2018) 8(1):12637. doi: 10.1038/s41598-018-30977-7
24. Jung CH, Ro SH, Cao J, Otto NM, Kim DH. mTOR regulation of autophagy. FEBS Lett (2010) 584(7):1287–95. doi: 10.1016/j.febslet.2010.01.017
25. Shao X, Lai D, Zhang L, Xu H. Induction of Autophagy and Apoptosis via PI3K/AKT/TOR Pathways by Azadirachtin A in Spodoptera litura Cells. Sci Rep (2016) 6(December 2015):1–12. doi: 10.1038/srep35482
26. Bourouis M, Mondin M, Dussert A, Leopold P. Control of basal autophagy rate by vacuolar peduncle. Trajkovic V, ed. PloS One (2019) 14(2):e0209759. doi: 10.1371/journal.pone.0209759
27. Rabanal-Ruiz Y, Otten EG, Krolchuk VI. mTORC1 as the main Gateway to autophagy. Essays Biochem (2017) 61:565–84. doi: 10.1042/EBC20170027
28. Cork GK, Thompson J, Slawson C. Real Talk; The Inter-play Between the mTOR, AMPK, and Hexosamine Biosynthetic Pathways in Cell Signaling. Front Endocrinol (2018) 522(9):1–9. doi: 10.1111/odi.12254
29. Klionsky DJ, Schulman BA. Dynamic regulation of macroautophagy by distinctive ubiquitin-like proteins. Nat Struct Mol Biol (2014) 21(4):336–45. doi: 10.1038/nsmb.2787
30. Zaza G, Granata S, Caletti C, Signorini L, Stallone G, Lupo A. mTOR Inhibition Role in Cellular Mechanisms. Transplantation (2018) 102(>2S Suppl 1):S3–S16. doi: 10.1097/TP.0000000000001806
31. Li X, He S, Ma B. Autophagy and autophagy-related proteins in cancer. Mol Cancer (2020) 19(1):12. doi: 10.1186/s12943-020-1138-4
32. Heras-Sandoval D, Pérez-Rojas JM, Pedraza-Chaverri J. Novel compounds for the modulation of mTOR and autophagy to treat neurodegenerative diseases. Cell Signal (2020) 65:109442. doi: 10.1016/j.cellsig.2019.109442
33. Qin L, Wang Z, Tao L, Wang Y. ER stress negatively regulates AKT/TSC/mTOR pathway to enhance autophagy. Autophagy (2010) 6(2):239–47. doi: 10.4161/auto.6.2.11062
34. Di Nardo A, Wertz MH, Kwiatkowski E, Tsai PT, Leech JD, Greene-Colozzi E, et al. Neuronal Tsc1/2 complex controls autophagy through AMPK-dependent regulation of ULK1. Hum Mol Genet (2014) 23(14):3865–74. doi: 10.1093/hmg/ddu101
35. Galluzzi L, Pietrocola F, Bravo-San Pedro JM, Amaravadi RK, Baehrecke EH, Cecconi F, et al. Autophagy in malignant transformation and cancer progression. EMBO J (2015) 34(7):856–80. doi: 10.15252/embj.201490784
36. Janku F, McConkey DJ, Hong DS, Kurzrock R. Autophagy as a target for anticancer therapy. Nat Rev Clin Oncol (2011) 8(9):528–39. doi: 10.1038/nrclinonc.2011.71
37. Kotani T, Kirisako H, Koizumi M, Ohsumi Y, Nakatogawa H. The Atg2-Atg18 complex tethers pre-autophagosomal membranes to the endoplasmic reticulum for autophagosome formation. Proc Natl Acad Sci (2018) 115(41):10363–8. doi: 10.1073/pnas.1806727115
38. Ktistakis NT, Tooze SA. Digesting the Expanding Mechanisms of Autophagy. Trends Cell Biol (2016) 26(8):624–35. doi: 10.1016/j.tcb.2016.03.006
39. Ge L, Melville D, Zhang M, Schekman R. The ER–Golgi intermediate compartment is a key membrane source for the LC3 lipidation step of autophagosome biogenesis. Elife (2013) 2:1–23. doi: 10.7554/eLife.00947
40. Wei Y, Liu M, Li X, Liu J, Li H. Origin of the Autophagosome Membrane in Mammals. BioMed Res Int (2018) 2018:1–9. doi: 10.1155/2018/1012789
41. Lamb CA, Yoshimori T, Tooze SA. The autophagosome: origins unknown, biogenesis complex. Nat Rev Mol Cell Biol (2013) 14(12):759–74. doi: 10.1038/nrm3696
42. Ge L, Baskaran S, Schekman R, Hurley JH. The protein-vesicle network of autophagy. Curr Opin Cell Biol (2014) 29(1):18–24. doi: 10.1016/j.ceb.2014.02.005
43. Decuypere JP, Parys JB, Bultynck G. Regulation of the Autophagic Bcl-2/Beclin 1 Interaction. Cells (2012) 1(3):284–312. doi: 10.3390/cells1030284
44. Wen J, Mai Z, Zhao M, Wang X, Chen T. Full anti-apoptotic function of Bcl-XL complexed with Beclin-1 verified by live-cell FRET assays. Biochem Biophys Res Commun (2019) 511(3):700–4. doi: 10.1016/j.bbrc.2019.02.107
45. Nakajima S, Aikawa C, Nozawa T, Minowa-Nozawa A, Toh H, Nakagawa I. Bcl-xL Affects Group A Streptococcus-Induced Autophagy Directly, by Inhibiting Fusion between Autophagosomes and Lysosomes, and Indirectly, by Inhibiting Bacterial Internalization via Interaction with Beclin 1-UVRAG. Trajkovic V, ed. PloS One (2017) 12(1):e0170138. doi: 10.1371/journal.pone.0170138
46. Into T, Inomata M, Takayama E, Takigawa T. Autophagy in regulation of Toll-like receptor signaling. Cell Signal (2012) 24(6):1150–62. doi: 10.1016/j.cellsig.2012.01.020
47. Marquez RT, Xu L. Bcl-2:Beclin 1 complex: multiple, mechanisms regulating autophagy/apoptosis toggle switch. Am J Cancer Res (2012) 2(2):214–21. doi: 2156-6976/ajcr0000098ajcr0000098
48. Grasso D, Ropolo A, Vaccaro MI. Autophagy in Cell Fate and Diseases. Cell Death - Autophagy, Apoptosis and Necrosis (2015) 1:3–26. doi: 10.5772/61553
49. Fujita N, Itoh T, Omori H, Fukuda M, Noda T, Yoshimori T. The Atg16L Complex Specifies the Site of LC3 Lipidation for Membrane Biogenesis in Autophagy. Riezman H, ed. Mol Biol Cell (2008) 19(5):2092–100. doi: 10.1091/mbc.e07-12-1257
50. Winer H, Fraiberg M, Abada A, Dadosh T, Tamim-Yecheskel BC, Elazar Z. Autophagy differentially regulates TNF receptor Fn14 by distinct mammalian Atg8 proteins. Nat Commun (2018) 9(1):3744. doi: 10.1038/s41467-018-06275-1
51. Schaaf MBE, Keulers TG, Vooijs MA, Rouschop KMA. LC3/GABARAP family proteins: autophagy-(un)related functions. FASEB J (2016) 30(12):3961–78. doi: 10.1096/fj.201600698R
52. Brier LW, Ge L, Stjepanovic G, Thelen AM, Hurley J, Schekman R. Regulation of LC3 Lipidation by the Autophagy-Specific Class III Phosphatidylinositol-3 Kinase Complex. Mol Biol Cell (2019) 30(9):1051–128. doi: 10.1091/mbc.E18-11-0743
53. Moscat J, Karin M, Diaz-Meco MT. p62 in Cancer: Signaling Adaptor Beyond Autophagy. Cell (2016) 167(3):606–9. doi: 10.1016/j.cell.2016.09.030
54. Lau A, Wang XJ, Zhao F, Villeneuve NF, Wu T, Jiang T, et al. A Noncanonical Mechanism of Nrf2 Activation by Autophagy Deficiency: Direct Interaction between Keap1 and p62. Mol Cell Biol (2010) 30(13):3275–85. doi: 10.1128/MCB.00248-10
55. Valencia T, Kim JY, Abu-Baker S, Moscat-Pardos J, Ahn CS, Reina-Campos M, et al. Metabolic Reprogramming of Stromal Fibroblasts through p62-mTORC1 Signaling Promotes Inflammation and Tumorigenesis. Cancer Cell (2014) 26(1):121–35. doi: 10.1016/j.ccr.2014.05.004
56. Zhao YG, Zhang H. Autophagosome maturation: An epic journey from the ER to lysosomes. J Cell Biol (2019) 218(3):757–70. doi: 10.1083/jcb.201810099
57. Parzych KR, Klionsky DJ. An Overview of Autophagy: Morphology, Mechanism, and Regulation. Antioxid Redox Signal (2014) 20(3):460–73. doi: 10.1089/ars.2013.5371
58. Jager S. Role for Rab7 in maturation of late autophagic vacuoles. J Cell Sci (2004) 117(20):4837–48. doi: 10.1242/jcs.01370
59. Itakura E, Mizushima N. Characterization of autophagosome formation site by a hierarchical analysis of mammalian Atg proteins. Autophagy (2010) 6(6):764–76. doi: 10.4161/auto.6.6.12709
60. Gatica D, Lahiri V, Klionsky DJ. Cargo recognition and degradation by selective autophagy. Nat Cell Biol (2018) 20(3):233–42. doi: 10.1038/s41556-018-0037-z
61. Jing K, Lim K. Why is autophagy important in human diseases? Exp Mol Med (2012) 44(2):69. doi: 10.3858/emm.2012.44.2.028
62. Yang Y, Klionsky DJ. Autophagy and disease: unanswered questions. Cell Death Differ (2020) 27(3):858–71. doi: 10.1038/s41418-019-0480-9
63. Zachari M, Gudmundsson SR, Li Z, Manifava M, Shah R, Smith M, et al. Selective Autophagy of Mitochondria on a Ubiquitin-Endoplasmic-Reticulum Platform. Dev Cell (2019) 50(5):627–43.e5. doi: 10.1016/j.devcel.2019.06.016
64. Germain K, Kim PK. Pexophagy: A Model for Selective Autophagy. Int J Mol Sci (2020) 21(2):578. doi: 10.3390/ijms21020578
65. Cebollero E, Reggiori F, Kraft C. Reticulophagy and Ribophagy: Regulated Degradation of Protein Production Factories. Int J Cell Biol (2012) 2012:1–9. doi: 10.1155/2012/182834
66. Nakatogawa H, Mochida K. Reticulophagy and nucleophagy: New findings and unsolved issues. Autophagy (2015) 11(12):2377–8. doi: 10.1080/15548627.2015.1106665
67. Singh R, Cuervo AM. Lipophagy: Connecting Autophagy and Lipid Metabolism. Int J Cell Biol (2012) 2012:1–12. doi: 10.1155/2012/282041
68. Bauckman KA, Owusu-Boaitey N, Mysorekar IU. Selective autophagy: Xenophagy. Methods (2015) 75:120–7. doi: 10.1016/j.ymeth.2014.12.005
69. Lamark T, Johansen T. Aggrephagy: Selective Disposal of Protein Aggregates by Macroautophagy. Int J Cell Biol (2012) 2012:1–21. doi: 10.1155/2012/736905
70. Doherty J, Baehrecke EH. Life, death and autophagy. Nat Cell Biol (2018) 20(10):1110–7. doi: 10.1038/s41556-018-0201-5
71. Nah J, Zablocki D, Sadoshima J. Autosis. JACC Basic to Transl Sci (2020) 5(8):857–69. doi: 10.1016/j.jacbts.2020.04.014
72. Swart C, Du Toit A, Loos B. Autophagy and the invisible line between life and death. Eur J Cell Biol (2016) 95(12):598–610. doi: 10.1016/j.ejcb.2016.10.005
73. Kriel J, Loos B. The good, the bad and the autophagosome: exploring unanswered questions of autophagy-dependent cell death. Cell Death Differ (2019) 26(4):640–52. doi: 10.1038/s41418-018-0267-4
74. Sridhar S, Botbol Y, MacIan F, Cuervo AM. Autophagy and disease: Always two sides to a problem. J Pathol (2012) 226(2):255–73. doi: 10.1002/path.3025
75. Santana-Codina N, Mancias JD, Kimmelman AC. The Role of Autophagy in Cancer. Annu Rev Cancer Biol (2017) 1(1):19–39. doi: 10.1146/annurev-cancerbio-041816-122338
76. Hanahan D, Weinberg RA. Hallmarks of Cancer: The Next Generation. Cell (2011) 144(5):646–74. doi: 10.1016/j.cell.2011.02.013
78. Das CK, Mandal M, Kögel D. Pro-survival autophagy and cancer cell resistance to therapy. Cancer Metastasis Rev (2018) 37(4):749–66. doi: 10.1007/s10555-018-9727-z
79. Singh SS, Vats S, Chia AY-Q, Tan TZ, Deng S, Ong MS, et al. Dual role of autophagy in hallmarks of cancer. Oncogene (2018) 37(9):1142–58. doi: 10.1038/s41388-017-0046-6
80. Wang K, Klionsky DJ. Mitochondria removal by autophagy. Autophagy (2011) 7(3):297–300. doi: 10.4161/auto.7.3.14502
81. Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H, et al. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature (1999) 402(6762):672–6. doi: 10.1038/45257
82. Erlich S, Mizrachy L, Segev O, Lindenboim L, Zmira O, Aldi-Harel S, et al. Differential Interactions Between Beclin 1 and Bcl-2 Family Members. Autophagy (2007) 2007:3(6):561–8. doi: 10.4161/auto.4713
83. Kang R, Zeh HJ, Lotze MT, Tang D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ (2011) 18(4):571–80. doi: 10.1038/cdd.2010.191
84. Toton E, Lisiak N, Sawicka P, Rybczynska M. Beclin-1 and its role as a target for anticancer therapy. J Physiol Pharmacol (2014) 65(4):459–67.
85. Hill SM, Wrobel L, Rubinsztein DC. Post-translational modifications of Beclin 1 provide multiple strategies for autophagy regulation. Cell Death Differ (2019) 26(4):617–29. doi: 10.1038/s41418-018-0254-9
86. Ávalos Y, Canales J, Bravo-Sagua R, Criollo A, Lavandero S, Quest AFG. Tumor Suppression and Promotion by Autophagy. BioMed Res Int (2014) 2014:1–15. doi: 10.1155/2014/603980
87. Eliopoulos AG, Havaki S, Gorgoulis VG. DNA Damage Response and Autophagy: A Meaningful Partnership. Front Genet (2016) 7:204(NOV). doi: 10.3389/fgene.2016.00204
88. Rodriguez-Rocha H, Garcia-Garcia A, Panayiotidis MI, Franco R. DNA damage and autophagy. Mutat Res Mol Mech Mutagen (2011) 711(1-2):158–66. doi: 10.1016/j.mrfmmm.2011.03.007
89. Yun C, Lee S. The Roles of Autophagy in Cancer. Int J Mol Sci (2018) 19(11):3466. doi: 10.3390/ijms19113466
90. Takamura A, Komatsu M, Hara T, Sakamoto A, Kishi C, Waguri S, et al. Autophagy-deficient mice develop multiple liver tumors. Genes Dev (2011) 25(8):795–800. doi: 10.1101/gad.2016211
91. Kimmelman AC. The dynamic nature of autophagy in cancer. Genes Dev (2011) 25(19):1999–2010. doi: 10.1101/gad.17558811
92. Mariño G, Salvador-Montoliu N, Fueyo A, Knecht E, Mizushima N, López-Otín C. Tissue-specific Autophagy Alterations and Increased Tumorigenesis in Mice Deficient in Atg4C/Autophagin-3. J Biol Chem (2007) 282(25):18573–83. doi: 10.1074/jbc.M701194200
93. Cianfanelli V, Fuoco C, Lorente M, Salazar M, Quondamatteo F, Gherardini PF, et al. AMBRA1 links autophagy to cell proliferation and tumorigenesis by promoting c-Myc dephosphorylation and degradation. Nat Cell Biol (2015) 17(1):20–30. doi: 10.1038/ncb3072
94. Maiuri MC, Galluzzi L, Morselli E, Kepp O, Malik SA, Kroemer G. Autophagy regulation by p53. Curr Opin Cell Biol (2010) 22(2):181–5. doi: 10.1016/j.ceb.2009.12.001
95. Luo Z, Zang M, Guo W. AMPK as a metabolic tumor suppressor: control of metabolism and cell growth. Futur Oncol (2010) 6(3):457–70. doi: 10.2217/fon.09.174
96. Tasdemir E, Maiuri MC, Morselli E, Criollo A, D’Amelio M, Djavaheri-Mergny M, et al. A dual role of p53 in the control of autophagy. Autophagy (2008) 4(6):810–4. doi: 10.4161/auto.6486
97. Mrakovcic M, Fröhlich LF. p53-Mediated Molecular Control of Autophagy in Tumor Cells. Biomolecules (2018) 8(2):14. doi: 10.3390/biom8020014
98. Rosenfeldt MT, O’Prey J, Morton JP, Nixon C, MacKay G, Mrowinska A, et al. p53 status determines the role of autophagy in pancreatic tumour development. Nature (2013) 504(7479):296–300. doi: 10.1038/nature12865
99. White E. Autophagy and p53. Cold Spring Harb Perspect Med (2016) 6(4):a026120. doi: 10.1101/cshperspect.a026120
100. Kenzelmann Broz D, Spano Mello S, Bieging KT, Jiang D, Dusek RL, Brady CA, et al. Global genomic profiling reveals an extensive p53-regulated autophagy program contributing to key p53 responses. Genes Dev (2013) 27(9):1016–31. doi: 10.1101/gad.212282.112
101. Eby KG, Rosenbluth JM, Mays DJ, Marshall CB, Barton CE, Sinha S, et al. ISG20L1 is a p53 family target gene that modulates genotoxic stress-induced autophagy. Mol Cancer (2010) 9(1):95. doi: 10.1186/1476-4598-9-95
102. Mowers EE, Sharifi MN, Macleod KF. Autophagy in cancer metastasis. Oncogene (2017) 36(12):1619–30. doi: 10.1038/onc.2016.333
103. Yang ZJ, Chee CE, Huang S, Sinicrope FA. The Role of Autophagy in Cancer: Therapeutic Implications. Mol Cancer Ther (2011) 10(9):1533–41. doi: 10.1158/1535-7163.MCT-11-0047
104. Simon T, Gagliano T, Giamas G. Direct Effects of Anti-Angiogenic Therapies on Tumor Cells: VEGF Signaling. Trends Mol Med (2017) 23(3):282–92. doi: 10.1016/j.molmed.2017.01.002
105. Salabei JK, Cummins TD, Singh M, Jones SP, Bhatnagar A, Hill BG. PDGF-mediated autophagy regulates vascular smooth muscle cell phenotype and resistance to oxidative stress. Biochem J (2013) 451(3):375–88. doi: 10.1042/BJ20121344
106. Bharath LP, Cho JM, Park S-K, Ruan T, Li Y, Mueller R, et al. Endothelial Cell Autophagy Maintains Shear Stress–Induced Nitric Oxide Generation via Glycolysis-Dependent Purinergic Signaling to Endothelial Nitric Oxide Synthase. Arterioscler Thromb Vasc Biol (2017) 37(9):1646–56. doi: 10.1161/ATVBAHA.117.309510
107. Qu X. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest (2003) 112(12):1809–20. doi: 10.1172/JCI200320039
108. Das CK, Banerjee I, Mandal M. Pro-survival autophagy: An emerging candidate of tumor progression through maintaining hallmarks of cancer. Semin Cancer Biol (2019) 66:59–74. doi: 10.1016/j.semcancer.2019.08.020
109. Bykov VJN, Eriksson SE, Bianchi J, Wiman KG. Targeting mutant p53 for efficient cancer therapy. Nat Rev Cancer (2018) 18(2):89–102. doi: 10.1038/nrc.2017.109
110. Huo Y, Cai H, Teplova I, Bowman-Colin C, Chen G, Price S, et al. Autophagy Opposes p53-Mediated Tumor Barrier to Facilitate Tumorigenesis in a Model of PALB2-Associated Hereditary Breast Cancer. Cancer Discov (2013) 3(8):894–907. doi: 10.1158/2159-8290.CD-13-0011
111. Moscat J, Diaz-Meco MT. p62: a versatile multitasker takes on cancer. Trends Biochem Sci (2012) 37(6):230–6. doi: 10.1016/j.tibs.2012.02.008
112. Li L, Shen C, Nakamura E, Ando K, Signoretti S, Beroukhim R, et al. SQSTM1 Is a Pathogenic Target of 5q Copy Number Gains in Kidney Cancer. Cancer Cell (2013) 24(6):738–50. doi: 10.1016/j.ccr.2013.10.025
113. Umemura A, He F, Taniguchi K, Nakagawa H, Yamachika S, font-Burgada J, et al. p62, Upregulated during Preneoplasia, Induces Hepatocellular Carcinogenesis by Maintaining Survival of Stressed HCC-Initiating Cells. Cancer Cell (2016) 29(6):935–48. doi: 10.1016/j.ccell.2016.04.006
114. Zhang J, Yang S, Xu B, Wang T, Zheng Y, Liu F, et al. p62 functions as an oncogene in colorectal cancer through inhibiting apoptosis and promoting cell proliferation by interacting with the vitamin D receptor. Cell Prolif (2019) 52(3):e12585. doi: 10.1111/cpr.12585
115. Ren F, Shu G, Liu G, Liu D, Zhou J, Yuan L, et al. Knockdown of p62/sequestosome 1 attenuates autophagy and inhibits colorectal cancer cell growth. Mol Cell Biochem (2014) 385(1-2):95–102. doi: 10.1007/s11010-013-1818-0
116. Schmukler E, Kloog Y, Pinkas-Kramarski R. Ras and autophagy in cancer development and therapy. Oncotarget (2014) 5(3):577–86. doi: 10.18632/oncotarget.1775
117. Lock R, Kenific CM, Leidal AM, Salas E, Debnath J. Autophagy-dependent production of secreted factors facilitates oncogenic RAS-Driven invasion. Cancer Discov (2014) 4(4):466–79. doi: 10.1158/2159-8290.CD-13-0841
118. Guo JY, Karsli-Uzunbas G, Mathew R, Aisner SC, Kamphorst JJ, Strohecker AM, et al. Autophagy suppresses progression of K-ras-induced lung tumors to oncocytomas and maintains lipid homeostasis. Genes Dev (2013) 27(13):1447–61. doi: 10.1101/gad.219642.113
119. Ling J, Kang Y, Zhao R, Xia Q, Lee D-F, Chang Z, et al. KrasG12D-Induced IKK2/β/NF-κB Activation by IL-1α and p62 Feedforward Loops Is Required for Development of Pancreatic Ductal Adenocarcinoma. Cancer Cell (2012) 21(1):105–20. doi: 10.1016/j.ccr.2011.12.006
120. Sosa MS, Bragado P, Debnath J, Aguirre-Ghiso JA. Regulation of Tumor Cell Dormancy by Tissue Microenvironments and Autophagy. In: Enderling H, Almog N, Hlatky L, editors. Advances in Experimental Medicine and Biology, vol. 734 . New York, NY: Springer New York (2013). p. 73–89. doi: 10.1007/978-1-4614-1445-2_5
121. Lorin S, Hamaï A, Mehrpour M, Codogno P. Autophagy regulation and its role in cancer. Semin Cancer Biol (2013) 23(5):361–79. doi: 10.1016/j.semcancer.2013.06.007
122. Paoli P, Giannoni E, Chiarugi P. Anoikis molecular pathways and its role in cancer progression. Biochim Biophys Acta - Mol Cell Res (2013) 1833(12):3481–98. doi: 10.1016/j.bbamcr.2013.06.026
123. Buchheit CL, Weigel KJ, Schafer ZT. Cancer cell survival during detachment from the ECM: multiple barriers to tumour progression. Nat Rev Cancer (2014) 14(9):632–41. doi: 10.1038/nrc3789
124. Zhi X, Zhong Q. Autophagy in cancer. F1000Prime Rep (2015) 7(February):1–12. doi: 10.12703/P7-18
125. Swartz MA, Iida N, Roberts EW, Sangaletti S, Wong MH, Yull FE, et al. Tumor Microenvironment Complexity: Emerging Roles in Cancer Therapy: Figure 1. Cancer Res (2012) 72(10):2473–80. doi: 10.1158/0008-5472.CAN-12-0122
126. Camuzard O, Santucci-Darmanin S, Carle GF, Pierrefite-Carle V. Autophagy in the crosstalk between tumor and microenvironment. Cancer Lett (2020) 490:143–53. doi: 10.1016/j.canlet.2020.06.015
127. Roma-Rodrigues C, Mendes R, Baptista P, Fernandes A. Targeting Tumor Microenvironment for Cancer Therapy. Int J Mol Sci (2019) 20(4):840. doi: 10.3390/ijms20040840
128. Ye J, Wu D, Wu P, Chen Z, Huang J. The cancer stem cell niche: cross talk between cancer stem cells and their microenvironment. Tumor Biol (2014) 35(5):3945–51. doi: 10.1007/s13277-013-1561-x
129. Kalluri R, Zeisberg M. Fibroblasts in cancer. Nat Rev Cancer (2006) 6(5):392–401. doi: 10.1038/nrc1877
130. Folkerts H, Hilgendorf S, Vellenga E, Bremer E, Wiersma VR. The multifaceted role of autophagy in cancer and the microenvironment. Med Res Rev (2019) 39(2):517–60. doi: 10.1002/med.21531
131. Guo JY, Teng X, Laddha SV, Ma S, Van Nostrand SC, Yang Y, et al. Autophagy provides metabolic substrates to maintain energy charge and nucleotide pools in Ras-driven lung cancer cells. Genes Dev (2016) 30(15):1704–17. doi: 10.1101/gad.283416.116
132. Ngabire D, Kim G-D. Autophagy and Inflammatory Response in the Tumor Microenvironment. Int J Mol Sci (2017) 18(9):2016. doi: 10.3390/ijms18092016
133. Hui L, Chen Y. Tumor microenvironment: Sanctuary of the devil. Cancer Lett (2015) 368(1):7–13. doi: 10.1016/j.canlet.2015.07.039
134. Anthony BA, Link DC. Regulation of hematopoietic stem cells by bone marrow stromal cells. Trends Immunol (2014) 35(1):32–7. doi: 10.1016/j.it.2013.10.002
135. Guan J-L, Simon AK, Prescott M, Menendez JA, Liu F, Wang F, et al. Autophagy in stem cells. Autophagy (2013) 9(6):830–49. doi: 10.4161/auto.24132
136. Ito K, Ito K. Hematopoietic stem cell fate through metabolic control. Exp Hematol (2018) 64:1–11. doi: 10.1016/j.exphem.2018.05.005
137. Salemi S, Yousefi S, Constantinescu MA, Fey MF, Simon H-U. Autophagy is required for self-renewal and differentiation of adult human stem cells. Cell Res (2012) 22(2):432–5. doi: 10.1038/cr.2011.200
138. Liu Q, Chen L, Atkinson JM, Claxton DF, Wang H-G. Atg5-dependent autophagy contributes to the development of acute myeloid leukemia in an MLL-AF9-driven mouse model. Cell Death Dis (2016) 7(9):e2361–1. doi: 10.1038/cddis.2016.264
139. Jacquel A, Obba S, Boyer L, Dufies M, Robert G, Gounon P, et al. Autophagy is required for CSF-1–induced macrophagic differentiation and acquisition of phagocytic functions. Blood (2012) 119(19):4527–31. doi: 10.1182/blood-2011-11-392167
140. Jacquel A, Benikhlef N, Paggetti J, Lalaoui N, Guery L, Dufour EK, et al. Colony-stimulating factor-1–induced oscillations in phosphatidylinositol-3 kinase/AKT are required for caspase activation in monocytes undergoing differentiation into macrophages. Blood (2009) 114(17):3633–41. doi: 10.1182/blood-2009-03-208843
141. Zhang Y, Morgan MJ, Chen K, Choksi S, Liu Z. Induction of autophagy is essential for monocyte-macrophage differentiation. Blood (2012) 119(12):2895–905. doi: 10.1182/blood-2011-08-372383
142. Modi J, Menzie-Suderam J, Xu H, Trujillo P, Medley K, Marshall ML, et al. Mode of action of granulocyte-colony stimulating factor (G-CSF) as a novel therapy for stroke in a mouse model. J BioMed Sci (2020) 27(1):19. doi: 10.1186/s12929-019-0597-7
143. Martinez-Outschoorn UE, Whitaker-Menezes D, Lin Z, Flomenberg N, Howell A, Pestell RG, et al. Cytokine production and inflammation drive autophagy in the tumor microenvironment. Cell Cycle (2011) 10(11):1784–93. doi: 10.4161/cc.10.11.15674
144. Goswami KK, Ghosh T, Ghosh S, Sarkar M, Bose A, Baral R. Tumor promoting role of anti-tumor macrophages in tumor microenvironment. Cell Immunol (2017) 316(April):1–10. doi: 10.1016/j.cellimm.2017.04.005
145. Martinez-Outschoorn UE, Trimmer C, Lin Z, Whitaker-Menezes D, Chiavarina B, Zhou J, et al. Autophagy in cancer associated fibroblasts promotes tumor cell survival. Cell Cycle (2010) 9(17):3515–33. doi: 10.4161/cc.9.17.12928
146. Capparelli C, Guido C, Whitaker-Menezes D, Bonuccelli G, Balliet R, Pestell TG, et al. Autophagy and senescence in cancer-associated fibroblasts metabolically supports tumor growth and metastasis, via glycolysis and ketone production. Cell Cycle (2012) 11(12):2285–302. doi: 10.4161/cc.20718
147. Grasso D, Garcia MN, Iovanna JL. Autophagy in Pancreatic Cancer. Int J Cell Biol (2012) 2012:1–7. doi: 10.1155/2012/760498
148. Mowers EE, Sharifi MN, Macleod KF. Functions of autophagy in the tumor microenvironment and cancer metastasis. FEBS J (2018) 285(10):1751–66. doi: 10.1111/febs.14388
149. Endo S, Nakata K, Ohuchida K, Takesue S, Nakayama H, Abe T, et al. Autophagy Is Required for Activation of Pancreatic Stellate Cells, Associated With Pancreatic Cancer Progression and Promotes Growth of Pancreatic Tumors in Mice. Gastroenterology (2017) 152(6):1492–1506.e24. doi: 10.1053/j.gastro.2017.01.010
150. Schaaf MB, Houbaert D, Meçe O, Agostinis P. Autophagy in endothelial cells and tumor angiogenesis. Cell Death Differ (2019) 26(4):665–79. doi: 10.1038/s41418-019-0287-8
151. Welti J, Loges S, Dimmeler S, Carmeliet P. Recent molecular discoveries in angiogenesis and antiangiogenic therapies in cancer. J Clin Invest (2013) 123(8):3190–200. doi: 10.1172/JCI70212
152. Carmeliet P, Jain RK. Angiogenesis in cancer and other diseases. Nature (2000) 407(6801):249–57. doi: 10.1038/35025220
153. Filippi I, Saltarella I, Aldinucci C, Carraro F, Ria R, Vacca A, et al. Different Adaptive Responses to Hypoxia in Normal and Multiple Myeloma Endothelial Cells. Cell Physiol Biochem (2018) 46(1):203–12. doi: 10.1159/000488423
154. Chandra A, Rick J, Yagnik G, Aghi MK. Autophagy as a mechanism for anti-angiogenic therapy resistance. Semin Cancer Biol (2020) 66(January):75–88. doi: 10.1016/j.semcancer.2019.08.031
155. Maes H, Olmeda D, Soengas MS, Agostinis P. Vesicular trafficking mechanisms in endothelial cells as modulators of the tumor vasculature and targets of antiangiogenic therapies. FEBS J (2016) 283(1):25–38. doi: 10.1111/febs.13545
156. Maes H, Rubio N, Garg AD, Agostinis P. Autophagy: shaping the tumor microenvironment and therapeutic response. Trends Mol Med (2013) 19(7):428–46. doi: 10.1016/j.molmed.2013.04.005
157. Ma Y, Galluzzi L, Zitvogel L, Kroemer G. Autophagy and Cellular Immune Responses. Immunity (2013) 39(2):211–27. doi: 10.1016/j.immuni.2013.07.017
158. Caron E, Vincent K, Fortier M, Laverdure J-P, Bramoullé A, Hardy M-P, et al. The MHC I immunopeptidome conveys to the cell surface an integrative view of cellular regulation. Mol Syst Biol (2011) 7(1):533. doi: 10.1038/msb.2011.68
159. Michaud M, Martins I, Sukkurwala AQ, Adjemian S, Ma Y, Pellegatti P, et al. Autophagy-Dependent Anticancer Immune Responses Induced by Chemotherapeutic Agents in Mice. Science (80- ) (2011) 334(6062):1573–7. doi: 10.1126/science.1208347
160. Wildenberg ME, Vos ACW, Wolfkamp SCS, Duijvestein M, Verhaar AP, Te Velde AA, et al. Autophagy Attenuates the Adaptive Immune Response by Destabilizing the Immunologic Synapse. Gastroenterology (2012) 142(7):1493–503.e6. doi: 10.1053/j.gastro.2012.02.034
161. Jia W, He Y-W. Temporal Regulation of Intracellular Organelle Homeostasis in T Lymphocytes by Autophagy. J Immunol (2011) 186(9):5313–22. doi: 10.4049/jimmunol.1002404
162. Fiegl D, Kägebein D, Liebler-Tenorio EM, Weisser T, Sens M, Gutjahr M, et al. Amphisomal Route of MHC Class I Cross-Presentation in Bacteria-Infected Dendritic Cells. J Immunol (2013) 190(6):2791–806. doi: 10.4049/jimmunol.1202741
163. Vesely MD, Kershaw MH, Schreiber RD, Smyth MJ. Natural Innate and Adaptive Immunity to Cancer. Annu Rev Immunol (2011) 29(1):235–71. doi: 10.1146/annurev-immunol-031210-101324
164. Lapaquette P, Guzzo J, Bretillon L, Bringer M-A. Cellular and Molecular Connections between Autophagy and Inflammation. Mediators Inflamm (2015) 2015:1–13. doi: 10.1155/2015/398483
165. Vidya MK, Kumar VG, Sejian V, Bagath M, Krishnan G, Bhatta R. Toll-like receptors: Significance, ligands, signaling pathways, and functions in mammals. Int Rev Immunol (2018) 37(1):20–36. doi: 10.1080/08830185.2017.1380200
166. Hontelez S, Sanecka A, Netea MG, van Spriel AB, Adema GJ. Molecular view on PRR cross-talk in antifungal immunity. Cell Microbiol (2012) 14(4):467–74. doi: 10.1111/j.1462-5822.2012.01748.x
167. Sharma V, Verma S, Seranova E, Sarkar S, Kumar D. Selective Autophagy and Xenophagy in Infection and Disease. Front Cell Dev Biol (2018) 6:147(NOV). doi: 10.3389/fcell.2018.00147
168. Delgado MA, Elmaoued RA, Davis AS, Kyei G, Deretic V. Toll-like receptors control autophagy. EMBO J (2008) 27(7):1110–21. doi: 10.1038/emboj.2008.31
169. Shi C-S, Kehrl JH. MyD88 and Trif Target Beclin 1 to Trigger Autophagy in Macrophages. J Biol Chem (2008) 283(48):33175–82. doi: 10.1074/jbc.M804478200
170. Weichhart T, Hengstschläger M, Linke M. Regulation of innate immune cell function by mTOR. Nat Rev Immunol (2015) 15(10):599–614. doi: 10.1038/nri3901
171. Hamon MA, Quintin J. Innate immune memory in mammals. Semin Immunol (2016) 28(4):351–8. doi: 10.1016/j.smim.2016.05.003
172. Mah LY, Ryan KM. Autophagy and Cancer. Cold Spring Harb Perspect Biol (2012) 4(1):a008821–a008821. doi: 10.1101/cshperspect.a008821
173. Wieczorek M, Abualrous ET, Sticht J, Álvalo-Benito M, Stolzenberg S, Noé F, et al. Major Histocompatibility Complex (MHC) Class I and MHC Class II Proteins: Conformational Plasticity in Antigen Presentation. Front Immunol (2017) 8:292(MAR). doi: 10.3389/fimmu.2017.00292
174. Li B, Lei Z, Lichty BD, Li D, Zhang G-M, Feng Z-H, et al. Autophagy facilitates major histocompatibility complex class I expression induced by IFN-γ in B16 melanoma cells. Cancer Immunol Immunother (2010) 59(2):313–21. doi: 10.1007/s00262-009-0752-1
175. Crotzer VL, Blum JS. Autophagy and adaptive immunity. Immunology (2010) 131(1):no–o. doi: 10.1111/j.1365-2567.2010.03321.x
176. You L, Jin S, Zhu L, Qian W. Autophagy, autophagy-associated adaptive immune responses and its role in hematologic malignancies. Oncotarget (2017) 8(7):12374–88. doi: 10.18632/oncotarget.13583
177. Puleston DJ, Simon AK. Autophagy in the immune system. Immunology (2014) 141(1):1–8. doi: 10.1111/imm.12165
178. Mortensen M, Ferguson DJP, Edelmann M, Kessler B, Morten KJ, Komatsu M, et al. Loss of autophagy in erythroid cells leads to defective removal of mitochondria and severe anemia in vivo. Proc Natl Acad Sci (2010) 107(2):832–7. doi: 10.1073/pnas.0913170107
179. Miller BC, Zhao Z, Stephenson LM, Cadwell K, Pua HH, Lee HK, et al. The autophagy gene ATG5 plays an essential role in B lymphocyte development. Autophagy (2008) 4(3):309–14. doi: 10.4161/auto.5474
180. Rybstein MD, Bravo-San Pedro JM, Kroemer G, Galluzzi L. The autophagic network and cancer. Nat Cell Biol (2018) 20(3):243–51. doi: 10.1038/s41556-018-0042-2
181. Monkkonen T, Debnath J. Inflammatory signaling cascades and autophagy in cancer. Autophagy (2018) 14(2):190–8. doi: 10.1080/15548627.2017.1345412
182. Nakamura K, Smyth MJ. Targeting cancer-related inflammation in the era of immunotherapy. Immunol Cell Biol (2017) 95(4):325–32. doi: 10.1038/icb.2016.126
183. Deretic V, Jiang S, Dupont N. Autophagy intersections with conventional and unconventional secretion in tissue development, remodeling and inflammation. Trends Cell Biol (2012) 22(8):397–406. doi: 10.1016/j.tcb.2012.04.008
184. Nawas AF, Mistry R, Narayanan S, Thomas-Jardin SE, Ramachandran J, Ravichandran J, et al. IL-1 induces p62/SQSTM1 and autophagy in ERα + /PR + BCa cell lines concomitant with ERα and PR repression, conferring an ERα – /PR – BCa-like phenotype. J Cell Biochem (2019) 120(2):1477–91. doi: 10.1002/jcb.27340
185. Li TT, Zhu D, Mou T, Guo Z, Pu JL, Chen QS, et al. IL-37 induces autophagy in hepatocellular carcinoma cells by inhibiting the PI3K/AKT/mTOR pathway. Mol Immunol (2017) 87(1):132–40. doi: 10.1016/j.molimm.2017.04.010
186. Chio IIC, Tuveson DA. ROS in Cancer: The Burning Question. Trends Mol Med (2017) 23(5):411–29. doi: 10.1016/j.molmed.2017.03.004
187. Hoesel B, Schmid JA. The complexity of NF-κB signaling in inflammation and cancer. Mol Cancer (2013) 12(1):86. doi: 10.1186/1476-4598-12-86
188. Duran A, Linares JF, Galvez AS, Wikenheiser K, Flores JM, Diaz-Meco MT, et al. The Signaling Adaptor p62 Is an Important NF-κB Mediator in Tumorigenesis. Cancer Cell (2008) 13(4):343–54. doi: 10.1016/j.ccr.2008.02.001
189. Schwartz L, Supuran C, Alfarouk K. The Warburg Effect and the Hallmarks of Cancer. Anticancer Agents Med Chem (2017) 17(2):164–70. doi: 10.2174/1871520616666161031143301
190. Pavlides S, Tsirigos A, Migneco G, Whitaker-Menezes D, chiavarina B, Flomenberg N, et al. The autophagic tumor stroma model of cancer. Cell Cycle (2010) 9(17):3485–505. doi: 10.4161/cc.9.17.12721
191. Xing Y, Zhao S, Zhou BP, Mi J. Metabolic reprogramming of the tumour microenvironment. FEBS J (2015) 282(20):3892–8. doi: 10.1111/febs.13402
192. Wei H, Wei S, Gan B, Peng X, Zou W, Guan J-L. Suppression of autophagy by FIP200 deletion inhibits mammary tumorigenesis. Genes Dev (2011) 25(14):1510–27. doi: 10.1101/gad.2051011
193. Karvela M, Baquero P, Kuntz EM, Mukhopadhyay A, Mitchell R, Allan EK, et al. ATG7 regulates energy metabolism, differentiation and survival of Philadelphia-chromosome-positive cells. Autophagy (2016) 12(6):936–48. doi: 10.1080/15548627.2016.1162359
194. Correia M, Pinheiro P, Batista R, Soares P, Sobrinho-Simões M, Máximo V. Etiopathogenesis of oncocytomas. Semin Cancer Biol (2017) 47:82–94. doi: 10.1016/j.semcancer.2017.06.014
195. Amaravadi R, Kimmelman AC, White E. Recent insights into the function of autophagy in cancer. Genes Dev (2016) 30(17):1913–30. doi: 10.1101/gad.287524.116
196. Vara-Perez M, Felipe-Abrio B, Agostinis P. Mitophagy in Cancer: A Tale of Adaptation. Cells (2019) 8(5):493. doi: 10.3390/cells8050493
197. Semenza GL. Hypoxia-inducible factor 1: Regulator of mitochondrial metabolism and mediator of ischemic preconditioning. Biochim Biophys Acta - Mol Cell Res (2011) 1813(7):1263–8. doi: 10.1016/j.bbamcr.2010.08.006
198. Sowter HM, Ratcliffe PJ, Watson P, Greenberg AH, Harris AL. HIF-1-dependent regulation of hypoxic induction of the cell death factors BNIP3 and NIX in human tumors. Cancer Res (2001) 61(18):6669–73.
199. Lyons A, Coleman M, Riis S, Favre C, O’Flanagan CH, Zhdanov AV, et al. Insulin-like growth factor 1 signaling is essential for mitochondrial biogenesis and mitophagy in cancer cells. J Biol Chem (2017) 292(41):16983–98. doi: 10.1074/jbc.M117.792838
200. Agnihotri S, Golbourn B, Huang X, Remke M, Younger S, Cairns RA, et al. PINK1 Is a Negative Regulator of Growth and the Warburg Effect in Glioblastoma. Cancer Res (2016) 76(16):4708–19. doi: 10.1158/0008-5472.CAN-15-3079
201. Chang HW, Kim MR, Lee HJ, Lee HJ, Kim GC, Lee YS, et al. p53/BNIP3-dependent mitophagy limits glycolytic shift in radioresistant cancer. Oncogene (2019) 38(19):3729–42. doi: 10.1038/s41388-019-0697-6
202. Hoshino A, Ariyoshi M, Okawa Y, Kaimoto S, Uchihashi M, Fukai K, et al. Inhibition of p53 preserves Parkin-mediated mitophagy and pancreatic -cell function in diabetes. Proc Natl Acad Sci (2014) 111(8):3116–21. doi: 10.1073/pnas.1318951111
203. Mariño G, Pietrocola F, Eisenberg T, Kong Y, Malik SA, Andryushkova A, et al. Regulation of Autophagy by Cytosolic Acetyl-Coenzyme A. Mol Cell (2014) 53(5):710–25. doi: 10.1016/j.molcel.2014.01.016
204. Lee JV, Berry CT, Kim K, Sen P, Kim T, Carrer A, et al. Acetyl-CoA promotes glioblastoma cell adhesion and migration through Ca 2+ –NFAT signaling. Genes Dev (2018) 32(7-8):497–511. doi: 10.1101/gad.311027.117
205. Schnittert J, Bansal R, Prakash J. Targeting Pancreatic Stellate Cells in Cancer. Trends Cancer (2019) 5(2):128–42. doi: 10.1016/j.trecan.2019.01.001
206. Sousa CM, Biancur DE, Wang X, Halbrook CJ, Sherman MH, Zhang L, et al. Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature (2016) 536(7617):479–83. doi: 10.1038/nature19084
207. Kim J, Gao P, Liu Y-C, Semenza GL, Dang CV. Hypoxia-Inducible Factor 1 and Dysregulated c-Myc Cooperatively Induce Vascular Endothelial Growth Factor and Metabolic Switches Hexokinase 2 and Pyruvate Dehydrogenase Kinase 1. Mol Cell Biol (2007) 27(21):7381–93. doi: 10.1128/MCB.00440-07
208. Nagao A, Kobayashi M, Koyasu S, Chow CCT, Harada H. HIF-1-Dependent Reprogramming of Glucose Metabolic Pathway of Cancer Cells and Its Therapeutic Significance. Int J Mol Sci (2019) 20(2):238. doi: 10.3390/ijms20020238
209. Dang CV. The Interplay Between MYC and HIF in the Warburg Effect. Ernst Schering Found Symp Proc (2008) 8:35–53. doi: 10.1007/2789_2008_088
210. Xiong J, Wang L, Fei X-C, Jiang X-F, Zheng Z, Zhao Y, et al. MYC is a positive regulator of choline metabolism and impedes mitophagy-dependent necroptosis in diffuse large B-cell lymphoma. Blood Cancer J (2017) 7(7):e582–2. doi: 10.1038/bcj.2017.61
211. Cianfanelli V, Fuoco C, Lorente M, Salazar M, Quondamatteo F, Gherardini PF, et al. Erratum: Corrigendum: AMBRA1 links autophagy to cell proliferation and tumorigenesis by promoting c-Myc dephosphorylation and degradation. Nat Cell Biol (2015) 17(5):706–6. doi: 10.1038/ncb3171
212. GARGINI R, GARCÍA-ESCUDERO V, IZQUIERDO M, WANDOSELL F. Oncogene-mediated tumor transformation sensitizes cells to autophagy induction. Oncol Rep (2016) 35(6):3689–95. doi: 10.3892/or.2016.4699
213. Poillet-Perez L, Xie X, Zhan L, Yang Y, Sharp DW, Hu ZS, et al. Autophagy maintains tumour growth through circulating arginine. Nature (2018) 563(7732):569–73. doi: 10.1038/s41586-018-0697-7
214. Saha S, Panigrahi DP, Patil S, Bhutia SK. Autophagy in health and disease: A comprehensive review. BioMed Pharmacother (2018) 104(February):485–95. doi: 10.1016/j.biopha.2018.05.007
215. Perot BP, Ingersoll MA, Albert ML. The impact of macroautophagy on CD8 + T-cell-mediated antiviral immunity. Immunol Rev (2013) 255(1):40–56. doi: 10.1111/imr.12096
216. Abdoli A, Alirezaei M, Mehrbod P, Forouzanfar F. Autophagy: The multi-purpose bridge in viral infections and host cells. Rev Med Virol (2018) 28(4):e1973. doi: 10.1002/rmv.1973
217. Rey-Jurado E, Riedel CA, González PA, Bueno SM, Kalergis AM. Contribution of autophagy to antiviral immunity. FEBS Lett (2015) 589(22):3461–70. doi: 10.1016/j.febslet.2015.07.047
218. Tang D, Kang R, Coyne CB, Zeh HJ, Lotze MT. PAMPs and DAMPs: signal 0s that spur autophagy and immunity. Immunol Rev (2012) 249(1):158–75. doi: 10.1111/j.1600-065X.2012.01146.x
219. Wang Z, Li C. Xenophagy in innate immunity: A battle between host and pathogen. Dev Comp Immunol (2020) 109(March):103693. doi: 10.1016/j.dci.2020.103693
220. Wu M-F, Chen S-T, Hsieh S-L. Distinct regulation of dengue virus-induced inflammasome activation in human macrophage subsets. J BioMed Sci (2013) 20(1):36. doi: 10.1186/1423-0127-20-36
221. Ojha CR, Rodriguez M, Karuppan MKM, Lapierre J, Kashanchi F, El-Hage N. Toll-like receptor 3 regulates Zika virus infection and associated host inflammatory response in primary human astrocytes. Jin D-Y, ed. PloS One (2019) 14(2):e0208543. doi: 10.1371/journal.pone.0208543
222. Li Y, Wang L-X, Yang G, Hao F, Urba WJ, Hu H-M. Efficient Cross-presentation Depends on Autophagy in Tumor Cells. Cancer Res (2008) 68(17):6889–95. doi: 10.1158/0008-5472.CAN-08-0161
223. Hanada T, Noda NN, Satomi Y, Ichimura Y, Fujioka Y, Takao T, et al. The Atg12-Atg5 Conjugate Has a Novel E3-like Activity for Protein Lipidation in Autophagy. J Biol Chem (2007) 282(52):37298–302. doi: 10.1074/jbc.C700195200
224. Shaw PJ, Lamkanfi M, Kanneganti T-D. NOD-like receptor (NLR) signaling beyond the inflammasome. Eur J Immunol (2010) 40(3):624–7. doi: 10.1002/eji.200940211
225. Elinav E, Strowig T, Henao-Mejia J, Flavell RA. Regulation of the Antimicrobial Response by NLR Proteins. Immunity (2011) 34(5):665–79. doi: 10.1016/j.immuni.2011.05.007
226. Caruso R, Warner N, Inohara N, Núñez G. NOD1 and NOD2: Signaling, Host Defense, and Inflammatory Disease. Immunity (2014) 41(6):898–908. doi: 10.1016/j.immuni.2014.12.010
227. Philpott DJ, Sorbara MT, Robertson SJ, Croitoru K, Girardin SE. NOD proteins: regulators of inflammation in health and disease. Nat Rev Immunol (2014) 14(1):9–23. doi: 10.1038/nri3565
228. Xi J, Yan M, Li S, Song H, Liu L, Shen Z, et al. NOD1 activates autophagy to aggravate hepatic ischemia-reperfusion injury in mice. J Cell Biochem (2019) 120(6):10605–12. doi: 10.1002/jcb.28349
229. Negroni A, Colantoni E, Vitali R, Palone F, Pierdomenico M, Constanzo M, et al. NOD2 induces autophagy to control AIEC bacteria infectiveness in intestinal epithelial cells. Inflamm Res (2016) 65(10):803–13. doi: 10.1007/s00011-016-0964-8
230. Osorio F, Reis e Sousa C. Myeloid C-type Lectin Receptors in Pathogen Recognition and Host Defense. Immunity (2011) 34(5):651–64. doi: 10.1016/j.immuni.2011.05.001
231. Pahari S, Negi S, Aqdas M, Arnett E, Schlesinger LS, Agrewala JN. Induction of autophagy through CLEC4E in combination with TLR4: an innovative strategy to restrict the survival of Mycobacterium tuberculosis. Autophagy (2020) 16(6):1021–43. doi: 10.1080/15548627.2019.1658436
232. Rodriguez KR, Bruns AM, Horvath CM. MDA5 and LGP2: Accomplices and Antagonists of Antiviral Signal Transduction. J Virol (2014) 88(15):8194–200. doi: 10.1128/JVI.00640-14
233. Subramani S, Malhotra V. Non-autophagic roles of autophagy-related proteins. EMBO Rep (2013) 14(2):143–51. doi: 10.1038/embor.2012.220
234. Ahlers LRH, Goodman AG. Nucleic Acid Sensing and Innate Immunity: Signaling Pathways Controlling Viral Pathogenesis and Autoimmunity. Curr Clin Microbiol Rep (2016) 3(3):132–41. doi: 10.1007/s40588-016-0043-5
235. Tian Y, Wang M-L, Zhao J. Crosstalk between Autophagy and Type I Interferon Responses in Innate Antiviral Immunity. Viruses (2019) 11(2):132. doi: 10.3390/v11020132
236. Liang Q, Seo GJ, Choi YJ, Kwak M-J, Ge J, Rodgers MA, et al. Crosstalk between the cGAS DNA Sensor and Beclin-1 Autophagy Protein Shapes Innate Antimicrobial Immune Responses. Cell Host Microbe (2014) 15(2):228–38. doi: 10.1016/j.chom.2014.01.009
237. Randow F, Münz C. Autophagy in the regulation of pathogen replication and adaptive immunity. Trends Immunol (2012) 33(10):475–87. doi: 10.1016/j.it.2012.06.003
238. Van Kaer L, Parekh VV, Postoak JL, Wu L. Role of autophagy in MHC class I-restricted antigen presentation. Mol Immunol (2019) 113(October):2–5. doi: 10.1016/j.molimm.2017.10.021
239. Mintern JD, Macri C, Villadangos JA. Modulation of antigen presentation by intracellular trafficking. Curr Opin Immunol (2015) 34(Mhc I):16–21. doi: 10.1016/j.coi.2014.12.006
240. Joffre OP, Segura E, Savina A, Amigorena S. Cross-presentation by dendritic cells. Nat Rev Immunol (2012) 12(8):557–69. doi: 10.1038/nri3254
241. Noubade R, Majri-Morrison S, Tarbell KV. Beyond cDC1: Emerging Roles of DC Crosstalk in Cancer Immunity. Front Immunol (2019) 10:1014(MAY). doi: 10.3389/fimmu.2019.01014
242. Merzougui N, Kratzer R, Saveanu L, van Endert P. A proteasome-dependent, TAP-independent pathway for cross-presentation of phagocytosed antigen. EMBO Rep (2011) 12(12):1257–64. doi: 10.1038/embor.2011.203
243. Uhl M, Kepp O, Jusforgues-Saklani H, Vicencio J-M, Kroemer G, Albert ML. Autophagy within the antigen donor cell facilitates efficient antigen cross-priming of virus-specific CD8+ T cells. Cell Death Differ (2009) 16(7):991–1005. doi: 10.1038/cdd.2009.8
244. Münz C. Autophagy Beyond Intracellular MHC Class II Antigen Presentation. Trends Immunol (2016) 37(11):755–63. doi: 10.1016/j.it.2016.08.017
245. Schelhaas M. Viruses and cancer: molecular relations and perspectives. Biol Chem (2017) 398(8):815–6. doi: 10.1515/hsz-2017-0176
246. Kvansakul M, Hinds MG. Structural biology of the Bcl-2 family and its mimicry by viral proteins. Cell Death Dis (2013) 4(11):e909–9. doi: 10.1038/cddis.2013.436
247. Wimmer P, Schreiner S. Viral mimicry to usurp ubiquitin and sumo host pathways. Viruses (2015) 7(9):4854–77. doi: 10.3390/v7092849
248. Wong HH, Sanyal S. Manipulation of autophagy by (+) RNA viruses. Semin Cell Dev Biol (2020) 101(March):3–11. doi: 10.1016/j.semcdb.2019.07.013
249. Bird SW, Maynard ND, Covert MW, Kirkegaard K. Nonlytic viral spread enhanced by autophagy components. Proc Natl Acad Sci (2014) 111(36):13081–6. doi: 10.1073/pnas.1401437111
250. Jackson WT, Giddings TH, Taylor MP, Mulinyawe S, Rabinovitch M, Kopito RR, et al. Subversion of Cellular Autophagosomal Machinery by RNA Viruses. Sugden B, ed. PloS Biol (2005) 3(5):e156. doi: 10.1371/journal.pbio.0030156
251. Alirezaei M, Flynn CT, Wood MR, Harkins S, Whitton JL. Coxsackievirus can exploit LC3 in both autophagy-dependent and -independent manners in vivo. Autophagy (2015) 11(8):1389–407. doi: 10.1080/15548627.2015.1063769
252. Mohamud Y, Shi J, Qu J, Poon T, Xue YC, Deng H, et al. Enteroviral Infection Inhibits Autophagic Flux via Disruption of the SNARE Complex to Enhance Viral Replication. Cell Rep (2018) 22(12):3292–303. doi: 10.1016/j.celrep.2018.02.090
253. Yoon SY, Ha YE, Choi JE, Ahn J, Leen H, Kweon H-S, et al. Coxsackievirus B4 Uses Autophagy for Replication after Calpain Activation in Rat Primary Neurons. J Virol (2008) 82(23):11976–8. doi: 10.1128/JVI.01028-08
254. Lee Y-R, Wang P-S, Wang J-R, Liu H-S. Enterovirus 71-induced autophagy increases viral replication and pathogenesis in a suckling mouse model. J BioMed Sci (2014) 21(1):80. doi: 10.1186/s12929-014-0080-4
255. Klein KA, Jackson WT. Human Rhinovirus 2 Induces the Autophagic Pathway and Replicates More Efficiently in Autophagic Cells. J Virol (2011) 85(18):9651–4. doi: 10.1128/JVI.00316-11
256. O’Donnell V, Pacheco JM, LaRocco M, Burrage T, Jackson W, Rodríguez LL, et al. Foot-and-mouth disease virus utilizes an autophagic pathway during viral replication. Virology (2011) 410(1):142–50. doi: 10.1016/j.virol.2010.10.042
257. Hou L, Ge X, Xin L, Zhou L, Guo X, Yang H. Nonstructural proteins 2C and 3D are involved in autophagy as induced by the encephalomyocarditis virus. Virol J (2014) 11(1):156. doi: 10.1186/1743-422X-11-156
258. Heaton NS, Randall G. Dengue Virus-Induced Autophagy Regulates Lipid Metabolism. Cell Host Microbe (2010) 8(5):422–32. doi: 10.1016/j.chom.2010.10.006
259. Zhang J, Lan Y, Li MY, Lamers MM, Fusade-Boyer M, Klemm E, et al. Flaviviruses Exploit the Lipid Droplet Protein AUP1 to Trigger Lipophagy and Drive Virus Production. Cell Host Microbe (2018) 23(6):819–31.e5. doi: 10.1016/j.chom.2018.05.005
260. Offerdahl DK, Dorward DW, Hansen BT, Bloom ME. Cytoarchitecture of Zika virus infection in human neuroblastoma and Aedes albopictus cell lines. Virology (2017) 501(November 2016):54–62. doi: 10.1016/j.virol.2016.11.002
261. Cortese M, Goellner S, Acosta EG, Neufeldt CJ, Oleksiuk O, Lampe M, et al. Ultrastructural Characterization of Zika Virus Replication Factories. Cell Rep (2017) 18(9):2113–23. doi: 10.1016/j.celrep.2017.02.014
262. de Haan CAM, Molinari M, Reggiori F. Autophagy-independent LC3 function in vesicular traffic. Autophagy (2010) 6(7):994–6. doi: 10.4161/auto.6.7.13309
263. Sun Y, Yu S, Ding N, Meng C, Meng S, Zhang S, et al. Autophagy Benefits the Replication of Newcastle Disease Virus in Chicken Cells and Tissues. J Virol (2014) 88(1):525–37. doi: 10.1128/JVI.01849-13
264. Prentice E, Jerome WG, Yoshimori T, Mizushima N, Denison MR. Coronavirus Replication Complex Formation Utilizes Components of Cellular Autophagy. J Biol Chem (2004) 279(11):10136–41. doi: 10.1074/jbc.M306124200
265. Krejbich-Trotot P, Gay B, Li-Pat-Yuen G, Hoarau J-J, Jaffar-Bandjee M-C, Briant L, et al. Chikungunya triggers an autophagic process which promotes viral replication. Virol J (2011) 8(1):432. doi: 10.1186/1743-422X-8-432
266. Sharma M, Bhattacharyya S, Sharma KB, Chauhan S, Asthana S, Abdin MZ, et al. Japanese encephalitis virus activates autophagy through XBP1 and ATF6 ER stress sensors in neuronal cells. J Gen Virol (2017) 98(5):1027–39. doi: 10.1099/jgv.0.000792
267. Dales S, Eggers HJ, Tamm I, Palade GE. Electron microscopic study of the formation of poliovirus. Virology (1965) 26(3):379–89. doi: 10.1016/0042-6822(65)90001-2
268. Jackson WT. Poliovirus-induced changes in cellular membranes throughout infection. Curr Opin Virol (2014) 9:67–73. doi: 10.1016/j.coviro.2014.09.007
269. Corona Velazquez A, Corona AK, Klein KA, Jackson WT. Poliovirus induces autophagic signaling independent of the ULK1 complex. Autophagy (2018) 14(7):1201–13. doi: 10.1080/15548627.2018.1458805
270. Wang H, Li K, Ma L, Wu S, Hu J, Yan H, et al. Berberine inhibits enterovirus 71 replication by downregulating the MEK/ERK signaling pathway and autophagy. Virol J (2017) 14(1):2. doi: 10.1186/s12985-016-0674-4
271. Rozières A, Viret C, Faure M. Autophagy in Measles Virus Infection. Viruses (2017) 9(12):359. doi: 10.3390/v9120359
272. Chiramel A, Brady N, Bartenschlager R. Divergent Roles of Autophagy in Virus Infection. Cells (2013) 2(1):83–104. doi: 10.3390/cells2010083
273. Joubert P-E, Meiffren G, Grégoire IP, Guillemette P, Clémence R, Flacher M, et al. Autophagy Induction by the Pathogen Receptor CD46. Cell Host Microbe (2009) 6(4):354–66. doi: 10.1016/j.chom.2009.09.006
274. Richetta C, Grégoire IP, Verlhac P, Azocar O, Baguet J, Flacher M, et al. Sustained Autophagy Contributes to Measles Virus Infectivity. Deretic V, ed. PloS Pathog (2013) 9(9):e1003599. doi: 10.1371/journal.ppat.1003599
276. Moir S, Fauci AS. B-cell responses to HIV infection. Immunol Rev (2017) 275(1):33–48. doi: 10.1111/imr.12502
277. Yarchoan R, Uldrick TS. HIV-Associated Cancers and Related Diseases. Longo DL, ed. N Engl J Med (2018) 378(11):1029–41. doi: 10.1056/NEJMra1615896
278. Wood NH, Feller L. The malignant potential of HIV-associated Kaposi sarcoma. Cancer Cell Int (2008) 8(1):14. doi: 10.1186/1475-2867-8-14
279. Lu D-Y, Wu H-Y, Yarla NS, Xu B, Ding J, Lu T-R. HAART in HIV/AIDS Treatments: Future Trends. Infect Disord Drug Targets (2018) 18(1):15–22. doi: 10.2174/1871526517666170505122800
280. Tang S-W, Ducroux A, Jeang K-T, Neuveut C. Impact of cellular autophagy on viruses: Insights from hepatitis B virus and human retroviruses. J BioMed Sci (2012) 19(1):92. doi: 10.1186/1423-0127-19-92
281. Castro-Gonzalez S, Shi Y, Colomer-Lluch M, Song Y, Mowery K, Almodovar S, et al. HIV-1 Nef counteracts autophagy restriction by enhancing the association between BECN1 and its inhibitor BCL2 in a PRKN-dependent manner. Autophagy (2020) 00(00):1–25. doi: 10.1080/15548627.2020.1725401
283. Wang X, Gao Y, Tan J, Devadas K, Ragupathy V, Takeda K, et al. HIV-1 and HIV-2 infections induce autophagy in Jurkat and CD4+ T cells. Cell Signal (2012) 24(7):1414–9. doi: 10.1016/j.cellsig.2012.02.016
284. Neher RA, Dyrdak R, Druelle V, Hodcroft EB, Albert J. Potential impact of seasonal forcing on a SARS-CoV-2 pandemic. Swiss Med Wkly (2020) 150(March):w20224. doi: 10.4414/smw.2020.20224
285. Raoult D, Zumla A, Locatelli F, Ippolito G, Kroemer G. Coronavirus infections: Epidemiological, clinical and immunological features and hypotheses. Cell Stress (2020) 4(4):66–75. doi: 10.15698/cst2020.04.216
286. Carmona-Gutierrez D, Bauer MA, Zimmermann A, Kainz K, Hofer SJ, Kroemer G, et al. Digesting the crisis: autophagy and coronaviruses. Microb Cell (2020) 2020:7(5):119–128. doi: 10.15698/mic2020.05.715
287. Petrova VN, Russell CA. The evolution of seasonal influenza viruses. Nat Rev Microbiol (2018) 16(1):47–60. doi: 10.1038/nrmicro.2017.118
288. Paulino J, Pang X, Hung I, Zhou H-X, Cross TA. Influenza A M2 Channel Clustering at High Protein/Lipid Ratios: Viral Budding Implications. Biophys J (2019) 116(6):1075–84. doi: 10.1016/j.bpj.2019.01.042
289. Beale R, Wise H, Stuart A, Ravenhill BJ, Digard P, Randow F. A LC3-Interacting Motif in the Influenza A Virus M2 Protein Is Required to Subvert Autophagy and Maintain Virion Stability. Cell Host Microbe (2014) 15(2):239–47. doi: 10.1016/j.chom.2014.01.006
290. Joubert P-E, Werneke SW, de la Calle C, Florence G-B, Alessandra G, Lucie P, et al. Chikungunya virus–induced autophagy delays caspase-dependent cell death. J Exp Med (2012) 209(5):1029–47. doi: 10.1084/jem.20110996
291. Jordan TX, Randall G. Manipulation or capitulation: virus interactions with autophagy. Microbes Infect (2012) 14(2):126–39. doi: 10.1016/j.micinf.2011.09.007
292. Zompi S, Harris E. Animal Models of Dengue Virus Infection. Viruses (2012) 4(1):62–82. doi: 10.3390/v4010062
293. Heaton NS, Randall G. Dengue Virus and Autophagy. Viruses (2011) 3(8):1332–41. doi: 10.3390/v3081332
294. Musso D, Ko AI, Baud D. Zika Virus Infection — After the Pandemic. Longo DL, ed. N Engl J Med (2019) 381(15):1444–57. doi: 10.1056/NEJMra1808246
295. Chiramel AI, Best SM. Role of autophagy in Zika virus infection and pathogenesis. Virus Res (2018) 254:34–40. doi: 10.1016/j.virusres.2017.09.006
296. Fishbein A, Hammock BD, Serhan CN, Panigrahy D. Carcinogenesis: Failure of resolution of inflammation? Pharmacol Ther (2020) 61(18):107670. doi: 10.1016/j.pharmthera.2020.107670
297. Kakavandi E, Shahbahrami R, Goudarzi H, Eslami G, Faghihloo E. Anoikis resistance and oncoviruses. J Cell Biochem (2018) 119(3):2484–91. doi: 10.1002/jcb.26363
298. Tornesello ML, Annunziata C, Tornesello AL, Buonaguro L, Buonaguro FM. Human Oncoviruses and p53 Tumor Suppressor Pathway Deregulation at the Origin of Human Cancers. Cancers (Basel) (2018) 10(7):213. doi: 10.3390/cancers10070213
299. Mesri EA, Feitelson MA, Munger K. Human Viral Oncogenesis: A Cancer Hallmarks Analysis. Cell Host Microbe (2014) 15(3):266–82. doi: 10.1016/j.chom.2014.02.011
300. Mui U, Haley C, Tyring S. Viral Oncology: Molecular Biology and Pathogenesis. J Clin Med (2017) 6(12):111. doi: 10.3390/jcm6120111
301. Krump NA, You J. Molecular mechanisms of viral oncogenesis in humans. Nat Rev Microbiol (2018) 16(11):684–98. doi: 10.1038/s41579-018-0064-6
302. Tornesello ML, Buonaguro L, Buonaguro FM. An overview of new biomolecular pathways in pathogen-related cancers. Future Oncol (2015) 11(11):1625–39. doi: 10.2217/fon.15.87
303. Akram N, Imran M, Noreen M, Ahmed F, Atif M, Fatima Z, et al. Oncogenic Role of Tumor Viruses in Humans. Viral Immunol (2017) 30(1):20–7. doi: 10.1089/vim.2016.0109
304. Jackson WT. Viruses and the autophagy pathway. Virology (2015) 479-480:450–6. doi: 10.1016/j.virol.2015.03.042
305. Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature (2011) 469(7330):323–35. doi: 10.1038/nature09782
306. Haley CT, Mui UN, Vangipuram R, Rady PL, Tyring SK. Human oncoviruses: Mucocutaneous manifestations, pathogenesis, therapeutics, and prevention. J Am Acad Dermatol (2019) 81(1):1–21. doi: 10.1016/j.jaad.2018.09.062
307. Ringehan M, McKeating JA, Protzer U. Viral hepatitis and liver cancer. Philos Trans R Soc B Biol Sci (2017) 372(1732):20160274. doi: 10.1098/rstb.2016.0274
308. Cao J, Li D. Searching for human oncoviruses: Histories, challenges, and opportunities. J Cell Biochem (2018) 119(6):4897–906. doi: 10.1002/jcb.26717
309. Herbein G. The Human Cytomegalovirus, from Oncomodulation to Oncogenesis. Viruses (2018) 10(8):408. doi: 10.3390/v10080408
310. Yamagishi M, Fujikawa D, Watanabe T, Uchimaru K. HTLV-1-Mediated Epigenetic Pathway to Adult T-Cell Leukemia–Lymphoma. Front Microbiol (2018) 9:1686(JUL). doi: 10.3389/fmicb.2018.01686
311. Vescovo T, Pagni B, Piacentini M, Fimia GM, Antonioli M. Regulation of Autophagy in Cells Infected With Oncogenic Human Viruses and Its Impact on Cancer Development. Front Cell Dev Biol (2020) 8:47(February). doi: 10.3389/fcell.2020.00047
312. McElwee M, Vijayakrishnan S, Rixon F, Bhella D. Structure of the herpes simplex virus portal-vertex. Sugden B, ed. PloS Biol (2018) 16(6):e2006191. doi: 10.1371/journal.pbio.2006191
313. Agelidis AM, Shukla D. Cell entry mechanisms of HSV: what we have learned in recent years. Future Virol (2015) 10(10):1145–54. doi: 10.2217/fvl.15.85
314. Jha H, Banerjee S, Robertson E. The Role of Gammaherpesviruses in Cancer Pathogenesis. Pathogens (2016) 5(1):18. doi: 10.3390/pathogens5010018
315. Chang Y, Moore PS, Weiss RA. Human oncogenic viruses: nature and discovery. Philos Trans R Soc B Biol Sci (2017) 372(1732):20160264. doi: 10.1098/rstb.2016.0264
316. Cirone M. EBV and KSHV Infection Dysregulates Autophagy to Optimize Viral Replication, Prevent Immune Recognition and Promote Tumorigenesis. Viruses (2018) 10(11):599. doi: 10.3390/v10110599
317. Chang Y, Cesarman E, Pessin M, Lee F, Culpepper J, Knowles D, et al. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi’s sarcoma. Science (80- ) (1994) 266(5192):1865–9. doi: 10.1126/science.7997879
318. Calabrò ML, Sarid R. Human Herpesvirus 8 And Lymphoproliferative Diseases. Mediterr J Hematol Infect Dis (2018) 10(1):e2018061. doi: 10.4084/mjhid.2018.061
319. De Paoli P, Carbone A. Kaposi’s Sarcoma Herpesvirus: twenty years after its discovery. Eur Rev Med Pharmacol Sci (2016) 20(7):1288–94.
320. Wakeman BS, Izumiya Y, Speck SH. Identification of Novel Kaposi’s Sarcoma-Associated Herpesvirus Orf50 Transcripts: Discovery of New RTA Isoforms with Variable Transactivation Potential. Jung JU, ed. J Virol (2017) 91(1):1–23. doi: 10.1128/JVI.01434-16
321. Cesarman E, Damania B, Krown SE, Martin J, Bower M, Whitby D. Kaposi sarcoma. Nat Rev Dis Primers (2019) 5(1):9. doi: 10.1038/s41572-019-0060-9
322. Veettil M, Bandyopadhyay C, Dutta D, Chandran B. Interaction of KSHV with Host Cell Surface Receptors and Cell Entry. Viruses (2014) 6(10):4024–46. doi: 10.3390/v6104024
323. Giffin L, Damania B. Chapter Two-KSHV: Pathways to Tumorigenesis and Persisten Infection. Advances in Virus Research, 1st ed, vol. 88. . Elsevier Inc (2014). p. 111–59. doi: 10.1016/B978-0-12-800098-4.00002-7
324. Rosario SA, Santiago GE, Mesri EA, Verdun RE. Kaposi’s Sarcoma-Associated Herpesvirus-Encoded Viral IL-6 (vIL-6) Enhances Immunoglobulin Class-Switch Recombination. Front Microbiol (2018) 9:3119(December). doi: 10.3389/fmicb.2018.03119
325. Choi Y, Bowman JW, Jung JU. Autophagy during viral infection — a double-edged sword. Nat Rev Microbiol (2018) 16(6):341–54. doi: 10.1038/s41579-018-0003-6
326. Ueda K. KSHV Genome Replication and Maintenance in Latency. Adv Exp Med Biol (2018) 1045:299–320. doi: 10.1007/978-981-10-7230-7_14
327. Wang L. The Kaposi’s Sarcoma-Associated Herpesvirus (KSHV/HHV-8) K1 Protein Induces Expression of Angiogenic and Invasion Factors. Cancer Res (2004) 64(8):2774–81. doi: 10.1158/0008-5472.CAN-03-3653
328. Leidal AM, Cyr DP, Hill RJ, Lee PWK, McCormick C. Subversion of Autophagy by Kaposi’s Sarcoma-Associated Herpesvirus Impairs Oncogene-Induced Senescence. Cell Host Microbe (2012) 11(2):167–80. doi: 10.1016/j.chom.2012.01.005
329. Anders PM, Zhang Z, Bhende PM, Giffin L, Damania B. The KSHV K1 Protein Modulates AMPK Function to Enhance Cell Survival. Schulz TF, ed. PloS Pathog (2016) 12(11):e1005985. doi: 10.1371/journal.ppat.1005985
330. Yin H, Shao S, Jiang X, Xie P, Sun W, Yu T. Interactions between Autophagy and DNA Viruses. Viruses (2019) 11(9):776. doi: 10.3390/v11090776
331. He M, Zhang W, Bakken T, Schutten M, Toth Z, Jung JU, et al. Cancer Angiogenesis Induced by Kaposi Sarcoma-Associated Herpesvirus Is Mediated by EZH2. Cancer Res (2012) 72(14):3582–92. doi: 10.1158/0008-5472.CAN-11-2876
332. Granato M, Santarelli R, Filardi M, Gonnrlls R, Farina A, Torrisi MR, et al. The activation of KSHV lytic cycle blocks autophagy in PEL cells. Autophagy (2015) 11(11):1978–86. doi: 10.1080/15548627.2015.1091911
333. Santarelli R, Gonnella R, Di Giovenale G, Cuomo L, Capobianchi A, Granato M, et al. STAT3 activation by KSHV correlates with IL-10, IL-6 and IL-23 release and an autophagic block in dendritic cells. Sci Rep (2015) 4(1):4241. doi: 10.1038/srep04241
334. Yan L, Majerciak V, Zheng Z-M, Lan K. Towards Better Understanding of KSHV Life Cycle: from Transcription and Posttranscriptional Regulations to Pathogenesis. Virol Sin (2019) 34(2):135–61. doi: 10.1007/s12250-019-00114-3
335. Broussard G, Damania B. Regulation of KSHV Latency and Lytic Reactivation. Viruses (2020) 12(9):1034. doi: 10.3390/v12091034
336. De Leo A, Chen H-S, Hu C-CA, Lieberman PM. Deregulation of KSHV latency conformation by ER-stress and caspase-dependent RAD21-cleavage. Flemington EK, ed. PloS Pathog (2017) 13(8):e1006596. doi: 10.1371/journal.ppat.1006596
337. Guito J, Lukac D. KSHV Reactivation and Novel Implications of Protein Isomerization on Lytic Switch Control. Viruses (2015) 7(1):72–109. doi: 10.3390/v7010072
338. Pringle ES, Robinson C-A, McCormick C. Kaposi’s Sarcoma-Associated Herpesvirus Lytic Replication Interferes with mTORC1 Regulation of Autophagy and Viral Protein Synthesis. Jung JU, ed. J Virol (2019) 93(21):1–22. doi: 10.1128/JVI.00854-19
339. Wen H-J, Yang Z, Zhou Y, Wood C. Enhancement of Autophagy during Lytic Replication by the Kaposi’s Sarcoma-Associated Herpesvirus Replication and Transcription Activator. J Virol (2010) 84(15):7448–58. doi: 10.1128/JVI.00024-10
340. Mariggiò G, Koch S, Zhang G, Weidner-Glunde M, Rϋckert J, Kati S, et al. Kaposi Sarcoma Herpesvirus (KSHV) Latency-Associated Nuclear Antigen (LANA) recruits components of the MRN (Mre11-Rad50-NBS1) repair complex to modulate an innate immune signaling pathway and viral latency. Robertson ES, ed. PloS Pathog (2017) 13(4):e1006335. doi: 10.1371/journal.ppat.1006335
341. Gilardini Montani MS, Falcinelli L, Santarelli R, Romeo MA, Granato M, Faggioni A, et al. Kaposi Sarcoma Herpes Virus (KSHV) infection inhibits macrophage formation and survival by counteracting Macrophage Colony-Stimulating Factor (M-CSF)-induced increase of Reactive Oxygen Species (ROS), c-Jun N-terminal kinase (JNK) phosphorylation and auto. Int J Biochem Cell Biol (2019) 114(June):105560. doi: 10.1016/j.biocel.2019.06.008
342. Cai Q, Chen K, Young KH. Epstein–Barr virus-positive T/NK-cell lymphoproliferative disorders. Exp Mol Med (2015) 47(1):e133–3. doi: 10.1038/emm.2014.105
343. Shinozaku-Ushiku A, Kunita A, Fukuyama M. Update on Epstein-Barr virus and gastric cancer (Review). Int J Oncol (2015) 46(4):1421–34. doi: 10.3892/ijo.2015.2856
344. Stanfield BA, Luftig MA. Recent advances in understanding Epstein-Barr virus. F1000Research (2017) 6(0):386. doi: 10.12688/f1000research.10591.1
345. Pujals A, Favre L, Pioche-Durieu C, Robert A, Meurice G, Le Gentil M, et al. Constitutive autophagy contributes to resistance to TP53-mediated apoptosis in Epstein-Barr virus-positive latency III B-cell lymphoproliferations. Autophagy (2015) 11(12):2275–87. doi: 10.1080/15548627.2015.1115939
346. Farrell PJ. Epstein–Barr Virus and Cancer. Annu Rev Pathol Mech Dis (2019) 14(1):29–53. doi: 10.1146/annurev-pathmechdis-012418-013023
347. Ascherio A, Munger KL. Epstein–Barr Virus Infection and Multiple Sclerosis: A Review. J Neuroimmune Pharmacol (2010) 5(3):271–7. doi: 10.1007/s11481-010-9201-3
348. Chen J. Roles of the PI3K/Akt pathway in Epstein-Barr virus-induced cancers and therapeutic implications. World J Virol (2012) 1(6):154. doi: 10.5501/wjv.v1.i6.154
349. Fang W-L, Huang K-H, Lan Y-T, Lin C-H, Chang S-C, Chen M-H, et al. Mutations in PI3K/AKT pathway genes and amplifications of PI3KCA are associated with patterns of recurrence in gastric cancers. Oncotarget (2016) 7(5):6201–20. doi: 10.18632/oncotarget.6641
350. Cen O, Longnecker R. Latent Membrane Protein 2 (LMP2). Epstein Barr Virus Volume 2. Curr Top Microbiol Immunol (2015) 391:151–80. doi: 10.1007/978-3-319-22834-1_5
351. Fish K, Sora RP, Schaller SJ, Longnecker R, Ikeda M. EBV latent membrane protein 2A orchestrates p27kip1 degradation via Cks1 to accelerate MYC-driven lymphoma in mice. Blood (2017) 130(23):2516–26. doi: 10.1182/blood-2017-07-796821
352. Smatti MK, Al-Sadeq DW, Ali NH, Pintus G, Abou-Saleh H, Nasrallah GK. Epstein–Barr Virus Epidemiology, Serology, and Genetic Variability of LMP-1 Oncogene Among Healthy Population: An Update. Front Oncol (2018) 8:211(JUN):1–16. doi: 10.3389/fonc.2018.00211
353. Hurwitz SN, Cheerathodi MR, Nkosi D, York SB, Meckes DG. Tetraspanin CD63 Bridges Autophagic and Endosomal Processes To Regulate Exosomal Secretion and Intracellular Signaling of Epstein-Barr Virus LMP1. Longnecker RM, ed. J Virol (2017) 92(5):1–21. doi: 10.1128/JVI.01969-17
354. Bhattacharjee S, Bose P, Patel K, Roy SG, Gain C, Gowda H, et al. Transcriptional and epigenetic modulation of autophagy promotes EBV oncoprotein EBNA3C induced B-cell survival. Cell Death Dis (2018) 9(6):605. doi: 10.1038/s41419-018-0668-9
355. De Leo A, Colavita F, Ciccosanti F, Fimia GM, Lieberman PM, Mattia E. Inhibition of autophagy in EBV-positive Burkitt’s lymphoma cells enhances EBV lytic genes expression and replication. Cell Death Dis (2015) 6(9):e1876–6. doi: 10.1038/cddis.2015.156
356. Nowag H, Guhl B, Thriene K, Romao S, Ziegler U, Dengjel J, et al. Macroautophagy Proteins Assist Epstein Barr Virus Production and Get Incorporated Into the Virus Particles. EBioMedicine (2014) 1(2-3):116–25. doi: 10.1016/j.ebiom.2014.11.007
357. Granato M, Santarelli R, Farina A, Gonnela R, Lotti LV, Faggioni A, et al. Epstein-Barr Virus Blocks the Autophagic Flux and Appropriates the Autophagic Machinery To Enhance Viral Replication. J Virol (2014) 88(21):12715–26. doi: 10.1128/JVI.02199-14
358. Hung C-H, Chen L-W, Wang W-H, Chang P-J, Chiu Y-F, Hung C-C, et al. Regulation of Autophagic Activation by Rta of Epstein-Barr Virus via the Extracellular Signal-Regulated Kinase Pathway. J Virol (2014) 88(20):12133–45. doi: 10.1128/JVI.02033-14
359. Khanim F, Mackett M, Young LS, Dawson J, Meseda CA, Dawson C. BHRF1, a viral homologue of the Bcl-2 oncogene, is conserved at both the sequence and functional level in different Epstein-Barr virus isolates. J Gen Virol (1997) 78(11):2987–99. doi: 10.1099/0022-1317-78-11-2987
360. Marshall WL, Yim C, Gustafson E, Graf T, Sage DR, Hanify K, et al. Epstein-Barr Virus Encodes a Novel Homolog of the bcl-2 Oncogene That Inhibits Apoptosis and Associates with Bax and Bak. J Virol (1999) 73(6):5181–5. doi: 10.1128/JVI.73.6.5181-5185.1999
361. Altmann M, Hammerschmidt W. Epstein-Barr Virus Provides a New Paradigm: A Requirement for the Immediate Inhibition of Apoptosis. Sugden B, ed. PloS Biol (2005) 3(12):e404. doi: 10.1371/journal.pbio.0030404
362. Henderson S, Huen D, Rowe M, Dawson C, Johnson G, Rickinson A. Epstein-Barr virus-coded BHRF1 protein, a viral homologue of Bcl-2, protects human B cells from programmed cell death. Proc Natl Acad Sci U S A (1993) 90(18):8479–83. doi: 10.1073/pnas.90.18.8479
363. Bellows DS, Howell M, Pearson C, Hazlewood SA, Hardwick JM. Epstein-Barr Virus BALF1 Is a BCL-2-Like Antagonist of the Herpesvirus Antiapoptotic BCL-2 Proteins. J Virol (2002) 76(5):2469–79. doi: 10.1128/jvi.76.5.2469-2479.2002
364. Shao Z, Borde C, Quignon F, Escargueil A, Maréchal V. Epstein–Barr Virus BALF0 and BALF1 Modulate Autophagy. Viruses (2019) 11(12):1099. doi: 10.3390/v11121099
365. Plotkin SA, Boppana SB. Vaccination against the human cytomegalovirus. Vaccine (2019) 37(50):7437–42. doi: 10.1016/j.vaccine.2018.02.089
366. Lussignol M, Esclatine A. Cytomegalovirus and Autophagy. Immunology (2018) 1:9–21. doi: 10.1016/B978-0-12-809819-6.00002-2
367. Joseph GP, McDermott R, Baryshnikova MA, Cobbs CS, Ulasov IV. Cytomegalovirus as an oncomodulatory agent in the progression of glioma. Cancer Lett (2017) 384:79–85. doi: 10.1016/j.canlet.2016.10.022
368. Chaumorcel M, Lussignol M, Mouna L, Cavignac Y, Fahie K, Cotte-Laffitte J, et al. The Human Cytomegalovirus Protein TRS1 Inhibits Autophagy via Its Interaction with Beclin 1. J Virol (2012) 86(5):2571–84. doi: 10.1128/JVI.05746-11
369. Mouna L, Hernandez E, Bonte D, Brost R, Amazit L, Delgui LR, et al. Analysis of the role of autophagy inhibition by two complementary human cytomegalovirus BECN1/Beclin 1-binding proteins. Autophagy (2016) 12(2):327–42. doi: 10.1080/15548627.2015.1125071
370. Mlera L, Moy M, Maness K, Tran LN, Goodrum FD. The Role of the Human Cytomegalovirus UL133-UL138 Gene Locus in Latency and Reactivation. Viruses (2020) 12(7):714. doi: 10.3390/v12070714
371. Tey SK, Khanna R. Autophagy mediates transporter associated with antigen processing- independent presentation of viral epitopes through MHC class I pathway. Blood (2012) 120(5):994–1004. doi: 10.1182/blood-2012-01-402404
372. Loi M, Müller A, Steinbach K, Niven J, Barreira da Silva R, Paul P, et al. Macroautophagy Proteins Control MHC Class I Levels on Dendritic Cells and Shape Anti-viral CD8 + T Cell Responses. Cell Rep (2016) 15(5):1076–87. doi: 10.1016/j.celrep.2016.04.002
373. Reddehase MJ, Lemmermann NAW. Cellular reservoirs of latent cytomegaloviruses. Med Microbiol Immunol (2019) 208(3-4):391–403. doi: 10.1007/s00430-019-00592-y
374. Møller R, Schwarz TM, Noriega VM, Panis M, Sachs D, Tortorella D, et al. miRNA-mediated targeting of human cytomegalovirus reveals biological host and viral targets of IE2. Proc Natl Acad Sci (2018) 115(5):1069–74. doi: 10.1073/pnas.1719036115
375. Li M, Ball CB, Collins G, Hu Q, Luse DS, Price DH, et al. Human cytomegalovirus IE2 drives transcription initiation from a select subset of late infection viral promoters by host RNA polymerase II. Glaunsinger BA, ed. PloS Pathog (2020) 16(4):e1008402. doi: 10.1371/journal.ppat.1008402
376. Zhang X, Xi T, Zhang L, Bi Y, Huang Y, Lu Y, et al. The role of autophagy in human cytomegalovirus IE2 expression. J Med Virol 2020) 25:jmv.26357. doi: 10.1002/jmv.26357
377. Martin J, Maldonado J, Mueller J, Zhang W, Mansky L. Molecular Studies of HTLV-1 Replication: An Update. Viruses (2016) 8(2):31. doi: 10.3390/v8020031
378. Poiesz BJ, Ruscetti FW, Gazdar AF, Bunn PA, Minna JD, Gallo RC. Detection and isolation of type C retrovirus particles from fresh and cultured lymphocytes of a patient with cutaneous T-cell lymphoma. Proc Natl Acad Sci (1980) 77(12):7415–9. doi: 10.1073/pnas.77.12.7415
379. Futsch N, Prates G, Mahieux R, Casseb J, Dutartre H. Cytokine Networks Dysregulation during HTLV-1 Infection and Associated Diseases. Viruses (2018) 10(12):691. doi: 10.3390/v10120691
380. Schierhout G, McGregor S, Gessain A, Einsiedel L, Martinello M, Kaldor J. Association between HTLV-1 infection and adverse health outcomes: a systematic review and meta-analysis of epidemiological studies. Lancet Infect Dis (2020) 20(1):133–43. doi: 10.1016/S1473-3099(19)30402-5
381. Gessain A, Cassar O. Epidemiological Aspects and World Distribution of HTLV-1 Infection. Front Microbiol (2012) 3:388(NOV). doi: 10.3389/fmicb.2012.00388
382. Tang S-W, Chen C-Y, Klase Z, Zane L, Jeang K-T. The Cellular Autophagy Pathway Modulates Human T-Cell Leukemia Virus Type 1 Replication. J Virol (2013) 87(3):1699–707. doi: 10.1128/JVI.02147-12
383. Chen L, Liu D, Zhang Y, Zhang H, Cheng H. The autophagy molecule Beclin 1 maintains persistent activity of NF-κB and Stat3 in HTLV-1-transformed T lymphocytes. Biochem Biophys Res Commun (2015) 465(4):739–45. doi: 10.1016/j.bbrc.2015.08.070
384. Nakahata S, Syahrul C, Nakatake A, Sakamoto K, Yoshihama M, Nishikata I, et al. Clinical significance of soluble CADM1 as a novel marker for adult T-cell leukemia/lymphoma. Haematologica (2020) 106(2):532–42. doi: 10.3324/haematol.2019.234096
385. Sarkar B, Nishikata I, Nakahata S, Ichikawa T, Shiraga T, Saha HR, et al. Degradation of p47 by autophagy contributes to CADM1 overexpression in ATLL cells through the activation of NF-κB. Sci Rep (2019) 9(1):3491. doi: 10.1038/s41598-019-39424-7
386. Mukai R, Ohshima T. HTLV-1 HBZ positively regulates the mTOR signaling pathway via inhibition of GADD34 activity in the cytoplasm. Oncogene (2014) 33(18):2317–28. doi: 10.1038/onc.2013.181
387. Baratella M, Forlani G, Accolla RS. HTLV-1 HBZ Viral Protein: A Key Player in HTLV-1 Mediated Diseases. Front Microbiol (2017) 8:2615(DEC). doi: 10.3389/fmicb.2017.02615
388. Ren T, Takahashi Y, Liu X, Loughran TP, Sun S-C, Wang H-G, et al. HTLV-1 Tax deregulates autophagy by recruiting autophagic molecules into lipid raft microdomains. Oncogene (2015) 34(3):334–45. doi: 10.1038/onc.2013.552
389. Manns MP, Buti M, Gane E, Pawlotsky J-M, Razavi H, Terrault N, et al. Hepatitis C virus infection. Nat Rev Dis Primers (2017) 3(1):17006. doi: 10.1038/nrdp.2017.6
390. Balakrishnan M, Glover MT, Kanwal F. Hepatitis C and Risk of Nonhepatic Malignancies. Clin Liver Dis (2017) 21(3):543–54. doi: 10.1016/j.cld.2017.03.009
391. Pol S, Vallet-Pichard A, Hermine O. Extrahepatic cancers and chronic HCV infection. Nat Rev Gastroenterol Hepatol (2018) 15(5):283–90. doi: 10.1038/nrgastro.2017.172
392. Paul D, Hoppe S, Saher G, Krijnse-Locker J, Bartenschlager R. Morphological and Biochemical Characterization of the Membranous Hepatitis C Virus Replication Compartment. J Virol (2013) 87(19):10612–27. doi: 10.1128/JVI.01370-13
393. Ferraris P, Beaumont E, Uzbekov R, Brand D, Gaillard J, Blanchard E, et al. Sequential biogenesis of host cell membrane rearrangements induced by hepatitis C virus infection. Cell Mol Life Sci (2013) 70(7):1297–306. doi: 10.1007/s00018-012-1213-0
394. Vescovo T, Refolo G, Romagnoli A, Ciccosanti F, Corazzari M, Alonzi T, et al. Autophagy in HCV Infection: Keeping Fat and Inflammation at Bay. BioMed Res Int (2014) 2014:1–10. doi: 10.1155/2014/265353
395. Fahmy AM, Labonté P. The autophagy elongation complex (ATG5-12/16L1) positively regulates HCV replication and is required for wild-type membranous web formation. Sci Rep (2017) 7(1):40351. doi: 10.1038/srep40351
396. Wang L, James Ou J. Hepatitis C virus and autophagy. Biol Chem (2015) 396(11):1215–22. doi: 10.1515/hsz-2015-0172
397. Jassey A, Liu C-H, Changou C, Richardson C, Hsu H-Y, Lin L-T. Hepatitis C Virus Non-Structural Protein 5A (NS5A) Disrupts Mitochondrial Dynamics and Induces Mitophagy. Cells (2019) 8(4):290. doi: 10.3390/cells8040290
398. Kim S-J, Syed GH, Siddiqui A. Hepatitis C Virus Induces the Mitochondrial Translocation of Parkin and Subsequent Mitophagy. Ou JJ, ed. PloS Pathog (2013) 9(3):e1003285. doi: 10.1371/journal.ppat.1003285
399. Seto W-K, Lo Y-R, Pawlotsky J-M, Yuen M-F. Chronic hepatitis B virus infection. Lancet (2018) 392(10161):2313–24. doi: 10.1016/S0140-6736(18)31865-8
400. Herrscher C, Roingeard P, Blanchard E. Hepatitis B Virus Entry into Cells. Cells (2020) 9(6):1486. doi: 10.3390/cells9061486
401. Schinzari V, Barnaba V, Piconese S. Chronic hepatitis B virus and hepatitis C virus infections and cancer: synergy between viral and host factors. Clin Microbiol Infect (2015) 21(11):969–74. doi: 10.1016/j.cmi.2015.06.026
402. Seeger C, Mason WS. Molecular biology of hepatitis B virus infection. Virology (2015) 479-480:672–86. doi: 10.1016/j.virol.2015.02.031
403. Ringelhan M, Heikenwalder M, Protzer U. Direct Effects of Hepatitis B Virus-Encoded Proteins and Chronic Infection in Liver Cancer Development. Dig Dis (2013) 31(1):138–51. doi: 10.1159/000347209
404. Li J, Liu Y, Wang Z, Liu K, Wang Y, Liu J, et al. Subversion of Cellular Autophagy Machinery by Hepatitis B Virus for Viral Envelopment. J Virol (2011) 85(13):6319–33. doi: 10.1128/JVI.02627-10
405. Tang H, Da L, Mao Y, Li Y, Li D, Xu Z, et al. Hepatitis B virus X protein sensitizes cells to starvation-induced autophagy via up-regulation of beclin 1 expression. Hepatology (2009) 49(1):60–71. doi: 10.1002/hep.22581
406. Bouchard MJ, Schneider RJ. The Enigmatic X Gene of Hepatitis B Virus. J Virol (2004) 78(23):12725–34. doi: 10.1128/JVI.78.23.12725-12734.2004
407. Zhang H-T, Chen GG, Hu B-G, Zhang Z-Y, Yun J-P, He M-L, et al. Hepatitis B virus x protein induces autophagy via activating death-associated protein kinase. J Viral Hepat (2014) 21(9):642–9. doi: 10.1111/jvh.12191
408. Liu B, Fang M, Hu Y, Huang B, Li N, Chang C, et al. Hepatitis B virus X protein inhibits autophagic degradation by impairing lysosomal maturation. Autophagy (2014) 10(3):416–30. doi: 10.4161/auto.27286
409. Zhou T, Jin M, Ding Y, Zhang Y, Sun Y, Huang S, et al. Hepatitis B virus dampens autophagy maturation via negative regulation of Rab7 expression. Biosci Trends (2016) 10(4):244–50. doi: 10.5582/bst.2016.01049
410. Kim S-J, Khan M, Quan J, Till A, Subramani S, Siddiqui A. Hepatitis B Virus Disrupts Mitochondrial Dynamics: Induces Fission and Mitophagy to Attenuate Apoptosis. Luo G, ed. PloS Pathog (2013) 9(12):e1003722. doi: 10.1371/journal.ppat.1003722
411. Wang X, Lin Y, Kemper T, Chen J, Yuan Z, Liu S, et al. AMPK and Akt/mTOR signalling pathways participate in glucose-mediated regulation of hepatitis B virus replication and cellular autophagy. Cell Microbiol (2020) 22(2):1–16. doi: 10.1111/cmi.13131
412. Chen L, Ming X, Li W, Bi M, Yan B, Wang X, et al. The microRNA-155 mediates hepatitis B virus replication by reinforcing SOCS1 signalling–induced autophagy. Cell Biochem Funct (2020) 38(4):436–42. doi: 10.1002/cbf.3488
413. Wu S-Y. Autophagy and microRNA in hepatitis B virus-related hepatocellular carcinoma. World J Gastroenterol (2016) 22(1):176. doi: 10.3748/wjg.v22.i1.176
414. Ballestar E, Li T. New insights into the epigenetics of inflammatory rheumatic diseases. Nat Rev Rheumatol (2017) 13(10):593–605. doi: 10.1038/nrrheum.2017.147
415. Liu D, Li P, Guo J, Li L-L, Guo B, Jiao H-H, et al. Exosomes derived from HBV−associated liver cancer promote chemoresistance by upregulating chaperone−mediated autophagy. Oncol Lett (2018) 17(1):323–31. doi: 10.3892/ol.2018.9584
416. Moens U. Human Polyomaviruses and Papillomaviruses. Int J Mol Sci (2018) 19(8):2360. doi: 10.3390/ijms19082360
417. Moens U, Rasheed K, Abdulsalam I, Sveinbjørnsson B. The Role of Merkel Cell Polyomavirus and Other Human Polyomaviruses in Emerging Hallmarks of Cancer. Viruses (2015) 7(4):1871–901. doi: 10.3390/v7041871
418. Liu W, MacDonald M, You J. Merkel cell polyomavirus infection and Merkel cell carcinoma. Curr Opin Virol (2016) 20:20–7. doi: 10.1016/j.coviro.2016.07.011
419. Grundhoff A, Fischer N. Merkel cell polyomavirus, a highly prevalent virus with tumorigenic potential. Curr Opin Virol (2015) 14:129–37. doi: 10.1016/j.coviro.2015.08.010
420. Wendzicki JA, Moore PS, Chang Y. Large T and small T antigens of Merkel cell polyomavirus. Curr Opin Virol (2015) 11:38–43. doi: 10.1016/j.coviro.2015.01.009
421. Borchert S, Czech-Sioli M, Neumann F, Schmidt C, Wimmer P, Dobner T, et al. High-Affinity Rb Binding, p53 Inhibition, Subcellular Localization, and Transformation by Wild-Type or Tumor-Derived Shortened Merkel Cell Polyomavirus Large T Antigens. J Virol (2014) 88(6):3144–60. doi: 10.1128/JVI.02916-13
422. Berrios C, Padi M, Keibler MA, Park DE, Molla V, Cheng J, et al. Merkel Cell Polyomavirus Small T Antigen Promotes Pro-Glycolytic Metabolic Perturbations Required for Transformation. Kalejta RF, ed. PloS Pathog (2016) 12(11):e1006020. doi: 10.1371/journal.ppat.1006020
423. Shuda M, Arora R, Kwun HJ, Feng H, Sarid R, Fernández-Figueras M-T, et al. Human Merkel cell polyomavirus infection I. MCV T antigen expression in Merkel cell carcinoma, lymphoid tissues and lymphoid tumors. Int J Cancer (2009) 125(6):1243–9. doi: 10.1002/ijc.24510
424. Verhaegen ME, Mangelberger D, Harms PW, Vozheiko TD, Weich JW, Wilbert DM, et al. Merkel Cell Polyomavirus Small T Antigen Is Oncogenic in Transgenic Mice. J Invest Dermatol (2015) 135(5):1415–24. doi: 10.1038/jid.2014.446
425. Kumar S, Xie H, Shi H, Gao J, Juhlin CC, Bjornhagen V, et al. Merkel cell polyomavirus oncoproteins induce microRNAs that suppress multiple autophagy genes. Int J Cancer (2020) 146(6):1652–66. doi: 10.1002/ijc.32503
426. Schiffman M, Castle PE, Jeronimo J, Rodriguez AC, Wacholder S. Human papillomavirus and cervical cancer. Lancet (2007) 370(9590):890–907. doi: 10.1016/S0140-6736(07)61416-0
427. Berman TA, Schiller JT. Human papillomavirus in cervical cancer and oropharyngeal cancer: One cause, two diseases. Cancer (2017) 123(12):2219–29. doi: 10.1002/cncr.30588
428. DiGiuseppe S, Bienkowska-Haba M, Sapp M. Human Papillomavirus Entry: Hiding in a Bubble. Tsai B, ed. J Virol (2016) 90(18):8032–5. doi: 10.1128/JVI.01065-16
429. Mattoscio D, Medda A, Chiocca S. Human Papilloma Virus and Autophagy. Int J Mol Sci (2018) 19(6):1775. doi: 10.3390/ijms19061775
430. Kim YC, Guan KL. MTOR: A pharmacologic target for autophagy regulation. J Clin Invest (2015) 125(1):25–32. doi: 10.1172/JCI73939
431. Papinski D, Kraft C. Regulation of Autophagy By Signaling Through the Atg1/ULK1 Complex. J Mol Biol (2016) 428(9):1725–41. doi: 10.1016/j.jmb.2016.03.030
432. Sapp M, Bienkowska-Haba M. Viral entry mechanisms: human papillomavirus and a long journey from extracellular matrix to the nucleus. FEBS J (2009) 276(24):7206–16. doi: 10.1111/j.1742-4658.2009.07400.x
433. Pinidis P, Tsikouras P, Iatrakis G, Zervoudis S, Koukouli Z, Bothou A, et al. Human Papilloma Virus’ Life Cycle and Carcinogenesis. Maedica (Buchar) (2016) 11(1):48–54.
434. Belleudi F, Nanni M, Raffa S, Torrisi MR. HPV16 E5 deregulates the autophagic process in human keratinocytes. Oncotarget (2015) 6(11):9370–86. doi: 10.18632/oncotarget.3326
435. Mattoscio D, Casadio C, Miccolo C, Maffini F, Raimondi A, Tacchetti C, et al. Autophagy regulates UBC9 levels during viral-mediated tumorigenesis. Raab-Traub N, ed. PloS Pathog (2017) 13(3):e1006262. doi: 10.1371/journal.ppat.1006262
436. Dayyani F, Etzel CJ, Liu M, Ho C-H, Lippman SM, Tsao AS. Meta-analysis of the impact of human papillomavirus (HPV) on cancer risk and overall survival in head and neck squamous cell carcinomas (HNSCC). Head Neck Oncol (2010) 2(1):15. doi: 10.1186/1758-3284-2-15
Keywords: autophagy, human diseases, cancer cells, oncoviruses, cell survival, cell death
Citation: Suares A, Medina MV and Coso O (2021) Autophagy in Viral Development and Progression of Cancer. Front. Oncol. 11:603224. doi: 10.3389/fonc.2021.603224
Received: 05 September 2020; Accepted: 12 January 2021;
Published: 08 March 2021.
Edited by:
Daniel Hector Grasso, Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET), ArgentinaReviewed by:
Alfredo Criollo, University of Chile, ChileCopyright © 2021 Suares, Medina and Coso. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Omar Coso, b2Nvc29AZmJtYy5mY2VuLnViYS5hcg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.