 Carmine De Angelis1*
Carmine De Angelis1* Carmela Nardelli2
Carmela Nardelli2 Paola Concolino3
Paola Concolino3 Martina Pagliuca1
Martina Pagliuca1 Mario Setaro4Elisa De Paolis3Pietro De Placido1Valeria Forestieri1Giovanni Luca Scaglione4Annalisa Ranieri2,4Barbara Lombardo2,4
Mario Setaro4Elisa De Paolis3Pietro De Placido1Valeria Forestieri1Giovanni Luca Scaglione4Annalisa Ranieri2,4Barbara Lombardo2,4 Lucio Pastore2,4Sabino De Placido1†Ettore Capoluongo2,4†
Lucio Pastore2,4Sabino De Placido1†Ettore Capoluongo2,4†- 1Department of Clinical Medicine and Surgery, University of Naples Federico II, Naples, Italy
- 2Department of Molecular Medicine and Medical Biotechnologies, University of Naples Federico II, Naples, Italy
- 3Molecular and Genomic Diagnostics Unit, Fondazione Policlinico Universitario A. Gemelli Istituto di Ricovero e Cura a Carattere Scientifico, Rome, Italy
- 4CEINGE-Biotecnologie Avanzate, Naples, Italy
The partner and localizer of BRCA2 (PALB2) is a major BRCA2 binding partner that participates in homologous recombination repair in response to DNA double-strand breaks. Germline alterations of the PALB2 gene have recently been associated with a high risk of developing breast cancer. We investigated a 37-year-old Caucasian woman with breast cancer and family history of breast cancer using targeted next generation sequencing. A novel heterozygous deletion involving exons 5 and 6 was found in the PALB2 gene, and resulted in the production of a truncated PALB2 protein. These findings expand the mutational spectra of PALB2-associated breast cancer, and may improve the mutation-based screening and genetic diagnosis of breast cancer.
Background
Breast cancer is the most frequently diagnosed cancer and the leading cause of death among women worldwide (1). Most breast cancers are sporadic, whereas up to 10% are hereditary. Hereditary breast cancers tend to develop earlier in life than non-inherited (sporadic) cases and are more likely to develop in both breasts. In addition, multiple cancer diagnoses in consecutive generations and/or within the same subject, and transmission of gene alterations increase the risk of certain types of cancers to offspring (2). The most common cause of hereditary breast cancer is an inherited germline pathogenic variants (PVs) in the high-penetrant cancer predisposition genes BRCA1 and BRCA2 (3–5). A recent study on a large prospective cohort showed that the cumulative breast cancer risk to age 80 years was 72% for BRCA1 and 69% for BRCA2 carriers, and the cumulative risk for contralateral breast cancer 20 years after breast cancer diagnosis was 40% for BRCA1 and 26% for BRCA2 carriers (5). Advances in DNA sequencing techniques have helped to identify additional breast cancer susceptibility genes (4, 6). Among these genes, germline PVs in the Partner and localizer of BRCA2 (PALB2) gene appear to confer the highest risks (7). PALB2 is located on chromosome 16p12.2, and encodes a protein which is essential for repair of double-strand DNA breaks by DNA homologous recombination (HR). The PALB2 protein acts as a linking hub of a macromolecular complex including BRCA1 and BRCA2 and facilitates the function of RAD51, a protein vital for strand invasion during HR. PALB2 also interacts directly with and stabilizes BRCA2 during formation of the RAD51 nucleoprotein filament (8–11). Notably, PALB2 is essential to enable BRCA2 to perform its repair functions, which occur via HR, DNA double-strand break repair and S-phase DNA damage checkpoint control (8, 11). Not only PALB2 protein interacts with BRCA2 thanks to the domain at the C-terminus of PALB2, but it also binds to BRCA1 with its coiled-coil motif at the N-terminus. The disruption of this complex may result in efficiency decline of HR repair (12). In addition to promoting HR, PALB2 reduces the oxidative stress through its interaction with KEAP1. KEAP1, under normal condition, binds and inhibits the antioxidant transcription factor NRF2 leading to its degradation. PALB2 interacts competitively with KEAP1 causing NRF2 accumulation, thus regulating cellular redox homeostasis (13). Biallelic mutations in PALB2 result in a subtype of Fanconi anemia, which is a rare autosomal recessive syndrome characterized by genome instability, early bone marrow failure, growth abnormalities and increased cancer susceptibility. Otherwise, when PALB2 mutations occur as monoallelic, they are associated with predisposition to breast and other cancers (11).
TP53, PTEN, SKT11, CDH1 and NF1 are syndromic genes causing also relevant/high-risk for breast cancer. Inherited PVs in CHEK2, ATM, BARD1, BRIP1, and RAD51D were associated with moderate risks of breast cancer (6, 14, 15). Recently, FANCM, which encodes for a DNA translocase, has been suggested as a novel breast cancer predisposition gene, with greater effects for the ER-negative and triple-negative breast cancer subtypes (16, 17).
At clinical level, when germline PVs are identified in breast cancer susceptibility genes, primary or secondary prevention programs can be implemented, and tailored treatments initiated. Primary breast cancer prevention measures should be implemented through an accurate risk assessment. Primary prevention strategies are represented by chemoprevention with the selective estrogen receptor modulators (tamoxifen and raloxifen) or aromatase inhibitors (anastrozole and exemestane) (18) and by bilateral risk-reducing mastectomy (19). Secondary breast cancer prevention consists in screening programs based on clinical breast examination, mammogram, and contrast enhanced magnetic resonance imaging, aiming to detect pre-cancerous lesions and initial stage tumors. Here, we report a novel large germline deletion in PALB2 in a young breast cancer patient who had a family history for different cancer types.
Case Presentation
In October 2017, a 37-year-old Caucasian pre-menopausal woman without any relevant medical history felt a palpable lump in her left breast. Mammography combined with breast ultrasonography followed by fine-needle aspiration led to a diagnosis of breast cancer. In March 2018, she underwent a quadrantectomy and axillary lymph node dissection. Pathology exmination revealed a 31 mm, high-grade, luminal B-like invasive ductal carcinoma (estrogen receptor-positive, progesteron receptor-positive, HER2-negative, and Ki-67 40%). Two of 3 axillary lymph nodes were found to be metastatic. Bone scan, chest X-ray, and liver ultrasound examination did not identify distant metastases. In April 2018, the patient was started on adjuvant chemotherapy with 5-fluororacil 500 mg/mq + epirubicin 100 mg/mq + cyclophosphamide 500 mg/mq for 3 cycles every 21 days, followed by docetaxel 100 mg/mq for 3 cycles every 21 days. The patient then received adjuvant radiation therapy on the residual mammary parenchyma (total dose: 60 Gy) and adjuvant endocrine therapy (ET) was started with exemestane 25 mg daily plus leuprorelin 11,25 mg every 12 weeks. In November 2018, the patient was enrolled in an open-label, randomized, phase III study (NCT03155997) of abemaciclib plus standard ET (Figure 1).
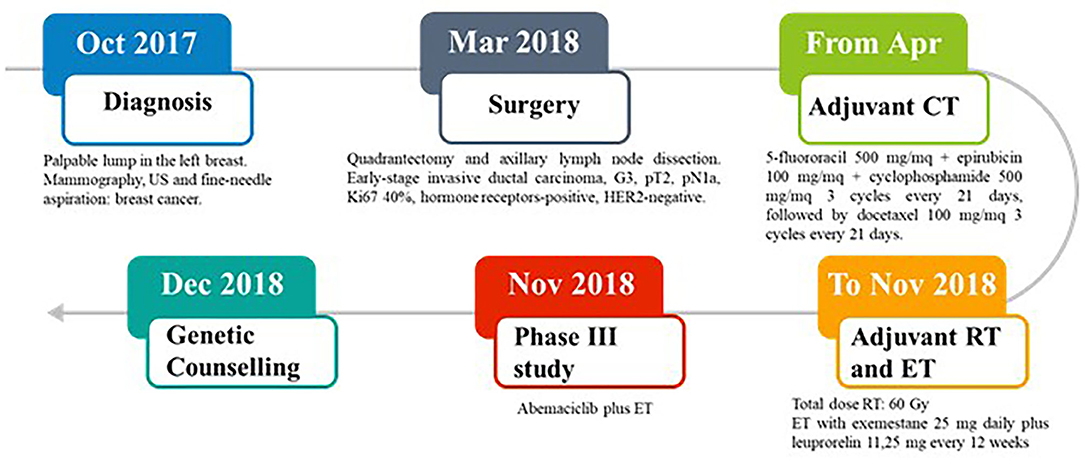
Figure 1. Case report timeline.
In December 2018, the patient was referred to the Hereditary Cancer Genetics Clinic at the Department of Clincial Medicine and Surgery of the University Federico II in Naples, Italy, where she underwent genetic counseling. Family history revealed that her mother was diagnosed with breast cancer at the age of 60 and papillary thyroid carcinoma at the age of 61, that her paternal grandmother died at the age of 36 from breast cancer, whereas her father sucummbed to lung cancer at the age of 39, and her maternal grandfather died of colorectal cancer at the age of 74 (Figure 2). Genetic risk was assessed based on the medical and surgical history of the proband, the number of affected relatives, their age at dignosis and the degree of relationships among them. Since germline testing criteria were met (20), we looked for BRCA1 and BRCA2 gene PVs. Next-generation sequencing did not detect any pathogenic or likely PVs. Similarly, no deletions or duplications in exons of BRCA1 and BRCA2 genes were identified by multiplex ligation-dependent probe amplification (MLPA). Given the patient's personal and family history suggestive of inherited susceptibility, the patient underwent multi-gene testing with the Devyser HBOC NGS kit (Devyser, Hägersten, Sweden). The sequencing reaction was carried out on the Illumina MiSeq System (Illumina, San Diego, CA, USA). Output data were analyzed using the CE- IVD Amplicom Suite Software v. 1.0 (SmartSeq, Novara, Italy). As shown in Figure 3A, the software predicted the presence of a deletion in exons 5 and 6 of PALB2 gene. To verify the presence of this deletion, multiplex ligation-dependent probe amplification (MLPA) was performed (Figure 3B) by means of SALSA MLPA Probemix P260 PALB2-RAD50-RAD51C-RAD51D according to the manufacturer's instructions. In detail, four normal samples, previously screened by NGS in duplicate, served as reference samples for the MLPA test. Five μL of the proband's DNA were run in duplicate.
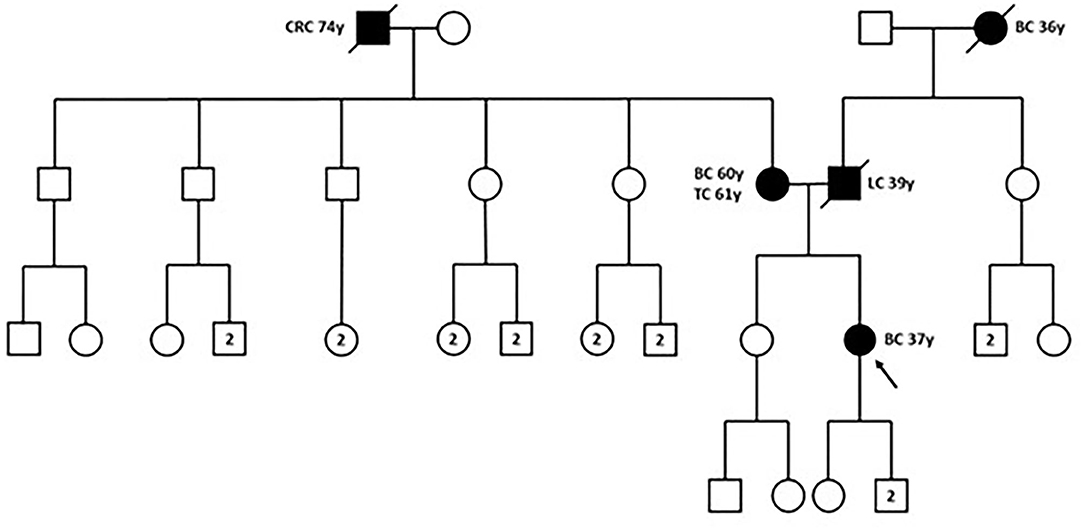
Figure 2. Family pedigree. The proband is indicated by black arrow. BC, breast cancer; CRC, colorectal cancer; LC, lung cancer; TC, thyroid cancer; y, years-old. Circle, female; square, male; filled symbols, individuals with cancer diagnosis; cross-hatched symbols, affected individuals already deceased; numbers in the circles/squares, amount of people of that gender.
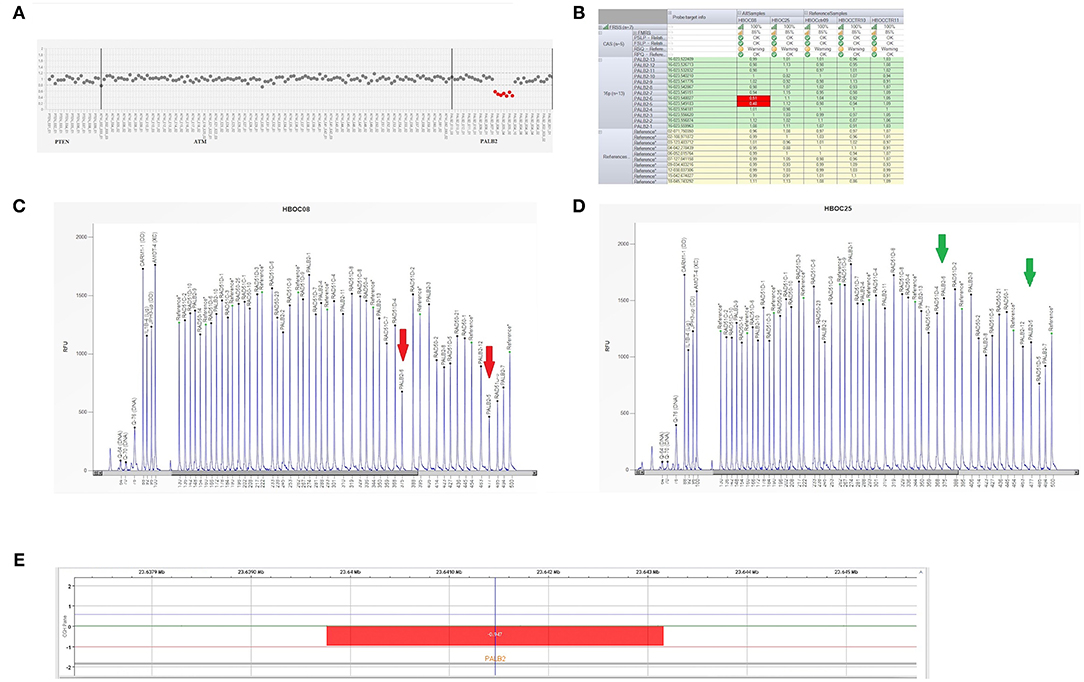
Figure 3. Genetic analysis of a PALB2 gene. (A) Plot of PALB2 amplicons corresponding to exon 5 and 6 which fell to about of 0.5 value of coverage. CNV analysis by Amplicon Suite Software for woman carrying the deletion of exon 5 and 6 of PALB2. Amplicon coverage: the coverage of the remaining PALB2 exons was within the normal range (0.8–1.2), with a homogeneous pattern of distribution of each amplicon. (B) Drop of probe signals (red cells) of peak height area related to PALB2 exons 5 and 6. (C,D) MLPA electropherogram from patient carrying the deletion of PALB2 exon 5 and 6. Red arrows (C) indicate the deletions exon 5 and exon 6, respectively, as compared to the normal samples (green arrows) (D). (E) CGH array profile of chromosome 16 from the Agilent 1X1M high resolution array revealed a heterozygous deletion in 16p12.2 region, of ~3.39 kb, involving PALB2 gene.
As shown in Figures 3C,D, the Q-Fragment plot of our sample confirmed the presence of a deletion in exons 5 and 6. To determine the extent of this deletion, we looked for large scale rearrangements using a high-resolution comparative genomic hybridization-array (a-CGH) (21). Genomic DNA was analyzed with the CGH 1 × 1M Microarray (Agilent Technologies, Santa Clara, CA, USA). The following [GRCh37] chr16p12.2 (23,639,765-23,643,159)x1 was investigated: results showed a 3.39 Kb deletion including the PALB2 gene (chr16p12.2: 23,614,483-23,652,678), as obtained using the probes A_16_P20429174, A_16_P40587478, A_16_P20429183. As shown in Figure 3E, the deletion probably involved a 3.39 Kb region included between intron 4 and 6 causing the loss of exons 5 and 6.
To characterize the breakpoint region, specific PCR primers (Del4F 5′- aagactccactgactatctc-3′ and Del7R 5′-catcctgatgaaccactcatg-3′), including a larger region respect to that identified by probes used by CGH and amplifying a PCR product of 6.692 bp from the wild-type DNA, were designed. PCR reactions were performed using a long-range PCR kit (Expand Long Template PCR System, Roche Applied Science). Sequencing was performed using a BigDye Terminator Cycle Sequencing Kit v3.1 (Thermo Fisher Scientific) and an ABI 3500 Genetic Analyser (Thermo Fisher Scientific). Results were analyzed with the SeqScape v2.5 software package (Thermo Fisher Scientific) using NG_007406.1 reference. Total RNA was isolated from peripheral blood lymphocytes with TRIzol reagent (Thermo Fisher Scientific, Inc., Waltham, MA, USA). Synthesis of complementary DNA (cDNA) was performed with SuperScript II Reverse Transcriptase (Thermo Fisher Scientific) using DNAase-treated RNA in the presence of random primers and RNAaseOUT (Thermo Fisher Scientific). cDNA amplification was performed using the following primers: R4delF 5′-aggaagaagtcacctcacac-3′, and R7delR 5′-catcttcgcaagcagttatg-3′. Sequencing was performed using a BigDye Terminator Cycle Sequencing Kit v3.1 (Thermo Fisher Scientific) and an ABI 3500 Genetic Analyser (Thermo Fisher Scientific). Results were analyzed with the SeqScape v2.5 software package (Thermo Fisher Scientific) using NM_024675.3 reference.
In the patient, two PCR products of 6.692bp and 1.086 bp respectively, were obtained (Figure 4A). The 1.086 bp fragment, containing the expected deletion, was cut out and isolated from agarose gel and sequenced using PCR primers Del4F and Del7R. This fragment, containing the breakpoint region, showed a wild-type sequence until to the nucleotide g.13536C (NG_007406.1) of PALB2 gene intron 4. The following sequence corresponded to the PALB2 intron 6 starting from the g.19143G nucleotide (NG_007406.1) (Figure 4B). We report the novel PALB2 rearrangement in agreement with the recommended HGVS nomenclature: NG_007406:g.13536_19143del. The new deletion involves 5.607 bp of the PALB2 gene, a larger deletion than the expected one of 3.39kb identified by the CGH, and includes part of the intron 4, exon 5, intron 5, exon 6 and part of the intron 6. Two sequences of 43 nucleotides, occurring in the same orientation (100% homology) within intron 4 and 6, suggested that the deletion is probably the result of a homologous recombination event (Figure 4C). A single PCR fragment of 1.052 bp was obtained from the cDNA control, while two fragments of 1.052 and 152 bp were amplified using cDNA patient. PCR product of 152 bp, containing the expected deletion, was cut out and isolated from agarose gel, sequenced with Del4F and Del7R primers, and analyzed. Sequencing analysis revealed a wild-type sequence until to the nucleotide c.1884G (NM_024675.3) of PALB2 gene in exon 4. The following sequence corresponded to the PALB2 exon 7 starting from the nucleotide c.2687A (NM_024675.3:c.1884_2687). The PALB2 exons 5-6 deletion, involving 803bp, disrupts the reading frame of the mRNA producing a pre-mature stop codon and a truncated protein of 581 amino acids [NP_078951.2:p. (Gly562GlufsTer21)] (Figure 4D).
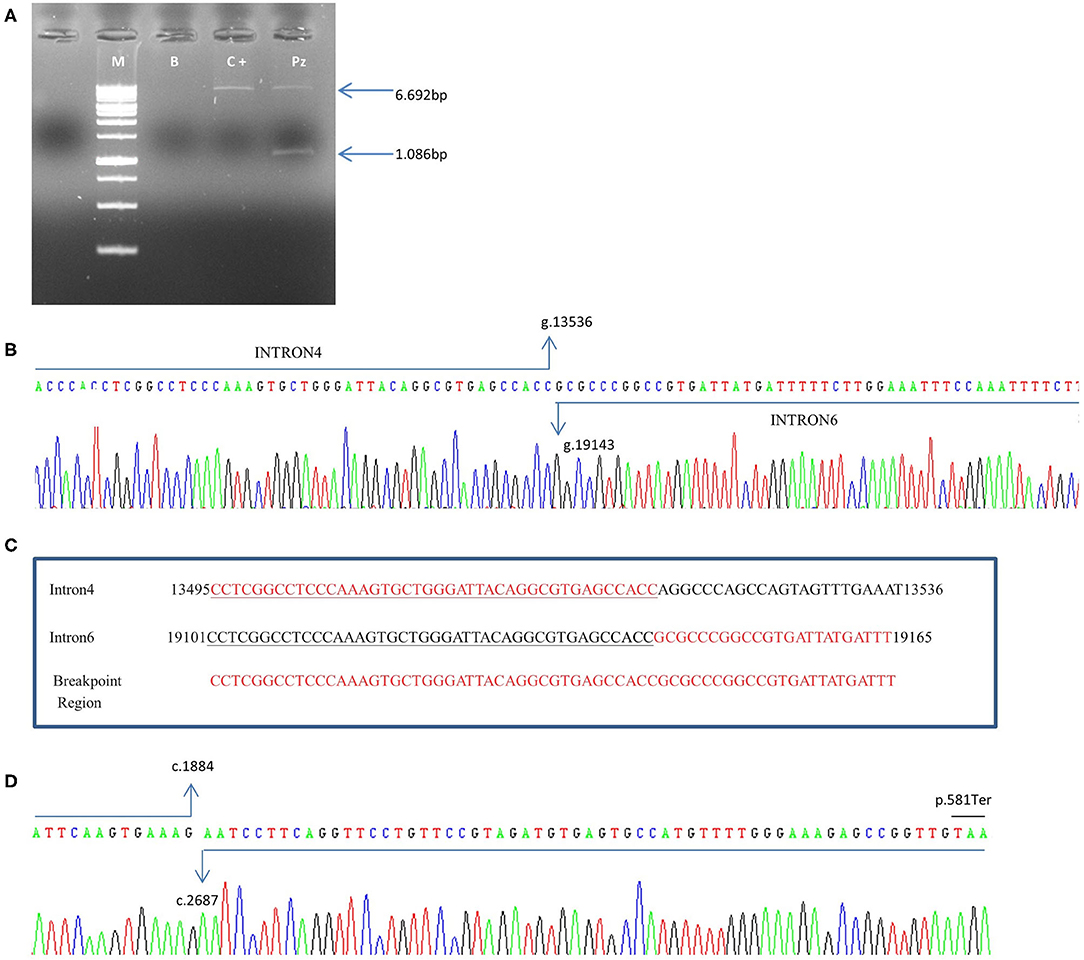
Figure 4. Characterization of exon 5–6 deletion in the PALB2 gene. (A) Genomic DNA was amplified using specific deletion primers (Del4F and Del7R). The mutant allele gives rise to a specific 1.086-bp fragment. M: marker, B: blank, C+: wild-type control, Pz: patient. (B) The electropherogram of the 1.086 bp PCR fragment, containing the deletion breakpoint, showed a wild-type sequence until the nucleotide g.13536C (NG_007406.1) of PALB2 intron 4. The following sequence corresponded to the PALB2 intron 6 starting from the g.19143G nucleotide (NG_007406.1). (C) An homologous sequence of 43 nucleotides (underlined nucleotides), identified at breakpoint region, represents the cause of the rearrangement. (D) Electropherogram showing the sequence of the PCR product of 152 bp obtained from cDNA patient. Sequencing analysis revealed a wild-type sequence until the nucleotide c.1884G (NM_024675.3) of PALB2 gene in exon 4. The following sequence corresponded to the PALB2 exon 7 starting from the nucleotide c.2687A (NM_024675.3). A premature stop codon produces a truncated protein of 581 amino acids.
Discussion
Over the past years, several studies have shown that germline loss-of-function variants in the PALB2 gene may confer an increased lifetime risk of breast, pancreatic, ovarian and other cancers. Rahman and colleagues identified PALB2 monoallelic truncating variants in ~1% of patients with hereditary breast cancer (9) [as confirmed by Fernandes et al. (22)], whose risk of developing breast cancer was 2.3-fold higher than the risk observed amid controls. Subsequently, population-based screenings of PALB2 PVs revealed a 2- to 30-fold increase in the risk of breast cancer (23–27). The analysis conducted by Antoniou and colleagues revealed that the relative risk for individuals with deleterious PALB2 PVs was 8–9 before the age of 40 and around 5 after the age of 60 years. Thus, the cumulative breast cancer risk for female carriers of PALB2 variants is 14% by the age of 50 years and up to 35% in women above the age of 70. Furthermore, PALB2 is the most frequently altered gene (1.2%) in non BRCA1/2 mutated male breast cancer patients, accordingly the risk of MBC increases from 9.63 to 17.30-fold in presence of PALB2 pathogenetic variants (28). A study conducted in Poland in 2015 (29) evaluated the clinical outcomes of 116 PALB2 mutation carriers among 12,529 women with breast cancer, and found that PALB2 variants increased the risk of death from breast cancer. Indeed, 10-year survival was 48% in PALB2 mutation carriers with breast cancer vs. 74.7% in non-mutation carriers with breast cancer and 72% in BRCA1 mutated patients. In addition, survival differed in relation to tumor size. Indeed, 10-year survival was 82.4% in case of tumors smaller than 2 centimeters vs. 32.4% in case of tumors ranging from 2 to 4.9 centimeters (29). Notably, individuals with PALB2 PVs more frequently had a triple-negative status (30%) vs. a frequency ranging between 12 and 17% in unselected patients with breast cancer (30). Yang et al. (7) analyzed 524 families from 21 countries who carried pathogenetic PALB2 variants. They identified an association between mutated PALB2 and female breast cancer (RR = 7.18), male breast cancer (RR = 7.34), ovarian cancer (RR = 2.91), and pancreatic cancer (RR = 2.37). The association between PALB2 germline alterations and risk of developing pancreatic cancer has been also described by other groups. In 2009, Jones and colleagues carried out exomic sequencing of PALB2 gene in 96 familial pancreatic cancer (FPC) patients in USA, thus identifying 3 truncating PVs producing different stop codons (31). Three more PALB2 germline PVs producing truncated protein were identified among 81 European FPC family, each of these three family had also history of breast cancer (none of the patients was carrier of BRCA2 mutation) (32). Furthermore, in a Japanese study, two out of 54 patients with pancreatic ductal adenocarcinoma carried deleterious variants of PALB2 gene (33). In 2019, Janssen and colleagues provided a comprehensive catalog of PALB2 gene predicted pathogenic or likely PVs published. They included 984 described cancer cases distributed over 146 PALB2 predicted PVs. They observed that 911 (92.5%) cases were described in breast cancer patients, 49 (5.0%) cases in ovarian cancer patients, and 24 (2.4%) cases in pancreatic cancer patients. They also found that exons 2, 3 and 1 of PALB2 gene showed the highest mutation rates (6.7, 5.8, 5.2%, respectively) (18).
Being a carrier of PALB2 PVs has therapeutic and surveillance implications. As recommended by guidelines for the management of hereditary breast cancer of the American Society of Clinical Oncology, the American Society for Radiation Oncology and the Society of Surgical Oncology (34), the decision whether to apply local therapy or to perform a contralateral risk-reducing mastectomy in patients with breast cancer should not be based only on the presence of an alteration in a moderate-penetrance breast cancer gene, such as PALB2. If indicated, breast-conserving therapy may be a treatment option, followed by high-risk breast screening of remaining tissue with annual mammography and breast magnetic resonance imaging. Mammography and breast MRI should be alternated at 6 month intervals (34). Given the lack of data regarding the risk of developing contralateral breast cancer from moderate-penetrance genes, additional factors such as age at diagnosis and family history should be taken into account in selected patients (34). This would provide the best therapeutic option such as nipple-sparing mastectomy, bilateral mastectomy or contralateral risk-reducing mastectomy in patient previously undergone unilateral mastectomy. Despite some studies reported an association between PALB2 PVs and the diagnosis of ovarian cancer (35–37), to date evidence are not sufficient to support risk-reducing salpingo-oophorectomy (20, 38). Similarly, there are no solid data regarding the systemic treatment options in PALB2 carriers with breast cancer. Although PALB2 encodes a protein involved in DNA double-strand break repair carried out by BRCA2, tailored therapies for breast cancer patients carrying PVs in genes other than BRCA1/2 have not yet been established. However, the ongoing Olaparib Expanded phase II trial evaluated the effect of the poly ADP-ribose polymerase (PARP) inhibitor olaparib monotherapy in metastatic breast cancer patients with germline (other than BRCA1/2) or somatic PVs in DNA damage response (DDR)-pathway genes. In this trial, response to olaparib was observed mostly in patients with somatic BRCA1/2 or germline PALB2 PVs but not with ATM or CHEK2 PVs. Among patients with gPALB2 PVs, the overall response rate was 82% (90% CI, 53% to 96%), the clinical benefit rate was 100% (90% CI, 74–100%), and median progression-free survival (PFS) was 13.3 months (90% CI, 12 months to NA) (39). Other clinical trials are evaluating the potential role of PARP inhibitors in the treatment of PALB2 PVs carriers with breast cancer (13, 41).
To the best of our knowledge, this is the first time that PALB2 deletion involving a 5.6 Kb region, between intron 4 and 6 and causing the loss of exons 5 and 6, is reported as associated to a hereditary breast cancer. In addition, we also established the exact breakpoints of this new arrangement and estimated that introduces a pre-mature stop codon in PALB2 mRNA resulting in the production of a truncated protein of 581 amino acids.
Conclusions
Here we report a hitherto unknown PALB2 5.6-kilobase deletion involving exons 5 and 6 and the neighboring introns in a breast cancer patient. This case is paradigmatic of the clinical relevance of an in-depth evaluation of genetic risk, especially in patients with history highly suggestive of a hereditary syndrome. Consequently, patients without BRCA1/2 alterations should be offered NGS multi-gene panel testing when personal and/or family history is suggestive for hereditary syndrome (40).
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding authors.
Ethics Statement
The studies involving human participants were reviewed and approved by Ethics Committee of the University of Naples Federico II. The patients/participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author Contributions
All authors of this research paper have directly participated in the planning, execution, or analysis of the study. All authors were involved in manuscript writing and reviewing, gave their final approval and agreed to be accountable for all aspects of the work.
Conflict of Interest
CDA was a consultant/advisory board member for Novartis, Eli Lilly, and Pfizer. SDP had declared honoraria from Roche, Pfizer, Astra-Zeneca, Novartis, Celgene, Eli Lilly, Amgen and Eisai.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We gratefully thank Jean Ann Gilder (Scientific Communication srl., Naples, Italy) for editing the article.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin. (2020) 70:7–30. doi: 10.3322/caac.21590
2. Foulkes WD. Inherited susceptibility to common cancers. N Engl J Med. (2008) 359:2143–53. doi: 10.1056/NEJMra0802968
3. Easton DF, Pharoah PDP, Antoniou AC, Tischkowitz M, Tavtigian SV, Nathanson KL, et al. Gene-panel sequencing and the prediction of breast-cancer risk. N Engl J Med. (2015) 372:2243–57. doi: 10.1056/NEJMsr1501341
4. Nielsen FC, van Overeem Hansen T, Sørensen CS. Hereditary breast and ovarian cancer: new genes in confined pathways. Nat Rev Cancer. (2016) 16:599–612. doi: 10.1038/nrc.2016.72
5. Kuchenbaecker KB, Hopper JL, Barnes DR, Phillips K-A, Mooij TM, Roos-Blom M-J, et al. Risks of breast, ovarian, and contralateral breast cancer for BRCA1 and BRCA2 mutation carriers. JAMA. (2017) 317:2402–16. doi: 10.1001/jama.2017.7112
6. Couch FJ, Shimelis H, Hu C, Hart SN, Polley EC, Na J, et al. Associations between cancer predisposition testing panel genes and breast cancer. JAMA Oncol. (2017) 3:1190–6. doi: 10.1001/jamaoncol.2017.0424
7. Yang X, Leslie G, Doroszuk A, Schneider S, Allen J, Decker B, et al. Cancer risks associated with germline PALB2 pathogenic variants: an international study of 524 families. J Clin Oncol. (2020) 38:674–85. doi: 10.1200/JCO.19.01907
8. Xia B, Sheng Q, Nakanishi K, Ohashi A, Wu J, Christ N, et al. Control of BRCA2 cellular and clinical functions by a nuclear partner, PALB2. Mol Cell. (2006) 22:719–29. doi: 10.1016/j.molcel.2006.05.022
9. Rahman N, Seal S, Thompson D, Kelly P, Renwick A, Elliott A, et al. PALB2, which encodes a BRCA2-interacting protein, is a breast cancer susceptibility gene. Nat Genet. (2007) 39:165–7. doi: 10.1038/ng1959
10. Buisson R, Dion-Côté A-M, Coulombe Y, Launay H, Cai H, Stasiak AZ, et al. Cooperation of breast cancer proteins PALB2 and piccolo BRCA2 in stimulating homologous recombination. Nat Struct Mol Biol. (2010) 17:1247–54. doi: 10.1038/nsmb.1915
11. Wu S, Zhou J, Zhang K, Chen H, Luo M, Lu Y, et al. Molecular mechanisms of PALB2 function and its role in breast cancer management. Front Oncol. (2020) 10:301. doi: 10.3389/fonc.2020.00301
12. Foo TK, Tischkowitz M, Simhadri S, Boshari T, Zayed N, Burke KA, et al. Compromised BRCA1-PALB2 interaction is associated with breast cancer risk. Oncogene. (2017) 36:4161–70. doi: 10.1038/onc.2017.46
13. Ducy M, Sesma-Sanz L, Guitton-Sert L, Lashgari A, Gao Y, Brahiti N, et al. The tumor suppressor PALB2: inside out. Trends Biochem Sci. (2019) 44:226–40. doi: 10.1016/j.tibs.2018.10.008
14. Vrdoljak E, Marschner N, Zielinski C, Gligorov J, Cortes J, Puglisi F, et al. Final results of the TANIA randomised phase III trial of bevacizumab after progression on first-line bevacizumab therapy for HER2-negative locally recurrent/metastatic breast cancer. Ann Oncol Off J Eur Soc Med Oncol. (2016) 27:2046–52. doi: 10.1093/annonc/mdw316
15. Yang X, Song H, Leslie G, Engel C, Hahnen E, Auber B, et al. Ovarian and breast cancer risks associated with pathogenic variants in RAD51C and RAD51D. J Natl Cancer Inst. (2020) 112:1242–50. doi: 10.1093/jnci/djaa030
16. Neidhardt G, Hauke J, Ramser J, Groß E, Gehrig A, Müller CR, et al. Association between loss-of-function mutations within the FANCM gene and early-onset familial breast cancer. JAMA Oncol. (2017) 3:1245–8. doi: 10.1001/jamaoncol.2016.5592
17. Figlioli G, Bogliolo M, Catucci I, Caleca L, Lasheras SV, Pujol R, et al. The FANCM:p.Arg658* truncating variant is associated with risk of triple-negative breast cancer. NPJ Breast Cancer. (2019) 5:38. doi: 10.1038/s41523-019-0127-5
18. Janssen B, Bellis S, Koller T, Tischkowitz M, Liau S-S. A systematic review of predicted pathogenic PALB2 variants: an analysis of mutational overlap between epithelial cancers. J Hum Genet. (2020) 65:199–205. doi: 10.1038/s10038-019-0680-7
19. Carbine NE, Lostumbo L, Wallace J, Ko H. Risk-reducing mastectomy for the prevention of primary breast cancer. Cochrane Database Syst Rev. (2018) 4:CD002748. doi: 10.1002/14651858.CD002748.pub4
20. Pilarski R, Berry MP, Jude S, Buys SS, Dickson P, Domchek SM, et al. NCCN Guidelines Version 1.2020 Genetic/Familial High-Risk Assessment: Breast, Ovarian, and Pancreatic. NCCN (2019).
21. Sanna V, Ceglia C, Tarsitano M, Lombardo B, Coppola A, Zarrilli F, et al. Aberrant F8 gene intron 1 inversion with concomitant duplication and deletion in a severe hemophilia A patient from Southern Italy. J Thromb Haemost. (2013) 11:195–7. doi: 10.1111/jth.12061
22. Fernandes PH, Saam J, Peterson J, Hughes E, Kaldate R, Cummings S, et al. Comprehensive sequencing of PALB2 in patients with breast cancer suggests PALB2 mutations explain a subset of hereditary breast cancer. Cancer. (2014) 120:963–7. doi: 10.1002/cncr.28504
23. Erkko H, Xia B, Nikkilä J, Schleutker J, Syrjäkoski K, Mannermaa A, et al. A recurrent mutation in PALB2 in Finnish cancer families. Nature. (2007) 446:316–9. doi: 10.1038/nature05609
24. Southey MC, Teo ZL, Dowty JG, Odefrey FA, Park DJ, Tischkowitz M, et al. A PALB2 mutation associated with high risk of breast cancer. Breast Cancer Res. (2010) 12:R109. doi: 10.1186/bcr2796
25. Southey MC, Goldgar DE, Winqvist R, Pylkäs K, Couch F, Tischkowitz M, et al. PALB2, CHEK2 and ATM rare variants and cancer risk: data from COGS. J Med Genet. (2016) 53:800–11. doi: 10.1136/jmedgenet-2016-103839
26. Decker B, Allen J, Luccarini C, Pooley KA, Shah M, Bolla MK, et al. Rare, protein-truncating variants in ATM, CHEK2 and PALB2, but not XRCC2, are associated with increased breast cancer risks. J Med Genet. (2017) 54:732–41. doi: 10.1136/jmedgenet-2017-104588
27. Momozawa Y, Iwasaki Y, Parsons MT, Kamatani Y, Takahashi A, Tamura C, et al. Germline pathogenic variants of 11 breast cancer genes in 7,051 Japanese patients and 11,241 controls. Nat Commun. (2018) 9:1–7. doi: 10.1038/s41467-018-06581-8
28. Rizzolo P, Zelli V, Silvestri V, Valentini V, Zanna I, Bianchi S, et al. Insight into genetic susceptibility to male breast cancer by multigene panel testing: results from a multicenter study in Italy. Int J Cancer. (2019) 145:390–400. doi: 10.1002/ijc.32106
29. Cybulski C, Kluzniak W, Huzarski T, Wokołorczyk D, Kashyap A, Jakubowska A, et al. Clinical outcomes in women with breast cancer and a PALB2 mutation: a prospective cohort analysis. Lancet Oncol. (2015) 16:638–44. doi: 10.1016/S1470-2045(15)70142-7
30. Foulkes WD, Smith IE, Reis-Filho JS. Triple-negative breast cancer. N Engl J Med. (2010) 363:1938–48. doi: 10.1056/NEJMra1001389
31. Jones S, Hruban RH, Kamiyama M, Borges M, Zhang X, Parsons DW, et al. Exomic sequencing identifies PALB2 as a pancreatic cancer susceptibility gene. Science. (2009) 324:217. doi: 10.1126/science.1171202
32. Slater EP, Langer P, Niemczyk E, Strauch K, Butler J, Habbe N, et al. PALB2 mutations in European familial pancreatic cancer families. Clin Genet. (2010) 78:490–4. doi: 10.1111/j.1399-0004.2010.01425.x
33. Takai E, Yachida S, Shimizu K, Furuse J, Kubo E, Ohmoto A, et al. Germline mutations in Japanese familial pancreatic cancer patients. Oncotarget. (2016) 7:74227–35. doi: 10.18632/oncotarget.12490
34. Tung NM, Boughey JC, Pierce LJ, Robson ME, Bedrosian I, Dietz JR, et al. Management of hereditary breast cancer: American society of clinical oncology, American society for radiation oncology, and society of surgical oncology guideline. J Clin Oncol. (2020) 38:2080–106. doi: 10.1200/JCO.20.00299
35. Walsh T, Casadei S, Lee MK, Pennil CC, Nord AS, Thornton AM, et al. Mutations in 12 genes for inherited ovarian, fallopian tube, and peritoneal carcinoma identified by massively parallel sequencing. Proc Natl Acad Sci USA. (2011) 108:18032–7. doi: 10.1073/pnas.1115052108
36. Kanchi KL, Johnson KJ, Lu C, McLellan MD, Leiserson MDM, Wendl MC, et al. Integrated analysis of germline and somatic variants in ovarian cancer. Nat Commun. (2014) 5:3156. doi: 10.1038/ncomms4156
37. Norquist BM, Harrell MI, Brady MF, Walsh T, Lee MK, Gulsuner S, et al. Inherited mutations in women with ovarian carcinoma. JAMA Oncol. (2016) 2:482–90. doi: 10.1001/jamaoncol.2015.5495
38. Ramus SJ, Song H, Dicks E, Tyrer JP, Rosenthal AN, Intermaggio MP, et al. Germline mutations in the BRIP1, BARD1, PALB2, and NBN genes in women with ovarian cancer. J Natl Cancer Inst. (2015) 107:djv214. doi: 10.1093/jnci/djv214
39. Tung NM, Robson ME, Ventz S, Santa-Maria CA, Nanda R, Marcom PK, et al. TBCRC 048: phase II study of olaparib for metastatic breast cancer and mutations in homologous recombination-related genes. J Clin Oncol. (2020) 38:4274–82. doi: 10.1200/JCO.20.02151
40. Kurian AW, Hughes E, Handorf EA, Gutin A, Allen B, Hartman A-R, et al. Breast and ovarian cancer penetrance estimates derived from germline multiple-gene sequencing results in women. JCO Precis Oncol. (2017) 2017:1–12. doi: 10.1200/PO.16.00066
41. Clinical Trials for PALB2- My Cancer Genome. Available online at: https://www.mycancergenome.org/content/gene/palb2/
Keywords: hereditary breast cancer, PALB2, breast-cancer risk, deletion, surveillance
Citation: De Angelis C, Nardelli C, Concolino P, Pagliuca M, Setaro M, De Paolis E, De Placido P, Forestieri V, Scaglione GL, Ranieri A, Lombardo B, Pastore L, De Placido S and Capoluongo E (2021) Case Report: Detection of a Novel Germline PALB2 Deletion in a Young Woman With Hereditary Breast Cancer: When the Patient's Phenotype History Doesn't Lie. Front. Oncol. 11:602523. doi: 10.3389/fonc.2021.602523
Received: 03 September 2020; Accepted: 05 February 2021;
Published: 24 February 2021.
Edited by:
Sergio Giannattasio, Consiglio Nazionale delle Ricerche, ItalyReviewed by:
Paolo Peterlongo, IFOM - The FIRC Institute of Molecular Oncology, ItalyAlvaro Galli, Italian National Research Council, Italy
Copyright © 2021 De Angelis, Nardelli, Concolino, Pagliuca, Setaro, De Paolis, De Placido, Forestieri, Scaglione, Ranieri, Lombardo, Pastore, De Placido and Capoluongo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Carmine De Angelis, carmine.deangelis1@unina.it
†These authors share last authorship