 Caixia Han
Caixia Han Xuefeng Gao
Xuefeng Gao Yonghui Li
Yonghui Li Li Zhang
Li Zhang Li Yu
Li Yu
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 13 April 2021
Sec. Hematologic Malignancies
Volume 11 - 2021 | https://doi.org/10.3389/fonc.2021.579881
The occurrence of gene mutation is a major contributor to the initiation and propagation of acute myeloid leukemia (AML). Accumulating evidence suggests that genes encoding cohesin subunits have a high prevalence of mutations in AML, especially in the t(8;21) subtype. Therefore, it is important to understand how cohesin mutations contribute to leukemogenesis. However, the fundamental understanding of cohesin mutation in clonal expansion and myeloid transformation in hematopoietic cells remains ambiguous. Previous studies briefly introduced the cohesin mutation in AML; however, an in-depth summary of mutations in AML was not provided, and the correlation between cohesin and AML1-ETO in t (8;21) AML was also not analyzed. By summarizing the major findings regarding the cohesin mutation in AML, this review aims to define the characteristics of the cohesin complex mutation, identify its relationships with co-occurring gene mutations, assess its roles in clonal evolution, and discuss its potential for the prognosis of AML. In particular, we focus on the function of cohesin mutations in RUNX1-RUNX1T1 fusion.
It has become apparent that acute myeloid leukemia (AML) is induced by the cooperative action of deregulated genes that alter cell proliferation and differentiation. As compared with other AML cytogenetic groups, patients with t(8; 21) AML had a relatively favorable prognosis, and most of them (>85%) achieved complete remission (CR) with high-dose cytosine arabinoside (Ara-C). However, approximately 30% of the t(8; 21) AML patients relapsed within 1 year, and the overall survival at 5 years was close to 51% (1, 2). Thus, it is imperative to identify the molecular mechanisms that lead to this disease.
Chromosomal translocations and/or gene mutations are the most common chromosome abnormalities in AML. In particular, AML with chromosomal rearrangements t(8;21) (q22;q22) can trigger the generation of the aberrant oncogenic fusion protein AML1-ETO (3, 4). However, AML1-ETO alone is not sufficient to induce the onset of leukemia, and additional genetic/epigenetic abnormalities are required (5, 6). To date, mutations in multiple driver genes have been identified in AML patients with AML1-ETO fusion (7–9), further indicating that AML1-ETO required additional genetic/epigenetic abnormalities to induce t(8;21) leukemogenesis. Recently, investigations, through genome-wide sequencing, revealed recurrent mutations in the cohesin complex genes in AML patients, especially in the t(8;21) subtype (10–13). However, only few studies have investigated the role of cohesin mutation in the pathogenesis of AML.
Cohesin with a ring shape is composed of the following four core members: structural maintenance of chromosomes (SMC3 and SMC1A), RAD cohesin complex component (RAD21), and cohesin subunit SA (STAG1/STAG2) (Figure 1A). During the cell cycle, cohesin facilitates the establishment of cohesion by assisting several additional subunits, including NIPBL, MAU2, WAPL, PDS5A, PDS5B, and sororin (Figure 1B) (14–17). Therefore, the ring-shaped cohesin can entrap the sister chromatids, and it regulates the separation of sister chromatids, DNA replication, and repair of the damaged double-strand DNA during cell cycle progression (18–22). Cohesin can exert its functions in increased DNA accessibility for transcription factors (TFs) owing to its cellular memory that promotes re-establishment of TF clusters in the early M phase (23). Cohesin complex can also interact with transcriptional repressor CTCF, promoters, mediators, enhancers, initiation and elongation forms of RNA polymerase II (RNAPII) or TFs to regulate chromatin architecture and gene expression (19, 24–28). Cohesin facilitates the promoter and distant conserved regulatory elements (CREs) interactions by “looping out” the intervening chromatin segment to activate or inhibit the gene expression (29, 30). Furthermore, cohesin drives the assembly of the gene enhancer/promoter complex through enhancer-promoter looping, and it produces multiple alternative splicing forms to mediate differential control of space- and stage- specific developmental programs (30–32). Mutation of cohesin can strongly affect the enhancer-promoter interaction (33), therefore altering normal gene expression. Growing evidence indicates that cohesin function deficiency is caused by cohesin mutations (11, 12, 34), suggesting a possible tumor-suppressor function of cohesin in AML. Herein, we review the major findings regarding the cohesin complex mutation, and to discuss the potential molecular functions of cohesin especially in t(8;21) AML by investigating the published references.
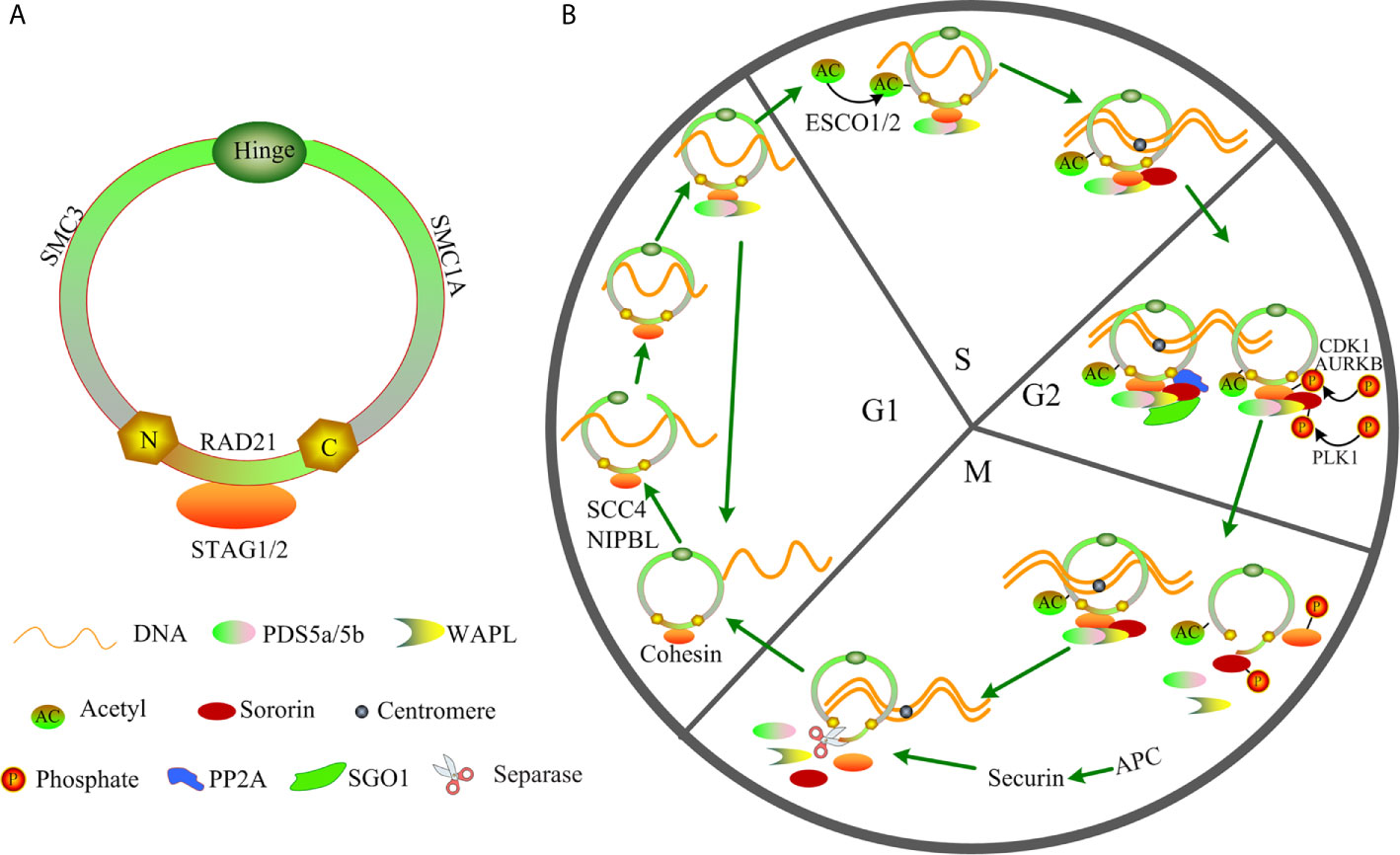
Figure 1 Structure and function of cohesin in the cell cycle. (A) Structure of cohesin. (B) Function of cohesin in the cell cycle.
The data of cohesin subunits mutations were collected from 16 articles. The percentage of cohesin mutations with different characteristics is displayed in Table 1, and detailed data are presented in Supplement Table 1.
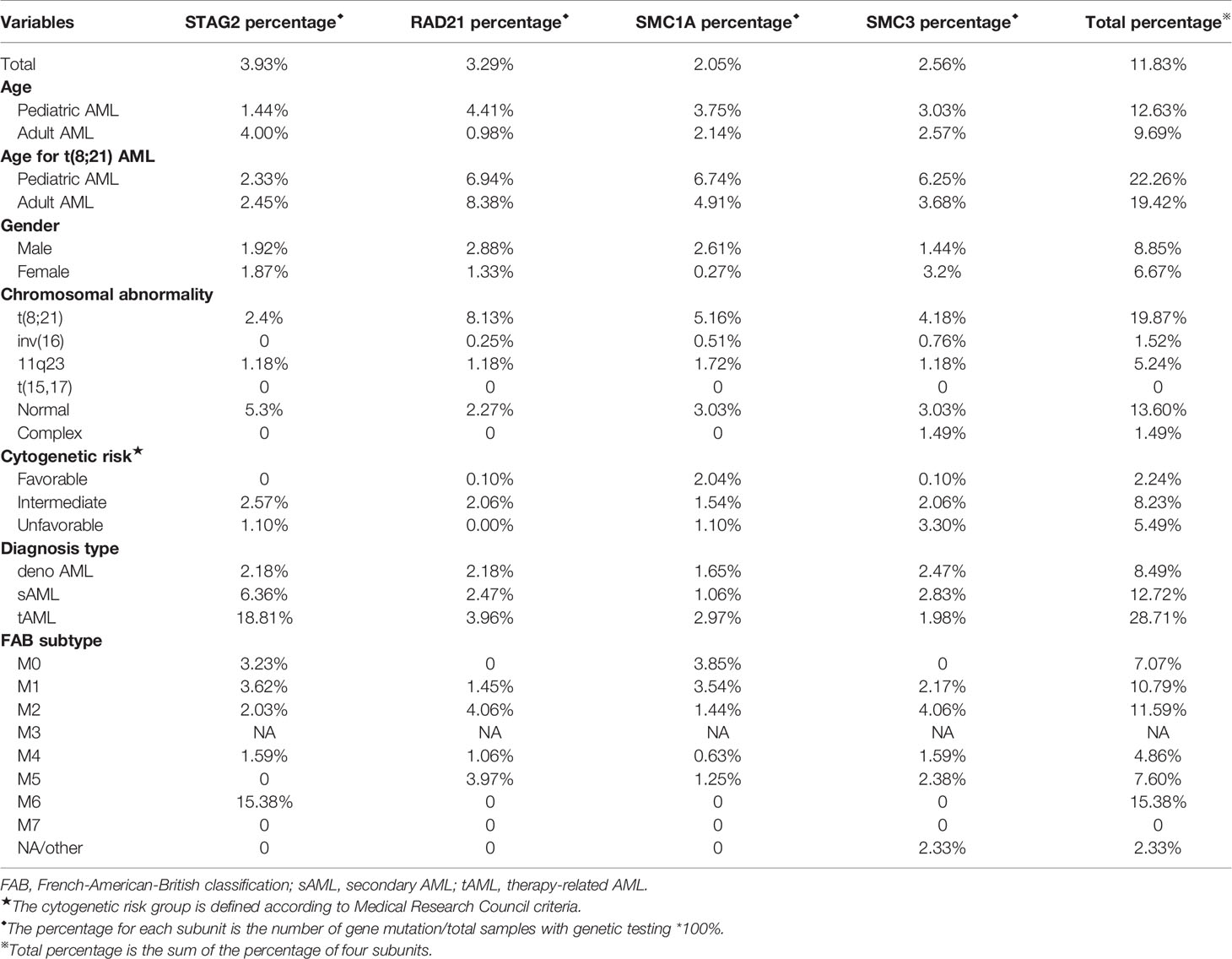
Table 1 Characteristics of AML patients with cohesin mutation.
As shown in Table 1, cohesin subunit mutations were harbored by a total of 11.88% of AML patients including all cytogenetic groups (Table 1). Similar to our study, other studies demonstrated that the most frequently mutated cohesin subunit was STAG2 (3.93%), especially in cytogenetically normal AML (CN-AML) and adult AML (12, 35). Cohesin mutation was found in 19.87% of t(8;21) AML cases, which was much higher than that in the other cytogenetic subgroups, and was rare or none mutation in t(15;17), inv16 and complex chromosomal abnormality groups. Interestingly, the incidence and profile of cohesin gene mutations in t(8;21) AML were different from those in other types of AML (Table 1). The frequency of RAD21 mutation (about 8.08%) was higher than the other cohesin compound gene mutations in t(8;21) AML. The mutation frequency of STAG2 (2.4%) was at the bottom of the list for t(8;21) AML. The frequencies of mutations in SMC1A and SMC3 were found to be 5.7% and 4.3%, respectively, in the t(8;21) cases, which were also higher than those in the other AML subtypes. Thus, we posit that RAD21, SMC1A, and SMC3 are more likely to be mutated in t(8;21) AML.
We further analyzed cohesin gene mutation data by comparing between pediatric and adult AML and found that the frequencies of cohesin mutations in childhood AML was higher than adult AML, while RAD21 mutations rarely occurred in adult AML, except for pediatric t(8; 21) AML. STAG2 was the most commonly mutated gene in adult AML. Besides, in agreement with our results, recent studies by Metzeler et al., demonstrated that STAG2 also tended to be more frequent in secondary AML (sAML) compared with de novo AML (35). For different FAB types, RAD21 mutations were mainly found in FAB M2 and M5. SMC3 were frequently identified in FAB M2. Because the number of AML patients in FAB M0 and M7 were limited, the proportion of STAG2 and SMC1A mutation was questionable. AML patients with intermediate risk cytogenetics but neither with favorable cytogenetics nor with unfavorable cytogenetics had the highest percentage of STAG2 and RAD21 mutation, and accord with the previous result (11). Intriguingly, the frequency of STAG2, RAD21, and SMC3 mutations in male had little difference from that in female and each type of mutation occur in about 1% to 3% of AML patients. Although SMC1A was located on the X-chromosome, the percentage of SMC1A mutation in male is higher than that in female.
The presence of a mutation in cohesin is highly specific for t(8;21) AML, so we next asked whether there were specific mutant form and hotspot in the mutated cohesin gene. We found that the majority of STAG2 and RAD21 mutations were nonsense and frameshift in all the AML subtypes (Figure 2A), suggesting that cohesin mutations resulted in a decrease or loss of the cohesin function. Almost all SMC1A and SMC3 mutations were found to be missense, which was consistent with previous studies (35–37).
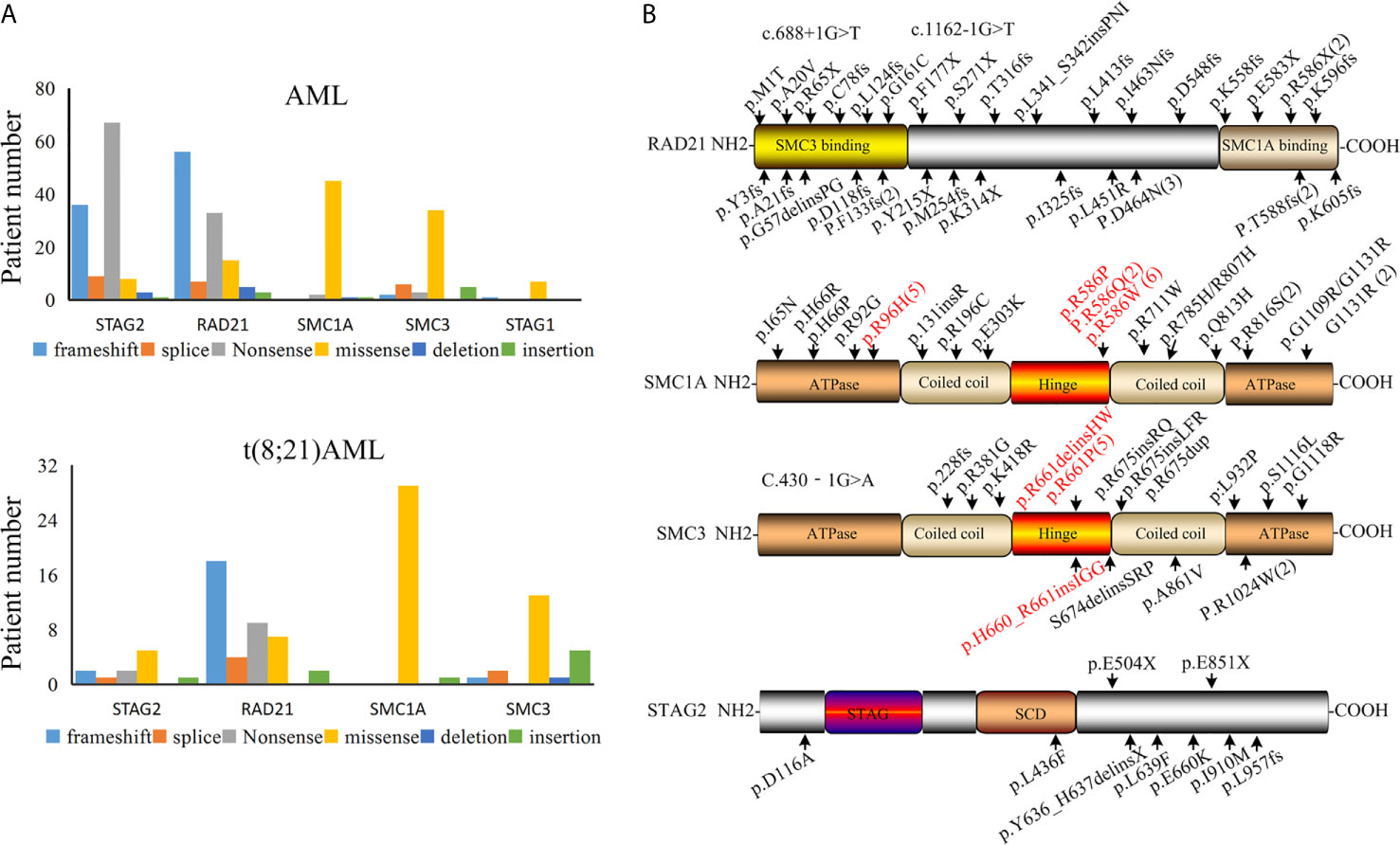
Figure 2 Cohesin mutation in patients with AML. (A) Various types of cohesin mutations. (B) Location of mutations in the cohesin complex in t(8;21) AML.
By summarizing the positions for the identified cohesin mutation genes that reported in t(8;21) AML patients (Figure 2B), we found that the mutation sites of RAD21 were present over the whole coding section. A mutated site was found in each domain of the RAD21, but no mutational hotspots were identified. It was found that RAD21 was rapidly cleaved by caspase-3 and -7 at the Asp279 site in apoptosis process (38). RAD21 is not only cleaved at Arg172 and Arg450 sites for sister chromatid separation, but also for the completion of cytokinesis (39). However, no mutations were found in these sites among t(8;21) AML patients based on our analysis, suggesting that the mutated RAD21 might not be involved in apoptosis induced by diverse stimuli or chromatid separation in the development AML1-ETO driven leukemogenesis.
The C-terminal plays an important role in the functions of STAG2 protein, as it is the location of many phosphorylation sites and potentially the nuclear localization signal (40, 41). The majority of STAG2 mutations in t(8;21) AML occur in the C-terminal and potentially result in loss of the phosphorylation site and the nuclear localization signal. These findings may explain why most of the STAG2 mutations were found in the C-terminal of STAG2 in t(8;21) AML patients. It was noted that mutated SMC1A was located at the codons R586 site in the hinge domain among from nine patients and was at the codons R96 site in adenosine triphosphatase heads among five patients in 30 cases with t(8;21) AML. In the data of 38 t(8;21) AML patients, seven mutated SMC3 genes were located at the codons R661 site in the hinge domain (Figure 2B). These mutation sites should be considered in functional analyses of these mutations.
Many leukemia genes are found to be infrequently mutated, and AML patients typically have more than one driver mutation (42, 43). Further, cohesin mutations are frequently found with other concurrent gene mutations. The mutational cohesin complex was often to be enriched or mutually antagonistic in specific molecular subgroups of cancer, which implied that mutations in the cohesin gene were more oncogenic in particular cytogenetics. Cohesin mutation were often observed in AML patients with t(8;21), but they were mutually exclusive in AML patients with favorable-risk cytogenetics, complex chromosomal abnormality, inv (16), and t(15;17) (11, 13, 37, 44–48), which also found in Table 1. Mutations in the gene of the cohesin complex rarely co-occurred with the spliceosome-complex, IDH1, IDH2, and TET2 and TP53 mutation (11, 13, 37). What is more, data from the Cancer Genome Atlas (TCGA) Research Network revealed that seldom AML patients carrying mutations in the cohesin complex had mutations in the IDH1 and IDH2 epigenetic modifiers, myeloid transcription factors, or NRAS (10).
Several studies have shown that mutations in the cohesin complex genes are often found in NPM1 mutation and CEBPAbi AML cases (11, 12, 37, 49, 50). Besides, lower expression of SMC1A was observed in patients with NPM1 mutation (51). But STAG2 mutation less frequently co-occurred with NPM1, and more frequently with RUNX1 (35, 37). It was worth noting that cohesin and NPM1 interact with the CCCTC-binding factor (CTCF) at the insulator sites through which they are involved in the insulator function (52, 53). Given the significant correlation between cohesin and NPM1 mutation, it is suggested they may have functional relevance in transcriptional regulation of the pathogenesis and mechanism in AML.
Contradictory results have been reported regarding the correlation between genes in the cohesin complex and other mutations. Mutations in Ras-family oncogenes including ASXL1 or BCOR, and FLT3-ITD were enriched in leukemia patients carrying the cohesin mutation (12, 37). However, No correlation was found between the cohesin complex and FLT3-ITD in AML (11).
The prognostic significance of cohesin mutations in AML remains controversial, and there are many factors that affect the prognostic significance of cohesin mutations. Tsai et al. suggested that cohesin mutations were independent favorable factors for a higher overall survival (OS) and disease-free survival (DFS) in de novo AML (37). Regardless of the cytogenetics, OS of adult AML patients harboring cohesin genes mutations tended to be higher than that of those without mutations (54). However, in contrast to these results, other studies found no survival differences were identified between mutant and wild cohesin in OS, DFS, and RFS at the different prognostic groups, such as total AML, pediatric AML, de novo AML (11, 12, 35, 54, 55). Despite cohesin gene mutation more frequently identified in sAML, CN-AML, and AML patients with NMP1 mutation, cohesin gene mutations had no significant implication on OS, RFS, and CR (11, 56). In addition, the outcome of allogeneic transplantation was not influenced by the presence of mutations in the cohesin complex (11). However, lower SMC3 protein levels were significantly associated with European Leukemia Net (ELN) adverse risk group classification (57). Thus the relationship between cohesin gene mutation and the prognostic is conflict.
Though there is a high frequency of cohesin gene mutation in t(8;21) AML, Faber et al. found that recurrent cohesin mutations did not affect the prognostic results of t(8;21) AML in 10 pediatric and 6 adult patients (58). Besides, AML patients with RAD21 mutation did not show statistical significance in OS and risk for relapse compared to those with wild RAD21 in the t(8;21) AML (13). However, recent works have shown that a mutation in cohesin genes was an independent poor prognostic factor for RFS and OS in patients with RUNX1-RUNX1T1 leukemia (43, 46). Among patients who had t(8;21) AML with tyrosine kinase (TK) mutations, those who had co-occurrent mutations in cohesin genes also had worse prognosis with a high 5-year cumulative incidence of relapse (CIR) and a higher risk of relapse (44). This result indicates that mutated genes function cooperatively in these events.
These findings all emphasize that cohesin have different functions under various clinical features in the biological mechanism. Taken together, the AML subtype, karyotype, age, and co-mutated genes should be considered together with cohesin mutation as prognostic factors. In particular, it is worth evaluating the prognostic significance of cohesin mutations in t(8;21) AML.
Previous studies indicated that the occurrence of mutations in cohesin genes was considered as pre-leukemic and early genetic events in human leukemogenesis (18, 59, 60). During the progression from refractory anemia with excess blasts (RAEB) to sAML, cohesin mutations were observed only in patients with sAML, which implied that the occurrence of cohesin mutations represented a late event in the process of AML transformation (12). Mutations in SMC1A, SMC3 and STAG2 might be the initiating events in the pathogenesis of AML (55, 61). STAG2 mutation was identified as a secondary-type mutation in secondary and de novo AML (35). In addition, STAG2 mutation was found to occur in the early stage of AML, whereas RAD21 and SMC1A mutations occurred relatively late in adult AML (37). In the clonal architecture of t(8;21) AML, SMC3, and SMC1A mutations occurred at diagnosis, while RAD21 and STAG2 mutations appeared at relapse (13, 45). Hence, more systematic investigations are needed to decipher the evolution of cohesin gene mutations in AML.
Mutated cohesin complex genes are almost mutually exclusive (34), suggesting the changes in one subunit may affect the entire complex function. Interestingly, gene expression in the entire cohesin complex was usually decreased in AML patients with cohesin mutations (12). Normal sister chromatid cohesion was found in AML patients with cohesin mutations. Moreover, no significant difference was found in the number of chromosome abnormalities between cohesin-mutated and non-mutated patients (34). Genomic stability was not affected when cohesin levels were reduced by 80% in yeast and Drosophila embryos (62, 63). Nevertheless, no correlation between was found transcription and translation of cohesin subunits was found in 48 AML patients analyzed in a quantitative manner (57).
A lower protein level of SMC3 was found to be associated with numerical chromosome abnormalities and complex karyotypes (57). In mice, bone marrow aplasia with premature sister chromatid separation was caused by biallelic loss of SMC3 and chromatin structure and hematopoietic stem cell (HSC) function were controlled by cohesin in a dose-dependent manner (64). Given the above information, some cohesin mutations may have primarily affected its repair function and could not be selected clonally, leading to chromosomal instability and aneuploidy. Mitotic disorder or cell death occurred due to complete loss of the cohesin function, thus the majority of cohesin mutations were heterozygous in AML. Though genes encoding STAG2 and SMC1A were located on the X-chromosome, the homologous protein may be partially compensated for the loss of STAG2 function caused by its alterations (65, 66). Given the results of other study, SMC1A mutations are neither significantly associated with X loss nor with gender in CBF leukemia patients (46), which indicated that homologous protein of SMC1A might exists in leukemia patients.
In early hematopoiesis and myeloid differentiation, self-renewal of hematopoietic stem and progenitor cells (HSPCs) was enhanced, and differentiation of HSPCs was impaired as a result of cohesin mutation or deletion (60, 66–68). STAG2 loss decreased the transcription of lineage-specification genes, thereby leading to an altered hematopoietic function in a mouse model (66). However, STAG2 knockdown caused an increase in HSC-specific genes in primary human CD34+ cells (67). In HSPCs with the cohesin complex mutation, knockdown of ERG, GATA2, or RUNX1 can revert the differentiation block caused by cohesin mutation (69). The cohesin complex directly recruits and/or stabilizes the binding of Polycomb Repressive Complex 1 (PRC1) to active genes (70). The expression of self-renewal genes Hoxa7 and Hoxa9 in HSPCs increased through derepression of Polycomb Repressive Complex 2 (PRC2) caused by depletion of the cohesin subunit Rad21 (71). In addition, cohesin deficiency in hematopoietic progenitor cells could not induce acute phenotypes through a series of shRNA mouse models (60). Self-renewal capacity of HSPCs increased as a result of SMC3 haploinsufficiency, but SMC3 knockdown together with FLT3-ITD could lead to the development of AML (64). Thus, heterozygous mutations may depend on the cooperation with other mutations for causing leukemia.
Previous studies suggested that some hematopoietic transcription factors such as TAL1 and ERG, are regulated by cohesin binding to distal enhancers or promoters (72–75). A recent research has shown that Stag2 and Runx1 play an important role in regulating chromatin looping and transcription in hematopoiesis, and cohesin-Runx1 deficiency can induce myeloid-skewed expansion of HPSCs and myelodysplastic syndromes (MDS) (76). In mouse and zebrafish, cohesin binds to runx1 enhancer region, which is located within the intron between the P1 (distal) and P2 (proximal) promoters of Runx1 (32, 77, 78). Similarly, cohesin subunits are found in conserved regulatory elements (CRE)/promoter of Runx1 in the human leukemia K562 cell line (32). It plays an indispensable role in terms of regulating Runx1 expression in a specific subpopulation of hematopoietic progenitors and cohesin together with CTCF determines the spatial distribution of runx1 transcripts in the zebrafish embryo (32, 79). Monoallelic loss of Rad21 resulted in a decrease in the transcription of runx1 and micro-injection of runx1 mRNA can rescue the defects caused by RAD21 mutations in differentiated blood cells of the zebrafish model (80). Nevertheless, RNAi-mediated cohesin knockdown was found to enhance RUNX1 transcription in the leukemia HL60 cell line (32). Moreover, Mazzola et al. found there was no significant correlation between the expression of RAD21 and RUNX1 in both megakaryocytes derived from healthy donors and megakaryocytes derived from adult AML patients (80). These evidences indicate that cohesin performs different functions in regulating RUNX1 expression in different species.
Cohesin mutations were prevalent in AML with RUNX1 abnormality (12), especially in RUNX1-RUX1T1. Significant growth suppression of the Kasumi-1 cell (harboring t(8;21) and RAD21 frameshift mutation p.K330PfsX6) was induced by forced expression of wild-type RAD21 (34). These results suggest that there is potential synergy between cohesin and AML1-ETO in hematopoiesis and leukemogenesis. Furthermore, AML patients with cohesin mutations expressed significantly higher levels of AE9a than those without cohesin mutations (81). AML1-ETO blind to the promoter and distal elements including those of itself, and it interacts with other hematopoietic transcription factors, such as ERG, FLI1, TAL1, and RUNX1, in this region (82). Importantly, the promoter can interact with short or long distances enhancers to activate gene expression, independent of the position and orientation (83). Given the fact that TFs are mainly enriched in enhancers to control gene expression (84), cohesin can eliminate the spatial distance of promoter and enhancer by promoting chromatin loop formation (85–87). Therefore, we postulate that AML1-ETO recruits hematopoietic TFs to the enhancer. Then, the mediator complex and RNA polymerase II (Pol II) blind to the TFs and promoters respectively, in a gene-specific manner. Cohesin is loaded onto the promoter and enhancer that contribute to gene regulation (Figure 3). RUNX1 and AML1-ETO share the same sequences before exon 5; thus, cohesin may also be involved in the regulation of AML1-ETO expression.

Figure 3 Interactions of cohesin and AML1-ETO.
In this review, we summarized and analyzed cohesin mutations in AML, especially in t(8; 21) subtype, from previous studies. The conclusion and outlook are as follows:
1. We found each subunit of cohesin mutated in different characteristics with different percentage. RAD21, SMC1A, and SMC3 mutations were remarkably higher in t(8; 21) than the other AML subtypes. STAG2 mutation occurred chiefly on adult AML and sAML. For FAB subtype, RAD21 and SMC3 are mainly found in FAB M2. SMC1A was common found in male. Such differences arise because each individual member of the cohesin complex has different influence and function in different characteristics of AML by different ways and mechanisms.
2. Most of the current studies confirmed that cohesin can act as a growth regulator of HSCs. Whether cohesin gene mutation leads to protein functional activation or inactivation is unclear. Although mutated cohesin perturbed the balance between self-renewal and differentiation, no evidence demonstrated that the impaired cohesin function could independently lead to AML, suggesting that cooperative genetic/epigenetic alterations are involved in AML development. So far, co-mutations of cohesin with other genes are still controversial and the biological functions of cohesin are not yet fully understood in AML.
3. The clinical outcomes of cohesin mutations are associated with the AML type, karyotype, and co-mutated gene. No specific therapeutic strategies targeting cohesin mutated AML have been described. In addition, most of the studies indicated that cohesin mutations are pre-leukemic and early genetic events in human leukemogenesis. Considering the four core subunits of cohesin associated with different prognosis and mutation frequencies, their specific functions in AML should be also investigated in depth.
4. The mechanism with respect to cohesion subunits mutation is still unestablished in AML, especially in the t(8; 21) AML. How cohesin subunits mutation cooperates with AML1-ETO, thereby promoting leukemic transformation should be illustrated in future studies. Further understanding of the relationship between cohesin and AML1-ETO may provide unprecedented insights into the pathogenesis of AML.
Conducting research on above questions would hopefully provide significant progress in diagnosis classification, prognostic stratification, and therapeutic strategies to determine AML risks in the near future.
CH wrote the manuscript. XG and YL revised the manuscript critically. JZ, EY, and LZ prepared the figures and revisions. LY contributed to study design, writing, and editing. All authors contributed to the article and approved the submitted version.
This work was supported by “Major New Drug Development Project” from Ministry of Science and Technology of China (2019ZX09201002003), Natural Science Foundation of Shenzhen University General Hospital (SUGH2020QD008), State Key Program of National Natural Science of China (82030076), National Natural Science Foundation of China (82070161, 81970151, 81870134, and 82000161), Beijing Natural Science Foundation (7202186), Shenzhen Science and Technology investigation project (JCYJ 20190808123005569 and JCYJ 20190808163601776), China Postdoctoral Science Foundation (2018M640824), and Science and Technology Foundation of Shenzhen (JCYJ20200109113810154).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2021.579881/full#supplementary-material
1. Marcucci G, Mrózek K, Ruppert AS, Maharry K, Kolitz JE, Moore JO, et al. Prognostic factors and outcome of core binding factor acute myeloid leukemia patients with t (8,21) differ from those of patients with inv(16): a Cancer and Leukemia Group B study. J Clin Oncol (2005) 23:5705–17. doi: 10.1200/JCO.2005.15.610
2. Hospital MA, Prebet T, Bertoli S, Thomas X, Tavernier E, Braun T, et al. Core-binding factor acute myeloid leukemia in first relapse: a retrospective study from the French AML intergroup. Blood (2014) 124:1312–9. doi: 10.1182/blood-2014-01-549212
3. van der Kouwe E, Staber PB. RUNX1-ETO: attacking the epigenome for genomic instable leukemia. Int J Mol Sci (2019) 20:350. doi: 10.3390/ijms20020350
4. Al-Harbi S, Aljurf M, Mohty M, Almohareb F, Ahmed SOI. An update on the molecular pathogenesis and potential therapeutic targeting of AML with t (8,21)(q22;q22.1); RUNX1-RUNX1T1. Blood Adv (2020) 4:229–38. doi: 10.1182/bloodadvances.2019000168
5. Yuan Y, Zhou L, Miyamoto T, Iwasaki H, Harakawa N, Hetherington CJ, et al. AML1-ETO expression is directly involved in the development of acute myeloid leukemia in the presence of additional mutations. Proc Natl Acad Sci USA (2001) 98:10398–403. doi: 10.1073/pnas.171321298
6. Höllein A, Nadarajah N, Meggendorfer M, Jeromin S, Kern W, Haferlach C, et al. Molecular characterization of aml with runx1-runx1t1 at diagnosis and relapse reveals net loss of co-mutations. Hemasphere (2019) 3:e178. doi: 10.1097/HS9.0000000000000178
7. Krauth MT, Eder C, Alpermann T, Bacher U, Nadarajah N, Kern W, et al. High number of additional genetic lesions in acute myeloid leukemia with t (8,21)/RUNX1-RUNX1T1: frequency and impact on clinical outcome. Leukemia (2014) 28:1449–58. doi: 10.1038/leu.2014.4
8. Hou HA, Liu CY, Kuo YY, Chou WC, Tsai CH, Lin CC, et al. Splicing factor mutations predict poor prognosis in patients with de novo acute myeloid leukemia. Oncotarget (2016) 7:9084–101. doi: 10.18632/oncotarget.7000
9. Itzykson R, Duployez N, Fasan A, Decool G, Marceau-Renaut A, Meggendorfer M, et al. Clonal Interference of Signaling Mutations Worsens Prognosis in Core-Binding Factor Acute Myeloid Leukemia. Blood (2018) 132:187–96. doi: 10.1182/blood-2018-03-837781
10. Cancer Genome Atlas Research Network, Ley TJ, Miller C, Ding L, Raphael BJ, Mungall AJ, et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. New Engl J Med (2013) 368:2059–74. doi: 10.1056/NEJMoa1301689
11. Thol F, Bollin R, Gehlhaar M, Walter C, Dugas M, Suchanek KJ, et al. Mutations in the cohesin complex in acute myeloid leukemia: clinical and prognostic implications. Blood (2014) 123:914–20. doi: 10.1182/blood-2013-07-518746
12. Thota S, Viny AD, Makishima H, Spitzer B, Radivoyevitch T, Przychodzen B, et al. Genetic alterations of the cohesin complex genes in myeloid malignancies. Blood (2014) 124:1790–8. doi: 10.1182/blood-2014-04-567057
13. Christen F, Hoyer K, Yoshida K, Hou HA, Waldhueter N, Heuser M, et al. Genomic landscape and clonal evolution of acute myeloid leukemia with t (8, 21): an international study on 331 patients. Blood (2019) 133:1140–51. doi: 10.1182/blood-2018-05-852822
14. Solomon DA, Kim JS, Waldman T. Mutational inactivation of STAG2 causes aneuploidy in human cancer. Science (2011) 333:1039–43. doi: 10.1126/science.1203619
15. Losada A. Cohesin in cancer: chromosome segregation and beyond. Nat Rev Cancer (2014) 14:389–93. doi: 10.1038/nrc3743
16. Litwin I, Wysocki R. New insights into cohesin loading. Curr Genet (2018) 64(1):53–61. doi: 10.1007/s00294-017-0723-6
17. Dauban L, Montagne R, Thierry A, Lazar-Stefanita L, Bastié N, Gadal O, et al. Regulation of Cohesin-Mediated Chromosome Folding by Eco1 and Other Partners. Mol Cell (2020) 77:1279–93.e4. doi: 10.1016/j.molcel.2020.01.019
18. Cuartero S, Innes AJ, Merkenschlager M. Towards a Better Understanding of Cohesin Mutations in AML. Front Oncol (2019) 9:867. doi: 10.3389/fonc.2019.00867
19. Kagey MH, Newman JJ, Bilodeau S, Zhan Y, Orlando D, Am van Berkumm NL, et al. Mediator and cohesin connect gene expression and chromatin architecture. Nature (2010) 467:430–5. doi: 10.1038/nature09380
20. Guillou E, Ibarra A, Coulon V, Casado-Vela J, Rico D, Casal I, et al. Cohesin organizes chromatin loops at DNA replication factories. Genes Dev (2010) 24:2812–22. doi: 10.1101/gad.608210
21. McAleenan A, Clemente-Blanco A, Cordon-Preciado V, Sen N, Esteras M, Jarmuz A, et al. Post-replicative repair involves separase-dependent removal of the kleisin subunit of cohesin. Nature (2012) 493:250–4. doi: 10.1038/nature11630
22. Panigrahi AK, Pati D. Higher-order orchestration of hematopoiesis: Is cohesin a new player? Exp Hematol (2012) 40:967–73. doi: 10.1016/j.exphem
23. Yan J, Enge M, Whitington T, Dave K, Liu J, Sur I, et al. Transcription factor binding in human cells occurs in dense clusters formed around cohesin anchor sites. Cell (2013) 154:801–13. doi: 10.1016/j.cell.2013.07.034
24. Guo Y, Monahan K, Wu H, Gertz J, Varley KE, Li W, et al. CTCF/cohesin-mediated DNA looping is required for protocadherin alpha promoter choice. Proc Natl Acad Sci USA (2012) 109:21081–6. doi: 10.1073/pnas.1219280110
25. Haarhuis JHI, van der Weide RH, Blomen VA, Yáñez-Cuna JO, Amendola M, van Ruiten MS, et al. The cohesin release factor wapl restricts chromatin loop extension. Cell (2017) 169:693–707. doi: 10.1016/j.cell.2017.04.013
26. Schwarzerm W, Abdennur N, Goloborodko A, Pekowska A, Fudenberg G, Loe-Mie Y, et al. Two independent modes of chromatin organization revealed by cohesin removal. Nature (2017) 551:51–6. doi: 10.1038/nature24281
27. Rao SS, Huang SC, Glenn St Hilaire B, Engreitz JM, Perez EM, Kieffer-Kwon KR, et al. Cohesin loss eliminates all loop domains. Cell (2017) 171:305–20. doi: 10.1016/j.cell.2017.09.026
28. Bintu B, Mateo LJ, Su JH, Sinnott-Armstrong NA, Parker M, Kinrot S, et al. Super-resolution chromatin tracing reveals domains and cooperative interactions in single cells. Science (2018) 362:eaau1783. doi: 10.1126/science.aau1783
29. Wendt KS, Yoshida K, Itoh T, Bando M, Koch B, Schirghuber E, et al. Cohesin mediates transcriptional insulation by ccctc-binding factor. Nature (2008) 451:796–801. doi: 10.1038/nature06634
30. Canzio D, Nwakeze CL, Horta A, Rajkumar SM, Coffey EL, Duffy EE, et al. Antisense lncrna transcription mediates dna demethylation to drive stochastic protocadherin α promoter choice. Cell (2019) 177:639–53.e15. doi: 10.1016/j.cell.2019.03.008
31. Zhu Z, Wang X. Roles of cohesin in chromosome architecture and gene expression. Semin Cell Dev Biol (2018) 90:187–93. doi: 10.1016/j.semcdb.2018.08.004
32. Marsman J, O’Neill AC, Kao BR, Rhodes JM, Meier M, Antony J, et al. Cohesin and CTCF differentially regulate spatiotemporal runx1 expression during zebrafish development. Biochim Biophys Acta (2014) 1839:50–61. doi: 10.1016/j.bbagrm.2013.11.007
33. Fagnocchi L, Poli V, Zippo A. Enhancer reprogramming in tumor progression: a new route towards cancer cell plasticity. Cell Mol Life Sci (2018) 75:2537–55. doi: 10.1007/s00018-018-2820-1
34. Kon A, Shih LY, Minamino M, Sanada M, Shiraishi Y, Nagata Y, et al. Recurrent mutations in multiple components of the cohesin complex in myeloid neoplasms. Nat Genet (2013) 45:1232–7. doi: 10.1038/ng.2731
35. Metzeler KH, Herold T, Rothenberg-Thurley M, Amler S, Sauerland MC, Görlich D, et al. Spectrum and prognostic relevance of driver gene mutations in acute myeloid leukemia. Blood (2016) 128:686–98. doi: 10.1182/blood-2016-01-693879
36. Papaemmanuil E, Gerstung M, Bullinger L, Gaidzik VI, Paschka P, Roberts ND, et al. Genomic classification and prognosis in acute myeloid leukemia. New Engl J Med (2016) 374:2209–21. doi: 10.1056/NEJMoa1516192
37. Tsai CH, Hou HA, Tang JL, Kuo YY, Chiu YC, Lin CC, et al. Prognostic impacts and dynamic changes of cohesin complex gene mutations in de novo acute myeloid leukemia. Blood Cancer (2017) 7:663. doi: 10.1038/s41408-017-0022-y
38. Pati D, Zhang N, Plon SE. Linking sister chromatid cohesion and apoptosis: role of rad21. Mol Cell Bio (2002) 22:8267–77. doi: 10.1128/mcb.22.23.8267-8277.2002
39. Hauf S, Waizenegger IC, Peters JM. Cohesin Cleavage by Separase Required for Anaphase and Cytokinesis in Human Cells. Science (2001) 293:1320–3. doi: 10.1126/science.1061376
40. Hauf S, Roitinger E, Koch B, Dittrich CM, Mechtler K, Peters JM. Dissociation of cohesin from chromosome arms and loss of arm cohesion during early mitosis depends on phosphorylation of SA2. PloS Biol (2005) 3:e69. doi: 10.1371/journal.pbio.0030069
41. Solomon DA, Kim JS, Waldman T. Cohesin gene mutations in tumorigenesis: from discovery to clinical significance. BMB Rep (2014) 47:299–310. doi: 10.5483/bmbrep.2014.47.6.092
42. Ding L, Ley T, Larson DE, Miller CA, Koboldt DC, Welch JS, et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature (2012) 481:506–10. doi: 10.1038/nature10738
43. Prieto-Conde MI, Jiménez C, García-Álvarez M, Ramos F, Medina A, Cuello R, et al. Identification of relapse-associated gene mutations by next-generation sequencing in low-risk acute myeloid leukaemia patients. Br J Haematol (2020) 189:718–30. doi: 10.1111/bjh.16420
44. Duployez N, Marceau-Renaut A, Boissel N, Petit A, Bucci M, Geffroy S, et al. Comprehensive mutational profiling of core binding factor acute myeloid leukemia. Blood (2016) 127:2451–9. doi: 10.1182/blood-2015-12-688705
45. Sood R, Hansen NF, Donovan FX, Carrington B, Bucci D, Maskeri B, et al. Somatic mutational landscape of AML with inv (16) or t (8,21) identifies patterns of clonal evolution in relapse leukemia. Leukemia (2016) 30:501–4. doi: 10.1038/leu.2015.141
46. Opatz S, Bamopoulos SA, Metzeler KH, Herold T, Ksienzyk B, Bräundl K, et al. The clinical mutatome of core binding factor leukemia. Leukemia (2020) 34:1553–62. doi: 10.1038/s41375-019-0697-0
47. Madan V, Shyamsunder P, Han L, Mayakonda A, Nagata Y, Sundaresan J, et al. Comprehensive mutational analysis of primary and relapse acute promyelocytic leukemia. Leukemia (2016) 30:1672–81. doi: 10.1038/leu.2016.69
48. Andersson AK, Ma J, Wang J, Chen X, Gedman AL, Dang J, et al. The landscape of somatic mutations in infant MLL-rearranged acute lymphoblastic leukemias. Nat Genet (2015) 47:330–7. doi: 10.1038/ng.3230
49. Lavallée VP, Krosl J, Lemieux S, Boucher G, Gendron P, Pabst C, et al. Chemo-genomic interrogation of CEBPA mutated AML reveals recurrent CSF3R mutations and subgroup sensitivity to JAK inhibitors. Blood (2016) 127:3054–61. doi: 10.1182/blood-2016-03-705053
50. Konstandin NP, Pastore F, Herold T, Dufour A, Rothenberg-Thurley M, Hinrichsen T, et al. Genetic heterogeneity of cytogenetically normal AML with mutations of CEBPA. Blood Adv (2018) 2:2724–31. doi: 10.1182/bloodadvances.2018016840
51. Gadewal N, Kumar R, Aher S, Gardane A, Gaur T, Varma AK, et al. miRNA-mRNA Profiling Reveals Prognostic Impact of SMC1A Expression in Acute Myeloid Leukemia. Oncol Res (2020) 28:321–30. doi: 10.3727/096504020X15816752427321
52. Lee BK, Iyer VR. Genome-wide studies of CCCTC-binding factor (CTCF) and cohesin provide insight into chromatin structure and regulation. J Bio Chem (2012) 287:30906–13. doi: 10.1074/jbc.R111.324962
53. Zlatanova J, Caiafa P. CTCF and its protein partners: divide and rule? J Cell Sci (2009) 122:1275–84. doi: 10.1242/jcs.039990
54. Kihara R, Nagata Y, Kiyoi H, Kato T, Yamamoto E, Suzuki K, et al. Comprehensive analysis of genetic alterations and their prognostic impacts in adult acute myeloid leukemia patients. Leukemia (2014) 28:1586–95. doi: 10.1038/leu.2014.55
55. Shiba N, Yoshida K, Shiraishi Y, Okuno Y, Yamato G, Hara Y, et al. Whole-exome sequencing reveals the spectrum of gene mutations and the clonal evolution patterns in paediatric acute myeloid leukaemia. Brit J Haematol (2016) 175:476–89. doi: 10.1111/bjh.14247
56. Lindsley RC, Mar BG, Mazzola E, Grauman PV, Shareef S, Allen SL, et al. Acute myeloid leukemia ontogeny is defined by distinct somatic mutations. Blood (2015) 125:1367–76. doi: 10.1182/blood-2014-11-610543
57. Kraft B, Lombard J, Kirsch M, Wuchter P, Bugert P, Hielscher T, et al. SMC3 protein levels impact on karyotype and outcome in acute myeloid leukemia. Leukemia (2018) 33:795–9. doi: 10.1038/s41375-018-0287-6
58. Faber ZJ, Chen X, Gedman AL, Boggs K, Cheng J, Ma J, et al. The genomic landscape of core-binding factor acute myeloid leukemias. Nat Genet (2016) 48:1551–6. doi: 10.1038/ng.3709
59. Corces-Zimmerman MR, Majeti R. Pre-leukemic evolution of hematopoietic stem cells: the importance of early mutations in leukemogenesis. Leukemia (2014) 28:2276–82. doi: 10.1038/leu.2014.211
60. Mullenders J, Aranda-Orgilles B, Lhoumaud P, Keller M, Pae J, Wang K, et al. Cohesin loss alters adult hematopoietic stem cell homeostasis, leading to myeloproliferative neoplasms. J Exp Med (2015) 212:1833–50. doi: 10.1084/jem.20151323
61. Welch JS, Ley TJ, Link DC, Miller CA, Larson DE, Koboldt DC, et al. The origin and evolution of mutations in acute myeloid leukemia. Cell (2012) 150:264–78. doi: 10.1016/j.cell.2012.06.023
62. Heidinger-Pauli JM, Mert O, Davenport C, Guacci V, Koshland D. Systematic reduction of cohesin differentially affects chromosome segregation, condensation, and DNA repair. Curr Biol (2010) 20:957–63. doi: 10.1016/j.cub.2010.04.018
63. Carvalhal S, Tavares A, Santos MB, Mirkovic M, Oliveira RA. A quantitative analysis of cohesin decay in mitotic fidelity. J Cell Biol (2018) 217:3343–53. doi: 10.1083/jcb.201801111
64. Viny AD, Ott CJ, Spitzer B, Rivas M, Meydan C, Papalexi E, et al. Dose-dependent role of the cohesin complex in normal and malignant hematopoiesis. J Exp Med (2015) 212:1819–32. doi: 10.1084/jem.20151317
65. Benedetti L, Cereda M, Monteverde L, Desai N, Ciccarelli FD. Synthetic lethal interaction between the tumour suppressor STAG2 and its paralog STAG1. Oncotarget (2017) 8:37619–32. doi: 10.18632/oncotarget.16838
66. Viny AD, Bowmanm RL, Lium Y, Lavallée VP, Eisman SE, Xiao W, et al. Cohesin Members Stag1 and Stag2 Display Distinct Roles in Chromatin Accessibility and Topological Control of HSC Self-Renewal and Differentiation. Cell Stem Cell (2019) 25:682–696.e8. doi: 10.1016/j.stem.2019.08.003
67. Galeev R, Baudet A, Kumar P, Rundberg Nilsson A, Nilsson B, Soneji S, et al. Genome-wide RNAi screen identifies cohesin genes as modifiers of renewal and differentiation in human HSCs. Cell Rep (2016) 14:2988–3000. doi: 10.1016/j.celrep.2016.02.082
68. Sasca D, Yun H, Giotopoulos G, Szybinski J, Evan T, Wilson NK, et al. Cohesin-dependent regulation of gene expression during differentiation is lost in cohesin-mutated myeloid malignancies. Blood (2019) 134:2195–208. doi: 10.1182/blood.2019001553
69. Mazumdar C, Shen Y, Xavy S, Zhao F, Reinisch A, Li R, et al. Leukemia-associated cohesin mutants dominantly enforce stem cell programs and impair human hematopoietic progenitor differentiation. Cell Stem Cell (2015) 17:675–88. doi: 10.1016/j.stem.2015.09.017
70. Schaaf CA, Misulovin Z, Gause M, Koenig A, Gohara DW, Watson A, et al. Cohesin and polycomb proteins functionally interact to control transcription at silenced and active genes. PloS Genet (2013) 9:e1003560. doi: 10.1371/journal.pgen.1003560
71. Fisher JB, Peterson J, Reimer M, Stelloh C, Pulakanti K, Gerbec ZJ, et al. The cohesin subunit Rad21 is a negative regulator of hematopoietic self-renewal through epigenetic repression of Hoxa7 and Hoxa9. Leukemia (2017) 31:712–9. doi: 10.1038/leu.2016.240
72. Zhou Y, Kurukuti S, Saffrey P, Vukovic M, Michie AM, Strogantsev R, et al. Chromatin looping defines expression of TAL1, its flanking genes, and regulation in T-ALL. Blood (2013) 122:4199–209. doi: 10.1182/blood-2013-02-483875
73. Diffner E, Beck D, Gudgin E, Thoms JA, Knezevic K, Pridans C, et al. Activity of a Heptad of Transcription Factors Is Associated With Stem Cell Programs and Clinical Outcome in Acute Myeloid Leukemia. Blood (2013) 121:2289–300. doi: 10.1182/blood-2012-07-446120
74. Cuartero S, Weiss FD, Dharmalingam G, Guo Y, Ing-Simmons E, Masella S, et al. Control of inducible gene expression links cohesin to hematopoietic progenitor self-renewal anddifferentiation. Nat Immunol (2018) 19:932–41. doi: 10.1038/s41590-018-0184-1
75. Rao S. Closing the loop on cohesin in hematopoiesis. Blood (2019) 134:2123–5. doi: 10.1182/blood.2019003279
76. Ochi Y, Kon A, Sakata T, Nakagawa MM, Nakazawa N, Kakuta M, et al. Combined Cohesin-Runx1 Deficiency Synergistically Perturbs Chromatin Looping and Causes Myelodysplastic Syndromes. Cancer Discovery (2020) 10:836–53. doi: 10.1158/2159-8290.CD-19-0982
77. Nottingham WT, Jarratt A, Burgess M, Speck CL, Cheng JF, Prabhakar S, et al. Runx1-mediated hematopoietic stem-cell emergence is controlled by a Gata/Ets/SCL-regulated enhancer. Blood (2007) 110:4188–97. doi: 10.1182/blood-2007-07-100883
78. Ng CEL, Yokomizo T, Yamashita N, Cirovic B, Jin H, Wen Z, et al. A runx1 intronic enhancer marks hemogenic endothelial cells and hematopoietic stem cells. Stem Cells (2010) 28:1869–81. doi: 10.1002/stem.507
79. Horsfield JA, Anagnostou SH, Hu JK, Cho KH, Geisler R, Lieschke G, et al. Cohesin-dependent regulation of runx genes. Development (2007) 134:2639–49. doi: 10.1242/dev.002485
80. Mazzola M, Pezzotta A, Fazio G, Rigamonti A, Pistocchi A. Dysregulation of nipbl leads to impaired runx1 expression and haematopoietic defects. J Cell Mol Med (2020) 24:6272–82. doi: 10.1111/jcmm.15269
81. Kawashima N, Akashi A, Nagata Y, Kihara R, Ishikawa Y, Asou N, et al. Clinical significance of ASXL2 and ZBTB7A mutations and C-terminally truncated RUNX1-RUNX1T1 expression in AML patients with t (8,21) enrolled in the JALSG AML201 study. Ann Hematol (2019) 98:83–91. doi: 10.1007/s00277-018-3492-5
82. Mandoli A, Singh AA, Prange KHM, Tijchon E, Oerlemans M, Dirks R, et al. The Hematopoietic Transcription Factors RUNX1 and ERG Prevent AML1-ETO Oncogene Overexpression and Onset of the Apoptosis Program in t(8;21) AML. Cell Rep (2016) 17:2087–100. doi: 10.1016/j.celrep.2016.08.082
83. Banerji J, Rusconi S, Schaffner W. Expression of a beta-globin gene is enhanced by remote SV40 DNA sequences. Cell (1981) 27:299–308. doi: 10.1016/0092-8674(81)90413-x
84. Calo E, Wysocka J. Modification of enhancer chromatin: what, how, and why? Mol Cell (2013) 49:825–37. doi: 10.1016/j.molcel.2013.01.038
85. Xu Z, Wei G, Chepelev I, Zhao K, Felsenfeld G. Mapping of INS promoter interactions reveals its role in long-range regulation of SYT8 transcription. Nat Struct Mol Biol (2011) 18:372–8. doi: 10.1038/nsmb.1993
86. Ing-Simmons E, Seitan VC, Faure AJ, Flicek P, Carroll T, Dekker J, et al. Spatial enhancer clustering and regulation of enhancer-proximal genes by cohesin. Genome Res (2015) 25:504–13. doi: 10.1101/gr.184986.114
Keywords: cohesin mutation, acute myeloid leukemia, AML1-ETO, hematopoietic stem and progenitor cells, leukemogenesis
Citation: Han C, Gao X, Li Y, Zhang J, Yang E, Zhang L and Yu L (2021) Characteristics of Cohesin Mutation in Acute Myeloid Leukemia and Its Clinical Significance. Front. Oncol. 11:579881. doi: 10.3389/fonc.2021.579881
Received: 03 July 2020; Accepted: 18 March 2021;
Published: 13 April 2021.
Edited by:
Naval Daver, University of Texas MD Anderson Cancer Center, United StatesReviewed by:
Walter Fiedler, University Medical Center Hamburg-Eppendorf, GermanyCopyright © 2021 Han, Gao, Li, Zhang, Yang, Zhang and Yu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Li Yu, bGl5dTMwMUB2aXAuMTYzLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.