 Hrishi Varayathu
Hrishi Varayathu Vinu Sarathy
Vinu Sarathy Beulah Elsa Thomas
Beulah Elsa Thomas Suhail Sayeed Mufti1
Suhail Sayeed Mufti1
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 28 May 2021
Sec. Pharmacology of Anti-Cancer Drugs
Volume 11 - 2021 | https://doi.org/10.3389/fonc.2021.559161
This article is part of the Research Topic Targeted Immunotherapy for Cancer View all 23 articles
Immune checkpoint inhibitor therapy has revolutionized the field of cancer immunotherapy. Even though it has shown a durable response in some solid tumors, several patients do not respond to these agents, irrespective of predictive biomarker (PD-L1, MSI, TMB) status. Multiple preclinical, as well as early-phase clinical studies are ongoing for combining immune checkpoint inhibitors with anti-cancer and/or non-anti-cancer drugs for beneficial therapeutic interactions. In this review, we discuss the mechanistic basis behind the combination of immune checkpoint inhibitors with other drugs currently being studied in early phase clinical studies including conventional chemotherapy drugs, metronomic chemotherapy, thalidomide and its derivatives, epigenetic therapy, targeted therapy, inhibitors of DNA damage repair, other small molecule inhibitors, anti-tumor antibodies hormonal therapy, multiple checkpoint Inhibitors, microbiome therapeutics, oncolytic viruses, radiotherapy, drugs targeting myeloid-derived suppressor cells, drugs targeting Tregs, drugs targeting renin-angiotensin system, drugs targeting the autonomic nervous system, metformin, etc. We also highlight how translational research strategies can help better understand the true therapeutic potential of such combinations.
Immunotherapy is often thought to be a recent discovery. However, if we look at the beginnings of cancer immunotherapy, the first scientific attempts were made Fehleisen and Busch, German physicians who noted significant tumor regression post erysipelas infection. William Bradley Coley, the father of immunotherapy first tried to use the immune system in 1981 for the treatment of bone cancer. His work was largely ignored for more than fifty years when the field of immunology took off with the discovery of T cell existence and their role in immunity. In the 1970s, Donald L. Morton treated melanoma patients with Bacillus Calmette-Guerin (BCG) and 91% of patients treated had disease regression. Then in the 1980s, the first immunotherapy cancer treatment IL-2 was approved for the treatment of melanoma and kidney cancer by FDA (1). The most significant breakthrough in immunotherapy came through James Allison & Tasuku Honjo who discovered CTLA4 and PDL1 respectively as therapeutic targets. That research work has further led to the successful development of new checkpoint inhibitors, Dendritic cell therapy, CAR-T cells, and other adoptive cell therapies, oncolytic viruses which have all brought hope to the future of immunotherapy (2).
PD1 and CTLA-4 are the two distinct inhibitory receptors on T cells, which are targeted by monoclonal antibodies for providing a durable antitumor immune response. The efficacy of immune checkpoint inhibitors (ICI) was first noted in metastatic melanoma with anti-tumor immune response and significant improvement in overall survival (OS) of patients received ipilimumab an anti-CTLA4 antibody (3). This led to the accelerated clinical studies using anti-PD1/PDL-1 antibodies and regulatory approval in other malignancies including few hematological malignancies, non-small cell lung cancer (NSCLC), and melanoma (4, 5). Currently, six ICIs have been approved by the US FDA, of which five ICIs also received market authorization by the European Medicines Agency (EMA).
Even though these therapies have shown survival benefits with reduced incidence of drug resistance or adverse effects than conventional chemotherapy, not all cancer patients are benefitted from these drugs (6). The ability of the tumor microenvironment (TME) to evade immune responses and thereby generating immunosuppression/immune escape is one of the prime challenges that remain (7). This tumor-derived local environment is known as the immunosuppressive TME which ultimately suppresses the antitumor immune response. Clinical trial data analysis of ICI reveals mainly three kinds of patient response: (a) patients who do not respond initially (innate resistance); (b) those who respond at first but fail subsequently (acquired resistance); and (c) those continuously respond from initially to later stages (8, 9). Antigen experienced T-cell activation and proliferation are entailed for the generation of an effective antitumor response. The difference in response is because of the insufficient generation and activity of anti-tumor CD8 T-cells (10). Impaired processing and presentation of neo antigens are other causes for the lack of activation of tumor-reactive T cells. Multiple other factors like type of cancer, tumor heterogeneity, multiple lines of treatment, and the immunosuppressive TME generated due to tumor-related and non- tumor related factors result in poor response to ICIs (11). Another important obstacle in reduced ICI efficacy is tumor associated hypoxia due to the structural organization of stroma. This can results in altering the pharmacokinetics of the drug by reducing the target drug disposition. Hypoxia can potentiate tumor invasion, stemness, and metastatic capacity mainly through the activation of hypoxia-inducible factor-1α– mediated hepatocyte growth factor/c-Met pathway (12, 13). Immunosuppressive microenvironment-associated macrophages enhance tumor hypoxia and further reduce the efficacy of ICIs (14). These results highlight the necessity of combination therapies that can improve the tumor immunogenicity, changing the immunosuppressive TME and targeting other pathways which potentially inhibit the activation of T cells. There are many drugs including cytotoxic as well as non-anti-cancer drugs which have shown promising results in potentiating the efficacy of ICIs.
We collected 250 published studies that had used the combination of ICIs and other therapies. From the collected studies duplicates were removed and further assessed for eligibility as shown in Figure 1. The review covered all countries with a web-based search of PubMed/Google Scholar published from 2005 to 2019. A further search was conducted in ClinicalTrials.gov to check for the available clinical trials. A few of the clinical trials were further explored by contacting the study teams or information gathered from recent press releases for the study.
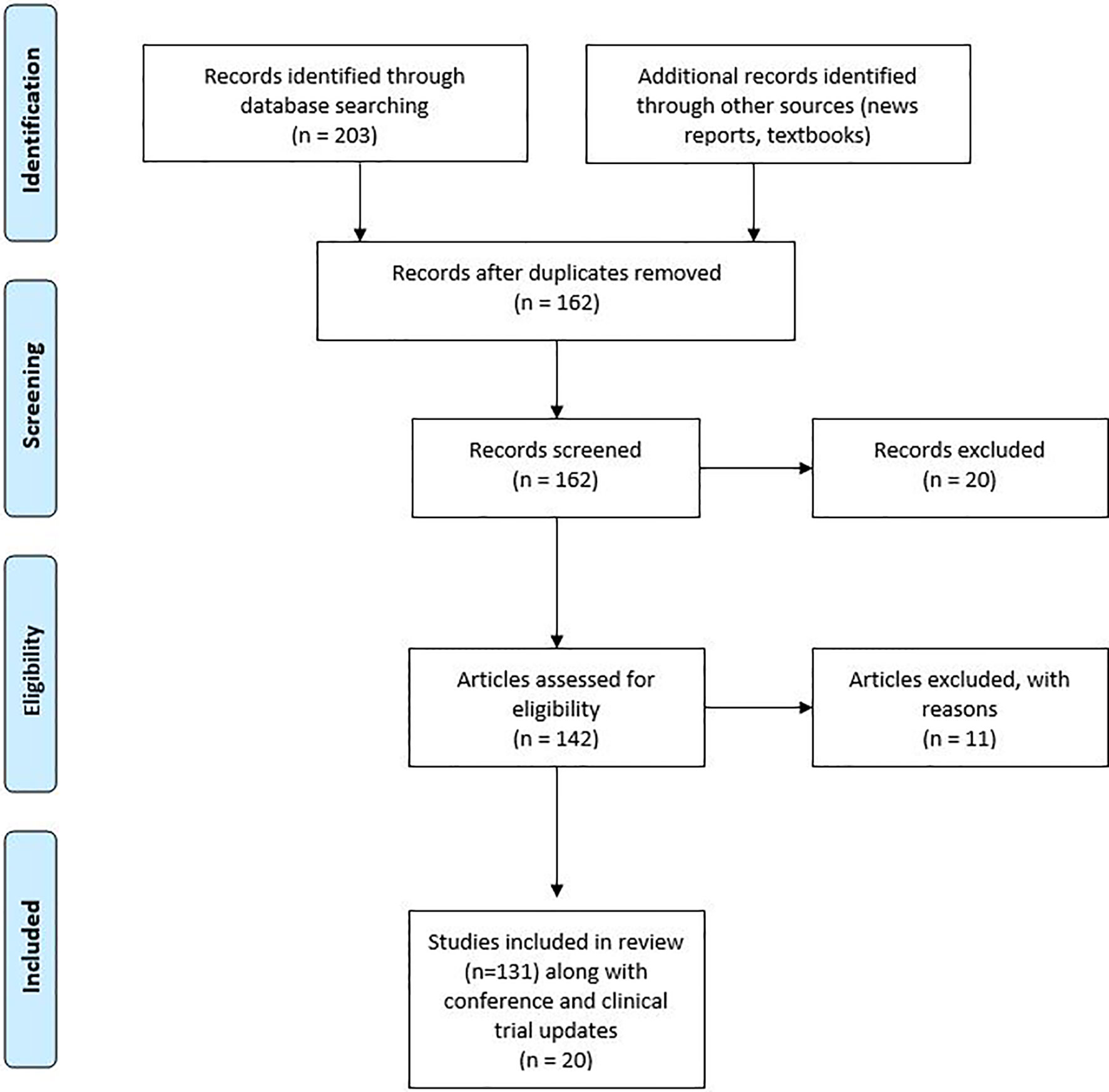
Figure 1 Research strategy with PRISMA flow diagram.
Original primary research such as experimental, observational, and qualitative studies which are written in English and published in peer-reviewed journals are included in the review. Some of the review articles were also selected if they contained outcomes of previous exploratory studies. Search terms were used to find specific studies related to the subject (Table 1). Articles that define the mechanism behinds the use of drugs as adjuncts with ICIs were considered eligible.

Table 1 Search terms used for the collection of articles.
We mainly focused on the mechanisms and rationale of using an adjunct with ICIs for the combinations already in clinical trials. Adjuncts used only in preclinical studies with no currently available clinical data have been excluded from the review.
Conventional chemotherapy drugs such as anthracyclines, cyclophosphamide, Cisplatin, Oxaliplatin, gemcitabine, temozolomide, and paclitaxel are observed to enhance tumor immunity at lesser doses. These drugs at normal dosage and schedule produce immunogenic cell death (ICD) which results in the tumor antigen release and danger signal generation from dying cancer cells known as damage-associated molecular patterns (DAMPs), which in turn provide tumor-targeting immune responses (15). The immunogenicity of tumor cells is modulated by these drugs via various mechanisms comprising (1) enhanced tumor antigen expression and presentation; (2) down regulating coinhibitory molecules (B7-H1/PDL1) and upregulating costimulatory molecules (B7-1) expressed on the surface of tumor cells which in turn increases the function of effector T-cell; (3) granzyme and perforin-dependent mechanisms increasing T-cell facilitated tumor cell lysis; and (4) decreasing MDSCs and Tregs infiltration in the TME.
In mouse immunization experiments using tumor cells pretreated with chemotherapeutic agents such as mitoxantrone and doxorubicin, effective cancer regression via CRT (calreticulin) expression and HMGB1 secretion was observed (16, 17). These findings have been confirmed in clinical studies which show that CRT expression is crucial for better prognosis in cancer patients (18). Many studies over the past decade have shown that different chemotherapy regimens also promote differentiation of Th1/Th2 and amplify the proliferation of T-lymphocytes in solid cancers (renal cell carcinoma, colon cancer, and ovarian cancer) (19, 20).
Various clinical trials such as KEYNOTE-189, IMPower-132 which used ICI combination with chemotherapy in non-squamous NSCLC have shown significant benefits.
After a median follow-up of 18.7 months, the combination of pembrolizumab and chemotherapy resulted in a longer OS compared to chemotherapy alone, with a median OS of 22.0 months versus 10.7 months (hazard ratio [HR], 0.56; 95% CI, 0.45-0.70, P.00001). At 9.0 months versus 4.9 months, respectively, progression-free survival (PFS) was also substantially increased as compared to placebo plus chemotherapy (HR, 0.48; 95% CI, 0.40-0.58, P <.00001).
Recent findings from KEYNOTE-062 which used first-line ICI chemotherapy combination in advanced gastric or gastroesophageal junction (G/GEJ) adenocarcinoma was non-inferior to chemotherapy and showed a slight improvement in OS. Pembrolizumab plus chemotherapy resulted in a median OS of 12.5 months (95% CI, 10.8-13.9) versus 11.1 months (95% CI, 9.2-12.8) with chemotherapy (HR 0.85; 95% CI, 0.70-1.03; p = 0.046) (21–23).
The second interim analysis of IM passion 130 trial that assessed the efficacy and safety of atezolizumab plus nab-paclitaxel in patients with unresectable, locally advanced, or metastatic triple-negative breast cancer (TNBC) reported that median OS analysis in patients with PD-L1 immune cell-positive tumors was 25 months (95% CI 19.6–30.7) in atezolizumab + nab-paclitaxel arm and 18 months (13.6–20.1) in placebo + nab-paclitaxel arm(stratified HR 0.71, 0.54–0.94]) (24).
In March 2020, FDA approved Durvalumab chemotherapy (Etoposide + Cisplatin or carboplatin) combination as a first-line treatment for extensive-stage small cell lung cancer (ES-SLC). In untreated previously ES-SLC, CASPIAN trial showed a median OS of 13 months (95% CI: 11.5, 14.8) in the durvalumab plus chemotherapy group compared with 10.3 months (95% CI: 9.3, 11.2) in the chemotherapy alone arm (hazard ratio 0.73; 95% CI: 0.59, 0.91; p=0.0047). However, median PFS was almost the same in the combination arm (5.1months) vs chemotherapy alone (5.4months) arm (25).
Metronomic therapy, an alternate approach for chemotherapy administration encompasses continuous administration of lesser doses of cytotoxic agents. Various studies have reported significant anti-tumor immune responses, lower therapeutic resistance, and a decrease in tumor vascularization (26–28). Chemotherapeutic drugs such as methotrexate, cyclophosphamide, etoposide, vinblastine, and paclitaxel are used widely as metronomic chemotherapy in various cancers. Even though high dose chemotherapy target cancer cells and are immunosuppressive, high-frequency low dose chemotherapy is considered to be immunostimulatory by targeting the supporting tumor stroma. Cancer cells in tumors cause the surrounding stroma to release additional angiogenesis stimulators. Angiogenesis in the tumor is a process initiated by growth factors and tumor-associated endothelial cells (TEC). When exposed to cancer chemotherapeutic agents at much lower concentrations than those needed to cause tumor cell damage, TECs lose functionality. This property of low-dose chemotherapy such as vinblastine, doxorubicin, paclitaxel, and carboplatin is documented in many studies (29, 30). This can reduce the tumor-resistant clones, by giving combination with other treatment modalities (31).
Chemotherapy administered metronomically reduces the number of immunosuppressive regulatory T cells (Tregs) (32–34), promote antigen-presenting cell maturations (35), improves the activity of dendritic cells (DCs) (36) and, chiefly enhances the activation and function of cytotoxic CD8+ T cells and NK cells. Moreover, it also targets other TME immunosuppressive components referred to as myeloid-derived suppressor cells (MDSCs) (37–40). MDSCs are a diverse group of myeloid immune cells characterized by their immature state and ability to suppress T cells. This particular property of metronomic chemotherapy can be used for the modulation of the efficacy of ICIs and many studies have reported positive results. It is proposed that metronomic chemotherapy promotes tumor-specific immune activation; concurrent administration of ICIs could maintain the T cells in an activated state. Combination of low dose cyclophosphamide and oxaliplatin sensitized tumor ICI which are initially refractory by increasing CTLs/Treg ratio in the TME in a murine model of lung adenocarcinoma (41). Similar results were obtained in the murine colorectal cancer model where oxaliplatin augmented the amounts of CTLs and activated dendritic cells (DCs) potentiating the efficacy of anti-PD-L1 therapy (42). Enhanced infiltration of CD8+ and CD4+FoxP3T along with suppression of the CD4+ CD25+ FoxP3+ regulatory T cell function have seen after low dose cyclophosphamide with an anti-PD1 agent and improved tumor-free survival in a model of cervical cancer (43, 44). There are clinical studies ongoing, combining a metronomic dose of cyclophosphamide along with ICIs as depicted in Table 2.
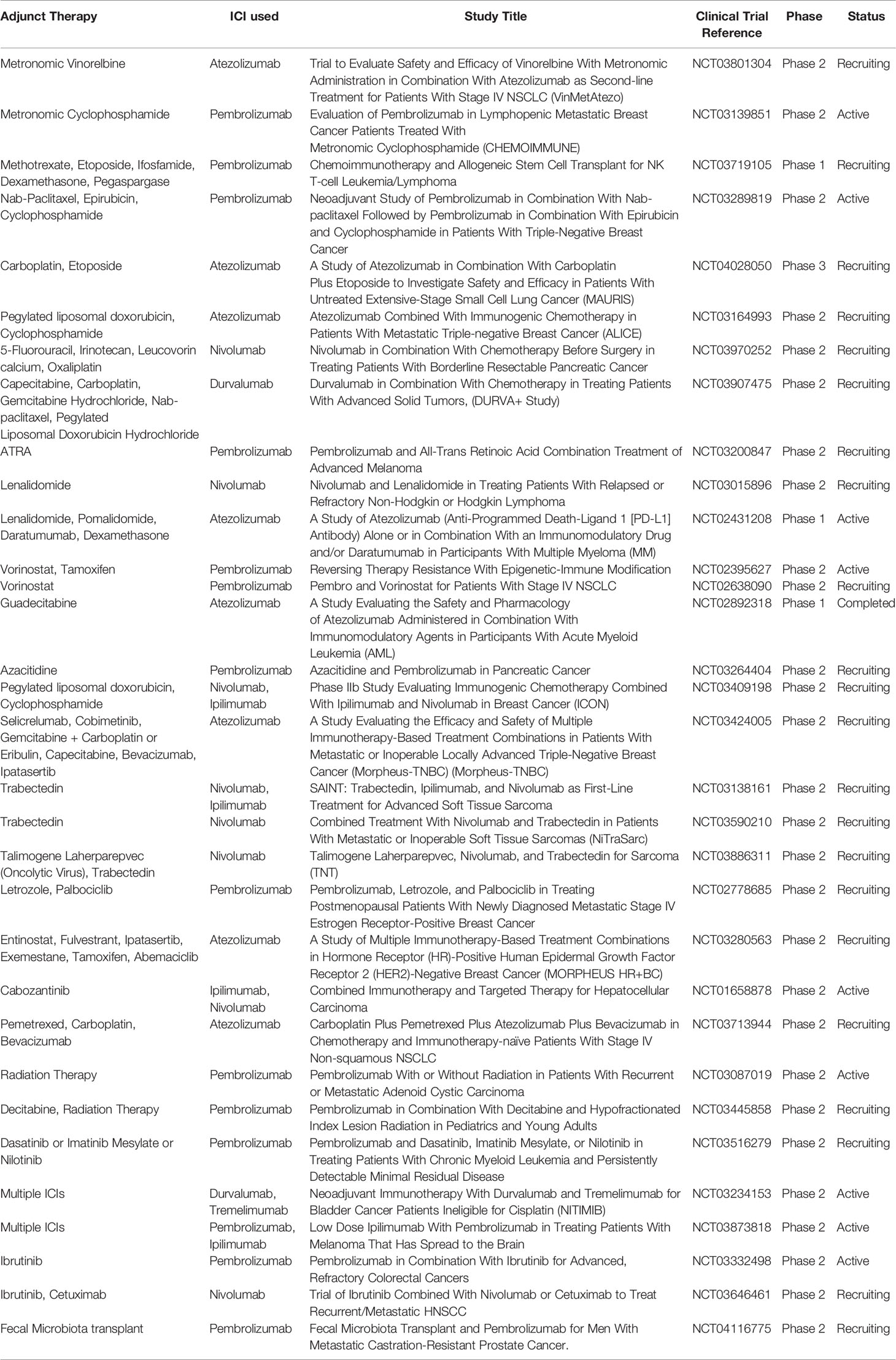
Table 2 Few clinical trials listed in Clinical trials.gov which uses combination strategies with ICIs as of 17-04-2020.
Thalidomide was first used for its direct anti-tumor actions on myeloma cells by resulting in cell cycle arrest and anti-angiogenic effect. Thalidomide derivatives such as lenalidomide, pomalidomide were then classified as immunomodulatory agents as they stimulate T cells and natural killer cells (NK) to secrete IL-2 and IFNγ and inhibit Tregs. This resulted in the amplification of specific immunity in myeloma (45, 46). Lenalidomide in combination with dexamethasone and proteasome inhibitor (Bortezomib and Carfilzomib) is considered as a standard chemotherapy regimen for multiple myeloma. Some studies show that the immunostimulatory effects of lenalidomide are hampered by the concurrent use of dexamethasone (46). The immunomodulatory effects of these drugs can be exploited to enable an enhanced action with ICIs. An invitro cell line study suggests good results with the combination of ICIs and thalidomide derivatives in anti-leukemic therapy (47).
Despite these effects, FDA on July 2017 placed a clinical hold on three clinical trials of the programmed death 1 (PD-1) inhibitor pembrolizumab in combination with lenalidomide or pomalidomide for patients with multiple myeloma: KEYNOTE-183, KEYNOTE-185, and KEYNOTE-023 because of increasing reports of death. However, there are similar clinical trials ongoing with the combination of ICIs and lenalidomide for other indications such as lymphoma (47–49). In KEYNOTE-023 responses were durable in the overall study population which includes lenalidomide refractory patients with a median duration of response (DOR) of 18.7 months, Objective response rate (ORR) of 44%. Median PFS was 7.2 months and median OS was not reached (50). The median PFS was 27.6 months with a median follow-up of 32.2 months in a two-year update of a Phase II trial of Pembrolizumab, Lenalidomide, and dexamethasone as post autologous stem cell transplant consolidation in multiple myeloma. Similarly, Badros et al. reported ORR-60%, DOR 14.7 months, median PFS 17.4 months, Median OS not reached with Pembrolizumab, pomalidomide and dexamethasone combination and they also reported long term remissions after discontinuing Pembrolizumab, 57% have ongoing response without any further treatment with a median survival of 18 months (51, 52).
Hypomethylating agents such as azacytidine and decitabine are mainly used in the treatment of myelodysplastic syndromes (MDS) and acute myeloid leukemia (AML). Due to the availability of advanced genomic studies, it is evident that hypermethylation of tumor suppressor genes in the promoter region is one of the pathways of carcinogenesis (53). The most common and stable of epigenetic alterations in cancer is methylation of DNA at CpG islands. Hypermethylation tends to limit ICIs by blocking endogenous interferon responses required for cancer cell recognition whereas global hypomethylation marks in the expression of inhibitory cytokines and PD-L1, supplemented by epithelial-mesenchymal variations contribute to immunosuppression. The drivers of these opposing states of methylation are not stated well. DNA methylation is also essential in the ‘exhaustion’ of cytotoxic T cells that occurs as a result of tumor progression (54). The feasibility of targeting this mechanism with hypomethylating agents is being widely studied now. Other than this, the collective data of studies suggests that hypomethylating agents are known to promote the expression of cancer-specific antigens and major histocompatibility complex (MHC) results in improved immunologic recognition of cancer cells and enhanced anti-tumor responses (55–57). This property is exploited By Brahmer J et al. and when six patients in a clinical trial of epigenetic therapy for advanced treatment-resistant NSCLC were switched to an immunotherapy trial targeting the PD-1/PD-L1 immune tolerance checkpoint, they saw a substantial improvement. Three of the six patients had long-term partial responses to immunotherapy, ranging from 14 to 26 months, while the other two had stable disease for 8.25 and 8.5 months, respectively (4, 5, 58). Various clinical studies are ongoing to check the efficacy and toxicity of this combination therapy in various cancers (Table 2).
As discussed above, in cancers that are immune to immunotherapy, epigenetic inhibitors may be used as adjunctive therapy or as a replacement for immunotherapy (sole agents of immune modification). In the TME, epigenetic changes are normal, resulting in a variety of gene expression changes and tumor escape. Recently, HDAC inhibitors have shown potential anti-cancer properties such as cell cycle arrest, anti-angiogenesis, activation of both intrinsic and extrinsic apoptotic pathways, autophagy, and modulation of immune responses. HDACs are classified mainly into 4 categories (Class I, Class II, Class III, Class IV) based on their structure, which is further subdivided into Zinc dependent (Class I, Class II, Class IV) and NAD+ dependent (Class III) for their enzymatic activity (59). Modulation of the immune response can be utilized for improving the outcome of ICIs. HDACis can change the expression of immune system upregulaters including MHC and costimulatory molecules, which affects antigen presentation and thus T cell activation. It has also been observed that naïve T cell functionality is stimulated by HDAC6i and Tregs are targeted by class II HDACis. The Class I HDACis target adaptive immunity by enhancing natural killer and CD8 cells functionality. Several non-selective HDACis such as vorinostat, belinostat, trichostatin A are being tried in clinical trials (60). HDAC I, II, and IV are inhibited by Pan-HDACi such as valproic acid, vorinostat, and panobinostat. MS275 (a class I HDAC inhibitor), MC1568 (a class III HDACi), sirtinol (a class II HDACi), and Nexturastat A (HDAC6i), among many others, are currently being used to reduce the toxic effects associated with pan-HDAC inhibition. The wide range of biological effects may be due to each inhibitor’s particular chemical structure and mechanistic profiles (61).
In a B16F10 syngeneic murine model study by Wood DM et al. HDAC inhibitor treatment resulted in rapid upregulation of histone acetylation of the PD-L1 gene leading to improved and persistent gene expression. The effectiveness of combining HDAC inhibitor with PD-1 blockade for the treatment of melanoma was studied in a murine B16F10 model and compared to control and single-agent therapies, they had a slower tumor development and a higher survival rates (62).
Jhanelle et al. Phase 1/1b study of pembrolizumab plus vorinostat in advanced/metastatic NSCLC concluded that Pembrolizumab plus vorinostat was well tolerated and demonstrated preliminary anti-tumor activity despite progression on prior ICI treatment. Of 30 patients (6 ICI-naive and 24 ICI-pretreated) were evaluable for response, 4 (13%) had a partial response, 16 (53%) had SD, and 10 (33%) had PD for a DCR of 67% without any dose-limiting toxicities (63).
CDK inhibitors are a class of naturally occurring molecules that belong to the Cyclin-dependent kinase inhibitors family INK4 and specifically inhibit the CDK4/6 proteins. By dephosphorylating the retinoblastoma tumor suppressor protein, CDK4/6 inhibitors block the G1 to S phase transition of the cell cycle, resulting in cell cycle arrest in tumor cells. Teo ZL et al. showed that a combination of CDK4/6 and PD1 blockade resulted in intensifying tumor growth inhibition. Increased antigen presentation by tumor cells, stimulation of effector T lymphocyte activation, and decreased proliferation of immunosuppressive Treg cells are among the various mechanisms suggested (64, 65). The immune checkpoint pathway is majorly implicated with the increased immune response upon inhibition of CDK4/6 inhibition. The results of a phase Ib clinical trial combining abemaciclib and pembrolizumab in ER-positive HER2-negative women at a 16-week interim study, MBC demonstrated that this combination is secure, with an ORR of 14.3% (66).
In the JPCE trial, 25 hormone receptor-positive Her2 negative metastatic breast cancer patients were treated with a combination of Abemaciclib and Pembrolizumab. In this trial, the partial response (PR) rate at 16 weeks was 14.3% compared to 6.8% seen in the MONARCH-1 trial which used single-agent abemaciclib. There were no complete responses in either trial. In both experiments, the incidence of stable disease was 60.7% in JPCE and 60.6% in MONARCH-1. In the JPCE experiment, fewer patients developed a progressive disease (17.9% and 25.0%). At the early assessment, the disease control rate (DCR) in JPCE was 75%, while in MONARCH-1 it was 67.4%. As of 17-04-2020, the study is still ongoing and results are yet to publish (66).
Anti-angiogenic drugs work by inhibiting platelet-derived growth factor receptors (PDGFRs) and fibroblast growth factor receptors, which target vascular endothelial growth factor (VEGF)/VEGF2 or other small molecules involved in angiogenic and proliferative pathways (FGFRs). Preclinical studies have shown that abnormal tumor vasculature promotes immunosuppressive TME, which can be reversed with anti-angiogenic therapies (67, 68). Inflammatory mediators such as cytokines and immune cells promote and control angiogenesis, which in turn may affect the microenvironment (Figure 2) (69). Anti-angiogenic agents can thus activate the immune system, and immunotherapy can also have an anti-angiogenic effect, therefore they can act synergistically on the tumor.
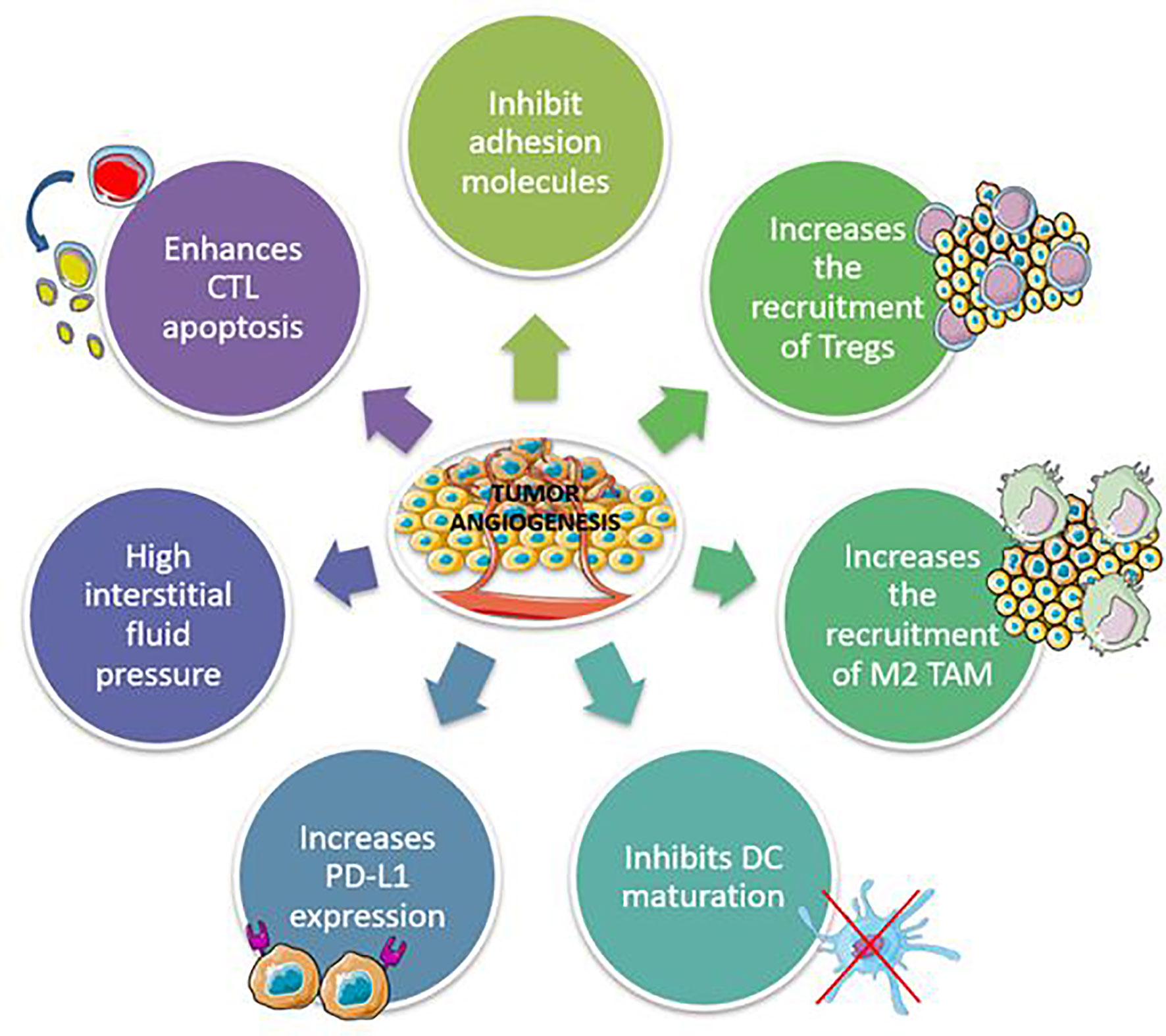
Figure 2 Mechanisms involved in the development of immunosuppressive tumor microenvironment by tumor angiogenesis. CTL, Cytotoxic T lymphocytes; TAM, Tumor-associated macrophages.
Results of the phase 3 IMpower150 (NCT02366143) study reported in 2018 showed significant improvement in OS of patients treated with a combination of Atezolizumab, Bevacizumab along with chemotherapy in treatment-naïve metastatic non-squamous non–small-cell lung cancer patients. A total of 800 patients were given either atezolizumab plus bevacizumab plus carboplatin plus paclitaxel therapy (ABCP group) or bevacizumab plus carboplatin plus paclitaxel therapy (BCCP group) (BCP group). The study found that the ABCP community had slightly longer progression-free survival (PFS) and overall survival (OS) (median PFS of ABCP vs. BCP: 8.3 vs. 6.8 months; hazard ratio: 0.61, 95% CI: 0.52 to 0.72) (median OS of ABCP vs. BCP: 19.2 vs. 14.7 months; hazard ratio: 0.78, 95% CI: 0.52 to 0.72). CI ranges from 0.64 to 0.96) (70).
Keynote-426 (NCT02853331), an open-label, phase 3 trial, which randomly assigned 861 patients with previously untreated advanced clear-cell renal-cell carcinoma to receive pembrolizumab (200 mg) intravenously once every 3 weeks plus axitinib (5 mg) orally twice daily (432 patients) and single-agent sunitinib (50 mg) orally once daily for the first 4 weeks of each 6-week cycle (429 patients) which disclose that after a median follow-up of 12.8 months, the estimated %age of patients who were alive at 12 months was 89.9% in the pembrolizumab–axitinib group and 78.3% in the sunitinib group (hazard ratio for death, 0.53; 95% confidence interval [CI], 0.38 to 0.74; P<0.0001) and median progression-free survival was 15.1 months in the pembrolizumab–axitinib group and 11.1 months in the sunitinib group (hazard ratio for disease progression or death, 0.69; 95% CI, 0.57 to 0.84; P<0.001) (71).
Synthetic lethal impact on cancer cells with a dearth in homologous recombination (HR) is induced by Poly ADP-ribose polymerase (PARP) inhibition. In addition, PARP inhibitors (PARPi) can enhance Th1-skewing immunity and modulate the immune microenvironment, as well as promote the priming of anti-cancer immune responses (72). As the immunological effect of PARPi is multifaceted, it might favor an increased efficacy of ICI treatment and boost the cancer-immunity cycle. Olaparib is a first-in-class PARP inhibitor for the treatment of adult patients with germline BRCA mutated advanced ovarian cancer, breast cancer, fallopian tube cancer, and peritoneal cancer. As discussed the immunological effects of PARP inhibitors are multifaceted (73), which includes
1. PARPi-mediated catastrophic DNA damage; elevated neoantigens and tumor immunogenicity are generated by the accumulation of chromosome rearrangements
2. PARPi-mediated DNA damage could enhance the recruitment and infiltration of T cells into the tumor via activating the cGAS-STING pathway
3. PARPi-induced double-strand breaks could directly upregulate PD-L1 by the ATM-ATRChk1 pathway which is independent of the IFN pathway
All these mechanisms reprogram the immune microenvironment to an immune-supportive environment. The maximal benefit of these above-mentioned immunomodulatory mechanisms of PARPi can be achieved in combination with ICIs. Various studies are ongoing, including preclinical as well as clinical trials using the combination of PARP inhibitor + anti-VEGF + ICIs and a combination of PARP inhibitor + checkpoint kinase1 (CHK) + ICIs in cancer (74, 75).
In metastatic ovarian cancer, a recent ESMO update of the MEDIOLA phase I/II open-label, multicenter analysis evaluating the combination of Olaparib and durvalumab in patients with advanced solid tumors who harbor BRCA mutations revealed a median PFS of 15.4 months. Median OS data was not mature yet. 83.5% of patients were still alive after a median follow-up of 23.7 months (76).
Several multiple tyrosine kinase inhibitors have shown enhanced action with ICIs. Sorafenib, regorafenib, and Lenvatinib are some of the drugs which have demonstrated superior effect with ICIs in hepatocellular carcinoma. Hepatocellular carcinomas generally arise from an immunosuppressive environment (77). Since it must interact with a variety of foreign antigens, the liver suppresses immune responses to toxins and antigens draining from the enteric circulation. Because of the progressive nature of the disease and the tolerogenic characteristics of the liver, the HCC TME has immunosuppressive characteristics (77–79). HCC exploits this immune tolerance to initiate and promote HCC carcinogenesis and progression. Even though the TME is highly immunosuppressive there are reports which show that ICIs have benefits in the treatment of HCC (78–80). Most of these successful attempts of ICI usage in HCC were demonstrated in combination with multiple tyrosine kinase inhibitors such as sorafenib and regorafenib or when pretreated with these small molecule inhibitors (78–80). However, the exact mechanisms of their additive activity are yet to be proven.
Other small molecule inhibitors include drugs which target PI3K and MAPK pathway. Both the PI3K-Akt-mTOR and the RAS/RAF/MEK/MAPK pathways have been implicated in the control of PD-L1 expression in previous studies (81, 82). Vemurafenib, a BRAF inhibitor, can enhance T cell antigen expression in melanoma and thus stimulate T cell immune responses. By the expression of anti-apoptotic proteins, the PI3K-Akt-mTOR pathway is also involved in resistance to T cell-mediated killing. A more recent study reported better tumor immune infiltration and more anti-tumor immune control in a CD8 T-cell-dependent way when the dual blockade of BRAF and MEK combined with PD-1 inhibitors (83). The experimental evidence presented above may be used to justify clinical trials of BRAF and/or MEK inhibition in conjunction with ICIs. Many studies are ongoing to test these hypotheses and some of the study results are promising. In a study, the selective targeting of PI3K–γ with a small molecule inhibitor IPI-549 (NCT02637531) showed that it could restore the TME and overcome resistance to ICIs by inducing cytotoxic T cell-mediated tumor regression (84). This discovery paves the way for the appealing technique of combining PI3Kis and ICIs to prevent immune checkpoint blockade resistance. One or two agents targeting the PI3KAkt- mTOR pathway could be a possible combination partner for immune checkpoint blockade (85), but the most effective combination is not defined.
Cabozantinib a multikinase inhibitor whose targets include MET, AXL, and VEGFR2 in combination with Nivolumab showed better response rate, delayed disease progression, and extended survival in the CheckMate -9ER trial (86). CheckMate -9ER (NCT03141177) is an open-label, randomized, multi-national Phase 3 trial that investigates Cabozantinib+Nivolumab combination in treatment naïve advanced or metastatic renal cell carcinoma. However, 3 drug combinations of Cabozantinib+Nivolumab+Ipilimumab for the same indication were discontinued.
Phase 1/2 (Checkmate-040) study of doublet therapy (Cabozantinib+Nivolumab) in advanced HCC reported 19% ORR, 75% DCR and median PFS 5.4 months. DOR was 8.3 months with a median OS of 21.5 months in the doublet. Interestingly, the triplet therapy (Cabozantinib+Nivolumab+Ipilimumab) showed comparatively better ORR (29%), DCR (83%), median PFS (6.8 months), and both DOR and median survival were not reached in triplet therapy. However, triplet therapy is associated with an increased risk of treatment-related toxicity (Grade 3 or 4) compared to doublet (71% vs 47%) (87).
Bruton’s tyrosine kinase (BTK), bone marrow-expressed kinase (BMX), redundant resting lymphocyte kinase (RLK), and IL-2 inducible T-Cell kinase are all members of the Tec kinase family (ITK). BTK is primarily expressed on B cells, though not exclusively, and ITK is primarily expressed on T cells (88). Ibrutinib is a small molecule that not only targets BTK, which is required for malignant B cell survival, but also blocks ITK, which shifts the Th1/Th2 balance and improves antitumor immune activity. The combination of ibrutinib anti-tumor activity and ICIs showed synergism in preclinical models and its positive therapeutic outcome does not result from the direct action against tumor cells, but rather from their activity on the immune system (89). Ibrutinib activates CD8+ T cells, lowers MDSC cytokine secretion, and modifies the inhibitory activity of immune checkpoint molecules like the PD1/PD-L1 and CTLA-4 axis on tumor-infiltrating lymphocytes (89, 90). Many clinical trials are testing this hypothesis and most of them are being tried in lymphoma patients. However, there is limited data in solid tumors and one study showed no benefits of adding ibrutinib along with ICIs (durvalumab) in pretreated solid tumors such as pancreatic cancer (ORR- 2%, Median OS- 4.2 months), breast cancer(ORR- 3%, Median OS- 4.2 months) and NSCLC (ORR- 0%, Median OS- 7.9 months) (91).
Fulvestrant is a selective ER down regulator (SERDs), a class of ERα antagonists. Fulvestrant treatment aids in overcoming various resistance mechanisms by ERα down-regulation. Several preclinical studies have proposed various mechanisms by which SERDs exert an anti-tumor response. It alters the TME by increasing Th1 immune response, decreasing Tregs, and inhibiting estrogen-mediated cell proliferation pathways (92, 93). Results of recent translational research indicate that SERDs with strong anti-estrogen activity such as Fulvestrant and potentially other anti-estrogens (94) can augment the action of ICIs to inhibit breast cancer progression. As a result, combining anti-estrogens with ICI may be an effective treatment technique for both ER-positive and potentially ER-negative or treatment-resistant breast cancers, greatly increasing the use and life-extending effects of these drugs to improve patient survival (95).
Recently, many studies are focusing on the combination of immunotherapy agents which target multiple pathways in cancer. It is proposed that targeting multiple checkpoints can increase the activity of each other and thereby overcoming each monotherapy’s limitations. The combination of CTLA-4 and PD 1/PD-L1 blockade has exhibited antitumor efficacy in preclinical models (96). The logic of combining multiple checkpoint inhibitors is that they have different mechanisms of action, with anti – CTLA-4 mainly acting in the lymph node compartment which is responsible for restoring the induction and proliferation of activated T cells, and with anti –PD-1 mainly acting at the periphery of tumor site, preventing the neutralization of cytotoxic T cells by PD-L1 expressing tumor and plasmacytoid dendritic cells in the TME (97).
In the RCT checkmate 067 trial in advanced melanoma, patients with PD-L1-positive tumors had the same median progression-free survival (14 months) when they were given nivolumab alone or in combination. However, in patients with PD-L1–negative tumors, ipilimumab plus nivolumab had a longer progression-free survival (11.2 months) than nivolumab alone (5.3 months). The major disadvantage of combination immunotherapy is the increase in grade 3 or grade 4 toxicities. The updated data in 2017 reported that only patients with no expression of PD-L1 benefited from the combination and the recent update in 2019 added that the %age of patients experiencing a complete response continued to increase, with complete response rates at five years of 22% for combination therapy and the proportion of patients alive and treatment-free was 74% in nivolumab/ipilimumab combination (98).
Based on ORR and DOR in phase I/II CheckMate-040 trials, FDA has approved Nivolumab-Ipilimumab combination in Hepatocellular carcinoma in March-2020. In the study, 16 of 49 patients (33%) treated with nivolumab in combination with ipilimumab responded to treatment after a minimum of 28 months of follow-up (95% CI, 20-48). Four patients (8%) had a full response (CR) and 12 people (24%) had a partial response (PR). DORs ranged from 4.6-30.5 months, with 88% lasting at least 6 months, 56% lasting at least 12 months, and 31% lasting at least 24 months (99, 100).
Updated result of Phase 3 Checkmate-214 trial (Ipilimumab+Nicolumab) in treatment naïve renal cell carcinoma after 42-month median follow-up showed very promising result with CR in 10% patients, OS rate and ORR as 52% and 42% respectively and DOR was not reached (101).
FDA granted fast track designation for investigation of the combination of Balstilimab PD-1 inhibitor and Zalifrelimab a CTLA-4 inhibitor for the treatment of patients with relapsed or refractory metastatic cervical cancer. The FDA designates an investigational drug for fast track review in order to speed up the production of drugs that treat severe or life-threatening conditions and address an unmet medical need. Combination of Balstilimab/Zalifrelimab exhibit 26.5% objective response rates (ORR) vs 11.4% in Balstilimab monotherapy (102).
Microorganisms live on our skin, in our lungs, and in our intestines, accounting for a large portion of our cellular, metabolic, and genetic mass (103). The microbiota is the collective term for the species that make up this population, while the microbiome is the collective term for their genetic material. Inflammatory bowel disease, autism, asthma, and obesity have all been linked to the intimate relationship between microbiota composition, metabolic function, and immune system development and regulation (104). When a combination of broad-spectrum antibiotics (i.e., ampicillin plus colistin plus streptomycin) and single-agent imipenem elicited antitumor effects of CTLA-4 monoclonal antibody (mAb) as a result of microbiota impairment, the relationship between microbiota and ICIs was demonstrated. Antibiotic use between two months before and two months after the start of immunotherapy has been linked to a worse prognosis in patients treated with anti–PD-1/PD-L1 mAb (105). It is suggested that microbiota can influence dendritic cell (DC) maturation and activation. Several studies have reported the influence of bacteria such as Akkermansia mucinphila, Bacteriodetes, and Firmicutes in augmenting the response of ICIs. Creating a favorable microbiome using fecal transplantation in patients on ICIs is also being widely studied.
Anti–cytotoxic T-lymphocyte antigen 4 (anti-CTLA-4) immunotherapy causes mucosal damage and the translocation of Burkholderiales and Bacteroidales bacteria, which promote anti-commensal immunity as an adjuvant to anti-tumor immunity and are necessary for a positive response to therapy. Anti–PD-1/PD-L1 therapy, which does not harm the gut epithelia, requires pre–existing antitumor immunity, which is particularly effective in mice with intestinal Bifidobacterium spp (106) as portrayed in Figure 3.
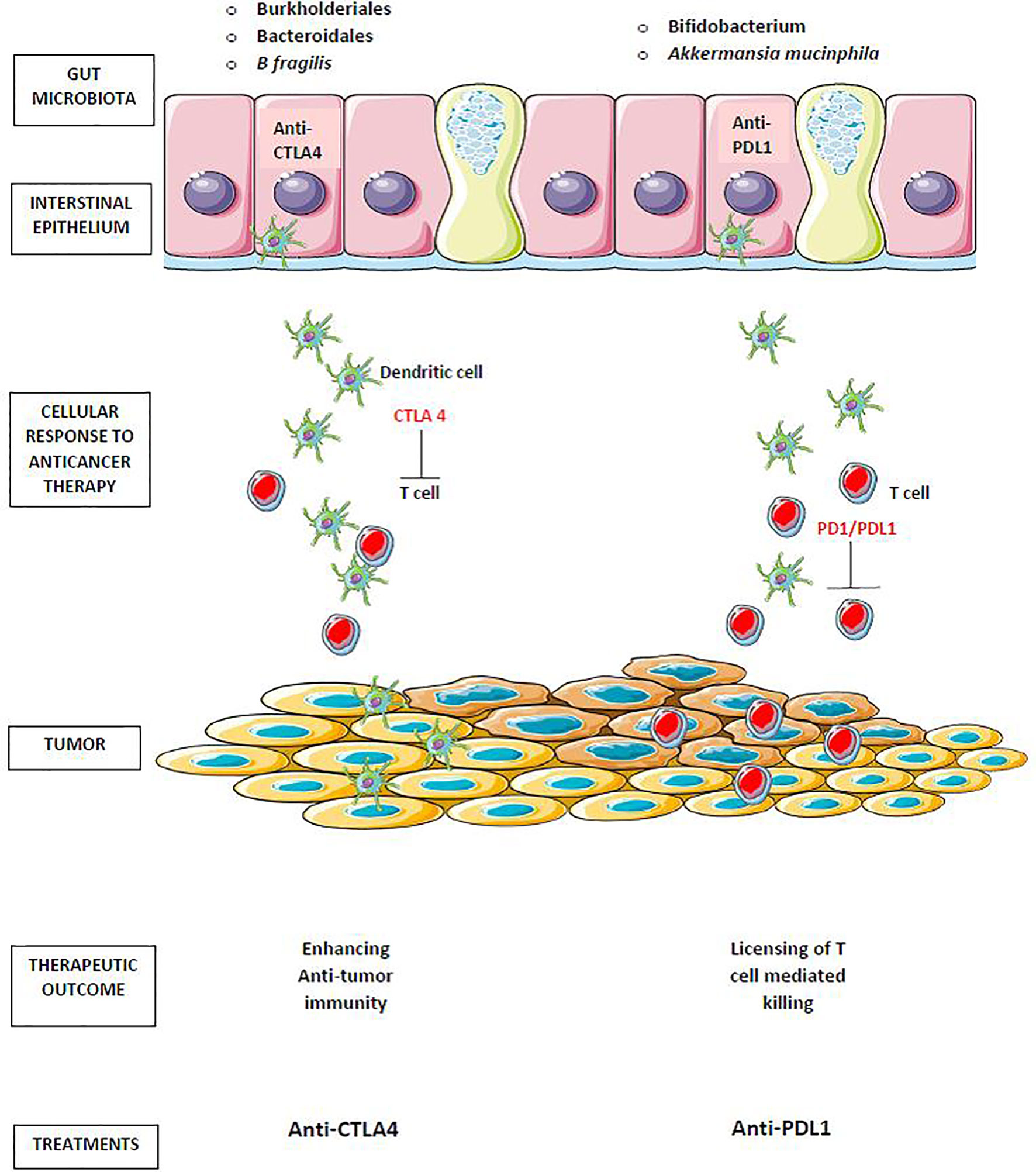
Figure 3 Gut microbiota in cancer immunotherapy. Translocation of Burkholderiales and Bacteroidales, enhances antitumor immunity of anti–cytotoxic T-lymphocyte antigen 4 (anti-CTLA-4). The antitumor effect of anti–PD-1/PD-L1 therapy enhanced by the preexisting antitumor immunity that is particularly effective in mice harboring intestinal Bifidobacterium spp and Akkermansia mucinphila.
Oncolytic viruses also appear to be an additional therapeutic agent for the management of cancer. These agents specifically target and kill cancer cells leaving the normal cells undamaged. Oncolytic viral infection triggers anti-cancer immune responses, which boost the effectiveness of checkpoint inhibition (107). Oncolytic viruses elicit anti-tumor immunity by promoting DC maturation and function. Improved DC maturation and function is because of the ability of oncolytic viruses in transferring genes encoding IFN-α, GM-CSF, and other cytokines. Breakdown of tumor cells induced by the oncolytic virus results in the release of DAMPS which further facilitates anti-tumor immune response. DAMPS include cell surface proteins, membrane proteins, and nucleic acid (108). FDA has approved an intralesional oncolytic immune therapy known as Talimogene Laherparepvec (T-VEC) for stage3b and stage 4 melanoma. A phase II trial comparing a combination of ipilimumab and T-VEC with ipilimumab monotherapy showed better ORR in the combination arm (39% vs. 18%; p < 0.02). Phase1b study of T-Vec combination with Pembrolizumab in melanoma patients reported no dose-limiting toxicity with an ORR of 62% and a CRR (complete response rate) of 33% (109). Many studies including a phase III trial is underway (110). Increased PD-L1 expression and distant inflammation from the injection sites are reported after T-VEC from the analysis performed before the administration of anti-PD1 antibodies. Similarly, several other oncolytic viruses are being evaluated under clinical studies in combination with ICIs as well as a single agent. HF 10 virus included in the HSV family, Tasadenoturev (DNX-2401), members of the Poxviridae family (Pexa-Vec), Coxsackieviruses (CVA 21) are some of the oncolytic viruses which have shown promising results in combination with ICIs in melanoma, GBM, liver cancer, etc. (111–119).
It is known that radiation induces DNA damage-induced apoptosis and recent evidence suggests that it stimulates tumor antigen release and exerts an immune-mediated tumor response (120). Radiotherapy has been stated to enhance immunosuppressive TME via (1) Treg proliferation, M2 polarization of TAMs, and MDSC accumulation by increasing HIF 1 alpha transcription (2) activating latent TGF beta in the tumor, which converts CD4+ T-cells to Tregs and polarizes TAMs into M2 phenotypes (121, 122). Some studies demonstrate the abscopal effect after radiation therapy distinctly from the treatment effects of immunotherapy alone (123). The abscopal effect is described as a phenomenon in the management of metastatic cancer by which localized radiation activates systemic antitumor effects and eradicates distant metastasis. There are several Phase 1 and Phase 2 clinical trials are ongoing in patients with metastatic NSCLC, metastatic head, and neck cancer, and metastatic castration-resistant prostate cancer, etc. (124–126).
Many other drugs are found to have an additive or synergistic effect with ICIs such as Inducible T cell Costimulatory (ICOS) Receptor Agonist, Nectin 4 directed therapies, and anti-IL-6 vaccine. Enfortumab vedotin, a Nectin-4-directed antibody and microtubule inhibitor conjugate, was recently granted accelerated approval by the FDA for the treatment of adult patients with locally advanced or metastatic urothelial cancer who have previously obtained a programmed death receptor-1 (PD-1) or programmed death-ligand 1 (PD-L1) inhibitor and platinum-containing chemotherapy in the neoadjuvant/adjuvant, locally advanced or metastatic setting. A recent study shows that the combination of Enfortumab Vedotin and pembrolizumab demonstrates encouraging efficacy with a tolerable and manageable safety profile in an early phase trial for locally advanced or metastatic urothelial carcinoma. A phase 1b study reported by C J Hoimes et al. states that of the 29 enrolled patients, preliminary confirmed ORR per RECIST 1.1 was 62% by investigators, including a 14% CR rate. The DCR was 90%. (NCT03288545) (127).
GSK609 is an anti-Inducible T cell Co-Stimulator (ICOS) receptor agonist antibody that is used to treat cancers with various histologies. The first in man Phase 1 studies are ongoing as a single agent and along with Pembrolizumab. In patients with previously treated, PD-1/L1 naive HNSCC, preliminary evidence suggests that the combination of GSK609 and pembrolizumab has promising antitumor activity and a manageable safety profile (NCT02723955) (128).
Promising results are reported with anti-Her2 antibody (trastuzumab) in combination with ICIs in patients with HER2-positive metastatic esophagogastric cancer. A study reported a median PFS of 13·0 months (95% CI 8·6 to not reached), and a median OS of 27·3 months with a median DOR of 9.4 months (129). Phase 3 Keynote-811 trial is ongoing and expected to get a detailed result in combining pembrolizumab and trastuzumab (130). Similarly, clinical trials are ongoing to evaluate the response of other anti-tumor antibodies such as rituximab (anti-CD20) in combination with pembrolizumab in recurrent follicular lymphoma and DLBCL (NCT02446457) and cetuximab (anti EGFR)+ ICI combination in recurrent/metastatic HNSCC (NCT03494322) and RAS wild type metastatic colorectal cancer (NCT04561336).
A very recent study published by Chan LC et al. found that JAK1, activated by IL-6 phosphorylates Tyr112 of programmed death-ligand 1 (PD-L1), causing STT3A, an endoplasmic reticulum-associated N-glycosyltransferase, to catalyze PD-L1 glycosylation and maintain PD-L1 stability. In animal models, targeting IL-6 with an IL-6 antibody resulted in synergistic T cell killing when paired with anti–T cell immunoglobulin mucin-3 (anti–Tim-3) therapy. In hepatocellular carcinoma patient tumor tissues, there was a positive association between IL-6 and PD-L1 expression. These findings point to the use of anti–IL-6 and anti–Tim-3 as a marker-guided therapeutic strategy (131).
There are a few drugs that are not used in cancer therapy however have been widely studied in combination with ICIs. The ultimate aim of using these drugs as adjuncts to ICIs is to create an immune stimulatory TME. Most of these studies are still in the preclinical setup but some of them are in clinical studies and show favorable outcomes (Table 3).

Table 3 Few clinical trials listed in Clinical trials.gov which uses non-cancer drugs combination strategies with ICIs as of 17-04-2020.
The key targets in the TME are considered to be MDSCs. Research demonstrates that diminished immunotherapy efficacy is associated with the presence of MDSCs in the TME (132, 133). MDSCs play a vital role in the immunosuppression of TME. These cells possess a strong immunosuppressive potential by representing a heterogeneous population of immature myeloid cells. Antitumor reactivity of NK cells and T cells have also been inhibited by MDSCs. They also encourage angiogenesis, the formation of pre-metastatic niches, and the recruitment of other immunosuppressive cells including regulatory T cells (134). Many commonly prescribed drugs such as rosiglitazone, cimetidine, tadalafil, etc. have shown preclinical benefits in targeting MDSCs (135). Even though the mechanism behind these drugs is different, the goal is to create an immune stimulatory TME inhibiting MDSCs. Examples of these classes of drugs with their proposed mechanism of action include:
● Rosiglitazone: Reduction of early MDSC accumulation
● PDE5 antagonist (Tadalafil and sildenafil): Reduction of MDSC levels, reduction of arginase and iNOS production
● Vitamin D: Stimulate differentiation of immature myeloid cells into DCs
● Amiloride: Inhibit MDSC suppressive capacity via reduced exosome secretion in preclinical studies
● Cimetidine: Reduction of MDSC expansion by induction of apoptosis and inhibition of NOS and ARG-I expression in preclinical studies
Paricalcitol a synthetic vitamin D analog has been used along with ICIs in some of the pancreatic clinical trials and showed a favorable response compared to conventional therapies. In a phase 2 trial using paricalcitol in combination with nivolumab, gemcitabine, nab-paclitaxel, and cisplatin in patients with untreated metastatic pancreatic ductal adenocarcinoma (PDAC), of the 25 patients enrolled thus far, OS has been recorded as 15·3 months, with an underlying ORR of 83% (NCT02754726).
A J Lugibuhl et al. conducted a study using Tadalafil Nivolumab combination in preoperative Head and Neck squamous cell cancer (HNSCC) showed 50% of patients having a pathologic treatment effect (TE) > 21% in 4 weeks. In the context of PD-1 blockade, tadalafil enhanced T lymphocyte and myeloid cell infiltration. 45 patients were treated with the combination regimen, and a preliminary assessment of pathologic treatment effect was performed on post-treatment specimens, revealing that 25% of patients had no treatment effect (TE), 25% had 1-20% TE, 41% had 21-99% TE, and 9% had CR (NCT03238365) (136).
Regulatory T cells (Tregs) play a pivotal role in maintaining immune homeostasis and self-tolerance. They also play a negative role in inducing efficacious anti-tumor response. There is substantial evidence that depleting Tregs or inhibiting Treg function improves antitumor effects (137, 138). The suppressive activity of Treg cells is exhibited by various mechanisms including secretion of inhibitory cytokines such as TGF beta, IL10, and IL35; inhibition of APC maturation through the CTLA-4 pathway; and expression of granzyme and perforin which kills effector T-cells (139, 140). TGFβ inhibitors and IDO inhibitors are currently explored along with checkpoint inhibitors in various cancer models such as melanoma, NSCLC, head, and neck, renal and bladder cancer, etc.
Transforming growth factor (TGF) is a cytokine that induces Tregs and mediates immunosuppression in the TME. Therefore the strategy of blocking the recruitment of Tregs by TGFβ inhibitors has shown benefits in some of the preclinical trials (141). T-cell sequestration away from the tumor mass is caused by the expression of a fibroblast TGF response signature (F-TBRS) in peritumoral fibroblasts. Immune checkpoint blockade with PD-1/PD-L1 inhibitors is ineffective in the absence of physical proximity between cytotoxic T cells and tumor cells. Pharmacological inhibition of TGFβ reverses such immune exclusion and promotes CD8+ T cell penetration into tumors. TGF inhibition combined with immune checkpoint blockade maximizes tumor regression, with several tumor-bearing mice showing complete remission (142–145). Active TGF-signaling in the peritumoral stroma, especially in patients with low tumor-infiltrating T cells in the tumor parenchyma, resulted in a lack of response to atezolizumab (anti-PD-L1 mAb) in metastatic urothelial cancer, according to a study (146). Galunisertib a TGFβ inhibitor is in various clinical trials in combination with ICIs in NSCLC, pancreatic cancer, urothelial cancer, etc. Besides, galunisertib is also in a clinical trial for advanced refractory solid tumors along with ICIs (NCT02423343).
In response to interferon-gamma, tumor cells and MDSCs express IDO. Recent research has found that IDO activity is important for FoxP3 Treg activity (147) and MDSCs, leading to suppression of the activity of T cells and NK cells (148). Several IDO inhibitors are being investigated in combination with CTLA-4 and PD-1 inhibition based on promising preclinical results. Epacadostat is an example of an IDO inhibitor that is being studied along with ICIs in clinical trials. COX-2 inhibitors which are another class of drugs have been found to have potential use as an adjunct with ICIs to treat immunosuppressive tumors that constitutively express indoleamine 2,3-dioxygenase (IDO1) (149–151). In the absence of IDO activity, recent studies have discovered that Tryptophan 2, 3-dioxygenase (TDO), another essential enzyme in the kynurenine pathway, plays a compensatory role. As a result, a dual inhibitor of IDO and TDO is currently being investigated in preclinical studies to achieve maximum inhibition of the kynurenine pathway and alleviate tumor immune suppression (152).
RAS components are also expressed in many cell types of the TME, such as endothelial cells, fibroblasts, monocytes, macrophages, neutrophils, dendritic cells, and T cells (153, 154). Growth and dissemination can be facilitated or hindered by RAS signaling in these cells which have been shown to affect cell proliferation, migration, invasion, metastasis, apoptosis, immunomodulation, angiogenesis, cancer-associated inflammation, and tumor fibrosis/desmoplasia (155, 156). The angiotensin II and angiotensin receptor (AngII/AT1R) axis regulates the tumor stroma and contributes to an immunosuppressive microenvironment through various mechanisms (Figure 4).
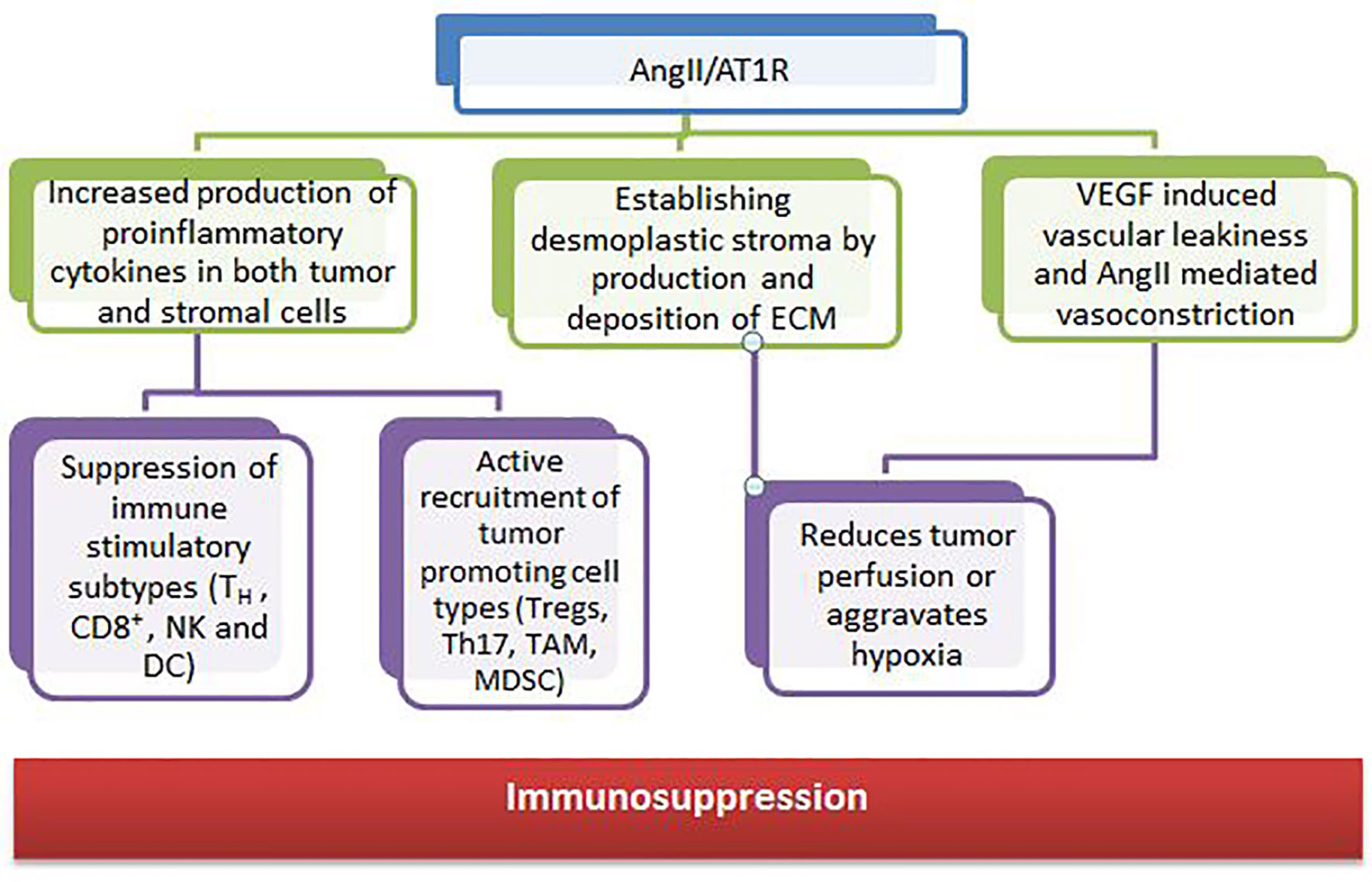
Figure 4 Contribution of angiotensin II and angiotensin receptor to the tumor microenvironment.
Several clinical studies have also shown that RASi may have favorable effects in a wide range of malignancies. The gain in survival is tumor type– and stage-dependent and ranged from 3 months (advanced NSCLC) to more than 25 months (metastatic RCC) in retrospective studies (157). Drugs such as losartan are already in clinical trials as an adjunct with ICIs in many cancers.
Lately, the role of the nervous system, in particular, the sympathetic nervous system has been observed in immune response regulation. Normally, the response of acute stress is beneficial whereas chronic stress is detrimental due to the suppression of effector immune cell activity while enhancing the immunosuppressive cell activity (158). Beta-blockers have been shown to have significant benefits when used along with ICIs. Data have shown that decreasing adrenergic stress by housing mice at thermoneutrality or treating mice housed with β-blockers at cooler temperatures reverses immunosuppression and remarkably increases responses to ICIs (159). Various pre-clinical, as well as clinical studies, have shown improved efficacy when ICIs are used along with a β blocker (159–161).
Metformin has shown antitumor properties in various studies (162, 163). Metformin prevents gluconeogenesis in the liver by regulating the adenosine monophosphate-activated protein kinase (AMPK)/liver kinase B1 (LKB1) pathway. The AMPK/LKB1 pathway controls the cell cycle by regulating protein synthesis and cell proliferation by adjusting the amount of energy needed by the cells (164). Cancerous cells are inhibited and apoptosis is induced as a result of this regulation of the cell cycle and proliferation. Metformin was found to target CD8+ tumor-infiltrating leukocytes in a sample (TILs). Metformin also prevents the production of immune tolerance to cancer cells by inhibiting the synthesis of unfolded proteins, activating the immune response to cancer cells, inhibiting the expression of CD39/73 on MDSCs, and inhibiting the expression of CD39/73 on MDSCs. Metformin prevents exhaustion of CD8+ TILs, thus increasing production of various cytokines such as TNF-α, TNF-γ, IL-2 and reversing immunosuppression. Some studies in NSCLC have reported that metformin exhibits cytotoxic effects as well (165–167). Preclinical and clinical studies are ongoing to explore the efficacy of metformin as an adjunct to ICIs.
An interim safety analysis of Durvalumab Metformin combination in Head and Neck squamous cell carcinoma proved safe in part 1 of the study which contains only 6 patients and enrollment for part 2 with more patients is currently ongoing (NCT03618654) (168).
Various other modalities are being explored to achieve maximum benefit from ICI therapy. Newer checkpoint targets are being analyzed such as LAG-3, TIM-3, TIGIT, VISTA, or B7/H3. Moreover, stimulatory checkpoint pathway agonists such as GITR, 4-1BB, OX40, ICOS, CD40, or molecules targeting TME components like TLR or IDO are under investigation (169, 170). Inhibiting multiple pathways along with ICIs is another ongoing topic of research. Combination of multiple checkpoint inhibitors and triplet or doublet therapy with adjunct drugs are being clinically validated. Improving TME to produce an immune stimulatory environment is being widely explored and drugs targeting these immune suppressive cells such as MDSC, TAM, etc. are being developed (Figures 5 and 6). Targeting mast cells is another approach that is currently being studied. Some of the recent studies have reported that intra-tumoral mast cell levels rose as the tumor progressed, and this independently predicted a shorter OS (171). It was found that CXCL12-CXCR4 chemotaxis results in the accumulation of these tumor-infiltrating mast cells. Intra-tumoral mast cells induced by cancer strongly express PD-L1 in both dose-dependent and time-dependent manner (172). This mechanism can be explored in the future to develop adjunct drugs with ICIs.
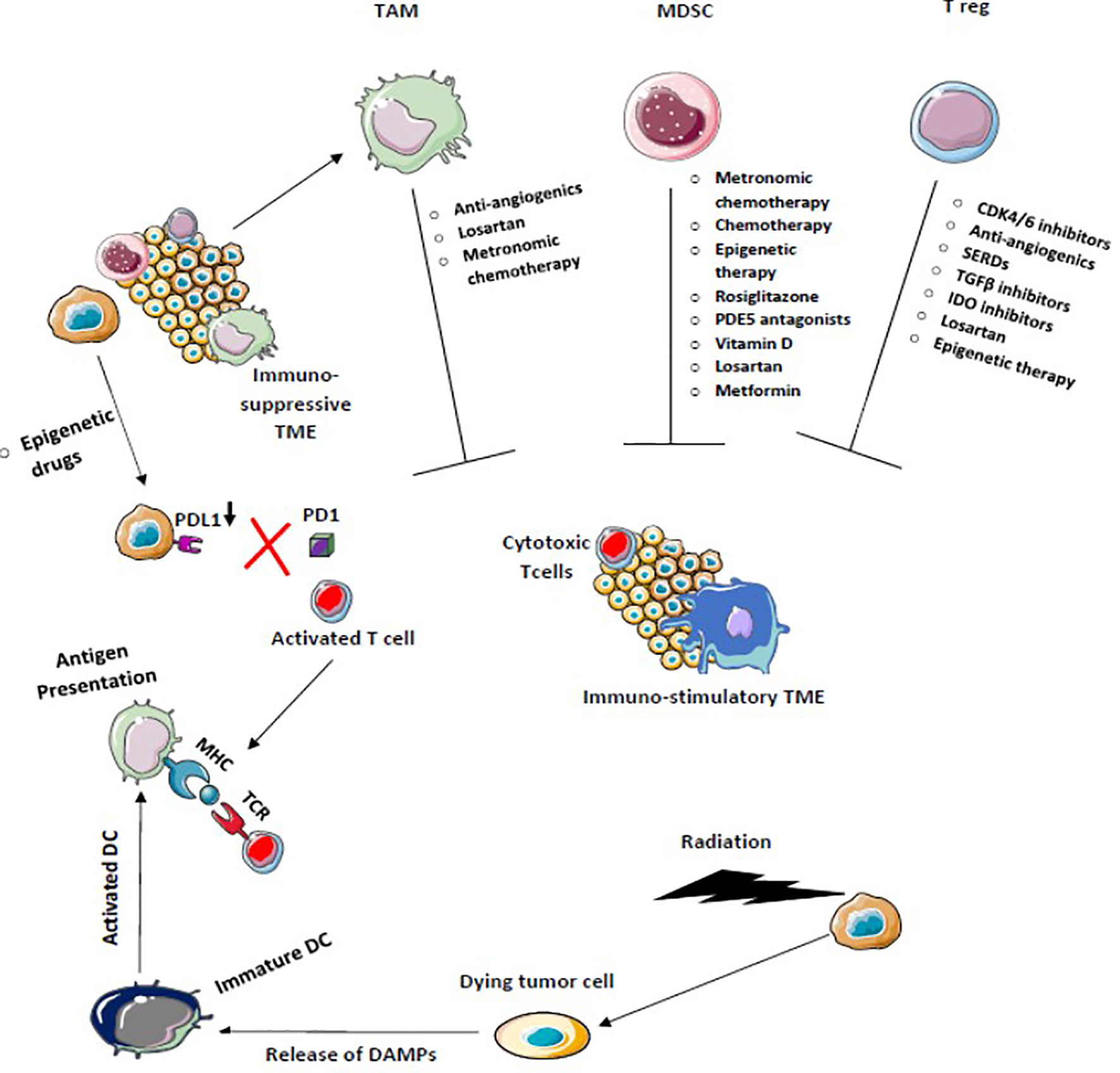
Figure 5 Chemotherapy, Radiotherapy, and Epigenetic therapy which facilitates immunogenic cell death increase antigen presentation enhances cross-priming of dendritic cells, and decreases PDL-1 expression on tumor cells activates the antitumor immune response. Drugs that target Tregs, tumor-associated macrophages (TAM), and myeloid-derived suppressor cells block immunosuppression and enhance T effector T cell function to convert immunosuppressive tumor microenvironment into the immune-stimulatory environment.
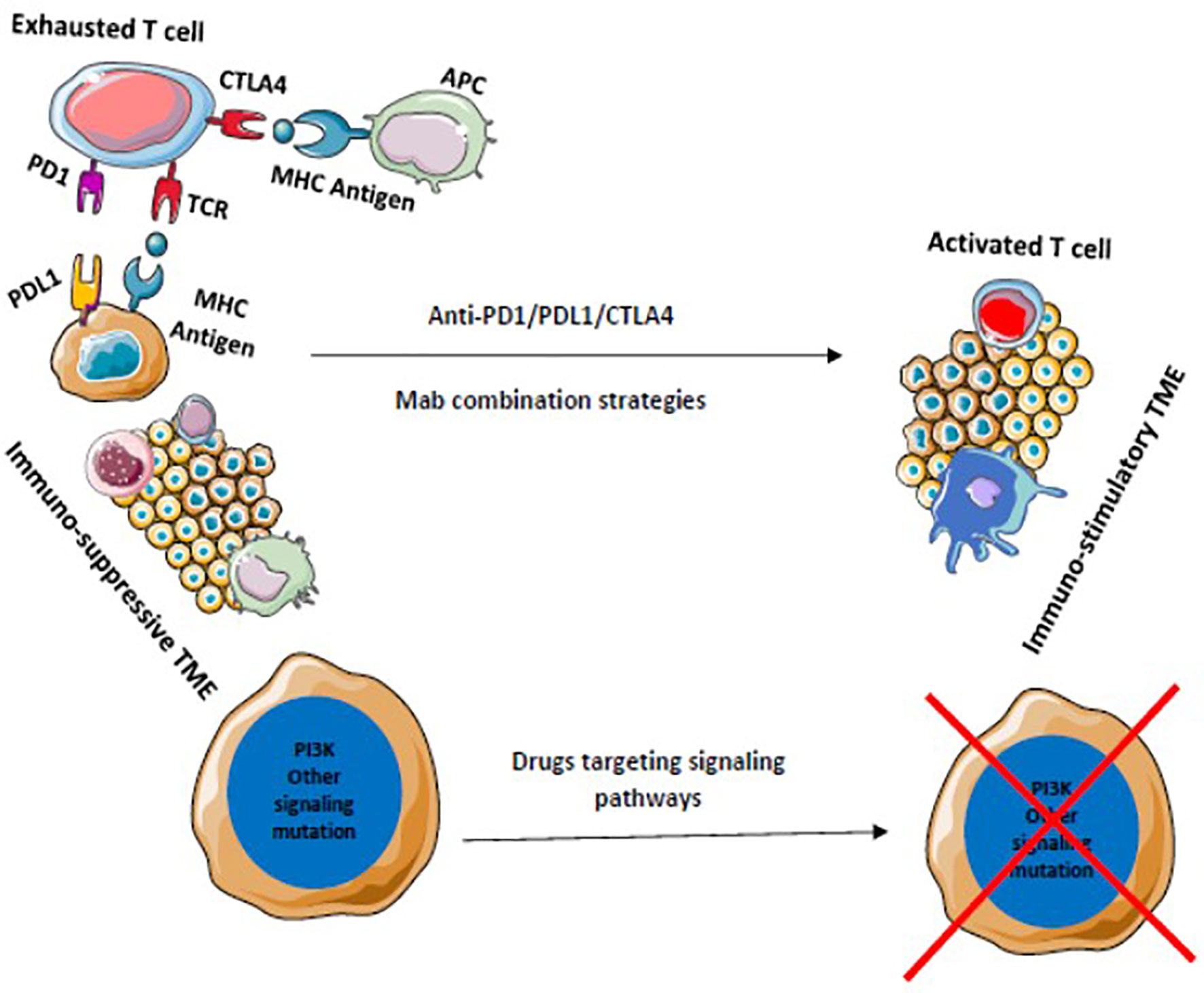
Figure 6 Drugs that target multiple signaling pathways kill tumor cells and stimulates immune cells to provide a durable adaptive immune response. Blocking of multiple immune checkpoints stimulates immune responses against tumors.
As the spectrum of combinatorial immunotherapy strategies widens, the demand for robust preclinical models is at an all-time high. Combination strategies with ICIs require stalwart preclinical models for precise repurposing of the partner drugs. Murine models generally used previously may not be helpful in immuno-oncology because of fundamental differences in the immune composition between mice and humans. The introduction of a patient-derived humanized xenograft model (Hu-PDX) is a very promising strategy in immuno-oncology. Hu-PDXs are mouse cancer models where tissue or cells from a patient’s tumor are implanted into a humanized mouse which contains an immune system identical to humans (173). This knowledge of the Hu-PDX model further propelled research in immuno-oncology and enlightened the concept of co-clinical trial. In a co-clinical trial model, the clinical trial is coupled with a preclinical trial which provides valuable information to the corresponding clinical trial (174). Simultaneous treatment of the Hu-PDX model of a patient enrolled in a clinical trial with new agents helps in further understanding of underlying mechanisms, evaluating novel combination strategies, and identification of potential biomarkers. These advantages of co-clinical trials will ensure the swift transition of preclinical data to the clinic which will be of great value to precision medicine and immuno-oncology.
Besides Hu-PDX models, patient-derived tumor three-dimensional organoid models are also an exciting proposition for co-clinical trials (175). Organoids (and cancer-derived organoids) are three-dimensional tissue-like cellular clusters made from tissue or tumor-specific stem cells that imitate in vivo (tumor) characteristics such as cell heterogeneity (176). The major limitation is the inability to create a tumor-associated immune system in these models. Despite these limitations, organoid cultures accurately mimic the in vivo response of patient-derived xenograft models, as demonstrated in recent studies using bladder cancer-derived organoids by Pauli et al. and Lee et al. (177, 178).
Our review shows that the majority of the adjuncts, which vary from chemotherapy, targeted therapy, radiation, immune-modulators, or even other forms of immunotherapy when combined with ICIs could provide far superior results than previously achieved. Adjunct drugs/modalities with favorable and unfavorable responses are distinctly illustrated in our review.
Maike Trommer, Sin Yuin Yeo et al. showed abscopal effects of RT in up to 29% of the patients when combined with anti PD1 immune check point inhibition (123). Huaqin Yuan et al. showed that Axitinib which is a known antiangiogenic agent could have an immune effect through its inhibition of MDSCs, STAT 3 pathway, and stimulation of CD8 T cells. This effect of Axitinib could enable us to use it as an adjunct along with checkpoint inhibition (179). A R Folgueras et al. showed that epigenetic modification could enhance immune surveillance in epithelial tumors. They found that HDAC inhibition could enhance the immunogenicity of epithelial tumors (180).
Similarly, David Roulois, Helen Loo Yau et al. found that hypomethylating agents may mount an antitumor response by inducing viral mimicry by endogenous transcripts (181). Recent studies have even used a triple-drug combination to augment antitumor immunity. Alexandra S. Zimmer, Erin Nichols et al. found a combination of PD1 inhibitor, Parp inhibitor, and antiangiogenic agent to be well tolerated and effective in a phase 1 study (75). Joseph A. Califano, Zubair Khan et al. found that even non-cancer drugs like tadalafil can have an immune-stimulating effect. Their work showed that tadalafil can reverse the resistance to immune recognition in head and neck cancer when patients were administered daily tadalafil (182).
Even though ICI’s have shown tremendous promise especially in tumors refractory to standard treatments, their response rates are rarely beyond 15-20% (183). Translational research is of utmost value in this scenario and may help to further harness and improve the potential of immunotherapy.
Firstly, standard immune biomarkers like PDL1 levels, MSI status may not hold good when adjuncts are being used along with ICIs. Hence the need to develop newer biomarkers such as tumor mutational burden, tumor-infiltrating lymphocytes, miRNA studies, establishing ‘foreignness’ of neoantigens, HLA variations, N/L ratio, etc. so that a larger %age of patients can reap the benefits of ICIs. Artificial intelligence, neural networks, virtual clinical trials, and patient-derived xenograft models may be required to determine which patients may benefit from ICIs, to learn about the variation in the durability of response among patients, and identify those who may benefit from re-challenge of ICIs.
Secondly, the dose of adjuncts to be administered with ICIs would need to be determined. Ideally, an immune stimulatory’ dose is required which may or may not be the same as the standard dose used otherwise. Once proof of concept is established, the ICI-adjunct combination could be used in tumor agnostic/basket trials thus further tapping into unknown potentials of ICIs. Thirdly, although the mechanisms of ICI-adjunct combinations have been broadly defined, the exact additive/synergistic mechanisms need to be determined through well-constructed trials and translational research before they can become part of standard treatment. Fourthly, adverse effects of ICI-adjunct combination could be a lot greater than when either of the agents is used singly. Moreover, failure of combination strategies in the first-line may result in a lack of availability of drugs for second-line use. Hence proper analysis of adverse events and determining risk: benefit ratio would be required before such combinations become part of routine practice.
Finally, the emerging role of the microbiome and its effect on ICI combinations needs to be addressed and evaluated as it may play a major role in immune-modulation and effectiveness of such combinations. The above-addressed points all form a major portion of T0 and T1 phases of translational research which could greatly widen our understanding and applicability of ICIs and their combinations.
The efficacy of ICIs is highly variable and only a few patients are benefited from them. To address this issue there are many ongoing translational studies to target the resistant mechanisms of ICIs. The main principle is to enhance the TME to an immune stimulatory type from an immune-suppressive one. In this review, we have evaluated available literature which included drugs used as adjuncts in clinical trials along with ICIs. Many drugs have shown significant benefits and some have shown a mixture of both favorable and unfavorable outcomes. There is conflicting evidence for most of the above-mentioned adjuncts such as metformin, Vit D, etc. From the available adjuncts, the selection of the best adjunct for the patients on ICI treatment is still a dilemma. Not only the selection of adjuncts but also the dose for the desired property is also questionable. It might require a dose that is different than used normally and needs to be further evaluated. Therefore, the need of conducting co-clinical trials or translational studies in this field is immense. We believe that recent advances in preclinical models such as humanized xenograft models, invitro modeling using 3D bioprinting, etc. may give further insight into this problem.
HV: Data collection, scientific writing, proof reading, and concept. VS: Scientific writing, proof reading, and concept. BT: Data collection and scientific writing. SM: Concept, proof reading, and scientific writing. RN: Proof reading and project overseer. All authors contributed to the article and approved the submitted version.
All authors are affiliated with the company HealthCare Global Enterprises Ltd.
1. Brief History of Cancer Immunotherapy Approaches and Theories. Available at: https://www.healio.com/hematology-oncology/learn-immuno-oncology/cancer-and-the-immune-system-history-and-theory/brief-history-of-cancer-immunotherapy-approaches-and-theories.
2. Dobosz P, Dzieciątkowski T. The Intriguing History of Cancer Immunotherapy. Front Immunol (2019) 17(10):2965. doi: 10.3389/fimmu.2019.02965
3. Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved Survival With Ipilimumab in Patients With Metastatic Melanoma. N Engl J Med (2010) 363(8):711–23. doi: 10.1056/NEJMoa1003466
4. Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, Activity, and Immune Correlates of Anti–PD-1 Antibody in Cancer. N Engl J Med (2012) 366(26):2443–54. doi: 10.1056/NEJMoa1200690
5. Brahmer JR, Tykodi SS, Chow LQM, Hwu W-J, Topalian SL, Hwu P, et al. Safety and Activity of Anti–PD-L1 Antibody in Patients With Advanced Cancer. N Engl J Med (2012) 366(26):2455–65. doi: 10.1056/NEJMoa1200694
6. Rotte A, Bhandaru M, Zhou Y, McElwee KJ. Immunotherapy of Melanoma: Present Options and Future Promises. Cancer Metastasis Rev (2015) 34(1):115–28. doi: 10.1007/s10555-014-9542-0
7. Becker JC, Andersen MH, Schrama D, Thor Straten P. Immune-Suppressive Properties of the Tumor Microenvironment. Cancer Immunol Immunother (2013) 62(7):1137–48. doi: 10.1007/s00262-013-1434-6
8. Pitt JM, Vétizou M, Daillère R, Roberti MP, Yamazaki T, Routy B, et al. Resistance Mechanisms to Immune-Checkpoint Blockade in Cancer: Tumor-Intrinsic and -Extrinsic Factors. Immunity (2016) 44:1255–69. doi: 10.1016/j.immuni.2016.06.001
9. O’donnell JS, Long GV, Scolyer RA, Teng WL, Smyth MJ. Resistance to PD1/PDL1 Checkpoint Inhibition. Cancer Treat Rev (2016) 52:71–81. doi: 10.1016/j.ctrv.2016.11.007
10. Marincola FM, Jaffee EM, Hickljn DJ, Ferrone S. Escape of Human Solid Tumors From T-Cell Recognition: Molecular Mechanisms and Functional Significance. Adv Immunol (2000) 74):181–273. doi: 10.1016/S0065-2776(08)60911-6
11. Fares CM, Van Allen EM, Drake CG, Allison JP, Hu-Lieskovan S. Mechanisms of Resistance to Immune Checkpoint Blockade: Why Does Checkpoint Inhibitor Immunotherapy Not Work for All Patients? Am Soc Clin Oncol Educ B (2019) 39):147–64. doi: 10.1200/EDBK_240837
12. Ide T, Kitajima Y, Miyoshi A, Ohtsuka T, Mitsuno M, Ohtaka K, et al. The Hypoxic Environment in Tumor-Stromal Cells Accelerates Pancreatic Cancer Progression Via the Activation of Paracrine Hepatocyte Growth Factor/c-Met Signaling. Ann Surg Oncol (2007) 14(9):2600–7. doi: 10.1245/s10434-007-9435-3
13. Kitajima Y, Ide T, Ohtsuka T, Miyazaki K. Induction of Hepatocyte Growth Factor Activator Gene Expression Under Hypoxia Activates the Hepatocyte Growth Factor/c-Met System Via Hypoxia Inducible Factor-1 in Pancreatic Cancer. Cancer Sci (2008) 99(7):1341–7. doi: 10.1111/j.1349-7006.2008.00828.x
14. Jeong H, Kim S, Hong BJ, Lee CJ, Kim YE, Bok S, et al. Tumor-Associated Macrophages Enhance Tumor Hypoxia and Aerobic Glycolysis. Cancer Res (2019) 79(4):795–806. doi: 10.1158/0008-5472.CAN-18-2545
15. Emens LA, Middleton G. The Interplay of Immunotherapy and Chemotherapy: Harnessing Potential Synergies. Cancer Immunol Res (2015) 3(5):436–43. doi: 10.1158/2326-6066.CIR-15-0064
16. Tesniere A, Schlemmer F, Boige V, Kepp O, Martins I, Ghiringhelli F, et al. Immunogenic Death of Colon Cancer Cells Treated With Oxaliplatin. Oncogene (2010) 29(4):482–91. doi: 10.1038/onc.2009.356
17. Lu X, Ding Z-C, Cao Y, Liu C, Habtetsion T, Yu M, et al. Alkylating Agent Melphalan Augments the Efficacy of Adoptive Immunotherapy Using Tumor-Specific CD4 + T Cells. J Immunol (2015) 194(4):2011–21. doi: 10.4049/jimmunol.1401894
18. Fucikova J, Truxova I, Hensler M, Becht E, Kasikova L, Moserova I, et al. Calreticulin Exposure by Malignant Blasts Correlates With Robust Anticancer Immunity and Improved Clinical Outcome in AML Patients. Blood (2016) 128(26):3113–24. doi: 10.1182/blood-2016-08-731737
19. Galluzzi L, Buqué A, Kepp O, Zitvogel L, Kroemer G. Immunological Effects of Conventional Chemotherapy and Targeted Anticancer Agents. Cancer Cell (2015) 28:690–714. doi: 10.1016/j.ccell.2015.10.012
20. Yan Y, Kumar AB, Finnes H, Markovic SN, Park S, Dronca RS, et al. Combining Immune Checkpoint Inhibitors With Conventional Cancer Therapy. Front Immunol (2018) 9:1739. doi: 10.3389/fimmu.2018.01739
21. Gadgeel SM, Garassino MC, Esteban E, Speranza G, Felip E, Hochmair MJ, et al. Keynote-189: Updated OS and Progression After the Next Line of Therapy (PFS2) With Pembrolizumab (Pembro) Plus Chemo With Pemetrexed and Platinum vs Placebo Plus Chemo for Metastatic Nonsquamous NSCLC. J Clin Oncol (2019) 37(15_suppl):9013. doi: 10.1200/JCO.2019.37.15_suppl.9013
22. Barlesi F, Nishio M, Cobo M, Steele N, Paramonov V, Parente B, et al. Impower132: Efficacy of Atezolizumab (Atezo) + Carboplatin (Carbo)/Cisplatin (Cis) + Pemetrexed (Pem) as 1L Treatment in Key Subgroups With Stage I. OncologyPRO. Available at: https://oncologypro.esmo.org/Meeting-Resources/ESMO-2018-Congress/IMpower132-efficacy-of-atezolizumab-atezo-carboplatin-carbo-cisplatin-cis-pemetrexed-pem-as-1L-treatment-in-key-subgroups-with-stage-IV-non-squamous-non-small-cell-lung-cancer-NSCLC.
23. 2019 ASCO. Keynote-062: Pembrolizumab With or Without Chemotherapy vs Chemotherapy in Advanced Gastric or GEJ Adenocarcinoma. The ASCO Post. (2019) Available at: https://www.ascopost.com/News/60099.
24. Schmid P, Rugo HS, Adams S, Schneeweiss A, Barrios CH, Iwata H, et al. Atezolizumab Plus Nab-Paclitaxel as First-Line Treatment for Unresectable, Locally Advanced or Metastatic Triple-Negative Breast Cancer (Impassion130): Updated Efficacy Results From a Randomised, Double-Blind, Placebo-Controlled, Phase 3 Trial. Lancet Oncol (2020) 21(1):44–59. doi: 10.1016/S1470-2045(19)30689-8
25. FDA Approves Durvalumab for Extensive-Stage Small Cell Lung Cancer. FDA. Available at: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-durvalumab-extensive-stage-small-cell-lung-cancer.
26. Allegrini G, Di Desidero T, Barletta MT, Fioravanti A, Orlandi P, Canu B, et al. Clinical, Pharmacokinetic and Pharmacodynamic Evaluations of Metronomic UFT and Cyclophosphamide Plus Celecoxib in Patients With Advanced Refractory Gastrointestinal Cancers. Angiogenesis (2012) 15(2):275–86. doi: 10.1007/s10456-012-9260-6
27. Khan OA, Blann AD, Payne MJ, Middleton MR, Protheroe AS, Talbot DC, et al. Continuous Low-Dose Cyclophosphamide and Methotrexate Combined With Celecoxib for Patients With Advanced Cancer. Br J Cancer (2011) 104(12):1822–7. doi: 10.1038/bjc.2011.154
28. Robison NJ, Campigotto F, Chi SN, Manley PE, Turner CD, Zimmerman MA, et al. A Phase II Trial of a Multi-Agent Oral Antiangiogenic (Metronomic) Regimen in Children With Recurrent or Progressive Cancer. Pediatr Blood Cancer (2014) 61(4):636–42. doi: 10.1002/pbc.24794
29. Bocci G, Kerbel RS. Pharmacokinetics of Metronomic Chemotherapy: A Neglected But Crucial Aspect. Nat Rev Clin Oncol (2016) 13(11):659–73. doi: 10.1038/nrclinonc.2016.64
30. Kareva I, Waxman DJ, Klement GL. Metronomic Chemotherapy: An Attractive Alternative to Maximum Tolerated Dose Therapy That can Activate Anti-Tumor Immunity and Minimize Therapeutic Resistance. Cancer Lett (2015) 358:100–6. doi: 10.1016/j.canlet.2014.12.039
31. Kareva I. A Combination of Immune Checkpoint Inhibition With Metronomic Chemotherapy as a Way of Targeting Therapy-Resistant Cancer Cells. Int J Mol Sci (2017) 18(10):2134. doi: 10.3390/ijms18102134
32. Banissi C, Ghiringhelli F, Chen L, Carpentier AF. Treg Depletion With a Low-Dose Metronomic Temozolomide Regimen in a Rat Glioma Model. Cancer Immunol Immunother (2009) 58(10):1627–34. doi: 10.1007/s00262-009-0671-1
33. Generali D, Bates G, Berruti A, Brizzi MP, Campo L, Bonardi S, et al. Immunomodulation of FOXP3+ Regulatory T Cells by the Aromatase Inhibitor Letrozole in Breast Cancer Patients. Clin Cancer Res (2009) 15(3):1046–51. doi: 10.1158/1078-0432.CCR-08-1507
34. Ghiringhelli F, Menard C, Puig PE, Ladoire S, Roux S, Martin F, et al. Metronomic Cyclophosphamide Regimen Selectively Depletes CD4 +CD25+ Regulatory T Cells and Restores T and NK Effector Functions in End Stage Cancer Patients. Cancer Immunol Immunother (2007) 56(5):641–8. doi: 10.1007/s00262-006-0225-8
35. Tanaka H, Matsushima H, Mizumoto N, Takashima A. Classification of Chemotherapeutic Agents Based on Their Differential In Vitro Effects on Dendritic Cells. Cancer Res (2009) 69(17):6978–86. doi: 10.1158/0008-5472.CAN-09-1101
36. Nars MS, Kaneno R. Immunomodulatory Effects of Low Dose Chemotherapy and Perspectives of Its Combination With Immunotherapy. Int J Cancer (2013) 132:2471–8. doi: 10.1002/ijc.27801
37. Ma J, Waxman DJ. Dominant Effect of Antiangiogenesis in Combination Therapy Involving Cyclophosphamide and Axitinib. Clin Cancer Res (2009) 15(2):578–88. doi: 10.1158/1078-0432.CCR-08-1174
38. Highfill SL, Cui Y, Giles AJ, Smith JP, Zhang H, Morse E, et al. Disruption of CXCR2-Mediated MDSC Tumor Trafficking Enhances Anti-PD1 Efficacy. Sci Transl Med (2014) 6(237):237ra67. doi: 10.1126/scitranslmed.3007974
39. Tu N, Trinh T, Zhou J-M, Gilvary DL, Coppola D, Wei S, et al. Abstract 3666: Chemotherapeutic Sensitivity of Myeloid-Derived Suppressor Cells During Cancer Therapy Is Dictated by Selective Expression of Clusterin. In Am Assoc Cancer Res (AACR) (2017) 77(13 Suppl):3666–6. doi: 10.1158/1538-7445.AM2017-3666
40. Munn DH, Bronte V. Immune Suppressive Mechanisms in the Tumor Microenvironment. Curr Opin Immunol (2016) 39:1–6. doi: 10.1016/j.coi.2015.10.009
41. Pfirschke C, Engblom C, Rickelt S, Cortez-Retamozo V, Garris C, Pucci F, et al. Immunogenic Chemotherapy Sensitizes Tumors to Checkpoint Blockade Therapy. Immunity (2016) 44(2):343–54. doi: 10.1016/j.immuni.2015.11.024
42. Song W, Shen L, Wang Y, Liu Q, Goodwin TJ, Li J, et al. Synergistic and Low Adverse Effect Cancer Immunotherapy by Immunogenic Chemotherapy and Locally Expressed PD-L1 Trap. Nat Commun (2018) 9(1):1–11. doi: 10.1038/s41467-018-04605-x
43. Merlano MC, Merlotti AM, Licitra L, Denaro N, Fea E, Galizia D, et al. Activation of Immune Responses in Patients With Relapsed-Metastatic Head and Neck Cancer (CONFRONT Phase I-II Trial): Multimodality Immunotherapy With Avelumab, Short-Course Radiotherapy, and Cyclophosphamide. Clin Transl Radiat Oncol (2018) 12:47–52. doi: 10.1016/j.ctro.2018.08.001
44. Cui S. Immunogenic Chemotherapy Sensitizes Renal Cancer to Immune Checkpoint Blockade Therapy in Preclinical Models. Med Sci Monit (2017) 23:3360–6. doi: 10.12659/MSM.902426
45. Luptakova K, Rosenblatt J, Glotzbecker B, Mills H, Stroopinsky D, Kufe T, et al. Lenalidomide Enhances Anti-Myeloma Cellular Immunity. Cancer Immunol Immunother (2013) 62(1):39–49. doi: 10.1007/s00262-012-1308-3
46. Hsu AK, Quach H, Tai T, Prince HM, Harrison SJ, Trapani JA, et al. The Immunostimulatory Effect of Lenalidomide on NK-Cell Function Is Profoundly Inhibited by Concurrent Dexamethasone Therapy. Blood (2011) 117(5):1605–13. doi: 10.1182/blood-2010-04-278432
47. Görgün G, Samur MK, Cowens KB, Paula S, Bianchi G, Anderson JE, et al. Lenalidomide Enhances Immune Checkpoint Blockade-Induced Immune Response in Multiple Myeloma. Clin Cancer Res (2015) 21(20):4617–8. doi: 10.1158/1078-0432.CCR-15-0200
48. Bansal M, Siegel DS, Ahn J, Feinman R, Vesole DH, Richter JR, et al. Two Year Update of Phase II Trial of Pembrolizumab, Lenalidomide and Dexamethasone as Post-Autologous Stem Cell Transplant Consolidation in Patients With High-Risk Multiple Myeloma. Blood (2019) 134(Supplement_1):1911–1. doi: 10.1182/blood-2019-125155
49. Paper: Pembrolizumab in Combination With Lenalidomide and Low-Dose Dexamethasone for Relapsed/Refractory Multiple Myeloma (Rrmm): Keynote-023. Available at: https://ash.confex.com/ash/2015/webprogramscheduler/Paper85801.html.
50. Mateos M, Orlowski RZ, Ocio EM, Rodríguez-Otero P, Reece D, Moreau P, et al. Pembrolizumab Combined With Lenalidomide and Low-Dose Dexamethasone for Relapsed or Refractory Multiple Myeloma: Phase I KEYNOTE -023 Study. Br J Haematol (2019) 186(5):e117–21. doi: 10.1111/bjh.15946
51. Badros A, Hyjek E, Ma N, Lesokhin A, Dogan A, Rapoport AP, et al. Pembrolizumab, Pomalidomide, and Low-Dose Dexamethasone for Relapsed/Refractory Multiple Myeloma. Blood (2017) 130(10):1189–97. doi: 10.1182/blood-2017-03-775122
52. Badros AZ, Ma N, Rapoport AP, Lederer E, Lesokhin AM. Long-Term Remissions After Stopping Pembrolizumab for Relapsed or Refractory Multiple Myeloma. Blood Adv (2019) 3:1658–60. doi: 10.1182/bloodadvances.2019000191
53. Luo N, Nixon MJ, Gonzalez-Ericsson PI, Sanchez V, Opalenik SR, Li H, et al. DNA Methyltransferase Inhibition Upregulates MHC-I to Potentiate Cytotoxic T Lymphocyte Responses in Breast Cancer. Nat Commun (2018) 9(1):1–11. doi: 10.1038/s41467-017-02630-w
54. Emran A, Chatterjee A, Rodger EJ, Tiffen JC, Gallagher SJ, Eccles MR, et al. Targeting DNA Methylation and EZH2 Activity to Overcome Melanoma Resistance to Immunotherapy. Trends Immunol (2019) 40:328–44. doi: 10.1016/j.it.2019.02.004
55. Jazirehi AR, Nazarian R, Torres-Collado AX, Economou JS. Aberrant Apoptotic Machinery Confers Melanoma Dual Resistance to BRAF(V600E) Inhibitor and Immune Effector Cells: Immunosensitization by a Histone Deacetylase Inhibitor. Am J Clin Exp Immunol (2014) 3(1):43–56.
56. Chiappinelli KB, Strissel PL, Desrichard A, Li H, Henke C, Akman B, et al. Inhibiting DNA Methylation Causes an Interferon Response in Cancer Via Dsrna Including Endogenous Retroviruses. Cell (2015) 162(5):974–86. doi: 10.1016/j.cell.2015.07.011
57. Roulois D, Loo Yau H, Singhania R, Wang Y, Danesh A, Shen SY, et al. Dna-Demethylating Agents Target Colorectal Cancer Cells by Inducing Viral Mimicry by Endogenous Transcripts. Cell (2015) 27162(5):961–73. doi: 10.1016/j.cell.2015.07.056
58. Brahmer JR, Horn L, Antonia SJ, Spigel DR, Gandhi L, Sequist LV, et al. Survival and Long-Term Follow-Up of the Phase I Trial of Nivolumab (Anti-PD-1; BMS-936558; Ono-4538) in Patients (pts) With Previously Treated Advanced Non-Small Cell Lung Cancer (NSCLC). J Clin Oncol (2013) 31(15_suppl):8030. doi: 10.1200/jco.2013.31.15_suppl.8030
59. Xu WS, Parmigiani RB, Marks PA. Histone Deacetylase Inhibitors: Molecular Mechanisms of Action. Oncogene (2007) 26:5541–52. doi: 10.1038/sj.onc.1210620
60. Banik D, Moufarrij S, Villagra A. Immunoepigenetics Combination Therapies: An Overview of the Role of HDACs in Cancer Immunotherapy. Int J Mol Sci (2019) 20(9):2241. doi: 10.3390/ijms20092241
61. Kroesen M, Gielen PR, Brok IC, Armandari I, Hoogerbrugge PM, Adema GJ. HDAC Inhibitors and Immunotherapy; A Double Edged Sword? Oncotarget (2014) 5(16):6558–72. doi: 10.18632/oncotarget.2289
62. Woods DM, Sodré AL, Villagra A, Sarnaik A, Sotomayor EM, Weber J. HDAC Inhibition Upregulates PD-1 Ligands in Melanoma and Augments Immunotherapy With PD-1 Blockade. Cancer Immunol Res (2015) 3(12):1375–85. doi: 10.1158/2326-6066.CIR-15-0077-T
63. Gray JE, Saltos A, Tanvetyanon T, Haura EB, Creelan B, Antonia SJ, et al. Phase I/Ib Study of Pembrolizumab Plus Vorinostat in Advanced/Metastatic non–Small Cell Lung Cancer. Clin Cancer Res (2019) 25(22):6623–32. doi: 10.1158/1078-0432.CCR-19-1305
64. Goel S, Decristo MJ, Watt AC, Brinjones H, Sceneay J, Li BB, et al. CDK4/6 Inhibition Triggers Anti-Tumour Immunity. Nature (2017) 548(7668):471–5. doi: 10.1038/nature23465
65. Deng J, Wang ES, Jenkins RW, Li S, Dries R, Yates K, et al. CDK4/6 Inhibition Augments Antitumor Immunity by Enhancing T-Cell Activation. Cancer Discov (2018) 8(2):216–33. doi: 10.1158/2159-8290.CD-17-0915
66. Rugo H, Kabos P, Dickler M, John W, Smith I, Lu Y, et al. Abstract P1-09-01: A Phase 1b Study of Abemaciclib Plus Pembrolizumab for Patients With Hormone Receptor-Positive (HR+), Human Epidermal Growth Factor Receptor 2-Negative (HER2-) Metastatic Breast Cancer (MBC). Am Assoc Cancer Res (AACR) (2018) 38(15 suppl):P1–09. doi: 10.1158/1538-7445.SABCS17-P1-09-01
67. Farsaci B, Donahue RN, Coplin MA, Grenga I, Lepone LM, Molinolo AA, et al. Immune Consequences of Decreasing Tumor Vasculature With Antiangiogenic Tyrosine Kinase Inhibitors in Combination With Therapeutic Vaccines. Cancer Immunol Res (2014) 2(11):1090–102. doi: 10.1158/2326-6066.CIR-14-0076
68. Huang Y, Yuan J, Righi E, Kamoun WS, Ancukiewicz M, Nezivar J, et al. Vascular Normalizing Doses of Antiangiogenic Treatment Reprogram the Immunosuppressive Tumor Microenvironment and Enhance Immunotherapy. Proc Natl Acad Sci USA (2012) 109(43):17561–6. doi: 10.1073/pnas.1215397109
69. Manegold C, Dingemans AMC, Gray JE, Nakagawa K, Nicolson M, Peters S, et al. The Potential of Combined Immunotherapy and Antiangiogenesis for the Synergistic Treatment of Advanced NSCLC. J Thoracic Oncol (2017) 12:194–207. doi: 10.1016/j.jtho.2016.10.003
70. Socinski MA, Jotte RM, Cappuzzo F, Orlandi FJ, Stroyakovskiy D, Nogami N, et al. Overall Survival (OS) Analysis of IMpower150, a Randomized Ph 3 Study of Atezolizumab (Atezo) + Chemotherapy (Chemo) ± Bevacizumab (Bev) vs Chemo + Bev in 1L Nonsquamous (NSQ) NSCLC. J Clin Oncol (2018) 36(15_suppl):9002–2. doi: 10.1200/JCO.2018.36.15_suppl.9002
71. Rini BI, Plimack ER, Stus V, Gafanov R, Hawkins R, Nosov D, et al. Pembrolizumab Plus Axitinib Versus Sunitinib for Advanced Renal-Cell Carcinoma. N Engl J Med (2019) 380(12):1116–27. doi: 10.1056/NEJMoa1816714
72. Stewart RA, Pilie PG, Yap TA. Development of PARP and Immune-Checkpoint Inhibitor Combinations. Cancer Res (2018) 78:6717–25. doi: 10.1158/0008-5472.CAN-18-2652
73. Li A, Yi M, Qin S, Chu Q, Luo S, Wu K. Prospects for Combining Immune Checkpoint Blockade With PARP Inhibition. J Hematol Oncol (2019) 12(1):1–2. doi: 10.1186/s13045-019-0784-8
74. Sen T, Rodriguez BL, Chen L, Della Corte CM, Morikawa N, Fujimoto J, et al. Targeting DNA Damage Response Promotes Antitumor Immunity Through STING-mediated T-Cell Activation in Small Cell Lung Cancer. Cancer Discov (2019) 9(5):646–61. doi: 10.1158/2159-8290.CD-18-1020
75. Zimmer AS, Nichols E, Cimino-Mathews A, Peer C, Cao L, Lee M-J, et al. A Phase I Study of the PD-L1 Inhibitor, Durvalumab, in Combination With a PARP Inhibitor, Olaparib, and a VEGFR1–3 Inhibitor, Cediranib, in Recurrent Women’s Cancers With Biomarker Analyses. J Immunother Cancer (2019) 7(1):1–8. doi: 10.1186/s40425-019-0680-3
76. Penson RT, Drew Y, de Jonge MJ, Hong SH, Park YH, Wolfer A, et al. Mediola: A Phase I/II Trial of Olaparib (PARP Inhibitor) in Combination With Durvalumab (anti-PD-L1 Antibody) in Pts With Advanced Solid Tumours –. OncologyPRO. Available at: https://oncologypro.esmo.org/Meeting-Resources/ESMO-2018-Congress/MEDIOLA-A-Phase-I-II-trial-of-olaparib-PARP-inhibitor-in-combination-with-durvalumab-anti-PD-L1-antibody-in-pts-with-advanced-solid-tumours-new-ovarian-cancer-cohorts.
77. Chan T, Wiltrout RH, Weiss JM. Immunotherapeutic Modulation of the Suppressive Liver and Tumor Microenvironments. Int Immunopharmacol (2011) 11:879–89. doi: 10.1016/j.intimp.2010.12.024
78. Joerger M, Güller U, Bastian S, Driessen C, von Moos R. Prolonged Tumor Response Associated With Sequential Immune Checkpoint Inhibitor Combination Treatment and Regorafenib in a Patient With Advanced Pretreated Hepatocellular Carcinoma. J Gastrointest Oncol (2019) 10(2):373–8. doi: 10.21037/jgo.2018.11.04
79. El-Khoueiry AB, Sangro B, Yau T, Crocenzi TS, Kudo M, Hsu C, et al. Nivolumab in Patients With Advanced Hepatocellular Carcinoma (CheckMate 040): An Open-Label, Non-Comparative, Phase 1/2 Dose Escalation and Expansion Trial. Lancet (2017) 389(10088):2492–502. doi: 10.1016/S0140-6736(17)31046-2
80. Zhu AX, Finn RS, Edeline J, Cattan S, Ogasawara S, Palmer D, et al. 【Keynote-224試験】 ソラフェニブ抵抗性の切除不能肝細胞癌に対するペンブロリズマブ療法の第ii相試験. Lancet Oncol (2018) 19(7):940–52. doi: 10.1016/S1470-2045(18)30351-6
81. Jiang X, Zhou J, Giobbie-Hurder A, Wargo J, Hodi FS. The Activation of MAPK in Melanoma Cells Resistant to BRAF Inhibition Promotes PD-L1 Expression That Is Reversible by MEK and PI3K Inhibition. Clin Cancer Res (2013) 19(3):598–609. doi: 10.1158/1078-0432.CCR-12-2731
82. Atefi M, Avramis E, Lassen A, Wong DJL, Robert L, Foulad D, et al. Effects of MAPK and PI3K Pathways on PD-L1 Expression in Melanoma. Clin Cancer Res (2014) 20(13):3446–57. doi: 10.1158/1078-0432.CCR-13-2797
83. Vanneman M, Dranoff G. Combining Immunotherapy and Targeted Therapies in Cancer Treatment. Nat Rev Cancer (2012) 12:237–51. doi: 10.1038/nrc3237
84. De Henau O, Rausch M, Winkler D, Campesato LF, Liu C, Cymerman DH, et al. Overcoming Resistance to Checkpoint Blockade Therapy by Targeting PI3Kγ in Myeloid Cells. Nature (2016) 539(7629):443–7. doi: 10.1038/nature20554
85. Peng W, Chen JQ, Liu C, Malu S, Creasy C, Tetzlaff MT, et al. Loss of PTEN Promotes Resistance to T Cell–Mediated Immunotherapy. Cancer Discov (2016) 6(2):202–16. doi: 10.1158/2159-8290.CD-15-0283
86. Opdivo-Cabometyx Combo Extended Survival in Advanced Kidney Cancer. Available at: https://immuno-oncologynews.com/2020/04/28/opdivo-cabometyx-combo-extends-survival-increases-response-rates-advanced-kidney-cancer-phase-3-trial/?utm_source=IO+News&utm_campaign=a6d281b789-RSS_WEEKLY_EMAIL_CAMPAIGN_NON-US&utm_medium=email&utm_term=0_f04c303b86-a6d281b789-73533625.
87. Selected Abstracts on Novel Treatments in Colon, Hepatocellular, and Biliary Tract Cancers. The ASCO Post. Available at: https://www.ascopost.com/issues/march-25-2020/selected-abstracts-on-novel-treatments-in-colon-hepatocellular-and-biliary-tract-cancers/.
88. Dubovsky JA, Beckwith KA, Natarajan G, Woyach JA, Jaglowski S, Zhong Y, et al. Ibrutinib Is an Irreversible Molecular Inhibitor of ITK Driving a Th1-Selective Pressure in T Lymphocytes. Blood (2013) 122(15):2539–49. doi: 10.1182/blood-2013-06-507947
89. Hude I, Sasse S, Engert A, Bröckelmann PJ. The Emerging Role of Immune Checkpoint Inhibition in Malignant Lymphoma. Haematologica (2017) 102:30–42. doi: 10.3324/haematol.2016.150656
90. Sagiv-Barfi I, Kohrt HEK, Czerwinski DK, Ng PP, Chang BY, Levy R. Therapeutic Antitumor Immunity by Checkpoint Blockade Is Enhanced by Ibrutinib, an Inhibitor of Both BTK and ITK. Proc Natl Acad Sci USA (2015) 112(9):E966–72. doi: 10.1073/pnas.1500712112
91. Hong D, Rasco D, Veeder M, Luke JJ, Chandler J, Balmanoukian A, et al. A Phase 1b/2 Study of the Bruton Tyrosine Kinase Inhibitor Ibrutinib and the PD-L1 Inhibitor Durvalumab in Patients With Pretreated Solid Tumors. Oncol (2019) 97(2):102–11. doi: 10.1159/000500571
92. Svoronos N, Perales-Puchalt A, Allegrezza MJ, Rutkowski MR, Payne KK, Tesone AJ, et al. Tumor Cell–Independent Estrogen Signaling Drives Disease Progression Through Mobilization of Myeloid-Derived Suppressor Cells. Cancer Discov (2017) 7(1):72–85. doi: 10.1158/2159-8290.CD-16-0502
93. Gabrilovich DI. Myeloid-Derived Suppressor Cells. Cancer Immunol Res (2017) 5(1):3–8. doi: 10.1158/2326-6066.CIR-16-0297
94. Zhao L, Huang S, Mei S, Yang Z, Xu L, Zhou N, et al. Pharmacological Activation of Estrogen Receptor Beta Augments Innate Immunity to Suppress Cancer Metastasis. Proc Natl Acad Sci USA (2018) 115(16):E3673–81. doi: 10.1073/pnas.1803291115
95. Márquez-Garbán DC, Deng G, Comin-Anduix B, Garcia AJ, Xing Y, Chen HW, et al. Antiestrogens in Combination With Immune Checkpoint Inhibitors in Breast Cancer Immunotherapy. J Steroid Biochem Mol Biol (2019) 193:105415. doi: 10.1016/j.jsbmb.2019.105415
96. Curran MA, Montalvo W, Yagita H, Allison JP. PD-1 and CTLA-4 Combination Blockade Expands Infiltrating T Cells and Reduces Regulatory T and Myeloid Cells Within B16 Melanoma Tumors. Proc Natl Acad Sci USA (2010) 107(9):4275–80. doi: 10.1073/pnas.0915174107
97. Eggermont AMM, Crittenden M, Wargo J. Combination Immunotherapy Development in Melanoma. Am Soc Clin Oncol Educ B (2018) 38):197–207. doi: 10.1200/EDBK_201131
98. Five-Year Outcomes for Opdivo (Nivolumab) in Combination With Yervoy (Ipilimumab) Demonstrate Durable Long-Term Survival Benefits in Patients With Advanced Melanoma. BMS Newsroom. Available at: https://news.bms.com/press-release/corporatefinancial-news/five-year-outcomes-opdivo-nivolumab-combination-yervoy-ipilimu.
99. U.s. Food and Drug Administration Accepts for Priority Review Bristol-Myers Squibb’s Application for Opdivo (Nivolumab) Plus Yervoy (Ipilimumab) Combination for Patients With Previously Treated Advanced Hepatocellular Carcinoma. Business Wire. Available at: https://www.businesswire.com/news/home/20191111005122/en.
100. Yau T, Kang Y-K, Kim T-Y, El-Khoueiry AB, Santoro A, Sangro B, et al. Nivolumab (NIVO) + Ipilimumab (IPI) Combination Therapy in Patients (Pts) With Advanced Hepatocellular Carcinoma (Ahcc): Results From CheckMate 040. J Clin Oncol (2019) 37(15_suppl):4012–2. doi: 10.1200/JCO.2019.37.15_suppl.4012
101. Opdivo (Nivolumab) Plus Yervoy (Ipilimumab) Demonstrates Continued Survival Benefit At 42-Month Follow-up in Patients With Previously Untreated Advanced or Metastatic Renal Cell Carcinoma. BMS Newsroom. Available at: https://news.bms.com/press-release/corporatefinancial-news/opdivo-nivolumab-plus-yervoy-ipilimumab-demonstrates-continued.
102. Agenus Announces Positive Interim Data From Balstilimab and Zalifrelimab Clinical Trials in Second-Line Cervical Cancer (2020). Available at: https://investor.agenusbio.com/2020-02-20-Agenus-Announces-Positive-Interim-Data-from-Balstilimab-and-Zalifrelimab-Clinical-Trials-in-Second-Line-Cervical-Cancer.
103. Huttenhower C, Gevers D, Knight R, Abubucker S, Badger JH, Chinwalla AT, et al. Structure, Function and Diversity of the Healthy Human Microbiome. Nature (2012) 486(7402):207–14. doi: 10.1038/nature11234
104. Schwartz DJ, Rebeck ON, Dantas G. Complex Interactions Between the Microbiome and Cancer Immune Therapy. Crit Rev Clin Lab Sci (2019) 56(8):567–85. doi: 10.1080/10408363.2019.1660303
105. Routy B, Le Chatelier E, Derosa L, Duong CPM, Alou MT, Daillère R, et al. Gut Microbiome Influences Efficacy of PD-1-based Immunotherapy Against Epithelial Tumors. Sci (80- ) (2018) 359(6371):91–7. doi: 10.1126/science.aan3706
106. Spanogiannopoulos P, Bess EN, Carmody RN, Turnbaugh PJ. The Microbial Pharmacists Within Us: A Metagenomic View of Xenobiotic Metabolism. Nat Rev Microbiol (2016) 14:273–87. doi: 10.1038/nrmicro.2016.17
107. Sivanandam V, LaRocca CJ, Chen NG, Fong Y, Warner SG. Oncolytic Viruses and Immune Checkpoint Inhibition: The Best of Both Worlds. Mol Ther - Oncolytics (2019) 13:93–106. doi: 10.1016/j.omto.2019.04.003
108. LaRocca CJ, Warner SG. Oncolytic Viruses and Checkpoint Inhibitors: Combination Therapy in Clinical Trials. Clin Transl Med (2018) 7(1):1–3. doi: 10.1186/s40169-018-0214-5
109. Ribas A, Dummer R, Puzanov I, VanderWalde A, Andtbacka RHI, Michielin O, et al. Oncolytic Virotherapy Promotes Intratumoral T Cell Infiltration and Improves Anti-PD-1 Immunotherapy. Cell (2017) 170(6):1109–19. doi: 10.1016/j.cell.2017.08.027
110. Pembrolizumab With or Without Talimogene Laherparepvec or Talimogene Laherparepvec Placebo in Unresected Melanoma (Keynote-034) - Full Text View. ClinicalTrials.gov.
111. Andtbacka RHI, Ross MI, Agarwala SS, Taylor MH, Vetto JT, Neves RI, et al. Final Results of a Phase II Multicenter Trial of HF10, a Replication-Competent HSV-1 Oncolytic Virus, and Ipilimumab Combination Treatment in Patients With Stage IIIB-IV Unresectable or Metastatic Melanoma. J Clin Oncol (2017) 35(15_suppl):9510–0. doi: 10.1200/JCO.2017.35.15_suppl.9510
112. Combination Adenovirus + Pembrolizumab to Trigger Immune Virus Effects - Full Text View. ClinicalTrials.gov.
113. Haddad D. Genetically Engineered Vaccinia Viruses as Agents for Cancer Treatment, Imaging, and Transgene Delivery. Front Oncol (2017) 77:96. doi: 10.3389/fonc.2017.00096
114. Anthoney A, Samson A, West E, Turnbull SJ, Scott K, Tidswell E, et al. Single Intravenous Preoperative Administration of the Oncolytic Virus Pexa-Vec to Prime Anti-Tumor Immunity. J Clin Oncol (2018) 36(15_suppl):3092–2. doi: 10.1200/JCO.2018.36.15_suppl.3092
115. A Phase I/Ii Study of Pexa-Vec Oncolytic Virus in Combination With Immune Checkpoint Inhibition in Refractory Colorectal Cancer - Full Text View. ClinicalTrials.gov.
116. Immunization Strategy With Intra-Tumoral Injections of Pexa-Vec With Ipilimumab in Metastatic / Advanced Solid Tumors. - Full Text View. ClinicalTrials.gov.
117. Intratumoral CAVATAK (CVA21) and Ipilimumab in Patients With Advanced Melanoma (Vla-013 MITCI) - Full Text View. ClinicalTrials.gov.
118. Silk AW, Kaufman H, Gabrail N, Mehnert J, Bryan J, Norrell J, et al. Abstract CT026: Phase 1b Study of Intratumoral Coxsackievirus A21 (C V A 21) and Systemic P Emb R Olizumab in a Dvanced Melanoma Patients: Interim Results of the CAPRA Clinical Trial. Am Assoc Cancer Res (AACR) (2017) 77(13 Suppl):CT026–6. doi: 10.1158/1538-7445.AM2017-CT026
119. Mahalingam D, Fountzilas C, Moseley JL, Noronha N, Cheetham K, Dzugalo A, et al. A Study of REOLYSIN in Combination With Pembrolizumab and Chemotherapy in Patients (pts) With Relapsed Metastatic Adenocarcinoma of the Pancreas (MAP). J Clin Oncol (2017) 35(15_suppl):e15753–3. doi: 10.1200/JCO.2017.35.15_suppl.e15753
120. Hanna GG, Coyle VM, Prise KM. Immune Modulation in Advanced Radiotherapies: Targeting Out-of-Field Effects. Cancer Lett (2015) 368:246–51. doi: 10.1016/j.canlet.2015.04.007
121. Frey B, Rückert M, Deloch L, Rühle PF, Derer A, Fietkau R, et al. Immunomodulation by Ionizing Radiation—Impact for Design of Radio-Immunotherapies and for Treatment of Inflammatory Diseases. Immunol Rev Blackwell Publishing Ltd (2017) 280:231–48. doi: 10.1111/imr.12572
122. Menon H, Ramapriyan R, Cushman TR, Verma V, Kim HH, Schoenhals JE, et al. Role of Radiation Therapy in Modulation of the Tumor Stroma and Microenvironment. Front Immunol (2019) 10:193. doi: 10.3389/fimmu.2019.00193
123. Trommer M, Yeo SY, Persigehl T, Bunck A, Grüll H, Schlaak M, et al. Abscopal Effects in Radio-Immunotherapy-Response Analysis of Metastatic Cancer Patients With Progressive Disease Under anti-PD-1 Immune Checkpoint Inhibition. Front Pharmacol (2019) 10:511. doi: 10.3389/fphar.2019.00511
124. Golden EB, Demaria S, Schiff PB, Chachoua A, Formenti SC. An Abscopal Response to Radiation and Ipilimumab in a Patient With Metastatic Non-Small Cell Lung Cancer. Cancer Immunol Res (2013) 1(6):365–72. doi: 10.1158/2326-6066.CIR-13-0115
125. Slovin SF, Higano CS, Hamid O, Tejwani S, Harzstark A, Alumkal JJ, et al. Ipilimumab Alone or in Combination With Radiotherapy in Metastatic Castration-Resistant Prostate Cancer: Results From an Open-Label, Multicenter Phase I/Ii Study. Ann Oncol (2013) 24(7):1813–21. doi: 10.1093/annonc/mdt107
126. Ling DC, Bakkenist CJ, Ferris RL, Clump DA. Role of Immunotherapy in Head and Neck Cancer. Semin Radiat Oncol (2018) 28:12–6. doi: 10.1016/j.semradonc.2017.08.009
127. Hoimes CJ, Rosenberg JE, Srinivas S, Petrylak DP, Milowsky M, Merchan JR, et al. Ev-103: Initial Results of Enfortumab Vedotin Plus Pembrolizumab for Locally Advanced or Metastatic Urothelial Carcinoma. OncologyPRO. Available at: https://oncologypro.esmo.org/Meeting-Resources/ESMO-2019-Congress/EV-103-Initial-results-of-enfortumab-vedotin-plus-pembrolizumab-for-locally-advanced-or-metastatic-urothelial-carcinoma.
128. Rischin D, Groenland SL, Lim AM, Martin-Liberal J, Moreno V, Perez JT, et al. Inducible T Cell Costimulatory (Icos) Receptor Agonist, GSK3359609 (GSK609) Alone and in Combination With Pembrolizumab (Pembro): Preliminary Resul. OncologyPRO. Available at: https://oncologypro.esmo.org/Meeting-Resources/ESMO-2019-Congress/Inducible-T-cell-Costimulatory-ICOS-Receptor-Agonist-GSK3359609-GSK609-alone-and-in-combination-with-Pembrolizumab-pembro-preliminary-results-from-INDUCE-1-expansion-cohorts-EC-in-Head-and-Neck-Squamous-Cell-Carcinoma-HNSCC.
129. Janjigian YY, Maron SB, Chatila WK, Millang B, Chavan SS, Alterman C, et al. First-Line Pembrolizumab and Trastuzumab in HER2-positive Oesophageal, Gastric, or Gastro-Oesophageal Junction Cancer: An Open-Label, Single-Arm, Phase 2 Trial. Lancet Oncol (2020) 21(6):821–31. doi: 10.1016/S1470-2045(20)30169-8
130. Narita Y, Kadowaki S, Muro K. Immune Checkpoint Inhibitor Plus Anti-HER2 Therapy: A New Standard for HER2-positive Oesophagogastric Cancer? Lancet Oncol (2020) 21:741–3. doi: 10.1016/S1470-2045(20)30208-4
131. Chan LC, Li CW, Xia W, Hsu JM, Lee HH, Cha JH, et al. Il-6/JAK1 Pathway Drives PD-L1 Y112 Phosphorylation to Promote Cancer Immune Evasion. J Clin Invest (2019) 1129(8):3324–38. doi: 10.1172/JCI126022
132. Wang Z, Liu Y, Zhang Y, Shang Y, Gao Q. MDSC-Decreasing Chemotherapy Increases the Efficacy of Cytokine-Induced Killer Cell Immunotherapy in Metastatic Renal Cell Carcinoma and Pancreatic Cancer. Oncotarget (2016) 7(4):4760–9. doi: 10.18632/oncotarget.6734
133. Weber R, Fleming V, Hu X, Nagibin V, Groth C, Altevogt P, et al. Myeloid-Derived Suppressor Cells Hinder the Anti-Cancer Activity of Immune Checkpoint Inhibitors. Front Immunol (2018) 99:1310. doi: 10.3389/fimmu.2018.01310
134. Tesi RJ. MDSC; the Most Important Cell You Have Never Heard of. Trends Pharmacol Sci (2019) 40:4–7. doi: 10.1016/j.tips.2018.10.008
135. Fleming V, Hu X, Weber R, Nagibin V, Groth C, Altevogt P, et al. Targeting Myeloid-Derived Suppressor Cells to Bypass Tumor-Induced Immunosuppression. Front Immunol (2018) 99:398. doi: 10.3389/fimmu.2018.00398
136. Luginbuhl AJ, Johnson JM, Harshyne L, Tuluc M, Mardekian S, Leiby BE, et al. A Window of Opportunity Trial of Preoperative Nivolumab With or Without Tadalafil in Squamous Cell Carcinoma of the Head and Neck (SCCHN): Safety. OncologyPRO. Available at: https://oncologypro.esmo.org/Meeting-Resources/ESMO-2019-Congress/A-window-of-opportunity-trial-of-preoperative-nivolumab-with-or-without-tadalafil-in-squamous-cell-carcinoma-of-the-head-and-neck-SCCHN-safety-clinical-and-correlative-outcomes.
137. Yan S, Zhang Y, Sun B. The Function and Potential Drug Targets of Tumour-Associated Tregs for Cancer Immunotherapy. Sci China Life Sci (2019) 62:179–86. doi: 10.1007/s11427-018-9428-9
138. Furukawa A, Wisel SA, Tang Q. Impact of Immune-Modulatory Drugs on Regulatory T Cell. Transplantation (2016) 100:2288–300. doi: 10.1097/TP.0000000000001379
139. König M, Rharbaoui F, Aigner S, Dälken B, Schüttrumpf J. Tregalizumab – A Monoclonal Antibody to Target Regulatory T Cells. Front Immunol (2016) 7:11. doi: 10.3389/fimmu.2016.00011
140. Ohue Y, Nishikawa H. Regulatory T (Treg) Cells in Cancer: Can Treg Cells be a New Therapeutic Target? Cancer Sci (2019) 110:2080–9. doi: 10.1111/cas.14069
141. Hanks BA, Holtzhausen A, Evans K, Heid M, Blobe GC. Combinatorial TGF-β Signaling Blockade and anti-CTLA-4 Antibody Immunotherapy in a Murine BRAF V600e -PTEN-/- Transgenic Model of Melanoma. J Clin Oncol (2014) 32(15_suppl):3011–1. doi: 10.1200/jco.2014.32.15_suppl.3011
142. Tauriello DVF, Palomo-Ponce S, Stork D, Berenguer-Llergo A, Badia-Ramentol J, Iglesias M, et al. Tgfβ Drives Immune Evasion in Genetically Reconstituted Colon Cancer Metastasis. Nature (2018) 554(7693):538–43. doi: 10.1038/nature25492
143. Holmgaard RB, Schaer DA, Li Y, Castaneda SP, Murphy MY, Xu X, et al. Targeting the TGFβ Pathway With Galunisertib, a TGFβRI Small Molecule Inhibitor, Promotes Anti-Tumor Immunity Leading to Durable, Complete Responses, as Monotherapy and in Combination With Checkpoint Blockade. J Immunother Cancer (2018) 6(1):1–5. doi: 10.1186/s40425-018-0356-4
144. Knudson KM, Hicks KC, Luo X, Chen JQ, Schlom J, Gameiro SR. M7824, a Novel Bifunctional Anti-PD-L1/TGFβ Trap Fusion Protein, Promotes Anti-Tumor Efficacy as Monotherapy and in Combination With Vaccine. Oncoimmunology (2018) 7(5):e1426519. doi: 10.1080/2162402X.2018.1426519
145. Principe DR, Park A, Dorman MJ, Kumar S, Viswakarma N, Rubin J, et al. TGFb Blockade Augments PD-1 Inhibition to Promote T-cell–mediated Regression of Pancreatic Cancer. Mol Cancer Ther (2019) 18(3):613–20. doi: 10.1158/1535-7163.MCT-18-0850
146. Mariathasan S, Turley SJ, Nickles D, Castiglioni A, Yuen K, Wang Y, et al. TGFβ Attenuates Tumour Response to PD-L1 Blockade by Contributing to Exclusion of T Cells. Nature (2018) 554(7693):544–8. doi: 10.1038/nature25501
147. Spranger S, Koblish HK, Horton B, Scherle PA, Newton R, Gajewski TF. Mechanism of Tumor Rejection With Doublets of CTLA-4, PD-1/PD-L1, or IDO Blockade Involves Restored IL-2 Production and Proliferation of CD8+ T Cells Directly Within the Tumor Microenvironment. J Immunother Cancer (2014) 2(1):1–4. doi: 10.1186/2051-1426-2-3
148. Holmgaard RB, Zamarin D, Munn DH, Wolchok JD, Allison JP. Indoleamine 2,3-Dioxygenase Is a Critical Resistance Mechanism in Antitumor T Cell Immunotherapy Targeting CTLA-4. J Exp Med (2013) 210(7):1389–402. doi: 10.1084/jem.20130066
149. Adding COX-2 Inhibition to Checkpoint Inhibitors Could Treat IDO1-Expressing Cancers. Available at: https://www.genengnews.com/topics/drug-discovery/adding-cox-2-inhibition-to-checkpoint-inhibitors-could-treat-ido1-expressing-cancers/.
150. Cox-2 Inhibitors may Reverse Ido1-Mediated Immunosuppression in Some Cancers. The ASCO Post. Available at: https://www.ascopost.com/News/57861.
151. Labadie BW, Bao R, Luke JJ. Reimagining IDO Pathway Inhibition in Cancer Immunotherapy Via Downstream Focus on the Tryptophan–Kynurenine–Aryl Hydrocarbon Axis. Clin Cancer Res (2019) 25:1462–71. doi: 10.1158/1078-0432.CCR-18-2882
152. Kim C, Kim JH, Kim JS, Chon HJ, Kim J-H. A Novel Dual Inhibitor of IDO and TDO, CMG017, Potently Suppresses the Kynurenine Pathway and Overcomes Resistance to Immune Checkpoint Inhibitors. J Clin Oncol (2019) 37(15_suppl):e14228–8. doi: 10.1200/JCO.2019.37.15_suppl.e14228
153. Cortez-Retamozo V, Etzrodt M, Newton A, Ryan R, Pucci F, Sio SW, et al. Angiotensin II Drives the Production of Tumor-Promoting Macrophages. Immunity (2013) 38(2):296–308. doi: 10.1016/j.immuni.2012.10.015
154. Lin C, Datta V, Okwan-Duodu D, Chen X, Fuchs S, Alsabeh R, et al. Angiotensin-Converting Enzyme Is Required for Normal Myelopoiesis. FASEB J (2011) 25(4):1145–55. doi: 10.1096/fj.10-169433
155. George AJ, Thomas WG, Hannan RD. The Renin–Angiotensin System and Cancer: Old Dog, New Tricks. Nat Rev Cancer (2010) 10(11):745–59. doi: 10.1038/nrc2945
156. Bader M. Tissue Renin-Angiotensin-Aldosterone Systems: Targets for Pharmacological Therapy. Annu Rev Pharmacol Toxicol (2010) 50(1):439–65. doi: 10.1146/annurev.pharmtox.010909.105610
157. Pinter M, Jain RK. Targeting the Renin-Angiotensin System to Improve Cancer Treatment: Implications for Immunotherapy. Sci Trans Med Am Assoc Advancement Sci; (2017) 9(410):1–11. doi: 10.1126/scitranslmed.aan5616
158. Kamiya A, Hayama Y, Kato S, Shimomura A, Shimomura T, Irie K, et al. Genetic Manipulation of Autonomic Nerve Fiber Innervation and Activity and its Effect on Breast Cancer Progression. Nat Neurosci (2019) 22(8):1289–305. doi: 10.1038/s41593-019-0430-3
159. Wrobel LJ, Bod L, Lengagne R, Kato M, Prévost-Blondel A, Gal FA Le. Propranolol Induces a Favourable Shift of Anti-Tumor Immunity in a Murine Spontaneous Model of Melanoma. Oncotarget (2016) 7(47):77825–37. doi: 10.18632/oncotarget.12833
160. Patel VG, Oh WK, Galsky MD, Liaw BC-H, Tsao C-K. Effect of Concurrent Beta-Blocker (BB) Use in Patients Receiving Immune Checkpoint Inhibitors for Metastatic Urothelial (mUC) and Renal Cell Carcinomas (mRCC). J Clin Oncol (2019) 37(7_suppl):467–7. doi: 10.1200/JCO.2019.37.7_suppl.467
161. Kokolus KM, Zhang Y, Sivik JM, Schmeck C, Zhu J, Repasky EA, et al. Beta Blocker Use Correlates With Better Overall Survival in Metastatic Melanoma Patients and Improves the Efficacy of Immunotherapies in Mice. Oncoimmunology (2018) 7(3):e1405205. doi: 10.1080/2162402X.2017.1405205
162. Sahra IB, Marchand-Brustel YL, Tanti JF, Bost F. Metformin in Cancer Therapy: A New Perspective for an Old Antidiabetic Drug? Mol Cancer Ther (2010) 9:1092–9. doi: 10.1158/1535-7163.MCT-09-1186
163. Bodmer M, Meier C, Krähenbühl S, Jick SS, Meier CR. Long-Term Metformin Use Is Associated With Decreased Risk of Breast Cancer. Diabetes Care (2010) 33(6):1304–8. doi: 10.2337/dc09-1791
164. Zakikhani M, Dowling R, Fantus IG, Sonenberg N, Pollak M. Metformin Is an AMP Kinase-Dependent Growth Inhibitor for Breast Cancer Cells. Cancer Res (2006) 66(21):10269–73. doi: 10.1158/0008-5472.CAN-06-1500
165. Eikawa S, Nishida M, Mizukami S, Yamazaki C, Nakayama E, Udono H. Immune-Mediated Antitumor Effect by Type 2 Diabetes Drug, Metformin. Proc Natl Acad Sci USA (2015) 112(6):1809–14. doi: 10.1073/pnas.1417636112
166. Levy A, Doyen J. Metformin for non-Small Cell Lung Cancer Patients: Opportunities and Pitfalls. Crit Rev Oncol/Hematol (2018) 125:41–7. doi: 10.1016/j.critrevonc.2018.03.001
167. Song CW, Lee H, Dings RPM, Williams B, Powers J, Dos ST, et al. Metformin Kills and Radiosensitizes Cancer Cells and Preferentially Kills Cancer Stem Cells. Sci Rep (2012) 2(1):1–9. doi: 10.1038/srep00362
168. Richa T, Johnson JM, Cognetti DM, Argiris A, Luginbuhl A, Zinner R, et al. Window of Opportunity for Durvalumab (MEDI4736) Plus Metformin Trial in Squamous Cell Carcinoma of the Head and Neck (SCCHN): Interim Safety Analysis. OncologyPRO. Available at: https://oncologypro.esmo.org/Meeting-Resources/ESMO-2019-Congress/Window-of-Opportunity-for-Durvalumab-MEDI4736-plus-Metformin-Trial-in-Squamous-Cell-Carcinoma-of-the-Head-and-Neck-SCCHN-interim-safety-analysis.
169. Marin-Acevedo JA, Dholaria B, Soyano AE, Knutson KL, Chumsri S, Lou Y. Next Generation of Immune Checkpoint Therapy in Cancer: New Developments and Challenges. J Hematol Oncol (2018) 1111(1):1–20. doi: 10.1186/s13045-018-0582-8
170. Zappasodi R, Sirard C, Li Y, Budhu S, Abu-Akeel M, Liu C, et al. Rational Design of anti-GITR-based Combination Immunotherapy. Nat Med (2019) 25:759–66. doi: 10.1038/s41591-019-0420-8
171. Kaesler S, Wölbing F, Kempf WE, Skabytska Y, Köberle M, Volz T, et al. Targeting Tumor-Resident Mast Cells for Effective Anti-Melanoma Immune Responses. JCI Insight (2019) 4(19):e125057. doi: 10.1172/jci.insight.125057
172. Lv Y, Zhao Y, Wang X, Chen N, Mao F, Teng Y, et al. Increased Intratumoral Mast Cells Foster Immune Suppression and Gastric Cancer Progression Through TNF-α-PD-L1 Pathway. J Immunother Cancer (2019) 7(1):54. doi: 10.1186/s40425-019-0530-3
173. Meraz IM, Majidi M, Meng F, Shao RP, Ha MJ, Neri S, et al. An Improved Patient-Derived Xenograft Humanized Mouse Model for Evaluation of Lung Cancer Immune Responses. Cancer Immunol Res (2019) 7(8):1267–79. doi: 10.1158/2326-6066.CIR-18-0874
174. Koga Y, Ochiai A. Systematic Review of Patient-Derived Xenograft Models for Preclinical Studies of Anti-Cancer Drugs in Solid Tumors. Cells (2019) 8(5):418. doi: 10.3390/cells8050418
175. Vlachogiannis G, Hedayat S, Vatsiou A, Jamin Y, Fernández-Mateos J, Khan K, et al. Patient-Derived Organoids Model Treatment Response of Metastatic Gastrointestinal Cancers Europe PMC Funders Group. Sci (80- ) (2018) 359(6378):920–6. doi: 10.1126/science.aao2774
176. Di Modugno F, Colosi C, Trono P, Antonacci G, Ruocco G, Nisticò P. 3D Models in the New Era of Immune Oncology: Focus on T Cells, CAF and ECM. J Exp Clin Cancer Res (2019) 38:1–14. doi: 10.1186/s13046-019-1086-2
177. Lee SH, Hu W, Matulay JT, Silva MV, Owczarek TB, Kim K, et al. Tumor Evolution and Drug Response in Patient-Derived Organoid Models of Bladder Cancer. Cell (2018) 173(2):515–28.e17. doi: 10.1016/j.cell.2018.03.017
178. Pauli C, Hopkins BD, Prandi D, Shaw R, Fedrizzi T, Sboner A, et al. Personalized In Vitro and In Vivo Cancer Models to Guide Precision Medicine. Cancer Discov (2017) 7(5):462–77. doi: 10.1158/2159-8290.CD-16-1154
179. Yuan H, Cai P, Li Q, Wang W, Sun Y, Xu Q, et al. Axitinib Augments Antitumor Activity in Renal Cell Carcinoma Via STAT3-dependent Reversal of Myeloid-Derived Suppressor Cell Accumulation. BioMed Pharmacother (2014) 68(6):751–6. doi: 10.1016/j.biopha.2014.07.002
180. López-Soto A, Folgueras AR, Seto E, Gonzalez S. HDAC3 Represses the Expression of NKG2D Ligands ULBPs in Epithelial Tumour Cells: Potential Implications for the Immunosurveillance of Cancer. Oncogene (2009) 28(25):2370–82. doi: 10.1038/onc.2009.117
181. Roulois D, Yau HL, De Carvalho DD. Pharmacological DNA Demethylation: Implications for Cancer Immunotherapy. Oncoimmunology (2016) 5(3):e1090077. doi: 10.1080/2162402X.2015.1090077
182. Califano JA, Khan Z, Noonan KA, Rudraraju L, Zhang Z, Wang H, et al. Tadalafil Augments Tumor Specific Immunity in Patients With Head and Neck Squamous Cell Carcinoma. Clin Cancer Res (2015) 21(1):30–8. doi: 10.1158/1078-0432.CCR-14-1716
183. Immunotherapy: Precision Medicine in Action. Johns Hopkins Medicine. Available at: https://www.hopkinsmedicine.org/inhealth/old-template/policy-briefs/immunotherapy-precision-medicine-action-policy-brief.html.
Keywords: translational research, immune checkpoint inhibitors (ICI), preclinical model, clinical trials, combination strategies, immunotherapy adjuncts
Citation: Varayathu H, Sarathy V, Thomas BE, Mufti SS and Naik R (2021) Combination Strategies to Augment Immune Check Point Inhibitors Efficacy - Implications for Translational Research. Front. Oncol. 11:559161. doi: 10.3389/fonc.2021.559161
Received: 05 May 2020; Accepted: 30 April 2021;
Published: 28 May 2021.
Edited by:
William Valentine Williams, BriaCell Therapeutics Corp., United StatesReviewed by:
Jacques Barbet, Arronax, FranceCopyright © 2021 Varayathu, Sarathy, Thomas, Mufti and Naik. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hrishi Varayathu, aHJpc2hpdmFyYXlhdGh1QGdtYWlsLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.