 Ruochen Liu1,2,3
Ruochen Liu1,2,3 Pengfei Shi
Pengfei Shi Zhongze Wang
Zhongze Wang Hongjuan Cui
Hongjuan Cui- 1State Key Laboratory of Silkworm Genome Biology, College of Sericulture, Textile and Biomass Sciences, Southwest University, Chongqing, China
- 2Cancer Center, Reproductive Medicine Center, Medical Research Institute, Southwest University, Chongqing, China
- 3NHC Key Laboratory of Birth Defects and Reproductive Health (Chongqing Key Laboratory of Birth Defects and Reproductive Health, Chongqing Population and Family Planning Science and Technology Research Institute), Chongqing, China
MYCN, a member of MYC proto-oncogene family, encodes a basic helix-loop-helix transcription factor N-MYC. Abnormal expression of N-MYC is correlated with high-risk cancers and poor prognosis. Initially identified as an amplified oncogene in neuroblastoma in 1983, the oncogenic effect of N-MYC is expanded to multiple neuronal and nonneuronal tumors. Direct targeting N-MYC remains challenge due to its “undruggable” features. Therefore, alternative therapeutic approaches for targeting MYCN-driven tumors have been focused on the disruption of transcription, translation, protein stability as well as synthetic lethality of MYCN. In this review, we summarize the latest advances in understanding the molecular mechanisms of MYCN dysregulation in cancers.
Introduction
N-MYC is a transcription factor of the MYC oncogene family. This gene family of humans consists of three members, namely, MYCC, MYCN, MYCL, which encodes C-MYC, N-MYC, and L-MYC protein respectively (“MYC” was used to indicate all three genes in this review). The first identified MYC gene was MYCC as a homolog of an avian retroviral gene v-myc, then MYCN in neuroblastoma and MYCL in lung cancer (1–3). These proteins show similar structure with the highest homology in five short stretches called MYC boxes 1 to 4 at the N terminus and in the basic helix-loop-helix-leucine-zipper (bHLH-LZ) domain at the C terminus (Figure 1A) (6–9). The former enables MYC to interact with different effector proteins including TRRAP and P400 which mediate chromatin remodeling and modification (10, 11), the latter allows MYC to form a heterodimer with partner proteins that also contain a bHLH-LZ domain, such as MAX. MYC/MAX heterodimer bind to the target motif called E-box with the consensus sequence of CAC(G/A)TG to regulate the expression of targeted genes (Figure 1B). In addition, MYC can also bind to targeted sequences that show deviation from or no similarity to the E-box, suggesting the association of MYC to chromatin can be instructed by other factors (12, 13). For example, MYC can invade promoter regions of active genes and cause global transcriptional amplification (Figure 1C) (4, 14, 15). The two different action modes of MYC seem conflicting, i.e., gene-specific regulation model versus global gene activation model. The third model, gene-specific affinity model, in which the affinity of promoters for MYC is different and relies on the MYC levels and the interaction of MYC with core promoter-binding factors, such as WDR5 (Figure 1D), has been proposed to reconcile the action modes of MYC (5, 16). MYC proteins affect transcription of a large number of genes and thus regulate fundamental cellular processes, including proliferation, metabolism, apoptosis, differentiation, and immune surveillance (17–21).
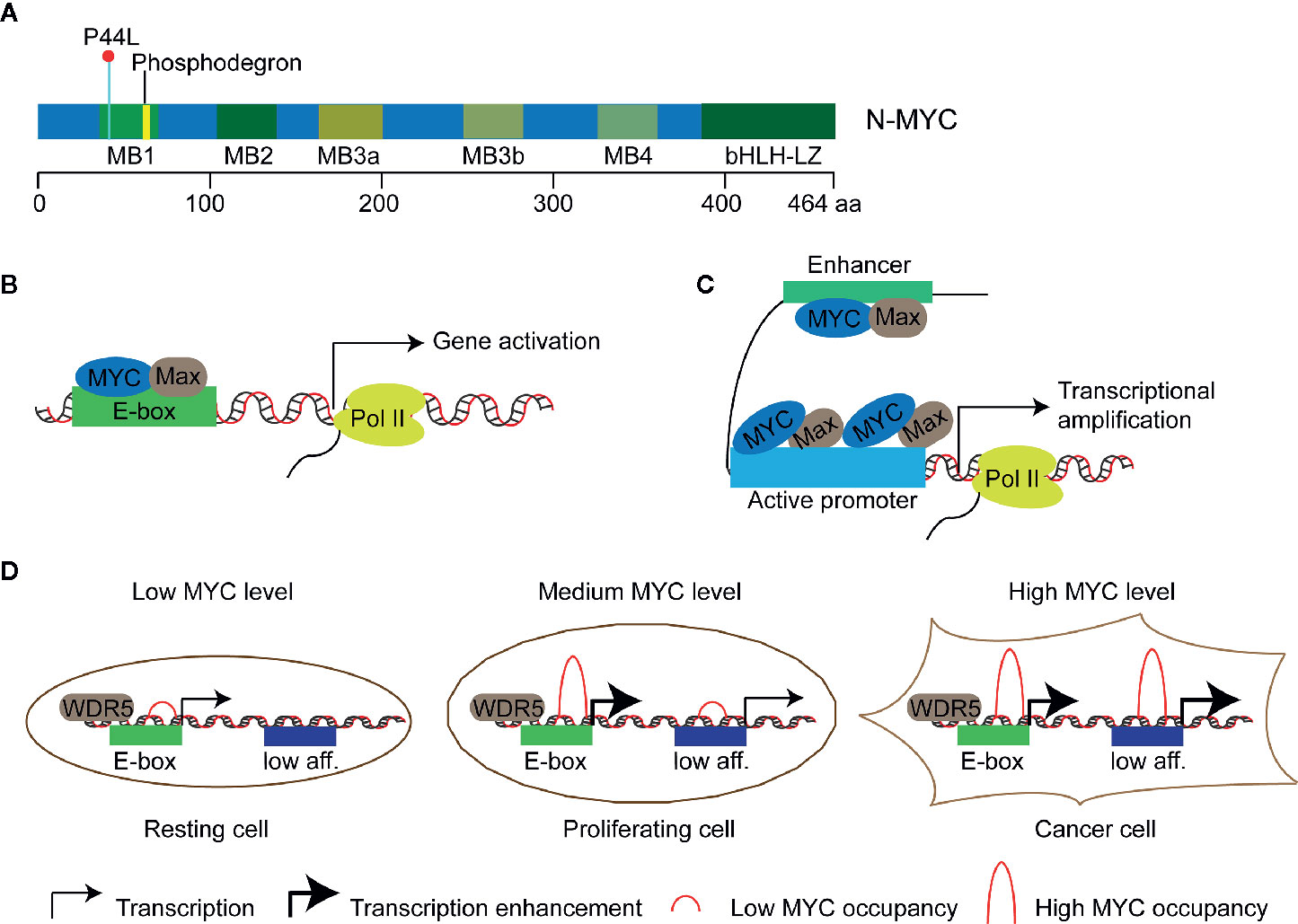
Figure 1 Models of transcriptional regulation of target genes by MYC proteins. (A) Schematic diagram of N-MYC protein structure. Five highly conserved stretched called MYC boxes 1 to 4 (MB) and the basic helix-loop-helix-leucine-zipper (bHLH-LZ) domain at the C terminus are shown. The recurrent somatic mutation P44L and the putative N-MYC phosphodegron are shown in cyanine and yellow respectively. (B) Gene-specific regulation model: MYC/Max dimer binds and regulates a subset of genes with E-boxes in their promoters. (C) Global gene activation model: MYC accumulates in the promoter regions of active genes independent of E-box and leads to transcriptional amplification in cancer cells with high level of MYC proteins (4). (D) Gene-specific affinity model: high-affinity binding sites, such as those with E-boxes and WDR5 (WD-repeat protein 5) binding, are already fully occupied by MYC at physiological MYC protein level (medium level) in proliferating cells; low-affinity (low aff.) binding sites can be occupied by MYC at oncogenic MYC protein level (high level) in cancer cells (5).
With evolutionarily conserved domains, the three MYC proteins share certain extent of functional redundancy. For instance, when N-MYC is expressed from the MYCC locus, it can rescue development, cellular growth, and differentiation in MYCC deficient mice (22). On the other hand, C-MYC, N-MYC, and L-MYC have their own unique features. Enhanced expression of different MYC paralogs induces tumors with different biological characteristics in medulloblastoma (23, 24), prostate cancer (25), and lung cancer (26). Furthermore, the amplification of MYC genes is mutually exclusive, and the switch of gene expression among the members is associated with cell lineage shift, tumor progression, and treatment resistance (27, 28). Different collaborative proteins of MYC paralogs help to demarcate a unique subset of responsive genes, which could partially explain the distinct biological functions among MYC members. For example, N-MYC interacts with TWIST1 at enhancers to activate developmental genes important to neuroblastoma tumorigenesis, while TCF3 (E2A) is selectively required for progression of C-MYC driven myeloma (15). In this mini-review, we focus on N-MYC-driven tumors. Since discovered in 1983 in neuroblastoma (1, 3), the oncogenic effect of N-MYC has been demonstrated both in various neuronal [e.g., glioblastoma (29), medulloblastoma (30), astrocytoma (31)], and nonneuronal [e.g., prostate cancers (32), breast cancers (33), hematologic malignancies (34), pancreatic tumors (35), Wilms tumors (36), hepatocellular carcinoma (37), rhabdomyosarcoma (38), ovarian cancers (39)] tumors. Specifically, this mini-review summarizes the latest advances in the regulation network of N-MYC expression (Figure 2) and the related therapeutic targets for MYCN-driven tumors.
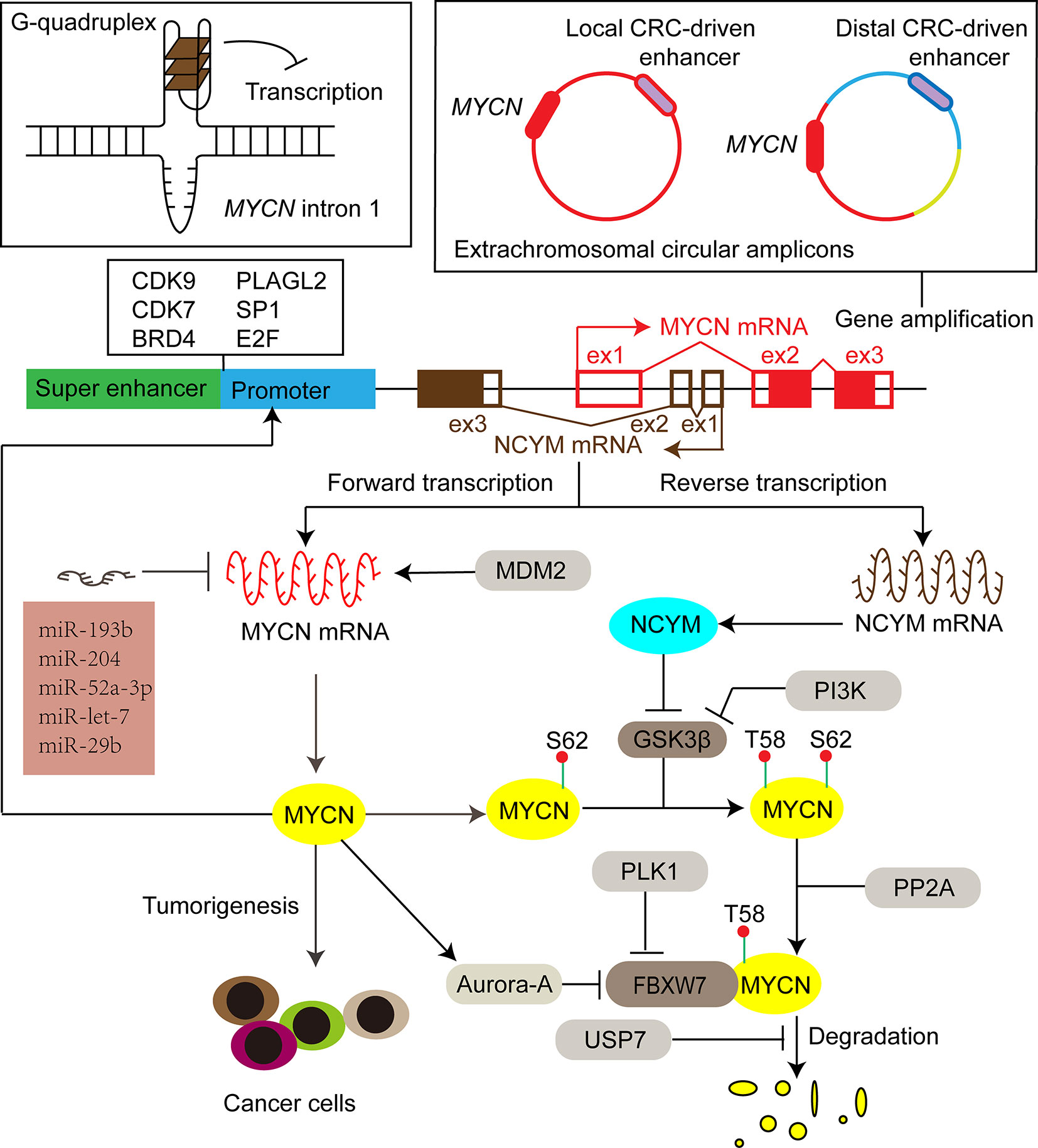
Figure 2 The expression of MYCN is activated or repressed at DNA, mRNA and protein levels by different factors, including secondary DNA structure, enhancers, transcription factors, miRNAs, ubiquitination-dependent proteasome degradation machinery and its cis-antisense gene NCYM. Filled red and brown boxes indicate translated regions of MYCN and NCYM respectively, while the blank counterparts represent untranslated regions. CRC core regulatory circuitry, CDK9 cyclin-dependent kinase 9, CDK7 cyclin-dependent kinase 7, BRD4 bromodomain-containing 4, PLAGL2 pleiomorphic adenoma gene-like 2, SP1 specific protein 1, GSK3β glycogen synthase kinase 3β, PI3K phosphoinositide 3-kinase, FBXW7 F-box and WD repeat domain-containing 7, PP2A protein phosphatase 2A, PLK1 polo-like kinase 1, USP7 ubiquitin-specific protease 7, MDM2 murine double minute 2.
Molecular Mechanisms of MYCN Dysregulation and the Therapeutic Targets
The tissue specificity and strength of MYC gene expression are under tight control in normal circumstances. Studies of mice show that the expression of MYCN is high during early developmental stages and in specific tissues including forebrain, hindbrain, and kidney of newborn mice, while MYCC is broadly expressed throughout the tissues and the developmental stages analyzed. Clinical observation of MYCN amplification in human neuroblastoma firstly pointed out the potential association between MYCN gene and tumorigenesis (1, 3). Although amplified DNAs encompassing MYCN are more than 100 kb and can include adjacent co-amplified genes, MYCN has emerged as the only consistently amplified gene (40). Using transgenic animal models, multiple studies establish that N-MYC overexpression is a driver of cancers. For example, targeted expression of human N-MYC causes neuroblastoma in transgenic mice and zebrafish (41, 42). Neuroblastomas with enhanced expression of N-MYC without MYCN amplification are known to be similarly high-risk and poor prognosis (43). Recent studies show that high N-MYC protein and RNA levels could be better biomarkers than MYCN gene amplification in predicting the prognosis of neuroblastoma patients (44, 45), underscoring the importance of aberrant expression of N-MYC in tumor progression. Here, we discuss mechanisms of MYCN dysregulation at DNA, mRNA and protein levels, and corresponding therapeutic targets.
Gene Amplification of MYCN
Gene amplification is a frequent mechanism that can cause proto-oncogene overexpression. It is a process that involves unscheduled DNA replication, recombination and/or formation of extrachromosomal DNA, leading to a selective increase of gene copy number up to several hundred (40). The occurrence of proto-oncogene amplification can be detected by the presence of “double minutes” or “homogeneously staining chromosomal regions”. MYCN was the first discovered paradigm of proto-oncogene amplification and is an important bio-marker to stratify clinical risk. It was initially detected in about 20% to 25% of neuroblastoma, then at a much lower incidence in small cell lung cancer, retinoblastoma, hepatocellular carcinoma, malignant gliomas, and peripheral neuroectodermal tumors (46, 47). Amplification of MYCN has been recognized as a consequence of genomic instability and occurs sporadically (48). Overexpression of N-MYC initiates tumorigenesis by preventing the normal physiological process of neural crest cell death in TH-MYCN transgenic mice in which human MYCN is under the control of a tyrosine hydroxylase (TH) promoter, and the formation of neuroblastoma involves further changes of the persisting embryonal neural crest cells, including MYCN amplification (49). In addition, MYCN amplification is associated with advanced neuroblastomas, suggesting that the amplification is a late event during the tumorigenesis (49–51)
Although multiple replication-based mechanisms, such as double rolling-circle replication, have been proposed to explain gene amplification, the important factors that induce and regulate MYCN amplification remain to be completely investigated (52–55). Proto-oncoprotein c-MYB transcription factor is implicated in the regulation of cell growth and proliferation of neuroblastoma (56). The functional ortholog of Drosophila melanogaster, Dm-Myb, is directly implicated in the site-specific DNA replication, leading to amplification of the chromosomal loci with the chorion gene cluster (57). Aygun and Altungoz showed that c-MYB is involved in the control of MYCN amplification in MYCN-amplified neuroblastoma cell lines (58). Specifically, the MYCN gene dosage is increased upon knockdown of c-MYB expression, which may be associated with the elevated expression of geminin protein that causes a shift from genomic DNA replication to MYCN amplification (58–60). Recent sequencing studies indicate that the structure of extrachromosomal MYCN amplicons are shaped by enhancer sequences (61, 62). Specifically, Helmsauer et al. reported two distinct classes of extrachromosomal circular MYCN amplicons: the first class co-amplifies a local core regulatory circuitry (CRC)-driven enhancer; the second class shows a complex chimeric structure with a distal CRC-driven enhancer instead of the local enhancer (Figure 2) (61). Long inverted repeats and microhomology are significantly associated with boundary regions of the MYCN amplicon units, and thus might also be involved in the initiation or regulation of MYCN amplification (55, 58). Elucidating the mechanisms of MYCN amplification may bring about new therapeutic strategies targeting MYCN amplification to treat MYCN-driven tumors.
Although the amplified genes tend to overexpress, gene amplification not necessarily leads to high level of gene expression. In fact, there is inconsistency between MYCN gene dosage, mRNA and protein levels, and clinical outcomes (44, 63). For example, low DNA dosage but high RNA level is detected in some neuroblastoma samples, while high DNA dosage but low RNA level in some other samples (45). Additional MYCN gene copies may also suppress their own expression (58). Genome-wide analysis in humans and some model organisms revealed that genes in copy number variation regions are expressed at lower and more variable levels than genes mapped elsewhere (64). Alternatively, as in plants, repeated genes may suffer from homology-dependent gene silencing that involves DNA methylation or histone modification (65, 66). Consistently, only a weak positive correlation of MYCN expression with copy number is detected in Wilms tumor, while a strong negative correlation of MYCN expression with DNA methylation level at specific loci is observed (67). Importantly, transcriptional and posttranscriptional regulation determines the final level of N-MYC protein in both MYCN amplified and non-amplified tumors. For instance, enhancer hijacking that repositions a super enhancer close to the affected genes through chromosomal translocation accounts for the high level of C-MYC or N-MYC expression in some neuroblastoma cells without MYCC amplification or without a high MYCN copy number, respectively (68, 69).
Regulation of MYCN Transcription
Super Enhancer and Transcription Factors
A general feature of MYC genes is their transcriptional regulation by upstream super enhancers (SEs) (70). SE regions are occupied by abundant transcription factors, cofactors, and chromatin regulators, thereby promoting transcription of MYC genes (71). Specifically, H3K27 acetylation (H3K27ac), a marker of active enhancers and promoters, is enriched in the SE regions and recognized by BRD4 of bromodomain and extra-terminal domain (BET) protein family that recruits positive transcription elongation factor b (P-TEFb) to the promoters to phosphorylate RNA polymerase II, and thus facilitates transcriptional initiation, pause release and elongation (72–74). BET inhibitors, such as JQ1 and OTX015, can displace the BRD4 oncoprotein from chromatin (75), which potently represses MYCN transcription in neuroblastoma cell lines and effectively reduces neuroblastoma cell viability in vitro and in vivo (76, 77). It has been reported that the toxic effects of BET inhibitors depend on p53 (78). The combination of MDM2 (an E3-ubiquitin ligase involved in proteasomal degradation of p53) inhibitor (CGM097) and OTX015 results in p53 activation and decreased expression of MYC proteins, which synergistically promotes neuroblastoma cell death (79). A recent study shows that triple-negative breast cancer (TNBC) cells with high expression of MYCN are also sensitive to BET inhibitors (80). Furthermore, combined BET and MEK inhibition synergistically represses the growth of MYCN-expressing patient-derived xenograft TNBC tumors (80).
Besides BET proteins, transcriptional cyclin-dependent kinases (CDKs) are recruited to SEs, especially CDK7, a catalytic subunit of the transcription factor IIH complex (TFIIH), and CDK9, a kinase subunit of P-TEFb (81, 82). These CDKs regulate the transcriptional cycle of RNA polymerase II via phosphorylating the C-terminal domain of the polymerase, which enhances expression of SE-associated oncogenes, such as MYCN (83–85). A covalent inhibitor of CDK7, THZ1, selectively targets MYCN-amplified neuroblastoma cells, leading to global suppression of MYCN-dependent transcriptional amplification and sustained growth inhibition of tumors in a mouse model of neuroblastoma (85). CYC065 (fadraciclib), a clinical inhibitor of CDK9 and CDK2 (a major regulator of apoptotic cell death), selectively targets MYCN-amplified neuroblastoma through a loss of MYCN transcription and growth arrest, followed by sensitizing cells for apoptosis as a result of CDK2 inhibition (86). Furthermore, the combined use of CYC065 with temozolomide (a reference therapy for relapsed neuroblastoma), leads to long-term repression of neuroblastoma growth in vivo (86).
Recent studies reveal that several super-enhancer harboring transcription factors including HAND2, ISL1, PHOX2B, GATA3, and TBX2 constitute a CRC that is essential for the MYCN expression and the survival of MYCN-amplified neuroblastoma cells (61, 87). BRD4 and CDK7 inhibitors synergistically repress the expression of all the CRC transcription factors and N-MYC, which inhibits neuroblastoma cell growth (87). Knockdown of each CRC transcription factors also suppresses the expression of MYCN (87). Interestingly, the CRC-driven enhancers (local or distal) are associated with extrachromosomal circular MYCN amplicons (Figure 2) (61), underscoring the role of the CRC transcription factors in the regulation of MYCN expression.
Other transcription factors, such as specific protein 1 (SP1) (88), E2F (89), and pleiomorphic adenoma gene-like 2 (PLAGL2) (90), participate in the regulation of MYCN expression. The three transcription factors directly bind to the cognate binding sites in the MYCN promoter, contributing to MYCN activation. Moreover, N-MYC regulates PLAGL2 transcription through five N-MYC-binding E-boxes in the PLAGL2 promoter region, forming a positively regulatory loop between the two transcription factors, which is crucial for expression of each other in neuroblastoma tumors (90). Lipid desaturation-associated endoplasmic reticulum (ER) stress inhibits MYCN expression via upregulating the transcriptional repressor ATF3 in hepatocellular carcinoma cells (91). Since these transcription factors including SP1, E2F2, and PLAGL2 are involved in the regulation of MYCN expression, they mediate the effects of metabolic change and pharmacological treatment on MYCN expression and MYCN-driven tumors (92). Aldehyde dehydrogenase family 18 member A1 (ALDH18A1) is a key enzyme for the synthesis of proline from glutamate and plays important role in the proliferation, self-renewal, and tumorigenicity of neuroblastoma cells (93). ALDH18A1 promotes the transcription of MYCN via the miR-29b/SP1 regulatory loop. ALDH18A1-specific inhibitor, YG1702, inhibits MYCN expression and attenuates the growth of human neuroblastoma (93). All-trans retinoic acids have been used for neuroblastoma therapy for decades by inhibiting the expression of MYCN and inducing the neuronal differentiation of neuroblastoma cells (94–96). Loss of E2F binding or suppression of PLAGL2 expression mediates the negative regulation of MYCN expression by retinoic acid (89, 90). Acyclic retinoid dampens MYCN gene expression and suppresses cell proliferation of MYCN-overexpressed hepatocellular carcinoma cells, at least in part by ER stress-induced ATF3 signaling pathway (91).
G-Quadruplex
Another feature of MYC genes is their transcriptional regulation by non-B DNA structures including single-stranded bubbles, Z-DNA, and G-quadruplexes (97). G-quadruplexes are four-stranded DNA secondary structures and consist of stacked G-quartets that formed by the assembly of four Hoogsteen hydrogen-bonded guanines in guanine-rich regions of DNA. A G-quadruplex forming sequence lies in the promoter of MYCC gene (98) and in intron 1 of MYCN gene (99) respectively. This sequence exists in equilibrium between transcriptionally active forms (double helical and single stranded) and a silenced form (G-quadruplex), which controls up to 90% of MYCC transcription (100). Thus, targeting MYC expression through G-quadruplex stabilization becomes an attractive candidate for the treatment of MYC-driven tumors. Cationic porphyrin TMPyP4 is a small molecule able to stabilize G-quadruplex structure and efficiently repress MYCC transcription, which establishes the principle that MYC transcription can be controlled by ligand-mediated G-quadruplex stabilization (98). A cell penetrating thiazole peptide, TH3, shows improved targeting specificity to MYCC G-quadruplex over other tested G-quadruplexes (100). This peptide down-regulates MYCC expression in cancer cells and reduces proliferative activities by inducing S phase cell cycle arrest and apoptosis (100). Nucleolin is a protein involved in the folding the G-quadruplex (101). Quarfloxin (CX-3543), a fluoroquinolone-based antitumor agent, can inhibit MYCC expression by redistribution of nucleolin from the nucleolus to the nucleoplasm to bind to MYCC G-quadruplex (102). Treating neuroblastoma cells with quarfloxin represses N-MYC expression and causes G2-cell cycle arrest and apoptosis (103). The most profound anti-tumor effects of quarfloxin are associated with MYCN amplification (103), implying the above drugs that target MYCC G-quadruplex can also be used to target MYCN G-quadruplex for treatment of MYCN-driven tumors.
Posttranscriptional Regulation of MYCN mRNA
Along with transcription factors, noncoding RNAs including long noncoding RNA (lncRNAs) and microRNAs (miRNAs) are involved in the regulatory network of MYCN expression. miR-506-3p is a potent differentiation inducer and a strong repressor of MYCN expression in neuroblastoma cells by targeting PLAGL2 transcription factor (90, 104). miR-204 directly binds MYCN mRNA, represses MYCN expression, and inhibits a subnetwork of oncogenes that strongly correlate with MYCN-amplified neuroblastoma and poor patient outcome (105). miR-193b targets several important oncogenes including MYCN and is expressed at low levels in neuroblastoma cell lines (106). MYCN mRNA is a direct target of miR-520c-3p in cholangiocarcinoma, and transcription factor SP1-induced lncRNA HOXD-AS1 enhances MYCN expression through competitively binding to miR-520c-3p, which associates with lymph node invasion, advanced TNM stage and poor prognosis (107). A miRNA network, consisting of miR-29b, miR-29a, and miR-193b, mediates posttranscriptional regulation of the MYCN expression by ALDH18A1 (93, 108). miRNA let-7 is a strong negative regulator of MYCN expression and can inhibit proliferation and clonogenic growth of MYCN-amplified neuroblastoma cells (108). LIN28B, an RNA-binding protein and a suppressor of microRNA biogenesis, selectively blocks the biogenesis of let-7 miRNA, consequently leading to increased MYCN expression in neuroblastoma cells (109). These results indicate that MYCN is targeted by several miRNAs. Increased expression of these miRNAs inhibits cell proliferation and tumorigenesis (105). Furthermore, miR-506-3p has been reported to mediate the antitumor effect of retinoic acid in neuroblastoma cells (90). These results underscore the potential of miRNA-based anticancer therapy. Interestingly, the E3-ubiquitin ligase MDM2 increases the MYCN mRNA stability and translation by binding to AU-rich elements of the 3′ UTR of MYCN mRNA through its C-terminal RING domain (110). RNAi-mediated knockdown of MDM2 leads to remarkable suppression of neuroblastoma cell growth and induction of cell death through a p53-independent pathway (110).
Regulation of MYCN Translation
Efficient translation guarantees the oncogenic level of N-MYC protein. N-MYC has been shown to promote the expression of many genes involved in ribosome biogenesis and protein synthesis (111), suggesting N-MYC contributes to its own overexpression by enhancing the capacity of translation. The N-MYC protein level is decreased as a result of ribosome biogenesis inhibition (103). Mammalian target of rapamycin (mTOR) is a serine/threonine protein kinase that controls initiation of protein translation (112). mTOR directly phosphorylates and inactivates eukaryotic translation initiation factor 4E (eIF4E)-binding protein 1 (4E-BP1), which leads to activation of eIF4E and thus promotes cap-dependent translation of mRNAs including MYC family (112). Pharmacological inhibition of the AKT/mTOR pathway reduces N-MYC level and exhibits therapeutic efficacy in MYCN-amplified neuroblastoma (113, 114).
Regulation of N-MYC Stability
After translation, the stability and activity of N-MYC protein are tightly controlled by ubiquitination-dependent proteasome degradation that is a brake in the MYCN-driven cancers. The degradation of the N-MYC proto-oncoprotein in neural stem/progenitor cells is required for the arrest of proliferation and the start of differentiation. Two E3 ubiquitin ligases FBXW7 and HUWE1 ubiquitinate N-MYC through Lys 48-mediated linkages and target it for destruction by the proteasome (115, 116). The recognition of N-MYC by FBXW7 involves several sequential reactions, i.e., phosphorylation on Ser62 by CDK1 (117), phosphorylation on Thr58 by glycogen synthase kinase 3β (GSK3β), dephosphorylation of Ser62 by protein phosphatase 2A (PP2A) (118), which facilitates the Thr58 phosphorylated N-MYC binding with FBXW7 (116).
Dysregulation of the degradation process will cause the accumulation of N-MYC protein to the oncogenic level. Aurora-A, a member of the Aurora kinase family, is identified in an shRNA screen of genes that are highly expressed in MYCN-amplified neuroblastoma cells and contributes to the stabilization of N-MYC (119). Mechanistically, the catalytic domain of Aurora-A interacts directly with N-MYC through binding sites that flank either side of MYC box 1 which contains the phosphodegron (Thr58) recognized by FBXW7, thereby preventing the binding of FBXW7 with N-MYC substrate (120). Furthermore, the expression of Aurora-A is increased in the MYCN-amplified neuroblastoma, suggesting a potential feed-forward loop that improves the stability of both proteins (121). Two Aurora-A kinase activity inhibitors, MLN8054 and MLN8237, disrupt the Aurora-A/N-MYC complex and promote FBXW7-mediated degradation of N-MYC, which correlates with tumor regression and prolonged survival in a mouse model of MYCN-driven neuroblastoma (122, 123). MLN8237 destabilizes N-MYC and synergizes with BCL2/BCLxL inhibitor (venetoclax or navitoclax) to kill MYCN-amplified tumor cells including neuroblastoma and rhabdomyosarcoma (124, 125). Since the degradation of N-MYC is regulated in part by a kinase-independent function of Aurora-A, CD532, a conformation-disrupting inhibitor of Aurora-A, acts as a more potent N-MYC inhibitor than the kinase activity inhibitor MLN8237 in neuroblastoma (126).
Polo-like kinase 1 (PLK1), a serine/threonine kinase that promotes G2/M-phase transformation, has an elevated expression level in high-risk neuroblastoma and is associated with poor prognosis of patients (127). PLK1 interacts with and phosphorylates FBXW7, promoting auto polyubiquitination and proteasomal degradation of FBXW7, which counteracts FBXW7-mediated degradation of N-MYC (128). In turn, stabilized N-MYC directly enhances the transcription of PLK1, forming a positive feedforward regulatory loop that reinforces the progress of MYCN-driven cancers. Inhibitors of PLK1, such as BI6727 and BI2356, preferentially trigger apoptosis of MYCN-amplified neuroblastoma and small cell lung cancer, and this therapeutic efficacy is synergistically enhanced by combined use with antagonists of anti-apoptotic B cell lymphoma 2 (BCL2) (128). UME103 and 9b, two novel dual PLK1 and BRD4 inhibitors, show better antitumor activity by inhibiting the transcription of MYCN gene and promoting the degradation of N-MYC protein (129, 130).
Ubiquitin-specific protease 7 (USP7) regulates the stability and activity of N-MYC in neuroblastoma (131). USP7 directly binds to N-MYC, deubiquitinates it, which preventing degradation of N-MYC by the 26S proteasome. The expression of USP7 is enhanced in patients of neuroblastoma with poorer prognosis. A small molecular inhibitor of USP’s deubiquitinase activity, P22077, destabilizes N-MYC, thereby markedly repressing the growth of MYCN-amplified human neuroblastoma cell lines in xenograft mouse models (131). Novel, selective inhibitors of USP7, USP7-055, and USP7-797, have been developed recently for tumor therapy including MYCN-amplified neuroblastoma (132).
NCYM, a cis-Antisense Gene of MYCN
An interesting feature of MYCN gene is its cis-antisense transcript called NCYM. NCYM was initially recognized as a large non-coding RNA (133, 134), while recent studies indicate it encodes a de novo evolved protein that promotes tumor progression (135). The transcription of NCYM begins from intron 1 of the MYCN gene in the opposite direction to that of the MYCN, ultimately generating NCYM protein with 109 amino acids (Figure 2) (135). As a cis-antisense gene of MYCN, NCYM is always co-amplified with MYCN (136). Both coding and noncoding transcripts of NCYM contribute to higher N-MYC expression. NCYM stabilizes N-MYC protein by inhibiting the activity of GSK3β, thereby preventing phosphodegron-mediated N-MYC degradation (135). Noncoding transcript variants of NCYM may reinforce MYCN translation via expelling exon 1b through alternative splicing or promoter shift (136). MYCN stimulates transcription of both NCYM and MYCN, forming a positive regulatory loop and leading to high expression of both genes (137).
NVP-BEZ235, a dual inhibitor of both phosphoinositide 3-kinase (PI3K) and mTOR, promotes the degradation of N-MYC by GSK3β activation and effectively decreases tumor burden in the MYCN transgenic mouse. In contrast, NVP-BEZ235 cannot prolong the survival of the MYCN/NCYM double transgenic mice (135). This might be related to the N-MYC-independent functions of NCYM, e.g., NCYM-mediated inhibition of GSK3β also lead to the stabilization of β-catenin, which promotes bladder cancer progression (138); NCYM promotes generation of MYC-nicks, cytoplasmic cleavage products of N-MYC and C-MYC, which inhibits apoptosis and enhances cancer cell migration (139). TAp63, an isoform of p63 protein and a p53 family protein, suppresses MYCN/NCYM bidirectional transcription, repressing neuroblastoma growth (140). Thus, the implication of NCYM gene in MYCN-driven tumors increases complexity and contributes to treatment resistance.
Somatic Mutation of MYCN
In addition to deregulated expression of N-MYC due to gene amplification or dysregulation at mRNA and protein levels, a recurrent somatic mutation, proline 44 to leucine (P44L) (Figure 1A), is identified in various tumors (141), including, glioma (142), neoplastic cysts of the pancreas (143), medulloblastoma (144), neuroblastoma (145), Wilms tumor (67), skin basal cell carcinoma (146), T-lineage acute lymphoblastic leukemia (147), NUT midline carcinoma (148), Ovarian mesonephric-like adenocarcinoma (149). Notably, P44L mutation of N-MYC has occurred in 1.7% of high-risk neuroblastoma without MYCN amplification (145). Since the frequent occurrence of P44L switch in different cancers, this mutation has long been assumed as an activating one, but it has not been functionally or biochemically characterized until recently (28). KE Mengwasser compared the function of P44L mutant with the wild type N-MYC in terms of promoting proliferation, and they found that P44L N-MYC mutant displayed 2- and 4.5-fold higher log2-fold-change in pancreas cells and breast cells, respectively (150). Similarly, Liu et al. observed a modest but significantly shorter latency for the induction of highly penetrant T-lineage leukemia in P44L N-MYC expressing cells than that of wild-type N-MYC expressing cells (147). These evidences solidly confirm that P44L N-MYC is indeed an activating mutation.
Mechanistically, as P44L mutation site locates adjacent to the conserved phosphor-degron sites recognized by E3 ubiquitin ligases FBXW7 and HUWE1 (Figure 1A), a hypothesis was proposed in which P44L mutation could perturb the interaction between these ligases and N-MYC substrate, therefore, prevented N-MYC degradation and enhanced oncogenicity (147). Consistently, Liu et al. show that the degradation of the N-MYC protein is significantly delayed in the P44L mutated type than that of the wild type after the cells are treated with cycloheximide to block protein translation (147). However, Bonilla et al. display that the interacting with FBXW7 is not affected by the P44L mutation, instead, the autoubiquitination of FBXW7 is increased in the presence of P44L mutation, suggesting a different mechanism for the enhanced stability of P44L N-MYC (146). Furthermore, the P44L mutation is associated with increased mRNA levels of MYCN in neuroblastoma (145). A previous study shows that MYCN can be directly recruited to the intron1 region of its own gene which contains two putative E-box sites and thus promotes its own transcription in neuroblastoma cells (151). Considering this positive auto-regulatory loop, it is possible that P44L mutation enhances MYCN mRNA level through the auto-activating mechanism with the more stable form of N-MYC protein.
Synthetic Lethal Interaction With Deregulated MYCN
The concept of synthetic lethality means targeting specific targets including proteins and metabolites that are essential for the viability of tumor cells with specific physiology, such as N-MYC overexpression. This strategy can kill cancer cells only while spares normal counterpart. For instance, checkpoint kinase 1 (CHK1) is a key player in the DNA damage checkpoint control, and inhibition of CHK1 sensitizes cells to additional genomic instability (152). Overexpression of N-MYC causes replication stress and DNA damage by the ectopic replication-fork firing, which results in remarkably higher sensitivity of N-MYC overexpressing tumors to CHK1 inhibition, and thereby CKH1 inhibition is synthetic lethal with N-MYC overexpression (153, 154). Similarly, we demonstrate that N-MYC sensitizes neuroblastoma cells to apoptosis induced by various death ligand or DNA-damaging drugs (155, 156). These results indicate targeting DNA repair system or drugs causing DNA damage could be synthetic lethal in MYCN-driven tumors. Recent studies reveal various strategies based on N-MYC-mediated synthetic lethality, including glutaminase inhibition or glutamine deprivation (157), BCL2 inhibition (125), eliminating SKP2 complexes (158), kinesin spindle protein (KSP) inhibition (159), G9a inhibition (160), poly (ADP-ribose) polymerase (PARP) inhibition (161, 162).
Conclusion and Perspectives
Here we describe the regulatory network of MYCN expression (Figure 2). Multiple mechanisms can cause abnormal level of N-MYC, including gene amplification, enhanced transcription, translation and protein stability. Various therapeutic targets have been found to address N-MYC overexpression based on knowledge of these regulatory mechanisms. However, strategies that globally inhibiting gene expression (such as inhibiting CDK7 and BDR4) has not yet convincingly demonstrated that these inhibitors specifically target tumors with high N-MYC level, nor have these inhibitors reached advanced stages in clinical trials (16). Although directly and specifically targeting N-MYC has not yet been available, promise remains in developing new approaches to effectively treat MYCN-driven tumors. For examples, short interfering RNA (siRNA)-mediated silence of MYCN induces neurogenesis and inhibits proliferation in neuroblastoma models resistant to retinoic acid (163). Clinical applications of siRNA are developing and the first siRNA-based drug Patisiran (Onpattro) was approved for clinical use to treat transthyretin amyloidosis by the U.S. Food and Drug Administration (FDA) in 2018 (164). In addition, Yoda et al. identify a pyrrole-imidazole polyamide, MYCN-A3, able to directly target MYCN amplicons, which specifically reduces copy number and suppresses gene expression of MYCN (165).
Author Contributions
RL wrote the manuscript. PS drew the cartoon figures. ZW and CY collected the articles. HC provided the idea and revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This research was supported by the National Key Research and Development Program of China (2016YFC1302204, 2017YFC1308601), the National Natural Science Foundation of China (81872071, 81672502), the Natural Science Foundation of Chongqing (cstc2019jcyj-zdxmX0033), and Chongqing University Innovation Team Building Program funded projects (CXTDX201601010).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Kohl NE, Kanda N, Schreck RR, Bruns G, Latt SA, Gilbert F, et al. Transposition and amplification of oncogene-related sequences in human neuroblastomas. Cell (1983) 35:359–67. doi: 10.1016/0092-8674(83)90169-1
2. Nau MM, Brooks BJ, Battey J, Sausville E, Gazdar AF, Kirsch IR, et al. L-myc, a new myc-related gene amplified and expressed in human small cell lung cancer. Nature (1985) 318:69–73. doi: 10.1038/318069a0
3. Schwab M, Alitalo K, Klempnauer K-H, Varmus HE, Bishop JM, Gilbert F, et al. Amplified DNA with limited homology to myc cellular oncogene is shared by human neuroblastoma cell lines and a neuroblastoma tumour. Nature (1983) 305:245–8. doi: 10.1038/305245a0
4. Lin CY, Lovén J, Rahl PB, Paranal RM, Burge CB, Bradner JE, et al. Transcriptional amplification in tumor cells with elevated c-Myc. Cell (2012) 151:56–67. doi: 10.1016/j.cell.2012.08.026
5. Lorenzin F, Benary U, Baluapuri A, Walz S, Jung LA, von Eyss B, et al. Different promoter affinities account for specificity in MYC-dependent gene regulation. eLife (2016) 5:e15161. doi: 10.7554/eLife.15161
6. Kohl NE, Legouy E, DePinho RA, Nisen PD, Smith RK, Gee CE, et al. Human N-myc is closely related in organization and nucleotide sequence to c-myc. Nature (1986) 319:73–7. doi: 10.1038/319073a0
7. Legouy E, DePinho R, Zimmerman K, Collum R, Yancopoulos G, Mitsock L, et al. Structure and expression of the murine L-myc gene. EMBO J (1987) 6:3359–66. doi: 10.1002/j.1460-2075.1987.tb02657.x
8. Sarid J, Halazonetis TD, Murphy W, Leder P. Evolutionarily conserved regions of the human c-myc protein can be uncoupled from transforming activity. Proc Natl Acad Sci (1987) 84:170–3. doi: 10.1073/pnas.84.1.170
9. Stanton LW, Schwab M, Bishop JM. Nucleotide sequence of the human N-myc gene. Proc Natl Acad Sci (1986) 83:1772–6. doi: 10.1073/pnas.83.6.1772
10. McMahon SB, Van Buskirk HA, Dugan KA, Copeland TD, Cole MD. The novel ATM-related protein TRRAP is an essential cofactor for the c-Myc and E2F oncoproteins. Cell (1998) 94:363–74. doi: 10.1016/S0092-8674(00)81479-8
11. Fuchs M, Gerber J, Drapkin R, Sif S, Ikura T, Ogryzko V, et al. The p400 complex is an essential E1A transformation target. Cell (2001) 106:297–307. doi: 10.1016/S0092-8674(01)00450-0
12. Blackwell TK, Huang J, Ma A, Kretzner L, Alt FW, Eisenman RN, et al. Binding of myc proteins to canonical and noncanonical DNA sequences. Mol Cell Biol (1993) 13:5216–24. doi: 10.1128/MCB.13.9.5216
13. Guo J, Li T, Schipper J, Nilson KA, Fordjour FK, Cooper JJ, et al. Sequence specificity incompletely defines the genome-wide occupancy of Myc. Genome Biol (2014) 15:482. doi: 10.1186/s13059-014-0482-3
14. Nie Z, Hu G, Wei G, Cui K, Yamane A, Resch W, et al. c-Myc is a universal amplifier of expressed genes in lymphocytes and embryonic stem cells. Cell (2012) 151:68–79. doi: 10.1016/j.cell.2012.08.033
15. Zeid R, Lawlor MA, Poon E, Reyes JM, Fulciniti M, Lopez MA, et al. Enhancer invasion shapes MYCN-dependent transcriptional amplification in neuroblastoma. Nat Genet (2018) 50:515–23. doi: 10.1038/s41588-018-0044-9
16. Baluapuri A, Wolf E, Eilers M. Target gene-independent functions of MYC oncoproteins. Nat Rev Mol Cell Biol (2020) 21:255–67. doi: 10.1038/s41580-020-0215-2
17. Meyer N, Penn LZ. Reflecting on 25 years with MYC. Nat Rev Cancer (2008) 8:976–90. doi: 10.1038/nrc2231
19. Yoshida GJ. Emerging roles of Myc in stem cell biology and novel tumor therapies. J Exp Clin Cancer Res (2018) 37:173. doi: 10.1186/s13046-018-0964-3
20. Yang L, Shi P, Zhao G, Xu J, Peng W, Zhang J, et al. Targeting cancer stem cell pathways for cancer therapy. Signal Transduct Target Ther (2020) 5:8. doi: 10.1038/s41392-020-0110-5
21. Izumi H, Kaneko Y, Nakagawara A. The role of MYCN in symmetric vs. asymmetric cell division of human neuroblastoma cells. Front Oncol (2020) 10:570815. doi: 10.3389/fonc.2020.570815
22. Malynn BA, de Alboran IM, O’Hagan RC, Bronson R, Davidson L, DePinho RA, et al. N-myc can functionally replace c-myc in murine development, cellular growth, and differentiation. Genes Dev (2000) 14:1390–9. doi: 10.1101/gad.14.11.1390
23. Kawauchi D, Robinson G, Uziel T, Gibson P, Rehg J, Gao C, et al. A mouse model of the most aggressive subgroup of human medulloblastoma. Cancer Cell (2012) 21:168–80. doi: 10.1016/j.ccr.2011.12.023
24. Vo BT, Wolf E, Kawauchi D, Gebhardt A, Rehg JE, Finkelstein D, et al. The interaction of Myc with Miz1 defines medulloblastoma subgroup identity. Cancer Cell (2016) 29:5–16. doi: 10.1016/j.ccell.2015.12.003
25. Beltran H, Prandi D, Mosquera JM, Benelli M, Puca L, Cyrta J, et al. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat Med (2016) 22:298–305. doi: 10.1038/nm.4045
26. Dammert MA, Brägelmann J, Olsen RR, Böhm S, Monhasery N, Whitney CP, et al. MYC paralog-dependent apoptotic priming orchestrates a spectrum of vulnerabilities in small cell lung cancer. Nat Commun (2019) 10:3485. doi: 10.1038/s41467-019-11371-x
27. Massó-Vallés D, Beaulieu M-E, Soucek L. MYC, MYCL, and MYCN as therapeutic targets in lung cancer. Expert Opin Ther Targets (2020) 24:101–14. doi: 10.1080/14728222.2020.1723548
28. Rickman DS, Schulte JH, Eilers M. The expanding world of N-MYC–driven tumors. Cancer Discov (2018) 8:150–63. doi: 10.1158/2159-8290.CD-17-0273
29. Bjerke L, Mackay A, Nandhabalan M, Burford A, Jury A, Popov S, et al. Histone H3.3 mutations drive pediatric glioblastoma through upregulation of MYCN. Cancer Discov (2013) 3:512–9. doi: 10.1158/2159-8290.CD-12-0426
30. Pfister S, Remke M, Benner A, Mendrzyk F, Toedt G, Felsberg J, et al. Outcome prediction in pediatric medulloblastoma based on DNA copy-number aberrations of chromosomes 6q and 17q and the MYC and MYCN loci. J Clin Oncol (2009) 27:1627–36. doi: 10.1200/JCO.2008.17.9432
31. Garson JA, McIntyre PG, Kemshead JT. N-MYC amplification in malignant astrocytoma. Lancet (1985) 326:718–9. doi: 10.1016/S0140-6736(85)92950-2
32. Berger A, Brady NJ, Bareja R, Robinson B, Conteduca V, Augello MA, et al. N-Myc–mediated epigenetic reprogramming drives lineage plasticity in advanced prostate cancer. J Clin Invest (2019) 129:3924–40. doi: 10.1172/JCI127961
33. Mizukami Y, Nonomura A, Takizawa T, Noguchi M, Michigishi T, Nakamura S, et al. N-myc protein expression in human breast carcinoma: prognostic implications. Anticancer Res (1995) 15:2899–905. doi: 10.1007/BF02307090
34. Hirvonen H, Hukkanen V, Salmi TT, Pelliniemi T-T, Alitalo R. L-myc and N-myc in hematopoietic malignancies. Leuk Lymphoma (1993) 11:197–205. doi: 10.3109/10428199309086996
35. Yang HW, Kutok JL, Lee NH, Piao HY, Fletcher CDM, Kanki JP, et al. Targeted expression of human MYCN selectively causes pancreatic neuroendocrine tumors in transgenic zebrafish. Cancer Res (2004) 64:7256–62. doi: 10.1158/0008-5472.CAN-04-0931
36. Williams RD, Al-Saadi R, Natrajan R, Mackay A, Chagtai T, Little S, et al. Molecular profiling reveals frequent gain of MYCN and anaplasia-specific loss of 4q and 14q in Wilms tumor. Genes Chromosomes Cancer (2011) 50:982–95. doi: 10.1002/gcc.20907
37. Qin X-Y, Suzuki H, Honda M, Okada H, Kaneko S, Inoue I, et al. Prevention of hepatocellular carcinoma by targeting MYCN-positive liver cancer stem cells with acyclic retinoid. Proc Natl Acad Sci (2018) 115:4969–74. doi: 10.1073/pnas.1802279115
38. Driman D, Thorner PS, Greenberg ML, Chilton-MacNeill S, Squire J. MYCN gene amplification in rhabdomyosarcoma. Cancer (1994) 73:2231–7. doi: 10.1002/1097-0142(19940415)73:8<2231::AID-CNCR2820730832>3.0.CO;2-E
39. Baratta MG, Schinzel AC, Zwang Y, Bandopadhayay P, Bowman-Colin C, Kutt J, et al. An in-tumor genetic screen reveals that the BET bromodomain protein, BRD4, is a potential therapeutic target in ovarian carcinoma. Proc Natl Acad Sci (2015) 112:232–7. doi: 10.1073/pnas.1422165112
40. Schwab M. MYCN in neuronal tumours. Cancer Lett (2004) 204:179–87. doi: 10.1016/S0304-3835(03)00454-3
41. Weiss WA, Aldape K, Mohapatra G, Feuerstein BG, Bishop JM. Targeted expression of MYCN causes neuroblastoma in transgenic mice. EMBO J (1997) 16:2985–95. doi: 10.1093/emboj/16.11.2985
42. Zhu S, Lee J-S, Guo F, Shin J, Perez-Atayde AR, Kutok JL, et al. Activated ALK collaborates with MYCN in neuroblastoma pathogenesis. Cancer Cell (2012) 21:362–73. doi: 10.1016/j.ccr.2012.02.010
43. Valentijn LJ, Koster J, Haneveld F, Aissa RA, van Sluis P, Broekmans MEC, et al. Functional MYCN signature predicts outcome of neuroblastoma irrespective of MYCN amplification. Proc Natl Acad Sci (2012) 109:19190–5. doi: 10.1073/pnas.1208215109
44. Chang H-H, Tseng Y-F, Lu M-Y, Yang Y-L, Chou S-W, Lin D-T, et al. MYCN RNA levels determined by quantitative in situ hybridization is better than MYCN gene dosages in predicting the prognosis of neuroblastoma patients. Mod Pathol (2020) 33:531–40. doi: 10.1038/s41379-019-0410-x
45. Yang Yi, Zhao J, Zhang Y, Yv Bo, Wang J, Feng H, et al. MYCN Fish combined with immunohistochemistry is a more prognostic factor for neuroblastoma. Res Square (2020). doi: 10.21203/rs.3.rs-62365/v1
46. Yoshida GJ. Beyond the Warburg effect: N-Myc contributes to metabolic reprogramming in cancer cells. Front Oncol (2020) 10:791. doi: 10.3389/fonc.2020.00791
47. Schwab M. Oncogene amplification in solid tumors. Semin Cancer Biol (1999) 9:319–25. doi: 10.1006/scbi.1999.0126
48. Schwab M, Corvi R, Amler LC. N-MYC oncogene amplification: a consequence of genomic instability in human neuroblastoma. Neuroscientist (1995) 1:277–85. doi: 10.1177/107385849500100505
49. Hansford LM, Thomas WD, Keating JM, Burkhart CA, Peaston AE, Norris MD, et al. Mechanisms of embryonal tumor initiation: distinct roles for MycN expression and MYCN amplification. Proc Natl Acad Sci U S A (2004) 101:12664–9. doi: 10.1073/pnas.0401083101
50. Brodeur G, Seeger R, Schwab M, Varmus H, Bishop J. Amplification of N-myc in untreated human neuroblastomas correlates with advanced disease stage. Science (1984) 224:1121–4. doi: 10.1126/science.6719137
51. Kohl N, Gee C, Alt F. Activated expression of the N-myc gene in human neuroblastomas and related tumors. Science (1984) 226:1335–7. doi: 10.1126/science.6505694
52. Tower J. Developmental gene amplification and origin regulation. Annu Rev Genet (2004) 38:273–304. doi: 10.1146/annurev.genet.37.110801.143851
53. Watanabe T, Tanabe H, Horiuchi T. Gene amplification system based on double rolling-circle replication as a model for oncogene-type amplification. Nucleic Acids Res (2011) 39:e106–6. doi: 10.1093/nar/gkr442
54. Amler LC, Schwab M. Amplified N-myc in human neuroblastoma cells is often arranged as clustered tandem repeats of differently recombined DNA. Mol Cell Biol (1989) 9:4903–13. doi: 10.1128/MCB.9.11.4903
55. Nevim A. Biological and genetic features of neuroblastoma and their clinical importance. Curr Pediatr Rev (2018) 14:73–90. doi: 10.2174/1573396314666180129101627
56. Raschellà G, Negroni A, Skorski T, Pucci S, Nieborowska-Skorska M, Romeo A, et al. Inhibition of proliferation by c-myb antisense RNA and oligodeoxynucleotides in transformed neuroectodermal cell lines. Cancer Res (1992) 52:4221–6. doi: 10.1097/00002820-199215040-00008
57. Beall EL, Manak JR, Zhou S, Bell M, Lipsick JS, Botchan MR. Role for a Drosophila Myb-containing protein complex in site-specific DNA replication. Nature (2002) 420:833–7. doi: 10.1038/nature01228
58. Aygun N, Altungoz O. MYCN is amplified during S phase, and c−myb is involved in controlling MYCN expression and amplification in MYCN−amplified neuroblastoma cell lines. Mol Med Rep (2019) 19:345–61. doi: 10.3892/mmr.2018.9686
59. McGarry TJ, Kirschner MW. Geminin, an inhibitor of DNA replication, is degraded during mitosis. Cell (1998) 93:1043–53. doi: 10.1016/S0092-8674(00)81209-X
60. Albertson DG. Gene amplification in cancer. Trends Genet (2006) 22:447–55. doi: 10.1016/j.tig.2006.06.007
61. Helmsauer K, Valieva ME, Ali S, Chamorro González R, Schöpflin R, Röefzaad C, et al. Enhancer hijacking determines extrachromosomal circular MYCN amplicon architecture in neuroblastoma. Nat Commun (2020) 11:5823. doi: 10.1038/s41467-020-19452-y
62. Morton AR, Dogan-Artun N, Faber ZJ, MacLeod G, Bartels CF, Piazza MS, et al. Functional enhancers shape extrachromosomal oncogene amplifications. Cell (2019) 179:1330–41.e13. doi: 10.1016/j.cell.2019.10.039
63. Cohn SL, London WB, Huang D, Katzenstein HM, Salwen HR, Reinhart T, et al. MYCN expression is not prognostic of adverse outcome in advanced-stage neuroblastoma with nonamplified MYCN. J Clin Oncol (2000) 18:3604–13. doi: 10.1200/JCO.2000.18.21.3604
64. Henrichsen CN, Chaignat E, Reymond A. Copy number variants, diseases and gene expression. Hum Mol Genet (2009) 18:R1–8. doi: 10.1093/hmg/ddp011
65. Law JA, Jacobsen SE. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat Rev Genet (2010) 11:204–20. doi: 10.1038/nrg2719
66. Birchler JA, Pal Bhadra M, Bhadra U. Making noise about silence: repression of repeated genes in animals. Curr Opin Genet Dev (2000) 10:211–6. doi: 10.1016/S0959-437X(00)00065-4
67. Williams RD, Chagtai T, Alcaide-German M, Apps J, Wegert J, Popov S, et al. Multiple mechanisms of MYCN dysregulation in Wilms tumour. Oncotarget (2015) 6:7232–43. doi: 10.18632/oncotarget.3377
68. Zimmerman MW, Durbin AD, He S, Oppel F, Shi H, Tao T, et al. Retinoic acid rewires the adrenergic core regulatory circuitry of neuroblastoma but can be subverted by enhancer hijacking of MYC or MYCN. bioRxiv (2020), 2020.07.23.218834. doi: 10.1101/2020.07.23.218834
69. Zimmerman MW, Liu Y, He S, Durbin AD, Abraham BJ, Easton J, et al. c-MYC drives a subset of high-risk pediatric neuroblastomas and is activated through mechanisms including enhancer hijacking and focal enhancer amplification. Cancer Discov (2018) 8:320–35. doi: 10.1158/2159-8290.CD-17-0993
70. Lovén J, Hoke HA, Lin CY, Lau A, Orlando DA, Vakoc CR, et al. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell (2013) 153:320–34. doi: 10.1016/j.cell.2013.03.036
71. Hnisz D, Abraham BJ, Lee T II, Lau A, Saint-André V, Sigova AA, et al. Super-enhancers in the control of cell identity and disease. Cell (2013) 155:934–47. doi: 10.1016/j.cell.2013.09.053
72. Kouskouti A, Talianidis I. Histone modifications defining active genes persist after transcriptional and mitotic inactivation. EMBO J (2005) 24:347–57. doi: 10.1038/sj.emboj.7600516
73. Sims RJ, Belotserkovskaya R, Reinberg D. Elongation by RNA polymerase II: the short and long of it. Genes Dev (2004) 18:2437–68. doi: 10.1101/gad.1235904
74. Yang Z, He N, Zhou Q. Brd4 recruits P-TEFb to chromosomes at late mitosis to promote G1 gene expression and cell cycle progression. Mol Cell Biol (2008) 28:967–76. doi: 10.1128/MCB.01020-07
75. Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, et al. Selective inhibition of BET bromodomains. Nature (2010) 468:1067–73. doi: 10.1038/nature09504
76. Henssen A, Althoff K, Odersky A, Beckers A, Koche R, Speleman F, et al. Targeting MYCN-driven transcription by BET-bromodomain inhibition. Clin Cancer Res (2016) 22:2470–81. doi: 10.1158/1078-0432.CCR-15-1449
77. Puissant A, Frumm SM, Alexe G, Bassil CF, Qi J, Chanthery YH, et al. Targeting MYCN in neuroblastoma by BET bromodomain inhibition. Cancer Discov (2013) 3:308–23. doi: 10.1158/2159-8290.CD-12-0418
78. Mazar J, Gordon C, Naga V, Westmoreland TJ. The killing of human neuroblastoma cells by the small molecule JQ1 occurs in a p53-dependent manner. Anti-Cancer Agents Med Chem (2020) 20:1613–25. doi: 10.2174/1871520620666200424123834
79. Maser T, Zagorski J, Kelly S, Ostrander A, Goodyke A, Nagulapally A, et al. The MDM2 inhibitor CGM097 combined with the BET inhibitor OTX015 induces cell death and inhibits tumor growth in models of neuroblastoma. Cancer Med (2020) 9:8144–58. doi: 10.1002/cam4.3407
80. Schafer JM, Lehmann BD, Gonzalez-Ericsson PI, Marshall CB, Beeler JS, Redman LN, et al. Targeting MYCN-expressing triple-negative breast cancer with BET and MEK inhibitors. Sci Transl Med (2020) 12:eaaw8275. doi: 10.1126/scitranslmed.aaw8275
81. Shiekhattar R, Mermelstein F, Fisher RP, Drapkin R, Dynlacht B, Wessling HC, et al. Cdk-activating kinase complex is a component of human transcription factor TFIIH. Nature (1995) 374:283–7. doi: 10.1038/374283a0
82. Peng J, Marshall NF, Price DH. Identification of a cyclin subunit required for the function of Drosophila P-TEFb. J Biol Chem (1998) 273:13855–60. doi: 10.1074/jbc.273.22.13855
83. Fisher RP. Secrets of a double agent: CDK7 in cell-cycle control and transcription. J Cell Sci (2005) 118:5171–80. doi: 10.1242/jcs.02718
84. Larochelle S, Amat R, Glover-Cutter K, Sansó M, Zhang C, Allen JJ, et al. Cyclin-dependent kinase control of the initiation-to-elongation switch of RNA polymerase II. Nat Struct Mol Biol (2012) 19:1108–15. doi: 10.1038/nsmb.2399
85. Chipumuro E, Marco E, Christensen CL, Kwiatkowski N, Zhang T, Hatheway CM, et al. CDK7 inhibition suppresses super-enhancer-linked oncogenic transcription in MYCN-driven cancer. Cell (2014) 159:1126–39. doi: 10.1016/j.cell.2014.10.024
86. Poon E, Liang T, Jamin Y, Walz S, Kwok C, Hakkert A, et al. Orally bioavailable CDK9/2 inhibitor shows mechanism-based therapeutic potential in MYCN-driven neuroblastoma. J Clin Invest (2020) 130:5875–92. doi: 10.1172/JCI134132
87. Durbin AD, Zimmerman MW, Dharia NV, Abraham BJ, Iniguez AB, Weichert-Leahey N, et al. Selective gene dependencies in MYCN-amplified neuroblastoma include the core transcriptional regulatory circuitry. Nat Genet (2018) 50:1240–6. doi: 10.1038/s41588-018-0191-z
88. Tuthill MC, Wada RK, Arimoto JM, Sugino CN, Kanemaru KK, Takeuchi KK, et al. N-myc oncogene expression in neuroblastoma is driven by Sp1 and Sp3. Mol Genet Metab (2003) 80:272–80. doi: 10.1016/S1096-7192(03)00133-1
89. Strieder V, Lutz W. E2F proteins regulate MYCN expression in neuroblastomas. J Biol Chem (2003) 278:2983–9. doi: 10.1074/jbc.M207596200
90. Zhao Z, Shelton SD, Oviedo A, Baker AL, Bryant CP, Omidvarnia S, et al. The PLAGL2/MYCN/miR-506-3p interplay regulates neuroblastoma cell fate and associates with neuroblastoma progression. J Exp Clin Cancer Res (2020) 39:41. doi: 10.1186/s13046-020-1531-2
91. Qin X-Y, Su T, Yu W, Kojima S. Lipid desaturation-associated endoplasmic reticulum stress regulates MYCN gene expression in hepatocellular carcinoma cells. Cell Death Dis (2020) 11:66. doi: 10.1038/s41419-020-2257-y
92. Zhao E, Hou J, Cui H. Serine–glycine-one-carbon metabolism: vulnerabilities in MYCN-amplified neuroblastoma. Oncogenesis (2020) 9:14. doi: 10.1038/s41389-020-0200-9
93. Guo Y-F, Duan J-J, Wang J, Li L, Wang D, Liu X-Z, et al. Inhibition of the ALDH18A1-MYCN positive feedback loop attenuates MYCN-amplified neuroblastoma growth. Sci Transl Med (2020) 12:eaax8694. doi: 10.1126/scitranslmed.aax8694
94. Thiele CJ, Reynolds CP, Israel MA. Decreased expression of N-myc precedes retinoic acid-induced morphological differentiation of human neuroblastoma. Nature (1985) 313:404–6. doi: 10.1038/313404a0
95. Matthay KK, Reynolds CP, Seeger RC, Shimada H, Adkins ES, Haas-Kogan D, et al. Long-term results for children with high-risk neuroblastoma treated on a randomized trial of myeloablative therapy followed by 13-cis-retinoic acid: a children’s oncology group study. J Clin Oncol (2009) 27:1007–13. doi: 10.1200/JCO.2007.13.8925
96. Veal GJ, Errington J, Rowbotham SE, Illingworth NA, Malik G, Cole M, et al. Adaptive dosing approaches to the individualization of 13-Cis-retinoic acid (Isotretinoin) treatment for children with high-risk neuroblastoma. Clin Cancer Res (2013) 19:469–79. doi: 10.1158/1078-0432.CCR-12-2225
97. Levens D. “You don’t muck with MYC”. Genes Cancer (2010) 1:547–54. doi: 10.1177/1947601910377492
98. Siddiqui-Jain A, Grand CL, Bearss DJ, Hurley LH. Direct evidence for a G-quadruplex in a promoter region and its targeting with a small molecule to repress c-MYC transcription. Proc Natl Acad Sci (2002) 99:11593–8. doi: 10.1073/pnas.182256799
99. Trajkovski M, Webba da Silva M, Plavec J. Unique structural features of interconverting monomeric and dimeric G-quadruplexes adopted by a sequence from the intron of the N-myc gene. J Am Chem Soc (2012) 134:4132–41. doi: 10.1021/ja208483v
100. Dutta D, Debnath M, Müller D, Paul R, Das T, Bessi I, et al. Cell penetrating thiazole peptides inhibit c-MYC expression via site-specific targeting of c-MYC q-quadruplex. Nucleic Acids Res (2018) 46:5355–65. doi: 10.1093/nar/gky385
101. González V, Guo K, Hurley L, Sun D. Identification and characterization of nucleolin as a c-myc G-quadruplex-binding protein. J Biol Chem (2009) 284:23622–35. doi: 10.1074/jbc.M109.018028
102. Brooks TA, Hurley LH. Targeting MYC expression through G-quadruplexes. Genes Cancer (2010) 1:641–9. doi: 10.1177/1947601910377493
103. Hald Ø.H., Olsen L, Gallo-Oller G, Elfman LHM, Løkke C, Kogner P, et al. Inhibitors of ribosome biogenesis repress the growth of MYCN-amplified neuroblastoma. Oncogene (2019) 38:2800–13. doi: 10.1038/s41388-018-0611-7
104. Zhao Z, Ma X, Shelton SD, Sung DC, Li M, Hernandez D, et al. A combined gene expression and functional study reveals the crosstalk between N-Myc and differentiation-inducing microRNAs in neuroblastoma cells. Oncotarget (2016) 7:79372–87. doi: 10.18632/oncotarget.12676
105. Ooi CY, Carter DR, Liu B, Mayoh C, Beckers A, Lalwani A, et al. Network modeling of microRNA–mRNA interactions in neuroblastoma tumorigenesis identifies miR-204 as a direct inhibitor of MYCN. Cancer Res (2018) 78:3122–34. doi: 10.1158/0008-5472.CAN-17-3034
106. Roth SA, Hald Ø.H., Fuchs S, Løkke C, Mikkola I, Flægstad T, et al. MicroRNA-193b-3p represses neuroblastoma cell growth via downregulation of Cyclin D1, MCL-1 and MYCN. Oncotarget (2018) 9:18160–79. doi: 10.18632/oncotarget.24793
107. Li J, Jiang X, Li Z, Huang L, Ji D, Yu L, et al. SP1-induced HOXD-AS1 promotes malignant progression of cholangiocarcinoma by regulating miR-520c-3p/MYCN. Aging (Albany NY) (2020) 12:16304–25. doi: 10.18632/aging.103660
108. Buechner J, Tømte E, Haug BH, Henriksen JR, Løkke C, Flægstad T, et al. Tumour-suppressor microRNAs let-7 and mir-101 target the proto-oncogene MYCN and inhibit cell proliferation in MYCN-amplified neuroblastoma. Br J Cancer (2011) 105:296–303. doi: 10.1038/bjc.2011.220
109. Molenaar JJ, Domingo-Fernández R, Ebus ME, Lindner S, Koster J, Drabek K, et al. LIN28B induces neuroblastoma and enhances MYCN levels via let-7 suppression. Nat Genet (2012) 44:1199–206. doi: 10.1038/ng.2436
110. Gu L, Zhang H, He J, Li J, Huang M, Zhou M. MDM2 regulates MYCN mRNA stabilization and translation in human neuroblastoma cells. Oncogene (2012) 31:1342–53. doi: 10.1038/onc.2011.343
111. Boon K, Caron HN, van Asperen R, Valentijn L, Hermus M-C, van Sluis P, et al. N-myc enhances the expression of a large set of genes functioning in ribosome biogenesis and protein synthesis. EMBO J (2001) 20:1383–93. doi: 10.1093/emboj/20.6.1383
112. Bjornsti M-A, Houghton PJ. The TOR pathway: a target for cancer therapy. Nat Rev Cancer (2004) 4:335–48. doi: 10.1038/nrc1362
113. Chen H, Liu H, Qing G. Targeting oncogenic Myc as a strategy for cancer treatment. Signal Transduct Target Ther (2018) 3:5. doi: 10.1038/s41392-018-0008-7
114. Dong Y, Gong W, Hua Z, Chen B, Zhao G, Liu Z, et al. Combination of rapamycin and MK-2206 induced cell death via autophagy and necroptosis in MYCN-amplified neuroblastoma cell lines. Front Pharmacol (2020) 11:31–1. doi: 10.3389/fphar.2020.00031
115. Zhao X, Heng JI-T, Guardavaccaro D, Jiang R, Pagano M, Guillemot F, et al. The HECT-domain ubiquitin ligase Huwe1 controls neural differentiation and proliferation by destabilizing the N-Myc oncoprotein. Nat Cell Biol (2008) 10:643–53. doi: 10.1038/ncb1727
116. Welcker M, Orian A, Jin J, Grim JA, Harper JW, Eisenman RN, et al. The Fbw7 tumor suppressor regulates glycogen synthase kinase 3 phosphorylation-dependent c-Myc protein degradation. Proc Natl Acad Sci (2004) 101:9085–90. doi: 10.1073/pnas.0402770101
117. Sjostrom SK, Finn G, Hahn WC, Rowitch DH, Kenney AM. The Cdk1 complex plays a prime role in regulating N-Myc phosphorylation and turnover in neural precursors. Dev Cell (2005) 9:327–38. doi: 10.1016/j.devcel.2005.07.014
118. Yeh E, Cunningham M, Arnold H, Chasse D, Monteith T, Ivaldi G, et al. A signalling pathway controlling c-Myc degradation that impacts oncogenic transformation of human cells. Nat Cell Biol (2004) 6:308–18. doi: 10.1038/ncb1110
119. Otto T, Horn S, Brockmann M, Eilers U, Schüttrumpf L, Popov N, et al. Stabilization of N-Myc is a critical function of Aurora A in human neuroblastoma. Cancer Cell (2009) 15:67–78. doi: 10.1016/j.ccr.2008.12.005
120. Richards MW, Burgess SG, Poon E, Carstensen A, Eilers M, Chesler L, et al. Structural basis of N-Myc binding by Aurora-A and its destabilization by kinase inhibitors. Proc Natl Acad Sci (2016) 113:13726–31. doi: 10.1073/pnas.1610626113
121. Shang X, Burlingame SM, Okcu MF, Ge N, Russell HV, Egler RA, et al. Aurora A is a negative prognostic factor and a new therapeutic target in human neuroblastoma. Mol Cancer Ther (2009) 8:2461–9. doi: 10.1158/1535-7163.MCT-08-0857
122. Brockmann M, Poon E, Berry T, Carstensen A, Deubzer HE, Rycak L, et al. Small molecule inhibitors of Aurora-A induce proteasomal degradation of N-Myc in childhood neuroblastoma. Cancer Cell (2013) 24:75–89. doi: 10.1016/j.ccr.2013.05.005
123. Manfredi MG, Ecsedy JA, Meetze KA, Balani SK, Burenkova O, Chen W, et al. Antitumor activity of MLN8054, an orally active small-molecule inhibitor of Aurora A kinase. Proc Natl Acad Sci (2007) 104:4106–11. doi: 10.1073/pnas.0608798104
124. Ommer J, Selfe JL, Wachtel M, O’Brien EM, Laubscher D, Roemmele M, et al. Aurora A kinase inhibition destabilizes PAX3-FOXO1 and MYCN and synergizes with navitoclax to induce rhabdomyosarcoma cell death. Cancer Res (2020) 80:832–42. doi: 10.1158/0008-5472.CAN-19-1479
125. Ham J, Costa C, Sano R, Lochmann TL, Sennott EM, Patel NU, et al. Exploitation of the apoptosis-primed state of MYCN-amplified neuroblastoma to develop a potent and specific targeted therapy combination. Cancer Cell (2016) 29:159–72. doi: 10.1016/j.ccell.2016.01.002
126. Gustafson WC, Meyerowitz JG, Nekritz EA, Chen J, Benes C, Charron E, et al. Drugging MYCN through an allosteric transition in Aurora kinase A. Cancer Cell (2014) 26:414–27. doi: 10.1016/j.ccr.2014.07.015
127. Pajtler KW, Sadowski N, Ackermann S, Althoff K, Schoenbeck K, Batzke K, et al. The GSK461364 PLK1 inhibitor exhibits strong antitumoral activity in preclinical neuroblastoma models. Oncotarget (2017) 8:6730–41. doi: 10.18632/oncotarget.14268
128. Xiao D, Yue M, Su H, Ren P, Jiang J, Li F, et al. Polo-like kinase-1 regulates Myc stabilization and activates a feedforward circuit promoting tumor cell survival. Mol Cell (2016) 64:493–506. doi: 10.1016/j.molcel.2016.09.016
129. Timme N, Han Y, Liu S, Yosief HO, Garcia HD, Bei Y, et al. Small-molecule dual PLK1 and BRD4 inhibitors are active against preclinical models of pediatric solid tumors. Trans Oncol (2020) 13:221–32. doi: 10.1016/j.tranon.2019.09.013
130. Wang N-Y, Xu Y, Xiao K-J, Zuo W-Q, Zhu Y-X, Hu R, et al. Design, synthesis, and biological evaluation of 4,5-dihydro-1,2,4 triazolo 4,3-f pteridine derivatives as novel dual-PLK1/BRD4 inhibitors. Eur J Med Chem (2020) 191:112152. doi: 10.1016/j.ejmech.2020.112152
131. Tavana O, Li D, Dai C, Lopez G, Banerjee D, Kon N, et al. HAUSP deubiquitinates and stabilizes N-Myc in neuroblastoma. Nat Med (2016) 22:1180–6. doi: 10.1038/nm.4180
132. Ohol YM, Sun MT, Cutler G, Leger PR, Hu DX, Biannic B, et al. Novel, selective inhibitors of USP7 uncover multiple mechanisms of antitumor activity in vitro and in vivo. Mol Cancer Ther (2020) 19:1970–80. doi: 10.1158/1535-7163.MCT-20-0184
133. Armstrong B, Krystal G. Isolation and characterization of complementary DNA for N-cym, a gene encoded by the DNA strand opposite to N-myc. Cell Growth Differ (1992) 3:385–90. doi: 10.1103/PhysRevE.73.031911
134. Krystal GW, Armstrong BC, Battey JF. N-myc mRNA forms an RNA-RNA duplex with endogenous antisense transcripts. Mol Cell Biol (1990) 10:4180–91. doi: 10.1128/MCB.10.8.4180
135. Suenaga Y, Islam SMR, Alagu J, Kaneko Y, Kato M, Tanaka Y, et al. NCYM, a cis-antisense gene of MYCN, encodes a de novo evolved protein that inhibits GSK3β resulting in the stabilization of MYCN in human neuroblastomas. PloS Genet (2014) 10:e1003996. doi: 10.1371/journal.pgen.1003996
136. Suenaga Y, Nakatani K, Nakagawara A. De novo evolved gene product NCYM in the pathogenesis and clinical outcome of human neuroblastomas and other cancers. Jpn J Clin Oncol (2020) 50:839–46. doi: 10.1093/jjco/hyaa097
137. Kaneko Y, Suenaga Y, Islam SMR, Matsumoto D, Nakamura Y, Ohira M, et al. Functional interplay between MYCN, NCYM, and OCT4 promotes aggressiveness of human neuroblastomas. Cancer Sci (2015) 106:840–7. doi: 10.1111/cas.12677
138. Zhu X, Li Y, Zhao S, Zhao S. LSINCT5 activates Wnt/β-catenin signaling by interacting with NCYM to promote bladder cancer progression. Biochem Biophys Res Commun (2018) 502:299–306. doi: 10.1016/j.bbrc.2018.05.076
139. Shoji W, Suenaga Y, Kaneko Y, Islam SMR, Alagu J, Yokoi S, et al. NCYM promotes calpain-mediated Myc-nick production in human MYCN-amplified neuroblastoma cells. Biochem Biophys Res Commun (2015) 461:501–6. doi: 10.1016/j.bbrc.2015.04.050
140. Suenaga Y, Yamamoto M, Sakuma T, Sasada M, Fukai F, Ohira M, et al. TAp63 represses transcription of MYCN/NCYM gene and its high levels of expression are associated with favorable outcome in neuroblastoma. Biochem Biophys Res Commun (2019) 518:311–8. doi: 10.1016/j.bbrc.2019.08.052
141. Forbes SA, Bindal N, Bamford S, Cole C, Kok CY, Beare D, et al. COSMIC: mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res (2010) 39:D945–50. doi: 10.1093/nar/gkq929
142. McLendon R, Friedman A, Bigner D, Van Meir EG, Brat DJ, Mastrogianakis GM, et al. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature (2008) 455:1061–8. doi: 10.1038/nature07385
143. Wu J, Jiao Y, Dal Molin M, Maitra A, de Wilde RF, Wood LD, et al. Whole-exome sequencing of neoplastic cysts of the pancreas reveals recurrent mutations in components of ubiquitin-dependent pathways. Proc Natl Acad Sci (2011) 108:21188–93. doi: 10.1073/pnas.1118046108
144. Jones DTW, Jäger N, Kool M, Zichner T, Hutter B, Sultan M, et al. Dissecting the genomic complexity underlying medulloblastoma. Nature (2012) 488:100–5. doi: 10.1038/nature11284
145. Pugh TJ, Morozova O, Attiyeh EF, Asgharzadeh S, Wei JS, Auclair D, et al. The genetic landscape of high-risk neuroblastoma. Nat Genet (2013) 45:279–84. doi: 10.1038/ng.2529
146. Bonilla X, Parmentier L, King B, Bezrukov F, Kaya G, Zoete V, et al. Genomic analysis identifies new drivers and progression pathways in skin basal cell carcinoma. Nat Genet (2016) 48:398–406. doi: 10.1038/ng.3525
147. Liu Y, Easton J, Shao Y, Maciaszek J, Wang Z, Wilkinson MR, et al. The genomic landscape of pediatric and young adult T-lineage acute lymphoblastic leukemia. Nat Genet (2017) 49:1211–8. doi: 10.1038/ng.3909
148. Liao S, Maertens O, Cichowski K, Elledge SJ. Genetic modifiers of the BRD4-NUT dependency of NUT midline carcinoma uncovers a synergism between BETis and CDK4/6is. Genes Dev (2018) 32:1188–200. doi: 10.1101/gad.315648.118
149. Dundr P, Gregová M, Němejcová K, Bártů M, Hájková N, Hojný J, et al. Ovarian mesonephric-like adenocarcinoma arising in serous borderline tumor: a case report with complex morphological and molecular analysis. Diagn Pathol (2020) 15:91. doi: 10.1186/s13000-020-01012-z
150. Mengwasser KE. Genetic screening approaches to cancer driver characterization and synthetic lethal target discovery. Graduate School of Arts & Sciences, Harvard University (2018).
151. Suenaga Y, Kaneko Y, Matsumoto D, Hossain MS, Ozaki T, Nakagawara A. Positive auto-regulation of MYCN in human neuroblastoma. Biochem Biophys Res Commun (2009) 390:21–6. doi: 10.1016/j.bbrc.2009.09.044
152. Furnari B, Rhind N, Russell P. Cdc25 mitotic inducer targeted by Chk1 DNA damage checkpoint kinase. Science (1997) 277:1495–7. doi: 10.1126/science.277.5331.1495
153. Cole KA, Huggins J, Laquaglia M, Hulderman CE, Russell MR, Bosse K, et al. RNAi screen of the protein kinome identifies checkpoint kinase 1 (CHK1) as a therapeutic target in neuroblastoma. Proc Natl Acad Sci (2011) 108:3336–41. doi: 10.1073/pnas.1012351108
154. Höglund A, Nilsson LM, Muralidharan SV, Hasvold LA, Merta P, Rudelius M, et al. Therapeutic implications for the induced levels of Chk1 in Myc-expressing cancer cells. Clin Cancer Res (2011) 17:7067–79. doi: 10.1158/1078-0432.CCR-11-1198
155. Li T, Wang L, Ke X-X, Gong X-Y, Wan J-H, Hao X-W, et al. DNA-damaging drug-induced apoptosis sensitized by N-myc in neuroblastoma cells. Cell Biol Int (2012) 36:331–7. doi: 10.1042/CBI20110231
156. Cui H, Li T, Ding H-F. Linking of N-Myc to death receptor machinery in neuroblastoma cells. J Biol Chem (2005) 280:9474–81. doi: 10.1074/jbc.M410450200
157. Qing G, Li B, Vu A, Skuli N, Zandra W, Liu X, et al. ATF4 regulates MYC-mediated neuroblastoma cell death upon glutamine deprivation. Cancer Cell (2012) 22:631–44. doi: 10.1016/j.ccr.2012.09.021
158. Aubry A, Yu T, Bremner R. Preclinical studies reveal MLN4924 is a promising new retinoblastoma therapy. Cell Death Discovery (2020) 6:2. doi: 10.1038/s41420-020-0237-8
159. Hansson K, Radke K, Aaltonen K, Saarela J, Manas A, Sjolund J, et al. Therapeutic targeting of KSP in preclinical models of high-risk neuroblastoma. Sci Trans Med (2020) 12:eaba4434. doi: 10.1126/scitranslmed.aba4434
160. Bellamy J, Szemes M, Melegh Z, Dallosso A, Kollareddy M, Catchpoole D, et al. Increased efficacy of histone methyltransferase G9a inhibitors against MYCN-amplified neuroblastoma. Front Oncol (2020) 10:14. doi: 10.3389/fonc.2020.00818
161. King D, Li XD, Almeida GS, Kwok C, Gravells P, Harrison D, et al. MYCN expression induces replication stress and sensitivity to PARP inhibition in neuroblastoma. Oncotarget (2020) 11:2020. 2141–2159. doi: 10.18632/oncotarget.27329
162. Southgate HED, Chen L, Tweddle DA, Curtin NJ. ATR inhibition potentiates PARP inhibitor cytotoxicity in high risk neuroblastoma cell lines by multiple mechanisms. Cancers (2020) 12:1095. doi: 10.3390/cancers12051095
163. Maeshima R, Moulding D, Stoker AW, Hart SL. MYCN silencing by RNAi induces neurogenesis and suppresses proliferation in models of neuroblastoma with resistance to retinoic acid. Nucleic Acid Ther (2020) 30:237–48. doi: 10.1089/nat.2019.0831
164. Adams D, Gonzalez-Duarte A, O’Riordan WD, Yang C-C, Ueda M, Kristen AV, et al. Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis. N Engl J Med (2018) 379:11–21. doi: 10.1056/NEJMoa1716153
Keywords: MYCN, cancer, gene amplification, G-quadruplex, NCYM, super enhancer, synthetic lethality
Citation: Liu R, Shi P, Wang Z, Yuan C and Cui H (2021) Molecular Mechanisms of MYCN Dysregulation in Cancers. Front. Oncol. 10:625332. doi: 10.3389/fonc.2020.625332
Received: 02 November 2020; Accepted: 18 December 2020;
Published: 03 February 2021.
Edited by:
Atsushi Takatori, Chiba Cancer Center, JapanReviewed by:
Enrique Hernandez-Lemus, Instituto Nacional de Medicina Genómica (INMEGEN), MexicoHeike Laman, University of Cambridge, United Kingdom
Shunpei Satoh, Saitama Cancer Center, Japan
Copyright © 2021 Liu, Shi, Wang, Yuan and Cui. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hongjuan Cui, aGN1aUBzd3UuZWR1LmNu