 Zhihui Liu
Zhihui Liu Samuel S. Chen
Samuel S. Chen Saki Clarke
Saki Clarke Veronica Veschi
Veronica Veschi Carol J. Thiele
Carol J. Thiele
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 08 February 2021
Sec. Molecular and Cellular Oncology
Volume 10 - 2020 | https://doi.org/10.3389/fonc.2020.623679
This article is part of the Research Topic Molecular Mechanisms and Treatment of MYCN-driven Tumors View all 14 articles
The deregulation of the MYC family of oncogenes, including c-MYC, MYCN and MYCL occurs in many types of cancers, and is frequently associated with a poor prognosis. The majority of functional studies have focused on c-MYC due to its broad expression profile in human cancers. The existence of highly conserved functional domains between MYCN and c-MYC suggests that MYCN participates in similar activities. MYC encodes a basic helix-loop-helix-leucine zipper (bHLH-LZ) transcription factor (TF) whose central oncogenic role in many human cancers makes it a highly desirable therapeutic target. Historically, as a TF, MYC has been regarded as “undruggable”. Thus, recent efforts focus on investigating methods to indirectly target MYC to achieve anti-tumor effects. This review will primarily summarize the recent progress in understanding the function of MYCN. It will explore efforts at targeting MYCN, including strategies aimed at suppression of MYCN transcription, destabilization of MYCN protein, inhibition of MYCN transcriptional activity, repression of MYCN targets and utilization of MYCN overexpression dependent synthetic lethality.
MYCN is a member of the MYC family of oncogenes, which also includes c-MYC and MYCL (1). MYCN was first reported in 1983 as an amplified gene homologous to v-myc in human neuroblastoma (2, 3). Like c-MYC, the MYCN gene encodes a basic helix-loop-helix-leucine zipper (bHLH-LZ) protein named N-Myc or MYCN. MYCN and c-MYC exhibit high-structural homology, including highly conserved Myc boxes (MB) and a BR-HLH-LZ motif (1, 4, 5). Both MYCN and c-MYC heterodimerize with MAX to bind to an enhancer-box (E-box) sequence with a consensus CAC(C/A)TG motif to regulate gene transcription (1, 4–6). MYCN and c-MYC differ in their expression patterns and regulation. While c-MYC is ubiquitously and highly expressed in most rapidly proliferating cells throughout development and in adult tissues, MYCN is preferentially expressed in neural tissues including the forebrain and hindbrain, as well as pre-B cells, cells in the intestine, heart and kidney during embryogenesis (5, 7). Tissue-specific conditional deletions demonstrated that c-MYC is necessary for the development and growth of specific hematopoietic cell lineages, crypt progenitor cells in the intestine and many other types of cells where c-MYC is expressed (8). MYCN but not c-MYC is essential during neurogenesis for the rapid expansion of progenitor cells and the inhibition of neuronal differentiation (9). Importantly, investigations at a gross level indicate that Mycn can substitute for c-Myc in murine development (10). For example, transgenic expression of Mycn from the c-Myc locus (c-MycN/N) rescues the embryonic lethality associated with the loss of c-Myc. Unlike c-Myc gene that is expressed throughout lymphocyte development, Mycn is only expressed in the precursor stage lymphocyte of development (10, 11), but Mycn can replace all c-Myc functions required for lymphocyte development in the c-MycN/N mice (10). However, subtle differences between c-MycN/N and normal mice were observed, such as the observation of periodic skeletal muscle dystrophy in some newborn c-MycN/N mice (10). This indicates a general functional similarity between these TFs in regulating certain lineages of murine cell growth and differentiation during embryogenesis and late development.
As TFs, both MYCN and c-MYC directly regulate transcription of genes that are involved in the control of cell growth, the cell cycle, proliferation, survival, apoptosis, pluripotency, self-renewal, DNA replication, RNA biology, metabolism, metastasis, angiogenesis and immune surveillance to play an oncogenic role (5, 12). Most studies indicate that MYC regulates differential gene transcription in the majority of cell types and model systems (4, 12–14). However, instead of regulating differential gene transcription, it has been shown that in B cells, the high levels of c-MYC expression results in a global increase in mRNA levels during the mitogenic stimulation of early B cells (15). Similarly, the expression of high levels of c-MYC in tumor cells leads to an increase in total levels of transcripts in each cell (16). These studies conclude that high levels of c-MYC amplify transcriptional output (15, 16). Further studies and analyses of existing data reveal that MYC dependent changes in global RNA levels may occur only when the cells are cultured under special conditions and/or after prolonged MYC activation. It is possible that a feedback effect from MYC-induced physiological and metabolic changes contributes to a global RNA amplification (4, 12, 13). MYCN interacts with Transcription Factor IIIC complex (TFIIIC), DNA topoisomerase II alpha (TOP2A) and the cohesion complex component RAD21, but in S phase, Aurora-A kinase displaces these interactors from MYCN to block MYCN-dependent promoter release of RNA polymerase II to suppress MYCN-dependent gene transcription (17). MYCN recruits BRCA1 to promoter-proximal regions, stabilizing mRNA de-capping complexes. This enables MYCN to suppress R-loop formation in promoter-proximal regions and prevent MYCN-dependent accumulation of stalled RNAPII, thus, enhancing MYCN transcriptional activation (18). The discovery of this non-canonical transcriptional function of MYCN may explain the discrepancy between universal binding and the small effects on relative and/or absolute mRNA levels of most genes that are bound by the MYC proteins (4, 18).
This review discusses MYCN genetic alterations in different types of cancers, the structure and transcriptional function of MYCN and the strategies used to target MYCN indirectly.
Deregulation of MYCN occurs in both pediatric cancers and adult cancers. MYCN amplification has been found in pediatric cancers including neuroblastoma, rhabdomyosarcoma, medulloblastoma, Wilms tumor and retinoblastoma. Amplification of the MYCN oncogene is present in 18–20% of all neuroblastomas (40% of high-risk neuroblastomas) and is an adverse prognostic factor (19–23). In alveolar rhabdomyosarcoma, amplification of MYCN is present in 25% of cases and overexpression of MYCN occurs in 55% of cases (24, 25). Amplification of MYCN is observed in 5–10% of medulloblastomas and is associated with poor prognosis (26–28). Copy number gains that include the MYCN locus are detected in 12.7% of Wilms tumors and 30.4% of diffuse anaplastic Wilms tumors, and MYCN gain is associated with poorer relapse-free and overall survival (29). In retinoblastomas, MYCN amplification is present in <5% of patients, and MYCN gain is associated with poor prognosis (30, 31). In adult cancers, amplification of MYCN is present in 40% of neuroendocrine prostate cancers and 5% of prostate adenocarcinomas (32), 15%-20% of small-cell lung cancers (33, 34) and 17.5% of basal cell carcinomas (35). Overexpression of MYCN is present in a subset of T-cell acute lymphoblastic leukemias (36), glioblastoma multiforme (37, 38) and breast cancer (39). Importantly, the amplification or overexpression of MYCN in the majority of these adult cancers is found to be associated with a poor prognosis.
To investigate whether MYCN functions as an oncogenic driver, genetically engineered mouse models (GEMM) have been generated to express MYCN in specific cell lineages. The transgenic expression of MYCN in the neural crest lineage of mice or zebrafish alone, or in combination with LMO1 or activated ALK gives rise to neuroblastomas (40–44). The transgenic expression of MYCN in murine luminal prostate epithelial cells in combination with Pten knockout results in a GEMM model with neuroendocrine prostate cancer formation (45). Mice transplanted with bone marrow expressing MYCN developed clonal and transplantable acute myeloid leukemias (46). When neural stem cells (NSCs) from different brain regions are transduced with a protein stabilizing MYCN(T58A) mutation and transplanted into their homotypic regions they give rise to distinct tumors. The transplantation of forbrain MYCN(T58A) NSCs gives rise to gliomas (47), while cerebellum and brain stem MYCN(T58A) NSCs transplants give rise to medulloblastoma and primitive neuroectodermal tumors (47). The enforced expression of MYCN in primary cerebellar granule neuron precursors isolated from Ink4c(-/-), p53(-/-) mice also results in medulloblastomas when transplanted into the brains of immunocompromised mice (48). These studies demonstrate that MYCN functions as an oncogene and is capable of driving tumor formation in cells with different lineage specific genetic programs to give rise to distinct tumor types. Thus, the inhibition of MYCN will be an important anti-tumor therapeutic strategy in many different human cancers with aberrantly over-expressed MYCN.
MYCN is composed of 464 amino acids (AA) with several functional domains (Figure 1) derived from sequence homology to known c-MYC protein functional domains (NCBI reference number of MYCN, NP_001280157.1 verse c-MYC, CAA25015.2) and mutagenesis analyses (1, 6). The N-terminal transcriptional regulation domain contains two highly conserved regions known as Myc Homology Box (MB), MBI and MBII. The central region contains 3 more MBs with a nuclear localization signal that overlaps MBIV. The C-terminal basic region (BR) is involved in DNA binding while the HLH-LZ heterodimerizes with Max (1). Many critical proteins regulating various biological processes do not have unique structures, but contain intrinsically disordered regions (IDRs), making this structural region extremely dynamic (49, 50). IDRs are involved in modulation of the specificity or affinity of protein binding interactions (49, 50). The IDRs in a protein can undergo characteristic disorder-to-order transitions upon interactions with specific binding partners and/or through post-translational modifications (49). Using a protein intrinsic disorder region prediction tool PONDR (http://www.pondr.com/) (51) to analyze MYCN, we find that the majority of MYCN residues tend to form broad disordered regions (Figure 1), which indicates that MYCN has the potential to bind to many different partners. Moreover, the intrinsically disordered character of MYCN suggests that using it as a direct drug target would be challenging due to its structural flexibility.
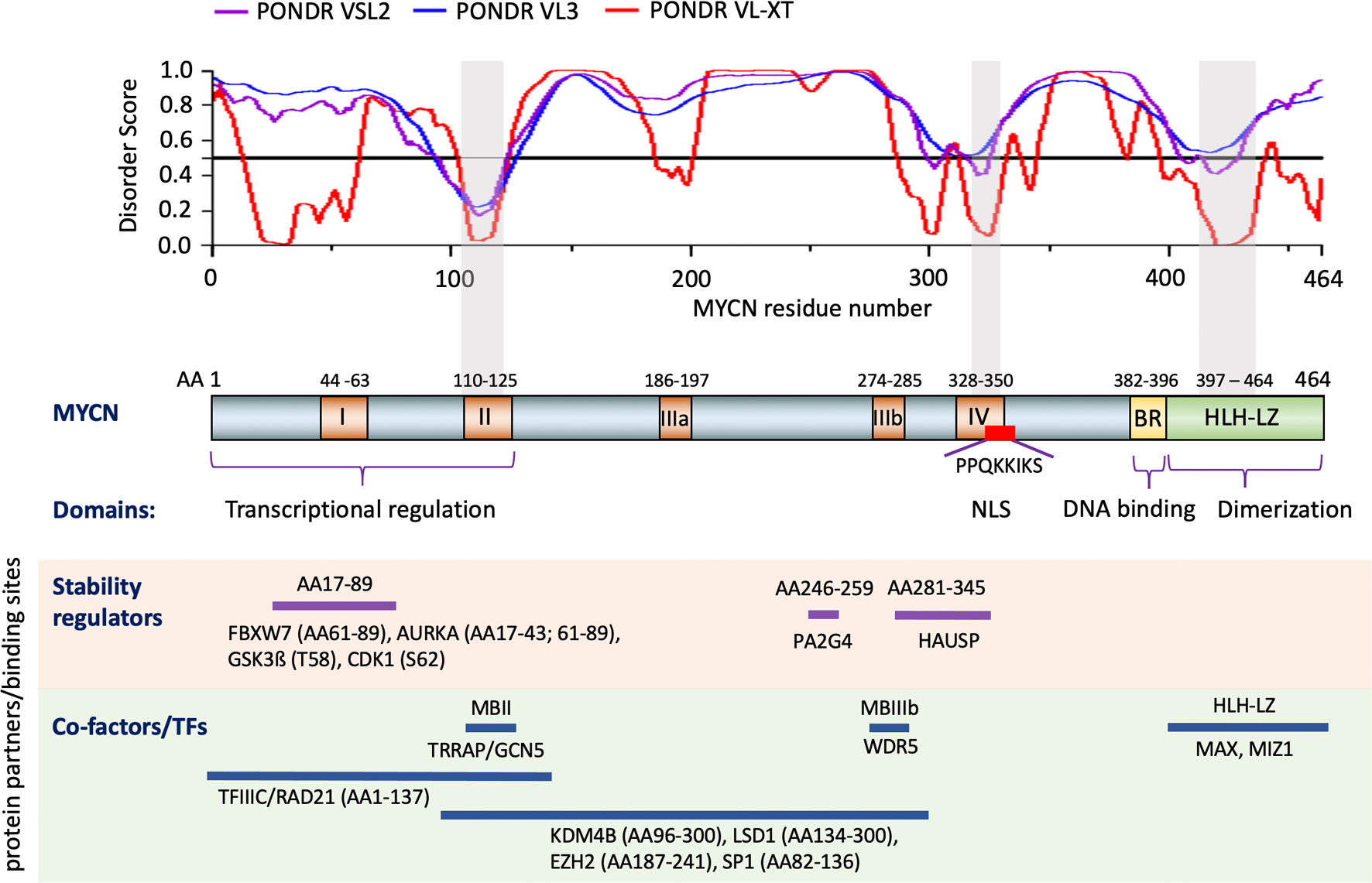
Figure 1 Structure and functional domains of MYCN. Three predictors of the intrinsically disordered region prediction tool PONDR are used to identify intrinsically disordered regions of MYCN (top section). Functional domains of MYCN defined by comparing c-MYC and mutagenesis assay (middle section). Examples of known MYCN protein partners and the regions of MYCN that contributed to the interaction (bottom section). Notes: Color boxes on the MYCN protein diagram: brown box, Myc homology Box (MB) I-IV; yellow box, Basic Region (BR); green box, Helix-Loop-Helix-Leucine Zipper (HLH-LZ); red box, nuclear localization signal (NLS). Gray shade box on the disorder score graph and MYCN protein diagram, regions of MYCN with relatively low disorder score. AA: amino acid.
Critical regions within MYC/MYCN proteins have been implicated in regulating protein stability. Residues Ser62 (S62) and Thr58 (T58) within MBI are critical phosphorylation sites for MYC/MYCN protein stability during cell cycle progression. As growth factors stimulate cell progression through the cell cycle, protein stability is tightly regulated. Phosphorylation at S62 of MYCN protein is mediated via CDK1, which stabilizes MYCN and primes T58 for phosphorylation by GSK3β. GSK3β is repressed by phosphatidylinositol 3-kinase (PI3K) and AKT kinase signaling (52–57). Dephosphorylation of MYC-S62 via protein phosphatase 2A (PP2A) enables E3 ligase FBXW7 binding to phosphorylated MYC-T58, targeting it for ubiquitination and subsequent degradation by the proteasome (58, 59). The regulation of MYCN protein stability is cell-cycle dependent. In normal neuronal progenitors, CDK1 phosphorylates MYCN protein at S62 in G1-phase. As cells enter M-phase, signaling by growth factors declines leading to activation of GSK3β enabling phosphorylation of MYCN(T58) which leads to its degradation (52). Two additional ubiquitin ligases, TRIM32 and HUWE1, are involved in regulation of MYCN degradation. During late M-phase, the ubiquitin ligase TRIM32 is bound to the mitotic spindle pole apparatus in conjunction with MYCN, contributing to its ubiquitination and degradation (60). HUWE1, a HECT-domain E3 ubiquitin ligase, binds to MYCN and primes it for MYCN-K48-linked polyubiquitination and proteasomal-mediated degradation (61, 62).
MYCN protein degradation is antagonized through interactions with different proteins at distinct MYCN regions (Figure 1). Aurora A kinase (AURKA) associates with the mitotic spindle poles and interacts with the N-terminus of MYCN in cells over-expressing MYCN and in this way interferes with FBXW7-mediated degradation leading to MYCN stabilization (17, 56, 63). The ubiquitin-specific protease HAUSP binds to a partially overlapping region of MBIII and MBIV in MYCN, but not c-MYC (Figure 1), to specifically deubiquitinate MYCN, which results in MYCN protein stabilization (64). The proliferation-associated 2AG4 protein (PA2G4) directly binds to and stabilizes MYCN by protecting it from ubiquitin-mediated proteasomal degradation (65). Co-immunoprecipitation results show that a MYCN deletion mutant (AA82–254) binds strongly to PA2G4, and further studies show that a 14 AA MYCN oligopeptide (AA246–259) sequence contributes to this protein-protein interaction (65). Additionally, MYCN has been found to be methylated at R160, R238 and R242; protein arginine methyltransferase 5 (PRMT5) physically interacts with MYCN and increases MYCN protein stability, possibly by methylating MYCN at R242 (66). The identification of different signaling pathways and proteins regulating MYCN protein stability provides additional modes for indirect targeting of MYCN.
TFs, co-repressors and co-activators interact with different regions of MYCN (Figure 1), enabling MYCN to activate or repress gene transcription. MYC family proteins directly interact with MAX through HLH-LZ to form a heterodimer and activate transcription by binding to E-box elements (1, 6). Activation involves the recruitment of multiple coactivators and protein complexes to E-box elements. The TIP60 acetyltransferase complex and the histone acetyltransferase GCN5 are bound to MYC indirectly through the TRRAP adaptor protein that interacts with MBII of the MYC protein (67–70). Two other proteins, TIP48 and TIP49, found in the TIP60 complex, are involved in chromatin remodeling and bind to the N-terminus of MYCN (67). Recent studies show that target gene recognition by c-MYC and MYCN depends on its interaction with the histone H3-K4-methyl-associated protein WDR5 and their interaction is mediated through the MBIIIb region of c-MYC (71, 72). MYCN interacts with the H3K9me3/me2 demethylase KDM4B through the region between AA96-300 and when overexpressed MYCN recruits KDM4B to E-box regions to decrease H3K9me3 levels (73). Thus, MYCN may also activate gene transcription by relieving transcriptional repression. Moreover, MYCN AA1-137 also interacts with TFIIIC and RAD21 to regulate the pause release of RNA Polymerase II (17). BRCA1 interacts with MYCN and enables MYCN to suppress R-loop formation in promoter-proximal regions, thus enhancing transcription (18). When MYCN functions as a transcriptional repressor, it interacts with SP1 and MIZ1 to repress gene transcription (74–77). The region between MYCN AA82-136 that includes the MBII domain specifically interacts with SP1 in pull down experiments, whereas MYCN AA400-464 that includes the HLH-LZ domain interacts with MIZ1 (74). These MYCN/SP1/MIZ1 interactions repress gene transcription by recruiting HDAC1 (74). MYCN, via MBIII, associates with EZH2, a methyltransferase and member of the polycomb repressor complex 2, to suppress gene transcription (78). Similarly, MYCN physically binds lysine-specific histone demethylase 1A (KDM1A/LSD1) through MBIII to repress gene transcription (79). The above studies show how MYCN interacts with the epigenome to regulate gene transcription.
Many mechanisms have been identified to be involved in the transcriptional regulation of MYCN (Figure 2). Soon after the discovery of the MYCN gene, it was found that retinoic acid (RA) treatment of NB cells resulted in a down-regulation of MYCN expression at the mRNA level, and this preceded cell cycle arrest and implementation of a differentiation program (80). This indicated that MYCN down-regulation, at least partially, contributes to the biological effect of RA on NB cells. A classic RA response element (RARE) was not implicated in RA regulation of MYCN transcription, as studies showed that RA exerts its effects across multiple regulatory regions within the MYCN promoter, distally or even on different chromosomes (81). Retinoid repression of MYCN transcription was a major motivation for the inclusion of 13 cis-retinoic acid during the consolidation phase of treatment for high-risk neuroblastomas (82). Retinoid regulation of MYCN represents one of the first strategies developed to target MYCN gene transcription and provides an example of indirect targeting of MYCN.
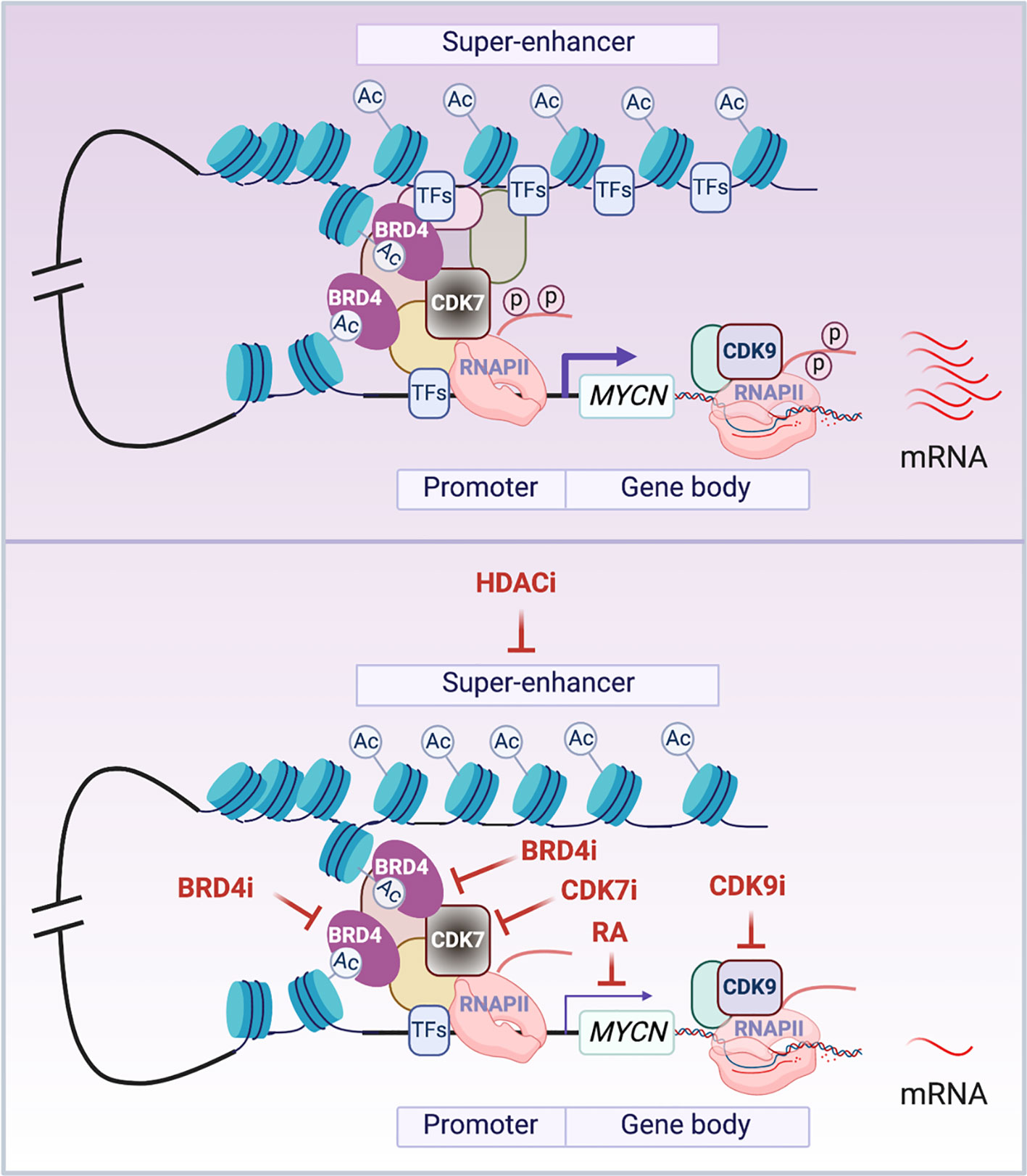
Figure 2 Transcriptional regulation of MYCN. The schematic illustrates the presumed looping between the super-enhancer (SE) and the promoter of MYCN gene. In cancer cells, MYCN is driven by SEs that are marked by stretches of acetylated lysine 27 of histone 3 (H3K27Ac). BRD4 is a chromatin ‘reader’ that binds to acetylated lysine residues (AcK) and activates MYCN transcription. CDK7 is a TFIIH subunit that phosphorylates the carboxy-terminal domain of RNA Pol II (RNAPII) to initiate MYCN gene transcription. CDK9 is a pTEFb subunit that phosphorylates the carboxy-terminal domain of RNAPII to regulate MYCN transcriptional elongation. The enrichment or activation of these components of the transcriptional machinery in cancer cells results in aberrantly elevated transcription of MYCN (top panel). The treatment of cells with HDAC inhibitors (HDACi) inactivates MYCN SEs possibly through disrupting normal looping and depleting transcription factors (TFs) that bind to the SEs; BRD4 inhibitors (BRD4i) impact the ‘reader’ function of BRD4 to inactivate MYCN gene transcription; CDK7 inhibitors (CDK7i) and CDK9 inhibitors (CDK9i) treatment impedes the phosphorylation of RNAPII to inhibit MYCN gene transcription initiation and elongation; RA treatment inactivates MYCN transcription in a RA response element independent manner (bottom panel). Notes: circled ‘Ac’ represents H3K27Ac; circled ‘p’ represents phosphate at the RNAPII tail.
Gene transcription is mediated by cis-regulatory elements such as enhancers and promoters. Enhancers are distal regulatory elements in the genome that play an important role in driving cell-type-specific gene expression and are frequently mis-regulated in cancer (83, 84). Super-enhancers (SEs) are composed of a cluster of enhancers that are central to the maintenance of cell identity in normal development and disease (85). SEs were found to be associated with various oncogenic molecules including both c-MYC and MYCN; this makes them putative therapeutic targets for cancer therapy (86–89). Histone deacetylases (HDACs) have an important function in regulating both DNA packaging in chromatin and gene transcription. Treatment of NB cells with HDAC inhibitors such as MS-275, BL1521 or SAHA resulted in a decrease in MYCN mRNA levels accompanied by cell apoptosis (90–92). Although not directly demonstrated, recent studies have shown that HDAC inhibition results in enhancer remodeling and suppression of oncogenic SEs possibly through disruption of normal chromatin-looping and TFs depletion on the SEs (93, 94). This may be involved in the HDAC inhibitor mediated repression of MYCN transcription (Figure 2).
MYC-driven tumors are especially sensitive to inhibition of BET bromodomain containing proteins (BRD1–4) (95). BRD4 belongs to family of proteins that contain variable numbers of bromodomains and a central ET domain and function as chromatin “readers” by binding to acetylated lysine residues (Figure 2). BRD4 has also been implicated in regulating RNA-PolII transcriptional activity (96). BET inhibitors downregulate c-MYC transcription, suppress MYC-dependent target genes and inhibit myeloma cell proliferation (95). An unbiased screen of 673 genetically characterized tumor-derived cell lines shows that neuroblastoma cell lines with MYCN amplification are more sensitive to JQ1 treatment compared to MYCN-wild-type tumors. BRD4 knock-down phenocopied these effects, indicating that BRD4 functions as a transcriptional regulator of MYCN. Importantly, BET bromodomain-mediated inhibition of MYCN suppresses neuroblastoma growth both in vitro and in vivo (97). Similarly, OTX015 (Oncoethix), a small molecule that prevents BRD2/3/4 from binding to acetylated histones, also represses MYCN transcription. This study showed that BRD4 binds to super-enhancers (SEs) and MYCN target genes, while OTX015 treatment disrupts BRD4 binding and transcription of MYCN as well as its target genes (98), which is consistent with the finding that bromodomain inhibitor treatment selectively inhibits oncogenes by disrupting their SEs (99). Importantly, bromodomain and extra-terminal domain inhibitor (BETi), GSK525762, is under phase I clinical trial for solid tumors including NB (100). In addition to MYC, many tumor-associated genes such as RUNX1, FOSL2, BCL3 and ID2 are driven by SEs in diverse tumor types (99). However, SEs drive many cell identity genes essential for normal cell development, such as Oct4, Sox2 and Nanog in embryonic stem cells (101), so as with many cytotoxic agents a therapeutic window is needed when using BETi for the treatment of cancer patients to minimize side effects. Recent studies showed that the combination of a bromodomain inhibitor with a CDK7 inhibitor, an AURKA inhibitor or an HDAC inhibitor is significantly more effective in suppressing MYCN-driven NB tumor growth than either drug alone (88, 102, 103). This highlights the importance of combinatorial therapeutic approaches for cancer treatment.
To regulate gene transcription, the RNA polymerase II (RNAPII) transcription initiation apparatus needs to be recruited to promoters by specific DNA binding transcription factors. Promoter-proximal pausing of RNAPII is a post-initiation regulatory event, and c-Myc plays a key role in release of Pol II at many actively transcribed genes in ES cells (104). Cyclin-dependent kinases (CDKs) are important in regulating the transcription cycle of RNAPII. The TFIIH subunit CDK7 and the pTEFb subunit CDK9 phosphorylate the carboxy-terminal domain of RNAPII, facilitating efficient transcriptional initiation, pause release and elongation. This suggests that the inhibition of these CDKs would be expected to block MYC-driven transcriptional amplification. Indeed, THZ1, a covalent inhibitor of CDK7, was found to selectively target MYCN-amplified NB cells, leading to global repression of MYCN-dependent transcriptional amplification and reductions in expression of SE-associated oncogenic drivers including MYCN itself and suppression of NB tumor xenograft growth (87). CYC065, an inhibitor of CDK9 and CDK2, was found to selectively target MYCN-amplified NB cells by leading to a selective loss of nascent MYCN transcription (105). These studies indicate that the inhibition of CDK7 or CDK9 can be exploited to disrupt aberrant MYCN-driven transcription and to repress MYCN gene transcription as a therapeutic for MYCN-driven cancers (Figure 2).
DNA G-quadruplexes (G4s) are noncanonical DNA structures that are formed by guanine-rich DNA sequences. They often occur in the promoter regions of oncogenes and regulate their expression (106–108). An early study identified a specific G4 structure formed in the c-MYC promoter region. Although a cationic porphyrin TmPyP4, which binds non-selectively to G4s in vitro was able to inhibit the transcription of c-MYC (108), a recent study identified a small molecule DC-34 that more specifically binds to the c-MYC G4 in vitro. In a G4-dependent mechanism, DC-34 plays a more potent and selective role in downregulating MYC gene transcription compared to other G4 containing oncogenes in leukemia cells. Moreover, the treatment of cancer cells with DC-34 results in a G0-G1 arrest and a reduction of cell viability (106). Although not yet reported, the identification and targeting of G4s within the MYCN promoter and regulatory regions would be another approach to inhibit MYCN gene transcription.
Many TFs have been validated as oncogenes in human cancers and their dysregulated transcriptional programs result in a high dependency of cancer cells on these gene expression regulators (109). Importantly, RA, inhibitors of HDACs, BET bromodomain containing proteins, CDK7, CDK9 and small molecules that bind to G4s have been demonstrated to be effective for the treatment of many types of cancers by targeting their dysregulated transcriptional programs (109, 110). Thus, targeting MYCN at branch points involved in its oncogenic regulation of transcription (Figure 2) is an important therapeutic approach for MYCN-driven cancers.
MYCN is a short-lived protein whose stability is tightly regulated by different signaling pathways that target it for ubiquitin-mediated degradation by the proteasome (52, 55, 111) (Figure 3). A major signaling pathway affecting MYCN protein stability occurs upon activation of PI3K. PI3K activates Akt which phosphorylates GSK3ß, suppressing GSK3ß kinase activity. This results in decreased phosphorylation of MYCN-T58 which is critical for targeted degradation by the proteasome (55) (Figure 3). As expected, inhibitors of PI3K destabilize the MYCN protein and suppress tumor growth in the TH-MYCN GEMM NB model (53, 112). In NB cells, AURKA interacts with MYCN by interfering with the FBXW7 subunit of the ubiquitin protein ligase complex to impede MYCN ubiquitination and subsequent degradation (56) (Figure 3). Treatment with AURKA inhibitors decreases MYCN protein levels resulting in suppression of NB tumor growth, making AURKA a suitable target for MYCN-driven cancers (32, 113–117). Due to the promising preclinical results, the oral AURKA inhibitor MLN8237 is under clinical evaluation for multiple cancers including relapsed NB (118). PLK1 is a serine/threonine kinase formally known as the polo-like kinase. The PLK1 inhibitor BI 2356 exhibits strong antitumor activity in NB cells in vitro and in vivo (119). PLK1 does not directly bind to the MYCN protein. Rather, it increases MYCN protein stability by destabilizing the FBXW7 ubiquitin ligase complex to counteract FBXW7-mediated degradation of MYCN (120) (Figure 3). Importantly, MYCN-amplified tumor cells in neuroblastoma and small cell lung cancer are more sensitive to treatment with PLK1 inhibitors than tumors with normal MYCN copy number, indicating that PLK1 inhibitors are potential therapeutics for MYCN-overexpressing cancers (120).
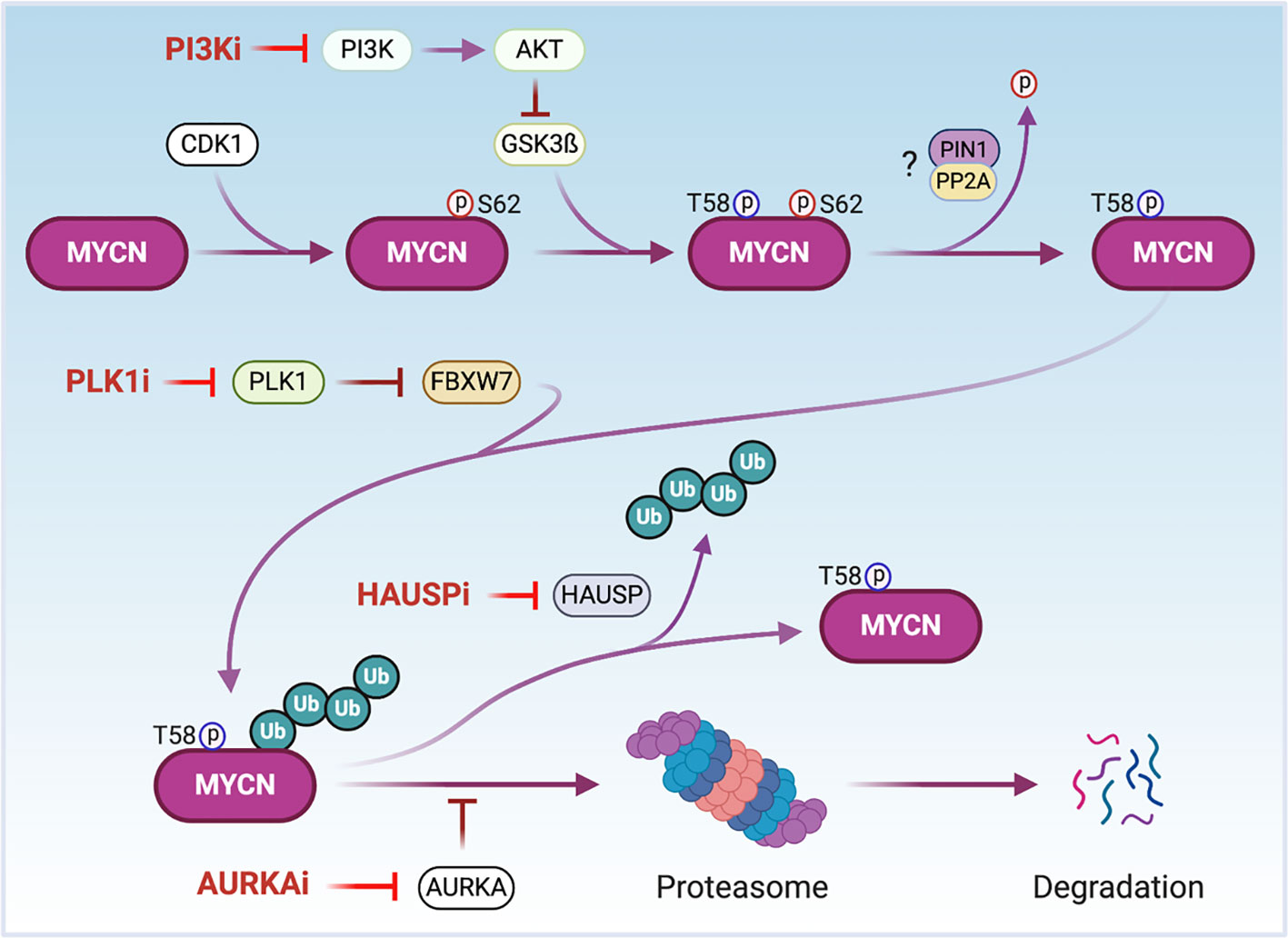
Figure 3 The regulation of MYCN protein stability. CDK1 phosphorylates MYCN at serine 62 (S62) to stabilize MYCN and prime threonine 58 (T58) for phosphorylation via GSK3β. AKT phosphorylates GSK3ß inactivating its kinase. After dephosphorylation of S62 possibly through PIN1/PP2A, MYCN is poly-ubiquitinated by the ubiquitin ligase FBXW7 and undergoes proteolytic degradation via the proteasome. AURKA binds to and stabilizes phosphorylated and poly-ubiquitinated MYCN to protect MYCN from degradation. PLK1 destabilizes FBXW7 to counteract FBXW7-mediated degradation of MYCN. The ubiquitin-specific protease HAUSP deubiquitinates MYCN to stabilize it. Thus, the treatment of cells with PI3K, AURKA, PLK1 or HAUSP inhibitors (PI3Ki, AURKAi, PLKi or HAUSPi) leads MYCN proteasomal degradation. Notes: circled ‘p’ represents phosphate; circled ‘Ub’ represents ubiquitin.
Components of the proteasome targeting and degrading system contribute to MYCN protein regulation. The ubiquitin-specific peptidase HAUSP (also known as USP7) binds to and deubiquitinates MYCN leading to its stabilization (64) (Figure 3). HAUSP is highly expressed in tumors from NB patients with poor prognoses. Silencing of HAUSP expression in NB cells destabilizes MYCN and results in an inhibition of MYCN mediated functions. Importantly, the HAUSP inhibitor P22077 markedly suppresses the growth of MYCN-amplified human neuroblastoma cell lines in xenograft mouse models (64).
Although first identified as an RNA binding protein, the proliferation associated 2G4 protein, PA2G4, directly binds and stabilizes MYCN by protecting MYCN from proteasomal degradation (65). When PA2G4 is silenced in NB cells using siRNAs or a small molecule inhibitor WS6, MYCN protein levels are markedly reduced (65). WS6 treatment of NB cell lines completely blocked PA2G4-MYCN protein binding, and this competitive chemical inhibition results in a delay of tumorigenesis in the TH-MYCN NB mouse model (65).
Protein methylation is a post-translational modification recently identified to regulate protein stability. The protein arginine methyltransferase 5 (PRMT5) interacts with both MYC and MYCN proteins (66, 121). Silencing of PRMT5 in MYCN‐overexpressing NB cells or MYC-driven medulloblastoma cells leads to a decrease in MYCN and MYC protein levels and cell growth inhibition (66, 121). Tandem mass spectrometry analysis of immunoprecipitated MYCN protein in NB cells reveals several potential sites of arginine dimethylation on MYCN protein, suggesting that MYCN may be methylated by PRMT5 as a protection from proteasomal degradation (66). Treatment with the PRMT5 inhibitor EPZ015666 results in a decrease of MYC protein levels and medulloblastoma cell growth, which suggests that PRMT5 inhibitors are potential therapeutics for MYC- and MYCN-driven cancers.
We have described critical regions that are needed for MYCN interactions with its cofactors/coregulators in the previous section and in Figure 1. As a TF, MYCN cooperates with other TFs to bind to DNA and recruit cofactors/coregulators to activate or repress gene transcription, making these protein partners potential targets to disrupt the transcriptional activity of MYCN (Figure 4). The enzymatic activity of many co-regulators makes them attractive drug targets.
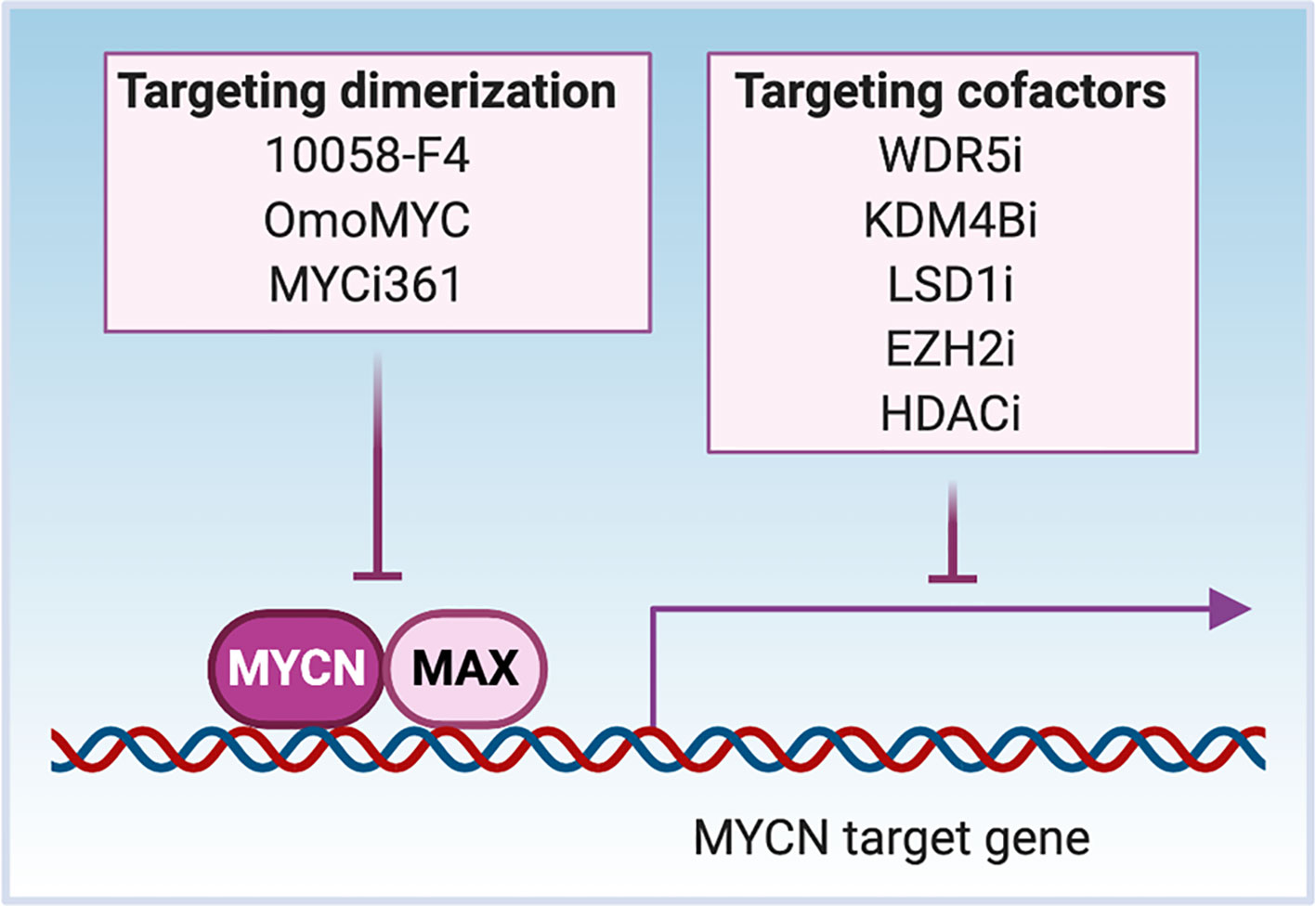
Figure 4 Targeting MYCN transcriptional activity. MYCN heterodimerizes with MAX to bind to the cis-genomic elements in DNA. MYCN interacts with cofactors WDR5 and KDM4B to activate gene transcription, while interacts with LSD1, EZH2 and HADCs to repress gene transcription through affecting chromatin status. Inhibitors such as 10058-F4, OmoMYC and MYCi361 disrupt the dimerization between MYCN and MAX to inhibit the DNA binding of MYCN. The treatment of cells with MYCN cofactor inhibitors (WDR5i, KDM4Bi, LSD1i, EZH2i or HDACi) inactivates MYCN transcriptional activity through regional epigenetic modification and/or opening or closing chromatin.
The first identified mechanism through which MYCN functions as a TF is via heterodimerization with MAX. MYC-MAX complexes recognize E-box DNA sequences, and binding of the heterodimer to gene promoters activates transcription of downstream MYCN-related genes. Small-molecule inhibitors of MYC-MAX dimerization illustrate the importance of dimerization to MYC function (Figure 4). For instance, the peptidomimetic compound IIA4B20 exerts a strong inhibitory effect on MYC-MAX dimerization and DNA binding to functionally inhibit MYC-induced fibroblast transformation (122). The compound 10058-F4 binds to AA402-409 of MYC, which disrupts MYC-MAX dimerization of either c-MYC or MYCN. The treatment of MYCN-amplified NB cells with 10058-F4 leads to neural differentiation (123–125). Another known inhibitor of MYC-MAX dimerization is OmoMYC, a c-MYC derived mutant bHLH-LZ domain protein generated by substituting four amino acids within the c-MYC leucine zipper. When overexpressed, OmoMYC competes with MAX for binding to either c-MYC or MYCN and prevents MYC/MYCN proteins from binding to E-boxes and activating transcription (126, 127). The recently discovered MYC inhibitor 361 (MYCi361) binds to the HLH region of the MYC protein (AA366-378), disrupts MYC/MAX heterodimerization, enhances degradation of both MYC and MYCN, and suppresses MYC-dependent tumor cell growth in vitro and in vivo (128). The asymmetric polycyclic lactam, KI-MS2-008 stabilizes MAX homodimers, resulting in decreased MYC protein levels (129). Treatment of cancer cells with KI-MS2-008 suppresses MYC-dependent tumor growth in vivo. This is another example whereby altering the ability of MAX to dimerize with MYC functionally targets MYC (129). It will be interesting to evaluate whether either of these approaches affects MAX/MYCN interactions to inhibit the growth of MYCN-driven cancers.
MYCN has been shown to recruit several druggable cofactors with methylase and demethylase activity to regulate gene transcription, and cofactor inhibition provides a way to indirectly target MYCN (Figure 4). The histone H3K4 methyltransferase complex subunit WDR5 forms a protein complex with MYCN at the MDM2 promoter that results in histone H3K4 trimethylation and activation of MDM2 transcription (72). Treatment of NB cells with the WDR5 antagonist OICR9429 reduces MYCN/WDR5 complex formation and the expression of MYCN target genes, resulting in the inhibition of cell growth (72). When MYCN is overexpressed, it interacts with the H3K9me3/me2 demethylase KDM4B and recruits KDM4B to E-box containing regions to decrease H3K9me3 levels (73). Functional studies demonstrate that KDM4B acts as a MYCN co-activator to regulate MYCN signature genes. Knockdown of KDM4B decreases NB cell proliferation in vitro and NB xenograft growth in vivo, which provides proof-of-concept for the potential therapeutic efficacy of inhibiting KDM4B to target oncogenic MYCN signaling in cancers (73). MYCN has also been found to recruit co-repressors to suppress gene transcription. MYCN associates with EZH2, a methyltransferase and a member of the polycomb repressor complex 2 (PRC2) to repress the NB tumor suppressor gene CLU through a bivalent modification of the chromatin at the CLU promoter (78). The prevalence of this activity has not been evaluated. In MYCN-amplified tumors, MYCN increases levels of EZH2 and components of the PRC2 complex leading to increased activity of PRC2-mediated transcriptional repression primarily of differentiation associated genes. Genomic or pharmacologic inhibition of EZH2 suppresses NB growth in vitro and in vivo (130–132). MYCN also binds the lysine-specific histone demethylase 1A (KDM1A/LSD1) to repress gene transcription. LSD1 co-localizes with MYCN on promoter regions of CLU and CDKN1A, and the treatment with an LSD1 inhibitor restores the expression of these genes and suppresses NB cell growth (79). c-MYC interacts with histone methyltransferase EHMT2 to repress gene transcription, and knockdown of EHMT2 results in decreased tumor volume (133). EHMT2 is essential in NB cells and inhibition of EHMT2 using BIX-01294 decreased proliferation of NB cells and induced apoptosis (132, 134).
Acetylation and deacetylation of histones are key regulatory features of gene transcription and are potential targets that regulate MYCN transcriptional activity. MYCN recruits many HDACs (HDAC1, HDAC2 and HDAC5) to repress gene transcription (74, 135, 136). The histone acetyltransferase, GCN5, binds to MYC and MYCN proteins (67–69). In vitro luciferase assays show that MYC recruits GCN5 to activate gene transcription (70); however, few GCN5 specific inhibitors are available and have limited testing in NB cells (137).
As a TF, MYCN regulates many target genes but the critical ones that mediate MYCN tumor initiating functions are not clear. One of the MYCN downstream targets that is under clinical evaluation is ornithine decarboxylase 1 (ODC1), the rate-limiting enzyme involved in polyamine synthesis (138, 139). In neuroblastoma, the expression levels of MYCN are strongly correlated with those of ODC1, and high levels of ODC1 driven by MYCN amplification and overexpression are strongly associated with poor clinical outcome in NB patients (138). Treatment of TH-MYCN transgenic mice with the ODC inhibitor α-difluoromethylornithine (DFMO) prevents oncogenesis in hemizygous mice, while delaying tumor development in homozygous mice. Transient Odc ablation in hemizygous TH-MYCN mice permanently prevented tumor onset. This work indicates that ODC mediates an oncogenic function of MYCN that is important in tumor initiation and demonstrates the therapeutic potential of polyamine depletion strategies in NB (138, 139). A recent Phase II study of single agent DFMO as maintenance therapy in NB showed increased survival compared to historical controls for high-risk NB patients (140, 141).
The FACT (facilitates chromatin transcription) complex is another potential MYCN downstream target that is druggable. FACT facilitates transcriptional elongation on chromatin templates by binding and displacing the H2A/H2B dimer from nucleosomes, a process that is believed to be required for RNA polymerase II to pass through a nucleosomal barrier (142). MYC is confirmed to interact with a component of the FACT complex, the transcription elongation factor SSRP1 (4). SiRNA knockdown experiments demonstrate that expression of FACT and MYCN is controlled in a forward feedback loop, which drives MYCN transcription and protein stability (143). Inhibition of FACT using the small molecule curaxin compound CBL0137 results in a decrease of MYCN and SSRP1 expression, as well as a markedly reduced NB tumor initiation and progression in the TH-MYCN mice especially when combined with standard chemotherapy (143).
Synthetic dosage lethality (SDL) is a genetic interaction in which the alteration of one gene, combined with the reduction in function of a second gene, results in lethality (144). SDL is an attractive therapy for cancer because inhibition of such a gene will only induce cell death in cells carrying the specific gene alteration. MYCN activates both proliferative and apoptotic cellular responses. Whether it promotes a net proliferative response is dependent on cooperating apoptotic factors such as the antiapoptotic protein BCL2 (145, 146). It has been demonstrated that MYCN-amplified neuroblastoma cells are highly sensitive to BCL2 inhibitors ABT-263 (navitoclax) and ABT-199 (venetoclax) (147). When screening for enhancers of ABT-199 sensitivity in MYCN-amplified NB, researchers found that the Aurora Kinase A inhibitor (alisertib) cooperates with ABT-199 to induce widespread apoptosis. This drug combination was more effective in killing MYCN-amplified NB cells in vitro and in vivo than either compound alone (147). Moreover, in MYCN-amplified NB, Polo-Like Kinase 1 (PLK1) and MYCN create a positive, feedforward activation loop essential for maintaining their high levels of expression (120). BCL2 antagonists have been shown to synergize with inhibitors of PLK1, such as BI6727 or BI2356 and may be an effective drug combination for NB over-expressing MYCN (120).
One of the mechanisms through which MYCN exerts its tumorigenic effect in NB is to activate transcription of genes involved in proliferation, including checkpoint kinase 1 (CHK1), an important regulator of the G1/S and G2/M checkpoints. This mechanism may contribute to the ability of MYCN-amplified NB tumors to become refractory to standard chemotherapy (148). Conversely, tumor cells lacking DNA damage checkpoints during tumorigenesis or during cytotoxic therapy are highly sensitive to additional genomic instability (149). MYCN induces replication stresses and DNA damage through excessive replication-fork firing. MYCN-overexpressing tumors are more sensitive to CHK1 inhibition (150, 151). Another cell cycle related synthetic lethality protein identified in MYCN-amplified NB is cyclin-dependent kinase 2 (CDK2) (152). Knockdown of CDK2 or treatment with the CDK2 inhibitor roscovitine induces apoptosis in MYCN-amplified neuroblastoma cell lines but not in those with MYCN single copy. Thus, inhibition of CDK2 is synthetically lethal to NB cells with overexpressed MYCN (152).
NB arising in adolescents and young adults is frequently associated with loss of function mutations in the alpha thalassemia X-linked (ATRX) gene (153, 154). Interestingly, ATRX mutations and MYCN amplification have never been observed in the same NB tumor, suggesting a potential synthetic lethal condition (153, 154). Doxycycline-induced overexpression of MYCN in ATRX-mutant NB cell lines showed a marked loss of tumor cells. Moreover, in the LSL-MYCN GEM of NB tumors failed to develop when LSL-MYCN : Dbh-iCre NB mice were crossed with ATRXflox mice demonstrating synthetic lethality between mutant ATRX and high levels of MYCN (154). This is an example of rare synthetic lethality between an inactivated tumor suppressor and an activated oncogene. MYCN has been shown to play an apoptotic role in cancer cells under certain circumstances (155). Thus, it is possible that under the stress of DNA replication, when ATRX is inactivated, high levels of MYCN induce an apoptotic cellular response. Therefore, ATRX targeting may be a therapeutic approach in MYCN-amplified NB tumors. Alternative strategies that increase MYCN protein levels may lead to an SDL situation in ATRX-mutant NB cells. Increasing MYCN levels may be achieved by interfering with critical components in the MYCN protein degradation pathway, such as HUWE1. HUWE1 ubiquitinates and directs MYCN degradation to the proteasome (61). Knockdown of the HUWE1 gene impedes MYCN degradation and increases MYCN protein levels in NB cells (61). HUWE1 inhibitors such as BI8622 and BI8626 have been generated, but not tested in this situation.
Complementing experimental approaches to the identification of SDL in tumor cells, a recent computational approach utilized accumulating tumor genomic data to identify candidate SDL networks in various cancers (156). Synthetic lethality or ‘oncogene addiction’ offers an attractive therapeutic strategy for MYCN-driven cancers. Both bioinformatic analysis and high throughput drug screening can be used to identify novel-druggable synthetic lethal genes to which MYCN expressing cells are ‘addicted’.
Once considered undruggable, recent advances in chemistry and chemical genomics have begun to directly target transcription factors. Covalent reaction with their protein targets through cysteine residues is a known mechanism for many covalent drugs (157). A recent in vitro study that screened a library of cysteine-reactive covalent ligands, consisting of acrylamides and chloroacetamides, identified EN4. EN4 directly and covalently modifies the pure full-length c-MYC protein at cystine 171 (C171) of its intrinsically disordered region (158). In cells EN4 targets MYC interfering with MYC transcriptional activity. This reactive C171 on c-MYC is not conserved in MYCN. However, a similar screening approach could be used to identify small molecules that target cysteine residues in MYCN.
Proteolysis targeting chimeras (PROTACs) induced protein degradation is a recently developed therapeutic strategy, especially for undruggable targets (159). PROTACs are composed of three chemical elements: 1) a ligand binding to a target protein, 2) a ligand binding to E3 ubiquitin ligase, and 3) a linker for conjugating these two ligands (159). The small molecules 10058-F4, 7954-0035-G5, 10074-G5, JKY-2-169, MYCi361 and MYCi975 have been shown to bind to the HLH domain of MYC protein with some binding to MYCN protein as well (125, 128, 160). It may be possible to use these small molecules to develop PROTACs reagents to directly target and degrade the MYC/MYCN proteins (Figure 5). Intrinsically disordered region analysis of MYCN indicates that the MB II domain of MYCN is the most ordered region of MYCN having less flexibility and interacts with TRRAP (Figure 1). A small molecule or peptide screen to identify binders to the MBII region of MYCN would be another strategy to identify components needed to construct a MYCN PROTAC.
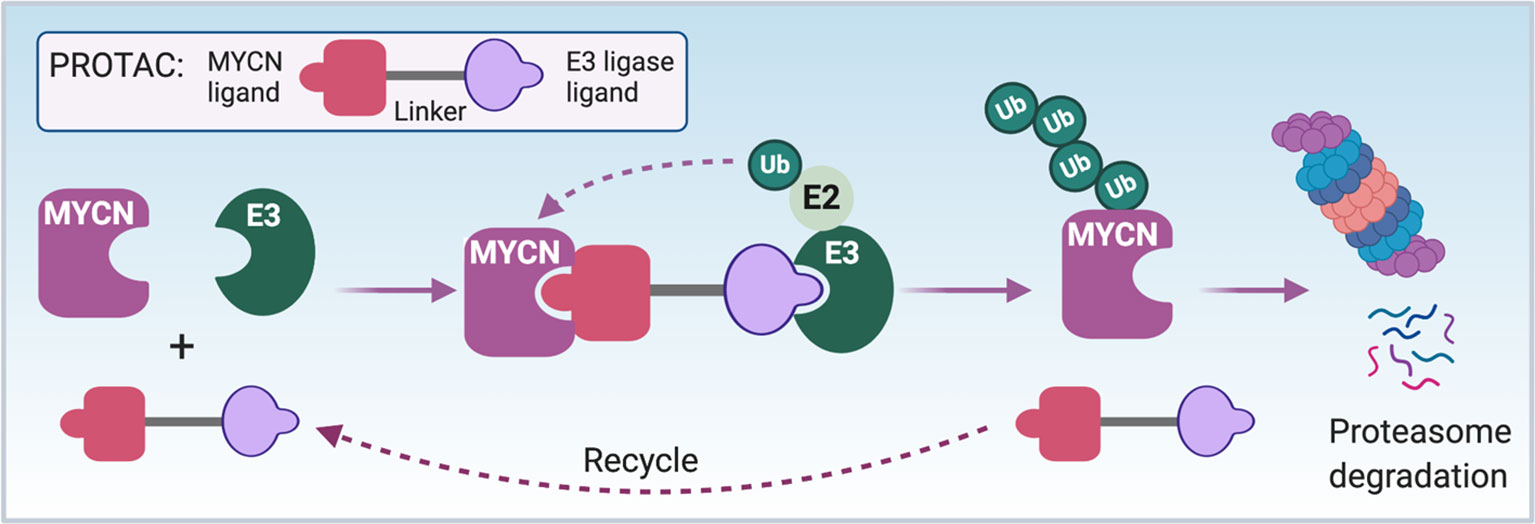
Figure 5 PROTAC strategy to directly target MYCN. The schematic illustrates the mode of action of a proteolysis targeting chimera (PROTAC) targeting MYCN. First of all, a bio-conjugatable analog of a MYCN binding ligand (such as modified 10058-F4 or MYCi361) will be conjugated to E3 ubiquitin ligase binding ligand through a linker to synthesize a MYCN PROTAC. The formation of MYCN-PROTAC-E3 ubiquitin ligase complex will result in a transfer of ubiquitin (Ub) to the lysine residues of MYCN by E2 ubiquitin-conjugating enzyme. Afterwards, the PROTAC will be released and reutilized, and the poly-ubiquitinated MYCN will undergo proteasome degradation. Notes: circled ‘Ub’ represents ubiquitin.
MiRNA are small RNA molecules that regulate their target gene expression at the post-transcriptional level and via their effects on the epigenetic machinery. Many miRNAs such as miR-34a, miR-375, miR-393-5p and let-7 are found to inhibit MYCN mRNA translation or target MYCN mRNA for degradation to suppress tumor cell growth (161–164). With more and more effective drug delivery systems for small interference RNA (siRNA) and miRNA being developed (165), directly targeting MYCN mRNA using miRNAs or siRNAs is a another approach for the treatment of MYCN-driven tumors. Indeed, the recent clinical findings which showed the systemic administration of a next generation antisense targeting the STAT3 TF decreased nuclear STAT3 levels in tumors are proof of principle that direct mRNA targeting of a transcription factor is feasible (166).
Targeting DNA amplification is another possible way to directly target MYCN. The genome-editing tool CRISPR-Cas9 is able to cut DNA at a targeted location and lead to cancer cell death if the targeted regions contain copy number gains. Whether this is clinically translatable is unknown. Pyrrole-imidazole (PI) polyamides when conjugated with DNA-alkylating agents could induce sequence-specific DNA alkylation to suppress target gene expression. A recent study showed that a MYCN-targeting PI- polyamide, MYCN-A3, binds to and alkylates DNA within the MYCN transcript, resulting in a decrease in MYCN copy number, downregulation of MYCN expression and suppression of NB growth in vitro and in xenografts (167). This indicates that the direct targeting of amplified MYCN at a genomic level is feasible. However, the feasibility of developing targeting approaches in pre-clinical models is only the first and sometimes the easiest step in the drug development pipeline.
The oncogenic amplification and/or overexpression of MYC family genes occur in most human cancers, making MYC family oncogenes one of the most sought-after therapeutic targets. Here we specifically reviewed multiple pharmacological approaches to target MYCN by interfering with pathways that MYCN uses to drive oncogenesis. Indirect inhibitors of MYCN, such as the BET bromodomain inhibitor, the CDK7 inhibitor, the AURKA inhibitor, the HAUSP inhibitor and the ODC inhibitor have clearly shown benefit in suppressing MYCN-amplified tumor growth in the preclinical studies, and a few of these inhibitors including bromodomain inhibitor GSK525762, AURKA inhibitor MLN8237 and ODC inhibitor DFMO are being evaluated in the clinic for MYCN-driven cancers. Once again it is possible that combinatorial strategies that integrate these new approaches with standard chemo- and immunotherapy will lead to improved tumor control with less toxicity for patients.
ZL and CT conceptualized and wrote the manuscript while SSC, SC, and VV wrote sections of the manuscript. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We apologize to our respected colleagues whose work we were unable to cite due to space constraints. We thank Tiffany Prout, our administrative assistant for her support. LZ, SSC, SC and CJT acknowledge support from the Center for Cancer Research, the Intramural Research Program of the National Cancer Institute. VV is a research fellow funded by European Union FESR FSE, PON Ricerca e Innovazione 2014–2020. Some of the figures were created with BioRender.com.
1. Meyer N, Penn LZ. Reflecting on 25 years with MYC. Nat Rev Cancer (2008) 8(12):976–90. doi: 10.1038/nrc2231
2. Kohl NE, Kanda N, Schreck RR, Bruns G, Latt SA, Gilbert F, et al. Transposition and amplification of oncogene-related sequences in human neuroblastomas. Cell (1983) 35(2 Pt 1):359–67. doi: 10.1016/0092-8674(83)90169-1
3. Schwab M, Alitalo K, Klempnauer KH, Varmus HE, Bishop JM, Gilbert F, et al. Amplified DNA with limited homology to myc cellular oncogene is shared by human neuroblastoma cell lines and a neuroblastoma tumour. Nature (1983) 305(5931):245–8. doi: 10.1038/305245a0
4. Baluapuri A, Wolf E, Eilers M. Target gene-independent functions of MYC oncoproteins. Nat Rev Mol Cell Biol (2020) 21(5):255–67. doi: 10.1038/s41580-020-0215-2
5. Huang M, Weiss WA. Neuroblastoma and MYCN. Cold Spring Harb Perspect Med (2013) 3(10):a014415. doi: 10.1101/cshperspect.a014415
6. Wenzel A, Schwab M. The mycN/max protein complex in neuroblastoma. Short review. Eur J Cancer (1995) 31A(4):516–9. doi: 10.1016/0959-8049(95)00060-V
7. Beltran H. The N-myc Oncogene: Maximizing its Targets, Regulation, and Therapeutic Potential. Mol Cancer Res (2014) 12(6):815–22. doi: 10.1158/1541-7786.MCR-13-0536
8. Muncan V, Sansom OJ, Tertoolen L, Phesse TJ, Begthel H, Sancho E, et al. Rapid loss of intestinal crypts upon conditional deletion of the Wnt/Tcf-4 target gene c-Myc. Mol Cell Biol (2006) 26(22):8418–26. doi: 10.1128/MCB.00821-06
9. Knoepfler PS, Cheng PF, Eisenman RN. N-myc is essential during neurogenesis for the rapid expansion of progenitor cell populations and the inhibition of neuronal differentiation. Genes Dev (2002) 16(20):2699–712. doi: 10.1101/gad.1021202
10. Malynn BA, de Alboran IM, O’Hagan RC, Bronson R, Davidson L, DePinho RA, et al. N-myc can functionally replace c-myc in murine development, cellular growth, and differentiation. Genes Dev (2000) 14(11):1390–9.
11. Smith RK, Zimmerman K, Yancopoulos GD, Ma A, Alt FW, et al. Transcriptional down-regulation of N-myc expression during B-cell development. Mol Cell Biol (1992) 12(4):1578–84. doi: 10.1128/MCB.12.4.1578
12. Kress TR, Sabo A, Amati B. MYC: connecting selective transcriptional control to global RNA production. Nat Rev Cancer (2015) 15(10):593–607. doi: 10.1038/nrc3984
13. Sabo A, Kress TR, Pelizzola M, de Pretis S, Gorski MM, Tesi A, et al. Selective transcriptional regulation by Myc in cellular growth control and lymphomagenesis. Nature (2014) 511(7510):488–92. doi: 10.1038/nature13537
14. Walz S, Lorenzin F, Morton J, Wiese KE, von Eyss B, Herold S, et al. Activation and repression by oncogenic MYC shape tumour-specific gene expression profiles. Nature (2014) 511(7510):483–7. doi: 10.1038/nature13473
15. Nie Z, Hu G, Wei G, Cui K, Yamane A, Resch W, et al. c-Myc is a universal amplifier of expressed genes in lymphocytes and embryonic stem cells. Cell (2012) 151(1):68–79. doi: 10.1016/j.cell.2012.08.033
16. Lin CY, Loven J, Rahl PB, Paranal RM, Burge CB, Bradner JE, et al. Transcriptional amplification in tumor cells with elevated c-Myc. Cell (2012) 151(1):56–67. doi: 10.1016/j.cell.2012.08.026
17. Buchel G, Carstensen A, Mak KY, Roeschert I, Leen E, Sumara O, et al. Association with Aurora-A Controls N-MYC-Dependent Promoter Escape and Pause Release of RNA Polymerase II during the Cell Cycle. Cell Rep (2017) 21(12):3483–97. doi: 10.1016/j.celrep.2017.11.090
18. Herold S, Kalb J, Buchel G, Ade CP, Baluapuri A, Xu J, et al. Recruitment of BRCA1 limits MYCN-driven accumulation of stalled RNA polymerase. Nature (2019) 567(7749):545–9. doi: 10.1038/s41586-019-1030-9
19. Seeger RC, Brodeur GM, Sather H, Dalton A, Siegel SE, Wong KY, et al. Association of multiple copies of the N-myc oncogene with rapid progression of neuroblastomas. N Engl J Med (1985) 313(18):1111–6. doi: 10.1056/NEJM198510313131802
20. Brodeur GM, Seeger RC, Schwab M, Varmus HE, Bishop JM. Amplification of N-myc in untreated human neuroblastomas correlates with advanced disease stage. Science (1984) 224(4653):1121–4. doi: 10.1126/science.6719137
21. Schwab M, Varmus HE, Bishop JM, Grzeschik KH, Naylor SL, Sakaguchi AY, et al. Chromosome localization in normal human cells and neuroblastomas of a gene related to c-myc. Nature (1984) 308(5956):288–91. doi: 10.1038/308288a0
22. Campbell K, Naranjo A, Hibbitts E, Gastier-Foster JM, Bagatell R, Irwin MS, et al. Association of heterogeneous MYCN amplification with clinical features, biological characteristics and outcomes in neuroblastoma: A report from the Children’s Oncology Group. Eur J Cancer (2020) 133:112–9. doi: 10.1016/j.ejca.2020.04.007
23. Campbell K, Gastier-Foster JM, Mann M, Naranjo AH, Van Ryn C, Bagatell R, et al. Association of MYCN copy number with clinical features, tumor biology, and outcomes in neuroblastoma: A report from the Children’s Oncology Group. Cancer (2017) 123(21):4224–35. doi: 10.1002/cncr.30873
24. Tonelli R, McIntyre A, Camerin C, Walters ZS, Di Leo K, Selfe J, et al. Antitumor activity of sustained N-myc reduction in rhabdomyosarcomas and transcriptional block by antigene therapy. Clin Cancer Res (2012) 18(3):796–807. doi: 10.1158/1078-0432.CCR-11-1981
25. Williamson D, Lu YJ, Gordon T, Sciot R, Kelsey A, Fisher C, et al. Relationship between MYCN copy number and expression in rhabdomyosarcomas and correlation with adverse prognosis in the alveolar subtype. J Clin Oncol (2005) 23(4):880–8. doi: 10.1200/JCO.2005.11.078
26. Swartling FJ, Grimmer MR, Hackett CS, Northcott PA, Fan QW, Goldenberg DD, et al. Pleiotropic role for MYCN in medulloblastoma. Genes Dev (2010) 24(10):1059–72. doi: 10.1101/gad.1907510
27. Aldosari N, Bigner SH, Burger PC, Becker L, Kepner JL, Friedman HS, et al. MYCC and MYCN oncogene amplification in medulloblastoma. A fluorescence in situ hybridization study on paraffin sections from the Children’s Oncology Group. Arch Pathol Lab Med (2002) 126(5):540–4. doi: 10.1043/0003-9985(2002)126<0540:MAMOAI>2.0.CO;2
28. Gessi M, von Bueren A, Treszl A, zur Muhlen A, Hartmann W, Warmuth-Metz M, et al. MYCN amplification predicts poor outcome for patients with supratentorial primitive neuroectodermal tumors of the central nervous system. Neuro Oncol (2014) 16(7):924–32. doi: 10.1093/neuonc/not302
29. Williams RD, Chagtai T, Alcaide-German M, Apps J, Wegert J, Popov S, et al. Multiple mechanisms of MYCN dysregulation in Wilms tumour. Oncotarget (2015) 6(9):7232–43. doi: 10.18632/oncotarget.3377
30. Lee WH, Murphree AL, Benedict WF. Expression and amplification of the N-myc gene in primary retinoblastoma. Nature (1984) 309(5967):458–60. doi: 10.1038/309458a0
31. Rushlow DE, Mol BM, Kennett JY, Yee S, Pajovic S, Theriault BL, et al. Characterisation of retinoblastomas without RB1 mutations: genomic, gene expression, and clinical studies. Lancet Oncol (2013) 14(4):327–34. doi: 10.1016/S1470-2045(13)70045-7
32. Beltran H, Rickman DS, Park K, Chae SS, Sboner A, MacDonald TY, et al. Molecular characterization of neuroendocrine prostate cancer and identification of new drug targets. Cancer Discovery (2011) 1(6):487–95. doi: 10.1158/2159-8290.CD-11-0130
33. Nau MM, Brooks BJ Jr, Carney DN, Gazdar AF, Battey JF, Sausville EA, et al. Human small-cell lung cancers show amplification and expression of the N-myc gene. Proc Natl Acad Sci USA (1986) 83(4):1092–6. doi: 10.1073/pnas.83.4.1092
34. Funa K, Steinholtz L, Nou E, Bergh J. Increased expression of N-myc in human small cell lung cancer biopsies predicts lack of response to chemotherapy and poor prognosis. Am J Clin Pathol (1987) 88(2):216–20. doi: 10.1093/ajcp/88.2.216
35. Freier K, Flechtenmacher C, Devens F, Hartschuh W, Hofele C, Lichter P, et al. Recurrent NMYC copy number gain and high protein expression in basal cell carcinoma. Oncol Rep (2006) 15(5):1141–5. doi: 10.3892/or.15.5.1141
36. Astolfi A, Vendemini F, Urbini M, Melchionda F, Masetti R, Franzoni M, et al. MYCN is a novel oncogenic target in pediatric T-cell acute lymphoblastic leukemia. Oncotarget (2014) 5(1):120–30. doi: 10.18632/oncotarget.1337
37. Bjerke L, Mackay A, Nandhabalan M, Burford A, Jury A, Popov S, et al. Histone H3.3. mutations drive pediatric glioblastoma through upregulation of MYCN. Cancer Discovery (2013) 3(5):512–9. doi: 10.1158/2159-8290.CD-12-0426
38. Hodgson JG, Yeh RF, Ray A, Wang NJ, Smirnov I, Yu M, et al. Comparative analyses of gene copy number and mRNA expression in glioblastoma multiforme tumors and xenografts. Neuro Oncol (2009) 11(5):477–87. doi: 10.1215/15228517-2008-113
39. Mizukami Y, Nonomura A, Takizawa T, Noguchi M, Michigishi T, Nakamura S, et al. N-myc protein expression in human breast carcinoma: prognostic implications. Anticancer Res (1995) 15(6B):2899–905.
40. Weiss WA, Aldape K, Mohapatra G, Feuerstein BG, Bishop JM. Targeted expression of MYCN causes neuroblastoma in transgenic mice. EMBO J (1997) 16(11):2985–95. doi: 10.1093/emboj/16.11.2985
41. Althoff K, Beckers A, Bell E, Nortmeyer M, Thor T, Sprussel A, et al. A Cre-conditional MYCN-driven neuroblastoma mouse model as an improved tool for preclinical studies. Oncogene (2015) 34(26):3357–68. doi: 10.1038/onc.2014.269
42. Zhu S, Lee JS, Guo F, Shin J, Perez-Atayde AR, Kutok JL, et al. Activated ALK collaborates with MYCN in neuroblastoma pathogenesis. Cancer Cell (2012) 21(3):362–73. doi: 10.1016/j.ccr.2012.02.010
43. Zhu S, Zhang X, Weichert-Leahey N, Dong Z, Zhang C, Lopez G, et al. LMO1 Synergizes with MYCN to Promote Neuroblastoma Initiation and Metastasis. Cancer Cell (2017) 32(3):310–23.e5. doi: 10.1016/j.ccell.2017.08.002
44. Heukamp LC, Thor T, Schramm A, De Preter K, Kumps C, De Wilde B, et al. Targeted expression of mutated ALK induces neuroblastoma in transgenic mice. Sci Transl Med (2012) 4(141):141ra91. doi: 10.1126/scitranslmed.3003967
45. Dardenne E, Beltran H, Benelli M, Gayvert K, Berger A, Puca L, et al. N-Myc Induces an EZH2-Mediated Transcriptional Program Driving Neuroendocrine Prostate Cancer. Cancer Cell (2016) 30(4):563–77. doi: 10.1016/j.ccell.2016.09.005
46. Kawagoe H, Kandilci A, Kranenburg TA, Grosveld GC. Overexpression of N-Myc rapidly causes acute myeloid leukemia in mice. Cancer Res (2007) 67(22):10677–85. doi: 10.1158/0008-5472.CAN-07-1118
47. Swartling FJ, Savov V, Persson AI, Chen J, Hackett CS, Northcott PA, et al. Distinct neural stem cell populations give rise to disparate brain tumors in response to N-MYC. Cancer Cell (2012) 21(5):601–13. doi: 10.1016/j.ccr.2012.04.012
48. Zindy F, Uziel T, Ayrault O, Calabrese C, Valentine M, Rehg JE, et al. Genetic alterations in mouse medulloblastomas and generation of tumors de novo from primary cerebellar granule neuron precursors. Cancer Res (2007) 67(6):2676–84. doi: 10.1158/0008-5472.CAN-06-3418
49. Wright PE, Dyson HJ. Intrinsically disordered proteins in cellular signalling and regulation. Nat Rev Mol Cell Biol (2015) 16(1):18–29. doi: 10.1038/nrm3920
50. Oldfield CJ, Dunker AK. Intrinsically disordered proteins and intrinsically disordered protein regions. Annu Rev Biochem (2014) 83:553–84. doi: 10.1146/annurev-biochem-072711-164947
51. Obradovic Z, Peng K, Vucetic S, Radivojac P, Brown CJ, Dunker AK. Predicting intrinsic disorder from amino acid sequence. Proteins (2003) 53 Suppl 6:566–72. doi: 10.1002/prot.10532
52. Sjostrom SK, Finn G, Hahn WC, Rowitch DH, Kenney AM. The Cdk1 complex plays a prime role in regulating N-myc phosphorylation and turnover in neural precursors. Dev Cell (2005) 9(3):327–38. doi: 10.1016/j.devcel.2005.07.014
53. Chesler L, Schlieve C, Goldenberg DD, Kenney A, Kim G, McMillan A, et al. Inhibition of phosphatidylinositol 3-kinase destabilizes Mycn protein and blocks malignant progression in neuroblastoma. Cancer Res (2006) 66(16):8139–46. doi: 10.1158/0008-5472.CAN-05-2769
54. Pulverer BJ, Fisher C, Vousden K, Littlewood T, Evan G, Woodgett JR. Site-specific modulation of c-Myc cotransformation by residues phosphorylated in vivo. Oncogene (1994) 9(1):59–70.
55. Kenney AM, Widlund HR, Rowitch DH. Hedgehog and PI-3 kinase signaling converge on Nmyc1 to promote cell cycle progression in cerebellar neuronal precursors. Development (2004) 131(1):217–28. doi: 10.1242/dev.00891
56. Otto T, Horn S, Brockmann M, Eilers U, Schuttrumpf L, Popov N, et al. Stabilization of N-Myc is a critical function of Aurora A in human neuroblastoma. Cancer Cell (2009) 15(1):67–78. doi: 10.1016/j.ccr.2008.12.005
57. Sears R, Nuckolls F, Haura E, Taya Y, Tamai K, Nevins JR, et al. Multiple Ras-dependent phosphorylation pathways regulate Myc protein stability. Genes Dev (2000) 14(19):2501–14. doi: 10.1101/gad.836800
58. Welcker M, Orian A, Jin J, Grim JE, Harper JW, Eisenman RN, et al. The Fbw7 tumor suppressor regulates glycogen synthase kinase 3 phosphorylation-dependent c-Myc protein degradation. Proc Natl Acad Sci USA (2004) 101(24):9085–90. doi: 10.1073/pnas.0402770101
59. Yada M, Hatakeyama S, Kamura T, Nishiyama M, Tsunematsu R, Imaki H, et al. Phosphorylation-dependent degradation of c-Myc is mediated by the F-box protein Fbw7. EMBO J (2004) 23(10):2116–25. doi: 10.1038/sj.emboj.7600217
60. Izumi H, Kaneko Y. Trim32 facilitates degradation of MYCN on spindle poles and induces asymmetric cell division in human neuroblastoma cells. Cancer Res (2014) 74(19):5620–30. doi: 10.1158/0008-5472.CAN-14-0169
61. Zhao X, Heng JI, Guardavaccaro D, Jiang R, Pagano M, Guillemot F, et al. The HECT-domain ubiquitin ligase Huwe1 controls neural differentiation and proliferation by destabilizing the N-Myc oncoprotein. Nat Cell Biol (2008) 10(6):643–53. doi: 10.1038/ncb1727
62. King B, Boccalatte F, Moran-Crusio K, Wolf E, Wang J, Kayembe C, et al. The ubiquitin ligase Huwe1 regulates the maintenance and lymphoid commitment of hematopoietic stem cells. Nat Immunol (2016) 17(11):1312–21. doi: 10.1038/ni.3559
63. Richards MW, Burgess SG, Poon E, Carstensen A, Eilers M, Chesler L, et al. Structural basis of N-Myc binding by Aurora-A and its destabilization by kinase inhibitors. Proc Natl Acad Sci USA (2016) 113(48):13726–31. doi: 10.1073/pnas.1610626113
64. Tavana O, Li D, Dai C, Lopez G, Banerjee D, Kon N, et al. HAUSP deubiquitinates and stabilizes N-Myc in neuroblastoma. Nat Med (2016) 22(10):1180–6. doi: 10.1038/nm.4180
65. Koach J, Holien JK, Massudi H, Carter DR, Ciampa OC, Herath M, et al. Drugging MYCN Oncogenic Signaling through the MYCN-PA2G4 Binding Interface. Cancer Res (2019) 79(21):5652–67. doi: 10.1158/0008-5472.CAN-19-1112
66. Park JH, Szemes M, Vieira GC, Melegh Z, Malik S, Heesom KJ, et al. Protein arginine methyltransferase 5 is a key regulator of the MYCN oncoprotein in neuroblastoma cells. Mol Oncol (2015) 9(3):617–27. doi: 10.1016/j.molonc.2014.10.015
67. Wood MA, McMahon SB, Cole MD. An ATPase/helicase complex is an essential cofactor for oncogenic transformation by c-Myc. Mol Cell (2000) 5(2):321–30. doi: 10.1016/S1097-2765(00)80427-X
68. McMahon SB, Wood MA, Cole MD. The essential cofactor TRRAP recruits the histone acetyltransferase hGCN5 to c-Myc. Mol Cell Biol (2000) 20(2):556–62. doi: 10.1128/MCB.20.2.556-562.2000
69. Park J, Kunjibettu S, McMahon SB, Cole MD. The ATM-related domain of TRRAP is required for histone acetyltransferase recruitment and Myc-dependent oncogenesis. Genes Dev (2001) 15(13):1619–24. doi: 10.1101/gad.900101
70. Liu X, Tesfai J, Evrard YA, Dent SY, Martinez E. c-Myc transformation domain recruits the human STAGA complex and requires TRRAP and GCN5 acetylase activity for transcription activation. J Biol Chem (2003) 278(22):20405–12. doi: 10.1074/jbc.M211795200
71. Thomas LR, Wang Q, Grieb BC, Phan J, Foshage AM, Sun Q, et al. Interaction with WDR5 promotes target gene recognition and tumorigenesis by MYC. Mol Cell (2015) 58(3):440–52. doi: 10.1016/j.molcel.2015.02.028
72. Sun Y, Bell JL, Carter D, Gherardi S, Poulos RC, Milazzo G, et al. WDR5 Supports an N-Myc Transcriptional Complex That Drives a Protumorigenic Gene Expression Signature in Neuroblastoma. Cancer Res (2015) 75(23):5143–54. doi: 10.1158/0008-5472.CAN-15-0423
73. Yang J, AlTahan AM, Hu D, Wang Y, Cheng PH, Morton CL, et al. The role of histone demethylase KDM4B in Myc signaling in neuroblastoma. J Natl Cancer Inst (2015) 107(6):djv080. doi: 10.1093/jnci/djv080
74. Iraci N, Diolaiti D, Papa A, Porro A, Valli E, Gherardi S, et al. A SP1/MIZ1/MYCN repression complex recruits HDAC1 at the TRKA and p75NTR promoters and affects neuroblastoma malignancy by inhibiting the cell response to NGF. Cancer Res (2011) 71(2):404–12. doi: 10.1158/0008-5472.CAN-10-2627
75. Gartel AL, Ye X, Goufman E, Shianov P, Hay N, Najmabadi F, et al. Myc represses the p21(WAF1/CIP1) promoter and interacts with Sp1/Sp3. Proc Natl Acad Sci USA (2001) 98(8):4510–5. doi: 10.1073/pnas.081074898
76. Staller P, Peukert K, Kiermaier A, Seoane J, Lukas J, Karsunky H, et al. Repression of p15INK4b expression by Myc through association with Miz-1. Nat Cell Biol (2001) 3(4):392–9. doi: 10.1038/35070076
77. Liu T, Tee AE, Porro A, Smith SA, Dwarte T, Liu PY, et al. Activation of tissue transglutaminase transcription by histone deacetylase inhibition as a therapeutic approach for Myc oncogenesis. Proc Natl Acad Sci USA (2007) 104(47):18682–7. doi: 10.1073/pnas.0705524104
78. Corvetta D, Chayka O, Gherardi S, D’Acunto CW, Cantilena S, Valli E, et al. Physical interaction between MYCN oncogene and polycomb repressive complex 2 (PRC2) in neuroblastoma: functional and therapeutic implications. J Biol Chem (2013) 288(12):8332–41. doi: 10.1074/jbc.M113.454280
79. Amente S, Milazzo G, Sorrentino MC, Ambrosio S, Di Palo G, Lania L, et al. Lysine-specific demethylase (LSD1/KDM1A) and MYCN cooperatively repress tumor suppressor genes in neuroblastoma. Oncotarget (2015) 6(16):14572–83. doi: 10.18632/oncotarget.3990
80. Thiele CJ, Reynolds CP, Israel MA. Decreased expression of N-myc precedes retinoic acid-induced morphological differentiation of human neuroblastoma. Nature (1985) 313(6001):404–6. doi: 10.1038/313404a0
81. Kanemaru KK, Tuthill MC, Takeuchi KK, Sidell N, Wada RK. Retinoic acid induced downregulation of MYCN is not mediated through changes in Sp1/Sp3. Pediatr Blood Cancer (2008) 50(4):806–11. doi: 10.1002/pbc.21273
82. Reynolds CP, Matthay KK, Villablanca JG, Maurer BJ. Retinoid therapy of high-risk neuroblastoma. Cancer Lett (2003) 197(1-2):185–92. doi: 10.1016/S0304-3835(03)00108-3
83. Calo E, Wysocka J. Modification of enhancer chromatin: what, how, and why? Mol Cell (2013) 49(5):825–37. doi: 10.1016/j.molcel.2013.01.038
84. Hamdan FH, Johnsen SA. Perturbing Enhancer Activity in Cancer Therapy. Cancers (Basel) (2019) 11(5). doi: 10.3390/cancers11050634
85. Sengupta S, George RE. Super-Enhancer-Driven Transcriptional Dependencies in Cancer. Trends Cancer (2017) 3(4):269–81. doi: 10.1016/j.trecan.2017.03.006
86. Shin HY. Targeting Super-Enhancers for Disease Treatment and Diagnosis. Mol Cells (2018) 41(6):506–14. doi: 10.14348/molcells.2018.2297
87. Chipumuro E, Marco E, Christensen CL, Kwiatkowski N, Zhang T, Hatheway CM, et al. CDK7 inhibition suppresses super-enhancer-linked oncogenic transcription in MYCN-driven cancer. Cell (2014) 159(5):1126–39. doi: 10.1016/j.cell.2014.10.024
88. Durbin AD, Zimmerman MW, Dharia NV, Abraham BJ, Iniguez AB, Weichert-Leahey N, et al. Selective gene dependencies in MYCN-amplified neuroblastoma include the core transcriptional regulatory circuitry. Nat Genet (2018) 50(9):1240–6. doi: 10.1038/s41588-018-0191-z
89. Zimmerman MW, Liu Y, He S, Durbin AD, Abraham BJ, Easton J, et al. MYC Drives a Subset of High-Risk Pediatric Neuroblastomas and Is Activated through Mechanisms Including Enhancer Hijacking and Focal Enhancer Amplification. Cancer Discovery (2018) 8(3):320–35. doi: 10.1158/2159-8290.CD-17-0993
90. de Ruijter AJ, Kemp S, Kramer G, Meinsma RJ, Kaufmann JO, Caron HN, et al. The novel histone deacetylase inhibitor BL1521 inhibits proliferation and induces apoptosis in neuroblastoma cells. Biochem Pharmacol (2004) 68(7):1279–88. doi: 10.1016/j.bcp.2004.05.010
91. Cortes C, Kozma SC, Tauler A, Ambrosio S. MYCN concurrence with SAHA-induced cell death in human neuroblastoma cells. Cell Oncol (Dordr) (2015) 38(5):341–52. doi: 10.1007/s13402-015-0233-9
92. Jaboin J, Wild J, Hamidi H, Khanna C, Kim CJ, Robey R, et al. MS-27-275, an inhibitor of histone deacetylase, has marked in vitro and in vivo antitumor activity against pediatric solid tumors. Cancer Res (2002) 62(21):6108–15.
93. Sanchez GJ, Richmond PA, Bunker EN, Karman SS, Azofeifa J, Garnett AT, et al. Genome-wide dose-dependent inhibition of histone deacetylases studies reveal their roles in enhancer remodeling and suppression of oncogenic super-enhancers. Nucleic Acids Res (2018) 46(4):1756–76. doi: 10.1093/nar/gkx1225
94. Gryder BE, Pomella S, Sayers C, Wu XS, Song Y, Chiarella AM, et al. Histone hyperacetylation disrupts core gene regulatory architecture in rhabdomyosarcoma. Nat Genet (2019) 51(12):1714–22. doi: 10.1038/s41588-019-0534-4
95. Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, Jacobs HM, et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell (2011) 146(6):904–17. doi: 10.1016/j.cell.2011.08.017
96. Donati B, Lorenzini E, Ciarrocchi A. BRD4 and Cancer: going beyond transcriptional regulation. Mol Cancer (2018) 17(1):164. doi: 10.1186/s12943-018-0915-9
97. Puissant A, Frumm SM, Alexe G, Bassil CF, Qi J, Chanthery YH, et al. Targeting MYCN in neuroblastoma by BET bromodomain inhibition. Cancer Discovery (2013) 3(3):308–23. doi: 10.1158/2159-8290.CD-12-0418
98. Henssen A, Althoff K, Odersky A, Beckers A, Koche R, Speleman F, et al. Targeting MYCN-Driven Transcription By BET-Bromodomain Inhibition. Clin Cancer Res (2016) 22(10):2470–81. doi: 10.1158/1078-0432.CCR-15-1449
99. Loven J, Hoke HA, Lin CY, Lau A, Orlando DA, Vakoc CR, et al. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell (2013) 153(2):320–34. doi: 10.1016/j.cell.2013.03.036
100. Piha-Paul SA, Hann CL, French CA, Cousin S, Brana I, Cassier PA, et al. Phase 1 Study of Molibresib (GSK525762), a Bromodomain and Extra-Terminal Domain Protein Inhibitor, in NUT Carcinoma and Other Solid Tumors. JNCI Cancer Spectr (2020) 4(2):pkz093. doi: 10.1093/jncics/pkz093
101. Whyte WA, Orlando DA, Hnisz D, Abraham BJ, Lin CY, Kagey MH, et al. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell (2013) 153(2):307–19. doi: 10.1016/j.cell.2013.03.035
102. Felgenhauer J, Tomino L, Selich-Anderson J, Bopp E, Shah N. Dual BRD4 and AURKA Inhibition Is Synergistic against MYCN-Amplified and Nonamplified Neuroblastoma. Neoplasia (2018) 20(10):965–74. doi: 10.1016/j.neo.2018.08.002
103. Shahbazi J, Liu PY, Atmadibrata B, Bradner JE, Marshall GM, Lock RB, et al. The Bromodomain Inhibitor JQ1 and the Histone Deacetylase Inhibitor Panobinostat Synergistically Reduce N-Myc Expression and Induce Anticancer Effects. Clin Cancer Res (2016) 22(10):2534–44. doi: 10.1158/1078-0432.CCR-15-1666
104. Rahl PB, Lin CY, Seila AC, Flynn RA, McCuine S, Burge CB, et al. c-Myc regulates transcriptional pause release. Cell (2010) 141(3):432–45. doi: 10.1016/j.cell.2010.03.030
105. Poon E, Liang T, Jamin Y, Walz S, Kwok C, Hakkert A, et al. Orally bioavailable CDK9/2 inhibitor shows mechanism-based therapeutic potential in MYCN-driven neuroblastoma. J Clin Invest (2020). doi: 10.1172/JCI134132
106. Calabrese DR, Chen X, Leon EC, Gaikwad SM, Phyo Z, Hewitt WM, et al. Chemical and structural studies provide a mechanistic basis for recognition of the MYC G-quadruplex. Nat Commun (2018) 9(1):4229. doi: 10.1038/s41467-018-06315-w
107. Spiegel J, Adhikari S, Balasubramanian S. The Structure and Function of DNA G-Quadruplexes. Trends Chem (2020) 2(2):123–36. doi: 10.1016/j.trechm.2019.07.002
108. Siddiqui-Jain A, Grand CL, Bearss DJ, Hurley LH. Direct evidence for a G-quadruplex in a promoter region and its targeting with a small molecule to repress c-MYC transcription. Proc Natl Acad Sci USA (2002) 99(18):11593–8. doi: 10.1073/pnas.182256799
109. Bradner JE, Hnisz D, Young RA. Transcriptional Addiction in Cancer. Cell (2017) 168(4):629–43. doi: 10.1016/j.cell.2016.12.013
110. Bushweller JH. Targeting transcription factors in cancer - from undruggable to reality. Nat Rev Cancer (2019) 19(11):611–24. doi: 10.1038/s41568-019-0196-7
111. Bonvini P, Nguyen P, Trepel J, Neckers LM. In vivo degradation of N-myc in neuroblastoma cells is mediated by the 26S proteasome. Oncogene (1998) 16(9):1131–9. doi: 10.1038/sj.onc.1201625
112. Cage TA, Chanthery Y, Chesler L, Grimmer M, Knight Z, Shokat K, et al. Downregulation of MYCN through PI3K Inhibition in Mouse Models of Pediatric Neural Cancer. Front Oncol (2015) 5:111. doi: 10.3389/fonc.2015.00111
113. Yang Y, Ding L, Zhou Q, Fen L, Cao Y, Sun J, et al. Silencing of AURKA augments the antitumor efficacy of the AURKA inhibitor MLN8237 on neuroblastoma cells. Cancer Cell Int (2020) 20:9. doi: 10.1186/s12935-019-1072-y
114. Brockmann M, Poon E, Berry T, Carstensen A, Deubzer HE, Rycak L, et al. Small molecule inhibitors of aurora-a induce proteasomal degradation of N-myc in childhood neuroblastoma. Cancer Cell (2013) 24(1):75–89. doi: 10.1016/j.ccr.2013.05.005
115. Zormpas-Petridis K, Poon E, Clarke M, Jerome NP, Boult JKR, Blackledge MD, et al. Noninvasive MRI Native T1 Mapping Detects Response to MYCN-targeted Therapies in the Th-MYCN Model of Neuroblastoma. Cancer Res (2020) 80(16):3424–35. doi: 10.1158/0008-5472.CAN-20-0133
116. Ommer J, Selfe JL, Wachtel M, O’Brien EM, Laubscher D, Roemmele M, et al. Aurora A Kinase Inhibition Destabilizes PAX3-FOXO1 and MYCN and Synergizes with Navitoclax to Induce Rhabdomyosarcoma Cell Death. Cancer Res (2020) 80(4):832–42. doi: 10.1158/0008-5472.CAN-19-1479
117. Gustafson WC, Meyerowitz JG, Nekritz EA, Chen J, Benes C, Charron E, et al. Drugging MYCN through an allosteric transition in Aurora kinase A. Cancer Cell (2014) 26(3):414–27. doi: 10.1016/j.ccr.2014.07.015
118. DuBois SG, Marachelian A, Fox E, Kudgus RA, Reid JM, Groshen S, et al. Phase I Study of the Aurora A Kinase Inhibitor Alisertib in Combination With Irinotecan and Temozolomide for Patients With Relapsed or Refractory Neuroblastoma: A NANT (New Approaches to Neuroblastoma Therapy) Trial. J Clin Oncol (2016) 34(12):1368–75. doi: 10.1200/JCO.2015.65.4889
119. Ackermann S, Goeser F, Schulte JH, Schramm A, Ehemann V, Hero B, et al. Polo-like kinase 1 is a therapeutic target in high-risk neuroblastoma. Clin Cancer Res (2011) 17(4):731–41. doi: 10.1158/1078-0432.CCR-10-1129
120. Xiao D, Yue M, Su H, Ren P, Jiang J, Li F, et al. Polo-like Kinase-1 Regulates Myc Stabilization and Activates a Feedforward Circuit Promoting Tumor Cell Survival. Mol Cell (2016) 64(3):493–506. doi: 10.1016/j.molcel.2016.09.016
121. Chaturvedi NK, Mahapatra S, Kesherwani V, Kling MJ, Shukla M, Ray S, et al. Role of protein arginine methyltransferase 5 in group 3 (MYC-driven) Medulloblastoma. BMC Cancer (2019) 19(1):1056. doi: 10.1186/s12885-019-6291-z
122. Berg T, Cohen SB, Desharnais J, Sonderegger C, Maslyar DJ, Goldberg J, et al. Small-molecule antagonists of Myc/Max dimerization inhibit Myc-induced transformation of chicken embryo fibroblasts. Proc Natl Acad Sci USA (2002) 99(6):3830–5. doi: 10.1073/pnas.062036999
123. Wang H, Hammoudeh DI, Follis AV, Reese BE, Lazo JS, Metallo SJ, et al. Improved low molecular weight Myc-Max inhibitors. Mol Cancer Ther (2007) 6(9):2399–408. doi: 10.1158/1535-7163.MCT-07-0005
124. Muller I, Larsson K, Frenzel A, Oliynyk G, Zirath H, Prochownik EV, et al. Targeting of the MYCN protein with small molecule c-MYC inhibitors. PloS One (2014) 9(5):e97285. doi: 10.1371/journal.pone.0097285
125. Follis AV, Hammoudeh DI, Wang H, Prochownik EV, Metallo SJ. Structural rationale for the coupled binding and unfolding of the c-Myc oncoprotein by small molecules. Chem Biol (2008) 15(11):1149–55. doi: 10.1016/j.chembiol.2008.09.011
126. Soucek L, Helmer-Citterich M, Sacco A, Jucker R, Cesareni G, Nasi S. Design and properties of a Myc derivative that efficiently homodimerizes. Oncogene (1998) 17(19):2463–72. doi: 10.1038/sj.onc.1202199
127. Savino M, Annibali D, Carucci N, Favuzzi E, Cole MD, Evan GI, et al. The action mechanism of the Myc inhibitor termed Omomyc may give clues on how to target Myc for cancer therapy. PloS One (2011) 6(7):e22284. doi: 10.1371/journal.pone.0022284
128. Han H, Jain AD, Truica MI, Izquierdo-Ferrer J, Anker JF, Lysy B, et al. Small-Molecule MYC Inhibitors Suppress Tumor Growth and Enhance Immunotherapy. Cancer Cell (2019) 36(5):483–497 e15. doi: 10.1016/j.ccell.2019.10.001
129. Struntz NB, Chen A, Deutzmann A, Wilson RM, Stefan E, Evans HL, et al. Stabilization of the Max Homodimer with a Small Molecule Attenuates Myc-Driven Transcription. Cell Chem Biol (2019) 26(5):711–723 e14. doi: 10.1016/j.chembiol.2019.02.009
130. Wang C, Liu Z, Woo CW, Li Z, Wang L, Wei JS, et al. EZH2 Mediates epigenetic silencing of neuroblastoma suppressor genes CASZ1, CLU, RUNX3, and NGFR. Cancer Res (2012) 72(1):315–24. doi: 10.1158/0008-5472.CAN-11-0961
131. Chen L, Alexe G, Dharia NV, Ross L, Iniguez AB, Conway AS, et al. CRISPR-Cas9 screen reveals a MYCN-amplified neuroblastoma dependency on EZH2. J Clin Invest (2018) 128(1):446–62. doi: 10.1172/JCI90793
132. Veschi V, Liu Z, Voss TC, Ozbun L, Gryder B, Yan C, et al. Epigenetic siRNA and Chemical Screens Identify SETD8 Inhibition as a Therapeutic Strategy for p53 Activation in High-Risk Neuroblastoma. Cancer Cell (2017) 31(1):50–63. doi: 10.1016/j.ccell.2016.12.002
133. Tu WB, Shiah YJ, Lourenco C, Mullen PJ, Dingar D, Redel C, et al. MYC Interacts with the G9a Histone Methyltransferase to Drive Transcriptional Repression and Tumorigenesis. Cancer Cell (2018) 34(4):579–95.e8. doi: 10.1016/j.ccell.2018.09.001
134. Lu Z, Tian Y, Salwen HR, Chlenski A, Godley LA, Raj JU, et al. Histone-lysine methyltransferase EHMT2 is involved in proliferation, apoptosis, cell invasion, and DNA methylation of human neuroblastoma cells. Anticancer Drugs (2013) 24(5):484–93. doi: 10.1097/CAD.0b013e32835ffdbb
135. Lodrini M, Oehme I, Schroeder C, Milde T, Schier MC, Kopp-Schneider A, et al. MYCN and HDAC2 cooperate to repress miR-183 signaling in neuroblastoma. Nucleic Acids Res (2013) 41(12):6018–33. doi: 10.1093/nar/gkt346
136. Sun Y, Liu PY, Scarlett CJ, Malyukova A, Liu B, Marshall GM, et al. Histone deacetylase 5 blocks neuroblastoma cell differentiation by interacting with N-Myc. Oncogene (2014) 33(23):2987–94. doi: 10.1038/onc.2013.253
137. Secci D, Carradori S, Bizzarri B, Bolasco A, Ballario P, Patramani Z, et al. Synthesis of a novel series of thiazole-based histone acetyltransferase inhibitors. Bioorg Med Chem (2014) 22(5):1680–9. doi: 10.1016/j.bmc.2014.01.022
138. Hogarty MD, Norris MD, Davis K, Liu X, Evageliou NF, Hayes CS, et al. ODC1 is a critical determinant of MYCN oncogenesis and a therapeutic target in neuroblastoma. Cancer Res (2008) 68(23):9735–45. doi: 10.1158/0008-5472.CAN-07-6866
139. Gamble LD, Hogarty MD, Liu X, Ziegler DS, Marshall G, Norris MD, et al. Polyamine pathway inhibition as a novel therapeutic approach to treating neuroblastoma. Front Oncol (2012) 2:162. doi: 10.3389/fonc.2012.00162
140. Sholler GLS, Ferguson W, Bergendahl G, Bond JP, Neville K, Eslin D, et al. Maintenance DFMO Increases Survival in High Risk Neuroblastoma. Sci Rep (2018) 8(1):14445. doi: 10.1038/s41598-018-32659-w
141. Lewis EC, Kraveka JM, Ferguson W, Eslin D, Brown VI, Bergendahl G, et al. A subset analysis of a phase II trial evaluating the use of DFMO as maintenance therapy for high-risk neuroblastoma. Int J Cancer (2020) 147(11):3152–9. doi: 10.1002/ijc.33044
142. Belotserkovskaya R, Oh S, Bondarenko VA, Orphanides G, Studitsky VM, Reinberg D. FACT facilitates transcription-dependent nucleosome alteration. Science (2003) 301(5636):1090–3. doi: 10.1126/science.1085703
143. Carter DR, Murray J, Cheung BB, Gamble L, Koach J, Tsang J, et al. Therapeutic targeting of the MYC signal by inhibition of histone chaperone FACT in neuroblastoma. Sci Transl Med (2015) 7(312):312ra176. doi: 10.1126/scitranslmed.aab1803
144. O’Neil NJ, Bailey ML, Hieter P. Synthetic lethality and cancer. Nat Rev Genet (2017) 18(10):613–23. doi: 10.1038/nrg.2017.47
145. Fulda S, Lutz W, Schwab M, Debatin KM. MycN sensitizes neuroblastoma cells for drug-induced apoptosis. Oncogene (1999) 18(7):1479–86. doi: 10.1038/sj.onc.1202435
146. Strasser A, Harris AW, Bath ML, Cory S. Novel primitive lymphoid tumours induced in transgenic mice by cooperation between myc and bcl-2. Nature (1990) 348(6299):331–3. doi: 10.1038/348331a0
147. Ham J, Costa C, Sano R, Lochmann TL, Sennott EM, Patel NU, et al. Exploitation of the Apoptosis-Primed State of MYCN-Amplified Neuroblastoma to Develop a Potent and Specific Targeted Therapy Combination. Cancer Cell (2016) 29(2):159–72. doi: 10.1016/j.ccell.2016.01.002
148. Cole KA, Huggins J, Laquaglia M, Hulderman CE, Russell MR, Bosse K, et al. RNAi screen of the protein kinome identifies checkpoint kinase 1 (CHK1) as a therapeutic target in neuroblastoma. Proc Natl Acad Sci USA (2011) 108(8):3336–41. doi: 10.1073/pnas.1012351108
149. Hoglund A, Nilsson LM, Muralidharan SV, Hasvold LA, Merta P, Rudelius M, et al. Therapeutic implications for the induced levels of Chk1 in Myc-expressing cancer cells. Clin Cancer Res (2011) 17(22):7067–79. doi: 10.1158/1078-0432.CCR-11-1198
150. Chen H, Liu H, Qing G. Targeting oncogenic Myc as a strategy for cancer treatment. Signal Transduct Target Ther (2018) 3:5. doi: 10.1038/s41392-018-0008-7
151. Dominguez-Sola D, Ying CY, Grandori C, Ruggiero L, Chen B, Li M, et al. Non-transcriptional control of DNA replication by c-Myc. Nature (2007) 448(7152):445–51. doi: 10.1038/nature05953
152. Molenaar JJ, Ebus ME, Geerts D, Koster J, Lamers F, Valentijn LJ, et al. Inactivation of CDK2 is synthetically lethal to MYCN over-expressing cancer cells. Proc Natl Acad Sci USA (2009) 106(31):12968–73. doi: 10.1073/pnas.0901418106
153. Cheung NK, Zhang J, Lu C, Parker M, Bahrami A, Tickoo SK, et al. Association of age at diagnosis and genetic mutations in patients with neuroblastoma. JAMA (2012) 307(10):1062–71. doi: 10.1001/jama.2012.228
154. Zeineldin M, Federico S, Chen X, Fan Y, Xu B, Stewart E, et al. MYCN amplification and ATRX mutations are incompatible in neuroblastoma. Nat Commun (2020) 11(1):913.
155. Petroni M, Veschi V, Gulino A, Giannini G. Molecular mechanisms of MYCN-dependent apoptosis and the MDM2-p53 pathway: an Achille’s heel to be exploited for the therapy of MYCN-amplified neuroblastoma. Front Oncol (2012) 2:141. doi: 10.3389/fonc.2012.00141
156. Jerby-Arnon L, Pfetzer N, Waldman YY, McGarry L, James D, Shanks E, et al. Predicting cancer-specific vulnerability via data-driven detection of synthetic lethality. Cell (2014) 158(5):1199–209. doi: 10.1016/j.cell.2014.07.027
157. Lanning BR, Whitby LR, Dix MM, Douhan J, Gilbert AM, Hett EC, et al. A road map to evaluate the proteome-wide selectivity of covalent kinase inhibitors. Nat Chem Biol (2014) 10(9):760–7. doi: 10.1038/nchembio.1582
158. Boike L, Cioffi AG, Majewski FC, Co J, Henning NJ, Jones MD, et al. Discovery of a Functional Covalent Ligand Targeting an Intrinsically Disordered Cysteine within MYC. Cell Chem Biol (2020). doi: 10.1016/j.chembiol.2020.09.001
159. Gao H, Sun X, Rao Y. PROTAC Technology: Opportunities and Challenges. ACS Med Chem Lett (2020) 11(3):237–40. doi: 10.1021/acsmedchemlett.9b00597
160. Jung KY, Wang H, Teriete P, Yap JL, Chen L, Lanning ME, et al. Perturbation of the c-Myc-Max protein-protein interaction via synthetic alpha-helix mimetics. J Med Chem (2015) 58(7):3002–24. doi: 10.1021/jm501440q
161. Wei JS, Song YK, Durinck S, Chen QR, Cheuk AT, Tsang P, et al. The MYCN oncogene is a direct target of miR-34a. Oncogene (2008) 27(39):5204–13. doi: 10.1038/onc.2008.154
162. Yasukawa K, Liew LC, Hagiwara K, Hironaka-Mitsuhashi A, Qin XY, Furutani Y, et al. MicroRNA-493-5p-mediated repression of the MYCN oncogene inhibits hepatic cancer cell growth and invasion. Cancer Sci (2020) 111(3):869–80. doi: 10.1111/cas.14292
163. Zhang H, Liu T, Yi S, Gu L, Zhou M. Targeting MYCN IRES in MYCN-amplified neuroblastoma with miR-375 inhibits tumor growth and sensitizes tumor cells to radiation. Mol Oncol (2015) 9(7):1301–11. doi: 10.1016/j.molonc.2015.03.005
164. Molenaar JJ, Domingo-Fernandez R, Ebus ME, Lindner S, Koster J, Drabek K, et al. LIN28B induces neuroblastoma and enhances MYCN levels via let-7 suppression. Nat Genet (2012) 44(11):1199–206. doi: 10.1038/ng.2436
165. Singh A, Trivedi P, Jain NK. Advances in siRNA delivery in cancer therapy. Artif Cells Nanomed Biotechnol (2018) 46(2):274–83. doi: 10.1080/21691401.2017.1307210
166. Lau YK, Ramaiyer M, Johnson DE, Grandis JR. Targeting STAT3 in Cancer with Nucleotide Therapeutics. Cancers (Basel) (2019) 11(11). doi: 10.3390/cancers11111681
Keywords: MYCN, Super-enhancer (SE), cofactor, cancer, pediatric cancer, MYC, transcription factor
Citation: Liu Z, Chen SS, Clarke S, Veschi V and Thiele CJ (2021) Targeting MYCN in Pediatric and Adult Cancers. Front. Oncol. 10:623679. doi: 10.3389/fonc.2020.623679
Received: 30 October 2020; Accepted: 14 December 2020;
Published: 08 February 2021.
Edited by:
Yusuke Suenaga, Chiba Cancer Center, JapanReviewed by:
Robert Eisenman, Fred Hutchinson Cancer Research Center, United StatesCopyright © 2021 Liu, Chen, Clarke, Veschi and Thiele. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhihui Liu, bGl1emhpaHVAbWFpbC5uaWguZ292; Carol J. Thiele, dGhpZWxlY0BtYWlsLm5paC5nb3Y=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.