 Elisabet Cuyàs
Elisabet Cuyàs Sara Verdura
Sara Verdura Begoña Martin-Castillo2,3
Begoña Martin-Castillo2,3 Javier A. Menendez
Javier A. Menendez- 1Girona Biomedical Research Institute, Girona, Spain
- 2Program Against Cancer Therapeutic Resistance (ProCURE), Metabolism & Cancer Group, Catalan Institute of Oncology, Girona, Spain
- 3Unit of Clinical Research, Catalan Institute of Oncology, Girona, Spain
Metabolism can directly drive or indirectly enable an aberrant chromatin state of cancer cells. The physiological and molecular principles of the metabolic link to epigenetics provide a basis for pharmacological modulation with the anti-diabetic biguanide metformin. Here, we briefly review how metabolite-derived chromatin modifications and the metabolo-epigenetic machinery itself are both amenable to modification by metformin in a local and a systemic manner. First, we consider the capacity of metformin to target global metabolic pathways or specific metabolic enzymes producing chromatin-modifying metabolites. Second, we examine its ability to directly or indirectly fine-tune the activation status of chromatin-modifying enzymes. Third, we envision how the interaction between metformin, diet and gut microbiota might systemically regulate the metabolic inputs to chromatin. Experimental and clinical validation of metformin’s capacity to change the functional outcomes of the metabolo-epigenetic link could offer a proof-of-concept to therapeutically test the metabolic adjustability of the epigenomic landscape of cancer.
Introduction
Global changes in the topology of the epigenetic landscape are a hallmark of cancer (1–3). Metabolism can directly drive or indirectly enable an aberrant chromatin state of cancer cells (4–9). Elucidating pharmacological and dietary interventions that can fine-tune the molecular cross-talk between metabolism and the chromatin state is an active and rapidly growing area in cancer research (10–12).
More than 60 years after its introduction as a first-line therapy for managing type 2 diabetes, the biguanide metformin is emerging as an archetypal drug capable of targeting most of the epigenetic traits of cancer (13–16) (Figure 1). The role of metformin as a remodeler of the epigenetic landscape largely relies on its multi-faceted capacity to link metabolic signals to chromatin status. The ability of metformin to modify the functional outcomes of the metabolo-epigenetic link might serve as a proof-of-concept to therapeutically test the metabolic adjustability of the epigenomic landscape in cancer biology. Here, we briefly review how the biochemical principles of metabolite-derived chromatin modifications and the metabolo-epigenetic machinery itself are both amenable to regulation by metformin in a local and systemic manner.
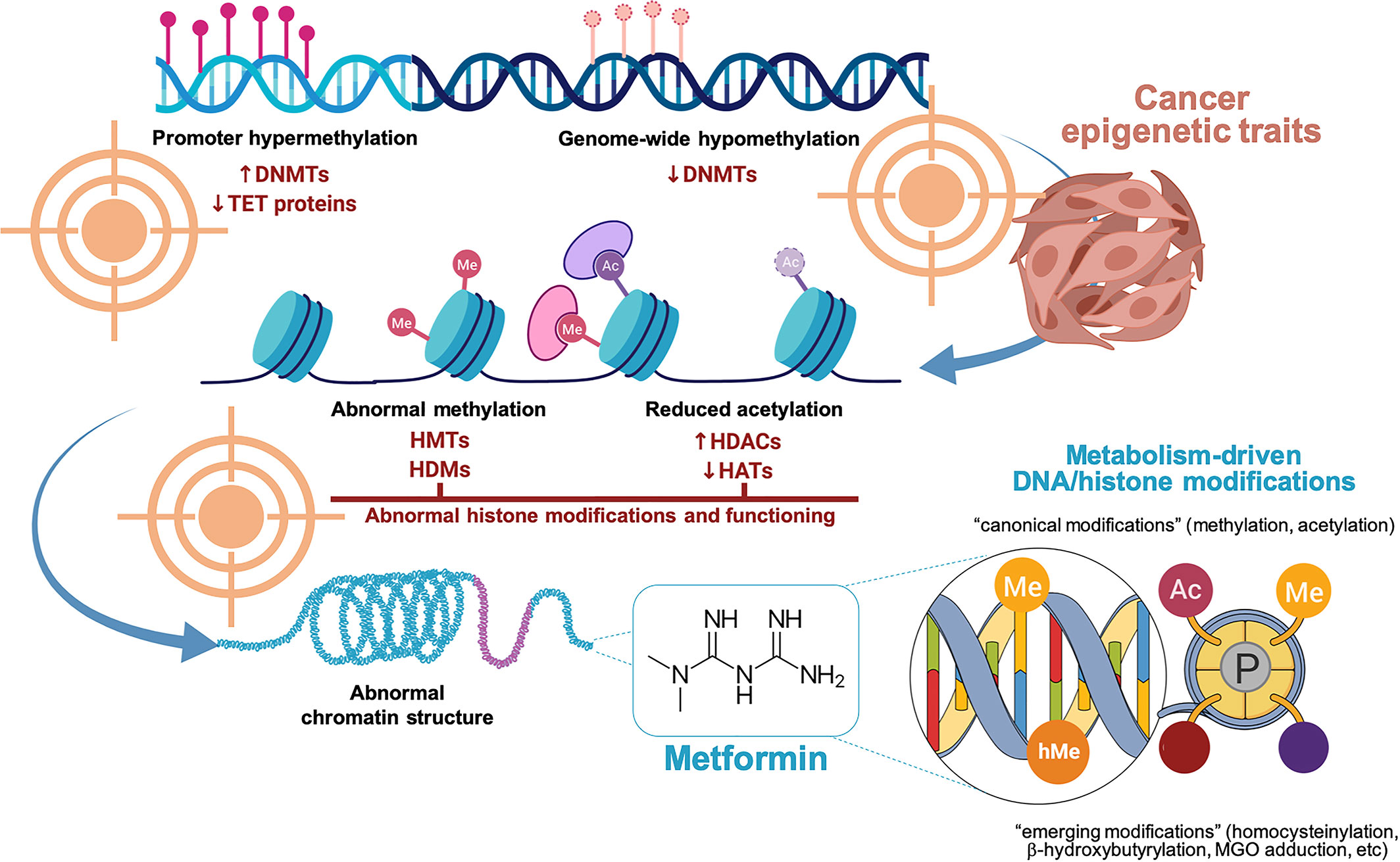
Figure 1 Metformin and the epigenetic landscape in cancer biology. The epigenetic traits in human cancers may lead to at least four five major phenotypes, namely: DNA promoter hypermethylation, genome-wide DNA hypomethylation, abnormal histone modifications (e.g., acetylation, methylation), and abnormal chromatin structure. We are accumulating ever-growing evidence that metformin can reverse several of the metabolism-related DNA/histone modifications underlying cancer biology including abnormal DNA methylation patterns, reduced histone acetylation, and abnormal histone methylation marking. It remains unclear whether and how metformin could influence the high-level architecture of chromosomes. Created with BioRender.com.
Metformin and Chromatin-Modifying Metabolites
Many cellular metabolic pathways related to central-carbon/tricarboxylic acid cycle, one-carbon/methionine metabolism, acetate metabolism, ketogenesis, and redox balance produce metabolites that are used as substrates and cofactors by chromatin-modifying enzymes (4–12). The physiological concentrations of these metabolites are commensurate with the catalytic efficiency of some chromatin-modifying enzymes (e.g., methyltransferases and acetyltransferases, among many others). Perturbations in the abundance of chromatin-modifying metabolites can therefore influence the activities of certain chromatin-remodeling enzymes and, consequently, the types and/or levels of “classical” (methylation, acetylation) and “emerging” (e.g., lactylation, butyrylation, homocysteinylation, etc) epigenetic modifications (4–8, 12). Metformin can fine-tune the relative sensitivity of epigenetic regulators to metabolic alterations by modifying the functioning of global metabolic pathways or specific metabolic enzymes producing chromatin-modifying metabolites (Figure 2).
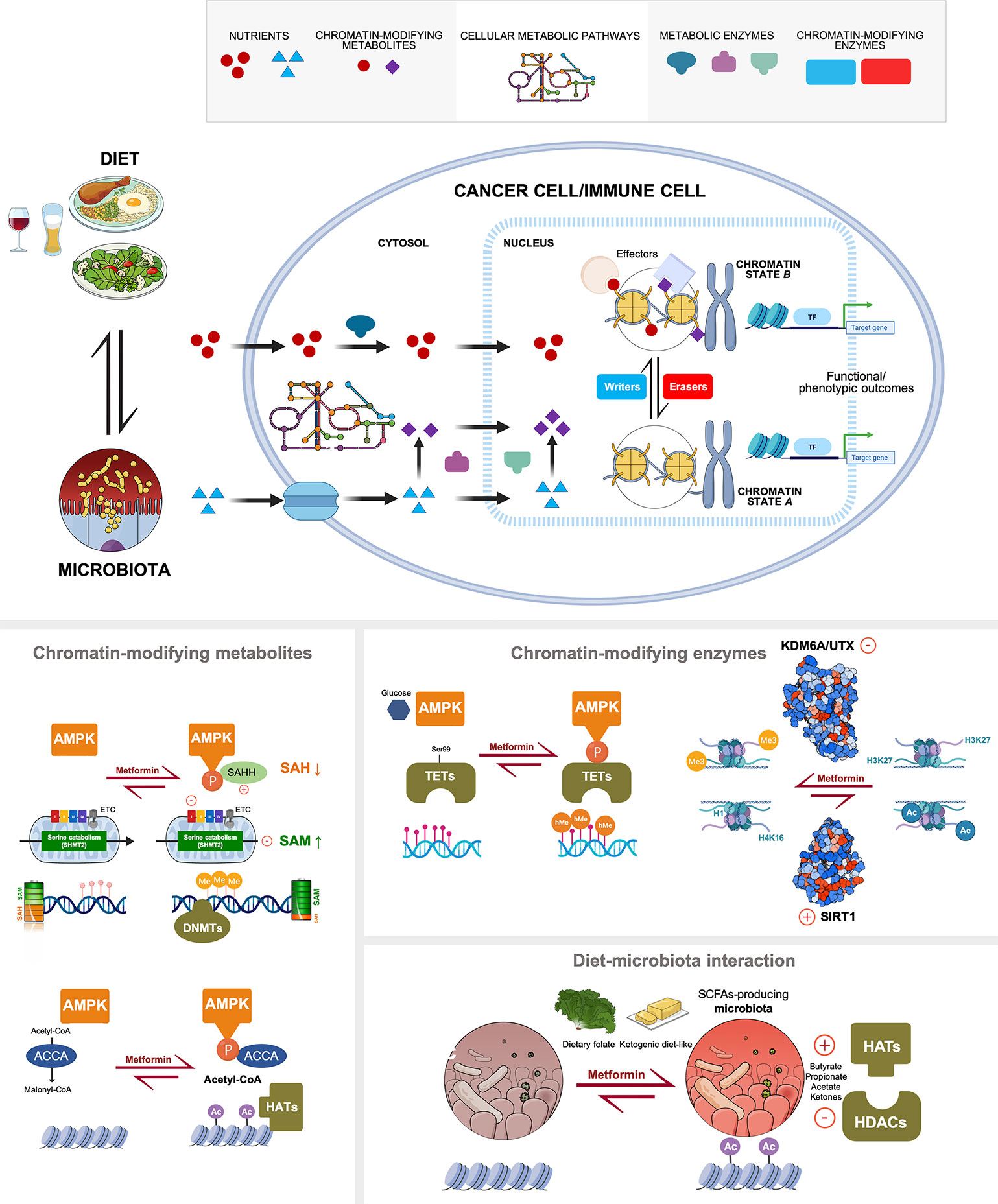
Figure 2 Multi-layered regulation of the cancer metabolo-epigenetic link by metformin. Top. Nutrients and metabolites derived from the bidirectional interaction between diet and human microbiome are directly utilized or internally processed by cellular metabolic pathways (either globally in the cytosol or locally in the nucleus) to modify chromatin. The catalytic dependence of chromatin-modifying enzymes on the abundance of substrates or activity modulators (cofactors) dictates, at least in part, both the type and rate of chromatin modifications. The metabolic regulation of epigenetic markings lastly determine the expression of genes related to functional outcomes (e.g., stemness/differentiation, immune suppression/activation, etc) that are relevant not only at the cancer cell-autonomous level but also to a variety of immune cell types. Bottom. All the physiological and molecular principles of the local and systemic metabolic link to cancer epigenetics can be targeted with the anti-diabetic biguanide metformin, namely: global metabolic pathways or specific metabolic enzymes producing chromatin-modifying metabolites, activation status of chromatin-modifying enzymes, and the interaction between diet and gut microbiota. Created with BioRender.com.
Metformin-driven changes in the metabolic pathways producing chromatin-modifying metabolites can be orchestrated in parallel with mitochondrial complex I (mCI) inhibition and its inhibitory action on the mitochondrial electron transport chain (ETC), and can be attributed both to its AMP-activated protein kinase (AMPK)-dependent and -independent actions. There are numerous examples that illustrate the ability of metformin to alter the pools of metabolic intermediates acting as activity modulators of a variety of epigenome-modifying enzymes, including nicotinamide adenine dinucleotide (NAD+) and β-hydroxyglutarate for histone deacetylases (HDACs), acetyl-CoA for histone acetyltransferases (HATs), α-ketoglutarate/succinate for histone/DNA demethylases (HDMs), and s-adenosyl-methionine/homocysteine (SAM/SAH) for DNA/histone methyltransferases (DNMTs/HMTs), among others (15, 17–28). The ability of metformin to tweak the SAM : SAH ratio-dependent methylation capacity exemplifies how it can regulate molecular inputs to chromatin via AMPK-dependent and -independent regulatory actions on central epigenetic metabolites. Metformin-induced AMPK activation positively modulates the activity of SAH hydrolase (SAHH) (29), to decrease the availability of SAH, a potent feedback inhibitor of SAM-dependent DNA and histone methyltransferases. In an AMPK-independent manner, the metformin-driven disruption of the coupling between mCI activity and serine hydroxymethytransferase (SHMT2)-driven mitochondrial serine catabolism increases the contribution of one-carbon units to SAM from folate stores (30). The targeting of core “hub” metabolites with epigenetic properties, such as SAM and SAH, supports the notion that metformin can activate epigenetic cancer-protection programs by promoting genome-wide DNA methylation events including methylation-regulated motility of retrotransposable elements such as long interspersed nuclear element-1 (LINE-1) or hypermethylation of tumor-promoting pathway genes (15, 29–31). Yet, we should acknowledge that there is a paucity of mechanistic studies examining a causal relationship between metformin-induced alterations in specific metabolites, regulation of the epigenomic landscape in concert with metabolite-associated chromatin modifiers, and epigenetic control of cancer-related functional outcomes.
Metformin and Chromatin-Modifying Enzymes
Beyond promoting fluctuations in the abundance of metabolites that can positively or negatively influence the activities of certain chromatin-modifying enzymes, metformin can modulate the levels of specific epigenetic modifications by indirectly or directly targeting the activation status of core epigenetic enzymes (Figure 2).
Perhaps the most obvious way in which metformin can target epigenetic enzymes is via the multifunctional kinase effects of AMPK (32–35). By inhibiting mCI and oxidative phosphorylation, metformin increases the AMP : ATP ratio, which induces the activation of AMPK. In turn, AMPK can directly regulate the phosphorylation/activation status of some DNA methyltransferases isoforms (e.g., DNMT1), which catalyze the methylation of cytosine-guanine dinucleotides (CpG), in addition to the ten-eleven translocation hydroxylases (TET), which catalyze the conversion of 5-methylcytosine to unmethylated cytosine. AMPK activation also induces the phosphorylation of histones/histone-modifying enzymes that play a major role in the regulation of the structure and function of nucleosomes (36), namely: histone H2B to promote chromatin relaxation, HDACs to increase histone acetylation, polycomb repressive complex 2 (PRC2) to decrease histone methylation, poly [ADP-ribose] polymerase 1 (PARP1) to increase histone ribosylation, and O-linked β-N-acetylglucosamine (O-GlcNAc) transferase to inhibit histone O-GlcNAcylation. There are already some encouraging examples of how metformin-induced AMPK activation regulates the activity of DNA methyltransferases/demethylases and histone-modifying enzymes to translate the metformin-driven transient decrease in cellular energy status into epigenetically-regulated transcriptional events. At the level of DNA methylation, metformin can protect the AMPK-mediated phosphorylation of the TET2 demethylating enzyme, which increases TET2 stability and 5-hydroxymethylcytosine (5 hmC) levels, thus impeding the ability of certain metabolic environments (e.g., high glucose state) to aberrantly reprogram the epigenome, thereby maintaining genomic stability (37). At the level of histone post-translational modifications, metformin-induced AMPK activation has been shown to phosphorylate the histone methyltransferase EZH2 (enhancer of zeste homolog 2), leading to the attenuated methyltransferase activity (and oncogenic function) of PRC2 (polycomb repressive complex 2) (38).
One should acknowledge that the aforementioned ability of metformin to regulate the substrate/cofactor availability of chromatin-modifying metabolites adds an additional layer of regulatory complexity to its capacity to drive histone modifications. For instance, metformin treatment reduces intracellular succinate levels and activates AMPK, both of which can independently mediate the activation of the lysine-demethylase 2 A (KDM2A) to repress ribosomal RNA (rRNA) transcription and cell proliferation (24). Moreover, certain epigenetic enzymes appear to behave as metformin-binding proteins, pointing to the possibility that metformin can directly modulate their chromatin-modifying activities without the involvement of mCI and AMPK. Metformin-induced activation of AMPK can indirectly activate the class III HDAC SIRT1 (Sirtuin 1) by increasing the NAD+/NADH ratio (17, 39, 40). Intriguingly, the AMPK-independent behavior of metformin as a bona fide SIRT1 agonist might also rely on its ability to directly improve the catalytic efficiency of SIRT1 operating under conditions of low NAD+ (41). Although metformin-induced inhibition of catalysis by isolated mCI, which is believed to be the primary target of metformin, requires concentrations as high as 20–100 mmol/L (42, 43), the physiological relevance of the direct binding of metformin to chromatin modifiers such as SIRT1 (41) and KDM6A/UTX (44) at millimolar concentrations in excess of the therapeutic levels achieved in human patients remains unknown.
Metformin and Diet/Microbiota-Associated Epigenetic Reprogramming
Environmental factors including diet and microbiota can regulate HMTs and HATs by modulating the intracellular pools of their substrates SAM and acetyl-CoA, respectively. Dietary methionine and other nutrients feeding into one-carbon and methionine metabolism (e.g., folate, vitamin B, and choline) can modulate the levels of SAM and SAH to reprogram the landscape of DNA and histone methylation (10, 45–47). Metabolic adaptation to calorie restriction, fasting, the intake of a ketogenic diet, and exercise, can generate ketone bodies through the breakdown of fatty acids and ketogenic amino acids (48–54). The ketone body β-hydroxybutyrate (β-OHB) can elicit multi-faceted chromatin modifications by inhibiting class I HDACs to cause global upregulation of histone acetylation (55), while also serving as a substrate for β-hydroxybutyrylation, which associates with the upregulation of starvation-responsive metabolic pathways (56). The ketone body acetoacetate can suppress the adduction of histones by methyglyoxal (MGO) (57), a reactive dicarbonyl sugar metabolite that induces disease-related changes in chromatin architecture and the epigenetic landscape (58, 59).
The bidirectional interaction between diet and gut microbiota can drive microbiota metabolomes to alter systemic metabolic physiology and influence epigenetic programs (60–62) (Figure 2). Microbiotal activity on dietary fiber can enhance circulating levels of short-chain fatty acids (SCFAs) such as a formate [C1], acetate [C2], propionate [C3], butyrate/isobutyrate [C4], crotonate [C4], and valerate/isovalerate [C5], which can feed the intracellular pool of acetyl-CoA and/or promote the inhibition of HDACs to augment histone acetylation and crotonylation (63–66). Lesser-explored diet/gut microbiota interactions might involve the colonization of specific microbiota species competing with host cells for epigenome metabolites, such as choline-consuming strains reducing the serum levels of the methyl donor SAM, and consequently promoting changes to global DNA methylation (67). Formaldehyde, a formate-related one-carbon benign/genotoxic molecule at the crossroad between cellular and whole-body metabolism, coupled with the societal use of alcohol consumption (68, 69), might alter epigenetic regulation as a source of pathological formyl-lysine that is refractory to HDACs (70, 71).
Early evidence of the role of metformin in modulating the gut microbiota involved the modulation of microbial folate, leading to methionine restriction in C. elegans (72). The addition of metformin to an established anti-breast cancer therapeutic regimen was found to induce a fasting- and antifolate-modification of systemic host metabolism that included a significant increase in the circulating levels of the ketone body β-OHB and of homocysteine (28, 73), a surrogate marker of the availability of methyl groups (SAM) via one-carbon/methionine metabolism. In diabetic individuals, metformin can also shift gut microbiota composition through the enrichment of predominantly SCFA-producing bacteria (74, 75). Metformin is also known to significantly reduce systemic MGO levels, not only by operating as an efficient scavenger (76) but by also by increasing the activity of glyoxalase 1, the major route of MGO detoxification (77). The ability of metformin to fine-tune the shaping influence of environmental factors on the epigenomic landscape might go beyond the bidirectional cross-talk between diet and microbiota (Figure 2). In this regard, the reduction of circulating formate as a consequence of metformin-induced disruption of the coupling between mCI activity and mitochondrial serine catabolism (78, 79) and the recently described formaldehyde-scavenging capacity of metformin (80), add new metabolic facets likely involved in the systemic effects of metformin on the cancer-driving metabolo-epigenetic link. Yet, it remains an open question whether systemic metabolic adaptation to metformin through modulation of methionine/one-carbon metabolism, which might be secondary to reduced vitamin B12 and/or folate levels (81), or via nutritional modulation of the gut microbiota [e.g., decreasing the availability of branched-chain amino acids (82)], suffices to induce distinct epigenetic responses capable of regulating cancer-relevant phenotypic outcomes.
Metformin-Targeted Metabolo-Epigenetics Axis in Cancer Prevention and Treatment: Are We There Yet?
We are accumulating more and more evidence that metformin can dynamically reshape the epigenomic landscape of cancer. First, metformin can target global metabolic pathways or specific metabolic enzymes producing chromatin-modifying metabolites. Second, metformin can indirectly or directly modify the activation status of chromatin-modifying enzymes. Third, metformin can interact with diet/gut microbiota to systemically regulate the metabolic inputs to chromatin. However, despite the exciting progress in deciphering how metformin influences metabolically regulated epigenetic marks, we should acknowledge that mechanistic understanding of how it can reverse cancer-relevant functional outcomes [e.g., phenotypic plasticity and cell fate decisions in heterogenous cancer populations, metabolism-epigenome axes controlling anti-tumor immunity and immunotherapy efficacy (83–87)] by interfering with the metabolic regulation of the epigenome remains unsatisfactory.
Important future questions for the field include: Can metformin impact the distribution of metabolites between the nuclear and non-nuclear compartment by changing metabolite consumption on and release from chromatin (88)? Can metformin reconduct the same core metabolic pathways or metabolites to establish distinct epigenetic signatures of the heterogenous tumor microenvironment at the single cell level (89)? Can metabolic regulation of higher-order genomic organizers elicit a metformin-driven regulation of the three-dimensional architecture of chromosomes (90–92)? These remain exciting areas of investigation. While decoding the molecular intimacy of the metabolo-epigenetic “language” of metformin in cancer biology, it remains true that the experimental and clinical validation of its capacity to alter the functional outcomes of the metabolo-epigenetic link could provide a proof-of-concept to therapeutically test the metabolic adjustability of the epigenomic landscape in cancer prevention and treatment.
Author Contributions
JM conceived the scope of the manuscript, as well as for drafting the manuscript. EC, SV, and BM-C helped with final formatting of the manuscript, schematics and literature search and selection. All authors contributed to the article and approved the submitted version.
Funding
Work in the Menendez laboratory is supported by the Spanish Ministry of Science and Innovation (grants SAF2016-80639-P and PID2019-10455GB-I00, Plan Nacional de l+D+I, founded by the European Regional Development Fund, Spain) and by an unrestricted research grant from the Fundació Oncolliga Girona (Lliga catalana d’ajuda al malalt de càncer, Girona). EC is a recipient of a research contract “Miguel Servet” (CP20/00003) from the Instituto de Salud Carlos III, Spanish Ministry of Science and Innovation (Spain).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank Dr. Kenneth McCreath for editorial support.
References
1. Flavahan WA, Gaskell E, Bernstein BE. Epigenetic plasticity and the hallmarks of cancer. Science (2017) 357:eaal2380. doi: 10.1126/science.aal2380
2. Nebbioso A, Tambaro FP, Dell’Aversana C, Altucci L. Cancer epigenetics: Moving forward. PLoS Genet (2018) 14:e1007362. doi: 10.1371/journal.pgen.1007362
3. Chatterjee A, Rodger EJ, Eccles MR. Epigenetic drivers of tumourigenesis and cancer metastasis. Semin Cancer Biol (2018) 51:149–59. doi: 10.1016/j.semcancer.2017.08.004
4. Menendez JA, Alarcón T. Metabostemness: a new cancer hallmark. Front Oncol (2014) 4:262. doi: 10.3389/fonc.2014.00262
5. Johnson C, Warmoes MO, Shen X, Locasale JW. Epigenetics and cancer metabolism. Cancer Lett (2015) 356:309–14. doi: 10.1016/j.canlet.2013.09.043
6. Menendez JA. The Metaboloepigenetic Dimension of Cancer Stem Cells: Evaluating the Market Potential for New Metabostemness-Targeting Oncology Drugs. Curr Pharm Des (2015) 21:3644–53. doi: 10.2174/1381612821666150710150327
7. Mentch SJ, Locasale JW. One-carbon metabolism and epigenetics: understanding the specificity. Ann N Y Acad Sci (2016) 1363:91–8. doi: 10.1111/nyas.12956
8. Reid MA, Dai Z, Locasale JW. The impact of cellular metabolism on chromatin dynamics and epigenetics. Nat Cell Biol (2017) 19:1298–306. doi: 10.1038/ncb3629
9. Gao X, Reid MA, Kong M, Locasale JW. Metabolic interactions with cancer epigenetics. Mol Aspects Med (2017) 54:50–7. doi: 10.1016/j.mam.2016.09.001
10. Sanderson SM, Gao X, Dai Z, Locasale JW. Methionine metabolism in health and cancer: a nexus of diet and precision medicine. Nat Rev Cancer (2019) 19:625–37. doi: 10.1038/s41568-019-0187-8
11. Bose S, Allen AE, Locasale JW. The Molecular Link from Diet to Cancer Cell Metabolism. Mol Cell (2020) 78:1034–44. doi: 10.1016/j.molcel.2020.05.018
12. Dai Z, Ramesh V, Locasale JW. The evolving metabolic landscape of chromatin biology and epigenetics. Nat Rev Genet (2020) 21:737–53. doi: 10.1038/s41576-020-0270-8
13. Yu X, Mao W, Zhai Y, Tong C, Liu M, Ma L, et al. Anti-tumor activity of metformin: from metabolic and epigenetic perspectives. Oncotarget (2017) 8:5619–28. doi: 10.18632/oncotarget.13639
14. Bridgeman SC, Ellison GC, Melton PE, Newsholme P, Mamotte CDS. Epigenetic effects of metformin: From molecular mechanisms to clinical implications. Diabetes Obes Metab (2018) 20:1553–62. doi: 10.1111/dom.13262
15. Menendez JA. Metformin: Sentinel of the Epigenetic Landscapes That Underlie Cell Fate and Identity. Biomolecules (2020) 10:780. doi: 10.3390/biom10050780
16. Kulkarni AS, Gubbi S, Barzilai N. Benefits of Metformin in Attenuating the Hallmarks of Aging. Cell Metab (2020) 32:15–30. doi: 10.1016/j.cmet.2020.04.001
17. Cantó C, Gerhart-Hines Z, Feige JN, Lagouge M, Noriega L, Milne JC, et al. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature (2009) 458:1056–60. doi: 10.1038/nature07813
18. Gui DY, Sullivan LB, Luengo A, Hosios AM, Bush LN, Gitego N, et al. Environment Dictates Dependence on Mitochondrial Complex I for NAD+ and Aspartate Production and Determines Cancer Cell Sensitivity to Metformin. Cell Metab (2016) 24:716–27. doi: 10.1016/j.cmet.2016.09.006
19. Benjamin D, Robay D, Hindupur SK, Pohlmann J, Colombi M, El-Shemerly MY, et al. Dual Inhibition of the Lactate Transporters MCT1 and MCT4 Is Synthetic Lethal with Metformin due to NAD+ Depletion in Cancer Cells. Cell Rep (2018) 25:3047–58.e4. doi: 10.1016/j.celrep.2018.11.043
20. Alshawi A, Agius L. Low metformin causes a more oxidized mitochondrial NADH/NAD redox state in hepatocytes and inhibits gluconeogenesis by a redox-independent mechanism. J Biol Chem (2019) 294:2839–53. doi: 10.1074/jbc.RA118.006670
21. Galdieri L, Gatla H, Vancurova I, Vancura A. Activation of AMP-activated Protein Kinase by Metformin Induces Protein Acetylation in Prostate and Ovarian Cancer Cells. J Biol Chem (2016) 291:25154–66. doi: 10.1074/jbc.M116.742247
22. Yang Q, Liang X, Sun X, Zhang L, Fu X, Rogers CJ, et al. AMPK/α-Ketoglutarate Axis Dynamically Mediates DNA Demethylation in the Prdm16 Promoter and Brown Adipogenesis. Cell Metab (2016) 24:542–54. doi: 10.1016/j.cmet.2016.08.010
23. Hohnholt MC, Blumrich EM, Waagepetersen HS, Dringen R. The antidiabetic drug metformin decreases mitochondrial respiration and tricarboxylic acid cycle activity in cultured primary rat astrocytes. J Neurosci Res (2017) 95:2307–20. doi: 10.1002/jnr.24050
24. Tanaka Y, Konishi A, Obinata H, Tsuneoka M. Metformin activates KDM2A to reduce rRNA transcription and cell proliferation by dual regulation of AMPK activity and intracellular succinate level. Sci Rep (2019) 9:18694. doi: 10.1038/s41598-019-55075-0
25. Jenkins Y, Sun TQ, Li Y, Markovtsov V, Uy G, Gross L, et al. Global metabolite profiling of mice with high-fat diet-induced obesity chronically treated with AMPK activators R118 or metformin reveals tissue-selective alterations in metabolic pathways. BMC Res Notes (2014) 7:674. doi: 10.1186/1756-0500-7-674
26. Schuler KM, Rambally BS, DiFurio MJ, Sampey BP, Gehrig PA, Makowski L, et al. Antiproliferative and metabolic effects of metformin in a preoperative window clinical trial for endometrial cancer. Cancer Med (2015) 4:161–73. doi: 10.1002/cam4.353
27. Liu X, Romero IL, Litchfield LM, Lengyel E, Locasale JW. Metformin Targets Central Carbon Metabolism and Reveals Mitochondrial Requirements in Human Cancers. Cell Metab (2016) 24:728–39. doi: 10.1016/j.cmet.2016.09.005
28. Cuyàs E, Fernández-Arroyo S, Buxó M, Pernas S, Dorca J, Álvarez I, et al. Metformin induces a fasting- and antifolate-mimicking modification of systemic host metabolism in breast cancer patients. Aging (Albany NY) (2019) 11:2874–88. doi: 10.18632/aging.101960
29. Zhong T, Men Y, Lu L, Geng T, Zhou J, Mitsuhashi A, et al. Metformin alters DNA methylation genome-wide via the H19/SAHH axis. Oncogene (2017) 36:2345–54. doi: 10.1038/onc.2016.391
30. Cuyàs E, Fernández-Arroyo S, Verdura S, García RÁ, Stursa J, Werner L, et al. Metformin regulates global DNA methylation via mitochondrial one-carbon metabolism. Oncogene (2018) 37:963–70. doi: 10.1038/onc.2017.367
31. Wu P, Tang Y, Fang X, Xie C, Zeng J, Wang W, et al. Metformin Suppresses Hypopharyngeal Cancer Growth by Epigenetically Silencing Long Non-coding RNA SNHG7 in FaDu Cells. Front Pharmacol (2019) 10:143. doi: 10.3389/fphar.2019.00143
32. Salminen A, Kauppinen A, Kaarniranta K. AMPK/Snf1 signaling regulates histone acetylation: Impact on gene expression and epigenetic functions. Cell Signal (2016) 28:887–95. doi: 10.1016/j.cellsig.2016.03.009
33. Marin TL, Gongol B, Zhang F, Martin M, Johnson DA, Xiao H, et al. AMPK promotes mitochondrial biogenesis and function by phosphorylating the epigenetic factors DNMT1, RBBP7, and HAT1. Sci Signal (2017) 10:eaaf7478. doi: 10.1126/scisignal.aaf7478
34. Fiedler EC, Shaw RJ. AMPK Regulates the Epigenome through Phosphorylation of TET2. Cell Metab (2018) 28:534–6. doi: 10.1016/j.cmet.2018.09.015
35. Wang YP, Lei QY. Metabolic recoding of epigenetics in cancer. Cancer Commun (Lond) (2018) 38:25. doi: 10.1186/s40880-018-0302-3
36. Gongol B, Sari I, Bryant T, Rosete G, Marin T. AMPK: An Epigenetic Landscape Modulator. Int J Mol Sci (2018) 19:3238. doi: 10.3390/ijms19103238
37. Wu D, Hu D, Chen H, Shi G, Fetahu IS, Wu F, et al. Glucose-regulated phosphorylation of TET2 by AMPK reveals a pathway linking diabetes to cancer. Nature (2018) 559:637–41. doi: 10.1038/s41586-018-0350-5
38. Wan L, Xu K, Wei Y, Zhang J, Han T, Fry C, et al. Phosphorylation of EZH2 by AMPK Suppresses PRC2 Methyltransferase Activity and Oncogenic Function. Mol Cell (2018) 69:279–91.e5. doi: 10.1016/j.molcel.2017.12.024
39. Madeo F, Carmona-Gutierrez D, Hofer SJ, Kroemer G. Caloric Restriction Mimetics against Age-Associated Disease: Targets, Mechanisms, and Therapeutic Potential. Cell Metab (2019) 29:592–610. doi: 10.1016/j.cmet.2019.01.018
40. Price NL, Gomes AP, Ling AJ, Duarte FV, Martin-Montalvo A, North BJ, et al. SIRT1 is required for AMPK activation and the beneficial effects of resveratrol on mitochondrial function. Cell Metab (2012) 15:675–90. doi: 10.1016/j.cmet.2012.04.003
41. Cuyàs E, Verdura S, Llorach-Parés L, Fernández-Arroyo S, Joven J, Martin-Castillo B, et al. Metformin Is a Direct SIRT1-Activating Compound: Computational Modeling and Experimental Validation. Front Endocrinol (Lausanne) (2018) 9:657. doi: 10.3389/fendo.2018.00657
42. Bridges HR, Jones AJ, Pollak MN, Hirst J. Effects of metformin and other biguanides on oxidative phosphorylation in mitochondria. Biochem J (2014) 462:475–87. doi: 10.1042/BJ20140620
43. Bridges HR, Sirviö VA, Agip AN, Hirst J. Molecular features of biguanides required for targeting of mitochondrial respiratory complex I and activation of AMP-kinase. BMC Biol (2016) 14:65. doi: 10.1186/s12915-016-0287-9
44. Cuyàs E, Verdura S, Llorach-Pares L, Fernández-Arroyo S, Luciano-Mateo F, Cabré N, et al. Metformin directly targets the H3K27me3 demethylase KDM6A/UTX. Aging Cell (2018) 17:e12772. doi: 10.1111/acel.12772
45. Dai Z, Mentch SJ, Gao X, Nichenametla SN, Locasale JW. Methionine metabolism influences genomic architecture and gene expression through H3K4me3 peak width. Nat Commun (2018) 9:1955. doi: 10.1038/s41467-018-04426-y
46. Mentch SJ, Mehrmohamadi M, Huang L, Liu X, Gupta D, Mattocks D, et al. Histone Methylation Dynamics and Gene Regulation Occur through the Sensing of One-Carbon Metabolism. Cell Metab (2015) 22:861–73. doi: 10.1016/j.cmet.2015.08.024
47. Gao X, Sanderson SM, Dai Z, Reid MA, Cooper DE, Lu M, et al. Dietary methionine influences therapy in mouse cancer models and altershuman metabolism. Nature (2019) 572:397–401. doi: 10.1038/s41586-019-1437-3
48. Wang T, Tsui B, Kreisberg JF, Robertson NA, Gross AM, Yu MK, et al. Epigenetic aging signatures in mice livers are slowed by dwarfism, calorie restriction and rapamycin treatment. Genome Biol (2017) 18:57. doi: 10.1186/s13059-017-1186-2
49. Newman JC, Verdin E. Ketone bodies as signaling metabolites. Trends Endocrinol Metab (2014) 25:42–52. doi: 10.1016/j.tem.2013.09.002
50. Puchalska P, Crawford PA. Multi-dimensional Roles of Ketone Bodies in Fuel Metabolism, Signaling, and Therapeutics. Cell Metab (2017) 25:262–84. doi: 10.1016/j.cmet.2016.12.022
51. Di Francesco A, Di Germanio C, Bernier M, de Cabo R. A time to fast. Science (2018) 362:770–5. doi: 10.1126/science.aau2095
52. Sleiman SF, Henry J, Al-Haddad R, El Hayek L, Abou Haidar E, Stringer T, et al. Exercise promotes the expression of brain derived neurotrophic factor (BDNF) through the action of the ketone body β-hydroxybutyrate. Elife (2016) 5:e15092. doi: 10.7554/eLife.15092
53. Benjamin JS, Pilarowski GO, Carosso GA, Zhang L, Huso DL, Goff LA, et al. A ketogenic diet rescues hippocampal memory defects in a mouse model of Kabuki syndrome. Proc Natl Acad Sci USA (2017) 114:125–30. doi: 10.1073/pnas.1611431114
54. Tognini P, Murakami M, Liu Y, Eckel-Mahan KL, Newman JC, Verdin E, et al. Distinct Circadian Signatures in Liver and Gut Clocks Revealed by Ketogenic Diet. Cell Metab (2017) 26:523–38.e5. doi: 10.1016/j.cmet.2017.08.015
55. Shimazu T, Hirschey MD, Newman J, He W, Shirakawa K, Le Moan N, et al. Suppression of oxidative stress by β-hydroxybutyrate, an endogenous histone deacetylase inhibitor. Science (2013) 339:211–4. doi: 10.1126/science.1227166
56. Xie Z, Zhang D, Chung D, Tang Z, Huang H, Dai L, et al. Metabolic Regulation of Gene Expression by Histone Lysine β-Hydroxybutyrylation. Mol Cell (2016) 62:194–206. doi: 10.1016/j.molcel.2016.03.036
57. Salomón T, Sibbersen C, Hansen J, Britz D, Svart MV, Voss TS, et al. Ketone Body Acetoacetate Buffers Methylglyoxal via a Non-enzymatic Conversion during Diabetic and Dietary Ketosis. Cell Chem Biol (2017) 24:935–43.e7. doi: 10.1016/j.chembiol.2017.07.012
58. Zheng Q, Osunsade A, David Y. Protein arginine deiminase 4 antagonizes methylglyoxal-induced histone glycation. Nat Commun (2020) 11:3241. doi: 10.1038/s41467-020-17066-y
59. Zheng Q, Omans ND, Leicher R, Osunsade A, Agustinus AS, Finkin-Groner E, et al. Reversible histone glycation is associated with disease-related changes in chromatin architecture. Nat Commun (2019) 10:1289. doi: 10.1038/s41467-019-09192-z
60. Rangan P, Choi I, Wei M, Navarrete G, Guen E, Brandhorst S, et al. Fasting-Mimicking Diet Modulates Microbiota and Promotes Intestinal Regeneration to Reduce Inflammatory Bowel Disease Pathology. Cell Rep (2019) 26:2704–19.e6. doi: 10.1016/j.celrep.2019.02.019
61. David LA, Maurice CF, Carmody RN, Gootenberg DB, Button JE, Wolfe BE, et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature (2014) 505:559–63. doi: 10.1038/nature12820
62. Blacher E, Bashiardes S, Shapiro H, Rothschild D, Mor U, Dori-Bachash M, et al. Potential roles of gut microbiome and metabolites in modulating ALS in mice. Nature (2019) 572:474–80. doi: 10.1038/s41586-019-1443-5
63. Fellows R, Denizot J, Stellato C, Cuomo A, Jain P, Stoyanova E, et al. Microbiota derived short chain fatty acids promote histone crotonylation in the colon through histone deacetylases. Nat Commun (2018) 9:105. doi: 10.1038/s41467-017-02651-5
64. Chang PV, Hao L, Offermanns S, Medzhitov R. The microbial metabolite butyrate regulates intestinal macrophage function via histone deacetylase inhibition. Proc Natl Acad Sci USA (2014) 111:2247–52. doi: 10.1073/pnas.1322269111
65. den Besten G, van Eunen K, Groen AK, Venema K, Reijngoud DJ, Bakker BM. The role of short-chain fatty acids in the interplay between diet, gut microbiota, and host energy metabolism. J Lipid Res (2013) 54:2325–40. doi: 10.1194/jlr.R036012
66. Donohoe DR, Holley D, Collins LB, Montgomery SA, Whitmore AC, Hillhouse A, et al. A gnotobiotic mouse model demonstrates that dietary fiber protects against colorectal tumorigenesis in a microbiota- and butyrate-dependent manner. Cancer Discov (2014) 4:1387–97. doi: 10.1158/2159-8290.CD-14-0501
67. Romano KA, Martinez-Del Campo A, Kasahara K, Chittim CL, Vivas EI, Amador-Noguez D, et al. Metabolic, Epigenetic, and Transgenerational Effects of Gut Bacterial Choline Consumption. Cell Host Microbe (2017) 22:279–90.e7. doi: 10.1016/j.chom.2017.07.021
68. Pontel LB, Rosado IV, Burgos-Barragan G, Garaycoechea JI, Yu R, Arends MJ, et al. Endogenous Formaldehyde Is a Hematopoietic Stem Cell Genotoxin and Metabolic Carcinogen. Mol Cell (2015) 60:177–88. doi: 10.1016/j.molcel.2015.08.020
69. Burgos-Barragan G, Wit N, Meiser J, Dingler FA, Pietzke M, Mulderrig L, et al. Mammals divert endogenous genotoxic formaldehyde into one-carbon metabolism. Nature (2017) 548:549–54. doi: 10.1038/nature23481
70. Edrissi B, Taghizadeh K, Moeller BC, Yu R, Kracko D, Doyle-Eisele M, et al. N6-Formyllysine as a Biomarker of Formaldehyde Exposure: Formation and Loss of N6-Formyllysine in Nasal Epithelium in Long-Term, Low-Dose Inhalation Studies in Rats. Chem Res Toxicol (2017) 30:1572–6. doi: 10.1021/acs.chemrestox.7b00075
71. Edrissi B, Taghizadeh K, Dedon PC. Quantitative analysis of histone modifications: formaldehyde is a source of pathological n(6)-formyllysine that is refractory to histone deacetylases. PLoS Genet (2013) 9:e1003328. doi: 10.1371/journal.pgen.1003328
72. Cabreiro F, Au C, Leung KY, Vergara-Irigaray N, Cochemé HM, Noori T, et al. Metformin retards aging in C. elegans by altering microbial folate and methionine metabolism. Cell (2013) 153:228–39. doi: 10.1016/j.cell.2013.02.035
73. Corominas-Faja B, Quirantes-Piné R, Oliveras-Ferraros C, Vazquez-Martin A, Cufí S, Martin-Castillo B, et al. Metabolomic fingerprint reveals that metformin impairs one-carbon metabolism in a manner similar to the antifolate class of chemotherapy drugs. Aging (Albany NY) (2012) 4:480–98. doi: 10.18632/aging.100472
74. de la Cuesta-Zuluaga J, Mueller NT, Corrales-Agudelo V, Velásquez-Mejía EP, Carmona JA, Abad JM, et al. Metformin Is Associated With Higher Relative Abundance of Mucin-Degrading Akkermansia muciniphila and Several Short-Chain Fatty Acid-Producing Microbiota in the Gut. Diabetes Care (2017) 40:54–62. Principio del formulario Final del formulario. doi: 10.2337/dc16-1324
75. Rosario D, Benfeitas R, Bidkhori G, Zhang C, Uhlen M, Shoaie S, et al. Understanding the Representative Gut Microbiota Dysbiosis in Metformin-Treated Type 2 Diabetes Patients Using Genome-Scale Metabolic Modeling. Front Physiol (2018) 9:775. doi: 10.3389/fphys.2018.00775
76. Kinsky OR, Hargraves TL, Anumol T, Jacobsen NE, Dai J, Snyder SA, et al. Metformin Scavenges Methylglyoxal To Form a Novel Imidazolinone Metabolite in Humans. Chem Res Toxicol (2016) 29:227–34. doi: 10.1021/acs.chemrestox.5b00497
77. Kender Z, Fleming T, Kopf S, Torzsa P, Grolmusz V, Herzig S, et al. Effect of metformin on methylglyoxal metabolism in patients with type 2 diabetes. Exp Clin Endocrinol Diabetes (2014) 122:316–9. doi: 10.1055/s-0034-1371818
78. Meiser J, Tumanov S, Maddocks O, Labuschagne CF, Athineos D, Van Den Broek N, et al. Serine one-carbon catabolism with formate overflow. Sci Adv (2016) 2:e1601273. doi: 10.1126/sciadv.1601273
79. Pietzke M, Arroyo SF, Sumpton D, Mackay GM, Martin-Castillo B, Camps J, et al. Stratification of cancer and diabetes based on circulating levels of formate and glucose. Cancer Metab (2019) 7:3. doi: 10.1186/s40170-019-0195-x
80. Mai X, Zhou F, Lin P, Lin S, Gao J, Ma Y, et al. Metformin scavenges formaldehyde and attenuates formaldehyde-induced bovine serum albumin crosslinking and cellular DNA damage. Environ Toxicol (2020) 35:1170–8. doi: 10.1002/tox.22982
81. Aroda VR, Edelstein SL, Goldberg RB, Knowler WC, Marcovina SM, Orchard TJ, et al. Long-term Metformin Use and Vitamin B12 Deficiency in the Diabetes Prevention Program Outcomes Study. J Clin Endocrinol Metab (2016) 101:1754–61. doi: 10.1210/jc.2015-3754
82. Sonnet D S, O’Leary M N, Gutierrez M A, Nguyen S M, Mateen S, Hsu Y, et al. Metformin inhibits Branched Chain Amino Acid (BCAA) derived ketoacidosis and promotes metabolic homeostasis in MSUD. Sci Rep (2016) 6:28775. doi: 10.1038/srep28775
83. Verdura S, Cuyàs E, Martin-Castillo B, Menendez JA. Metformin as an archetype immuno-metabolic adjuvant for cancer immunotherapy. Oncoimmunology (2019) 8:e1633235. doi: 10.1080/2162402X.2019.1633235
84. Zhang X, Liu J, Cao X. Metabolic control of T-cell immunity via epigenetic mechanisms. Cell Mol Immunol (2018) 15:203–5. doi: 10.1038/cmi.2017.115
85. Yerinde C, Siegmund B, Glauben R, Weidinger C. Metabolic Control of Epigenetics and Its Role in CD8(+) T CellDifferentiation and Function. Front Immunol (2019) 10:2718. doi: 10.3389/fimmu.2019.02718
86. Arpaia N, Campbell C, Fan X, Dikiy S, van der Veeken J, deRoos P, et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature (2013) 504:451–5. doi: 10.1038/nature12726
87. Li X, Wenes M, Romero P, Huang SC, Fendt SM, Ho PC. Navigating metabolic pathways to enhance antitumour immunity and immunotherapy. Nat Rev Clin Oncol (2019) 16:425–41. doi: 10.1038/s41571-019-0203-7
88. Boon R, Silveira GG, Mostoslavsky R. Nuclear metabolism and the regulation of the epigenome. Nat Metab (2020) 12:1190–203. doi: 10.1038/s42255-020-00285-4
89. Xiao Z, Dai Z, Locasale JW. Metabolic landscape of the tumor microenvironment at single cell resolution. Nat Commun (2019) 10:3763. doi: 10.1038/s41467-019-11738-0
90. Morris JP4, Yashinskie JJ, Koche R, Chandwani R, Tian S, CC C, et al. a-ketoglutarate links p53 to cell fate during tumour suppression. Nature (2019) 573:595–9. doi: 10.1038/s41586-019-1577-5
91. Baksh SC, Finley LWS. Metabolic coordination of cell fate by α-ketoglutarate-dependent dioxygenases. Trends Cell Biol (2020) 31:24-3. doi: 10.1016/j.tcb.2020.09.010
Keywords: metformin, metabolism, epigenetics, chromatin, cancer, diet, microbiota
Citation: Cuyàs E, Verdura S, Martin-Castillo B and Menendez JA (2021) Metformin: Targeting the Metabolo-Epigenetic Link in Cancer Biology. Front. Oncol. 10:620641. doi: 10.3389/fonc.2020.620641
Received: 23 October 2020; Accepted: 17 December 2020;
Published: 02 February 2021.
Edited by:
Fatima Baltazar, University of Minho, PortugalReviewed by:
Barbara Marengo, University of Genoa, ItalyElisabetta Benedetti, University of L’Aquila, Italy
Copyright © 2021 Cuyàs, Verdura, Martin-Castillo and Menendez. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Javier A. Menendez, am1lbmVuZGV6QGlkaWJnaS5vcmc=