
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Oncol. , 15 February 2021
Sec. Cancer Immunity and Immunotherapy
Volume 10 - 2020 | https://doi.org/10.3389/fonc.2020.617385
This article is part of the Research Topic Impact of the Glioma Microenvironment on Antitumor Immunity View all 14 articles
 Thamiris Becker Scheffel1,2
Thamiris Becker Scheffel1,2 Nathália Grave1,3
Nathália Grave1,3 Pedro Vargas1,3Fernando Mendonça Diz1Liliana Rockenbach1,3
Pedro Vargas1,3Fernando Mendonça Diz1Liliana Rockenbach1,3 Fernanda Bueno Morrone1,2,3*
Fernanda Bueno Morrone1,2,3*Glioblastoma is the most malignant and lethal subtype of glioma. Despite progress in therapeutic approaches, issues with the tumor immune landscape persist. Multiple immunosuppression pathways coexist in the tumor microenvironment, which can determine tumor progression and therapy outcomes. Research in immune checkpoints, such as the PD-1/PD-L1 axis, has renewed the interest in immune-based cancer therapies due to their ability to prevent immunosuppression against tumors. However, PD-1/PD-L1 blockage is not completely effective, as some patients remain unresponsive to such treatment. The production of adenosine is a major obstacle for the efficacy of immune therapies and is a key source of innate or adaptive resistance. In general, adenosine promotes the pro-tumor immune response, dictates the profile of suppressive immune cells, modulates the release of anti-inflammatory cytokines, and induces the expression of alternative immune checkpoint molecules, such as PD-1, thus maintaining a loop of immunosuppression. In this context, this review aims to depict the complexity of the immunosuppression in glioma microenvironment. We primarily consider the PD-1/PD-L1 axis and adenosine pathway, which may be critical points of resistance and potential targets for tumor treatment strategies.
Cancer is characterized by genetic instability and heterogeneity in the tumor microenvironment (TME). Currently, one of the major challenges in cancer treatment is to block the multifaceted network of tumor mechanisms that cause immunosuppression and resistance to cell death (1, 2).
Gliomas are the most aggressive primary brain tumors in adults, and are of different genetic, phenotypic, and pathological subtypes, depending on the glial lineage from which they arise (3). Glioblastoma multiforme (GBM) is the most malignant subtype of diffuse glioma, and remains the most lethal among brain tumors (3, 4). Similar to other malignances, genetic and phenotypic variability within GBM present problems for the treatment of these tumors (5, 6).
Despite advances in modern medicine, the prognosis for malignant glioma patients remains just over a year. Therefore, several avenues, such as tumor resistance, need to be explored to improve therapeutic approaches (7, 8). Tumor resistance is related to redundant and synergic immunosuppressive pathways coexisting in the TME. Malignant and host cells create a specific niche, where cellular interactions shape the profile of cytokines and chemokines, favoring pro-tumoral activities (9).
Recent evidence has shown that tumors are proficient at evading immunostimulatory responses and resisting standard therapy by producing adenosine (ADO) and upregulating molecules like programmed cell death 1 (PD-1) that function as immune checkpoints (9, 10). Therefore, this review aims to depict the complexity of the immune system in the glioma microenvironment, including the role of the PD-1/PD-L1 axis and adenosine pathway in the maintenance of immunosuppression and resistance to glioma treatments.
The TME has been described as a regulator of tumor progression as well as a mediator of successful therapy. The complexity of tumor niche is shaped by a variable combination of stromal cells, endothelial cells, fibroblasts, cancer stem cells and immune system. Specially, stromal and cancer stem cells have been described by a significant involvement on glioma initiation, maintenance, and progression. In fact, cancer stem cells can suppress cytotoxic responses and modulate immune and endothelial cell functions, suggesting an important role of these cells on immunosuppressive tumor site (11, 12). Importantly, studies have demonstrated an increasing significance of the immune infiltrate and its products in the process of tumor malignancy (10, 13, 14). In GBM, resident microglia and macrophages represent up to one-third of the tumor mass and may have pro-tumorigenic functions (15).
Microglial cells are considered “plastic” due to their ability to change their functions based on environment. These cells may exhibit pro-inflammatory (M1) or immunosuppressive (M2) functions (15–17). All macrophages produce several cytokines such as tumor necrosis factor (TNF) and interleukins (IL-1, IL-6, IL-8, and IL-12), which influence the generation of effector cells and activation of lymphocytes (14). Previous data has shown that the interaction between glioma and microglia is very complex and may not be beneficial for tumor resolution. Indeed, microglia cells co-cultured with glioma cells lack phagocytic ability against tumor cells (16).
Immunosuppression in gliomas involves dynamic crosstalk between tumor and stromal cells, tumor-associated macrophages (TAMs), microglia, regulatory T cells (Tregs), and tumor-infiltrating lymphocytes (TIL) (17–19). Generally, the number of CD4+ lymphocytes is lower than that of CD8+ lymphocytes in a GBM environment, but it has been observed that the numbers of both CD4+ and CD8+ cells increase with tumor grade (20). Despite the presence of these lymphocytes in the GBM microenvironment, effector T cells do not function properly and M2 macrophages are unable to promote CD4+ and CD8+ polarized immune responses, which are important for the regulation of Tregs (21).
The release of chemokines such as C-C motif chemokine ligand 2 (CCL2) is critical for the recruitment of Tregs and myeloid derived suppressor cells (MDSCs). MDSCs alter the TME and suppress immune responses by blocking CD8+ cells and inhibiting the function of natural killer cells (NK) (9, 19, 22). NK cells express death receptor ligands, which can induce caspase-dependent apoptosis in target cells, and can thus kill cancer cells (23). This cytotoxic action is limited by GBM-HLA-G expression, which protects tumors from T cells and NK-mediated killing. Moreover, NK cells are reduced in GBM patients (23, 24).
Studies have shown influences of effector and regulator T cells on the prognosis of cancer patients. For example, Tregs play a significant role in the immune response in the TME since they mediate immunotolerance by suppressing the function of effector T cells (9, 23). GBM patients showed an increased proportion of Tregs among CD4+ cells, contributing to the reduced immune response (25). In addition, the removal of the Treg fraction from patients with GBM rescues T cell proliferation and pro-inflammatory cytokine production to standard levels. This reveals the critical role of Tregs in glioma-mediated immunosuppression (26).
Interest in immune-based treatments of cancer has been renewed after the discovery of immune checkpoint inhibitors. Recently, the co-Nobel Prize in Physiology or Medicine was awarded to Tasuko Honjo, who showed the negative regulation of T cells mediated by the PD-1 pathway (27, 28). Thus, the expression and activity of immunological checkpoints have emerged as the main immunosuppressive mechanisms in gliomas (29, 30).
The transmembrane co-receptor PD-1 (CD279), encoded by the PDCD1 gene, belongs to the family of immunoglobulins and is expressed predominantly by activated T lymphocytes (31). PD-1 is often activated by PD-L1 (B7-H1; CD274), one of the ligands known to be expressed by antigen presenting cells (APCs), B lymphocytes, and parenchymal cells. PD-L2 (B7-DC; CD273) is another ligand for PD-1 and is expressed by fewer cells than PD-L1 (31–33). In normal conditions, PD-1/PD-L engagement occurs controlling a prolonged activation of immune system, often avoiding autoimmunity processes. It is known that PD-1 interaction provides T-cell inhibitory signals. PD-1/PD-L engagement during TCR stimulation leads to tyrosine phosphorylation of the PD-1 cytoplasmic tail on high affinity sites for SH2 domain-containing phosphatase (SHP-2 and SHP-1), resulting in the dephosphorylation of proximal signaling molecules which decrease T cell proliferation and survival by attenuate PI3K and Akt pathways (31, 34).
Importantly, expression of PD-L1 has been detected in glioma (35–37). Moreover, PD-L1 expression in tumor cells is related to levels of malignancy, and high PD-L1 expression is associated with greater invasiveness and aggressiveness of GBM cells (38, 39). Studies have shown heterogeneity of PD-L1 expression in tumor mass such that greater expression is seen at the edges of the tumor than in the core. This could also facilitate immune evasion and invasiveness of gliomas (38, 40).
The expression of PD-L1 in the TME is regulated mainly by cytokine and receptor antigen signaling (31). Interferon gamma (IFN-γ) is the major PD-L1 regulation factor in tumor cells and reflects ongoing antitumor immune activity. In addition, oncogenic mutations, such as loss of phosphatase and tensin homolog (PTEN) in glioma, can activate PD-L1 expression in tumor cells (31, 41, 42).
The PD-1/PD-L1 pathway has been appropriated by tumor cells to resist antitumor responses and facilitate tumor survival (42, 43). Influenced by hypoxia, cytokines, and oncogenes, GBM cells express PD-L1, which engages with the PD-1 receptor primarily on T cells and attenuates its functions, effectively reducing the antitumor activity of these cells (42).
A subset of lymphocytes (Tregs) has emerged as a critical target in cancer therapy. Tregs express both PD-1 and PD-L1, and the generation, immunosuppression, and interaction of Tregs with effector T cells could be, at least in part, modulated by PD-1/PD-L1 binding (44, 45). Francisco et al. have shown that PD-L1 can induce and maintain the expression of FOXP3 in induced Tregs, suggesting that PD-L1 may control Treg plasticity (46).
GBM cells were also able to upregulate PD-L1 expression in tumor-infiltrating macrophages via modulation of IL-10 signaling (29). Macrophages may express PD-1 and PD-L1 (47). PD-1 positive TAMs exhibit decreased phagocytic potential and PD-1 blockade improves macrophage functionalities, besides reducing tumor growth in mouse models of cancer (48).
The use of PD-1 inhibitors is becoming an effective strategy for the treatment of cancer, and several preclinical and clinical studies have been conducted for GBM (30, 49). In fact, immune checkpoint inhibitors may reverse the immunosuppressive condition and restore dysfunctional or “exhausted” T cell function in cancer (39). However, some patients remain unresponsive to PD-1/PD-L1 blockade. Therefore, fresh clinical trials to evaluate tumor resistance in PD-1/PD-L1 immunotherapy in GBM patients are required (39, 50).
Adenosine 5′-triphosphate (ATP) is the main energy molecule produced by cellular respiration. It has multiple release routes and is involved in practically all cellular responses (51). It is known that during cancer growth and progression, ATP and its main metabolite, ADO, are actively secreted or generated in the extracellular space, and accumulate to high levels in the TME (52–54).
Physiologically, extracellular ATP (eATP) functions as a “danger” signal alerting the immune system to the presence of inflammation, and is crucial for inflammasome activation and the concomitant release of cytokines (54, 55). These effects are mediated via P2 receptors, which are subdivided into two subfamilies: P2X ionotropic ion channel receptors (P2X1-7) and P2Y G-protein-coupled receptors (P2Y1, 2, 4, 6, 11, 12, 13, 14) (53–55). These purinergic receptors display distinct agonist affinity and specificity, affecting both tumor and immune cells, depending on the eATP levels available in the TME (56). Different innate and adaptive immune responses are generated through activation of P2 receptors by eATP (Table 1). Particularly, the participation of P2X7 in inflammation is extensive, and has been better characterized compared to that of other P2 receptors (54, 55, 71–74). The direct role of P2X7 in carcinogenesis is still controversial, but it is known that cell growth or death is triggered according to the cell type that expresses P2X7 and their activation level (75).
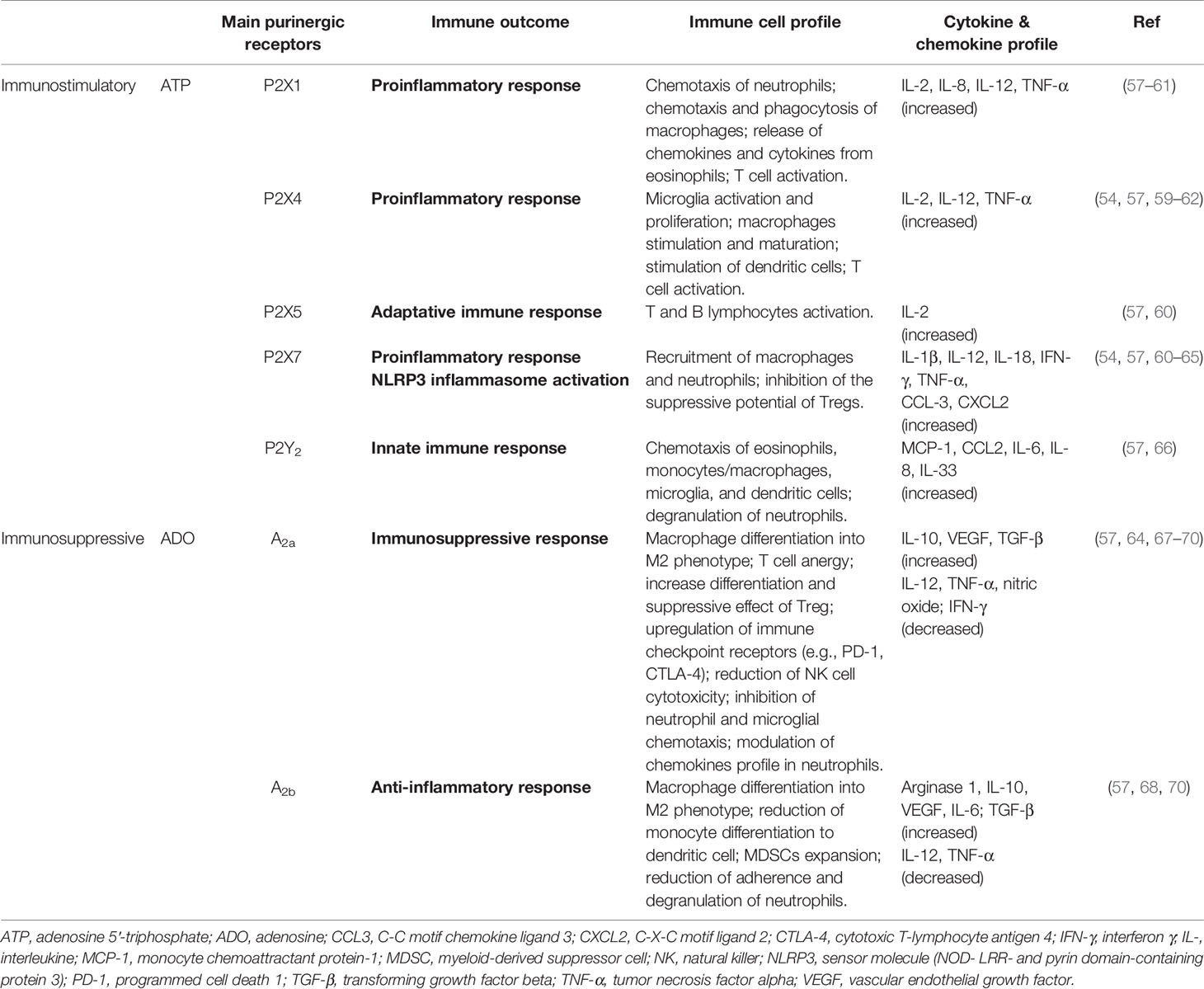
Table 1 Functional immune responses triggered by nucleotides and nucleosides actions in glioblastoma microenvironment.
P2 receptors are assumed to be inactive in normal physiological conditions, where ATP-dependent signaling should be at baseline levels. Ectoenzymes, such as NTPDase1 (CD39) and ecto-5′-nucleotidase (CD73), maintain levels of extracellular ATP, which is crucial to avoid P2 receptor desensitization (76).
In the TME, eATP is quickly hydrolyzed to AMP by CD39 of TILs which is then efficiently converted to the immunosuppressant ADO by CD73 expressed in glioma cells (77). ATP hydrolysis drives the immune response to collaborate with tumor growth, making the CD39/CD73 axis an important regulator of immune effector function. This is a hallmark of cancer (78–81). Interestingly, CD39 inhibition can restore TIL function, and a single nucleotide polymorphism has been identified that may predict dysfunctional CD39+ expression in TILs in some solid tumors (81).
The suppressive role of ADO in the TME is primarily mediated by cytotoxicity, anti-inflammatory cytokine production, and restriction of immune cell infiltration (79). Adenosine effects are mediated by P1 receptors (A1, A2a, A2b, and A3). Interestingly, the pro-tumoral effects of ADO occur mainly through A2 receptors, as depicted in Table 1. Physiologically, ADO orchestrates tissue recovery after initial inflammation, which involves the decrease of M1 phenotype, cell proliferation, and angiogenesis. This sets the stage for tumor growth. Hence, the ADO signaling pathway may be an important therapeutic target (79, 80, 82, 83).
Typically, tumor growth involves disruption of the surrounding microenvironment, in which extracellular nucleotides might confer immunomodulatory properties that are critical for driving glioma immune escape. One of the main mechanisms of tumor immune evasion is the generation of high levels of ADO mediated by excessive activity of ectonucleotidases (83–85).
An effective immunosuppressive environment is maintained when the actions of ADO are synergistic or additive to other immunosuppressive mechanisms. There is growing evidence that immunosuppressive proteins, such as PD-1 and PD-L1, can be increased in the TME by the same mechanism that is implicated in hypoxia-mediated adenosinergic immunosuppression (86). Extracellular ADO increases in hypoxic conditions, concomitant with upregulation of CD39 and CD73. In addition, the oxygen deprivation in the tumor core is related to the upregulation of immunoregulatory mechanisms such as PD-L1 expression in glioma cells, making them resistant to T cell-dependent cytotoxicity (87).
Notably, it was suggested that ADO also induces increase in PD-1 levels (88) because ADO signaling may positively regulate TGF-β levels. TGF-β is mainly involved in stopping effector T cell activation and stimulating the activity of antigen presenting cells that express PD-1 (89). In the presence of TGF-β, CD4+ cell activation may predominantly generate inducible Tregs (90). These cells primarily express CD39, while GBM cells express high levels of CD73, suggesting that cancer and immune cells can cooperate to promote local adenosinergic immunosuppression. Accordingly, a vicious cycle is formed, favoring the upregulation of the PD-1/PD-L1 axis that maintains a complex synergism between the ADO pathway and immune checkpoint axis (77, 91, 92).
Additionally, ADO is involved in macrophage activation, predominantly via A2a (A2aR) and A2b receptors (A2bR). A2bR stimulation during macrophage differentiation could skew macrophages toward the M2 phenotype. M2 macrophages can express immunoregulatory molecules such as arginase, TGF-β, and PD-1/PD-L1 proteins, resulting in the downregulation of cellular immune responses (93).
Overall, the multifaceted role of ADO in tumor immune evasion is seen in its promotion of pro-tumor rather than antitumor immune responses, dictation of Treg function, inhibition of effector T cells, modulation of anti-inflammatory cytokines, and induction of immune checkpoints as illustrated in Figure 1 (83, 84, 86, 88, 89, 94).
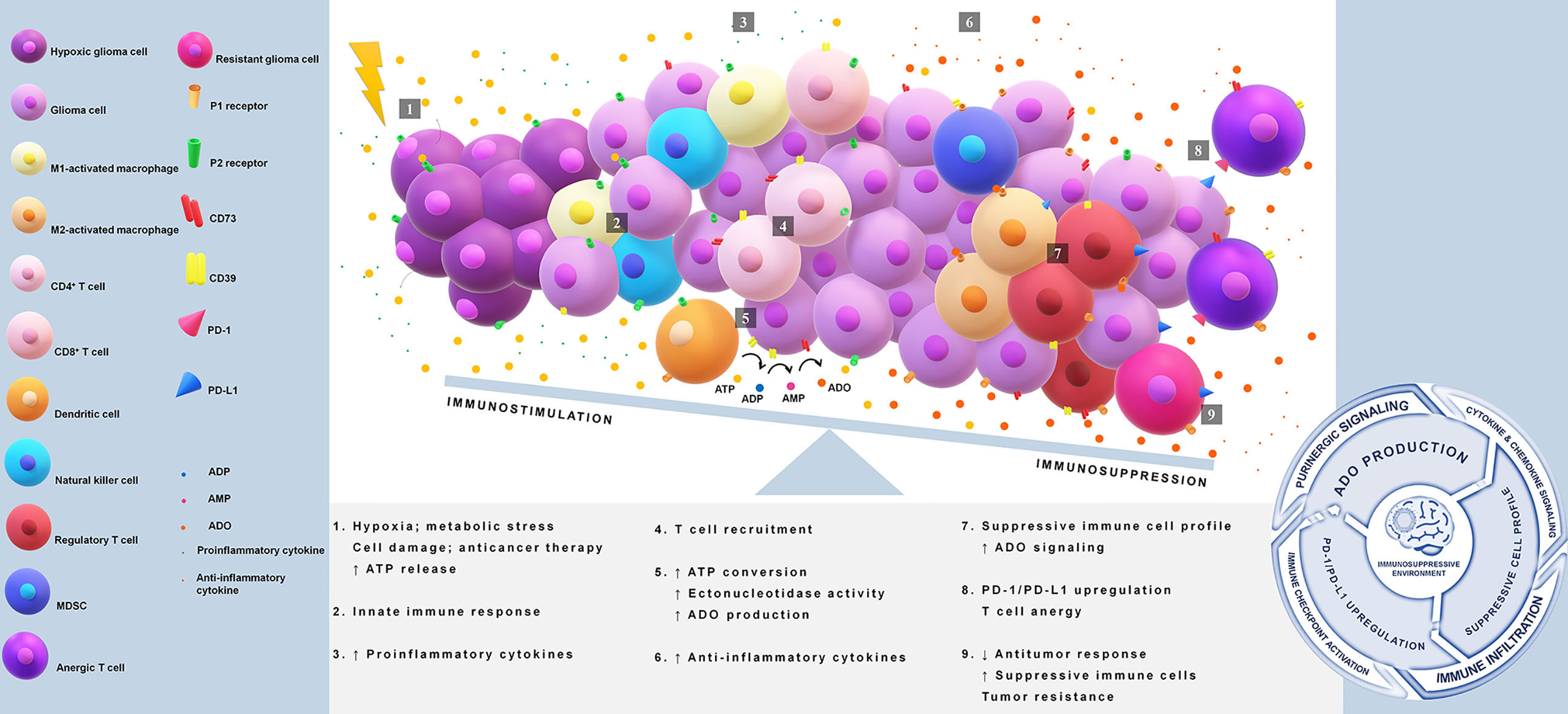
Figure 1 Immunosuppression in glioblastoma via PD-1/PD-L1 axis and adenosine pathway. Tumor core acquires reduction in the oxygen supply causing a release of high amounts of ATP. This nucleotide acts as a damage-associated molecular pattern (DAMP) and starts immune activation. Extracellular ATP binds to P2 receptors and triggers proinflammatory responses through the induction of cytokines and chemokines. A disbalance in the ATP concentration gradient leads to an upregulation of CD39/CD73 axis, favoring adenosine production. Adenosine is a key molecule that initiates a suppressive immune cell infiltration and drives the activation of PD-1/PD-L1 axis. The immunosuppressive loop is maintained indirectly by ATP release and adenosine signaling, which avoids antitumor defenses, promotes immunosuppressive cell profile, and induces upregulation of immune checkpoints. ATP, adenosine 5′-triphosphate; ADO, adenosine; CD39 or ectonucleoside triphosphate diphosphohydrolase 1, cluster of differentiation 39; CD73 or ecto-5′-nucleotidase, cluster of differentiation 73; DAMP, damage-associated molecular pattern; MDSC, myeloid-derived suppressor cells; PD-1, programmed cell death 1; PD-L1, programmed cell death ligand 1.
Taken together, the ADO pathway and the PD-1/PD-L1 axis may act synergistically to modify the TME, favoring tumor progression. Based on this landscape, the GBM standard treatment should be multimodal, involving maximal surgical removal followed by radiotherapy (RT) and/or temozolomide (TMZ). Despite such treatments, refractoriness is often observed (95, 96).
TMZ and RT have several immune modulatory effects on the TME. In addition to immune activation, RT and TMZ therapy may even worsen the immunosuppressive system in GBM. This is because both interventions induce immunogenic cell death, and consequently release immunogenic factors such as ATP (97, 98). ATP binding to P2X7 purinergic receptor is a signal that primes the immune system against tumor (99). However, glioma therapy also increases the expression of CD39/CD73. Hence, it is possible that ADO rapidly rises in the TME. RT also stimulates TGF-β and chemokines that promote the recruitment of immunosuppressive cells; therefore, the activity of the PD-1/PD-L1 axis increases. In addition to Tregs recruitment, RT-induced ATP release also can be related to Treg differentiation from naïve CD4+ cell via A2bR (100, 101).
Interestingly, some studies have shown irradiation-induced PD-L1 expression through an IFN-dependent pathway (102). Xia et al. (90) showed that under RT, PD-L1 expression in GBM cells is greater than that observed without radiation, and that the inhibition of PD-L1 increased radio-sensitivity in these cells (90). High PD-L1 expression was also associated with high numbers of M2 macrophages and Tregs, and low CD8+ cells in the TME, favoring high levels of ADO. Consequently, the immunosuppressive TME resulting from PD-L1-induction could be an important mechanism of tumor radio-resistance (103).
Recently, there has been a surge in the research and development of immunotherapies in cancer, using the PD-1/PD-L1 axis blockade as a strategy to reduce tumor immune evasion (103). Anti-PD-1 immunotherapy has been shown to be successful in prolonging responses in only a fraction of patients (36, 47). There is a subset of them who fail to overcome the immunosuppression, even they can mount an antitumor response. Consequently, the focus of research has changed toward uncovering intrinsic factors that contribute to treatment failures. Currently, the ADO pathway is considered a barrier for the efficacy of immunotherapies (103, 104).
As seen in some solid tumors, alternative immunomodulatory molecules, including CD39, CD73 and A2aR, are upregulated in response to anti-PD-1 monoclonal antibody (mAb) (88, 104). Beavis et al. showed that CD73+ tumor cells restrict anti-PD-1 efficacy, and that this effect was relieved by concomitant treatment with an A2aR antagonist (104). Li et al. demonstrated that CD39 inhibition sensitizes tumor-resistant models to anti-PD1, and that blocking CD39 activity is associated with the enrichment of cytotoxic T cells in the TME and upregulation of inflammatory markers on these infiltrates (105).
Various clinical trials that evaluate purinergic modulation therapy along with anti-PD-1 mAb or anti-PD-L1 mAb are currently active or in the recruitment phase for multiple cancer types (Supplementary Table S1). In fact, simultaneous therapy using PD-1 inhibitors and targeting the adenosine pathway was more effective in improving survival, reducing tumor growth, and limiting metastasis than single therapy in some types of cancer (106–108). Furthermore, there is a rising range of anti-CD73 mAbs being tested in combination with other immunotherapies, generating encouraging results (100, 101, 109).
GBM is one of the most immunologically “cold” tumors among all cancers. The PD1/PD-L1 target characterizes a potential strategy for conversion of the “cold” GBM microenvironment into a “hot” microenvironment to enhance the immune response to antitumor immunotherapy (110). Therefore, anti-PD-1/PD-L1 is an emerging therapeutic possibility in gliomas (111). Since PD-1/PD-L1 blockades do not significantly promote global survivor in patients with recurrent GBM compared with standard therapy, clinical trials are exploring association between anti-PD-1/PD-L1 mAb with standard radio/chemotherapy and bevacizumab or new therapies such as genetically engineered T cells and vaccines (39, 111, 112). Most studies are undergoing clinical trials evaluation and the results still have not provided decisive conclusions (Supplementary Table S2).
Overall, the study of alterations in “purinoma” caused by immune checkpoint inhibitors would likely provide insights for the development of interventions to overcome the immunosuppressive glioma environment and boost immune responses generated by immunotherapies.
The environment surrounding tumors directly impacts their progression. Multiple redundant and compensatory pro-tumor pathways coexist in the TME and are closely related to the success of therapeutic treatments. Immune checkpoint inhibitors help in cancer treatment, though it is not effective in some patients. Thus, immunosuppression remains a major obstacle to therapeutic success. Studies on the relationship between purinergic signaling and inflammation show that the ADO pathway and PD-1/PD-L1 axis have a close relationship and act together to create a favorable environment for tumor immune evasion. The eATP-adenosine axis has a specific role in pro-tumor immune responses including upregulating the PD-1/PD-L1 axis. The ADO pathway has been identified as the main compensatory route involved in the maintenance of immunosuppression in patients using anti-PD-1 immunotherapy, through a drop in innate or adaptive immunity. Therefore, future research should focus on concomitant disruption of the ADO pathway and PD-1/PD-L1 axis to avoid cancer resistance.
TS, NG, PV, FD, and LR critically appraised the literature and wrote. FM reviewed and approved final version of the manuscript.
The authors thank Coordenação de Aperfeiçoamento de Pessoal de Nivel Superior–Brasil [CAPES, Finance Code 001] and Conselho Nacional de Desenvolvimento Cientı́fico e Tecnológico [CNPq, project no. 310317/2018-5] for financial support.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2020.617385/full#supplementary-material
1. Hanahan D, Weinberg RA. Hallmarks of Cancer: The Next Generation. Cell (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
2. Zugazagoitia J, Guedes C, Ponce S, Ferrer I, Molina-Pinelo S, Paz-Ares L. Current Challenges in Cancer Treatment. Clin Ther (2016) 38:1551–66. doi: 10.1016/j.clinthera.2016.03.026
3. Gerlinger M, Swanton C. How Darwinian models inform therapeutic failure initiated by clonal heterogeneity in cancer medicine. Br J Cancer (2010) 103:1139–43. doi: 10.1038/sj.bjc.6605912
4. Pawlowska E, Szczepanska J, Szatkowska M, Blasiak J. An Interplay between Senescence, Apoptosis and Autophagy in Glioblastoma Multiforme-Role in Pathogenesis and Therapeutic Perspective. Int J Mol Sci (2018) 19(3):889–908. doi: 10.3390/ijms19030889
5. Ostrom QT, Gittleman H, Xu J, Kromer C, Wolinsky Y, Kruchko C, et al. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2009-2013. Neuro-Oncol (2016) 18:v1–v75. doi: 10.1093/neuonc/now207
6. Shergalis A, Bankhead A, Luesakul U, Muangsin N, Neamati N. Current Challenges and Opportunities in Treating Glioblastoma. Pharmacol Rev (2018) 70:412–45. doi: 10.1124/pr.117.014944
7. Fridman WH, Zitvogel L, Sautès-Fridman C, Kroemer G. The immune contexture in cancer prognosis and treatment. Nat Rev Clin Oncol (2017) 14:717–34. doi: 10.1038/nrclinonc.2017.101
8. Martinez-Lage M, Lynch TM, Bi Y, Cocito C, Way GP, Pal S, et al. Immune landscapes associated with different glioblastoma molecular subtypes. Acta Neuropathol Commun (2019) 7:203. doi: 10.1186/s40478-019-0803-6
9. Li G, Qin Z, Chen Z, Xie L, Wang R, Zhao H. Tumor Microenvironment in Treatment of Glioma. Open Med (2017) 12:247–51. doi: 10.1515/med-2017-0035
10. Strepkos D, Markouli M, Klonou A, Piperi C, Papavassiliou AG. Insights in the immunobiology of glioblastoma. J Mol Med (2020) 98:1–10. doi: 10.1007/s00109-019-01835-4
11. Hamerlik P. Cancer Stem Cells and Glioblastoma. In: Sedo A, Mentlein R, editors. Glioma Cell Biology. Vienna: Springer Vienna. (2014) p. 3–22. doi: 10.1007/978-3-7091-1431-5_1
12. Ma Q, Long W, Xing C, Chu J, Luo M, Wang HY, et al. Cancer Stem Cells and Immunosuppressive Microenvironment in Glioma. Front Immunol (2018) 9:2924–41. doi: 10.3389/fimmu.2018.02924
13. Locarno CV, Simonelli M, Carenza C, Capucetti A, Stanzani E, Lorenzi E, et al. Role of myeloid cells in the immunosuppressive microenvironment in gliomas. Immunobiology (2020) 225:151853. doi: 10.1016/j.imbio.2019.10.002
14. Guadagno E, Presta I, Maisano D, Donato A, Pirrone CK, Cardillo G, et al. Role of Macrophages in Brain Tumor Growth and Progression. Int J Mol Sci (2018) 19(4):1005–24. doi: 10.3390/ijms19041005
15. Lee KY. M1 and M2 polarization of macrophages: a mini-review. Med Biol Sci Eng (2019) 2:1–5. doi: 10.30579/mbse.2019.2.1.1
16. Voisin P, Bouchaud V, Merle M, Diolez P, Duffy L, Flint K, et al. Microglia in Close Vicinity of Glioma Cells: Correlation Between Phenotype and Metabolic Alterations. Front Neuroenerget (2010) 2:131–46. doi: 10.3389/fnene.2010.00131
17. Watters JJ, Schartner JM, Badie B. Microglia function in brain tumors. J Neurosci Res (2005) 81:447–55. doi: 10.1002/jnr.20485
18. Kim R, Emi M, Tanabe K. Cancer immunosuppression and autoimmune disease: beyond immunosuppressive networks for tumour immunity. Immunology (2006) 119:254–64. doi: 10.1111/j.1365-2567.2006.02430.x
19. Sayour EJ, McLendon P, McLendon R, De Leon G, Reynolds R, Kresak J, et al. Increased proportion of FoxP3+ regulatory T cells in tumor infiltrating lymphocytes is associated with tumor recurrence and reduced survival in patients with glioblastoma. Cancer Immunol Immunother CII (2015) 64:419–27. doi: 10.1007/s00262-014-1651-7
20. Dubinski D, Wölfer J, Hasselblatt M, Schneider-Hohendorf T, Bogdahn U, Stummer W, et al. CD4+ T effector memory cell dysfunction is associated with the accumulation of granulocytic myeloid-derived suppressor cells in glioblastoma patients. Neuro-Oncol (2016) 18:807–18. doi: 10.1093/neuonc/nov280
21. Sica A, Larghi P, Mancino A, Rubino L, Porta C, Totaro MG, et al. Macrophage polarization in tumour progression. Semin Cancer Biol (2008) 18:349–55. doi: 10.1016/j.semcancer.2008.03.004
22. Iwami K, Natsume A, Wakabayashi T. Cytokine networks in glioma. Neurosurg Rev (2011) 34:253–63. doi: 10.1007/s10143-011-0320-y
23. Zhang X, Safonova A, Rao A, Amankulor N. Role of natural killer cells in isocitrate dehydrogenase 1/2 mutant glioma pathogenesis and emerging therapies. Glioma (2019) 2:133. doi: 10.4103/glioma.glioma_10_19
24. Gieryng A, Pszczolkowska D, Walentynowicz KA, Rajan WD, Kaminska B. Immune microenvironment of gliomas. Lab Invest J Tech Methods Pathol (2017) 97:498–518. doi: 10.1038/labinvest.2017.19
25. Woroniecka KI, Rhodin KE, Chongsathidkiet P, Keith KA. Fecci PE. T-cell Dysfunction in Glioblastoma: Applying a New Framework. Clin Cancer Res Off J Am Assoc Cancer Res (2018) 24:3792–802. doi: 10.1158/1078-0432.CCR-18-0047
26. Wainwright DA, Dey M, Chang A, Lesniak MS. Targeting Tregs in Malignant Brain Cancer: Overcoming IDO. Front Immunol (2013) 4:116. doi: 10.3389/fimmu.2013.00116
27. Ishida Y, Agata Y, Shibahara K, Honjo T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J (1992) 11:3887–95. doi: 10.1002/j.1460-2075.1992.tb05481.x
28. Ledford H, Else H, Warren M. Cancer immunologists scoop medicine Nobel prize. Nature (2018) 562:20–1. doi: 10.1038/d41586-018-06751-0
29. Bloch O, Crane CA, Kaur R, Safaee M, Rutkowski MJ, Parsa AT. Gliomas promote immunosuppression through induction of B7-H1 expression in tumor-associated macrophages. Clin Cancer Res Off J Am Assoc Cancer Res (2013) 19:3165–75. doi: 10.1158/1078-0432.CCR-12-3314
30. Preusser M, Lim M, Hafler DA, Reardon DA, Sampson JH. Prospects of immune checkpoint modulators in the treatment of glioblastoma. Nat Rev Neurol (2015) 11:504–14. doi: 10.1038/nrneurol.2015.139
31. Francisco LM, Sage PT, Sharpe AH. The PD-1 Pathway in Tolerance and Autoimmunity. Immunol Rev (2010) 236:219–42. doi: 10.1111/j.1600-065X.2010.00923.x
32. Flies DB, Chen L. The new B7s: playing a pivotal role in tumor immunity. J Immunother Hagerstown Md 1997 (2007) 30:251–60. doi: 10.1097/CJI.0b013e31802e085a
33. Pedoeem A, Azoulay-Alfaguter I, Strazza M, Silverman GJ, Mor A. Programmed death-1 pathway in cancer and autoimmunity. Clin Immunol Orlando Fla (2014) 153:145–52. doi: 10.1016/j.clim.2014.04.010
34. Patsoukis N, Wang Q, Strauss L, Boussiotis VA. Revisiting the PD-1 pathway. Sci Adv (2020) 6:2712–25. doi: 10.1126/sciadv.abd2712
35. Wintterle S, Schreiner B, Mitsdoerffer M, Schneider D, Chen L, Meyermann R, et al. Expression of the B7-related molecule B7-H1 by glioma cells: a potential mechanism of immune paralysis. Cancer Res (2003) 63:7462–7.
36. Berghoff AS, Kiesel B, Widhalm G, Rajky O, Ricken G, Wöhrer A, et al. Programmed death ligand 1 expression and tumor-infiltrating lymphocytes in glioblastoma. Neuro-Oncol (2015) 17:1064–75. doi: 10.1093/neuonc/nou307
37. Nduom EK, Wei J, Yaghi NK, Huang N, Kong L-Y, Gabrusiewicz K, et al. PD-L1 expression and prognostic impact in glioblastoma. Neuro-Oncol (2016) 18:195–205. doi: 10.1093/neuonc/nov172
38. Yao Y, Tao R, Wang X, Wang Y, Mao Y, Zhou LF. B7-H1 is correlated with malignancy-grade gliomas but is not expressed exclusively on tumor stem-like cells. Neuro-Oncol (2009) 11:757–66. doi: 10.1215/15228517-2009-014
39. Xue S, Hu M, Iyer V, Yu J. Blocking the PD-1/PD-L1 pathway in glioma: a potential new treatment strategy. J Hematol OncolJ Hematol Oncol (2017) 10:81–91. doi: 10.1186/s13045-017-0455-6
40. Wilmotte R, Burkhardt K, Kindler V, Belkouch M-C, Dussex G, de Tribolet N, et al. B7-homolog 1 expression by human glioma: a new mechanism of immune evasion. Neuroreport (2005) 16:1081–5. doi: 10.1097/00001756-200507130-00010
41. Parsa AT, Waldron JS, Panner A, Crane CA, Parney IF, Barry JJ, et al. Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nat Med (2007) 13:84–8. doi: 10.1038/nm1517
42. Broekman ML, Maas SLN, Abels ER, Mempel TR, Krichevsky AM, Breakefield XO. Multidimensional communication in the microenvirons of glioblastoma. Nat Rev Neurol (2018) 14:482–95. doi: 10.1038/s41582-018-0025-8
43. Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol (2008) 26:677–704. doi: 10.1146/annurev.immunol.26.021607.090331
44. Wang L, Pino-Lagos K, de Vries VC, Guleria I, Sayegh MH, Noelle RJ. Programmed death 1 ligand signaling regulates the generation of adaptive Foxp3+CD4+ regulatory T cells. Proc Natl Acad Sci USA (2008) 105:9331–6. doi: 10.1073/pnas.0710441105
45. Gianchecchi E, Fierabracci A. Inhibitory Receptors and Pathways of Lymphocytes: The Role of PD-1 in Treg Development and Their Involvement in Autoimmunity Onset and Cancer Progression. Front Immunol (2018) 9:2374. doi: 10.3389/fimmu.2018.02374
46. Francisco LM, Salinas VH, Brown KE, Vanguri VK, Freeman GJ, Kuchroo VK, et al. PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J Exp Med (2009) 206:3015–29. doi: 10.1084/jem.20090847
47. Cai J, Qi Q, Qian X, Han J, Zhu X, Zhang Q, et al. The role of PD-1/PD-L1 axis and macrophage in the progression and treatment of cancer. J Cancer Res Clin Oncol (2019) 145:1377–85. doi: 10.1007/s00432-019-02879-2
48. Gordon SR, Maute RL, Dulken BW, Hutter G, George BM, McCracken MN, et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature (2017) 545:495–9. doi: 10.1038/nature22396
49. Schalper KA, Rodriguez-Ruiz ME, Diez-Valle R, López-Janeiro A, Porciuncula A, Idoate MA, et al. Neoadjuvant nivolumab modifies the tumor immune microenvironment in resectable glioblastoma. Nat Med (2019) 25:470–6. doi: 10.1038/s41591-018-0339-5
50. Zou W, Wolchok JD, Chen L. PD-L1 (B7-H1) and PD-1 Pathway Blockade for Cancer Therapy: Mechanisms, Response Biomarkers and Combinations. Sci Transl Med (2016) 8:328rv4. doi: 10.1126/scitranslmed.aad7118
51. Nath S, Villadsen J. Oxidative phosphorylation revisited. Biotechnol Bioeng (2015) 112:429–37. doi: 10.1002/bit.25492
52. Burnstock G, Di Virgilio F. Purinergic signalling and cancer. Purinerg Signal (2013) 9:491–540. doi: 10.1007/s11302-013-9372-5
53. Borea PA, Gessi S, Merighi S, Vincenzi F, Varani K. Pharmacology of Adenosine Receptors: The State of the Art. Physiol Rev (2018) 98:1591–625. doi: 10.1152/physrev.00049.2017
54. Di Virgilio F, Sarti AC, Falzoni S, De Marchi E, Adinolfi E. Extracellular ATP and P2 purinergic signalling in the tumour microenvironment. Nat Rev Cancer (2018) 18:601–18. doi: 10.1038/s41568-018-0037-0
55. Eltzschig HK, Sitkovsky MV, Robson SC. Purinergic signaling during inflammation. N Engl J Med (2012) 367:2322–33. doi: 10.1056/NEJMra1205750
56. Volonté C, Amadio S, D’Ambrosi N, Colpi M, Burnstock G. P2 receptor web: Complexity and fine-tuning. Pharmacol Ther (2006) 112:264–80. doi: 10.1016/j.pharmthera.2005.04.012
57. Burnstock G, Boeynaems J-M. Purinergic signalling and immune cells. Purinerg Signal (2014) 10:529–64. doi: 10.1007/s11302-014-9427-2
58. Lecut C, Frederix K, Johnson DM, Deroanne C, Thiry M, Faccinetto C, et al. P2X1 ion channels promote neutrophil chemotaxis through Rho kinase activation. J Immunol Baltim Md 1950 (2009) 183:2801–9. doi: 10.4049/jimmunol.0804007
59. Woehrle T, Yip L, Elkhal A, Sumi Y, Chen Y, Yao Y, et al. Pannexin-1 hemichannel–mediated ATP release together with P2X1 and P2X4 receptors regulate T-cell activation at the immune synapse. Blood (2010) 116:3475–84. doi: 10.1182/blood-2010-04-277707
60. Jacob F, Novo CP, Bachert C, Van Crombruggen K. Purinergic signaling in inflammatory cells: P2 receptor expression, functional effects, and modulation of inflammatory responses. Purinerg Signal (2013) 9:285–306. doi: 10.1007/s11302-013-9357-4
61. Mostofa AGM, Punganuru SR, Madala HR, Al-Obaide M, Srivenugopal KS. The Process and Regulatory Components of Inflammation in Brain Oncogenesis. Biomolecules (2017) 7(2):34–67. doi: 10.3390/biom7020034
62. Burnstock G. P2X ion channel receptors and inflammation. Purinerg Signal (2016) 12:59–67. doi: 10.1007/s11302-015-9493-0
63. Hide I, Tanaka M, Inoue A, Nakajima K, Kohsaka S, Inoue K, et al. Extracellular ATP triggers tumor necrosis factor-alpha release from rat microglia. J Neurochem (2000) 75:965–72. doi: 10.1046/j.1471-4159.2000.0750965.x
64. Ferrari D, McNamee EN, Idzko M, Gambari R, Eltzschig HK. Purinergic Signaling During Immune Cell Trafficking. Trends Immunol (2016) 37:399–411. doi: 10.1016/j.it.2016.04.004
65. Kan LK, Williams D, Drummond K, O’Brien T, Monif M. The role of microglia and P2X7 receptors in gliomas. J Neuroimmunol (2019) 332:138–46. doi: 10.1016/j.jneuroim.2019.04.010
66. Rayah A, Kanellopoulos JM, Di Virgilio F. P2 receptors and immunity. Microbes Infect Inst Pasteur (2012) 14:1254–62. doi: 10.1016/j.micinf.2012.07.006
67. Campos-Contreras ADR, Díaz-Muñoz M, Vázquez-Cuevas FG. Purinergic Signaling in the Hallmarks of Cancer. Cells (2020) 9:1612–36. doi: 10.3390/cells9071612
68. Di Virgilio F, Vuerich M. Purinergic signaling in the immune system. Auton Neurosci (2015) 191:117–23. doi: 10.1016/j.autneu.2015.04.011
69. Leone RD, Lo Y-C, Powell JD. A2aR antagonists: Next generation checkpoint blockade for cancer immunotherapy. Comput Struct Biotechnol J (2015) 13:265–72. doi: 10.1016/j.csbj.2015.03.008
70. Vigano S, Alatzoglou D, Irving M, Ménétrier-Caux C, Caux C, Romero P, et al. Targeting Adenosine in Cancer Immunotherapy to Enhance T-Cell Function. Front Immunol (2019) 10:925–55. doi: 10.3389/fimmu.2019.00925
71. De Marchi E, Orioli E, Pegoraro A, Sangaletti S, Portararo P, Curti A, et al. The P2X7 receptor modulates immune cells infiltration, ectonucleotidases expression and extracellular ATP levels in the tumor microenvironment. Oncogene (2019) 38:3636–50. doi: 10.1038/s41388-019-0684-y
72. Wiley JS, Sluyter R, Gu BJ, Stokes L, Fuller SJ. The human P2X7 receptor and its role in innate immunity. Tissue Antigens (2011) 78:321–32. doi: 10.1111/j.1399-0039.2011.01780.x
73. Rassendren F, Buell GN, Virginio C, Collo G, North RA, Surprenant A. The permeabilizing ATP receptor, P2X7. Cloning and expression of a human cDNA. J Biol Chem (1997) 272:5482–6. doi: 10.1074/jbc.272.9.5482
74. Monif M, Reid CA, Powell KL, Smart ML, Williams DA. The P2X7 receptor drives microglial activation and proliferation: a trophic role for P2X7R pore. J Neurosci Off J Soc Neurosci (2009) 29:3781–91. doi: 10.1523/JNEUROSCI.5512-08.2009
75. de Andrade Mello P, Coutinho-Silva R, Savio LEB. Multifaceted Effects of Extracellular Adenosine Triphosphate and Adenosine in the Tumor-Host Interaction and Therapeutic Perspectives. Front Immunol (2017) 8:1526. doi: 10.3389/fimmu.2017.01526
76. Di Virgilio F. Purinergic mechanism in the immune system: A signal of danger for dendritic cells. Purinerg Signal (2005) 1:205–9. doi: 10.1007/s11302-005-6312-z
77. Xu S, Shao Q-Q, Sun J-T, Yang N, Xie Q, Wang D-H, et al. Synergy between the ectoenzymes CD39 and CD73 contributes to adenosinergic immunosuppression in human malignant gliomas. Neuro-Oncol (2013) 15:1160–72. doi: 10.1093/neuonc/not067
78. Bastid J, Regairaz A, Bonnefoy N, Déjou C, Giustiniani J, Laheurte C, et al. Inhibition of CD39 enzymatic function at the surface of tumor cells alleviates their immunosuppressive activity. Cancer Immunol Res (2015) 3:254–65. doi: 10.1158/2326-6066.CIR-14-0018
79. Vijayan D, Young A, Teng MWL, Smyth MJ. Targeting immunosuppressive adenosine in cancer. Nat Rev Cancer (2017) 17:709–24. doi: 10.1038/nrc.2017.86
80. Hammami A, Allard D, Allard B, Stagg J. Targeting the adenosine pathway for cancer immunotherapy. Semin Immunol (2019) 42:101304. doi: 10.1016/j.smim.2019.101304
81. Gallerano D, Ciminati S, Grimaldi A, Piconese S, Cammarata I, Focaccetti C, et al. Genetically driven CD39 expression shapes human tumor-infiltrating CD8+ T-cell functions. (2020) Int J Cancer 147(9):2597–610. doi: 10.1002/ijc.33131
82. Haskó G, Linden J, Cronstein B, Pacher P. Adenosine receptors: therapeutic aspects for inflammatory and immune diseases. Nat Rev Drug Discovery (2008) 7:759–70. doi: 10.1038/nrd2638
83. Antonioli L, Blandizzi C, Pacher P, Haskó G. Immunity, inflammation and cancer: a leading role for adenosine. Nat Rev Cancer (2013) 13:842–57. doi: 10.1038/nrc3613
84. Stagg J, Smyth MJ. Extracellular adenosine triphosphate and adenosine in cancer. Oncogene (2010) 29:5346–58. doi: 10.1038/onc.2010.292
85. Beavis PA, Stagg J, Darcy PK, Smyth MJ. CD73: a potent suppressor of antitumor immune responses. Trends Immunol (2012) 33:231–7. doi: 10.1016/j.it.2012.02.009
86. Ohta A, Kini R, Ohta A, Subramanian M, Madasu M, Sitkovsky M. The development and immunosuppressive functions of CD4+ CD25+ FoxP3+ regulatory T cells are under influence of the adenosine-A2A adenosine receptor pathway. Front Immunol (2012) 3:190–202. doi: 10.3389/fimmu.2012.00190
87. Ohta A. Oxygen-dependent regulation of immune checkpoint mechanisms. Int Immunol (2018) 30(8):335–43. doi: 10.1093/intimm/dxy038
88. Allard B, Pommey S, Smyth MJ, Stagg J. Targeting CD73 Enhances the Antitumor Activity of Anti-PD-1 and Anti-CTLA-4 mAbs. Clin Cancer Res (2013) 19:5626–35. doi: 10.1158/1078-0432.CCR-13-0545
89. Zarek PE, Huang C-T, Lutz ER, Kowalski J, Horton MR, Linden J, et al. A2A receptor signaling promotes peripheral tolerance by inducing T-cell anergy and the generation of adaptive regulatory T cells. Blood (2008) 111:251–9. doi: 10.1182/blood-2007-03-081646
90. Xia W, Zhu J, Tang Y, Wang X, Wei X, Zheng X, et al. PD-L1 Inhibitor Regulates the miR-33a-5p/PTEN Signaling Pathway and Can Be Targeted to Sensitize Glioblastomas to Radiation. Front Oncol (2020) 10:821–33. doi: 10.3389/fonc.2020.00821
91. Bavaresco L, Bernardi A, Braganhol E, Cappellari AR, Rockenbach L, Farias PF, et al. The role of ecto-5’-nucleotidase/CD73 in glioma cell line proliferation. Mol Cell Biochem (2008) 319:61–8. doi: 10.1007/s11010-008-9877-3
92. Ceruti S, Abbracchio M. Adenosine Signaling in Glioma Cells. Adv Exp Med Biol 986:13–30. doi: 10.1007/978-3-030-30651-9_2
93. Csóka B, Selmeczy Z, Koscsó B, Németh ZH, Pacher P, Murray PJ, et al. Adenosine promotes alternative macrophage activation via A2A and A2B receptors. FASEB J Off Publ Fed Am Soc Exp Biol (2012) 26:376–86. doi: 10.1096/fj.11-190934
94. Sek K, Mølck C, Stewart GD, Kats L, Darcy PK, Beavis PA. Targeting Adenosine Receptor Signaling in Cancer Immunotherapy. Int J Mol Sci (2018) 19:3837–60. doi: 10.3390/ijms19123837
95. Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJB, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med (2005) 352:987–96. doi: 10.1056/NEJMoa043330
96. Fernandes C, Costa A, Osório L, Lago RC, Linhares P, Carvalho B, et al. Current Standards of Care in Glioblastoma Therapy, in: Glioblastoma. Brisbane (AU): Codon Publications. Available at: http://www.ncbi.nlm.nih.gov/books/NBK469987/ (Accessed August 6, 2020).
97. Martins I, Tesniere A, Kepp O, Michaud M, Schlemmer F, Senovilla L, et al. Chemotherapy induces ATP release from tumor cells. Cell Cycle (2009) 8:3723–8. doi: 10.4161/cc.8.22.10026
98. Golden EB, Frances D, Pellicciotta I, Demaria S, Helen Barcellos-Hoff M, Formenti SC. Radiation fosters dose-dependent and chemotherapy-induced immunogenic cell death. Oncoimmunology (2014) 3:e28518. doi: 10.4161/onci.28518
99. Ma Y, Kepp O, Ghiringhelli F, Apetoh L, Aymeric L, Locher C, et al. Chemotherapy and radiotherapy: cryptic anticancer vaccines. Semin Immunol (2010) 22:113–24. doi: 10.1016/j.smim.2010.03.001
100. Hay CM, Sult E, Huang Q, Mulgrew K, Fuhrmann SR, McGlinchey KA, et al. Targeting CD73 in the tumor microenvironment with MEDI9447. Oncoimmunology (2016) 5(8):e1208875–85. doi: 10.1080/2162402X.2016.1208875
101. Siu LL, Burris H, Le DT, Hollebecque A, Steeghs N, Delord J-P, et al. Abstract CT180: Preliminary phase 1 profile of BMS-986179, an anti-CD73 antibody, in combination with nivolumab in patients with advanced solid tumors. Cancer Res (2018) 78:CT180–0. doi: 10.1158/1538-7445.AM2018-CT180
102. Dovedi SJ, Adlard AL, Lipowska-Bhalla G, McKenna C, Jones S, Cheadle EJ, et al. Acquired resistance to fractionated radiotherapy can be overcome by concurrent PD-L1 blockade. Cancer Res (2014) 74:5458–68. doi: 10.1158/0008-5472.CAN-14-1258
103. Jang B-S, Kim IA. A Radiosensitivity Gene Signature and PD-L1 Status Predict Clinical Outcome of Patients with Glioblastoma Multiforme in The Cancer Genome Atlas Dataset. Cancer Res Treat Off J Korean Cancer Assoc (2020) 52:530–42. doi: 10.4143/crt.2019.440
104. Beavis PA, Milenkovski N, Henderson MA, John LB, Allard B, Loi S, et al. Adenosine Receptor 2A Blockade Increases the Efficacy of Anti-PD-1 through Enhanced Antitumor T-cell Responses. Cancer Immunol Res (2015) 3:506–17. doi: 10.1158/2326-6066.CIR-14-0211
105. Li X-Y, Moesta AK, Xiao C, Nakamura K, Casey M, Zhang H, et al. Targeting CD39 in cancer reveals an extracellular ATP and inflammasome driven tumor immunity. Cancer Discovery (2019) 9(12):1754–73. doi: 10.1158/2159-8290.CD-19-0541
106. Mittal D, Young A, Stannard K, Yong M, Teng MWL, Allard B, et al. Antimetastatic effects of blocking PD-1 and the adenosine A2A receptor. Cancer Res (2014) 74:3652–8. doi: 10.1158/0008-5472.CAN-14-0957
107. Leone RD, Sun I-M, Oh M-H, Sun I-H, Wen J, Englert J, et al. Inhibition of the adenosine A2a receptor modulates expression of T cell coinhibitory receptors and improves effector function for enhanced checkpoint blockade and ACT in murine cancer models. Cancer Immunol Immunother CII (2018) 67:1271–84. doi: 10.1007/s00262-018-2186-0
108. Steingold JM, Hatfield SM. Targeting Hypoxia-A2A Adenosinergic Immunosuppression of Antitumor T Cells During Cancer Immunotherapy. Front Immunol (2020) 11:570041–48. doi: 10.3389/fimmu.2020.570041
109. Barnhart BC, Sega E, Yamniuk A, Hatcher S, Lei M, Ghermazien H, et al. Abstract 1476: A therapeutic antibody that inhibits CD73 activity by dual mechanisms. Cancer Res (2016) 76:1476–6. doi: 10.1158/1538-7445.AM2016-1476
110. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer (2012) 12:252–64. doi: 10.1038/nrc3239
111. Shu C, Li Q. Current advances in PD-1/PD-L1 axis-related tumour-infiltrating immune cells and therapeutic regimens in glioblastoma. Crit Rev Oncol Hematol (2020) 151:102965. doi: 10.1016/j.critrevonc.2020.102965
Keywords: glioma, immunosuppression, adenosine, PD-1/PD-L1, tumor microenvironment
Citation: Scheffel TB, Grave N, Vargas P, Diz FM, Rockenbach L and Morrone FB (2021) Immunosuppression in Gliomas via PD-1/PD-L1 Axis and Adenosine Pathway. Front. Oncol. 10:617385. doi: 10.3389/fonc.2020.617385
Received: 14 October 2020; Accepted: 23 December 2020;
Published: 15 February 2021.
Edited by:
Payal Watchmaker, University of California, San Francisco, United StatesReviewed by:
Zoltan Vereb, University of Szeged, HungaryCopyright © 2021 Scheffel, Grave, Vargas, Diz, Rockenbach and Morrone. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fernanda Bueno Morrone, ZmVybmFuZGEubW9ycm9uZUBwdWNycy5icg==; ZmJtb3Jyb25lQGdtYWlsLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.