 Aukie Hooglugt
Aukie Hooglugt Miesje M. van der Stoel
Miesje M. van der Stoel Reinier A. Boon2,3,4
Reinier A. Boon2,3,4 Stephan Huveneers
Stephan Huveneers- 1Department of Medical Biochemistry, Amsterdam Cardiovascular Sciences, Amsterdam UMC, University of Amsterdam, Amsterdam, Netherlands
- 2Department of Physiology, Amsterdam Cardiovascular Sciences, Amsterdam UMC, VU University Medical Center, Amsterdam, Netherlands
- 3German Center for Cardiovascular Research (DZHK), Partner Site Rhein-Main, Berlin, Germany
- 4Institute of Cardiovascular Regeneration, Goethe University, Frankfurt am Main, Germany
Solid tumors are dependent on vascularization for their growth. The hypoxic, stiff, and pro-angiogenic tumor microenvironment induces angiogenesis, giving rise to an immature, proliferative, and permeable vasculature. The tumor vessels promote tumor metastasis and complicate delivery of anti-cancer therapies. In many types of tumors, YAP/TAZ activation is correlated with increased levels of angiogenesis. In addition, endothelial YAP/TAZ activation is important for the formation of new blood and lymphatic vessels during development. Oncogenic activation of YAP/TAZ in tumor cell growth and invasion has been studied in great detail, however the role of YAP/TAZ within the tumor endothelium remains insufficiently understood, which complicates therapeutic strategies aimed at targeting YAP/TAZ in cancer. Here, we overview the upstream signals from the tumor microenvironment that control endothelial YAP/TAZ activation and explore the role of their downstream targets in driving tumor angiogenesis. We further discuss the potential for anti-cancer treatments and vascular normalization strategies to improve tumor therapies.
Introduction
It is estimated that solid tumors can grow to a size of approximately 2 mm3 without being vascularized (1). For further growth, tumors require blood vessels that deliver oxygen and nutrients. Tumors use several mechanisms for neovascularization, including angiogenesis, vessel co-option, vascular mimicry, trans-differentiation of cancer cells into endothelial cells (ECs), and through the recruitment of endothelial progenitor cells (2, 3). Angiogenesis, the formation of new vessels from pre-existing ones, is essential for tumor progression and growth and is promoted by pro-angiogenic signals secreted by the tumor cells and the tumor microenvironment (TME) (4, 5). The TME consists of cancer-associated fibroblast (CAFs), mesenchymal stromal cells, immune cells, ECs, as well as extracellular matrix (ECM) components, growth factors and cytokines (6–8). The tumor cells together with the TME, generate a hypoxic, acidic and inflammatory environment that further drives tumor angiogenesis, tumor growth and contributes to drug resistance (8, 9).
The tumor vasculature is morphologically different compared to the normal blood vessels (5, 9–11). Tumor angiogenesis gives rise to a dense and disorganized vessel network, in which the vessels are immature, dilated, hyperpermeable, and lack the support of pericytes or normal basement membrane. Tumor vessels are further characterized by irregular blood flow, which fails to supply sufficient oxygen and nutrients to the tumor tissue (12, 13). The tumor vessels contribute to malignancy by maintaining the hypoxic, acidic, and inflammatory environment, thereby fueling a vicious cycle that prevents normalization of the tumor vasculature and maintains a pro-metastatic environment (14, 15).
Targeting of tumor angiogenesis as treatment for cancer has been extensively investigated in pre-clinical and clinical settings. The first commercially available anti-angiogenic drug, Bevacizumab improves survival of patients with metastatic colorectal cancer by targeting of vascular endothelial growth factor (VEGF) (16). Currently, several VEGF-targeting FDA-approved drugs are used as anti-angiogenic cancer treatments, including for gastrointestinal cancer, glioblastoma, non-small lung carcinoma, breast cancer, and renal cancer (17, 18). Patient studies have shown increased survival after combining anti-VEGF therapy with chemotherapy (17, 19, 20). Unfortunately, long term administration of anti-VEGF treatments raises therapy resistance (21, 22) and major reductions in tumor blood vessels are not achieved, likely due to the activation of alternative neovascularization events in tumors (23, 24). Furthermore, pre-clinical in vivo studies have shown that after termination of anti-VEGF treatments, the tumor vessels rapidly return to an angiogenic and disorganized state (25).
Anti-angiogenic agents are often prescribed in high doses, which effectively leads to tumor vessel pruning and consequently decreases drug delivery to the TME (26). Moreover, local hypoxia in the TME, such as induced by vascular regression, enhances tumor invasiveness, chemo- and immunotherapy resistance, and metastasis (27). Hypoxia also induces expression of alternative angiogenic cytokines and compensatory mechanisms of neovascularization, further limiting the anti-angiogenic potential of anti-VEGF treatments (2, 28). Alternative angiogenic pathways are suspected to enhance tumor invasion and metastasis in response to anti-VEGF treatment (29).
Upon anti-angiogenic treatments, there is typically a short “window” during which vascular normalization is achieved, restoring normal blood vessel function and reducing hypoxia in the TME (21, 30–32). During this time frame, radiation- and immunotherapies were found to be most effective (33, 34). Because high doses and long-term treatment of anti-angiogenic drugs promote hypoxia in the tumor tissue, it is thought that lowering of drug dosage may reduce the levels of angiogenic factors and normalize the tumor vasculature accordingly (30). Experimental tumorigenesis studies using low doses or short-term treatments of anti-angiogenic drugs, indeed observed increased functional blood vessels, improved immunotherapy efficacy, and reduced metastatic activity of tumor cells (26, 35, 36). The discovery of additional therapeutic strategies are pursued to try to overcome anti-angiogenic resistance and to better control normalization of tumor vessels (37, 38).
The oncogene Yes-associated protein (YAP) and its paralogue Transcriptional Co-Activator With PDZ-binding Motif (TAZ or WWTR1) have been considered as attractive pharmacological targets, as they are highly activated in many forms of cancers and contribute to tumor growth and invasion (39). YAP/TAZ are also well known for their regulatory role during physiological and developmental angiogenesis and have recently gained attention in the context of endothelial-driven tumor angiogenesis (40–42). In this review we aim to understand how YAP/TAZ signaling affects the (tumor) endothelium. We will further discuss the potential mechanisms of YAP/TAZ activation by the TME and the downstream transcriptional program of YAP/TAZ that controls angiogenesis.
Molecular Regulation of YAP/TAZ Activity
In general, in normal quiescent adherent cells YAP/TAZ are inhibited by the Hippo pathway and located in the cytoplasm. Upon various activating signals YAP/TAZ translocate toward the nucleus and act as transcriptional co-factors for the regulation of tissue homeostasis and organ growth (43, 44). In addition, YAP/TAZ respond to mechanical stimuli derived from cell spreading, contact inhibition, cytoskeletal contractility, ECM stiffness and fluid shear forces (45, 46).
The Hippo signaling pathway consists of a phosphorylation cascade with several effectors. Serine/threonine kinases 3 and 4 (STK3 and STK4, also called MST1/2) interact with the scaffolding protein Salvador family WW-domain-containing-protein-1 (SAV1). If Hippo signaling is turned “on”, the MST-SAV1 complex phosphorylates MOB kinase activator 1A and 1B (MOB1A and MOB1B). This leads to an interaction of MOB1A and MOB1B with large tumor suppressor kinase 1 and 2 (LATS1/2) (47, 48). Once in complex with MOB1A/1B, LATS1/2 become autophosphorylated and phosphorylated by MST1/2 (47, 49). In turn, active LATS1/2 kinases phosphorylate YAP on 5 serine residues (S61, S109, S127, S164, and S381) and TAZ on 4 serine residues (S66, S89, S117, S311) (50). Serine phosphorylated YAP/TAZ bind to 14-3-3 proteins, which sequesters the proteins in the cytoplasm or targets YAP/TAZ for ubiquitin-mediated proteasomal degradation (51, 52). Alternatively, YAP/TAZ can be sequestered in the cytoplasm by Angiomotin (AMOT) proteins that interact with YAP/TAZ or Hippo pathway effectors. If Hippo signaling is turned “off”, YAP/TAZ act as transcriptional co-activators in the nucleus, where they primarily interact with TEA domain family member (TEAD) transcriptional factors to regulate genes involved in proliferation, migration and survival (53). The Hippo pathway crosstalks with major signaling routes that control tissue remodeling and growth, including the Wnt/β-catenin, TGFβ, and Notch pathways (54–56).
YAP/TAZ are also activated in various force-dependent manners (43, 45, 51). Upon cytoskeletal-driven cellular adaptations, such as during ECM stiffening, shear stress sensing or upon G-protein-coupled receptor (GPCR) signaling, the AMOT proteins enhance their interaction with F-actin, allowing YAP/TAZ to translocate toward the nucleus (57–60). ECM stiffening also remodels the integrin-based focal adhesions (FA). Cell adhesion promotes activation of Focal Adhesion Kinase (FAK) and SRC tyrosine kinases, that impinge on the Hippo pathway through direct activation of YAP/TAZ and inhibition of LATS1/2 and MOB1 through FAK/Rac and SRC/PI3K signaling (51, 61). The stiffness-sensing integrin receptors transduce forces to the cytoskeleton-anchored nucleus, opening the nuclear pores and driving YAP/TAZ nuclear translocation (46). Mechanical forces at cell-cell junctions also control YAP/TAZ, as strain on epithelial monolayers induce β-catenin and YAP1 nuclear localization (62, 63). Moreover, high tension inferred at the junctional cadherin-catenin complex, triggers the interaction of α-catenin with TRIP6 and LIMD1, which recruit LATS1/2 to the junctions and inhibit their kinase activity, leading to nuclear translocation of YAP/TAZ (64–66). The various Hippo and mechanotransduction pathways that regulate YAP/TAZ have been elegantly discussed previously (45, 63, 67, 68). These molecular pathways have been investigated in great detail in normal epithelia or tumor cells and are expected to be responsible for YAP/TAZ regulation in the endothelium as well (45).
The Role of YAP/TAZ in Developmental Angiogenesis
Angiogenesis is driven by endothelial proliferation, collective cell migration, and cellular rearrangements (69–71). Angiogenic stimuli, such as VEGF-A and FGF2, activate the ECs to promote the formation of endothelial tip cells that migrate toward the angiogenic cue. Tip cells are followed by proliferative endothelial stalk cells, which shape the developing sprouts and the vascular lumen (2, 72, 73). The tip and stalk cell rearrangements are regulated through feedback loops between VEGF and Dll4/Notch signaling (74).
YAP/TAZ are activated in the sprouting ECs of the developing vasculature in the mouse retina (75). YAP/TAZ are expressed in the entire retinal vasculature, but they reside in the endothelial cytoplasm in the central vascular region, while YAP/TAZ are mainly nuclear in sprouting ECs and the remodeling vascular plexus (76–78). Especially, TAZ nuclear localization is prominent in spouting ECs of the developing retinal vasculature (76, 77). Importantly, the activation of YAP/TAZ in ECs is crucial for angiogenesis (75, 77). The nuclear translocation of endothelial YAP/TAZ is regulated by VEGF signaling, VE-cadherin-based adherens junctions, and cytoskeletal remodeling (45, 75, 79, 80). The VE-cadherin complex sequesters YAP/TAZ and (force-dependent) remodeling of the cell-cell junctions leads to YAP/TAZ activation (81, 82). VEGF-A signaling stimulates YAP/TAZ through cytoskeletal remodeling and inactivation of the Hippo effectors LATS1/2 (79). In turn, active endothelial YAP/TAZ induce a downstream transcriptional program which regulates proliferation, actin cytoskeleton contractility, cell adhesion, and collective cell migration (76, 77, 79, 80, 83).
The importance of YAP/TAZ function for vascular development has been studied by several groups using (inducible) endothelial-specific YAP/TAZ double knock out mouse models (76, 77, 79, 83). Endothelial-specific depletion of YAP/TAZ reduces the number of tip cells and angiogenic sprouts, and leads to excessive vessel crossing in the developing vasculature of the mouse retina (76, 77, 79, 83). Moreover, once the vasculature in the YAP/TAZ endothelial-specific knockout mice makes it to the stage of larger vessels, the vessels turn out to be leaky due to perturbation of endothelial cell-cell junctions (76, 77). Interestingly, knocking out only the YAP or TAZ genes from the endothelium resulted in mild vascular defects, indicating that YAP and TAZ have redundant functions and can compensate for each other in the endothelium (77).
Endothelial-specific overexpression of YAP or TAZ induced retinal vessel growth through increased angiogenic sprouting (77, 84). Interestingly, endothelial-specific YAP overexpression did not affect the vasculature of quiescent tissue in adult mice (84). Constitutive activation of YAP/TAZ, induced by knockout of LATS kinases or overexpression of an active mutant of YAP or TAZ, resulted in endothelial hypersprouting in vivo (77, 83). In vitro it was found that overexpression of an active form of YAP promotes hypersprouting via the angiogenic growth factor angiopoietin-2 (Ang2) signaling (75).
In agreement with the knockout mouse models that demonstrate an important role for YAP/TAZ in vascular development, depleting YAP/TAZ from zebrafish resulted in embryonic lethality due to severe developmental and vascular malformations (85, 86). YAP1 null mutant zebrafish showed a drastic reduction in transcriptional activity of TEAD2, while in TAZ null mutant zebrafish TEAD2 transcriptional activity was unaffected (86), suggesting that YAP is the major transcriptional regulator for vascular development in zebrafish. YAP1 null mutant zebrafish showed increased vessel regression and lumen stenosis, suggesting an important role for YAP1 in lumen maintenance in response to blood flow (86). Moreover, truncation of the cranial and ocular vasculature is observed (85). By contrast, TAZ null mutants did not display clear vascular defects (85). Transgenic mutant zebrafish in which the binding of the YAP/TAZ-TEAD complex to the DNA has been prevented, display altered vascular remodeling (87). Interestingly, expression of constitutively active YAP (YAP-5SA), TAZ (TAZ-4SA), or TEAD mutants initially promote vessel sprouting, but the sprouts fail to anastomose or stabilize at later stages (85). In summary, the regulation of endothelial YAP/TAZ activity is critical during vascular development and both the down- and upregulation of YAP/TAZ activity leads to aberrant sprouting angiogenesis and blood vessel formation.
After new blood vessels are formed during development, angiogenic growth factor levels drop and vessels mature through the stabilization of cell-cell junctions and the recruitment of mural pericytes. In mature vessels, ECs are quiescent (88–90) and endothelial YAP/TAZ are inactivated (79). During wound healing in physiological and pathological conditions, YAP/TAZ are activated on demand to induce angiogenesis (53). The TME is somewhat comparable to the tissue of an inflamed wound (91), but in tumors endothelial YAP/TAZ remain activated and its vasculature does not evolve into a mature state (79) (Figure 1).
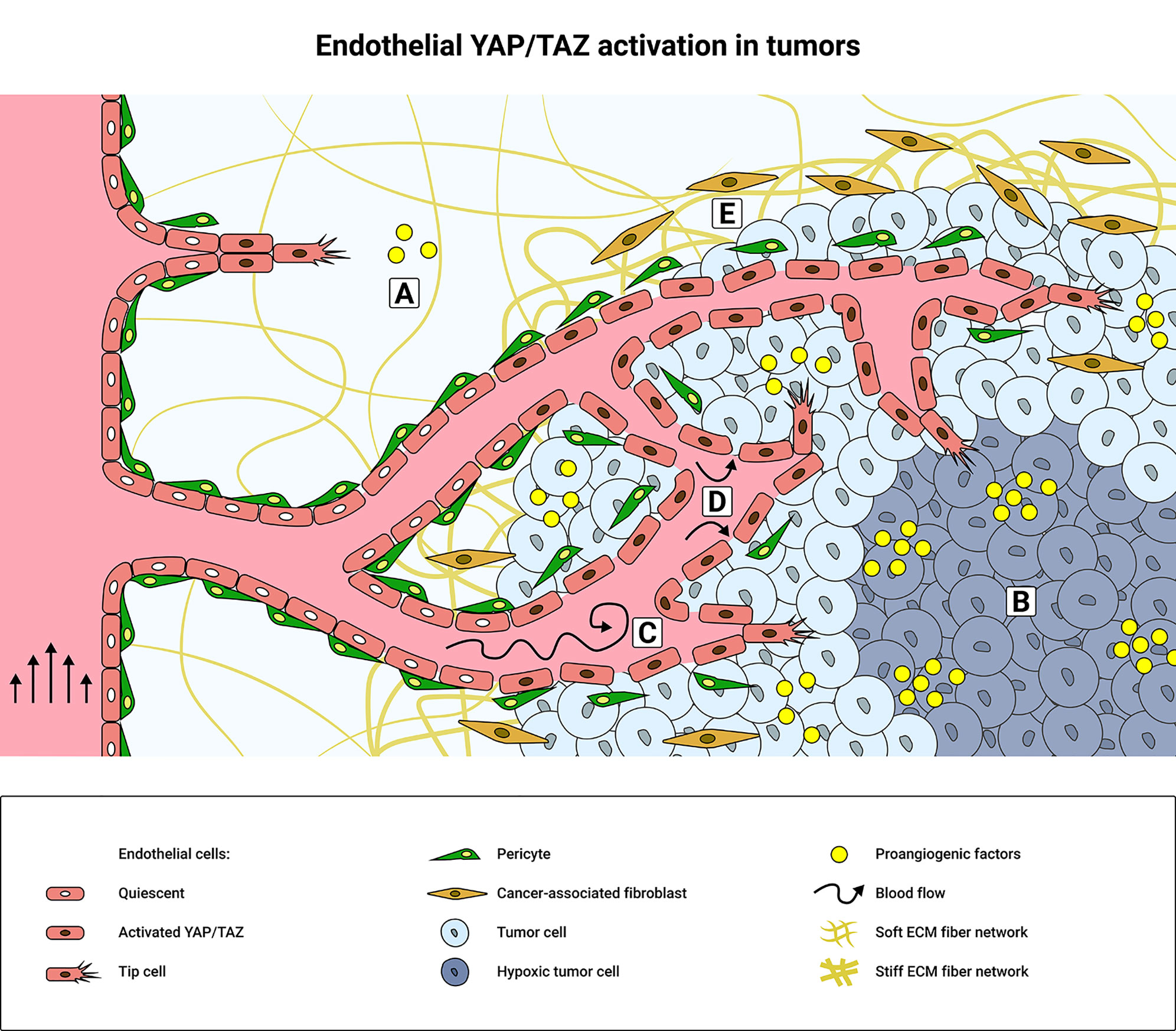
Figure 1 Schematic overview of tumor microenvironment factors that promote endothelial YAP/TAZ activity. (A) During physiological angiogenesis, pro-angiogenic cytokines and growth factors activate YAP/TAZ signaling in endothelial cells (ECs) through GPCRs and RTKs, such as VEGFR2 or Tie2. (B) Hypoxia induces secretion of pro-angiogenic factors to promote tumor angiogenesis and consequently YAP/TAZ activity in the tumor microenvironment (TME). Elevated levels of pro-angiogenic factors promote the formation of a dense and disorganized tumor vasculature, resulting in disturbed blood flow patterns (C) and increased interstitial fluid pressure (D) that mechanically activate YAP/TAZ. The integrity of the tumor endothelium is weakened due to decreased cell-cell interactions between the ECs and/or pericytes. YAP/TAZ are further mechanically activated in the TME by ECM stiffening (E), which is promoted by the cancer-associated fibroblasts (CAFs).
YAP/TAZ Activation in Tumor Angiogenesis
Many types of cancer are accompanied by increased levels and activity of YAP/TAZ, including breast, pancreatic, liver and colorectal cancer (39, 92–94). Increased expression or activation of YAP/TAZ in cancer is associated with poor prognosis and reduced survival (93, 95). Increased YAP/TAZ levels are often observed both in tumor and stromal cells, including the CAFs, ECs, immune cells, and pericytes (96–99). YAP/TAZ activation in the TME promotes tumor growth, metastasis and angiogenesis (95, 97–99). For instance, in glioblastoma, high expression of TAZ in tumor endothelium is correlated with increased blood vessel density and tumor malignancy (100). There is an intricate connection between YAP/TAZ activation in the TME and the tumor vasculature.
Oncogenic activation of YAP/TAZ in tumor cells drives the ECs toward a pro-angiogenic state (Figure 1). Conditioned medium from (YAP positive) breast cancer cells induced endothelial YAP activation, which in turn promoted tumor angiogenesis (101). Also, ECs treated with conditioned medium from cholangiocarcinoma cells containing a constitutively active YAP mutation (YAP S127A), showed increased tube formation capacity in vitro (102), suggesting enhanced endothelial activity. Moreover, YAP activation in mesenchymal stromal cells has been shown to enhance the crosstalk between gastric cancer cells and the tumor endothelium (103). Importantly, transplantations of Lewis Lung carcinoma allografts in transgenic mice with endothelial-specific YAP overexpression, resulted in an increase in tumor size and tumor vasculature (84). Vice versa, YAP knockdown in renal cell carcinoma, inhibited the angiogenic capacity of ECs via paracrine VEGF signaling (104). Overall, these findings emphasize the critical involvement of endothelial YAP/TAZ signaling during tumor angiogenesis following the interplay between tumor tissue and the ECs. Of note, YAP/TAZ activation does not always increase tumor angiogenesis and differences between tumor types should be considered. For instance, in angiosarcoma, a rare type of cancer derived from the vasculature, inhibition of PECAM-1 raised YAP levels, but decreased the tubulogenic potential of the angiosarcoma cells (105).
Tumor Microenvironmental Factors That Activate YAP/TAZ
In solid tumors many microenvironmental properties have changed compared to in healthy tissue, for example increased interstitial fluid pressure (IFP), inflammation and ECM stiffness. These TME properties are key drivers of YAP/TAZ and promote the immature characteristics of the tumor vasculature (43, 106) (Figure 1).
Hypoxia
One of the prominent angiogenic features of solid tumors is their hypoxic condition. Hypoxia stabilizes hypoxia inducible transcription factor 1α (HIF1α) in tumor cells, initiating the transcription and secretion of pro-angiogenic factors, such as VEGF and Ang2 (107). Also in ECs, HIF1α promotes the transcription of autocrine pro-angiogenic molecules and matrix metalloproteases (MMPs) (40). Importantly, the endothelial-specific depletion of HIF1α resulted in reduced tumor growth and angiogenesis in experimental Lewis lung carcinoma (108). The onset of hypoxia in the retinal vasculature is known to inhibit Hippo pathway effectors and activates endothelial YAP (109). In turn, YAP is able to interact with HIF1α to sustain HIF1α signaling (110). In hypoxic colorectal cancer cells, HIF1α induces the transcription of GPCR5A, which in turn activates YAP to promote cell survival (111). Of note, in hepatocellular carcinoma cells, hypoxia was shown to activate YAP through a HIF1α independent manner (112). The intricate crosstalk between YAP/TAZ and HIF1α signaling in cancer has recently been overviewed (113).
Hypoxia also induces the activation of Signal Transducer and Activator of Transcription-3 (STAT3), which forms a complex with YAP in ECs to drive expression of angiogenic factors VEGF and Ang2 (84, 109) (Figure 2). Furthermore, the transcription factor SNAIL is a direct target of HIF1α, and hypoxia-induced expression of SNAIL promotes endothelial to mesenchymal transition (EndoMT) (114). YAP has been shown to induce EndoMT during cardiac development by upregulating expression of SNAIL (115). YAP activation potentially promotes hypoxia-induced EndoMT in the TME by facilitating SNAIL expression. Taken together, the hypoxic conditions within tumors activate YAP/TAZ-dependent programs that promote tumor vascularization. Notably, the described crosstalk between YAP/TAZ and HIF1α may differ between tissues, because in the hypoxic environment of the bone marrow, endothelial YAP/TAZ function to inhibit angiogenesis (116).
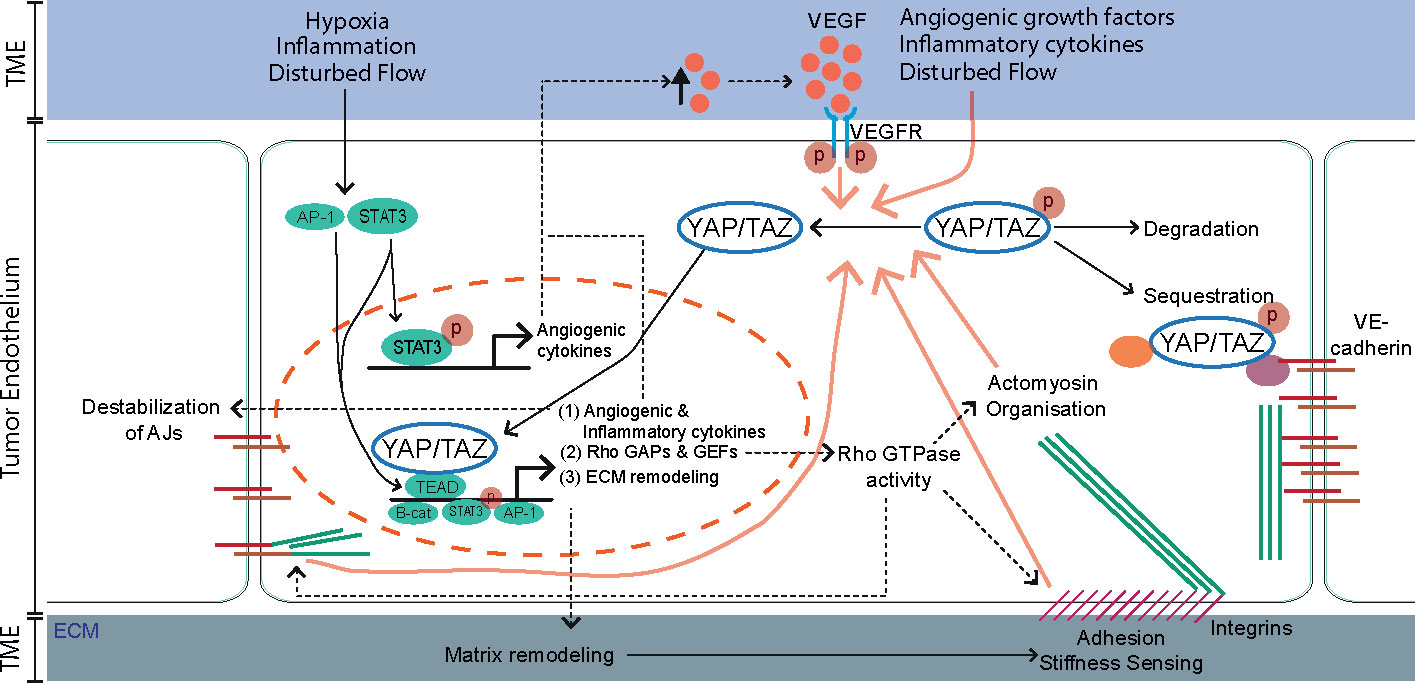
Figure 2 The tumor microenvironment (TME) and downstream transcriptional targets of YAP/TAZ engage in a positive feedback program that sustains YAP/TAZ activity in the endothelium. In quiescent endothelial cells YAP/TAZ are kept inactive and sequestered in the cytoplasm via interaction with 14-3-3 proteins, Angiomotin (AMOT) proteins or the VE-cadherin complex. Alternatively, inactive YAP/TAZ can be targeted for ubiquitin-mediated degradation. External cues from the tumor microenvironment (TME), including pro-angiogenic growth factors, inflammatory cytokines, stiff extracellular matrix (ECM), hypoxia, and disturbed blood flow activate YAP/TAZ leading to their translocation to the nucleus (Figure 1). In parallel, multiple TME factors activate the transcription factors STAT3 and AP-1. Within the nucleus, YAP/TAZ interacts with several transcription factors, most notably the TEA domain family members (TEADs), but also with STAT3 and β-catenin to induce transcription of downstream target genes. Activated YAP/TAZ induce the expression of angiogenic and inflammatory cytokines, Rho GAPs and GEFs and extracellular matrix (ECM) remodeling proteins that engage in a positive feedback system that sustains YAP/TAZ activity. (1) angiogenic and inflammatory cytokines maintain the pro-angiogenic and inflammatory TME. Moreover, the angiogenic effectors of YAP/TAZ (such as Ang2 and VEGF) destabilize VE-cadherin-based adherens junctions (AJs), further promoting the activity of YAP/TAZ. (2) Rho GAPs and GEFs control the level of Rho-GTPase activities, crucial switches that organize the actomyosin cytoskeleton, AJs and integrin-based focal adhesions (FA). In turn, the FAs and actomyosin cytoskeleton transduce forces from stiff ECM and maintain the nuclear translocation of YAP/TAZ. (3) ECM remodeling proteins are secreted and remodel the TME in favor of YAP/TAZ activity.
Angiogenic Growth Factors and Cytokines
A second important TME property that promotes endothelial YAP/TAZ activity and angiogenesis is the increased presence of angiogenic and inflammatory cytokines (i.e., VEGF, TGFβ and TNFα). Key is the (hypoxia-driven) VEGF production by tumor cells, ECs, and CAFs (117, 118). VEGF interaction with VEGF receptor 2 (VEGFR2) induces downstream signaling toward SRC kinases, PI3K/Akt, and MEK/ERK. These signaling pathways inactivate the Hippo pathway effectors MST1/2 and LATS1/2, leading to activation of endothelial YAP/TAZ (79, 119). The stimulation of ECs by VEGF also remodels the actin cytoskeleton and endothelial cell-cell junctions (120, 121), which further modulates YAP/TAZ activity (79). Actomyosin contractility is a well-established regulator of YAP/TAZ activity in a variety of cell types, acting directly on YAP/TAZ or via Hippo pathway effectors (45, 46, 122). In summary, VEGF-VEGFR2 signaling can induce endothelial YAP activation through various pathways, directly by interrupting the Hippo signaling cascade, or indirectly by its effect on cell-cell junctions and the actin cytoskeleton. Similar mechanisms may take place in the tumor endothelium.
Another important growth factor that affects YAP/TAZ signaling in tumors is TGFβ. Hypoxia together with TGFβ-mediated SMAD transcriptional activation induces complex formation between Zyxin, LATS2 and SIAH2 (123). The latter being an E3 ligase that mediates LATS2 ubiquitination and degradation (123). Furthermore, SMAD2 can interact with YAP via the RASS1FA scaffold protein, which helps to retain the SMAD2/YAP complex in the cytoplasm in quiescent cells (124). TGFβ induces the degradation of RASS1FA, which enables the SMAD2/YAP complex to translocate to the nucleus, leading to transcription of their target genes (124). YAP plays a crucial role in the nuclear translocation of SMADs (125). Most of these molecular mechanisms have been studied in epithelial or tumor cells. Yet, the tumor ECs express high levels of Endoglin (126), a BMP9 receptor that is part of the TGFβ receptor complex and an important receptor for endothelial migration and angiogenesis (127–129). BMP9-endoglin signaling induces YAP/SMAD nuclear translocation driving the expression of inflammatory genes in ECs (130), implying that endothelial YAP-SMAD may regulate tumor angiogenesis. Taken together, there are multiple angiogenic growth factors and cytokines within the TME that control YAP/TAZ and contribute to the immature and proliferative tumor vasculature.
Tumor Microenvironment Stiffness
Overall, solid tumors are stiffer in comparison to normal tissue (106). The stiffening is caused by increased collagen deposition and cross-linking (131). In tumors, ECM stiffening promotes sprouting angiogenesis and gives rise to a dense and hyperpermeable vasculature (106). Integrin adhesion receptors sense the increased ECM stiffness and induce the assembly of integrin-based FAs (132, 133). In turn, FAK/SRC signaling downstream of integrins inhibits LATS1/2 and activates YAP/TAZ (51, 134–136). The activation of endothelial FAK through phosphorylation of its autophosphorylated Y397 residue is crucial for tumor angiogenesis and tumor progression (137). Interestingly, targeting of endothelial FAK activity improved the efficacy of DNA-damaging chemotherapeutics, providing proof-of-principle for normalization of tumor vasculature as an adjuvant approach in cancer therapies (138). Importantly, integrin-mediated stiffness-sensing also activates YAP/TAZ independently of the Hippo pathway through activation of Rho-GTPases and actomyosin contractility (45, 46). YAP/TAZ activation, in turn, regulates FA turnover (139), a feedback mechanism that is crucial for endothelial collective migration and angiogenic sprouting (80, 140). Taken together, it is likely that direct stiffness-sensing through the tumor endothelium promotes endothelial YAP/TAZ activation and tumor angiogenesis. To prove this concept more experimental work is needed.
In addition, TME stiffening and tumor cell contractility promote MMP activity (141). MMP activity and subsequent matrix degradation controls endothelial sprouting through the stiff ECM (131). Vice versa, inhibition of lysyl oxidase, a matrix cross-linking enzyme involved in tumor stiffening, was shown to reduce the tumor stiffness and tumor vasculature in a mouse mammary tumor model (131). It was recently reported that metastasis-associated fibroblasts promote TME stiffening and angiogenesis in particular in liver metastases from colorectal cancer (142). Intriguingly, inhibition of renin-angiotensin signaling in combination with the anti-VEGF drug Bevacizumab, inactivated tumor endothelial YAP and reduced the fibroblast-induced metastatic TME stiffness and tumor vasculature (142). Moreover, the activation of YAP in CAFs is known to increase ECM stiffening, tumor cell invasion and tumor angiogenesis (97). Thus CAFs participate in a self-sustaining YAP-dependent feed forward loop that aggravates tumorigenesis, through a mechanism in which tumor stiffening and angiogenesis take central roles (Figure 1).
Interstitial Fluid Pressure and Blood Flow
Tumor vessels are disorganized and hyperpermeable, which increases interstitial fluid pressure (IFP) and leads to disturbed blood flow (143), altogether hindering drug delivery to tumors (144). The high level of IFP also stretches the blood vessels and promotes metastasis (145–148). Mechanical stretching of tumor stromal cells, such as fibroblasts and ECs, is a well-known driver of YAP/TAZ nuclear translocation (46, 77, 149, 150). Consequently, IFP likely activates endothelial YAP/TAZ signaling in the TME. The tumor vasculature is also poorly perfused and the blood flow is often turbulent (13, 151). Disturbed flow patterns activate endothelial YAP/TAZ and trigger proliferative and inflammatory responses in vitro (152–154). In the mouse aortic arch, a vascular area exposed to disturbed blood flow, YAP phosphorylation was lower and its nuclear localization higher, as in comparison to the thoracic aorta, an area exposed to laminar blood flow (153). Consequently, disturbed haemodynamics activate endothelial YAP/TAZ and we speculate that they sustain YAP/TAZ signaling in the tumor vasculature.
In summary, the pro-angiogenic, inflammatory, hypoxic, and stiff TME activates endothelial YAP/TAZ and promotes tumor angiogenesis. A major challenge is to tackle the essential events that sustain this oncogenic vascular environment. Investing in pre-clinical engineered tumor vascular models in which the TME cues can be manipulated in a defined manner are expected to greatly propel research aimed at developing anti-angiogenic therapies for cancer (155).
Downstream Effectors of YAP/TAZ in Tumor Angiogenesis
To date, the mechanisms through which YAP/TAZ control (tumor) angiogenesis remains unclear. Active YAP/TAZ act as co-factors and bind to transcription factors to modulate gene expression (156). YAP/TAZ both contain a TEAD-binding domain and one WW (TAZ) or two WW domains (YAP) that mediate the interaction with transcription factors (157). The interaction of YAP/TAZ with the transcriptional enhancer factor domain (TEAD) family has been considered as the primary axis through which YAP/TAZ regulate transcription. TEAD-dependent transcription include most of the classical YAP/TAZ target genes, including CTGF, CYR61, and ANKRD1 (158–160). The YAP-TEAD complex is important for tumorigenesis: mutating the TEAD binding domain in YAP suppresses its oncogenic capacity in cancer cells (159, 161). The ability of tumor tissue to promote tumor angiogenesis through expression of angiogenic factors occurs in a TEAD-dependent manner (102, 162). Furthermore, pharmacological inhibition of the YAP-TEAD complex with verteporfin suppresses tumor growth in pancreatic cancer, by inhibiting the proliferation of pancreatic ductal adenocarcinoma cells and through inhibition of the angiogenic activity of associated tumor ECs (162).
The WW domains of YAP/TAZ bind to proline-rich sequences such as the PPXY motif, found in a variety of transcription factors, and they mediate the interaction with SMADs, AMOTs, ErbB4, β-catenin, RUNXs, and p73 (163). Mutations in the WW domain perturb YAP-controlled transcriptional programs and reduce its oncogenic capacity (161). While not yet investigated directly, the WW domains of YAP/TAZ are likely to modulate tumor angiogenesis, as YAP/TAZ WW-binding proteins, such as SMADs and AMOTs, have readily been linked to angiogenesis (164, 165).
Promoting gene expression through TEAD-binding or the WW domains are not the only mechanisms through which YAP/TAZ may regulate tumor angiogenesis. For instance TBX5, a transcription factor that lacks a PPXY-motif, can interact with YAP/TAZ via its carboxyl-terminus (166). Moreover, SMAD2/3 can bind the coiled-coil region of YAP/TAZ (167). Multiple YAP/TAZ interactors can synergize to promote gene expression. For instance, ErbB4 binds to YAP through the WW domain (168), which promotes the binding of TEAD to YAP. In this way, ErbB4 regulates the expression of the canonical YAP/TAZ-TEAD targets, including CTGF, CYR61, and ANKRD1 (169). AP-1 is a transcription factor present in most of YAP/TAZ-TEAD genomic binding sites and its presence greatly enhances oncogenic growth induced by active YAP/TAZ. Conversely, AP-1 inactivation inhibits YAP/TAZ-driven proliferation and tumorigenesis (170, 171). AP-1 does not directly bind to YAP/TAZ, but it controls TEAD-dependent gene expression in a cis-regulatory fashion (170–172) (Figure 2). Interestingly, TRPS1 also controls YAP/TAZ through regulatory elements, but decreases YAP transcriptional activity by recruiting co-repressor complexes (173). This shows that YAP/TAZ is capable of inducing gene expression, not only through direct interactions with transcription factors, but also through regulators which are in close proximity of nuclear YAP/TAZ. Which of these transcriptional regulators are activated in tumor ECs is currently still unclear.
Canonical YAP/TAZ Effectors in Tumor Angiogenesis
Tumor ECs express a different transcriptional program compared to normal ECs (174). Many genes which are upregulated during physiological angiogenesis, are also upregulated in ECs in the TME (175, 176); for example increased VEGF-VEGFR2 signaling (177, 178). In addition, the expression of various genes involved in the interaction of ECs with immune cells are downregulated, suggesting that tumor-associated ECs play an important role in the immunotherapeutic resistance of the TME (179, 180). To understand how YAP/TAZ influences tumor angiogenesis, we need to consider the (potential) function of their downstream transcriptional targets.
Activation of YAP/TAZ induce the transcription of a canonical set of genes involved in proliferation, migration and cytoskeletal rearrangement and suppresses genes related to apoptosis in a variety of cell types (67, 94). In Table 1 we summarize the currently known YAP/TAZ target genes and their possible involvement in (tumor) angiogenesis (Table 1). These genes were included as part of the endothelial YAP/TAZ target gene signature if detected in a minimum of three independent YAP/TAZ transcriptome studies, and have been confirmed at least once in an endothelial context (77, 79, 153, 158, 244, 245). For an overview of all YAP/TAZ target genes from these studies, see Supplemental Table 1.

Table 1 Selection of YAP/TAZ-regulated genes involved in (tumor) angiogenesis.
The well-established YAP/TAZ transcriptional targets connective tissue growth factor (CTGF), Cysteine-rich angiogenic inducer 61 (CYR61) and Ankyrin Repeat Domain 1 (ANKRD1) were upregulated in all of the above-mentioned transcriptome studies. CTGF and CYR61 are part of the CCN protein family (246). CTGF is a known mediator of fibrosis in a wide range of diseases (247). During tumor progression, CTGF plays a role in ECM deposition and promotes proliferation and epithelial to mesenchymal transition (EMT) (159, 248). Stromal expression of CTGF increases micro-vessel density in prostate cancer xenografts (249). CTGF was found to promote angiogenesis by inducing VEGF-A secretion of TGFβ-stimulated fibroblasts (250). CTGF also promotes tumor angiogenesis through regulation of Ang2 (251).
The YAP/TAZ target CYR61 was demonstrated to promote angiogenesis and improve tissue perfusion in ischemic models (252). CYR61 null mice are embryonically lethal caused by vascular defects (253). CYR61 is expressed in angiogenic ECs at sites of neovascularization (254). Increased expression of CYR61 is found in many forms of cancer and is linked to an increase of size and vascularization of tumors (255–257). CYR61 promotes tumor angiogenesis through its interaction with integrin αvβ3, which in turn regulates endothelial adhesions, migration, proliferation, and activates VEGFR2 (154).
ANKRD1 is a transcriptional effector of YAP/TAZ. ANKRD1 has been shown to have an anti-inflammatory role through inhibition of NF-κB (182). ANKRD1 may act as a co-activator of the tumor suppressor p53, and the presence of p53 maintains the expression levels of ANKRD1 through a positive feedback loop (183, 258). In cancer ANKRD1 can be epigenetically inactivated (258), which may explain how such a well-established YAP/TAZ target does not attenuate YAP/TAZ-mediated tumorigenesis. ANKRD1 has been proposed to mediate angiogenesis through MMP-mediated ECM remodeling (184). ANKRD1 promotes angiogenesis after acute wounding of mouse skin (259, 260), but currently little is known about the potential role of ANKRD1 in tumor angiogenesis.
Endothelial-Specific YAP/TAZ Angiogenic Effectors
YAP/TAZ also induce the expression of endothelial-specific genes to modulate the tumor vasculature. While not always being referred to as YAP/TAZ effectors in the literature, these pro-angiogenic proteins are readily investigated for their role in tumor angiogenesis and their potential as therapeutic targets.
Ephrin-Eph System
The Ephrin receptor genes EphA2-4 and EphB4 and Ephrin genes EFNB2-3 were found to be downstream targets of endothelial YAP/TAZ in developmental angiogenesis (79). Ephrins and Eph receptors are upregulated in almost all tumors and are considered as promising targets for cancer therapy (261). Interestingly, in YAP/TAZ transcriptome studies focusing on expression in cancer cell lines, the Ephrin family does not seem to be a prominent transcriptional target of YAP/TAZ (158, 244, 245), which indicates that Ephrins may be specifically derived from YAP/TAZ activation in the tumor endothelium.
The Eph receptors and Ephrin ligands are crucial for vascular specification during development via their signaling toward Rho-GTPases, cytoskeletal remodeling, and cell migration (262). Expression of the EphA2 receptor has been reported to promote tumor size and vascular density (263, 264). Genetic silencing or blocking activation of the EphA receptors attenuated tumor angiogenesis and decreased tumor vessel density (265, 266). Signaling through EphrinB2 and EphB4 has been directly associated with tumor angiogenesis and with tumor resistance to anti-angiogenic therapy (267, 268). Inhibition of EphB4 signaling through the use of soluble ligands, reduced the growth and vascularization of tumors (269). Another study showed that EphB4 suppresses sprouting angiogenesis and induces circumferential growth of blood vessels in tumor xenografts (270). Furthermore, the Ephrins and their receptors differ in expression levels between tumor types. For instance, EphA2 and EphB2 both promote tumor angiogenesis, but EphA2 is upregulated in prostate cancer, while EphB2 is downregulated (261). Before considering Ephrin signaling as therapeutic target to normalize the tumor vasculature, more understanding is needed of the downstream mechanisms of Eph receptors, as they induce vascularization in some cancers, but restrict tumor growth in others.
Angiopoietin-Tie System
Ang2 is a downstream transcriptional target of YAP/TAZ in ECs (75, 79). The ligands Angiopoietin-1 (Ang1) and Ang2, bind to Tie receptors and control the angiogenic activation of ECs to finely tune vascular development and homeostasis (271, 272). The Ang2 antagonist Ang1 is an important regulator of vessel maturation and Ang1-Tie2 signaling induces endothelial quiescence (272). Conversely, interaction of Ang2 with Tie2, results in disruption of EC monolayer integrity, making ECs more responsive to inflammatory and pro-angiogenic cytokines (273–275). Ang2 is produced by ECs and signals in an autocrine manner (274). Ang2 expression is induced in response to inflammatory cytokines, hypoxia, and haemodynamic forces (275, 276). Ang2 is highly expressed in ECs of remodeling vessels, indicating an important role for Ang2 in angiogenesis (275, 277). Interestingly, Ang2 may exert both pro- and anti-angiogenic functions. In the presence of VEGF, Ang2 has a pro-angiogenic effect on ECs, while in the absence of VEGF, apoptosis, and vessel regression is induced by Ang2 (278–281). Ang1 binding to Tie2 induces Tie2 autophosphorylation and downstream PI3K/Akt and ERK signaling pathways and inhibits NF-κB activation (271, 273). Moreover, Ang1-Tie2 signaling inhibits Ang2 expression, maintaining endothelial quiescence (273). Upregulation of Ang2 competes with Ang1-Tie2 signaling, resulting in destabilization of the vascular endothelium (273).
Ang2 is described to be a mediator of YAP-induced angiogenesis in mouse retinal vasculature (75, 84). Ang2 levels correlate with YAP activation in sprouting vessels in the retina (75). Supplementation of Ang2 rescues angiogenic defects in the retinal vasculature of YAP/TAZ knockdown mouse and marks Ang2 as a prominent downstream effector of YAP/TAZ-regulated angiogenesis (75). Blocking Ang2 was able to inhibit endothelial YAP-induced angiogenic sprouting (84). Ang2 is upregulated in several types of cancer and is a mediator of tumor angiogenesis (282). Interestingly, YAP and Ang2 association is also observed in the tumor vessels of melanoma (75). In astrocytomas, Ang2 upregulation was correlated with increased vascular growth and an abnormal tumor vasculature (283). Furthermore, angiogenic tumor vessels of human squamous cell carcinoma and skin carcinogenesis xenografts showed an upregulation of Ang2 (284). Interestingly, Ang1 overexpression inhibits tumor growth in these cancer models. The amount of vascularization was unchanged, however more pericyte coverage of the tumor vessels was observed, pointing toward vessel maturation (284, 285). Alternatively, in tumor ECs, Ang2 has been described to induce pro-angiogenic effects by triggering integrin adhesion signaling (286). Ang2 inhibition and Tie2 activation in experimental glioma models resulted in normalization of the tumor vessels, reduced hypoxia and acidosis in the TME, and reduced tumor growth (287). In experimental glioblastoma models, a combination of VEGF- and Ang2-inhibition, was also found to induce tumor vessel normalization (288). Overall, these findings indicate that the YAP/TAZ effector Ang2 might be a promising therapeutic target in cancer.
Fibroblast Growth Factor 2
Fibroblast growth factor 2 (FGF2) is a well-defined YAP/TAZ target expressed in both tumor cells and ECs (77, 79, 158, 244, 245). FGF2 signals through the FGF receptor (FGFR) tyrosine kinase family and induces a broad range of cellular functions, including proliferation, migration, and angiogenesis. Tumor-secreted FGF2, in conjunction with VEGF, promotes tumor angiogenesis (190, 191). The FGF2 pathway is being considered as an important angiogenic pathway involved in bypassing tumor resistance to anti-angiogenic therapies that target VEGF signaling (289, 290). Inhibiting FGF2-FGFR signaling in mice indeed improved tumor sensitivity to anti-VEGF therapy, suggesting that therapeutic strategies that target both growth factors could form a stronger anti-angiogenic intervention in cancer (192).
Deleted-in-Liver-Cancer 1
We recently discovered that Deleted-in-liver-cancer-1 (DLC1), a Rho GAP protein, functions as a direct target of YAP/TAZ in ECs (140). DLC1 is a potential tumor suppressor in various cancer types (291, 292). DLC1 is recruited to integrin-based adhesions through binding to the FA proteins talin, tensin and/or FAK (293, 294). DLC1 knock out mice are embryonically lethal, and the depletion of DLC1 leads to vascular defects (197, 295). DLC1 expression is upregulated by ECM stiffening and angiogenic VEGF signaling (140, 296), and it functions as a prominent target of YAP/TAZ by driving endothelial FA turnover, collective cell migration and sprouting angiogenesis (140). Perturbation of YAP/TAZ signaling and DLC1 levels affect endothelial contact inhibition and promote the development of angiosarcoma (198, 297). Intriguingly, depletion of DLC1 from normal epithelial cells, resulted in increased production of VEGF and upregulation of active HIF1α, suggesting that the absence of DLC1 in oncogenic cells can drive angiogenesis in a paracrine fashion (298). Whether DLC1 is an important target of YAP/TAZ in the tumor endothelium remains to be addressed.
CXCL Chemokines
Various chemokine CXC family members (e.g., CXCL1, CXCL6, CXCL12) are regulated by YAP/TAZ in the endothelium (79, 153). CXCL1, CXCL2, CXCL3, and CXCL8 are part of an angiogenic subset of the CXC family that regulate chemotaxis and angiogenesis through the GPCR CXCR2 (299). In physiological context, CXCL1 primarily targets ECs and neutrophils (299). However, CXCL1 and CXCR2 are also upregulated in different tumor tissues and induce tumor angiogenesis (300–303). It is thought that the inflammatory cytokines contribute to the TME by recruiting inflammatory cells and inducing stromal cell senescence (304–307).
Are There Tumor Angiogenesis-Specific YAP/TAZ Effectors?
The YAP/TAZ-regulated transcriptome is tissue specific. In the endothelium, YAP/TAZ modulate angiogenesis by regulating angiogenic genes. Interestingly, Wang et al. compared RNA-sequencing data from endothelial-specific YAP/TAZ KO mice with that from VEGF-treated HUVECs. Intriguingly, the VEGF-regulated genes were enriched in the gene set that was downregulated upon YAP/TAZ depletion (79). This indicates that YAP/TAZ activation and VEGF signaling synergize to promote angiogenesis.
The transcriptome of tumor-associated ECs in human lung tumors were recently investigated at single-cell resolution (179, 180, 308). Tumor ECs upregulate genes involved in transcription, oxidative phosphorylation and glycolysis, whereas they suppress inflammatory genes. Interestingly, TEAD1 was found as one of the two transcription factors responsible for the tumor-associated endothelial phenotype (179). A large number of the upregulated genes in tumor-associated ECs as reported by Lambrechts et al. are also controlled by YAP/TAZ (Supplemental table 1). Furthermore, tumor-associated ECs strongly activate VEGF and Notch signaling (180), which is likely mediated by YAP/TAZ through crosstalk between these pathways. Treatment with anti-VEGF therapies converts the transcriptomic profile of tumor-associated ECs into a quiescent EC type (308), emphasizing that the activity of tumor ECs is sensitive to therapeutic interventions.
To address if tumor angiogenesis is promoted through specific genes, other than those employed during physiological angiogenesis, the gene expression profiles of ECs in resting liver, regenerating liver, and tumor-bearing liver were compared (233). This study confirmed the upregulation of established angiogenic genes involved in proliferation, such as Top2a, TK1, and Ki67. Incidentally, these genes have also been reported as downstream targets of YAP/TAZ (77, 79, 158, 244, 245). The study further identified two genes that were markedly upregulated during tumor angiogenesis and have been reported as YAP/TAZ targets, namely, SH2D5 (158) and Apelin (79).
SH2 domain containing protein 5, or SH2D5, is a transcriptional target of YAP/TAZ (158) and promotes tumor growth through interaction with transketolase, a regulator of the STAT3 signaling pathway (309). The STAT3 signaling pathway is essential during physiological and tumor angiogenesis (310). In tumors, sustained STAT3 signaling promotes VEGF expression and angiogenesis (311). Furthermore, STAT3 interacts with YAP to promote angiogenesis in a synergistic manner (109). By contrast, a recent paper described YAP to downregulate STAT3 activity and inhibit VEGF expression (312). By regulating STAT3 and VEGF signaling, SH2D5 might be involved in regulating YAP/TAZ activity during tumor angiogenesis.
Apelin is a secreted protein and its expression is regulated by endothelial YAP/TAZ (79). Apelin is required during vascular development (313) and controls initiation of angiogenesis (314). In tumors, increased levels of Apelin promote tumor angiogenesis (314–317). In addition, Apelin drives angiogenesis of lymphatic vessels (318). Interestingly, the GPCR of Apelin, is upregulated in ECs taking part in pathological angiogenesis (308). Because of the anti-angiogenic and anti-lymphangiogenic abilities of Apelin, Apelin has been proposed as a potential therapeutic target for tumor therapies (319).
YAP/TAZ Sustain Tumor Angiogenesis Through Feedback Mechanisms
Physiological angiogenesis and tumor angiogenesis are largely driven by common mechanisms (174, 233). The tumor vasculature is however very different in its organization and function compared to healthy vasculature (10). YAP/TAZ promote the formation of a disorganized and dense tumor vasculature network and simultaneously prevent vessel maturation and specification by sustaining angiogenic signaling (40). In this section we provide an overview of the potential mechanisms through which endothelial YAP/TAZ sustain angiogenic signaling in the TME.
Feedback Signals that Fine Tune YAP/TAZ
YAP/TAZ are regulated by genes that feedback on the upstream elements of the signaling pathway. For instance, the YAP/TAZ target NUAK2 sustains YAP/TAZ activity in breast cancer cells through inhibition of LATS1/2 (320). Pharmacological inhibition of NUAK2 reduces tumor growth in mice, indicating its activity is important to enforce tumorigenic YAP/TAZ signaling (320). By contrast, in collectively migrating ECs, NUAK2 provides negative feedback to YAP/TAZ by reducing actomyosin contractility (80), indicating that this YAP/TAZ target’s feedback function is tissue-dependent. Members of the AMOT family are known regulators of YAP/TAZ (321), and likely also play a role in regulating endothelial YAP/TAZ activity through feedback; since AMOT and AMOTL2 were identified as YAP/TAZ transcriptional targets in endothelial RNA-sequencing studies (77, 79). The p130 isoform of AMOT increases YAP transcriptional activity by binding to YAP and preventing LATS1-mediated YAP phosphorylation (322). In contrast, AMOTL2 inhibits YAP/TAZ activity by binding directly to YAP or to LATS1/2 kinases (57, 188). In most cancers the AMOT family promotes tumorigenesis, while in others its effect is inhibitory (321), indicating AMOT function to be tissue-specific. Altogether the involvement of the AMOT family in the regulation of YAP/TAZ activity and tumor angiogenesis is controversial and should be studied more closely in endothelial context. Interestingly, LATS1/2 kinases were found to phosphorylate AMOT and to inhibit angiogenesis in ECs, independent of YAP/TAZ transcriptional activity (165). These debated findings highlight the currently limited understanding behind the regulatory switches in the YAP/TAZ pathway.
YAP/TAZ activity is further defined by other transcriptional regulators. β-catenin binds to YAP/TAZ in the nucleus and the complex drives tumorigenesis in β-catenin-driven tumors (323). BIRC5 is one of the transcriptional targets of the β-catenin-YAP/TAZ complex that promotes tumorigenesis (323) and is also a target of endothelial YAP/TAZ (Supplemental table 1) (77). BIRC5 was found to upregulate VEGF expression in esophageal cancer cells (213), and may therefore potentially sustain tumor angiogenesis through VEGF signaling. The interaction of YAP/TAZ with β-catenin is regulated by the Wnt pathway: in the absence of Wnt activity, the APC/axin destruction complex degrades the β-catenin-YAP/TAZ complex (324, 325). Moreover, Wnt5a/b and Wnt3a promote YAP/TAZ activity through Rho-GTPase-mediated inactivation of LATS (326). YAP/TAZ, in turn, promote expression of DKK1, BMP4, and IGFBP4, inhibitors of the canonical Wnt/β-catenin signaling (326). Thus, the crosstalk events that take place between YAP/TAZ and Wnt signaling could be an interesting element in the sustained YAP/TAZ signaling in tumor growth and angiogenesis.
YAP/TAZ-Triggered Positive Feedback Loops
Angiogenic Growth Factor Signaling
In angiogenic tissue, YAP/TAZ generate positive feedback by driving a transcriptional response that sustains VEGF-VEGFR2 signaling (79). VEGF signaling itself activates endothelial YAP/TAZ and promotes YAP-dependent STAT3 activation (79, 84, 109). In turn, activated STAT3 elevates VEGF expression (109, 311). YAP/TAZ further enhance VEGF signaling through the YAP/TAZ target CRIM1, a transmembrane receptor that is highly expressed in angiogenic ECs (204). CRIM1 interacts with VEGF, and promotes VEGFR2 phosphorylation (204, 205).
Several YAP/TAZ effectors amplify VEGF signaling downstream of the VEGFR. For instance, the YAP/TAZ effector Rho GEF ECT2 is essential for VEGF-mediated activation of RhoA and endothelial migration (237, 327). In addition, increased VEGF signaling induces MMP expression, which is important for matrix degradation and basement membrane remodeling during angiogenesis (328). MMPs are also upregulated by the YAP/TAZ transcriptional program (329, 330), and may in turn modulate VEGF signaling by controlling VEGFR2 expression (331, 332).
Active endothelial YAP/TAZ drive the expression of other angiogenic cytokines as well, such as FGF2, CXCL1 and TGFβ-2 (see Supplemental table 1). TGFβ-2 activates the TGFβ receptor/SMAD signaling axis to promote angiogenesis, whereas TGFβ-1 promotes angiogenesis through VEGF expression (164). Thus, endothelial YAP/TAZ activation sustains tumor angiogenesis by enhancing VEGF signaling and related angiogenic factors.
TME Stiffening and Cellular Contractility
YAP/TAZ promote a transcriptional program that increases TME stiffening and intracellular contractility, which in turn reinforces YAP/TAZ signaling (67, 97). Activation of YAP/TAZ in fibroblasts is well known to promote deposition of ECM proteins, secretion of MMPs and cross-linking enzymes (78, 97). Within tumors, the CAFs synthesize fibronectin and collagens, which are key constituents of the stiff tumor tissue and modulate tumor angiogenesis (78, 97). ECM stiffening induces endothelial YAP/TAZ signaling to promote angiogenic sprouting (131, 140). In addition, ECM stiffening and YAP/TAZ signaling jointly induce EndoMT, during which ECs undergo mesenchymal transformation and start to actively participate in TME stiffening and vascular remodeling (333). Along that line, YAP/TAZ signaling in cholangiocarcinoma cells promotes expression and deposition of MFAP5, which is an component of the elastin fibrils in the ECM and promotes tumor vasculature formation (102). The YAP/TAZ target SERPINE1 is another secreted factor that correlates with tumor progression and modulates angiogenesis by competing with ECM proteins for binding to integrins (200, 201).
Finally, YAP/TAZ also regulate expression of genes that increase cytoskeletal contractility to mechanoactivate and further enforce their signaling. Endothelial YAP/TAZ induce expression of well-known modulators of intracellular tension, including the Ephrin-Eph system and the Rho-GTPase family and their regulators, such as the Rho GAP DLC1 and Rho GEF ECT2 (77, 79, 140).
Inflammatory Factors
Inflammatory cytokines in the TME activate YAP/TAZ and sustain angiogenesis (334–338). Inflammatory cytokines activate AP-1 (339), which promotes oncogenic growth in conjunction with YAP/TAZ (171). Expression of the YAP/TAZ effector CYR61 is upregulated by inflammatory cytokines such as IL-1 and TNF-α (340). CYR61 enhances VEGFR2 activity and consequently endothelial YAP/TAZ activity through an integrin αvβ3-VEGFR2-MAPK/PI3K-YAP/TAZ axis and enhanced STAT3 activation (154). However, the details of the reciprocal regulatory mechanism between CYR61 and YAP/TAZ need further investigations, as another study described CYR61 to negatively regulate YAP/TAZ activity (341).
Inflammatory stimuli also trigger the release of the YAP/TAZ target Ang2. Interestingly, blocking the function of Ang2, impaired the interaction between ECs and immune cells and reduced tumor neovascularization (285), suggesting that Ang2 is one of the targets of the YAP/TAZ pathway that might be amenable to normalize the tumor vasculature. Ang2-Tie2 signaling weakens the junctional integrity between ECs (273, 274). The destabilization of cell-cell contacts is known to activate YAP/TAZ via the inhibition of LATS1/2 (62, 63). Moreover, the destabilization of VE-cadherin-based cell-cell junctions activates YAP and induces Ang2 expression (342), pointing toward a positive feedback loop between endothelial YAP and Ang2 signaling.
In conclusion, various factors of the TME activate YAP/TAZ in the endothelium. Activated YAP/TAZ promote angiogenesis through a subset of downstream effectors, while other targets further aggravate the TME conditions. Figure 2 gives an overview of the positive feedback loops that likely sustain YAP/TAZ activation in the TME.
Outlook
The oncogenes YAP/TAZ are interesting targets for cancer therapy as they play an essential role during tumor vascularization. YAP/TAZ have readily been investigated as therapeutic targets for the tumor stroma. Therapeutic interventions have focused on inhibiting YAP/TAZ by targeting upstream Hippo effectors or the YAP/TAZ-TEAD interaction; such strategies have been reviewed in great detail in recent years (343–345). One of the major challenges of targeting strategies is that YAP/TAZ modulate multiple signaling pathways and that completely blocking YAP/TAZ signaling likely has large side effects on tissue homeostasis (95). In this review, we highlight how YAP/TAZ is able to maintain a hyperactive endothelial state within the TME and how this leads to aberrant tumor angiogenesis. Targeting the transcriptional downstream targets that reinforce YAP/TAZ activity in the endothelium may provide an interesting approach to normalize tumor vasculature and improve the efficacy of cancer therapies.
When determining the role of transcriptional targets downstream of YAP/TAZ in a biological process, such as tumor angiogenesis, there are a few things to consider. First, besides promoting gene expression, YAP/TAZ are capable of silencing genes through recruitment of inhibitory co-factors. Therefore YAP/TAZ effectively downregulate a significant number of target genes (173, 346). Secondly, YAP and TAZ each control unique (as well as overlapping) transcriptomes (158) and could therefore affect tumor angiogenesis differently. While YAP and TAZ are considered closely related paralogs, structural differences between YAP and TAZ proteins likely affect their specific transcriptional activities, which may be relevant for cancer subtypes. Finally, endothelial YAP was shown to have a cytoplasmic function, from where it regulates EC migration through interaction with CDC42 in the mouse retinal neovasculature (83). Hence, future research aimed to understand the cytoplasmic role of YAP/TAZ during tumor angiogenesis could provide important new insights.
Finally, different microRNAs (miRNAs) and long noncoding RNAs (lncRNAs) have been reported to regulate YAP/TAZ activity (347–350). Surprisingly, little is known regarding noncoding RNAs downstream of YAP/TAZ. In renal cell carcinoma, expression of lncRNA lncARSR is increased in a YAP/TEAD-dependent manner. In turn, the binding of lncARSR to YAP prevented LATS-mediated phosphorylation and activated YAP (351). TAZ upregulates the miRNAs miR-224 and miR-135, which promote tumorigenesis through inhibition of tumor suppressor SMAD4 (352) and suppression of LATS kinases (353), respectively. Furthermore, YAP was found to downregulate the lncRNA MT1DP, a tumor suppressor that inhibits YAP expression (354). A recent review of Tu et al. nicely summarizes the crosstalk between lncRNAs and YAP/TAZ during tumorigenesis (355). The role of noncoding RNAs as effectors of YAP/TAZ in tumor angiogenesis and their promise as therapeutic application remains a topic to address in the nearby future.
Here, we have described upstream mechanisms of YAP/TAZ activation in (tumor) ECs and provided an overview of the downstream transcriptional effectors of YAP/TAZ that participate in the development of tumor vasculature. Many YAP/TAZ downstream targets drive a stiff, pro-inflammatory, hypoxic TME, creating a self-sustained positive loop of YAP/TAZ activity and tumor angiogenesis. Interfering with the crucial events in such YAP/TAZ signal amplification steps are expected to put a brake on pathological angiogenesis in tumors and help to inhibit tumor growth and progression.
Author Contributions
All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This study was financially supported by the Netherlands Organization of Scientific Research (NWO-VIDI grant 016.156.327) and the Amsterdam Cardiovascular Sciences institute.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2020.612802/full#supplementary-material
Abbreviations
AMOT, Angiomotin; Ang1, Angiopoietin-1; Ang2, Angiopoietin-2; ANKRD1, Ankyrin repeat domain 1; CAFs, Cancer-associated fibroblast; CTGF, Connective tissue growth factor; CYR61, Cysteine-rich angiogenic inducer 61; ECM, Extracellular matrix; EndoMT, Endothelial to mesenchymal transition; ECs, Endothelial cells; FA, Focal adhesion; FAK, Focal Adhesion Kinase; FGFR, FGF receptor; FGF2, Fibroblast growth factor 2; GPCR, G-protein-coupled receptor; HIF1α, Hypoxia inducible transcription factor 1α; IFP, Interstitial fluid pressure; LATS, Large tumor suppressor kinase; MMPs, Matrix metalloproteases; RTK, Receptor tyrosine kinase; SAV1, Salvador family WW-domain-containing-protein-1; STAT3, Signal Transducer and Activator of Transcription-3; STK, Serine/threonine kinases; TAZ, Transcriptional Co-Activator With PDZ-binding Motif; TEAD, TEA domain family member; TME, Tumor microenvironment; VEGF, Vascular endothelial growth factor; VEGFR2, VEGF receptor 2; YAP, Yes-associated protein.
References
1. Folkman J. The role of angiogenesis in tumor growth. Semin Cancer Biol (1992) 3:65–71. doi: 10.1053/sonc.2002.37263
2. Lugano R, Ramachandran M, Dimberg A. Tumor angiogenesis: causes, consequences, challenges and opportunities. Cell Mol Life Sci (2019) 77:1745–70. doi: 10.1007/s00018-019-03351-7
3. Zuazo-Gaztelu I, Casanovas O. Unraveling the Role of Angiogenesis in Cancer Ecosystems. Front Oncol (2018) 8:248. doi: 10.3389/fonc.2018.00248
4. Hanahan D, Folkman J. Patterns and Emerging Mechanisms of the Angiogenic Switch during Tumorigenesis. Cell (1996) 86:353–64. doi: 10.1016/S0092-8674(00)80108-7
5. Carmeliet P, Jain RK. Angiogenesis in cancer and other disease. Nature (2000) 407:249–57. doi: 10.1038/35025220
6. Wang M, Zhao J, Zhang L, Wei F, Lian Y, Wu Y, et al. Role of tumor microenvironment in tumorigenesis. J Cancer (2017) 8:761–73. doi: 10.7150/jca.17648
7. Franco PIR, Rodrigues AP, de Menezes LB, Pacheco Miguel M. Tumor microenvironment components: Allies of cancer progression. Pathol - Res Pract (2020) 216:152729. doi: 10.1016/j.prp.2019.152729
8. Baghban R, Roshangar L, Jahanban-Esfahlan R, Seidi K, Ebrahimi-Kalan A, Jaymand M, et al. Tumor microenvironment complexity and therapeutic implications at a glance. Cell Commun Signal (2020) 18:59. doi: 10.1186/s12964-020-0530-4
9. Weis SM, Cheresh DA. Tumor angiogenesis: molecular pathways and therapeutic targets. Nat Med (2011) 17:1359–70. doi: 10.1038/nm.2537
10. Baluk P, Hashizume H, McDonald DM. Cellular abnormalities of blood vessels as targets in cancer. Curr Opin Genet Dev (2005) 15:102–11. doi: 10.1016/j.gde.2004.12.005
11. Hashizume H, Baluk P, Morikawa S, McLean JW, Thurston G, Roberge S, et al. Openings between Defective Endothelial Cells Explain Tumor Vessel Leakiness. Am J Pathol (2000) 156:1363–80. doi: 10.1016/S0002-9440(10)65006-7
12. Weis SM, Cheresh DA. Pathophysiological consequences of VEGF-induced vascular permeability. Nature (2005) 437:497–504. doi: 10.1038/nature03987
13. McDonald DM, Baluk P. Significance of Blood Vessel Leakiness in Cancer. Cancer Res (2002) 62:5381–5.
14. Goel S, Wong AH-K, Jain RK. Vascular normalization as a therapeutic strategy for malignant and nonmalignant disease. Cold Spring Harb Perspect Med (2012) 2:a006486. doi: 10.1101/cshperspect.a006486
15. Martin JD, Seano G, Jain RK. Normalizing Function of Tumor Vessels: Progress, Opportunities, and Challenges. Annu Rev Physiol (2019) 81:505–34. doi: 10.1146/annurev-physiol-020518-114700
16. Hicklin DJ, Ellis LM. Role of the vascular endothelial growth factor pathway in tumor growth and angiogenesis. J Clin Oncol (2005) 23:1011–27. doi: 10.1200/JCO.2005.06.081
17. Hurwitz H, Fehrenbacher L, Novotny W, Cartwright T, Hainsworth J, Heim W, et al. Bevacizumab plus Irinotecan, Fluorouracil, and Leucovorin for Metastatic Colorectal Cancer. N Engl J Med (2004) 350:2335–42. doi: 10.1056/NEJMoa032691
18. Miller K, Wang M, Gralow J, Dickler M, Cobleigh M, Perez EA, et al. Paclitaxel plus Bevacizumab versus Paclitaxel Alone for Metastatic Breast Cancer. N Engl J Med (2007) 357:2666–76. doi: 10.1056/NEJMoa072113
19. Sandler A, Gray R, Perry MC, Brahmer J, Schiller JH, Dowlati A, et al. Paclitaxel–Carboplatin Alone or with Bevacizumab for Non–Small-Cell Lung Cancer. N Engl J Med (2006) 355:2542–50. doi: 10.1056/NEJMoa061884
20. Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of angiogenesis. Nature (2011) 473:298–307. doi: 10.1038/nature10144
21. Ribatti D, Annese T, Ruggieri S, Tamma R, Crivellato E. Limitations of Anti-Angiogenic Treatment of Tumors. Transl Oncol (2019) 12:981–6. doi: 10.1016/j.tranon.2019.04.022
22. Goel S, Duda DG, Xu L, Munn LL, Boucher Y, Fukumura D, et al. Normalization of the Vasculature for Treatment of Cancer and Other Diseases. Physiol Rev (2011) 91:1071–121. doi: 10.1152/physrev.00038.2010
23. van Beijnum JR, Nowak-Sliwinska P, Huijbers EJM, Thijssen VL, Griffioen AW. The great escape; the hallmarks of resistance to antiangiogenic therapy. Pharmacol Rev (2015) 67:441–61. doi: 10.1124/pr.114.010215
24. Hillen F, Griffioen AW. Tumour vascularization: sprouting angiogenesis and beyond. Cancer Metastasis Rev (2007) 26:489–502. doi: 10.1007/s10555-007-9094-7
25. Mancuso MR, Davis R, Norberg SM, O’Brien S, Sennino B, Nakahara T, et al. Rapid vascular regrowth in tumors after reversal of VEGF inhibition. J Clin Invest (2006) 116:2610–21. doi: 10.1172/JCI24612
26. Huang Y, Yuan J, Righi E, Kamoun WS, Ancukiewicz M, Nezivar J, et al. Vascular normalizing doses of antiangiogenic treatment reprogram the immunosuppressive tumor microenvironment and enhance immunotherapy. Proc Natl Acad Sci USA (2012) 109:17561–6. doi: 10.1073/pnas.1215397109
27. Wilson WR, Hay MP. Targeting hypoxia in cancer therapy. Nat Rev Cancer (2011) 11:393–410. doi: 10.1038/nrc3064
28. Bergers G, Hanahan D. Modes of resistance to anti-angiogenic therapy. Nat Rev Cancer (2008) 8:592–603. doi: 10.1038/nrc2442
29. Pàez-Ribes M, Allen E, Hudock J, Takeda T, Okuyama H, Viñals F, et al. Antiangiogenic Therapy Elicits Malignant Progression of Tumors to Increased Local Invasion and Distant Metastasis. Cancer Cell (2009) 15:220–31. doi: 10.1016/j.ccr.2009.01.027
30. Jain RK. Antiangiogenesis Strategies Revisited: From Starving Tumors to Alleviating Hypoxia. Cancer Cell (2014) 26:605–22. doi: 10.1016/j.ccell.2014.10.006
31. Jain RK. Normalizing tumor vasculature with anti-angiogenic therapy: A new paradigm for combination therapy. Nat Med (2001) 7:987–9. doi: 10.1038/nm0901-987
32. Wong P-P, Bodrug N, Hodivala-Dilke KM. Exploring Novel Methods for Modulating Tumor Blood Vessels in Cancer Treatment. Curr Biol (2016) 26:R1161. doi: 10.1016/j.cub.2016.09.043
33. Winkler F, Kozin SV, Tong RT, Chae S-S, Booth MF, Garkavtsev I, et al. Kinetics of vascular normalization by VEGFR2 blockade governs brain tumor response to radiation: Role of oxygenation, angiopoietin-1, and matrix metalloproteinases. Cancer Cell (2004) 6:553–63. doi: 10.1016/j.ccr.2004.10.011
34. Fukumura D, Kloepper J, Amoozgar Z, Duda DG, Jain RK. Enhancing cancer immunotherapy using antiangiogenics: opportunities and challenges. Nat Rev Clin Oncol (2018) 15:325–40. doi: 10.1038/nrclinonc.2018.29
35. Mpekris F, Voutouri C, Baish JW, Duda DG, Munn LL, Stylianopoulos T, et al. Combining microenvironment normalization strategies to improve cancer immunotherapy. Proc Natl Acad Sci (2020) 117:3728–37. doi: 10.1073/pnas.1919764117
36. Steins A, Klaassen R, Jacobs I, Schabel MC, van Lier MGJTB, Ebbing EA, et al. Rapid stromal remodeling by short-term VEGFR2 inhibition increases chemotherapy delivery in esophagogastric adenocarcinoma. Mol Oncol (2020) 14:704–20. doi: 10.1002/1878-0261.12599
37. Eelen G, Treps L, Li X, Carmeliet P. Basic and Therapeutic Aspects of Angiogenesis Updated. Circ Res (2020) 127:310–29. doi: 10.1161/CIRCRESAHA.120.316851
38. Wong P-P, Demircioglu F, Ghazaly E, Alrawashdeh W, Stratford MRL, Scudamore CL, et al. Dual-Action Combination Therapy Enhances Angiogenesis while Reducing Tumor Growth and Spread. Cancer Cell (2015) 27:123–37. doi: 10.1016/j.ccell.2014.10.015
39. Zanconato F, Cordenonsi M, Piccolo S. YAP/TAZ at the Roots of Cancer. Cancer Cell (2016) 29:783–803. doi: 10.1016/j.ccell.2016.05.005
40. Azad T, Ghahremani M, Yang X. The Role of YAP and TAZ in Angiogenesis and Vascular Mimicry. Cells (2019) 8:407. doi: 10.3390/cells8050407
41. Park JA, Kwon Y-G. Hippo-YAP/TAZ signaling in angiogenesis. BMB Rep (2018) 51:157–62. doi: 10.5483/bmbrep.2018.51.3.016
42. Elaimy AL, Mercurio AM. Convergence of VEGF and YAP/TAZ signaling: Implications for angiogenesis and cancer biology. Sci Signal (2018) 11:eaau1165. doi: 10.1126/scisignal.aau1165
43. Panciera T, Azzolin L, Cordenonsi M, Piccolo S. Mechanobiology of YAP and TAZ in physiology and disease. Nat Rev Mol Cell Biol (2017) 18:758–70. doi: 10.1038/nrm.2017.87
44. Zhao B, Li L, Lei Q, Guan K-L. The Hippo-YAP pathway in organ size control and tumorigenesis: an updated version. Genes Dev (2010) 24:862–74. doi: 10.1101/gad.1909210
45. Dupont S, Morsut L, Aragona M, Enzo E, Giulitti S, Cordenonsi M, et al. Role of YAP/TAZ in mechanotransduction. Nature (2011) 474:179. doi: 10.1016/j.yexcr.2015.10.034
46. Elosegui-Artola A, Andreu I, Beedle AEM, Lezamiz A, Uroz M, Kosmalska AJ, et al. Force Triggers YAP Nuclear Entry by Regulating Transport across Nuclear Pores. Cell (2017) 171:1397–410. doi: 10.1016/j.cell.2017.10.008
47. Praskova M, Xia F, Avruch J. MOBKL1A/MOBKL1B Phosphorylation by MST1 and MST2 Inhibits Cell Proliferation. Curr Biol (2008) 18:311–21. doi: 10.1016/j.cub.2008.02.006
48. Meng Z, Moroishi T, Guan K-L. Mechanisms of Hippo pathway regulation. Genes Dev (2016) 30:1–17. doi: 10.1101/gad.274027.115
49. Chan EHY, Nousiainen M, Chalamalasetty RB, Schäfer A, Nigg EA, Silljé HHW. The Ste20-like kinase Mst2 activates the human large tumor suppressor kinase Lats1. Oncogene (2005) 24:2076–86. doi: 10.1038/sj.onc.1208445
50. He M, Zhou Z, Shah AA, Hong Y, Chen Q, Wan Y. New insights into posttranslational modifications of Hippo pathway in carcinogenesis and therapeutics. Cell Div (2016) 11:4. doi: 10.1186/s13008-016-0013-6
51. Dobrokhotov O, Samsonov M, Sokabe M, Hirata H. Mechanoregulation and pathology of YAP/TAZ via Hippo and non-Hippo mechanisms. Clin Transl Med (2018) 7:23. doi: 10.1186/s40169-018-0202-9
52. Zhao B, Wei X, Li W, Udan RS, Yang Q, Kim J, et al. Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev (2007) 21:2747–61. doi: 10.1101/gad.1602907
53. Boopathy GTK, Hong W. Role of Hippo Pathway-YAP/TAZ Signaling in Angiogenesis. Front Cell Dev Biol (2019) 7:49. doi: 10.3389/fcell.2019.00049
54. Totaro A, Castellan M, Di Biagio D, Piccolo S. Crosstalk between YAP/TAZ and Notch Signaling. Trends Cell Biol (2018) 28:560–73. doi: 10.1016/j.tcb.2018.03.001
55. Konsavage WM, Yochum GS. Intersection of Hippo / YAP and Wnt / b-catenin signaling pathways Hippo / YAP Signaling Pathway. Acta Biochim Biophys Sin Adv Access (2012) 45:71–9. doi: 10.1093/abbs/gms084.Intersection
56. Attisano L, Wrana JL. Signal integration in TGF-β, WNT, and Hippo pathways. F1000Prime Rep (2013) 5:1–8. doi: 10.12703/P5-17
57. Wang W, Huang J, Chen J. Angiomotin-like Proteins Associate with and Negatively Regulate YAP1. J Biol Chem (2011) 286:4364–70. doi: 10.1074/jbc.C110.205401
58. Zhao B, Li L, Lu Q, Wang LH, Liu CY, Lei Q, et al. Angiomotin is a novel Hippo pathway component that inhibits YAP oncoprotein. Genes Dev (2011) 25:51–63. doi: 10.1101/gad.2000111
59. Chan SW, Lim CJ, Chong YF, Pobbati AV, Huang C, Hong W. Hippo Pathway-independent Restriction of TAZ and YAP by Angiomotin. J Biol Chem (2011) 286:7018–26. doi: 10.1074/jbc.C110.212621
60. Sun S, Irvine KD. Cellular Organization and Cytoskeletal Regulation of the Hippo Signaling Network. Trends Cell Biol (2016) 26:694–704. doi: 10.1016/j.tcb.2016.05.003
61. Kim N-G, Gumbiner BM. Adhesion to fibronectin regulates Hippo signaling via the FAK-Src-PI3K pathway. J Cell Biol (2015) 210:503–15. doi: 10.1083/jcb.201501025
62. Benham-Pyle BW, Pruitt BL, Nelson WJ. Mechanical strain induces E-cadherin-dependent Yap1 and β-catenin activation to drive cell cycle entry. Sci (80- ) (2015) 348:1024–7. doi: 10.1126/science.aaa4559
63. Dasgupta I, McCollum D. Control of cellular responses to mechanical cues through YAP/TAZ regulation. J Biol Chem (2019) 294:17693–706. doi: 10.1074/jbc.REV119.007963
64. Venkatramanan S, Ibar C, Irvine KD. TRIP6 is required for tension at adherens junctions. bioRxiv (2020) 2020.04.19.049569. doi: 10.1101/2020.04.19.049569. 2020.04.19.049569.
65. Dutta S, Mana-Capelli S, Paramasivam M, Dasgupta I, Cirka H, Billiar K, et al. TRIP6 inhibits Hippo signaling in response to tension at adherens junctions. EMBO Rep (2018) 19:337–50. doi: 10.15252/embr.201744777
66. Ibar C, Kirichenko E, Keepers B, Enners E, Fleisch K, Irvine KD. Tension-dependent regulation of mammalian Hippo signaling through LIMD1. J Cell Sci (2018) 131:jcs214700. doi: 10.1242/jcs.214700
67. Totaro A, Panciera T, Piccolo S. YAP/TAZ upstream signals and downstream responses. Nat Cell Biol (2018) 20:888–99. doi: 10.1038/s41556-018-0142-z
68. Aragona M, Panciera T, Manfrin A, Giulitti S, Michielin F, Elvassore N, et al. A Mechanical Checkpoint Controls Multicellular Growth through YAP/TAZ Regulation by Actin-Processing Factors. Cell (2013) 154:1047–59. doi: 10.1016/j.cell.2013.07.042
69. Dejana E, Orsenigo F, Lampugnani MG. The role of adherens junctions and VE-cadherin in the control of vascular permeability. J Cell Sci (2008) 121:2115–22. doi: 10.1242/jcs.017897
70. Potente M, Gerhardt H, Carmeliet P. Basic and Therapeutic Aspects of Angiogenesis. Cell (2011) 146:873–87. doi: 10.1016/J.CELL.2011.08.039
71. Szymborska A, Gerhardt H. Hold Me, but Not Too Tight—Endothelial Cell–Cell Junctions in Angiogenesis. Cold Spring Harb Perspect Biol (2018) 10:a029223. doi: 10.1101/cshperspect.a029223
72. Gerhardt H, Golding M, Fruttiger M, Ruhrberg C, Lundkvist A, Abramsson A, et al. VEGF guides angiogenic sprouting utilizing endothelial tip cell filopodia. J Cell Biol (2003) 161:1163–77. doi: 10.1083/jcb.200302047
73. Chung AS, Lee J, Ferrara N. Targeting the tumour vasculature: insights from physiological angiogenesis. Nat Rev Cancer (2010) 10:505–14. doi: 10.1038/nrc2868
74. Jakobsson L, Bentley K, Gerhardt H. VEGFRs and Notch: a dynamic collaboration in vascular patterning. Biochem Soc Trans (2009) 37:1233–6. doi: 10.1042/BST0371233
75. Choi H-J, Zhang H, Park H, Choi KS, Lee HW, Agrawal V, et al. Yes-associated protein regulates endothelial cell contact-mediated expression of angiopoietin-2. Nat Commun (2015) 6:1–14. doi: 10.1038/ncomms7943
76. Kim J, Kim YH, Kim J, Park DY, Bae H, Lee D-H, et al. YAP/TAZ regulates sprouting angiogenesis and vascular barrier maturation. J Clin Invest (2017) 127:3441–61. doi: 10.1172/JCI93825
77. Neto F, Klaus-Bergmann A, Ong YT, Alt S, Vion A-C, Szymborska A, et al. YAP and TAZ regulate adherens junction dynamics and endothelial cell distribution during vascular development. Elife (2018) 7:e31037. doi: 10.7554/eLife.31037
78. Liu F, Lagares D, Choi KM, Stopfer L, Marinković A, Vrbanac V, et al. Mechanosignaling through YAP and TAZ drives fibroblast activation and fibrosis. Am J Physiol - Lung Cell Mol Physiol (2015) 308:L344–57. doi: 10.1152/ajplung.00300.2014
79. Wang X, Freire Valls A, Schermann G, Shen Y, Moya IM, Castro L, et al. YAP/TAZ Orchestrate VEGF Signaling during Developmental Angiogenesis. Dev Cell (2017) 42:462–78. doi: 10.1016/j.devcel.2017.08.002
80. Mason DE, Collins JM, Dawahare JH, Nguyen TD, Lin Y, Voytik-Harbin SL, et al. YAP and TAZ limit cytoskeletal and focal adhesion maturation to enable persistent cell motility. J Cell Biol (2019) 218:1369–89. doi: 10.1083/jcb.201806065
81. Giampietro C, Disanza A, Bravi L, Barrios-Rodiles M, Corada M, Frittoli E, et al. The actin-binding protein EPS8 binds VE-cadherin and modulates YAP localization and signaling. J Cell Biol (2015) 211:1177–92. doi: 10.1083/jcb.201501089
82. Bornhorst D, Xia P, Nakajima H, Dingare C, Herzog W, Lecaudey V, et al. Biomechanical signaling within the developing zebrafish heart attunes endocardial growth to myocardial chamber dimensions. Nat Commun (2019) 10:4113. doi: 10.1038/s41467-019-12068-x
83. Sakabe M, Fan J, Odaka Y, Liu N, Hassan A, Duan X, et al. YAP/TAZ-CDC42 signaling regulates vascular tip cell migration. Proc Natl Acad Sci USA (2017) 114:10918–23. doi: 10.1073/pnas.1704030114
84. He J, Bao Q, Zhang Y, Liu M, Lv H, Liu Y, et al. Yes-associated protein promotes angiogenesis via signal transducer and activator of transcription 3 in endothelial cells. Circ Res (2018) 122:591–605. doi: 10.1161/CIRCRESAHA.117.311950
85. Astone M, Lai JKH, Dupont S, Stainier DYR, Argenton F, Vettori A. Zebrafish mutants and TEAD reporters reveal essential functions for Yap and Taz in posterior cardinal vein development. Sci Rep (2018) 8:10189. doi: 10.1038/s41598-018-27657-x
86. Nakajima H, Yamamoto K, Agarwala S, Terai K, Fukui H, Fukuhara S, et al. Flow-Dependent Endothelial YAP Regulation Contributes to Vessel Maintenance. Dev Cell (2017) 40:523–36.e6. doi: 10.1016/J.DEVCEL.2017.02.019
87. Nagasawa-Masuda A, Terai K. Yap/Taz transcriptional activity is essential for vascular regression via Ctgf expression and actin polymerization. PloS One (2017) 12:e0174633. doi: 10.1371/journal.pone.0174633
88. Raza A, Franklin MJ, Dudek AZ. Pericytes and vessel maturation during tumor angiogenesis and metastasis. Am J Hematol (2010) 85:593–8. doi: 10.1002/ajh.21745
89. Jain RK. Molecular regulation of vessel maturation. Nat Med (2003) 9:685–93. doi: 10.1038/nm0603-685
90. Abraham S, Yeo M, Montero-Balaguer M, Paterson H, Dejana E, Marshall CJ, et al. VE-Cadherin-Mediated Cell-Cell Interaction Suppresses Sprouting via Signaling to MLC2 Phosphorylation. Curr Biol (2009) 19:668–74. doi: 10.1016/j.cub.2009.02.057
91. Dvorak HF. “Tumors: Wounds that do not heal.” New Engl J Med (1986) 314:144–9. doi: 10.1056/NEJM198107023050102
92. Moroishi T, Hansen CG, Guan K-L. The emerging roles of YAP and TAZ in cancer. Nat Rev Cancer (2015) 15:73–9. doi: 10.1038/nrc3876
93. Harvey KF, Zhang X, Thomas DM. The Hippo pathway and human cancer. Nat Rev Cancer (2013) 13:246–57. doi: 10.1038/nrc3458
94. Thompson BJ. YAP/TAZ: Drivers of Tumor Growth, Metastasis, and Resistance to Therapy. BioEssays (2020) 42:1900162. doi: 10.1002/bies.201900162
95. Warren JSA, Xiao Y, Lamar JM. YAP/TAZ Activation as a Target for Treating Metastatic Cancer. Cancers (Basel) (2018) 10:115. doi: 10.3390/cancers10040115
96. Zanconato F, Cordenonsi M, Piccolo S. YAP and TAZ: a signalling hub of the tumour microenvironment. Nat Rev Cancer (2019) 19:454–64. doi: 10.1038/s41568-019-0168-y
97. Calvo F, Ege N, Grande-Garcia A, Hooper S, Jenkins RP, Chaudhry SI, et al. Mechanotransduction and YAP-dependent matrix remodelling is required for the generation and maintenance of cancer-associated fibroblasts. Nat Cell Biol (2013) 15:637. doi: 10.1038/ncb2756
98. Pan Z, Tian Y, Cao C, Niu G. The emerging role of YAP/TAZ in tumor immunity. Mol Cancer Res (2019) 17:1777–86. doi: 10.1158/1541-7786.MCR-19-0375
99. Yang W, Yang S, Zhang F, Cheng F, Wang X, Rao J. Influence of the Hippo-YAP signalling pathway on tumor associated macrophages (TAMs) and its implications on cancer immunosuppressive microenvironment. Ann Transl Med (2020) 8:399. doi: 10.21037/atm.2020.02.11
100. Xu C, Mao L, Xiong J, Wen J, Wang Y, Geng D, et al. TAZ Expression on Endothelial Cells Is Closely Related to Blood Vascular Density and VEGFR2 Expression in Astrocytomas. J Neuropathol Exp Neurol (2019) 78:172–80. doi: 10.1093/jnen/nly122
101. Yan Y, Song Q, Yao L, Zhao L, Cai H. YAP Overexpression in Breast Cancer Cells Promotes Angiogenesis Through Activating Yap Signaling in Vascular Endothelial Cells. Researchsquare PREPRINT (Version 1) (2020). doi: 10.21203/rs.3.rs-27591/v1
102. Marti P, Stein C, Blumer T, Abraham Y, Dill MT, Pikiolek M, et al. YAP promotes proliferation, chemoresistance, and angiogenesis in human cholangiocarcinoma through TEAD transcription factors. Hepatology (2015) 62:1497–510. doi: 10.1002/hep.27992
103. Pan Z, Tian Y, Zhang B, Zhang X, Shi H, Liang Z, et al. YAP signaling in gastric cancer-derived mesenchymal stem cells is critical for its promoting role in cancer progression. Int J Oncol (2017) 51:1055–66. doi: 10.3892/ijo.2017.4101
104. Xu S, Zhang H, Chong Y, Guan B, Guo P. YAP Promotes VEGFA Expression and Tumor Angiogenesis Though Gli2 in Human Renal Cell Carcinoma. Arch Med Res (2019) 50:225–33. doi: 10.1016/j.arcmed.2019.08.010
105. Venkataramani V, Küffer S, Cheung KCP, Jiang X, Trümper L, Wulf GG, et al. CD31 Expression Determines Redox Status and Chemoresistance in Human Angiosarcomas. Clin Cancer Res (2018) 24:460–73. doi: 10.1158/1078-0432.CCR-17-1778
106. Zanotelli MR, Reinhart-King CA. Mechanical Forces in Tumor Angiogenesis. Adv Exp Med Biol (2018) 1092:91–112. doi: 10.1007/978-3-319-95294-9_6
107. Ajith TA. Current insights and future perspectives of hypoxia-inducible factor-targeted therapy in cancer. J Basic Clin Physiol Pharmacol (2018) 30:11–8. doi: 10.1515/jbcpp-2017-0167
108. Tang N, Wang L, Esko J, Giordano FJ, Huang Y, Gerber H-P, et al. Loss of HIF-1α in endothelial cells disrupts a hypoxia-driven VEGF autocrine loop necessary for tumorigenesis. Cancer Cell (2004) 6:485–95. doi: 10.1016/j.ccr.2004.09.026
109. Zhu M, Liu X, Wang Y, Chen L, Wang L, Qin X, et al. YAP via interacting with STAT3 regulates VEGF-induced angiogenesis in human retinal microvascular endothelial cells. Exp Cell Res (2018) 373:155–63. doi: 10.1016/j.yexcr.2018.10.007
110. Zhang X, Li Y, Ma Y, Yang L, Wang T, Meng X, et al. Yes-associated protein (YAP) binds to HIF-1α and sustains HIF-1α protein stability to promote hepatocellular carcinoma cell glycolysis under hypoxic stress. J Exp Clin Cancer Res (2018) 37:216. doi: 10.1186/s13046-018-0892-2
111. Greenhough A, Bagley C, Heesom KJ, Gurevich DB, Gay D, Bond M, et al. Cancer cell adaptation to hypoxia involves a HIF-GPRC5A-YAP axis. EMBO Mol Med (2018) 10:e8699. doi: 10.15252/emmm.201708699
112. Dai X-Y, Zhuang L-H, Wang D-D, Zhou T-Y, Chang L-L, Gai R-H, et al. Nuclear translocation and activation of YAP by hypoxia contributes to the chemoresistance of SN38 in hepatocellular carcinoma cells. Oncotarget (2016) 7:6933–47. doi: 10.18632/oncotarget.6903
113. Zhao C, Zeng C, Ye S, Dai X, He Q, Yang B, et al. Yes-associated protein (YAP) and transcriptional coactivator with a PDZ-binding motif (TAZ): a nexus between hypoxia and cancer. Acta Pharm Sin B (2019) 10:947–60. doi: 10.1016/j.apsb.2019.12.010
114. Xu X, Tan X, Tampe B, Sanchez E, Zeisberg M, Zeisberg EM. Snail Is a direct target of hypoxia-inducible factor 1α (HIF1α) in hypoxia-induced endothelial to mesenchymal transition of human coronary endothelial cells. J Biol Chem (2015) 290:16553–664. doi: 10.1074/jbc.M115.636944
115. Zhang H, Von Gise A, Liu Q, Hu T, Tian X, He L, et al. Yap1 Is required for endothelial to mesenchymal transition of the atrioventricular cushion. J Biol Chem (2014) 289:18681–92. doi: 10.1074/jbc.M114.554584
116. Sivaraj KK, Dharmalingam B, Mohanakrishnan V, Jeong H-W, Kato K, Schröder S, et al. YAP1 and TAZ negatively control bone angiogenesis by limiting hypoxia-inducible factor signaling in endothelial cells. Elife (2020) 9:e50770. doi: 10.7554/eLife.50770
117. Kugeratski FG, Atkinson SJ, Neilson LJ, Lilla S, Knight JRP, Serneels J, et al. Hypoxic cancer-associated fibroblasts increase NCBP2-AS2/HIAR to promote endothelial sprouting through enhanced VEGF signaling. Sci Signal (2019) 12:eaan8247. doi: 10.1126/scisignal.aan8247
118. Tamura R, Tanaka T, Akasaki Y, Murayama Y, Yoshida K, Sasaki H. The role of vascular endothelial growth factor in the hypoxic and immunosuppressive tumor microenvironment: perspectives for therapeutic implications. Med Oncol (2019) 37:2. doi: 10.1007/s12032-019-1329-2
119. Azad T, Janse van Rensburg HJ, Lightbody ED, Neveu B, Champagne A, Ghaffari A, et al. A LATS biosensor screen identifies VEGFR as a regulator of the Hippo pathway in angiogenesis. Nat Commun (2018) 9:1061. doi: 10.1038/s41467-018-03278-w
120. Weis SM, Cui J, Barnes L, Cheresh D. Endothelial barrier disruption by VEGF-mediated Src activity potentiates tumor cell extravasation and metastasis. J Cell Biol (2004) 167:223–9. doi: 10.1083/jcb.200408130
121. Gavard J, Gutkind JS. VEGF controls endothelial-cell permeability by promoting the β-arrestin-dependent endocytosis of VE-cadherin. Nat Cell Biol (2006) 8:1223–34. doi: 10.1038/ncb1486
122. Yu F-X, Zhao B, Panupinthu N, Jewell JL, Lian I, Wang LH, et al. Regulation of the Hippo-YAP pathway by G-protein-coupled receptor signaling. Cell (2012) 150:780–91. doi: 10.1016/j.cell.2012.06.037
123. Ma B, Cheng H, Gao R, Mu C, Chen L, Wu S, et al. Zyxin-Siah2–Lats2 axis mediates cooperation between Hippo and TGF-β signalling pathways. Nat Commun (2016) 7:11123. doi: 10.1038/ncomms11123
124. Pefani D-E, Pankova D, Abraham AG, Grawenda AM, Vlahov N, Scrace S, et al. TGF-β Targets the Hippo Pathway Scaffold RASSF1A to Facilitate YAP/SMAD2 Nuclear Translocation. Mol Cell (2016) 63:156–66. doi: 10.1016/j.molcel.2016.05.012
125. Varelas X, Sakuma R, Samavarchi-Tehrani P, Peerani R, Rao BM, Dembowy J, et al. TAZ controls Smad nucleocytoplasmic shuttling and regulates human embryonic stem-cell self-renewal. Nat Cell Biol (2008) 10:837–48. doi: 10.1038/ncb1748
126. ten Dijke P, Goumans M-J, Pardali E. Endoglin in angiogenesis and vascular diseases. Angiogenesis (2008) 11:79–89. doi: 10.1007/s10456-008-9101-9
127. Tual-Chalot S, Mahmoud M, Allinson KR, Redgrave RE, Zhai Z, Oh SP, et al. Endothelial Depletion of Acvrl1 in Mice Leads to Arteriovenous Malformations Associated with Reduced Endoglin Expression. PloS One (2014) 9:e98646. doi: 10.1371/journal.pone.0098646
128. Lebrin F, Goumans M-J, Jonker L, Carvalho RLC, Valdimarsdottir G, Thorikay M, et al. Endoglin promotes endothelial cell proliferation and TGF-beta/ALK1 signal transduction. EMBO J (2004) 23:4018–28. doi: 10.1038/sj.emboj.7600386
129. Park S, Dimaio TA, Liu W, Wang S, Sorenson CM, Sheibani N. Endoglin regulates the activation and quiescence of endothelium by participating in canonical and non-canonical TGF-β signaling pathways. J Cell Sci (2013) 126:1392–405. doi: 10.1242/jcs.117275
130. Young K, Tweedie E, Conley B, Ames J, FitzSimons M, Brooks P, et al. BMP9 Crosstalk with the Hippo Pathway Regulates Endothelial Cell Matricellular and Chemokine Responses. PloS One (2015) 10:e0122892–e0122892. doi: 10.1371/journal.pone.0122892
131. Bordeleau F, Mason BN, Lollis EM, Mazzola M, Zanotelli MR, Somasegar S, et al. Matrix stiffening promotes a tumor vasculature phenotype. Proc Natl Acad Sci USA (2017) 114:492–7. doi: 10.1073/pnas.1613855114
132. Schwartz MA, Ginsberg MH. Networks and crosstalk: integrin signalling spreads. Nat Cell Biol (2002) 4:E65–8. doi: 10.1038/ncb0402-e65
133. Kechagia JZ, Ivaska J, Roca-Cusachs P. Integrins as biomechanical sensors of the microenvironment. Nat Rev Mol Cell Biol (2019) 20:457–73. doi: 10.1038/s41580-019-0134-2
134. Wada KI, Itoga K, Okano T, Yonemura S, Sasaki H. Hippo pathway regulation by cell morphology and stress fibers. Development (2011) 138:3907–14. doi: 10.1242/dev.070987
135. Feng X, Arang N, Rigiracciolo DC, Lee JS, Yeerna H, Wang Z, et al. A Platform of Synthetic Lethal Gene Interaction Networks Reveals that the GNAQ Uveal Melanoma Oncogene Controls the Hippo Pathway through FAK. Cancer Cell (2019) 35:457–72. doi: 10.1016/j.ccell.2019.01.009
136. Lamar JM, Xiao Y, Norton E, Jiang Z-G, Gerhard GM, Kooner S, et al. SRC tyrosine kinase activates the YAP/TAZ axis and thereby drives tumor growth and metastasis. J Biol Chem (2019) 294:2302–17. doi: 10.1074/jbc.RA118.004364
137. Pedrosa A-R, Bodrug N, Gomez-Escudero J, Carter EP, Reynolds LE, Georgiou PN, et al. Tumor Angiogenesis Is Differentially Regulated by Phosphorylation of Endothelial Cell Focal Adhesion Kinase Tyrosines-397 and -861. Cancer Res (2019) 79:4371–86. doi: 10.1158/0008-5472.CAN-18-3934
138. Tavora B, Reynolds LE, Batista S, Demircioglu F, Fernandez I, Lechertier T, et al. Endothelial-cell FAK targeting sensitizes tumours to DNA-damaging therapy. Nature (2014) 514:112–6. doi: 10.1038/nature13541
139. Nardone G, Oliver-De La Cruz J, Vrbsky J, Martini C, Pribyl J, Skládal P, et al. YAP regulates cell mechanics by controlling focal adhesion assembly. Nat Commun (2017) 8:15321. doi: 10.1038/ncomms15321
140. van der Stoel M, Schimmel L, Nawaz K, van Stalborch A-M, de Haan A, Klaus-Bergmann A, et al. DLC1 is a direct target of activated YAP/TAZ that drives collective migration and sprouting angiogenesis. J Cell Sci (2020) 133:jcs239947. doi: 10.1242/jcs.239947
141. Haage A, Schneider IC. Cellular contractility and extracellular matrix stiffness regulate matrix metalloproteinase activity in pancreatic cancer cells. FASEB J (2014) 28:3589–99. doi: 10.1096/fj.13-245613
142. Shen Y, Wang X, Lu J, Salfenmoser M, Wirsik NM, Schleussner N, et al. Reduction of Liver Metastasis Stiffness Improves Response to Bevacizumab in Metastatic Colorectal Cancer. Cancer Cell (2020) 37:800–17. doi: 10.1016/j.ccell.2020.05.005
143. Boucher Y, Jain RK. Microvascular Pressure Is the Principal Driving Force for Interstitial Hypertension in Solid Tumors: Implications for Vascular Collapse. Cancer Res (1992) 52:5110–4.
144. Schaaf MB, Garg AD, Agostinis P. Defining the role of the tumor vasculature in antitumor immunity and immunotherapy. Cell Death Dis (2018) 9:115. doi: 10.1038/s41419-017-0061-0
145. Hofmann M, Guschel M, Bernd A, Bereiter-Hahn J, Kaufmann R, Tandi C, et al. Lowering of tumor interstitial fluid pressure reduces tumor cell proliferation in a xenograft tumor model. Neoplasia (2006) 8:89–95. doi: 10.1593/neo.05469
146. Haessler U, Teo JCM, Foretay D, Renaud P, Swartz MA. Migration dynamics of breast cancer cells in a tunable 3D interstitial flow chamber. Integr Biol (2011) 4:401–9. doi: 10.1039/c1ib00128k
147. Piotrowski-Daspit AS, Tien J, Nelson CM. Interstitial fluid pressure regulates collective invasion in engineered human breast tumors via Snail, vimentin, and E-cadherin. Integr Biol (Camb) (2016) 8:319–31. doi: 10.1039/c5ib00282f
148. Huang YL, Tung C, Zheng A, Kim BJ, Wu M. Interstitial flows promote amoeboid over mesenchymal motility of breast cancer cells revealed by a three dimensional microfluidic model. Integr Biol (2015) 7:1402–11. doi: 10.1039/c5ib00115c
149. Hoffman LM, Smith MA, Jensen CC, Yoshigi M, Blankman E, Ullman KS, et al. Mechanical stress triggers nuclear remodeling and the formation of transmembrane actin nuclear lines with associated nuclear pore complexes. Mol Biol Cell (2020) 31:1774–87. doi: 10.1091/mbc.E19-01-0027
150. Yamashiro Y, Thang BQ, Ramirez K, Shin SJ, Kohata T, Ohata S, et al. Matrix mechanotransduction mediated by thrombospondin-1/integrin/YAP signaling pathway in the remodeling of blood vessels. Proc Natl Acad Sci (2020) 117:9896–905. doi: 10.1073/pnas.1919702117
151. Gillies RJ, Schornack PA, Secomb TW, Raghunand N. Causes and effects of heterogeneous perfusion in tumors. Neoplasia (1999) 1:197–207. doi: 10.1038/sj.neo.7900037
152. Wang KC, Yeh YT, Nguyen P, Limqueco E, Lopez J, Thorossian S, et al. Flow-dependent YAP/TAZ activities regulate endothelial phenotypes and atherosclerosis. Proc Natl Acad Sci USA (2016) 113:11525–30. doi: 10.1073/pnas.1613121113
153. Wang L, Luo J-Y, Li B, Tian XY, Chen L-J, Huang Y, et al. Integrin-YAP/TAZ-JNK cascade mediates atheroprotective effect of unidirectional shear flow. Nature (2016) 540:579. doi: 10.1038/nature20602
154. Park M-H, Kim AK, Manandhar S, Oh S-Y, Jang G-H, Kang L, et al. CCN1 interlinks integrin and hippo pathway to autoregulate tip cell activity. Elife (2019) 8:e46012. doi: 10.7554/eLife.46012
155. Cui X, Morales R-TT, Qian W, Wang H, Gagner J-P, Dolgalev I, et al. Hacking macrophage-associated immunosuppression for regulating glioblastoma angiogenesis. Biomaterials (2018) 161:164–78. doi: 10.1016/j.biomaterials.2018.01.053
156. Kim M, Jang J, Bae S. DNA binding partners of YAP / TAZ. BMB Rep (2018) 51:126–33. doi: 10.5483/bmbrep.2018.51.3.015
157. Piccolo S, Dupont S, Cordenonsi M. The Biology of YAP/TAZ: Hippo Signaling and Beyond. Physiol Rev (2014) 94:1287–312. doi: 10.1152/physrev.00005.2014
158. Zhang H, Liu CY, Zha ZY, Zhao B, Yao J, Zhao S, et al. TEAD transcription factors mediate the function of TAZ in cell growth and epithelial-mesenchymal transition. J Biol Chem (2009) 284:13355–62. doi: 10.1074/jbc.M900843200
159. Zhao B, Ye X, Yu J, Li L, Li W, Li S, et al. TEAD mediates YAP-dependent gene induction and growth control. Genes Dev (2008) 22:1962–71. doi: 10.1101/gad.1664408
160. Stein C, Bardet AF, Roma G, Bergling S, Clay I, Ruchti A, et al. YAP1 Exerts Its Transcriptional Control via TEAD-Mediated Activation of Enhancers. PloS Genet (2015) 11:e1005465. doi: 10.1371/journal.pgen.1005465
161. Zhao B, Kim J, Ye X, Lai ZC, Guan KL. Both TEAD-binding and WW domains are required for the growth stimulation and oncogenic transformation activity of yes-associated protein. Cancer Res (2009) 69:1089–98. doi: 10.1158/0008-5472.CAN-08-2997
162. Wei H, Wang F, Wang Y, Li T, Xiu P, Zhong J, et al. Verteporfin suppresses cell survival, angiogenesis and vasculogenic mimicry of pancreatic ductal adenocarcinoma via disrupting the YAP-TEAD complex. Cancer Sci (2017) 108:478–87. doi: 10.1111/cas.13138
163. Callus BA, Finch-Edmondson ML, Fletcher S, Wilton SD. YAPping about and not forgetting TAZ. FEBS Lett (2019) 593:253–76. doi: 10.1002/1873-3468.13318
164. Bertolino P, Deckers M, Lebrin F, ten Dijke P. Transforming Growth Factor-beta Signal Transduction in Angiogenesis and Vascular Disorders. Chest (2006) 128:585S–90S. doi: 10.1378/chest.128.6
165. Dai X, She P, Chi F, Feng Y, Liu H, Jin D, et al. Phosphorylation of angiomotin by Lats1/2 kinases inhibits F-actin binding, cell migration, and angiogenesis. J Biol Chem (2013) 288:34041–51. doi: 10.1074/jbc.M113.518019
166. Murakami M, Nakagawa M, Olson EN, Nakagawa O. A WW domain protein TAZ is a critical coactivator for TBX5, a transcription factor implicated in Holt-Oram syndrome. Proc Natl Acad Sci USA (2005) 102:18034–9. doi: 10.1073/pnas.0509109102
167. Varelas X, Samavarchi-Tehrani P, Narimatsu M, Weiss A, Cockburn K, Larsen BG, et al. The Crumbs complex couples cell density sensing to Hippo-dependent control of the TGF-β-SMAD pathway. Dev Cell (2010) 19:831–44. doi: 10.1016/j.devcel.2010.11.012
168. Schuchardt BJ, Bhat V, Mikles DC, McDonald CB, Sudol M, Farooq A. Molecular Basis of the Binding of YAP Transcriptional Regulator to the ErbB4 Receptor Tyrosine Kinase. Biochimie (2014) 101:192–202. doi: 10.1016/j.biochi.2014.01.011
169. Haskins JW, Nguyen DX, Stern DF. Neuregulin 1-activated ERBB4 interacts with YAP to induce Hippo pathway target genes and promote cell migration. Sci Signal (2014) 7:ra116. doi: 10.1126/scisignal.2005770
170. Koo JH, Plouffe SW, Meng Z, Lee D-H, Yang D, Lim D-S, et al. Induction of AP-1 by YAP/TAZ contributes to cell proliferation and organ growth. Genes Dev (2020) 34:72–86. doi: 10.1101/gad.331546.119
171. Zanconato F, Forcato M, Battilana G, Azzolin L, Quaranta E, Bodega B, et al. Genome-wide association between YAP/TAZ/TEAD and AP-1 at enhancers drives oncogenic growth. Nat Cell Biol (2015) 17:1218–27. doi: 10.1038/ncb3216
172. Liu X, Li H, Rajurkar M, Li Q, Cotton JL, Ou J, et al. Tead and AP1 Coordinate Transcription and Motility. Cell Rep (2016) 14:1169–80. doi: 10.1016/j.celrep.2015.12.104
173. Elster D, Tollot M, Schlegelmilch K, Ori A, Rosenwald A, Sahai E. TRPS1 shapes YAP/TEAD-dependent transcription in breast cancer cells. Nat Commun (2018) 9:3115. doi: 10.1038/s41467-018-05370-7
174. Rohlenova K, Goveia J, García-Caballero M, Subramanian A, Kalucka J, Treps L, et al. Single-Cell RNA Sequencing Maps Endothelial Metabolic Plasticity in Pathological Angiogenesis. Cell Metab (2020) 31:862–77. doi: 10.1016/j.cmet.2020.03.009
175. Hida K, Maishi N, Annan DA, Hida Y. Contribution of Tumor Endothelial Cells in Cancer Progression. Int J Mol Sci (2018) 19:1272. doi: 10.3390/ijms19051272
176. Croix B, Rago C, Velculescu V, Traverso G, Romans KE, Montgomery E, et al. Genes Expressed in Human Tumor Endothelium. Sci (80- ) (2000) 289:1197–202. doi: 10.1126/science.289.5482.1197
177. Matsuda K, Ohga N, Hida Y, Muraki C, Tsuchiya K, Kurosu T, et al. Isolated tumor endothelial cells maintain specific character during long-term culture. Biochem Biophys Res Commun (2010) 394:947–54. doi: 10.1016/j.bbrc.2010.03.089
178. Bussolati B, Assenzio B, Deregibus MC, Camussi G. The proangiogenic phenotype of human tumor-derived endothelial cells depends on thrombospondin-1 downregulation via phosphatidylinositol 3-kinase/Akt pathway. J Mol Med (2006) 84:852–63. doi: 10.1007/s00109-006-0075-z
179. Lambrechts D, Wauters E, Boeckx B, Aibar S, Nittner D, Burton O, et al. Phenotype molding of stromal cells in the lung tumor microenvironment. Nat Med (2018) 24:1277–89. doi: 10.1038/s41591-018-0096-5
180. Kim N, Kim HK, Lee K, Hong Y, Cho JH, Choi JW, et al. Single-cell RNA sequencing demonstrates the molecular and cellular reprogramming of metastatic lung adenocarcinoma. Nat Commun (2020) 11:2285. doi: 10.1038/s41467-020-16164-1
181. Lau LF. CCN1 / CYR61 : the very model of a modern matricellular protein. Cell Mol Life Sci (2011) 1:3149–63. doi: 10.1007/s00018-011-0778-3
182. Liu X, Bauman WA, Cardozo C. ANKRD1 modulates inflammatory responses IN C2C12 myoblasts through feedback inhibition OF NF-κB signaling activity. Biochem Biophys Res Commun (2015) 464:208–13. doi: 10.1016/j.bbrc.2015.06.118
183. Kojic S, Nestorovic A, Rakicevic L, Belgrano A, Stankovic M, Divac A, et al. A novel role for cardiac ankyrin repeat protein Ankrd1 / CARP as a co-activator of the p53 tumor suppressor protein. Arch Biochem Biophys (2010) 502:60–7. doi: 10.1016/j.abb.2010.06.029
184. Almodovar-Garcia K, Kwon M, Samaras SE, Davidson JM. ANKRD1 Acts as a Transcriptional Repressor of MMP13 via the AP-1 Site. Mol Cell Biol (2014) 34:1500–11. doi: 10.1128/MCB.01357-13
185. Lee MS, Ghim J, Kim SJ, Yun YS, Yoo SA, Suh PG, et al. Functional interaction between CTGF and FPRL1 regulates VEGF-A-induced angiogenesis. Cell Signal (2015) 27:1439–48. doi: 10.1016/j.cellsig.2015.04.001
186. Mochizuki S, Tanaka R, Shimoda M, Onuma J, Fujii Y, Jinno H, et al. Connective tissue growth factor is a substrate of ADAM28. Biochem Biophys Res Commun (2010) 402:651–7. doi: 10.1016/j.bbrc.2010.10.077
187. Aguiar DP, Correa de Farias G, Branco de Sousa E, De Mattos Coelho-Aguiar J, Lobo JC, Casado PL, et al. New strategy to control cell migration and metastasis regulated by CCN2 CTGF. Cancer Cell Int (2014) 14:61. doi: 10.1186/1475-2867-14-61
188. Kim M, Kim M, Park S, Lee C, Lim D. Role of Angiomotin-like 2 mono-ubiquitination on YAP inhibition. EMBO Rep (2016) 17:64–78. doi: 10.15252/embr.201540809
189. Wang Y, Li Z, Xu P, Huang L, Tong J, Huang H, et al. Angiomotin-like2 Gene ( amotl2 ) Is Required for Migration and Proliferation of Endothelial Cells during Angiogenesis *. J Biol Chem (2011) 286:41095–104. doi: 10.1074/jbc.M111.296806
190. Compagni A, Wilgenbus P, Impagnatiello M, Cotten M, Christofori G. Fibroblast Growth Factors Are Required for Efficient Tumor Angiogenesis. Cancer Res (2000) 1:7163–9.
191. Presta M, Era PD, Mitola S, Moroni E, Ronca R, Rusnati M. Fibroblast growth factor / fibroblast growth factor receptor system in angiogenesis. Cytokine Growth Factor Rev (2005) 16:159–78. doi: 10.1016/j.cytogfr.2005.01.004
192. Incio J, Ligibel JA, Mcmanus DT, Suboj P, Jung K, Kawaguchi K, et al. Obesity promotes resistance to anti-VEGF therapy in breast cancer by up-regulating IL-6 and potentially FGF-2. Sci Transl Med (2018) 10:eaag0945. doi: 10.1126/scitranslmed.aag0945
193. Seth P, Lin Y, Hanai JI, Shivalingappa V, Duyao MP, Sukhatme VP. Magic roundabout, a tumor endothelial marker: Expression and signaling. Biochem Biophys Res Commun (2005) 332:533–41. doi: 10.1016/j.bbrc.2005.03.250
194. Park KW, Morrison CM, Sorensen LK, Jones CA, Rao Y, Bin CC, et al. Robo4 is a vascular-specific receptor that inhibits endothelial migration. Dev Biol (2003) 261:251–67. doi: 10.1016/S0012-1606(03)00258-6
195. Wang B, Xiao Y, Ding BB, Zhang N, Bin YX, Gui L, et al. Induction of tumor angiogenesis by Slit-Robo signaling and inhibition of cancer growth by blocking Robo activity. Cancer Cell (2003) 4:19–29. doi: 10.1016/S1535-6108(03)00164-8
196. Tavora B, Mederer T, Wessel KJ, Ruffing S, Sadjadi M, Missmahl M, et al. Tumoural activation of TLR3–SLIT2 axis in endothelium drives metastasis. Nature (2020) 586:299–304. doi: 10.1038/s41586-020-2774-y
197. Shih Y-P, Yuan SY, Lo SH. Down-regulation of DLC1 in endothelial cells compromises the angiogenesis process. Cancer Lett (2017) 398:46–51. doi: 10.1016/j.canlet.2017.04.004
198. Sánchez-Martín D, Otsuka A, Kabashima K, Ha T, Wang D, Qian X, et al. Effects of DLC1 Deficiency on Endothelial Cell Contact Growth Inhibition and Angiosarcoma Progression. JNCI J Natl Cancer Inst (2018) 110:390–9. doi: 10.1093/jnci/djx219
199. Shih Y-P, Sun P, Wang A, Lo SH. Tensin1 positively regulates RhoA activity through its interaction with DLC1. Biochim Biophys Acta (2015) 1853:3258–65. doi: 10.1016/j.bbamcr.2015.09.028
200. Kjøller L, Kanse SM, Kirkegaard T, Rodenburg KW, Rønne E, Goodman SL, et al. Plasminogen activator inhibitor-1 represses integrin-and vitronectin- mediated cell migration independently of its function as an inhibitor of plasminogen activation. Exp Cell Res (1997) 232:420–9. doi: 10.1006/excr.1997.3540
201. Degryse B, Orlando S, Resnati M, Rabbani SA, Blasi F. Urokinase/urokinase receptor and vitronectin/αvβ3 integrin induce chemotaxis and cytoskeleton reorganization through different signaling pathways. Oncogene (2001) 20:2032–43. doi: 10.1038/sj.onc.1204261
202. Subramanian R, Gondi CS, Lakka SS, Jutla A, Rao JS. siRNA-mediated simultaneous downregulation of uPA and its receptor inhibits angiogenesis and invasiveness triggering apoptosis in breast cancer cells. Int J Oncol (2006) 28:831–9. doi: 10.3892/ijo.28.4.831
203. Degryse B, Neels JG, Czekay RP, Aertgeerts K, Kamikubo YI, Loskutoff DJ. The low density lipoprotein receptor-related protein is a motogenic receptor for plasminogen activator inhibitor-1. J Biol Chem (2004) 279:22595–604. doi: 10.1074/jbc.M313004200
204. Fan J, Ponferrada VG, Sato T, Vemaraju S, Fruttiger M, Gerhardt H, et al. Crim1 maintains retinal vascular stability during development by regulating endothelial cell Vegfa autocrine signaling. Dev (2014) 141:448–59. doi: 10.1242/dev.097949
205. Wilkinson L, Gilbert T, Kinna G, Ruta LA, Pennisi D, Kett M, et al. Crim1KST264/KST264 mice implicate Crim1 in the regulation of vascular endothelial growth factor-A activity during glomerular vascular development. J Am Soc Nephrol (2007) 18:1697–708. doi: 10.1681/ASN.2006091012
206. Zeng H, Zhang Y, Yi Q, Wu Y, Wan R, Tang L. CRIM1, a newfound cancer-related player, regulates the adhesion and migration of lung cancer cells. Growth Factors (2015) 33:384–92. doi: 10.3109/08977194.2015.1119132
207. Glienke J, Sturz A, Menrad A, Thierauch K-H. CRIM1 is involved in endothelial cell capillary formation in vitro and is expressed in blood vessels in vivo. Mech Dev (2002) 119:165–75. doi: 10.1016/s0925-4773(02)00355-6
208. Li Y, Ye X, Tan C, Hongo JA, Zha J, Liu J, et al. Axl as a potential therapeutic target in cancer: Role of Axl in tumor growth, metastasis and angiogenesis. Oncogene (2009) 28:3442–55. doi: 10.1038/onc.2009.212
209. Xu MZ, Chan SW, Liu AM, Wong KF, Fan ST, Chen J, et al. AXL receptor kinase is a mediator of YAP-dependent oncogenic functions in hepatocellular carcinoma. Oncogene (2011) 30:1229–40. doi: 10.1038/onc.2010.504
210. Linger RMA, Keating AK, Earp HS, Graham DK. Taking aim at Mer and Axl receptor tyrosine kinases as novel therapeutic targets in solid tumors. Expert Opin Ther Targets (2010) 14:1073–90. doi: 10.1517/14728222.2010.515980
211. Holland SJ, Powell MJ, Franci C, Chan EW, Friera AM, Atchison RE, et al. Multiple roles for the receptor tyrosine kinase Axl in tumor formation. Cancer Res (2005) 65:9294–303. doi: 10.1158/0008-5472.CAN-05-0993
212. Gallicchio M, Mitola S, Valdembri D, Fantozzi R, Varnum B, Avanzi GC, et al. Inhibition of vascular endothelial growth factor receptor 2-mediated endothelial cell activation by Axl tyrosine kinase receptor. Blood (2005) 105:1970–6. doi: 10.1182/blood-2004-04-1469
213. Shang X, Liu G, Zhang Y, Tang P, Zhang H, Jiang H, et al. Downregulation of BIRC5 inhibits the migration and invasion of esophageal cancer cells by interacting with the PI3K/Akt signaling pathway. Oncol Lett (2018) 16:3373–9. doi: 10.3892/ol.2018.8986
214. Tu SP, Jiang XH, Lin MCM, Cui JT, Yang Y, Lum CT, et al. Suppression of Survivin Expression Inhibits in Vivo Tumorigenicity and Angiogenesis in Gastric Cancer. Cancer Res (2003) 63:7724–32.
215. Kawasaki H, Toyoda M, Shinohara H, Okuda J, Watanabe I, Yamamoto T, et al. Expression of survivin correlates with apoptosis, proliferation, and angiogenesis during human colorectal tumorigenesis. Cancer (2001) 91:2026–32. doi: 10.1002/1097-0142(20010601)91:11<2026::AID-CNCR1228>3.0.CO;2-E
216. O’Connor DS, Schechner JS, Adida C, Mesri M, Rothermel AL, Li F, et al. Control of apoptosis during angiogenesis by survivin expression in endothelial cells. Am J Pathol (2000) 156:393–8. doi: 10.1016/S0002-9440(10)64742-6
217. Wang P, Zhen H, Zhang J, Zhang W, Zhang R, Cheng X, et al. Survivin promotes glioma angiogenesis through vascular endothelial growth factor and basic fibroblast growth factor in vitro and in vivo. Mol Carcinog (2012) 51:586–95. doi: 10.1002/mc.20829
218. Zhou Y, Tan Z, Chen K, Wu W, Zhu J, Wu G, et al. Overexpression of SHCBP1 promotes migration and invasion in gliomas by activating the NF-κB signaling pathway. Mol Carcinog (2018) 57:1181–90. doi: 10.1002/mc.22834
219. Feng W, Li H, Xu K, Chen Y, Pan L, Mei Y, et al. SHCBP1 is over-expressed in breast cancer and is important in the proliferation and apoptosis of the human malignant breast cancer cell line. Mol Carcinog (2018) 57:1181–90. doi: 10.1016/j.gene.2016.04.046
220. Peng C, Zhao H, Song Y, Chen W, Wang X, Liu X, et al. SHCBP1 promotes synovial sarcoma cell metastasis via targeting TGF- β 1 / Smad signaling pathway and is associated with poor prognosis. Mol Carcinog (2018) 57:1181–90. doi: 10.1186/s13046-017-0616-z
221. Tao H-C, Wang H, Dai M, Gu C, Wang Q, Han Z, et al. Targeting SHCBP1 Inhibits Cell Proliferation in Human Hepatocellular Carcinoma Cells. Mol Carcinog (2018) 57:1181–90. doi: 10.7314/apjcp.2013.14.10.5645
222. Catela C, Kratsios P, Hede M, Lang F, Rosenthal N. Serum and glucocorticoid-inducible kinase 1 (SGK1) is necessary for vascular remodeling during angiogenesis. Dev Dyn (2010) 239:2149–60. doi: 10.1002/dvdy.22345
223. Zarrinpashneh E, Poggioli T, Sarathchandra P, Lexow J, Terracciano C, Lang F, et al. Ablation of SGK1 Impairs Endothelial Cell Migration and Tube Formation Leading to Decreased Neo-Angiogenesis Following Myocardial Infarction. PloS One (2013) 8:1–13. doi: 10.1371/journal.pone.0080268
224. Amato R, Antona LD, Porciatti G, Agosti V, Menniti M, Rinaldo C, et al. Sgk1 activates MDM2-dependent p53 degradation and affects cell proliferation, survival, and differentiation. J Mol Med (2009) 87:1221–39. doi: 10.1007/s00109-009-0525-5
225. Zhang L, Cui R, Cheng X, Du J. Antiapoptotic Effect of Serum and Glucocorticoid-Inducible Protein Kinase Is Mediated by Novel Mechanism Activating IκB Kinase. Cancer Res (2005) 65:457–64.
226. Buijs N, Oosterink JE, Jessup M, Schierbeek H, Stolz DB, Houdijk AP, et al. A new key player in VEGF-dependent angiogenesis in human hepatocellular carcinoma: dimethylarginine dimethylaminohydrolase 1. Angiogenesis (2017) 20:557–65. doi: 10.1007/s10456-017-9567-4
227. Zhang P, Xu X, Hu X, Wang H, Fassett J, Huo Y, et al. DDAH1 deficiency attenuates endothelial cell cycle progression and angiogenesis. PloS One (2013) 8:1–9. doi: 10.1371/journal.pone.0079444
228. Kami Reddy KR, Dasari C, Vandavasi S, Natani S, Supriya B, Jadav SS, et al. Novel Cellularly Active Inhibitor Regresses DDAH1 Induced Prostate Tumor Growth by Restraining Tumor Angiogenesis through Targeting DDAH1/ADMA/NOS Pathway. ACS Comb Sci (2019) 21:241–56. doi: 10.1021/acscombsci.8b00133
229. Trittmann JK, Almazroue H, Jin Y, Nelin LD. DDAH1 regulates apoptosis and angiogenesis in human fetal pulmonary microvascular endothelial cells. Physiol Rep (2019) 7:1–14. doi: 10.14814/phy2.14150
230. Reddy K, Reddy K, Dasari C, Duscharla D, Supriya B, Ram NS. Dimethylarginine dimethylaminohydrolase−1 (DDAH1) is frequently upregulated in prostate cancer, and its overexpression conveys tumor growth and angiogenesis by metabolizing asymmetric dimethylarginine (ADMA). Angiogenesis (2018) 21:79–94. doi: 10.1007/s10456-017-9587-0
231. Lange C, Mowat F, Sayed H, Mehad M, Duluc L, Piper S, et al. Dimethylarginine dimethylaminohydrolase-2 deficiency promotes vascular regeneration and attenuates pathological angiogenesis. Exp Eye Res (2016) 147:148–55. doi: 10.1016/j.exer.2016.05.007
232. Kostourou V, Robinson SP, Cartwright JE, Whitley G. Dimethylarginine dimethylaminohydrolase I enhances tumour growth and angiogenesis. Br J Cancer (2002) 87:673–80. doi: 10.1038/sj.bjc.6600518
233. Seaman S, Stevens J, Yang MY, Logsdon D, Graff-cherry C, Croix BS. Genes that Distinguish Physiological and Pathological Angiogenesis. Cancer Cell (2007) 11:539–54. doi: 10.1016/j.ccr.2007.04.017
234. Brockenbrough JS, Morihara JK, Hawes SE, Stern JE, Rasey JS, Wiens LW, et al. Thymidine kinase 1 and thymidine phosphorylase expression in non-small-cell lung carcinoma in relation to angiogenesis and proliferation. J Histochem Cytochem (2009) 57:1087–97. doi: 10.1369/jhc.2009.952804
235. Hisaka Y, Ieda M, Nakamura T, Kosai KI, Ogawa S, Fukuda K. Powerful and controllable angiogenesis by using gene-modified cells expressing human hepatocyte growth factor and thymidine kinase. J Am Coll Cardiol (2004) 43:1915–22. doi: 10.1016/j.jacc.2004.01.034
236. Nakagawa T, Li JH, Garcia G, Mu W, Piek E, Böttinger EP, et al. TGF-β induces proangiogenic and antiangiogenic factors via parallel but distinct Smad pathways. Kidney Int (2004) 66:605–13. doi: 10.1111/j.1523-1755.2004.00780.x
237. Nacak TG, Alajati A, Leptien K, Fulda C, Weber H, Miki T, et al. The BTB-kelch protein KLEIP controls endothelial migration and sprouting angiogenesis. Circ Res (2007) 100:1155–63. doi: 10.1161/01.RES.0000265844.56493.ac
238. Justilien V, Fields AP. Ect2 links the PKC-Par6α complex to Rac1 activation and cellular transformation. Oncogene (2009) 28:3597–607. doi: 10.1038/onc.2009.217
239. Saito S, Liu XF, Kamijo K, Raziuddin R, Tatsumoto T, Okamoto I, et al. Deregulation and Mislocalization of the Cytokinesis Regulator ECT2 Activate the Rho Signaling Pathways Leading to Malignant Transformation. J Biol Chem (2004) 279:7169–79. doi: 10.1074/jbc.M306725200
240. Tatsumoto T, Xie X, Blumenthal R, Okamoto I, Miki T. Human ECT2 is an exchange factor for Rho GTPases, phosphorylated in G2/M phases, and involved in cytokinesis. J Cell Biol (1999) 147:921–7. doi: 10.1083/jcb.147.5.921
241. Georgiou GK, Igglezou M, Sainis I, Vareli K, Batsis H, Briasoulis E, et al. Impact of breast cancer surgery on angiogenesis circulating biomarkers: A prospective longitudinal study. World J Surg Oncol (2013) 11:1–8. doi: 10.1186/1477-7819-11-213
242. Bahramsoltani M, Slosarek I, De Spiegelaere W, Plendl J. Angiogenesis and Collagen Type IV Expression in Different Endothelial Cell Culture Systems. J Vet Med Ser C Anat Histol Embryol (2014) 43:103–15. doi: 10.1111/ahe.12052
243. Nie XC, Wang JP, Zhu W, Xu XY, Xing YN, Yu M, et al. COL4A3 expression correlates with pathogenesis, pathologic behaviors, and prognosis of gastric carcinomas. Hum Pathol (2013) 44:77–86. doi: 10.1016/j.humpath.2011.10.028
244. Cordenonsi M, Zanconato F, Azzolin L, Forcato M, Rosato A, Frasson C, et al. The Hippo Transducer TAZ Confers Cancer Stem Cell-Related Traits on Breast Cancer Cells. Cell (2011) 147:759–72. doi: 10.1016/j.cell.2011.09.048
245. Hiemer SE, Zhang L, Kartha VK, Packer TS, Almershed M, Noonan V, et al. A YAP/TAZ-Regulated Molecular Signature is Associated with Oral Squamous Cell Carcinoma. Mol Cancer Res (2016) 13:617–38. doi: 10.1158/1541-7786.MCR-14-0580.A
246. Li J, Ye L, Owen S, Weeks Ping H, Zhang Z, Jiang GW. Emerging role of CCN family proteins in tumorigenesis and cancer metastasis (Review). Int J Mol Med (2015) 36:1451–63. doi: 10.3892/ijmm.2015.2390
247. Lipson KE, Wong C, Teng Y, Spong S. CTGF is a central mediator of tissue remodeling and fibrosis and its inhibition can reverse the process of fibrosis. Fibrogenesis Tissue Repair (2012) 5:S24–4. doi: 10.1186/1755-1536-5-S1-S24
248. Zhu X, Zhong J, Zhao Z, Sheng J, Wang J, Liu J, et al. Epithelial derived CTGF promotes breast tumor progression via inducing EMT and collagen I fibers deposition. Oncotarget (2015) 6:25320–38. doi: 10.18632/oncotarget.4659
249. Yang F, Tuxhorn JA, Ressler SJ, McAlhany SJ, Dang TD, Rowley DR. Stromal expression of connective tissue growth factor promotes angiogenesis and prostate cancer tumorigenesis. Cancer Res (2005) 65:8887–95. doi: 10.1158/0008-5472.CAN-05-1702
250. Sakai N, Nakamura M, Lipson KE, Miyake T, Kamikawa Y, Sagara A, et al. Inhibition of CTGF ameliorates peritoneal fibrosis through suppression of fibroblast and myofibroblast accumulation and angiogenesis. Sci Rep (2017) 7:1–13. doi: 10.1038/s41598-017-05624-2
251. Wang L-H, Tsai H-C, Cheng Y-C, Lin C-Y, Huang Y-L, Tsai C-H, et al. CTGF promotes osteosarcoma angiogenesis by regulating miR-543/angiopoietin 2 signaling. Cancer Lett (2017) 391:28–37. doi: 10.1016/j.canlet.2017.01.013
252. Fataccioli V, Abergel V, Wingertsmann L, Neuville P, Spitz E, Adnot S, et al. Stimulation of Angiogenesis by Cyr61 gene: A New Therapeutic Candidate. Hum Gene Ther (2002) 1470:1461–70. doi: 10.1089/10430340260185094
253. Mo F-E, Muntean AG, Chen C-C, Stolz DB, Watkins SC, Lau LF. CYR61 (CCN1) Is Essential for Placental Development and Vascular Integrity. Mol Cell Biol (2002) 22:8709–20. doi: 10.1128/MCB.22.24.8709-8720.2002
254. Chintala H, Krupska I, Yan L, Lau L, Grant M, Chaqour B. The matricellular protein CCN1 controls retinal angiogenesis by targeting VEGF, Src homology 2 domain phosphatase-1 and Notch signaling. Development (2015) 142:2364–74. doi: 10.1242/dev.121913
255. Tsai M-S, Bogart DF, Castañeda JM, Li P, Lupu R. Cyr61 promotes breast tumorigenesis and cancer progression. Oncogene (2002) 21:8178–85. doi: 10.1038/sj.onc.1205682
256. Babic AM, Kireeva ML, Kolesnikova TV, Lau LF. CYR61, a product of a growth factor-inducible immediate early gene, promotes angiogenesis and tumor growth. Proc Natl Acad Sci (1998) 95:6355–60. doi: 10.1073/pnas.95.11.6355
257. Xie D, Miller CW, O’Kelly J, Nakachi K, Sakashita A, Said JW, et al. Breast Cancer: Cyr61 is Overexpressed, Estrogen- Inducible, and associated with more Advanced Disease. J Biol Chem (2001) 276:14187–94. doi: 10.1074/jbc.M009755200
258. Jiménez AP, Traum A, Boettger T, Hackstein H, Richter AM, Dammann RH. The tumor suppressor RASSF1A induces the YAP1 target gene ANKRD1 that is epigenetically inactivated in human cancers and inhibits tumor growth. Oncotarget (2017) 8:88437–52. doi: 10.18632/oncotarget.18177
259. Samaras SE, Almodóvar-García K, Wu N, Yu F, Davidson JM. Global deletion of ankrd1 results in a wound-healing phenotype associated with dermal fibroblast dysfunction. Am J Pathol (2015) 185:96–109. doi: 10.1016/j.ajpath.2014.09.018
260. Shi Y, Reitmaier B, Regenbogen J, Slowey RM, Opalenik SR, Wolf E, et al. CARP, a cardiac ankyrin repeat protein, is up-regulated during wound healing and induces angiogenesis in experimental granulation tissue. Am J Pathol (2005) 166:303–12. doi: 10.1016/S0002-9440(10)62254-7
261. Héroult M, Schaffner F, Augustin HG. Eph receptor and ephrin ligand-mediated interactions during angiogenesis and tumor progression. Exp Cell Res (2006) 312:642–50. doi: 10.1016/j.yexcr.2005.10.028
262. Lisabeth EM, Falivelli G, Pasquale EB. Eph receptor signaling and ephrins. Cold Spring Harb Perspect Biol (2013) 5:a009159. doi: 10.1101/cshperspect.a009159
263. Ogawa K, Pasqualini R, Lindberg RA, Kain R, Freeman AL, Pasquale EB. The ephrin-A1 ligand and its receptor, EphA2, are expressed during tumor neovascularization. Oncogene (2000) 19:6043–52. doi: 10.1038/sj.onc.1204004
264. Shao Z, Zhang WF, Chen XM, Shang ZJ. Expression of EphA2 and VEGF in squamous cell carcinoma of the tongue: Correlation with the angiogenesis and clinical outcome. Oral Oncol (2008) 44:1110–7. doi: 10.1016/j.oraloncology.2008.01.018
265. Dobrzanski P, Hunter K, Jones-bolin S, Chang H, Robinson C, Pritchard S, et al. Antiangiogenic and Antitumor Efficacy of EphA2 Receptor Antagonist. Cancer Res (2004) 64:910–9. doi: 10.1158/0008-5472.can-3430-2
266. Brantley-Sieders DM, Caughron J, Hicks D, Pozzi A, Ruiz JC, Chen J. EphA2 receptor tyrosine kinase regulates endothelial cell migration and vascular assembly through phosphoinositide 3-kinase-mediated Rac1 GTPase activation. J Cell Sci (2004) 117:2037–49. doi: 10.1242/jcs.01061
267. Noren NK, Lu M, Freeman AL, Koolpe M, Pasquale EB. Interplay between EphB4 on tumor cells and vascular ephrin-B2 regulates tumor growth. Proc Natl Acad Sci (2004) 101:5583–8. doi: 10.1073/pnas.0401381101
268. Uhl C, Markel M, Broggini T, Nieminen M, Kremenetskaia I, Vajkoczy P, et al. EphB4 mediates resistance to antiangiogenic therapy in experimental glioma. Angiogenesis (2018) 21:873–81. doi: 10.1007/s10456-018-9633-6
269. Kertesz N, Krasnoperov V, Reddy R, Leshanski L, Kumar SR, Zozulya S, et al. The soluble extracellular domain of EphB4 (sEphB4) antagonizes EphB4-EphrinB2 interaction, modulates angiogenesis, and inhibits tumor growth. Blood (2006) 107:2330–8. doi: 10.1182/blood-2005-04-1655
270. Erber R, Eichelsbacher U, Powajbo V, Korn T, Djonov V, Lin J, et al. EphB4 controls blood vascular morphogenesis during postnatal angiogenesis. EMBO J (2006) 25:628–41. doi: 10.1038/sj.emboj.7600949
271. Thurston G. Role of Angiopoietins and Tie receptor tyrosine kinases in angiogenesis and lymphangiogenesis. Cell Tissue Res (2003) 314:61–8. doi: 10.1007/s00441-003-0749-6
272. Thomas M, Augustin HG. The role of the Angiopoietins in vascular morphogenesis. Angiogenesis (2009) 12:125. doi: 10.1007/s10456-009-9147-3
273. Fiedler U, Reiss Y, Scharpfenecker M, Grunow V, Koidl S, Thurston G, et al. Angiopoietin-2 sensitizes endothelial cells to TNF-α and has a crucial role in the induction of inflammation. Nat Med (2006) 12:235–9. doi: 10.1038/nm1351
274. Scharpfenecker M, Fiedler U, Reiss Y, Augustin HG. The Tie-2 ligand Angiopoietin-2 destabilizes quiescent endothelium through an internal autocrine loop mechanism. J Cell Sci (2005) 118:771–80. doi: 10.1242/jcs.01653
275. Augustin HG, Young Koh G, Thurston G, Alitalo K. Control of vascular morphogenesis and homeostasis through the angiopoietin–Tie system. Nat Rev Mol Cell Biol (2009) 10:165–77. doi: 10.1038/nrm2639
276. Oh H, Takagi H, Suzuma K, Otani A, Matsumura M, Honda Y. Hypoxia and Vascular Endothelial Growth Factor Selectively Up-regulate Angiopoietin-2 in Bovine Microvascular Endothelial Cells. J Biol Chem (1999) 274:15732–9. doi: 10.1074/jbc.274.22.15732
277. Maisonpierre PC, Suri C, Jones PF, Bartunkova S, Wiegand SJ, Radziejewski C, et al. Angiopoietin-2, a Natural Antagonist for Tie2 That Disrupts in vivo Angiogenesis. Sci (80- ) (1997) 277:55–60. doi: 10.1126/science.277.5322.55
278. Lobov IB, Brooks PC, Lang RA. Angiopoietin-2 displays VEGF-dependent modulation of capillary structure and endothelial cell survival in vivo. Proc Natl Acad Sci (2002) 99:11205–10. doi: 10.1073/pnas.172161899
279. Oshima Y, Oshima S, Nambu H, Kachi S, Takahashi K, Umeda N, et al. Different effects of angiopoietin-2 in different vascular beds in the eye: new vessels are most sensitive. FASEB J (2005) 19:963–5. doi: 10.1096/fj.04-2209fje
280. Oshima Y, Deering T, Oshima S, Nambu H, Reddy PS, Kaleko M, et al. Angiopoietin-2 enhances retinal vessel sensitivity to vascular endothelial growth factor. J Cell Physiol (2004) 199:412–7. doi: 10.1002/jcp.10442
281. Daly C, Pasnikowski E, Burova E, Wong V, Aldrich TH, Griffiths J, et al. Angiopoietin-2 functions as an autocrine protective factor in stressed endothelial cells. Proc Natl Acad Sci (2006) 103:15491–6. doi: 10.1073/pnas.0607538103
282. Shim WSN, Ho IAW, Wong PEH. Angiopoietin : A TIE ( d ) Balance in Tumor Angiogenesis. Mol Cancer Res (2007) 5:655–66. doi: 10.1158/1541-7786.MCR-07-0072
283. Zadeh G, Koushan K, Baoping Q, Shannon P, Guha A. Role of angiopoietin-2 in regulating growth and vascularity of astrocytomas. J Oncol (2010) 2010:659231. doi: 10.1155/2010/659231
284. Hawighorst T, Skobe M, Streit M, Hong Y-K, Velasco P, Brown LF, et al. Activation of the Tie2 Receptor by Angiopoietin-1 Enhances Tumor Vessel Maturation and Impairs Squamous Cell Carcinoma Growth. Am J Pathol (2002) 160:1381–92. doi: 10.1016/S0002-9440(10)62565-5
285. Mazzieri R, Pucci F, Moi D, Zonari E, Ranghetti A, Berti A, et al. Targeting the ANG2/TIE2 axis inhibits tumor growth and metastasis by impairing angiogenesis and disabling rebounds of proangiogenic myeloid cells. Cancer Cell (2011) 19:512–26. doi: 10.1016/j.ccr.2011.02.005
286. Felcht M, Luck R, Schering A, Seidel P, Srivastava K, Hu J, et al. Angiopoietin-2 differentially regulates angiogenesis through TIE2 and integrin signaling. J Clin Invest (2012) 122:1991–2005. doi: 10.1172/JCI58832
287. Park J-S, Kim I-K, Han S, Park I, Kim C, Bae J, et al. Normalization of Tumor Vessels by Tie2 Activation and Ang2 Inhibition Enhances Drug Delivery and Produces a Favorable Tumor Microenvironment. Cancer Cell (2016) 30:953–67. doi: 10.1016/j.ccell.2016.10.018
288. Peterson TE, Kirkpatrick ND, Huang Y, Farrar CT, Marijt KA, Kloepper J, et al. Dual inhibition of Ang-2 and VEGF receptors normalizes tumor vasculature and prolongs survival in glioblastoma by altering macrophages. Proc Natl Acad Sci (2016) 113:4470–5. doi: 10.1073/pnas.1525349113
289. Jászai J, Schmidt M. Trends and Challenges in Tumor Anti-Angiogenic Therapies. Cells (2019) 8:1102. doi: 10.3390/cells8091102
290. Al-Abd AM, Alamoudi AJ, Abdel-Naim AB, Neamatallah TA, Ashour OM. Anti-angiogenic agents for the treatment of solid tumors: Potential pathways, therapy and current strategies – A review. J Adv Res (2017) 8:591–605. doi: 10.1016/j.jare.2017.06.006
291. Durkin ME, Yuan B-Z, Zhou X, Zimonjic DB, Lowy DR, Thorgeirsson SS, et al. DLC-1:a Rho GTPase-activating protein and tumour suppressor. J Cell Mol Med (2007) 11:1185–207. doi: 10.1111/j.1582-4934.2007.00098.x
292. Liao Y-C, Lo SH. Deleted in liver cancer-1 (DLC-1): a tumor suppressor not just for liver. Int J Biochem Cell Biol (2008) 40:843–7. doi: 10.1016/j.biocel.2007.04.008
293. Li G, Du X, Vass WC, Papageorge AG, Lowy DR, Qian X. Full activity of the deleted in liver cancer 1 (DLC1) tumor suppressor depends on an LD-like motif that binds talin and focal adhesion kinase (FAK). Proc Natl Acad Sci (2011) 108:17129–34. doi: 10.1073/pnas.1112122108
294. Haining AWM, Rahikainen R, Cortes E, Lachowski D, Rice A, von Essen M, et al. Mechanotransduction in talin through the interaction of the R8 domain with DLC1. PloS Biol (2018) 16:1–20. doi: 10.1371/journal.pbio.2005599
295. Durkin ME, Avner MR, Huh C-G, Yuan B-Z, Thorgeirsson SS, Popescu NC. DLC-1, a Rho GTPase-activating protein with tumor suppressor function, is essential for embryonic development. FEBS Lett (2005) 579:1191–6. doi: 10.1016/j.febslet.2004.12.090
296. Schimmel L, van der Stoel M, Rianna C, van Stalborch AM, de Ligt A, Hoogenboezem M, et al. Stiffness-Induced Endothelial DLC-1 Expression Forces Leukocyte Spreading through Stabilization of the ICAM-1 Adhesome. Cell Rep (2018) 24:3115–24. doi: 10.1016/j.celrep.2018.08.045
297. Ritchey L, Ha T, Otsuka A, Kabashima K, Wang D, Wang Y, et al. DLC1 deficiency and YAP signaling drive endothelial cell contact inhibition of growth and tumorigenesis. Oncogene (2019) 38:7046–59. doi: 10.1038/s41388-019-0944-x
298. Shih Y-P, Liao Y-C, Lin Y, Lo SH. DLC1 negatively regulates angiogenesis in a paracrine fashion. Cancer Res (2010) 70:8270–5. doi: 10.1158/0008-5472.CAN-10-1174
299. Le Y, Zhou Y, Iribarren P, Wang JM. Chemokines and Chemokine Receptors : Their Manifold Roles in Homeostasis and Disease. Cell Mol Immunol (2004) 1:95–104.
300. Bandapalli OR, Ehrmann F, Ehemann V, Gaida M, Macher-goeppinger S, Wente M, et al. Cytokine Down-regulation of CXCL1 inhibits tumor growth in colorectal liver metastasis. Cytokine (2012) 57:46–53. doi: 10.1016/j.cyto.2011.10.019
301. Dhawan P, Richmond A. Role of CXCL1 in tumorigenesis of melanoma. J Leukoc Biol (2002) 72:9–18.
302. Wang D, Wang H, Brown J, Daikoku T, Ning W, Shi Q, et al. CXCL1 induced by prostaglandin E 2 promotes angiogenesis in colorectal cancer. J Exp Med (2006) 203:941–51. doi: 10.1084/jem.20052124
303. Wang Y, Liu J, Jiang Q, Deng J, Xu F, Chen X, et al. Human Adipose-Derived Mesenchymal Stem Cell- Secreted CXCL1 and CXCL8 Facilitate Breast Tumor Growth by Promoting Angiogenesis. Stem Cells (2017) 35:2060–70. doi: 10.1002/stem.2643
304. Miyake M, Goodison S, Urquidi V, Giacoia EG, Rosser CJ. Expression of CXCL1 in human endothelial cells induces angiogenesis through the CXCR2 receptor and the ERK1 / 2 and EGF pathways. Lab Invest (2013) 93:768–78. doi: 10.1038/labinvest.2013.71
305. Wei Z, Xia G, Wu Y, Chen W, Xiang Z, Schwarz RE, et al. CXCL1 promotes tumor growth through VEGF pathway activation and is associated with inferior survival in gastric cancer. Cancer Lett (2015) 359:335–43. doi: 10.1016/j.canlet.2015.01.033
306. Scapini P, Morini M, Tecchio C, Di Carlo E, Tanghetti E, Albini A, et al. CXCL1/Macrophage Inflammatory Protein-2-Induced Angiogenesis In Vivo Is Mediated by Neutrophil-Derived Vascular Endothelial Growth Factor-A. J Immunol (2004) 172:5034–40. doi: 10.4049/jimmunol.172.8.5034
307. Cai L, Xu S, Piao C, Qiu S, Li H, Du J. Adiponectin Induces CXCL1 Secretion From Cancer Cells and Promotes Tumor Angiogenesis by Inducing Stromal Fibroblast Senescence. Mol Carcinog (2016) 55:1796–806. doi: 10.1002/mc.22428
308. Goveia J, Rohlenova K, Taverna F, Treps L, Conradi L-C, Pircher A, et al. An Integrated Gene Expression Landscape Profiling Approach to Identify Lung Tumor Endothelial Cell Heterogeneity and Angiogenic Candidates. Cancer Cell (2020) 37:21–36. doi: 10.1016/j.ccell.2019.12.001
309. Zheng Y, Ming P, Zhu C, Si Y, Xu S, Chen A, et al. Hepatitis B virus X protein–induced SH2 domain– containing 5 (SH2D5) expression promotes hepatoma cell growth via an SH2D5–transketolase interaction. J Biol Chem (2019) 294:4815–27. doi: 10.1074/jbc.RA118.005739
310. Chen Z, Zhong CH. STAT3: A critical transcription activator in angiogenesis. Med Res Rev (2008) 28:185–200. doi: 10.1002/med.20101
311. Niu G, Wright KL, Huang M, Song L, Haura E, Turkson J, et al. Constitutive Stat3 activity up-regulates VEGF expression and tumor angiogenesis. Oncogene (2002) 21:2000–8. doi: 10.1038/sj.onc.1205260
312. Fan X, Shan X, Jiang S, Wang S, Zhang F, Tian Q, et al. YAP promotes endothelial barrier repair by repressing STAT3/VEGF signaling. Life Sci (2020) 256:117884. doi: 10.1016/j.lfs.2020.117884
313. Kidoya H, Takakura N. Biology of the apelin-APJ axis in vascular formation. J Biochem (2012) 152:125–31. doi: 10.1093/jb/mvs071
314. Kälin RE, Kretz MP, Meyer AM, Kispert A, Heppner FL, Brändli AW. Paracrine and autocrine mechanisms of apelin signaling govern embryonic and tumor angiogenesis. Dev Biol (2007) 305:599–614. doi: 10.1016/j.ydbio.2007.03.004
315. Muto J, Shirabe K, Yoshizumi T, Ikegami T, Aishima S, Ishigami K, et al. The Apelin–APJ System Induces Tumor Arteriogenesis in Hepatocellular Carcinoma. Anticancer Res (2014) 34:5313–20. doi: 10.1111/liv.12459
316. Sorli SC, Le Gonidec S, Knibiehler B, Audigier Y. Apelin is a potent activator of tumour neoangiogenesis. Oncogene (2007) 26:7692–9. doi: 10.1038/sj.onc.1210573
317. Berta J, Kenessey I, Dobos J, Tovari J, Klepetko W, Jan Ankersmit H, et al. Apelin expression in human non-small cell lung cancer: Role in angiogenesis and prognosis. J Thorac Oncol (2010) 5:1120–9. doi: 10.1097/JTO.0b013e3181e2c1ff
318. Berta J, Hoda MA, Laszlo V, Rozsas A, Garay T, Torok S, et al. Apelin promotes lymphangiogenesis and lymph node metastasis. Oncotarget (2014) 5:4426–37. doi: 10.18632/oncotarget.2032
319. Rayalam S A, Della-Fera M, Kasser T, Warren W, Baile C A. Emerging Role of Apelin as a Therapeutic Target in Cancer: A Patent Review. Recent Pat Anticancer Drug Discovery (2011) 6:367–72. doi: 10.2174/157489211796957856
320. Gill MK, Christova T, Zhang YY, Gregorieff A, Zhang L, Narimatsu M, et al. A feed forward loop enforces YAP/TAZ signaling during tumorigenesis. Nat Commun (2018) 9:3510. doi: 10.1038/s41467-018-05939-2
321. Lv M, Shen Y, Yang J, Li S, Wang B, Chen Z, et al. Angiomotin Family Members: Oncogenes or Tumor Suppressors? Int J Biol Sci (2017) 13:772–81. doi: 10.7150/ijbs.19603
322. Yi C, Shen Z, Stemmer-Rachamimov A, Dawany N, Troutman S, Showe LC, et al. The p130 isoform of angiomotin is required for yap-mediated hepatic epithelial cell proliferation and tumorigenesis. Sci Signal (2013) 6:ra77. doi: 10.1126/scisignal.2004060
323. Rosenbluh J, Nijhawan D, Cox AG, Li X, Neal JT, Schafer EJ, et al. β-Catenin-driven cancers require a YAP1 transcriptional complex for survival and tumorigenesis. Cell (2012) 151:1457–73. doi: 10.1016/j.cell.2012.11.026
324. Azzolin L, Panciera T, Soligo S, Enzo E, Bicciato S, Dupont S, et al. YAP/TAZ incorporation in the β-catenin destruction complex orchestrates the Wnt response. Cell (2014) 158:157–70. doi: 10.1016/j.cell.2014.06.013
325. Azzolin L, Zanconato F, Bresolin S, Forcato M, Basso G, Bicciato S, et al. Role of TAZ as mediator of wnt signaling. Cell (2012) 151:1443–56. doi: 10.1016/j.cell.2012.11.027
326. Park HW, Kim YC, Yu B, Moroishi T, Mo JS, Plouffe SW, et al. Alternative Wnt Signaling Activates YAP/TAZ. Cell (2015) 162:780–94. doi: 10.1016/j.cell.2015.07.013
327. Miyamura N, Nishina H. YAP regulates liver size and function. Cell Cycle (2018) 17:267–8. doi: 10.1080/15384101.2017.1407390
328. Heo S-H, Choi Y-J, Ryoo H-M, Cho J-Y. Expression profiling of ETS and MMP factors in VEGF-activated endothelial cells: Role of MMP-10 in VEGF-induced angiogenesis. J Cell Physiol (2010) 224:734–42. doi: 10.1002/jcp.22175
329. Nukuda A, Sasaki C, Ishihara S, Mizutani T, Nakamura K, Ayabe T, et al. Stiff substrates increase YAP-signaling-mediated matrix metalloproteinase-7 expression. Oncogenesis (2015) 4:1–11. doi: 10.1038/oncsis.2015.24
330. Tang Y, Rowe RG, Botvinick EL, Kurup A, Putnam AJ, Seiki M, et al. MT1-MMP-Dependent Control of Skeletal Stem Cell Commitment via a β1-Integrin/YAP/TAZ Signaling Axis. Dev Cell (2013) 25:402–16. doi: 10.1016/j.devcel.2013.04.011
331. Mazor R, Alsaigh T, Shaked H, Altshuler AE, Pocock ES, Kistler EB, et al. Matrix metalloproteinase-1-mediated up-regulation of vascular endothelial growth factor-2 in endothelial cells. J Biol Chem (2013) 288:598–607. doi: 10.1074/jbc.M112.417451
332. Quintero-Fabián S, Arreola R, Becerril-Villanueva E, Torres-Romero JC, Arana-Argáez V, Lara-Riegos J, et al. Role of Matrix Metalloproteinases in Angiogenesis and Cancer. Front Oncol (2019) 9:1370. doi: 10.3389/fonc.2019.01370
333. Souilhol C, Harmsen MC, Evans PC, Krenning G. Endothelial–mesenchymal transition in atherosclerosis. Cardiovasc Res (2018) 114:565–77. doi: 10.1093/cvr/cvx253
334. Numasaki M, Fukushi JI, Ono M, Narula SK, Zavodny PJ, Kudo T, et al. Interleukin-17 promotes angiogenesis and tumor growth. Blood (2003) 101:2620–7. doi: 10.1182/blood-2002-05-1461
335. Brat DJ, Bellail AC, Van Meir EG. The role of interleukin-8 and its receptors in gliomagenesis and tumoral angiogenesis. Neuro Oncol (2005) 7:122–33. doi: 10.1215/s1152851704001061
336. Gopinathan G, Milagre C, Pearce OMT, Reynolds LE, Hodivala-Dilke K, Leinster DA, et al. Interleukin-6 Stimulates Defective Angiogenesis. Cancer Res (2015) 75:3098–107. doi: 10.1158/0008-5472.CAN-15-1227
337. Romagnani P, Lasagni L, Annunziato F, Serio M, Romagnani S. CXC chemokines: the regulatory link between inflammation and angiogenesis. Trends Immunol (2004) 25:201–9. doi: 10.1016/j.it.2004.02.006
338. Wang S, Zhou L, Ling L, Meng X, Chu F, Zhang S, et al. The Crosstalk Between Hippo-YAP Pathway and Innate Immunity. Front Immunol (2020) 11:323. doi: 10.3389/fimmu.2020.00323
339. Kyriakis JM. Activation of the AP-1 transcription factor by inflammatory cytokines of the TNF family. Gene Expr J Liver Res (1999) 7:217–31.
340. Gashaw I, Stiller S, Böing C, Kimmig R, Winterhager E. Premenstrual regulation of the pro-angiogenic factor CYR61 in human endometrium. Endocrinology (2008) 149:2261–9. doi: 10.1210/en.2007-1568
341. Lee S, Ahad A, Luu M, Moon S, Caesar J, Cardoso WV, et al. CCN1–Yes-Associated Protein Feedback Loop Regulates Physiological and Pathological Angiogenesis. Mol Cell Biol (2019) 39:e00107–19. doi: 10.1128/mcb.00107-19
342. Choi H-J, Kwon Y-G. Roles of YAP in mediating endothelial cell junctional stability and vascular remodeling. BMB Rep (2015) 48:429–30. doi: 10.5483/bmbrep.2015.48.8.146
343. Nguyen CDK, Yi C. YAP/TAZ Signaling and Resistance to Cancer Therapy. Trends Cancer (2019) 5:283–96. doi: 10.1016/j.trecan.2019.02.010
344. Pobbati AV, Hong W. A combat with the YAP/TAZ-TEAD oncoproteins for cancer therapy. Theranostics (2020) 10:3622–35. doi: 10.7150/thno.40889
345. Santucci M, Vignudelli T, Ferrari S, Mor M, Scalvini L, Bolognesi ML, et al. The Hippo Pathway and YAP/TAZ–TEAD Protein–Protein Interaction as Targets for Regenerative Medicine and Cancer Treatment. J Med Chem (2015) 58:4857–73. doi: 10.1021/jm501615v
346. Kim M, Kim T, Randy JL, Lim D-S. Transcriptional Co-repressor Function of the Hippo Pathway Transducers YAP and TAZ. CellReports (2015) 11:270–82. doi: 10.1016/j.celrep.2015.03.015
347. Guan Z-B, Cao Y-S, Li Y, Tong W-N, Zhuo A-S. Knockdown of lncRNA GHET1 suppresses cell proliferation, invasion and LATS1/YAP pathway in non small cell lung cancer. Cancer Biomarkers (2018) 21:557–63. doi: 10.3233/CBM-170431
348. Wang Y, Cui M, Sun B, Liu FB, Zhang XD, Ye LH. MiR-506 suppresses proliferation of hepatoma cells through targeting YAP mRNA 3′UTR. Acta Pharmacol Sin (2014) 35:1207–14. doi: 10.1038/aps.2014.59
349. Liu P, Zhang H, Liang X, Ma H, Luan F, Wang B, et al. HBV preS2 promotes the expression of TAZ via miRNA-338-3p to enhance the tumorigenesis of hepatocellular carcinoma. Oncotarget (2015) 6:29048–59. doi: 10.18632/oncotarget.4804
350. Zhang M, Zhao Y, Zhang Y, Wang D, Gu S, Feng W, et al. LncRNA UCA1 promotes migration and invasion in pancreatic cancer cells via the Hippo pathway. Biochim Biophys Acta - Mol Basis Dis (2018) 1864:1770–82. doi: 10.1016/j.bbadis.2018.03.005
351. Qu L, Wu Z, Li Y, Xu Z, Liu B, Liu F, et al. A feed-forward loop between lncARSR and YAP activity promotes expansion of renal tumour-initiating cells. Nat Commun (2016) 7:1–14. doi: 10.1038/ncomms12692
352. Ma J, Huang K, Ma Y, Zhou M, Fan S. The TAZ-miR-224-SMAD4 axis promotes tumorigenesis in osteosarcoma. Cell Death Dis (2017) 8:1–16. doi: 10.1038/cddis.2016.468
353. Shen S, Huang K, Wu Y, Ma Y, Wang J, Qin F, et al. A miR-135b-TAZ positive feedback loop promotes epithelial–mesenchymal transition (EMT) and tumorigenesis in osteosarcoma. Cancer Lett (2017) 407:32–44. doi: 10.1016/j.canlet.2017.08.005
354. Yu W, Qiao Y, Tang X, Ma L, Wang Y, Zhang X, et al. Tumor suppressor long non-coding RNA, MT1DP is negatively regulated by YAP and Runx2 to inhibit FoxA1 in liver cancer cells. Cell Signal (2014) 26:2961–8. doi: 10.1016/j.cellsig.2014.09.011
Keywords: yes-associated protein (YAP), TAZ, tumor vasculature, endothelium, mechanotransduction, cancer, Angiogenic therapy, tumor angiogenesis
Citation: Hooglugt A, van der Stoel MM, Boon RA and Huveneers S (2021) Endothelial YAP/TAZ Signaling in Angiogenesis and Tumor Vasculature. Front. Oncol. 10:612802. doi: 10.3389/fonc.2020.612802
Received: 30 September 2020; Accepted: 07 December 2020;
Published: 04 February 2021.
Edited by:
Lucas Treps, VIB KU Leuven Center for Cancer Biology, BelgiumReviewed by:
Carmen Ruiz De Almodovar, Heidelberg University, GermanyJinlong He, Tianjin Medical University, China
Copyright © 2021 Hooglugt, van der Stoel, Boon and Huveneers. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stephan Huveneers, cy5odXZlbmVlcnNAYW1zdGVyZGFtdW1jLm5s
†These authors share first authorship