 Kai Yin
Kai Yin Xueli Xia
Xueli Xia Ke Rui
Ke Rui Tingting Wang4*
Tingting Wang4* Shengjun Wang
Shengjun Wang
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 15 December 2020
Sec. Cancer Immunity and Immunotherapy
Volume 10 - 2020 | https://doi.org/10.3389/fonc.2020.610104
This article is part of the Research Topic Manipulating the Immunological Tumor Microenvironment View all 57 articles
Colorectal cancer (CRC) remains a devastating human malignancy with poor prognosis. Of the various factors, immune evasion mechanisms play pivotal roles in CRC progression and impede the effects of cancer therapy. Myeloid-derived suppressor cells (MDSCs) constitute an immature population of myeloid cells that are typical during tumor progression. These cells have the ability to induce strong immunosuppressive effects within the tumor microenvironment (TME) and promote CRC development. Indeed, MDSCs have been shown to accumulate in both tumor-bearing mice and CRC patients, and may therefore become an obstacle for cancer immunotherapy. Consequently, numerous studies have focused on the characterization of MDSCs and their immunosuppressive capacity, as well as developing novel approaches to suppress MDSCs function with different approaches. Current therapeutic strategies that target MDSCs in CRC include inhibition of their recruitment and alteration of their function, alone or in combination with other therapies including chemotherapy, radiotherapy and immunotherapy. Herein, we summarize the recent roles and mechanisms of MDSCs in CRC progression. In addition, a brief review of MDSC-targeting approaches for potential CRC therapy is presented.
Colorectal cancer (CRC) remains the third most common cancer and the third leading cause of cancer-related deaths in males and females (1). Despite improvements in systemic treatments for advanced CRC in recent years, only 12–14% of patients with metastatic CRC survive for five years (2). Moreover, patients with advanced CRC develop resistance to chemotherapy, radiotherapy, immunotherapy and targeted drug therapy, which results in increasing challenges in treating CRC. Recently, different types of immune cells such as myeloid-derived suppressor cells (MDSCs), dendritic cells (DCs), tumor-associated macrophages (TAMs), natural killer (NK) cells, and regulatory T cells (Tregs) were shown to impact CRC progression (3, 4).
CRC patients’ responses to chemotherapy, radiation therapy, targeted drugs therapy and immunotherapy are affected by the tumor immune microenvironment (5, 6). Growing evidence has demonstrated that MDSCs accumulate and expand in the peripheral blood and tumor tissues, where they regulate host anti-tumor immune responses (7, 8). Once the MDSC population is expanded and activated in the immune system, it executes its numerous functions in tumor progression. MDSCs not only suppress anti-tumor immunity but also impede the efficacy of therapeutic agents for cancer treatment (9).
Immunosuppression is an important hallmark of most cancer growth and progression (10). In recent years, accumulating data have indicated that MDSCs, as one of the main immunosuppressive cell populations, are pivotal for cancer development (11). MDSCs represent a heterogeneous population of immature myeloid cells (IMCs) that fail to complete their differentiation into macrophages, DCs, or granulocytes (12). MDSCs consist of two large groups of cells: granulocytic or polymorphonuclear MDSCs (PMN-MDSCs) and monocytic MDSCs (M-MDSCs) (13). In general, the immunosuppressive function of MDSCs is regulated by multiple signaling pathways as well as interactions with several immune cell populations and mediators, which directly or indirectly suppress anti-tumor immunity and support cancer progression (7, 9). An increasing number of studies have focused on MDSCs, which are involved in regulation of the immune response in many types of cancer, but are poorly understood in CRC. It has been shown that different populations of MDSCs are observed in the peripheral blood and tumor tissue of CRC patients (14). A positive relationship between MDSCs and CRC progression including growth, metastasis, invasion, and angiogenesis has also been reported (15). Therapeutic agents targeting MDSCs have been proven to promote anti-tumor immunity and enhance the effects of immunotherapy against CRC. In this review, we discuss developments on the role of MDSCs in CRC: (1) MDSCs and their functional correlation with cancer, (2) MDSCs-mediated signaling pathways in CRC progression, and (3) MDSCs-targeting approaches for potential CRC treatment.
Under normal circumstances, IMCs do not have immunosuppressive functions and are believed to be constitutively present in healthy individuals. The generation of IMCs occurs in the bone marrow and is regulated by growth factors including granulocyte colony-stimulating factor (G-CSF), granulocyte macrophage colony-stimulating factor (GM-CSF), and macrophage colony-stimulating factor (M-CSF) (16, 17). During this process, IMCs migrate to the blood and various peripheral organs, where they differentiate into myeloid cells such as macrophages, neutrophils, and DCs. However, in pathological conditions including cancers, chronic infections and autoimmune diseases, IMC differentiation is impaired leading to an accumulation of MDSCs (18–20).
MDSCs are IMCs that expand during the growth and metastasis of malignant tumors and in inflammatory conditions. Their heterogeneity is tumor-dependent, and their phenotype and functions change with cancer progression (21). Recently, neutrophils were distinguished from MDSCs by the expression of the lectin-type oxidized LDL receptor 1 (LOX-1). LOX-1+ neutrophils have been shown to suppress T cells proliferation (22). These cells, also called tumor-associated neutrophils (TANs), were found in the tumor microenvironment (TME) and promote cancer cell migration and invasion (23). However, the main difference between MDSCs, neutrophils, and monocytes is their functions, with MDSCs having the potential to suppress immune activity.
There are two types of macrophages, namely, M1-like macrophage and M2-like macrophage. The macrophages found in the TME are known as TAMs and are predominantly M2 macrophages (24). TAMs are abundant in the microenvironment of CRC and are strong promoters of angiogenesis and lymphogenesis, thus contributing to tumor progression (25). Interferon gamma (IFN-γ), LPS, and GM-CSF induce polarization of M1 TAMs from monocytes, which are involved in antitumor immunity (26). In contrast, CSF-1, interleukin (IL)-4, IL-10, transforming growth factor beta (TGF)-β, and IL-13 contribute to M2 TAM polarization (27). M2 macrophages suppress cytotoxic T cell activities and attract Tregs, which promote tumor growth and immune escape (28). Furthermore, while M1 macrophages express CD64, suppressor of cytokine signaling 1 (SOCS1), indoleamine 2,3-dioxygenase (IDO) and chemokine (C-X-C motif) ligand 1 (CXCL1), M2 macrophages express mannose receptor C-type 1 (MRC1), CD23, and chemokine ligand 22 (CCL22) (29). Macrophages are highly plastic, and under certain physiological and pathological conditions, M1 macrophages can repolarize into M2 macrophages, and vice versa (30). However, the molecular mechanisms that regulate the macrophage polarization remain poorly understood.
Although TAMs and MDSCs are distinct cell types, they are not clearly distinguishable and have several characteristics in common. In the TME, cytokines and chemokines from tumor cells can influence normal myelopoiesis and increase the differentiation of M-MDSCs into PMN-MDSCs (31). M-MDSCs and inflammatory monocytes migrate to the tumor site via the CCL2/CCR2 pathways and differentiated into TAMs in response to various factors secreted by tumor cells (32). In addition, infiltrating MDSCs can also differentiate into TAMs through a combination of Toll-like receptor (TLR) and cytokine signaling (33, 34). MDSCs are phenotypically distinct from neutrophils, macrophages and monocytes. They can be divided into two major types based on their cell surface markers. In mice, PMN-MDSCs are defined as CD11b+Gr-1+Ly6GhighLy6Clow cells, whereas M-MDSCs are defined as CD11b+Gr-1+Ly6GlowLy6Chigh cells. The frequencies of PMN-MDSCs and M-MDSC subsets differ between tumors and organs in tumor-bearing mice with PMN-MDSCs accounting for 70–80% of MDSCs and M-MDSCs representing 20–30% (35). In humans, PMN-MDSCs are HLA-DR-CD11b+CD14−CD33+ (CD15+ or CD66+) cells and M-MDSCs are HLA-DRlow/−CD11b+CD14+CD15− cells (36). Other cell surface molecules can also be used to identify other subsets of MDSCs, such as CD115, CD80, and CD124 (37, 38). Moreover, M-MDSCs express inducible nitric oxide synthase (iNOS) and generate nitric oxide (NO), while PMN-MDSCs produce reactive oxygen species (ROS) and arginase-1 (39).
In human peripheral blood, early-stage MDSCs (e-MDSCs), which are comprised of more immature progenitors than conventional MDSCs, are defined by Lin− (including CD3, CD14, CD15, CD19, CD56) HLA-DR-CD33+. While e-MDSCs markers for murine cells have yet to be determined (36, 40–42), a subset of e-MDSCs with the phenotype of CD11b+Gr-1−F4/80−MHC-II− has been described in IL-6 high-expressing 4T1 mice mammary carcinoma models (43). Human fibrocytic MDSCs (F-MDSCs), which can be differentiated from umbilical cord blood (UCB) precursors, have been identified as a new MDSCs subset with fibrocytic phenotypes and immunosuppressive functions. They are defined as CD11blowCD11clowCD33+IL-4Ra+ (44, 45). In addition, PMN-MDSCs and M-MDSCs are also phenotypically different from neutrophils. Compared to neutrophils, PMN-MDSCs have fewer granules and low expression of CD16 and CD62L (36, 41, 42, 46). The phenotypes and markers of MDSCs are shown in Table 1.

Table 1 Summary of commonly expressed markers in mice and human MDSCs.
Cytokines can facilitate the recruitment and expansion of MDSCs in the TME. Tumor-induced factors, including prostaglandin E2 (PGE2), IL-6, IL-10, IL-1β, and transforming growth factor beta (TGF)-β have been shown to result in the recruitment and activation of MDSCs in the TME in malignant tumors (47). For example, IL-1β and IL-6 can induce the accumulation and activation of MDSCs at tumor sites (48–50). IL-1β not only promotes the accumulation of MDSCs but also induces expression of other molecules that are necessary for the expansion of MDSCs such as vascular endothelial growth factor (VEGF), IL-6 and GM-CSF (51). Other cytokines, such as IL-10 and TGF-β have the ability to generate MDSCs populations, as well as mediate their suppressive functions (52, 53). Thus, MDSCs are able to produce TGF-β and create a feedback loop that sustains their antitumor immunity. Furthermore, IL-17, which is secreted by Th17 cells, is overexpressed in cancer cells and promote MDSCs translocation from the periphery to the tumor sites (54).
Chemokines also influence MDSCs expansion and activation. Some of them, such as chemokine ligand 2 (CCL2) can interact with its corresponding receptor C-C chemokine receptor type 2 (CCR2) to promote chemotaxis to areas of inflammation (55, 56). IL-8 is released by cancer cells and binds to G protein-coupled receptors C-X-C motif chemokine receptor 1 and 2 (CXCR1 and CXCR2) on MDSCs (57). Moreover, the CCL3/CCR5 axis has been reported to induce the maintenance of immunosuppressive myeloid cells in tumor areas (55).
Hypoxia is commonly found in the TME and is recognized as an important factor that mediates MDSCs expansion. It has been shown that hypoxia inducible factor 1 alpha (HIF-1α) can induce the expression of ectonucleoside triphosphate diphosphohydrolase 2 (ENTPD2), an ectoenzyme on MDSCs, resulting in MDSCs expansion in the TME (40). In addition, hypoxia can upregulate VEGF and functional molecule expression and lead to MDSCs accumulation in both mice and patients with lung cancer (58, 59). This process is mediated by the VEGF receptor, which is expressed on MDSCs (58).
Notably, one of the features of MDSCs in TME is the immune suppressive function. MDSCs suppress the activity of immune cells through multiple mechanisms, including the generation of reactive oxygen and nitrogen species (RONS), the degradation of L-arginine, the production of immunosuppressive cytokines such as IL-10 and TGF-β, the inhibition of T cells, and the induction of other immunosuppressive cells (41, 60). Firstly, MDSCs can regulate anti-tumor immune responses through the production of RONS including NO and ROS (61, 62). MDSCs require activation of signal transducer and activator of transcription 3 (STAT3) and increase nicotinamide adenine dinucleotide phosphate (NADPH) oxidase activity to produce ROS (13, 41). However, NADPH oxidase may also synthesize reactive nitrogen species (RNS) like NO by metabolizing L-arginine (63). ROS are also activated via the STAT3 transcription factor and are associated with the metabolism of L-arginine (52, 64). Taken together, these data suggest that production of ROS, NO, and RNS are dependent on L-arginine metabolism. Furthermore, these factors can suppress T cell populations, thus rendering them incapable of facilitating an anti-cancer response (65). In the TME, S100A8/A9 has been shown to activate the production of ROS in a STAT3-dependent manner. This leads to nitration of the T cell receptor-alpha-beta (TCRαβ) chain, resulting in T cells that lack the ability to interact with the peptide antigen bound to the major histocompatibility complex class II (MHC-II) and are therefore unable to initiate an anti-cancer response (51). Similarly, iNOS released by MDSCs is an additional mechanism responsible for inducing oxidative stress in the TME. NO produced by iNOS can suppress the T cells’ response and induce T cell apoptosis via various mechanisms, including the inhibition of Janus kinase 3(JAK3), STAT5, and MHC-II expression (11, 13). Synergistically, S100A8/A9 also increases the production of iNOS through activation of STAT1 (66).
Secondly, MDSCs can suppress T cell activation and proliferation by depleting essential amino acids. MDSCs increase arginase-1 activity and induce T cell suppression via the depletion of L-arginine (11, 13, 67). The lack of L-arginine suppresses proliferation of activated T cells and decreases the expression of the T cell receptor-zeta (TCR-ζ) chain (68). As a result, arginase-1 leading to depletion of L-arginine in the TME suppresses the ability of the T cells to exert their anti-tumor functions (69). Indeed, MDSCs have the ability to inhibit T cell proliferation by regulating the G0/G1 phase of their cell cycle (70). Expression of IDO by MDSCs can also suppress T cell proliferation by decreasing tryptophan levels and producing cytotoxic metabolites (71). Furthermore, it was reported that chronic psychological stress can also lead to MDSCs accumulation in the bone marrow of Balb/c mice where they inhibit T cells proliferation (72).
Other mechanisms that result in MDSCs-induced T cell apoptosis have been described. For example, MDSCs can decrease expression level of B-cell lymphoma 2 (Bcl-2) expression and increase FAS (CD95 ligand) expression in T cells. Furthermore, MDSCs express galectin-9, which binds to T-cell immunoglobulin domain and mucin domain 3 (TIM3), an inhibitory surface molecule on lymphocytes, leading to decreased T cell viability (73, 74). Interestingly, different subtypes of MDSCs utilize different mechanisms to mediate immunosuppressive functions in the TME. M-MDSCs produce high levels of NO and immunosuppressive cytokines such as IL-10, which suppress both antigen-specific and non-specific T-cell responses (55). In contrast, PMN-MDSCs suppress T-cell responses by generating ROS based on an antigen-specific approach (75, 76).
Thirdly, MDSCs-mediated lymphocyte trafficking and viability are restricted. MDSCs can suppress T cell movement to the lymph nodes via down-regulation of L-selectin (CD62L) on the surface of T cells by increasing expression of disintegrin and metalloproteinase domain17 (ADAM17). MDSCs can also interrupt the migration of CD8+ T cells to tumor sites by peroxynitrite modification of CCL2 (77, 78). Finally, MDSCs can promote the induction of other immunosuppressive cells. MDSCs have been shown to induce the generation of FoxP3+ Treg cells in vivo through the production of IFN-γ, TGF-β and IL-10. This effect is independent of NO production (38). Furthermore, the CCR5 ligands CCL3, CCL4, and CCL5 were shown to promote CCR5+ Treg cell recruitment in mouse models of melanoma (74). It has been reported that CD14+HLA-DR–/low M-MDSCs could induce CD4+CD25+Foxp3+Treg cells when co-cultured with autologous T cells in hepatocellular carcinoma (HCC) patients (79). Moreover, F-MDSCs can inhibit T cell proliferation by releasing IDO and promoting the expansion of Tregs (80). With the exception of Tregs stimulation, MDSCs can also reverse macrophages to an M2-like phenotype with low IL-12 production, thereby promoting tumor growth (81). In addition, MDSCs impair NK cell function and cytotoxicity by suppressing the production of IFN-γ from NK cells and decreasing the expression of natural killer group 2 member D (NKG2D) (82). Induction of MDSCs in a tumor-bearing mouse model of lung cancer can lead to impairment of B cell differentiation and function though an IL-7 and STAT5-dependent manner (83). The MDSCs immunosuppressive mechanisms described above are outlined in Figure 1.
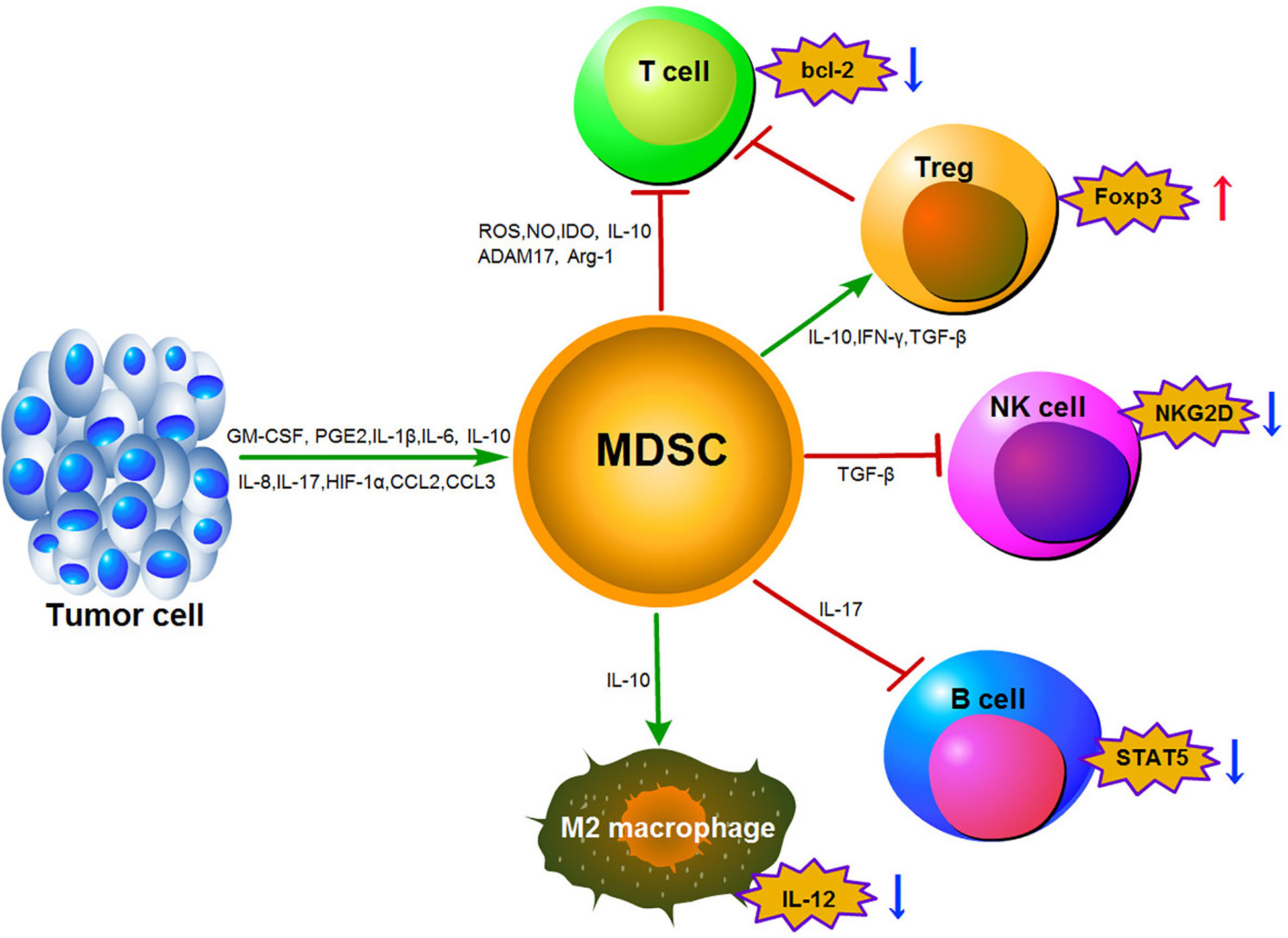
Figure 1 Main mechanisms of immunosuppression function mediated by MDSCs in the tumor microenvironment.
MDSCs play an important role in the immunosuppressive mechanism associated with CRC progression. Several studies have demonstrated that MDSCs in the peripheral blood and tumor tissues are associated with tumor stage, histological grade of cancer and lymph node metastases in CRC (84, 85). It has been reported that the peripheral blood of CRC patients contains a significantly increased percentage and absolute number of CD33+CD11b+HLA-DR−/low MDSCs compared with healthy donors. This increasement was closely correlated with clinical cancer stage and tumor metastasis but not primary tumor size. Interestingly, radical operation can significantly decrease the level of circulating MDSCs in CRC patients (76, 85). Moreover, the proportion of PMN-MDSCs and immature MDSCs (I-MDSCs) was found to be higher in the tumor tissues of CRC patients compared to tumor-adjacent tissues (86).
In a recent study, CD33+ MDSCs and Yes-associated protein 1 (YAP1) were identified as predictors for the prognosis of CRC patients. This study demonstrated that CD33+MDSCs numbers and YAP1 expression levels were increased in tumor tissues compared with those of tumor-adjacent tissues from the same CRC patients (87). Furthermore, CD33+CD11b+HLA-DR−/low myeloid cells were shown to be expanded in the peripheral blood of CRC patients, with the number of circulating MDSCs positively correlating with poor prognosis and low survival rates (88). In addition, a lower lymphocyte-to-monocyte ratio (LMR) was associated with poor prognosis in CRC patients, who were found to have higher levels of circulating MDSCs (89). Unresectable metastatic CRC patients with high M-MDSCs levels in their peripheral blood were also shown to have a significantly shorter progression-free survival (90). Interestingly, it was proven that although Tregs, Th17, and PMN-MDSCs were significantly increased in metastatic CRC, only high levels of PMN-MDSCs were associated with a poor prognosis for CRC patients (91).
The immunosuppressive function of MDSCs relies on the activation of different intracellular signaling pathways. Many studies indicate that MDSCs-associated signaling pathways are involved in CRC development. KRAS mutations, for example, are frequently observed in human CRC and correlate positively with tumor aggressiveness and metastasis (92–94). KRAS-mediated repression of interferon regulatory factor 2 (IRF2) was associated with high expression of CXCL3, which led to MDSCs migration to the TME through binding to CXCR2 on the MDSCs (95). Receptor-interacting protein kinase 3 (RIPK3) is essential for mucosal repair in CRC. It has been reported that reduction of RIPK3 in CRC cells induces expansion of MDSCs and increases cyclooxygenase-2 (COX-2) expression, which then catalyze PGE2 and enhance MDSCs function (96). Similarly, RIPK3 signaling in an I-MDSCs subset promoted intestinal tumor development in MC38 cell tumors by expanding IL17-producing T cells (97).
It has been suggested that down-regulation of mucin 1 (MUC1) in hematopoietic cells increases MDSCs expansion in inflammatory bowel disease (IBD) leading to the development of CRC (98). Moreover, MUC13 promotes colitis-associated colorectal tumors development through the β-catenin signaling pathway and increases MDSCs expansion in the TME (99). In addition, MDSCs can increase the expression levels of ROS and NO, which may result in DNA damage and promote tumor progression in CRC (100). Previous studies have also demonstrated that CRC cells secrete VEGF-A, which leads to TAMs induction and subsequent production of chemokine (C-X-C motif) ligand 1 (CXCL1) in the primary tumor. Increased CXCL1 in liver tissue was shown to recruit CXCR2-positive MDSCs to form a premetastatic niche in CRC (101). In addition, overexpression of CXCR4 has been found to play a crucial role in the invasion of CRC, as well as promoting the epithelial–mesenchymal transition (EMT) and infiltration of MDSCs in colonic tissue, accelerating colitis-associated and Apc mutation-driven colorectal tumorigenesis (102). Recently, Varun Sasidharan Nair et al. reported some genes associated with histone deacetylases (HDAC) activation, DNA methylation, Wnt and IL-6 signaling pathways are upregulated in CRC tumor infiltrating I-MDSCs, and propose that they could be exploited as potential targets for CRC therapy (86).
The JAK/STAT pathway is considered to be a major player in mediating immunosuppression (103, 104). MDSCs isolated from the spleen of CT26 cell-bearing mice exhibited inhibition of phosphorylation of STAT1 (p-STAT1) in response to IFN-α or IFN-γ (105). However, another study demonstrated that IFN-γ is not a key regulator of MDSCs and that targeting it would be unlikely to alter MDSCs accumulation or function in tumor-bearing mice (106). It has been reported that IL-6 activates expansion of MDSCs via the JAK2/STAT3/NF-κB signaling pathway, resulting in AOM/DSS-induced colon tumor development in G protein subunit alpha i1 (GNAI1) and GNAI3 (GNAI1;3) double-knockout (DKO) mice (107). It has also been shown that protease-activated receptor 2 (PAR2) deficiency significantly promotes tumor development in the AOM/DSS-induced colitis-associated colon cancer model through accumulation of MDSCs and enhancement of their immunosuppressive activity via STAT3-mediated ROS production (108). Previous studies have demonstrated that G-CSF could promote MDSCs’ survival and activation through the STAT3 signaling pathway in a mouse colitis-associated cancer model (109). Additionally, CCL2 was initially characterized as a cytokine that was found to be increased in CRC tissues and reported to enhance PMN-MDSCs’ function in a STAT3-mediated manner (110). Xu et al. demonstrated that sphingosine-1-phosphate receptor 1 (S1PR1) and STAT3 are elevated in human CRC tissues and propose that they recruit MDSCs through the S1PR1–STAT3–IL-6 axis to promote tumor growth and liver metastasis niche (111). In addition, STAT6 appears to promote expansion of MDSCs and contributes to intestinal tumor progression in ApcMin/+ mice (112). Finally, S100A8/A9 is another pro-inflammatory molecular that activates the STAT3 signaling pathway, which is responsible for maintaining the MDSCs suppressive function (113).
Of significance, a highly hypoxic environment leads to the accumulation and activation of MDSCs in CRC development. Hypoxia within the TME is associated with increased V-domain Ig suppressor of T-cell activation (VISTA) expression, which promotes MDSCs function. VISTA is highly expressed in the CRC microenvironment, while both VISTA and HIF-1α activity were found to be increased in a cohort of CRC patients (114). Notably, malignant tumors can potentially recruit MDSCs from the bone marrow by releasing exosomes. Previous studies have shown that hypoxia can induce MDSCs to secrete more exosomes in a HIF-1α dependent manner (115). The exosomal contents can reprogram the target cell and increase mobility of MDSCs to the tumor sites. Inhibition of S100A9 was found to suppress the susceptibility of mice to AOM/DSS-induced colitis-associated colon cancer (116).
The safety and efficacy of using MDSCs inhibition as a CRC therapy have been evaluated in an increasing number of studies (Table 2). Here we summarize novel preclinical approaches targeting MDSCs in CRC (Figure 2). Current treatments aim to deplete MDSCs, inhibit their immunosuppressive function, and block their expansion to the tumor site (128). Several studies have proved that depletion of the number of MDSCs and inhibition of their function in tumor tissue are an important strategy for CRC therapy. For example, targeting MDSCs with all-trans-retinoic acid (ATRA) has been shown to decrease their number and suppress their function in tumor bearing mice (117). Consequently, ATRA may consider being a novel immunotherapeutic protocols to target CRC in the future. Similarly, histamine dihydrochloride (HDC), a NADPH oxidase 2 (NOX2) inhibitor, has also been shown to inhibit tumor progression by reducing the accumulation of tumor MDSCs in MC-38 cell-bearing mice (118). Embelin (2,5-dihydroxy-3-undecyl-1,4-benzoquinone) is a non-peptidic small molecule inhibitor of X-linked inhibitor of apoptosis protein (XIAP). Wu et al. found that embelin can significantly reduce the accumulation number of MDSCs in the peripheral lymphoid organ and tumor tissue, and impair the immunosuppressive function of MDSCs by reducing the production of ROS and arginase-1 level in colitis-associated tumorgenesis (119). Naringin (4′,5,7-trihydroxyavanone-7-rhamnoglucoside), a major flavanone glycoside that occurs naturally in citrus fruits, inhibits the severity of colitis and CRC development through regulation of the MDSCs’ immunosuppressive function via the NF-κB/IL-6/STAT3 axis in colorectal tissues (120).

Table 2 Summary of preclinical studies analyzing the role of MDSCs in CRC and therapeutic agents.
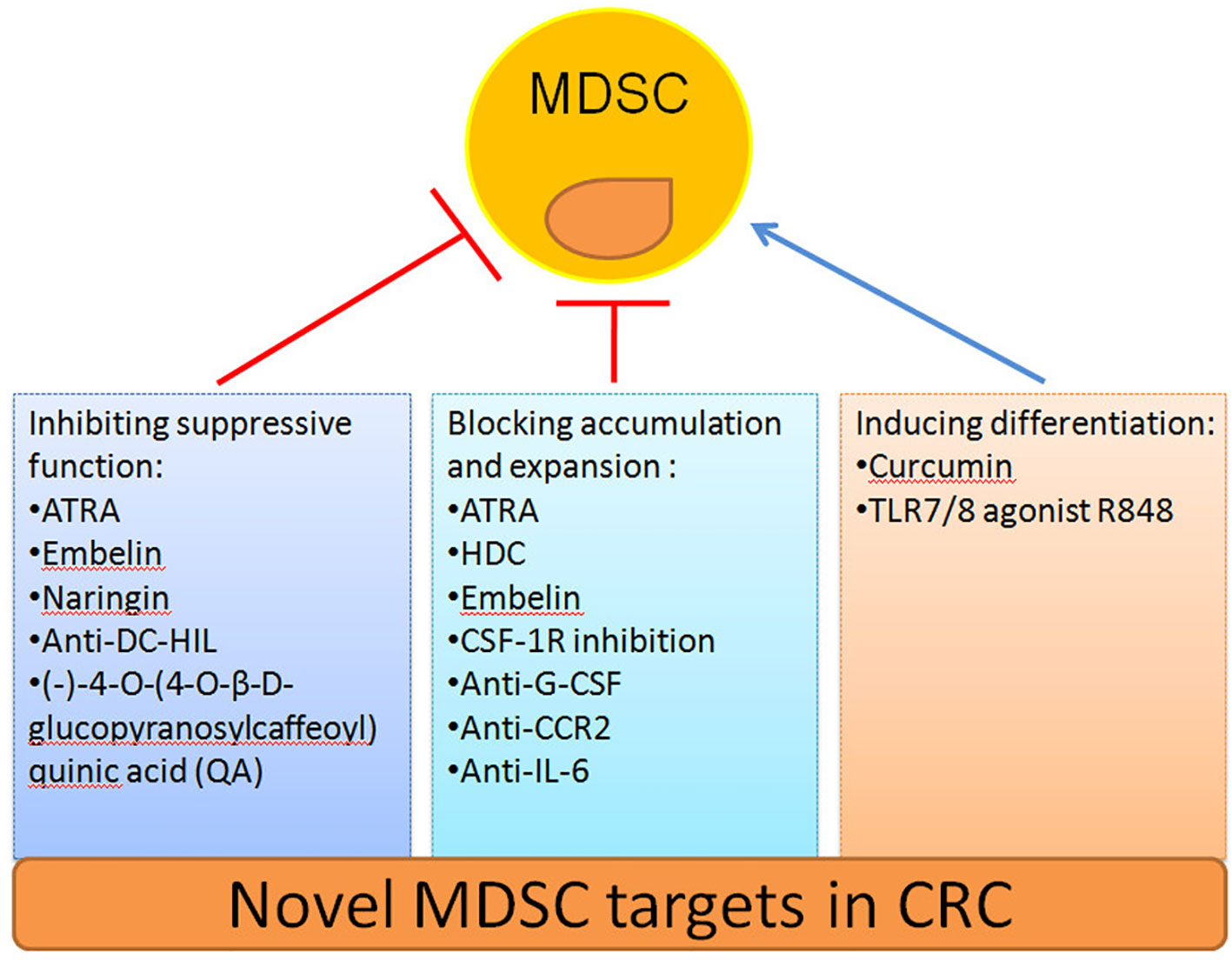
Figure 2 Novel strategies to target MDSCs in CRC.
Recruitment of tumor MDSCs is dependent on the receptor tyrosine kinase CSF-1R. Thus, inhibition of CSF-1R signaling can significantly block the number of tumor-infiltrating MDSCs number and enhance anti-tumor T cells responses in tumor bearing mice (121). In addition, blocking the immunosuppressive function of MDSCs can be achieved by targeting anti-G-CSF monoclonal antibody. Treatment with an anti-G-CSF monoclonal antibody reduces MDSCs accumulation and decreases the migration, proliferation, and functional maintenance of MDSCs and could therefore become a potential therapeutic agent for colitis-associated cancer (109). Previous results have indicated that DC-HIL+MDSCs are expanded in the blood of metastatic CRC patients. Since, anti-DC-HIL mAb treatment can suppress the function of MDSCs isolated from treated mice, functionally blocking DC-HIL on MDSCs could also be potentially beneficial in the treatment of metastatic CRC (122).
Other treatments include the induction of MDSCs differentiation alone or in combination with radiotherapy, chemotherapy, surgery or other kinds of immunotherapy to target CRC (129). Curcumin has been shown to inhibit the expansion of MDSCs, activate STAT3 and NF-κB in MDSCs, and polarize MDSCs toward a M1-like phenotype in CT26 cell-bearing mice (123). Recently, the TLR7/8 agonist R848, as a new immunologic adjuvant, was found to reverse the functional orientation of MDSCs towards M1 macrophages, suggesting that R848 may be a potential immunologic adjuvant in chemotherapy for oxaliplatin-resistant CRC (124). It has been reported that treatment with anti-CCR2 antibody can alleviate radiation-induced MDSCs infiltration in CRC tumor tissues by activation of the STING pathway (125). Hence, anti-CCR2 antibody treatment may improve radiotherapy for advanced CRC patients. A previous study indicated that IL-6 induces strong immunosuppression in the CRC microenvironment by recruiting MDSCs and impairing T cells infiltration. Interestingly, an anti-IL-6 and anti-PD-L1 combination treatment prolonged tumor-bearing mouse survival, providing a novel strategy to overcome anti-PD-L1 resistance in CRC (126). In addition, blocking the immunosuppressive function of MDSCs can also be achieved by targeting phosphatidylinositol 3-kinase (PI3K)δ/γ. Hence, (-)-4-O-(4-O-β-D-glucopyranosylcaffeoyl) quinic acid (QA), a selective small molecule inhibitor of PI3Kδ/γ, has the ability to reshape the tumor immune microenvironment and promote responses to anti-PD-1 treatment in a colon tumor model (127).
While there is an abundance of preclinical data supporting the theory that suppression of MDSCs could be a beneficial therapeutic tool. Several clinical studies have also indicated that inhibition of MDSCs is beneficial to CRC patients. Metastatic CRC patients treated with a first-line combination regimen of 5-FU, oxaliplatin, and bevacizumab (FOLFOX-bevacizumab) were associated with a better survival outcome. Furthermore, the FOLFOX-bevacizumab treatment was found to decrease the PMN-MDSC population in metastatic CRC patients (91). While docosahexaenoic acid (DHA) has been shown to inhibit caspase-1 activity in 5-fluorouracil (5-FU) treated MDSCs, a negative relationship between the DHA content in plasma and the induction of caspase-1 activity in MDSCs of CRC patients treated with 5-FU-based chemotherapy has been reported (130). Thus, these data provide new insights into the regulation of DHA and its potential benefit in 5-FU-based chemotherapy for CRC patients. Previous studies demonstrated that CD38 is a transmembrane receptor–ectoenzyme expressed by MDSCs in esophageal cancer and multiple myeloma (131, 132). Interestingly, a significant expansion of CD38+M-MDSCs were observed in PBMCs of CRC patients when compared with healthy donors, and CD38+M-MDSCs frequencies were significantly higher in CRC patients who had previously received any form of cancer treatment (surgery, chemotherapy or radiotherapy, targeted therapy, or a combination of these methods) when compared with treatment-naive patients (133). This study supported a method to target M-MDSCs with an anti-CD38 monoclonal antibody could be a valuable therapeutic tool for the treatment of metastatic CRC patients.
Growing evidence suggests that the immunosuppressive microenvironments in tumors result from the activation of MDSCs, PD-1/PD-L1 and cytotoxic T lymphocyte-associated antigen-4(CTLA-4) pathways (134). PD-1 has been found to bind to its ligand PD-L1 and then induce T cell anergy and apoptosis (135). CTLA-4 is another receptor expressed on the surface of T cells, which inhibits T cells activities by competing with CD28 to bind to the two T cell activation antigens, CD80 and CD86 (136). PD-1 and CTLA-4 are immune checkpoint proteins expressed on activated T cells (Figure 3). Blocking PD-L1 or CTLA-4 signaling has been shown to be beneficial for cancer patient survival. MDSCs express high levels of PD-L1, and this upregulation of PD-L1 has been associated with expression of S100A9 and HIF-1α (69, 137). It has been indicated that PD-L1 expression on MDSCs is increased in CRC patients and colon tumor bearing mice, suggesting that it may be a potent mediator of immunosuppression function (138, 139). Furthermore, PD-L1+MDSCs were significantly decreased after neutralization of IFN-γ in the TME (139). Interestingly, PD-L1+MDSCs are significantly increased in HCC patients, while M-CSF and VEGFA have been shown to induce PD-L1+MDSCs in vitro (140). Compared to responding patients, PMN-MDSCs also expressed high levels of PD-L1 in non-responding melanoma patients treated by ipilimumab (141).
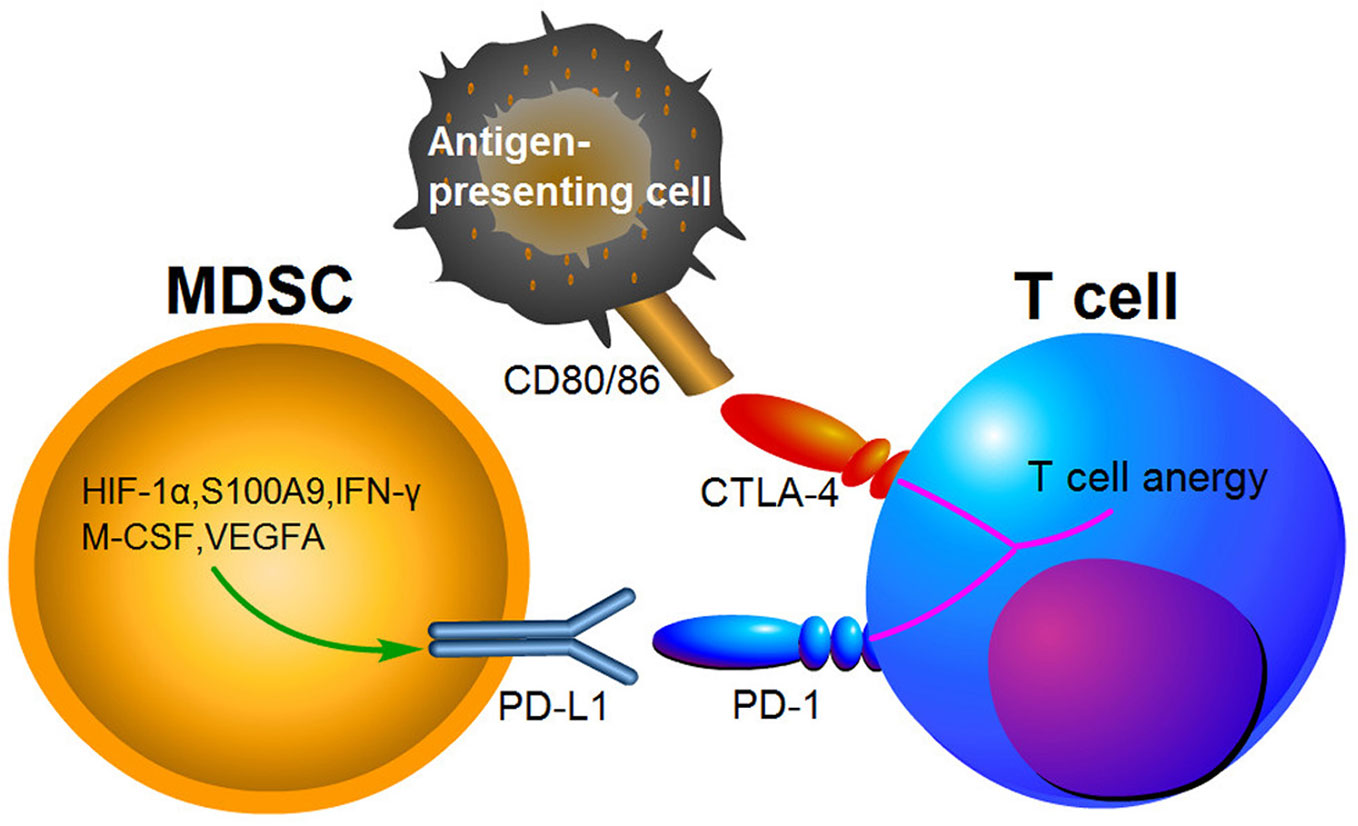
Figure 3 The expression of PD-L1 in MDSCs causing T cell anergy via binding to its respective receptor.
Several studies found that MDSCs and other molecules such as PD-L1 and CTLA-4 in tumor tissues are sensitive predictive markers for patients’ response to chemoradiotherapy for rectal cancer patients (142). In the latest study, it was demonstrated that blockade of CXCR2 on MDSCs can overcome resistance to anti-PD-1 therapy in CRC-expressing oncogenic KRAS (95). Similarly, MDSCs elimination can reverse resistance to anti-PD-L1 or combination of normo-fractionated radiotherapy plus immunotherapy in CRC (143). Moreover, HDC can reduce the accumulation of intratumoral MDSCs in colon tumor-bearing mice and improve the anti-tumor efficacy of the PD-1/PD-L1 checkpoint blockade (118). Interestingly, IL-6 blockade was also reported to reverse the anti-PD-L1 resistance and inhibit CRC growth by reducing the number of MDSCs (126). MUC13-deficient mice have fewer MDSCs and are sensitive to anti-PD-L1 therapy, suggesting that MUC13 may be useful for in the treatment of colitis-associated cancer (99). Additionally, it has been revealed that treatment using an anti-KIT monoclonal antibody in a mouse colon cancer model enhanced the anti-tumor activity of anti-CTLA-4 and anti-PD-1 therapy by selectively reducing the MDSCs population (144).
Several studies have shown that the increasing number of TAMs in tumor correlates with favorable 5-year overall survival (OS) for CRC patients (145, 146). Increased M2 macrophage numbers in the TME promote the initiation and growth of tumor. However, few strategies are currently available to modulate TAMs by repolarizing the M2 macrophages to become M1 macrophages. A recent study demonstrated that PD-L1+ T cells can engage PD-1+ macrophages, inducing an alternative M2-like program, which have effects on adaptive antitumor immunity (147). In addition, Wang et al. showed that CD30L deficiency promote the accumulation of MDSCs, increase the expression of PD-L1 on MDSCs and TAMs, and enhance immunosuppressive function in an AOM/DSS-induced CAC model, suggesting that CD30L/CD30 signaling could be a potential candidate target for immunotherapy in CAC (148).
In conclusion, numerous studies have documented the important role of immunosuppressive MDSCs in CRC development in mice and cancer patients. During CRC progression, MDSCs-mediated immunosuppressive activity is regulated by many different signaling pathways. MDSCs promote CRC progression by increasing cell proliferation, cancer stemness, enhancing tumor invasiveness and metastasis. Given that the mechanisms controlling expansion and activation of MDSCs in tumor tissues or in the peripheral blood are distinct, it is difficult to devise a therapeutic approach to reduce their numbers or arrest their function. Furthermore, monotherapies targeting MDSCs have shown promising but limited efficacy. Thus, it is important to elucidate novel mechanisms involving different stromal components and myeloid cells such as cancer associated fibroblasts (CAFs), TAMs and neutrophils. Further studies are required to strengthen the knowledge about MDSCs and to better understand the effects in combination with other therapies involving different immunotherapeutic approaches for CRC therapy.
KY drafted the manuscript. XX and KR discussed and revised the manuscript. TW and SW designed the study and provided critical suggestions. All authors contributed to the article and approved the submitted version.
This study was conducted with support from the Natural Science Foundation of Jiangsu (Grant No. BK20190242 and BK20170563), Jiangsu Province’s Key Medical Talents Program (Grant No.ZDRCB2016018), Research Project of Jiangsu Commission of Health (Grant No. K2019019).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
DCs, Dendritic cells; TAMs, tumor-associated macrophages; NK, natural killer; Tregs, regulatory T cells; UCB, umbilical cord blood; MDSCs, myeloid-derived suppressor cells; IMCs, immature myeloid cells; PMN-MDSCs, polymorphonuclear MDSCs; M-MDSCs, monocytic MDSCs; e-MDSCs, early-stage MDSCs; PBMCs, peripheral blood mononuclear cells; F-MDSCs, fibrocytic MDSCs; PGE2, prostaglandin E2; TGF-β, transforming growth factor beta; HIF-1α, hypoxia-inducible factor-1α; CRC, colorectal cancer; CCR2, C-C chemokine receptor type 2; iNOS, inducible nitric oxide synthase; CCL2, chemokine ligand 2; CXCR, C-X-C motif chemokine receptor; ENTPD2, ectonucleoside triphosphate diphosphohydrolase 2; RONS, reactive oxygen and nitrogen species; RNS, reactive nitrogen species; G-CSF, granulocyte colony-stimulating factor, GM-CSF, granulocytes macrophage colony-stimulating factor; M-CSF, macrophage colony-stimulating factor; STAT3, signal transducer and activator of transcription 3; NADPH, nicotinamide adenine dinucleotide phosphate; TME, tumor microenvironment; NF-κB, nuclear factor-κB; NO, nitric oxide; ROS, reactive oxygen species; VEGF, vascular endothelial growth factor; JAK3, Janus kinase 3; MHC, major histocompatibility complex; IDO, indoleamine 2,3dioxygenase; Bcl-2, B-cell lymphoma 2; TIM3, T-cell immunoglobulin domain and mucin domain 3; ADAM17, disintegrin and metalloproteinase domain17; IFN-γ, interferon gamma; HCC, hepatocellular carcinoma; IRF2, interferon regulatory factor 2; RIPK3, receptor-interacting protein kinase 3; COX-2, cyclooxygenase-2; IBD, inflammatory bowel disease; VISTA, V-domain Ig suppressor of T-cell activation; GNAI1, G protein subunit alpha i1; PAR2, protease-activated receptor 2; S1PR1, sphingosine-1-phosphate receptor 1; ATRA, all-trans-retinoic acid; HDC, histamine dihydrochloride; XIAP, X-linked inhibitor of apoptosis protein; PI3K, phosphatidylinositol 3-kinase; CTLA-4, cytotoxic T lymphocyte-associated antigen-4.
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin (2019) 69:7–34. doi: 10.3322/caac.21551
2. Siegel RL, Miller KD, Fedewa SA, Ahnen DJ, Meester RGS, Barzi A, et al. Colorectal cancer statistics, 2017. CA Cancer J Clin (2017) 67:177–93. doi: 10.3322/caac.21395
3. Le DT, Hubbard-Lucey VM, Morse MA, Heery CR, Dwyer A, Marsilje TH, et al. A Blueprint to Advance Colorectal Cancer Immunotherapies. Cancer Immunol Res (2017) 5:942–9. doi: 10.1158/2326-6066.CIR-17-0375
4. Fletcher R, Wang YJ, Schoen RE, Finn OJ, Yu J, Zhang L. Colorectal cancer prevention: Immune modulation taking the stage. Biochim Biophys Acta Rev Cancer (2018) 1869:138–48. doi: 10.1016/j.bbcan.2017.12.002
5. Gotwals P, Cameron S, Cipolletta D, Cremasco V, Crystal A, Hewes B, et al. Prospects for combining targeted and conventional cancer therapy with immunotherapy. Nat Rev Cancer (2017) 17:286–301. doi: 10.1038/nrc.2017.17
6. Roelands J, Kuppen PJK, Vermeulen L, Maccalli C, Decock J, Wang E, et al. Immunogenomic Classification of Colorectal Cancer and Therapeutic Implications. Int J Mol Sci (2017) 18:2229. doi: 10.3390/ijms18102229
7. Tian X, Zheng Y, Yin K, Ma J, Tian J, Zhang Y, et al. LncRNA AK036396 inhibits maturation and accelerates immunosuppression of polymorphonuclear myeloid-derived suppressor cells by enhancing the stability of ficolin B. Cancer Immunol Res (2020) 8:565–77. doi: 10.1158/2326-6066.CIR-19-0595
8. Zhang Y, Velez-Delgado A, Mathew E, Li D, Mendez FM, Flannagan K, et al. Myeloid cells are required for PD-1/PD-L1 checkpoint activation and the establishment of an immunosuppressive environment in pancreatic cancer. Gut (2017) 66:124–36. doi: 10.1136/gutjnl-2016-312078
9. Markowitz J, Brooks TR, Duggan MC, Paul BK, Pan X, Wei L, et al. Patients with pancreatic adenocarcinoma exhibit elevated levels of myeloid-derived suppressor cells upon progression of disease. Cancer Immunol Immunother (2015) 64:149–59. doi: 10.1007/s00262-014-1618-8
10. Cavallo F, De Giovanni C, Nanni P, Forni G, Lollini PL. 2011: the immune hallmarks of cancer. Cancer Immunol Immunother (2011) 60:319–26. doi: 10.1007/s00262-010-0968-0
11. Consonni FM, Porta C, Marino A, Pandolfo C, Mola S, Bleve A, et al. Myeloid-Derived Suppressor Cells: Ductile Targets in Disease. Front Immunol (2019) 10:949. doi: 10.3389/fimmu.2019.00949
12. Veglia F, Perego M, Gabrilovich D. Myeloid-derived suppressor cells coming of age. Nat Immunol (2018) 19:108–19. doi: 10.1038/s41590-017-0022-x
13. Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol (2009) 9:162–74. doi: 10.1038/nri2506
14. Fedorova L, Pilatova K, Selingerova I, Bencsikova B, Budinska E, Zwinsova B, et al. Circulating Myeloid-Derived Suppressor Cell Subsets in Patients with Colorectal Cancer - Exploratory Analysis of Their Biomarker Potential. Klin Onkol (2018) 31:88–92. doi: 10.14735/amko20182S88
15. Zhang Y, Xu J, Zhang N, Chen M, Wang H, Zhu D. Targeting the tumour immune microenvironment for cancer therapy in human gastrointestinal malignancies. Cancer Lett (2019) 458:123–35. doi: 10.1016/j.canlet.2019.05.017
16. Gabrilovich DI. Myeloid-Derived Suppressor Cells. Cancer Immunol Res (2017) 5:3–8. doi: 10.1158/2326-6066.CIR-16-0297
17. Solito S, Marigo I, Pinton L, Damuzzo V, Mandruzzato S, Bronte V. Myeloid-derived suppressor cell heterogeneity in human cancers. Ann N Y Acad Sci (2014) 1319:47–65. doi: 10.1111/nyas.12469
18. Tian J, Rui K, Hong Y, Wang X, Xiao F, Lin X, et al. Increased GITRL impairs the function of myeloid-derived suppressor cells and exacerbates primary Sjögren’s syndrome. J Immunol (2019) 202:1693–703. doi: 10.4049/jimmunol.1801051
19. Dorhoi A, Du Plessis N. Monocytic Myeloid-Derived Suppressor Cells in Chronic Infections. Front Immunol (2017) 8:1895. doi: 10.3389/fimmu.2017.01895
20. Meyer C, Sevko A, Ramacher M, Bazhin AV, Falk CS, Osen W, et al. Chronic inflammation promotes myeloid-derived suppressor cell activation blocking antitumor immunity in transgenic mouse melanoma model. Proc Natl Acad Sci USA (2011) 108:17111–6. doi: 10.1073/pnas.1108121108
21. Katoh H, Watanabe M. Myeloid-Derived Suppressor Cells and Therapeutic Strategies in Cancer. Mediators Inflammation (2015) 2015:159269. doi: 10.1155/2015/159269
22. Kumar V, Cheng P, Condamine T, Mony S, Languino LR, McCaffrey JC, et al. CD45 Phosphatase Inhibits STAT3 Transcription Factor Activity in Myeloid Cells and Promotes Tumor-Associated Macrophage Differentiation. Immunity (2016) 44:303–15. doi: 10.1016/j.immuni.2016.01.014
23. Sagiv JY, Michaeli J, Assi S, Mishalian I, Kisos H, Levy L, et al. Phenotypic diversity and plasticity in circulating neutrophil subpopulations in cancer. Cell Rep (2015) 10:562–73. doi: 10.1016/j.celrep.2014.12.039
24. Biswas SK, Gangi L, Paul S, Schioppa T, Saccani A, Sironi M, et al. A distinct and unique transcriptional program expressed by tumor-associated macrophages (defective NF-kappaB and enhanced IRF-3/STAT1 activation). Blood (2006) 107:2112–22. doi: 10.1182/blood-2005-01-0428
25. Barbera-Guillem E, Nyhus JK, Wolford CC, Friece CR, Sampsel JW. Vascular endothelial growth factor secretion by tumor-infiltrating macrophages essentially supports tumor angiogenesis, and IgG immune complexes potentiate the process. Cancer Res (2002) 62:7042–9.
26. Mantovani A, Marchesi F, Malesci A, Laghi L, Allavena P. Tumour-associated macrophages as treatment targets in oncology. Nat Rev Clin Oncol (2017) 14:399–416. doi: 10.1038/nrclinonc.2016.217
27. Biswas SK, Mantovani A. Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nat Immunol (2010) 11:889–96. doi: 10.1038/ni.1937
28. Calabretta E, d’Amore F, Carlo-Stella C. Immune and Inflammatory Cells of the Tumor Microenvironment Represent Novel Therapeutic Targets in Classical Hodgkin Lymphoma. Int J Mol Sci (2019) 20:5503. doi: 10.3390/ijms20215503
29. Martinez FO, Gordon S. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep (2014) 6:13. doi: 10.12703/P6-13
30. Yahaya MAF, Lila MAM, Ismail S, Zainol M, Afizan N. Tumour-Associated Macrophages (TAMs) in Colon Cancer and How to Reeducate Them. J Immunol Res (2019) 2019:2368249. doi: 10.1155/2019/2368249
31. Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science (2011) 331:1565–70. doi: 10.1126/science.1203486
32. Korbecki J, Kojder K, Barczak K, Siminska D, Gutowska I, Chlubek D, et al. Hypoxia Alters the Expression of CC Chemokines and CC Chemokine Receptors in a Tumor-A Literature Review. Int J Mol Sci (2020) 21:5647. doi: 10.3390/ijms21165647
33. Trellakis S, Bruderek K, Hutte J, Elian M, Hoffmann TK, Lang S, et al. Granulocytic myeloid-derived suppressor cells are cryosensitive and their frequency does not correlate with serum concentrations of colony-stimulating factors in head and neck cancer. Innate Immun (2013) 19:328–36. doi: 10.1177/1753425912463618
34. Mishalian I, Bayuh R, Levy L, Zolotarov L, Michaeli J, Fridlender ZG. Tumor-associated neutrophils (TAN) develop pro-tumorigenic properties during tumor progression. Cancer Immunol Immunother (2013) 62:1745–56. doi: 10.1007/s00262-013-1476-9
35. Jiang J, Guo W, Liang X. Phenotypes, accumulation, and functions of myeloid-derived suppressor cells and associated treatment strategies in cancer patients. Hum Immunol (2014) 75:1128–37. doi: 10.1016/j.humimm.2014.09.025
36. Youn JI, Collazo M, Shalova IN, Biswas SK, Gabrilovich DI. Characterization of the nature of granulocytic myeloid-derived suppressor cells in tumor-bearing mice. J Leukoc Biol (2012) 91:167–81. doi: 10.1189/jlb.0311177
37. Gallina G, Dolcetti L, Serafini P, De Santo C, Marigo I, Colombo MP, et al. Tumors induce a subset of inflammatory monocytes with immunosuppressive activity on CD8+ T cells. J Clin Invest (2006) 116:2777–90. doi: 10.1172/JCI28828
38. Huang B, Pan PY, Li Q, Sato AI, Levy DE, Bromberg J, et al. Gr-1+CD115+ immature myeloid suppressor cells mediate the development of tumor-induced T regulatory cells and T-cell anergy in tumor-bearing host. Cancer Res (2006) 66:1123–31. doi: 10.1158/0008-5472.CAN-05-1299
39. Cha YJ, Koo JS. Role of Tumor-Associated Myeloid Cells in Breast Cancer. Cells (2020) 9:1785. doi: 10.3390/cells9081785
40. Groth C, Hu X, Weber R, Fleming V, Altevogt P, Utikal J, et al. Immunosuppression mediated by myeloid-derived suppressor cells (MDSCs) during tumour progression. Br J Cancer (2019) 120:16–25. doi: 10.1038/s41416-018-0333-1
41. Bronte V, Brandau S, Chen SH, Colombo MP, Frey AB, Greten TF, et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat Commun (2016) 7:12150. doi: 10.1038/ncomms12150
42. Rodriguez PC, Ernstoff MS, Hernandez C, Atkins M, Zabaleta J, Sierra R, et al. Arginase I-producing myeloid-derived suppressor cells in renal cell carcinoma are a subpopulation of activated granulocytes. Cancer Res (2009) 69:1553–60. doi: 10.1158/0008-5472.CAN-08-1921
43. Zhang W, Jiang M, Chen J, Zhang R, Ye Y, Liu P, et al. SOCS3 Suppression Promoted the Recruitment of CD11b(+)Gr-1(-)F4/80(-)MHCII(-) Early-Stage Myeloid-Derived Suppressor Cells and Accelerated Interleukin-6-Related Tumor Invasion via Affecting Myeloid Differentiation in Breast Cancer. Front Immunol (2018) 9:1699. doi: 10.3389/fimmu.2018.01699
44. Gunaydin G, Kesikli SA, Guc D. Cancer associated fibroblasts have phenotypic and functional characteristics similar to the fibrocytes that represent a novel MDSC subset. Oncoimmunology (2015) 4:e1034918. doi: 10.1080/2162402X.2015.1034918
45. Mazza EM, Zoso A, Mandruzzato S, Bronte V, Serafini P, Inverardi L, et al. Gene expression profiling of human fibrocytic myeloid-derived suppressor cells (f-MDSCs). Genom Data (2014) 2:389–92. doi: 10.1016/j.gdata.2014.10.018
46. Condamine T, Ramachandran I, Youn JI, Gabrilovich DI. Regulation of tumor metastasis by myeloid-derived suppressor cells. Annu Rev Med (2015) 66:97–110. doi: 10.1146/annurev-med-051013-052304
47. Lechner MG, Liebertz DJ, Epstein AL. Characterization of cytokine-induced myeloid-derived suppressor cells from normal human peripheral blood mononuclear cells. J Immunol (2010) 185:2273–84. doi: 10.4049/jimmunol.1000901
48. Song X, Krelin Y, Dvorkin T, Bjorkdahl O, Segal S, Dinarello CA, et al. CD11b+/Gr-1+ immature myeloid cells mediate suppression of T cells in mice bearing tumors of IL-1beta-secreting cells. J Immunol (2005) 175:8200–8. doi: 10.4049/jimmunol.175.12.8200
49. Bunt SK, Yang L, Sinha P, Clements VK, Leips J, Ostrand-Rosenberg S. Reduced inflammation in the tumor microenvironment delays the accumulation of myeloid-derived suppressor cells and limits tumor progression. Cancer Res (2007) 67:10019–26. doi: 10.1158/0008-5472.CAN-07-2354
50. Wu CT, Hsieh CC, Lin CC, Chen WC, Hong JH, Chen MF. Significance of IL-6 in the transition of hormone-resistant prostate cancer and the induction of myeloid-derived suppressor cells. J Mol Med (Berl) (2012) 90:1343–55. doi: 10.1007/s00109-012-0916-x
51. Parker KH, Beury DW, Ostrand-Rosenberg S. Myeloid-Derived Suppressor Cells: Critical Cells Driving Immune Suppression in the Tumor Microenvironment. Adv Cancer Res (2015) 128:95–139. doi: 10.1016/bs.acr.2015.04.002
52. Umansky V, Blattner C, Gebhardt C, Utikal J. The Role of Myeloid-Derived Suppressor Cells (MDSC) in Cancer Progression. Vaccines (Basel) (2016) 4:36. doi: 10.3390/vaccines4040036
53. Lee CR, Lee W, Cho SK, Park SG. Characterization of Multiple Cytokine Combinations and TGF-beta on Differentiation and Functions of Myeloid-Derived Suppressor Cells. Int J Mol Sci (2018) 19:869. doi: 10.3390/ijms19030869
54. Guan X, Liu Z, Zhang J, Jin X. Myeloid-derived suppressor cell accumulation in renal cell carcinoma is correlated with CCL2, IL-17 and IL-18 expression in blood and tumors. Adv Clin Exp Med (2018) 27:947–53. doi: 10.17219/acem/70065
55. Kumar V, Patel S, Tcyganov E, Gabrilovich DI. The Nature of Myeloid-Derived Suppressor Cells in the Tumor Microenvironment. Trends Immunol (2016) 37:208–20. doi: 10.1016/j.it.2016.01.004
56. Ding AS, Routkevitch D, Jackson C, Lim M. Targeting Myeloid Cells in Combination Treatments for Glioma and Other Tumors. Front Immunol (2019) 10:1715. doi: 10.3389/fimmu.2019.01715
57. Alfaro C, Sanmamed MF, Rodriguez-Ruiz ME, Teijeira A, Onate C, Gonzalez A, et al. Interleukin-8 in cancer pathogenesis, treatment and follow-up. Cancer Treat Rev (2017) 60:24–31. doi: 10.1016/j.ctrv.2017.08.004
58. Ostrand-Rosenberg S, Fenselau C. Myeloid-Derived Suppressor Cells: Immune-Suppressive Cells That Impair Antitumor Immunity and Are Sculpted by Their Environment. J Immunol (2018) 200:422–31. doi: 10.4049/jimmunol.1701019
59. Zheng Y, Tian X, Wang T, Xia X, Cao F, Tian J, et al. Long noncoding RNA Pvt1 regulates the immunosuppression activity of granulocytic myeloid-derived suppressor cells in tumor-bearing mice. Mol Cancer (2019) 18:61. doi: 10.1186/s12943-019-0978-2
60. Cassetta L, Baekkevold ES, Brandau S, Bujko A, Cassatella MA, Dorhoi A, et al. Deciphering myeloid-derived suppressor cells: isolation and markers in humans, mice and non-human primates. Cancer Immunol Immunother (2019) 68:687–97. doi: 10.1007/s00262-019-02302-2
61. Brandau S, Moses K, Lang S. The kinship of neutrophils and granulocytic myeloid-derived suppressor cells in cancer: cousins, siblings or twins? Semin Cancer Biol (2013) 23:171–82. doi: 10.1016/j.semcancer.2013.02.007
62. Dumitru CA, Lang S, Brandau S. Modulation of neutrophil granulocytes in the tumor microenvironment: mechanisms and consequences for tumor progression. Semin Cancer Biol (2013) 23:141–8. doi: 10.1016/j.semcancer.2013.02.005
63. de Haas N, de Koning C, Spilgies L, de Vries IJ, Hato SV. Improving cancer immunotherapy by targeting the STATe of MDSCs. Oncoimmunology (2016) 5:e1196312. doi: 10.1080/2162402X.2016.1196312
64. Pinton L, Solito S, Damuzzo V, Francescato S, Pozzuoli A, Berizzi A, et al. Activated T cells sustain myeloid-derived suppressor cell-mediated immune suppression. Oncotarget (2016) 7:1168–84. doi: 10.18632/oncotarget.6662
65. Cimen Bozkus C, Elzey BD, Crist SA, Ellies LG, Ratliff TL. Expression of Cationic Amino Acid Transporter 2 Is Required for Myeloid-Derived Suppressor Cell-Mediated Control of T Cell Immunity. J Immunol (2015) 195:5237–50. doi: 10.4049/jimmunol.1500959
66. Sinha P, Okoro C, Foell D, Freeze HH, Ostrand-Rosenberg S, Srikrishna G. Proinflammatory S100 proteins regulate the accumulation of myeloid-derived suppressor cells. J Immunol (2008) 181:4666–75. doi: 10.4049/jimmunol.181.7.4666
67. Pawelec G, Verschoor CP, Ostrand-Rosenberg S. Myeloid-Derived Suppressor Cells: Not Only in Tumor Immunity. Front Immunol (2019) 10:1099. doi: 10.3389/fimmu.2019.01099
68. Rodriguez PC, Quiceno DG, Zabaleta J, Ortiz B, Zea AH, Piazuelo MB, et al. Arginase I production in the tumor microenvironment by mature myeloid cells inhibits T-cell receptor expression and antigen-specific T-cell responses. Cancer Res (2004) 64:5839–49. doi: 10.1158/0008-5472.CAN-04-0465
69. Noman MZ, Desantis G, Janji B, Hasmim M, Karray S, Dessen P, et al. PD-L1 is a novel direct target of HIF-1alpha, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J Exp Med (2014) 211:781–90. doi: 10.1084/jem.20131916
70. Fujimura T, Mahnke K, Enk AH. Myeloid derived suppressor cells and their role in tolerance induction in cancer. J Dermatol Sci (2010) 59:1–6. doi: 10.1016/j.jdermsci.2010.05.001
71. Yu J, Du W, Yan F, Wang Y, Li H, Cao S, et al. Myeloid-derived suppressor cells suppress antitumor immune responses through IDO expression and correlate with lymph node metastasis in patients with breast cancer. J Immunol (2013) 190:3783–97. doi: 10.4049/jimmunol.1201449
72. Jin J, Wang X, Wang Q, Guo X, Cao J, Zhang X, et al. Chronic psychological stress induces the accumulation of myeloid-derived suppressor cells in mice. PloS One (2013) 8:e74497. doi: 10.1371/journal.pone.0074497
73. Bronte V, Zanovello P. Regulation of immune responses by L-arginine metabolism. Nat Rev Immunol (2005) 5:641–54. doi: 10.1038/nri1668
74. Schlecker E, Stojanovic A, Eisen C, Quack C, Falk CS, Umansky V, et al. Tumor-infiltrating monocytic myeloid-derived suppressor cells mediate CCR5-dependent recruitment of regulatory T cells favoring tumor growth. J Immunol (2012) 189:5602–11. doi: 10.4049/jimmunol.1201018
75. Marvel D, Gabrilovich DI. Myeloid-derived suppressor cells in the tumor microenvironment: expect the unexpected. J Clin Invest (2015) 125:3356–64. doi: 10.1172/JCI80005
76. OuYang LY, Wu XJ, Ye SB, Zhang RX, Li ZL, Liao W, et al. Tumor-induced myeloid-derived suppressor cells promote tumor progression through oxidative metabolism in human colorectal cancer. J Transl Med (2015) 13:47. doi: 10.1186/s12967-015-0410-7
77. Molon B, Ugel S, Del Pozzo F, Soldani C, Zilio S, Avella D, et al. Chemokine nitration prevents intratumoral infiltration of antigen-specific T cells. J Exp Med (2011) 208:1949–62. doi: 10.1084/jem.20101956
78. Ostrand-Rosenberg S, Sinha P, Beury DW, Clements VK. Cross-talk between myeloid-derived suppressor cells (MDSC), macrophages, and dendritic cells enhances tumor-induced immune suppression. Semin Cancer Biol (2012) 22:275–81. doi: 10.1016/j.semcancer.2012.01.011
79. Zhao F, Korangy F, Greten TF. Cellular immune suppressor mechanisms in patients with hepatocellular carcinoma. Dig Dis (2012) 30:477–82. doi: 10.1159/000341695
80. Zoso A, Mazza EM, Bicciato S, Mandruzzato S, Bronte V, Serafini P, et al. Human fibrocytic myeloid-derived suppressor cells express IDO and promote tolerance via Treg-cell expansion. Eur J Immunol (2014) 44:3307–19. doi: 10.1002/eji.201444522
81. Beury DW, Parker KH, Nyandjo M, Sinha P, Carter KA, Ostrand-Rosenberg S. Cross-talk among myeloid-derived suppressor cells, macrophages, and tumor cells impacts the inflammatory milieu of solid tumors. J Leukoc Biol (2014) 96:1109–18. doi: 10.1189/jlb.3A0414-210R
82. Liu C, Yu S, Kappes J, Wang J, Grizzle WE, Zinn KR, et al. Expansion of spleen myeloid suppressor cells represses NK cell cytotoxicity in tumor-bearing host. Blood (2007) 109:4336–42. doi: 10.1182/blood-2006-09-046201
83. Wang Y, Schafer CC, Hough KP, Tousif S, Duncan SR, Kearney JF, et al. Myeloid-Derived Suppressor Cells Impair B Cell Responses in Lung Cancer through IL-7 and STAT5. J Immunol (2018) 201:278–95. doi: 10.4049/jimmunol.1701069
84. Sun HL, Zhou X, Xue YF, Wang K, Shen YF, Mao JJ, et al. Increased frequency and clinical significance of myeloid-derived suppressor cells in human colorectal carcinoma. World J Gastroenterol (2012) 18:3303–9. doi: 10.3748/wjg.v18.i25.3303
85. Zhang B, Wang Z, Wu L, Zhang M, Li W, Ding J, et al. Circulating and tumor-infiltrating myeloid-derived suppressor cells in patients with colorectal carcinoma. PloS One (2013) 8:e57114. doi: 10.1371/journal.pone.0057114
86. Sasidharan Nair V, Saleh R, Toor SM, Taha RZ, Ahmed AA, Kurer MA, et al. Transcriptomic profiling disclosed the role of DNA methylation and histone modifications in tumor-infiltrating myeloid-derived suppressor cell subsets in colorectal cancer. Clin Epigenet (2020) 12:13. doi: 10.1186/s13148-020-0808-9
87. Yang R, Cai TT, Wu XJ, Liu YN, He J, Zhang XS, et al. Tumour YAP1 and PTEN expression correlates with tumour-associated myeloid suppressor cell expansion and reduced survival in colorectal cancer. Immunology (2018) 155:263–72. doi: 10.1111/imm.12949
88. Toor SM, Syed Khaja AS, El Salhat H, Bekdache O, Kanbar J, Jaloudi M, et al. Increased Levels of Circulating and Tumor-Infiltrating Granulocytic Myeloid Cells in Colorectal Cancer Patients. Front Immunol (2016) 7:560. doi: 10.3389/fimmu.2016.00560
89. Shimura T, Shibata M, Gonda K, Hayase S, Sakamoto W, Okayama H, et al. Prognostic impact of preoperative lymphocyte-to-monocyte ratio in patients with colorectal cancer with special reference to myeloid-derived suppressor cells. Fukushima J Med Sci (2018) 64:64–72. doi: 10.5387/fms.2018-10
90. Tada K, Kitano S, Shoji H, Nishimura T, Shimada Y, Nagashima K, et al. Pretreatment Immune Status Correlates with Progression-Free Survival in Chemotherapy-Treated Metastatic Colorectal Cancer Patients. Cancer Immunol Res (2016) 4:592–9. doi: 10.1158/2326-6066.CIR-15-0298
91. Limagne E, Euvrard R, Thibaudin M, Rebe C, Derangere V, Chevriaux A, et al. Accumulation of MDSC and Th17 Cells in Patients with Metastatic Colorectal Cancer Predicts the Efficacy of a FOLFOX-Bevacizumab Drug Treatment Regimen. Cancer Res (2016) 76:5241–52. doi: 10.1158/0008-5472.CAN-15-3164
92. Wood LD, Parsons DW, Jones S, Lin J, Sjoblom T, Leary RJ, et al. The genomic landscapes of human breast and colorectal cancers. Science (2007) 318:1108–13. doi: 10.1126/science.1145720
93. Pereira AA, Rego JF, Morris V, Overman MJ, Eng C, Garrett CR, et al. Association between KRAS mutation and lung metastasis in advanced colorectal cancer. Br J Cancer (2015) 112:424–8. doi: 10.1038/bjc.2014.619
94. Vakiani E, Janakiraman M, Shen R, Sinha R, Zeng Z, Shia J, et al. Comparative genomic analysis of primary versus metastatic colorectal carcinomas. J Clin Oncol (2012) 30:2956–62. doi: 10.1200/JCO.2011.38.2994
95. Liao W, Overman MJ, Boutin AT, Shang X, Zhao D, Dey P, et al. KRAS-IRF2 Axis Drives Immune Suppression and Immune Therapy Resistance in Colorectal Cancer. Cancer Cell (2019) 35:559–72.e557. doi: 10.1016/j.ccell.2019.02.008
96. Yan G, Zhao H, Zhang Q, Zhou Y, Wu L, Lei J, et al. A RIPK3-PGE2 Circuit Mediates Myeloid-Derived Suppressor Cell-Potentiated Colorectal Carcinogenesis. Cancer Res (2018) 78:5586–99. doi: 10.1158/0008-5472.CAN-17-3962
97. Jayakumar A, Bothwell ALM. RIPK3-Induced Inflammation by I-MDSCs Promotes Intestinal Tumors. Cancer Res (2019) 79:1587–99. doi: 10.1158/0008-5472.CAN-18-2153
98. Poh TW, Madsen CS, Gorman JE, Marler RJ, Leighton JA, Cohen PA, et al. Downregulation of hematopoietic MUC1 during experimental colitis increases tumor-promoting myeloid-derived suppressor cells. Clin Cancer Res (2013) 19:5039–52. doi: 10.1158/1078-0432.CCR-13-0278
99. Sheng YH, Wong KY, Seim I, Wang R, He Y, Wu A, et al. MUC13 promotes the development of colitis-associated colorectal tumors via beta-catenin activity. Oncogene (2019) 38:7294–310. doi: 10.1038/s41388-019-0951-y
100. Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell (2010) 140:883–99. doi: 10.1016/j.cell.2010.01.025
101. Wang D, Sun H, Wei J, Cen B, DuBois RN. CXCL1 Is Critical for Premetastatic Niche Formation and Metastasis in Colorectal Cancer. Cancer Res (2017) 77:3655–65. doi: 10.1158/0008-5472.CAN-16-3199
102. Yu X, Wang D, Wang X, Sun S, Zhang Y, Wang S, et al. CXCL12/CXCR4 promotes inflammation-driven colorectal cancer progression through activation of RhoA signaling by sponging miR-133a-3p. J Exp Clin Cancer Res (2019) 38:32. doi: 10.1186/s13046-018-1014-x
103. Villarino AV, Kanno Y, O’Shea JJ. Mechanisms and consequences of Jak-STAT signaling in the immune system. Nat Immunol (2017) 18:374–84. doi: 10.1038/ni.3691
104. Trikha P, Carson WE,3. Signaling pathways involved in MDSC regulation. Biochim Biophys Acta (2014) 1846:55–65. doi: 10.1016/j.bbcan.2014.04.003
105. Mundy-Bosse BL, Lesinski GB, Jaime-Ramirez AC, Benninger K, Khan M, Kuppusamy P, et al. Myeloid-derived suppressor cell inhibition of the IFN response in tumor-bearing mice. Cancer Res (2011) 71:5101–10. doi: 10.1158/0008-5472.CAN-10-2670
106. Sinha P, Parker KH, Horn L, Ostrand-Rosenberg S. Tumor-induced myeloid-derived suppressor cell function is independent of IFN-gamma and IL-4Ralpha. Eur J Immunol (2012) 42:2052–9. doi: 10.1002/eji.201142230
107. Li ZW, Sun B, Gong T, Guo S, Zhang J, Wang J, et al. GNAI1 and GNAI3 Reduce Colitis-Associated Tumorigenesis in Mice by Blocking IL6 Signaling and Down-regulating Expression of GNAI2. Gastroenterology (2019) 156:2297–312. doi: 10.1053/j.gastro.2019.02.040
108. Ke Z, Wang C, Wu T, Wang W, Yang Y, Dai Y. PAR2 deficiency enhances myeloid cell-mediated immunosuppression and promotes colitis-associated tumorigenesis. Cancer Lett (2020) 469:437–46. doi: 10.1016/j.canlet.2019.11.015
109. Li W, Zhang X, Chen Y, Xie Y, Liu J, Feng Q, et al. G-CSF is a key modulator of MDSC and could be a potential therapeutic target in colitis-associated colorectal cancers. Protein Cell (2016) 7:130–40. doi: 10.1007/s13238-015-0237-2
110. Chun E, Lavoie S, Michaud M, Gallini CA, Kim J, Soucy G, et al. CCL2 Promotes Colorectal Carcinogenesis by Enhancing Polymorphonuclear Myeloid-Derived Suppressor Cell Population and Function. Cell Rep (2015) 12:244–57. doi: 10.1016/j.celrep.2015.06.024
111. Lin Q, Ren L, Jian M, Xu P, Li J, Zheng P, et al. The mechanism of the premetastatic niche facilitating colorectal cancer liver metastasis generated from myeloid-derived suppressor cells induced by the S1PR1-STAT3 signaling pathway. Cell Death Dis (2019) 10:693. doi: 10.1038/s41419-019-1922-5
112. Jayakumar A, Bothwell ALM. Stat6 Promotes Intestinal Tumorigenesis in a Mouse Model of Adenomatous Polyposis by Expansion of MDSCs and Inhibition of Cytotoxic CD8 Response. Neoplasia (2017) 19:595–605. doi: 10.1016/j.neo.2017.04.006
113. Zoller M. Janus-Faced Myeloid-Derived Suppressor Cell Exosomes for the Good and the Bad in Cancer and Autoimmune Disease. Front Immunol (2018) 9:137. doi: 10.3389/fimmu.2018.00137
114. Deng J, Li J, Sarde A, Lines JL, Lee YC, Qian DC, et al. Hypoxia-Induced VISTA Promotes the Suppressive Function of Myeloid-Derived Suppressor Cells in the Tumor Microenvironment. Cancer Immunol Res (2019) 7:1079–90. doi: 10.1158/2326-6066.CIR-18-0507
115. Tian X, Shen H, Li Z, Wang T, Wang S. Tumor-derived exosomes, myeloid-derived suppressor cells, and tumor microenvironment. J Hematol Oncol (2019) 12:84. doi: 10.1186/s13045-019-0772-z
116. Wang Y, Yin K, Tian J, Xia X, Ma J, Tang X, et al. Granulocytic Myeloid-Derived Suppressor Cells Promote the Stemness of Colorectal Cancer Cells through Exosomal S100A9. Adv Sci (Weinh) (2019) 6:1901278. doi: 10.1002/advs.201901278
117. Heine A, Flores C, Gevensleben H, Diehl L, Heikenwalder M, Ringelhan M, et al. Targeting myeloid derived suppressor cells with all-trans retinoic acid is highly time-dependent in therapeutic tumor vaccination. Oncoimmunology (2017) 6:e1338995. doi: 10.1080/2162402X.2017.1338995
118. Grauers Wiktorin H, Nilsson MS, Kiffin R, Sander FE, Lenox B, Rydstrom A, et al. Histamine targets myeloid-derived suppressor cells and improves the anti-tumor efficacy of PD-1/PD-L1 checkpoint blockade. Cancer Immunol Immunother (2019) 68:163–74. doi: 10.1007/s00262-018-2253-6
119. Wu T, Wang C, Wang W, Hui Y, Zhang R, Qiao L, et al. Embelin impairs the accumulation and activation of MDSCs in colitis-associated tumorigenesis. Oncoimmunology (2018) 7:e1498437. doi: 10.1080/2162402X.2018.1498437
120. Zhang YS, Wang F, Cui SX, Qu XJ. Natural dietary compound naringin prevents azoxymethane/dextran sodium sulfate-induced chronic colorectal inflammation and carcinogenesis in mice. Cancer Biol Ther (2018) 19:735–44. doi: 10.1080/15384047.2018.1453971
121. Holmgaard RB, Zamarin D, Lesokhin A, Merghoub T, Wolchok JD. Targeting myeloid-derived suppressor cells with colony stimulating factor-1 receptor blockade can reverse immune resistance to immunotherapy in indoleamine 2,3-dioxygenase-expressing tumors. EBioMedicine (2016) 6:50–8. doi: 10.1016/j.ebiom.2016.02.024
122. Kobayashi M, Chung JS, Beg M, Arriaga Y, Verma U, Courtney K, et al. Blocking Monocytic Myeloid-Derived Suppressor Cell Function via Anti-DC-HIL/GPNMB Antibody Restores the In Vitro Integrity of T Cells from Cancer Patients. Clin Cancer Res (2019) 25:828–38. doi: 10.1158/1078-0432.CCR-18-0330
123. Tu SP, Jin H, Shi JD, Zhu LM, Suo Y, Lu G, et al. Curcumin induces the differentiation of myeloid-derived suppressor cells and inhibits their interaction with cancer cells and related tumor growth. Cancer Prev Res (Phila) (2012) 5:205–15. doi: 10.1158/1940-6207.CAPR-11-0247
124. Liu Z, Xie Y, Xiong Y, Liu S, Qiu C, Zhu Z, et al. TLR 7/8 agonist reverses oxaliplatin resistance in colorectal cancer via directing the myeloid-derived suppressor cells to tumoricidal M1-macrophages. Cancer Lett (2020) 469:173–85. doi: 10.1016/j.canlet.2019.10.020
125. Liang H, Deng L, Hou Y, Meng X, Huang X, Rao E, et al. Host STING-dependent MDSC mobilization drives extrinsic radiation resistance. Nat Commun (2017) 8:1736. doi: 10.1038/s41467-017-01566-5
126. Li J, Xu J, Yan X, Jin K, Li W, Zhang R. Targeting Interleukin-6 (IL-6) Sensitizes Anti-PD-L1 Treatment in a Colorectal Cancer Preclinical Model. Med Sci Monit (2018) 24:5501–8. doi: 10.12659/MSM.907439
127. Lin H, Wu Y, Chen J, Huang S, Wang Y. (-)-4-O-(4-O-beta-D-glucopyranosylcaffeoyl) Quinic Acid Inhibits the Function of Myeloid-Derived Suppressor Cells to Enhance the Efficacy of Anti-PD1 against Colon Cancer. Pharm Res (2018) 35:183. doi: 10.1007/s11095-018-2459-5
128. Fleming V, Hu X, Weber R, Nagibin V, Groth C, Altevogt P, et al. Targeting Myeloid-Derived Suppressor Cells to Bypass Tumor-Induced Immunosuppression. Front Immunol (2018) 9:398. doi: 10.3389/fimmu.2018.00398
129. Seliger B. Combinatorial Approaches With Checkpoint Inhibitors to Enhance Anti-tumor Immunity. Front Immunol (2019) 10:999. doi: 10.3389/fimmu.2019.00999
130. Dumont A, de Rosny C, Kieu TL, Perrey S, Berger H, Fluckiger A, et al. Docosahexaenoic acid inhibits both NLRP3 inflammasome assembly and JNK-mediated mature IL-1beta secretion in 5-fluorouracil-treated MDSC: implication in cancer treatment. Cell Death Dis (2019) 10:485. doi: 10.1038/s41419-019-1723-x
131. Karakasheva TA, Waldron TJ, Eruslanov E, Kim SB, Lee JS, O’Brien S, et al. CD38-Expressing Myeloid-Derived Suppressor Cells Promote Tumor Growth in a Murine Model of Esophageal Cancer. Cancer Res (2015) 75:4074–85. doi: 10.1158/0008-5472.CAN-14-3639
132. Krejcik J, Casneuf T, Nijhof IS, Verbist B, Bald J, Plesner T, et al. Daratumumab depletes CD38+ immune regulatory cells, promotes T-cell expansion, and skews T-cell repertoire in multiple myeloma. Blood (2016) 128:384–94. doi: 10.1182/blood-2015-12-687749
133. Karakasheva TA, Dominguez GA, Hashimoto A, Lin EW, Chiu C, Sasser K, et al. CD38+ M-MDSC expansion characterizes a subset of advanced colorectal cancer patients. JCI Insight (2018) 3:e97022. doi: 10.1172/jci.insight.97022
134. Zumwalt TJ, Goel A. Immunotherapy of Metastatic Colorectal Cancer: Prevailing Challenges and New Perspectives. Curr Colorectal Cancer Rep (2015) 11:125–40. doi: 10.1007/s11888-015-0269-2
135. Sambi M, Bagheri L, Szewczuk MR. Current Challenges in Cancer Immunotherapy: Multimodal Approaches to Improve Efficacy and Patient Response Rates. J Oncol (2019) 2019:4508794. doi: 10.1155/2019/4508794
136. Sansom DM. CD28, CTLA-4 and their ligands: who does what and to whom? Immunology (2000) 101:169–77. doi: 10.1046/j.1365-2567.2000.00121.x
137. Cheng P, Eksioglu EA, Chen X, Kandell W, Le Trinh T, Cen L, et al. S100A9-induced overexpression of PD-1/PD-L1 contributes to ineffective hematopoiesis in myelodysplastic syndromes. Leukemia (2019) 33:2034–46. doi: 10.1038/s41375-019-0397-9
138. Yamauchi Y, Safi S, Blattner C, Rathinasamy A, Umansky L, Juenger S, et al. Circulating and Tumor Myeloid-derived Suppressor Cells in Resectable Non-Small Cell Lung Cancer. Am J Respir Crit Care Med (2018) 198:777–87. doi: 10.1164/rccm.201708-1707OC
139. Lu C, Redd PS, Lee JR, Savage N, Liu K. The expression profiles and regulation of PD-L1 in tumor-induced myeloid-derived suppressor cells. Oncoimmunology (2016) 5:e1247135. doi: 10.1080/2162402X.2016.1247135
140. Iwata T, Kondo Y, Kimura O, Morosawa T, Fujisaka Y, Umetsu T, et al. PD-L1(+)MDSCs are increased in HCC patients and induced by soluble factor in the tumor microenvironment. Sci Rep (2016) 6:39296. doi: 10.1038/srep39296
141. Gebhardt C, Sevko A, Jiang H, Lichtenberger R, Reith M, Tarnanidis K, et al. Myeloid Cells and Related Chronic Inflammatory Factors as Novel Predictive Markers in Melanoma Treatment with Ipilimumab. Clin Cancer Res (2015) 21:5453–9. doi: 10.1158/1078-0432.CCR-15-0676
142. Teng F, Meng X, Kong L, Mu D, Zhu H, Liu S, et al. Tumor-infiltrating lymphocytes, forkhead box P3, programmed death ligand-1, and cytotoxic T lymphocyte-associated antigen-4 expressions before and after neoadjuvant chemoradiation in rectal cancer. Transl Res (2015) 166:721–732 e721. doi: 10.1016/j.trsl.2015.06.019
143. Grapin M, Richard C, Limagne E, Boidot R, Morgand V, Bertaut A, et al. Optimized fractionated radiotherapy with anti-PD-L1 and anti-TIGIT: a promising new combination. J Immunother Cancer (2019) 7:160. doi: 10.1186/s40425-019-0634-9
144. Garton AJ, Seibel S, Lopresti-Morrow L, Crew L, Janson N, Mandiyan S, et al. Anti-KIT Monoclonal Antibody Treatment Enhances the Antitumor Activity of Immune Checkpoint Inhibitors by Reversing Tumor-Induced Immunosuppression. Mol Cancer Ther (2017) 16:671–80. doi: 10.1158/1535-7163.MCT-16-0676
145. Edin S, Wikberg ML, Dahlin AM, Rutegard J, Oberg A, Oldenborg PA, et al. The distribution of macrophages with a M1 or M2 phenotype in relation to prognosis and the molecular characteristics of colorectal cancer. PloS One (2012) 7:e47045. doi: 10.1371/journal.pone.0047045
146. Li J, Li L, Li Y, Long Y, Zhao Q, Ouyang Y, et al. Tumor-associated macrophage infiltration and prognosis in colorectal cancer: systematic review and meta-analysis. Int J Colorectal Dis (2020) 35:1203–10. doi: 10.1007/s00384-020-03593-z
147. Diskin B, Adam S, Cassini MF, Sanchez G, Liria M, Aykut B, et al. PD-L1 engagement on T cells promotes self-tolerance and suppression of neighboring macrophages and effector T cells in cancer. Nat Immunol (2020) 21:442–54. doi: 10.1038/s41590-020-0620-x
Keywords: myeloid-derived suppressor cells, colorectal cancer, cancer immunology, colorectal cancer immunotherapy, tumor microenvironment
Citation: Yin K, Xia X, Rui K, Wang T and Wang S (2020) Myeloid-Derived Suppressor Cells: A New and Pivotal Player in Colorectal Cancer Progression. Front. Oncol. 10:610104. doi: 10.3389/fonc.2020.610104
Received: 25 September 2020; Accepted: 10 November 2020;
Published: 15 December 2020.
Edited by:
Peng Qu, National Institutes of Health (NIH), United StatesReviewed by:
Anurag Singh, Tübingen University Hospital, GermanyCopyright © 2020 Yin, Xia, Rui, Wang and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kai Yin, anN5aW5rYWlAMTYzLmNvbQ==; Tingting Wang, MzI4ODY4OTM4OEBxcS5jb20=; Shengjun Wang, c2p3anNAdWpzLmVkdS5jbg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.