 Silvina Odete Bustos
Silvina Odete Bustos Fernanda Antunes
Fernanda Antunes Maria Cristina Rangel
Maria Cristina Rangel Roger Chammas*
Roger Chammas*
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 25 November 2020
Sec. Molecular and Cellular Oncology
Volume 10 - 2020 | https://doi.org/10.3389/fonc.2020.606436
This article is part of the Research Topic New Roles of Autophagy Pathways in Cancer View all 24 articles
The tumor microenvironment (TME) is a complex environment where cancer cells reside and interact with different types of cells, secreted factors, and the extracellular matrix. Additionally, TME is shaped by several processes, such as autophagy. Autophagy has emerged as a conserved intracellular degradation pathway for clearance of damaged organelles or aberrant proteins. With its central role, autophagy maintains the cellular homeostasis and orchestrates stress responses, playing opposite roles in tumorigenesis. During tumor development, autophagy also mediates autophagy-independent functions associated with several hallmarks of cancer, and therefore exerting several effects on tumor suppression and/or tumor promotion mechanisms. Beyond the concept of degradation, new different forms of autophagy have been described as modulators of cancer progression, such as secretory autophagy enabling intercellular communication in the TME by cargo release. In this context, the synthesis of senescence-associated secretory proteins by autophagy lead to a senescent phenotype. Besides disturbing tumor treatment responses, autophagy also participates in innate and adaptive immune signaling. Furthermore, recent studies have indicated intricate crosstalk between autophagy and the epithelial-mesenchymal transition (EMT), by which cancer cells obtain an invasive phenotype and metastatic potential. Thus, autophagy in the cancer context is far broader and complex than just a cell energy sensing mechanism. In this scenario, we will discuss the key roles of autophagy in the TME and surrounding cells, contributing to cancer development and progression/EMT. Finally, the potential intervention in autophagy processes as a strategy for cancer therapy will be addressed.
The autophagy process has been explored for almost 60 years, from morphological studies since early 70’s to molecular studies initiated in the 1990s (1–3). During this period, several studies were conducted to understand the genetic mechanisms of autophagy, leading to the discovery of autophagy-related genes (ATG) in yeast (4, 5). Subsequently, ATG homologs were identified in various organisms, and new ATG genes were described in mammals (6). Building upon these findings, efforts to delve into the molecular mechanisms involved in the degradation of intracellular constituents have grown rapidly. However, several issues remain unsolved regarding the molecular regulation of autophagy, its integration and control at the tissue and systemic levels and its role in cancer pathophysiology. Three main types of autophagy have been described, depending on the morphology and mechanisms: microautophagy, chaperone-mediated autophagy and the best characterized macroautophagy (hereafter referred as autophagy).
Autophagy is an evolutionarily conserved process responsible for removing intracellular molecular aggregates of misfolded proteins and damaged organelles, through the sequestration of these substrates in a double-membrane vesicle, which fuses with lysosomes, where degradation of the macromolecular machines or complexes takes place. Multiple proteins are involved in the sequential stages of autophagy consisting in initiation, elongation of isolated membranes decorated by microtubule-associated protein 1A/1B-light chain 3 (LC3) to form an autophagosome, and its fusion with the lysosome to cargo degradation. Autophagy is stimulated in physiological and pathological conditions regulating cell metabolism and homeostasis (7). In cancer cells, stressors, such as hypoxia or nutrient deprivation, induce autophagy to support the high energy demand of cells with dysregulated proliferation. Many tumor suppressor and oncogene products are elements of autophagy pathways, pointing to the relationship between autophagy and tumorigenesis. Moreover, it is well known that many cancer cells have high basal levels of autophagy. Although autophagy contributes to cancer promotion in advanced stages, it is also capable to inhibit tumor initiation in early stages (8, 9). The molecular circuitry controlling autophagy is therefore complex, as it can either induce cell-death or promote cell survival (Figure 1) (10, 11). Understanding the mechanisms for the protective role of autophagy in cancer is essential for the identification of novel targets to the control of resistance of tumors to treatment.
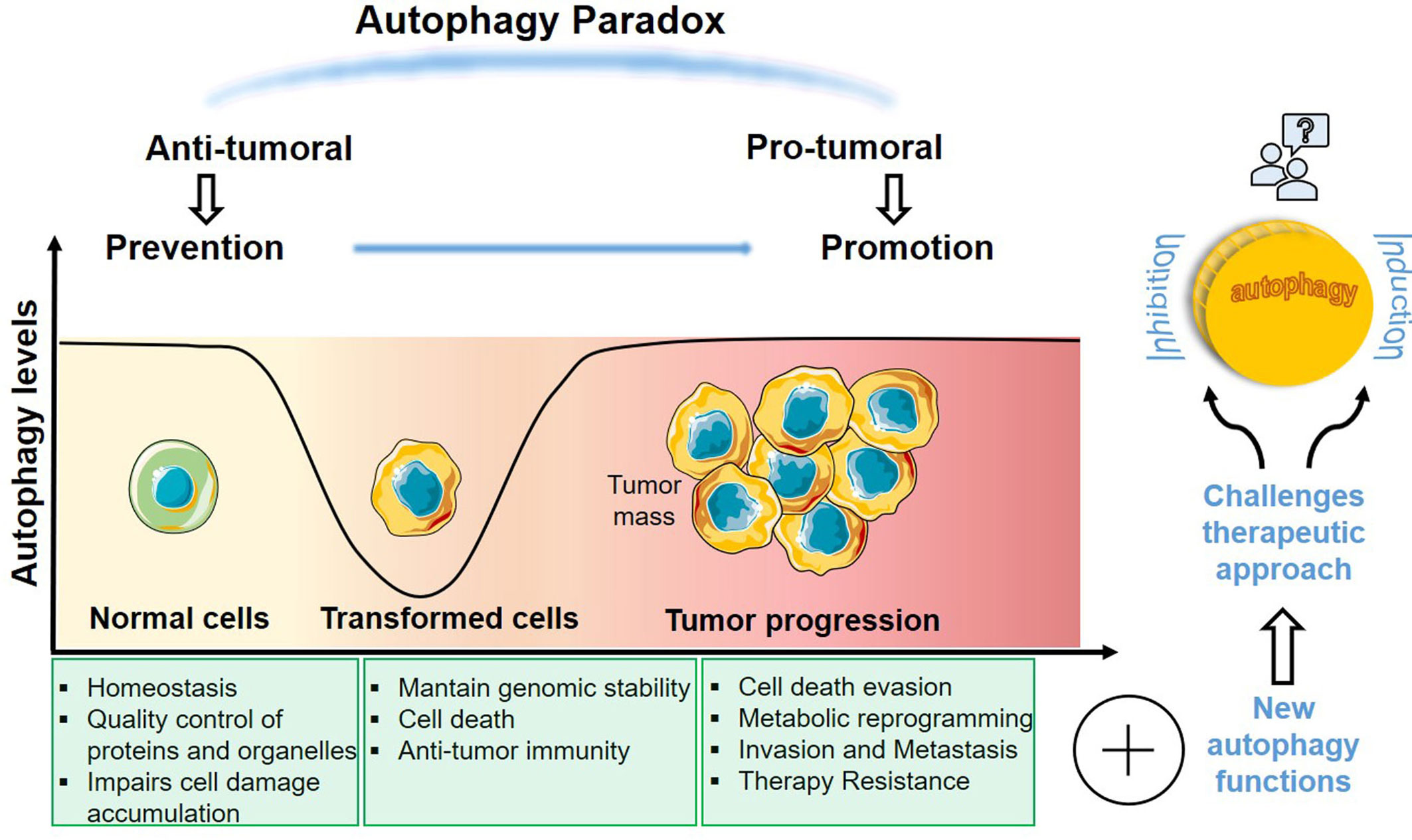
Figure 1 Dual role of autophagy in cancer. Autophagy is implicated in several stages of tumorigenesis executing different processes. The extensive and opposite functions in cancer makes autophagy an important target to develop new therapies. A deeper knowledge about this complex feature of autophagy in cancer research is essential to find more accurate therapeutic approaches.
The process of recycling cellular components performed by autophagy has been well characterized. Beyond self-eating and recycling damaged organelles, new roles for autophagy and the ATG genes have been ascribed (12, 13). Indeed, autophagy interferes in a wide range of cellular processes. Interestingly, components of autophagy can influence dynamic cellular processes and lead to tumor microenvironment (TME) reprogramming. Here, we discuss the novel roles of the autophagy machinery in tumor secretion, immune response, migration, and invasion capacity of tumor cells undergoing the epithelial to mesenchymal transition (EMT) (14, 15). These processes may occur simultaneously or not, affecting not only tumor cells, but also tumor microenvironmental components. These processes may also be interconnected and thus interfering with tumor progression, amplifying the roles of autophagy in tumor development and heterogeneity.
Among the diverse functions triggered by autophagy, “autophagy secretion” has received attention for its ability to alter the secretory profile of the tumor microenvironment, participating in the modulation of processes related to cancer progression (16, 17). Since the term autophagy secretion does not culminate in degradation into lysosomes, we adopt here, like some authors, the term autophagy-dependent secretion (ADS) (18). Nowadays, it is well established that some components of the autophagy route are involved in both conventional and unconventional secretion pathways (19). The conventional secretion route is the best-studied route for protein secretion; it can be regulated positively and negatively by autophagy components, for instance during protein recycling or through the selective clearance of secretory vesicles (20, 21). Unlike proteins exported through the conventional secretory pathway, the cargo delivered to the extracellular space or the plasma membrane by unconventional secretion (UPS) lacks the specific signal peptide and bypasses the classical Endoplasmic Reticulum (ER)-to-Golgi pathways of protein secretion (22). Usually, this pathway is activated by cellular stress, being an alternative route to proteins that use the conventional secretion (23).
Over the last few years, various studies have shown that autophagy takes part in the secretion of several proteins that critically contribute to tumor development. Among them are included different types of cargo, such as High Mobility Group Box 1 (HMGB1), IL1-β and other cytokines, immune mediators, and RNAs (18, 24, 25). Although the major complexes of classical autophagy and their molecular machinery have been clarified, novel and specific autophagy-dependent processes are still under investigation. Despite that, some evidence about the mechanism of intracellular traffic in ADS has emerged by the examination of unconventional secretion in yeast and the alternative route of IL1-β extracellular secretion (Table 1) (26, 49). Thereby, the results revealed that some markers involved in this pathway are shared with the classical autophagy program, but there are others exclusive to ADS. Findings of the machinery implicated in cargo selection and release have been suggested three different pathways.

Table 1 Evidence summary of studies related to molecular mechanisms and components of autophagy implicated in the three topics covered in this review: secretion, epithelial to mesenchymal transition (EMT) and immunity.
First, the ATG genes stimulate the generation of an intermediate membrane, not a regular autophagosome, required to LC3I lipidation (LC3II) and the cargo contained within the inner membrane is subsequently delivered extracellularly instead to the lysosomes (50). Second, leaderless proteins are translocated to the intermembrane space of an autophagosome and released directly by fusion with the plasma membrane or with multivesicular bodies (MVBs). The last process proposed consists of an MVB/amphisome intermediate (late endosome-MVB and autophagosome fusion) and the secretion of material in small extracellular vesicles (51).
Considering all strategies, recent studies have been shown that the ADS needs SNARE (soluble N-ethylmaleimide-sensitive fusion protein attachment protein receptors) proteins, as SEC22B, to prevent the fusion with lysosomes and drive the select target to the plasma membrane (52). Besides the regulation by ATG proteins, for instance, ATG5/ATG12/ATG16L1 complex and ATG3, the unconventional secretion also requires of cargo receptors as TRIM (Tripartite motif family) proteins, specifically TRIM16, as well as LC3, GRASP65 (Golgi reassembly-stacking protein)and the GTPase Rab8A of the Ras family, necessary for sorting the target to the plasma membrane (27, 53). Curiously, proteins implicated in extracellular vesicle secretion, like VCP and Rab7, were also found in autophagy pathways and conversely, key ATG proteins, such as ATG12/3 and ATG5, were identified as crucial regulators of exosome biogenesis (54–57). Thus, this data demonstrated a potential link between autophagy and extracellular vesicles (EVs) machinery, processes that contribute to cellular communication and signaling in the tumor microenvironment acting as modulators of tumor progression and aggressiveness. All data collected until now expose the relevance of the set of proteins released by ADS to contribute with some of the hallmarks of cancer (58).
Most of the proteins released by ADS can activate invasion and metastasis, induce resistance to cancer therapies and/or promote inflammation, helping tumor cells to mitigate stress. Such set includes cytokines that use unconventional routes, as TGF-β and IL1-β, both responsible for playing antagonistic roles within the tumor microenvironment, depending on the cellular context (59, 60). Despite that, many studies support their functions as tumor promotors influencing in the inflammatory response of immune cells and contributing to immune suppression, tumor growth, angiogenesis, and metastasis, as observed in breast cancer cells, where the secretion of IL1-β drives colonization of the bone microenvironment, establishing a metastatic niche and cell proliferation (61, 62).
Similar responses were observed in HMGB1 and ATP secretion. Extracellular HMGB1 induces pro-inflammatory cytokines and regulates other genes leading to cell migration and metastasis. HMGB1exerts pro-tumoral functions favoring prostate cancer cells survival and cancer progression (63). Simultaneously, HMGB1 is also responsible for autophagy induction. Regarding ATP, recent studies support its role in tumor survival by switching the ATP-gated receptor P2x to nfP2X7, a non-pore functional, that impairs the membrane permeability and the subsequent cell death (64). In addition, IL-6 and IL-8 secreted by autophagy are key determinants of the senescence-associated secretion phenotype (SASP), characteristic of senescence activated by DNA damage-mediated signals (65–67). High levels of both cytokines secreted by tumor cells and other cells such as cancer-associated fibroblasts (CAF) establish a senescent microenvironment and increase tumor aggressiveness showing a correlation with cancer progression and poor prognosis in many tumor types (68, 69). For example, in colorectal cancer, it was showed that peritoneal mesothelial cells control metastasis in SW480 cells and stimulate proliferation by the activation of senescence along with the secretion of mediators as IL-6 and IL-8 (70). Based on these findings and the prominent role of autophagy in cancer, various researchers have hypothesized a link between autophagy and senescence induction (71).
One of the first evidence of autophagy acting as an effector mechanism of senescence came from Young et al. (12), who demonstrated, in an oncogene-induced senescence model (OIS), that up-regulation of ATG genes induced autophagy and senescence, while the inhibition of ATG7 and ATG5 by shRNA delayed senescence. The OIS program is a dynamic process consisting of an initial phase of rapid proliferation and mTOR activation, a transition phase where diverse changes in morphology, signaling, translation and mTOR activity occur, culminating in a senescence phase, achieved by diverse senescence programs. Thus, autophagy is activated by stress, oncogenic stress, helping to shift the proliferative cell state to a senescent state through the fast protein remodeling and the synthesis/secretion of proteins as IL-6 and IL-8. Later, the same group demonstrated that autophagy is involved in IL-6, IL-8 secretion in a posttranslational manner since the mRNA levels remain stable in ATG knockdown cells. Secretion of these cytokines was further associated with a new type of autophagy called TOR- autophagy spatial coupling compartment (TASCC), which is located at the trans side of Golgi apparatus of senescent cells to accumulate autolysosomes, and mTOR1 facilitating the biosynthesis and secretion of proteins (20, 72). These secretion events were related to survival in tumor cells dependent on autophagy (73, 74). Moreover, several studies in different cell types endorsed the connection between these processes, but the mechanisms are not completely understood and occasionally contradictory, making it crucial to assess what type of autophagy program has been activated (75, 76). Collectively, there is evidence supporting pro-senescence and anti-senescence mechanisms induced by autophagy, including those promoting senescence under different conditions (77, 78).
As a pro-senescence program, a set of studies of Caparelli et al. (79–81), validated an autophagy-senescence transition (AST) process which consists of autophagy activation, metabolism alteration and the subsequent senescence induction in CAFs, responsible to promote tumor growth. They also showed that overexpression of CDK inhibitors (p16/p19/p21) was able to induce autophagy and senescence in CAFs and breast cancer cells favoring tumor promotion. Another study illustrated the notion that p53-mediated senescence is regulated by autophagy, which leads to the degradation of a p53 isoform capable of inhibiting the whole protein, and thereby inducing senescence (82). Likewise, the loss of p53 function can boost SASP in cells and promote tumor growth (83). However, the induction of senescence by wild type p53 has also been reported in different cellular contexts (84, 85). In a different approach, Knizhnik, and collaborators demonstrated that temozolomide triggers autophagy in glioma cells through the generation of DNA adducts, leading to senescence and not apoptosis, thus playing a role in cell survival rather than cell death (86). Besides, exposure of cancer cells to either chemotherapeutic agents or irradiation-induced autophagy is followed by cellular senescence. The entry to senescence has been described as a tumor suppressor mechanism limiting the replication of premalignant cells (75, 87). Although therapy-induced senescence has the intent to suppress cancer cell growth, senescent cells can also contribute with the survival of non-damaged neighboring cells. This protumoral effect of senescence, a bystander effect by SASP activation, may consequently stimulate invasion and tumor progression (88). Alternatively, studies in human fibroblasts showed that autophagy impairment by ATG7, ATG12 knockdown induces premature senescence mediated by activation of p53 and the generation of reactive oxidative species from dysfunctional mitochondria (89). In line with this, a work using a glioma model driven by oncogenic KRAS observed that autophagy inhibition using KRAS:shAtg7 cells predisposes cell to senescence, characterized by β-galactosidase activity and SASP markers (90). A similar outcome came from data of miR-212 in prostate cancer. Interestingly, the authors found that miR-212 is upregulated in benign regions compared with PCa tissues and responsible to negatively modulate autophagy, inducing premature senescence by inhibiting SIRT1. Thus, miR-212 controls senescence induction, acting as a tumor suppressor (91).
Over the last years, senescence has been considered an important process to fight cancer, encouraging the search for anti-cancer therapies based on the induction of cell senescence (92–94). However, studies based on therapy-induced senescence (TIS) indicated the emergence of adverse effects on cancer treatment (95, 96). Chemotherapy-induced SASP drives bone loss in breast cancer and its regulation by p38-MAPK-MK2 inhibition could preserve bone, improving the quality of life of patients (97). TIS may contribute to unwanted outcomes through the stimulation of inflammation by increased secretion of SASP factors, the induction of senescence-associated stemness phenotype or senescence cell scape and further proliferation recovery (98–100). Together, these findings attracted interest to autophagy-modulated senescence and the therapeutic responses associated with both processes since senescence has been implicated with maintaining tumor dormancy, and thus mediating cancer relapse (101, 102). Then, senescence has a potential pro-tumorigenic role supporting aggressiveness, survival responses and shorter recurrence-free survival in patients (103). Finally, regarding its pro-tumorigenic role, there is increasing evidence that SASP components are involved in the establishment of an immunosuppressive environment and in the induction of EMT in TME (104–107). Further studies addressing these novel functions of autophagy and senescence in the tumor microenvironment are warranted and may pave the way to novel targeted therapies that increase the efficacy of the existing cancer treatment modalities.
Besides being involved in the regulation of protein secretion and tumor cell immunogenicity, autophagy has also been implicated in the process of tumor cell invasion. One of the first associations between autophagy and the invasion process was evidenced by the capacity of epithelial cells to evade anoikis via autophagy, what enabled cancer cells migration and invasion (108). More recently, autophagy has been connected to epithelial-to-mesenchymal transition (EMT), a critical multistep process required for cancer cells to invade and metastasize (109, 110). During EMT, epithelial cells undergo profound molecular and biochemical changes to be transiently converted into mesenchymal cells to gain motility, invasiveness, stemness characteristics, and chemoresistance. Multiple embryonic signaling pathways cooperate in the initiation and progression of EMT, including TGFβ, WNT, Hedgehog, and Notch (109, 111).
Notably, there is a multifaceted link between autophagy-correlated and EMT-correlated signaling pathways, reflected by an intricate web of regulatory signaling pathways that converge on the regulation of EMT and autophagy, and that may alter the reciprocal equilibrium between these two processes (30). These pathways often activate EMT-transcription factors and are initially triggered by extracellular signals (112). Probably, the best characterized EMT inducer is TGFβ, known to trigger EMT through the activation of SMAD, PI3K/AKT, MAPK, and Rho-GTPases (112). During cancer progression, cells that undergo EMT require autophagy activation to survive the metastatic spreading. On the other hand, autophagy tends to inhibit the early phases of metastasis, contrasting the activation of the EMT mainly by selectively destabilizing crucial mediators of this process, such as TGFβ (Table 1). As part of the tumor-suppressive program dependent of TGFβ, it would promote autophagy in the early phases of tumor formation. On the other hand, later in tumor progression, TGFβ would restrain autophagy while inducing EMT and promoting metastatic spreading of cancer cells (30). Regarding TGFβ and the convergence of signaling pathways between both processes, it was identified recently that the autophagic activity mediated by the transcription factor EB regulates TGFβ signaling in melanoma. Blockage of the BRAFi-induced autophagy function led to an augment of EMT activation and metastasis by enhancing TGFβ signaling, which was responsible for driving tumor progression (113).
Based on the complex relationship between autophagy and EMT, controversies have emerged in the literature regarding the role of autophagy inhibition on EMT: while several studies implicate autophagy in the promotion of EMT, others have suggested the inverse, indicating that inhibition of autophagy could promote EMT and consequently induce cancer cell invasion. Although considerable evidence suggests that the inhibition of autophagy will improve cancer therapy and despite early phase clinical trials show promising results for the use of hydroxychloroquine for this purpose (114), others have highlighted possible undesirable effects of the inhibition of autophagy in cancer therapy (31, 115, 116). Supporting the beneficial effect of autophagy inhibition during cancer progression, there are several compounds and/or microenvironmental conditions that activate the EMT program, and can also induce an autophagic response in different types of cultured cancer and non-cancerous cells, impairing EMT. It has been suggested that EMT impairment could benefit the treatment efficacy of renal cell carcinoma with existing therapeutic regimen when combined to the autophagy inhibitor chloroquine, supporting the evidence that an EMT-like phenotype corresponds to a higher autophagic flux (15). In this regard, it has been also suggested that autophagy is required for EMT induction and metastasis in hepatoblastoma cells (117) and for TGFβ1-induced EMT in non-small-cell lung carcinoma cells (118). Additionally, autophagy induced by starvation was able to activate migration, invasion, and EMT marker expression upon rapamycin induction, and BECN1 knockdown reverted this phenotype. (119). Also, following mTOR signaling inhibition, which is known to induce autophagy, the migration, invasion and EMT marker expression were reduced in colorectal cancer cells (120). Moreover, autophagy is critical for hepatocellular carcinoma cells invasion through the induction of EMT and activation of TGF-β/Smad3-dependent signaling, which plays a key role in regulating autophagy-induced EMT (33), as well as is required for TGFβ2-induced EMT and reactive oxygen species (ROS) modulation in these cells (34). Another model of hepatocellular carcinoma revealed that inhibition of autophagy did not alter cell migration, invasion or EMT marker expression in vitro, however sensitized cells to anoikis and decreased lung metastases in vivo (121). Therefore, the role of autophagy in EMT seems context-dependent and indicates that the effects of autophagy inhibition in the establishment of metastasis are not necessarily due to its effects on EMT, but rather on its effects on other steps of the metastatic process or in the promotion of cell death. In this regard, autophagic stimulation of metastasis could be simply a consequence of its pro-survival activity against the apoptotic signals coming from changes in adhesion and cytoskeleton reorganization (121).
Ultimate evidence has indicated that autophagy activation could rather induce a reversion of the EMT phenotype and several anticancer compounds that induce autophagy also inhibit EMT (Table 2) (37, 122–129, 135). By its dual role in cancer, the effect of autophagy on EMT appears controversial and likely dependent on the cellular type and/or stage of tumor progression (130–134). Thus, at early stages of metastasis, autophagy could inhibit the EMT program mainly by destabilizing EMT crucial players. Later, metastatic cells could require sustained autophagy to survive environmental and metabolic stressful conditions encountered (30). Therefore, our efforts should be concentrated in selecting the precise approaches needed to stimulate or block autophagy in a time/context-dependent manner, to primarily suppress EMT and control cancer progression.
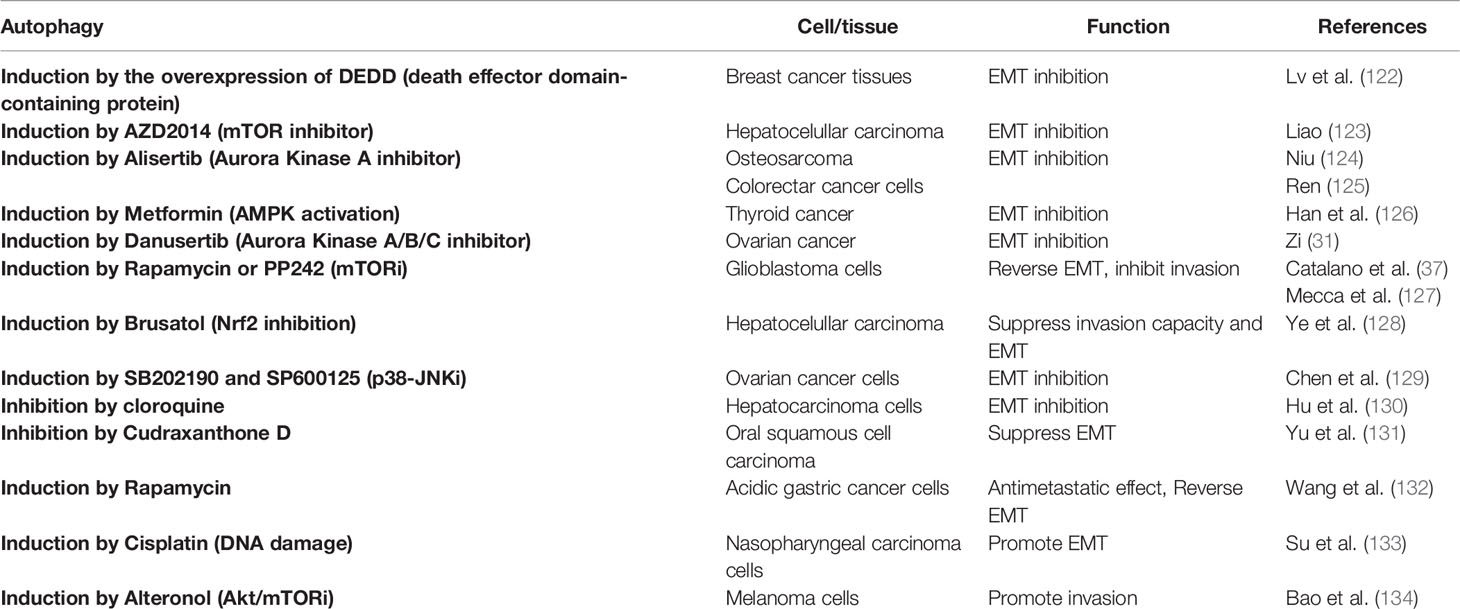
Table 2 Examples of autophagy modulation and its role in epithelial-mesenchymal transition (EMT) in cancer.
Once the transformation has occurred, autophagy can maintain cellular senescence to avoid the proliferation of transformed cells (109). Accumulating evidence indicates that autophagy is also a fundamental characteristic of stem cells, including cancer stem cells (CSCs). As CSCs are likely to play a central role in tumor dormancy, it appears that autophagy could contribute to the capacity of these cells to survive for extended periods of time in a dormant state and eventually give rise to recurrent tumors that are determinants of morbidity and mortality in cancer patients. Hence, once a tumor is established, tumor cells use autophagy as a survival mechanism to metabolic stress and hypoxia, to maintain tumor-related inflammation, CSCs survival and resistance to therapy.
The ‘reawakening’ of tumor cells at distant sites leading to the outgrowth of metastatic disease many years after primary tumors were treated has led to the concept of metastatic dormancy (136–138). Several studies have shown a role for autophagy in promoting cancer cells survival during dormancy (139). Autophagy may promote the dormancy of disseminated tumor cells simply by supplying key amino acids and other nutrients or, autophagy may play a more instructive role by eliminating mitochondria, modulating redox balance, and actively promoting the CSC state (136, 140, 141). However, it has been suggested that dormant tumor cells are CSCs that depend upon autophagy to survive at distant sites over extended periods of time to expand later as metastatic lesions composed of both CSCs and non-CSCs, representing the full heterogeneity of rapidly growing tumors (136, 139). Indeed, dormant disseminated cancer cells can survive for several years before recurring as extremely aggressive metastatic tumors. There are relevant observations providing insights into the connection between autophagy and dormancy. Despite the autophagy-associated dormancy has not been fully elucidated and some results seem controversial or related to specific phenomena, several studies recognize its crosstalk with cancer relapse (142). Findings in dormant breast cancer cells support that autophagy is crucial to promote their metastasis and survival, probably preventing the accumulation of ROS and damaged organelles (143). Additionally, increased unfolded protein response (UPR) markers have been found in dormant cells. Considering the established link between autophagy and UPR under stress conditions, it has been suggested that UPR-induced autophagy activation in dormant cells to sustain tumor survival (144, 145). Breast cancer stem cells (BCSCs), which undergo tumor initiation and unlimited self-renewal, also exhibit dormancy-associated phenotypes by upregulating autophagy during metastatic dormancy to survive environmental stress and nutrient poor conditions. Consequently, therapeutic targeting of autophagy is actively being pursued as an attractive strategy to alleviate metastatic disease and the recurrence of dormant BCSCs (146).
In conclusion, dormant cancer cells are especially dependent on autophagy for survival, which provides a rationale for combining autophagy inhibition with conventional therapeutic strategies to eliminate these cells and prevent subsequent metastatic outgrowth (147).
As mentioned before, autophagy has an important role in innate and adaptative immunity and can act in several steps of the immune response, leading to its activation or inhibition, depending the context, taking part in tumor immunosurveillance. Besides the ability to modulate the TME through its secretory function, autophagy regulates cellular components (natural killer (NK) cells, dendritic cells (DC), macrophages and lymphocytes (T and B cells)) of immune response, acting on differentiation, proliferation, activation, survival and homeostasis of these cells (Table 1). Moreover, autophagy acts on cytokines (interleukins (IL), interferons (IFN), transforming growth factors (TGF)) and antibodies production as well as phagocytosis. Interestingly, cytokines can act as autophagy stimulators or autophagy inhibitors (148).
Autophagy also has a role in tumor response to immunotherapy and a better understand of autophagy-modulation of innate and adaptative immune response could contribute to better strategies to circumvent immunotherapy resistance. For example, autophagy enhances antigen delivery to immune cells (antigen-presenting cells (APCs) and CD8+ cytotoxic T lymphocytes) and in this way can initiate an immune response against tumor cells and enhance immunotherapy efficacy. However, in the case of cancer development, autophagy is a double edge sword for immunity since it can inhibit immune response and attenuate immunotherapy outcomes (149, 150).
Innate immunity is the first defense of eukaryotic cells against invading pathogens and autophagy participates in the process with autophagy adaptor proteins that interact with pattern recognition receptors and activates immune response together with elimination of intracellular invaders (47). The activation of innate immune receptors as Toll-like receptors (TLRs) and nucleotide oligomerization domain-like receptors (NLRs) induces innate-immunity-mediated autophagy upregulation (150–152). TLRs interact with pathogens on the cell surface and usually are also expressed in cancer cells, inducing cytokines production together with NF-κB and MAPK pathways activation (151–153). TLRs are proposed to activate autophagy as demonstrated for TLR2 that enhances innate immunity through ERK and JNK signaling pathways after autophagy stimulation, and also boosts autophagy in glioma cells being correlated with poorer patients outcome (154, 155). For TLR4, the autophagy induction is mediated by TRIF (Toll-IL-1 receptor adapter-inducing IFN)/RIP1 (receptor-interacting protein)/p38-MAPK axis (156). TLR4 and TLR3 activation after LPS (lipopolysaccharides) treatment induces autophagy by TRIF pathway, which contributes to TRAF6 ubiquitination followed by MAPK and NF- κB activation and harmful cytokine production, leading to lung cancer cell migration and invasion (157). In p62 knockout cancer cells, stimulation of TLR4 induced activation of the TRAF6-BECN1-autophagy axis leads to cancer cell migration and invasion (158). Additionally, in patients with luminal breast cancer, higher levels of TLR4 and accumulation of LC3II were observed in CAFs. These features were associated with a more aggressive relapse and poorer prognosis in the cohort of patients studied (159). Taken together, TLR and autophagy activation can contribute to tumor development since it enhances survival and proliferation of cancer cells and also triggers the release of cytokines and immunosuppressive factors, contributing to immune evasion and tumor cell resistance (160).
NLR family members, such as NOD1 and NOD2, that recognize intracytoplasmic pathogens, can also activate NF-κB and MAPK pathways and produce immunosuppressive cytokines, as well as induce autophagy by recruiting ATG16L1 (161, 162). Both NOD1 and NOD2, altering the balance of anti- and pro-inflammatory cytokines, can modulate the risk of cancer development (163). For example, in triple negative breast cancer (TNBC), the expression of NOD1 and NOD2 is associated with cancer progression and a global proteome profiling of TNBC-derived cells overexpressing these receptors demonstrated disrupted immune-related pathways such as NF- κB and MAPK signaling and autophagy (164).
Autophagy participates in adaptative immune response such as thymus selection, lymphocyte development and homeostasis, antigen presentation and cytokine release, exerting anti-tumor effects (47, 165). Adaptative immunity occurs when extracellular or intracellular peptide epitopes are presented by APCs through the major histocompatibility complex (MHC) class I and II to CD8+ and CD4+ T cells, respectively. The interaction of antigen and T cell receptors triggers cellular (cytotoxic lymphocytes) and humoral (antibody-producing B cells) adaptative immune response (42, 166). The efficient antigen presentation requires proteasomal or lysosomal antigen degradation and delivery of resulting peptides to MHC molecules and this step can be enhanced by autophagy, for example, in APCs upon uptake of extracellular antigens (e.g. tumor antigens) and in antigen processing for MHC I cross- presentation. Autophagosomes facilitate intracellular trafficking of these antigens to endosomes to be degraded by cathepsins followed by peptide load onto MHC II molecules that mature and get translocated to the plasma membrane and present antigens to CD4+ T cells (42, 166). A non-canonical regulation of phagocytosis by ATG proteins can also be used to engulfment of extracellular antigens, which is known as LC3-associated phagocytosis (LAP), characterized by a single membrane vesicle decorated with LC3-II instead of double-membrane autophagosome as in autophagy (Table 1) (42, 167).
Autophagy has multiple roles on immune cells acting during their differentiation, proliferation, activation, and homeostasis maintenance and in this setting can also promote or inhibit tumor development (150). Dendritic cells link the innate and adaptative immune system as they are powerful professional APCs. Autophagy is involved in different DC functions both in physiological and pathological conditions (168). The inhibition of autophagy impacts the ability of DCs to process and present cytoplasmic antigens through the MHC II pathway and cytokines secretion, which increases their immunostimulatory phenotype (169–171). Macrophages are also APCs that require autophagy during the differentiation process in monocytes from the bone marrow into macrophages in tissue site (43, 172). Granulocyte-macrophage colony-stimulating factor (GM-CSF) is a signal to maturation and prevents monocytes apoptosis together with autophagy induction. When autophagy is downregulated either by BECN1 knockdown or pharmacological inhibition using 3-methyladenine (3-MA) and chloroquine, caspases are activated and cytokine production is prevented (43). Autophagy is also involved in macrophage polarization with its inhibition leading to the classical activation profile and augmented pro-inflammatory cytokines secretion, and its induction promoting macrophage alternative activation, resulting in increased production of anti-inflammatory cytokines (173).
T cells use basal autophagy to maintain organelle homeostasis and it can be induced after T cell antigen receptor (TCR) stimulation. Moreover impaired autophagy after deletion of ATG proteins (ATG3, ATG5, and ATG7), BECN1 or Vps34 can hinder T cell survival, proliferation, differentiation, and activation (150, 174–176). Autophagy proteins may also be involved in other functions besides autophagy as demonstrated for memory CD8+ T cell, in which UVRAG (ultraviolet radiation resistance-associated protein) deletion does not impair autophagy but affects proliferation (177). On CD4+ T cells, autophagy impairment after BECN1 deletion leads to apoptosis upon TCR stimulation (178). On the other hand, blockage of mTOR signaling after rapamycin treatment in effector CD8+ T cells can enhance memory CD8+ T cells in lymphoid tissue or inhibit them in mucosal tissue (179). In antigen-specific memory CD8+ T cells, deficient autophagy leads to the accumulation of damaged mitochondria and increased apoptosis (180). Moreover, mTOR status can also interfere with T cell differentiation since its induction lead to activated T cell to differentiate into Th cells and its downregulation together with AMPK induction cause naïve T cells differentiation into regulatory T (Treg) cells (181). The metabolic profile also influences the dependence on autophagy since cells as memory lymphocytes and Treg cells, that use more oxidative phosphorylation (OXPHOS), are more dependent on autophagy for homeostasis than effector T cells that use preferentially aerobic glycolysis (45, 181, 182). On Treg cells, impaired autophagy induces mTORC1 and MYC signaling pathways, contributing to apoptosis induction (182).
For B cells, autophagy participates during cell development to support extremely high metabolic demands for their differentiation and reaches maximal levels during the earliest stages of development and diminish as B cells mature. It is also important, at basal levels, to maintain peripheral B cell numbers as required to cell survival after LPS stimulation, as well as for IgM production after immunization. However, autophagy is not essential for transition of pro- to pre-B cell stages in the bone marrow and B cell activation after BCR stimulation (183). Mature B cells with impaired autophagy (Atg5-/-) accumulate damaged organelles and have enlarged endoplasmic reticulum together with ER stress, more antibody secretion and plasma cells apoptosis (184).
Autophagy is also required for NK cell differentiation, since it regulates the number and quality of mitochondria on proliferating NK cells and enhances memory NK cells in an ATG3 dependent manner (185). In invariant natural killer cells (iNKT), it is observed a high level of autophagy during iNKT cell thymic differentiation into memory cells to regulate mitochondrial content and ROS production. A conditional deletion of Atg7 gene in T-cell compartment blocked iNKT development and maturation, as well augmented its susceptibility to apoptosis (46). In another study, the deletion of Atg5 or Atg7 decreased iNKT mature cells and IL-4 and IFN-γ levels accompanied by an increase in apoptosis (186).
In neutrophils, autophagy deficiency has no impact on their morphology, migration, granular content, apoptosis or effector functions, but in autophagy-deficient mice, neutrophil proliferation and differentiation is augmented, indicating an inverse correlation between autophagy and neutrophil differentiation (187).
As mentioned previously, there are feedback loops between autophagy and different cytokines. For IL-1 (IL-1α and IL-1β), autophagy limits its secretion although it is observed autophagy induction by these cytokines, indicating a negative feedback mechanism (188, 189). The interferon family (IFN types I and II) also induces autophagy in epithelial, immune, and tumor cells (190, 191). IL-2, IL-12, and TGF-β also stimulate autophagy (192–194). On the other hand, IL-6 has an anti-autophagic effect in starvation-induced autophagy in U937 cells (195) but stimulates autophagy in B cells (196). IL-10 also inhibits starvation-induced autophagy in DCs (197).
Autophagy can also act in immune tolerance mediated by immunotherapy strategies since immunologic molecules such as indoleamine 2,3-dioxygenase (IDO), PD-1 and CTLA-4 can be regulated by autophagy pathways. IDO is found in tumor sites and has anti-tumor immunity effects through interference with a cytotoxic T-cell response, DC maturation and increase in Treg population, promoting immunologic tolerance and tumor development, but its production can be inhibited by autophagy stimulation (198, 199). PD-1 from tumor cell surface interacts with PD-L1 on T-cells and acts as an inhibitory checkpoint molecule, preventing recognition of tumor cells, suppressing T cell proliferation, development, and anti-tumor immunity. It has been reported that the interaction of PD1 with its ligand limits nutrients availability to nearby T-cells, promoting autophagy induction (200). Treatment with Sigma1 can induce autophagy in co-cultured T-cells and tumor cells, leading to degradation of PD-L1 and suppression of PD-1 and PD-L1 interaction, which could favor immunotherapy effects due to immune microenvironment modulation (201). However, the expression of PD-L1 affects several genes involved in mTOR signaling and autophagy. When PD-L1 is lost, it is observed autophagy upregulation and less sensitization to autophagy inhibitors to reduce tumor cell proliferation (202). CTLA-4 is another immune tolerance checkpoint and an effective target for tumor treatment. In human melanomas, over-expression of MAGE-A, a cancer-germline antigen, is associated with CTLA-4 blockade resistance and can downregulate autophagy, suggesting autophagy induction as a potential therapeutic approach to improve CTLA-4 inhibitors efficacy (203).
Increasing data suggest that autophagy can interfere with antitumor immunity together with tumor development and survival (149, 150). Knockdown of ATG5 in cancer cells was followed by increased induction of DC maturation, production of IL-6 and IFN-γ along with the proliferation of CD4+ and CD8+ T-cells after an immunogenic cell death inducer treatment (204). In Treg cells, autophagy is an important and active process to support their homeostasis contributing to their immunosuppressive profile. Suboptimal NK cell activity induces autophagy in surviving tumor cells, leading to treatment resistance (205). As mentioned before, many pro-inflammatory cytokines contribute to tumor growth, metastasis and can induce autophagy (206). Although the treatment with high-dose-IL-2 has antitumor effects it is limited by severe side effects, as multiorgan dysfunction that is accompanied by systemic autophagic syndrome induced by cytokines. In a murine model of metastatic liver tumor, the combined treatment of high-dose-IL2 and chloroquine increased antitumor effects along with decreased toxicity, increased long-term survival and enhanced infiltration of immune cells in liver (207). In a renal cell carcinoma model, autophagy inhibition also improved HDIL-2 anti-tumor effects due to apoptosis induction and immune system stimulation together with increased activity of DCs, T-cells, and NK cells (208). Inhibition of autophagy through 3-MA treatment also potentiates apoptosis induced by IL-24 in oral squamous cell carcinomas, demonstrating that autophagy inhibition can be explored as a promising approach to increase immunotherapy efficacy (209). The phytochemical shikonin can induce necroptosis accompanied by autophagy enhancement that directly contributes to DAMP upregulation in tumor cells. However, if autophagy flux is blocked by chloroquine treatment, there is an even greater upregulation of ectoDAMPS, resulting in DC activation. In the context of DC vaccines, the pretreatment of tumor cells with chloroquine and shikonin potentiated antimetastatic activity and reduced chemotherapy doses in vivo (210). Autophagy can also reduce immunotherapy effect by impairing cytotoxic T-lymphocyte (CTL)-mediated tumor cell lysis when autophagy is induced under hypoxia conditions, activating STAT3 signaling in target cells which in turn favors tumor cell survival, proliferation, and immune escape. If autophagy is blocked in this context and CTL-response is boosted with a vaccination strategy, vaccination efficacy is improved, leading to tumor regression in vivo (211). Hypoxia-induced autophagy also impairs NK cells function by degradation of NK-derived granzyme B in autophagosomes of hypoxic breast cancer cells, leading them less susceptible to NK killing and immunotherapy effects. However, if autophagy is blocked by deletion of BECN1, granzyme B levels are restored and favors tumor regression in vivo due to tumor cell death by NK-mediated lysis (212). Pancreatic ductal adenocarcinoma (PDAC) is known for immune checkpoint blockade resistance and frequently altered MHC-I expression that facilitates immune evasion trough NBR1 selective autophagy downregulation of MHC-I. If autophagy is inhibited, there is recover in MHC-I expression and augmented immunotherapy response along with enhanced T-cell immunity in tumor models in vivo (213). In a cohort of gastric cancer (GC) patients, the expression of CXCL10 has a positive correlation with patient prognosis and induces T lymphocyte migration and infiltration into the GC 3D cell culture model. It is also observed in basal conditions that GC cells have increased autophagy, and the knockdown of essential autophagy genes (Atg5 and Atg7) or their pharmacological block in these cells augmented CXCL10 expression under normal and hypoxic conditions facilitating T cell lymphocyte migration and potentiating tumor immunity (214). Recent studies demonstrated that autophagy activation in tumor cells is one of the main reasons for decreased antitumor immune response, reinforcing the concept of autophagy inhibition as a valuable approach to increase immunotherapy results. One of the autophagic proteins that have been recently described as drug targetable is Vps34, whose inhibition with genetic or pharmacological approaches decreased tumor growth along with increased mice survival due to infiltration of immune cells (NK, CD8+ and CD4+ T effector cells) within the tumor microenvironment, which could turn cold tumors into hot inflamed tumors to enable immunotherapy treatments. Moreover, the combined treatment of Vps34 inhibitor and anti-PD-L1/PD-1 in melanoma and colorectal cancer models prolonged mice survival and enhanced immunotherapy benefits (215). Impairment of autophagy with BECN1 ablation is also beneficial to increase NK infiltration and inhibit tumor growth in a melanoma tumor model. In addition, NK infiltration in the tumor microenvironment is mediated by CCL5 chemokine overexpression in autophagy-deficient cells trough c-Jun/JNK activation. Similar results were also obtained after deletion of other autophagic genes as Atg5 and SQSTM1/p62 and pharmacological inhibition by chloroquine. In conclusion, targeting autophagy may be a valuable approach to improve immunotherapy mediated by NK cells (216).
On the other hand, autophagy has also anti-tumoral effects since its induction contributes to a better response of helper T lymphocytes (HTLs) against head and neck squamous carcinoma cells and its inhibition decreases HTLs recognition of tumor cells (217). Tumors can act as antigen donor cells that require autophagy to form tumor-derived autophagosomes (Dribbles) which contain tumor-associated antigens. Dribbles can stimulate efficient cross-presentation of T-cells (218) and induce B cell activation along with cytokine release and antibody production (219) which can contribute to tumor control and elimination. For vaccination strategies, in contrast to whole-cell tumor vaccine, Dribbles prime T cells by enhancing costimulatory molecules as well as MHCI, and reduce tumor formation on hosts challenged with nonhomologous tumors, effect limited if there is depletion of the autophagic protein SQSTM1/p62 (220, 221). For efficient immunotherapy, tumor antigens should be immunogenic, essential only for tumor cells and overexpressed in tumors compared to normal tissue. Recently, it was demonstrated that SQSTM1/p62 fits all these requisites and a DNA-vaccine encoding this protein had antitumor and antimetastatic effect against several tumor models in dogs, suggesting that this can be a useful strategy for immunotherapy (222). Inactivation of ATG5 in non-small cell lung carcinoma favors carcinogenesis and its development is accelerated in KRASAtg5(fl/fl) autophagy-deficient mice. In these mice, a higher expression of ENTPD1/CD39 was observed, culminating in an immunosuppressive environment along with increased Treg infiltration that contributes to tumor development (223). As discussed previously, autophagy plays a role in monocytes/macrophages recruitment, what decreases infiltration in liver tissues accompanied by autophagy reduction and hepatocarcinogenesis (224). Immunotherapy can also be potentiated by autophagy. For instance, in murine tumor models, the treatment with chemotherapy or radiotherapy induced autophagy, which favored translocation of the mannose-6-phosphate receptor (MPR) from autophagosomes to tumor cells surface, rendering the cells more sensitive to granzyme B from activated CTLs, potentiating CTLs killing and immunotherapy (225, 226). In another example, autophagy elicited by alpha-tocopheryloxyacetic acid (α-TEA) on tumor cells improved cross-presentation of tumor antigens for MHCI and MHCII, which can be use as an adjuvant strategy to improve anti-tumor immune responses and strength immunotherapy (227, 228). Treatment of ovarian cancer models with farletuzumab, a humanized monoclonal antibody against folate receptor α, also induced autophagy and reduced proliferation, which was reversed by autophagy pharmacological blockage (229). Finally, studies indicate that although statins have no protective effect on breast cancer incidence, they can be used as adjuvant therapy to increase apoptosis and radio-sensitivity along with proliferation and invasion inhibition of cancer cells. Fluvastatin belongs to the statin family and when used in vitro to treat breast cancer cells, it induced autophagy but with impaired lysosome function which may contribute to cell death. Moreover, a decrease of pro-inflammatory cytokines, such as IL-6 and TNF-α, was observed along with autophagy consequent effect in tumor immunity (230).
Given the dual role of autophagy in cancer and its involvement in cancer therapeutic responses, the process of autophagy has been pointed as an important theme in cancer research (Figure 1). The interconnection of autophagy to the regulation of several biological processes in the TME indicates that autophagy has key roles in tumor progression. Thus, in addition to its primary function of degradation and recycling, most of the components of the autophagy machinery also mediate numerous non-autophagic functions. This suggests that autophagy operates in many ways, establishing an intricate network of signaling, along with other cell elements, integrating diverse signals within the TME and regulating the fate of cancer and other microenvironmental cells (Figure 2). Integrative approaches, which address the impact of autophagy inhibition in complex systems, are therefore necessary for the development of strategies that exploit the autophagy machinery as a target to control tumor growth, without impeding the generation of a long-lasting memory cytotoxic immune response or the induction of a stemness phenotype in residual cancer cells. Models of the intricated and dynamic network of cancer cells and the tumor microenvironmental cells are warranted for filtering compounds that may control tumor growth and increase the efficacy of many known therapeutic regimens.
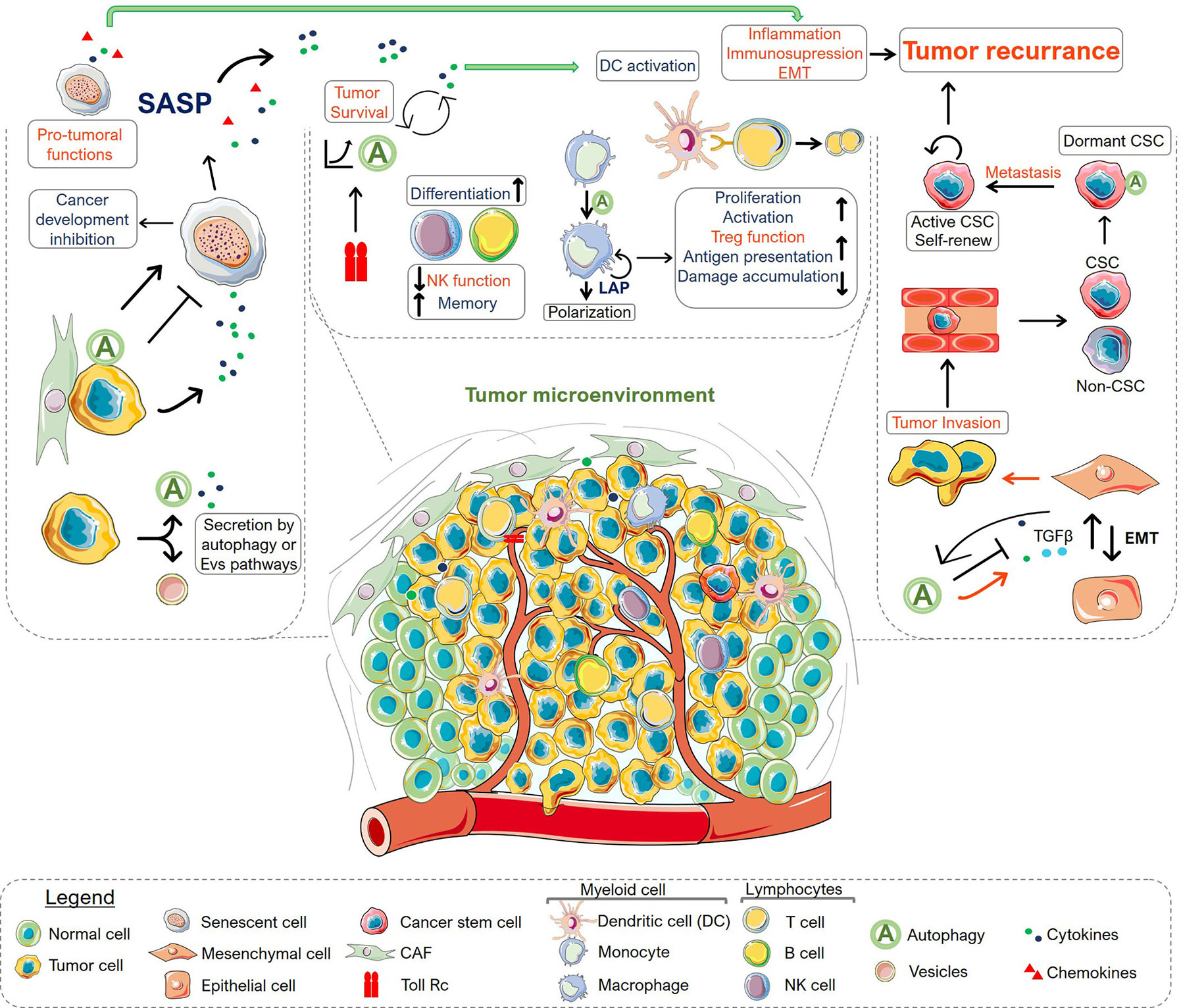
Figure 2 Overview of autophagy roles in the tumor microenvironment. The scheme summarizes the role of autophagy in secretion (left), immune system (middle), epithelial-mesenchymal transition (EMT) and tumor dormancy (right). There is an intricate and dynamic network of signaling circuits that drive tumor development and progression within the tumor microenvironment. The connectivity among various processes may regulate the fate of the microenvironment components, indicating the importance of viewing this as an emerging system, where the resulting interactions are larger than the sum of the individual parcels. Autophagy can act in many ways in different types of cells displaying anti-tumoral (shown in blue) or pro-tumoral functions (shown in red). Protein secretion by CAF or tumor cells can modulate cellular states inducing or inhibiting senescence, which ultimately can control tumor survival, immune cell response and interfere with the epithelial-mesenchymal transition, affecting tumor invasion capacity. In the context of the immune system, autophagy has a key role in immune cell differentiation, proliferation, activation and effector function, covering the range of homeostatic to reactive functions of the immune system. At the same time, autophagy is also connected with the innate immune response being controlled by receptors such as TLRs. Importantly, in advanced stages, the autophagy system in tumor cells is involved with EMT and the consequent ability of cancer cells to invade tissues and metastasize. The interplay among these functions contributes to tumor aggressiveness. Moreover, autophagy was also appointed as a characteristic of cancer stem cells (CSC) playing a central role in tumor dormancy. Altogether, the myriad of connected process regulated by autophagy in the TME modulate tumor response and may determine its regression or progression. Altogether, understanding the integrated mechanisms that regulate autophagy within the TME constitute a niche for development of novel strategies for combination therapy.
SB, FA, MR, and RC wrote the manuscript. SB drew the figures. SB and RC designed and edited the review. All authors contributed to the article and approved the submitted version.
The authors’ work has been funded by grants from Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP 2014/03742-0 and 2015/22814-5) and Conselho de Desenvolvimento Científico e Tecnológico (CNPq 305700/2017-0).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. de Duve C, Wattiaux R. Functions of lysosomes. Annu Rev Physiol (1966) 28:435–92. doi: 10.1146/annurev.ph.28.030166.002251
2. Arstila AU, Trump BF. Studies on cellular autophagocytosis. The formation of autophagic vacuoles in the liver after glucagon administration. Am J Pathol (1968) 53(5):687–733.
3. Bolender RP, Weibel ER. A morphometric study of the removal of phenobarbital-induced membranes from hepatocytes after cessation of threatment. J Cell Biol (1973) 56(3):746–61. doi: 10.1083/jcb.56.3.746
4. Tsukada M, Ohsumi Y. Isolation and characterization of autophagy-defective mutants of Saccharomyces cerevisiae. FEBS Lett (1993) 333(1-2):169–74. doi: 10.1016/0014-5793(93)80398-E
5. Harding TM, Morano KA, Scott SV, Klionsky DJ. Isolation and characterization of yeast mutants in the cytoplasm to vacuole protein targeting pathway. J Cell Biol (1995) 131(3):591–602. doi: 10.1083/jcb.131.3.591
6. Klionsky DJ, Cregg JM, Dunn WA, Emr SD, Sakai Y, Sandoval IV, et al. A unified nomenclature for yeast autophagy-related genes. Dev Cell (2003) 5(4):539–45. doi: 10.1016/S1534-5807(03)00296-X
7. Mathew R, Karantza-Wadsworth V, White E. Role of autophagy in cancer. Nat Rev Cancer (2007) 7(12):961–7. doi: 10.1038/nrc2254
8. Qu X, Yu J, Bhagat G, Furuya N, Hibshoosh H, Troxel A, et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest (2003) 112(12):1809–20. doi: 10.1172/JCI20039
9. Liang C, Feng P, Ku B, Dotan I, Canaani D, Oh BH, et al. Autophagic and tumour suppressor activity of a novel Beclin1-binding protein UVRAG. Nat Cell Biol (2006) 8(7):688–99. doi: 10.1038/ncb1426
10. Corazzari M, Fimia GM, Lovat P, Piacentini M. Why is autophagy important for melanoma? Molecular mechanisms and therapeutic implications. Semin Cancer Biol (2013) 23(5):337–43. doi: 10.1016/j.semcancer.2013.07.001
11. Huang YH, Lei J, Yi GH, Huang FY, Li YN, Wang CC, et al. Coroglaucigenin induces senescence and autophagy in colorectal cancer cells. Cell Prolif (2018) 51(4):e12451. doi: 10.1111/cpr.12451
12. Young AR, Narita M, Ferreira M, Kirschner K, Sadaie M, Darot JF, et al. Autophagy mediates the mitotic senescence transition. Genes Dev (2009) 23(7):798–803. doi: 10.1101/gad.519709
13. Galluzzi L, Green DR. Autophagy-Independent Functions of the Autophagy Machinery. Cell (2019) 177(7):1682–99. doi: 10.1016/j.cell.2019.05.026
14. Wu H, Huang S, Chen Z, Liu W, Zhou X, Zhang D. Hypoxia-induced autophagy contributes to the invasion of salivary adenoid cystic carcinoma through the HIF-1α/BNIP3 signaling pathway. Mol Med Rep (2015) 12(5):6467–74. doi: 10.3892/mmr.2015.4255
15. Singla M, Bhattacharyya S. Autophagy as a potential therapeutic target during epithelial to mesenchymal transition in renal cell carcinoma: An in vitro study. BioMed Pharmacother (2017) 94:332–40. doi: 10.1016/j.biopha.2017.07.070
16. Dupont N, Jiang S, Pilli M, Ornatowski W, Bhattacharya D, Deretic V. Autophagy-based unconventional secretory pathway for extracellular delivery of IL-1β. EMBO J (2011) 30(23):4701–11. doi: 10.1038/emboj.2011.398
17. Martins I, Wang Y, Michaud M, Ma Y, Sukkurwala AQ, Shen S, et al. Molecular mechanisms of ATP secretion during immunogenic cell death. Cell Death Differ (2014) 21(1):79–91. doi: 10.1038/cdd.2013.75
18. New J, Thomas SM. Autophagy-dependent secretion: mechanism, factors secreted, and disease implications. Autophagy (2019) 15(10):1682–93. doi: 10.1080/15548627.2019.1596479
19. Ponpuak M, Mandell MA, Kimura T, Chauhan S, Cleyrat C, Deretic V. Secretory autophagy. Curr Opin Cell Biol (2015) 35:106–16. doi: 10.1016/j.ceb.2015.04.016
20. Narita M, Young AR, Arakawa S, Samarajiwa SA, Nakashima T, Yoshida S, et al. Spatial coupling of mTOR and autophagy augments secretory phenotypes. Science (2011) 332(6032):966–70. doi: 10.1126/science.1205407
21. Lock R, Kenific CM, Leidal AM, Salas E, Debnath J. Autophagy-dependent production of secreted factors facilitates oncogenic RAS-driven invasion. Cancer Discovery (2014) 4(4):466–79. doi: 10.1158/2159-8290.CD-13-0841
22. Viotti C. ER to Golgi-Dependent Protein Secretion: The Conventional Pathway. Methods Mol Biol (2016) 1459:3–29. doi: 10.1007/978-1-4939-3804-9_1
23. Gee HY, Noh SH, Tang BL, Kim KH, Lee MG. Rescue of ΔF508-CFTR trafficking via a GRASP-dependent unconventional secretion pathway. Cell (2011) 146(5):746–60. doi: 10.1016/j.cell.2011.07.021
24. Cadwell K, Debnath J. Beyond self-eating: The control of nonautophagic functions and signaling pathways by autophagy-related proteins. J Cell Biol (2018) 217(3):813–22. doi: 10.1083/jcb.201706157
25. Rak J. L(C3)icensing of exosomes for RNA export. Nat Cell Biol (2020) 22(2):137–9. doi: 10.1038/s41556-020-0466-3
26. Duran JM, Anjard C, Stefan C, Loomis WF, Malhotra V. Unconventional secretion of Acb1 is mediated by autophagosomes. J Cell Biol (2010) 188(4):527–36. doi: 10.1083/jcb.200911154
27. Kimura T, Jia J, Kumar S, Choi SW, Gu Y, Mudd M, et al. Dedicated SNAREs and specialized TRIM cargo receptors mediate secretory autophagy. EMBO J (2017) 36(1):42–60. doi: 10.15252/embj.201695081
28. Noh SH, Gee HY, Kim Y, Piao H, Kim J, Kang CM, et al. Specific autophagy and ESCRT components participate in the unconventional secretion of CFTR. Autophagy (2018) 14(10):1761–78. doi: 10.1080/15548627.2018.1489479
29. Rabouille C, Malhotra V, Nickel W. Diversity in unconventional protein secretion. J Cell Sci (2012) 125(Pt 22):5251–5. doi: 10.1242/jcs.103630
30. Gugnoni M, Sancisi V, Gandolfi G, Manzotti G, Ragazzi M, Giordano D, et al. Cadherin-6 promotes EMT and cancer metastasis by restraining autophagy. Oncogene (2017) 36(5):667–77. doi: 10.1038/onc.2016.237
31. Zi D, Zhou ZW, Yang YJ, Huang L, Zhou ZL, He SM, et al. Danusertib Induces Apoptosis, Cell Cycle Arrest, and Autophagy but Inhibits Epithelial to Mesenchymal Transition Involving PI3K/Akt/mTOR Signaling Pathway in Human Ovarian Cancer Cells. Int J Mol Sci (2015) 16(11):27228–51. doi: 10.3390/ijms161126018
32. Wang H, Zhang C, Xu L, Zang K, Ning Z, Jiang F, et al. Bufalin suppresses hepatocellular carcinoma invasion and metastasis by targeting HIF-1α via the PI3K/AKT/mTOR pathway. Oncotarget (2016) 7(15):20193–208. doi: 10.18632/oncotarget.7935
33. Li J, Yang B, Zhou Q, Wu Y, Shang D, Guo Y, et al. Autophagy promotes hepatocellular carcinoma cell invasion through activation of epithelial-mesenchymal transition. Carcinogenesis (2013) 34(6):1343–51. doi: 10.1093/carcin/bgt063
34. Dash S, Sarashetti PM, Rajashekar B, Chowdhury R, Mukherjee S. TGF-β2-induced EMT is dampened by inhibition of autophagy and TNF-α treatment. Oncotarget (2018) 9(5):6433–49. doi: 10.18632/oncotarget.23942
35. Li S, Zhang HY, Du ZX, Li C, An MX, Zong ZH, et al. Induction of epithelial-mesenchymal transition (EMT) by Beclin 1 knockdown via posttranscriptional upregulation of ZEB1 in thyroid cancer cells. Oncotarget (2016) 7(43):70364–77. doi: 10.18632/oncotarget.12217
36. Cicchini M, Chakrabarti R, Kongara S, Price S, Nahar R, Lozy F, et al. Autophagy regulator BECN1 suppresses mammary tumorigenesis driven by WNT1 activation and following parity. Autophagy (2014) 10(11):2036–52. doi: 10.4161/auto.34398
37. Catalano M, D’Alessandro G, Lepore F, Corazzari M, Caldarola S, Valacca C, et al. Autophagy induction impairs migration and invasion by reversing EMT in glioblastoma cells. Mol Oncol (2015) 9(8):1612–25. doi: 10.1016/j.molonc.2015.04.016
38. Heckmann BL, Green DR. Correction: LC3-associated phagocytosis at a glance. J Cell Sci (2019) 132(5). doi: 10.1242/jcs.222984
39. Botbol Y, Guerrero-Ros I, Macian F. Key roles of autophagy in regulating T-cell function. Eur J Immunol (2016) 46(6):1326–34. doi: 10.1002/eji.201545955
40. Murera D, Arbogast F, Arnold J, Bouis D, Muller S, Gros F. CD4 T cell autophagy is integral to memory maintenance. Sci Rep (2018) 8(1):5951. doi: 10.1038/s41598-018-23993-0
41. Merkley SD, Chock CJ, Yang XO, Harris J, Castillo EF. Modulating T Cell Responses via Autophagy: The Intrinsic Influence Controlling the Function of Both Antigen-Presenting Cells and T Cells. Front Immunol (2018) 9:2914. doi: 10.3389/fimmu.2018.02914
42. Munz C. Autophagy proteins influence endocytosis for MHC restricted antigen presentation. Semin Cancer Biol (2019) 66:110–5. doi: 10.1016/j.semcancer.2019.03.005
43. Zhang Y, Morgan MJ, Chen K, Choksi S, Liu ZG. Induction of autophagy is essential for monocyte-macrophage differentiation. Blood (2012) 119(12):2895–905. doi: 10.1182/blood-2011-08-372383
44. Pua HH, Dzhagalov I, Chuck M, Mizushima N, He YW. A critical role for the autophagy gene Atg5 in T cell survival and proliferation. J Exp Med (2007) 204(1):25–31. doi: 10.1084/jem.20061303
45. Xu X, Araki K, Li S, Han JH, Ye L, Tan WG, et al. Autophagy is essential for effector CD8(+) T cell survival and memory formation. Nat Immunol (2014) 15(12):1152–61. doi: 10.1038/ni.3025
46. Salio M, Puleston DJ, Mathan TS, Shepherd D, Stranks AJ, Adamopoulou E, et al. Essential role for autophagy during invariant NKT cell development. Proc Natl Acad Sci USA (2014) 111(52):E5678–87. doi: 10.1073/pnas.1413935112
47. Clarke AJ, Simon AK. Autophagy in the renewal, differentiation and homeostasis of immune cells. Nat Rev Immunol (2019) 19(3):170–83. doi: 10.1038/s41577-018-0095-2
48. Sil P, Zhao F, Muse G, Wong S-W, Kolb J, DeGraff LM, et al. Non-canonical autophagy in dendritic cells restricts cross-presentation and anti-tumor immunity. bioRxiv (2019) 145(5):1389–405. doi: 10.1101/789867
49. Manjithaya R, Subramani S. Role of autophagy in unconventional protein secretion. Autophagy (2010) 6(5):650–1. doi: 10.4161/auto.6.5.12066
50. Zhang M, Kenny SJ, Ge L, Xu K, Schekman R. Translocation of interleukin-1β into a vesicle intermediate in autophagy-mediated secretion. Elife (2015) 4:e11205. doi: 10.7554/eLife.11205
51. Levine B, Kroemer G. Biological Functions of Autophagy Genes: A Disease Perspective. Cell (2019) 176(1-2):11–42. doi: 10.1016/j.cell.2018.09.048
52. Liu Y, Flanagan JJ, Barlowe C. Sec22p export from the endoplasmic reticulum is independent of SNARE pairing. J Biol Chem (2004) 279(26):27225–32. doi: 10.1074/jbc.M312122200
53. Chen YD, Fang YT, Cheng YL, Lin CF, Hsu LJ, Wang SY, et al. Exophagy of annexin A2 via RAB11, RAB8A and RAB27A in IFN-γ-stimulated lung epithelial cells. Sci Rep (2017) 7(1):5676. doi: 10.1038/s41598-017-06076-4
54. Galluzzi L, Baehrecke EH, Ballabio A, Boya P, Bravo-San Pedro JM, Cecconi F, et al. Molecular definitions of autophagy and related processes. EMBO J (2017) 36(13):1811–36. doi: 10.15252/embj.201796697
55. Kalra H, Simpson RJ, Ji H, Aikawa E, Altevogt P, Askenase P, et al. Vesiclepedia: a compendium for extracellular vesicles with continuous community annotation. PLoS Biol (2012) 10(12):e1001450. doi: 10.1371/journal.pbio.1001450
56. Xu J, Camfield R, Gorski SM. The interplay between exosomes and autophagy - partners in crime. J Cell Sci (2018) 131(15):jcs215210. doi: 10.1242/jcs.215210
57. Guo H, Chitiprolu M, Roncevic L, Javalet C, Hemming FJ, Trung MT, et al. Atg5 Disassociates the V. Dev Cell (2017) 43(6):716–30.e7. doi: 10.1016/j.devcel.2017.11.018
58. Singh SS, Vats S, Chia AY, Tan TZ, Deng S, Ong MS, et al. Dual role of autophagy in hallmarks of cancer. Oncogene (2018) 37(9):1142–58. doi: 10.1038/s41388-017-0046-6
59. Bent R, Moll L, Grabbe S, Bros M. Interleukin-1 Beta-A Friend or Foe in Malignancies? Int J Mol Sci (2018) 19(8):2155. doi: 10.3390/ijms19082155
60. Liu S, Chen S, Zeng J. TGF−β signaling: A complex role in tumorigenesis (Review). Mol Med Rep (2018) 17(1):699–704. doi: 10.3892/mmr.2017.7970
61. Fabregat I, Fernando J, Mainez J, Sancho P. TGF-beta signaling in cancer treatment. Curr Pharm Des (2014) 20(17):2934–47. doi: 10.2174/13816128113199990591
62. Tulotta C, Lefley DV, Freeman K, Gregory WM, Hanby AM, Heath PR, et al. Endogenous Production of IL1B by Breast Cancer Cells Drives Metastasis and Colonization of the Bone Microenvironment. Clin Cancer Res (2019) 25(9):2769–82. doi: 10.1158/1078-0432.CCR-18-2202
63. Lv DJ, Song XL, Huang B, Yu YZ, Shu FP, Wang C, et al. HMGB1 Promotes Prostate Cancer Development and Metastasis by Interacting with Brahma-Related Gene 1 and Activating the Akt Signaling Pathway. Theranostics (2019) 9(18):5166–82. doi: 10.7150/thno.33972
64. Gilbert SM, Oliphant CJ, Hassan S, Peille AL, Bronsert P, Falzoni S, et al. ATP in the tumour microenvironment drives expression of nfP2X. Oncogene (2019) 38(2):194–208. doi: 10.1038/s41388-018-0426-6
65. Cotzomi-Ortega I, Aguilar-Alonso P, Reyes-Leyva J, Maycotte P. Autophagy and Its Role in Protein Secretion: Implications for Cancer Therapy. Mediators Inflamm (2018) 2018:4231591. doi: 10.1155/2018/4231591
66. Coppé JP, Desprez PY, Krtolica A, Campisi J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol (2010) 5:99–118. doi: 10.1146/annurev-pathol-121808-102144
67. Ohtani N. Deciphering the mechanism for induction of senescence-associated secretory phenotype (SASP) and its role in aging and cancer development. J Biochem (2019) 166(4):289–95. doi: 10.1093/jb/mvz055
68. Ortiz-Montero P, Londoño-Vallejo A, Vernot JP. Senescence-associated IL-6 and IL-8 cytokines induce a self- and cross-reinforced senescence/inflammatory milieu strengthening tumorigenic capabilities in the MCF-7 breast cancer cell line. Cell Commun Signal (2017) 15(1):17. doi: 10.1186/s12964-017-0172-3
69. Kim YH, Choi YW, Lee J, Soh EY, Kim JH, Park TJ. Senescent tumor cells lead the collective invasion in thyroid cancer. Nat Commun (2017) 8:15208. doi: 10.1038/ncomms15208
70. Mikuła-Pietrasik J, Sosińska P, Maksin K, Kucińska MG, Piotrowska H, Murias M, et al. Colorectal cancer-promoting activity of the senescent peritoneal mesothelium. Oncotarget (2015) 6(30):29178–95. doi: 10.18632/oncotarget.4932
71. Slobodnyuk K, Radic N, Ivanova S, Llado A, Trempolec N, Zorzano A, et al. Autophagy-induced senescence is regulated by p38α signaling. Cell Death Dis (2019) 10(6):376. doi: 10.1038/s41419-019-1607-0
72. Canaud G, Brooks CR, Kishi S, Taguchi K, Nishimura K, Magassa S, et al. Cyclin G1 and TASCC regulate kidney epithelial cell G. Sci Transl Med (2019) 11(476). doi: 10.1126/scitranslmed.aav4754
73. Maycotte P, Gearheart CM, Barnard R, Aryal S, Mulcahy Levy JM, Fosmire SP, et al. STAT3-mediated autophagy dependence identifies subtypes of breast cancer where autophagy inhibition can be efficacious. Cancer Res (2014) 74(9):2579–90. doi: 10.1158/0008-5472.CAN-13-3470
74. Kraya AA, Piao S, Xu X, Zhang G, Herlyn M, Gimotty P, et al. Identification of secreted proteins that reflect autophagy dynamics within tumor cells. Autophagy (2015) 11(1):60–74. doi: 10.4161/15548627.2014.984273
75. Kang TW, Yevsa T, Woller N, Hoenicke L, Wuestefeld T, Dauch D, et al. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature (2011) 479(7374):547–51. doi: 10.1038/nature10599
76. Singh K, Matsuyama S, Drazba JA, Almasan A. Autophagy-dependent senescence in response to DNA damage and chronic apoptotic stress. Autophagy (2012) 8(2):236–51. doi: 10.4161/auto.8.2.18600
77. Perez-Neut M, Haar L, Rao V, Santha S, Lansu K, Rana B, et al. Activation of hERG3 channel stimulates autophagy and promotes cellular senescence in melanoma. Oncotarget (2016) 7(16):21991–2004. doi: 10.18632/oncotarget.7831
78. Cho YY, Kim DJ, Lee HS, Jeong CH, Cho EJ, Kim MO, et al. Autophagy and cellular senescence mediated by Sox2 suppress malignancy of cancer cells. PLoS One (2013) 8(2):e57172. doi: 10.1371/journal.pone.0057172
79. Capparelli C, Whitaker-Menezes D, Guido C, Balliet R, Pestell TG, Howell A, et al. CTGF drives autophagy, glycolysis and senescence in cancer-associated fibroblasts via HIF1 activation, metabolically promoting tumor growth. Cell Cycle (2012) 11(12):2272–84. doi: 10.4161/cc.20717
80. Capparelli C, Guido C, Whitaker-Menezes D, Bonuccelli G, Balliet R, Pestell TG, et al. Autophagy and senescence in cancer-associated fibroblasts metabolically supports tumor growth and metastasis via glycolysis and ketone production. Cell Cycle (2012) 11(12):2285–302. doi: 10.4161/cc.20718
81. Capparelli C, Chiavarina B, Whitaker-Menezes D, Pestell TG, Pestell RG, Hulit J, et al. CDK inhibitors (p16/p19/p21) induce senescence and autophagy in cancer-associated fibroblasts, “fueling” tumor growth via paracrine interactions, without an increase in neo-angiogenesis. Cell Cycle (2012) 11(19):3599–610. doi: 10.4161/cc.21884
82. Horikawa I, Fujita K, Jenkins LM, Hiyoshi Y, Mondal AM, Vojtesek B, et al. Autophagic degradation of the inhibitory p53 isoform Δ133p53α as a regulatory mechanism for p53-mediated senescence. Nat Commun (2014) 5:4706. doi: 10.1038/ncomms5706
83. Coppé JP, Patil CK, Rodier F, Sun Y, Muñoz DP, Goldstein J, et al. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol (2008) 6(12):2853–68. doi: 10.1371/journal.pbio.0060301
84. Zhu Y, Xu L, Zhang J, Hu X, Liu Y, Yin H, et al. Sunitinib induces cellular senescence via p53/Dec1 activation in renal cell carcinoma cells. Cancer Sci (2013) 104(8):1052–61. doi: 10.1111/cas.12176
85. Harajly M, Zalzali H, Nawaz Z, Ghayad SE, Ghamloush F, Basma H, et al. p53 Restoration in Induction and Maintenance of Senescence: Differential Effects in Premalignant and Malignant Tumor Cells. Mol Cell Biol (2016) 36(3):438–51. doi: 10.1128/MCB.00747-15
86. Knizhnik AV, Roos WP, Nikolova T, Quiros S, Tomaszowski KH, Christmann M, et al. Survival and death strategies in glioma cells: autophagy, senescence and apoptosis triggered by a single type of temozolomide-induced DNA damage. PLoS One (2013) 8(1):e55665. doi: 10.1371/journal.pone.0055665
87. Braig M, Lee S, Loddenkemper C, Rudolph C, Peters AH, Schlegelberger B, et al. Oncogene-induced senescence as an initial barrier in lymphoma development. Nature (2005) 436(7051):660–5. doi: 10.1038/nature03841
88. Huang YH, Yang PM, Chuah QY, Lee YJ, Hsieh YF, Peng CW, et al. Autophagy promotes radiation-induced senescence but inhibits bystander effects in human breast cancer cells. Autophagy (2014) 10(7):1212–28. doi: 10.4161/auto.28772
89. Kang HT, Lee KB, Kim SY, Choi HR, Park SC. Autophagy impairment induces premature senescence in primary human fibroblasts. PLoS One (2011) 6(8):e23367. doi: 10.1371/journal.pone.0023367
90. Gammoh N, Fraser J, Puente C, Syred HM, Kang H, Ozawa T, et al. Suppression of autophagy impedes glioblastoma development and induces senescence. Autophagy (2016) 12(9):1431–9. doi: 10.1080/15548627.2016.1190053
91. Ramalinga M, Roy A, Srivastava A, Bhattarai A, Harish V, Suy S, et al. MicroRNA-212 negatively regulates starvation induced autophagy in prostate cancer cells by inhibiting SIRT1 and is a modulator of angiogenesis and cellular senescence. Oncotarget (2015) 6(33):34446–57. doi: 10.18632/oncotarget.5920
92. Schmitt CA, Fridman JS, Yang M, Lee S, Baranov E, Hoffman RM, et al. A senescence program controlled by p53 and p16INK4a contributes to the outcome of cancer therapy. Cell (2002) 109(3):335–46. doi: 10.1016/S0092-8674(02)00734-1
94. Nardella C, Clohessy JG, Alimonti A, Pandolfi PP. Pro-senescence therapy for cancer treatment. Nat Rev Cancer (2011) 11(7):503–11. doi: 10.1038/nrc3057
95. Saleh T, Tyutynuk-Massey L, Cudjoe EK, Idowu MO, Landry JW, Gewirtz DA. Non-Cell Autonomous Effects of the Senescence-Associated Secretory Phenotype in Cancer Therapy. Front Oncol (2018) 8:164. doi: 10.3389/fonc.2018.00164
96. Patel NH, Sohal SS, Manjili MH, Harrell JC, Gewirtz DA. The Roles of Autophagy and Senescence in the Tumor Cell Response to Radiation. Radiat Res (2020) 194(autophagy and senescence):103–15. doi: 10.1667/RADE-20-00009
97. Yao Z, Murali B, Ren Q, Luo X, Faget DV, Cole T, et al. Therapy-Induced Senescence Drives Bone Loss. Cancer Res (2020) 80(5):1171–82. doi: 10.1158/0008-5472.CAN-19-2348
98. Demaria M, O’Leary MN, Chang J, Shao L, Liu S, Alimirah F, et al. Cellular Senescence Promotes Adverse Effects of Chemotherapy and Cancer Relapse. Cancer Discovery (2017) 7(2):165–76. doi: 10.1158/2159-8290.CD-16-0241
99. Milanovic M, Fan DNY, Belenki D, Däbritz JHM, Zhao Z, Yu Y, et al. Senescence-associated reprogramming promotes cancer stemness. Nature (2018) 553(7686):96–100. doi: 10.1038/nature25167
100. Saleh T, Tyutyunyk-Massey L, Murray GF, Alotaibi MR, Kawale AS, Elsayed Z, et al. Tumor cell escape from therapy-induced senescence. Biochem Pharmacol (2019) 162:202–12. doi: 10.1016/j.bcp.2018.12.013
101. Mosieniak G, Sliwinska MA, Alster O, Strzeszewska A, Sunderland P, Piechota M, et al. Polyploidy Formation in Doxorubicin-Treated Cancer Cells Can Favor Escape from Senescence. Neoplasia (2015) 17(12):882–93. doi: 10.1016/j.neo.2015.11.008
102. Wang B, Kohli J, Demaria M. Senescent Cells in Cancer Therapy: Friends or Foes? Trends Cancer (2020) 6(10):838–57. doi: 10.1016/j.trecan.2020.05.004
103. Eggert T, Wolter K, Ji J, Ma C, Yevsa T, Klotz S, et al. Distinct Functions of Senescence-Associated Immune Responses in Liver Tumor Surveillance and Tumor Progression. Cancer Cell (2016) 30(4):533–47. doi: 10.1016/j.ccell.2016.09.003
104. Ruhland MK, Loza AJ, Capietto AH, Luo X, Knolhoff BL, Flanagan KC, et al. Stromal senescence establishes an immunosuppressive microenvironment that drives tumorigenesis. Nat Commun (2016) 7:11762. doi: 10.1038/ncomms11762
105. Canino C, Mori F, Cambria A, Diamantini A, Germoni S, Alessandrini G, et al. SASP mediates chemoresistance and tumor-initiating-activity of mesothelioma cells. Oncogene (2012) 31(26):3148–63. doi: 10.1038/onc.2011.485
106. Goulet CR, Champagne A, Bernard G, Vandal D, Chabaud S, Pouliot F, et al. Cancer-associated fibroblasts induce epithelial-mesenchymal transition of bladder cancer cells through paracrine IL-6 signalling. BMC Cancer (2019) 19(1):137. doi: 10.1186/s12885-019-5353-6
107. Miao JW, Liu LJ, Huang J. Interleukin-6-induced epithelial-mesenchymal transition through signal transducer and activator of transcription 3 in human cervical carcinoma. Int J Oncol (2014) 45(1):165–76. doi: 10.3892/ijo.2014.2422
108. Fung C, Lock R, Gao S, Salas E, Debnath J. Induction of autophagy during extracellular matrix detachment promotes cell survival. Mol Biol Cell (2008) 19(3):797–806. doi: 10.1091/mbc.e07-10-1092
109. Rojas-Sanchez G, Cotzomi-Ortega I, Pazos-Salazar NG, Reyes-Leyva J, Maycotte P. Autophagy and Its Relationship to Epithelial to Mesenchymal Transition: When Autophagy Inhibition for Cancer Therapy Turns Counterproductive. Biol (Basel) (2019) 8(4):71. doi: 10.3390/biology8040071
110. Marcucci F, Ghezzi P, Rumio C. The role of autophagy in the cross-talk between epithelial-mesenchymal transitioned tumor cells and cancer stem-like cells. Mol Cancer (2017) 16(1):3. doi: 10.1186/s12943-016-0573-8
111. Chen HT, Liu H, Mao MJ, Tan Y, Mo XQ, Meng XJ, et al. Crosstalk between autophagy and epithelial-mesenchymal transition and its application in cancer therapy. Mol Cancer (2019) 18(1):101. doi: 10.1186/s12943-019-1030-2
112. Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol (2014) 15(3):178–96. doi: 10.1038/nrm3758
113. Li S, Song Y, Quach C, Guo H, Jang GB, Maazi H, et al. Transcriptional regulation of autophagy-lysosomal function in BRAF-driven melanoma progression and chemoresistance. Nat Commun (2019) 10(1):1693. doi: 10.1038/s41467-019-09634-8
114. Amaravadi RK, Kimmelman AC, Debnath J. Targeting Autophagy in Cancer: Recent Advances and Future Directions. Cancer Discovery (2019) 9(9):1167–81. doi: 10.1158/2159-8290.CD-19-0292
115. Subramani R, Gonzalez E, Arumugam A, Nandy S, Gonzalez V, Medel J, et al. Nimbolide inhibits pancreatic cancer growth and metastasis through ROS-mediated apoptosis and inhibition of epithelial-to-mesenchymal transition. Sci Rep (2016) 6:19819. doi: 10.1038/srep19819
116. Kocaturk NM, Akkoc Y, Kig C, Bayraktar O, Gozuacik D, Kutlu O. Autophagy as a molecular target for cancer treatment. Eur J Pharm Sci (2019) 134:116–37. doi: 10.1016/j.ejps.2019.04.011
117. Chen C, Liang QY, Chen HK, Wu PF, Feng ZY, Ma XM, et al. DRAM1 regulates the migration and invasion of hepatoblastoma cells via autophagy-EMT pathway. Oncol Lett (2018) 16(2):2427–33. doi: 10.3892/ol.2018.8937
118. Alizadeh J, Glogowska A, Thliveris J, Kalantari F, Shojaei S, Hombach-Klonisch S, et al. Autophagy modulates transforming growth factor beta 1 induced epithelial to mesenchymal transition in non-small cell lung cancer cells. Biochim Biophys Acta Mol Cell Res (2018) 1865(5):749–68. doi: 10.1016/j.bbamcr.2018.02.007
119. Shen H, Yin L, Deng G, Guo C, Han Y, Li Y, et al. Knockdown of Beclin-1 impairs epithelial-mesenchymal transition of colon cancer cells. J Cell Biochem (2018) 119(8):7022–31. doi: 10.1002/jcb.26912
120. Gulhati P, Bowen KA, Liu J, Stevens PD, Rychahou PG, Chen M, et al. mTORC1 and mTORC2 regulate EMT, motility, and metastasis of colorectal cancer via RhoA and Rac1 signaling pathways. Cancer Res (2011) 71(9):3246–56. doi: 10.1158/0008-5472.CAN-10-4058
121. Peng YF, Shi YH, Ding ZB, Ke AW, Gu CY, Hui B, et al. Autophagy inhibition suppresses pulmonary metastasis of HCC in mice via impairing anoikis resistance and colonization of HCC cells. Autophagy (2013) 9(12):2056–68. doi: 10.4161/auto.26398
122. Lv Q, Wang W, Xue J, Hua F, Mu R, Lin H, et al. DEDD interacts with PI3KC3 to activate autophagy and attenuate epithelial-mesenchymal transition in human breast cancer. Cancer Res (2012) 72(13):3238–50. doi: 10.1158/0008-5472.CAN-11-3832
123. Liao H, Huang Y, Guo B, Liang B, Liu X, Ou H, et al. Dramatic antitumor effects of the dual mTORC1 and mTORC2 inhibitor AZD2014 in hepatocellular carcinoma. Am J Cancer Res (2015) 5(1):125–39.
124. Niu NK, Wang ZL, Pan ST, Ding HQ, Au GH, He ZX, et al. Pro-apoptotic and pro-autophagic effects of the Aurora kinase A inhibitor alisertib (MLN8237) on human osteosarcoma U-2 OS and MG-63 cells through the activation of mitochondria-mediated pathway and inhibition of p38 MAPK/PI3K/Akt/mTOR signaling pathway. Drug Des Devel Ther (2015) 9:1555–84. doi: 10.2147/DDDT.S74197
125. Ren BJ, Zhou ZW, Zhu DJ, Ju YL, Wu JH, Ouyang MZ, et al. Alisertib Induces Cell Cycle Arrest, Apoptosis, Autophagy and Suppresses EMT in HT29 and Caco-2 Cells. Int J Mol Sci (2015) 17(1):41. doi: 10.3390/ijms17010041
126. Han B, Cui H, Kang L, Zhang X, Jin Z, Lu L, et al. Metformin inhibits thyroid cancer cell growth, migration, and EMT through the mTOR pathway. Tumour Biol (2015) 36(8):6295–304. doi: 10.1007/s13277-015-3315-4
127. Mecca C, Giambanco I, Donato R, Arcuri C. Targeting mTOR in Glioblastoma: Rationale and Preclinical/Clinical Evidence. Dis Markers (2018) 2018:9230479. doi: 10.1155/2018/9230479
128. Ye R, Dai N, He Q, Guo P, Xiang Y, Zhang Q, et al. Comprehensive anti-tumor effect of Brusatol through inhibition of cell viability and promotion of apoptosis caused by autophagy via the PI3K/Akt/mTOR pathway in hepatocellular carcinoma. BioMed Pharmacother (2018) 105:962–73. doi: 10.1016/j.biopha.2018.06.065
129. Chen X, Chen Y, Lin X, Su S, Hou X, Zhang Q, et al. The Drug Combination of SB202190 and SP600125 Significantly Inhibit the Growth and Metastasis of Olaparib-resistant Ovarian Cancer Cell. Curr Pharm Biotechnol (2018) 19(6):506–13. doi: 10.2174/1389201019666180713102656
130. Hu S, Wang L, Zhang X, Wu Y, Yang J, Li J. Autophagy induces transforming growth factor-β-dependent epithelial-mesenchymal transition in hepatocarcinoma cells through cAMP response element binding signalling. J Cell Mol Med (2018) 22(11):5518–32. doi: 10.1111/jcmm.13825
131. Yu SB, Kang HM, Park DB, Park BS, Kim IR. Cudraxanthone D Regulates Epithelial-Mesenchymal Transition by Autophagy Inhibition in Oral Squamous Cell Carcinoma Cell Lines. Evid Based Complement Alternat Med (2019) 2019:5213028. doi: 10.1155/2019/5213028
132. Wang Y, Sun L, Qiu W, Qi W, Qi Y, Liu Z, et al. Inhibiting Forkhead box K1 induces autophagy to reverse epithelial-mesenchymal transition and metastasis in gastric cancer by regulating Myc-associated zinc finger protein in an acidic microenvironment. Aging (Albany NY) (2020) 12(7):6129–50. doi: 10.18632/aging.103013
133. Su Z, Li G, Liu C, Ren S, Deng T, Zhang S, et al. Autophagy inhibition impairs the epithelial-mesenchymal transition and enhances cisplatin sensitivity in nasopharyngeal carcinoma. Oncol Lett (2017) 13(6):4147–54. doi: 10.3892/ol.2017.5963
134. Bao Y, Ding Z, Zhao P, Li J, Chen P, Zheng J, et al. Autophagy inhibition potentiates the anti-EMT effects of alteronol through TGF-β/Smad3 signaling in melanoma cells. Cell Death Dis (2020) 11(4):223. doi: 10.1038/s41419-020-2419-y
135. Lv LH, Wan YL, Lin Y, Zhang W, Yang M, Li GL, et al. Anticancer drugs cause release of exosomes with heat shock proteins from human hepatocellular carcinoma cells that elicit effective natural killer cell antitumor responses in vitro. J Biol Chem (2012) 287(19):15874–85. doi: 10.1074/jbc.M112.340588
136. Sosa MS, Bragado P, Aguirre-Ghiso JA. Mechanisms of disseminated cancer cell dormancy: an awakening field. Nat Rev Cancer (2014) 14(9):611–22. doi: 10.1038/nrc3793
137. McGowan PM, Kirstein JM, Chambers AF. Micrometastatic disease and metastatic outgrowth: clinical issues and experimental approaches. Future Oncol (2009) 5(7):1083–98. doi: 10.2217/fon.09.73
138. Giancotti FG. Mechanisms governing metastatic dormancy and reactivation. Cell (2013) 155(4):750–64. doi: 10.1016/j.cell.2013.10.029
139. Lu Z, Luo RZ, Lu Y, Zhang X, Yu Q, Khare S, et al. The tumor suppressor gene ARHI regulates autophagy and tumor dormancy in human ovarian cancer cells. J Clin Invest (2008) 118(12):3917–29. doi: 10.1172/JCI35512
140. Lum JJ, Bauer DE, Kong M, Harris MH, Li C, Lindsten T, et al. Growth factor regulation of autophagy and cell survival in the absence of apoptosis. Cell (2005) 120(2):237–48. doi: 10.1016/j.cell.2004.11.046
141. Galluzzi L, Vacchelli E, Bravo-San Pedro JM, Buqué A, Senovilla L, Baracco EE, et al. Classification of current anticancer immunotherapies. Oncotarget (2014) 5(24):12472–508. doi: 10.18632/oncotarget.2998
142. Aqbi HF, Tyutyunyk-Massey L, Keim RC, Butler SE, Thekkudan T, Joshi S, et al. Autophagy-deficient breast cancer shows early tumor recurrence and escape from dormancy. Oncotarget (2018) 9(31):22113–22. doi: 10.18632/oncotarget.25197
143. Vera-Ramirez L, Vodnala SK, Nini R, Hunter KW, Green JE. Autophagy promotes the survival of dormant breast cancer cells and metastatic tumour recurrence. Nat Commun (2018) 9(1):1944. doi: 10.1038/s41467-018-04070-6
144. Madden E, Logue SE, Healy SJ, Manie S, Samali A. The role of the unfolded protein response in cancer progression: From oncogenesis to chemoresistance. Biol Cell (2019) 111(1):1–17. doi: 10.1111/boc.201800050
145. Zhang Y, Qu X, Jiang L. An oasis in the desert of cancer chemotherapeutic resistance: The enlightenment from reciprocal crosstalk between signaling pathways of UPR and autophagy in cancers. BioMed Pharmacother (2017) 92:972–81. doi: 10.1016/j.biopha.2017.05.132
146. La Belle Flynn A, Calhoun BC, Sharma A, Chang JC, Almasan A, Schiemann WP. Autophagy inhibition elicits emergence from metastatic dormancy by inducing and stabilizing Pfkfb3 expression. Nat Commun (2019) 10(1):3668. doi: 10.1038/s41467-019-11640-9
147. Mowers EE, Sharifi MN, Macleod KF. Functions of autophagy in the tumor microenvironment and cancer metastasis. FEBS J (2018) 285(10):1751–66. doi: 10.1111/febs.14388
148. Harris J. Autophagy and cytokines. Cytokine (2011) 56(2):140–4. doi: 10.1016/j.cyto.2011.08.022
149. Ishimwe N, Zhang W, Qian J, Zhang Y, Wen L. Autophagy regulation as a promising approach for improving cancer immunotherapy. Cancer Lett (2020) 475:34–42. doi: 10.1016/j.canlet.2020.01.034
150. Jiang GM, Tan Y, Wang H, Peng L, Chen HT, Meng XJ, et al. The relationship between autophagy and the immune system and its applications for tumor immunotherapy. Mol Cancer (2019) 18(1):17. doi: 10.1186/s12943-019-0944-z
151. Pan H, Chen L, Xu Y, Han W, Lou F, Fei W, et al. Autophagy-associated immune responses and cancer immunotherapy. Oncotarget (2016) 7(16):21235–46. doi: 10.18632/oncotarget.6908
152. Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol (2010) 11(5):373–84. doi: 10.1038/ni.1863
153. Noh JY, Yoon SR, Kim TD, Choi I, Jung H. Toll-Like Receptors in Natural Killer Cells and Their Application for Immunotherapy. J Immunol Res (2020) 2020:2045860. doi: 10.1155/2020/2045860
154. Lu Z, Xie D, Chen Y, Tian E, Muhammad I, Chen X, et al. TLR2 mediates autophagy through ERK signaling pathway in Mycoplasma gallisepticum-infected RAW264.7 cells. Mol Immunol (2017) 87:161–70. doi: 10.1016/j.molimm.2017.04.013
155. Fang L, Shen Q, Wu H, He F, Ding P, Xu K, et al. TLR2 favors OVA-induced allergic airway inflammation in mice through JNK signaling pathway with activation of autophagy. Life Sci (2020) 256:117896. doi: 10.1016/j.lfs.2020.117896
156. Xu Y, Jagannath C, Liu XD, Sharafkhaneh A, Kolodziejska KE, Eissa NT. Toll-like receptor 4 is a sensor for autophagy associated with innate immunity. Immunity (2007) 27(1):135–44. doi: 10.1016/j.immuni.2007.05.022
157. Zhan Z, Xie X, Cao H, Zhou X, Zhang XD, Fan H, et al. Autophagy facilitates TLR4- and TLR3-triggered migration and invasion of lung cancer cells through the promotion of TRAF6 ubiquitination. Autophagy (2014) 10(2):257–68. doi: 10.4161/auto.27162
158. Kim MJ, Min Y, Im JS, Son J, Lee JS, Lee KY. p62 is Negatively Implicated in the TRAF6-BECN1 Signaling Axis for Autophagy Activation and Cancer Progression by Toll-Like Receptor 4 (TLR4). Cells (2020) 9(5). doi: 10.3390/cells9051142
159. Zhao XL, Lin Y, Jiang J, Tang Z, Yang S, Lu L, et al. High-mobility group box 1 released by autophagic cancer-associated fibroblasts maintains the stemness of luminal breast cancer cells. J Pathol (2017) 243(3):376–89. doi: 10.1002/path.4958
160. Braunstein MJ, Kucharczyk J, Adams S. Targeting Toll-Like Receptors for Cancer Therapy. Target Oncol (2018) 13(5):583–98. doi: 10.1007/s11523-018-0589-7
161. Velloso FJ, Trombetta-Lima M, Anschau V, Sogayar MC, Correa RG. NOD-like receptors: major players (and targets) in the interface between innate immunity and cancer. Biosci Rep (2019) 39(4). doi: 10.1042/BSR20181709
162. Travassos LH, Carneiro LA, Ramjeet M, Hussey S, Kim YG, Magalhaes JG, et al. Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat Immunol (2010) 11(1):55–62. doi: 10.1038/ni.1823
163. Kutikhin AG. Role of NOD1/CARD4 and NOD2/CARD15 gene polymorphisms in cancer etiology. Hum Immunol (2011) 72(10):955–68. doi: 10.1016/j.humimm.2011.06.003
164. Velloso FJ, Campos AR, Sogayar MC, Correa RG. Proteome profiling of triple negative breast cancer cells overexpressing NOD1 and NOD2 receptors unveils molecular signatures of malignant cell proliferation. BMC Genomics (2019) 20(1):152. doi: 10.1186/s12864-019-5523-6
165. Jin Y, Hong Y, Park CY, Hong Y. Molecular Interactions of Autophagy with the Immune System and Cancer. Int J Mol Sci (2017) 18(8):1694. doi: 10.3390/ijms18081694
166. Zhong Z, Sanchez-Lopez E, Karin M. Autophagy, Inflammation, and Immunity: A Troika Governing Cancer and Its Treatment. Cell (2016) 166(2):288–98. doi: 10.1016/j.cell.2016.05.051
167. Sanjuan MA, Dillon CP, Tait SW, Moshiach S, Dorsey F, Connell S, et al. Toll-like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature (2007) 450(7173):1253–7. doi: 10.1038/nature06421
168. Ghislat G, Lawrence T. Autophagy in dendritic cells. Cell Mol Immunol (2018) 15(11):944–52. doi: 10.1038/cmi.2018.2
169. Liu E, Van Grol J, Subauste CS. Atg5 but not Atg7 in dendritic cells enhances IL-2 and IFN-gamma production by Toxoplasma gondii-reactive CD4+ T cells. Microbes Infect (2015) 17(4):275–84. doi: 10.1016/j.micinf.2014.12.008
170. Lee HK, Mattei LM, Steinberg BE, Alberts P, Lee YH, Chervonsky A, et al. In vivo requirement for Atg5 in antigen presentation by dendritic cells. Immunity (2010) 32(2):227–39. doi: 10.1016/j.immuni.2009.12.006
171. Tao S, Drexler I. Targeting Autophagy in Innate Immune Cells: Angel or Demon During Infection and Vaccination? Front Immunol (2020) 11:460. doi: 10.3389/fimmu.2020.00460
172. Jacquel A, Obba S, Solary E, Auberger P. Proper macrophagic differentiation requires both autophagy and caspase activation. Autophagy (2012) 8(7):1141–3. doi: 10.4161/auto.20367
173. Liu K, Zhao E, Ilyas G, Lalazar G, Lin Y, Haseeb M, et al. Impaired macrophage autophagy increases the immune response in obese mice by promoting proinflammatory macrophage polarization. Autophagy (2015) 11(2):271–84. doi: 10.1080/15548627.2015.1009787
174. Jia W, Pua HH, Li QJ, He YW. Autophagy regulates endoplasmic reticulum homeostasis and calcium mobilization in T lymphocytes. J Immunol (2011) 186(3):1564–74. doi: 10.4049/jimmunol.1001822
175. Pua HH, Guo J, Komatsu M, He YW. Autophagy is essential for mitochondrial clearance in mature T lymphocytes. J Immunol (2009) 182(7):4046–55. doi: 10.4049/jimmunol.0801143
176. Willinger T, Flavell RA. Canonical autophagy dependent on the class III phosphoinositide-3 kinase Vps34 is required for naive T-cell homeostasis. Proc Natl Acad Sci U S A (2012) 109(22):8670–5. doi: 10.1073/pnas.1205305109
177. Afzal S, Hao Z, Itsumi M, Abouelkheer Y, Brenner D, Gao Y, et al. Autophagy-independent functions of UVRAG are essential for peripheral naive T-cell homeostasis. Proc Natl Acad Sci U S A (2015) 112(4):1119–24. doi: 10.1073/pnas.1423588112
178. Kovacs JR, Li C, Yang Q, Li G, Garcia IG, Ju S, et al. Autophagy promotes T-cell survival through degradation of proteins of the cell death machinery. Cell Death Differ (2012) 19(1):144–52. doi: 10.1038/cdd.2011.78
179. Sowell RT, Rogozinska M, Nelson CE, Vezys V, Marzo AL. Cutting edge: generation of effector cells that localize to mucosal tissues and form resident memory CD8 T cells is controlled by mTOR. J Immunol (2014) 193(5):2067–71. doi: 10.4049/jimmunol.1400074
180. Puleston DJ, Zhang H, Powell TJ, Lipina E, Sims S, Panse I, et al. Autophagy is a critical regulator of memory CD8(+) T cell formation. Elife (2014) 3:e03706. doi: 10.7554/eLife.03706
181. Michalek RD, Gerriets VA, Jacobs SR, Macintyre AN, MacIver NJ, Mason EF, et al. Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J Immunol (2011) 186(6):3299–303. doi: 10.4049/jimmunol.1003613
182. Wei J, Long L, Yang K, Guy C, Shrestha S, Chen Z, et al. Autophagy enforces functional integrity of regulatory T cells by coupling environmental cues and metabolic homeostasis. Nat Immunol (2016) 17(3):277–85. doi: 10.1038/ni.3365
183. Arnold J, Murera D, Arbogast F, Fauny JD, Muller S, Gros F. Autophagy is dispensable for B-cell development but essential for humoral autoimmune responses. Cell Death Differ (2016) 23(5):853–64. doi: 10.1038/cdd.2015.149
184. Pengo N, Scolari M, Oliva L, Milan E, Mainoldi F, Raimondi A, et al. Plasma cells require autophagy for sustainable immunoglobulin production. Nat Immunol (2013) 14(3):298–305. doi: 10.1038/ni.2524
185. O’Sullivan TE, Johnson LR, Kang HH, Sun JC. BNIP3- and BNIP3L-Mediated Mitophagy Promotes the Generation of Natural Killer Cell Memory. Immunity (2015) 43(2):331–42. doi: 10.1016/j.immuni.2015.07.012
186. Pei B, Zhao M, Miller BC, Vela JL, Bruinsma MW, Virgin HW, et al. Invariant NKT cells require autophagy to coordinate proliferation and survival signals during differentiation. J Immunol (2015) 194(12):5872–84. doi: 10.4049/jimmunol.1402154
187. Rozman S, Yousefi S, Oberson K, Kaufmann T, Benarafa C, Simon HU. The generation of neutrophils in the bone marrow is controlled by autophagy. Cell Death Differ (2015) 22(3):445–56. doi: 10.1038/cdd.2014.169
188. Harris J, Hartman M, Roche C, Zeng SG, O’Shea A, Sharp FA, et al. Autophagy controls IL-1beta secretion by targeting pro-IL-1beta for degradation. J Biol Chem (2011) 286(11):9587–97. doi: 10.1074/jbc.M110.202911
189. Peral de Castro C, Jones SA, Ni Cheallaigh C, Hearnden CA, Williams L, Winter J, et al. Autophagy regulates IL-23 secretion and innate T cell responses through effects on IL-1 secretion. J Immunol (2012) 189(8):4144–53. doi: 10.4049/jimmunol.1201946
190. Schmeisser H, Bekisz J, Zoon KC. New function of type I IFN: induction of autophagy. J Interferon Cytokine Res (2014) 34(2):71–8. doi: 10.1089/jir.2013.0128
191. Matsuzawa T, Kim BH, Shenoy AR, Kamitani S, Miyake M, Macmicking JD. IFN-gamma elicits macrophage autophagy via the p38 MAPK signaling pathway. J Immunol (2012) 189(2):813–8. doi: 10.4049/jimmunol.1102041
192. Kang R, Tang D, Lotze MT, Zeh Iii HJ. Autophagy is required for IL-2-mediated fibroblast growth. Exp Cell Res (2013) 319(4):556–65. doi: 10.1016/j.yexcr.2012.11.012
193. Lin Y, Kuang W, Wu B, Xie C, Liu C, Tu Z. IL-12 induces autophagy in human breast cancer cells through AMPK and the PI3K/Akt pathway. Mol Med Rep (2017) 16(4):4113–8. doi: 10.3892/mmr.2017.7114
194. Zhang C, Zhang X, Xu R, Huang B, Chen AJ, Li C, et al. TGF-beta2 initiates autophagy via Smad and non-Smad pathway to promote glioma cells’ invasion. J Exp Clin Cancer Res (2017) 36(1):162. doi: 10.1186/s13046-017-0628-8
195. Qin B, Zhou Z, He J, Yan C, Ding S. IL-6 Inhibits Starvation-induced Autophagy via the STAT3/Bcl-2 Signaling Pathway. Sci Rep (2015) 5:15701. doi: 10.1038/srep15701
196. Linnemann AK, Blumer J, Marasco MR, Battiola TJ, Umhoefer HM, Han JY, et al. Interleukin 6 protects pancreatic beta cells from apoptosis by stimulation of autophagy. FASEB J (2017) 31(9):4140–52. doi: 10.1096/fj.201700061RR
197. Martin C, Espaillat MP, Santiago-Schwarz F. IL-10 restricts dendritic cell (DC) growth at the monocyte-to-monocyte-derived DC interface by disrupting anti-apoptotic and cytoprotective autophagic molecular machinery. Immunol Res (2015) 63(1-3):131–43. doi: 10.1007/s12026-015-8700-y
198. Folgiero V, Miele E, Carai A, Ferretti E, Alfano V, Po A, et al. IDO1 involvement in mTOR pathway: a molecular mechanism of resistance to mTOR targeting in medulloblastoma. Oncotarget (2016) 7(33):52900–11. doi: 10.18632/oncotarget.9284
199. Mahoney KM, Rennert PD, Freeman GJ. Combination cancer immunotherapy and new immunomodulatory targets. Nat Rev Drug Discovery (2015) 14(8):561–84. doi: 10.1038/nrd4591
200. Robainas M, Otano R, Bueno S, Ait-Oudhia S. Understanding the role of PD-L1/PD1 pathway blockade and autophagy in cancer therapy. Onco Targets Ther (2017) 10:1803–7. doi: 10.2147/OTT.S132508
201. Maher CM, Thomas JD, Haas DA, Longen CG, Oyer HM, Tong JY, et al. Small-Molecule Sigma1 Modulator Induces Autophagic Degradation of PD-L1. Mol Cancer Res (2018) 16(2):243–55. doi: 10.1158/1541-7786.MCR-17-0166
202. Clark CA, Gupta HB, Sareddy G, Pandeswara S, Lao S, Yuan B, et al. Tumor-Intrinsic PD-L1 Signals Regulate Cell Growth, Pathogenesis, and Autophagy in Ovarian Cancer and Melanoma. Cancer Res (2016) 76(23):6964–74. doi: 10.1158/0008-5472.CAN-16-0258
203. Shukla SA, Bachireddy P, Schilling B, Galonska C, Zhan Q, Bango C, et al. Cancer-Germline Antigen Expression Discriminates Clinical Outcome to CTLA-4 Blockade. Cell (2018) 173(3):624–33. doi: 10.1016/j.cell.2018.03.026
204. Garg AD, Dudek AM, Ferreira GB, Verfaillie T, Vandenabeele P, Krysko DV, et al. ROS-induced autophagy in cancer cells assists in evasion from determinants of immunogenic cell death. Autophagy (2013) 9(9):1292–307. doi: 10.4161/auto.25399
205. Buchser WJ, Laskow TC, Pavlik PJ, Lin HM, Lotze MT. Cell-mediated autophagy promotes cancer cell survival. Cancer Res (2012) 72(12):2970–9. doi: 10.1158/0008-5472.CAN-11-3396
206. Sun K, Xu L, Jing Y, Han Z, Chen X, Cai C, et al. Autophagy-deficient Kupffer cells promote tumorigenesis by enhancing mtROS-NF-kappaB-IL1alpha/beta-dependent inflammation and fibrosis during the preneoplastic stage of hepatocarcinogenesis. Cancer Lett (2017) 388:198–207. doi: 10.1016/j.canlet.2016.12.004
207. Liang X, De Vera ME, Buchser WJ, Romo de Vivar Chavez A, Loughran P, Beer Stolz D, et al. Inhibiting systemic autophagy during interleukin 2 immunotherapy promotes long-term tumor regression. Cancer Res (2012) 72(11):2791–801. doi: 10.1158/0008-5472.CAN-12-0320
208. Lotze MT, Buchser WJ, Liang X. Blocking the interleukin 2 (IL2)-induced systemic autophagic syndrome promotes profound antitumor effects and limits toxicity. Autophagy (2012) 8(8):1264–6. doi: 10.4161/auto.20752
209. Li J, Yang D, Wang W, Piao S, Zhou J, Saiyin W, et al. Inhibition of autophagy by 3-MA enhances IL-24-induced apoptosis in human oral squamous cell carcinoma cells. J Exp Clin Cancer Res (2015) 34:97. doi: 10.1186/s13046-015-0211-0
210. Lin SY, Hsieh SY, Fan YT, Wei WC, Hsiao PW, Tsai DH, et al. Necroptosis promotes autophagy-dependent upregulation of DAMP and results in immunosurveillance. Autophagy (2018) 14(5):778–95. doi: 10.1080/15548627.2017.1386359
211. Noman MZ, Janji B, Kaminska B, Van Moer K, Pierson S, Przanowski P, et al. Blocking hypoxia-induced autophagy in tumors restores cytotoxic T-cell activity and promotes regression. Cancer Res (2011) 71(18):5976–86. doi: 10.1158/0008-5472.CAN-11-1094
212. Baginska J, Viry E, Berchem G, Poli A, Noman MZ, van Moer K, et al. Granzyme B degradation by autophagy decreases tumor cell susceptibility to natural killer-mediated lysis under hypoxia. Proc Natl Acad Sci U S A (2013) 110(43):17450–5. doi: 10.1073/pnas.1304790110
213. Yamamoto K, Venida A, Perera RM, Kimmelman AC. Selective autophagy of MHC-I promotes immune evasion of pancreatic cancer. Autophagy (2020) 2020:1–2. doi: 10.1080/15548627.2020.1769973
214. Meng Q, Zhang Y, Hu LG. Targeting Autophagy Facilitates T Lymphocyte Migration by Inducing the Expression of CXCL10 in Gastric Cancer Cell Lines. Front Oncol (2020) 10:886. doi: 10.3389/fonc.2020.00886
215. Noman MZ, Parpal S, Van Moer K, Xiao M, Yu Y, Viklund J, et al. Inhibition of Vps34 reprograms cold into hot inflamed tumors and improves anti-PD-1/PD-L1 immunotherapy. Sci Adv (2020) 6(18):eaax7881. doi: 10.1126/sciadv.aax7881
216. Mgrditchian T, Arakelian T, Paggetti J, Noman MZ, Viry E, Moussay E, et al. Targeting autophagy inhibits melanoma growth by enhancing NK cells infiltration in a CCL5-dependent manner. Proc Natl Acad Sci U.S.A. (2017) 114(44):E9271–E9. doi: 10.1073/pnas.1703921114
217. Kumai T, Matsuda Y, Ohkuri T, Oikawa K, Ishibashi K, Aoki N, et al. c-Met is a novel tumor associated antigen for T-cell based immunotherapy against NK/T cell lymphoma. Oncoimmunology (2015) 4(2):e976077. doi: 10.4161/2162402X.2014.976077
218. Li Y, Wang LX, Yang G, Hao F, Urba WJ, Hu HM. Efficient cross-presentation depends on autophagy in tumor cells. Cancer Res (2008) 68(17):6889–95. doi: 10.1158/0008-5472.CAN-08-0161
219. Li W, Zhou M, Ren H, Hu HM, Lu L, Cao M, et al. Tumor-derived autophagosomes (DRibbles) induce B cell activation in a TLR2-MyD88 dependent manner. PLoS One (2013) 8(1):e53564. doi: 10.1371/journal.pone.0053564
220. Twitty CG, Jensen SM, Hu HM, Fox BA. Tumor-derived autophagosome vaccine: induction of cross-protective immune responses against short-lived proteins through a p62-dependent mechanism. Clin Cancer Res (2011) 17(20):6467–81. doi: 10.1158/1078-0432.CCR-11-0812
221. Su H, Luo Q, Xie H, Huang X, Ni Y, Mou Y, et al. Therapeutic antitumor efficacy of tumor-derived autophagosome (DRibble) vaccine on head and neck cancer. Int J Nanomed (2015) 10:1921–30. doi: 10.2147/IJN.S74204
222. Gabai VL, Shifrin VI. Feasibility analysis of p62 (SQSTM1)-encoding DNA vaccine as a novel cancer immunotherapy. Int Rev Immunol (2014) 33(5):375–82. doi: 10.3109/08830185.2014.954699
223. Rao S, Yang H, Penninger JM, Kroemer G. Autophagy in non-small cell lung carcinogenesis: A positive regulator of antitumor immunosurveillance. Autophagy (2014) 10(3):529–31. doi: 10.4161/auto.27643
224. Lin H, Yan J, Wang Z, Hua F, Yu J, Sun W, et al. Loss of immunity-supported senescence enhances susceptibility to hepatocellular carcinogenesis and progression in Toll-like receptor 2-deficient mice. Hepatology (2013) 57(1):171–82. doi: 10.1002/hep.25991
225. Ramakrishnan R, Huang C, Cho HI, Lloyd M, Johnson J, Ren X, et al. Autophagy induced by conventional chemotherapy mediates tumor cell sensitivity to immunotherapy. Cancer Res (2012) 72(21):5483–93. doi: 10.1158/0008-5472.CAN-12-2236
226. Kim S, Ramakrishnan R, Lavilla-Alonso S, Chinnaiyan P, Rao N, Fowler E, et al. Radiation-induced autophagy potentiates immunotherapy of cancer via up-regulation of mannose 6-phosphate receptor on tumor cells in mice. Cancer Immunol Immunother (2014) 63(10):1009–21. doi: 10.1007/s00262-014-1573-4
227. Hahn T, Akporiaye ET. alpha-TEA as a stimulator of tumor autophagy and enhancer of antigen cross-presentation. Autophagy (2013) 9(3):429–31. doi: 10.4161/auto.22969
228. Li Y, Hahn T, Garrison K, Cui ZH, Thorburn A, Thorburn J, et al. The vitamin E analogue alpha-TEA stimulates tumor autophagy and enhances antigen cross-presentation. Cancer Res (2012) 72(14):3535–45. doi: 10.1158/0008-5472.CAN-11-3103
229. Wen Y, Graybill WS, Previs RA, Hu W, Ivan C, Mangala LS, et al. Immunotherapy targeting folate receptor induces cell death associated with autophagy in ovarian cancer. Clin Cancer Res (2015) 21(2):448–59. doi: 10.1158/1078-0432.CCR-14-1578
Keywords: tumor microenvironment, secretion, immune system, epithelial-mesenchymal transition, cancer, new autophagy functions
Citation: Bustos SO, Antunes F, Rangel MC and Chammas R (2020) Emerging Autophagy Functions Shape the Tumor Microenvironment and Play a Role in Cancer Progression - Implications for Cancer Therapy. Front. Oncol. 10:606436. doi: 10.3389/fonc.2020.606436
Received: 14 September 2020; Accepted: 22 October 2020;
Published: 25 November 2020.
Edited by:
Daniel Hector Grasso, Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET), ArgentinaReviewed by:
Daniela Laura Papademetrio, University of Buenos Aires, ArgentinaCopyright © 2020 Bustos, Antunes, Rangel and Chammas. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Silvina Odete Bustos, c2lsdmluYWJ2Z0BnbWFpbC5jb20=; Roger Chammas, cmNoYW1tYXNAdXNwLmJy
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.