 Wenjun Wang
Wenjun Wang Ling Bai†‡
Ling Bai†‡ Wei Li
Wei Li Jiuwei Cui
Jiuwei Cui
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol., 08 December 2020
Sec. Cancer Metabolism
Volume 10 - 2020 | https://doi.org/10.3389/fonc.2020.605154
This article is part of the Research TopicRegulation of Metabolic Rewiring in T-Cell MalignanciesView all 8 articles
Lipid metabolism reprograming, as a hallmark of malignancy, has received renewed interest in recent years in such areas as energy sources, cell membrane components, and signaling molecules involved in the rapid tumor growth and the adaptation to the tumor microenvironment. Lipid metabolism deregulation in cancer involves multiple aspects, including an increased lipid uptake, endogenous de novo fatty acid synthesis, fatty acid oxidation, and cholesterol accumulation, thereby promoting tumor growth and progression. Recent advances in the understanding of specific metabolic alterations in cancer reveal novel pathogenesis mechanisms and a growing number of drugs targeting lipid metabolism have been applied in anti-tumor therapy. Thus, this review discusses the lipid metabolic landscape of cancers and the interplay with oncogenic signaling, and summarizes potential therapeutic targets to improve the therapeutic efficiency in cancer patients, in order to provide more reference and thinking for the treatment of lipid metabolism of cancer patients.
Lipid metabolism reprograming is a hallmark of cancer and plays an important role in shaping the tumor microenvironment and cancer cell phenotype, contributing to the occurrence and development of tumors (1). Lipid metabolism of tumor cells can be used to store energy and act as a mediator for cell signaling cascades by utilizing carbon-based precursors produced by aerobic glycolysis to synthesize basic cell components necessary for proliferation (2). Thus, lipid metabolism reprogramming is an essential link in tumor metabolism.
Lipids include triglycerides (TGs), phospholipids (PLs), sphingolipids, and cholesterol, and can function as energy sources, cell membrane components, and precursors of molecules involved in multiple biological processes (such as steroid hormones, vitamins, bile acids, and eicosanoids). Changes in lipid metabolism can affect numerous cellular processes, including cell proliferation, differentiation, and motility (3). In addition, since cancer cells compete for oxygen and nutrients in a nutrient-limited microenvironment, they maintain their malignant potential by altering their metabolism and obtaining fatty acids (FAs).
Cancer cells rewire lipid metabolism through several mechanisms, including increased de novo synthesis and exogenous uptake of FAs, upregulated fatty acid oxidation (FAO), cholesterol accumulation, and induced cancer-associated adipose tissue. At the same time, cancer cells metabolic reprograming can also influence immune cells in a variety of ways, such as through metabolic competition, oncometabolites, and exosomes (4, 5). With the gradual understanding of the extensive roles of metabolism in cancer pathogenesis, the specific metabolic preferences of cancer cells have been exploited to limit cancer progression for clinical benefit, and some therapies have been tested in clinical trials addressing their efficacy in multiple cancers. Meanwhile, accumulating evidence indicates that metabolism-targeting combination treatment or short-term starvation can improve the immune therapy or chemotherapy efficacy (6). Based on the studies on lipid metabolism in pan-cancer, the most extensive changes in lipid metabolism pathways are FA metabolism, cholesterol metabolism, arachidonic acid metabolism, and peroxisome proliferator-activated receptor (PPAR) signal transduction (1). Therefore, this review mainly anchors new advances in FA metabolism and cholesterol metabolism in tumor cells, aiming to provide potential targets for novel cancer therapeutic options.
FAs are the crucial building blocks of several lipid species, and dysregulated FA metabolism is a vital component of lipid metabolism reprograming in cancer. Due to the critical roles of the FAs in the synthesis of the biological membrane, second messengers to transduce signals as well as vital energy sources (2). FA metabolism reprograming contributes to rapid proliferation and invasiveness of the tumor. Lipid metabolic reprograming in cancer cells includes several aspects, such as increased lipid uptake, de novo fatty acid synthesis (FAS), and FAO (Figure 1).
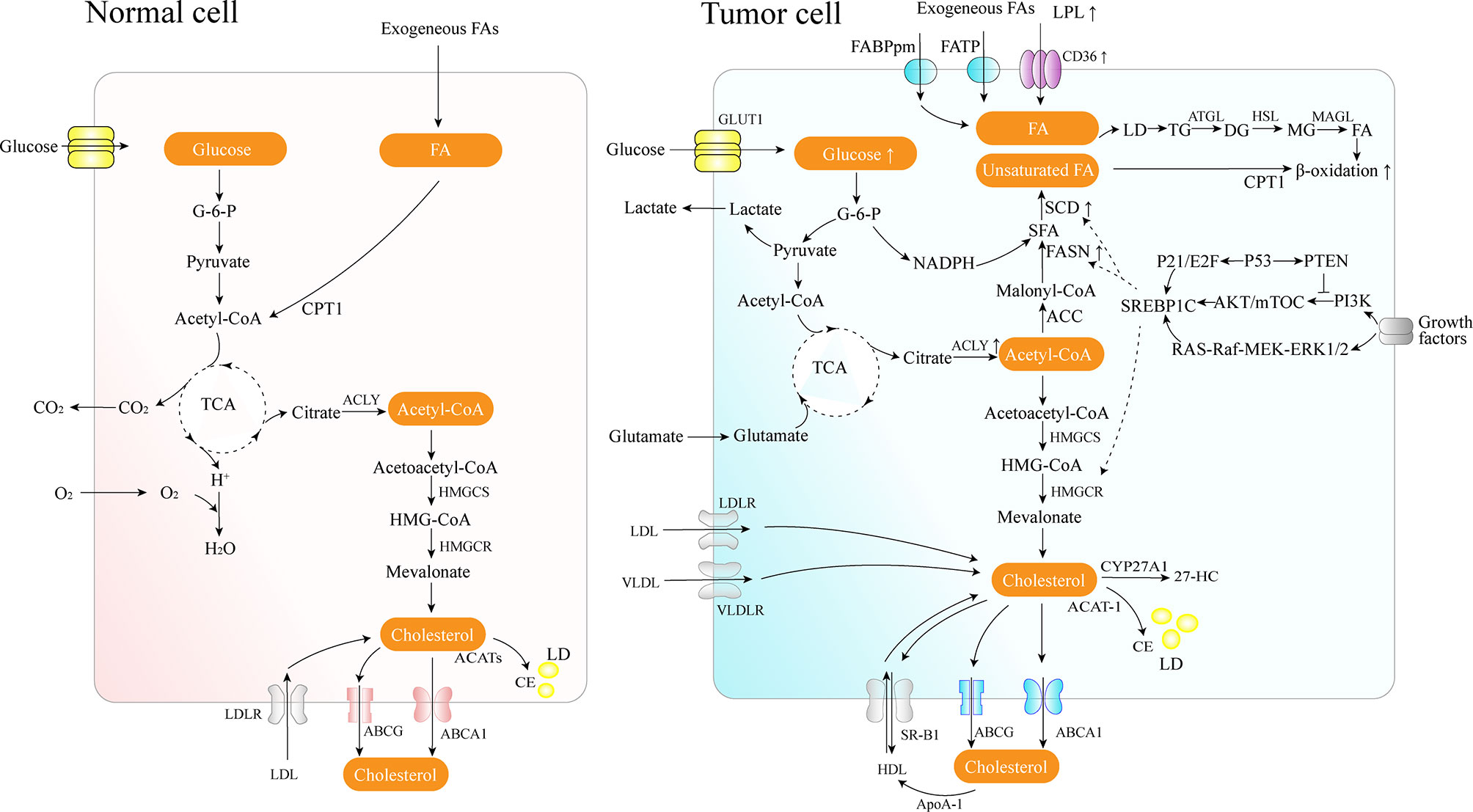
Figure 1 Lipid metabolism overview in normal and cancer cells. Cancer cells acquire diet-derived FA through LPL, CD36, FATPs, and FABPpm. Glucose is converted to acetyl-CoA by glycolysis and on to citrate through the TCA cycle in the mitochondria. The citrate is transported to the cytoplasm and converted back to acetyl-CoA by citrate lyase, which is used as the carbon source for the growing acyl chains. The pentose phosphate pathway from glycolysis generates NADPH. Cancer cells also develop effective de novo FAS machinery with an increase in the activity of key lipogenic enzymes. The surplus lipids (including excess FAs and cholesterol) in a cell exist in the form of neutral, inert biomolecules in the core of LDs. ATGL catalyzes the initial step of lipolysis, converting TGs to DGs; HSL is primarily responsible for the hydrolysis of DGs to MGs, and MAGL hydrolyzes MGs into FFA and glycerol. CPT1, as an outer mitochondrial membrane enzyme, translocates FA across the mitochondrial membranes and then the degradation of long-chain FAs occurs in the mitochondria. Cholesterol homeostasis involves the interplay between de novo synthesis (mevalonate pathway), uptake of dietary cholesterol, and removal of excess cholesterol from peripheral tissues. 27-HC is the metabolite substrate of cholesterol by CYP27A1 enzymes. SREBP-1 is activated through the PI3K/Akt/mTOR pathway and the Ras/Raf/MEK/ERK signaling pathway.
Tumor cells can use circulating free FAs (FFAs) as an energy supply by lipolysis for membrane biosynthesis or signaling processes. Lipoprotein lipase (LPL), CD36 (also known as a fatty acid translocase, FAT), fatty acid transport protein family (FATPs), and plasma membrane fatty acid-binding proteins (FABPpm) are used to acquire diet-derived FAs from the bloodstream by lipolysis in specific cancer cells, such as non-small cell lung cancer (NSCLC), triple-negative breast cancer, liposarcoma, prostate cancer (PCa), etc. (7, 8). LPL is a crucial enzyme secreted by extracellular lipolysis and bound to the luminal surface of capillary endothelial cells, and it can potentially be supplied by tumor cells or by nonmalignant cells in the tumor microenvironment (8, 9). The enzyme is responsible for the TGs hydrolysis from circulating chylomicrons and very-low-density lipoprotein (VLDL). LPL is frequently overexpressed in invasive cervical squamous cell carcinomas, and subsequently increases its invasiveness (10).
FAs released by circulating TGs hydrolysis can be taken up by cells via CD36 for exogenous FFAs uptake, and CD36 exhibits a high affinity for transporting long-chain FAs (8, 11). Moreover, in recent decades, numerous studies have concluded that CD36 plays a role in accelerating tumor growth, metastasis, regulating chemoresistance and radioresistance, modulating tumor immunity, etc. (11–13). Oleic acid, the principal lipid in olive oil, can upregulate the expression of CD36 and facilitate tumor development by activating the Src kinase and the downstream ERK1/2 pathway in a CD36-dependent manner (14). In gastric cancer, CD36 promotes the uptake of exogenous palmitic acid to induce metastasis via the AKT/GSK-3β/β-catenin signaling pathway; therefore, targeting CD36 might constitute a promising new therapeutical approach for peritoneal metastases (12). Elevated CD36 levels and the consequent elevated FFAs uptake may activate the Wnt and TGF-β signaling pathways, thereby inducing epithelial-mesenchymal transition (EMT), which is involved in cancer cell metastasis (15). In pancreatic ductal adenocarcinoma, CD36 can also enhance the expression of several anti-apoptotic proteins, contributing to the resistance to gemcitabine and poor prognosis (13).
On other hand, tumor hypoxia constrains oxygen-dependent stearoyl-CoA desaturase (SCD), resulting in an accumulation of saturated FA precursors (16). While, in hypoxic condition, increased uptake of exogenous unsaturated lipids also plays a vital role in bypassing the requirement for FA desaturation, thus alleviating saturated FA-induced toxicity and maintaining homeostasis in hypoxic cancer cells (16).
De novo FAS in the adult organism occurs mainly in the adipose tissue, liver, and the lactating breast, and people consuming an adequately balanced diet present little endogenous FAS (17). However, several cancer cells show high rates of de novo endogenous FAS (18). In addition, when tumors grow in reduced blood vessel density areas, access to lipids in the circulatory system is also reduced.
FAs are essential constituents of all biological membrane lipids and are crucial substrates for energy metabolism. Since tumor cell growth and division demand carbon, nitrogen, free energy, and reducing equivalents from glucose and glutamine, aerobic glycolysis can support the robust production of acetyl-CoA and nicotinamide adenine dinucleotide phosphate (NADPH) needed for FAS (19). Long-chain saturated FAs can be further modified by elongases or desaturases to form more complex FAs, which are used to synthesize various cellular lipids such as PLs, triglycerides, and cholesterol esters, or to acylate proteins (18). Cancer cells develop capable de novo FAS machinery with an increase in the activity of key lipogenic enzymes such as adenosine triphosphate (ATP)-citrate lyase (ACLY), acetyl-CoA carboxylase (ACC), CoA carboxylase (ACACA), fatty acid synthase (FASN), and SCD (18).
In the cytosol, ACLY converts mitochondria-derived citrate into acetyl-CoA, a vital building block for the endogenous biosynthesis of FAs and cholesterol. ACLY presents elevated levels of activity and expression in several types of cancers (20). In contrast, ACLY knockdown may trigger the activation of p53, thereby facilitating DNA damage-induced apoptosis in cancer cells (20). Moreover, the FAS pathway has two rate-limiting enzymes, ACC and FASN. ACC catalyzes the ATP-dependent carboxylation of acetyl-CoA, generating malonyl-CoA used for FA synthesis following the conversion of citrate and acetate to acetyl-CoA. FASN, downregulated in most normal human tissues, is the leading synthetic enzyme that catalyzes the NADPH-dependent condensation of malonyl-CoA and acetyl-CoA to produce the saturated FA palmitic acid. In contrast, FASN is often highly expressed in human cancers and represents a nearly universal phenotypic alteration in most human malignancies, such as breast, prostate, colon, ovary, endometrium, thyroid, esophagus, stomach, lung cancer, etc. (18, 21, 22). Additionally, a metabolic feature of PCa progression consist of increased rates of de novo FAS via overexpression of FASN, especially in metastatic castration-resistant prostate cancer (mCRPC) (23).
As master regulators of cholesterogenesis and lipogenesis, sterol regulatory element-binding proteins (SREBPs, including SREBP1a, SREBP1c, and SREBP2) transcriptionally activate a cascade of enzymes required for endogenous cholesterol, FAs, TGs, and phospholipid synthesis (24) (Table 1). In hepatocellular carcinoma (HCC), SREBP-1 promotes cancer cell proliferation and metastasis, and its levels negatively correlate with the HCC patient prognosis (25). Previous work has shown that SREBP-1 is activated through several mechanisms, including the phosphatidylinositol 3-kinase (PI3K)/Akt/mTOR pathway, Ras/Raf/MEK/extracellular signal-regulated kinase (ERK) signaling pathway (Ras/ERK pathway), and oncogenic BRAF signaling (26, 27). First, a lack of expression or mutation of the tumor suppressor gene, PTEN, has been clearly established in various types of tumors, and NADH accumulation in respiration-deficient cells leads to inactivation of PTEN and subsequent activation of the Akt survival pathway (28). It has previously been reported that p53-mediated transactivation can also increase PTEN levels, as it has a functional p53 binding site within its promoter (29). Akt-dependent lipogenesis requires a mammalian target of rapamycin (mTORC1) activity to regulate SREBP1 activity, inducing the increased expression of enzymes involved in lipid biosynthesis, including ACLY, FASN, and ACC (30). Furthermore, in glioblastoma cells, epidermal growth factor receptor (EGFR) can promote SREBP-1 cleavage and nuclear translocation through EGFR-PI3K-Akt signaling, without depending on mTORC1 activity (31). In addition, hypoxia also has an impact on the level of FASN expression in vivo: hypoxia significantly upregulates SREBP-1 through induction of Akt, which then firmly binds to the SREBP-binding site/E-box sequence on the FASN promoter (32). Second, in BRAFV600E-mutant melanoma, SREBP-1 is a crucial downstream target of BRAF signaling that induces lipogenesis and enhances membrane lipid saturation, promoting targeted therapy resistance (26). In addition, SREBP-1 is also known to regulate glucose and glutamine metabolic pathways, and SREBP-1 can also protect tumor cells by enhancing glycolytic activities (25).
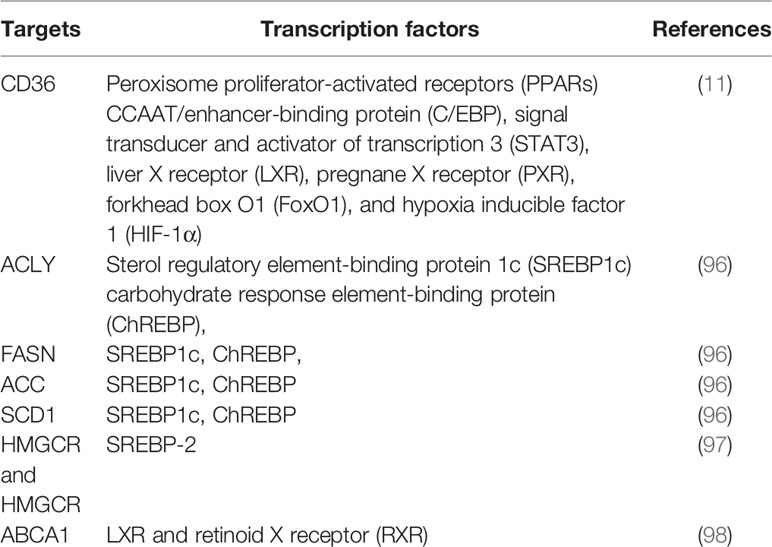
Table 1 Summary of transcription factors that regulate key enzymes in lipid metabolism.
An ever-growing body of experimental evidence supports the notion that the oncogenic nature of FASN-associated lipogenesis strictly depends on the activity and/or expression of important oncogenes and tumor suppressors, such as p53, MYC, HER2, and retinoblastoma (RB). MYC, a dominant oncogene, in collaboration with SREBP, induces lipogenesis both in vitro and in vivo, and plays a role in the initiation and maintenance of tumorigenic growth in MYC-driven cancers (33). Rueda–Rincon et al. reported that p53, an important tumor suppressor, can repress SREBP1 expression via the p21 (cyclin-dependent kinase inhibitor 1A)/Rb/E2F transcription factor pathway, resulting in SCD and a subsequent decrease in mono-unsaturation of phospholipid acyl chains (27). HER2, which is frequently overexpressed in breast cancer and other cancers, can stimulate the expression of FASN via the PI3K/Akt/mTOR and Ras/Raf/MAPK pathway (34, 35). The prolyl isomerase Pin1 can enhance FAS by regulating ACC1, FASN, and the SREBP-1, and it can also suppress AMPK phosphorylation to stabilize the ACC1 protein (36).
Humans lack the enzymes required for generating polyunsaturated FAs from saturated and mono-unsaturated species; however, SCD1, which introduces a double bond in the Δ9 position of saturated fatty acids (SFAs) to produce mono-unsaturated fatty acids (MUFAs), has an increased expression in various cancer cells and is involved in the promotion of cancer cell proliferation, migration, metastasis, and tumor growth (37, 38). Intensively proliferating cancer cells are distinguished by the higher demand for MUFAs, which are utilized mainly to synthesize new membrane-forming PLs, triacylglycerols, and cholesteryl esters (37). The unsaturated FAs promote the activation of NF-κB (nuclear factor κ-light-chain-enhancer of activated B cells), a well-known pro-tumorigenic driver, which in turn regulates the expression levels of SCD1 at transcriptional level (39). Single-cell imaging studies and mass spectrometry analysis showed that ovarian cancer stem-like cells (CSCs) (ALDH+/CD133+) had an higher degree of lipid unsaturation than non-CSCs (ALDH−/CD133−), mediated by lipid desaturases (including SCD1 and Δ6) (39). In turn, lipid desaturases play a role in the maintenance of cancer cell stemness (39). Furthermore, Lai et al. proposed a Wnt-SCD- lipoprotein receptor-related protein (LRP) loop in CSC-related tumor development (40). In rodent hepatic stellate cells and tumor-initiating stem cell-like cells, Wnt/β-catenin interacts with SREBP-1c in novel sterol-regulatory element sites to promote SCD1 expression, which amplifies the Wnt pathway via stabilization of low-density LRP5 and six mRNAs (40).
Long-chain FAs degradation occurs in mitochondria and is catalyzed by several carnitine acyl transferases, including carnitine palmitoyltransferase (CPT) 1, located in the outer membrane, and CPT2, located in the inner membrane, together with a carnitine-acylcarnitine translocase (CAT) (41). CPT1, as an outer mitochondrial membrane enzyme, catalyzes the rate-limiting step of β-oxidation by translocating FA across the mitochondrial membranes. Its activities vary according to tissue-specific needs in FA metabolism and energy expenditure. Immunohistochemistry staining has shown elevated expression levels of CPT1 in human glioma tissues (41).
Furthermore, tumor cells can acquire FAs through lipolysis to perform FA β-oxidation (also known as FAO), which further promotes cancer proliferation, survival, drug resistance, and stemness (42–44). FAO can be utilized to produce high levels of ATP and support the proliferation of triple-negative breast cancer, and glioma (41, 43). However, FAO is not a predominant oxidative substrate for ATP generation. Guppy and colleagues reported that approximately 10% glucose, 14% glutamine, 7% palmitate, 4% oleate, and 65% from unidentified sources contribute to the oxidative component in MCF-7 breast cancer cell line (45). Moreover, promyelocytic leukemia can modulate PPAR signaling and FAO, thereby contributing to the hematopoietic stem cell maintenance (44). At the same time, NANOG, a key regulator of cell reprograming, reduces mitochondrial oxidative phosphorylation (OXPHOS) and production of ROS and promotes FAO, contributing to the self-renewal ability and therapeutic of CSC (46).
Considering that newly synthesized FAs are rapidly incorporated into neutral and phospholipid stores, cancer cells are also required to possess a complementary lipolytic pathway to liberate stored FAs for metabolic and signaling purposes (47). During periods of hypoxia, FA delivery to the cells exceeds FA oxidation rates, which consumes significant amounts of oxygen, resulting in elevated mitochondrial ROS production and subsequent cell damage and apoptosis, known as lipotoxicity (48, 49). Therefore, FA oxidation switch-off combined with the storage of excess FAs in TG-LDs through inhibition of lipolysis would constitute a conceivable strategy for cancer cells in hypoxia. The surplus lipids (including excess FAs and cholesterol) in a cell exist in the form of neutral, inert biomolecules in the core of lipid droplets (LDs) (50), which are a hallmark of hypoxic cancer cells, and are released through a combination of lipolysis and a selective autophagic mechanism called lipophagy (48, 51–53). At the same time, when exposed in acid TME, autocrine TGF-β2 signaling is drived in cancer cells, thus further fueling FAs uptake and oxidation as well as formation of LDs (54). LDs accumulation is associated with a more aggressive cancer phenotype and increased migration via elevated levels of pro-oncogenic signaling lipids, particularly lysophosphatidylcholines, and activation of pro-oncogenic SRC kinase signaling (53). Moreover, prolonged nutrient deficiency or lipid overload tend to provoke the autophagy of cancer cells (55). Cancer cells can employ LDs to modulate autophagy through providing the lipid precursors for the formation of autophagic membranes or signaling that activates the autophagy genes expression (56–58). HER2 overexpression can upregulate the PPAR-γ, which promotes conversion and storage of excess FAs to TGs, thus allowing cells to avert cell death resulting from endogenous palmitate-related lipotoxicity (34). TGs can also sequester exogenous unsaturated FAs, particularly oleate (16). While, under serum- and O2-limited conditions, TGs can neutralize excess FA saturation through preferential release of unsaturated FAs to ameliorate stress (16).
Conversely, adipose triglyceride lipase (ATGL), hormone-sensitive lipase (HSL), and monoacylglycerol lipase (MAGL) provide a stream of intracellular FFAs that play important and critical roles in cancer cell proliferation and tumor progression by de-esterification.
ATGL has been found to be significantly reduced in a variety of human malignancies, including lung and pancreatic cancer. The role of ATGL in several cancer cells such as PCa and HCC is ambiguous, as contradicting evidence has been reported so far (59, 60). Liu et al. reported that ATGL is highly expressed in human HCC tissues and positively correlated with tumor size, predicting poor prognosis (61). At the same time, Di Leo et al. demonstrated that ATGL levels are inversely correlated with the proliferation rate of HCC-derived cell lines, which depend on intact ATGL enzymatic activity (62). Moreover, ATGL upregulation in breast cancer was associated with an enriched adipocyte tumor microenvironment (TME), contributing to the aggressiveness of high-grade tumors (63). Zhang et al. demonstrated that, in hypoxia, hypoxia-inducible gene 2 (HIG2), as a novel endogenous inhibitor of ATGL, mediates the lipolytic inhibition, promotes LD accumulation, attenuates ROS production, and enhances cancer cell survival (48). However, Di Leo et al. did not observe this mechanism in their model, suggesting that alternative mechanisms contributing to the control of proliferation need further research (62). In addition, long non-coding RNA NEAT1 expression is upregulated by hypoxia through hypoxia inducible factor (HIF)-2α in various types of cancers, and can disrupt hepatoma cells lipolysis via ATGL (61). Additionally, ATGL-mediated p53 acetylation by the PPARα/p300 axis is responsible for its inhibitory effect on glycolysis. In contrast, ATGL overexpression redirects HCC cell metabolism towards a less glycolytic phenotype via P53 and thereby is more resistant to glycolysis inhibitors (62). Liu et al. reported that ATGL contributed to the proliferation of HCC cells by upregulating AKT phosphorylation levels (64). Therefore, cancer cell metabolism is also regulated by direct crosstalk with tumor-surrounding stromal components (such as adipocytes) and hypoxia (63). On the other hand, ATGL may have a broad influence on cancer processes, such as redox homeostasis, inflammation, and autophagy, through PPARα signaling (59).
MAGL expression is highly elevated in human cancer cells and primary tumors, including PCa, neuroblastoma, HCC, colorectal, ovarian, endometrial cancers, etc. Aggressive cancer cells do indeed acquire the ability to liberate FFAs from neutral lipid stores because of the heightened expression of MAGL, which is the principal regulator of FFA levels (47). Zhang et al. confirmed that promoter methylation of large tumor suppressor kinase 1 (LATS1) resulted in the dysfunction of the Hippo signal pathway, which induced overexpression of MAGL in HCC (65). Moreover, Nomura et al. reported that MAGL regulates a host of secondary lipid metabolites that include essential signaling molecules, such as LPA and prostaglandin E2 (PGE2), which have been reported to promote cancer cell aggressiveness (47).
Accumulating evidence indicates that sphingolipids including sphingosine, ceramide, and sphingosine-1-phosphate, involve multi-layered aspects of cancer cell biology (66). Perturbation of sphingolipid homeostasis has been reported in various solid tumors and hematological malignancies (67). As the most abundant cellular sphingolipids, ceramides can exert pro-apoptotic effects as tumor-suppressing lipids and augment the efficacy of chemotherapeutics and targeted therapies (67–69). However, several enzymes (e.g., acid ceramidase, ceramide kinase, etc.) that accelerate the metabolism of ceramides, have been found to have an enhanced expression level in multiple tumors (67, 68, 70). Therefore, ceramide metabolism has been utilized to develop some drugs that target these vital enzymes thus inducing lethal ceramide accumulation and overcoming cancer therapy resistance (67).
Cholesterol, as an essential component of the cell membrane, alters the biophysical properties of the membrane by governing its fluidity and impacts various biochemical functions, including modulation of signaling pathways and intercellular communication, as an integral component of lipid rafts (71, 72). Cholesterol homeostasis involves the interplay between de novo synthesis, uptake of dietary cholesterol (low density lipoprotein – LDL or high density lipoprotein – HDL), and removal of excess cholesterol from peripheral tissues (73) (Figure 1). In cancer, the reprogramed cholesterol metabolism can provide a signal transduction platform and activate stemness and oncogenic signaling (such as the Hedgehog pathway, mTORC1) as a second messenger or component of lipid rafts, thereby contributing to cancer progression and invasion (74).
Since several tumor cells have been shown to present with an abnormal distribution of cellular cholesterol, such as clear cell renal cell carcinoma (ccRCC) and PCa bone metastases, some essential cholesterol transporters have been characterized as cancer-related factors (75, 76). As a transmembrane protein, the ATP-binding cassette transporter (ABCA1) is responsible for reverse cholesterol transport from the inner cell to the circulatory system. By stabilizing ABCA1, apolipoprotein A-I (ApoA-I) can recover the extracted cholesterol and synthesize HDL (77). The interaction of lipidated ApoA-I in discoid or more mature HDL particles with another transporter of the ABC family, ATP-binding cassette subfamily G member 1 (ABCG1), further contributes to reverse cholesterol transport. Finally, HDL particles binding to the scavenger receptor class B type 1 (SR-B1) transfer cholesterol down a cholesterol gradient (78). ABCA1 is significantly overexpressed, promotes EMT, and leads to increased invasiveness in advanced stages of colorectal cancer (CRC) (77). Reduced ApoA-1 mRNA and protein levels have been found in HCC compared to normal liver tissue, the main source of ApoA-I (78). ApoA-I exhibits tumor suppressive activity both in vitro and in animal studies, accompanied by anti-inflammatory and immune-modulating effects, which have been reviewed by Georgila et al. (78). However, conflicting conclusions are present in some cancers; for example, ApoA-I levels were reported to be positively associated with breast cancer risk (79). As a receptor for the uptake of HDL-associated cholesterol, elevated SR-B1 expression causes an increased uptake of HDL-cholesterol in ccRCC through the reduction in CpG islands methylation (75). Besides, sterol carrier protein 2, an intracellular cholesterol trafficking protein, can transport cholesterol from the cytoplasm to the plasma membrane (80). Then, the membrane cholesterol concentration induces the activation of PKA/SUFU/GLI1 signaling via the smoothened receptor, which is well-known as Hedgehog signaling, resulting in the inhibition of apoptosis and promotion of the cell cycle in pituitary adenomas (80).
Cholesterol is synthesized via the mevalonate pathway and regulated by its rate-limiting enzyme, 3-hydroxy-3-methylglutarylcoenzyme A reductase (HMGCR), which catalyzes the reduction of 3-hydroxy-3-methylglutarylcoenzyme A (HMG-CoA) to mevalonate. In glioblastoma patients, CSCs have been found to overexpress mevalonate pathway genes, and MYC can alter mevalonate metabolism (81). A meta-analysis encompassing 865 primary breast cancer patients reported that high HMGCR and additional mevalonate pathway genes mRNA levels correlated with poor patient prognosis and reduced survival (82). Mevalonate was found to constitute an escape mechanism of survival and growth in HER2+ breast cancer models resistant to anti-HER2 therapies, partly through the activation of downstream YAP (Yes-associated protein)/TAZ (transcriptional coactivator with PDZ-binding motif)-Survivin signaling particularly through farnesyl pyrophosphate/geranylgeranyl pyrophosphate (83). Regulation of HMGCR is influenced by a network of modulatory proteins, including SREBP2, SREBP cleavage activated protein (SCAP), and insulin-induced gene (Insigs). Oncogenic growth signaling, such as PI3K/AKT and RAS/MAPK, is triggered to maintain cholesterol homeostasis by activating SREBP-mediated cholesterol biosynthesis. SREBP2, transcribed from the SREBF2 gene, is the main transcription factor that activates the genes involved in mevalonate and cholesterol synthesis (84). Constitutively activated RAS/MAPK signaling also activates heat shock factor 1 (HSF1), thereby increasing the expression of cholesterol biosynthesis-related genes such as SREBF2, HMGCR, HMGCS1, etc. In contrast, HSF1 inhibition sensitizes HCC cells to the antiproliferative effects of simvastatin (85). Hypoxia also induces HIF-1α accumulation, which increases HMGCR levels and activity by stimulating its transcription (86). P53 can actively repress activation of SREBP-2 through transcriptionally upregulating the ABCA1 (87). In breast cancer cells, mutant p53, which is the most frequent target for mutation in tumors, contributes to the upregulation of the mevalonate pathway, through recruitment of many sterol biosynthesis genes, or probably through one or more of the SREBP proteins (88). Furthermore, recent research reported that radiation upregulated the expression of four enzymes in the cholesterol biosynthesis pathway and increased the cell cholesterol content (89).
Moreover, cholesterol is also related to therapeutic resistance. Cholesterol and sphingolipids are also components of caveolae as major structural lipids, accompanied by the defining protein components, caveolins, and cholesterol appears to modulate caveolin-1 expression through a steroid regulatory binding element present in the caveolin-1 promoter and SREBP-1 (90). Since caveolin-1 is crucial for cellular energy homeostasis in tyrosine kinase inhibitor (TKI)-resistant tumor cells by mediating glucose uptake via GLUT3, Azhar et al. demonstrated a link between elevated cellular cholesterol and TKI resistance in NSCLC, which is independent of EGFR mutation status (91).
The oxysterol 27-hydroxycholesterol (27-HC) is known as the metabolite substrate of cholesterol by cytochrome P450 family 27 subfamily A member 1 (CYP27A1) enzymes. An elevated level of serum cholesterol corresponds to a high level of serum 27-HC. As a selective endogenous estrogen receptor (ER) agonist and liver-X-receptor (LXR) agonist, 27-HC may contribute to ER-positive breast cancer growth and metastasis by inducing several EMT genes and disrupting constitutive p53 signaling in an MDM2-dependent manner (92, 93).
Furthermore, inside cells, the free cholesterol excess is esterified and stored as CE in LDs to prevent the toxicity generated by the over-accumulation of free cholesterol, mediated by acyl-CoA cholesterol acyltransferase (ACAT) encoded by Acat1 and Acat2 genes. Increased CE levels have been reported in breast cancer, leukemia, glioma, prostate, pancreatic cancer, renal cell carcinoma, etc. (94, 95).
The critical role of altered metabolic pathways in cancer cell proliferation, but not in most normal human tissues, has led to some proposed strategies for cancer-specific treatments by targeting these pathways. To date, several metabolic enzymes inhibitors, such as glycolysis inhibitors, have been studied in clinical trials as targeted cancer therapeutics (Table 2 and Figure 2).
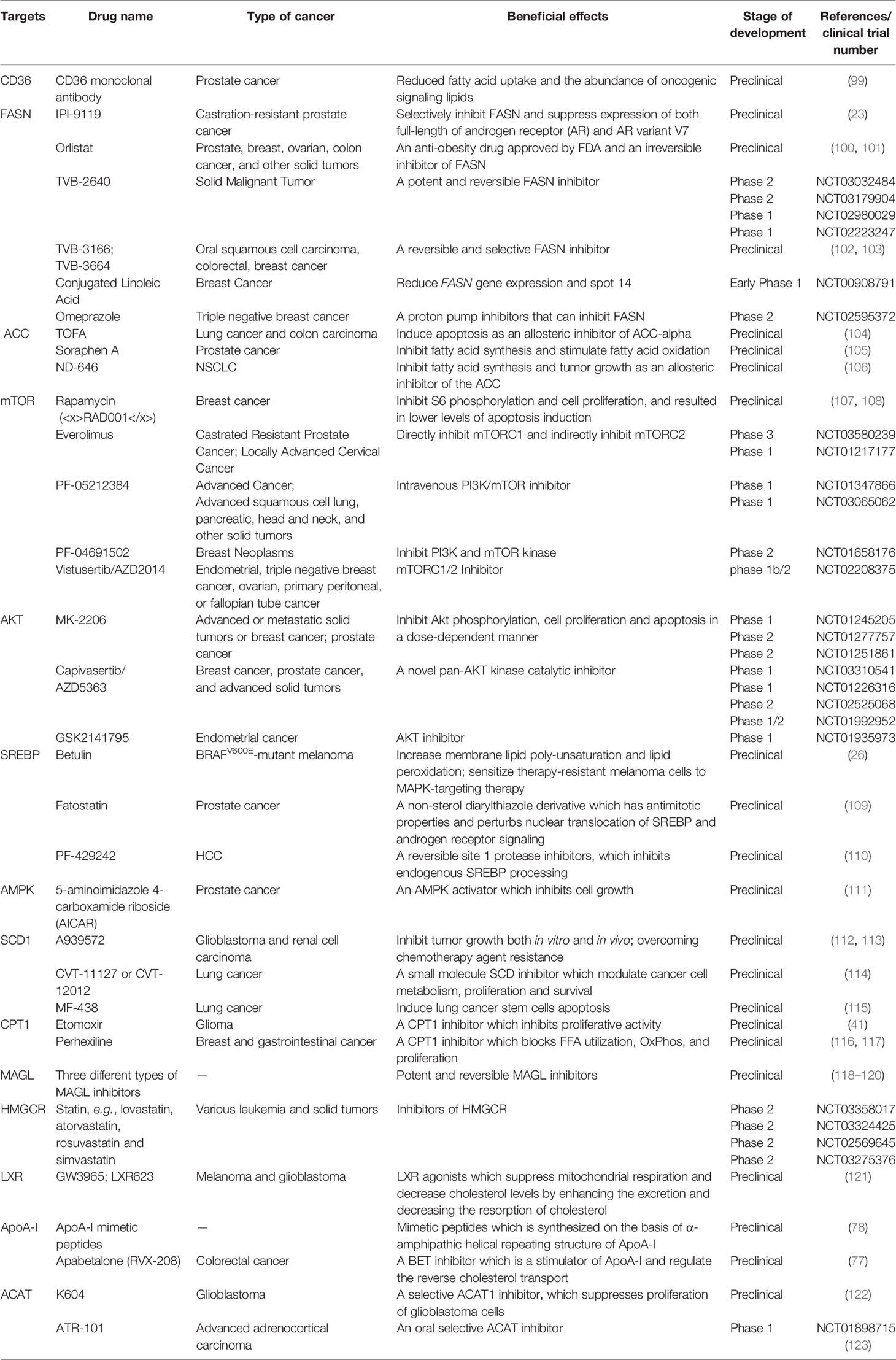
Table 2 Therapeutic perspectives in lipid metabolic pathways.
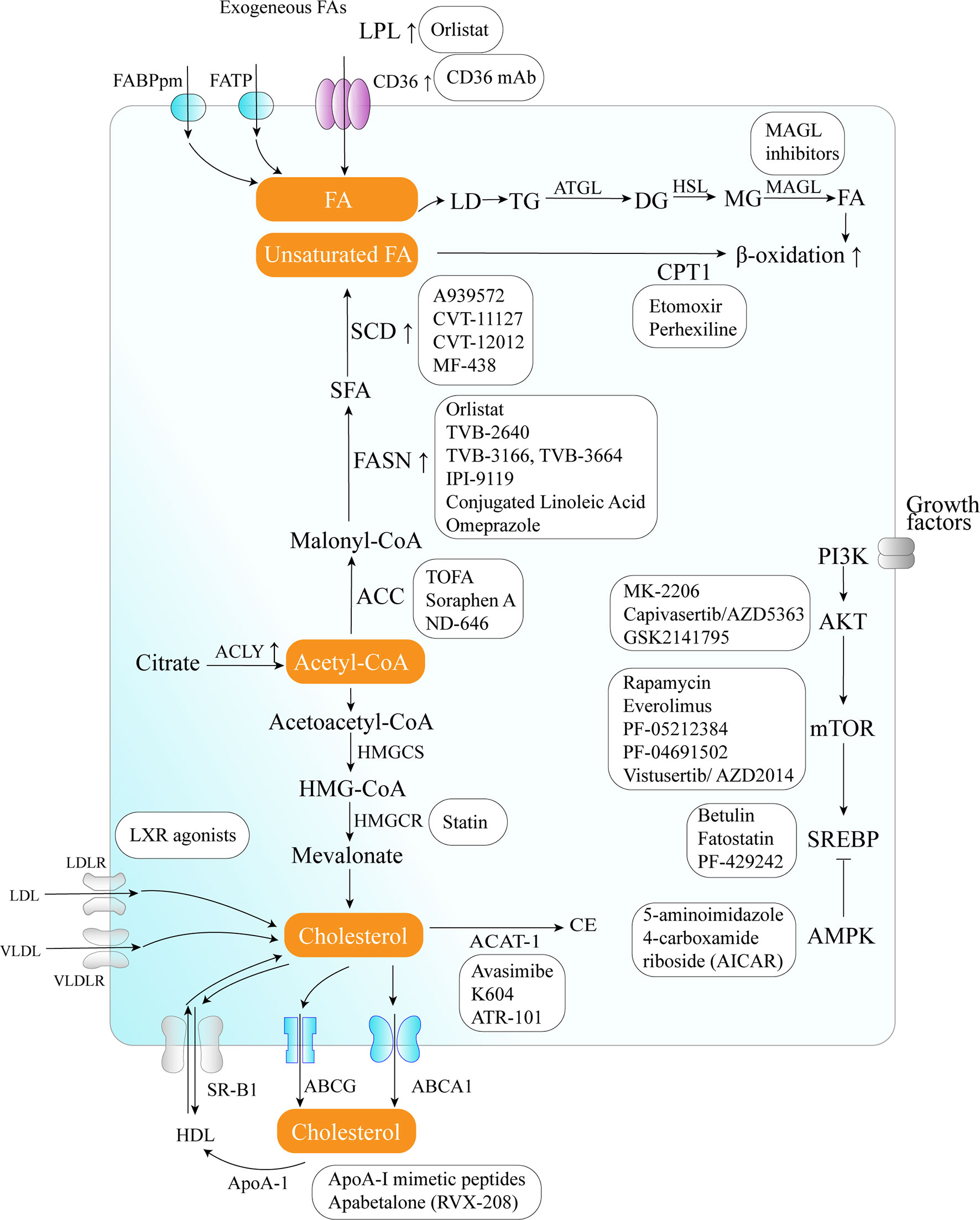
Figure 2 Therapeutic targets and anticancer drugs within the lipid metabolism pathway. The lipid metabolism-targeting therapies are shown as white boxes.
Orlistat, a compound that inhibits both LPL and FASN, may be used in tumors that are provided with LPL and express CD36 (8). However, the prolonged systemic suppression of LPL activity could result in hypertriglyceridemia and consequent pancreatitis, particularly if dietary fat intake is not curtailed. Furthermore, tumor cells may establish metastases in LPL-rich tissues, which can provide LPL to the nearby TME (8).
The potential of targeting oxLDL and related receptors (e.g., CD36 receptor) to reduce metastasis has been demonstrated in a variety of orthotopic cancer models (124). The direct impact of reducing oxLDL has never been investigated. At the same time, the use of anti-CD36 neutralizing antibodies causes almost complete inhibition of metastasis in immunodeficient or immunocompetent orthotopic mouse models of human oral cancer, with no side effects (125). CD36 monoclonal antibodies can reduce tumor growth in PCa patient-derived xenografts; at the same time, the combination with FASN inhibitor increases the efficacy of the CD36 blockade (99). In ovarian cancer, CD36 inhibition effectively reduced adhesion on collagen I matrices, which are abundantly expressed in the omental basement membrane where ovarian cancer cells preferentially attach (126). However, since CD36 also plays an important role in the myocardial metabolism of FAs (127), side effects caused by long-term inhibition of CD36 and the effect of direct targeting of oxLDL need further investigation.
The inhibition of different enzymes (especially ACC and FASN, SCD) and the AKT/mTOR/SREBP-1 pathway within the de novo FA biosynthetic pathway can block cancer cell growth. Several ACC inhibitors, including TOFA, Soraphen A, and ND-646, have shown high efficacy in disturbing FAS, inducing oxidative stress, and inhibiting tumor growth in lung, colon, and prostate cancer in preclinical studies (104–106).
FASN is an established therapeutic target. First-generation (e.g., orlistat and cerulenin) and next-generation (TVB-3166 and TVB-2640) FASN-targeting drugs have been developed. Orlistat (tetrahydrolipstatin), a tight-binding irreversible inhibitor of the FASN thioesterase domain, exhibits both in vitro and in vivo antitumor properties against melanoma, breast cancer, PCa cells, and oral tongue squamous cell carcinoma (100). However, first-generation FASN inhibitors such as orlistat have pharmacological characteristics (e.g., lack of selectivity, poor metabolic stability, low cell permeability, etc.) and display detrimental systemic side effects (e.g., anorexia) (102). In contrast, next-generation FASN inhibitors exhibit anti-tumor potential, higher specificity for FASN, and limited systemic toxicity in a preclinical study. TVB-2640 was included in a phase II clinical trial. In multiple phases, 1/2 FASN inhibitor studies have been conducted (e.g., NCT00908791 for breast cancer, NCT02980029 for colon cancer, etc.), but no results have been published. TVB-3166 and TVB-3664 have shown anti-tumor activity in oral squamous cell carcinoma, breast, and colorectal cancer preclinical studies (102, 103). Giorgia et al. developed a selective and potent FASN inhibitor (IPI-9119), which can significantly reduce cell growth and induce cell cycle arrest and apoptosis in PCa cells, reduced growth of AR-V7-driven CRPC xenografts, and human mCRPC-derived organoids and enhances the efficacy of enzalutamide in CRPC cells (23). Furthermore, proton pump inhibitors (PPIs) are also effective inhibitors of the thioesterase activity of FASN (128), and a PPI, omeprazole, has entered clinical trials in patients with triple-negative breast cancer (NCT02595372).
The importance of the AKT/mTOR/SREBP-1 pathway in regulating gene expression of critical enzymes has received interest as a target. To date, several clinical trials aimed at evaluating different AKT inhibitors such as MK-2206 (NCT01245205, NCT01277757, NCT01251861), Capivasertib/AZD5363 (NCT03310541, NCT01226316, NCT02525068, NCT01992952), and GSK2141795 (NCT01935973) in multiple cancers; however, the results of these studies have not been reported to date. A combination of the mTOR inhibitor everolimus and metformin seems to synergistically inhibit proliferation and colony formation of breast cancer cells with few side effects in vitro (129). As an allosteric inhibitor of mTOR, some rapamycin analogs have been approved by the US Food and Drug Administration to treat breast cancer (107). Since knockdown and inhibition of SREBP-1 thwarts the glucose uptake, glycolytic activity, and lipid metabolism of HCC cells, betulin, a SREBP-1 inhibitor, has been shown to enhance the sensitivity of HCC cells to sorafenib (a first-line antitumor agent for advanced HCC treatment) and to facilitate its antitumor effect in vivo, suggesting that a SREBP-1 inhibitor and sorafenib combination can be a novel therapeutic option (25). In therapy-resistant melanoma, the inhibition of SREBP-1 re-sensitizes resistant cells to BRAF-targeting therapy in vivo, partly through alterations of membrane polyunsaturation and subsequent lipid peroxidation (26). Since SREBPs also induce androgen receptor (AR) activity in addition to lipogenesis, fatostatin, a non-sterol diarylthiazole derivative, has been reported to perturb the nuclear translocation of SREBP and caspase-dependent programed cell death of prostate cancer cells in vitro (109). Furthermore, AMPK inhibits SREBP activation through interaction with and phosphorylates SREBP-1 and SREBP-2 (111). Two agents that activate AMPK, 5-amino-4-imidazolecarboxamide ribose (AICAR) and rosiglitazone, reduce the activity of ACC and the concentrations of FASN and ACC in PCa cells and inhibit the stimulatory effect of androgen on these parameters (111).
Moreover, SCD1 appears to be a potential anticancer therapeutic target (37). A combined pharmacological approach involving SCD1 may counteract cancer cell chemoresistance and enhance the therapeutic efficacy of commonly used chemotherapeutic drugs, such as SCD1 inhibitor (CVT-11127 or CVT-12012) + gefitinib (114), SCD1 inhibitor (A939572) + temozolomide (112), SCD1 inhibitor (A939572) + temsirolimus (113). However, modest effects have been achieved by targeting SCD, suggesting that alternative desaturation pathways reduce the cancer cell dependence on SCD-mediated desaturation. Indeed, some cancer cell lines exploit an alternative FA desaturation pathway that desaturates palmitic acid to the unusual FA sapient via fatty acid desaturase 2 (FADS2) (130). Moreover, since SCD1 plays a major role in the de novo synthesis of TGs, cholesterol esters, and wax esters required for normal skin and eyelid function, the targeted disruption of SCD1 in mice causes atrophy of sebaceous and meibomian glands and depletion of wax esters in the eyelid (131).
FA catabolism inhibition might be a promising anticancer strategy. A blinded, placebo-controlled preclinical study showed that etomoxir, a CPT1 inhibitor, slowed tumor growth and prolonged survival in a mouse model of malignant glioma (41). Similarly, another β-oxidation inhibitor, perhexiline, blocked FFA utilization, OxPhos, and cancer cell proliferation in vitro (116). Recently, perhexiline was reported to increase the sensitivity of gastrointestinal cancer cells to oxaliplatin. This suggests that inhibiting FA catabolism can be a promising therapeutic strategy in combination with conventional chemotherapy for patients with gastrointestinal cancers (117).
MAGL inhibition also decreases cyclin D1 and Bcl-2 expression, thereby inhibiting the proliferation and promoting tumor apoptosis or/and cell cycle arrest in endometrial cancer and CRC (132, 133). Three different types of MAGL inhibitors have been reported in the literature so far, which have been thoroughly reviewed by Carlotta et al. including (1): compounds that bind the enzyme covalently and irreversibly (2), compounds that bind it covalently and reversibly, and (3) compounds that bind it non-covalently (118). Irreversible covalent inhibitors are the most widespread class of MAGL inhibitors. In contrast, only recently, several reversible MAGL inhibitors have been reported to lack unwanted side effects from chronic treatment, including long-chain salicylketoxime derivatives and phenyl (piperazin-1-yl) methanone derivatives (118–120).
Cancer cells need cholesterol for growth and survival and decreasing intracellular cholesterol biosynthesis may be a potential therapeutic strategy. Statins, inhibitors of HMGCR, trigger a robust homeostatic feedback response that ensures the cells upregulate and restore the MVA pathway, which has been successfully exploited for over 20 years to control hypercholesterolemia (82). Statins are proven to benefit lung cancer patients receiving EGFR-TKI therapy with improved response rates, prolonged progression-free survival, and overall survival by lowering cellular cholesterol, inducing loss of Cav1 expression, and triggering apoptosis to suppress NSCLC cell growth (91). Apart from cholesterol inhibition, statins inhibit cholesterol-independent processes, including cellular proliferation and intracellular signaling. However, statins were not associated with a reduced risk of pancreatic cancer in clinical trials. One possible reason is that HMGCR is also required for downstream protein prenylation, a critical process for protein activation. Thus, the statin effect not only inhibits cholesterol synthesis, but also other pathways that may render toxicity to normal cells. This nonspecific toxicity is a possible reason for the limited anticancer outcome of statins in clinical trials (94). At the same time, statins seem to increase the risk of some cancers, such as breast cancer and non-melanoma skin cancers, in several case-control studies and prospective studies (134). There are some serious adverse effects of statins such as muscle symptoms (e.g., rhabdomyolysis), type 2 diabetes mellitus, neurological and neurocognitive conditions (e.g., hemorrhagic stroke), hepatotoxicity, renal toxicity, and so on (135). Further investigation should focus on the more accurate and specific metabolite effects produced in the MVA pathway in different tumor cells.
LXR agonists represent novel means to counteract cholesterol levels in tumor cells, including glioblastoma and melanoma, by enhancing the excretion (increase in ABCA1) and, at the same time, by decreasing the resorption of cholesterol (decrease in LDL receptor) (121). Since LXR agonists upregulate the expression of the pro-apoptotic Bcl-2 family member, Noxa, BH3 mimetics (ABT263 and ABT199), and LXR agonists synergistically reduce cellular viability by enhancing apoptosis, resulting in a synergistic anti-proliferative effect across solid tumor cells (121). In preclinical models of diffuse intrinsic pontine glioma, a small molecule, menin inhibitor MI-2, has been proven to disrupt cholesterol homeostasis by inhibiting the conversion of 2,3-oxidosqualene to lanosterol, which also activates LXR to increase cholesterol efflux (136).
Furthermore, some strategies aimed at directly augmenting ApoA-I (such as intravenous administration of autologous delipidated HDL, purified native ApoA-I, or recombinant ApoA-I Milano protein, etc.), or mimicking ApoA-I (such as ApoA-I mimetic peptides) functionality have already been used successfully in preclinical cancer studies (78).
In an orthotopic mouse model, cholesterol esterification inhibition disturbed cholesterol homeostasis by increasing the intracellular free cholesterol level, which was associated with elevated ER stress and eventually led to apoptosis and decreased tumor growth and metastasis (94). Several ACAT inhibitors, such as avasimibe, K604, and ATR-101, have been studied preclinically (123), and ATR-101 has entered clinical trials in patients with advanced adrenocortical carcinoma (NCT01898715).
Lipid metabolism not only composes the essential component of survival and proliferation in cancer cells, but also influences the crosstalk with immune cells in TME. With the gradual understanding of the mechanism of lipid metabolism in tumors, targeting the related enzymes and genes involved in the metabolism may provide an emerging approach for cancer treatment and restoration of tumor immunology.
However, not only tumor cells, but also immune cells, such as activated T cells, are considerable similarities in metabolic reprograming (137). The FA metabolic enzymes and synthetases are also involved in both cancer and normal whole-body metabolic homeostasis. For example, FASN expression is considered a metabolic marker of cell proliferation instead of merely a marker of malignancy (138). Therefore, by analyzing the difference of lipid content in tumor and normal tissue microenvironment, it is helpful to find more targeted lipid metabolism regulation strategies. At the same time, clarifying the specific mechanisms involved in the reprograming of lipid metabolism as well as their dual role in multiple tumor-associated signaling pathways may be better to identify therapeutic targets.
It is well known that some drugs have been used for metabolic diseases, such as FASN inhibitors, have been gradually developed for the field of anti-tumor therapy, and more overlap between metabolic diseases and cancer may be found in the future. So, additional investigations should carefully consider how to manage and combine targeting lipid metabolism or dietary interventions and other therapies to obtain the maximal clinical benefit.
WW and LB carried out the primary literature search, drafted and revised the manuscript, and participated in discussions. WL helped modify the manuscript. JC carried out the design of the research and literature analysis, drafted and revised the manuscript, and participated in discussions. All authors contributed to the article and approved the submitted version.
This work was supported by the National Natural Science Foundation of China (Grant 81672275 and 81874052 to JC; grant 81702589 to LB); Project of Jilin Provincial Department of Education (Grant JJKH20190020KJ to JC); Project of Department of Science and Technology of Jilin Province (Grants 20180101009JC and 20190303146SF to JC); Outstanding Talent Cultivation Program for Doctoral Students in Norman Bethune Health Science Center of Jilin University (Grant to LB).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
ATP, adenosine triphosphate; ACLY, ATP-citrate lyase; ACC, acetyl-CoA carboxylase; ACACA, CoA carboxylase; ATGL, adipose triglyceride lipase; ApoA-I, apolipoprotein A-I; ABCA1, ATP-binding cassette transporter; ACAT, acyl-CoA cholesterol acyltransferase; AR, androgen receptor; CSCs, cancer stem-like cells; CEs, cholesteryl esters; CPT, carnitine palmitoyltransferase; CAT, carnitine-acylcarnitine translocase; ccRCC, clear cell renal cell carcinoma; CRC, colorectal cancer; CYP27A1, Cytochrome P450 Family 27 Subfamily A Member 1; EMT, epithelial-mesenchymal transition; EGFR, epidermal growth factor receptor; ERK, extracellular signal-regulated kinases; ER, estrogen receptor; FAs, fatty acids; FAS, fatty acid synthesis; FASN, fatty acid synthase; FAO, fatty acid oxidation; FATPs, fatty acid transport protein family; FABPpm, plasma membrane fatty acid-binding proteins; HCC, hepatocellular carcinoma; HIF, hypoxia inducible factor; HSL, hormone-sensitive lipase; HDL, high density lipoprotein; HMGCR, 3-Hydroxy-3-methylglutarylcoenzyme A reductase; HMG-CoA, 3-Hydroxy-3-methylglutarylcoenzyme A; HSF1, heat shock factor 1; IGF, insulinlike growth factor; LPCAT1, lysophosphatidylcholine-acyltransferase 1; LPL, lipoprotein lipase; LDs, lipid droplets; LDL, low density lipoprotein; LXR, liver-X-receptor; MUFA, mono-unsaturated fatty acids; mCRPC, metastatic castration-resistant prostate cancer; MAGL, monoacylglycerol lipase; NADPH, nicotinamide adenine dinucleotide phosphate; NSCLC, non-small cell lung cancer; OXPHOS, oxidative phosphorylation; PLs, phospholipids; PI3K, phosphoinositide 3-kinase; PCa, prostate cancer; ROS, reactive oxygen species; SCD, stearoyl-CoA desaturase; SREBP, sterol regulatory element binding proteins; SFAs, saturated fatty acids; SCAP, SREBP cleavage activated protein; SR-B1, scavenger receptor class B type 1; TME, tumor microenvironment; TG, triglyceride; VLDL, very-low-density lipoprotein; 27-HC, 27-hydroxycholesterol.
1. Hao Y, Li D, Xu Y, Ouyang J, Wang Y, Zhang Y, et al. Investigation of lipid metabolism dysregulation and the effects on immune microenvironments in pan-cancer using multiple omics data. BMC Bioinf (2019) 20(Suppl 7):195. doi: 10.1186/s12859-019-2734-4
2. Lim JY, Kwan H. Roles of Lipids in Cancer. In: Advances in Lipid Metabolism. IntechOpen (2018). doi: 10.5772/intechopen.80788
3. Santos CR, Schulze A. Lipid metabolism in cancer. FEBS J (2012) 279(15):2610–23. doi: 10.1111/j.1742-4658.2012.08644.x
4. Yin Z, Bai L, Li W, Zeng T, Tian H, Cui J. Targeting T cell metabolism in the tumor microenvironment: an anti-cancer therapeutic strategy. J Exp Clin Cancer Res CR (2019) 38(1):403–. doi: 10.1186/s13046-019-1409-3
5. Chang CH, Qiu J, O’Sullivan D, Buck MD, Noguchi T, Curtis JD, et al. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell (2015) 162(6):1229–41. doi: 10.1016/j.cell.2015.08.016
6. Ajona D, Ortiz-Espinosa S, Lozano T, Exposito F, Calvo A, Valencia K, et al. Short-term starvation reduces IGF-1 levels to sensitize lung tumors to PD-1 immune checkpoint blockade. Nat Cancer (2020) 1(1):75–85. doi: 10.1038/s43018-019-0007-9
7. Maan M, Peters JM, Dutta M, Patterson AD. Lipid metabolism and lipophagy in cancer. Biochem Biophys Res Commun (2018) 504(3):582–9. doi: 10.1016/j.bbrc.2018.02.097
8. Kuemmerle NB, Rysman E, Lombardo PS, Flanagan AJ, Lipe BC, Wells WA, et al. Lipoprotein lipase links dietary fat to solid tumor cell proliferation. Mol Cancer Ther (2011) 10(3):427–36. doi: 10.1158/1535-7163.mct-10-0802
9. Klein JC, Moses K, Zelinskyy G, Sody S, Buer J, Lang S, et al. Combined toll-like receptor 3/7/9 deficiency on host cells results in T-cell-dependent control of tumour growth. Nat Commun (2017) 8:14600. doi: 10.1038/ncomms14600
10. Carter SA, Foster NA, Scarpini CG, Chattopadhyay A, Pett MR, Roberts I, et al. Lipoprotein lipase is frequently overexpressed or translocated in cervical squamous cell carcinoma and promotes invasiveness through the non-catalytic C terminus. Br J Cancer (2012) 107(4):739–47. doi: 10.1038/bjc.2012.301
11. Wang J, Li Y. CD36 tango in cancer: signaling pathways and functions. Theranostics (2019) 9(17):4893–908. doi: 10.7150/thno.36037
12. Pan J, Fan Z, Wang Z, Dai Q, Xiang Z, Yuan F, et al. CD36 mediates palmitate acid-induced metastasis of gastric cancer via AKT/GSK-3β/β-catenin pathway. J Exp Clin Cancer Res CR (2019) 38(1):52–. doi: 10.1186/s13046-019-1049-7
13. Kubo M, Gotoh K, Eguchi H, Kobayashi S, Iwagami Y, Tomimaru Y, et al. Impact of CD36 on Chemoresistance in Pancreatic Ductal Adenocarcinoma. Ann Surg Oncol (2019) 27(2):610–9. doi: 10.1245/s10434-019-07927-2
14. Yang P, Su C, Luo X, Zeng H, Zhao L, Wei L, et al. Dietary oleic acid-induced CD36 promotes cervical cancer cell growth and metastasis via up-regulation Src/ERK pathway. Cancer Lett (2018) 438:76–85. doi: 10.1016/j.canlet.2018.09.006
15. Nath A, Li I, Roberts LR, Chan C. Elevated free fatty acid uptake via CD36 promotes epithelial-mesenchymal transition in hepatocellular carcinoma. Sci Rep (2015) 5:14752. doi: 10.1038/srep14752
16. Ackerman D, Tumanov S, Qiu B, Michalopoulou E, Spata M, Azzam A, et al. Triglycerides Promote Lipid Homeostasis during Hypoxic Stress by Balancing Fatty Acid Saturation. Cell Rep (2018) 24(10):2596–605.e5. doi: 10.1016/j.celrep.2018.08.015
17. Ameer F, Scandiuzzi L, Hasnain S, Kalbacher H, Zaidi N. De novo lipogenesis in health and disease. Metabolism (2014) 63(7):895–902. doi: 10.1016/j.metabol.2014.04.003
18. Menendez JA, Lupu R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat Rev Cancer (2007) 7(10):763–77. doi: 10.1038/nrc2222
19. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science (2009) 324(5930):1029–33. doi: 10.1126/science.1160809
20. Lee J-H, Jang H, Lee S-M, Lee J-E, Choi J, Kim TW, et al. ATP-citrate lyase regulates cellular senescence via an AMPK- and p53-dependent pathway. FEBS J (2015) 282(2):361–71. doi: 10.1111/febs.13139
21. Kuhajda FP. Fatty-acid synthase and human cancer: new perspectives on its role in tumor biology. Nutrition (2000) 16(3):202–8. doi: 10.1016/S0899-9007(99)00266-X
22. Swinnen JV, Vanderhoydonc F, Elgamal AA, Eelen M, Vercaeren I, Joniau S, et al. Selective activation of the fatty acid synthesis pathway in human prostate cancer. Int J Cancer (2000) 88(2):176–9. doi: 10.1002/1097-0215(20001015)88:2<176::aid-ijc5>3.0.co;2-3
23. Zadra G, Ribeiro CF, Chetta P, Ho Y, Cacciatore S, Gao X, et al. Inhibition of de novo lipogenesis targets androgen receptor signaling in castration-resistant prostate cancer. Proc Natl Acad Sci USA (2019) 116(2):631–40. doi: 10.1073/pnas.1808834116
24. Eberle D, Hegarty B, Bossard P, Ferre P, Foufelle F. SREBP transcription factors: master regulators of lipid homeostasis. Biochimie (2004) 86(11):839–48. doi: 10.1016/j.biochi.2004.09.018
25. Yin F, Feng F, Wang L, Wang X, Li Z, Cao Y. SREBP-1 inhibitor Betulin enhances the antitumor effect of Sorafenib on hepatocellular carcinoma via restricting cellular glycolytic activity. Cell Death Dis (2019) 10(9):672. doi: 10.1038/s41419-019-1884-7
26. Talebi A, Dehairs J, Rambow F, Rogiers A, Nittner D, Derua R, et al. Sustained SREBP-1-dependent lipogenesis as a key mediator of resistance to BRAF-targeted therapy. Nat Commun (2018) 9(1):2500. doi: 10.1038/s41467-018-04664-0
27. Rueda-Rincon N, Bloch K, Derua R, Vyas R, Harms A, Hankemeier T, et al. p53 attenuates AKT signaling by modulating membrane phospholipid composition. Oncotarget (2015) 6(25):21240–54. doi: 10.18632/oncotarget.4067
28. Pelicano H, Xu RH, Du M, Feng L, Sasaki R, Carew JS, et al. Mitochondrial respiration defects in cancer cells cause activation of Akt survival pathway through a redox-mediated mechanism. J Cell Biol (2006) 175(6):913–23. doi: 10.1083/jcb.200512100
29. Stambolic V, MacPherson D, Sas D, Lin Y, Snow B, Jang Y, et al. Regulation of PTEN transcription by p53. Mol Cell (2001) 8(2):317–25. doi: 10.1016/s1097-2765(01)00323-9
30. Porstmann T, Santos CR, Griffiths B, Cully M, Wu M, Leevers S, et al. SREBP activity is regulated by mTORC1 and contributes to Akt-dependent cell growth. Cell Metab (2008) 8(3):224–36. doi: 10.1016/j.cmet.2008.07.007
31. Guo D, Prins RM, Dang J, Kuga D, Iwanami A, Soto H, et al. EGFR signaling through an Akt-SREBP-1-dependent, rapamycin-resistant pathway sensitizes glioblastomas to antilipogenic therapy. Sci Signal (2009) 2(101):ra82. doi: 10.1126/scisignal.2000446
32. Furuta E, Pai SK, Zhan R, Bandyopadhyay S, Watabe M, Mo YY, et al. Fatty acid synthase gene is up-regulated by hypoxia via activation of Akt and sterol regulatory element binding protein-1. Cancer Res (2008) 68(4):1003–11. doi: 10.1158/0008-5472.can-07-2489
33. Gouw AM, Margulis K, Liu NS, Raman SJ, Mancuso A, Toal GG, et al. The MYC Oncogene Cooperates with Sterol-Regulated Element-Binding Protein to Regulate Lipogenesis Essential for Neoplastic Growth. Cell Metab (2019) 30(3):556–72.e5. doi: 10.1016/j.cmet.2019.07.012
34. Menendez JA. Fine-tuning the lipogenic/lipolytic balance to optimize the metabolic requirements of cancer cell growth: molecular mechanisms and therapeutic perspectives. Biochim Biophys Acta (2010) 1801(3):381–91. doi: 10.1016/j.bbalip.2009.09.005
35. Kumar-Sinha C, Ignatoski KW, Lippman ME, Ethier SP, Chinnaiyan AM. Transcriptome analysis of HER2 reveals a molecular connection to fatty acid synthesis. Cancer Res (2003) 63(1):132–9.
36. Nakatsu Y, Yamamotoya T, Ueda K, Ono H, Inoue MK, Matsunaga Y, et al. Prolyl isomerase Pin1 in metabolic reprogramming of cancer cells. Cancer Lett (2019) 470:106–14. doi: 10.1016/j.canlet.2019.10.043
37. Tracz-Gaszewska Z, Dobrzyn P. Stearoyl-CoA Desaturase 1 as a Therapeutic Target for the Treatment of Cancer. Cancers (Basel) (2019) 11(7):948. doi: 10.3390/cancers11070948
38. Presler M, Wojtczyk-Miaskowska A, Schlichtholz B, Kaluzny A, Matuszewski M, Mika A, et al. Increased expression of the gene encoding stearoyl-CoA desaturase 1 in human bladder cancer. Mol Cell Biochem (2018) 447(1-2):217–24. doi: 10.1007/s11010-018-3306-z
39. Li J, Condello S, Thomes-Pepin J, Ma X, Xia Y, Hurley TD, et al. Lipid Desaturation Is a Metabolic Marker and Therapeutic Target of Ovarian Cancer Stem Cells. Cell Stem Cell (2017) 20(3):303–14.e5. doi: 10.1016/j.stem.2016.11.004
40. Lai KKY, Kweon SM, Chi F, Hwang E, Kabe Y, Higashiyama R, et al. Stearoyl-CoA Desaturase Promotes Liver Fibrosis and Tumor Development in Mice via a Wnt Positive-Signaling Loop by Stabilization of Low-Density Lipoprotein-Receptor-Related Proteins 5 and 6. Gastroenterology (2017) 152(6):1477–91. doi: 10.1053/j.gastro.2017.01.021
41. Lin H, Patel S, Affleck VS, Wilson I, Turnbull DM, Joshi AR, et al. Fatty acid oxidation is required for the respiration and proliferation of malignant glioma cells. Neuro-Oncology (2016) 19(1):43–54. doi: 10.1093/neuonc/now128
42. Carracedo A, Cantley LC, Pandolfi PP. Cancer metabolism: fatty acid oxidation in the limelight. Nat Rev Cancer (2013) 13(4):227–32. doi: 10.1038/nrc3483
43. Park J, Vithayathil S, Kumar S, Sung P, Dobrolecki L, Putluri V, et al. Fatty Acid Oxidation-Driven Src Links Mitochondrial Energy Reprogramming and Oncogenic Properties in Triple-Negative Breast Cancer. Cell Rep (2016) 14(9):2154–65. doi: 10.1016/j.celrep.2016.02.004
44. Ito K, Carracedo A, Weiss D, Arai F, Ala U, Avigan D, et al. A PML–PPAR-δ pathway for fatty acid oxidation regulates hematopoietic stem cell maintenance. Nat Med (2012) 18(9):1350–8. doi: 10.1038/nm.2882
45. Guppy M, Leedman P, Zu X, Russell V. Contribution by different fuels and metabolic pathways to the total ATP turnover of proliferating MCF-7 breast cancer cells. Biochem J (2002) 364(Pt 1):309–15. doi: 10.1042/bj3640309
46. Chen CL, Uthaya Kumar DB, Punj V, Xu J, Sher L, Tahara SM, et al. NANOG Metabolically Reprograms Tumor-Initiating Stem-like Cells through Tumorigenic Changes in Oxidative Phosphorylation and Fatty Acid Metabolism. Cell Metab (2016) 23(1):206–19. doi: 10.1016/j.cmet.2015.12.004
47. Nomura DK, Long JZ, Niessen S, Hoover HS, Ng SW, Cravatt BF. Monoacylglycerol lipase regulates a fatty acid network that promotes cancer pathogenesis. Cell (2010) 140(1):49–61. doi: 10.1016/j.cell.2009.11.027
48. Zhang X, Saarinen AM, Hitosugi T, Wang Z, Wang L, Ho TH, et al. Inhibition of intracellular lipolysis promotes human cancer cell adaptation to hypoxia. Elife (2017) 6. doi: 10.7554/eLife.31132
49. Duncan JG. Lipotoxicity: what is the fate of fatty acids? J Lipid Res (2008) 49(7):1375–6.e31132. doi: 10.1194/jlr.E800010-JLR200
50. Childs KS, Randall RE, Goodbourn S. LGP2 plays a critical role in sensitizing mda-5 to activation by double-stranded RNA. PloS One (2013) 8(5):e64202. doi: 10.1371/journal.pone.0064202
51. Young SG, Zechner R. Biochemistry and pathophysiology of intravascular and intracellular lipolysis. Genes Dev (2013) 27(5):459–84. doi: 10.1101/gad.209296.112
52. Schott MB, Weller SG, Schulze RJ, Krueger EW, Drizyte-Miller K, Casey CA, et al. Lipid droplet size directs lipolysis and lipophagy catabolism in hepatocytes. J Cell Biol (2019) 218(10):3320–35. doi: 10.1083/jcb.201803153
53. Tomin T, Fritz K, Gindlhuber J, Waldherr L, Pucher B, Thallinger GG, et al. Deletion of Adipose Triglyceride Lipase Links Triacylglycerol Accumulation to a More-Aggressive Phenotype in A549 Lung Carcinoma Cells. J Proteome Res (2018) 17(4):1415–25. doi: 10.1021/acs.jproteome.7b00782
54. Corbet C, Bastien E, Santiago de Jesus JP, Dierge E, Martherus R, Vander Linden C, et al. TGFβ2-induced formation of lipid droplets supports acidosis-driven EMT and the metastatic spreading of cancer cells. Nat Commun (2020) 11(1):454. doi: 10.1038/s41467-019-14262-3
55. Bosch M, Parton RG, Pol A. Lipid droplets, bioenergetic fluxes, and metabolic flexibility. Semin Cell Dev Biol (2020) S1084-9521(19):30052–7. doi: 10.1016/j.semcdb.2020.02.010
56. Dupont N, Chauhan S, Arko-Mensah J, Castillo E, Masedunskas A, Weigert R, et al. Neutral lipid stores and lipase PNPLA5 contribute to autophagosome biogenesis. Curr Biol CB (2014) 24(6):609–20. doi: 10.1016/j.cub.2014.02.008
57. Ogasawara Y, Tsuji T, Fujimoto T. Multifarious roles of lipid droplets in autophagy - Target, product, and what else? Semin Cell Dev Biol (2020). doi: 10.1016/j.semcdb.2020.02.013
58. Shpilka T, Welter E, Borovsky N, Amar N, Mari M, Reggiori F, et al. Lipid droplets and their component triglycerides and steryl esters regulate autophagosome biogenesis. EMBO J (2015) 34(16):2117–31. doi: 10.15252/embj.201490315
59. Vegliante R, Di Leo L, Ciccarone F, Ciriolo MR. Hints on ATGL implications in cancer: beyond bioenergetic clues. Cell Death Dis (2018) 9(3):316. doi: 10.1038/s41419-018-0345-z
60. Xie H, Heier C, Kien B, Vesely PW, Tang Z, Sexl V, et al. Adipose triglyceride lipase activity regulates cancer cell proliferation via AMP-kinase and mTOR signaling. Biochim Biophys Acta Mol Cell Biol Lipids (2020) 1865(9):158737. doi: 10.1016/j.bbalip.2020.158737
61. Liu X, Liang Y, Song R, Yang G, Han J, Lan Y, et al. Long non-coding RNA NEAT1-modulated abnormal lipolysis via ATGL drives hepatocellular carcinoma proliferation. Mol Cancer (2018) 17(1):90. doi: 10.1186/s12943-018-0838-5
62. Di Leo L, Vegliante R, Ciccarone F, Salvatori I, Scimeca M, Bonanno E, et al. Forcing ATGL expression in hepatocarcinoma cells imposes glycolytic rewiring through PPAR-alpha/p300-mediated acetylation of p53. Oncogene (2019) 38(11):1860–75. doi: 10.1038/s41388-018-0545-0
63. Wang YY, Attane C, Milhas D, Dirat B, Dauvillier S, Guerard A, et al. Mammary adipocytes stimulate breast cancer invasion through metabolic remodeling of tumor cells. JCI Insight (2017) 2(4):e87489. doi: 10.1172/jci.insight.87489
64. Liu M, Yu X, Lin L, Deng J, Wang K, Xia Y, et al. ATGL promotes the proliferation of hepatocellular carcinoma cells via the p-AKT signaling pathway. J Biochem Mol Toxicol (2019) 33(11):e22391. doi: 10.1002/jbt.22391
65. Zhang J, Liu Z, Lian Z, Liao R, Chen Y, Qin Y, et al. Monoacylglycerol Lipase: A Novel Potential Therapeutic Target and Prognostic Indicator for Hepatocellular Carcinoma. Sci Rep (2016) 6:35784. doi: 10.1038/srep35784
66. Hannun YA, Obeid LM. Sphingolipids and their metabolism in physiology and disease. Nat Rev Mol Cell Biol (2018) 19(3):175–91. doi: 10.1038/nrm.2017.107
67. Lewis AC, Wallington-Beddoe CT, Powell JA, Pitson SM. Targeting sphingolipid metabolism as an approach for combination therapies in haematological malignancies. Cell Death Discovery (2018) 4:4. doi: 10.1038/s41420-018-0075-0
68. Realini N, Palese F, Pizzirani D, Pontis S, Basit A, Bach A, et al. Acid Ceramidase in Melanoma: Expression, Localization, And Effects Of Pharmacological Inhibition. J Biol Chem (2016) 291(5):2422–34. doi: 10.1074/jbc.M115.666909
69. Huang W-C, Chen C-L, Lin Y-S, Lin C-F. Apoptotic Sphingolipid Ceramide in Cancer Therapy. J Lipids (2011) 2011:565316. doi: 10.1155/2011/565316
70. Lai M, La Rocca V, Amato R, Freer G, Pistello M. Sphingolipid/Ceramide Pathways and Autophagy in the Onset and Progression of Melanoma: Novel Therapeutic Targets and Opportunities. Int J Mol Sci (2019) 20(14):3436. doi: 10.3390/ijms20143436
71. Simons K, Ikonen E. Functional rafts in cell membranes. Nature (1997) 387(6633):569–72. doi: 10.1038/42408
72. Novak A, Binnington B, Ngan B, Chadwick K, Fleshner N, Lingwood CA. Cholesterol masks membrane glycosphingolipid tumor-associated antigens to reduce their immunodetection in human cancer biopsies. Glycobiology (2013) 23(11):1230–9. doi: 10.1093/glycob/cwt059
73. Sharma B, Gupta V, Dahiya D, Kumar H, Vaiphei K, Agnihotri N. Clinical relevance of cholesterol homeostasis genes in colorectal cancer. Biochim Biophys Acta Mol Cell Biol Lipids (2019) 1864(10):1314–27. doi: 10.1016/j.bbalip.2019.06.008
74. Ding X, Zhang W, Li S, Yang H. The role of cholesterol metabolism in cancer. Am J Cancer Res (2019) 9(2):219–27.
75. Kim J, Thompson B, Han S, Lotan Y, McDonald JG, Ye J. Uptake of HDL-cholesterol contributes to lipid accumulation in clear cell renal cell carcinoma. Biochim Biophys Acta Mol Cell Biol Lipids (2019) 1864(12):158525. doi: 10.1016/j.bbalip.2019.158525
76. Thysell E, Surowiec I, Hornberg E, Crnalic S, Widmark A, Johansson AI, et al. Metabolomic characterization of human prostate cancer bone metastases reveals increased levels of cholesterol. PloS One (2010) 5(12):e14175. doi: 10.1371/journal.pone.0014175
77. Aguirre-Portolés C, Feliu J, Reglero G, Ramírez de Molina A. ABCA1 overexpression worsens colorectal cancer prognosis by facilitating tumour growth and caveolin-1-dependent invasiveness, and these effects can be ameliorated using the BET inhibitor apabetalone. Mol Oncol (2018) 12(10):1735–52. doi: 10.1002/1878-0261.12367
78. Georgila K, Vyrla D, Drakos E. Apolipoprotein A-I (ApoA-I), Immunity, Inflammation and Cancer. Cancers (Basel) (2019) 11(8):1097. doi: 10.3390/cancers11081097
79. Martin LJ, Melnichouk O, Huszti E, Connelly PW, Greenberg CV, Minkin S, et al. Serum lipids, lipoproteins, and risk of breast cancer: a nested case-control study using multiple time points. J Natl Cancer Institute (2015) 107(5):djv032. doi: 10.1093/jnci/djv032
80. Ding X, Fan K, Hu J, Zang Z, Zhang S, Zhang Y, et al. SCP2-mediated cholesterol membrane trafficking promotes the growth of pituitary adenomas via Hedgehog signaling activation. J Exp Clin Cancer Res (2019) 38(1):404. doi: 10.1186/s13046-019-1411-9
81. Wang X, Huang Z, Wu Q, Prager BC, Mack SC, Yang K, et al. MYC-Regulated Mevalonate Metabolism Maintains Brain Tumor-Initiating Cells. Cancer Res (2017) 77(18):4947–60. doi: 10.1158/0008-5472.can-17-0114
82. Clendening JW, Pandyra A, Boutros PC, El Ghamrasni S, Khosravi F, Trentin GA, et al. Dysregulation of the mevalonate pathway promotes transformation. Proc Natl Acad Sci USA (2010) 107(34):15051–6. doi: 10.1073/pnas.0910258107
83. Sethunath V, Hu H, De Angelis C, Veeraraghavan J, Qin L, Wang N, et al. Targeting the Mevalonate Pathway to Overcome Acquired Anti-HER2 Treatment Resistance in Breast Cancer. Mol Cancer Res (2019) 17(11):2318–30. doi: 10.1158/1541-7786.mcr-19-0756
84. Mullen PJ, Yu R, Longo J, Archer MC, Penn LZ. The interplay between cell signalling and the mevalonate pathway in cancer. Nat Rev Cancer (2016) 16(11):718–31. doi: 10.1038/nrc.2016.76
85. Kang H, Oh T, Bahk YY, Kim GH, Kan SY, Shin DH, et al. HSF1 Regulates Mevalonate and Cholesterol Biosynthesis Pathways. Cancers (Basel) (2019) 11(9):1363. doi: 10.3390/cancers11091363
86. Pallottini V, Guantario B, Martini C, Totta P, Filippi I, Carraro F, et al. Regulation of HMG-CoA reductase expression by hypoxia. J Cell Biochem (2008) 104(3):701–9. doi: 10.1002/jcb.21757
87. Moon SH, Huang CH, Houlihan SL, Regunath K, Freed-Pastor WA, Morris J, et al. p53 Represses the Mevalonate Pathway to Mediate Tumor Suppression. Cell (2019) 176(3):564–80.e19. doi: 10.1016/j.cell.2018.11.011
88. Freed-Pastor WA, Mizuno H, Zhao X, Langerød A, Moon S-H, Rodriguez-Barrueco R, et al. Mutant p53 Disrupts Mammary Tissue Architecture via the Mevalonate Pathway. Cell (2012) 148(1):244–58. doi: 10.1016/j.cell.2011.12.017
89. Werner E, Alter A, Deng Q, Dammer EB, Wang Y, Yu DS, et al. Ionizing Radiation induction of cholesterol biosynthesis in Lung tissue. Sci Rep (2019) 9(1):12546. doi: 10.1038/s41598-019-48972-x
90. Smart EJ, Graf GA, McNiven MA, Sessa WC, Engelman JA, Scherer PE, et al. Caveolins, liquid-ordered domains, and signal transduction. Mol Cell Biol (1999) 19(11):7289–304. doi: 10.1128/mcb.19.11.7289
91. Ali A, Levantini E, Fhu CW, Teo JT, Clohessy JG, Goggi JL, et al. CAV1 - GLUT3 signaling is important for cellular energy and can be targeted by Atorvastatin in Non-Small Cell Lung Cancer. Theranostics (2019) 9(21):6157–74. doi: 10.7150/thno.35805
92. Raza S, Ohm JE, Dhasarathy A, Schommer J, Roche C, Hammer KDP, et al. The cholesterol metabolite 27-hydroxycholesterol regulates p53 activity and increases cell proliferation via MDM2 in breast cancer cells. Mol Cell Biochem (2015) 410(1):187–95. doi: 10.1007/s11010-015-2551-7
93. Nelson ER, Wardell SE, Jasper JS, Park S, Suchindran S, Howe MK, et al. 27-Hydroxycholesterol Links Hypercholesterolemia and Breast Cancer Pathophysiology. Science (2013) 342(6162):1094. doi: 10.1126/science.1241908
94. Li J, Gu D, Lee SS, Song B, Bandyopadhyay S, Chen S, et al. Abrogating cholesterol esterification suppresses growth and metastasis of pancreatic cancer. Oncogene (2016) 35(50):6378–88. doi: 10.1038/onc.2016.168
95. Wang J, Tan M, Ge J, Zhang P, Zhong J, Tao L, et al. Lysosomal acid lipase promotes cholesterol ester metabolism and drives clear cell renal cell carcinoma progression. Cell Prolif (2018) 51(4):e12452. doi: 10.1111/cpr.12452
96. Song Z, Xiaoli AM, Yang F. Regulation and Metabolic Significance of De Novo Lipogenesis in Adipose Tissues. Nutrients (2018) 10(10):1383. doi: 10.3390/nu10101383
97. Luo J, Yang H, Song B. Mechanisms and regulation of cholesterol homeostasis. Nat Rev Mol Cell Biol (2020) 21(4):225–45. doi: 10.1038/s41580-019-0190-7
98. Costet P, Luo Y, Wang N, Tall A. Sterol-dependent transactivation of the ABC1 promoter by the liver X receptor/retinoid X receptor. J Biol Chem (2000) 275(36):28240–5. doi: 10.1074/jbc.M003337200
99. Watt MJ, Clark AK, Selth LA, Haynes VR, Lister N, Rebello R, et al. Suppressing fatty acid uptake has therapeutic effects in preclinical models of prostate cancer. Sci Transl Med (2019) 11(478):eaau5758. doi: 10.1126/scitranslmed.aau5758
100. Agostini M, Almeida LY, Bastos DC, Ortega RM, Moreira FS, Seguin F, et al. The Fatty Acid Synthase Inhibitor Orlistat Reduces the Growth and Metastasis of Orthotopic Tongue Oral Squamous Cell Carcinomas. Mol Cancer Ther (2014) 13(3):585. doi: 10.1158/1535-7163.MCT-12-1136
101. Schcolnik-Cabrera A, Chavez-Blanco A, Dominguez-Gomez G, Taja-Chayeb L, Morales-Barcenas R, Trejo-Becerril C, et al. Orlistat as a FASN inhibitor and multitargeted agent for cancer therapy. Expert Opin Invest Drugs (2018) 27(5):475–89. doi: 10.1080/13543784.2018.1471132
102. Aquino IG, Bastos DC, Cuadra-Zelaya FJM, Teixeira IF, Salo T, Coletta RD, et al. Anticancer properties of the fatty acid synthase inhibitor TVB-3166 on oral squamous cell carcinoma cell lines. Arch Oral Biol (2020) 113:104707. doi: 10.1016/j.archoralbio.2020.104707
103. Zaytseva YY, Rychahou PG, Le AT, Scott TL, Flight RM, Kim JT, et al. Preclinical evaluation of novel fatty acid synthase inhibitors in primary colorectal cancer cells and a patient-derived xenograft model of colorectal cancer. Oncotarget (2018) 9(37):24787–800. doi: 10.18632/oncotarget.25361
104. Wang C, Xu C, Sun M, Luo D, Liao DF, Cao D. Acetyl-CoA carboxylase-alpha inhibitor TOFA induces human cancer cell apoptosis. Biochem Biophys Res Commun (2009) 385(3):302–6. doi: 10.1016/j.bbrc.2009.05.045
105. Beckers A, Organe S, Timmermans L, Scheys K, Peeters A, Brusselmans K, et al. Chemical inhibition of acetyl-CoA carboxylase induces growth arrest and cytotoxicity selectively in cancer cells. Cancer Res (2007) 67(17):8180–7. doi: 10.1158/0008-5472.can-07-0389
106. Svensson RU, Parker SJ, Eichner LJ, Kolar MJ, Wallace M, Brun SN, et al. Inhibition of acetyl-CoA carboxylase suppresses fatty acid synthesis and tumor growth of non-small-cell lung cancer in preclinical models. Nat Med (2016) 22(10):1108–19. doi: 10.1038/nm.4181
107. Woo SU, Sangai T, Akcakanat A, Chen H, Wei C, Meric-Bernstam F. Vertical inhibition of the PI3K/Akt/mTOR pathway is synergistic in breast cancer. Oncogenesis (2017) 6(10):e385. doi: 10.1038/oncsis.2017.86
108. Deng F, Ma YX, Liang L, Zhang P, Feng J. The pro-apoptosis effect of sinomenine in renal carcinoma via inducing autophagy through inactivating PI3K/AKT/mTOR pathway. BioMed Pharmacother (2018) 97:1269–74. doi: 10.1016/j.biopha.2017.11.064
109. Li X, Chen YT, Hu P, Huang WC. Fatostatin displays high antitumor activity in prostate cancer by blocking SREBP-regulated metabolic pathways and androgen receptor signaling. Mol Cancer Ther (2014) 13(4):855–66. doi: 10.1158/1535-7163.mct-13-0797
110. Hawkins JL, Robbins MD, Warren LC, Xia D, Petras SF, Valentine JJ, et al. Pharmacologic inhibition of site 1 protease activity inhibits sterol regulatory element-binding protein processing and reduces lipogenic enzyme gene expression and lipid synthesis in cultured cells and experimental animals. J Pharmacol Exp Ther (2008) 326(3):801–8. doi: 10.1124/jpet.108.139626
111. Xiang X, Saha AK, Wen R, Ruderman NB, Luo Z. AMP-activated protein kinase activators can inhibit the growth of prostate cancer cells by multiple mechanisms. Biochem Biophys Res Commun (2004) 321(1):161–7. doi: 10.1016/j.bbrc.2004.06.133
112. Dai S, Yan Y, Xu Z, Zeng S, Qian L, Huo L, et al. SCD1 Confers Temozolomide Resistance to Human Glioma Cells via the Akt/GSK3beta/beta-Catenin Signaling Axis. Front Pharmacol (2017) 8:960. doi: 10.3389/fphar.2017.00960
113. von Roemeling CA, Marlow LA, Wei JJ, Cooper SJ, Caulfield TR, Wu K, et al. Stearoyl-CoA desaturase 1 is a novel molecular therapeutic target for clear cell renal cell carcinoma. Clin Cancer Res (2013) 19(9):2368–80. doi: 10.1158/1078-0432.ccr-12-3249
114. Nashed M, Chisholm JW, Igal RA. Stearoyl-CoA desaturase activity modulates the activation of epidermal growth factor receptor in human lung cancer cells. Exp Biol Med (Maywood) (2012) 237(9):1007–17. doi: 10.1258/ebm.2012.012126
115. Pisanu ME, Maugeri-Sacca M, Fattore L, Bruschini S, De Vitis C, Tabbi E, et al. Inhibition of Stearoyl-CoA desaturase 1 reverts BRAF and MEK inhibition-induced selection of cancer stem cells in BRAF-mutated melanoma. J Exp Clin Cancer Res (2018) 37(1):318. doi: 10.1186/s13046-018-0989-7
116. Rodríguez-Enríquez S, Hernández-Esquivel L, Marín-Hernández A, El Hafidi M, Gallardo-Pérez JC, Hernández-Reséndiz I, et al. Mitochondrial free fatty acid β-oxidation supports oxidative phosphorylation and proliferation in cancer cells. Int J Biochem Cell Biol (2015) 65:209–21. doi: 10.1016/j.biocel.2015.06.010
117. Wang Y, Lu JH, Wang F, Wang YN, He MM, Wu QN, et al. Inhibition of fatty acid catabolism augments the efficacy of oxaliplatin-based chemotherapy in gastrointestinal cancers. Cancer Lett (2020) 473:74–89. doi: 10.1016/j.canlet.2019.12.036
118. Granchi C, Caligiuri I, Minutolo F, Rizzolio F, Tuccinardi T. A patent review of Monoacylglycerol Lipase (MAGL) inhibitors (2013-2017). Expert Opin Ther Pat (2017) 27(12):1341–51. doi: 10.1080/13543776.2018.1389899
119. Bononi G, Granchi C, Lapillo M, Giannotti M, Nieri D, Fortunato S, et al. Discovery of long-chain salicylketoxime derivatives as monoacylglycerol lipase (MAGL) inhibitors. Eur J Med Chem (2018) 157:817–36. doi: 10.1016/j.ejmech.2018.08.038
120. Poli G, Lapillo M, Jha V, Mouawad N, Caligiuri I, Macchia M, et al. Computationally driven discovery of phenyl(piperazin-1-yl)methanone derivatives as reversible monoacylglycerol lipase (MAGL) inhibitors. J Enzyme Inhib Med Chem (2019) 34(1):589–96. doi: 10.1080/14756366.2019.1571271
121. Nguyen TTT, Ishida CT, Shang E, Shu C, Torrini C, Zhang Y, et al. Activation of LXRbeta inhibits tumor respiration and is synthetically lethal with Bcl-xL inhibition. EMBO Mol Med (2019) 11(10):e10769. doi: 10.15252/emmm.201910769
122. Ohmoto T, Nishitsuji K, Yoshitani N, Mizuguchi M, Yanagisawa Y, Saito H, et al. K604, a specific acylCoA:cholesterol acyltransferase 1 inhibitor, suppresses proliferation of U251MG glioblastoma cells. Mol Med Rep (2015) 12(4):6037–42. doi: 10.3892/mmr.2015.4200
123. LaPensee CR, Mann JE, Rainey WE, Crudo V, Hunt SW, Hammer GD. ATR-101, a Selective and Potent Inhibitor of Acyl-CoA Acyltransferase 1, Induces Apoptosis in H295R Adrenocortical Cells and in the Adrenal Cortex of Dogs. Endocrinology (2016) 157(5):1775–88. doi: 10.1210/en.2015-2052
124. Bitorina AV, Oligschlaeger Y, Shiri-Sverdlov R, Theys J. Low profile high value target: The role of OxLDL in cancer. Biochim Biophys Acta Mol Cell Biol Lipids (2019) 1864(12):158518. doi: 10.1016/j.bbalip.2019.158518
125. Pascual G, Avgustinova A, Mejetta S, Martin M, Castellanos A, Attolini CS, et al. Targeting metastasis-initiating cells through the fatty acid receptor CD36. Nature (2017) 541(7635):41–5. doi: 10.1038/nature20791
126. Ladanyi A, Mukherjee A, Kenny HA, Johnson A, Mitra AK, Sundaresan S, et al. Adipocyte-induced CD36 expression drives ovarian cancer progression and metastasis. Oncogene (2018) 37(17):2285–301. doi: 10.1038/s41388-017-0093-z
127. Kim TT, Dyck JR. The role of CD36 in the regulation of myocardial lipid metabolism. Biochim Biophys Acta (2016) 1861(10):1450–60. doi: 10.1016/j.bbalip.2016.03.018
128. Fako VE, Wu X, Pflug B, Liu JY, Zhang JT. Repositioning proton pump inhibitors as anticancer drugs by targeting the thioesterase domain of human fatty acid synthase. J Med Chem (2015) 58(2):778–84. doi: 10.1021/jm501543u
129. Ariaans G, Jalving M, Vries EG, Jong S. Anti-tumor effects of everolimus and metformin are complementary and glucose-dependent in breast cancer cells. BMC Cancer (2017) 17(1):232. doi: 10.1186/s12885-017-3230-8
130. Vriens K, Christen S, Parik S, Broekaert D, Yoshinaga K, Talebi A, et al. Evidence for an alternative fatty acid desaturation pathway increasing cancer plasticity. Nature (2019) 566(7744):403–6. doi: 10.1038/s41586-019-0904-1
131. Miyazaki M, Man WC, Ntambi JM. Targeted disruption of stearoyl-CoA desaturase1 gene in mice causes atrophy of sebaceous and meibomian glands and depletion of wax esters in the eyelid. J Nutr (2001) 131(9):2260–8. doi: 10.1093/jn/131.9.2260
132. Ye L, Zhang B, Seviour EG, Tao KX, Liu XH, Ling Y, et al. Monoacylglycerol lipase (MAGL) knockdown inhibits tumor cells growth in colorectal cancer. Cancer Lett (2011) 307(1):6–17. doi: 10.1016/j.canlet.2011.03.007
133. Zhou F, Zhou J, Wang W, Zhang XJ, Ji YX, Zhang P, et al. Unexpected Rapid Increase in the Burden of NAFLD in China From 2008 to 2018: A Systematic Review and Meta-Analysis. Hepatology (2019) 70(4):1119–33. doi: 10.1002/hep.30702
134. Ravnskov U, Rosch PJ, McCully KS. Statins Do Not Protect Against Cancer: Quite the Opposite. J Clin Oncol (2015) 33(7):810–1. doi: 10.1200/JCO.2014.58.9564
135. Ward NC, Watts GF, Eckel RH. Statin Toxicity. Circ Res (2019) 124(2):328–50. doi: 10.1161/circresaha.118.312782
136. Phillips RE, Yang Y, Smith RC, Thompson BM, Yamasaki T, Soto-Feliciano YM, et al. Target identification reveals lanosterol synthase as a vulnerability in glioma. Proc Natl Acad Sci (2019) 116(16):7957. doi: 10.1073/pnas.1820989116
137. Raud B, McGuire PJ, Jones RG, Sparwasser T, Berod L. Fatty acid metabolism in CD8(+) T cell memory: Challenging current concepts. Immunol Rev (2018) 283(1):213–31. doi: 10.1111/imr.12655
Keywords: cancer, cancer metabolism, fatty acid catabolism, cholesterol, fatty acid synthesis, lipid uptake, tumor microenvironment
Citation: Wang W, Bai L, Li W and Cui J (2020) The Lipid Metabolic Landscape of Cancers and New Therapeutic Perspectives. Front. Oncol. 10:605154. doi: 10.3389/fonc.2020.605154
Received: 11 September 2020; Accepted: 02 November 2020;
Published: 08 December 2020.
Edited by:
Egidio Iorio, National Institute of Health (ISS), ItalyReviewed by:
Sofia Avnet, Rizzoli Orthopedic Institute (IRCCS), ItalyCopyright © 2020 Wang, Bai, Li and Cui. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jiuwei Cui, Y3VpandAamx1LmVkdS5jbg==
†ORCID: Wenjun Wang, orcid.org/0000-0003-1302-694X
Ling Bai, orcid.org/0000-0001-5085-8628
Wei Li, orcid.org/0000-0002-8627-1578
Jiuwei Cui, orcid.org/0000-0001-6496-7550
‡These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.