
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Oncol. , 25 January 2021
Sec. Molecular and Cellular Oncology
Volume 10 - 2020 | https://doi.org/10.3389/fonc.2020.604747
This article is part of the Research Topic The Role of Helicobacter pylori in Gastric Carcinogenesis View all 6 articles
 Iqra Jan1
Iqra Jan1 Rafiq A. Rather2Ifra Mushtaq2Ajaz A. Malik3Syed Besina4Abdul Basit Baba2Muzamil Farooq2Tahira Yousuf2
Rafiq A. Rather2Ifra Mushtaq2Ajaz A. Malik3Syed Besina4Abdul Basit Baba2Muzamil Farooq2Tahira Yousuf2 Bilal Rah2
Bilal Rah2 Dil Afroze1,2*
Dil Afroze1,2*Helicobacter pylori infection has been associated with the onset of gastric mucosal inflammation and is known to perturb the balance between T-regulatory (Treg) and T-helper 17 (Th17) cells which causes a spurt of interleukin 17 (IL17) and transforming growth factor-β (TGF-β) from Th17 and Treg cells within the gastric milieu. IL17 instigates a surge of interleukin 6 (IL6) from T-helper 1 (Th1) and T-helper 2 (Th2) cells. Further, H. pylori infection is known to stimulate the atypical DNA methylation in gastric mucosa. However, the precise role of cytokine signaling in induction of epigenetic modifications during gastric carcinogenesis is vaguely understood. In this study, patient samples from were examined using real-time polymerase chain reaction (qPCR), PCR, methylation-specific (MS)-PCR, and enzyme-linked immunosorbent assays. We found that H. pylori infection augments the production of interleukin 10 (IL10), IL6, and TGF-β in the gastric milieu and systemic circulation. Together with the IL6/IL10 mediated hyperactivation of the JAK/STAT pathway, H. pylori infection causes the inactivation of suppressor of cytokine signaling 1 (SOCS1) gene through the hypermethylation of the promoter region. This study signifies that H. pylori-mediated epigenetic silencing of SOCS1 in concert with inflammatory cytokines miffs hyperactivation of the JAK/STAT cascade during gastric carcinogenesis.
Gastric cancer arises as a consequence of series of genetic, epigenetic and metabolic alteration in the mucus-producing cells on the inner surface of the gastric mucosa. Gastric cancer cells express a plethora of growth factor and cytokine receptor systems that form a highly complex interaction network between cancer cells and stromal cells within tumor microenvironment and impart therapeutically-resistant phenotype to gastric tumors. Further, a very small population of cancer-stem cells (CSCs) exists within gastric tumor microenvironment which play fundamental role in tumor initiation, progression, metastasis and recurrence. Recently, mucosal gastric microbiota has received tremendous attention due to its effect on the growth of gastric cancer and cancer-associated signaling cascades (1). Because of the acidic pH within stomach, gastric mucosa is typically considered as an unfriendly environment for microbial growth. Nevertheless, gram-negative bacterium Helicobacter pylori successfully reside in the gastric mucosa and damages gastric epithelial cells (2). H. pylori protect itself from acidic pH by epithelial secretions of bicarbonate buffer system and urea and the ammonia created by urease action and by tight attachment to mucosal glycan receptors (3). Although short-term colonization of H. pylori in gastric mucosa is asymptomatic (4), approximately 10% of H. pylori-infected individuals develop peptic ulcers, 1 to 3% develop gastric adenocarcinoma and <0.1% develop mucosa associated lymphoid tissue (MALT) lymphoma (5). However, persistent colonization of H. pylori in gastric mucosa significantly enhances the probability of developing gastric ulcers, MALT lymphoma and gastric carcinoma (6). Notably, H. pylori infection mediated precancerous lesions are immunopathological in origin (7).
In response to H. pylori infection, a group of cytokines such as IL17, IL6 and transforming growth factor-β (TFG-β) are released from immune-sensitive cells like T-helper 1 (Th1), T-helper 2 (Th2), T-helper 17 (Th17) and T-regulatory (Treg) cells within gastric milieu. It has been recognized that IL17 produced by Th17 cells causes activation of Th1 and Th2 cells and subsequently causes release of IL6 from these cells in gastric milieu (8, 9). IL6 and TGF-β are considered as central mediators in H. pylori-associated pathogenesis and are capable of directly influencing JAK/STAT signaling cascade (10). Among the key genes that are involved in H. pylori pathogenicity, phosphoglucosamine mutase gene (glmM) and cytotoxin associated gene A (CagA) virulence genes are strong predictors of clinical outcomes (11). The interaction of virulence factors, host genetic factors and environmental factors contribute to H. pylori-associated pathogenesis. Although gastritis is induced by all strains of H. pylori, not all H. pylori strains are capable of causing the gastric carcinoma. Only the H. pylori strains containing Cag PAI (CagA+) increases the probability of developing gastric cancer (12–14). H. pylori-carrying CagA gene cause genetic instability in gastric mucosa cells, usually modulating activities of different genes (15).
The immune response to the H. pylori infection in the gastric mucosa is typically associated with the release of inflammatory and anti-inflammatory cytokines from different types of immune cells like Th1, Th2, Th17, macrophages, monocytes, mast cells and neutrophils. Many of these cytokines such as IL1, IL2, IL6, IL10, IL17, IL23, TNF-α, TGF-β, IFN-γ, CXCL12, and CXCL4 are secreted both at the site of infection and in the general blood circulation (16). Specific membrane receptors are expressed by gastric epithelial cells that help to transmit the signal carried by different cytokines to the cell interior (17). Cytokines act on broad range of cell types wherein they bind to their cognate membrane receptors and modulate downstream effectors in the signalling cascade (18). Many cytokines activate janus kinase 2 (JAK2) which in turn phosphorylates signal transducers and activators of transcription 3 (STAT3). Phosphorylated STAT3 enter the nucleus and bind specific regulatory sequences to activate or repress transcription of target genes. Besides acting as a transcription factor, STAT3 has recently been shown to act as a marker for gastric carcinoma progression (19). An important negative regulator of JAK/STAT signaling cascade is suppressor of cytokine signaling-1 (SOCS1) which interacts with phosphotyrosine residues on JAK kinases to interfere with the activation of STAT proteins or other signaling intermediates. Interestingly, inactivation of the SOCS1 has been proposed to be responsible for oncogenesis in gastric mucosa (20). IL6 and TFG-β are vital for regulating SOCS1 expression. It has been reported that hypermethylation of SOCS1 significantly retarded growth and proliferation of variety of gastric cancer cell lines (21). The present study has brought out that H. pylori-mediated epigenetic silencing of SOCS1 in concert with inflammatory cytokines causes hyperactivation of the JAK/STAT cascade during gastric cancer development.
In this study we enrolled fifty two gastric cancer patients whose disease status was confirmed by clinical and histopathological examination from the Department of General and Minimal Invasive Surgery, SKIMS, Srinagar, Kashmir. Sample collection was completed in accordance with norms and regulations of Institutional Ethics Committee (IEC-SKIMS). Ethical clearance for the use of human samples was obtained in accordance with the declaration of Helsinki. Only the gastric cancer patients with upfront gastrectomies were included in the study. All the enrolled patients gave informed consent for collecting the samples. The number of patient samples was calculated by open-Epi statistical software. The power of study was maintained at >80% and confidence interval was maintained at 95%.
The extraction of whole genomic DNA from gastric cancer and adjacent normal tissues was achieved by using phenol/chloroform-isoamyl (PCI) method. Briefly, tissue samples were homogenized and solubilized in DNA lysis buffer [Tris-HCl (10 mM, pH 8.0), NaCl (100 mM), EDTA (5 mM), triton X-100 (5%), SDS (0.25%), RNase A (200 μg/mL)] and incubated at 37°C overnight. After incubation, tissue lysate was treated with proteinase K (200 μg/mL) for one and a half hour. Subsequently, DNA was extracted with phenol/chloroform-isoamyl alcohol (25:24:1). The concentration of DNA was measured by Nano Drop 2000. A260/A280 ratio was used to ascertain the purity of DNA. The purity and quality of DNA was further determined by 2% agarose gel electrophoresis. After the extraction process, specific primers for glmM and CagA bacterial genes were used to determine the infection status of gastric cancer.
TRIzol reagent (Invitrogen) was used to extract total RNA from gastric cancer and adjacent normal tissues. The concentration of the total RNA used for the synthesis of cDNA was >1µg. The concentration of total RNA was determined by Nano Drop 2000. MMLV reverse transcriptase kit (Invitrogen) was used for the reverse transcription of RNA into cDNA. Next, we analyzed the real time quantitative expression of IL6, IL10, JAK2, STAT3, and β-actin using designed exon specific primers of each gene. β-actin was used as an internal control. In order to assess the purity and quality of RNA, small samples of RNA were electrophoresed on 2% agarose gel, stained with ethidium bromide and visualized under ultraviolet illumination. The quality of RT-PCR products was assessed using the same technique.
A solid phase immunoassay kit from “BOSTER Antibody & ELISA Experts” were used to measure serum cytokine level. Briefly, to measure cytokine levels in serum, standard and test samples were added to 96-well plate followed by the addition of biotinylated detection antibody. After this, wells were washed with tris-buffered saline (TBS) buffer and avidin–biotin–peroxidase complex (ABC-HRP) was added. Any unbound ABC-HRP can be washed off by TBS. Subsequently, 3,3’,5,5’-tetramethylbenzidine (TMB) substrate was added which is metabolized to a blue colored product by HRP enzyme. The reaction is stopped by the addition of acidic stop solution, resulting in change of color from blue to yellow. The optical density of yellow color is linearly proportional to the amount of cytokine present in the sample which can be measured at 450 nm using a plate reader. The biomarkers evaluated from the gastric cancer patient serum were IL6, IL10, TNF-α, and TGF-β.
MS-PCR analysis of SOCS-1 and STAT-3 gene promoters was carried out using methylation-specific primer sets. About 2–3 µl of the bisulphite converted DNA from each sample (including both gastric cancer and adjacent normal tissue) was used for the setting up of the MS-PCR. Universal methylated human DNA (Zymo Research, USA) was used as positive control for methylated alleles whereas DNA from normal healthy subjects was used as a control for unmethylated alleles. Water was used as a negative PCR control in both reactions.
Statistical analysis of numerical data was performed by using non-parametrical statistical analysis tools including Mann-Whitney U test and Kruskal-Wallis tests. Overall survival analysis was carried out by using Kaplan-Meir analysis. Software used was SPSS ver. 23.
In this study, a total of 52 histopathologically confirmed gastric cancer specimens and 52 adjacent noncancer tissues were obtained from General and Minimal Invasive Surgery, SKIMS, Srinagar, Kashmir, India to understand the mechanism of gastric carcinogenesis in ethnic Kashmiri population. First, we found that clinical characteristics such as age (> 50 years), gender, family history, active life style, and rural dwelling were significantly associated with the occurrence of gastric cancer (Table 1). Kashmiri population is often exposed to pesticides. Therefore, to exclude the involvement of pesticides in the development of gastric carcinoma, pesticide exposure was taken into consideration. However, pesticide exposure did not reveal any association with the development of gastric carcinoma. Our study revealed that most of the patients were above 50 years of age and male individuals with smoking habit appeared increasingly susceptible to the gastric carcinoma.
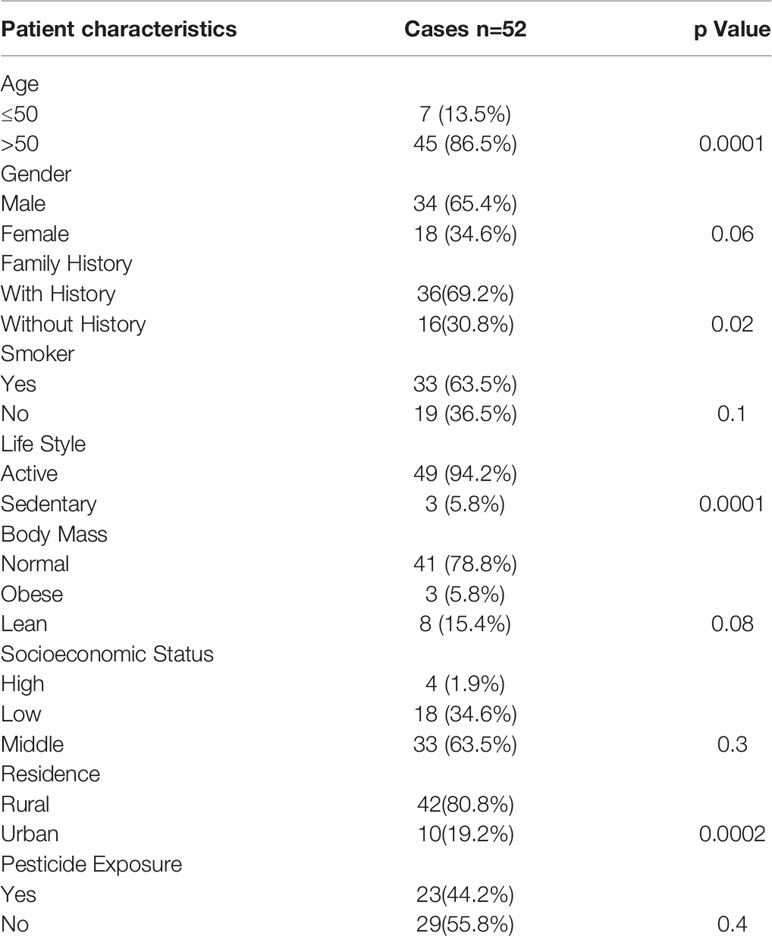
Table 1 Characteristics of H. pylori-infected gastric cancer patients.
Data from gastric cancer patients (n=52) was examined, and it was observed that abdominal pain was the most noticeable clinical complaint experienced by majority of gastric cancer patients. In fact, abdominal pain was the single most noted pain and therefore should not be ignored while examining a patient. However, the patients also revealed one or more of the clinical complaints such as vomiting, anorexia, generalized weakness, weight loss, dysphagia, dyspepsia, malena and upper gastrointestinal (UGI) bleed. The clinical complaints have been arranged in the ascending order of their occurrence among the enrolled patients. These clinical complaints are not mutually exclusive, but many of these may develop in a single individual patient (Supplementary Figure 1).
H. pylori affect the gastric epithelium of nearly 50% of world’s human population. Although a strong correlation exists between the H. pylori infection and gastric carcinoma, the exact mechanism of H. pylori-induced gastric carcinoma has not been precisely determined. CagA is an oncoprotein that plays an important role in gastric carcinogenesis whereas glmM is often used an indicative of the bacterial load. PCR analysis of CagA and glmM in gastric cancer tissues indicated that majority of gastric tumors are positive for CagA (~92%) and glmM (~67%) (Supplementary Figures 2 and 3). This discrepancy may be due to more prominent role of CagA in atropic gastritis which leads to increased risk of gastric carcinoma than glmM which is the responsible for chronic active gastritis only. Our results suggest that CagA oncoprotein (an indicative of bacterial virulence) plays a greater role in gastric carcinogenesis than glmM (an indicative of the bacterial load).
In the present study we found that presence of CagA was significantly associated with the aberrantly high ratio of IL6 and IL10. High IL6/IL10 ratio in H. pylori-infected patients indicated that pro-inflammatory cytokines play a fundamental role in gastric cancer development (Figure 1).
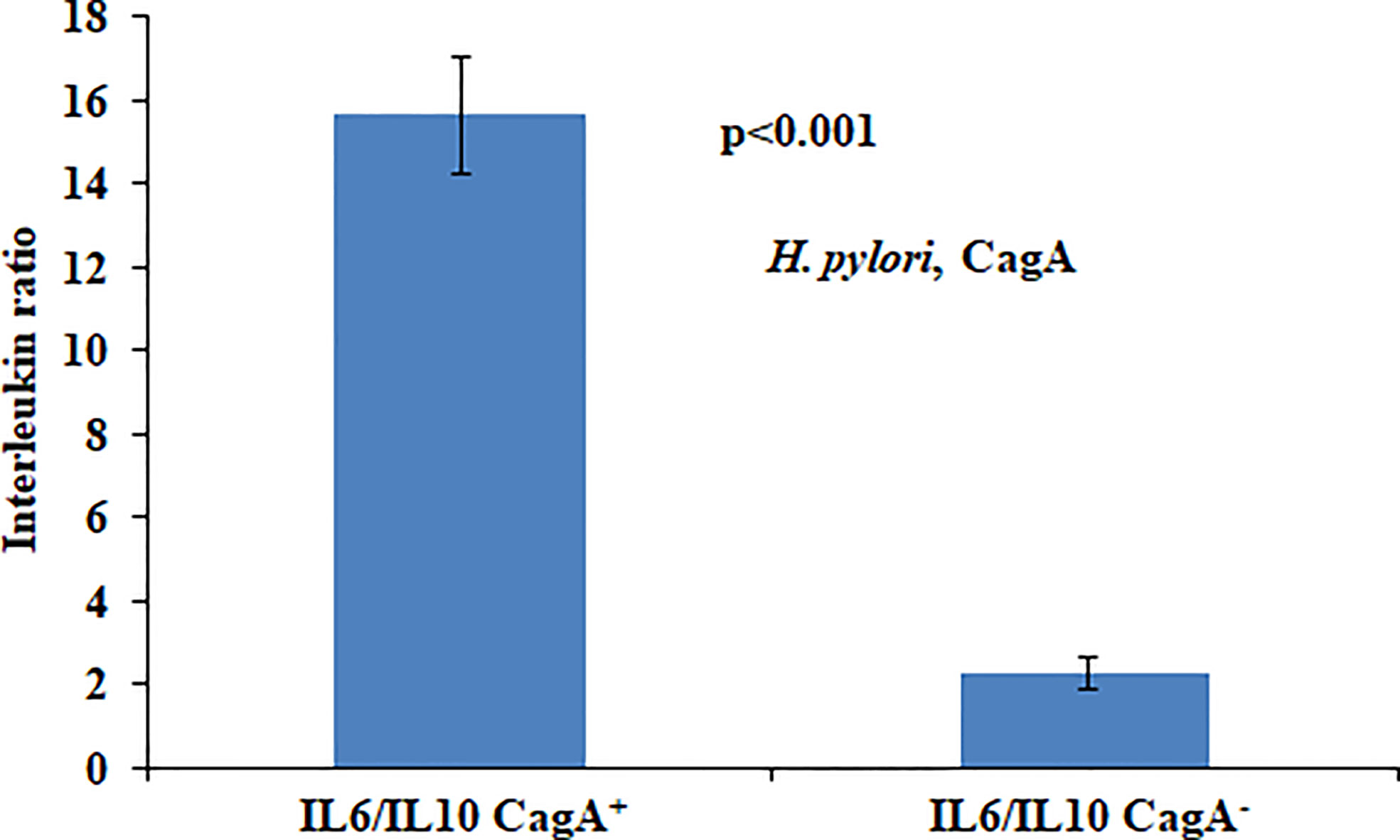
Figure 1 Ratio of circulatory cytokines (IL6/IL10) vis-à-vis local H. pylori infection status.
As discussed above, we found that that IL6 was substantially expressed compared to IL10 in H. pylori-infected tumor tissues. Since IL6 upregulates SOCS-1, which is a negative regulator of JAK-STAT signaling cascade, we next examined the methylation pattern of SOCS-1 gene promoter in tumor tissues. It is interesting to note that DNA methylation occurs when the cytosine in cytosine-guanine dinucleotides is converted into methyl-cytosine. Binding of transcription factors to such gene promoter regions is blocked due to their hypermethylation, thus rendering gene silencing or loss of expression. We found that SOCS-1 gene promoter was methylated in majority of tumor specimens (partially methylated in ~75% and fully methylated in ~15% of tumor specimens) (Supplementary Figure 4). SOCS-1 hypermethylation results in overactivation of JAK/STAT signaling cascade which in turn results abnormal proliferation of gastric mucosal cells. It is worthy to mention here that hypermethylation was also found in adjacent normal tissues suggesting that methylation may be involved during the early phases of gastric carcinogenesis as well.
On correlating patient characteristics with virulence marker of H. pylori infection (CagA and glmM), we found that male individuals were increasingly prone to H. pylori infection. Further, we analyzed that the patients admitted with clinical symptoms of generalized weakness, weight loss and malena were more susceptible to H. pylori-induced development of gastric cancer (Table 2). Furthermore, we found that H. pylori-infected patients acquired intestinal type gastric cancer and entered into metastatic stage III of cancer progression. In this study, we worked out that H. pylori infection leads to higher mRNA expression of IL-6, JAK-2 and STAT-3 at the local site of infection which better explains the progression of gastric cancer at the molecular level. We found that most of the patients (~86.5%) with H. pylori-infected gastric cancer tissues had hypermethylation of promoter region of SOCS-1 gene which could be a possible reason for the devastating nature of gastric cancer (Table 3). Besides the higher levels inflammatory cytokines at the local area of infection by H. pylori, we also evaluated that concentration of inflammatory cytokines like IL-6 and TGF-β was raised in the general circulation predicting the aggressive nature of gastric cancer.
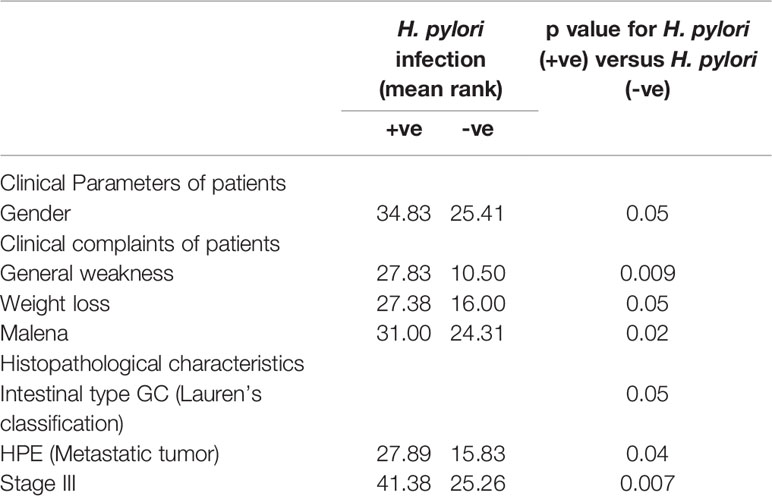
Table 2 Correlation of H. pylori infection with general characteristics/clinical complaints/histopathological characteristics of patients.
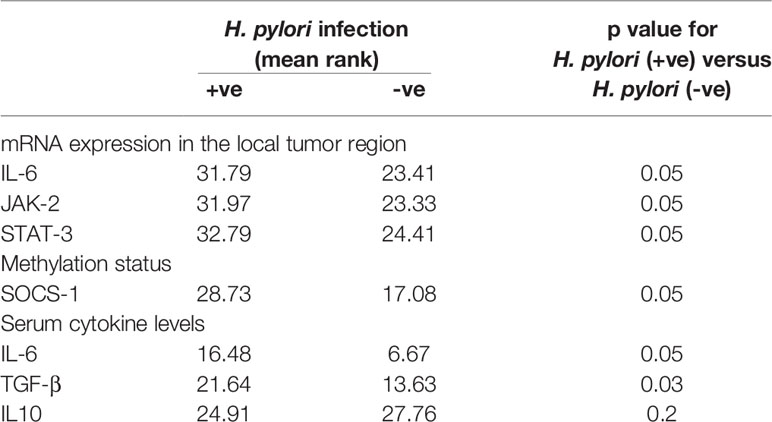
Table 3 Correlation of H. pylori infection with mRNA expression, methylation profile and serum cytokine levels of gastric cancer patients.
In the non-parametric analysis, absence of H. pylori infection was associated with better gastric cancer-specific survival. Our data revealed that the mean survival rank of H. pylori negative patients was ~22 while the mean survival rank of H. pylori positive patients was ~12 (Figure 2). Kaplan–Meier survival curves indicated that H. pylori infection can be a potential prognostic biomarker for gastric cancer.
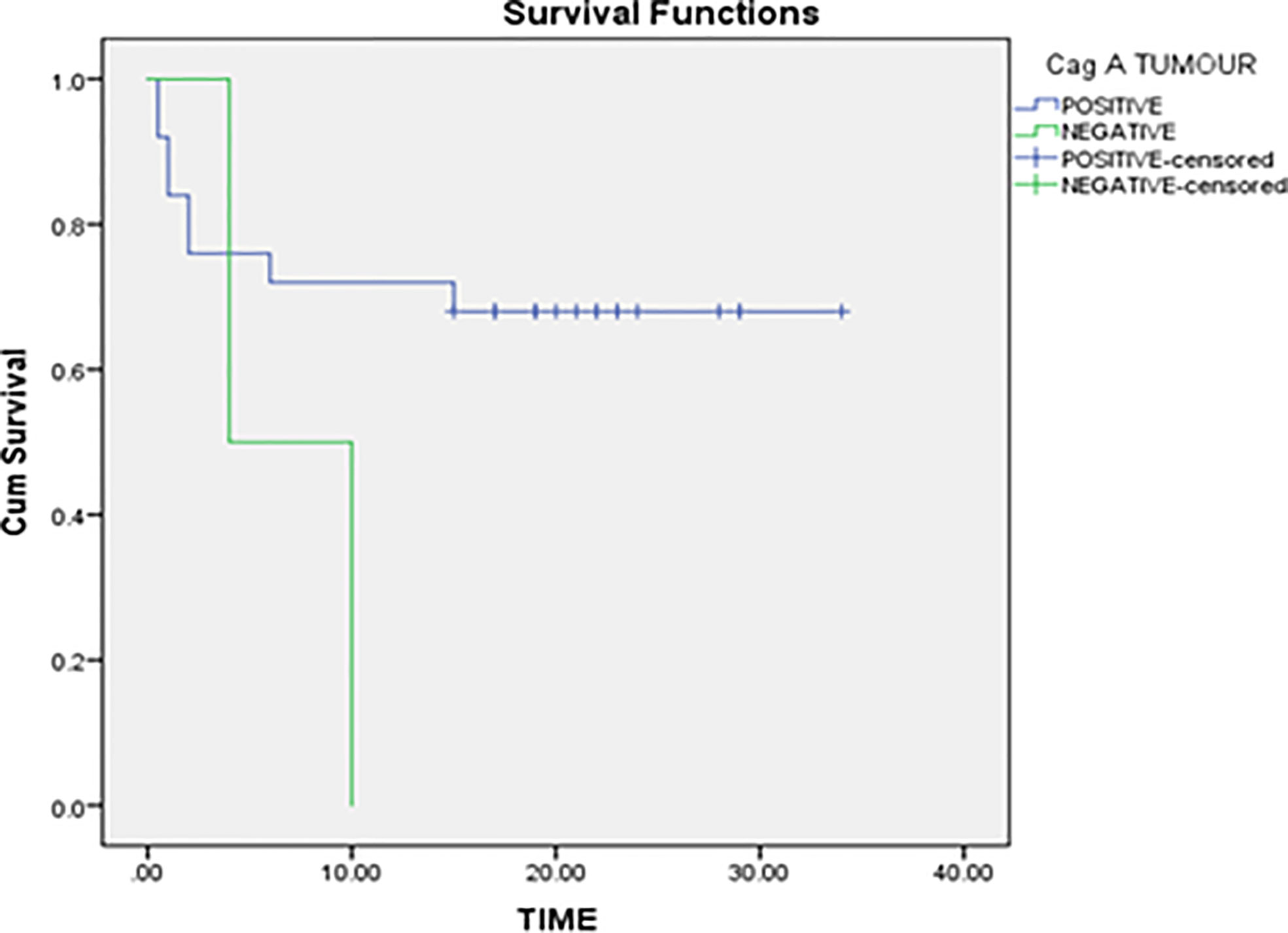
Figure 2 Correlation of H. pylori infection with overall survival of gastric cancer patients.
Gastric cancer is an intricate disease which develops as a consequence of complex interaction between environmental, habitual, dietary, genetic, and epigenetic factors. A proper analysis of different risk factors associated with gastric cancer development has helped a lot in understanding the possible mechanism of gastric carcinogenesis. A recently conducted meta-analysis revealed that H. pylori infection is the central reason for the growth of gastric cancer and affects males more aggressively than females (22).
H. pylori infection has been shown to trigger multiple motor neuron disorders which cause generalized weakness experienced by the H. pylori-infected gastric cancer patients (23). Apart from generalized weakness, the most common clinical complaints experienced by the H. pylori-infected gastric cancer patients, included dyspepsia, dysphagia, vomiting, malena, abdominal pain and weight loss. These findings are in agreement with our results. H. pylori-induced chronic active gastritis has been associated with the initiation of intestinal type of gastric adenocarcinoma. In our study, we found that gastritis-cum-abdominal pain was the most common complaint experienced by gastric cancer patients. Thus, functional abdominal pain of H. pylori-infected patients should not be ignored. In addition, we found that gastric cancers, that we studied, were majorly of intestinal-type and originated in the antropyloric region of the stomach. Further, it has been reported that H. pylori-infected gastric cancer patients usually reach to stage III of the disease (24). Using mouse models of gastric cancer, it has been reported that upregulation of IL-6 and activation of STAT-3 in H. pylori-infected gastric cancer ensues to activation of JAK/STAT signaling cascade. Several studies have been conducted to understand the underlying mechanism of JAK/STAT activation (25). Recently, it has been found that hyperactivation of JAK/STAT in H. pylori-infected gastric cancer patients occur via hypermethylation of promoter region of SOCS1 gene (21). This study is strongly in agreement with our finding. We evaluated that chronic H. pylori infection leads to the release of numerous inflammatory mediators including chemokines [IL8, macrophage chemotactic protein (MCP)-1, growth-regulated oncogene (GRO)-α] and cytokines [IL1-β, tumor necrosis factor (TNF)-α, IL6, IL12, interferon (IFN)-γ], which enter into the general blood circulation and exert a systemic effect (23).
In summary, hypermethylation of the promoter region of the SOCS-1 gene together with high production of endogenous IL6 and TGF-β leads to hyperactivation of JAK/STAT in gastric tumors. The increase in STAT3 activity concomitant with overexpression of IL6 and TGF-β suggests that aberrant activation of JAK-STAT signaling cascade is a key event for gastric carcinogenesis. Therefore, the release of inflammatory as well as regulatory cytokines by the H. pylori-infected mucosal immune cells at the local area as well as in the general circulation can be considered as an important trigger for the initiation and progression of gastric cancer. The present study has brought out that H. pylori-mediated cytokine signaling promotes gastric cancer development through hypermethylation of SOCS-1 gene promoter and aberrant activation of JAK-STAT cascade. Therefore, concurrent eradication of H. pylori and inhibition of JAK-STAT signaling cascade by applying specific JAK2 inhibitors may unravel new therapeutic strategies against the gastric cancer in ethnic Kashmiri population. This study provides us with ample clues regarding the different steps and factors which we can tackle therapeutically in order to stop this deadly disease from progression.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The studies involving human participants were reviewed and approved by Institutional Ethics Committee (IEC-SKIMS). The patients/participants provided their written informed consent to participate in this study.
IJ performed the experiments with the assistance from DA, RR, IM, AM, SB, BB, MF, TY, and BR. IJ, DA, and RR analyzed the data. DA acquired the funding and supervised the project. All authors contributed to the article and approved the submitted version.
This research was funded by the Science and Engineering Research Board (SERB), Department of Science and Technology, New Delhi, India (Grant Number: EMR/2016/004794) and intramural grants from SKIMS, Soura, Srinagar (India).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2020.604747/full#supplementary-material
Supplementary Figure 1 | Clinical complications of gastric cancer patients
Supplementary Figure 2 | PCR products of CagA gene in gastric cancer tissues along with their adjacent normal tissues run on 2% agarose gel.
Supplementary Figure 3 | PCR amplified products of GlmM gene in gastric cancer tissues along with their adjacent normal tissues run on 2% agarose gel.
Supplementary Figure 4 | MS-PCR products of SOCS1 in normal (methylated, unmethylated) and gastric cancer tissues (methylated & unmethylated). (A) Lane1-M(N), Lane2-UM(N), Lane3-M(N), Lane4-UM(N), Lane5-M(N), Lane6-UM(N), Lane7-M(N), Lane8-UM(N), Lane9-M(N), Lane10-UM(N), Lane11-M(N), Lane12-UM(N), Lane13-50bp ladder. (B) Lane1-M (N), Lane2-UM(N), Lane3-M(N), Lane4-UM(N), Lane5-M(N), Lane6-UM(N), Lane7-M(N), Lane8-UM (N), Lane9-M(N), Lane10-UM(N), Lane11-M(N), Lane12-UM(N), Lane13-50bp ladder. (C) Lane1-M(N), Lane2-UM(N), Lane3-M(N), Lane4-UM(N), Lane5-M(N), Lane6-UM(N), Lane7-M(N), Lane8-UM(N), Lane9-M(N), Lane10-UM(N), Lane11-M(N), Lane12-UM(N), Lane13-50bp ladde.
1. Liu X, Shao L, Liu X, Ji F, Mei Y, Cheng Y, et al. Alterations of gastric mucosal microbiota across different stomach microhabitats in a cohort of 276 patients with gastric cancer. EBioMedicine (2019) 40:336–48. doi: 10.1016/j.ebiom.2018.12.034
2. Blosse A, Lehours P, Wilson KT, Gobert AP. Helicobacter: Inflammation, immunology, and vaccines. Helicobacter (2018) 23:e12517. doi: 10.1111/hel.12517
3. Bik EM, Eckburg PB, Gill SR, Nelson KE, Purdom EA, Francois F, et al. Molecular analysis of the bacterial microbiota in the human stomach. Proc Natl Acad Sci (2006) 103:732–7. doi: 10.1073/pnas.0506655103
4. Peek RM, Blaser MJ. Helicobacter pylori and gastrointestinal tract adenocarcinomas. Nat Rev Cancer (2002) 2:28–37. doi: 10.1038/nrc703
5. Peek RM Jr, Crabtree JE. Helicobacter infection and gastric neoplasia. J Pathol (2006) 208:233–48. doi: 10.1002/path.1868
6. Müller A, Hitzler I, Kohler E, Engler DB, Sayi Yazgan A. The role of Th cell subsets in the control of Helicobacter infections and in T cell-driven gastric immunopathology. Front Immunol (2012) 3:142. doi: 10.3389/fimmu.2012.00142
7. Arnold IC, Lee JY, Amieva MR, Roers A, Flavell RA, Sparwasser T, et al. Tolerance rather than immunity protects from Helicobacter pylori–induced gastric preneoplasia. Gastroenterology (2011) 140:199–209. e8. doi: 10.1053/j.gastro.2010.06.047
8. Algood HMS, Allen SS, Washington MK, Peek RM, Miller GG, Cover TL. Regulation of gastric B cell recruitment is dependent on IL-17 receptor A signaling in a model of chronic bacterial infection. J Immunol (2009) 183:5837–46. doi: 10.4049/jimmunol.0901206
9. Sheh A, Lee CW, Masumura K, Rickman BH, Nohmi T, Wogan GN, et al. Mutagenic potency of Helicobacter pylori in the gastric mucosa of mice is determined by sex and duration of infection. Proc Natl Acad Sci (2010) 107:15217–22. doi: 10.1073/pnas.1009017107
10. Horvath CM. The Jak-STAT pathway stimulated by interleukin 6. Science’s STKE (2004) 2004:tr9–9. doi: 10.1126/stke.2602004tr9
11. Šebunova N, Štšepetova J, Sillakivi T, Mändar R. The prevalence of Helicobacter pylori in Estonian bariatric surgery patients. Int J Mol Sci (2018) 19:338. doi: 10.3390/ijms19020338
12. Beevers DG, Lip GY, Blann AD. Salt intake and Helicobacter pylori infection. J Hyperten (2004) 22:1475–7. doi: 10.1097/01.hjh.0000133736.77866.77
13. Torres J, Pérez-Pérez GI, Leal-Herrera Y, MUNoz O. Infection with CagA+ Helicobacter pylori strains as a possible predictor of risk in the development of gastric adenocarcinoma in Mexico. Int J Cancer (1998) 78:298–300. doi: 10.1002/(SICI)1097-0215(19981029)78:3<298::AID-IJC6>3.0.CO;2-Q
14. Vorobjova T, Nilsson I, Kull K, Maaroos H-I, Covacci A, Wadström T, et al. CagA protein seropositivity in a random sample of adult population and gastric cancer patients in Estonia. Eur J Gastroenterol Hepatol (1998) 10:41–6. doi: 10.1097/00042737-199801000-00008
15. Zhang W, Lu H, Graham DY. An update on Helicobacter pylori as the cause of gastric cancer. Gastrointest Tumors (2014) 1:155–65. doi: 10.1159/000365310
16. Niu Q, Zhu J, Yu X, Feng T, Ji H, Li Y, et al. Immune Response in H. pylori-Associated Gastritis and Gastric Cancer. Gastroenterol Res Pract (2020) 2020:9. doi: 10.1155/2020/9342563
17. Zhuang Y, Peng LS, Zhao YL, Shi Y, Mao XH, Chen W, et al. CD8+ T cells that produce interleukin-17 regulate myeloid-derived suppressor cells and are associated with survival time of patients with gastric cancer. Gastroenterology (2012) 143:951–962. e8. doi: 10.1053/j.gastro.2012.06.010
18. Bockstett K, DiPaolo R. Regulation of Gastric Carcinogenesis by Inflammatory Cytokines. Cell Mol Gastroentenol Hepatol (2017) 1:47–53. doi: 10.1016/j.jcmgh.2017.03.005
19. Pandey A, Tripathi SC, Shukla S, Mahata S, Vishnoi K, Misra SP, et al. Differentially localized survivin and STAT3 as markers of gastric cancer progression: Association with Helicobacter pylori. 1 (2018) 1:e1004. doi: 10.1002/cnr2.1004
20. Natatsuka R, Takahashi T, Serada S, Fujimoto M, Ookawara T, Nishida T, et al. Gene therapy with SOCS1 for gastric cancer induces G2/M arrest and has an antitumour effect on peritoneal carcinomatosis. Br J Cancer (2015) 113:433–42. doi: 10.1038/bjc.2015.229
21. To K, Chan M, Leung W, Ng E, Yu J, Bai A, et al. Constitutional activation of IL-6-mediated JAK/STAT pathway through hypermethylation of SOCS-1 in human gastric cancer cell line. Br J Cancer (2004) 91:1335–41. doi: 10.1038/sj.bjc.6602133
22. De Martel C, Parsonnet J. Helicobacter pylori infection and gender: a meta-analysis of population-based prevalence surveys. Digestive Dis Sci (2006) 51:2292–301. doi: 10.1007/s10620-006-9210-5
23. Álvarez-Arellano L, Maldonado-Bernal C. Helicobacter pylori and neurological diseases: Married by the laws of inflammation. World J Gastrointest Pathophysiol (2014) 5:400. doi: 10.4291/wjgp.v5.i4.400
24. Turanli S, Bozdogan N, Mersin H, Berberoglu U. The effect of helicobacter pylori on gastric cancer treated with adjuvant chemotherapy after curative resection. Indian J Surg (2015) 77:489–94. doi: 10.1007/s12262-015-1305-9
25. Ishii Y, Shibata W, Sugimori M, Kaneta Y, Kanno M, Sato T, et al. Activation of signal transduction and activator of transcription 3 signaling contributes to Helicobacter-associated gastric epithelial proliferation and inflammation. Gastroenterol Res Pract (2018) 2018:1–9. doi: 10.1155/2018/9050715
Keywords: mucosal inflammation, H. pylori, SOCS1, hypermethylation, carcinogenesis
Citation: Jan I, Rather RA, Mushtaq I, Malik AA, Besina S, Baba AB, Farooq M, Yousuf T, Rah B and Afroze D (2021) Helicobacter pylori Subdues Cytokine Signaling to Alter Mucosal Inflammation via Hypermethylation of Suppressor of Cytokine Signaling 1 Gene During Gastric Carcinogenesis. Front. Oncol. 10:604747. doi: 10.3389/fonc.2020.604747
Received: 10 September 2020; Accepted: 07 December 2020;
Published: 25 January 2021.
Edited by:
Ashok Kumar, University of Houston, United StatesReviewed by:
Alok Chandra Bharti, University of Delhi, IndiaCopyright © 2021 Jan, Rather, Mushtaq, Malik, Besina, Baba, Farooq, Yousuf, Rah and Afroze. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dil Afroze, YWZyb3plZGlsQGdtYWlsLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.