 Lingling Wang
Lingling Wang Shizhen Zhang
Shizhen Zhang Xiaochen Wang
Xiaochen Wang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol., 07 January 2021
Sec. Women's Cancer
Volume 10 - 2020 | https://doi.org/10.3389/fonc.2020.602416
This article is part of the Research TopicMetabolic Abnormalities and Breast Cancer: Challenges from Bench to BedsideView all 19 articles
Breast cancer is one of the most common malignancy among women worldwide. Metastasis is mainly responsible for treatment failure and is the cause of most breast cancer deaths. The role of metabolism in the progression and metastasis of breast cancer is gradually being emphasized. However, the regulatory mechanisms that conduce to cancer metastasis by metabolic reprogramming in breast cancer have not been expounded. Breast cancer cells exhibit different metabolic phenotypes depending on their molecular subtypes and metastatic sites. Both intrinsic factors, such as MYC amplification, PIK3CA, and TP53 mutations, and extrinsic factors, such as hypoxia, oxidative stress, and acidosis, contribute to different metabolic reprogramming phenotypes in metastatic breast cancers. Understanding the metabolic mechanisms underlying breast cancer metastasis will provide important clues to develop novel therapeutic approaches for treatment of metastatic breast cancer.
Breast cancer is the most common malignant tumor and the second capital reason for cancer death among women worldwide (1, 2). Metastatic breast cancer, not the primary tumor, is responsible for more than 90% cancer-related deaths (3). A SEER based study showed that for metastatic breast cancer patients: 30–60% have metastases in the bone, 21–32% in the lung, 15–32% in the liver and 4–10% in the brain. Moreover, the preferred metastatic sites appear to depend on the specific pathological subtypes of primary breast cancers (4).
Recently, increasing evidence point out that cancer is not only a genetic disease but also a metabolic disease, in which oncogenic signaling pathways participate in energy regulation and anabolism to support rapidly spreading tumors (5). In this sense, metabolic reprogramming is considered a hallmark of cancer (6, 7). Notably, metabolic reprogramming and its complex regulatory networks also affect the tumorigenesis and progression of breast cancer (8). Considered as a high heterogeneous disease, breast cancer includes four main intrinsic molecular subtypes: Luminal A, luminal B, HER2-positive, and triple-negative breast cancer (TNBC). Each subtype has different proliferation and metastasis capabilities, as well as metabolic genotypes and phenotypes (9–16) (Table 1). Specifically, TNBC cells possess particular metabolic traits characterized by high glycolysis and low mitochondrial respiration (22). HER2-positive tumors display higher glutamine metabolic activity and higher lipid metabolism than other subtypes (15, 20). Nevertheless, metabolic changes may not only be varied in different breast cancer subtypes, but also diverged relying on the interplay of cancer cells with the complex microenvironment (16, 23).
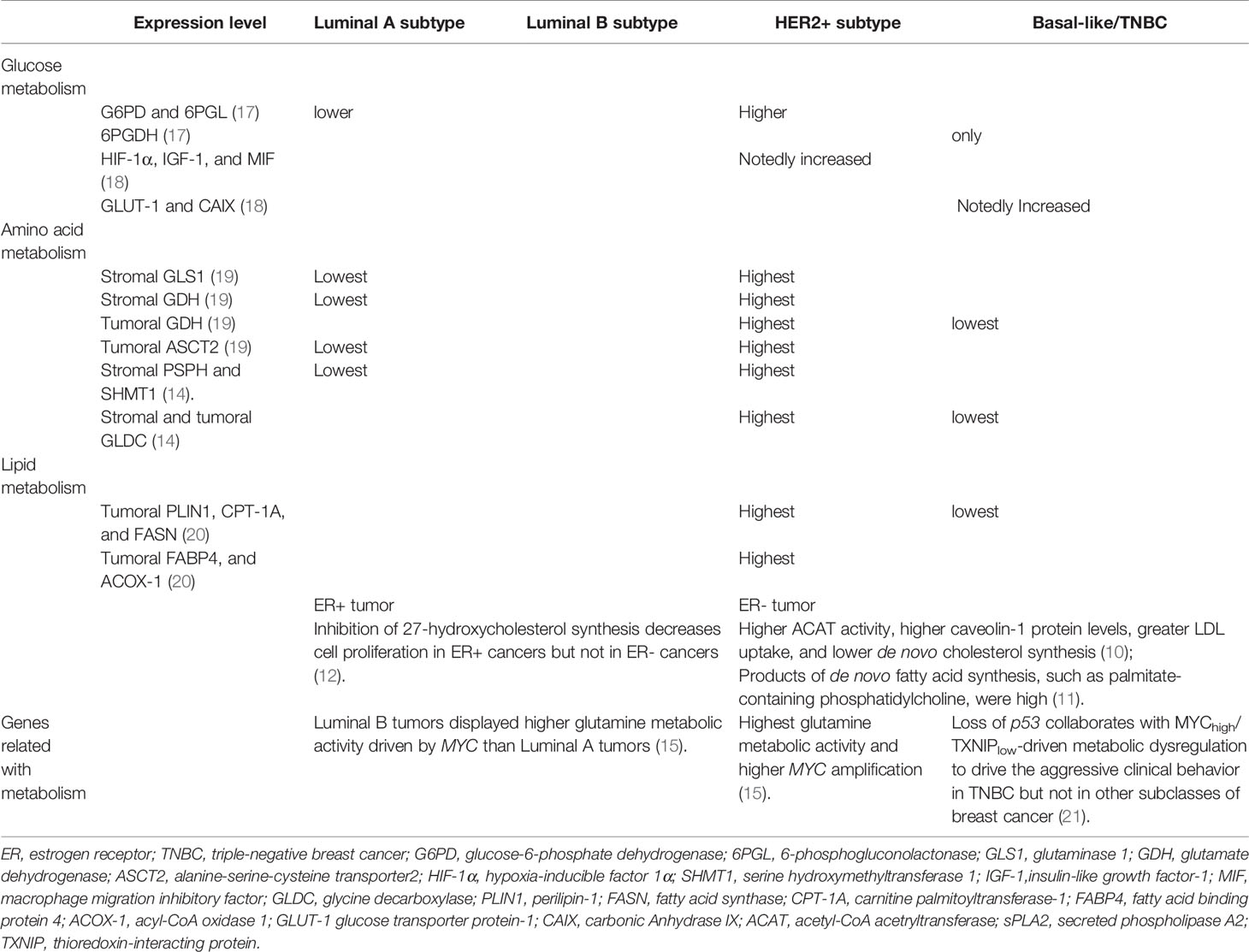
Table 1 Metabolic differences in different breast cancer subtypes.
This review addresses the current knowledge on the crosstalk between metabolic reprogramming and metastatic process in breast cancer. A better understanding of the metabolic mechanisms driving breast cancer metastasis may provide clues for discovering new anticancer therapeutics.
In response to external growth signals, normal cells in a rapidly proliferating state activate assorted signaling pathways to suppress oxidative phosphorylation (OXPHOS), and advance glycolysis and anabolic metabolism for cell growth. Cancer cells is able to hijack this mechanism to meet developmental needs even if there are no external signals (24) (Figure 1). Different from normal cells where glycolysis and OXPHOS are always negatively correlated, cancer cells possess these two modes coexisting to disparate degrees (25). Moreover, unlike normal cells, which mainly produce adenosine triphosphate (ATP) from glucose-derived pyruvate by OXPHOS through the TCA cycle, most cancer cells depend on glycolysis to generate energy even under aerobic conditions (26). It was found that tumors displayed dual metabolic natures that tumor cells could switch from the aerobic glycolysis back to OXPHOS phenotype upon lactic acidosis (27). Furthermore, some tumors exhibit two-compartment tumor metabolism, called the reverse Warburg effect or metabolic coupling, which indicates that glycolytic metabolism in the cancer-related stroma sustains the adjacent cancer cells. Such metabolic phenotype will contribute to chemotherapy resistance, and also explain the contradictory phenomenon of high mitochondrial respiration and low glycolysis rate in some tumor cells (28–30). Moreover, a large sample data study showed luminal subtype correlated with reverse-Warburg/null phenotypes that are metabolically inactive, while TNBC correlated with Warburg/mixed phenotypes that are metabolically active (31). Additionally, hypoxic environment in breast tumors brings about increased production of reactive oxygen species (ROS) (32), at the same time, induced hypoxia-inducible factor 1 (HIF-1) is able to boost glucose metabolism to maintain the redox homeostasis (33).
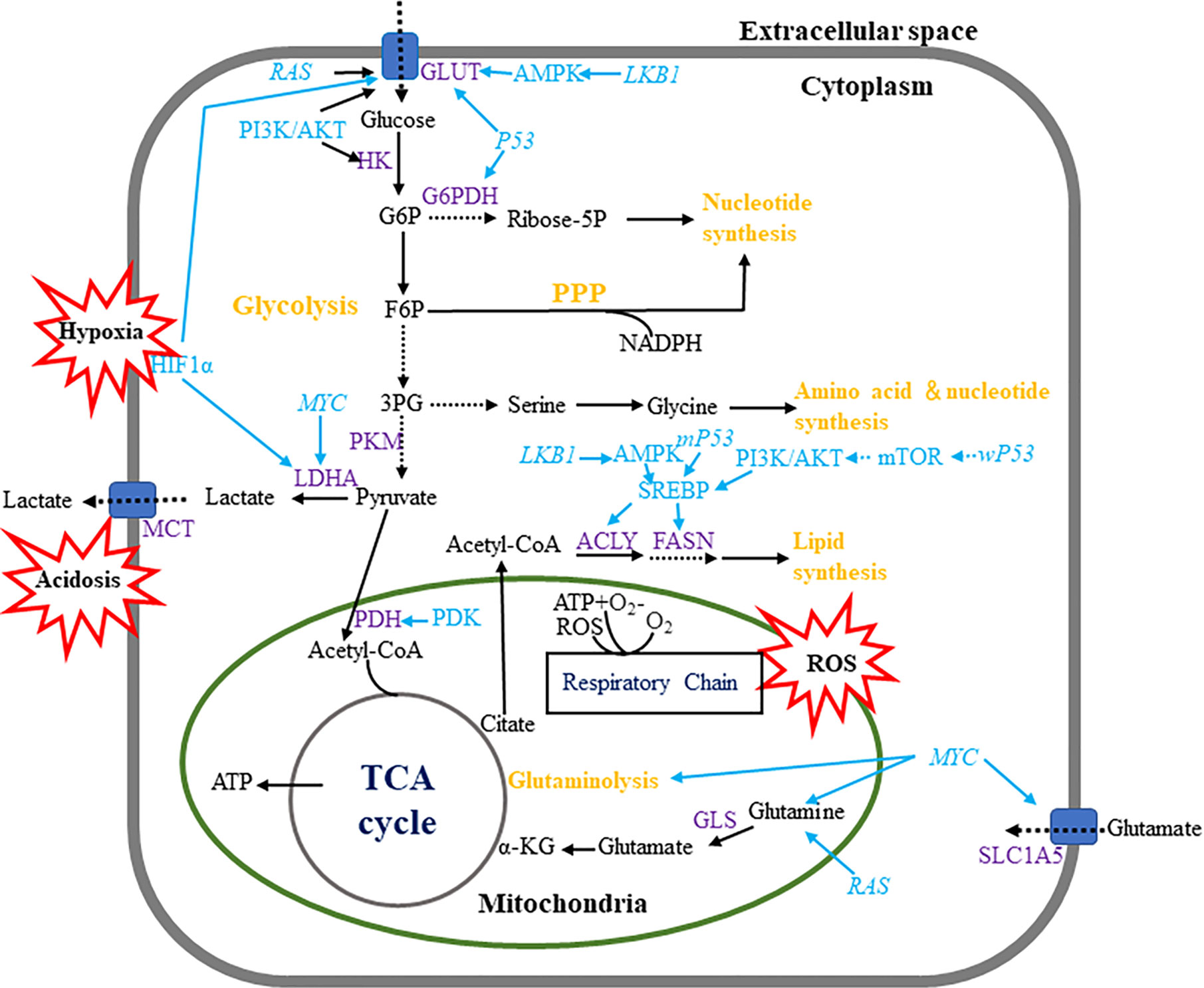
Figure 1 Metabolic pathways in breast cancer cells. Breast cancer cells enhance metabolism of glucose, amino acid lipid by regulating multiple metabolic pathways. Breast tumor cells mainly use aerobic glycolysis to produce ATP and utilize the pentose phosphate pathway to produce macromolecules such as NADPH. Wild-type and mutant P53 have contrary effects in monitoring fatty acid metabolism. Hypoxia, acidosis and ROS are regarded as important events which influence multiple metabolic pathways.
Glucose transport cross cell membrane through the glucose transporter proteins (GLUTs) and different GLUTs expression in breast cancers are related to dissimilar pathological grades and prognosis. GLUT1-5 and GLUT12 are functionally in breast cancer cells (34–37), and GLUT1 appears to play the most important role (38). Interestingly, TNBC had the highest expression of GLUT1 when contrasted with other subtypes, suggesting the highly active metabolic status in TNBC (18). Moreover, some critical glycolysis-related enzymes, such as hexokinase (HK) and lactate dehydrogenase-A (LDHA), are highly activated in breast cancer and related to cancer growth and progression (39, 40).
The pentose phosphate pathway (PPP) is another way of oxidative decomposition of glucose besides glycolysis and TCA cycle, which produces nicotinamide adenine dinucleotide phosphate (NADPH), ribose phosphate, fructose-6-phosphate (F6P) to make cancer cells satisfy their anabolic needs and respond to oxidative stress (41). Proteins involved in PPP are distinctively expressed in different molecular subtypes of breast cancers. For example, the expression of glucose-6-phosphate dehydrogenase (G6PD) and 6-phosphogluconolactonase (6PGL) were elevated, implying a more activated PPP in HER2 subtype than other subtypes of breast cancer (17). It has been suggested that the expression of G6PD and transketolase (TKT) are positively correlated to the decreased overall and relapse-free survival in breast cancer (42).
Glutamine and its metabolic intermediates such as antioxidants nicotinamide adenine dinucleotide (NADH), and glutathione (GSH), participate in energy supply, supplement glucose metabolism and help cells resist oxidative stress to uphold proliferation and progression of tumor cells (43, 44). Some cancer cells exhibit “glutamine addiction” that cannot survive in the absence of exogenous glutamine (45). More importantly, certain oncogenic transcription factors, such as c-MYC and RAS, can increase the cancer cell glutamine metabolic activity by upregulating glutamine transporters such as alanine-serine-cysteine transporter 2 (ASCT2) and enzymes participating in the conversion of glutamine-to-glutamate, such as glutaminase (GLS)-1 (46). For example, c-MYC activates the expression of ASCT2 and GLS-1 under the induction of lactic acid, leading to elevated glutamine uptake and catabolism in cancer cells (47). Notably, a metabolomic analysis indicted that breast tumor tissues had a higher glutamate‐to-glutamine ratio (GGR) than normal tissues, especially in estrogen receptor (ER) negative tumors and the GGR levels dramatically correlated with ER status and tumor grade (48). The glutamine metabolism-related proteins, such as GLS-1, glutamate dehydrogenase (GDH), and ASCT2 were found to be highly expressed in HER2-positive breast cancer than other subtypes, which indicated that HER2-positive breast cancer had the highest glutamine metabolism activity (19).
One-carbon metabolism, also known as network of folate utilization reactions, participates in multiple metabolic pathways such as amino acid biosynthesis and degradation, de novo nucleotide biosynthesis, and methylation and reductive metabolism (49). It has been widely accepted that one-carbon metabolism acts a pivotal part in supporting the high proliferative rate of tumor cells (50). Folate (vitamin B9), a carrier of one-carbon units, and other B vitamins, such as B6 and B12, take part widely in one-carbon metabolism, which is requisite for DNA biosynthesis and methylation (51). Although the relationship between folic acid intake and the risk of breast cancer is still controversial, a recent meta-analysis, analyzing 23 prospective studies, found that an increment of folate intake decreased the risk of ER-, ER-/PR-, premenopausal breast cancer and had the preventive effects against breast cancer in individuals with alcohol consumption (52).
In addition to glutamine, upregulation of serine/glycine metabolism closely connected with folate metabolism is relevant to high proliferation of tumor cells and poor prognosis of patients (50). Tryptophan and arginine are involved in the manipulation of immunity and tolerance, which are generally deregulated in cancers (53). The activity of arginase, the key enzyme catalyzing L-arginine, in breast tumor environments is strengthened, which generates an unfavorable milieu for T cell adaptability (54).
The fatty acids (FAs) and lipid metabolic programming also play significant parts in promoting breast cancer growth and progression (55). Cancer cells maintain a highly proliferative state by activating the uptake of exogenous lipids and lipoproteins, or by reinforcing de novo lipid and cholesterol biosynthesis, showing active lipid and cholesterol metabolisms (56). Moreover, tumor cells mostly rely on de novo fatty acid synthesis (FAS) to satisfy the augmented demand for membrane metabolism in favor of rapid growth and proliferation. The expression of fatty acid synthase (FASN), a key enzyme essential for the FAS, is elevated in breast cancer (57), and its upregulation appears to be connected with cancer development, recurrence and poor prognosis (58), suggesting the augmented FAS activity is important for breast cancer progression. Notably, FASN was found to be expressed highest in HER2-positive breast cancer and lowest in TNBC at both cell and tissue levels (20, 59). It has been assumed that a two-way regulatory system between FASN and HER2, the “HER2-FASN axis”, may enhance breast cancer proliferation, metastasis and chemoresistance (60). Sterol regulatory element-binding protein (SREBP)-1, a lipogenic transcription factor, can regulate FASN expression by binding with the FASN promoter site (61, 62). And phosphatidylinositol-3-kinase (PI3K)/AKT/mammalian target of rapamycin (mTOR) and mitogen-activated protein kinase (MAPK) signal transduction pathways are also likely to regulate FASN expression (63, 64). Under hypoxic conditions, FASN gene is upregulated due to the arousal of AKT and SREBP-1 in breast tumor cells (65). Inhibition of MAPK pathway and mTOR inhibitor rapamycin both can decrease FASN expression in breast cancer cells (66, 67).
Tumor metastasis is a sequential multi-step process, which includes local invasion, intravasation, migration through the lymphatics or blood vessels, extravasation and colonization giving rise to the formation of metastases in distant organs (68). Particularly, organ-specific colonization hinge on the dynamic and mutual interrelation between tumor cells and tumor microenvironment (TME), comprised by varieties of non-cancerous cells such as immune cells, endothelial cells, fibroblasts, adipocytes, together with extracellular matrix (ECM) and soluble factors (69). In addition to the linear metastasis model, breast cancers prefer to the parallel metastasis model, which means that breast cancer cells begin to spread in the early stages of tumor development (70), and the spread of cancer cells may be independent of the progression of the primary tumors (71). Studies have shown that the genetic changes of the bone marrow disseminated breast cancer cells are usually not identical to their corresponding primary tumors (72). Different breast cancer subtypes have been found to show different metastatic sites preference governed by different molecular mechanisms (73). The molecular characteristics of breast cancers and target tissues appear to confirm the organotropism of metastasis (74). All the breast cancer subtypes are apt to develop bone metastasis, luminal A subtype is regarded as a risk factor for recurrence in the bone (75), and luminal B subtype is more likely to have bone as a first relapse site when compared to other subtypes (76). Moreover, the incidence of luminal subtype tumors to have bone metastasis is much higher (80.5%) than HER2-positive tumors (55.6%) and basal-like tumors (41.7%) (77). While luminal B and basal-like subtypes present higher levels of lung-specific metastasis (78). Compared with the HER2-negative subtype, the HER2-positive subtype is more often observed with liver metastases (4). Another study showed that basal-like tumors had a higher rate of metastasis to the brain, lungs and distant lymph nodes, while the rate of liver and bone metastasis is much lower (79).
The step one of the metastasis is that tumor cells break away from the tumor bed and migrate from the stroma into the bloodstream (80). In order to leave the primary tumor and invade surrounding tissues, these tumor cells need to reduce their tight cell adhesion through undergoing epithelial-mesenchymal transitions (EMT) (81, 82). EMT is typified by loss of epithelial traits (including cell polarity and cell-cell junctions) and acquisition of mesenchymal traits (including fibroblastic spindle-shaped morphology) to increase the mobility of tumor cells. EMT also links to cancer metastasis with stem cell properties (83, 84). Moreover, the integrin-mediated adhesion and debonding interactions with matrix components is critical for regional migration. And the intratumoral blood vessels characterized by increased permeability allow cancer cells to enter the systemic circulation readily (85).
After escaping from the original tumor site to blood circulation, breast tumor cells begin to migrate to remote organs. The first obstacle encountered by circulating tumor cells is the blood vessel wall, especially endothelial cells. In some organs, such as bone marrow and liver, microvessels are composed of sinuses with strong permeability, which make cancer cells easier to break through (86). Whereas in most other organs, including brain, endothelial cells form a continuous barrier that prevents cancer cells from penetrating freely. Platelets and white blood cells can help tumor cells pass through the vasculature by forming complexes with tumor cells through L- or P-selectin (87, 88). As such, increased expression of selectin ligands by tumor cells is well connected with metastatic progression and bad prognosis (89). The induction of angiopoietin-like 4 (ANGPTL4) by transforming growth factor-beta (TGFβ)/small mother against decapentaplegic (SMAD) signaling pathway in cancer cells is reported to enhance their subsequent retention in the lungs and empower breast cancer cells to destroy lung capillary wall and form pulmonary metastases (90). Chemokines in target cell tissues can also induce directed cell migration, initiate signal pathways, and monitor cytoskeletal rearrangment and adhesion (91).
Adjusting to new environment is another hurdle for circulating tumor cells (CTCs) to form metastasis. Disseminated cancer cells will spring up in targeted tissues and organs through a way that is significantly different from their origins. Cancer cells must acquire new capabilities, especially the ability to interact with cells in the ECM and new microenvironment. Tumor cells form a two-way connection with circumferent stroma in the early stage of invasion and after that, tumor-stroma interaction helps the tumor to develop toward metastasis (6).
An appropriate microenvironment, namely, pre-metastatic niche, can be established in secondary tissues and organs before metastases occurring through a complicated mechanism by interaction between the primary tumors and organs stromal components (92). Kaplan et al. emphasized the role of tumor-mobilized bone marrow-derived cells (BMDCs) in developing a satisfactory microenvironment for lung metastatic colonization. The factors, such as vascular endothelial growth factor (VEGF), and placental growth factor (PlGF), released by the primary tumor act on the bone marrow mesenchymal stem cells to induce the BMDCs to reach the expected metastasis site before the disseminated tumor cells arrive (93). Hiratsuka et al. demonstrated that matrix metalloproteinase (MMP)-9 is particularly motivated in premetastatic lung endothelial cells and macrophages mediated by primary tumors via the VEGFR-1/Flt-1 pathway, which is important for lung metastasis (94). The integrin β1/α5/JNK/c-JUN signaling pathway in cancer cells is able to upregulate the higher matrix stiffness-induced lysyl oxidase like (LOXL)-2, then subsequently promote production of fibronectin, expression of MMP-9 and C-X-C motif chemokine ligand (CXCL)-12 and recruitment of BMDCs to encourage pre-metastatic niche establishment (95). Chemokines binding to specific receptors on the target cell membrane help to recruit immune cells into the tumor microenvironment, thereby managing immune surveillance, angiogenesis, invasion and metastasis (96). The CXCL-12/C-X-C motif chemokine receptor (CXCR)-4 axis provides a fit microenvironment before breast cancer bone metastasis formation (97). Carmen et al. suggested that HIF-1 is a crucial regulatory factor inducing breast cancer metastatic niche forming through activation of several elements of the lysyl oxidase (LOX) family, which catalyze collagen cross-linking in the lungs before BMDC recruitment (98). Dickkopf (DKK)-1 suppresses prostaglandin endoperoxide synthase (PTGS)-2-induced macrophage and neutrophil recruitment to lung metastases by antagonizing cancer cell non-classical WNT/Planar cell polarity (PCP)-RAC1-JNK signaling, whereas it encourages breast-to-bone metastasis by modulating classical WNT signaling of osteoblasts (99).
The site-specific metastasis of breast cancer is related to subtypes and divergent gene signatures of metastatic cancer cells. Functional studies have identified many key genes that mediate breast cancer organ-specific metastasis, and the expression of these genes in the primary tumor is likely to forecast the patient’s organ-specific metastasis (100, 101).
Bone is the most frequent site of breast cancer metastasis (73). Bone metastasis is usually connected with osteolytic-type lesions as a result of the overactive bone resorption mediated by osteoclasts (102). Integrin complexes, such as integrin αvβ3, α4β1 and α5, play important roles in the attraction and adhesion of breast tumor cells to the bone (103–105). Some clinical, genetic, and functional evidence suggest that the SMAD tumor suppressor pathway may diverted into potent pro-metastatic factor in breast cancer, and signaling through the SMAD pathway can facilitate breast cancer bone metastasis (106). Moreover, both hypoxia (via HIF-1α) and TGFβ signaling can independently stimulate the VEGF and CXCR4 expression to drive breast cancer bone metastases (107). In basal-like TN breast cancer, CCL20 promotes bone metastasis by raising the secretion of MMP-2/9 and increasing the receptor activator of nuclear factors-kappa B (NF-κB) ligand/osteoprotegerin ratio in breast cancer and osteoblastic cells (108).
The second most common metastatic site of breast cancer is the brain (73). Brain metastasis of breast cancer can be located in the parenchymal brain (around four-fifths) or in leptomeningeal region (109). CTCs need to break through the blood-brain barrier (BBB), interplay with the local microenvironment to survive, and then set up brain metastatic colonies. CD44, VEGF and CXCR4 can impair endothelial integrity to raise the transendothelial migration of tumor cells (110). Angiopoietin-2 (Ang-2) expression is elevated in brain microvascular endothelial cells (BMECs) and secreted Ang-2 can increase BBB permeability by disrupting tight junction protein structures between ZO-1 and Claudin-5 in TNBC models of brain metastasis (111). Cyclooxygenase (COX)-2, heparin-binding epidermal growth factor-like growth factor (HBEGF), and ST6GALNAC5 are all able to help tumor cell pass through the BBB (112). Additionally, astrocytes and microglia are related with brain metastases. Astrocytes-derived factors, such as MMP-2 and MMP-9, are able to enhance the migration and invasion of breast tumor cells, thus leading to brain metastasis (113). Similarly, microglia can also be stimulated by culturing with cancer cells, so that it boosts cancer cell colonization in a WNT-dependent manner (114).
Compared to other metastatic lesions, lung metastasis generally show phenotypes of aggressive growth and invasiveness (101). EGFR, COX2, MMP-1, and MMP-2 expressed in breast cancers jointly facilitate lung metastasis by promoting the angiogenesis, emancipating cancer cells into the circulation and breaking through lung capillaries (115). Studies have determined that compared with primary breast cancer, the degree of pyruvate carboxylase (PC)-dependent anaplerosis in lung metastasis of breast cancer is higher, as a result of responding to the lung microenvironment (116). Bone morphogenetic proteins (BMPs) secreted by lung resident cells can restrict cancer development by turning cancer cells into a dormant state, while Coco and GALNTs derived from lung metastatic breast tumor cells are able to inhibit the effect of BMPs and reactivate dormant tumor cells to seed in the lung, thereby leading to metastasis (117).
Breast cancer cells preferred to liver-specific homing display unique transcriptional profiling (118). The status of ER, progesterone receptor (PR) and HER2 between the primary and liver metastatic tumors of breast cancer can be changed after treatment (119). Development of breast cancer liver metastasis is reported to be associated with the activation of β-catenin-independent WNT signaling (120). A model for breast cancer liver metastasis was established involving diverse factors from breast tumor cells and the liver microenvironment such as integrin complexes, HIFs and LOX (121).
Breast cancer stem cells (BCSCs), a small number of cells with self-renewal and unlimited replication capabilities, have been shown in numerous cancer models to be involved in tumor development and metastatic dissemination. Moreover, the occurrence of BCSCs with the properties of stemness, EMT and drug resistibility, is the main cause for cancer recurrence and treatment failure (122). Multiple researches revealed that several signaling pathways, such as WNT/β-catenin and Notch, contribute significantly to the development of BCSCs (123). Devon A et al. showed that early metastatic breast cancer cells had unique stem-like gene expression characteristics and prefer to proliferate and differentiate to produce advanced metastatic disease at the single-cell level (124). Additionally, BCSCs isolated from primary human breast cancers possess the advanced metastasis potential and the CD70+ subpopulations appear to preferentially mediate lung-specific metastasis by enhancing self-renewal potential (125).
After colonizing the distant metastatic site, BCSC can enter into a metastatic dormant state, showing the halted proliferation and activated cellular stress response, while maintaining metabolic activity (126–128). The dormant phenotype is able to be reversed by manipulating of intrinsic and/or extrinsic factors and then the proliferative program restarts in vivo (129, 130). However, the biological mechanisms of cell dormancy and re-awakening are still elusive (131). The dormant state is regarded as a high risk of cancer recurrence and is supposedly limit the efficiency of chemotherapy. Targeting the metastatic dormancy, therefore, could be an promising treatment strategy to improve long-term control of cancer progression (132).
Metabolic plasticity is one of the important characteristics that distinguishes the tumor cells with high metastatic potentiality from non-metastatic tumor cells. Metastatic cancer cells always operate multiple metabolic pathways concurrently, thus they can adjust the application of diverse pathways according to their adaptive requirements (133, 134). Cancer cells are challenged by diverse environmental and cellular stresses during metastatic progression (135). Strikingly, cancer cells are capable of manipulating one or more metabolic pathways according to their stage in the metastatic cascade and the site they metastasize (133, 136–139). For instance, extracellular acidification by the release of CO2, lactic acid and other organic acids from metabolically vigorous tumor cells promotes intravasation of cells from the primary tumor. Once tumor cells enter the circulatory system, they produce NADPH and GSH through the PPP pathway to protect themselves from oxidative stress. The coordination of the metabolism between cancer cells and adjacent microenvironment is critical for successful colonization of distant sites and survival during dormancy. Most importantly, anabolic metabolism is reactivated in cancer cells to facilitate the growth of macro-metastatic tumors (140). Tumor metabolism reprogram also occurs when tumors progress in order to adapt to lack of sufficient blood supply. While during adaptation to environmental stress, such as cyclic hypoxia, tumor metabolism reprogram contributes to selection of drug-resistant and metastatic clones (141, 142).
Primary breast tumor cells exhibit metabolic heterogeneity and participate in different metabolic reprogramming according to metastatic sites (Figure 2). Liver-metastatic breast cancer cells display a distinct metabolic reprogramming characterized by accumulation of glucose-derived lactate and reduction in the TCA cycle and OXPHOS (138). In brain metastatic breast cancer, the significant metabolic changes are mainly the enhanced glycolysis, mitochondrial respiration and the PPP. Intriguingly, breast cancer cells metastasized to the brain are less sensitive to glucose deficiency (136), which may attribute to upregulation of glutamine and branched chain amino acid oxidation (143).
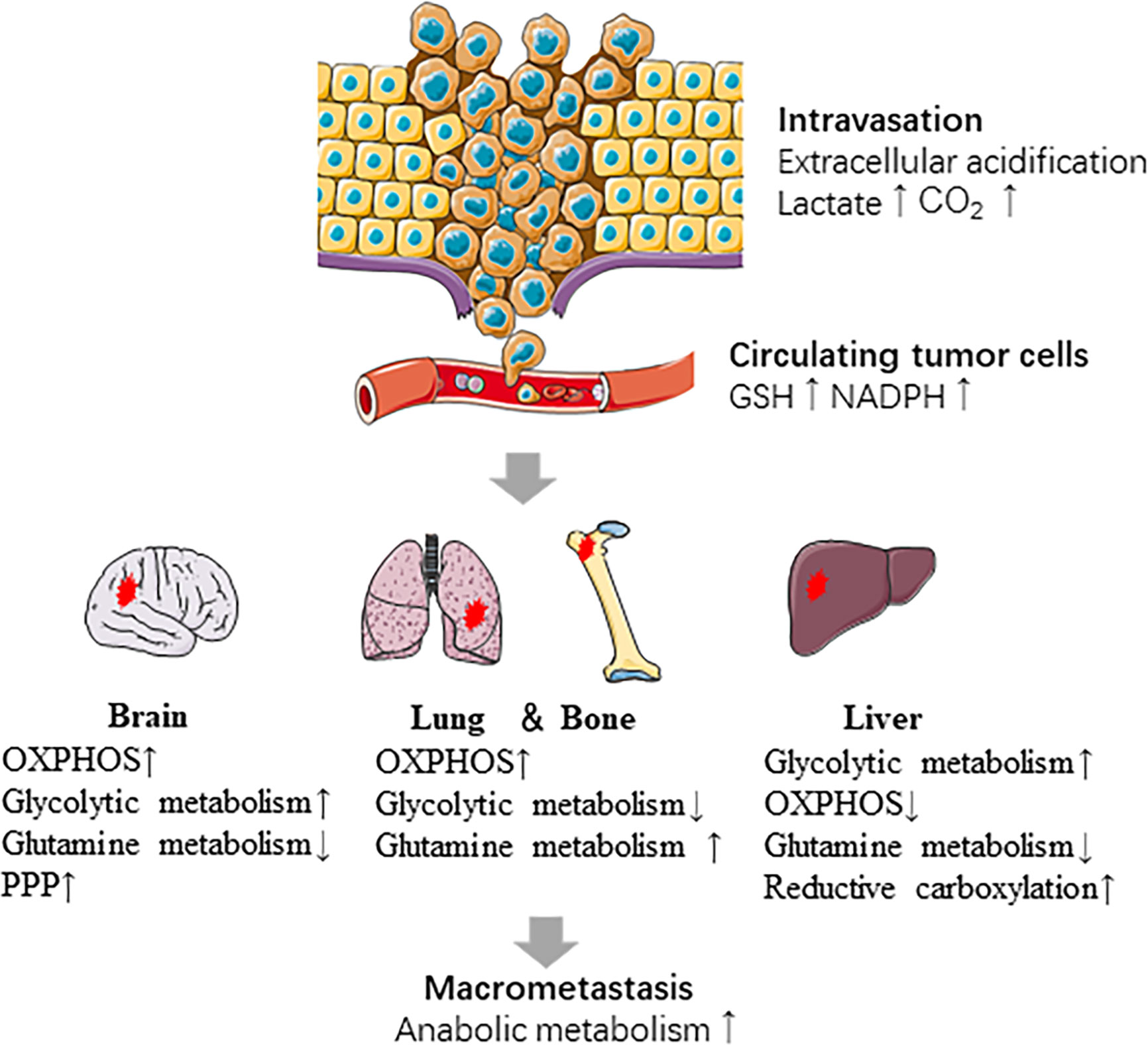
Figure 2 Metabolic reprogramming in the metastatic cascade. Metabolic reprogramming occurs at several steps of metastasis. The intravasation of cells from the primary tumor is promoted by extracellular acidification. CTCs survive in oxidative stress by producing NADPH and GSH. Cancer cells show different metabolic characteristics based on the sites which they metastasize. Last, anabolic metabolism is reactivated during macro-metastatic tumor proliferation.
Preference for metastatic sites is determined by many factors, including proximity to the primary tumor site and breast cancer subtype. Not only pro-metastatic genes in these subtypes, but also related metabolic mechanisms are closely related to the propensity of metastatic organs. Monica et al. utilized Raman spectroscopy (RS) and Multivariate Curve Resolution-Alternating Least Squares (MCR-ALS) analysis to study biochemical differences between metastasis tropisms in two TNBC cell lines and showed that bone metastasis tropism was characterized by the increase of amino acids and the decrease of mitochondrial signal, while high lipid and mitochondrial (cytochrome C and RNA) levels for lung metastasis (144). NETosis is an important neutrophil function that can promote liver metastasis of breast cancer and different pro-metastatic neutrophil populations are highly metabolically adaptable, which facilitates the formation of liver metastases (145). The products of pathologically deposited lipids can promote metastasis and nonalcoholic fatty liver disease (NAFLD) activates tumor-induced triglyceride lipolysis in juxtaposed hepatocytes, thereby promoting breast-to-liver metastasis (146). In addition to genetic tendency, metastatic cells that inhabit the brain are adaptive to crosstalk with many different brain residential cells (112, 147). The important role of Notch signaling in breast cancer brain metastasis has been recognized, and it has recently been considered to regulate metabolism (148, 149). Reactive astrocytes promote the metastatic growth of breast cancer stem-like cells by activating Notch signals in the brain and astrocyte-derived cytokines contribute to the metastatic brain specificity of breast cancer cells (150, 151). In addition to the similarity of certain metabolic signaling pathways such as the Wnt/β-catenin pathway, Heregulin-HER3-HER2 signaling and the EGFR/PI3K/Akt pathway, brain metastatic cancer cells also share certain metabolic characteristics with neuronal cells (152). Metastatic cells with neuron-like properties thrive in the brain microenvironment. For example, neurons typically catabolize gamma-aminobutyric acid (GABA) to create NADH to support biosynthetic processes and breast tumor cells with a GABAergic phenotype have a strong growth advantage in the brain by converting GABA to succinate to boost the citric acid cycle (153). Enzymes involved in lipid metabolism may also be the appropriate target to prevent the formation of brain metastases, because oncogenic lipid signaling can promote the metastasis of breast tumor cells to the brain by supporting cell survival, migration, and invasion (154).
TP53 mutations are very common in breast cancers, especially in triple-negative and HER2-positive subtypes (155). TP53 is recognized to mediate its tumor-suppressive functions by adjusting the expression of genes that promote cell cycle arrest, apoptosis, and senescence (156). Moreover, TP53 is able to suppress tumorigenesis by regulation of metabolism and reactive oxygen species (ROS) production (157). There are several mechanisms involved in TP53-mediated metabolic changes (158, 159). For example, wild-type TP53 is able to inhibit glycolysis by suppressing the expression of GLUT1, GLUT3, and GLUT4 (160, 161), and regulating the expression of enzymes involved in the glycolytic pathway, such as HK2 (162), phosphofructokinase 1 (PFK1) (163), phosphoglycerate mutase (PGM) (163), pyruvate dehydrogenase (PDH), parkin 2 (PARK2) (164), and pyruvate dehydrogenase kinase (PDK2) (165). TP53 also regulates mitochondrial respiration in cancer progression. TP53 loss results in downregulation of mitochondrial respiration and oxidative metabolism, which contribute to the Warburg effect in tumor cells, thus linking to tumor progression (166). Besides, by upregulating the cytochrome c oxidase 2 (SCO2) (167), TP53 initiates several transcriptional programs to promote the expression of genes related to mitochondrial biogenesis (168), such as apoptosis-inducing factor (AIF) (169, 170) and ferredoxin reductase (FDXR) (171). The expression of GLS-2 is also positively regulated by wild-type TP53, as such the conversion of glutamine-to-glutamate increases, which is requisite for supplement of NADPH and GSH (172, 173). In contrast, mutant TP53 has been proved to drive the glycolysis by activating the RhoA/ROCK/GLUT1 signaling cascade (164), repress the catabolic activities, such as fatty acid oxidation (FAO), by inhibiting 5′-AMP-activated protein kinase (AMPK) pathways, and enhance the anabolic processes, such as enhanced fatty acid synthesis (174).
Wild-type and mutant TP53 have contrary effects in managing the fatty acid metabolism. Wild-type TP53 hampers the shunt of the glucose carbon to anabolic pathways by binding to and inhibiting the G6PD, whereas mutant TP53 is unable to affect the G6PD activity (175). Moreover, wild-type TP53 appears to negatively control the mTOR pathway and the PPP, therefore governing fatty acid synthesis (175, 176). However, mutant TP53 enhances lipid synthesis through interacting with SREBPs (177). In particular, TP53 mutation connects with raised expression of genes involved in mevalonate pathway in human breast cancer, and most importantly, mutant TP53 upregulates these genes and activates the mevalonate pathway, which is indispensable to keep the malignant status of breast cancer (178).
Amplification of c-MYC and activation of its downstream effectors are related with high metastatic ability, endocrine resistance and poor disease outcome in breast tumors (179). The c-MYC pathway is well known to enhance the cancer cell growth and proliferation. Its role in the orchestration of metabolic pathways, which provides nutrients and other essential factors to motivate DNA replication and cell division, was recently identified. Specifically, MYC amplification mediates the glutamine-related metabolic rewiring in breast cancers, that promotes the excessive uptake of glutamine by inducing the expression of glutamine transporters and glutamine-metabolizing enzymes (180). Such MYC amplification-mediated molecular mechanism is specifically upregulated in the luminal B, HER2-positive, and TN breast cancers (15). Moreover, c-MYC activation links to TCA cycle overactivation in HER2-positive and TN breast tumors by increasing the uptake of serine, glycine, and tryptophan and the synthesis of one-carbon units (181).
Beside, c-MYC and other transcription factors, such as mTOR and HIF-1, can act synergistically to improve glycolysis and promote cancer proliferation (182, 183). c-MYC is also a direct target and a coregulator of ERα (184), it can act synergistically with ERα to induce breast cancer cell proliferation (185). Furthermore, ER regulates the glutamine metabolism by crosstalking with HER2 signaling in a way dependent on c-MYC in aromatase inhibitor-resistant breast cancer cells (186). Other studies have reported that c-MYC drives glucose metabolism in TNBC by inhibiting thioredoxin-interacting protein (TXNIP)—an inhibitor of glycolysis (21).
PI3K/AKT/mTOR pathway is an intracellular signaling pathway significant for cell cycle and metabolism involved in cancer progression (187). The activation of PI3K/Akt/mTOR pathway is able to enhance expression of genes related to glucose uptake and glycolysis through normoxic upregulation of HIF-1α (188–191). Activation of mTORC1 is also likely to be a latent mechanism driving the Warburg effect by upregulation of c-MYC (182, 183). Moreover, the PI3K/AKT/mTOR pathway can facilitate the expression of lipogenic genes in an SREBP-dependent manner (192), and mTORC1 has been regarded as a vital effector in advancing the trafficking or processing of SREBP to stimulate de novo lipogenesis (193). Activation of mTORC1 is adequate to provoke the expression of genes encoding the enzymes of both the oxidative and non-oxidative branches of the PPP, thus activating specific bioenergetic and anabolic cellular processes (194). The PI3K/AKT/mTOR pathway was recently showed to reduce oxidative stress and promote cell survival of breast epithelial cells segregated from the ECM by strengthening flux through the oxidative PPP (195)
PIK3CA mutation, which leads to increased PI3K activity, is the most common somatic mutation in breast cancer, and 36% of patients with HR+/HER2- breast cancer are PIK3CA mutated (196). It was suggested that crosstalk between the ER and PI3K/AKT/mTOR signaling pathway exists during breast cancer development (197). Estrogens stimulate PI3K/AKT/mTOR pathway to conduct the migratory and invasive features of ER tumors (198, 199). Reciprocally, mTOR signaling monitors the expression and activity of ERα (200). A recent study reported that PI3K pathway repression triggered the activity of the histone-lysine N-methyltransferase 2D (KMTD2), which leads to the activation of ER in breast cancer cells (201). Interestingly, reactivation of AKT/mTOR signaling by using small molecule PI3K antagonists activates the transport of energy-active mitochondria to the cortical cytoskeleton of cancer cells, therefore heightening tumor cell invasion (202). Although PI3K pathway inhibitors reduce cancer growth, they could accidentally increase tumor invasion by inducing reprograming of mitochondrial trafficking, OXPHOS, and promoting cell motility (203). Moreover, suppression of the mTOR-p70S6K axis is able to induce possessing of unique metabolic features, distinguished by high glucose uptake, incremental lactate production, and low mitochondrial respiration, in TNBC cells (22).
More than two-thirds of the breast cancer cases present as ERα-positive, and cancers with ERα-positive without HER2-positive is termed as luminal breast cancer (204). Luminal breast cancer appears to have a metabolic phenotype that balances the glycolysis and OXPHOS, while TNBC is more relying on OXPHOS (22). ER-positive tumors have lower levels of glycine, lactate, and glutamate (high glutamine) and lower GGR with lower levels of glutaminolysis, which suggest that ER is implicated in regulation of tumor metabolism (205). ER plays a central role in metabolic regulation through crosstalk with multiple pivotal regulators and pathways, such as TP53, c-MYC, HIF, Ras/Raf/MAPK and PI3K/AKT/mTOR pathway, enabling tumors to reprogram their metabolism to fit various kinds of environment (16). 17β-estradiol (E2) is capable of increasing the expression of insulin receptors and decreasing the lipogenic activity of lipoprotein lipase in adipose tissue by activating ERα (206). Moreover, E2 and ERα can regulate the metabolism reprogram based on glucose availability. In high glucose conditions, E2 enhances glycolysis via enhanced AKT kinase activity and suppresses TCA cycle activity, while in low extracellular glucose conditions, E2 stimulates the TCA cycle via the upregulation of PDH activity and suppresses glycolysis to satisfy the energy requirements of the tumor cell (207). Besides, a study employing the nuclear magnetic resonance spectroscopy illustrated that E2 appeared to induce glycolysis, whereas tamoxifen reduced it (208–210). Mechanically, E2 is able to transcriptionally upregulate GLUT1, thus promote glycolysis (210).
Contrary to ERα, ERβ is expressed in more than 50% of normal mammary epithelial cells, but less than 10% of tumor cells in invasive ductal carcinoma (211). In general, expression of ERβ is downregulated or lost in high grade breast tumors, but its relation to clinical outcome does not reach an agreed conclusion (212). In glucose metabolism, ERβ, similar to ERα, seems to enhance glycolysis while repress OXPHOS (213). Most importantly, ERβ is suggested to play a key role in regulating the metabolism of BCSCs, given several glycolysis-related pathways are upregulated in ERβ-activated mammospheres (214).
HER2-positive breast tumors generally exhibit a glycolytic phenotype (215, 216) and display the increased uptake levels of glutamine, glycine, creatinine, and succinate while a reduction in alanine levels (205). Moreover, the expression of FASN, carnitine perilipin-1 (PLIN1), and palmitoyltransferase-1A (CPT1A) are elevated in HER2-positive breast cancers (20). HER2 is involved in multiple signaling pathways that promote glucose utilization (216), regulate LDHA (40) and 6-phosphofructo-2-kinase (PFKFB3) expression levels (217), and induce lactate accumulation in tumors (218). Additionally, HER2 can be translocated to the mitochondria by the intercourse with mitochondrial heat shock protein-70 (mtHSP70), which negatively controls oxygen consumption and thus enhancing glycolysis (219). Inhibition of HER2 pathways by a dual novel EGFR/HER2 inhibitor, KU004, significantly inhibits the Warburg effect by downregulating HK2, thus decreasing cancer cell proliferation (220). Overactivated HER2 signaling results in increased HIF-1α and VEGF expression, which in turn activate the downstream kinase FKBP-rapamycin-associated protein (FRAP), therefore contributing to tumor progression by mediating angiogenesis and metabolic adaptation (221).
BRCA1-mutated breast tumors are usually phenotyped as aggressive, high-grade, aneuploidy tumors (222, 223), and with a worse prognosis (224). Loss of BRCA1 function caused by BRCA1 mutation results in the production of hydrogen peroxide in both epithelial breast tumor cells and adjacent stromal fibroblasts, which is able to promote the onset of a reactive glycolytic stroma, suggesting the metabolic phenotype of stromal cells in the TME may also be affected by BRCA1 mutation in tumor cells (225). Moreover, the BRCA1 loss mutation, like oncogene activation (RAS, NF-κB, TGF-β), in cancer cells will drive the initiation of metabolic symbiosis phenotype between tumor cells and fibroblasts in both primary and metastatic cancers (226).
PGC-1α is a transcriptional co-activator that actively participates in gene regulation of energy metabolism. Elevated expression of PGC-1α in breast cancer is well associated with the formation of distant metastases. Notably, breast cancer cells with higher levels of PGC-1α may preferentially metastasize to some specific tissues, such as lung and bone (139). Silencing of PGC-1α appears to suspend cancer cell invasive potential and attenuate metastasis (137). The invasive cancer cells particularly do favor mitochondrial respiration with augmented production of ATP. As such, the circulating and metastatic cancer cells upregulate the PGC-1α to facilitate oxygen consumption rate oxidative phosphorylation, and mitochondrial biogenesis to uphold metastasis (137).
RB1 is a tumor suppressor that is commonly disrupted in many human tumors, including breast cancer (227). RB1 deficiency is connected with cancer invasion and metastasis (228, 229). It is evaluated that RB1 and TP53 are lost together in 28–40% of human TNBCs, and RB/P53-double mutant mouse breast tumor cells exhibit more mesenchymal phenotypes than only P53-deficient cells (230, 231). RB1 loss links to increased mitochondrial OXPHOS, which links to enhanced anabolic metabolism and augmented cancer cell stemness and metastatic spread (232). Additionally, RB1 deficiency is able to enhance tumor metastasis by increasing OXPHOS to generate more ATP fueling for tumor invasion and cooperating oncogenic alterations to uphold EMT and metastasis (232).
AMPK is a universally expressed metabolic sensor, which can be phosphorylated and activated under some stress conditions, such as energy deprivation. Phosphorylated AMPK activates multiple downstream elements to regulate adaptive changes and maintain metabolic homeostasis, including glucose, lipid or protein metabolism. Recently, the latent roles of AMPK signaling in tumorigenesis and progression have been gradually revealed (233). Activated AMPK signaling regulates protein and lipid synthesis by inhibiting mTORC1 through activation of tuberous sclerosis complex 2 (TSC2) and phosphorylation of raptor (234–237). The chief activator of AMPK is the serine-threonine tumor suppressor kinase LKB1, which contributes to phosphorylation of AMPK to activate energy sensors (235, 238). As long as LKB1-AMPK signaling is activated, the regulation of the metabolic branch of mTOR signaling cannot be impaired in spite of the abnormal of PI3K/AKT or receptor tyrosine kinase signaling (237). LKB1 inactivation has recently been reported to drive tumor progression by cooperating with certain activating oncogene mutations in various models of cancer (239–242). Lysine demethylase 5B (also known as KDM5B) is upregulated in breast tumors and play an important role in lipid metabolic reprogramming (243). A recent study clearly demonstrated the knockdown of KDM5B reversed the EMT process to inhibit breast tumor cell migration by activating AMPK signaling-mediated lipid metabolism (244).
Hypoxia represents an important characteristic in the TME arising as a mismatch between cellular oxygen consumption and supply (245). About 25%–40% of invasive breast tumors display hypoxic situations (246). Hypoxia is able to regulate glycolysis, glycogen synthesis, lipid metabolism and oxidative phosphorylation, thus playing a vital role in tumor cell survival and growth during all stages of metastasis (247).
Hypoxia-inducible factors, including HIF-1α and HIF-2α, are main regulators in adaptation to hypoxia and nutrient deprivation during tumor progression (141). The activated HIFs is able to induce the expression of various gene products, such as glycolysis- and EMT program-associated molecules (CXCR4, SNAIL and TWIST), the induced pluripotency-associated transcription factors (OCT-3/4, NANOG, and SOX2), angiogenic factors (VEGF) and microRNAs, which are vital to self-renewal, survival, invasion, metastasis, angiogenesis, metabolic reprogram, and treatment resistance of cancer cells. Furthermore, elevated HIF-1α level is a predictive marker of early relapse and metastasis, and correlated with bad clinical outcome in human breast cancer (248–250). Inhibition of HIF-1 activity has a significant inhibitory effect on primary tumor proliferation and metastasis to lymph nodes and lungs in mice by orthotopic transplantation of TNBC (251, 252). Notably, HIF-1 mediates adaptive metabolic responses to hypoxia by enhancing glycolytic pathway, serine synthesis and one-carbon metabolism to promote mitochondrial antioxidant production (NADPH and GSH), and inhibiting the TCA cycle so as to diminish mitochondrial ROS production (247). HIF-1α has recently been shown to increase the expression levels of pro-collagen prolyl (P4HA1 and P4HA2) and lysyl (PLOD1 and PLOD2) hydroxylases in both tumor and stromal cells, thereby enhancing cancer cell alignment along collagen fibers, thereby promoting invasion and metastasis to lymph nodes and lungs (253–255). Hypoxia raises the proportion of BCSCs in a HIF-1α–dependent manner (256, 257), which will contribute to cancer metastasis. A recent study demonstrated that HIF-1α appeared to dynamically regulate glucose metabolism based on oxygen availability to prevent the risk of continuous incremental ROS production to keep redox homeostasis. This HIF-1α-induced effect is vital for induction of the BCSC phenotype in breast cancer when in response to hypoxia or cytotoxic chemotherapy (33). PDK1, a HIF-1α target that antagonizes the function of PDH, a main rate-limiting enzyme for pyruvate converting to acetyl-coA and entering the TCA cycle, has been reported to be a critical regulator of breast cancer metabolism and metastasis (138). Liver metastatic breast cancer cells are recognized to depend on the HIF-1/PDK1 axis for their metabolic reprogramming to accelerate their efficient colonization and proliferation in the liver (138). Some metabolic enzymes, such as succinate dehydrogenase (SDH), fumarate hydratase (FH), IDH and pyruvate kinase 2 (PKM2) are likely to activate HIF-1 pathway by stabilizing HIF-1α, therefore enhancing cancer metastasis (258).
Tumor cells can only survive within a narrow window of ROS levels. Inhibition of ROS clearance is a therapeutic approach (259), and on the contrary, prohibition of ROS enhances tumor metastasis (260). Among the detachment from ECM during the procedure of metastasis, cancer cells can undergo alterations in metabolic pathways harmful to survival, such as moderated glucose uptake, PPP flux, and ATP levels while promoting the producing of ROS (261). Antioxidant enzymes support survival of breast tumor cells deprived of ECM, implying that eliminating antioxidant enzyme activity in ECM-detached tumor cells may be an efficacious strategy to stop metastatic spreading (262). Additionally, the untransformed breast epithelial cells upregulate PDK4 to inhibit PDH and attenuate the flux of glycolytic carbon into mitochondrial oxidation, consequently suppressing anoikis (the absence of the home environment) upon detachment from ECM (261). By stimulating PDH in cancer cells to normalize glucose metabolism, it can restore their sensitivity to anoikis and weaken their metastatic potential, suggesting that PDKs are potential targets for anti-metastasis therapy (261). Another way to counter increased ROS production in breast cancer cells is to induce the expression of catalases, such as manganese superoxide dismutase (MnSOD). The expression of MnSOD is elevated in metastatic breast cancer, and its overexpression is correlated with histologic tumor grades (263). Isaac et al. has suggested that combined inhibition of endogenous antioxidant GSH and thioredoxin antioxidant pathways can produce a synergistic anti-cancer effect both in vitro and in vivo (264).
Lactate, the final product of glycolysis, is released from cells together with H+ ions by means of monocarboxylate transporters and hydrogen ion pumps, and the excess carbon dioxide produced in the process of mitochondrial metabolism diffuses into the extracellular space and is then converted into H + and HCO3- by carbonic anhydrase (265). In situations of metabolic stress, such as nutrient deprivation and hypoxia, these reactions are strengthened, leading to extracellular acidification and enhancing the proteolytic activity of MMPs. Afterwards, the ECM is remodeled, which facilitates tumor invasion (265, 266). It has been reported that extracellular lactate also increase tumor invasion and metastasis by facilitating the fibroblast expression of hyaluronan and CD44 (267). Besides, increased extracellular lactate induces tumor-associated stromal cells to secrete VEGF, thus reinforcing angiogenesis (268). The augment in extracellular lactate has also been reported to provide an immune-conducive environment for tumor cells by reducing the activation and function of dendrites and T cells (269, 270).
Cancer-asscociated fibroblasts (CAFs), the paramount stromal cells in breast tumor microenvironment, contribute to tumor progression through many mechanisms, such as releasing of assorted secretory proteins (e.g. TGF-β, IGF, and IL6), direct interplaying with tumor cells, regulating immune-response, ECM remodeling, and inducing cancer metabolic reprogramming (271). Breast cancer cells MCF-7 exhibited increased aerobic glycolysis when co-cultured with adjacent fibroblasts. Mechanically, the lactate produced by the CAFs can be used by cancer cells, thus enhancing aerobic glycolysis, which is called the “reverse Warburg effect” (28, 272). Similarly, metabolomic analysis showed that CAFs also produce glutamine and other metabolites that can be utilized by tumor cells (273). Subsequent researches demonstrated that co-culture with MCF-7 and CAFs resulted in promoted glutamine catabolism and inhibited glutamine synthesis in cancer cells, thereby promoting cancer cell growth and progression (274).
The “cancer-associated adipocytes (CAAs)” are generated by the transformation of tumor adjacent adipocytes (275). It has been reported that tumor-surrounding adipocytes exhibit an distinct phenotype comparing to normal adipocytes, which is characterized by upregulated beige/brown adipose markers and increased catabolism and the release of metabolites, including lactate, free fatty acids, pyruvate, and ketone bodies. Importantly, the tumor-adipocyte interaction can reprogram energy metabolism and foster tumor progression (276).
Accumulation of lipids is found in breast tumor cells when co-cultured with adipocytes (277). Tumor cells can switch from glycolysis to lipid-dependent energy production and also store excess lipids, which provides energy to support their expansion and metastasis (278). The ketone bodies produced and released by glycolytic fat cells are the ideal fuel for ATP production and they can be burned more efficiently than other mitochondrial substrates, even during hypoxia, potentially allowing the tumor grow when without adequate blood supply (279). It is worth noting that the co-existence of adipocytes and cancer cells enhances both ketogenesis in adipocytes and ketolytic activity in breast cancer cells (276). In addition, β-hydroxybutyrate secreted from adipocytes is able to induce several tumor-promoting genes in breast cells, and facilitate breast tumor cells malignant growth in vitro (280). Moreover, elevated ketone-specific gene expression is related with worse outcomes in breast cancer patients (281).
The local immune surveillance environment is increasingly recognized as a significant factor inhibiting tumor metastasis. Apart from fundamental competition for nutrients required by cancer cells and immune cells in TME, metabolic pathways change in tumor cells may influence tumor-infiltrating immune cells, and different immune cell subgroups in TME have specific metabolic characteristics (282). The limitation of glucose and amino acids within the TME can significantly affect the T cell response and the determinants of metabolic dysfunction and associated T cell exhaustion within the TME are also being explored. Studies have shown that cancer itself can cause effector T (Teff) cell metabolism disorders, and there is a negative correlation between the degree of glycolytic activity of cancer cells and the antitumor function of infiltrating T cells (282). A study has confirmed that the expression of glycolysis-related genes in tumor samples from patients with melanoma and non-small-cell lung cancer is negatively correlated with T cell infiltration, and that tumor glycolysis is related to the efficacy of adoptive T cell therapy (ACT), suggesting that the glycolytic pathway may be a candidate target for combined therapeutic intervention (283). Inhibition of cholesterol esterification in T cells by genetic ablation or pharmacological inhibition of ACAT1 (a key cholesterol esterase) can lead to potentiated effector function and enhanced proliferation of CD8(+) T cells by increasing plasma membrane cholesterol levels, which causes enhanced T-cell receptor clustering and signaling as well as more efficient formation of the immunological synapse, thereby controlling the growth and metastasis of mouse melanoma (284). However, such studies are still lacking in breast cancer. Tumor-derived myeloid-derived suppressor cells (MDSCs) are critical tumor immunosuppression components. Glycolysis restriction limited the development of MDSCs by inhibiting tumor expression of granulocyte colony-stimulating factor (G-CSF) and granulocyte macrophage colony-stimulating factor (GM-CSF), therefore enhanced T cell immunity, reduced tumor growth and metastasis, and prolonged survival in two TNBC mouse models (285). Interestingly, hypoxia through HIF-1α significantly changes the function of MDSC in TME and shifts its differentiation direction to tumor-associated macrophages (TAMs) (286). TAMs are well-known parts of breast cancer microenvironment and most TAMs within TME are closely related to the M2-like phenotype, which participate in almost all metastatic processes, including local invasion, blood vessel intravasation, extravasation at distant sites and metastatic cell growth (287, 288).The hypoxic areas in tumors are related to the accumulation of macrophages, which assist tumor progression by producing angiogenic factors, mitogenic factors and cytokines related to tumor metastasis (289–291). In addition, hypoxia can promote the differentiation and functional capabilities of immunosuppressive macrophages (292). Blockade of Eotaxin/Oncostatin M not only prevented hypoxic breast tumor cells from recruiting and polarizing macrophages towards the M2-like phenotype and hindered cancer progression in 4T1 breast cancer model but also improved the efficacy of antiangiogenic Bevacizumab, suggesting these two cytokines as novel targets for devising effective anticancer therapy (293). Lactic acid production by tumor cells, as a byproduct of aerobic or anaerobic glycolysis, has also been shown to play a vital role in the M2-like polarization of TAMs, which is mediated by HIF- 1α (294).
The metabolites produced by cancer cells may hinder the antitumor immune response by affecting different tumor infiltrating immune cells (295). Cholesterol metabolites, oxysterols, which act as endogenous regulators of lipid metabolism through the interaction with the nuclear Liver X Receptors-(LXR)α and LXRβ, aid tumor progression by inhibiting antitumor immune responses, and by recruiting proangiogenic and immunosuppressive neutrophils. A recent study showed that in the 4T1 breast cancer model the enzymatic depletion of oxysterols in primary tumors decreases the formation of lung metastases by regulating the levels of immune cells infiltrating the metastatic TME, and tumor-associated neutrophils are the main driving force of local immunosuppression (296). Another work also proved that by recruiting immunosuppressive neutrophils in the metastatic niche, oxysterol 27-HC played a role in promoting metastasis in breast cancer models (297).
Breast cancer patients who have not yet found metastasis are at high risk of metastasis, and those metastatic breast cancers are not curable due to lack of effective treatments. Early intervention in the early stage of distant metastasis, during the period of colonization and growth will be more beneficial to the survival of the patient. There are many promising drugs targeting altered metabolism pathways undergoing disparate stages of preclinical studies and clinical trials (Table 2). However, there is currently no clear conclusion of the clinical benefit of metabolic interfering drugs in the treatment of breast cancer. A Phase II clinical trial involving 164 patients was recently showed that in patients with HER2-metastatic breast cancer, addition of indoximod, the Indoleamine 2,3-dioxygenase 1 (IDO1) pathway inhibitor, to taxane did not improve PFS compared with taxane alone (300). The use of glucose metabolic inhibitors such as 2-deoxy-D-glucose (2-DG) and metformin in combination with chemotherapy has shown encouraging results in combating chemotherapy resistance (301). One patient with medullary breast cancer metastatic to lung and lymph nodes underwent extensive pretreatment (8 previous systemic treatment options) was reported to have a confirmed partial response (PR) with a duration of 65 days when treated with 45 mg/kg 2-DG every other week (298). Dichloroacetate (DCA) can enhance metformin-induced oxidative damage with simultaneous reduce of metformin promoted lactate production through PDK1 inhibition, suggesting the innovative combinations, such as metformin and DCA, will be promising in expanding breast cancer therapies (302). Nevertheless, due to the limited sample and the lack of evidence for benefit, further researches are needed.
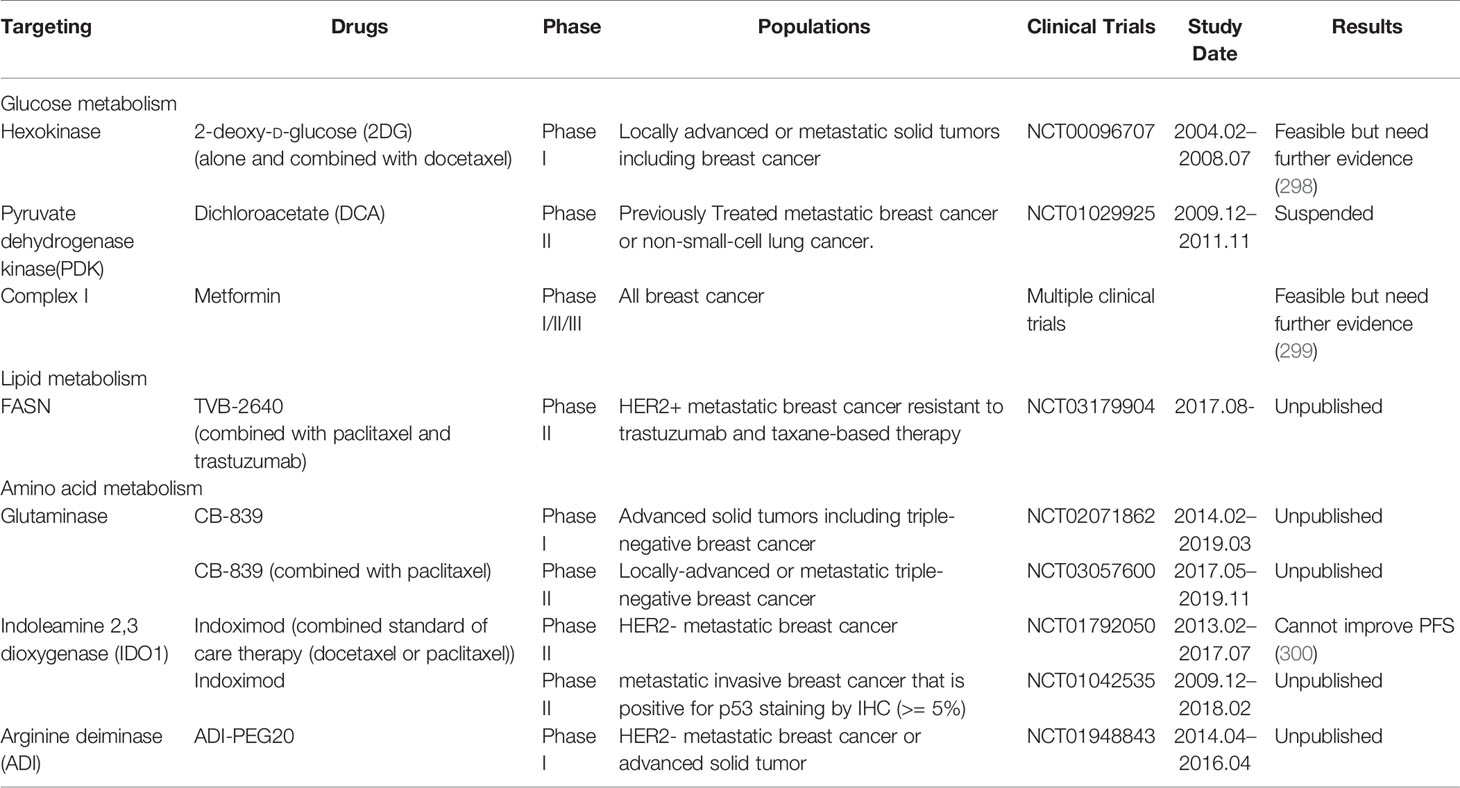
Table 2 Current metabolic interventions in metastatic breast cancer.
Metabolic inhibitors combined with checkpoint inhibitors holds promise to enhance the efficacy of immunotherapy and the relationship of tumor-intrinsic metabolism and successful immunotherapy is being explored. Tumor-imposed metabolic restrictions can mediate T cell hyporesponsiveness during cancer. Checkpoint blockade antibodies against CTLA-4, PD-1, and PD-L1, can restore glucose in tumor microenvironment, permitting T cell glycolysis and IFN-γ production, and blocking PD-L1 directly on tumors dampens glycolysis by inhibiting mTOR activity and decreasing expression of glycolysis enzymes (270). Because breast cancer immunotherapy is in the ascendant, understanding the metabolic dependence between infiltrating immune cells and cancer is an important direction for future research.
For ER-positive breast cancer patients, endocrine therapy is very beneficial, but some patients will develop endocrine therapy resistance. Whether endocrine therapy combined with metabolic therapy will achieve better results still needs a lot of preclinical studies and clinical trials to verify. It has been reported that trastuzumab resistant cells exhibit enhanced glycolysis phenotype, and glycolytic restraint is able to sensitize trastuzumab resistant HER2+ breast cancers to trastuzumab treatment (303). The TN/basal-like breast cancer lacks the therapeutic targets, and chemotherapy is currently the main treatment strategies. Based on the TNBC unique metabolic phenotype, there are many existing researches focus on the metabolic interference in chemotherapy resistance models and spontaneously metastatic preclinical models (304, 305). What is more, the metabolic characteristics of tumor cells and their microenvironment in different metastatic sites are different, therefore, the corresponding targeting treatment plans can also be considered in the future (306).
Although anti-cancer therapy targeting metabolism has achieved some gratifying results, it is still currently believed that this field has the following shortcomings for possible future breakthroughs: 1) The side effects of such drugs limit their clinical effects as the optimal dose window is hard to be determined; 2) Due to the extremely complex signaling pathways in the regulation of normal cellular biology, inhibition of a specific signaling pathway will definitely have feedback activation or upregulation of other alternative signaling pathways, therefore causing ultimately treatment failure; 3) To specifically target related mutations involved in metabolic pathways is challenging. 4) Accurate screening of the beneficiaries is the key to improve the drug effect, and is an urgent problem to be solved in the future.
Metabolic programming supports several steps of successful metastasis in breast cancer. Breast cancer cells exhibit different metabolic phenotypes in different metastasis sites. Both intrinsic factors, properties arising in the malignant cells, such as MYC amplification, PIK3CA, and TP53 mutations, and extrinsic factors, metabolic stresses imposed by the microenvironment, such as hypoxia, oxidative stress, acidosis, contribute to different metabolic programming phenotypes in metastatic breast cancer. More importantly, interfering with tumor metabolism to control tumor progression is a very promising approach in cancer treatment, although it is full of challenges. More researches are required to further discover the related genes and molecular mechanisms involved in metabolism reprograming during cancer progression, so that they can be used for targeting therapy in clinical practice in the future. We also look forward to further advances in approaches to judge and quantify metabolic phenotypes in human breast cancers in vivo, including metabolomics, metabolic imaging and isotope tracing studies, so that clinical oncologists will develop treatment strategies by matching the treatment to the patient-specific tumor metabolic characteristics.
LW wrote the first draft of the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by the Zhejiang Provincial Natural Science Foundation of China (grant no. Y19H160283).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin (2018) 68(6):394–424. doi: 10.3322/caac.21492
2. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin (2020) 70(1):7–30. doi: 10.3322/caac.21590
3. Chaffer CL, Weinberg RA. A perspective on cancer cell metastasis. Science (2011) 331(6024):1559–64. doi: 10.1126/science.1203543
4. Wu Q, Li J, Zhu S, Wu J, Chen C, Liu Q, et al. Breast cancer subtypes predict the preferential site of distant metastases: a SEER based study. Oncotarget (2017) 8(17):27990–6. doi: 10.18632/oncotarget.15856
5. Wishart DS. Is Cancer a Genetic Disease or a Metabolic Disease? EBioMedicine (2015) 2(6):478–9. doi: 10.1016/j.ebiom.2015.05.022
6. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144(5):646–74. doi: 10.1016/j.cell.2011.02.013
7. Pavlova NN, Thompson CB. The Emerging Hallmarks of Cancer Metabolism. Cell Metab (2016) 23(1):27–47. doi: 10.1016/j.cmet.2015.12.006
8. Long JP, Li XN, Zhang F. Targeting metabolism in breast cancer: How far we can go? World J Clin Oncol (2016) 7(1):122–30. doi: 10.5306/wjco.v7.i1.122
9. Alles MC, Gardiner-Garden M, Nott DJ, Wang Y, Foekens JA, Sutherland RL, et al. Meta-analysis and gene set enrichment relative to er status reveal elevated activity of MYC and E2F in the “basal” breast cancer subgroup. PloS One (2009) 4(3):e4710. doi: 10.1371/journal.pone.0004710
10. Antalis CJ, Arnold T, Rasool T, Lee B, Buhman KK, Siddiqui RA. High ACAT1 expression in estrogen receptor negative basal-like breast cancer cells is associated with LDL-induced proliferation. Breast Cancer Res Treat (2010) 122(3):661–70. doi: 10.1007/s10549-009-0594-8
11. Hilvo M, Denkert C, Lehtinen L, Muller B, Brockmoller S, Seppanen-Laakso T, et al. Novel theranostic opportunities offered by characterization of altered membrane lipid metabolism in breast cancer progression. Cancer Res (2011) 71(9):3236–45. doi: 10.1158/0008-5472.CAN-10-3894
12. Nelson ER, Wardell SE, Jasper JS, Park S, Suchindran S, Howe MK, et al. 27-Hydroxycholesterol links hypercholesterolemia and breast cancer pathophysiology. Science (2013) 342(6162):1094–8. doi: 10.1126/science.1241908
13. Brglez V, Pucer A, Pungercar J, Lambeau G, Petan T. Secreted phospholipases A(2)are differentially expressed and epigenetically silenced in human breast cancer cells. Biochem Biophys Res Commun (2014) 445(1):230–5. doi: 10.1016/j.bbrc.2014.01.182
14. Kim SK, Jung WH, Koo JS. Differential expression of enzymes associated with serine/glycine metabolism in different breast cancer subtypes. PloS One (2014) 9(6):e101004. doi: 10.1371/journal.pone.0101004
15. Craze ML, Cheung H, Jewa N, Coimbra NDM, Soria D, El-Ansari R, et al. MYC regulation of glutamine-proline regulatory axis is key in luminal B breast cancer. Br J Cancer (2018) 118(2):258–65. doi: 10.1038/bjc.2017.387
16. Kulkoyluoglu-Cotul E, Arca A, Madak-Erdogan Z. Crosstalk between Estrogen Signaling and Breast Cancer Metabolism. Trends Endocrinol Metab (2019) 30(1):25–38. doi: 10.1016/j.tem.2018.10.006
17. Choi J, Kim ES, Koo JS. Expression of Pentose Phosphate Pathway-Related Proteins in Breast Cancer. Dis Markers (2018) 2018:9369358. doi: 10.1155/2018/9369358
18. Choi J, Jung WH, Koo JS. Metabolism-related proteins are differentially expressed according to the molecular subtype of invasive breast cancer defined by surrogate immunohistochemistry. Pathobiology (2013) 80(1):41–52. doi: 10.1159/000339513
19. Kim S, Kim DH, Jung WH, Koo JS. Expression of glutamine metabolism-related proteins according to molecular subtype of breast cancer. Endocr Relat Cancer (2013) 20(3):339–48. doi: 10.1530/ERC-12-0398
20. Kim S, Lee Y, Koo JS. Differential expression of lipid metabolism-related proteins in different breast cancer subtypes. PloS One (2015) 10(3):e0119473. doi: 10.1371/journal.pone.0119473
21. Shen L, O’Shea JM, Kaadige MR, Cunha S, Wilde BR, Cohen AL, et al. Metabolic reprogramming in triple-negative breast cancer through Myc suppression of TXNIP. Proc Natl Acad Sci USA (2015) 112(17):5425–30. doi: 10.1073/pnas.1501555112
22. Pelicano H, Zhang W, Liu J, Hammoudi N, Dai J, Xu RH, et al. Mitochondrial dysfunction in some triple-negative breast cancer cell lines: role of mTOR pathway and therapeutic potential. Breast Cancer Res (2014) 16(5):434. doi: 10.1186/s13058-014-0434-6
23. Dias AS, Almeida CR, Helguero LA, Duarte IF. Metabolic crosstalk in the breast cancer microenvironment. Eur J Cancer (2019) 121:154–71. doi: 10.1016/j.ejca.2019.09.002
24. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science (2009) 324(5930):1029–33. doi: 10.1126/science.1160809
25. Sancho P, Barneda D, Heeschen C. Hallmarks of cancer stem cell metabolism. Br J Cancer (2016) 114(12):1305–12. doi: 10.1038/bjc.2016.152
26. Warburg O. On the origin of cancer cells. Science (1956) 123(3191):309–14. doi: 10.1126/science.123.3191.309
27. Wu H, Ying M, Hu X. Lactic acidosis switches cancer cells from aerobic glycolysis back to dominant oxidative phosphorylation. Oncotarget (2016) 7(26):40621–9. doi: 10.18632/oncotarget.9746
28. Pavlides S, Whitaker-Menezes D, Castello-Cros R, Flomenberg N, Witkiewicz AK, Frank PG, et al. The reverse Warburg effect: aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle (2009) 8(23):3984–4001. doi: 10.4161/cc.8.23.10238
29. Fu Y, Liu S, Yin S, Niu W, Xiong W, Tan M, et al. The reverse Warburg effect is likely to be an Achilles’ heel of cancer that can be exploited for cancer therapy. Oncotarget (2017) 8(34):57813–25. doi: 10.18632/oncotarget.18175
30. Wilde L, Roche M, Domingo-Vidal M, Tanson K, Philp N, Curry J, et al. Metabolic coupling and the Reverse Warburg Effect in cancer: Implications for novel biomarker and anticancer agent development. Semin Oncol (2017) 44(3):198–203. doi: 10.1053/j.seminoncol.2017.10.004
31. Choi J, Kim DH, Jung WH, Koo JS. Metabolic interaction between cancer cells and stromal cells according to breast cancer molecular subtype. Breast Cancer Res (2013) 15(5):R78. doi: 10.1186/bcr3472
32. Vaupel P, Hockel M, Mayer A. Detection and characterization of tumor hypoxia using pO2 histography. Antioxid Redox Signal (2007) 9(8):1221–35. doi: 10.1089/ars.2007.1628
33. Semenza GL. Hypoxia-inducible factors: coupling glucose metabolism and redox regulation with induction of the breast cancer stem cell phenotype. EMBO J (2017) 36(3):252–9. doi: 10.15252/embj.201695204
34. Rogers S, Docherty SE, Slavin JL, Henderson MA, Best JD. Differential expression of GLUT12 in breast cancer and normal breast tissue. Cancer Lett (2003) 193(2):225–33. doi: 10.1016/s0304-3835(03)00010-7
35. Godoy A, Ulloa V, Rodriguez F, Reinicke K, Yanez AJ, Garcia Mde L, et al. Differential subcellular distribution of glucose transporters GLUT1-6 and GLUT9 in human cancer: ultrastructural localization of GLUT1 and GLUT5 in breast tumor tissues. J Cell Physiol (2006) 207(3):614–27. doi: 10.1002/jcp.20606
36. Krzeslak A, Wojcik-Krowiranda K, Forma E, Jozwiak P, Romanowicz H, Bienkiewicz A, et al. Expression of GLUT1 and GLUT3 glucose transporters in endometrial and breast cancers. Pathol Oncol Res (2012) 18(3):721–8. doi: 10.1007/s12253-012-9500-5
37. Garrido P, Moran J, Alonso A, Gonzalez S, Gonzalez C. 17beta-estradiol activates glucose uptake via GLUT4 translocation and PI3K/Akt signaling pathway in MCF-7 cells. Endocrinology (2013) 154(6):1979–89. doi: 10.1210/en.2012-1558
38. Young CD, Lewis AS, Rudolph MC, Ruehle MD, Jackman MR, Yun UJ, et al. Modulation of glucose transporter 1 (GLUT1) expression levels alters mouse mammary tumor cell growth in vitro and in vivo. PloS One (2011) 6(8):e23205. doi: 10.1371/journal.pone.0023205
39. Hennipman A, van Oirschot BA, Smits J, Rijksen G, Staal GE. Glycolytic enzyme activities in breast cancer metastases. Tumour Biol (1988) 9(5):241–8. doi: 10.1159/000217568
40. Zhao YH, Zhou M, Liu H, Ding Y, Khong HT, Yu D, et al. Upregulation of lactate dehydrogenase A by ErbB2 through heat shock factor 1 promotes breast cancer cell glycolysis and growth. Oncogene (2009) 28(42):3689–701. doi: 10.1038/onc.2009.229
41. Patra KC, Hay N. The pentose phosphate pathway and cancer. Trends Biochem Sci (2014) 39(8):347–54. doi: 10.1016/j.tibs.2014.06.005
42. Benito A, Polat IH, Noe V, Ciudad CJ, Marin S, Cascante M. Glucose-6-phosphate dehydrogenase and transketolase modulate breast cancer cell metabolic reprogramming and correlate with poor patient outcome. Oncotarget (2017) 8(63):106693–706. doi: 10.18632/oncotarget.21601
43. DeBerardinis RJ, Cheng T. Q’s next: the diverse functions of glutamine in metabolism, cell biology and cancer. Oncogene (2010) 29(3):313–24. doi: 10.1038/onc.2009.358
44. Li T, Le A. Glutamine Metabolism in Cancer. Adv Exp Med Biol (2018) 1063:13–32. doi: 10.1007/978-3-319-77736-8_2
45. Wise DR, Thompson CB. Glutamine addiction: a new therapeutic target in cancer. Trends Biochem Sci (2010) 35(8):427–33. doi: 10.1016/j.tibs.2010.05.003
46. Eberhardy SR, Farnham PJ. c-Myc mediates activation of the cad promoter via a post-RNA polymerase II recruitment mechanism. J Biol Chem (2001) 276(51):48562–71. doi: 10.1074/jbc.M109014200
47. Perez-Escuredo J, Dadhich RK, Dhup S, Cacace A, Van Hee VF, De Saedeleer CJ, et al. Lactate promotes glutamine uptake and metabolism in oxidative cancer cells. Cell Cycle (2016) 15(1):72–83. doi: 10.1080/15384101.2015.1120930
48. Budczies J, Pfitzner BM, Gyorffy B, Winzer KJ, Radke C, Dietel M, et al. Glutamate enrichment as new diagnostic opportunity in breast cancer. Int J Cancer (2015) 136(7):1619–28. doi: 10.1002/ijc.29152
49. Tibbetts AS, Appling DR. Compartmentalization of Mammalian folate-mediated one-carbon metabolism. Annu Rev Nutr (2010) 30:57–81. doi: 10.1146/annurev.nutr.012809.104810
50. Newman AC, Maddocks ODK. One-carbon metabolism in cancer. Br J Cancer (2017) 116(12):1499–504. doi: 10.1038/bjc.2017.118
51. Williams KT, Schalinske KL. New insights into the regulation of methyl group and homocysteine metabolism. J Nutr (2007) 137(2):311–4. doi: 10.1093/jn/137.2.311
52. Zeng J, Wang K, Ye F, Lei L, Zhou Y, Chen J, et al. Folate intake and the risk of breast cancer: an up-to-date meta-analysis of prospective studies. Eur J Clin Nutr (2019) 73(12):1657–60. doi: 10.1038/s41430-019-0394-0
53. Mondanelli G, Ugel S, Grohmann U, Bronte V. The immune regulation in cancer by the amino acid metabolizing enzymes ARG and IDO. Curr Opin Pharmacol (2017) 35:30–9. doi: 10.1016/j.coph.2017.05.002
54. Cavdar Z, Onvural B, Guner G. Arginase in patients with breast cancer. Clin Chim Acta (2003) 338(1-2):171–2. doi: 10.1016/j.cccn.2003.09.002
55. Blucher C, Stadler SC. Obesity and Breast Cancer: Current Insights on the Role of Fatty Acids and Lipid Metabolism in Promoting Breast Cancer Growth and Progression. Front Endocrinol (Lausanne) (2017) 8:293:293. doi: 10.3389/fendo.2017.00293
56. Beloribi-Djefaflia S, Vasseur S, Guillaumond F. Lipid metabolic reprogramming in cancer cells. Oncogenesis (2016) 5:e189. doi: 10.1038/oncsis.2015.49
57. Santos CR, Schulze A. Lipid metabolism in cancer. FEBS J (2012) 279(15):2610–23. doi: 10.1111/j.1742-4658.2012.08644.x
58. Mashima T, Seimiya H, Tsuruo T. De novo fatty-acid synthesis and related pathways as molecular targets for cancer therapy. Br J Cancer (2009) 100(9):1369–72. doi: 10.1038/sj.bjc.6605007
59. Wang YY, Kuhajda FP, Li J, Finch TT, Cheng P, Koh C, et al. Fatty acid synthase as a tumor marker: its extracellular expression in human breast cancer. J Exp Ther Oncol (2004) 4(2):101–10.
60. Vazquez-Martin A, Ortega-Delgado FJ, Fernandez-Real JM, Menendez JA. The tyrosine kinase receptor HER2 (erbB-2): from oncogenesis to adipogenesis. J Cell Biochem (2008) 105(5):1147–52. doi: 10.1002/jcb.21917
61. Xiong S, Chirala SS, Wakil SJ. Sterol regulation of human fatty acid synthase promoter I requires nuclear factor-Y- and Sp-1-binding sites. Proc Natl Acad Sci USA (2000) 97(8):3948–53. doi: 10.1073/pnas.040574197
62. Donnelly C, Olsen AM, Lewis LD, Eisenberg BL, Eastman A, Kinlaw WB. Conjugated linoleic acid (CLA) inhibits expression of the Spot 14 (THRSP) and fatty acid synthase genes and impairs the growth of human breast cancer and liposarcoma cells. Nutr Cancer (2009) 61(1):114–22. doi: 10.1080/01635580802348666
63. Menendez JA, Lupu R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat Rev Cancer (2007) 7(10):763–77. doi: 10.1038/nrc2222
64. Kuhajda FP. AMP-activated protein kinase and human cancer: cancer metabolism revisited. Int J Obes (Lond) (2008) 32 Suppl 4:S36–41. doi: 10.1038/ijo.2008.121
65. Furuta E, Pai SK, Zhan R, Bandyopadhyay S, Watabe M, Mo YY, et al. Fatty acid synthase gene is up-regulated by hypoxia via activation of Akt and sterol regulatory element binding protein-1. Cancer Res (2008) 68(4):1003–11. doi: 10.1158/0008-5472.CAN-07-2489
66. Yang YA, Han WF, Morin PJ, Chrest FJ, Pizer ES. Activation of fatty acid synthesis during neoplastic transformation: role of mitogen-activated protein kinase and phosphatidylinositol 3-kinase. Exp Cell Res (2002) 279(1):80–90. doi: 10.1006/excr.2002.5600
67. Yan C, Wei H, Minjuan Z, Yan X, Jingyue Y, Wenchao L, et al. The mTOR inhibitor rapamycin synergizes with a fatty acid synthase inhibitor to induce cytotoxicity in ER/HER2-positive breast cancer cells. PloS One (2014) 9(5):e97697. doi: 10.1371/journal.pone.0097697
68. Pelon F, Bourachot B, Kieffer Y, Magagna I, Mermet-Meillon F, Bonnet I, et al. Cancer-associated fibroblast heterogeneity in axillary lymph nodes drives metastases in breast cancer through complementary mechanisms. Nat Commun (2020) 11(1):404. doi: 10.1038/s41467-019-14134-w
69. Ungefroren H, Sebens S, Seidl D, Lehnert H, Hass R. Interaction of tumor cells with the microenvironment. Cell Commun Signal (2011) 9:18. doi: 10.1186/1478-811X-9-18
70. Klein CA. Parallel progression of primary tumours and metastases. Nat Rev Cancer (2009) 9(4):302–12. doi: 10.1038/nrc2627
71. Slade MJ, Coombes RC. The clinical significance of disseminated tumor cells in breast cancer. Nat Clin Pract Oncol (2007) 4(1):30–41. doi: 10.1038/ncponc0685
72. Braun S, Vogl FD, Naume B, Janni W, Osborne MP, Coombes RC, et al. A pooled analysis of bone marrow micrometastasis in breast cancer. N Engl J Med (2005) 353(8):793–802. doi: 10.1056/NEJMoa050434
73. Chen W, Hoffmann AD, Liu H, Liu X. Organotropism: new insights into molecular mechanisms of breast cancer metastasis. NPJ Precis Oncol (2018) 2(1):4. doi: 10.1038/s41698-018-0047-0
74. Lu X, Kang Y. Organotropism of breast cancer metastasis. J Mammary Gland Biol Neoplasia (2007) 12(2-3):153–62. doi: 10.1007/s10911-007-9047-3
75. Huber KE, Carey LA, Wazer DE. Breast cancer molecular subtypes in patients with locally advanced disease: impact on prognosis, patterns of recurrence, and response to therapy. Semin Radiat Oncol (2009) 19(4):204–10. doi: 10.1016/j.semradonc.2009.05.004
76. Metzger-Filho O, Sun Z, Viale G, Price KN, Crivellari D, Snyder RD, et al. Patterns of Recurrence and outcome according to breast cancer subtypes in lymph node-negative disease: results from international breast cancer study group trials VIII and IX. J Clin Oncol (2013) 31(25):3083–90. doi: 10.1200/JCO.2012.46.1574
77. Savci-Heijink CD, Halfwerk H, Koster J, van de Vijver MJ. A novel gene expression signature for bone metastasis in breast carcinomas. Breast Cancer Res Treat (2016) 156(2):249–59. doi: 10.1007/s10549-016-3741-z
78. Smid M, Wang Y, Zhang Y, Sieuwerts AM, Yu J, Klijn JG, et al. Subtypes of breast cancer show preferential site of relapse. Cancer Res (2008) 68(9):3108–14. doi: 10.1158/0008-5472.CAN-07-5644
79. Kennecke H, Yerushalmi R, Woods R, Cheang MC, Voduc D, Speers CH, et al. Metastatic behavior of breast cancer subtypes. J Clin Oncol (2010) 28(20):3271–7. doi: 10.1200/JCO.2009.25.9820
80. Friedl P, Alexander S. Cancer invasion and the microenvironment: plasticity and reciprocity. Cell (2011) 147(5):992–1009. doi: 10.1016/j.cell.2011.11.016
81. Thiery JP. Epithelial-mesenchymal transitions in tumour progression. Nat Rev Cancer (2002) 2(6):442–54. doi: 10.1038/nrc822
82. Huang J, Li H, Ren G. Epithelial-mesenchymal transition and drug resistance in breast cancer (Review). Int J Oncol (2015) 47(3):840–8. doi: 10.3892/ijo.2015.3084
83. Weinberg RA. Twisted epithelial-mesenchymal transition blocks senescence. Nat Cell Biol (2008) 10(9):1021–3. doi: 10.1038/ncb0908-1021
84. Hass R, von der Ohe J, Ungefroren H. Potential Role of MSC/Cancer Cell Fusion and EMT for Breast Cancer Stem Cell Formation. Cancers (Basel) (2019) 11(10):1432. doi: 10.3390/cancers11101432
85. Carmeliet P, Jain RK. Angiogenesis in cancer and other diseases. Nature (2000) 407(6801):249–57. doi: 10.1038/35025220
86. Lorusso G, Ruegg C. New insights into the mechanisms of organ-specific breast cancer metastasis. Semin Cancer Biol (2012) 22(3):226–33. doi: 10.1016/j.semcancer.2012.03.007
87. Coupland LA, Parish CR. Platelets, selectins, and the control of tumor metastasis. Semin Oncol (2014) 41(3):422–34. doi: 10.1053/j.seminoncol.2014.04.003
88. Yadav A, Kumar B, Yu JG, Old M, Teknos TN, Kumar P. Tumor-Associated Endothelial Cells Promote Tumor Metastasis by Chaperoning Circulating Tumor Cells and Protecting Them from Anoikis. PloS One (2015) 10(10):e0141602. doi: 10.1371/journal.pone.0141602
89. Laubli H, Borsig L. Selectins promote tumor metastasis. Semin Cancer Biol (2010) 20(3):169–77. doi: 10.1016/j.semcancer.2010.04.005
90. Padua D, Zhang XH, Wang Q, Nadal C, Gerald WL, Gomis RR, et al. TGFbeta primes breast tumors for lung metastasis seeding through angiopoietin-like 4. Cell (2008) 133(1):66–77. doi: 10.1016/j.cell.2008.01.046
91. Yu PF, Huang Y, Xu CL, Lin LY, Han YY, Sun WH, et al. Downregulation of CXCL12 in mesenchymal stromal cells by TGFbeta promotes breast cancer metastasis. Oncogene (2017) 36(6):840–9. doi: 10.1038/onc.2016.252
92. Liu Y, Cao X. Characteristics and Significance of the Pre-metastatic Niche. Cancer Cell (2016) 30(5):668–81. doi: 10.1016/j.ccell.2016.09.011
93. Kaplan RN, Psaila B, Lyden D. Bone marrow cells in the ‘pre-metastatic niche’: within bone and beyond. Cancer Metastasis Rev (2006) 25(4):521–9. doi: 10.1007/s10555-006-9036-9
94. Hiratsuka S, Nakamura K, Iwai S, Murakami M, Itoh T, Kijima H, et al. MMP9 induction by vascular endothelial growth factor receptor-1 is involved in lung-specific metastasis. Cancer Cell (2002) 2(4):289–300. doi: 10.1016/s1535-6108(02)00153-8
95. Wu S, Zheng Q, Xing X, Dong Y, Wang Y, You Y, et al. Matrix stiffness-upregulated LOXL2 promotes fibronectin production, MMP9 and CXCL12 expression and BMDCs recruitment to assist pre-metastatic niche formation. J Exp Clin Cancer Res (2018) 37(1):99. doi: 10.1186/s13046-018-0761-z
96. Schwarz MK, Wells TN. New therapeutics that modulate chemokine networks. Nat Rev Drug Discovery (2002) 1(5):347–58. doi: 10.1038/nrd795
97. Wang J, Loberg R, Taichman RS. The pivotal role of CXCL12 (SDF-1)/CXCR4 axis in bone metastasis. Cancer Metastasis Rev (2006) 25(4):573–87. doi: 10.1007/s10555-006-9019-x
98. Wong CC, Gilkes DM, Zhang H, Chen J, Wei H, Chaturvedi P, et al. Hypoxia-inducible factor 1 is a master regulator of breast cancer metastatic niche formation. Proc Natl Acad Sci USA (2011) 108(39):16369–74. doi: 10.1073/pnas.1113483108
99. Zhuang X, Zhang H, Li X, Li X, Cong M, Peng F, et al. Differential effects on lung and bone metastasis of breast cancer by Wnt signalling inhibitor DKK1. Nat Cell Biol (2017) 19(10):1274–85. doi: 10.1038/ncb3613
100. Nevins JR, Potti A. Mining gene expression profiles: expression signatures as cancer phenotypes. Nat Rev Genet (2007) 8(8):601–9. doi: 10.1038/nrg2137
101. Blanco MA, Kang Y. Signaling pathways in breast cancer metastasis - novel insights from functional genomics. Breast Cancer Res (2011) 13(2):206. doi: 10.1186/bcr2831
102. Waning DL, Guise TA. Molecular mechanisms of bone metastasis and associated muscle weakness. Clin Cancer Res (2014) 20(12):3071–7. doi: 10.1158/1078-0432.CCR-13-1590
103. Lu X, Mu E, Wei Y, Riethdorf S, Yang Q, Yuan M, et al. VCAM-1 promotes osteolytic expansion of indolent bone micrometastasis of breast cancer by engaging alpha4beta1-positive osteoclast progenitors. Cancer Cell (2011) 20(6):701–14. doi: 10.1016/j.ccr.2011.11.002
104. Leblanc R, Lee SC, David M, Bordet JC, Norman DD, Patil R, et al. Interaction of platelet-derived autotaxin with tumor integrin alphaVbeta3 controls metastasis of breast cancer cells to bone. Blood (2014) 124(20):3141–50. doi: 10.1182/blood-2014-04-568683
105. Li XQ, Lu JT, Tan CC, Wang QS, Feng YM. RUNX2 promotes breast cancer bone metastasis by increasing integrin alpha5-mediated colonization. Cancer Lett (2016) 380(1):78–86. doi: 10.1016/j.canlet.2016.06.007
106. Kang Y, He W, Tulley S, Gupta GP, Serganova I, Chen CR, et al. Breast cancer bone metastasis mediated by the Smad tumor suppressor pathway. Proc Natl Acad Sci USA (2005) 102(39):13909–14. doi: 10.1073/pnas.0506517102
107. Dunn LK, Mohammad KS, Fournier PG, McKenna CR, Davis HW, Niewolna M, et al. Hypoxia and TGF-beta drive breast cancer bone metastases through parallel signaling pathways in tumor cells and the bone microenvironment. PloS One (2009) 4(9):e6896. doi: 10.1371/journal.pone.0006896
108. Lee SK, Park KK, Kim HJ, Park J, Son SH, Kim KR, et al. Human antigen R-regulated CCL20 contributes to osteolytic breast cancer bone metastasis. Sci Rep (2017) 7(1):9610. doi: 10.1038/s41598-017-09040-4
109. Scott BJ, Kesari S. Leptomeningeal metastases in breast cancer. Am J Cancer Res (2013) 3(2):117–26. doi: 10.1001/archeurol.2009.98
110. Lee BC, Lee TH, Avraham S, Avraham HK. Involvement of the chemokine receptor CXCR4 and its ligand stromal cell-derived factor 1alpha in breast cancer cell migration through human brain microvascular endothelial cells. Mol Cancer Res (2004) 2(6):327–38. doi: 10.1385/MB:27:2:169
111. Avraham HK, Jiang S, Fu Y, Nakshatri H, Ovadia H, Avraham S. Angiopoietin-2 mediates blood-brain barrier impairment and colonization of triple-negative breast cancer cells in brain. J Pathol (2014) 232(3):369–81. doi: 10.1002/path.4304
112. Witzel I, Oliveira-Ferrer L, Pantel K, Muller V, Wikman H. Breast cancer brain metastases: biology and new clinical perspectives. Breast Cancer Res (2016) 18(1):8. doi: 10.1186/s13058-015-0665-1
113. Wang L, Cossette SM, Rarick KR, Gershan J, Dwinell MB, Harder DR, et al. Astrocytes directly influence tumor cell invasion and metastasis in vivo. PloS One (2013) 8(12):e80933. doi: 10.1371/journal.pone.0080933
114. Pukrop T, Dehghani F, Chuang HN, Lohaus R, Bayanga K, Heermann S, et al. Microglia promote colonization of brain tissue by breast cancer cells in a Wnt-dependent way. Glia (2010) 58(12):1477–89. doi: 10.1002/glia.21022
115. Gupta GP, Nguyen DX, Chiang AC, Bos PD, Kim JY, Nadal C, et al. Mediators of vascular remodelling co-opted for sequential steps in lung metastasis. Nature (2007) 446(7137):765–70. doi: 10.1038/nature05760
116. Christen S, Lorendeau D, Schmieder R, Broekaert D, Metzger K, Veys K, et al. Breast Cancer-Derived Lung Metastases Show Increased Pyruvate Carboxylase-Dependent Anaplerosis. Cell Rep (2016) 17(3):837–48. doi: 10.1016/j.celrep.2016.09.042
117. Gao H, Chakraborty G, Lee-Lim AP, Mo Q, Decker M, Vonica A, et al. The BMP inhibitor Coco reactivates breast cancer cells at lung metastatic sites. Cell (2012) 150(4):764–79. doi: 10.1016/j.cell.2012.06.035
118. Kimbung S, Johansson I, Danielsson A, Veerla S, Egyhazi Brage S, Frostvik Stolt M, et al. Transcriptional Profiling of Breast Cancer Metastases Identifies Liver Metastasis-Selective Genes Associated with Adverse Outcome in Luminal A Primary Breast Cancer. Clin Cancer Res (2016) 22(1):146–57. doi: 10.1158/1078-0432.CCR-15-0487
119. Liu J, Deng H, Jia W, Zeng Y, Rao N, Li S, et al. Comparison of ER/PR and HER2 statuses in primary and paired liver metastatic sites of breast carcinoma in patients with or without treatment. J Cancer Res Clin Oncol (2012) 138(5):837–42. doi: 10.1007/s00432-012-1150-1
120. Bleckmann A, Conradi LC, Menck K, Schmick NA, Schubert A, Rietkotter E, et al. beta-catenin-independent WNT signaling and Ki67 in contrast to the estrogen receptor status are prognostic and associated with poor prognosis in breast cancer liver metastases. Clin Exp Metastasis (2016) 33(4):309–23. doi: 10.1007/s10585-016-9780-3
121. Ma R, Feng Y, Lin S, Chen J, Lin H, Liang X, et al. Mechanisms involved in breast cancer liver metastasis. J Transl Med (2015) 13:64. doi: 10.1186/s12967-015-0425-0
122. Chang JC. Cancer stem cells: Role in tumor growth, recurrence, metastasis, and treatment resistance. Medicine (Baltimore) (2016) 95(1 Suppl 1):S20–25. doi: 10.1097/MD.0000000000004766
123. Kotiyal S, Bhattacharya S. Breast cancer stem cells, EMT and therapeutic targets. Biochem Biophys Res Commun (2014) 453(1):112–6. doi: 10.1016/j.bbrc.2014.09.069
124. Lawson DA, Bhakta NR, Kessenbrock K, Prummel KD, Yu Y, Takai K, et al. Single-cell analysis reveals a stem-cell program in human metastatic breast cancer cells. Nature (2015) 526(7571):131–5. doi: 10.1038/nature15260
125. Liu L, Yin B, Yi Z, Liu X, Hu Z, Gao W, et al. Breast cancer stem cells characterized by CD70 expression preferentially metastasize to the lungs. Breast Cancer (2018) 25(6):706–16. doi: 10.1007/s12282-018-0880-6
126. Ponti D, Costa A, Zaffaroni N, Pratesi G, Petrangolini G, Coradini D, et al. Isolation and in vitro propagation of tumorigenic breast cancer cells with stem/progenitor cell properties. Cancer Res (2005) 65(13):5506–11. doi: 10.1158/0008-5472.CAN-05-0626
127. Aguirre-Ghiso JA. Models, mechanisms and clinical evidence for cancer dormancy. Nat Rev Cancer (2007) 7(11):834–46. doi: 10.1038/nrc2256
128. Carcereri de Prati A, Butturini E, Rigo A, Oppici E, Rossin M, Boriero D, et al. Metastatic Breast Cancer Cells Enter Into Dormant State and Express Cancer Stem Cells Phenotype Under Chronic Hypoxia. J Cell Biochem (2017) 118(10):3237–48. doi: 10.1002/jcb.25972
129. Wendt MK, Taylor MA, Schiemann BJ, Schiemann WP. Down-regulation of epithelial cadherin is required to initiate metastatic outgrowth of breast cancer. Mol Biol Cell (2011) 22(14):2423–35. doi: 10.1091/mbc.E11-04-0306
130. Gooding AJ, Zhang B, Jahanbani FK, Gilmore HL, Chang JC, Valadkhan S, et al. The lncRNA BORG Drives Breast Cancer Metastasis and Disease Recurrence. Sci Rep (2017) 7(1):12698. doi: 10.1038/s41598-017-12716-6
131. Goss PE, Chambers AF. Does tumour dormancy offer a therapeutic target? Nat Rev Cancer (2010) 10(12):871–7. doi: 10.1038/nrc2933
132. Giancotti FG. Mechanisms governing metastatic dormancy and reactivation. Cell (2013) 155(4):750–64. doi: 10.1016/j.cell.2013.10.029
133. Simoes RV, Serganova IS, Kruchevsky N, Leftin A, Shestov AA, Thaler HT, et al. Metabolic plasticity of metastatic breast cancer cells: adaptation to changes in the microenvironment. Neoplasia (2015) 17(8):671–84. doi: 10.1016/j.neo.2015.08.005
134. Gandhi N, Das GM. Metabolic Reprogramming in Breast Cancer and Its Therapeutic Implications. Cells (2019) 8(2):89. doi: 10.3390/cells8020089
135. Macintosh RL, Ryan KM. Autophagy in tumour cell death. Semin Cancer Biol (2013) 23(5):344–51. doi: 10.1016/j.semcancer.2013.05.006
136. Chen EI, Hewel J, Krueger JS, Tiraby C, Weber MR, Kralli A, et al. Adaptation of energy metabolism in breast cancer brain metastases. Cancer Res (2007) 67(4):1472–86. doi: 10.1158/0008-5472.CAN-06-3137
137. LeBleu VS, O’Connell JT, Gonzalez Herrera KN, Wikman H, Pantel K, Haigis MC, et al. PGC-1alpha mediates mitochondrial biogenesis and oxidative phosphorylation in cancer cells to promote metastasis. Nat Cell Biol (2014) 16(10):992–1003 1001-1015. doi: 10.1038/ncb3039
138. Dupuy F, Tabaries S, Andrzejewski S, Dong Z, Blagih J, Annis MG, et al. PDK1-Dependent Metabolic Reprogramming Dictates Metastatic Potential in Breast Cancer. Cell Metab (2015) 22(4):577–89. doi: 10.1016/j.cmet.2015.08.007
139. Andrzejewski S, Klimcakova E, Johnson RM, Tabaries S, Annis MG, McGuirk S, et al. PGC-1alpha Promotes Breast Cancer Metastasis and Confers Bioenergetic Flexibility against Metabolic Drugs. Cell Metab (2017) 26(5):778–87.e775. doi: 10.1016/j.cmet.2017.09.006
140. Faubert B, Solmonson A, DeBerardinis RJ. Metabolic reprogramming and cancer progression. Science (2020) 368(6487):eaaw5473. doi: 10.1126/science.aaw5473
141. Mimeault M, Batra SK. Hypoxia-inducing factors as master regulators of stemness properties and altered metabolism of cancer- and metastasis-initiating cells. J Cell Mol Med (2013) 17(1):30–54. doi: 10.1111/jcmm.12004
142. Verduzco D, Lloyd M, Xu L, Ibrahim-Hashim A, Balagurunathan Y, Gatenby RA, et al. Intermittent hypoxia selects for genotypes and phenotypes that increase survival, invasion, and therapy resistance. PloS One (2015) 10(3):e0120958. doi: 10.1371/journal.pone.0120958
143. Chen J, Lee HJ, Wu X, Huo L, Kim SJ, Xu L, et al. Gain of glucose-independent growth upon metastasis of breast cancer cells to the brain. Cancer Res (2015) 75(3):554–65. doi: 10.1158/0008-5472.CAN-14-2268
144. Marro M, Nieva C, de Juan A, Sierra A. Unravelling the Metabolic Progression of Breast Cancer Cells to Bone Metastasis by Coupling Raman Spectroscopy and a Novel Use of Mcr-Als Algorithm. Anal Chem (2018) 90(9):5594–602. doi: 10.1021/acs.analchem.7b04527
145. Hsu BE, Tabaries S, Johnson RM, Andrzejewski S, Senecal J, Lehuede C, et al. Immature Low-Density Neutrophils Exhibit Metabolic Flexibility that Facilitates Breast Cancer Liver Metastasis. Cell Rep (2019) 27(13):3902–3915 e3906. doi: 10.1016/j.celrep.2019.05.091
146. Li Y, Su X, Rohatgi N, Zhang Y, Brestoff JR, Shoghi KI, et al. Hepatic lipids promote liver metastasis. JCI Insight (2020) 5(17):e136215. doi: 10.1172/jci.insight.136215
147. Bos PD, Zhang XH, Nadal C, Shu W, Gomis RR, Nguyen DX, et al. Genes that mediate breast cancer metastasis to the brain. Nature (2009) 459(7249):1005–9. doi: 10.1038/nature08021
148. McGowan PM, Simedrea C, Ribot EJ, Foster PJ, Palmieri D, Steeg PS, et al. Notch1 inhibition alters the CD44hi/CD24lo population and reduces the formation of brain metastases from breast cancer. Mol Cancer Res (2011) 9(7):834–44. doi: 10.1158/1541-7786.MCR-10-0457
149. Bi P, Kuang S. Notch signaling as a novel regulator of metabolism. Trends Endocrinol Metab (2015) 26(5):248–55. doi: 10.1016/j.tem.2015.02.006
150. Sierra A, Price JE, Garcia-Ramirez M, Mendez O, Lopez L, Fabra A. Astrocyte-derived cytokines contribute to the metastatic brain specificity of breast cancer cells. Lab Invest (1997) 77(4):357–68.
151. Xing F, Kobayashi A, Okuda H, Watabe M, Pai SK, Pandey PR, et al. Reactive astrocytes promote the metastatic growth of breast cancer stem-like cells by activating Notch signalling in brain. EMBO Mol Med (2013) 5(3):384–96. doi: 10.1002/emmm.201201623
152. Ciminera AK, Jandial R, Termini J. Metabolic advantages and vulnerabilities in brain metastases. Clin Exp Metastasis (2017) 34(6-7):401–10. doi: 10.1007/s10585-017-9864-8
153. Neman J, Termini J, Wilczynski S, Vaidehi N, Choy C, Kowolik CM, et al. Human breast cancer metastases to the brain display GABAergic properties in the neural niche. Proc Natl Acad Sci USA (2014) 111(3):984–9. doi: 10.1073/pnas.1322098111
154. Mishra P, Ambs S. Metabolic Signatures of Human Breast Cancer. Mol Cell Oncol (2015) 2(3):e992217. doi: 10.4161/23723556.2014.992217
155. Sandhu R, Rein J, D’Arcy M, Herschkowitz JI, Hoadley KA, Troester MA. Overexpression of miR-146a in basal-like breast cancer cells confers enhanced tumorigenic potential in association with altered p53 status. Carcinogenesis (2014) 35(11):2567–75. doi: 10.1093/carcin/bgu175
156. Vousden KH, Prives C. Blinded by the Light: The Growing Complexity of p53. Cell (2009) 137(3):413–31. doi: 10.1016/j.cell.2009.04.037
157. Li T, Kon N, Jiang L, Tan M, Ludwig T, Zhao Y, et al. Tumor suppression in the absence of p53-mediated cell-cycle arrest, apoptosis, and senescence. Cell (2012) 149(6):1269–83. doi: 10.1016/j.cell.2012.04.026
158. Vousden KH, Ryan KM. p53 and metabolism. Nat Rev Cancer (2009) 9(10):691–700. doi: 10.1038/nrc2715
159. Zhang C, Liu J, Liang Y, Wu R, Zhao Y, Hong X, et al. Tumour-associated mutant p53 drives the Warburg effect. Nat Commun (2013) 4:2935. doi: 10.1038/ncomms3935
160. Schwartzenberg-Bar-Yoseph F, Armoni M, Karnieli E. The tumor suppressor p53 down-regulates glucose transporters GLUT1 and GLUT4 gene expression. Cancer Res (2004) 64(7):2627–33. doi: 10.1158/0008-5472.can-03-0846
161. Kawauchi K, Araki K, Tobiume K, Tanaka N. p53 regulates glucose metabolism through an IKK-NF-kappaB pathway and inhibits cell transformation. Nat Cell Biol (2008) 10(5):611–8. doi: 10.1038/ncb1724
162. Mathupala SP, Heese C, Pedersen PL. Glucose catabolism in cancer cells. The type II hexokinase promoter contains functionally active response elements for the tumor suppressor p53. J Biol Chem (1997) 272(36):22776–80. doi: 10.1074/jbc.272.36.22776
163. Bensaad K, Tsuruta A, Selak MA, Vidal MN, Nakano K, Bartrons R, et al. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell (2006) 126(1):107–20. doi: 10.1016/j.cell.2006.05.036
164. Zhang C, Lin M, Wu R, Wang X, Yang B, Levine AJ, et al. Parkin, a p53 target gene, mediates the role of p53 in glucose metabolism and the Warburg effect. Proc Natl Acad Sci USA (2011) 108(39):16259–64. doi: 10.1073/pnas.1113884108
165. Contractor T, Harris CR. p53 negatively regulates transcription of the pyruvate dehydrogenase kinase Pdk2. Cancer Res (2012) 72(2):560–7. doi: 10.1158/0008-5472.CAN-11-1215
166. Hsu CC, Tseng LM, Lee HC. Role of mitochondrial dysfunction in cancer progression. Exp Biol Med (Maywood) (2016) 241(12):1281–95. doi: 10.1177/1535370216641787
167. Matoba S, Kang JG, Patino WD, Wragg A, Boehm M, Gavrilova O, et al. p53 regulates mitochondrial respiration. Science (2006) 312(5780):1650–3. doi: 10.1126/science.1126863
168. Kamp WM, Wang PY, Hwang PM. TP53 mutation, mitochondria and cancer. Curr Opin Genet Dev (2016) 38:16–22. doi: 10.1016/j.gde.2016.02.007
169. Vahsen N, Cande C, Briere JJ, Benit P, Joza N, Larochette N, et al. AIF deficiency compromises oxidative phosphorylation. EMBO J (2004) 23(23):4679–89. doi: 10.1038/sj.emboj.7600461
170. Stambolsky P, Weisz L, Shats I, Klein Y, Goldfinger N, Oren M, et al. Regulation of AIF expression by p53. Cell Death Differ (2006) 13(12):2140–9. doi: 10.1038/sj.cdd.4401965
171. Hwang PM, Bunz F, Yu J, Rago C, Chan TA, Murphy MP, et al. Ferredoxin reductase affects p53-dependent, 5-fluorouracil-induced apoptosis in colorectal cancer cells. Nat Med (2001) 7(10):1111–7. doi: 10.1038/nm1001-1111
172. Hu W, Zhang C, Wu R, Sun Y, Levine A, Feng Z. Glutaminase 2, a novel p53 target gene regulating energy metabolism and antioxidant function. Proc Natl Acad Sci USA (2010) 107(16):7455–60. doi: 10.1073/pnas.1001006107
173. Suzuki S, Tanaka T, Poyurovsky MV, Nagano H, Mayama T, Ohkubo S, et al. Phosphate-activated glutaminase (GLS2), a p53-inducible regulator of glutamine metabolism and reactive oxygen species. Proc Natl Acad Sci USA (2010) 107(16):7461–6. doi: 10.1073/pnas.1002459107
174. Zhou G, Wang J, Zhao M, Xie TX, Tanaka N, Sano D, et al. Gain-of-function mutant p53 promotes cell growth and cancer cell metabolism via inhibition of AMPK activation. Mol Cell (2014) 54(6):960–74. doi: 10.1016/j.molcel.2014.04.024
175. Jiang P, Du W, Wang X, Mancuso A, Gao X, Wu M, et al. p53 regulates biosynthesis through direct inactivation of glucose-6-phosphate dehydrogenase. Nat Cell Biol (2011) 13(3):310–6. doi: 10.1038/ncb2172
176. Feng Z, Levine AJ. The regulation of energy metabolism and the IGF-1/mTOR pathways by the p53 protein. Trends Cell Biol (2010) 20(7):427–34. doi: 10.1016/j.tcb.2010.03.004
177. Raghow R, Yellaturu C, Deng X, Park EA, Elam MB. SREBPs: the crossroads of physiological and pathological lipid homeostasis. Trends Endocrinol Metab (2008) 19(2):65–73. doi: 10.1016/j.tem.2007.10.009
178. Freed-Pastor WA, Mizuno H, Zhao X, Langerod A, Moon SH, Rodriguez-Barrueco R, et al. Mutant p53 disrupts mammary tissue architecture via the mevalonate pathway. Cell (2012) 148(1–2):244–58. doi: 10.1016/j.cell.2011.12.017
179. Sengupta S, Biarnes MC, Jordan VC. Cyclin dependent kinase-9 mediated transcriptional de-regulation of cMYC as a critical determinant of endocrine-therapy resistance in breast cancers. Breast Cancer Res Treat (2014) 143(1):113–24. doi: 10.1007/s10549-013-2789-2
180. Yue M, Jiang J, Gao P, Liu H, Qing G. Oncogenic MYC Activates a Feedforward Regulatory Loop Promoting Essential Amino Acid Metabolism and Tumorigenesis. Cell Rep (2017) 21(13):3819–32. doi: 10.1016/j.celrep.2017.12.002
181. Locasale JW. Serine, glycine and one-carbon units: cancer metabolism in full circle. Nat Rev Cancer (2013) 13(8):572–83. doi: 10.1038/nrc3557
182. West MJ, Stoneley M, Willis AE. Translational induction of the c-myc oncogene via activation of the FRAP/TOR signalling pathway. Oncogene (1998) 17(6):769–80. doi: 10.1038/sj.onc.1201990
183. Gordan JD, Thompson CB, Simon MC. HIF and c-Myc: sibling rivals for control of cancer cell metabolism and proliferation. Cancer Cell (2007) 12(2):108–13. doi: 10.1016/j.ccr.2007.07.006
184. Cheng AS, Jin VX, Fan M, Smith LT, Liyanarachchi S, Yan PS, et al. Combinatorial analysis of transcription factor partners reveals recruitment of c-MYC to estrogen receptor-alpha responsive promoters. Mol Cell (2006) 21(3):393–404. doi: 10.1016/j.molcel.2005.12.016
185. Wang C, Mayer JA, Mazumdar A, Fertuck K, Kim H, Brown M, et al. Estrogen induces c-myc gene expression via an upstream enhancer activated by the estrogen receptor and the AP-1 transcription factor. Mol Endocrinol (2011) 25(9):1527–38. doi: 10.1210/me.2011-1037
186. Chen Z, Wang Y, Warden C, Chen S. Cross-talk between ER and HER2 regulates c-MYC-mediated glutamine metabolism in aromatase inhibitor resistant breast cancer cells. J Steroid Biochem Mol Biol (2015) 149:118–27. doi: 10.1016/j.jsbmb.2015.02.004
187. Fruman DA, Rommel C. PI3K and cancer: lessons, challenges and opportunities. Nat Rev Drug Discov (2014) 13(2):140–56. doi: 10.1038/nrd4204
188. Semenza GL, Roth PH, Fang HM, Wang GL. Transcriptional regulation of genes encoding glycolytic enzymes by hypoxia-inducible factor 1. J Biol Chem (1994) 269(38):23757–63. doi: 10.1016/0092-8674(94)90283-6
189. Zhong H, Chiles K, Feldser D, Laughner E, Hanrahan C, Georgescu MM, et al. Modulation of hypoxia-inducible factor 1alpha expression by the epidermal growth factor/phosphatidylinositol 3-kinase/PTEN/AKT/FRAP pathway in human prostate cancer cells: implications for tumor angiogenesis and therapeutics. Cancer Res (2000) 60(6):1541–5. doi: 10.1002/cyto.990020515
190. Hudson CC, Liu M, Chiang GG, Otterness DM, Loomis DC, Kaper F, et al. Regulation of hypoxia-inducible factor 1alpha expression and function by the mammalian target of rapamycin. Mol Cell Biol (2002) 22(20):7004–14. doi: 10.1128/mcb.22.20.7004-7014.2002
191. Hu CJ, Wang LY, Chodosh LA, Keith B, Simon MC. Differential roles of hypoxia-inducible factor 1alpha (HIF-1alpha) and HIF-2alpha in hypoxic gene regulation. Mol Cell Biol (2003) 23(24):9361–74. doi: 10.1128/mcb.23.24.9361-9374.2003
192. Porstmann T, Griffiths B, Chung YL, Delpuech O, Griffiths JR, Downward J, et al. PKB/Akt induces transcription of enzymes involved in cholesterol and fatty acid biosynthesis via activation of SREBP. Oncogene (2005) 24(43):6465–81. doi: 10.1038/sj.onc.1208802
193. Porstmann T, Santos CR, Griffiths B, Cully M, Wu M, Leevers S, et al. SREBP activity is regulated by mTORC1 and contributes to Akt-dependent cell growth. Cell Metab (2008) 8(3):224–36. doi: 10.1016/j.cmet.2008.07.007
194. Duvel K, Yecies JL, Menon S, Raman P, Lipovsky AI, Souza AL, et al. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol Cell (2010) 39(2):171–83. doi: 10.1016/j.molcel.2010.06.022
195. Schafer ZT, Grassian AR, Song L, Jiang Z, Gerhart-Hines Z, Irie HY, et al. Antioxidant and oncogene rescue of metabolic defects caused by loss of matrix attachment. Nature (2009) 461(7260):109–13. doi: 10.1038/nature08268
196. McCarthy AM, Kumar NP, He W, Regan S, Welch M, Moy B, et al. Different associations of tumor PIK3CA mutations and clinical outcomes according to aspirin use among women with metastatic hormone receptor positive breast cancer. BMC Cancer (2020) 20(1):347. doi: 10.1186/s12885-020-06810-8
197. Araki K, Miyoshi Y. Mechanism of resistance to endocrine therapy in breast cancer: the important role of PI3K/Akt/mTOR in estrogen receptor-positive, HER2-negative breast cancer. Breast Cancer (2018) 25(4):392–401. doi: 10.1007/s12282-017-0812-x
198. Hou X, Zhao M, Wang T, Zhang G. Upregulation of estrogen receptor mediates migration, invasion and proliferation of endometrial carcinoma cells by regulating the PI3K/AKT/mTOR pathway. Oncol Rep (2014) 31(3):1175–82. doi: 10.3892/or.2013.2944
199. Zhou K, Sun P, Zhang Y, You X, Li P, Wang T. Estrogen stimulated migration and invasion of estrogen receptor-negative breast cancer cells involves an ezrin-dependent crosstalk between G protein-coupled receptor 30 and estrogen receptor beta signaling. Steroids (2016) 111:113–20. doi: 10.1016/j.steroids.2016.01.021
200. Alayev A, Salamon RS, Berger SM, Schwartz NS, Cuesta R, Snyder RB, et al. mTORC1 directly phosphorylates and activates ERalpha upon estrogen stimulation. Oncogene (2016) 35(27):3535–43. doi: 10.1038/onc.2015.414
201. Toska E, Osmanbeyoglu HU, Castel P, Chan C, Hendrickson RC, Elkabets M, et al. PI3K pathway regulates ER-dependent transcription in breast cancer through the epigenetic regulator KMT2D. Science (2017) 355(6331):1324–30. doi: 10.1126/science.aah6893
202. Caino MC, Altieri DC. Cancer cells exploit adaptive mitochondrial dynamics to increase tumor cell invasion. Cell Cycle (2015) 14(20):3242–7. doi: 10.1080/15384101.2015.1084448
203. Caino MC, Ghosh JC, Chae YC, Vaira V, Rivadeneira DB, Faversani A, et al. PI3K therapy reprograms mitochondrial trafficking to fuel tumor cell invasion. Proc Natl Acad Sci USA (2015) 112(28):8638–43. doi: 10.1073/pnas.1500722112
204. Hammond ME, Hayes DF, Wolff AC, Mangu PB, Temin S. American society of clinical oncology/college of american pathologists guideline recommendations for immunohistochemical testing of estrogen and progesterone receptors in breast cancer. J Oncol Pract (2010) 6(4):195–7. doi: 10.1200/JOP.777003
205. Cao MD, Lamichhane S, Lundgren S, Bofin A, Fjosne H, Giskeodegard GF, et al. Metabolic characterization of triple negative breast cancer. BMC Cancer (2014) 14:941. doi: 10.1186/1471-2407-14-941
206. Clegg DJ, Brown LM, Woods SC, Benoit SC. Gonadal hormones determine sensitivity to central leptin and insulin. Diabetes (2006) 55(4):978–87. doi: 10.2337/diabetes.55.04.06.db05-1339
207. O’Mahony F, Razandi M, Pedram A, Harvey BJ, Levin ER. Estrogen modulates metabolic pathway adaptation to available glucose in breast cancer cells. Mol Endocrinol (2012) 26(12):2058–70. doi: 10.1210/me.2012-1191
208. Neeman M, Degani H. Metabolic studies of estrogen- and tamoxifen-treated human breast cancer cells by nuclear magnetic resonance spectroscopy. Cancer Res (1989) 49(3):589–94.
209. Furman E, Rushkin E, Margalit R, Bendel P, Degani H. Tamoxifen induced changes in MCF7 human breast cancer: in vitro and in vivo studies using nuclear magnetic resonance spectroscopy and imaging. J Steroid Biochem Mol Biol (1992) 43(1-3):189–95. doi: 10.1016/0960-0760(92)90207-y
210. Rivenzon-Segal D, Boldin-Adamsky S, Seger D, Seger R, Degani H. Glycolysis and glucose transporter 1 as markers of response to hormonal therapy in breast cancer. Int J Cancer (2003) 107(2):177–82. doi: 10.1002/ijc.11387
211. Huang B, Warner M, Gustafsson JA. Estrogen receptors in breast carcinogenesis and endocrine therapy. Mol Cell Endocrinol (2015) 418 Pt 3:240–4. doi: 10.1016/j.mce.2014.11.015
212. Murphy LC, Leygue E. The role of estrogen receptor-beta in breast cancer. Semin Reprod Med (2012) 30(1):5–13. doi: 10.1055/s-0031-1299592
213. Manente AG, Valenti D, Pinton G, Jithesh PV, Daga A, Rossi L, et al. Estrogen receptor beta activation impairs mitochondrial oxidative metabolism and affects malignant mesothelioma cell growth in vitro and in vivo. Oncogenesis (2013) 2:e72. doi: 10.1038/oncsis.2013.32
214. Ma R, Karthik GM, Lovrot J, Haglund F, Rosin G, Katchy A, et al. Estrogen Receptor beta as a Therapeutic Target in Breast Cancer Stem Cells. J Natl Cancer Inst (2017) 109(3):1–14. doi: 10.1093/jnci/djw236
215. Zhang D, Tai LK, Wong LL, Chiu LL, Sethi SK, Koay ES. Proteomic study reveals that proteins involved in metabolic and detoxification pathways are highly expressed in HER-2/neu-positive breast cancer. Mol Cell Proteomics (2005) 4(11):1686–96. doi: 10.1074/mcp.M400221-MCP200
216. Walsh AJ, Cook RS, Manning HC, Hicks DJ, Lafontant A, Arteaga CL, et al. Optical metabolic imaging identifies glycolytic levels, subtypes, and early-treatment response in breast cancer. Cancer Res (2013) 73(20):6164–74. doi: 10.1158/0008-5472.CAN-13-0527
217. O’Neal J, Clem A, Reynolds L, Dougherty S, Imbert-Fernandez Y, Telang S, et al. Inhibition of 6-phosphofructo-2-kinase (PFKFB3) suppresses glucose metabolism and the growth of HER2+ breast cancer. Breast Cancer Res Treat (2016) 160(1):29–40. doi: 10.1007/s10549-016-3968-8
218. Castagnoli L, Iorio E, Dugo M, Koschorke A, Faraci S, Canese R, et al. Intratumor lactate levels reflect HER2 addiction status in HER2-positive breast cancer. J Cell Physiol (2019) 234(2):1768–79. doi: 10.1002/jcp.27049
219. Ding Y, Liu Z, Desai S, Zhao Y, Liu H, Pannell LK, et al. Receptor tyrosine kinase ErbB2 translocates into mitochondria and regulates cellular metabolism. Nat Commun (2012) 3:1271. doi: 10.1038/ncomms2236
220. Tian C, Yuan Z, Xu D, Ding P, Wang T, Zhang L, et al. Inhibition of glycolysis by a novel EGFR/HER2 inhibitor KU004 suppresses the growth of HER2+ cancer. Exp Cell Res (2017) 357(2):211–21. doi: 10.1016/j.yexcr.2017.05.019
221. Laughner E, Taghavi P, Chiles K, Mahon PC, Semenza GL. HER2 (neu) signaling increases the rate of hypoxia-inducible factor 1alpha (HIF-1alpha) synthesis: novel mechanism for HIF-1-mediated vascular endothelial growth factor expression. Mol Cell Biol (2001) 21(12):3995–4004. doi: 10.1128/MCB.21.12.3995-4004.2001
222. Foulkes WD, Stefansson IM, Chappuis PO, Begin LR, Goffin JR, Wong N, et al. Germline BRCA1 mutations and a basal epithelial phenotype in breast cancer. J Natl Cancer Inst (2003) 95(19):1482–5. doi: 10.1093/jnci/djg050
223. Stefansson OA, Jonasson JG, Johannsson OT, Olafsdottir K, Steinarsdottir M, Valgeirsdottir S, et al. Genomic profiling of breast tumours in relation to BRCA abnormalities and phenotypes. Breast Cancer Res (2009) 11(4):R47. doi: 10.1186/bcr2334
224. Rakha EA, El-Sheikh SE, Kandil MA, El-Sayed ME, Green AR, Ellis IO. Expression of BRCA1 protein in breast cancer and its prognostic significance. Hum Pathol (2008) 39(6):857–65. doi: 10.1016/j.humpath.2007.10.011
225. Martinez-Outschoorn UE, Balliet R, Lin Z, Whitaker-Menezes D, Birbe RC, Bombonati A, et al. BRCA1 mutations drive oxidative stress and glycolysis in the tumor microenvironment: implications for breast cancer prevention with antioxidant therapies. Cell Cycle (2012) 11(23):4402–13. doi: 10.4161/cc.22776
226. Lisanti MP, Martinez-Outschoorn UE, Sotgia F. Oncogenes induce the cancer-associated fibroblast phenotype: metabolic symbiosis and “fibroblast addiction” are new therapeutic targets for drug discovery. Cell Cycle (2013) 12(17):2723–32. doi: 10.4161/cc.25695
227. Zacksenhaus E, Liu JC, Jiang Z, Yao Y, Xia L, Shrestha M, et al. Transcription Factors in Breast Cancer-Lessons From Recent Genomic Analyses and Therapeutic Implications. Adv Protein Chem Struct Biol (2017) 107:223–73. doi: 10.1016/bs.apcsb.2016.10.003
228. Kim KJ, Godarova A, Seedle K, Kim MH, Ince TA, Wells SI, et al. Rb suppresses collective invasion, circulation and metastasis of breast cancer cells in CD44-dependent manner. PloS One (2013) 8(12):e80590. doi: 10.1371/journal.pone.0080590
229. Knudsen ES, McClendon AK, Franco J, Ertel A, Fortina P, Witkiewicz AK. RB loss contributes to aggressive tumor phenotypes in MYC-driven triple negative breast cancer. Cell Cycle (2015) 14(1):109–22. doi: 10.4161/15384101.2014.967118
230. Jiang Z, Jones R, Liu JC, Deng T, Robinson T, Chung PE, et al. RB1 and p53 at the crossroad of EMT and triple-negative breast cancer. Cell Cycle (2011) 10(10):1563–70. doi: 10.4161/cc.10.10.15703
231. Jones RA, Robinson TJ, Liu JC, Shrestha M, Voisin V, Ju Y, et al. RB1 deficiency in triple-negative breast cancer induces mitochondrial protein translation. J Clin Invest (2016) 126(10):3739–57. doi: 10.1172/JCI81568
232. Zacksenhaus E, Shrestha M, Liu JC, Vorobieva I, Chung PED, Ju Y, et al. Mitochondrial OXPHOS Induced by RB1 Deficiency in Breast Cancer: Implications for Anabolic Metabolism, Stemness, and Metastasis. Trends Cancer (2017) 3(11):768–79. doi: 10.1016/j.trecan.2017.09.002
233. Cheng J, Zhang T, Ji H, Tao K, Guo J, Wei W. Functional characterization of AMP-activated protein kinase signaling in tumorigenesis. Biochim Biophys Acta (2016) 1866(2):232–51. doi: 10.1016/j.bbcan.2016.09.006
234. Corradetti MN, Inoki K, Bardeesy N, DePinho RA, Guan KL. Regulation of the TSC pathway by LKB1: evidence of a molecular link between tuberous sclerosis complex and Peutz-Jeghers syndrome. Genes Dev (2004) 18(13):1533–8. doi: 10.1101/gad.1199104
235. Lizcano JM, Goransson O, Toth R, Deak M, Morrice NA, Boudeau J, et al. LKB1 is a master kinase that activates 13 kinases of the AMPK subfamily, including MARK/PAR-1. EMBO J (2004) 23(4):833–43. doi: 10.1038/sj.emboj.7600110
236. Sarbassov DD, Ali SM, Kim DH, Guertin DA, Latek RR, Erdjument-Bromage H, et al. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr Biol (2004) 14(14):1296–302. doi: 10.1016/j.cub.2004.06.054
237. Shaw RJ, Bardeesy N, Manning BD, Lopez L, Kosmatka M, DePinho RA, et al. The LKB1 tumor suppressor negatively regulates mTOR signaling. Cancer Cell (2004) 6(1):91–9. doi: 10.1016/j.ccr.2004.06.007
238. Shaw RJ, Kosmatka M, Bardeesy N, Hurley RL, Witters LA, DePinho RA, et al. The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc Natl Acad Sci USA (2004) 101(10):3329–35. doi: 10.1073/pnas.0308061100
239. Morton JP, Jamieson NB, Karim SA, Athineos D, Ridgway RA, Nixon C, et al. LKB1 haploinsufficiency cooperates with Kras to promote pancreatic cancer through suppression of p21-dependent growth arrest. Gastroenterology (2010) 139(2):586–97, 597 e581-6. doi: 10.1053/j.gastro.2010.04.055
240. Garcia-Martinez JM, Wullschleger S, Preston G, Guichard S, Fleming S, Alessi DR, et al. Effect of PI3K- and mTOR-specific inhibitors on spontaneous B-cell follicular lymphomas in PTEN/LKB1-deficient mice. Br J Cancer (2011) 104(7):1116–25. doi: 10.1038/bjc.2011.83
241. Andrade-Vieira R, Xu Z, Colp P, Marignani PA. Loss of LKB1 expression reduces the latency of ErbB2-mediated mammary gland tumorigenesis, promoting changes in metabolic pathways. PloS One (2013) 8(2):e56567. doi: 10.1371/journal.pone.0056567
242. Liu Y, Marks K, Cowley GS, Carretero J, Liu Q, Nieland TJ, et al. Metabolic and functional genomic studies identify deoxythymidylate kinase as a target in LKB1-mutant lung cancer. Cancer Discov (2013) 3(8):870–9. doi: 10.1158/2159-8290.CD-13-0015
243. Han M, Xu W, Cheng P, Jin H, Wang X. Histone demethylase lysine demethylase 5B in development and cancer. Oncotarget (2017) 8(5):8980–91. doi: 10.18632/oncotarget.13858
244. Zhang ZG, Zhang HS, Sun HL, Liu HY, Liu MY, Zhou Z. KDM5B promotes breast cancer cell proliferation and migration via AMPK-mediated lipid metabolism reprogramming. Exp Cell Res (2019) 379(2):182–90. doi: 10.1016/j.yexcr.2019.04.006
245. Schito L, Rey S. Hypoxic pathobiology of breast cancer metastasis. Biochim Biophys Acta Rev Cancer (2017) 1868(1):239–45. doi: 10.1016/j.bbcan.2017.05.004
246. Lundgren K, Holm C, Landberg G. Hypoxia and breast cancer: prognostic and therapeutic implications. Cell Mol Life Sci (2007) 64(24):3233–47. doi: 10.1007/s00018-007-7390-6
247. Zhang T, Suo C, Zheng C, Zhang H. Hypoxia and Metabolism in Metastasis. Adv Exp Med Biol (2019) 1136:87–95. doi: 10.1007/978-3-030-12734-3_6
248. Bos R, van der Groep P, Greijer AE, Shvarts A, Meijer S, Pinedo HM, et al. Levels of hypoxia-inducible factor-1alpha independently predict prognosis in patients with lymph node negative breast carcinoma. Cancer (2003) 97(6):1573–81. doi: 10.1002/cncr.11246
249. Gruber G, Greiner RH, Hlushchuk R, Aebersold DM, Altermatt HJ, Berclaz G, et al. Hypoxia-inducible factor 1 alpha in high-risk breast cancer: an independent prognostic parameter? Breast Cancer Res (2004) 6(3):R191–198. doi: 10.1186/bcr775
250. Generali D, Berruti A, Brizzi MP, Campo L, Bonardi S, Wigfield S, et al. Hypoxia-inducible factor-1alpha expression predicts a poor response to primary chemoendocrine therapy and disease-free survival in primary human breast cancer. Clin Cancer Res (2006) 12(15):4562–8. doi: 10.1158/1078-0432.CCR-05-2690
251. Cancer Genome Atlas N. Comprehensive molecular portraits of human breast tumours. Nature (2012) 490(7418):61–70. doi: 10.1038/nature11412
252. Zhang H, Wong CC, Wei H, Gilkes DM, Korangath P, Chaturvedi P, et al. HIF-1-dependent expression of angiopoietin-like 4 and L1CAM mediates vascular metastasis of hypoxic breast cancer cells to the lungs. Oncogene (2012) 31(14):1757–70. doi: 10.1038/onc.2011.365
253. Gilkes DM, Bajpai S, Chaturvedi P, Wirtz D, Semenza GL. Hypoxia-inducible factor 1 (HIF-1) promotes extracellular matrix remodeling under hypoxic conditions by inducing P4HA1, P4HA2, and PLOD2 expression in fibroblasts. J Biol Chem (2013) 288(15):10819–29. doi: 10.1074/jbc.M112.442939
254. Gilkes DM, Bajpai S, Wong CC, Chaturvedi P, Hubbi ME, Wirtz D, et al. Procollagen lysyl hydroxylase 2 is essential for hypoxia-induced breast cancer metastasis. Mol Cancer Res (2013) 11(5):456–66. doi: 10.1158/1541-7786.MCR-12-0629
255. Gilkes DM, Chaturvedi P, Bajpai S, Wong CC, Wei H, Pitcairn S, et al. Collagen prolyl hydroxylases are essential for breast cancer metastasis. Cancer Res (2013) 73(11):3285–96. doi: 10.1158/0008-5472.CAN-12-3963
256. Conley SJ, Gheordunescu E, Kakarala P, Newman B, Korkaya H, Heath AN, et al. Antiangiogenic agents increase breast cancer stem cells via the generation of tumor hypoxia. Proc Natl Acad Sci USA (2012) 109(8):2784–9. doi: 10.1073/pnas.1018866109
257. Xiang L, Gilkes DM, Hu H, Takano N, Luo W, Lu H, et al. Hypoxia-inducible factor 1 mediates TAZ expression and nuclear localization to induce the breast cancer stem cell phenotype. Oncotarget (2014) 5(24):12509–27. doi: 10.18632/oncotarget.2997
258. Morin A, Letouze E, Gimenez-Roqueplo AP, Favier J. Oncometabolites-driven tumorigenesis: From genetics to targeted therapy. Int J Cancer (2014) 135(10):2237–48. doi: 10.1002/ijc.29080
259. Gorrini C, Harris IS, Mak TW. Modulation of oxidative stress as an anticancer strategy. Nat Rev Drug Discov (2013) 12(12):931–47. doi: 10.1038/nrd4002
260. Piskounova E, Agathocleous M, Murphy MM, Hu Z, Huddlestun SE, Zhao Z, et al. Oxidative stress inhibits distant metastasis by human melanoma cells. Nature (2015) 527(7577):186–91. doi: 10.1038/nature15726
261. Kamarajugadda S, Stemboroski L, Cai Q, Simpson NE, Nayak S, Tan M, et al. Glucose oxidation modulates anoikis and tumor metastasis. Mol Cell Biol (2012) 32(10):1893–907. doi: 10.1128/MCB.06248-11
262. Davison CA, Durbin SM, Thau MR, Zellmer VR, Chapman SE, Diener J, et al. Antioxidant enzymes mediate survival of breast cancer cells deprived of extracellular matrix. Cancer Res (2013) 73(12):3704–15. doi: 10.1158/0008-5472.CAN-12-2482
263. Kamarajugadda S, Cai Q, Chen H, Nayak S, Zhu J, He M, et al. Manganese superoxide dismutase promotes anoikis resistance and tumor metastasis. Cell Death Dis (2013) 4:e504. doi: 10.1038/cddis.2013.20
264. Harris IS, Treloar AE, Inoue S, Sasaki M, Gorrini C, Lee KC, et al. Glutathione and thioredoxin antioxidant pathways synergize to drive cancer initiation and progression. Cancer Cell (2015) 27(2):211–22. doi: 10.1016/j.ccell.2014.11.019
265. Svastova E, Hulikova A, Rafajova M, Zat’ovicova M, Gibadulinova A, Casini A, et al. Hypoxia activates the capacity of tumor-associated carbonic anhydrase IX to acidify extracellular pH. FEBS Lett (2004) 577(3):439–45. doi: 10.1016/j.febslet.2004.10.043
266. Martinez-Zaguilan R, Seftor EA, Seftor RE, Chu YW, Gillies RJ, Hendrix MJ. Acidic pH enhances the invasive behavior of human melanoma cells. Clin Exp Metastasis (1996) 14(2):176–86. doi: 10.1007/BF00121214
267. Stern R, Shuster S, Neudecker BA, Formby B. Lactate stimulates fibroblast expression of hyaluronan and CD44: the Warburg effect revisited. Exp Cell Res (2002) 276(1):24–31. doi: 10.1006/excr.2002.5508
268. Constant JS, Feng JJ, Zabel DD, Yuan H, Suh DY, Scheuenstuhl H, et al. Lactate elicits vascular endothelial growth factor from macrophages: a possible alternative to hypoxia. Wound Repair Regener (2000) 8(5):353–60. doi: 10.1111/j.1524-475x.2000.00353.x
269. Fischer K, Hoffmann P, Voelkl S, Meidenbauer N, Ammer J, Edinger M, et al. Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood (2007) 109(9):3812–9. doi: 10.1182/blood-2006-07-035972
270. Chang CH, Qiu J, O’Sullivan D, Buck MD, Noguchi T, Curtis JD, et al. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell (2015) 162(6):1229–41. doi: 10.1016/j.cell.2015.08.016
271. Ishii G, Ochiai A, Neri S. Phenotypic and functional heterogeneity of cancer-associated fibroblast within the tumor microenvironment. Adv Drug Deliv Rev (2016) 99(Pt B):186–96. doi: 10.1016/j.addr.2015.07.007
272. Martinez-Outschoorn UE, Balliet RM, Rivadeneira DB, Chiavarina B, Pavlides S, Wang C, et al. Oxidative stress in cancer associated fibroblasts drives tumor-stroma co-evolution: A new paradigm for understanding tumor metabolism, the field effect and genomic instability in cancer cells. Cell Cycle (2010) 9(16):3256–76. doi: 10.4161/cc.9.16.12553
273. Pavlides S, Tsirigos A, Migneco G, Whitaker-Menezes D, Chiavarina B, Flomenberg N, et al. The autophagic tumor stroma model of cancer: Role of oxidative stress and ketone production in fueling tumor cell metabolism. Cell Cycle (2010) 9(17):3485–505. doi: 10.4161/cc.9.17.12721
274. Ko YH, Lin Z, Flomenberg N, Pestell RG, Howell A, Sotgia F, et al. Glutamine fuels a vicious cycle of autophagy in the tumor stroma and oxidative mitochondrial metabolism in epithelial cancer cells: implications for preventing chemotherapy resistance. Cancer Biol Ther (2011) 12(12):1085–97. doi: 10.4161/cbt.12.12.18671
275. Dirat B, Bochet L, Dabek M, Daviaud D, Dauvillier S, Majed B, et al. Cancer-associated adipocytes exhibit an activated phenotype and contribute to breast cancer invasion. Cancer Res (2011) 71(7):2455–65. doi: 10.1158/0008-5472.CAN-10-3323
276. Wu Q, Li J, Li Z, Sun S, Zhu S, Wang L, et al. Exosomes from the tumour-adipocyte interplay stimulate beige/brown differentiation and reprogram metabolism in stromal adipocytes to promote tumour progression. J Exp Clin Cancer Res (2019) 38(1):223. doi: 10.1186/s13046-019-1210-3
277. Nieman KM, Kenny HA, Penicka CV, Ladanyi A, Buell-Gutbrod R, Zillhardt MR, et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat Med (2011) 17(11):1498–503. doi: 10.1038/nm.2492
278. Pascual G, Avgustinova A, Mejetta S, Martin M, Castellanos A, Attolini CS, et al. Targeting metastasis-initiating cells through the fatty acid receptor CD36. Nature (2017) 541(7635):41–5. doi: 10.1038/nature20791
279. Martinez-Outschoorn UE, Lin Z, Whitaker-Menezes D, Howell A, Lisanti MP, Sotgia F. Ketone bodies and two-compartment tumor metabolism: stromal ketone production fuels mitochondrial biogenesis in epithelial cancer cells. Cell Cycle (2012) 11(21):3956–63. doi: 10.4161/cc.22136
280. Huang CK, Chang PH, Kuo WH, Chen CL, Jeng YM, Chang KJ, et al. Adipocytes promote malignant growth of breast tumours with monocarboxylate transporter 2 expression via beta-hydroxybutyrate. Nat Commun (2017) 8:14706. doi: 10.1038/ncomms14706
281. Argiles JM, Busquets S, Stemmler B, Lopez-Soriano FJ. Cancer cachexia: understanding the molecular basis. Nat Rev Cancer (2014) 14(11):754–62. doi: 10.1038/nrc3829
282. Leone RD, Powell JD. Metabolism of immune cells in cancer. Nat Rev Cancer (2020) 20(9):516–31. doi: 10.1038/s41568-020-0273-y
283. Cascone T, McKenzie JA, Mbofung RM, Punt S, Wang Z, Xu C, et al. Increased Tumor Glycolysis Characterizes Immune Resistance to Adoptive T Cell Therapy. Cell Metab (2018) 27(5):977–87.e974. doi: 10.1016/j.cmet.2018.02.024
284. Yang W, Bai Y, Xiong Y, Zhang J, Chen S, Zheng X, et al. Potentiating the antitumour response of CD8(+) T cells by modulating cholesterol metabolism. Nature (2016) 531(7596):651–5. doi: 10.1038/nature17412
285. Li W, Tanikawa T, Kryczek I, Xia H, Li G, Wu K, et al. Aerobic Glycolysis Controls Myeloid-Derived Suppressor Cells and Tumor Immunity via a Specific CEBPB Isoform in Triple-Negative Breast Cancer. Cell Metab (2018) 28(1):87–103 e106. doi: 10.1016/j.cmet.2018.04.022
286. Corzo CA, Condamine T, Lu L, Cotter MJ, Youn JI, Cheng P, et al. HIF-1alpha regulates function and differentiation of myeloid-derived suppressor cells in the tumor microenvironment. J Exp Med (2010) 207(11):2439–53. doi: 10.1084/jem.20100587
287. Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell (2010) 141(1):39–51. doi: 10.1016/j.cell.2010.03.014
288. Qiu SQ, Waaijer SJH, Zwager MC, de Vries EGE, van der Vegt B, Schroder CP. Tumor-associated macrophages in breast cancer: Innocent bystander or important player? Cancer Treat Rev (2018) 70:178–89. doi: 10.1016/j.ctrv.2018.08.010
289. Murdoch C, Lewis CE. Macrophage migration and gene expression in response to tumor hypoxia. Int J Cancer (2005) 117(5):701–8. doi: 10.1002/ijc.21422
290. Casazza A, Laoui D, Wenes M, Rizzolio S, Bassani N, Mambretti M, et al. Impeding macrophage entry into hypoxic tumor areas by Sema3A/Nrp1 signaling blockade inhibits angiogenesis and restores antitumor immunity. Cancer Cell (2013) 24(6):695–709. doi: 10.1016/j.ccr.2013.11.007
291. Henze AT, Mazzone M. The impact of hypoxia on tumor-associated macrophages. J Clin Invest (2016) 126(10):3672–9. doi: 10.1172/JCI84427
292. Ferrante CJ, Pinhal-Enfield G, Elson G, Cronstein BN, Hasko G, Outram S, et al. The adenosine-dependent angiogenic switch of macrophages to an M2-like phenotype is independent of interleukin-4 receptor alpha (IL-4Ralpha) signaling. Inflammation (2013) 36(4):921–31. doi: 10.1007/s10753-013-9621-3
293. Tripathi C, Tewari BN, Kanchan RK, Baghel KS, Nautiyal N, Shrivastava R, et al. Macrophages are recruited to hypoxic tumor areas and acquire a pro-angiogenic M2-polarized phenotype via hypoxic cancer cell derived cytokines Oncostatin M and Eotaxin. Oncotarget (2014) 5(14):5350–68. doi: 10.18632/oncotarget.2110
294. Colegio OR, Chu NQ, Szabo AL, Chu T, Rhebergen AM, Jairam V, et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature (2014) 513(7519):559–63. doi: 10.1038/nature13490
295. Russo V, Protti MP. Tumor-derived factors affecting immune cells. Cytokine Growth Factor Rev (2017) 36:79–87. doi: 10.1016/j.cytogfr.2017.06.005
296. Moresco MA, Raccosta L, Corna G, Maggioni D, Soncini M, Bicciato S, et al. Enzymatic Inactivation of Oxysterols in Breast Tumor Cells Constraints Metastasis Formation by Reprogramming the Metastatic Lung Microenvironment. Front Immunol (2018) 9:2251. doi: 10.3389/fimmu.2018.02251
297. Baek AE, Yu YA, He S, Wardell SE, Chang CY, Kwon S, et al. The cholesterol metabolite 27 hydroxycholesterol facilitates breast cancer metastasis through its actions on immune cells. Nat Commun (2017) 8(1):864. doi: 10.1038/s41467-017-00910-z
298. Raez LE, Papadopoulos K, Ricart AD, Chiorean EG, Dipaola RS, Stein MN, et al. A phase I dose-escalation trial of 2-deoxy-D-glucose alone or combined with docetaxel in patients with advanced solid tumors. Cancer Chemother Pharmacol (2013) 71(2):523–30. doi: 10.1007/s00280-012-2045-1
299. Faria J, Negalha G, Azevedo A, Martel F. Metformin and Breast Cancer: Molecular Targets. J Mammary Gland Biol Neoplasia (2019) 24(2):111–23. doi: 10.1007/s10911-019-09429-z
300. Mariotti V, Han H, Ismail-Khan R, Tang SC, Dillon P, Montero AJ, et al. Effect of Taxane Chemotherapy With or Without Indoximod in Metastatic Breast Cancer: A Randomized Clinical Trial. JAMA Oncol (2020). doi: 10.1001/jamaoncol.2020.5572
301. Varghese E, Samuel SM, Liskova A, Samec M, Kubatka P, Busselberg D. Targeting Glucose Metabolism to Overcome Resistance to Anticancer Chemotherapy in Breast Cancer. Cancers (Basel) (2020) 12(8):2252. doi: 10.3390/cancers12082252
302. Haugrud AB, Zhuang Y, Coppock JD, Miskimins WK. Dichloroacetate enhances apoptotic cell death via oxidative damage and attenuates lactate production in metformin-treated breast cancer cells. Breast Cancer Res Treat (2014) 147(3):539–50. doi: 10.1007/s10549-014-3128-y
303. Zhao Y, Liu H, Liu Z, Ding Y, Ledoux SP, Wilson GL, et al. Overcoming trastuzumab resistance in breast cancer by targeting dysregulated glucose metabolism. Cancer Res (2011) 71(13):4585–97. doi: 10.1158/0008-5472.CAN-11-0127
304. Arroyo-Crespo JJ, Arminan A, Charbonnier D, Deladriere C, Palomino-Schatzlein M, Lamas-Domingo R, et al. Characterization of triple-negative breast cancer preclinical models provides functional evidence of metastatic progression. Int J Cancer (2019) 145(8):2267–81. doi: 10.1002/ijc.32270
305. Sirois I, Aguilar-Mahecha A, Lafleur J, Fowler E, Vu V, Scriver M, et al. A Unique Morphological Phenotype in Chemoresistant Triple-Negative Breast Cancer Reveals Metabolic Reprogramming and PLIN4 Expression as a Molecular Vulnerability. Mol Cancer Res (2019) 17(12):2492–507. doi: 10.1158/1541-7786.MCR-19-0264
Keywords: breast cancer, metabolism, metastasis, molecular mechanisms, metabolic phenotypes, glycolysis, hypoxia
Citation: Wang L, Zhang S and Wang X (2021) The Metabolic Mechanisms of Breast Cancer Metastasis. Front. Oncol. 10:602416. doi: 10.3389/fonc.2020.602416
Received: 03 September 2020; Accepted: 23 November 2020;
Published: 07 January 2021.
Edited by:
Xiaosong Chen, Shanghai Jiao Tong University, ChinaReviewed by:
Yingying Xu, The First Affiliated Hospital of China Medical University, ChinaCopyright © 2021 Wang, Zhang and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shizhen Zhang, emhhbmdzaGl6aGVuQHpqdS5lZHUuY24=; Xiaochen Wang, d2FuZ3hpYW9jaGVuQHpqdS5lZHUuY24=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.