 Lu Liu1,2
Lu Liu1,2 Ganlu Wang
Ganlu Wang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 23 December 2020
Sec. Cancer Immunity and Immunotherapy
Volume 10 - 2020 | https://doi.org/10.3389/fonc.2020.597468
Human leukocyte antigen-G (HLA-G) is a non-classical major histocompatibility complex class I (MHC I) molecule, and under physiological conditions, its expression is strictly restricted to the maternal–fetal interface and immune-privileged organs where HLA-G is expected to contribute to establishment and maintenance of immune tolerance. However, the expression of HLA-G has been found in various types of tumors, and the level of its expression frequently correlates with high-grade histology and poor prognosis, raising the possibility that it may play a negative role in tumor immunity. ILT2 and ILT4, present on a broad of immune cells, have been identified as the main receptors engaging HLA-G, and their interactions have been found to allow the conversion of effectors like NK cells and T cells to anergic or unresponsive state, activated DCs to tolerogenic state, and to drive the differentiation of T cells toward suppressive phenotype. Therefore, tumors can employ HLA-G to modulate the phenotype and function of immune cells, allowing them to escape immune attack. In this review, we discuss the mechanism underlying HLA-G expression and function, its role played in each step of the tumor-immunity cycle, as well as the potential to target it for therapeutic benefit.
Human leukocyte antigen-G (HLA-G) is a member of the nonclassical major histocompatibility complex (MHC) class I family. Its expression is initially described as restricted to the fetal–maternal interface, where it protects the fetus from the NK cell-mediated lysis (1). Therefore, the presence of HLA-G on cytotrophoblasts is proposed as a mechanism employed by the fetal to establish and maintain maternal–fetal immune tolerance. Subsequently, it was learned that inhibitory receptors, ILT2 and ILT4, were responsible for HLA-G-mediated inhibitory effect on NK cells (2). Numerous studies reveal that ILT2 and ILT 4 are broadly expressed on a wide range of immune cells, such as NK, T, B cells, and DCs, raising the possibility that HLA-G may exert immunosuppressive function on these cells (3). A series of studies provided strong experimental support for this idea, namely, the interaction of HLA-G with these inhibitory receptors can inhibit proliferation and cytotoxicity of T cells, and modulate the activity of DCs, neutrophils, macrophages, and B cells (4–10). In addition, HLA-G is reported to bind CD8 and KIR2DL4, resulting in the apoptosis of activated CD8+ T cells and the inhibition of NK-mediated cytotoxicity, respectively (1, 11).
Currently, HLA-G has been receiving increased attention, since it has been detected in numerous pathological conditions, particularly in tumors, and is expected to have a direct implication in the development of these diseases (12, 13). In this review, we will discuss how HLA-G expression is regulated under normal or abnormal conditions, and highlight the role of HLA-G played in tumor escape as well as the potential to target it for therapeutic benefit.
The HLA-G gene is located within the MHC class I locus on the short arm of chromosome 6 (p21.31), one of the most polymorphic regions in the human genome (14). However, HLA-G shows only minimal variation, with 80 alleles encoding 21 protein variants, far less than that of classical MHC class I genes (The International Immunogenetics Database-IMGT/HLA, database version 3.41.0) (15).
The exon/intron organization of HLA-G gene is identical to that of classical MHC class I molecules, which is composed of eight exons and seven introns (14). Because a termination code is located at the second codon of exon 6 of HLA-G gene, most of exon 6 and all of exons 7 and 8 would not be translated into protein (Figure 1). HLA-G gene can encode seven isoforms by alternative splicing of primary transcripts, including four membrane-bound (HLA-G1, HLA-G2, HLA-G3, and HLA-G4) and three soluble isoforms (HLA-G5, HLA-G6, and HLA-G7). Soluble HLA-G (sHLA-G) can also be generated through proteolytic cleavage of membrane-bound isoforms. HAL-G1 and HLA-G5 share an identical extracellular structure with classical MHC class I molecules: a heavy chain of three globular domains noncovalently bound to beta-2-microglobulin (β2M) and a peptide (16). The other isoforms do not bind β2M due to lack of one or two extracellular globular domains. It is now recognized that HLA-G spontaneously dimerizes through the unpaired cysteine in its α1 domain (17). Several lines of study reveal that HLA-G dimers bind to ILT receptors with a higher affinity and their inhibitory function is more efficiently than that of monomers (18).
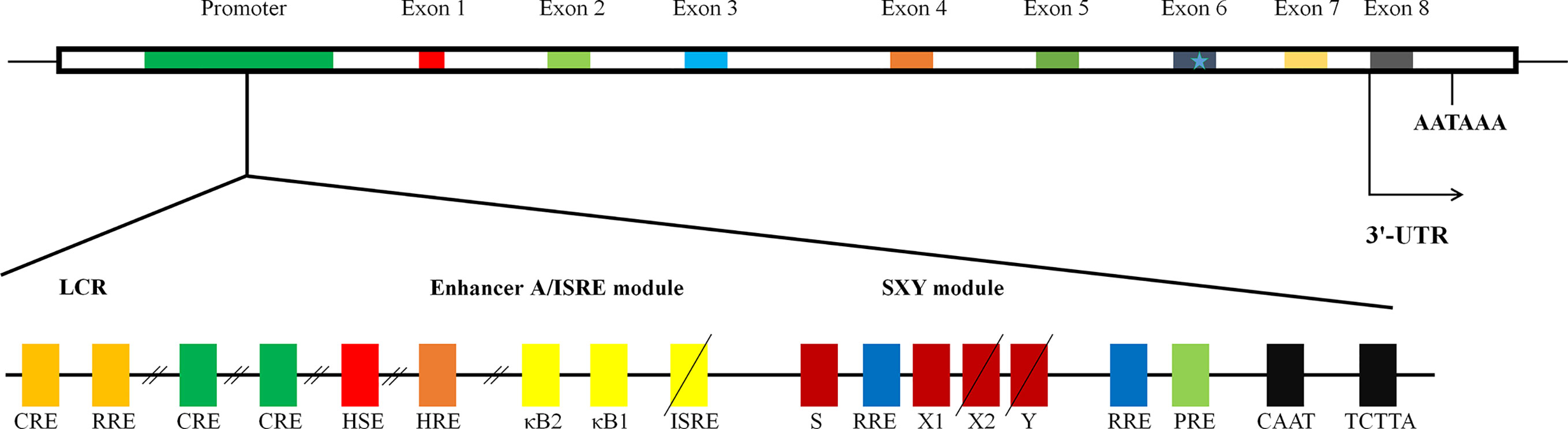
Figure 1 The structure of gene of HLA-G. The schematic diagram of HLA-G gene. It is composed of eight exons and seven introns. Many regulatory elements have been identified and characterized to regulate HLA-G expression. Three conserved cis-regulatory elements, enhancer A, the interferon-stimulated regulatory element (ISRE), and SXY module, mediating the transactivation of classical MHC class I genes; locus control region (LCR), located at least 1.2 kb upstream and crucial for spatio-temporal transcription; heat shock element (HSE), binding heat shock factor 1 in response to heat-shock or arsenate treatments; progesterone response element (PRE), mediating the up-regulation of HLA-G by progesterone during pregnancy; hypoxia response element (HRE), bound by hypoxia-inducible factor in response to hypoxia commonly observed in a majority of malignant tumors; three cAMP response elements (CRE), crucial for basal level of promoter activity of HLA-G; an additional functional ISRE, located at position −746 and capable of transactivating HLA-G following IFN-β treatment; three ras response elements (RREs), served as the binding site for transcriptional repressive factor RREB 1 (ras responsive element binding 1).
Physiologically, the expression of HLA-G is strictly restricted to fetal tissues such as amniotic cells, erythroid precursors, and cytotrophoblasts, and in adults, to immune-privileged organs, including thymus, pancreatic islets, endothelial cell precursors, and erythroblasts (19). HLA-G expression can also be induced during inflammatory-associated diseases such as cancer, transplantation, autoimmune disease, and infection (12, 13).
Given the important role in immune tolerance, HLA-G expression is tightly regulated at both the transcription and post-transcription levels. Like classical MHC class I genes, HLA-G promoter presents a CCAAT box and an unusual TATA element, TCTTA, controlling basal regulation (Figure 1). There are three conserved cis-regulatory elements in the MHC class I proximal promoter, namely enhancer A, the interferon-stimulated regulatory element (ISRE), and SXY module, by which transactivation of classical MHC class I genes is finely mediated (20–22). However, HLA-G gene exhibits nucleotide sequence variations, mutations, and/or deletions in those regions, rendering HLA-G unresponsive to NF-κB, interferon regulatory factor 1, and class II transactivator DNA-binding factors (22). Indeed, significant difference in expression distribution and function of HLA-G from those of classical MHC class I molecules suggests the existence of alternative mechanisms beyond the principal MHC class I transactivation mechanisms. To date, several active regulatory elements have been described (21). These include locus control region (LCR), heat shock element (HSE), progesterone response element (PRE), hypoxia response element (HRE), three cAMP response elements, an additional functional ISRE, and three ras response elements, by which the expression of HLA-G is mediated in response to specific signals from environment, by which HLA-G expression is regulated in response to specific environment cues, such as hypoxia and progesterone (Figure 1) (21).
Numerous studies have revealed that sequence polymorphism at 5’ upstream regulatory region (5’ URR) and 3’ untranslated region (3’UTR) is also associated with the expression of HLA-G (23–25). In contrast to classical MHC, HLA-G genes is shown to be well conserved in the coding region, but its 5’ URR and 3’UTR display a high level of polymorphism (24, 25). Currently, three variable sites in the 3’UTR, including the 14-base pair insertion/deletion (14-bp INS/DEL), +3142C/G and +3187A/G, have been reported to affect HLA-G expression by modifying mRNA stability or microRNA binding sties as well as the pattern of alternative splicing (26, 27). By analyzing the data from the 1000 Genomes project, 32 polymorphic sites have been identified within 5’URR, of which some—for example, variable site at positions −762 (between a CRE and ISRE)—are close to known regulatory elements and may somehow influence the binding of transcription factors (28). Notably, because polymorphism observed at non-coding regions of HLA-G can affect its expression pattern, it seems to be a genetic factor implicated in the cancer susceptibility. 14-bp INS/DEL, the most extensively studied polymorphism of HLA-G, has been shown to have a strong association with many tumor types, particularly breast and liver cancer, and is generally indicative of an increased risk of cancer (29).
HLA-G gene transcription activity could also be controlled by cis-acting epigenetic mechanisms involving DNA methylation and histone acetylation (30, 31). It has been reported that CpG methylation was associated with the HLA-G silencing in cultured cell lines of various origins and this methylation-mediated repression could be reversed by demethylating treatment for all cell-lines studied (30). Mouillot et al. have demonstrated that the level of histone acetylation in HLA-G promoter of HLA-G-expressing cells is significantly higher than that of HLA-G-deficient cells (31). In addition, several microRNAs, including the miR-152 family (miR-148a, miR-148b, and miR-152) and miR-133, have been characterized to regulate HLA-G expression at a post-translational level (32).
Although HLA-G shows high degree of sequence and structure similarities with classical MHC class I molecules, its main function is not in antigen presentation (18). It is well accepted that the primary function of HLA-G is to serve as an inhibitory ligand for immunocompetent cells, contributing to the establishment and maintenance of tolerance. HLA-G-induced tolerance mainly operates in two mechanisms. The first comprises direct suppression of effector cells: HLA-G directly binds to inhibitory receptors ILT2 and ILT4 that are widely expressed on NK cells, T cells, B cells, and neutrophils in which they mediate negative signaling that counteract immune activation, such as inhibiting T cell proliferation, cytotoxicity, and secretion of cytotoxic mediators (4–10). Moreover, it has been found that HLA-G can induce a shift in the expression of surface proteins present on NK, T, B cells, and antigen-presenting cells (APCs), such that immunosuppressive effects of HLA-G are amplified and maintained. For example, HLA-G significantly up-regulated the expression of its inhibitory receptors, ILT2, ILT3, ILT4, and KIR2DL4 on a broad range of immune cells, and down-regulated the expression of chemokine receptors, CCR2, CXCR3, and CXCR5 on T cells and CXCR4 and CXCR5 on germinal center B cells, which are crucial for the migration, differentiation, and function of immune cells (33–35). A second category of tolerogenic mechanisms comprises the induction of DCs tolerization, which, in turn, renders primed effector cells unresponsive, or promoted the development of regulatory cells. It has been shown that the treatment of human monocyte-derived DCs with HLA-G generated tolerogenic DCs, with a decrease in the level of expression of MHC class II and costimulatory molecules CD80 and CD86 (8). Stimulation T cells with HLA-G-treated DCs could favor to convert naïve CD4+ and CD8+ T cells to CD4+CD25+CTLA4+ and CD8+CD28- regulatory T cells, respectively, rather than effector T cells (8).
In addition, HLA-G can drive macrophages reprogramming to a M2 phenotype, as indicated by up-regulated CD163 and IDO-1, and down-regulated CD86, and these M2-like macrophages suppress T cell responses, such as inhibition of IFN-γ production and proliferation (10, 36). Besides inhibiting effector function of T cells, M2 macrophages are viewed as obligate partners for tumor cell due to secreting a wide range of bioactive mediators, such as VEGF and MMP-9, to promote tumor migration, invasion, and metastasis. Therefore, HLA-G could profoundly alter the phenotype and functional activity of immune cells, and thus may be involved in regulating multiple aspects of the immune response during tumor development.
ctopic expression of “tissue-restricted” HLA-G has been detected in many types of malignancies and is frequently associated with advanced tumor stage and poor prognosis in multiple cohorts of patients with cancer (13, 37). Table 1 summarizes the studies on the diagnostic and prognostic potential of HLA-G in malignancies (38–74). HLA-G functions as a tolerogenic molecule and contributes to establish an immunosuppressive milieu, enabling tumors to develop without challenge. This may explain why overexpression of HLA-G in tumors correlates with high-grade histology and poor prognosis in patients with breast, lung, ovarian, and pancreatic cancer.
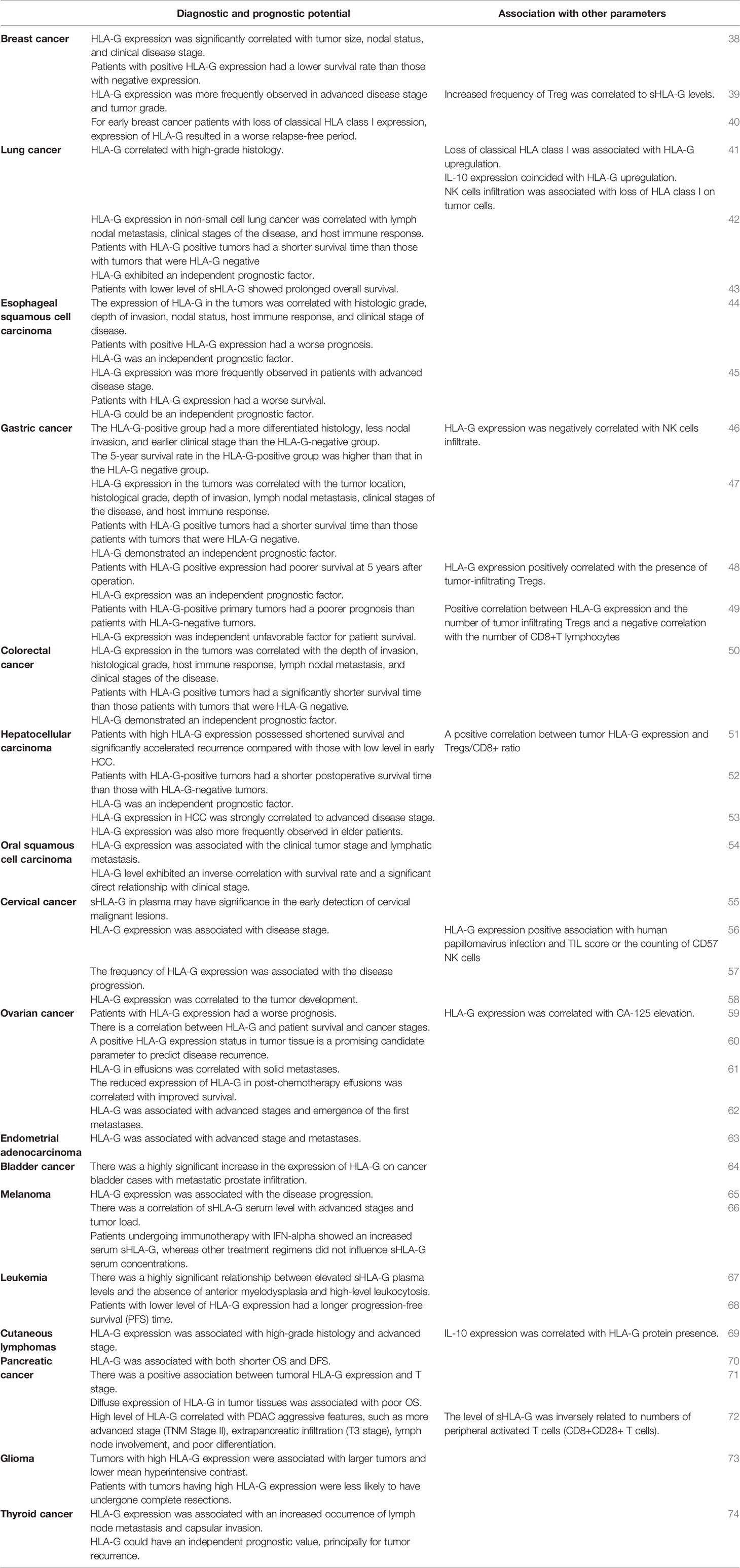
Table 1 Clinical studies involving diagnostic and prognostic significance of HLA-G in cancer.
T cells are presumed to be key effectors in cancer immunosurveillance, but to perform this role, they must be properly activated by DCs, home to tumor, and recognize and respond to their target. HLA-G can prevent all of the above requirements for T cell immunosurveillance by directly inhibiting DC maturation, T cell proliferation, and T cell-mediated cytotoxicity, as well as by inducting naïve T cell differentiation into regulatory T cells and recruiting immunosuppressive cells into tumor (Figure 2).
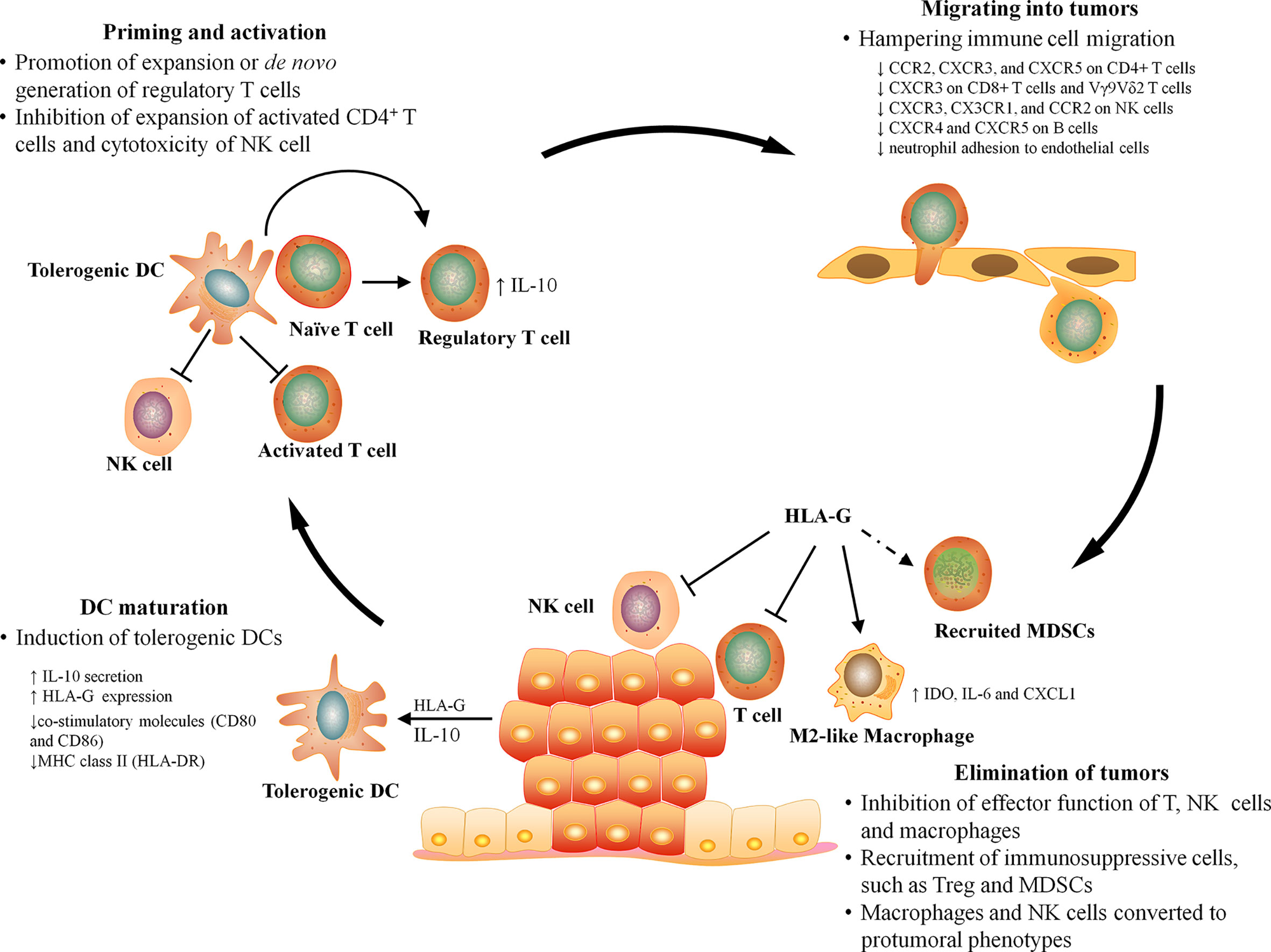
Figure 2 The effect of HLA-G on antitumor immunity. The antitumor immunity can be broadly divided into four major stages, DC maturation, T cell priming, leukocyte infiltration, and elimination. HLA-G can influence every stage of antitumor immune responses by inhibiting DC maturation, inducing T cell tolerance, hampering the migration of leukocyte, and suppressing the effector function and recruiting immunosuppressive cells at tumor sites.
T cell-mediated immunosurveillance is initiated in draining lymph nodes where naïve T lymphocytes differentiate into cytotoxic T lymphocytes upon encountering mature DCs that present tumor-derived antigens with proper co-stimulation signals. A series of studies by Horuzsko group clearly revealed that HLA-G can induce the decrease in the level of expression of MHC class II (HLA-DR) and co-stimulatory molecules (CD80 and CD86) in human monocyte-derived DCs, all of which are required for T cell priming and activation (8, 75). Functional assays further confirmed that these HLA-G treated DCs not only display a reduced capacity to present antigen on MHC II and activate CD4+ T cells, but also preferentially promote the differentiation of naïve CD4+ T cells into immunosuppressive/regulatory cells (8). Therefore, HLA-G can convert DCs into tolerogenic phenotype. Gregori et al. identified and characterized a novel subset of tolerogenic DCs, termed DC-10, which could also express HLA-G. DC-10 is present in vivo, and can be inducible in vitro from monocytes in the presence of IL-10 (76). Notably, when cultured in the presence of IL-10, DCs up-regulate the expression of HLA-G and its receptor, and produce more IL-10, resulting in a positive feedback loop.
HLA-G-expressing DCs have been detected in many types of tumor in vivo, such as in lung, breast, and ovarian cancer and melanoma, and correlated with a poor clinical outcome (77). In vitro, Grange et al. found that CD105+ cancer stem cells (CSCs) inhibited the maturation of DCs in a cell contact independent manner when they were cultured together (78). HLA-G released by CSCs was thought to be responsible for this inhibitory effect because the addition of blocking antibodies for HLA-G permitted monocyte-derived DCs to differentiate into mature state. These data, together with experimental studies described above, argue that HLA-G alone or in combination with IL-10 could drive DCs development toward a tolerogenic state at tumor sites.
Tolerogenic DCs is viewed as a crucial factor responsible for tumor immune escape. It has been shown that the interaction of naïve T cells with tolerogenic DCs can result in the induction of peripheral T cell tolerance in a T cell-intrinsic (e.g., antigen-specific anergy and deletion) or -extrinsic (e.g., via T regulatory cells or cytokines) fashion (79). The effect of HLA-G treated or positive DCs on T cell differentiation, expansion, and effector function has been extensively studied in vitro (8, 75, 76). Ristich et al. demonstrated that HLA-G treated DCs changed the cytokine profile (e.g., reducing the production of IL-2 and INF-γ, and increasing IL-10) and inhibited the expansion of activated CD4+ T cells and thus rendered T cell anergic (8). LeMaoult et al. also demonstrated that HLA-G-transfected DCs (HLA-G+ DCs) not only induced CD4+ T cell anergy but also caused the differentiation of CD4+ T cells into suppressive cells, as indicated by unresponsiveness to allostimulation in mixed lymphocyte reactions (80). Further phenotypic and functional analysis revealed that these HLA-G+ DCs primed T cell were CD4low Foxp3- or CD8low Foxp3-, and exerted immunosuppressive function through the actions of IL-10 (81).
To effectively control the growth of cancer cells, T cells must gain access to, and function within the tumor microenvironment. Migration of lymphocytes into tissues is a complex process that requires highly coordinated interactions, involving chemokine receptors and chemokines, and integrins, selectins, and their respective ligands. A role of HLA-G in regulating chemokine receptor repertoires in T cell was first described by Morandi et al. who demonstrated that HLA-G can significantly down-regulate the expression of CCR2, CXCR3, and CXCR5 on CD4+ T cells, and CXCR3 on CD8+ T cells and Vγ9Vδ2 T cells (81). Similar result has been found in NK cells, sHLA-G inducing CXCR3, CX3CR1, and CCR2 down-regulation in NK cells (82). Increasing evidence has suggested that these chemokine receptors are crucial for the migration of effector T and NK cells toward inflamed tissues. For example, CXCR3-deficient NK cells fail to migrate toward tumor following activation (83). Additionally, in the germinal centers (GCs) that are the main sites where activated B cells differentiate into memory B cells and plasma cells, CXCR4 and CXCR5 expressions on B cells were significantly down-regulated following exposure to sHLA-G, which limited B cell development by dampening B cell trafficking in GCs (35). It has been reported that HLA-G expressed on endothelial cells inhibited NK cells adhesion and transmigration, two key events which occurred during activated NK cells infiltrating from blood into inflamed sites (84). Mociornita et al. also found that in the model of cardiac allograft vasculopathy, HLA-G is overexpressed in vascular endothelial following everolimus treatment, which leads to the inhibition of HCASMC proliferation and of TNFα-stimulated neutrophil adhesion to endothelial cells at all concentrations (85). However, HLA-G has little effect on the migration of control T cells. Given that a high level of serum sHLA-G was frequently detected in patients with cancer, the importance of HLA-G in hampering the migration of NK and T cells into tumor microenvironment is becoming increasingly appreciated.
It has been well-established that HLA-G can directly bind to the inhibitory receptor, ILT-2 and ILT-4, on activated T cells, resulting in dramatically diminishing their proliferative and cytotoxic activities (4–6). Besides direct inhibitory actions, HLA-G can recruit suppressive cells to inflammatory site, by which effector functions of T cells are suppressed. According to the study of Agaugué et al, they developed an HLA-G+ xenotumor model by injecting melanoma tumor cells (M8) or M8 transfected with HLA-G into immunocompetent mice, and found that M8 expressing HLA-G grew rapidly, whereas M8 were rejected immediately in vivo (86). Analysis of the immune responses following the tumor progression released that HLA-G promoted accumulation and suppressive activity of MDSCs in HLA-G+ tumor-bearing mice through engagement of the paired immunoglobulin like receptor-B (PIR-B), the homolog of human ILTs. Therefore, HLA-G can inhibit the antitumor T cell responses in a direct and/or indirect manner.
Generally, innate immune cells, such as NK cells and macrophages, function as the first line resistance against transformed cells, and activate a much more potent T cell response against tumors. HLA-G is initially characterized as a suppressive regulator for NK cells to induce maternal-fetal tolerance by inhibiting their cytotoxic activity (1). For tumors, ectopic expression of “tissue-restricted” HLA-G is essential for evading NK-mediated killing, since malignant transformation of cells is frequently associated with the loss of expression of MHC class I molecules, rendering them directly susceptible to NK-mediated lysis, as proposed by the “missing self hypothesis” (87). It has been reported that in lung cancer and classical Hodgkin’s lymphoma, a frequent focal or complete loss of HLA class I molecules associated with HLA-G protein expression (41, 88). In addition to exerting direct inhibitory action on NK cells, HLA-G is found to impair NK/DC crosstalk, resulting in a slight inhibition of NK cell cytotoxicity (89). Several studies have revealed an important role of HLA-G in promoting vascular remodeling by activating uterine NK cells that secrete pro-angiogenic factors during early progeny (90, 91). This finding raises the possibility that HLA-G may alter the functional properties of tumor-infiltrating NK cells in a microenvironment similar to that of embryo and thus contribute to the angiogenesis at the tumor sites.
Similarly, HLA-G has long been known to be a potent negative regulator of macrophages (36). Moreover, it has been shown that HLA-G can drive macrophage polarization toward a protumoral phenotype, characterized by increased expression IDO, IL-6, and CXCL1, which could result in the inhibition of T cell response and the tumor progression; that is, HLA-G-polarized macrophages act as tumor-associated macrophages at tumor sites (36, 37). It has been found that in breast cancer and clear cell renal cell carcinoma, macrophages bearing ILT2 and ILT4 receptor, respectively, are present around HLA-G-positive tumor cells, which cooperatively establish an immune-tolerant microenvironment (92, 93). Therefore, HLA-G not only exerts immunosuppressive effects on macrophages and NK cells but also drives reprogramming into tumor-promoting cells.
Currently, no clear explanation for the ectopic expression of “tissue-restricted” HLA-G in various types of tumors exists. Because no genetic alternation that is heavily implicated in the upregulation of HLA-G gene, such as copy number gain or translocation, is detected in cancer genome, elevated HLA-G promoter activity is thought to be crucial for its expression in tumors. As described above, there are several cis-regulatory elements present in the promoter region of HLA-G gene which may respond to specific signals from the tumor microenvironment (Figure 1). For example, HRE can be bound by hypoxia-inducible factor in response to hypoxia commonly observed in a majority of malignant tumors; an additional functional ISR that is located at position −746 is capable of transactivating HLA-G following administration of IFNs for immunotherapy of malignant diseases; PRE has been reported to mediate the up-regulation of HLA-G by progesterone implicated in breast cancer progression; HSE can bind heat shock factor 1 in response to hypoxic stress and induce HLA-G gene transcription. Therefore, tumor microenvironmental factors, such as hypoxia, stress, hormones, and inflammation, have been proposed to activate HLA-G expression in tumor cells. It has been observed that HLA-G expression is gradually downregulated or even lost during long-term culture of primary tumor cells, which provides direct evidence for this hypothesis (41). A series of in vitro studies using tumor models have investigated the effect of these microenvironmental factors on HLA-G expression of tumor cells. Mouillot et al. found that hypoxia can activate HLA-G gene transcription in HLA-G-negative cell lines (94). He et al. demonstrated that HLA-G expression in breast cancer MCF-7 cells was upregulated by progesterone but was inhibited by its antagonist (38). Additionally, analysis of HLA-G expression in malignant lesions reveals that upregulation of HLA-G expression frequently correlates with high inflammatory infiltration, which in turn releases a variety of cytokines (91). In vitro studies have shown that GM-CSF and IFN-γ secreted by the infiltrating cytotoxic T cells can enhance HLA-G expression in tumor cells. In contrast, by systematically testing the effect of various cytokines in the choriocarcinoma cell line JEG-3, Persson et al. recently found that cytokines studied had no or adverse effect on HLA-G expression (95). These conflicting results suggest that HLA-G modulation by cytokines may be a cell-type specific phenomenon; JEG-3 decreases the production of HLA-G when exposed to the persistence of cytokines with high concentration.
Many immunosuppressive mechanisms are evolved by the host immune system to protect tissue damage caused by excessive or inappropriate immune activation, but they also provide opportunities for tumor to evade antitumor immune responses. Physiologically, HLA-G expression is strictly restricted to the maternal–fetal interface and immune-privileged organs where it is expected to contribute to establishment and maintenance of immune tolerance. However, overexpression of HLA-G is frequently detected in patients with cancers and thus is viewed as a commonly immunosuppressive strategy employed by tumor to counteract effective immune responses by manipulating the phenotype and function of immune cells, such as DCs, macrophages, and NK and T cells (Table 2). As we are now recognizing, HLA-G influences almost every stage of antitumor immune responses, such as T cell priming by DCs, and infiltration and function of effector cells at tumor (Figure 2). Therefore, HLA-G may represent an attractive target for therapeutic intervention. Several preclinical studies have provided some evidence that blocking HLA-G/ILTs signaling with antibody or down-regulating HLA-G expression with RNA interference can restore function of immune cells and prevent tumor reoccurrence (37, 96, 97). Currently, a clinical trial in phase I (NCT04485013) is underway targeting HLA-G by TTX-80, a monoclonal antibody, for patients with HLA-G-positive advanced cancers. Dumont et al. recently demonstrated the potential of combination of HLA-G and PD-1/PD-L1 blockade to confer a greater benefit to cancer patients, particularly for those with nonresponsiveness to anti-PD-1/PD-L1 (98). Therefore, combination blocking HLA-G with other immune checkpoints (PD-1/PD-L1 or CTLA-4) represents an inspiring strategy for cancer treatment, which may help overcome the resistance routinely developing in patients treated with a single immunotherapy. Currently, to bring HLA-G blockade therapy into clinical reality, several problems have yet to be solved, such as identifying predictive biomarkers for assessing the therapeutic effectiveness and elucidating the exact ways in which environmental factors and/or genetic changes regulate the expression of HLA-G in tumor or immune cells.
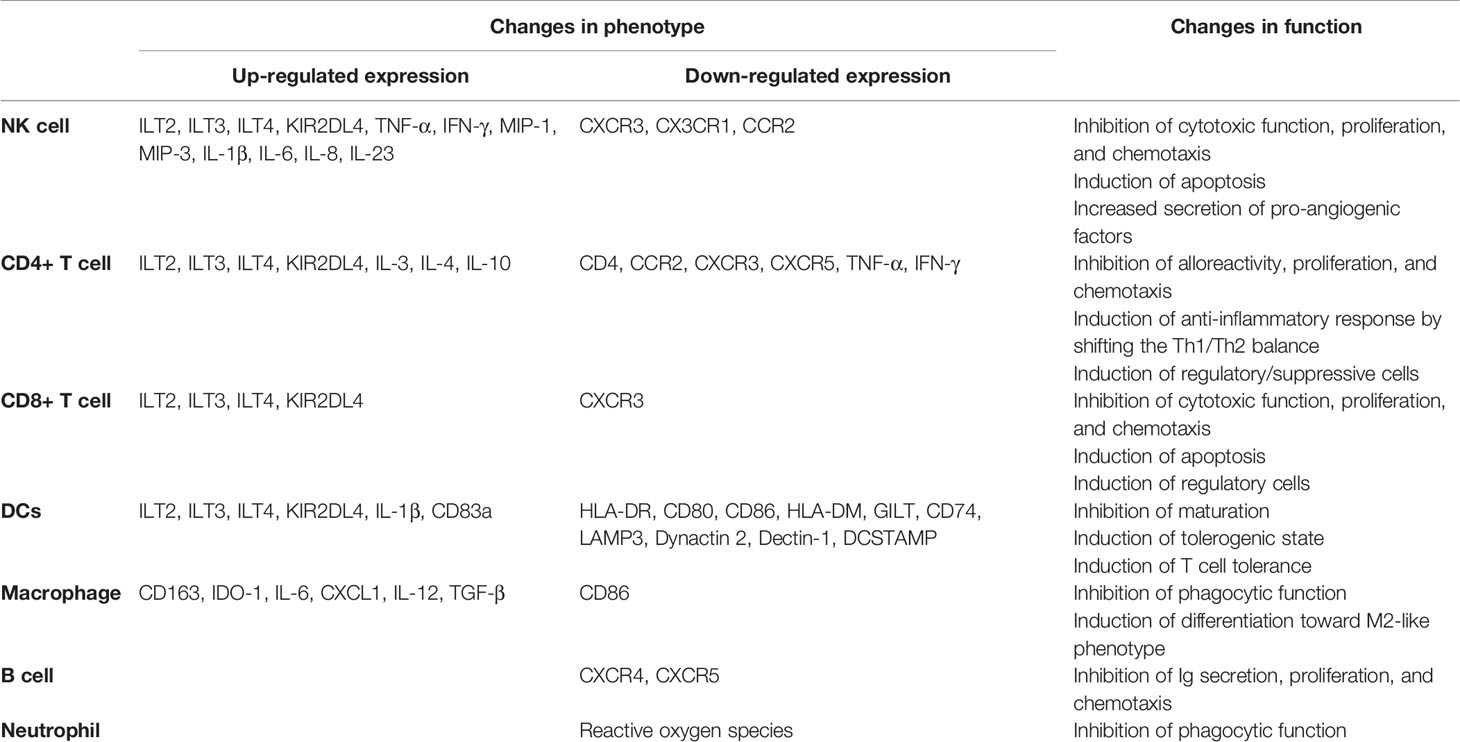
Table 2 Significant changes in phenotype and function of immune cells induced by HLA-G.
GW designed work. LL and LW wrote the paper. LZ and CH contributed to critical revision of the manuscript. All authors contributed to the article and approved the submitted version.
This study was supported by Shenzhen Foundation of Science and Technology (JCYJ20170307094549868 and JCYJ20180305123929814), Science and Technology Bureau of Baoan (2018JD124), and Funds for Young Scholar of Shenzhen Baoan People’s Hospital (2018A003).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Rouas-Freiss N, Gonçalves RM, Menier C, Dausset J, Carosella ED. Direct Evidence to Support the Role of HLA-G in Protecting the Fetus From Maternal Uterine Natural Killer Cytolysis. Proc Natl Acad Sci USA (1997) 94:11520–5. doi: 10.1073/pnas.94.21.11520
2. Pazmany L, Mandelboim O, Valés-Gómez M, Davis DM, Reyburn HT, Strominger JL. Protection From Natural Killer Cell-Mediated Lysis by HLA-G Expression on Target Cells. Science (1996) 274:792–5. doi: 10.1126/science.274.5288.792
3. Hirayasu K, Arase H. Functional and genetic diversity of leukocyte immunoglobulin-like receptor and implication for disease associations. J Hum Genet (2015) 60:703–8. doi: 10.1038/jhg.2015.64
4. Le Gal FA, Riteau B, Sedlik C, Khalil-Daher I, Menier C, Dausset J, et al. HLA-G-mediated inhibition of antigen-specific cytotoxic T lymphocytes. Int Immunol (1999) 11:1351–6. doi: 10.1093/intimm/11.8.1351
5. Bahri R, Hirsch F, Josse A, Rouas-Freiss N, Bidere N, Vasquez A, et al. Soluble HLA-G inhibits cell cycle progression in human alloreactive T lymphocytes. J Immunol (2006) 176:1331–9. doi: 10.4049/jimmunol.176.3.1331
6. Riteau B, Menier C, Khalil-Daher I, Sedlik C, Dausset J, Rouas-Freiss N, et al. HLA-G inhibits the allogeneic proliferative response. J Reprod Immunol (1999) 43:203–11. doi: 10.1016/s0165-0378(99)00034-0
7. Riteau B, Rouas-Freiss N, Menier C, Paul P, Dausset J, Carosella ED. HLA-G2, -G3, and -G4 isoforms expressed as nonmature cell surface glycoproteins inhibit NK and antigen-specific CTL cytolysis. J Immunol (2001) 166:5018–26. doi: 10.4049/jimmunol.166.8.5018
8. Ristich V, Liang S, Zhang W, Wu J, Horuzsko A. Tolerization of dendritic cells by HLA-G. Eur J Immunol (2005) 35:1133–42. doi: 10.1002/eji.200425741
9. Baudhuin J, Migraine J, Faivre V, Loumagne L, Lukaszewicz AC, Payen D, et al. Exocytosis acts as a modulator of the ILT4-mediated inhibition of neutrophil functions. Proc Natl Acad Sci USA (2013) 110:17957–62. doi: 10.1073/pnas.1221535110
10. Lombardelli L, Aguerre-Girr M, Logiodice F, Kullolli O, Casart Y, Polgar B, et al. HLA-G5 induces IL-4 secretion critical for successful pregnancy through differential expression of ILT2 receptor on decidual CD4+ T cells and macrophages. J Immunol (2013) 191:3651–62. doi: 10.4049/jimmunol.1300567
11. Contini P, Ghio M, Poggi A, Filaci G, Indiveri F, Ferrone S, et al. Soluble HLA-A,-B,-C and -G molecules induce apoptosis in T and NK CD8+ cells and inhibit cytotoxic T cell activity through CD8 ligation. Eur J Immunol (2003) 33:125–34. doi: 10.1002/immu.200390015
12. Carosella ED, Favier B, Rouas-Freiss N, Moreau P, Lemaoult J. Beyond the increasing complexity of the immunomodulatory HLA-G molecule. Blood (2008) 111:4862–70. doi: 10.1182/blood-2007-12-127662
13. Rouas-Freiss N, Moreau P, LeMaoult J, Carosella ED. The dual role of HLA-G in cancer. J Immunol Res (2014) 2014:359748. doi: 10.1155/2014/359748
14. Heinrichs H, Orr HT. HLA non-A,B,C class I genes: their structure and expression. Immunol Res (1990) 9:265–74. doi: 10.1007/BF02935526
15. Robinson J, Barker DJ, Georgiou X, Cooper MA, Flicek P, Marsh S. IPD-IMGT/HLA Database. Nucleic Acids Res (2020) 48:D948–55. doi: 10.1093/nar/gkz950
16. Morales PJ, Pace JL, Platt JS, Langat DK, Hunt JS. Synthesis of beta(2)-microglobulin-free, disulphide-linked HLA-G5 homodimers in human placental villous cytotrophoblast cells. Immunology (2007) 122:179–88. doi: 10.1111/j.1365-2567.2007.02623.x
17. Gonen-Gross T, Achdout H, Gazit R, Hanna J, Mizrahi S, Markel G, et al. Complexes of HLA-G protein on the cell surface are important for leukocyte Ig-like receptor-1 function. J Immunol (2003) 171:1343–51. doi: 10.4049/jimmunol.171.3.1343
18. Shiroishi M, Kuroki K, Ose T, Rasubala L, Shiratori I, Arase H, et al. Efficient leukocyte Ig-like receptor signaling and crystal structure of disulfide-linked HLA-G dimer. J Biol Chem (2006) 281:10439–47. doi: 10.1074/jbc
19. Carosella ED, HoWangYin KY, Favier B, LeMaoult J. HLA-G-dependent suppressor cells: Diverse by nature, function, and significance. Hum Immunol (2008) 69:700–7. doi: 10.1016/j.humimm.2008.08.280
20. Gobin SJ, van den Elsen PJ. Transcriptional regulation of the MHC class Ib genes HLA-E, HLA-F, and HLA-G. Hum Immunol (2000) 61:1102–7. doi: 10.1016/s0198-8859(00)00198-1
21. Moreau P, Flajollet S, Carosella ED. Non-classical transcriptional regulation of HLA-G: an update. J Cell Mol Med (2009) 13:2973–89. doi: 10.1111/j.1582-4934.2009.00800.x
22. van den Elsen PJ. Expression regulation of major histocompatibility complex class I and class II encoding genes. Front Immunol (2011) 2:48. doi: 10.3389/fimmu.2011.00048
23. Larsen MH, Hviid TV. Human leukocyte antigen-G polymorphism in relation to expression, function, and disease. Hum Immunol (2009) 70:1026–34. doi: 10.1016/j.humimm.2009.07.015
24. Hviid TV, Sørensen S, Morling N. Polymorphism in the regulatory region located more than 1.1 kilobases 5’ to the start site of transcription, the promoter region, and exon 1 of the HLA-G gene. Hum Immunol (1999) 60:1237–44. doi: 10.1016/s0198-8859(99)00130-5
25. Castelli EC, Mendes-Junior CT, Deghaide NH, de Albuquerque RS, Muniz YC, Simões RT, et al. The genetic structure of 3’untranslated region of the HLA-G gene: polymorphisms and haplotypes. Genes Immun (2010) 11:134–41. doi: 10.1038/gene.2009.74
26. Rousseau P, Le Discorde M, Mouillot G, Marcou C, Carosella ED, Moreau P. The 14 bp deletion-insertion polymorphism in the 3’ UT region of the HLA-G gene influences HLA-G mRNA stability. Hum Immunol (2003) 64:1005–10. doi: 10.1016/j.humimm.2003.08.347
27. Manaster I, Goldman-Wohl D, Greenfield C, Nachmani D, Tsukerman P, Hamani Y, et al. MiRNA-mediated control of HLA-G expression and function. PLoS One (2012) 7:e33395. doi: 10.1371/journal.pone.0033395
28. Castelli EC, Veiga-Castelli LC, Yaghi L, Moreau P, Donadi EA. Transcriptional and posttranscriptional regulations of the HLA-G gene. J Immunol Res (2014) 2014:73408. doi: 10.1155/2014/734068
29. Dias FC, Castelli EC, Collares CV, Moreau P, Donadi EA. The Role of HLA-G Molecule and HLA-G Gene Polymorphisms in Tumors, Viral Hepatitis, and Parasitic Diseases. Front Immunol (2015) 6:9. doi: 10.3389/fimmu.2015.00009
30. Moreau P, Mouillot G, Rousseau P, Marcou C, Dausset J, Carosella ED. HLA-G gene repression is reversed by demethylation. Proc Natl Acad Sci USA (2003) 100:1191–6. doi: 10.1073/pnas.0337539100
31. Mouillot G, Marcou C, Rousseau P, Rouas-Freiss N, Carosella ED, Moreau P. HLA-G gene activation in tumor cells involves cis-acting epigenetic changes. Int J Cancer (2005) 113:928–36. doi: 10.1002/ijc.20682
32. Zhu XM, Han T, Wang XH, Li YH, Yang HG, Luo YN, et al. Overexpression of miR-152 leads to reduced expression of human leukocyte antigen-G and increased natural killer cell mediated cytolysis in JEG-3 cells. Am J Obstet Gynecol (2010) 202:592.e1–e7. doi: 10.1016/j.ajog.2010.03.002
33. LeMaoult J, Zafaranloo K, Le Danff C, Carosella ED. HLA-G up-regulates ILT2, ILT3, ILT4, and KIR2DL4 in antigen presenting cells, NK cells, and T cells. FASEB J (2005) 19:662–4. doi: 10.1096/fj.04-1617fje
34. Morandi F, Ferretti E, Bocca P, Prigione I, Raffaghello L, Pistoia V. A novel mechanism of soluble HLA-G mediated immune modulation: downregulation of T cell chemokine receptor expression and impairment of chemotaxis. PLoS One (2010) 5(7):e11763. doi: 10.1371/journal.pone.0011763
35. Naji A, Menier C, Morandi F, Agaugué S, Maki G, Ferretti E, et al. Binding of HLA-G to ITIM-bearing Ig-like transcript 2 receptor suppresses B cell responses. J Immunol (2014) 192:1536–46. doi: 10.4049/jimmunol.1300438
36. Lee CL, Guo Y, So KH, Vijayan M, Guo Y, Wong VH, et al. Soluble human leukocyte antigen G5 polarizes differentiation of macrophages toward a decidual macrophage-like phenotype. Hum Reprod (2015) 30:2263–74. doi: 10.1093/humrep/dev196
37. Lin A, Yan WH. Heterogeneity of HLA-G Expression in Cancers: Facing the Challenges. Front Immunol (2018) 9:2164. doi: 10.3389/fimmu.2018.02164
38. He X, Dong DD, Yie SM, Yang H, Cao M, Ye SR, et al. HLA-G expression in human breast cancer: implications for diagnosis and prognosis, and effect on allocytotoxic lymphocyte response after hormone treatment in vitro. Ann Surg Oncol (2010) 17:1459–69. doi: 10.1245/s10434-009-0891-9
39. Chen HX, Lin A, Shen CJ, Zhen R, Chen BG, Zhang X, et al. Upregulation of human leukocyte antigen-G expression and its clinical significance in ductal breast cancer. Hum Immunol (2010) 71:892–8. doi: 10.1016/j.humimm.2010.06.009
40. de Kruijf EM, Sajet A, van Nes JG, Natanov R, Putter H, Smit VT, et al. HLA-E and HLA-G expression in classical HLA class I-negative tumors is of prognostic value for clinical outcome of early breast cancer patients. J Immunol (2010) 185:7452–9. doi: 10.4049/jimmunol.1002629
41. Urosevic M, Kurrer MO, Kamarashev J, Mueller B, Weder W, Burg G, et al. Human leukocyte antigen G up-regulation in lung cancer associates with high-grade histology, human leukocyte antigen class I loss and interleukin-10 production. Am J Pathol (2001) 159:817–24. doi: 10.1016/S0002-9440(10)61756-7
42. Yie SM, Yang H, Ye SR, Li K, Dong DD, Lin XM. Expression of human leucocyte antigen G (HLA-G) is associated with prognosis in non-small cell lung cancer. Lung Cancer (2007) 58:267–74. doi: 10.1016/j.lungcan.2007.06.011
43. Schütt P, Schütt B, Switala M, Bauer S, Stamatis G, Opalka B, et al. Prognostic relevance of soluble human leukocyte antigen-G and total human leukocyte antigen class I molecules in lung cancer patients. Hum Immunol (2010) 71:489–95. doi: 10.1016/j.humimm.2010.02.015
44. Yie SM, Yang H, Ye SR, Li K, Dong DD, Lin XM. Expression of HLA-G is associated with prognosis in esophageal squamous cell carcinoma. Am J Clin Pathol (2007) 128:1002–9. doi: 10.1309/JNCW1QLDFB6AM9WE
45. Lin A, Zhang X, Zhou WJ, Ruan YY, Xu DP, Wang Q, et al. Human leukocyte antigen-G expression is associated with a poor prognosis in patients with esophageal squamous cell carcinoma. Int J Cancer (2011) 129:1382–90. doi: 10.1002/ijc.25807
46. Ishigami S, Natsugoe S, Miyazono F, Nakajo A, Tokuda K, Matsumoto M, et al. HLA-G expression in gastric cancer. Anticancer Res (2006) 26:2467–72.
47. Yie SM, Yang H, Ye SR, Li K, Dong DD, Lin XM. Expression of human leukocyte antigen G (HLA-G) correlates with poor prognosis in gastric carcinoma. Ann Surg Oncol (2007) 14:2721–9. doi: 10.1245/s10434-007-9464-y
48. Du L, Xiao X, Wang C, Zhang X, Zheng N, Wang L, et al. Human leukocyte antigen-G is closely associated with tumor immune escape in gastric cancer by increasing local regulatory T cells. Cancer Sci (2011) 102:1272–80. doi: 10.1111/j.1349-7006.2011.01951.x
49. Tuncel T, Karagoz B, Haholu A, Ozgun A, Emirzeoglu L, Bilgi O, et al. Immunoregulatory function of HLA-G in gastric cancer. Asian Pac J Cancer Prev (2013) 14:7681–4. doi: 10.7314/apjcp.2013.14.12.7681
50. Ye SR, Yang H, Li K, Dong DD, Lin XM, Yie SM. Human leukocyte antigen G expression: as a significant prognostic indicator for patients with colorectal cancer. Mod Pathol (2007) 20:375–83. doi: 10.1038/modpathol.3800751
51. Cai MY, Xu YF, Qiu SJ, Ju MJ, Gao Q, Li YW, et al. Human leukocyte antigen-G protein expression is an unfavorable prognostic predictor of hepatocellular carcinoma following curative resection. Clin Cancer Res (2009) 15:4686–93. doi: 10.1158/1078-0432.CCR-09-0463
52. Wang Y, Ye Z, Meng XQ, Zheng SS. Expression of HLA-G in patients with hepatocellular carcinoma. Hepatobiliary Pancreat Dis Int (2011) 10:158–63. doi: 10.1016/s1499-3872(11)60025-8
53. Lin A, Chen HX, Zhu CC, Zhang X, Xu HH, Zhang JG, et al. Aberrant human leucocyte antigen-G expression and its clinical relevance in hepatocellular carcinoma. J Cell Mol Med (2010) 14:2162–71. doi: 10.1111/j.1582-4934.2009.00917.x
54. Imani R, Seyedmajidi M, Ghasemi N, Moslemi D, Shafaee S, Bijani A. HLA-G Expression is Associated with an Unfavorable Prognosis of Oral Squamous Cell Carcinoma. Asian Pac J Cancer Prev (2018) 19:2527–33. doi: 10.22034/APJCP.2018.19.9.2527
55. Zheng N, Wang CX, Zhang X, Du LT, Zhang J, Kan SF, et al. Up-regulation of HLA-G expression in cervical premalignant and malignant lesions. Tissue Antigens (2011) 77:218–24. doi: 10.1111/j.1399-0039.2010.01607.x
56. Dong DD, Yang H, Li K, Xu G, Song LH, Fan XL, et al. Human leukocyte antigen-G (HLA-G) expression in cervical lesions: association with cancer progression, HPV 16/18 infection, and host immune response. Reprod Sci (2010) 17:718–23. doi: 10.1177/1933719110369183
57. Li XJ, Zhang X, Lin A, Ruan YY, Yan WH. Human leukocyte antigen-G (HLA-G) expression in cervical cancer lesions is associated with disease progression. Hum Immunol (2012) 73:946–9. doi: 10.1016/j.humimm.2012.07.041
58. Miranda LN, Reginaldo FP, Souza DM, Soares CP, Silva TG, Rocha KB, et al. Greater expression of the human leukocyte antigen-G (HLA-G) and interleukin-17 (IL-17) in cervical intraepithelial neoplasia: analytical cross-sectional study. Sao Paulo Med J (2015) 133:336–42. doi: 10.1590/1516-3180.2013.7170009
59. Jung YW, Kim YT, Kim SW, Kim S, Kim JH, Cho NH, et al. Correlation of human leukocyte antigen-G (HLA-G) expression and disease progression in epithelial ovarian cancer. Reprod Sci (2009) 16:1103–11. doi: 10.1177/1933719109342131
60. Babay W, Ben Yahia H, Boujelbene N, Zidi N, Laaribi AB, Kacem D, et al. Clinicopathologic significance of HLA-G and HLA-E molecules in Tunisian patients with ovarian carcinoma. Hum Immunol (2018) 79:463–70. doi: 10.1016/j.humimm.2018.02.012
61. Davidson B, Elstrand MB, McMaster MT, Berner A, Kurman RJ, Risberg B, et al. HLA-G expression in effusions is a possible marker of tumor susceptibility to chemotherapy in ovarian carcinoma. Gynecol Oncol (2005) 96:42–7. doi: 10.1016/j.ygyno.2004.09.049
62. Menier C, Prevot S, Carosella ED, Rouas-Freiss N. Human leukocyte antigen-G is expressed in advanced-stage ovarian carcinoma of high-grade histology. Hum Immunol (2009) 70:1006–9. doi: 10.1016/j.humimm.2009.07.021
63. Barrier BF, Kendall BS, Sharpe-Timms KL, Kost ER. Characterization of human leukocyte antigen-G (HLA-G) expression in endometrial adenocarcinoma. Gynecol Oncol (2006) 103:25–30. doi: 10.1016/j.ygyno.2006.01.045
64. El-Chennawi FA, Auf FA, El-Diasty AM, El-Daim MA, El-Sherbiny SM, Ali A, et al. Expression of HLA-G in cancer bladder. Egypt J Immunol (2005) 12:57–64.
65. Ibrahim EC, Aractingi S, Allory Y, Borrini F, Dupuy A, Duvillard P, et al. Analysis of HLA antigen expression in benign and malignant melanocytic lesions reveals that upregulation of HLA-G expression correlates with malignant transformation, high inflammatory infiltration and HLA-A1 genotype. Int J Cancer (2004) 108:243–50. doi: 10.1002/ijc.11456
66. Ugurel S, Rebmann V, Ferrone S, Tilgen W, Grosse-Wilde H, Reinhold U. Soluble human leukocyte antigen–G serum level is elevated in melanoma patients and is further increased by interferon-alpha immunotherapy. Cancer (2001) 92:369–76. doi: 10.1002/1097-0142(20010715)92:2<369::aid-cncr1332>3.0.co;2-u
67. Gros F, Sebti Y, de Guibert S, Branger B, Bernard M, Fauchet R, et al. Soluble HLA-G molecules increase during acute leukemia, especially in subtypes affecting monocytic and lymphoid lineages. Neoplasia (2006) 8:223–30. doi: 10.1593/neo.05703
68. Nückel H, Rebmann V, Dürig J, Dührsen U, Grosse-Wilde H. HLA-G expression is associated with an unfavorable outcome and immunodeficiency in chronic lymphocytic leukemia. Blood (2005) 105:1694–8. doi: 10.1182/blood-2004-08-3335
69. Urosevic M, Willers J, Mueller B, Kempf W, Burg G, Dummer R. HLA-G protein up-regulation in primary cutaneous lymphomas is associated with interleukin-10 expression in large cell T-cell lymphomas and indolent B-cell lymphomas. Blood (2002) 99:609–17. doi: 10.1182/blood.v99.2.609
70. Hiraoka N, Ino Y, Hori S, Yamazaki-Itoh R, Naito C, Shimasaki M, et al. Expression of classical human leukocyte antigen class I antigens, HLA-E and HLA-G, is adversely prognostic in pancreatic cancer patients. Cancer Sci (2020) 111:3057–70. doi: 10.1111/cas.14514
71. Zhou L, Niu ZY, Liang ZY, Zhou WX, You L, Wang MY, et al. HLA-G impairs host immune response and predicts poor prognosis in pancreatic cancer. Am J Transl Res (2015) 7:2036–44.
72. Xu YF, Lu Y, Cheng H, Jiang J, Xu J, Long J, et al. High Expression of Human Leukocyte Antigen-G is Associated with a Poor Prognosis in Patients with PDAC. Curr Mol Med (2015) 15:360–7. doi: 10.2174/1566524015666150401102218
73. Wang Y, Fan X, Li H, Lin Z, Bao H, Li S, et al. Tumor border sharpness correlates with HLA-G expression in low-grade gliomas. J Neuroimmunol (2015) 282:1–6. doi: 10.1016/j.jneuroim.2015.02.013
74. Nunes LM, Ayres FM, Francescantonio IC, Saddi VA, Avelino MA, Alencar R, et al. Association between the HLA-G molecule and lymph node metastasis in papillary thyroid cancer. Hum Immunol (2013) 74:447–51. doi: 10.1016/j.humimm.2012.12.012
75. Liang S, Horuzsko A. Mobilizing dendritic cells for tolerance by engagement of immune inhibitory receptors for HLA-G. Hum Immunol (2003) 64:1025–32. doi: 10.1016/j.humimm.2003.08.348
76. Gregori S, Tomasoni D, Pacciani V, Scirpoli M, Battaglia M, Magnani CF, et al. Differentiation of type 1 T regulatory cells (Tr1) by tolerogenic DC-10 requires the IL-10-dependent ILT4/HLA-G pathway. Blood (2010) 116:935–44. doi: 10.1182/blood-2009-07-234872
77. Amiot L, Ferrone S, Grosse-Wilde H, Seliger B. Biology of HLA-G in cancer: a candidate molecule for therapeutic intervention? Cell Mol Life Sci (2011) 68:417–31. doi: 10.1007/s00018-010-0583-4
78. Grange C, Tapparo M, Tritta S, Deregibus MC, Battaglia A, Gontero P, et al. Role of HLA-G and extracellular vesicles in renal cancer stem cell-induced inhibition of dendritic cell differentiation. BMC Cancer (2015) 15:1009. doi: 10.1186/s12885-015-2025-z
79. van den Broek M. Dendritic cells break bonds to tolerize. Immunity (2007) 27:544–6. doi: 10.1016/j.immuni.2007.10.004
80. LeMaoult J, Krawice-Radanne I, Dausset J, Carosella ED. HLA-G1-expressing antigen-presenting cells induce immunosuppressive CD4+ T cells. Proc Natl Acad Sci USA (2004) 101:7064–9. doi: 10.1073/pnas.0401922101
81. Naji A, Le Rond S, Durrbach A, Krawice-Radanne I, Creput C, Daouya M, et al. CD3+CD4low and CD3+CD8low are induced by HLA-G: novel human peripheral blood suppressor T-cell subsets involved in transplant acceptance. Blood (2007) 110:3936–48. doi: 10.1182/blood-2007-04-083139
82. Morandi F, Ferretti E, Castriconi R, Dondero A, Petretto A, Bottino C, et al. Soluble HLA-G dampens CD94/NKG2A expression and function and differentially modulates chemotaxis and cytokine and chemokine secretion in CD56bright and CD56dim NK cells. Blood (2011) 118:5840–50. doi: 10.1182/blood-2011-05-352393
83. Kim J, Kim JS, Lee HK, Kim HS, Park EJ, Choi JE, et al. CXCR3-deficient natural killer cells fail to migrate to B16F10 melanoma cells. Int Immunopharmacol (2018) 63:66–73. doi: 10.1016/j.intimp.2018.07.026
84. Dorling A, Monk NJ, Lechler R II. HLA-G inhibits the transendothelial migration of human NK cells. Eur J Immunol (2000) 30:586–93. doi: 10.1002/1521-4141(200002)30:2<586::AID-IMMU586>3.0.CO;2-Y
85. Mociornita AG, Adamson MB, Tumiati LC, Ross HJ, Rao V, Delgado DH. Effects of everolimus and HLA-G on cellular proliferation and neutrophil adhesion in an in vitro model of cardiac allograft vasculopathy. Am J Transplant (2018) 18:3038–44. doi: 10.1111/ajt.15015
86. Agaugué S, Carosella ED, Rouas-Freiss N. Role of HLA-G in tumor escape through expansion of myeloid-derived suppressor cells and cytokinic balance in favor of Th2 versus Th1/Th17. Blood (2011) 117:7021–31. doi: 10.1182/blood-2010-07-294389
87. Kärre K. Natural killer cell recognition of missing self. Nat Immunol (2008) 9(5):477–80. doi: 10.1038/ni0508-477
88. Diepstra A, Poppema S, Boot M, Visser L, Nolte IM, Niens M, et al. HLA-G protein expression as a potential immune escape mechanism in classical Hodgkin’s lymphoma. Tissue Antigens (2008) 71:219–26. doi: 10.1111/j.1399-0039.2008.01005.x
89. Gros F, Cabillic F, Toutirais O, Maux AL, Sebti Y, Amiot L. Soluble HLA-G molecules impair natural killer/dendritic cell crosstalk via inhibition of dendritic cells. Eur J Immunol (2008) 38:742–9. doi: 10.1002/eji.200736918
90. Rajagopalan S, Bryceson YT, Kuppusamy SP, Geraghty DE, van der Meer A, Joosten I, et al. Activation of NK cells by an endocytosed receptor for soluble HLA-G. PLoS Biol (2006) 4:e9. doi: 10.1371/journal.pbio.0040009
91. Rajagopalan S. HLA-G-mediated NK cell senescence promotes vascular remodeling: implications for reproduction. Cell Mol Immunol (2014) 11:460–6. doi: 10.1038/cmi.2014.53
92. Lefebvre S, Antoine M, Uzan S, McMaster M, Dausset J, Carosella ED, et al. Specific activation of the non-classical class I histocompatibility HLA-G antigen and expression of the ILT2 inhibitory receptor in human breast cancer. J Pathol (2002) 196:266–74. doi: 10.1002/path.1039
93. García M, Palma MB, Verine J, Miriuka S, Inda AM, Errecalde AL, et al. The immune-checkpoint HLA-G/ILT4 is involved in the regulation of VEGF expression in clear cell renal cell carcinoma. BMC Cancer (2020) 20:624. doi: 10.1186/s12885-020-07113-8
94. Mouillot G, Marcou C, Zidi I, Guillard C, Sangrouber D, Carosella ED, et al. Hypoxia modulates HLA-G gene expression in tumor cells. Hum Immunol (2007) 68:277–85. doi: 10.1016/j.humimm.2006.10.016
95. Persson G, Bork J, Isgaard C, Larsen TG, Bordoy AM, Bengtsson MS, et al. Cytokine stimulation of the choriocarcinoma cell line JEG-3 leads to alterations in the HLA-G expression profile. Cell Immunol (2020) 352:104110. doi: 10.1016/j.cellimm.2020.104110
96. Porto IO, Mendes-Junior CT, Felício LP, Georg RC, Moreau P, Donadi EA, et al. MicroRNAs targeting the immunomodulatory HLA-G gene: a new survey searching for microRNAs with potential to regulate HLA-G. Mol Immunol (2015) 65:230–41. doi: 10.1016/j.molimm.2015.01.030
97. Carosella ED, Ploussard G, LeMaoult J, Desgrandchamps F. A Systematic Review of Immunotherapy in Urologic Cancer: Evolving Roles for Targeting of CTLA-4, PD-1/PD-L1, and HLA-G. Eur Urol (2015) 68:267–79. doi: 10.1016/j.eururo.2015.02.032
Keywords: human leukocyte antigen-G, immune cells, phenotype, function, tumor
Citation: Liu L, Wang L, Zhao L, He C and Wang G (2020) The Role of HLA-G in Tumor Escape: Manipulating the Phenotype and Function of Immune Cells. Front. Oncol. 10:597468. doi: 10.3389/fonc.2020.597468
Received: 21 August 2020; Accepted: 16 November 2020;
Published: 23 December 2020.
Edited by:
Fabio Malavasi, University of Turin, ItalyReviewed by:
Eric Vivier, INSERM U1104 Centre d’immunologie de Marseille-Luminy (CIML), FranceCopyright © 2020 Liu, Wang, Zhao, He and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ganlu Wang, Z2FubHV3YW5nMTFAMTYzLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.