 Eric Grignano
Eric Grignano Rudy Birsen
Rudy Birsen Nicolas Chapuis
Nicolas Chapuis Didier Bouscary
Didier Bouscary
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Oncol. , 18 September 2020
Sec. Cancer Metabolism
Volume 10 - 2020 | https://doi.org/10.3389/fonc.2020.586530
This article is part of the Research Topic Metabolic Rewiring in Leukemias View all 8 articles
Despite its crucial importance in numerous physiological processes, iron also causes oxidative stress and damage which can promote the growth and proliferation of leukemic cells. Iron metabolism is strictly regulated and the related therapeutic approaches to date have been to restrict iron availability to tumor cells. However, since a new form of iron-catalyzed cell death has been described, termed ferroptosis, and subsequently better understood, iron excess is thought to represent an opportunity to selectively kill leukemic cells and spare normal hematopoietic cells, based on their differential iron needs. This review summarizes the physiology of iron metabolism and its deregulation in leukemia, the known ferrotoposis pathways, and therapeutic strategies to target the altered iron metabolism in leukemia for the purposes of initiating ferroptosis in these cancer cells.
Iron is a crucial nutrient for the enzymes that are involved in ATP production (mitochondrial chain complex), DNA synthesis (ribonucleotide reductase), oxygen transport, antioxidant defense (peroxidase and catalase), oxygen sensing factors (hypoxia-inducible factor – HIF – and prolyl hydroxylases), and many others. The ability of iron to gain and lose electrons between its oxidized Fe3+-ferric- and Fe2+-ferrous- forms enables it to participate in free radical-generating reactions. Among these processes is the Fenton reaction, in which a ferrous iron donates an electron to hydrogen peroxide to yield hydroxyl radicals, thereby inducing highly reactive oxygen species (ROS) (1). The aberrant accumulation of iron and subsequent excess ROS levels generate oxidative stress, induces damage to DNA, proteins or lipids and even causing cell death. Significantly, these oxidative effects of iron can contribute to oncogenesis and iron is thought to be essential for the development of cancer (2).
Acute myeloid leukemia (AML) results from oligoclonal proliferations arising from the transformation of immature myeloid hematopoietic cells, characterized by a differentiation blockage and acquisition of a survival advantage and proliferation gain. AML displays high heterogeneity at the phenotypic, genetic and molecular levels (3). Recurrent molecular abnormalities resulting from mutations or translocations define the prognostic subgroups of AML, which vary in their sensitivity to conventional treatments such as chemotherapy and bone marrow transplantation (4). The prognosis for AML remains largely poor but new therapeutic strategies are emerging, stemming from better pathophysiological knowledge. Lastly, the association of the BCL2-BH3 mimetic venetoclax and demethylating agents (azacytidine or decitabine) directly targets the leukemic stem cell (LSC) compartment via oxidative metabolism perturbation and has shown compelling effectiveness in elderly patients with AML (5, 6).
To enable growth, leukemic cells exhibit an increased iron demand compared with normal cells (7). Accumulating evidence indicates that targeting iron homeostasis can induce differentiation and apoptosis in some leukemic cells (8–10), and these studies almost exclusively focused on intra-leukemic iron depletion by chelating agents. However, this iron dependency may also render cancer cells more vulnerable to iron-mediated cell death, referred to as ferroptosis (11), in which iron metabolism plays a key role. Hence, strategies aimed at targeting leukemic cells by increasing the intracellular iron pool could constitute a new therapeutic approach in AML. We therefore highlight the phenomenon of iron metabolism deregulation in AML in this review and focus on the opportunity of targeting this process to improve AML treatment.
Iron homeostasis is a highly tuned process. Dietary iron (predominantly in the form of Fe3+) is absorbed in the duodenum via divalent metal transporter 1 (DMT1) after the action of a ferric reductase, duodenal cytochrome b (Dcytb), which converts Fe3+ to Fe2+. Alternatively, heme iron is imported into enterocytes through an unknown mechanism and is thereafter, degraded by heme oxygenase 1 (HO-1), liberating Fe2+. Iron exits the basolateral surfaces of the enterocytes across the sole iron efflux pump yet known, ferroportin (FPN), along with hephaestin which oxidizes Fe3+ to Fe2+, and then is loaded onto transferrin (Tf). The holo-transferrin complex Tf – Fe3+ circulates in the plasma to deliver iron to its sites of utilization, mostly to the bone marrow to enable erythropoiesis. After binding to transferrin receptor 1 (TFR1), the Tf-Fe3+/TFR1 complex is subsequently taken into cells by receptor-mediated endocytosis, reduced to Fe2+ by the six-transmembrane epithelial antigen of prostate 3 (STEAP3), and transported into the cytosol by DMT1. The Tf/TFR1 complex is finally recycled to the cell surface. Recycling from senescent red blood cells by macrophages is by far the main source of iron, whereas dietary intake is marginal and compensates for iron losses only. The released Fe2+ constitutes the labile iron pool (LIP) and is either stored in ferritin (FTH) or used for metabolic needs, or even exported into the circulation via FPN after being re-oxidized by ceruloplasmin (12).
The predominant pathway for iron output into the cytosol from ferritin is controlled by nuclear receptor coactivator 4 (NCOA4)-mediated selective autophagy, whereby NCOA4 binds ferritin to traffic it to the lysosome where it is degraded and iron is then released for use by the cell (13). Non-stored cytoplasmic iron is either transported to mitochondria for heme synthesis or incorporated in Fe-S clusters. It can serve as a cofactor for several enzymes, such as prolyl hydroxylase 2 (PHD2), lipoxygenases (LOX), and others. At the systemic level, the iron balance is maintained via a strictly regulated absorption of dietary iron in the duodenum, which is mainly achieved by the ferroportin – hepcidin regulatory axis (14). The peptide hormone hepcidin, secreted primarily by hepatocytes, is the cardinal regulator of iron homeostasis. In conditions of excess iron, hepcidin binds to FPN and promotes its phosphorylation and subsequent lysosomal degradation in enterocytes and macrophages, thus blocking the delivery of iron into the circulation (15). At the cellular level, iron homeostasis is regulated at the post-transcriptional level by the binding of regulatory proteins IRP1 and IRP2 to the iron-responsive elements (IREs) within mRNAs. The IREs are hairpin structures located in 5′or 3′ untranslated regions (UTRs) of mRNAs and control either transcript stability or the initiation of translation. The role of the IRPs is to adapt, according to the cellular iron availability, and control the expression of proteins that regulate the import (TFR1, DMT1), storage (FTH), and export (FPN) of intracellular iron (16).
The normal mechanisms of iron handling are altered in cancer cells in order to facilitate tumor growth. Evidence for iron overload involvement in promoting cancer came some time ago from in vivo experiments (17), epidemiological studies (18), and cancer susceptibility in hemochromatosis-affected patients (19), but the underlying biological mechanisms have been more precisely understood for a decade (2). In particular, because of the high red blood cell transfusion needs in cancer patients due to normal erythropoiesis impairment and chemotherapy-induced anemia, iron excess is a common finding in leukemic patients. The organism response consists of limiting iron bioavailability, referred to as “the withholding response,” and therapeutic developments to date that are based on iron metabolism have mostly focussed on enabling this withholding response.
Iron is crucial for normal hematopoiesis since TFR1 inhibition and subsequent iron depletion impair the proliferation and differentiation of hematopoietic precursor cells, and reduce the regeneration potential of hematopoietic stem cells (HSCs) (20). Notably however, excess iron and ROS catalytic production also promote the malignant transformation of HSCs through nicotinamide adenine dinucleotide phosphate oxidases (NOX) and the subsequent depletion of glutathione (GSH) (21). DNA damage and double-strand breaks induced by ROS in myelodysplastic syndromes, a pre-leukemic disorder, may foster the transformation process in AML (22). On the other hand, HSC aging is associated with increasing ROS production, impaired HSC self-renewal and regeneration (23), and decreased erythropoiesis trough growth differentiation factor-11 (GDF11) secretion (24). However, in particular stress contexts, ROS promote short-term HSC repopulation, motility and differentiation (25), thus acting as a kind of bone marrow microenvironment messenger. Moreover, in monocytic AML, blasts produce high amounts of ROS via NADP oxidase (NOX) and thereby induce apoptosis in adjacent NK cells, and in CD4 and CD8 T-cells, through poly-ADP-ribose polymerase-1 (PARP-1), and a subsequent ineffective anti-leukemic adaptive response (26). Iron overload also disrupts the T-lymphocyte Th1/Th2 ratio and promotes regulatory T-cell production (Treg) (27).
Iron markers of intake, storage and export are commonly concurring to iron overload. TFR1 expression, known as CD71, is generally increased in leukemic cells compared to normal counterparts and its level may be directly correlated to the degree of differentiation as AML, whether minimally differentiated or not, has the highest level of CD71 expression (28). The prognostic significance of this observation is debatable however as even if high levels of TFR1 are associated with complex karyotypes and other features such as c-kit receptor expression (CD117), the survival outcomes seem to be unaffected (29). Notably, transferrin receptor 2 (TFR2) is also upregulated in different AML subtypes such as AML1, AML2, and erythroleukemia (AML6) (30, 31), and TFR2 α-subtype expression may be positively associated with a favorable prognosis (32). TFR2 is mostly present at the surface of hepatocytes and erythroid cells and acts as an iron sensor and hepcidin modulator (33). Since both TFR1 and TFR2 increase intracellular iron, the former, by increasing iron uptake and the latter via hepcidin upregulation through HFE binding, the prognosis discrepancies between each isoform likely imply an iron-independent pathway for TFR2. It is worth mentioning also that other forms of non-transferrin bound iron may penetrate cancer cells, such as for instance via lipocalin 2 (LCN2), also referred as neutrophil gelatinase-associated lipocalin (NGAL), whose expression is detected in leukemic cells, but at a lower rates than in normal HSC counterparts (34, 35). LCN2 overexpression at the transcriptional level is associated with a better prognosis, especially in normal karyotype AML with FLT3 wild-type expression (36).
Regarding the iron storage counterpart, serum ferritin is also frequently increased in leukemic patients and is a pejorative factor in overall and relapse-free survival in chemotherapy-treated patients (37–40) as well as in patients undergoing allogeneic stem cell transplantation (41). Ferritin comprises 24 polypeptide subunits of heavy chain (FTH and light chain (FTL). In an inflammation context, FTH appears to be a NF-κB downstream effector that suppresses TNFα-driven apoptosis via the inhibition of Jun N-terminal kinase (JNK) (42). In AML patients, a gene expression signature associated with FTH overexpression encompasses NF-κB and pro-oxidant pathways, leading to chemotherapy resistance (37).
Dysregulation of the ferroportin-hepcidin axis is a common feature in AML, entailing reduced iron efflux. Notably, low ferroportin expression in AML seems to correlate with improved outcomes and greater chemotherapy sensitivity, and is consistently found in core binding factor (CBF) AML subsets (43). Hepcidin expression varies heterogeneously, depending on the amount of iron, and the degree of anemia and inflammation. In AML patients, positive correlations have been shown between hepcidin and both ferritin and IL-6, whereas a high EPO level and anemia are associated with lower levels of hepcidine (44). Notably however, despite the expected high levels of hepcidin under conditions of inflammation and overexpression of ferritin, balancing strategies to counteract anemia such as erythroblast secretion of erythroferrone (ERFE) can foster hepcidin downregulation (45). An example of this is seen in myelodysplastic syndromes with an SF3B1 mutation (46). In response to systemic limitations in iron availability, autocrine secretion of hepcidin to degrade FPN has been described in several cancer models, although not yet shown in AML (47–49).
The ability of tumor cells to fuel iron availability that enables sustained proliferation, ROS accumulation and evasion of adaptative host immunity has prompted research into iron chelation strategies. Moreover, such strategies became of paramount interest in the context of iterative blood transfusions in leukemic patients, in whom chronic iron overload leads to secondary hemochromatosis responsible for cardiac, hepatic, or endocrinal damage.
Hence, iron chelators such as deferoxamine (DFO) and deferasirox (DFX), by lowering LIP and hampering ROS production and iron-dependent enzymes such as ribonucleotide reductase (50–53), exert anti-leukemic activity. Interestingly, as mentioned earlier, FTH effects on the NF-κB pathway and the subsequent inhibition of both TNF-JNK signaling pathway-mediated apoptosis and iron chelators have been shown to restore JNK and mitogen-activated protein kinase (MAPK) pathways (54, 55). This ability, along with the use of other differentiating agents, may overcome the impaired cellular differentiation in AML (8). Likewise, iron chelators may interfere with hypoxia-induced pathways such as that controlled by HIF1α (56) or with the REDD1 – mammalian target of rapamycin (mTOR) pathway (57). Iron chelators can also sensitize leukemic cells to conventional chemotherapy drugs (58, 59) or to demethylating agents such as decitabine in vitro (60), although the data are conflicting (61).
Similarly, strategies aiming at modulating factors involved in iron metabolism such as TFR1, have shown promising efficacy since the early 1990's (62, 63). To date, several strategies have been developed for targeting TFR1, including the use of its natural ligand Tf, targeting peptides, anti-TFR1 monoclonal antibodies, and antibody fragments (scFv) (64). As an example of this, the A24 monoclonal antibody that competitively binds to TFR1, inhibits Tf binding to TFR1, thus leading to TFR1 endocytosis and its downregulation at the cellular surface, and has shown proven efficacy against T-NHL (65, 66). Such a strategy raises concerns however due to high dependency of erythropoiesis on TFR1 (67). As well, clinical studies have reported only moderate and transient efficacy against hematological malignancies (68), which can be partly explained by the induced immunogenicity (69). Hence, TFR1-targeted drugs have been developed as a delivery system, harnessing the overexpression of TFR1 in leukemic cells. For instance, TFR1-targeted lipid nanoparticles containing an antisense oligonucleotide (ASO) against the antiapoptotic protein Bcl-2 have been developed (70, 71), as well as TFR1 conjugated to the autophagy activator compound artemisinin (72). In addition, TFR1 has been conjugated to nanoparticles bearing chemotherapeutics that are designed to pass through the blood-brain barrier (73). Ferritin is also a potential candidate to shelter targeted drugs (74), such as doxorubicin (75), in order to produce a longer circulation half-life and a higher tumor uptake. Another intriguing drug delivery mechanism relies on a pro-drug strategy via trioxolane conjugation, which takes advantage of the higher amounts of iron in malignancies. In this mechanism, a trioxolane-conjugated drug will react with Fe2+ in the tumor microenvironment to activate drug release (76).
Despite the many clues that support iron chelation as an anti-tumor strategy in leukemia and the advances in this field, another way to exploit the increased iron needs in leukemic cells is to consider the vulberability that this creates. Indeed, cancer cells are far more susceptible to iron-catalyzed necrosis, referred to as ferroptosis. Hence, strategies aimed at increasing LIP in cancer cells may overcome the limited antioxidant defenses, thus entailing ferroptosis.
Ferroptosis is a form of programed cell death, that differs from apoptosis, necroptosis or pyroptosis in terms of the characteristic morphological changes i.e., mitochondria with decreased crista, condensed membranes and rupture of the outer membrane, and an intact nucleus (11). Biologically, ferroptosis is characterized by the iron-catalyzed peroxidation of polyunsaturated fatty acids (PUFAs) containing phospholipids (PLs), thereby producing lipid ROS. Arachidonic acid (AA) or adrenic acid (AdA) are key membrane phospholipids that are processed by acyl-CoA synthase 4 (ACSL4) and lysophosphatidylcholine acyltransferase 3 (LPCAT3) to produce their esterification and generate phosphatidylethanolamine (PE)-AA/AdA that undergoes further oxididation to phospholipid hydroperoxides (PE-AA/AdA-OOH) by the lipoxygenases (LOXs) (77). Excessive Fe2+ fuels electron-driven lipid peroxidation (LOOH) from an alkoxyl or an hydroxyl radical via the Fenton reaction in the presence of peroxide (H2O2). These reactions are not mutually exclusive and can operate together, especially since the LOXs are iron-containing enzymes (78). Meanwhile, lipid ROS can also be formed spontaneously by autoxidation enzyme-catalyzed processes (79). Once formed, lipid peroxides can diffuse across lipid bilayers in a self-feedback loop manner and compromise membrane integrity (80). To prevent the lethal accumulation of lipid ROS, several protective mechanisms are involved. One is glutathione peroxidase 4 (GPX4), which was shown to constitutively hydrolyze lipid hydroperoxides (LOOH) into inactive redox alcohol radicals (LOH), thereby protecting the cell from ferroptosis. GPX4 is a selenocysteine enzyme that uses glutathione (GSH) as a cofactor and can be covalently inhibited by molecules such as (1S,3R)-RSL3 (RSL3), or indirectly blocked by the small molecules FIN56 or FINO2 through a still elusive mechanism (81). Moreover, GSH synthesis requires the rate-limiting amino acid cysteine, which is imported in its oxidized form cystine from the extracellular space via the sodium-independent cystine/glutamate antiporter system (consisting of two subunits SLC7A11 and SLC3A2). Inhibitors of system , such as the oncogenic RAS-selective lethal small molecule erastin, sulfasalazine or sorafenib, subsequently triggers ferroptosis in different cellular contexts (11, 82). In contrast, several lipophilic radical-trapping agents (RTA), such as ferrostatin-1 (Fer-1), liproxtatin-1, or α-tocopherol, acting as ROS-lipid scavengers, are able to halt ferroptosis. Mitochondria can play a key role in ferroptosis depending on the context i.e., whilst they are crucial in the case of cysteine deprivation, they may not be required under conditions of GPX4 inhibition (83). Moreover, CDGSH iron sulfur domain 1 (CISD1), an iron-containing outer mitochondrial membrane protein, modulates mitochondrial iron uptake and respiratory capacity and mitigates ferroptosis (84). Interestingly, ferroptosis can also modulate tumorigenic immune defenses as immunotherapy-activation of CD8+ T cells by checkpoint inhibitors such as anti-PD1 or anti-CTLA4 enhances ferroptosis-specific lipid peroxidation in tumor cells via system downregulation (85). Figure 1 summarizes the currently understood biology of ferroptosis, including its mechanisms and links with iron metabolism.
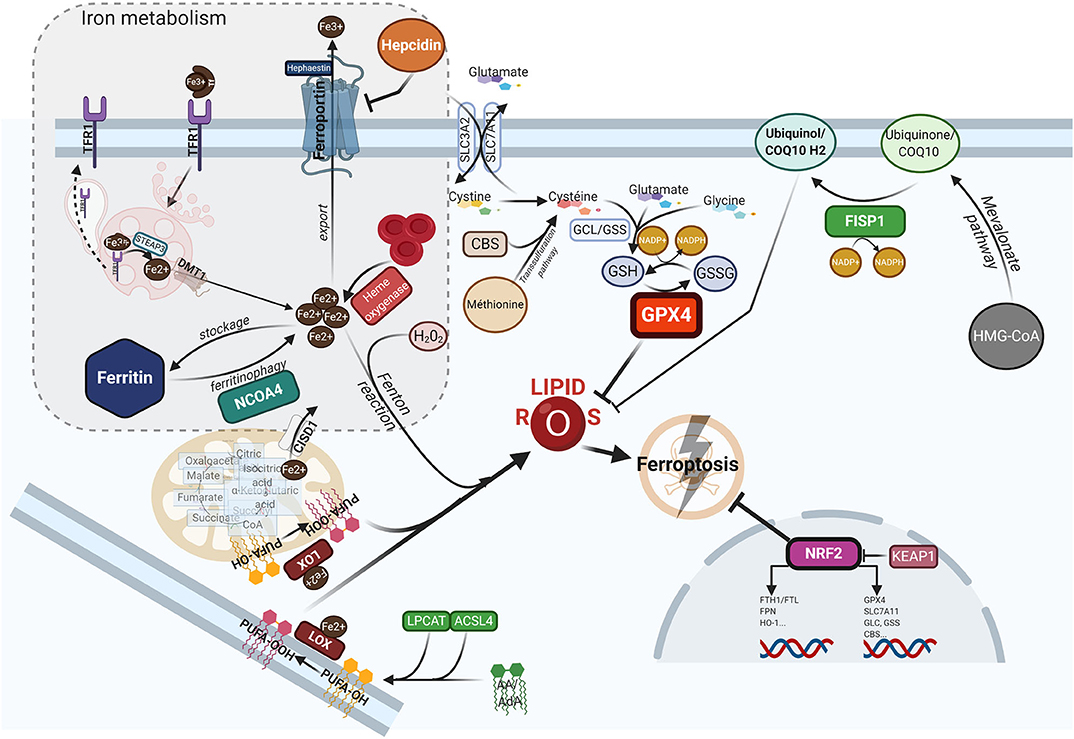
Figure 1. The ferroptotic cascade. Accumulation of free iron is a key initiator of ferroptosis. After loading onto Tf, the Fe3+/Tf complex binds to TFR1, and iron is thereafter released into endocytosis vesicles under lower pH conditions, where Fe3+ is reduced into the ferrous form Fe2+ under the action of ferrireductase STEAP3. Iron then exits into the cytosol via DMT1 to form the labile iron pool (LIP). The LIP represents a very slight per se, whereas most iron is either stored in FTH, either used as a cofactor for several iron-containing enzymes, or for the synthesis of heme. Excess iron is carried in the extracellular space through FPN after being reoxydized by hephaestin. FTH autophagic degradation, a process termed ferritinophagy, is mediated by NCOA4, a cargo receptor that binds to the heavy chain of FTH delivering it to the early-stage autophagosome, inducing the subsequent release of iron into the cytosol. Iron release from heme through HO-1 is another way to increase the LIP. Inside mitochondria, CISD1 modulates mitochondrial iron uptake and respiratory capacity and mitigates ferroptosis. The hormone hepcidin, mostly secreted by hepatocytes in the systemic circulation, triggers ferroportin lysosomal degradation, thus hindering iron exit. AA or AdA are key membrane phospholipids esterified by ACSL4 and LPCAT to generate PE-AA/ AdA, and further oxidized to phospholipid hydroperoxides (PE-AA/AdA-OOH) by LOXs. Free iron can also interact with ROS, specifically hydrogen peroxide, to form hydroxyl/peroxyl (LOH/LOOH) toxic radicals via the Fenton reaction. Therefore, iron can then abstract a hydrogen atom from PUFAs, forming a lipid radical which promptly reacts with oxygen to generate a lipid peroxide (PUFA-OOH). In a steady state, lipid peroxides and their degradation products are neutralized by GSH-based redox reactions. The xCT antiporter (consisting of two subunits SLC7A11 and SLC3A2) exports glutamine and imports cystine into the cell. Inside the cell, cystine is reduced to cysteine by cystine reductase and then GCL and GSS add L-glutamate and glycine respectively, to produce GSH. Another way to generate cysteine is via the trans-sulfuration pathway converting methionine in homocystein, and later cysteine by CBS. Many redox enzymes use GSH, including GPX4, which reduces reactive lipid peroxydes to their alcohol counterparts. Additionally, CoQ10, a byproduct of the mevalonate pathway, acts as a complemental RTA to mitigate ferroptosis. CoQ10 is reduced to CoQ1O-H2 by FSP1, enabling its activity. GPX4 and SLC7A11 are target genes of the master antioxidant regulator NRF2, as well as the enzymes contributing to GSH synthesis, GCL, GSS, and CBL. NRF2 additionally mediates iron metabolism by upregulating the transcriptional level of FTH, FPN, and HO-1. In the absence of redox stress, NRF2 is downregulated by the E3-ubiquitine ligase KEAP1. If the key redox regulator is genetically disrupted or pharmacologically inhibited, lipid peroxides and their degradation products accumulate, and thereby initiate ferroptosis through a yet unknown mechanism involving membrane destabilization, cytoskeletal changes, and cell death. AA, arachidonic acid; AdA, adrenic acid; ACSL4, acyi-CoA synthetase long chain family member 4; CBS, cystathionine-13-synthase; CISD1, CDGSH iron sulfur domain 1; CoQ10, coenzyme Q10/Ubiquinone-10; DMTl, divalent metal transporter 1; FSP1, ferroptosis-suppressor-protein 1; FPN, ferroportin; FTH, ferritin; GCL, glutamate-cysteine ligase; GPX4, glutathione peroxidase 4; GSH, glutathione; GSS, glutathione synthetase; HO-1, heme oxygenase 1; KEAP1, Kelch-like ECH-associated protein 1; LOXs, lipoxygenases; LPCAT, lysophosphatidylcholine acyltransferase; NCOA4, nuclear receptor coactivator 4; NRF2, nuclear factor (erythroid-derived 2)-like 2; PE, phosphatidylethanolamine; PUFA, polyunsaturated fatty acids; RTA, radical trapping agent; STEAP3, six-transmembrane epithelial antigen of the prostate 3; TFR1, transferrin receptor 1.
A hallmark of ferroptosis is the requirement for iron, supported by the fact that iron chelation by deferoxamine (DFO) prevents the experimental induction of ferroptosis (Dixon, cell 2012), although it cannot do so once ferroptosis is launched (86). In addition, transferrin has been shown to be critical for this pathway since its deprivation dramatically decreases ferroptosis (gao, mol cell 2015), as well as the silencing of iron metabolism master regulator 2 (IREB2) (11). TFR1 endocytosis recycling inhibition by heat-shock protein beta-1 (HSPB1) overexpression and subsequent iron intake inhibition also alleviates ferroptosis (87). On the other hand, precluding TFR1 and FTH degradation by proteasome inhibitors such as bortezomib along with iron overload can cooperate to trigger ferroptosis (88). Likewise, DMT1 has been shown to be up-regulated upon ferroptosis induction (89). Ferritinophagy is another key component of the iron supply mechanism regulating ferroptosis. Indeed, autophagy inhibitors or NCOA4 cargo receptor genetic deletion suppress ferroptosis (90), whilst ferritin overexpression consistently mitigates ferroptosis in a neuronal model (91). Dihydroartemisinin (DHA), a semi-synthetic derivative of artemisinin known for its anti-tumor properties, was shown to induce ferritinophagy by regulating the activity of the AMP-activated protein kinase AMPK/mTOR/p70S6K pathway, thereupon triggering ferroptotic cell death (92). Interestingly, mitochondrial function is critical for iron metabolism and subsequent ferroptosis. Indeed, mitochondrial iron intake inhibition by CDGSH iron sulfur domain 1 (CISD1) overexpression markedly inhibits ferroptosis (84), as does the overexpression of iron-sulfur cluster assembly enzyme (ISCU), a mitochondrial protein which plays a crucial role in Fe-S cluster biogenesis (92).
Ferroptosis has gained considerable interest as a potential widespread anti-cancer strategy, although most of the research thus far has been restricted to in vitro or murine in vivo models (93–98). GPX4 dependency and ferroptosis susceptibility can be acquired by cells in a therapy-resistant cell state (99), as for instance cells undergoing an epithelial-mesenchymal transition (EMT) (100). Hence, targeting leukemic cells via ferroptosis represents a promising new treatment approach, though very few data are available in AML models as yet. Recently, ferumoxytol (Feraheme©), an FDA approved intravenous iron nanoparticle for treating iron deficiency in patients with chronic kidney disease, showed anti-leukemic effects in vitro and in murine models bearing leukemia cells with low FPN expression (101). Iron salophen complexes, a kind of chemically engineered transition-metal complex, display in vitro anti-leukemic properties via ferroptosis or necroptosis (102). Notably, GPX transcription level in AML is associated with a worse prognosis and correlates with the reponse to oxidative stress, inflammation and the immmune response (103). Ferroptosis inducers such as erastin can also enhance the anticancer capacity of conventional chemotherapeutics like cytarabine or doxorubicin (104) in RAS-mutated AML cell lines. Moreover, erastin can boost the activity of more recent compounds such as APR-246, a p53-mutant reactivating compound in cancer (105). Erastin-induced ferroptosis in AML cell lines has been shown to involve high mobility group box 1 (HMGB1) cytosolic translocation, the RAS-JNK/p38 pathway and subsequent TFR1 up-regulation (106). Furthermore, autophagy-mediated ferritin degradation and iron subsequent supply play a key role in ferroptosis, explaining at least in part the anti-tumor activity of dihydroartemisin via the activation of the AMPK-mTOR signaling pathway (92). Recently, iron has shown the capacity to catalyze the oxidative demethylation of protein residues and nucleobases in association as a cofactor of demethylases like PHF8, which is a great interest in AML since demethylating agents have proven efficacy in this context (107). A putative model of the iron metabolism modulation that triggers ferroptosis is provided in Figure 2.
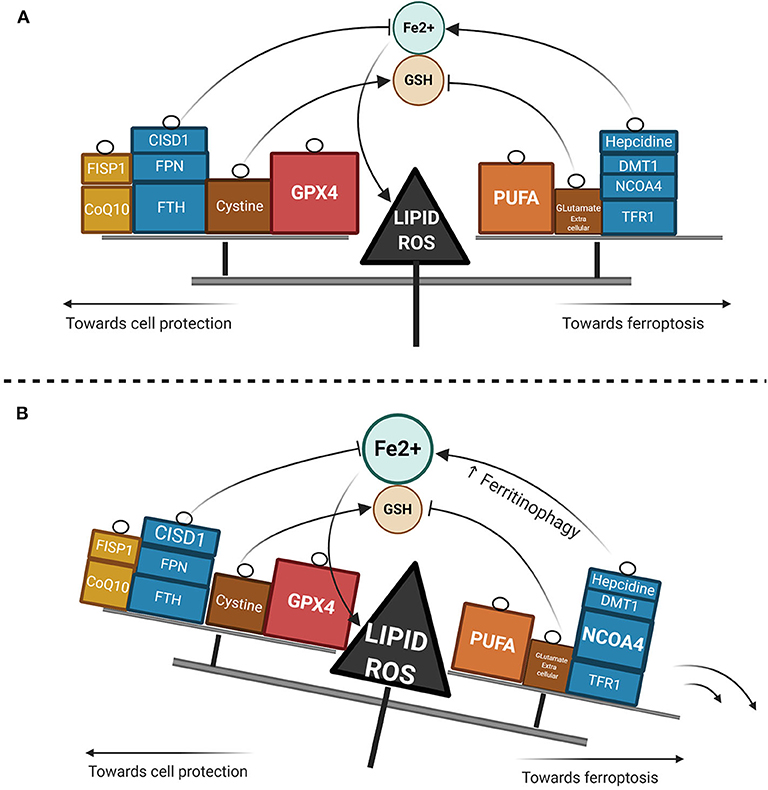
Figure 2. Lipid ROS homeostasis. (A) Schema showing enzymatic or substrate modulators of lipid ROS. The size of the box is proportional to the functional importance of the modulator. (B) Theorical imbalance of lipid ROS homeostasis, in this example caused by enhanced ferritinophagy through NCOA4 upregulation.
Recent insights into iron metabolism along with the recent discovery of ferroptosis have opened new avenues in the field of anti-tumor therapies. Metabolism rewiring between glycolysis and mitochondrial oxphos respiration in tumors has proven to be an anchor for new and effective therapeutic combination strategies such as the association of the BCL2-BH3 mimetic and hypomethylating agents, which are able to specifically target the LSC compartment. Additionally, iron metabolism dysregulation toward increased iron demands is a specific feature of tumor cells which has been considered almost exclusively until now as an opportunity to test iron deprivation approaches against the tumor cells, whether than using it as an Achilles heel. Opportunities to tailor new strategies that also utilize ferroptosis will arise in the coming years.
Because non-targeted iron supplementation could increase tumorigenicity and cause deleterious systemic effects, feeding tumor cells with iron or reshaping iron metabolism mechanisms to trigger ferroptosis will be a complex problem to solve. First, because of the heterogeneous sensitivity to iron-catalyzed necrosis among cancers, thorough analyses using in vitro and murine AML models are required to assess the feasibility of such strategies. Data are currently lacking in this regard for hematological malignancies and specifically for AML, thus hindering further therapeutic perspectives. Second, in addition to the iron supplementation or iron metabolism actor modulation, combination strategies using ferroptosis inducers are probably desirable to lower the sensitivity threshold of cancer cells to ferroptosis and thus avoid iron-mediated off-target damage. This point is of major interest since great efforts have been made to identify and develop ferroptosis triggers over the past decade. Hence, a widespread introduction of iron-based treatments into the mainstream oncology arsenal in a personalized fashion and with the purpose of inducing iron-catalyzed death is a promising anti-cancer strategy.
EG and DB contributed conception and design of the review. RB and NC provided critcial revisions to the content. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
AA, arachidonic acid; AdA, adrenic Acid; ACSL4, acyl-CoA synthetase long chain family member 4; AML, acute myeloid leukemia; DMT1, divalent Metal (Ion) Transporter 1; FTH, ferritin; FPN, ferroportin; GPX4, gluthatione peroxydase 4; GSH, glutathione; HSC, hematopoietic stem cell; IRE, iron responsive element; IRP, iron regulatory protein; LIP, labile iron pool; LOX, lipoxygenase; LPCAT, lysophosphatidylcholine acyltransferase 1; LSC, leukemic stem cell; NCOA4, nuclear receptor coactivator 4; PE, phosphatidylethanolamine; PL, phospholipids; PUFAs, polyunsaturated fatty acids; ROS, reactive oxygen species; Tf, transferrin; TFR1, transferrin receptor 1.
1. Lambeth JD, Neish AS. Nox enzymes and new thinking on reactive oxygen: a double-edged sword revisited. Annu Rev Pathol. (2014) 9:119–45. doi: 10.1146/annurev-pathol-012513-104651
2. Torti SV, Torti FM. Iron and cancer: more ore to be mined. Nat Rev Cancer. (2013) 13:342–55. doi: 10.1038/nrc3495
3. Bullinger L, Döhner K, Döhner H. Genomics of acute myeloid leukemia diagnosis and pathways. J Clin Oncol Off J Am Soc Clin Oncol. (2017) 35:934–46. doi: 10.1200/JCO.2016.71.2208
4. Papaemmanuil E, Gerstung M, Bullinger L, Gaidzik VI, Paschka P, Roberts ND, et al. Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med. (2016) 374:2209–21. doi: 10.1056/NEJMoa1516192
5. Jones CL, Stevens BM, D'Alessandro A, Reisz JA, Culp-Hill R, Nemkov T, et al. Inhibition of amino acid metabolism selectively targets human leukemia stem cells. Cancer Cell. (2018) 34:724–40.e4. doi: 10.1016/j.ccell.2018.10.005
6. Pollyea DA, Stevens BM, Jones CL, Winters A, Pei S, Minhajuddin M, et al. Venetoclax with azacitidine disrupts energy metabolism and targets leukemia stem cells in patients with acute myeloid leukemia. Nat Med. (2018) 24:1859–66. doi: 10.1038/s41591-018-0233-1
7. Eberhard Y, McDermott SP, Wang X, Gronda M, Venugopal A, Wood TE, et al. Chelation of intracellular iron with the antifungal agent ciclopirox olamine induces cell death in leukemia and myeloma cells. Blood. (2009) 114:3064–73. doi: 10.1182/blood-2009-03-209965
8. Callens C, Coulon S, Naudin J, Radford-Weiss I, Boissel N, Raffoux E, et al. Targeting iron homeostasis induces cellular differentiation and synergizes with differentiating agents in acute myeloid leukemia. J Exp Med. (2010) 207:731–50. doi: 10.1084/jem.20091488
9. Jeon S-R, Lee J-W, Jang P-S, Chung N-G, Cho B, Jeong D-C. Anti-leukemic properties of deferasirox via apoptosis in murine leukemia cell lines. Blood Res. (2015) 50:33–9. doi: 10.5045/br.2015.50.1.33
10. Roth M, Will B, Simkin G, Narayanagari S, Barreyro L, Bartholdy B, et al. Eltrombopag inhibits the proliferation of leukemia cells via reduction of intracellular iron and induction of differentiation. Blood. (2012) 120:386–94. doi: 10.1182/blood-2011-12-399667
11. Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. (2012) 149:1060–72. doi: 10.1016/j.cell.2012.03.042
12. Pantopoulos K, Porwal SK, Tartakoff A, Devireddy L. Mechanisms of mammalian iron homeostasis. Biochemistry. (2012) 51:5705–24. doi: 10.1021/bi300752r
13. Mancias JD, Wang X, Gygi SP, Harper JW, Kimmelman AC. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature. (2014) 509:105–9. doi: 10.1038/nature13148
14. Rishi G, Wallace DF, Subramaniam VN. Hepcidin: regulation of the master iron regulator. Biosci Rep. (2015) 35:e00192. doi: 10.1042/BSR20150014
15. Nemeth E, Tuttle MS, Powelson J, Vaughn MB, Donovan A, Ward DM, et al. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science. (2004) 306:2090–3. doi: 10.1126/science.1104742
16. Anderson CP, Shen M, Eisenstein RS, Leibold EA. Mammalian iron metabolism and its control by iron regulatory proteins. Biochim Biophys Acta. (2012) 1823:1468–83. doi: 10.1016/j.bbamcr.2012.05.010
17. Greenberg G. Sarcoma after intramuscular iron injection. Br Med J. (1976) 1:1508–9. doi: 10.1136/bmj.1.6024.1508-a
18. Fonseca-Nunes A, Jakszyn P, Agudo A. Iron and cancer risk—a systematic review and meta-analysis of the epidemiological evidence. Cancer Epidemiol Prev Biomark. (2014) 23:12–31. doi: 10.1158/1055-9965.EPI-13-0733
19. Kowdley KV. Iron, hemochromatosis, and hepatocellular carcinoma. Gastroenterology. (2004) 127:S79–86. doi: 10.1016/j.gastro.2004.09.019
20. Wang S, He X, Wu Q, Jiang L, Chen L, Yu Y, et al. Transferrin receptor 1-mediated iron uptake plays an essential role in hematopoiesis. Haematologica. (2019) 105:2071–82. doi: 10.3324/haematol.2019.224899
21. Hole PS, Zabkiewicz J, Munje C, Newton Z, Pearn L, White P, et al. Overproduction of NOX-derived ROS in AML promotes proliferation and is associated with defective oxidative stress signaling. Blood. (2013) 122:3322–30. doi: 10.1182/blood-2013-04-491944
22. Rassool FV, Gaymes TJ, Omidvar N, Brady N, Beurlet S, Pla M, et al. Reactive oxygen species, DNA damage, and error-prone repair: a model for genomic instability with progression in myeloid leukemia? Cancer Res. (2007) 67:8762–71. doi: 10.1158/0008-5472.CAN-06-4807
23. Samimi A, Khodayar MJ, Alidadi H, Khodadi E. The dual role of ROS in hematological malignancies: stem cell protection and cancer cell metastasis. Stem Cell Rev Rep. (2020) 16:262–75. doi: 10.1007/s12015-019-09949-5
24. Jin X, He X, Cao X, Xu P, Xing Y, Sui S, et al. Iron overload impairs normal hematopoietic stem and progenitor cells through reactive oxygen species and shortens survival in myelodysplastic syndrome mice. Haematologica. (2018) 103:1627–34. doi: 10.3324/haematol.2018.193128
25. Ludin A, Gur-Cohen S, Golan K, Kaufmann KB, Itkin T, Medaglia C, et al. Reactive oxygen species regulate hematopoietic stem cell self-renewal, migration and development, as well as their bone marrow microenvironment. Antioxid Redox Signal. (2014) 21:1605–19. doi: 10.1089/ars.2014.5941
26. Aurelius J, Thorén FB, Akhiani AA, Brune M, Palmqvist L, Hansson M, et al. Monocytic AML cells inactivate antileukemic lymphocytes: role of NADPH oxidase/gp91 (phox) expression and the PARP-1/PAR pathway of apoptosis. Blood. (2012) 119:5832–7. doi: 10.1182/blood-2011-11-391722
27. Chen J, Lu W-Y, Zhao M-F, Cao X-L, Jiang Y-Y, Jin X, et al. Reactive oxygen species mediated T lymphocyte abnormalities in an iron-overloaded mouse model and iron-overloaded patients with myelodysplastic syndromes. Ann Hematol. (2017) 96:1085–95. doi: 10.1007/s00277-017-2985-y
28. Liu Q, Wang M, Hu Y, Xing H, Chen X, Zhang Y, et al. Significance of CD71 expression by flow cytometry in diagnosis of acute leukemia. Leuk Lymphoma. (2014) 55:892–8. doi: 10.3109/10428194.2013.819100
29. Wu B, Shi N, Sun L, Liu L. Clinical value of high expression level of CD71 in acute myeloid leukemia. Neoplasma. (2016) 63:809–15. doi: 10.4149/neo_2016_519
30. Kawabata H, Nakamaki T, Ikonomi P, Smith RD, Germain RS, Koeffler HP. Expression of transferrin receptor 2 in normal and neoplastic hematopoietic cells. Blood. (2001) 98:2714–9. doi: 10.1182/blood.V98.9.2714
31. Kollia P, Samara M, Stamatopoulos K, Belessi C, Stavroyianni N, Tsompanakou A, et al. Molecular evidence for transferrin receptor 2 expression in all FAB subtypes of acute myeloid leukemia. Leuk Res. (2003) 27:1101–03. doi: 10.1016/S0145-2126(03)00100-0
32. Nakamaki T, Kawabata H, Saito B, Matsunawa M, Suzuki J, Adachi D, et al. Elevated levels of transferrin receptor 2 mRNA, not transferrin receptor 1 mRNA, are associated with increased survival in acute myeloid leukaemia. Br J Haematol. (2004) 125:42–9. doi: 10.1111/j.1365-2141.2004.04866.x
33. Pagani A, Vieillevoye M, Nai A, Rausa M, Ladli M, Lacombe C, et al. Regulation of cell surface transferrin receptor-2 by iron-dependent cleavage and release of a soluble form. Haematologica. (2015) 100:458–65. doi: 10.3324/haematol.2014.118521
34. Bouchet S, Bauvois B. Neutrophil gelatinase-associated lipocalin (NGAL), pro-matrix metalloproteinase-9 (pro-MMP-9) and their complex pro-MMP-9/NGAL in leukaemias. Cancers. (2014) 6:796–812. doi: 10.3390/cancers6020796
35. Candido S, Maestro R, Polesel J, Catania A, Maira F, Signorelli SS, et al. Roles of neutrophil gelatinase-associated lipocalin (NGAL) in human cancer. Oncotarget. (2014) 5:1576–94. doi: 10.18632/oncotarget.1738
36. Yang W-C, Lin P-M, Yang M-Y, Liu Y-C, Chang C-S, Chou W-C, et al. Higher lipocalin 2 expression may represent an independent favorable prognostic factor in cytogenetically normal acute myeloid leukemia. Leuk Lymphoma. (2013) 54:1614–25. doi: 10.3109/10428194.2012.749402
37. Bertoli S, Paubelle E, Bérard E, Saland E, Thomas X, Tavitian S, et al. Ferritin heavy/light chain (FTH1/FTL) expression, serum ferritin levels, and their functional as well as prognostic roles in acute myeloid leukemia. Eur J Haematol. (2019) 102:131–42. doi: 10.1111/ejh.13183
38. Ihlow J, Gross S, Sick A, Schneider T, Flörcken A, Burmeister T, et al. AML: high serum ferritin at initial diagnosis has a negative impact on long-term survival. Leuk Lymphoma. (2019) 60:69–77. doi: 10.1080/10428194.2018.1461860
39. Lebon D, Vergez F, Bertoli S, Harrivel V, De Botton S, Micol J-B, et al. Hyperferritinemia at diagnosis predicts relapse and overall survival in younger AML patients with intermediate-risk cytogenetics. Leuk Res. (2015) 39:818–21. doi: 10.1016/j.leukres.2015.05.001
40. Tachibana T, Andou T, Tanaka M, Ito S, Miyazaki T, Ishii Y, et al. Clinical significance of serum ferritin at diagnosis in patients with acute myeloid leukemia: a YACHT multicenter retrospective study. Clin Lymphoma Myeloma Leuk. (2018) 18:415–21. doi: 10.1016/j.clml.2018.03.009
41. Artz AS, Logan B, Zhu X, Akpek G, Bufarull RM, Gupta V, et al. The prognostic value of serum C-reactive protein, ferritin, and albumin prior to allogeneic transplantation for acute myeloid leukemia and myelodysplastic syndromes. Haematologica. (2016) 101:1426–33. doi: 10.3324/haematol.2016.145847
42. Pham CG, Bubici C, Zazzeroni F, Papa S, Jones J, Alvarez K, et al. Ferritin heavy chain upregulation by NF-kappaB inhibits TNFalpha-induced apoptosis by suppressing reactive oxygen species. Cell. (2004) 119:529–42. doi: 10.1016/j.cell.2004.10.017
43. Gasparetto M, Pei S, Minhajuddin M, Stevens B, Smith CA, Seligman P. Low ferroportin expression in AML is correlated with good risk cytogenetics, improved outcomes and increased sensitivity to chemotherapy. Leuk Res. (2019) 80:1–10. doi: 10.1016/j.leukres.2019.02.011
44. Cheng P-P, Sun Z-Z, Jiang F, Tang Y-T, Jiao X-Y. Hepcidin expression in patients with acute leukaemia. Eur J Clin Invest. (2012) 42:517–25. doi: 10.1111/j.1365-2362.2011.02608.x
45. Arezes J, Foy N, McHugh K, Sawant A, Quinkert D, Terraube V, et al. Erythroferrone inhibits the induction of hepcidin by BMP6. Blood. (2018) 132:1473–7. doi: 10.1182/blood-2018-06-857995
46. Bondu S, Alary A-S, Lefèvre C, Houy A, Jung G, Lefebvre T, et al. A variant erythroferrone disrupts iron homeostasis in SF3B1-mutated myelodysplastic syndrome. Sci Transl Med. (2019) 11:eaav5467. doi: 10.1126/scitranslmed.aav5467
47. Pinnix ZK, Miller LD, Wang W, D'Agostino R, Kute T, Willingham MC, et al. Ferroportin and iron regulation in breast cancer progression and prognosis. Sci Transl Med. (2010) 2:43ra56. doi: 10.1126/scitranslmed.3001127
48. Tesfay L, Clausen KA, Kim JW, Hegde P, Wang X, Miller LD, et al. Hepcidin regulation in prostate and its disruption in prostate cancer. Cancer Res. (2015) 75:2254–63. doi: 10.1158/0008-5472.CAN-14-2465
49. Zhou Q, Chen J, Feng J, Wang J. E4BP4 promotes thyroid cancer proliferation by modulating iron homeostasis through repression of hepcidin. Cell Death Dis. (2018) 9:987. doi: 10.1038/s41419-018-1001-3
50. Abdel-Wahab O, Levine RL. Metabolism and the leukemic stem cell. J Exp Med. (2010) 207:677–80. doi: 10.1084/jem.20100523
51. Cooper CE, Lynagh GR, Hoyes KP, Hider RC, Cammack R, Porter JB. The relationship of intracellular iron chelation to the inhibition and regeneration of human ribonucleotide reductase. J Biol Chem. (1996) 271:20291–9. doi: 10.1074/jbc.271.34.20291
52. Furukawa T, Naitoh Y, Kohno H, Tokunaga R, Taketani S. Iron deprivation decreases ribonucleotide reductase activity and DNA synthesis. Life Sci. (1992) 50:2059–65. doi: 10.1016/0024-3205(92)90572-7
53. Tataranni T, Agriesti F, Mazzoccoli C, Ruggieri V, Scrima R, Laurenzana I, et al. The iron chelator deferasirox affects redox signalling in haematopoietic stem/progenitor cells. Br J Haematol. (2015) 170:236–46. doi: 10.1111/bjh.13381
54. Messa E, Carturan S, Maffè C, Pautasso M, Bracco E, Roetto A, et al. Deferasirox is a powerful NF-kappaB inhibitor in myelodysplastic cells and in leukemia cell lines acting independently from cell iron deprivation by chelation and reactive oxygen species scavenging. Haematologica. (2010) 95:1308–16. doi: 10.3324/haematol.2009.016824
55. Yu Y, Richardson DR. Cellular iron depletion stimulates the JNK and p38 MAPK signaling transduction pathways, dissociation of ASK1-thioredoxin, and activation of ASK1. J Biol Chem. (2011) 286:15413–27. doi: 10.1074/jbc.M111.225946
56. Shapira S, Raanani P, Samara A, Nagler A, Lubin I, Arber N, et al. Deferasirox selectively induces cell death in the clinically relevant population of leukemic CD34+CD38- cells through iron chelation, induction of ROS, and inhibition of HIF1α expression. Exp Hematol. (2019) 70:55–69.e4. doi: 10.1016/j.exphem.2018.10.010
57. Ohyashiki JH, Kobayashi C, Hamamura R, Okabe S, Tauchi T, Ohyashiki K. The oral iron chelator deferasirox represses signaling through the mTOR in myeloid leukemia cells by enhancing expression of REDD1. Cancer Sci. (2009) 100:970–7. doi: 10.1111/j.1349-7006.2009.01131.x
58. Leardi A, Caraglia M, Selleri C, Pepe S, Pizzi C, Notaro R, et al. Desferioxamine increases iron depletion and apoptosis induced by ara-C of human myeloid leukaemic cells. Br J Haematol. (1998) 102:746–52. doi: 10.1046/j.1365-2141.1998.00834.x
59. Yalcintepe L, Halis E. Modulation of iron metabolism by iron chelation regulates intracellular calcium and increases sensitivity to doxorubicin. Bosn J Basic Med Sci. (2016) 16:14–20. doi: 10.17305/bjbms.2016.576
60. Li N, Chen Q, Gu J, Li S, Zhao G, Wang W, et al. Synergistic inhibitory effects of deferasirox in combination with decitabine on leukemia cell lines SKM-1, THP-1, and K-562. Oncotarget. (2017) 8:36517–30. doi: 10.18632/oncotarget.16583
61. Chang Y-C, Lo W-J, Huang Y-T, Lin C-L, Feng C-C, Lin H-T, et al. Deferasirox has strong anti-leukemia activity but may antagonize theanti-leukemia effect of doxorubicin. Leuk Lymphoma. (2017) 58:1–12. doi: 10.1080/10428194.2017.1280604
62. Taetle R, Castagnola J, Mendelsohn J. Mechanisms of growth inhibition by anti-transferrin receptor monoclonal antibodies. Cancer Res. (1986) 46:1759–63.
63. White S, Taetle R, Seligman PA, Rutherford M, Trowbridge IS. Combinations of anti-transferrin receptor monoclonal antibodies inhibit human tumor cell growth in vitro and in vivo: evidence for synergistic antiproliferative effects. Cancer Res. (1990) 50:6295–301.
64. Daniels TR, Bernabeu E, Rodríguez JA, Patel S, Kozman M, Chiappetta DA, et al. The transferrin receptor and the targeted delivery of therapeutic agents against cancer. Biochim Biophys Acta. (2012) 1820:291–317. doi: 10.1016/j.bbagen.2011.07.016
65. Callens C, Moura IC, Lepelletier Y, Coulon S, Renand A, Dussiot M, et al. Recent advances in adult T-cell leukemia therapy: focus on a new anti-transferrin receptor monoclonal antibody. Leukemia. (2008) 22:42–8. doi: 10.1038/sj.leu.2404958
66. Moura IC, Lepelletier Y, Arnulf B, England P, Baude C, Beaumont C, et al. A neutralizing monoclonal antibody (mAb A24) directed against the transferrin receptor induces apoptosis of tumor T lymphocytes from ATL patients. Blood. (2004) 103:1838–45. doi: 10.1182/blood-2003-07-2440
67. Sauvage CA, Mendelsohn JC, Lesley JF, Trowbridge IS. Effects of monoclonal antibodies that block transferrin receptor function on the in vivo growth of a syngeneic murine leukemia. Cancer Res. (1987) 47:747–53.
68. Brooks D, Taylor C, Dos Santos B, Linden H, Houghton A, Hecht TT, et al. Phase Ia trial of murine immunoglobulin A antitransferrin receptor antibody 42/6. Clin Cancer Res Off J Am Assoc Cancer Res. (1995) 1:1259–65.
69. Daniels-Wells TR, Penichet ML. Transferrin receptor 1: a target for antibody-mediated cancer therapy. Immunotherapy. (2016) 8:991–4. doi: 10.2217/imt-2016-0050
70. Yang X, Koh CG, Liu S, Pan X, Santhanam R, Yu B, et al. Transferrin receptor-targeted lipid nanoparticles for delivery of an antisense oligodeoxyribonucleotide against Bcl-2. Mol Pharm. (2009) 6:221–230. doi: 10.1021/mp800149s
71. Yuan Y, Zhang L, Cao H, Yang Y, Zheng Y, Yang X. A polyethylenimine-containing and transferrin-conjugated lipid nanoparticle system for antisense oligonucleotide delivery to AML. BioMed Res Int. (2016) 2016:1287128. doi: 10.1155/2016/1287128
72. Oh S, Kim BJ, Singh NP, Lai H, Sasaki T. Synthesis and anti-cancer activity of covalent conjugates of artemisinin and a transferrin-receptor targeting peptide. Cancer Lett. (2009) 274:33–9. doi: 10.1016/j.canlet.2008.08.031
73. Li S, Amat D, Peng Z, Vanni S, Raskin S, De Angulo G, et al. Transferrin conjugated nontoxic carbon dots for doxorubicin delivery to target pediatric brain tumor cells. Nanoscale. (2016) 8:16662–9. doi: 10.1039/C6NR05055G
74. Jutz G, van Rijn P, Santos Miranda B, Böker A. Ferritin: a versatile building block for bionanotechnology. Chem Rev. (2015) 115:1653–701. doi: 10.1021/cr400011b
75. Zhen Z, Tang W, Chen H, Lin X, Todd T, Wang G, et al. RGD-modified apoferritin nanoparticles for efficient drug delivery to tumors. ACS Nano. (2013) 7:4830–7. doi: 10.1021/nn305791q
76. Spangler B, Fontaine SD, Shi Y, Sambucetti L, Mattis AN, Hann B, et al. A novel tumor-activated prodrug strategy targeting ferrous iron is effective in multiple preclinical cancer models. J Med Chem. (2016) 59:11161–70. doi: 10.1021/acs.jmedchem.6b01470
77. Kagan VE, Mao G, Qu F, Angeli JPF, Doll S, Croix CS, et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol. (2017) 13:81–90. doi: 10.1038/nchembio.2238
78. Xu S, Mueser TC, Marnett LJ, Funk MO. Crystal structure of 12-lipoxygenase catalytic-domain-inhibitor complex identifies a substrate-binding channel for catalysis. Structure. (2012) 20:1490–7. doi: 10.1016/j.str.2012.06.003
79. Conrad M, Pratt DA. The chemical basis of ferroptosis. Nat Chem Biol. (2019) 15:1137–47. doi: 10.1038/s41589-019-0408-1
80. Agmon E, Solon J, Bassereau P, Stockwell BR. Modeling the effects of lipid peroxidation during ferroptosis on membrane properties. Sci Rep. (2018) 8:5155. doi: 10.1038/s41598-018-23408-0
81. Forcina GC, Dixon SJ. GPX4 at the crossroads of lipid homeostasis and ferroptosis. Proteomics. (2019) 19:e1800311. doi: 10.1002/pmic.201800311
82. Dixon SJ, Patel DN, Welsch M, Skouta R, Lee ED, Hayano M, et al. Pharmacological inhibition of cystine–glutamate exchange induces endoplasmic reticulum stress and ferroptosis. eLife. (2014) 3:e02523. doi: 10.7554/eLife.02523
83. Gao M, Yi J, Zhu J, Minikes AM, Monian P, Thompson CB, et al. Role of mitochondria in ferroptosis. Mol Cell. (2019) 73:354–363.e3. doi: 10.1016/j.molcel.2018.10.042
84. Yuan H, Li X, Zhang X, Kang R, Tang D. CISD1 inhibits ferroptosis by protection against mitochondrial lipid peroxidation. Biochem Biophys Res Commun. (2016) 478:838–44. doi: 10.1016/j.bbrc.2016.08.034
85. Wang W, Green M, Choi JE, Gijón M, Kennedy PD, Johnson JK, et al. CD8+ T cells regulate tumour ferroptosis during cancer immunotherapy. Nature. (2019) 569:270–4. doi: 10.1038/s41586-019-1170-y
86. Tang HM, Tang HL. Cell recovery by reversal of ferroptosis. Biol Open. (2019) 8:bio043182. doi: 10.1242/bio.043182
87. Sun X, Ou Z, Xie M, Kang R, Fan Y, Niu X, et al. HSPB1 as a novel regulator of ferroptotic cancer cell death. Oncogene. (2015) 34:5617–25. doi: 10.1038/onc.2015.32
88. Bordini J, Morisi F, Cerruti F, Cascio P, Camaschella C, Ghia P, et al. Iron causes lipid oxidation and inhibits proteasome function in multiple myeloma cells: a proof of concept for novel combination therapies. Cancers. (2020) 12:970. doi: 10.3390/cancers12040970
89. Yu H, Yang C, Jian L, Guo S, Chen R, Li K, et al. Sulfasalazine-induced ferroptosis in breast cancer cells is reduced by the inhibitory effect of estrogen receptor on the transferrin receptor. Oncol Rep. (2019) 42:826–38. doi: 10.3892/or.2019.7189
90. Hou W, Xie Y, Song X, Sun X, Lotze MT, Zeh HJ, et al. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy. (2016) 12:1425–8. doi: 10.1080/15548627.2016.1187366
91. Wang Y-Q, Chang S-Y, Wu Q, Gou Y-J, Jia L, Cui Y-M, et al. The protective role of mitochondrial ferritin on erastin-induced ferroptosis. Front Aging Neurosci. (2016) 8:308. doi: 10.3389/fnagi.2016.00308
92. Du J, Wang T, Li Y, Zhou Y, Wang X, Yu X, et al. DHA inhibits proliferation and induces ferroptosis of leukemia cells through autophagy dependent degradation of ferritin. Free Radic Biol Med. (2019) 131:356–69. doi: 10.1016/j.freeradbiomed.2018.12.011
93. Badgley MA, Kremer DM, Maurer HC, DelGiorno KE, Lee H-J, Purohit V, et al. Cysteine depletion induces pancreatic tumor ferroptosis in mice. Science. (2020) 368:85–9. doi: 10.1126/science.aaw9872
94. Belavgeni A, Bornstein SR, von Mässenhausen A, Tonnus W, Stumpf J, Meyer C, Otmarh E, et al. Exquisite sensitivity of adrenocortical carcinomas to induction of ferroptosis. Proc Natl Acad Sci USA. (2019) 116:22269–74. doi: 10.1073/pnas.1912700116
95. Doll S, Proneth B, Tyurina YY, Panzilius E, Kobayashi S, Ingold I, et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol. (2017) 13:91–8. doi: 10.1038/nchembio.2239
96. Miess H, Dankworth B, Gouw AM, Rosenfeldt M, Schmitz W, Jiang M, et al. The glutathione redox system is essential to prevent ferroptosis caused by impaired lipid metabolism in clear cell renal cell carcinoma. Oncogene. (2018) 37:5435–50. doi: 10.1038/s41388-018-0315-z
97. Sun X, Ou Z, Chen R, Niu X, Chen D, Kang R, et al. Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology. (2016) 63:173–84. doi: 10.1002/hep.28251
98. Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell. (2014) 156:317–31. doi: 10.1016/j.cell.2013.12.010
99. Viswanathan VS, Ryan MJ, Dhruv HD, Gill S, Eichhoff OM, Seashore-Ludlow B, et al. Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature. (2017) 547:453–7. doi: 10.1038/nature23007
100. Liu H, Schreiber SL, Stockwell BR. Targeting dependency on the GPX4 lipid peroxide repair pathway for cancer therapy. Biochemistry. (2018) 57:2059–60. doi: 10.1021/acs.biochem.8b00307
101. Trujillo-Alonso V, Pratt EC, Zong H, Lara-Martinez A, Kaittanis C, Rabie MO, et al. FDA-approved ferumoxytol displays anti-leukaemia efficacy against cells with low ferroportin levels. Nat Nanotechnol. (2019) 14:616–22. doi: 10.1038/s41565-019-0406-1
102. Sagasser J, Ma BN, Baecker D, Salcher S, Hermann M, Lamprecht J, et al. A new approach in cancer treatment: discovery of chlorido[N,N′-disalicylidene-1,2-phenylenediamine]iron (III) complexes as ferroptosis inducers. J Med Chem. (2019) 62:8053–61. doi: 10.1021/acs.jmedchem.9b00814
103. Wei J, Xie Q, Liu X, Wan C, Wu W, Fang K, et al. Identification the prognostic value of glutathione peroxidases expression levels in acute myeloid leukemia. Ann Transl Med. (2020) 8:678. doi: 10.21037/atm-20-3296
104. Yu Y, Xie Y, Cao L, Yang L, Yang M, Lotze MT, et al. The ferroptosis inducer erastin enhances sensitivity of acute myeloid leukemia cells to chemotherapeutic agents. Mol Cell Oncol. (2015) 2:e1054549. doi: 10.1080/23723556.2015.1054549
105. Liu DS, Duong CP, Haupt S, Montgomery KG, House CM, Azar WJ, et al. Inhibiting the system xC-/glutathione axis selectively targets cancers with mutant-p53 accumulation. Nat Commun. (2017) 8:14844. doi: 10.1038/ncomms14844
106. Ye F, Chai W, Xie M, Yang M, Yu Y, Cao L, et al. HMGB1 regulates erastin-induced ferroptosis via RAS-JNK/p38 signaling in HL-60/NRASQ61L cells. Am J Cancer Res. (2019) 9:730–39.
Keywords: acute myeloid leukemia, iron, reactive oxygen species, ferroptosis, ferritinophagy
Citation: Grignano E, Birsen R, Chapuis N and Bouscary D (2020) From Iron Chelation to Overload as a Therapeutic Strategy to Induce Ferroptosis in Leukemic Cells. Front. Oncol. 10:586530. doi: 10.3389/fonc.2020.586530
Received: 23 July 2020; Accepted: 18 August 2020;
Published: 18 September 2020.
Edited by:
Nelida Ines Noguera, University of Rome Tor Vergata, ItalyReviewed by:
Xia Xiao, Tianjin First Central Hospital, ChinaCopyright © 2020 Grignano, Birsen, Chapuis and Bouscary. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Eric Grignano, ZXJpYy5ncmlnbmFub0BpbnNlcm0uZnI=; Didier Bouscary, ZGlkaWVyLmJvdXNjYXJ5QGFwaHAuZnI=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.