 Jane Koo
Jane Koo Masanori Hayashi
Masanori Hayashi Michael R. Verneris
Michael R. Verneris Alisa B. Lee-Sherick
Alisa B. Lee-Sherick
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 14 December 2020
Sec. Pediatric Oncology
Volume 10 - 2020 | https://doi.org/10.3389/fonc.2020.581107
This article is part of the Research Topic The Characteristics of Pediatric Soft Tissue Sarcomas: Recent Advances in Management and Treatment View all 10 articles
For many pediatric sarcoma patients, multi-modal therapy including chemotherapy, radiation, and surgery is sufficient to cure their disease. However, event-free and overall survival rates for patients with more advanced disease are grim, necessitating the development of novel therapeutic approaches. Within many pediatric sarcomas, the normal immune response, including recognition and destruction of cancer cells, is lost due to the highly immune suppressive tumor microenvironment (TME). In this setting, tumor cells evade immune detection and capitalize on the immune suppressed microenvironment, leading to unchecked proliferation and metastasis. Recent preclinical and clinical approaches are aimed at understanding this immune suppressive microenvironment and employing cancer immunotherapy in an attempt to overcome this, by renewing the ability of the immune system to recognize and destroy cancer cells. While there are several factors that drive the attenuation of immune responses in the sarcoma TME, one of the most remarkable are tumor associated macrophage (TAMs). TAMs suppress immune cytolytic function, promote tumor growth and metastases, and are generally associated with a poor prognosis in most pediatric sarcoma subtypes. In this review, we summarize the mechanisms underlying TAM-facilitated immune evasion and tumorigenesis and discuss the potential therapeutic application of TAM-focused drugs in the treatment of pediatric sarcomas.
Pediatric sarcomas are a heterogenous group of tumors that comprise approximately 10% of all childhood cancers (1–5). While sarcomas also occur in adults, the prevalence of subtypes is strikingly unique for the pediatric population. The most common bony pediatric sarcomas are osteosarcoma and Ewing sarcoma (EWS), while rhabdomyosarcoma (RMS) is the most common pediatric soft tissue sarcoma. Other rarer sarcoma subtypes such as synovial sarcoma, leiomyosarcoma, and liposarcomas can occur in children, but are more common in adult patients (6). The cornerstone of treatment typically involves an intensive multi-modality approach including cytotoxic chemotherapy, surgery, and radiation. Over the last five decades, survival improvements have resulted from incremental adjustments to current therapy; however, very few new therapies have been shown to positively improve pediatric sarcomas outcomes (7–9).
For patients with metastatic RMS, the 3-year overall survival (OS) and event-free survival (EFS) are 34 and 27% respectively (10, 11). Survival rates for metastatic osteosarcoma and EWS are similarly dismal with 5-year survival rates reported between 20–30% and 30–40%, respectively (10, 11). Current therapies are highly toxic and associated with many short- and long-term side effects resulting in considerable life-long morbidities (12–15). Alternative approaches, such as immunotherapy, are desperately needed to both improve cure rates and to minimize long-term side effects. Therapeutic approaches that direct the immune system to recognize and destroy tumor cells are currently being trialed in patients with relapsed/refractory sarcomas.
To better understand the potential benefit of immunotherapy in pediatric sarcomas, certain biologic and mutational differences between pediatric and adult sarcomas warrant emphasis. In contrast to adult sarcomas, pediatric sarcomas are generally characterized by a low mutational burden, specific chromosomal translocations that encode “driver mutations,” and low somatic copy number alterations in some sarcoma subtypes (16–23). Higher mutational burden and presence of complex genomic aberrations that occur in adult patients may increase the presence and immune recognition of sarcoma neoantigens. This is further compounded by the observation that the pediatric adaptive immune system tends to be more plastic and may account for variations in individual responses to immunotherapy (24, 25). Additionally, there is higher marrow cellularity and more robust hematopoiesis in children compared to adult patients, exemplified by faster immune reconstitution in children following chemotherapy (26–28). Therefore, these unique differences in sarcoma biology and immune function between adult and pediatric patients likely affect responses to immunotherapy. Furthermore, before cellular immunotherapy can be fully leveraged for pediatric sarcomas an understanding of TAMs within the sarcoma TME is required.
The cellular composition of the TME is broadly comprised of tumor cells, non-malignant stromal cells, blood vessels, and immune cells. Stromal cells produce extracellular matrix (ECM) proteins and matricellular proteins that provide structural support and mediate signaling for cellular movement. The immune components of the sarcoma TME, including innate immune cells [neutrophils, TAMs, natural killer (NK) cells, dendritic cells (DCs)] and adaptive immune cells (B and T lymphocytes), can vary vastly with respect to sarcoma subtype, primary tumor location, genetic or mutational burden and previous therapy exposure. TAMs are one of at least four myeloid subpopulations derived from tumor-associated myeloid cells (TAMCs) that also include myeloid-derived suppressor cells (MDSCs), tumor-associated neutrophils (TANs), and angiogenic monocytes expressing angiopoietin-2 (TIE-2) (29–31). Cellular immunotherapeutic approaches have largely tested adopted transfer of activated and/or antigen specific T cells; however, efficacy of these cells can be significantly dampened by cells that exert immune regulatory function, including TAMs, regulatory T cells (Tregs), and mesenchymal stem cells (MSCs). For the purposes of this review, we focus on TAMs.
Several studies have demonstrated a strong correlation between macrophage infiltration, sarcoma tumor progression, and patient survival, highlighting TAMs as potential immunotherapeutic targets in pediatric sarcoma (32–36). In addition to phagocytosis of necrotic tumor cells, which decreases the presence of tumor antigen and subsequent immunogenic T cell response, TAMs have been shown to display a wide variety of immunosuppressive and tumor-promoting functions. For instance, increased proportion of TAMs has been shown to render chimeric antigen receptor T cell immunotherapy ineffective (37). However, TAM number and density in pediatric sarcomas do not explain the entirety of their importance in facilitating tumor progression, and the immune cell profiles in pediatric sarcomas vary across tumor subtypes.
Macrophages play critical roles in innate immunity including: phagocytosis, clearance of apoptotic debris, lymphocyte recruitment (38, 39), antigen presentation (40, 41), wound healing (42), and tissue homeostasis (43, 44). Thus, they both promote inflammatory responses as well as facilitate resolution. In the setting of cancer, macrophages have a response that is seemingly antithetical to the whole organism, as they drive immune tolerance and facilitate cancer progression (45).
Various clinically applicable techniques are in development to identify, quantify, and characterize sarcoma-associated TAMs. Such techniques include immunohistochemistry, single cell RNA sequencing, fluorescent magnetic nanoparticle labeling, and even non-invasive imaging including magnetic resonance imaging (MRI), given that T2* signal enhancement on MR images significantly correlated with TAM density in sarcoma patients (32, 46–48). Depending on their local microenvironments, TAMs can display phenotypic and functional heterogeneity, which is best understood through the concept of macrophage polarization (see next paragraph) (49). However, this dichotomous polarization paradigm is largely oversimplified, and there is a broad range of macrophage polarization phenotypes in vivo (50). While TAMs are the largest population of infiltrating immune cells within pediatric sarcomas and TAM infiltration into the tumor can be linked with worse prognosis, the density of TAMs within the tumor does not necessarily provide the full scope of how they influence the TME (34, 51).
The M1/M2 polarization spectrum was developed to explain macrophage phenotype and function in response to inflammation or infection. In the setting of inflammation, M1 macrophages (classically activated macrophages) migrate to sites of infection, phagocytose infected cells and serve as antigen presenting cells (APCs) and produce T helper cell type 1 (Th1) or pro-inflammatory cytokines, promoting T cell activation. In contrast, M2 (alternatively activated) macrophages promote tissue repair through efferocytosis, a phagocytic process in which antigen are cleared, antigen presentation is diminished, and T helper cell type 2 (Th2) cytokines are produced. This process also promotes immune tolerance to autologous (or “self”) tissue. Macrophage plasticity and polarization in the sarcoma TME is also critical for the progression or regression of these tumors (Figure 1).
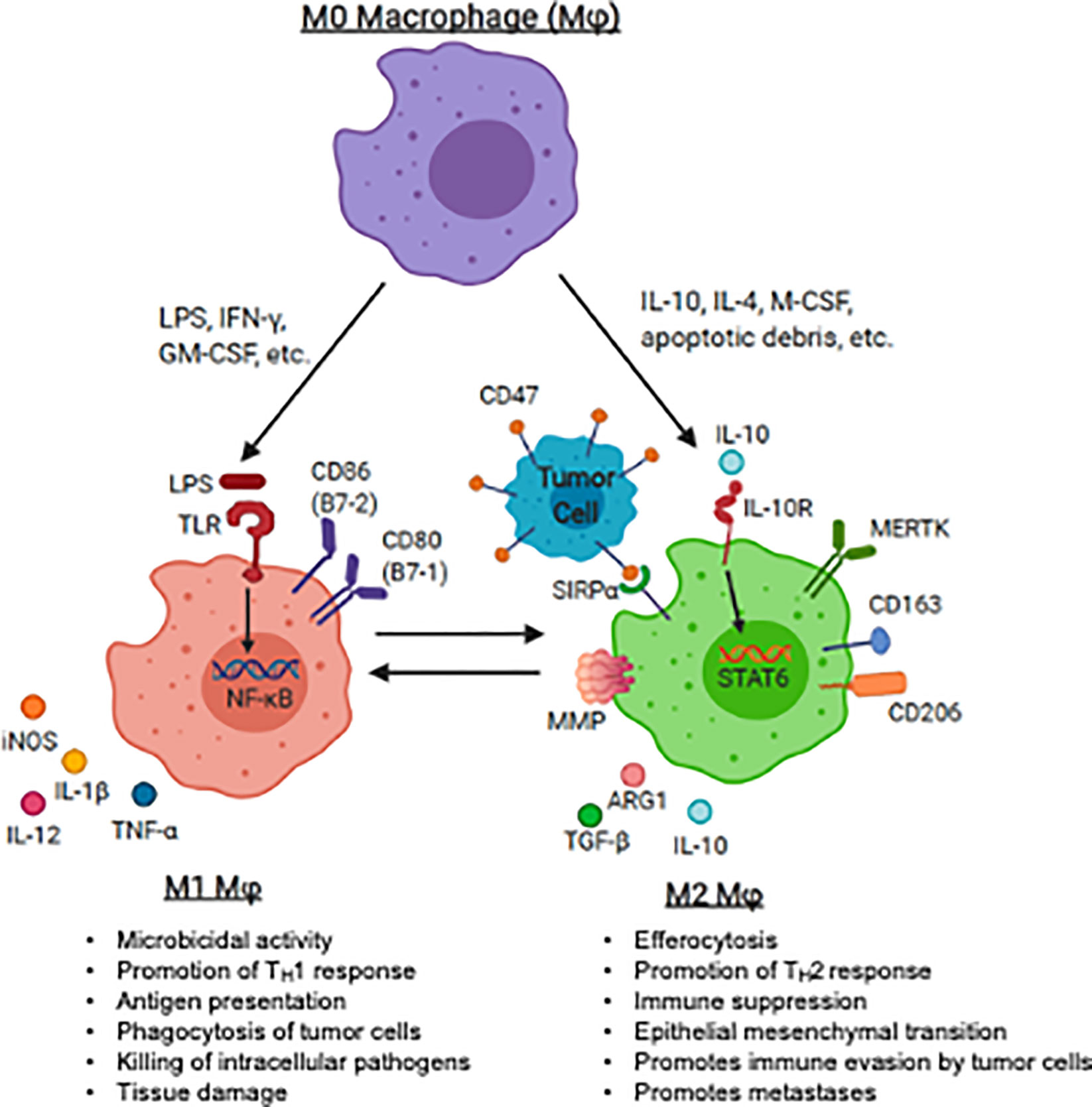
Figure 1 Macrophage polarization and plasticity within the pediatric sarcoma tumor microenvironment. The panel represents recognized M1 (anti-tumoral) and M2 (tumor-promoting) agonists that induce the induction of M1 and M2 markers by human macrophages. The major canonical functions of M1 macrophages and M2 macrophages are also described. LPS, lipopolysaccharide, IFN-γ, interferon-gamma; GM-CSF, granulocyte macrophage-colony stimulating factor; IL-4, interleukin 4; IL-10, interleukin-10; IL-13, interleukin 13; M-CSF, macrophage colony stimulating factor; TLR, toll-like receptor; TNF-α, tumor necrosis factor-alpha; IL-1β, interleukin 1 beta; PD-L1, programmed death ligand 1; PD-L2, programmed death ligand 2; MMP, matrix metalloprotease; MERTK, Mer receptor tyrosine kinase; TGF-β, transforming growth factor beta. Image created with biorender.com.
Following exposure to damage- or pathogen-associated molecular patterns (DAMPs or PAMPs), such as bacterial lipopolysaccharides (LPS), nucleic acids, and other microbial ligands, toll-like receptor (TLR) are triggered and M1 polarize macrophage (52–54). TLR ligation initiates a signaling cascade involving the innate immune signal transduction adaptor MYD88, interleukin 1 receptor associated kinase 4 (IRAK4), tumor necrosis factor associated factor 6 (TRAF6) and inhibitor of nuclear factor kappa B kinase subunit beta (IKK-β) which ultimately activates nuclear factor kappa B (NF-κB), one of the central regulators of inflammatory cytokine production. Translocation of NF-κB into the nucleus leads to transcription of Th1 genes, such as tumor necrosis factor-α (TNF-α), interleukin (IL)-12, IL-1β, and IL-6, leading to expansion of effector T cells (55–60). Activated T cells produce pro-inflammatory cytokines (e.g., interferon gamma (IFN- γ), granulocyte colony-stimulating factor (GM-CSF)) further perpetuating macrophage M1 activation (61–63). Additionally, GM-CSF is a potent driver of antibody-dependent cell-mediated cytotoxicity (ADCC) and antibody-dependent cellular phagocytosis (ADCP), a cell mediated immune defense whereby immune effector cells destroy antibody coated target cells (64–66). M1 macrophages generally express high levels of surface molecules for antigen presentation (e.g., major histocompatibility complex-II (MHC-II)), augment T cell activation (e.g., CD80, CD86), and further promote self-activation (e.g., TLR2, TLR4) (67). M1 macrophages produce cytokines to amplify T cell activation, such as IL-1β, IL-6, IL-12, IL-18 and TNF-α (68, 69).
Alternative activation, or M2 polarization, is thought to occur after exposure to cytokines such as IL-4, IL-10, macrophage colony-stimulating factor (M-CSF) and transforming growth factor beta (TGF-β) (70, 71), and/or apoptotic cellular debris which promote the resolution of inflammation and wound-healing. M2 macrophages may be identified by the up-regulation of surface markers that promote clearance of apoptotic debris, such as mannose receptor C-type 1 (MMR, CD206) and CD163 (63, 72–76). M2 macrophages may produce T cell suppressive cytokines such as TGF-β and IL-10 (77). In response to local cytokine milieu, alternatively activated macrophages also up-regulate inhibitory checkpoint ligands, such as programmed death 1 ligand 1 (PD-L1) and programmed death 1 ligand 2 (PD-L2), which inhibit T cell effector function (78, 79). Many of the above pathways have been or are being considered for targeting to either augment immunity or inhibit the counter-regulatory activity known to occur in malignancy. A summary of therapeutic strategies targeting TAMs in the pediatric sarcoma TME is summarized in Figure 2.
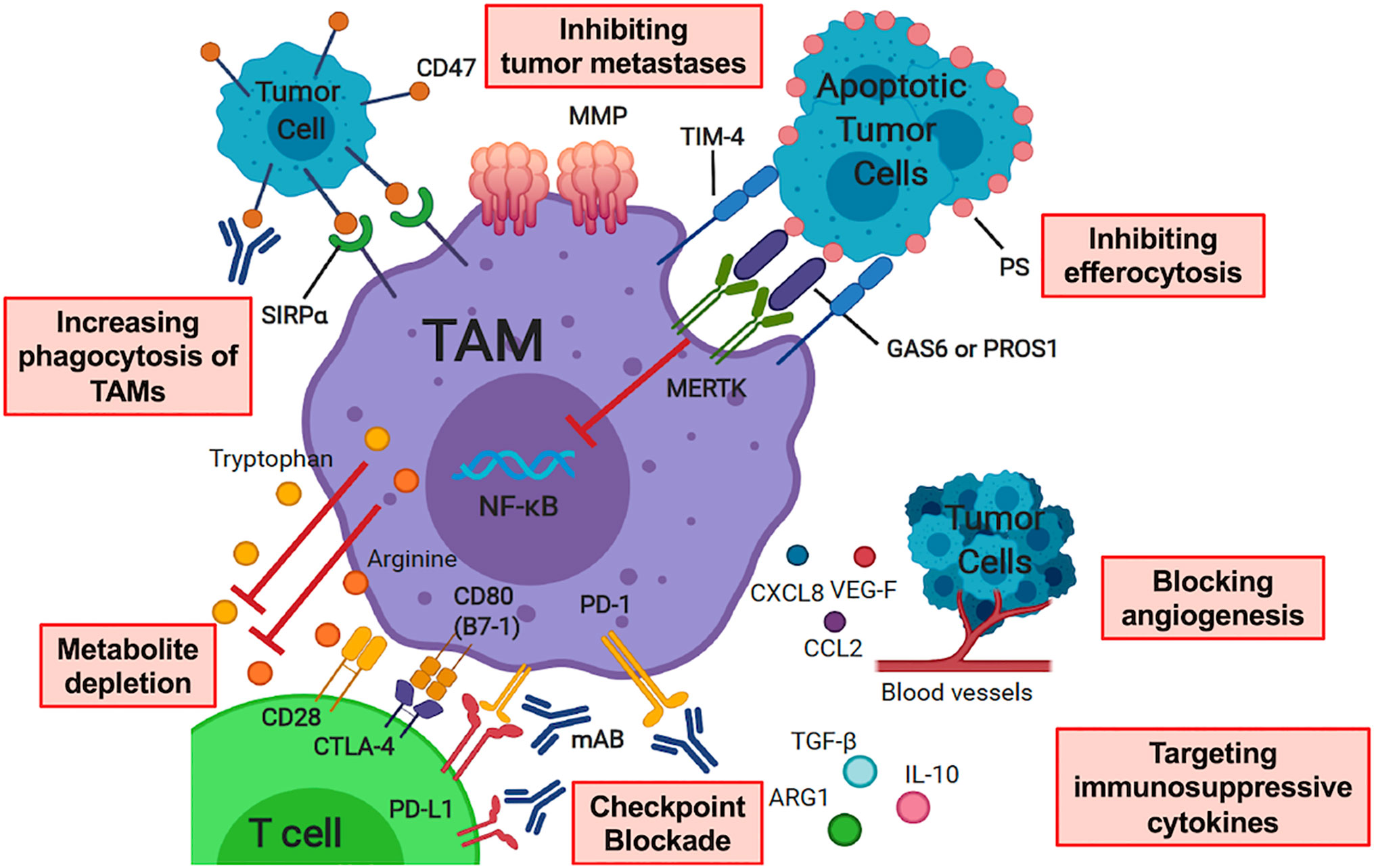
Figure 2 Therapeutic Strategies Targeting Tumor-Associated Macrophages in the Pediatric Sarcoma Microenvironment. Therapy modalities include increasing phagocytosis of TAMs, inhibiting tumor metastases, inhibiting efferocytosis, checkpoint blockade, altering macrophage polarization through targeting immunosuppressive cytokines, metabolite depletion and blocking angiogenesis. TAM, tumor-associated macrophage; SIRPα, signal-regulatory protein alpha; MMP, matrix metalloprotease; PS, phosphatidylserine; TIM-4, T Cell Immunoglobulin And Mucin Domain Containing 4; MERTK, Mer receptor tyrosine kinase; PROS1, protein S; GAS6, growth arrest-specific 6; CTLA-4, cytotoxic T-lymphocyte-associated protein 4; PD-1, programmed cell death protein 1; PD-L1, programmed death ligand 1; mAB, monoclonal antibody; IL-10, interleukin 10; TGF-β, transforming growth factor beta; ARG1, arginase 1; VEG-F, vascular endothelial growth factor; CXCL8, C-X-C motif chemokine ligand 8; CCL2, C-C motif chemokine ligand 2. Image created with biorender.com
Manipulating macrophage polarization in the TME toward M1 activation status has been evaluated using TLR agonists. Muramyl TriPeptide-PhosphatidylEthanolamine encapsulated into liposomes (L-MTP-PE) has been proposed as an adjuvant therapy for osteosarcoma patients. It is a synthetic analog of muramyl dipeptide (MD), a peptidoglycan that is found in bacterial cell walls. L-MTP-PE has been demonstrated to activate TLR4 on macrophages and monocytes and upregulate their tumoricidal functions through increased type 1 cytokine production (such as TNF-α, IL-1, IL-6, IL-8, IL-12, and nitric oxide (NO)) (80, 81). A preclinical evaluation of L-MTP-PE combined with zoledronic acid (ZA) in murine models of osteosarcoma showed that the two drugs significantly inhibited tumor growth and development of metastases (82). Phase I and II clinical trials evaluating L-MTP-PE in pediatric osteosarcoma patients showed acceptable toxicity, and even enhanced macrophage-mediated tumoricidal activity, but had variable results in prolongation of OS and EFS (see Table 1) (80, 90, 91). In a follow-up randomized phase III trial [Intergroup (INT)-0133] by the Children’s Oncology Group (COG) for patients with osteosarcoma addition of L-MTP-PE to standard chemotherapy showed no difference in 5-year OS or EFS. When patients with metastatic disease were analyzed separately, L-MTP-PE had improved survival compared (53 vs 40%); however, but the study was not powered to detect a significant difference between the two arms (92). L-MTP-PE is not currently approved by the United States Food and Drug Administration (FDA) (102) though the European Medicines Agency granted L-MTP-PE an indication as an adjuvant treatment of osteosarcoma in 2009.
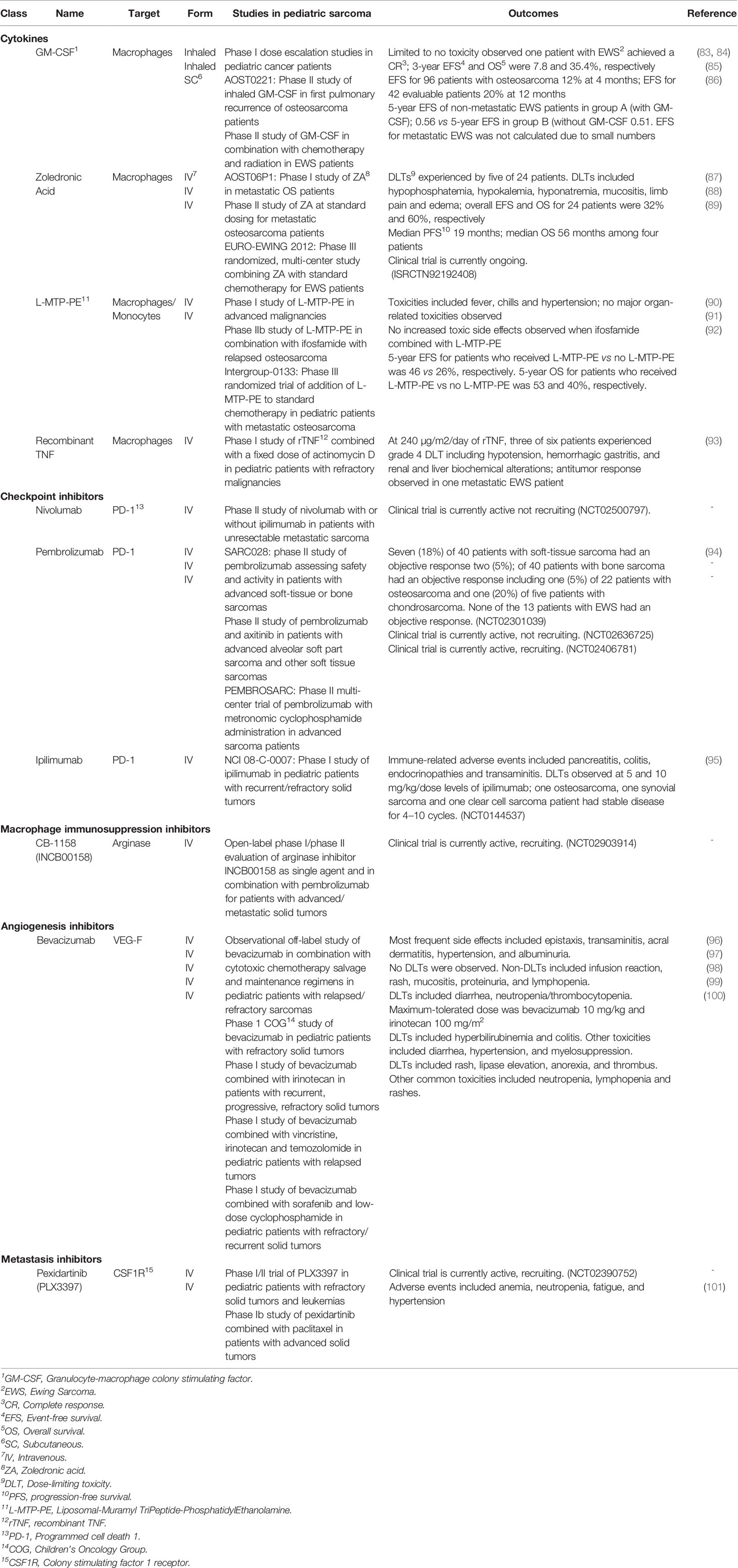
Table 1 Current macrophage targeted therapies for the treatment of pediatric sarcomas.
Administration of exogenous cytokines to reverse TAM M2 polarization may be an effective immunotherapeutic strategy for pediatric sarcomas. GM-CSF is a myeloid growth factor that stimulates the differentiation of hematopoietic progenitor cells into granulocytes and monocytes with subsequent type 1 cytokine mRNA expression, such as IL-1β, IL-6 and TNF (103). GM-CSF has been successfully incorporated into the standard therapy of high-risk neuroblastoma patients receiving antibody therapy (104). Knowing that the lungs are a common site for pulmonary metastasis, aerosolized GM-CSF has been tested and while it is safe (83–85, 105), it did not improve outcomes for patients with advanced sarcomas (83–85, 105). Similarly, subcutaneous GM-CSF was assessed in a phase II study for 18 pediatric patients with EWS after radiation with no significant difference in 5-year EFS between the treatment group and controls (see Table 1) (86).
Alternative methods of delivering intra-tumoral M1 polarizing cytokines have been developed with the goal of minimizing global toxicities associated with exogenous cytokine administration. Innovative methods, such as adoptive transfer of macrophages harboring a soft discoidal particle (“backpack”) that contains the cytokine payload, have been described. In recently published research, phagocytosis-resistant IFN-γ secreting macrophage “backpacks” is composed of external polymer layers sandwiching an IFN-γ core and a cell-adhesive layer which avidly binds to bone marrow derived macrophages. Adoptive transfer of macrophages carrying IFN-γ secreting backpacks into solid tumors maintained their M1 phenotype despite the immunosuppressive TME and also repolarized endogenous M2 TAMs toward an M1 phenotype. This was also associated with decreased tumor volume and lung metastases in vivo (106). Further studies into the function, feasibility, and toxicity of these and similar alternative delivery methods are needed as they seem promising as a means of avoiding systemic administration of exogenous cytokines.
Efferocytosis is a tolerogenic phagocytotic process characterized by clearance of auto-antigen (or “self”) present on apoptotic cells and suppressing T cell activation. Physiologically, efferocytosis is thought to be critical in the maintenance of self-tolerance and the prevention of autoimmunity. However, in the TME, the otherwise normal TAM process of efferocytosis diminishes immunity through phagocytic clearance of tumor antigen and suppression of T cell cytolytic function, thereby creating a TME supportive of immune evasion and subsequent tumor survival and metastasis. This may be especially relevant in settings of high cell turnover, such as malignancies which are characterized by spontaneous apoptosis due to a myriad of circumstances associated with cancer. Therefore, interfering in the multiple steps involved in efferocytosis may be a novel therapeutic approach with the potential for therapeutic benefit.
Efferocytosis of apoptotic debris is a series of coordinated events, including chemotaxis, recognition and binding of the apoptotic particle, and ingestion. This first step of the sequence includes the secretion of chemoattractant “find-me” signals by a dying cell, including lysophosphatidylcholine (LPC) (107, 108), sphingosine-1-phosphate (S1P) (109), C-X3-C motif chemokine ligand 1 (CX3CL1) (110, 111) and nucleotides (112). Intracellular LPC and S1P are released by apoptotic cells (109, 113), while CX3CL1 is a membrane-associated protein which is cleaved by matrix metalloproteases (MMPs) during inflammation, releasing the soluble protein that acts as a chemokine (114). Nucleotides, specifically adenosine triphosphate (ATP) and uridine-5′-triphosphate (UTP) are released into the extracellular space following caspase-dependent activation. These molecules are recognized by receptors on monocytes and macrophages and result in migration to the area of cellular damage (110, 112, 115). Preclinical studies in RMS and osteosarcoma tumors support the importance of these mechanisms in pediatric sarcoma and have confirmed upregulated expression of bioactive lipids such as S1P, LPC, and lysophosphatidic acid (an LPC cleavage product) in bone marrow extracts (a common site of sarcoma metastasis) by mass spectrometry following radiation and chemotherapy (116, 117). Further clinical studies are required to evaluate the utility of these “find-me” signals as prognostic biomarkers or therapeutic targets for pediatric sarcomas.
Tumor cells may evade immune-mediated attack through downregulation of “eat-me” signals. “Eat-me” signals, such as phosphatidylserine (PS) and calreticulin (CRT), are externalized on dying cell surfaces, tagging them for removal by phagocytes. PS is a phospholipid normally localized to the inner membrane of the lipid bilayer in healthy cells; however, during apoptosis, PS accumulates on the cell surface. Similarly, CRT is also exposed on the cell surface during apoptotic stress. CRT interacts with PS and binds the complement C1q protein that serves as both bridging molecule and a PS-binding protein. CRT then binds the SRF-1 endocytic receptor found on macrophages to facilitate phagocytosis of apoptotic cells (118, 119). It is also known that macrophages can utilize their own CRT to enhance phagocytosis of tumor cells (120). Preclinical studies have shown that high expression of PS on EWS tumors increased their sensitivity to tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) mediated cell death (121). Additional studies incubating alveolar and embryonal RMS cells with doxorubicin demonstrated enhanced CRT expression and increased phagocytosis of these RMS cells (4). Other studies are examining the use of “eat-me” signals, specifically CRT, as potential prognostic biomarkers in osteosarcoma (122).
To counter-balance PS or CRT expression, malignant cells may evade macrophage phagocytosis through the expression of “don’t eat-me” receptors. Healthy cells express “don’t eat-me” receptors CD47 and CD31 to avoid unwarranted phagocytic clearance (123, 124). CD47, the prototypical “don’t eat-me” signal, is a membrane protein of the immunoglobulin (Ig) superfamily, present on most cells of the body. Ligation of CD47 with the ssignal regulatory protein alpha (SIRPα) protein on macrophages leads to phosphorylation of immunoreceptor tyrosine-based inhibition (ITIM) motifs and a significant inhibitory signaling cascade, characterized by the downstream protooncogene SRC, protein tyrosine phosphatase non-receptor type 6 (PTPN6), and protein tyrosine phosphatase non-receptor type 11 (PTPN11) phosphatases, which inhibit the buildup of myosin-IIA, and prevent the cellular structural changes needed for phagocytosis (2, 125–130). Activation of SIRPα has also been found to mediate M2 macrophage polarization, through regulation of the Notch signaling pathway (131–133). Conversely, when the SIRPα is blocked, TAMs portend a M1 phenotype (133).
Previous work in experimental models of hematologic and solid malignancies have identified CD47 and SIRPα as potential therapeutic targets, whereby blocking this axis (predominantly using anti-CD47 mAb) demonstrated increased phagocytosis of cancer cells by macrophages (2, 4, 5, 126, 128, 134) and M1 polarization (131). Increased phagocytosis of human RMS cells was observed in vitro when macrophages were treated with anti-CD47 monoclonal antibody (4). In murine studies of osteosarcoma, CD47 blockade decreased tumor progression, increased macrophage infiltration into the tumor, and increased overall survival (2, 5). Currently, there are no open clinical trials targeting the CD47-SIRPα pathway for pediatric sarcomas. However, in adults, there are several open clinical trials evaluating the safety profile and efficacy of anti-CD47 monoclonal antibody (Hu5F9-G4) in a variety of solid and hematologic malignancies (NCT02953509, NCT03248479, NCT03922477, NCT03869190).
Simply put, phagocytosis occurs when the balance of “eat me” signals is greater than the “don’t eat me” signals. Recognition of “eat-me” signals by professional phagocytes occurs through multiple receptors, such as TYRO3, AXL, and MERTK receptor tyrosine kinases and the T-cell immunoglobulin and mucin domain (TIM) receptor family (TIM-3 and TIM-4). Of these, MERTK is the prototypic efferocytosis receptor, given its involvement in the recognition, tethering and engulfment of apoptotic cells, and subsequent generation of immune tolerance through M2 polarization and T cell suppression (135–138). Following apoptotic cell ingestion, MERTK phosphorylation suppresses NF-κB nuclear translocation, leading to diminished type 1 cytokine production (e.g. TNF-α and IL-12) (139–141). Conversely, inhibition of MERTK in preclinical studies has shown decreased leukemia-associated macrophage expression of inhibitory checkpoint ligands, including PD-L1 and PD-L2 (discussed below), demonstrating its role in immune tolerance (142). Drugs targeting MERTK have been developed as agents for both reversing cancer progression and cancer immune evasion (143). Pre-clinical studies of MERTK inhibitors in murine solid tumor models have shown decreased tumor growth and increased CTL infiltration (144), while others demonstrated a more profound effect when MerTK inhibition is used in combination with radiation therapy (145).
TYRO3, AXL, and MERTK receptors do not bind to PS directly, rather they use the plasma circulating and locally secreted molecules, protein S (PROS1) and growth arrest specific 6 (GAS6) to provide a bridge to PS. PROS1 binds more specifically to MERTK and GAS6 binds to MERTK, TYRO3, and AXL (146–150). PROS1 and GAS6 are elevated in EWS tumor patient samples, providing increased ligand for efferocytosis to occur (151, 152). Antibodies acting as ligand sinks to bind and inactivate these bridging molecules have been evaluated in preclinical studies but are not yet clinically available (153–155).
TIM family of proteins, TIM-3 and TIM-4, act as PS receptors on macrophages to facilitate the clearance of apoptotic cells (135, 156, 157). On macrophages, TIM-4 works in conjunction with MERTK to mediate tethering and binding of apoptotic cells (156, 158) (159–161). TIM-3, a known co-inhibitory receptor on T cells, is also expressed on antigen presenting cells such as macrophages, aids in the binding and phagocytosis of apoptotic cells through the FG loop in the immunoglobulin variable region (IgV) domain (136, 162–164). Co-expression of TIM-3 with other immune checkpoints such as lymphocyte activating 3 (LAG3) and PD-1 on T cells has been observed in sarcoma patient samples (165); however, its expression on TAMs in sarcoma has not been explored. TIM-3 antibodies are being clinically tested and may be useful in both augmenting T cell activation, as well as diminishing the tolerogenic effects of efferocytosis (166, 167).
Bisphosphonates are a class of drugs designed to inhibit osteoclast activity to prevent loss of bone density in osteoporosis; however, they also suppress macrophage phagocytosis, decrease macrophage recruitment to tumor sites, and increase apoptosis of tumor cells (168, 169). Zoledronic acid is a nitrogen-containing bisphosphate with anti-tumor activity including decreased tumor volume and bone growth in primary EWS tumors, decreased recruitment of TAMs into the tumor stroma in murine sarcoma and carcinoma models (170–172), and reduction in bone metastases EWS after administration of ZA in murine in vivo models (171). When combined with ifosfamide, ZA exhibited synergistic effects against tumor growth and progression in a soft tissue tumor model. These promising clinical results have led to the evaluation of ZA in pediatric sarcomas (see Table 1). In a phase I study of high-grade metastatic osteosarcoma patients, ZA was well tolerated when administered concurrently with multi-agent chemotherapy (87). One small clinical study evaluated the anti-tumor efficacy of ZA at standard dosing for four patients with advanced stage osteosarcoma with encouraging progression-free survival (PFS) results (88). ZA in combination with standard chemotherapy for EWS patients is currently being evaluated in a multicenter phase III randomized controlled trial (Euro-EWING2012) (89).
The physical interaction during antigen presentation between macrophage and T cells plays an integral role in T cell-mediated activation and tumor cell cytolysis. Antigen presentation involves three steps, which have been described as different signals. Signal 1 is the binding of peptide-loaded MHC on antigen presenting cells (APCs), such as macrophages, to antigen-specific T cell receptors (TCRs). Signal 2 is the engagement of costimulatory ligands with their cognate receptors on T cells. Conversely, binding of inhibitory ligands (on APCs) with their cognate receptors on T cells inhibits T cell activation. Signal 3 is the secretion of cytokines by APCs which modify or amplify T cell response (165).
Signal 1, consisting of the MHC-peptide-TCR complex, on its own is insufficient to activate T cells and may generate a tolerogenic response. However, concomitant engagement of adhesion receptors and co-stimulatory ligands (on APCs) and receptors (on T cells), known as Signal 2, creates an immunologic connection between T cells and macrophages (165). Co-stimulatory ligands and cognate receptors are divided into two major groups: CD28/B7 receptor family and TNF/tumor necrosis factor receptor (TNFR) family.
CD28 is a T cell costimulatory receptor that transmits activating intracellular signals when it binds costimulatory ligands CD80 (B7-1) and CD86 (B7-2) on macrophages and other APCs (173, 174). In contrast to the CD28 stimulatory effects on T cells, ligation of inhibitory B7 receptors, including programmed death 1 (PD-1) and cytotoxic T-lymphocyte associated protein 4 (CTLA-4), can promote T cell suppression and/or dysfunction (175, 176). PD-1 has two known B7 ligands on macrophages, including PD-L1 and PD-L2 (177). PD-L1 is often upregulated in tumor infiltrating immune cells including macrophages (178–180). In fact, in pediatric sarcoma patient samples with greater PD-L1 expression, there was higher macrophage and DC infiltration, and a worse outcome (181–183). The treatment of murine and human macrophages with anti-PD-L1 antibodies promotes their proliferation and activation (184). Blockade of the PD-L1/PD-1 axis also enhances macrophage-mediated anti-tumor activity through efferocytosis. Although blockade of this ligand/receptor binding is typically studied for its effects on T cell function, preclinical models of PD-L1/PD-1 blockade using BALB/c Rag2−/−γc−/− mice (which do not have functional T cells) showed TAM-mediated efferocytosis and clearance of tumor cells (185). Disruption of the PD-1/PD-L1 axis in osteosarcoma demonstrated decreased lung metastases, reduced numbers of tumor-promoting TAMs, and increased anti-tumor M1 macrophages in the absence of T cells (183). While anti-PD-L1 or anti-PD-L2 agents have not yet been evaluated in pediatric sarcomas, in a murine model of osteosarcoma nivolumab (an anti-PD-1 monoclonal antibody) increased tumor infiltrating CD4+ and CD8+ T cells with greater cytotoxic potential (i.e., granzyme B and IFN-γ production) and less lung metastases (186). PD-1 blockade using pembrolizumab in the SARC028 phase II study (see Table 1; NCT02301039) demonstrated an objective partial response (based on Response Evaluation Criteria in Solid Tumors (RECIST)) in only one osteosarcoma patient out of 22 patients and stable disease in six other patients. Of note, there were no responses in EWS patients, who typically had a low mutational burden (94). Correlative analysis of patient samples from the SARC028 study showed that pembrolizumab responders were more likely to have higher densities of activated CD8+CD3+PD-1+ T cells and increased percentages of PD-L1+ TAMs pre-treatment compared to non-responders. Pre-treatment analysis of tumors from responders also demonstrated higher densities of effector memory cytotoxic T cells and regulatory T cells compared to non-responders (187). Given the relatively mutated response of PD-1 axis blockade as monotherapy in pediatric sarcomas, combination strategies with other immune-targeted agents are currently being evaluated in clinical trials (188).
The TNFR family is the other major group of co-stimulatory molecules, which includes CD40, tumor necrosis factor receptor superfamily member 4 (TNFRSF4 or CD134), tumor necrosis factor receptor superfamily member 9 (TNFRSF9 or 4-1BB), and CD27 (165, 189, 190). The co-stimulatory receptor CD40 is a transmembrane protein expressed on monocytes, macrophages, and other antigen presenting cells (191). Its ligand, CD40 ligand (CD40L), is primarily expressed on activated T and B lymphocytes, monocytes and platelets (165). CD40 agonist monoclonal antibodies (mAbs) promote TAM M2 to M1 polarization, leading to increased production of nitric oxide and type 1 cytokines (such as IL-1, IL-12, and TNF-α), and activation of cytotoxic activity of CD8+ T cells (192–195). CD40 agonism as monotherapy in advanced solid tumors had limited anti-tumor activity (196); however, use of a CD40 agonist in combination with PD-L1 and CTLA-4 blockade (see below) has shown extended survival in murine solid tumor models (197) (NCT02636725, NCT02332668).
CTLA-4 (also known as CD152) is part of the B7/CD28 family that also inhibits T cell cytotoxic function. CTLA-4 suppresses T cell activation when engaged with its respective ligands, B7-1 (CD80) and B7-2 (CD86) through inhibition of T cell receptor (TCR) signal transduction (198). Sarcoma patients have T cells with high CTLA-4 expression within the tumor and peripheral blood (199, 200). Phase I and II studies of ipilimumab, a CTLA-4 blocking mAbs, in pediatric patients with advanced solid tumors (including sarcomas) showed tolerability but no objective clinical or radiologic responses as monotherapy (NCT01445379) (95, 201). Combination therapies utilizing CTLA-4 and PD-L1 mAb blockade in early phase clinical studies have shown synergistic slowing of disease progression and extended PFS in adults with metastatic or unresectable sarcomas; however, they have not been tested in children (NCT02500797) (202, 203).
Efferocytosis modulates the immune system beyond the regulation of engulfment and co-stimulation in the sarcoma TME. Intracellular signal transduction in efferocytosis favors production of tumor-permissive cytokines such as TGF-β and IL-10 consistent with the otherwise physiologic role of this process in immune tolerance, wound healing, and tissue homeostasis (204–206). TGF-β drives immunosuppressive responses in both the innate and adaptive immune systems. Within the innate immune system, TGF-β secreted by macrophages further skews cells toward an M2 alternative activation status, inhibits cytotoxic and cytokine producing activity of NK cells, and decreases migration and increases apoptosis of dendritic cells (204, 207). In the adaptive immune response, TGF-β promotes CD4+ T cells to differentiate into Th2 cells and inhibits CD8+ T cells antitumor activity by downregulating cytolytic genes such as granzyme B and Fas ligand (FasL), thereby reducing antitumor response (208, 209).
One method of overriding the potently suppressive TAM cytokine production is administration of exogenous type 1 cytokines (210, 211). Interferons and IL-2 are such powerful type 1 stimulators of the immune system. Dinutuximab (ch14.18, a mAb against tumor-associated disialoganglioside GD2) has demonstrated activity against neuroblastoma cells; however, administration of the mAb alone was insufficient to prevent tumor progression, thus both GM-CSF and IL-2 were added to augment efficacy. The addition of IL-2 and GM-CSF to dinutuximab greatly enhanced ADCC by M1 macrophages; however, systemic IL-2 administration was found to have significant toxicity in patients, thus this treatment regimen may still need to be further optimized (104, 212), but could conceptually be applied to other pediatric solid tumors considering the GD2 is also expressed by sarcoma (213, 214).
TNF-α is another type 1 cytokine that has been studied for its effects on augmenting activating antigen presentation within the sarcoma TME. It is produced by classically activated macrophages and lymphocytes and was thought to be a potential immunotherapeutic agent. The majority of exogenous TNF-α administration in preclinical studies was used to mimic chronic inflammation, and thus results were not as favorable as predicted. For instance, a preclinical study of osteosarcoma demonstrated that TNF-α administration promoted the de-differentiation of osteosarcoma cells toward a primitive state, which significantly contributed to tumor growth and progression. Furthermore, blocking TNF-α using a soluble receptor (etanercept) to diminish chronic inflammation inhibited osteosarcoma tumor growth (215). Systemic administration of recombinant TNF-α with chemotherapy in an early Children’s Cancer Group (CCG) phase I study was limited due to systemic toxicities and an inability to dose escalate (93). It has been suggested that further administration of cytokines may need to be targeted to the sarcoma microenvironment rather than systemic administration. As above regarding polarizing macrophages, innovative methods of cytokines delivery will be necessary to allow effective administration without the significant systemic toxicities.
Other mechanisms of TAM-induced immunosuppression leading to T cell dysfunction in the TME include breakdown of key metabolites, such as L-arginine and L-tryptophan, which are necessary for T cell activation and proliferation. TAMs produced and secrete arginase 1 (ARG1) and indoleamine 2,3-dioxygenase 1/2 (IDO 1/2), enzymes that catalyze and breakdown L-arginine and L-tryptophan respectively. Breakdown of these metabolites diminishes effector T cell function, thereby increasing the likelihood of cancer cell immune escape (216, 217). In fact, in a study of checkpoint inhibition in adult sarcoma patients where the response rates were lower than expected, the tumor samples had high infiltration of IDO1-expressing TAMs leading to the speculation that elimination of the suppressive TAMs is also needed (NCT02406781) (218). Supplementation with L-arginine in combination with a PD-1/PD-L1 inhibitor in a murine model of both localized and metastatic osteosarcoma increased tumor infiltrating lymphocytes and prolonged survival compared to controls (219). The use of ARG1 targeted small-molecule inhibitors demonstrated reversal of TAM-mediated immunosuppression including production of inflammatory cytokines, CTL, and NK cell tumor infiltration, T cell proliferation, expression of IFN-inducible genes, and restored cytolytic T cell function against solid malignancies in vitro and in vivo (220). While there are currently no pediatric clinical trials investigating the use of targeted agents against ARG1 and IDO 1/2, there are several studies in adult patients. Additionally, there is an open-label phase 1/2 study investigating an arginase inhibitor (INCB00158) as single or combination therapy with other immune checkpoint therapy in adult patients with advanced/metastatic solid tumors (NCT02903914).
When tumors reach a certain size, an “angiogenic switch” occurs in which mechanisms are triggered to promote angiogenesis, the formation of high-density vasculature, to increase tumor nutrient supply and improve waste removal (221). TAMs can hasten blood vessel growth through the release of pro-angiogenic factors such as vascular endothelial growth factor (VEGF). Other cytokines released by TAMs such as TGF-β, C-C motif chemokine ligand 2 (CCL2), C-X-C motif chemokine ligand 8 (CXCL8), and M-CSF further promote pro-angiogenic functions of macrophages (222–225). On the contrary, M1 polarization of TAMs results in inhibition of angiogenesis through the upregulation of anti-angiogenic factors (such as CXCL8 and IFN-β) (226).
VEGF-A is a pro-angiogenic cytokine released by TAMs (227), and has been studied in pediatric sarcomas given that angiogenesis is a critical step in solid tumor progression (228). EWS xenograft models have also showed delayed tumor progression with anti-VEGF directed therapies; however, rebound tumor growth occurred after therapy was discontinued, suggesting single agent VEGF-directed therapy may have limited success in the treatment of pediatric sarcomas (229, 230).
Once angiogenesis has been established, this allows for further tumor progression and metastasis. Metastasis is a complex multi-step process, which starts with tumor cells migrating and intravasating into the vasculature, circulating in the blood stream, eventual extravasation at target organs, and subsequent invasion and growth to establish disease. This complex process requires not only circulating tumor cells, but also requires the close cooperation of perivascular cells, endothelial cells, as well a variety of immune cells including macrophages.
CSF-1 is a chemokine that stimulates macrophage motility/migration, maturation, and survival, and has been implicated in metastasis. Its contribution to metastasis formation was demonstrated in a mammary cancer model where paracrine secretion of CSF-1 by tumor cells stimulated TAMs to migrate and provide a tract for tumors cells to follow along and invade normal tissue and vasculature (223). Congruent with this, immunohistochemistry examination of soft tissue tumor patient samples showed increased expression of CSF-1 (M-CSF) and colony stimulating factor receptor (CSF1R) in more aggressive, higher histologic grade tumors (231). Additionally, CSF-1 mediated mobilization of macrophages and other hematopoietic stem and progenitor cells (HSPCs) are thought to be integral to the formation of the pre-metastatic niche for sarcoma cells at distant sites in the body. In an embryonal RMS murine model, HSPCs were found to be elevated in the peripheral blood during formation of the pre-metastatic niche and contributed to tumor-promoting immunosuppressive myeloid subsets at metastatic sites. Similarly, peripheral blood samples from RMS patients had elevated circulating HSPCs, and patients at greatest risk of metastases had the highest levels of circulating HSPCs at the time of diagnosis (232). Because CSF-1 is essential for TAM migration and maturation, strategic targeting of its receptor (CSF1R) has been explored (233–235). Mice bearing CSF-1 negative neuroblastoma xenografts showed decreased TAM infiltration and angiogenesis, compared to mice with CSF-1 expressing xenografts. Inhibition of CSF1R in neuroblastoma decreased TAM infiltration, improved T cell function, and decreased tumor progression compared to controls (236, 237).
Furthermore, metastasis-associated macrophages (MAMs) are recruited to tumor sites through C-C motif chemokine ligand 2 (CCL2) secretion from tumor cells, a chemokine that mediates monocyte migration from bone marrow to tissue sites through interaction with the macrophage CCL2 receptor, C-C motif chemokine receptor 2 (CCR2) (238). These MAMs secrete additional CCL2 to further augment TAM recruitment to metastatic sites, and CCL3 which instigates tumor seeding at distant sites (239). In vivo anti-CCL2 antibody treatment reduced the number of MAMs at metastatic sites and reduced overall tumor burden in breast cancer models (240, 241). Collectively, these studies demonstrate macrophages play roles in the development of metastases and soft tissue infiltration and are potential targets in pediatric sarcomas.
In the clinical setting, attempts have been made to combine therapies targeting tumor angiogenesis and metastasis. For example, bevacizumab (anti-VEGF-A mAb) previously has been combined with conventional chemotherapy backbones, such as vincristine, irinotecan, and temozolamide (VIT), gemcitabine, docetaxel, or low dose cyclophosphamide and sorafenib have shown limited results, producing only stable disease or partial response in a subset of patients with refractory/relapsed disease (see Table 1) (96–100, 242, 243).
Additionally for metastasis targeted therapies, there is limited data on combining such drugs with conventional therapies like chemotherapy. Preclinical evaluations demonstrate that combination of CSF1R inhibition after radiation therapy may more effectively decrease tumor volume (244). A majority of clinical trials studying CSF1R inhibitors are in very early clinical trial phases either as monotherapy or combination therapies for the treatment of relapsed/refractory sarcomas. For example, in a phase 1 clinical trial using a CSF1R small molecule inhibitor, pexidartinib (PLX3397) in pediatric patients with refractory solid tumors (including sarcomas) and leukemias showed tolerability, and the expansion cohort is still ongoing (NCT02390752; see Table 1) (101). Some trials utilizing monoclonal antibodies directed at CSF-1/CSF1R in adults exhibited limited anti-tumor activity (NCT01346358) (245, 246).
The development and clinical use of chimeric antigen receptor (CAR) T cell therapy for the treatment of relapsed/refractory acute lymphoblastic leukemia has provided a promising new therapy option for some patients (247, 248). CAR T cell therapy for pediatric sarcomas has been centered on the development of a CAR directed against GD-2, which is overexpressed on pediatric sarcoma patient samples, with an especially high predominance on osteosarcoma primary and metastatic lesions (249). However, despite the efficacy of CAR therapy in treating hematological malignancies, use of CAR T cell therapy in sarcomas has been more challenging. This is partly due to the difficulty of T cell homing, tumor penetration, and the presence of inhibitory cell subsets in the microenvironment, including infiltrating TAMs that inhibit T cell function.
To overcome such challenges, researchers have explored methods of inhibiting infiltrating suppressor myeloid cells (including TAMs and MDSCs) alongside CAR platforms. In preclinical models, use of all-trans retinoic acid—which differentiated infiltrating myeloid cells, lessening their suppressor function—was found to significantly increase the efficacy of GD-2 directed CAR T cells in pediatric sarcoma models (249). Similar solid tumor models with high levels of TAM or MDSC infiltration found that inhibition of CSF1R increased the efficacy of adoptively transferred T cells (250). As an alternative strategy to mitigate the T cell suppressive effects of TAMs, some groups have engineered their CAR T cells to express cytokines that will lead to TAM M1 repolarization, including IL-12 and IL-18 (251, 252).
Interestingly, the idea of inhibiting TAMs has also been evaluated using a CAR T cell directed against the TAMs themselves. In preclinical models, CAR T cells directed against folate receptor β (FRβ), which is highly expressed by M2 macrophages, lead to cytolysis of M2 macrophages; however, this has not yet been assessed in pediatric sarcomas (253).
Though unrelated to targeting TAMs in pediatric sarcoma, it is notable that some groups are looking into harnessing the infiltrative properties of macrophages by engineering CAR-Macrophages (CAR-Ms). This therapy could be used to direct phagocytic anti-tumor immunity against tumor antigen expressing cells (e.g. human epidermal growth factor receptor (HER2), mesothelin) or use alongside CAR T cell therapies to improve T cell penetration into the sarcoma through ECM breakdown (254, 255).
Given the rising interest in cellular therapy to treat malignancies, targeting TAMs and their closely related MDSC populations in the TME will become increasingly important. Similar approaches to inhibiting TAMs in combination with immunotherapy in development include use of bi- and tri-valent T cell engagers (BiTEs, TriTEs) to deplete CD206 and FRβ expressing TAMs, inhibition of CXCR2 alongside T cell immunotherapy (e.g. nivolumab), or TAM repolarization (to an M1 phenotype) using tyrosine kinase inhibitors (256)
Research on TAM-targeting CAR T cells, TAM repolarizing agents or harnessing effector function of CAR-Ms is rapidly evolving. Further work is required to study the potential use of TAM inhibition in conjunction with immunotherapy in sarcomas to further boost anti-tumor immunity.
It is evident there is remarkable growth in the field of oncologic immunotherapy originating from overall improved understanding of the interaction of cancer cells, TME, and the host immune system. To enhance responses against pediatric sarcomas, new immunotherapy targets and rational combinations of existing immunotherapeutic agents are being investigated. As one of the major components of the TME, TAMs play an intricate role in the regulation of immune suppression within the tumor microenvironment, augmenting angiogenesis, and promoting tumor metastasis formation. All of these are growing areas of research for potential targets in the treatment of pediatric sarcomas. In this review, we discussed the numerous roles TAMs play in driving the immunosuppressive, tumor-promoting environment in the TME, as well as in promoting metastasis, and how this may be reversed in pediatric sarcomas. TAMs are an emerging novel target that has the potential to circumvent immune evasion and hopefully improve survival for pediatric sarcoma patients.
JK and AL-S conducted extensive literature review on this subject. JK AL-S, MV, and MH wrote, critically revised and edited the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by the National Institutes of Health (5K08CA222699-03, AL-S), The V Foundation (AL-S), Hyundai Hope on Wheels (MH, MV, and AL-S), the St. Baldrick’s Scholar Award (MH) and the National Pediatric Cancer Foundation Research Grant (MH).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Williams RF, Fernandez-Pineda I, Gosain A. Pediatric Sarcomas. Surg Clin North Am (2016) 96(5):1107–25. doi: 10.1016/j.suc.2016.05.012
2. Xu JF, Pan XH, Zhang SJ, Zhao C, Qiu BS, Gu HF, et al. CD47 blockade inhibits tumor progression human osteosarcoma in xenograft models. Oncotarget (2015) 6(27):23662–70. doi: 10.18632/oncotarget.4282
3. Edris B, Weiskopf K, Volkmer AK, Volkmer JP, Willingham SB, Contreras-Trujillo H, et al. Antibody therapy targeting the CD47 protein is effective in a model of aggressive metastatic leiomyosarcoma. Proc Natl Acad Sci U S A (2012) 109(17):6656–61. doi: 10.1073/pnas.1121629109
4. Herrmann D, Seitz G, Fuchs J, Armeanu-Ebinger S. Susceptibility of rhabdomyosarcoma cells to macrophage-mediated cytotoxicity. Oncoimmunology (2012) 1(3):279–86. doi: 10.4161/onci.18612
5. Mohanty S, Aghighi M, Yerneni K, Theruvath JL, Daldrup-Link HE. Improving the efficacy of osteosarcoma therapy: combining drugs that turn cancer cell ‘don’t eat me’ signals off and ‘eat me’ signals on. Mol Oncol (2019) 13(10):2049–61. doi: 10.1002/1878-0261.12556
6. Ferrari A, Sultan I, Huang TT, Rodriguez-Galindo C, Shehadeh A, Meazza C, et al. Soft tissue sarcoma across the age spectrum: a population-based study from the Surveillance Epidemiology and End Results database. Pediatr Blood Cancer (2011) 57(6):943–9. doi: 10.1002/pbc.23252
7. Crist WM, Anderson JR, Meza JL, Fryer C, Raney RB, Ruymann FB, et al. Intergroup rhabdomyosarcoma study-IV: results for patients with nonmetastatic disease. J Clin Oncol (2001) 19(12):3091–102. doi: 10.1200/JCO.2001.19.12.3091
8. Arndt CA, Stoner JA, Hawkins DS, Rodeberg DA, Hayes-Jordan AA, Paidas CN, et al. Vincristine, actinomycin, and cyclophosphamide compared with vincristine, actinomycin, and cyclophosphamide alternating with vincristine, topotecan, and cyclophosphamide for intermediate-risk rhabdomyosarcoma: children’s oncology group study D9803. J Clin Oncol (2009) 27(31):5182–8. doi: 10.1200/JCO.2009.22.3768
9. Stevens MC, Rey A, Bouvet N, Ellershaw C, Flamant F, Habrand JL, et al. Treatment of nonmetastatic rhabdomyosarcoma in childhood and adolescence: third study of the International Society of Paediatric Oncology–SIOP Malignant Mesenchymal Tumor 89. J Clin Oncol (2005) 23(12):2618–28. doi: 10.1200/JCO.2005.08.130
10. Oberlin O, Rey A, Lyden E, Bisogno G, Stevens MC, Meyer WH, et al. Prognostic factors in metastatic rhabdomyosarcomas: results of a pooled analysis from United States and European cooperative groups. J Clin Oncol (2008) 26(14):2384–9. doi: 10.1200/JCO.2007.14.7207
11. Howlader N NA, Krapcho M, Miller D, Brest A, Yu M, Ruhl J, et al eds. SEER Cancer Statistics Review, 1975-2016. National Cancer Institute DCCPS, Surveillance Research Program (2018). Available at: https://seer.cancer.gov/archive/csr/1975_2016/#citation. updated posted to the SEER web site, April 2019.
12. Paulino AC. Late effects of radiotherapy for pediatric extremity sarcomas. Int J Radiat Oncol Biol Phys (2004) 60(1):265–74. doi: 10.1016/j.ijrobp.2004.02.001
13. Brown TR, Vijarnsorn C, Potts J, Milner R, Sandor GG, Fryer C. Anthracycline induced cardiac toxicity in pediatric Ewing sarcoma: a longitudinal study. Pediatr Blood Cancer (2013) 60(5):842–8. doi: 10.1002/pbc.24404
14. Heyn R, Raney RB Jr., Hays DM, Tefft M, Gehan E, Webber B, et al. Late effects of therapy in patients with paratesticular rhabdomyosarcoma. Intergroup Rhabdomyosarcoma Study Committee. J Clin Oncol (1992) 10(4):614–23. doi: 10.1200/JCO.1992.10.4.614
15. Rich DC, Corpron CA, Smith MB, Black CT, Lally KP, Andrassy RJ. Second malignant neoplasms in children after treatment of soft tissue sarcoma. J Pediatr Surg (1997) 32(2):369–72. doi: 10.1016/S0022-3468(97)90213-X
16. Pinto A, Dickman P, Parham D. Pathobiologic markers of the ewing sarcoma family of tumors: state of the art and prediction of behaviour. Sarcoma (2011) 2011:856190. doi: 10.1155/2011/856190
17. Barr FG, Galili N, Holick J, Biegel JA, Rovera G, Emanuel BS. Rearrangement of the PAX3 paired box gene in the paediatric solid tumour alveolar rhabdomyosarcoma. Nat Genet (1993) 3(2):113–7. doi: 10.1038/ng0293-113
18. Clark J, Rocques PJ, Crew AJ, Gill S, Shipley J, Chan AM, et al. Identification of novel genes, SYT and SSX, involved in the t(X;18)(p11.2;q11.2) translocation found in human synovial sarcoma. Nat Genet (1994) 7(4):502–8. doi: 10.1038/ng0894-502
19. Chmielecki J, Bailey M, He J, Elvin J, Vergilio JA, Ramkissoon S, et al. Genomic Profiling of a Large Set of Diverse Pediatric Cancers Identifies Known and Novel Mutations across Tumor Spectra. Cancer Res (2017) 77(2):509–19. doi: 10.1158/0008-5472.CAN-16-1106
20. Shern JF, Chen L, Chmielecki J, Wei JS, Patidar R, Rosenberg M, et al. Comprehensive genomic analysis of rhabdomyosarcoma reveals a landscape of alterations affecting a common genetic axis in fusion-positive and fusion-negative tumors. Cancer Discov (2014) 4(2):216–31. doi: 10.1158/2159-8290.CD-13-0639
21. Tirode F, Surdez D, Ma X, Parker M, Le Deley MC, Bahrami A, et al. Genomic landscape of Ewing sarcoma defines an aggressive subtype with co-association of STAG2 and TP53 mutations. Cancer Discov (2014) 4(11):1342–53. doi: 10.1158/2159-8290.CD-14-0622
22. Crompton BD, Stewart C, Taylor-Weiner A, Alexe G, Kurek KC, Calicchio ML, et al. The genomic landscape of pediatric Ewing sarcoma. Cancer Discov (2014) 4(11):1326–41. doi: 10.1158/2159-8290.CD-13-1037
23. Sayles LC, Breese MR, Koehne AL, Leung SG, Lee AG, Liu HY, et al. Genome-Informed Targeted Therapy for Osteosarcoma. Cancer Discov (2019) 9(1):46–63. doi: 10.1158/2159-8290.CD-17-1152
24. Shearer WT, Rosenblatt HM, Gelman RS, Oyomopito R, Plaeger S, Stiehm ER, et al. Lymphocyte subsets in healthy children from birth through 18 years of age: the Pediatric AIDS Clinical Trials Group P1009 study. J Allergy Clin Immunol (2003) 112(5):973–80. doi: 10.1016/j.jaci.2003.07.003
25. Sheu TT, Chiang BL, Yen JH, Lin WC. Premature CD4+ T cell aging and its contribution to lymphopenia-induced proliferation of memory cells in autoimmune-prone non-obese diabetic mice. PLoS One (2014) 9(2):e89379. doi: 10.1371/journal.pone.0089379
26. Sturgeon P. Volumetric and microscopic pattern of bone marrow in normal infants and children. III. Histologic pattern. Pediatrics (1951) 7(6):774–81. doi: 10.1111/ijlh.12073
27. Ogawa T, Kitagawa M, Hirokawa K. Age-related changes of human bone marrow: a histometric estimation of proliferative cells, apoptotic cells, T cells, B cells and macrophages. Mech Ageing Dev (2000) 117(1-3):57–68. doi: 10.1016/S0047-6374(00)00137-8
28. Rego EM, Garcia AB, Viana SR, Falcao RP. Age-related changes of lymphocyte subsets in normal bone marrow biopsies. Cytometry (1998) 34(1):22–9. doi: 10.1002/(SICI)1097-0320(19980215)34:1<22::AID-CYTO4>3.0.CO;2-G
29. De Palma M, Venneri MA, Galli R, Sergi Sergi L, Politi LS, Sampaolesi M, et al. Tie2 identifies a hematopoietic lineage of proangiogenic monocytes required for tumor vessel formation and a mesenchymal population of pericyte progenitors. Cancer Cell (2005) 8(3):211–26. doi: 10.1016/j.ccr.2005.08.002
30. Elpek KG, Cremasco V, Shen H, Harvey CJ, Wucherpfennig KW, Goldstein DR, et al. The tumor microenvironment shapes lineage, transcriptional, and functional diversity of infiltrating myeloid cells. Cancer Immunol Res (2014) 2(7):655–67. doi: 10.1158/2326-6066.CIR-13-0209
31. Movahedi K, Guilliams M, Van den Bossche J, Van den Bergh R, Gysemans C, Beschin A, et al. Identification of discrete tumor-induced myeloid-derived suppressor cell subpopulations with distinct T cell-suppressive activity. Blood (2008) 111(8):4233–44. doi: 10.1182/blood-2007-07-099226
32. Fujiwara T, Fukushi J, Yamamoto S, Matsumoto Y, Setsu N, Oda Y, et al. Macrophage infiltration predicts a poor prognosis for human ewing sarcoma. Am J Pathol (2011) 179(3):1157–70. doi: 10.1016/j.ajpath.2011.05.034
33. Nabeshima A, Matsumoto Y, Fukushi J, Iura K, Matsunobu T, Endo M, et al. Tumour-associated macrophages correlate with poor prognosis in myxoid liposarcoma and promote cell motility and invasion via the HB-EGF-EGFR-PI3K/Akt pathways. Br J Cancer (2015) 112(3):547–55. doi: 10.1038/bjc.2014.637
34. Stahl D, Gentles AJ, Thiele R, Gutgemann I. Prognostic profiling of the immune cell microenvironment in Ewing s Sarcoma Family of Tumors. Oncoimmunology (2019) 8(12):e1674113. doi: 10.1080/2162402X.2019.1674113
35. Chen L, Oke T, Siegel N, Cojocaru G, Tam AJ, Blosser RL, et al. The Immunosuppressive Niche of Soft Tissue Sarcomas is Sustained by Tumor Associated Macrophages and Characterized by Intratumoral Tertiary Lymphoid Structures. Clin Cancer Res (2020) 26:4018–30. doi: 10.1158/1078-0432.CCR-19-3416
36. Raj SK, Kooshki M, Winters M, Russell GB, Miller LD, Laurini JA, et al. Prognostic implications of tumor associated macrophages (TAMs) in soft tissue sarcoma. J Clin Oncol (2019) 37(15_suppl):e22548–e. doi: 10.1200/JCO.2019.37.15_suppl.e22548
37. Stroncek DF, Ren J, Lee DW, Tran M, Frodigh SE, Sabatino M, et al. Myeloid cells in peripheral blood mononuclear cell concentrates inhibit the expansion of chimeric antigen receptor T cells. Cytotherapy (2016) 18(7):893–901. doi: 10.1016/j.jcyt.2016.04.003
38. Hume DA. The mononuclear phagocyte system. Curr Opin Immunol (2006) 18(1):49–53. doi: 10.1016/j.coi.2005.11.008
39. Hirayama D, Iida T, Nakase H. The Phagocytic Function of Macrophage-Enforcing Innate Immunity and Tissue Homeostasis. Int J Mol Sci (2017) 19(1):92. doi: 10.3390/ijms19010092
40. Hohl TM, Rivera A, Lipuma L, Gallegos A, Shi C, Mack M, et al. Inflammatory monocytes facilitate adaptive CD4 T cell responses during respiratory fungal infection. Cell Host Microbe (2009) 6(5):470–81. doi: 10.1016/j.chom.2009.10.007
41. Kim TS, Braciale TJ. Respiratory dendritic cell subsets differ in their capacity to support the induction of virus-specific cytotoxic CD8+ T cell responses. PLoS One (2009) 4(1):e4204. doi: 10.1371/journal.pone.0004204
42. Kim SY, Nair MG. Macrophages in wound healing: activation and plasticity. Immunol Cell Biol (2019) 97(3):258–67. doi: 10.1111/imcb.12236
43. Ramachandran P, Pellicoro A, Vernon MA, Boulter L, Aucott RL, Ali A, et al. Differential Ly-6C expression identifies the recruited macrophage phenotype, which orchestrates the regression of murine liver fibrosis. Proc Natl Acad Sci U S A (2012) 109(46):E3186–95. doi: 10.1073/pnas.1119964109
44. Deng B, Wehling-Henricks M, Villalta SA, Wang Y, Tidball JG. IL-10 triggers changes in macrophage phenotype that promote muscle growth and regeneration. J Immunol (2012) 189(7):3669–80. doi: 10.4049/jimmunol.1103180
45. Galdiero MR, Garlanda C, Jaillon S, Marone G, Mantovani A. Tumor associated macrophages and neutrophils in tumor progression. J Cell Physiol (2013) 228(7):1404–12. doi: 10.1002/jcp.24260
46. Lee CH, Espinosa I, Vrijaldenhoven S, Subramanian S, Montgomery KD, Zhu S, et al. Prognostic significance of macrophage infiltration in leiomyosarcomas. Clin Cancer Res (2008) 14(5):1423–30. doi: 10.1158/1078-0432.CCR-07-1712
47. Leimgruber A, Berger C, Cortez-Retamozo V, Etzrodt M, Newton AP, Waterman P, et al. Behavior of endogenous tumor-associated macrophages assessed in vivo using a functionalized nanoparticle. Neoplasia (2009) 11(5):459–68, 2 p following 68. doi: 10.1593/neo.09356
48. Aghighi M, Theruvath AJ, Pareek A, Pisani LL, Alford R, Muehe AM, et al. Magnetic Resonance Imaging of Tumor-Associated Macrophages: Clinical Translation. Clin Cancer Res (2018) 24(17):4110–8. doi: 10.1158/1078-0432.CCR-18-0673
49. Rahman N, Pervin M, Kuramochi M, Karim MR, Izawa T, Kuwamura M, et al. M1/M2-macrophage Polarization-based Hepatotoxicity in d-galactosamine-induced Acute Liver Injury in Rats. Toxicol Pathol (2018) 46(7):764–76. doi: 10.1177/0192623318801574
50. Dancsok AR, Gao D, Lee AF, Steigen SE, Blay JY, Thomas DM, et al. Tumor-associated macrophages and macrophage-related immune checkpoint expression in sarcomas. Oncoimmunology (2020) 9(1):1747340. doi: 10.1080/2162402X.2020.1747340
51. Kather JN, Horner C, Weis CA, Aung T, Vokuhl C, Weiss C, et al. CD163+ immune cell infiltrates and presence of CD54+ microvessels are prognostic markers for patients with embryonal rhabdomyosarcoma. Sci Rep (2019) 9(1):9211. doi: 10.1038/s41598-019-45551-y
52. Choo MK, Sano Y, Kim C, Yasuda K, Li XD, Lin X, et al. TLR sensing of bacterial spore-associated RNA triggers host immune responses with detrimental effects. J Exp Med (2017) 214(5):1297–311. doi: 10.1084/jem.20161141
53. Krieg AM, Yi AK, Matson S, Waldschmidt TJ, Bishop GA, Teasdale R, et al. CpG motifs in bacterial DNA trigger direct B-cell activation. Nature (1995) 374(6522):546–9. doi: 10.1038/374546a0
54. Chan MP, Onji M, Fukui R, Kawane K, Shibata T, Saitoh S, et al. DNase II-dependent DNA digestion is required for DNA sensing by TLR9. Nat Commun (2015) 6:5853. doi: 10.1038/ncomms6853
55. Gordon S. Alternative activation of macrophages. Nat Rev Immunol (2003) 3(1):23–35. doi: 10.1038/nri978
56. Kuper C, Beck FX, Neuhofer W. Toll-like receptor 4 activates NF-kappaB and MAP kinase pathways to regulate expression of proinflammatory COX-2 in renal medullary collecting duct cells. Am J Physiol Renal Physiol (2012) 302(1):F38–46. doi: 10.1152/ajprenal.00590.2010
57. Curtsinger JM, Schmidt CS, Mondino A, Lins DC, Kedl RM, Jenkins MK, et al. Inflammatory cytokines provide a third signal for activation of naive CD4+ and CD8+ T cells. J Immunol (1999) 162(6):3256–62. doi: 10.1016/j.coi.2010.02.013
58. Keppler SJ, Rosenits K, Koegl T, Vucikuja S, Aichele P. Signal 3 cytokines as modulators of primary immune responses during infections: the interplay of type I IFN and IL-12 in CD8 T cell responses. PLoS One (2012) 7(7):e40865. doi: 10.1371/journal.pone.0040865
59. Liu CP, Zhang X, Tan QL, Xu WX, Zhou CY, Luo M, et al. NF-kappaB pathways are involved in M1 polarization of RAW 264.7 macrophage by polyporus polysaccharide in the tumor microenvironment. PLoS One (2017) 12(11):e0188317. doi: 10.1371/journal.pone.0188317
60. Tugal D, Liao X, Jain MK. Transcriptional control of macrophage polarization. Arterioscler Thromb Vasc Biol (2013) 33(6):1135–44. doi: 10.1161/ATVBAHA.113.301453
61. de Gaetano M, Crean D, Barry M, Belton O. M1- and M2-Type Macrophage Responses Are Predictive of Adverse Outcomes in Human Atherosclerosis. Front Immunol (2016) 7:275. doi: 10.3389/fimmu.2016.00275
62. Verreck FA, de Boer T, Langenberg DM, Hoeve MA, Kramer M, Vaisberg E, et al. Human IL-23-producing type 1 macrophages promote but IL-10-producing type 2 macrophages subvert immunity to (myco)bacteria. Proc Natl Acad Sci U S A (2004) 101(13):4560–5. doi: 10.1073/pnas.0400983101
63. Verreck FA, de Boer T, Langenberg DM, van der Zanden L, Ottenhoff TH. Phenotypic and functional profiling of human proinflammatory type-1 and anti-inflammatory type-2 macrophages in response to microbial antigens and IFN-gamma- and CD40L-mediated costimulation. J Leukoc Biol (2006) 79(2):285–93. doi: 10.1189/jlb.0105015
64. Ragnhammar P, Frodin JE, Trotta PP, Mellstedt H. Cytotoxicity of white blood cells activated by granulocyte-colony-stimulating factor, granulocyte/macrophage-colony-stimulating factor and macrophage-colony-stimulating factor against tumor cells in the presence of various monoclonal antibodies. Cancer Immunol Immunother (1994) 39(4):254–62. doi: 10.1007/BF01525989
65. Charak BS, Agah R, Mazumder A. Granulocyte-macrophage colony-stimulating factor-induced antibody-dependent cellular cytotoxicity in bone marrow macrophages: application in bone marrow transplantation. Blood (1993) 81(12):3474–9. doi: 10.1182/blood.V81.12.3474.bloodjournal81123474
66. Young DA, Lowe LD, Clark SC. Comparison of the effects of IL-3, granulocyte-macrophage colony-stimulating factor, and macrophage colony-stimulating factor in supporting monocyte differentiation in culture. Analysis of macrophage antibody-dependent cellular cytotoxicity. J Immunol (1990) 145(2):607–15.
67. Yao Y, Xu XH, Jin L. Macrophage Polarization in Physiological and Pathological Pregnancy. Front Immunol (2019) 10:792. doi: 10.3389/fimmu.2019.00792
68. Foss DL, Zilliox MJ, Murtaugh MP. Differential regulation of macrophage interleukin-1 (IL-1), IL-12, and CD80-CD86 by two bacterial toxins. Infect Immun (1999) 67(10):5275–81. doi: 10.1128/IAI.67.10.5275-5281.1999
69. Moganti K, Li F, Schmuttermaier C, Riemann S, Kluter H, Gratchev A, et al. Hyperglycemia induces mixed M1/M2 cytokine profile in primary human monocyte-derived macrophages. Immunobiology (2017) 222(10):952–9. doi: 10.1016/j.imbio.2016.07.006
70. Stein M, Keshav S, Harris N, Gordon S. Interleukin 4 potently enhances murine macrophage mannose receptor activity: a marker of alternative immunologic macrophage activation. J Exp Med (1992) 176(1):287–92. doi: 10.1084/jem.176.1.287
71. Van Dyken SJ, Locksley RM. Interleukin-4- and interleukin-13-mediated alternatively activated macrophages: roles in homeostasis and disease. Annu Rev Immunol (2013) 31:317–43. doi: 10.1146/annurev-immunol-032712-095906
72. Trombetta AC, Soldano S, Contini P, Tomatis V, Ruaro B, Paolino S, et al. A circulating cell population showing both M1 and M2 monocyte/macrophage surface markers characterizes systemic sclerosis patients with lung involvement. Respir Res (2018) 19(1):186. doi: 10.1186/s12931-018-0891-z
73. Lee SJ, Evers S, Roeder D, Parlow AF, Risteli J, Risteli L, et al. Mannose receptor-mediated regulation of serum glycoprotein homeostasis. Science (2002) 295(5561):1898–901. doi: 10.1126/science.1069540
74. Halpern MS, Deery DT, Flores LJ, Fujita DJ, Mason WS. Differential reactivity of serum antibody from tumor-bearing 15I5 X 7(2) chickens for cross-reactive species of endogenous retroviral envelope glycoprotein. Virology (1983) 124(2):318–29. doi: 10.1016/0042-6822(83)90348-3
75. Nguyen TT, Schwartz EJ, West RB, Warnke RA, Arber DA, Natkunam Y. Expression of CD163 (hemoglobin scavenger receptor) in normal tissues, lymphomas, carcinomas, and sarcomas is largely restricted to the monocyte/macrophage lineage. Am J Surg Pathol (2005) 29(5):617–24. doi: 10.1097/01.pas.0000157940.80538.ec
76. Buechler C, Ritter M, Orso E, Langmann T, Klucken J, Schmitz G. Regulation of scavenger receptor CD163 expression in human monocytes and macrophages by pro- and antiinflammatory stimuli. J Leukoc Biol (2000) 67(1):97–103. doi: 10.1002/jlb.67.1.97
77. Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol (2002) 23(11):549–55. doi: 10.1016/S1471-4906(02)02302-5
78. Yamazaki T, Akiba H, Iwai H, Matsuda H, Aoki M, Tanno Y, et al. Expression of programmed death 1 ligands by murine T cells and APC. J Immunol (2002) 169(10):5538–45. doi: 10.4049/jimmunol.169.10.5538
79. Loke P, Allison JP. PD-L1 and PD-L2 are differentially regulated by Th1 and Th2 cells. Proc Natl Acad Sci U S A (2003) 100(9):5336–41. doi: 10.1073/pnas.0931259100
80. Kleinerman ES, Erickson KL, Schroit AJ, Fogler WE, Fidler IJ. Activation of tumoricidal properties in human blood monocytes by liposomes containing lipophilic muramyl tripeptide. Cancer Res (1983) 43(5):2010–4.
81. Asano T, McWatters A, An T, Matsushima K, Kleinerman ES. Liposomal muramyl tripeptide up-regulates interleukin-1 alpha, interleukin-1 beta, tumor necrosis factor-alpha, interleukin-6 and interleukin-8 gene expression in human monocytes. J Pharmacol Exp Ther (1994) 268(2):1032–9. doi: 10.1007/BF01517165
82. Biteau K, Guiho R, Chatelais M, Taurelle J, Chesneau J, Corradini N, et al. L-MTP-PE and zoledronic acid combination in osteosarcoma: preclinical evidence of positive therapeutic combination for clinical transfer. Am J Cancer Res (2016) 6(3):677–89.
83. Anderson PM, Markovic SN, Sloan JA, Clawson ML, Wylam M, Arndt CA, et al. Aerosol granulocyte macrophage-colony stimulating factor: a low toxicity, lung-specific biological therapy in patients with lung metastases. Clin Cancer Res (1999) 5(9):2316–23.
84. Arndt CA, Koshkina NV, Inwards CY, Hawkins DS, Krailo MD, Villaluna D, et al. Inhaled granulocyte-macrophage colony stimulating factor for first pulmonary recurrence of osteosarcoma: effects on disease-free survival and immunomodulation. a report from the Children’s Oncology Group. Clin Cancer Res (2010) 16(15):4024–30. doi: 10.1158/1078-0432.CCR-10-0662
85. Lagmay JP, Krailo MD, Dang H, Kim A, Hawkins DS, Beaty O,3, et al. Outcome of Patients With Recurrent Osteosarcoma Enrolled in Seven Phase II Trials Through Children’s Cancer Group, Pediatric Oncology Group, and Children’s Oncology Group: Learning From the Past to Move Forward. J Clin Oncol (2016) 34(25):3031–8. doi: 10.1200/JCO.2015.65.5381
86. Luksch R, Massimino M, Cefalo G, Lombardi F, Ferrari A, Casanova M, et al. Effects of recombinant human granulocyte-macrophage colony-stimulating factor in an intensive treatment program for children with Ewing’s sarcoma. Haematologica (2001) 86(7):753–60. doi: 10.3324/%25x
87. Goldsby RE, Fan TM, Villaluna D, Wagner LM, Isakoff MS, Meyer J, et al. Feasibility and dose discovery analysis of zoledronic acid with concurrent chemotherapy in the treatment of newly diagnosed metastatic osteosarcoma: a report from the Children’s Oncology Group. Eur J Cancer (2013) 49(10):2384–91. doi: 10.1016/j.ejca.2013.03.018
88. Conry RM, Rodriguez MG, Pressey JG. Zoledronic acid in metastatic osteosarcoma: encouraging progression free survival in four consecutive patients. Clin Sarcoma Res (2016) 6:6. doi: 10.1186/s13569-016-0046-2
89. Anderton J, Moroz V, Marec-Berard P, Gaspar N, Laurence V, Martin-Broto J, et al. International randomised controlled trial for the treatment of newly diagnosed EWING sarcoma family of tumours - EURO EWING 2012 Protocol. Trials (2020) 21(1):96. doi: 10.1186/s13063-019-4026-8
90. Creaven PJ, Cowens JW, Brenner DE, Dadey BM, Han T, Huben R, et al. Initial clinical trial of the macrophage activator muramyl tripeptide-phosphatidylethanolamine encapsulated in liposomes in patients with advanced cancer. J Biol Response Mod (1990) 9(5):492–8.
91. Kleinerman ES, Meyers PA, Raymond AK, Gano JB, Jia SF, Jaffe N. Combination therapy with ifosfamide and liposome-encapsulated muramyl tripeptide: tolerability, toxicity, and immune stimulation. J Immunother Emphasis Tumor Immunol (1995) 17(3):181–93. doi: 10.1097/00002371-199504000-00007
92. Chou AJ, Kleinerman ES, Krailo MD, Chen Z, Betcher DL, Healey JH, et al. Addition of muramyl tripeptide to chemotherapy for patients with newly diagnosed metastatic osteosarcoma: a report from the Children’s Oncology Group. Cancer (2009) 115(22):5339–48. doi: 10.1002/cncr.24566
93. Seibel NL, Dinndorf PA, Bauer M, Sondel PM, Hammond GD, Reaman GH. Phase I study of tumor necrosis factor-alpha and actinomycin D in pediatric patients with cancer: a Children’s Cancer Group study. J Immunother Emphasis Tumor Immunol (1994) 16(2):125–31. doi: 10.1097/00002371-199408000-00006
94. Tawbi HA, Burgess M, Bolejack V, Van Tine BA, Schuetze SM, Hu J, et al. Pembrolizumab in advanced soft-tissue sarcoma and bone sarcoma (SARC028): a multicentre, two-cohort, single-arm, open-label, phase 2 trial. Lancet Oncol (2017) 18(11):1493–501. doi: 10.1016/S1470-2045(17)30624-1
95. Merchant MS, Wright M, Baird K, Wexler LH, Rodriguez-Galindo C, Bernstein D, et al. Phase I Clinical Trial of Ipilimumab in Pediatric Patients with Advanced Solid Tumors. Clin Cancer Res (2016) 22(6):1364–70. doi: 10.1158/1078-0432.CCR-15-0491
96. Slotkin EK, Magnan HD, Meyers PA, Chou AJ, Ambati SR, Wexler LH. Off-label use of bevacizumab in relapsed and refractory pediatric sarcoma patients: The Memorial Sloan Kettering Cancer Center Experience. J Clin Oncol (2016) 34(15_suppl):10569–. doi: 10.1200/JCO.2016.34.15_suppl.10569
97. Glade Bender JL, Adamson PC, Reid JM, Xu L, Baruchel S, Shaked Y, et al. Phase I trial and pharmacokinetic study of bevacizumab in pediatric patients with refractory solid tumors: a Children’s Oncology Group Study. J Clin Oncol (2008) 26(3):399–405. doi: 10.1200/JCO.2007.11.9230
98. Okada K, Yamasaki K, Tanaka C, Fujisaki H, Osugi Y, Hara J. Phase I study of bevacizumab plus irinotecan in pediatric patients with recurrent/refractory solid tumors. Jpn J Clin Oncol (2013) 43(11):1073–9. doi: 10.1093/jjco/hyt124
99. Venkatramani R, Malogolowkin M, Davidson TB, May W, Sposto R, Mascarenhas L. A phase I study of vincristine, irinotecan, temozolomide and bevacizumab (vitb) in pediatric patients with relapsed solid tumors. PLoS One (2013) 8(7):e68416. doi: 10.1371/journal.pone.0068416
100. Navid F, Baker SD, McCarville MB, Stewart CF, Billups CA, Wu J, et al. Phase I and clinical pharmacology study of bevacizumab, sorafenib, and low-dose cyclophosphamide in children and young adults with refractory/recurrent solid tumors. Clin Cancer Res (2013) 19(1):236–46. doi: 10.1158/1078-0432.CCR-12-1897
101. Wesolowski R, Sharma N, Reebel L, Rodal MB, Peck A, West BL, et al. Phase Ib study of the combination of pexidartinib (PLX3397), a CSF-1R inhibitor, and paclitaxel in patients with advanced solid tumors. Ther Adv Med Oncol (2019) 11:1758835919854238. doi: 10.1177/1758835919854238
102. Roberts SS, Chou AJ, Cheung NK. Immunotherapy of Childhood Sarcomas. Front Oncol (2015) 5:181. doi: 10.3389/fonc.2015.00181
103. Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity (2014) 41(1):14–20. doi: 10.1016/j.immuni.2014.06.008
104. Yu AL, Gilman AL, Ozkaynak MF, London WB, Kreissman SG, Chen HX, et al. Anti-GD2 antibody with GM-CSF, interleukin-2, and isotretinoin for neuroblastoma. N Engl J Med (2010) 363(14):1324–34. doi: 10.1056/NEJMoa0911123
105. Worth LL, Lafleur EA, Jia SF, Kleinerman ES. Fas expression inversely correlates with metastatic potential in osteosarcoma cells. Oncol Rep (2002) 9(4):823–7. doi: 10.3892/or.9.4.823
106. Shields CW, Evans MA, Wang LL-W, Baugh N, Iyer S, Wu D, et al. Cellular backpacks for macrophage immunotherapy. Sci Adv (2020) 6(18):eaaz6579. doi: 10.1126/sciadv.aaz6579
107. Lee HJ, Ko HJ, Song DK, Jung YJ. Lysophosphatidylcholine Promotes Phagosome Maturation and Regulates Inflammatory Mediator Production Through the Protein Kinase A-Phosphatidylinositol 3 Kinase-p38 Mitogen-Activated Protein Kinase Signaling Pathway During Mycobacterium tuberculosis Infection in Mouse Macrophages. Front Immunol (2018) 9:920. doi: 10.3389/fimmu.2018.00920
108. Kao YC, Owosho AA, Sung YS, Zhang L, Fujisawa Y, Lee JC, et al. BCOR-CCNB3 Fusion Positive Sarcomas: A Clinicopathologic and Molecular Analysis of 36 Cases With Comparison to Morphologic Spectrum and Clinical Behavior of Other Round Cell Sarcomas. Am J Surg Pathol (2018) 42(5):604–15. doi: 10.1097/PAS.0000000000000965
109. Gude DR, Alvarez SE, Paugh SW, Mitra P, Yu J, Griffiths R, et al. Apoptosis induces expression of sphingosine kinase 1 to release sphingosine-1-phosphate as a “come-and-get-me” signal. FASEB J (2008) 22(8):2629–38. doi: 10.1096/fj.08-107169
110. Truman LA, Ford CA, Pasikowska M, Pound JD, Wilkinson SJ, Dumitriu IE, et al. CX3CL1/fractalkine is released from apoptotic lymphocytes to stimulate macrophage chemotaxis. Blood (2008) 112(13):5026–36. doi: 10.1182/blood-2008-06-162404
111. Tsai WH, Shih CH, Feng SY, Li IT, Chang SC, Lin YC, et al. CX3CL1(+) microparticles mediate the chemoattraction of alveolar macrophages toward apoptotic acute promyelocytic leukemic cells. Cell Physiol Biochem (2014) 33(3):594–604. doi: 10.1159/000358637
112. Elliott MR, Chekeni FB, Trampont PC, Lazarowski ER, Kadl A, Walk SF, et al. Nucleotides released by apoptotic cells act as a find-me signal to promote phagocytic clearance. Nature (2009) 461(7261):282–6. doi: 10.1038/nature08296
113. Peter C, Waibel M, Radu CG, Yang LV, Witte ON, Schulze-Osthoff K, et al. Migration to apoptotic “find-me” signals is mediated via the phagocyte receptor G2A. J Biol Chem (2008) 283(9):5296–305. doi: 10.1074/jbc.M706586200
114. Bazan JF, Bacon KB, Hardiman G, Wang W, Soo K, Rossi D, et al. A new class of membrane-bound chemokine with a CX3C motif. Nature (1997) 385(6617):640–4. doi: 10.1038/385640a0
115. Sokolowski JD, Chabanon-Hicks CN, Han CZ, Heffron DS, Mandell JW. Fractalkine is a “find-me” signal released by neurons undergoing ethanol-induced apoptosis. Front Cell Neurosci (2014) 8:360. doi: 10.3389/fncel.2014.00360
116. Schneider G, Sellers ZP, Abdel-Latif A, Morris AJ, Ratajczak MZ. Bioactive lipids, LPC and LPA, are novel prometastatic factors and their tissue levels increase in response to radio/chemotherapy. Mol Cancer Res (2014) 12(11):1560–73. doi: 10.1158/1541-7786.MCR-14-0188
117. Engel N, Adamus A, Schauer N, Kuhn J, Nebe B, Seitz G, et al. Synergistic Action of Genistein and Calcitriol in Immature Osteosarcoma MG-63 Cells by SGPL1 Up-Regulation. PLoS One (2017) 12(1):e0169742. doi: 10.1371/journal.pone.0169742
118. Berwin B, Delneste Y, Lovingood RV, Post SR, Pizzo SV. SREC-I, a type F scavenger receptor, is an endocytic receptor for calreticulin. J Biol Chem (2004) 279(49):51250–7. doi: 10.1074/jbc.M406202200
119. Paidassi H, Tacnet-Delorme P, Verneret M, Gaboriaud C, Houen G, Duus K, et al. Investigations on the C1q-calreticulin-phosphatidylserine interactions yield new insights into apoptotic cell recognition. J Mol Biol (2011) 408(2):277–90. doi: 10.1016/j.jmb.2011.02.029
120. Feng M, Chen JY, Weissman-Tsukamoto R, Volkmer JP, Ho PY, McKenna KM, et al. Macrophages eat cancer cells using their own calreticulin as a guide: roles of TLR and Btk. Proc Natl Acad Sci U S A (2015) 112(7):2145–50. doi: 10.1073/pnas.1424907112
121. Van Valen F, Fulda S, Truckenbrod B, Eckervogt V, Sonnemann J, Hillmann A, et al. Apoptotic responsiveness of the Ewing’s sarcoma family of tumours to tumour necrosis factor-related apoptosis-inducing ligand (TRAIL). Int J Cancer (2000) 88(2):252–9. doi: 10.1002/1097-0215(20001015)88:2<252::AID-IJC17>3.0.CO;2-U
122. Zhang XH, Zhang Y, Xie WP, Sun DS, Zhang YK, Hao YK, et al. Expression and significance of calreticulin in human osteosarcoma. Cancer Biomark (2017) 18(4):405–11. doi: 10.3233/CBM-160266
123. Schurch CM, Forster S, Bruhl F, Yang SH, Felley-Bosco E, Hewer E. The “don’t eat me” signal CD47 is a novel diagnostic biomarker and potential therapeutic target for diffuse malignant mesothelioma. Oncoimmunology (2017) 7(1):e1373235. doi: 10.1080/2162402X.2017.1373235
124. Brown S, Heinisch I, Ross E, Shaw K, Buckley CD, Savill J. Apoptosis disables CD31-mediated cell detachment from phagocytes promoting binding and engulfment. Nature (2002) 418(6894):200–3. doi: 10.1038/nature00811
125. Liu X, Pu Y, Cron K, Deng L, Kline J, Frazier WA, et al. CD47 blockade triggers T cell-mediated destruction of immunogenic tumors. Nat Med (2015) 21(10):1209–15. doi: 10.1038/nm.3931
126. Chao MP, Alizadeh AA, Tang C, Myklebust JH, Varghese B, Gill S, et al. Anti-CD47 antibody synergizes with rituximab to promote phagocytosis and eradicate non-Hodgkin lymphoma. Cell (2010) 142(5):699–713. doi: 10.1016/j.cell.2010.07.044
127. Iribarren K, Buque A, Mondragon L, Xie W, Levesque S, Pol J, et al. Anticancer effects of anti-CD47 immunotherapy in vivo. Oncoimmunology (2019) 8(3):1550619. doi: 10.1080/2162402X.2018.1550619
128. Jaiswal S, Jamieson CH, Pang WW, Park CY, Chao MP, Majeti R, et al. CD47 is upregulated on circulating hematopoietic stem cells and leukemia cells to avoid phagocytosis. Cell (2009) 138(2):271–85. doi: 10.1016/j.cell.2009.05.046
129. Chao MP, Weissman IL, Majeti R. The CD47-SIRPalpha pathway in cancer immune evasion and potential therapeutic implications. Curr Opin Immunol (2012) 24(2):225–32. doi: 10.1016/j.coi.2012.01.010
130. Tsai RK, Discher DE. Inhibition of “self” engulfment through deactivation of myosin-II at the phagocytic synapse between human cells. J Cell Biol (2008) 180(5):989–1003. doi: 10.1083/jcb.200708043
131. Lin Y, Zhao JL, Zheng QJ, Jiang X, Tian J, Liang SQ, et al. Notch Signaling Modulates Macrophage Polarization and Phagocytosis Through Direct Suppression of Signal Regulatory Protein alpha Expression. Front Immunol (2018) 9:1744. doi: 10.3389/fimmu.2018.01744
132. Huang F, Zhao JL, Wang L, Gao CC, Liang SQ, An DJ, et al. miR-148a-3p Mediates Notch Signaling to Promote the Differentiation and M1 Activation of Macrophages. Front Immunol (2017) 8:1327. doi: 10.3389/fimmu.2017.01327
133. Wang YC, He F, Feng F, Liu XW, Dong GY, Qin HY, et al. Notch signaling determines the M1 versus M2 polarization of macrophages in antitumor immune responses. Cancer Res (2010) 70(12):4840–9. doi: 10.1158/0008-5472.CAN-10-0269
134. Advani R, Flinn I, Popplewell L, Forero A, Bartlett NL, Ghosh N, et al. CD47 Blockade by Hu5F9-G4 and Rituximab in Non-Hodgkin’s Lymphoma. N Engl J Med (2018) 379(18):1711–21. doi: 10.1056/NEJMoa1807315
135. Santiago C, Ballesteros A, Martinez-Munoz L, Mellado M, Kaplan GG, Freeman GJ, et al. Structures of T cell immunoglobulin mucin protein 4 show a metal-Ion-dependent ligand binding site where phosphatidylserine binds. Immunity (2007) 27(6):941–51. doi: 10.1016/j.immuni.2007.11.008
136. DeKruyff RH, Bu X, Ballesteros A, Santiago C, Chim YL, Lee HH, et al. T cell/transmembrane, Ig, and mucin-3 allelic variants differentially recognize phosphatidylserine and mediate phagocytosis of apoptotic cells. J Immunol (2010) 184(4):1918–30. doi: 10.4049/jimmunol.0903059
137. Dransfield I, Zagorska A, Lew ED, Michail K, Lemke G. Mer receptor tyrosine kinase mediates both tethering and phagocytosis of apoptotic cells. Cell Death Dis (2015) 6:e1646. doi: 10.1038/cddis.2015.18
138. Shibata T, Habiel DM, Coelho AL, Kunkel SL, Lukacs NW, Hogaboam CM. Axl receptor blockade ameliorates pulmonary pathology resulting from primary viral infection and viral exacerbation of asthma. J Immunol (2014) 192(8):3569–81. doi: 10.4049/jimmunol.1302766
139. Sen P, Wallet MA, Yi Z, Huang Y, Henderson M, Mathews CE, et al. Apoptotic cells induce Mer tyrosine kinase-dependent blockade of NF-kappaB activation in dendritic cells. Blood (2007) 109(2):653–60. doi: 10.1182/blood-2006-04-017368
140. Camenisch TD, Koller BH, Earp HS, Matsushima GK. A novel receptor tyrosine kinase, Mer, inhibits TNF-alpha production and lipopolysaccharide-induced endotoxic shock. J Immunol (1999) 162(6):3498–503.
141. Wallet MA, Sen P, Flores RR, Wang Y, Yi Z, Huang Y, et al. MerTK is required for apoptotic cell-induced T cell tolerance. J Exp Med (2008) 205(1):219–32. doi: 10.1084/jem.20062293
142. Lee-Sherick AB, Jacobsen KM, Henry CJ, Huey MG, Parker RE, Page LS, et al. MERTK inhibition alters the PD-1 axis and promotes anti-leukemia immunity. JCI Insight (2018) 3(21):e97941. doi: 10.1172/jci.insight.97941
143. Myers KV, Amend SR, Pienta KJ. Targeting Tyro3, Axl and MerTK (TAM receptors): implications for macrophages in the tumor microenvironment. Mol Cancer (2019) 18(1):94. doi: 10.1186/s12943-019-1022-2
144. Cook RS, Jacobsen KM, Wofford AM, DeRyckere D, Stanford J, Prieto AL, et al. MerTK inhibition in tumor leukocytes decreases tumor growth and metastasis. J Clin Invest (2013) 123(8):3231–42. doi: 10.1172/JCI67655
145. Crittenden MR, Baird J, Friedman D, Savage T, Uhde L, Alice A, et al. Mertk on tumor macrophages is a therapeutic target to prevent tumor recurrence following radiation therapy. Oncotarget (2016) 7(48):78653–66. doi: 10.18632/oncotarget.11823
146. Stitt TN, Conn G, Gore M, Lai C, Bruno J, Radziejewski C, et al. The anticoagulation factor protein S and its relative, Gas6, are ligands for the Tyro 3/Axl family of receptor tyrosine kinases. Cell (1995) 80(4):661–70. doi: 10.1016/0092-8674(95)90520-0
147. Lew ED, Oh J, Burrola PG, Lax I, Zagorska A, Traves PG, et al. Differential TAM receptor-ligand-phospholipid interactions delimit differential TAM bioactivities. Elife (2014) 3:e03385. doi: 10.7554/eLife.03385
148. Shiozawa Y, Pedersen EA, Patel LR, Ziegler AM, Havens AM, Jung Y, et al. GAS6/AXL axis regulates prostate cancer invasion, proliferation, and survival in the bone marrow niche. Neoplasia (2010) 12(2):116–27. doi: 10.1593/neo.91384
149. Caraux A, Lu Q, Fernandez N, Riou S, Di Santo JP, Raulet DH, et al. Natural killer cell differentiation driven by Tyro3 receptor tyrosine kinases. Nat Immunol (2006) 7(7):747–54. doi: 10.1038/ni1353
150. Waizenegger JS, Ben-Batalla I, Weinhold N, Meissner T, Wroblewski M, Janning M, et al. Role of Growth arrest-specific gene 6-Mer axis in multiple myeloma. Leukemia (2015) 29(3):696–704. doi: 10.1038/leu.2014.236
151. Carrera Silva EA, Chan PY, Joannas L, Errasti AE, Gagliani N, Bosurgi L, et al. T cell-derived protein S engages TAM receptor signaling in dendritic cells to control the magnitude of the immune response. Immunity (2013) 39(1):160–70. doi: 10.1016/j.immuni.2013.06.010
152. Fleuren ED, Hillebrandt-Roeffen MH, Flucke UE, Te Loo DM, Boerman OC, van der Graaf WT, et al. The role of AXL and the in vitro activity of the receptor tyrosine kinase inhibitor BGB324 in Ewing sarcoma. Oncotarget (2014) 5(24):12753–68. doi: 10.18632/oncotarget.2648
153. Kimani SG, Kumar S, Bansal N, Singh K, Kholodovych V, Comollo T, et al. Small molecule inhibitors block Gas6-inducible TAM activation and tumorigenicity. Sci Rep (2017) 7:43908. doi: 10.1038/srep43908
154. Che Mat MF, Abdul Murad NA, Ibrahim K, Mohd Mokhtar N, Wan Ngah WZ, Harun R, et al. Silencing of PROS1 induces apoptosis and inhibits migration and invasion of glioblastoma multiforme cells. Int J Oncol (2016) 49(6):2359–66. doi: 10.3892/ijo.2016.3755
155. Abboud-Jarrous G, Priya S, Maimon A, Fischman S, Cohen-Elisha M, Czerninski R, et al. Protein S drives oral squamous cell carcinoma tumorigenicity through regulation of AXL. Oncotarget (2017) 8(8):13986–4002. doi: 10.18632/oncotarget.14753
156. Wong K, Valdez PA, Tan C, Yeh S, Hongo JA, Ouyang W. Phosphatidylserine receptor Tim-4 is essential for the maintenance of the homeostatic state of resident peritoneal macrophages. Proc Natl Acad Sci U S A (2010) 107(19):8712–7. doi: 10.1073/pnas.0910929107
157. Kobayashi N, Karisola P, Pena-Cruz V, Dorfman DM, Jinushi M, Umetsu SE, et al. TIM-1 and TIM-4 glycoproteins bind phosphatidylserine and mediate uptake of apoptotic cells. Immunity (2007) 27(6):927–40. doi: 10.1016/j.immuni.2007.11.011
158. Shakhov AN, Rybtsov S, Tumanov AV, Shulenin S, Dean M, Kuprash DV, et al. SMUCKLER/TIM4 is a distinct member of TIM family expressed by stromal cells of secondary lymphoid tissues and associated with lymphotoxin signaling. Eur J Immunol (2004) 34(2):494–503. doi: 10.1002/eji.200324590
159. Nishi C, Yanagihashi Y, Segawa K, Nagata S. MERTK tyrosine kinase receptor together with TIM4 phosphatidylserine receptor mediates distinct signal transduction pathways for efferocytosis and cell proliferation. J Biol Chem (2019) 294(18):7221–30. doi: 10.1074/jbc.RA118.006628
160. Nishi C, Toda S, Segawa K, Nagata S. Tim4- and MerTK-mediated engulfment of apoptotic cells by mouse resident peritoneal macrophages. Mol Cell Biol (2014) 34(8):1512–20. doi: 10.1128/MCB.01394-13
161. Yanagihashi Y, Segawa K, Maeda R, Nabeshima YI, Nagata S. Mouse macrophages show different requirements for phosphatidylserine receptor Tim4 in efferocytosis. Proc Natl Acad Sci U S A (2017) 114(33):8800–5. doi: 10.1073/pnas.1705365114
162. Gleason MK, Lenvik TR, McCullar V, Felices M, O’Brien MS, Cooley SA, et al. Tim-3 is an inducible human natural killer cell receptor that enhances interferon gamma production in response to galectin-9. Blood (2012) 119(13):3064–72. doi: 10.1182/blood-2011-06-360321
163. Avery L, Filderman J, Szymczak-Workman AL, Kane LP. Tim-3 co-stimulation promotes short-lived effector T cells, restricts memory precursors, and is dispensable for T cell exhaustion. Proc Natl Acad Sci U S A (2018) 115(10):2455–60. doi: 10.1073/pnas.1712107115
164. Nakayama M, Akiba H, Takeda K, Kojima Y, Hashiguchi M, Azuma M, et al. Tim-3 mediates phagocytosis of apoptotic cells and cross-presentation. Blood (2009) 113(16):3821–30. doi: 10.1182/blood-2008-10-185884
165. Dancsok AR, Setsu N, Gao D, Blay JY, Thomas D, Maki RG, et al. Expression of lymphocyte immunoregulatory biomarkers in bone and soft-tissue sarcomas. Mod Pathol (2019) 32(12):1772–85. doi: 10.1038/s41379-019-0312-y
166. Sakuishi K, Apetoh L, Sullivan JM, Blazar BR, Kuchroo VK, Anderson AC. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J Exp Med (2010) 207(10):2187–94. doi: 10.1084/jem.20100643
167. Zhou Q, Munger ME, Veenstra RG, Weigel BJ, Hirashima M, Munn DH, et al. Coexpression of Tim-3 and PD-1 identifies a CD8+ T-cell exhaustion phenotype in mice with disseminated acute myelogenous leukemia. Blood (2011) 117(17):4501–10. doi: 10.1182/blood-2010-10-310425
168. Moreau MF, Guillet C, Massin P, Chevalier S, Gascan H, Basle MF, et al. Comparative effects of five bisphosphonates on apoptosis of macrophage cells in vitro. Biochem Pharmacol (2007) 73(5):718–23. doi: 10.1016/j.bcp.2006.09.031
169. Rogers TL, Holen I. Tumour macrophages as potential targets of bisphosphonates. J Transl Med (2011) 9:177. doi: 10.1186/1479-5876-9-177
170. Coscia M, Quaglino E, Iezzi M, Curcio C, Pantaleoni F, Riganti C, et al. Zoledronic acid repolarizes tumour-associated macrophages and inhibits mammary carcinogenesis by targeting the mevalonate pathway. J Cell Mol Med (2010) 14(12):2803–15. doi: 10.1111/j.1582-4934.2009.00926.x
171. Zhou Z, Guan H, Duan X, Kleinerman ES. Zoledronic acid inhibits primary bone tumor growth in Ewing sarcoma. Cancer (2005) 104(8):1713–20. doi: 10.1002/cncr.21383
172. Odri GA, Dumoucel S, Picarda G, Battaglia S, Lamoureux F, Corradini N, et al. Zoledronic acid as a new adjuvant therapeutic strategy for Ewing’s sarcoma patients. Cancer Res (2010) 70(19):7610–9. doi: 10.1158/0008-5472.CAN-09-4272
173. Parker D. CD80/CD86 signaling contributes to the proinflammatory response of Staphylococcus aureus in the airway. Cytokine (2018) 107:130–6. doi: 10.1016/j.cyto.2018.01.016
174. Subauste CS, de Waal Malefyt R, Fuh F. Role of CD80 (B7.1) and CD86 (B7.2) in the immune response to an intracellular pathogen. J Immunol (1998) 160(4):1831–40.
175. Kuang DM, Zhao Q, Peng C, Xu J, Zhang JP, Wu C, et al. Activated monocytes in peritumoral stroma of hepatocellular carcinoma foster immune privilege and disease progression through PD-L1. J Exp Med (2009) 206(6):1327–37. doi: 10.1084/jem.20082173
176. Ojalvo LS, King W, Cox D, Pollard JW. High-density gene expression analysis of tumor-associated macrophages from mouse mammary tumors. Am J Pathol (2009) 174(3):1048–64. doi: 10.2353/ajpath.2009.080676
177. Calles A, Liao X, Sholl LM, Rodig SJ, Freeman GJ, Butaney M, et al. Expression of PD-1 and Its Ligands, PD-L1 and PD-L2, in Smokers and Never Smokers with KRAS-Mutant Lung Cancer. J Thorac Oncol (2015) 10(12):1726–35. doi: 10.1097/JTO.0000000000000687
178. Hino R, Kabashima K, Kato Y, Yagi H, Nakamura M, Honjo T, et al. Tumor cell expression of programmed cell death-1 ligand 1 is a prognostic factor for malignant melanoma. Cancer (2010) 116(7):1757–66. doi: 10.1002/cncr.24899
179. Nomi T, Sho M, Akahori T, Hamada K, Kubo A, Kanehiro H, et al. Clinical significance and therapeutic potential of the programmed death-1 ligand/programmed death-1 pathway in human pancreatic cancer. Clin Cancer Res (2007) 13(7):2151–7. doi: 10.1158/1078-0432.CCR-06-2746
180. Liu Y, Zugazagoitia J, Ahmed FS, Henick BS, Gettinger SN, Herbst RS, et al. Immune Cell PD-L1 Colocalizes with Macrophages and Is Associated with Outcome in PD-1 Pathway Blockade Therapy. Clin Cancer Res (2020) 26(4):970–7. doi: 10.1158/1078-0432.CCR-19-1040
181. Koirala P, Roth ME, Gill J, Piperdi S, Chinai JM, Geller DS, et al. Immune infiltration and PD-L1 expression in the tumor microenvironment are prognostic in osteosarcoma. Sci Rep (2016) 6:30093. doi: 10.1038/srep30093
182. Kim C, Kim EK, Jung H, Chon HJ, Han JW, Shin KH, et al. Prognostic implications of PD-L1 expression in patients with soft tissue sarcoma. BMC Cancer (2016) 16:434. doi: 10.1186/s12885-016-2451-6
183. Dhupkar P, Gordon N, Stewart J, Kleinerman ES. Anti-PD-1 therapy redirects macrophages from an M2 to an M1 phenotype inducing regression of OS lung metastases. Cancer Med (2018) 7(6):2654–64. doi: 10.1002/cam4.1518
184. Hartley GP, Chow L, Ammons DT, Wheat WH, Dow SW. Programmed Cell Death Ligand 1 (PD-L1) Signaling Regulates Macrophage Proliferation and Activation. Cancer Immunol Res (2018) 6(10):1260–73. doi: 10.1158/2326-6066.CIR-17-0537
185. Gordon SR, Maute RL, Dulken BW, Hutter G, George BM, McCracken MN, et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature (2017) 545(7655):495–9. doi: 10.1038/nature22396
186. Zheng B, Ren T, Huang Y, Sun K, Wang S, Bao X, et al. PD-1 axis expression in musculoskeletal tumors and antitumor effect of nivolumab in osteosarcoma model of humanized mouse. J Hematol Oncol (2018) 11(1):16. doi: 10.1186/s13045-018-0580-x
187. Keung EZ, Burgess M, Salazar R, Parra ER, Rodrigues-Canales J, Bolejack V, et al. Correlative Analyses of the SARC028 Trial Reveal an Association Between Sarcoma-Associated Immune Infiltrate and Response to Pembrolizumab. Clin Cancer Res (2020) 26(6):1258–66. doi: 10.1158/1078-0432.CCR-19-1824
188. Davis KL, Fox E, Merchant MS, Reid JM, Kudgus RA, Liu X, et al. Nivolumab in children and young adults with relapsed or refractory solid tumours or lymphoma (ADVL1412): a multicentre, open-label, single-arm, phase 1-2 trial. Lancet Oncol (2020) 21(4):541–50. doi: 10.1016/S1470-2045(20)30023-1
189. Collins AV, Brodie DW, Gilbert RJ, Iaboni A, Manso-Sancho R, Walse B, et al. The interaction properties of costimulatory molecules revisited. Immunity (2002) 17(2):201–10. doi: 10.1016/S1074-7613(02)00362-X
190. Ito T, Wang YH, Duramad O, Hori T, Delespesse GJ, Watanabe N, et al. TSLP-activated dendritic cells induce an inflammatory T helper type 2 cell response through OX40 ligand. J Exp Med (2005) 202(9):1213–23. doi: 10.1084/jem.20051135
191. Carbone E, Ruggiero G, Terrazzano G, Palomba C, Manzo C, Fontana S, et al. A new mechanism of NK cell cytotoxicity activation: the CD40-CD40 ligand interaction. J Exp Med (1997) 185(12):2053–60. doi: 10.1084/jem.185.12.2053
192. Mantovani A, Sica A. Macrophages, innate immunity and cancer: balance, tolerance, and diversity. Curr Opin Immunol (2010) 22(2):231–7. doi: 10.1016/j.coi.2010.01.009
193. Georgoudaki AM, Prokopec KE, Boura VF, Hellqvist E, Sohn S, Ostling J, et al. Reprogramming Tumor-Associated Macrophages by Antibody Targeting Inhibits Cancer Progression and Metastasis. Cell Rep (2016) 15(9):2000–11. doi: 10.1016/j.celrep.2016.04.084
194. Burger D, Molnarfi N, Gruaz L, Dayer JM. Differential induction of IL-1beta and TNF by CD40 ligand or cellular contact with stimulated T cells depends on the maturation stage of human monocytes. J Immunol (2004) 173(2):1292–7. doi: 10.4049/jimmunol.173.2.1292
195. Bullens DM, Kasran A, Thielemans K, Bakkus M, Ceuppens JL. CD40L-induced IL-12 production is further enhanced by the Th2 cytokines IL-4 and IL-13. Scand J Immunol (2001) 53(5):455–63. doi: 10.1046/j.1365-3083.2001.00900.x
196. Vonderheide RH, Flaherty KT, Khalil M, Stumacher MS, Bajor DL, Hutnick NA, et al. Clinical activity and immune modulation in cancer patients treated with CP-870,893, a novel CD40 agonist monoclonal antibody. J Clin Oncol (2007) 25(7):876–83. doi: 10.1200/JCO.2006.08.3311
197. Zippelius A, Schreiner J, Herzig P, Muller P. Induced PD-L1 expression mediates acquired resistance to agonistic anti-CD40 treatment. Cancer Immunol Res (2015) 3(3):236–44. doi: 10.1158/2326-6066.CIR-14-0226
198. Seidel JA, Otsuka A, Kabashima K. Anti-PD-1 and Anti-CTLA-4 Therapies in Cancer: Mechanisms of Action, Efficacy, and Limitations. Front Oncol (2018) 8:86. doi: 10.3389/fonc.2018.00086
199. Contardi E, Palmisano GL, Tazzari PL, Martelli AM, Fala F, Fabbi M, et al. CTLA-4 is constitutively expressed on tumor cells and can trigger apoptosis upon ligand interaction. Int J Cancer (2005) 117(4):538–50. doi: 10.1002/ijc.21155
200. Hingorani P, Maas ML, Gustafson MP, Dickman P, Adams RH, Watanabe M, et al. Increased CTLA-4(+) T cells and an increased ratio of monocytes with loss of class II (CD14(+) HLA-DR(lo/neg)) found in aggressive pediatric sarcoma patients. J Immunother Cancer (2015) 3:35. doi: 10.1186/s40425-015-0082-0
201. Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med (2013) 369(2):122–33. doi: 10.1056/NEJMoa1302369
202. Lussier DM, Johnson JL, Hingorani P, Blattman JN. Combination immunotherapy with alpha-CTLA-4 and alpha-PD-L1 antibody blockade prevents immune escape and leads to complete control of metastatic osteosarcoma. J Immunother Cancer (2015) 3:21. doi: 10.1186/s40425-015-0067-z
203. D’Angelo SP, Mahoney MR, Van Tine BA, Atkins J, Milhem MM, Jahagirdar BN, et al. Nivolumab with or without ipilimumab treatment for metastatic sarcoma (Alliance A091401): two open-label, non-comparative, randomised, phase 2 trials. Lancet Oncol (2018) 19(3):416–26. doi: 10.1016/S1470-2045(18)30006-8
204. DeRyckere D, Lee-Sherick AB, Huey MG, Hill AA, Tyner JW, Jacobsen KM, et al. UNC2025, a MERTK Small-Molecule Inhibitor, Is Therapeutically Effective Alone and in Combination with Methotrexate in Leukemia Models. Clin Cancer Res (2016) 23:1481–92. doi: 10.1158/1078-0432.CCR-16-1330
205. Arlauckas SP, Garren SB, Garris CS, Kohler RH, Oh J, Pittet MJ, et al. Arg1 expression defines immunosuppressive subsets of tumor-associated macrophages. Theranostics (2018) 8(21):5842–54. doi: 10.7150/thno.26888
206. Brouckaert G, Kalai M, Krysko DV, Saelens X, Vercammen D, Ndlovu MN, et al. Phagocytosis of necrotic cells by macrophages is phosphatidylserine dependent and does not induce inflammatory cytokine production. Mol Biol Cell (2004) 15(3):1089–100. doi: 10.1091/mbc.e03-09-0668
207. Ito M, Minamiya Y, Kawai H, Saito S, Saito H, Nakagawa T, et al. Tumor-derived TGFbeta-1 induces dendritic cell apoptosis in the sentinel lymph node. J Immunol (2006) 176(9):5637–43. doi: 10.4049/jimmunol.176.9.5637
208. Maeda H, Shiraishi A. TGF-beta contributes to the shift toward Th2-type responses through direct and IL-10-mediated pathways in tumor-bearing mice. J Immunol (1996) 156(1):73–8.
209. Thomas DA, Massague J. TGF-beta directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell (2005) 8(5):369–80. doi: 10.1016/j.ccr.2005.10.012
210. Watkins SK, Egilmez NK, Suttles J, Stout RD. IL-12 rapidly alters the functional profile of tumor-associated and tumor-infiltrating macrophages in vitro and in vivo. J Immunol (2007) 178(3):1357–62. doi: 10.4049/jimmunol.178.3.1357
211. Watkins SK, Li B, Richardson KS, Head K, Egilmez NK, Zeng Q, et al. Rapid release of cytoplasmic IL-15 from tumor-associated macrophages is an initial and critical event in IL-12-initiated tumor regression. Eur J Immunol (2009) 39(8):2126–35. doi: 10.1002/eji.200839010
212. Ladenstein R, Potschger U, Valteau-Couanet D, Luksch R, Castel V, Yaniv I, et al. Interleukin 2 with anti-GD2 antibody ch14.18/CHO (dinutuximab beta) in patients with high-risk neuroblastoma (HR-NBL1/SIOPEN): a multicentre, randomised, phase 3 trial. Lancet Oncol (2018) 19(12):1617–29. doi: 10.1016/S1470-2045(18)30578-3
213. Dobrenkov K, Ostrovnaya I, Gu J, Cheung IY, Cheung NK. Oncotargets GD2 and GD3 are highly expressed in sarcomas of children, adolescents, and young adults. Pediatr Blood Cancer (2016) 63(10):1780–5. doi: 10.1002/pbc.26097
214. Kailayangiri S, Altvater B, Meltzer J, Pscherer S, Luecke A, Dierkes C, et al. The ganglioside antigen G(D2) is surface-expressed in Ewing sarcoma and allows for MHC-independent immune targeting. Br J Cancer (2012) 106(6):1123–33. doi: 10.1038/bjc.2012.57
215. Mori T, Sato Y, Miyamoto K, Kobayashi T, Shimizu T, Kanagawa H, et al. TNFalpha promotes osteosarcoma progression by maintaining tumor cells in an undifferentiated state. Oncogene (2014) 33(33):4236–41. doi: 10.1038/onc.2013.545
216. Mondanelli G, Iacono A, Allegrucci M, Puccetti P, Grohmann U. Immunoregulatory Interplay Between Arginine and Tryptophan Metabolism in Health and Disease. Front Immunol (2019) 10:1565. doi: 10.3389/fimmu.2019.01565
217. Bronte V, Murray PJ. Understanding local macrophage phenotypes in disease: modulating macrophage function to treat cancer. Nat Med (2015) 21(2):117–9. doi: 10.1038/nm.3794
218. Toulmonde M, Penel N, Adam J, Chevreau C, Blay JY, Le Cesne A, et al. Use of PD-1 Targeting, Macrophage Infiltration, and IDO Pathway Activation in Sarcomas: A Phase 2 Clinical Trial. JAMA Oncol (2018) 4(1):93–7. doi: 10.1001/jamaoncol.2017.1617
219. He X, Lin H, Yuan L, Li B. Combination therapy with L-arginine and alpha-PD-L1 antibody boosts immune response against osteosarcoma in immunocompetent mice. Cancer Biol Ther (2017) 18(2):94–100. doi: 10.1080/15384047.2016.1276136
220. Steggerda SM, Bennett MK, Chen J, Emberley E, Huang T, Janes JR, et al. Inhibition of arginase by CB-1158 blocks myeloid cell-mediated immune suppression in the tumor microenvironment. J Immunother Cancer (2017) 5(1):101. doi: 10.1186/s40425-017-0308-4
221. Hanahan D, Christofori G, Naik P, Arbeit J. Transgenic mouse models of tumour angiogenesis: the angiogenic switch, its molecular controls, and prospects for preclinical therapeutic models. Eur J Cancer (1996) 32A(14):2386–93. doi: 10.1016/S0959-8049(96)00401-7
222. Eubank TD, Galloway M, Montague CM, Waldman WJ. Marsh CB. M-CSF induces vascular endothelial growth factor production and angiogenic activity from human monocytes. J Immunol (2003) 171(5):2637–43. doi: 10.4049/jimmunol.171.5.2637
223. Wyckoff J, Wang W, Lin EY, Wang Y, Pixley F, Stanley ER, et al. A paracrine loop between tumor cells and macrophages is required for tumor cell migration in mammary tumors. Cancer Res (2004) 64(19):7022–9. doi: 10.1158/0008-5472.CAN-04-1449
224. Jeon SH, Chae BC, Kim HA, Seo GY, Seo DW, Chun GT, et al. Mechanisms underlying TGF-beta1-induced expression of VEGF and Flk-1 in mouse macrophages and their implications for angiogenesis. J Leukoc Biol (2007) 81(2):557–66. doi: 10.1189/jlb.0806517
225. Martin D, Galisteo R, Gutkind JS. CXCL8/IL8 stimulates vascular endothelial growth factor (VEGF) expression and the autocrine activation of VEGFR2 in endothelial cells by activating NFkappaB through the CBM (Carma3/Bcl10/Malt1) complex. J Biol Chem (2009) 284(10):6038–42. doi: 10.1074/jbc.C800207200
226. Rolny C, Mazzone M, Tugues S, Laoui D, Johansson I, Coulon C, et al. HRG inhibits tumor growth and metastasis by inducing macrophage polarization and vessel normalization through downregulation of PlGF. Cancer Cell (2011) 19(1):31–44. doi: 10.1016/j.ccr.2010.11.009
227. Lewis JS, Landers RJ, Underwood JC, Harris AL, Lewis CE. Expression of vascular endothelial growth factor by macrophages is up-regulated in poorly vascularized areas of breast carcinomas. J Pathol (2000) 192(2):150–8. doi: 10.1002/1096-9896(2000)9999:9999<::AID-PATH687>3.0.CO;2-G
228. Lee TH, Bolontrade MF, Worth LL, Guan H, Ellis LM, Kleinerman ES. Production of VEGF165 by Ewing’s sarcoma cells induces vasculogenesis and the incorporation of CD34+ stem cells into the expanding tumor vasculature. Int J Cancer (2006) 119(4):839–46. doi: 10.1002/ijc.21916
229. Dalal S, Berry AM, Cullinane CJ, Mangham DC, Grimer R, Lewis IJ, et al. Vascular endothelial growth factor: a therapeutic target for tumors of the Ewing’s sarcoma family. Clin Cancer Res (2005) 11(6):2364–78. doi: 10.1158/1078-0432.CCR-04-1201
230. DuBois SG, Marina N, Glade-Bender J. Angiogenesis and vascular targeting in Ewing sarcoma: a review of preclinical and clinical data. Cancer (2010) 116(3):749–57. doi: 10.1002/cncr.24844
231. Richardsen E, Sorbye SW, Crowe JP, Yang JL, Busund LT. Expression of M-CSF and CSF-1R is correlated with histological grade in soft tissue tumors. Anticancer Res (2009) 29(10):3861–6.
232. Giles AJ, Reid CM, Evans JD, Murgai M, Vicioso Y, Highfill SL, et al. Activation of Hematopoietic Stem/Progenitor Cells Promotes Immunosuppression Within the Pre-metastatic Niche. Cancer Res (2016) 76(6):1335–47. doi: 10.1158/0008-5472.CAN-15-0204
233. Pyonteck SM, Akkari L, Schuhmacher AJ, Bowman RL, Sevenich L, Quail DF, et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat Med (2013) 19(10):1264–72. doi: 10.1038/nm.3337
234. Strachan DC, Ruffell B, Oei Y, Bissell MJ, Coussens LM, Pryer N, et al. CSF1R inhibition delays cervical and mammary tumor growth in murine models by attenuating the turnover of tumor-associated macrophages and enhancing infiltration by CD8(+) T cells. Oncoimmunology (2013) 2(12):e26968. doi: 10.4161/onci.26968
235. Yan D, Kowal J, Akkari L, Schuhmacher AJ, Huse JT, West BL, et al. Inhibition of colony stimulating factor-1 receptor abrogates microenvironment-mediated therapeutic resistance in gliomas. Oncogene (2017) 36(43):6049–58. doi: 10.1038/onc.2017.261
236. Abraham D, Zins K, Sioud M, Lucas T, Schafer R, Stanley ER, et al. Stromal cell-derived CSF-1 blockade prolongs xenograft survival of CSF-1-negative neuroblastoma. Int J Cancer (2010) 126(6):1339–52. doi: 10.1002/ijc.24859
237. Mao Y, Eissler N, Blanc KL, Johnsen JI, Kogner P, Kiessling R. Targeting Suppressive Myeloid Cells Potentiates Checkpoint Inhibitors to Control Spontaneous Neuroblastoma. Clin Cancer Res (2016) 22(15):3849–59. doi: 10.1158/1078-0432.CCR-15-1912
238. Sierra-Filardi E, Nieto C, Dominguez-Soto A, Barroso R, Sanchez-Mateos P, Puig-Kroger A, et al. CCL2 shapes macrophage polarization by GM-CSF and M-CSF: identification of CCL2/CCR2-dependent gene expression profile. J Immunol (2014) 192(8):3858–67. doi: 10.4049/jimmunol.1302821
239. Kitamura T, Qian BZ, Soong D, Cassetta L, Noy R, Sugano G, et al. CCL2-induced chemokine cascade promotes breast cancer metastasis by enhancing retention of metastasis-associated macrophages. J Exp Med (2015) 212(7):1043–59. doi: 10.1084/jem.20141836
240. Kitamura T, Pollard JW. Therapeutic potential of chemokine signal inhibition for metastatic breast cancer. Pharmacol Res (2015) 100:266–70. doi: 10.1016/j.phrs.2015.08.004
241. Qian BZ, Li J, Zhang H, Kitamura T, Zhang J, Campion LR, et al. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature (2011) 475(7355):222–5. doi: 10.1038/nature10138
242. Shaked Y, Kerbel RS. Antiangiogenic strategies on defense: on the possibility of blocking rebounds by the tumor vasculature after chemotherapy. Cancer Res (2007) 67(15):7055–8. doi: 10.1158/0008-5472.CAN-07-0905
243. Hingorani P, Eshun F, White-Collins A, Watanabe M. Gemcitabine, docetaxel, and bevacizumab in relapsed and refractory pediatric sarcomas. J Pediatr Hematol Oncol (2012) 34(7):524–7. doi: 10.1097/MPH.0b013e31826b9d25
244. Xu J, Escamilla J, Mok S, David J, Priceman S, West B, et al. CSF1R signaling blockade stanches tumor-infiltrating myeloid cells and improves the efficacy of radiotherapy in prostate cancer. Cancer Res (2013) 73(9):2782–94. doi: 10.1158/0008-5472.CAN-12-3981
245. Blay JY, El Sayadi H, Thiesse P, Garret J, Ray-Coquard I. Complete response to imatinib in relapsing pigmented villonodular synovitis/tenosynovial giant cell tumor (PVNS/TGCT). Ann Oncol (2008) 19(4):821–2. doi: 10.1093/annonc/mdn033
246. Papadopoulos KP, Gluck L, Martin LP, Olszanski AJ, Tolcher AW, Ngarmchamnanrith G, et al. First-in-Human Study of AMG 820, a Monoclonal Anti-Colony-Stimulating Factor 1 Receptor Antibody, in Patients with Advanced Solid Tumors. Clin Cancer Res (2017) 23(19):5703–10. doi: 10.1158/1078-0432.CCR-16-3261
247. Lee DW, Kochenderfer JN, Stetler-Stevenson M, Cui YK, Delbrook C, Feldman SA, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet (2015) 385(9967):517–28. doi: 10.1016/S0140-6736(14)61403-3
248. Gardner RA, Finney O, Annesley C, Brakke H, Summers C, Leger K, et al. Intent-to-treat leukemia remission by CD19 CAR T cells of defined formulation and dose in children and young adults. Blood (2017) 129(25):3322–31. doi: 10.1182/blood-2017-02-769208
249. Long AH, Highfill SL, Cui Y, Smith JP, Walker AJ, Ramakrishna S, et al. Reduction of MDSCs with All-trans Retinoic Acid Improves CAR Therapy Efficacy for Sarcomas. Cancer Immunol Res (2016) 4(10):869–80. doi: 10.1158/2326-6066.CIR-15-0230
250. Zhang P, Zhao S, Wu C, Li J, Li Z, Wen C, et al. Effects of CSF1R-targeted chimeric antigen receptor-modified NK92MI & T cells on tumor-associated macrophages. Immunotherapy (2018) 10(11):935–49. doi: 10.2217/imt-2018-0012
251. Chmielewski M, Kopecky C, Hombach AA, Abken H. IL-12 release by engineered T cells expressing chimeric antigen receptors can effectively Muster an antigen-independent macrophage response on tumor cells that have shut down tumor antigen expression. Cancer Res (2011) 71(17):5697–706. doi: 10.1158/0008-5472.CAN-11-0103
252. Chmielewski M, Abken H. CAR T Cells Releasing IL-18 Convert to T-Bet(high) FoxO1(low) Effectors that Exhibit Augmented Activity against Advanced Solid Tumors. Cell Rep (2017) 21(11):3205–19. doi: 10.1016/j.celrep.2017.11.063
253. Lynn RC, Matsuyama T, Powell DJ. Targeting FRβ+ tumor associated macrophages with car T cells in ovarian cancer. J ImmunoTher Cancer (2015) 3(Suppl 2):P32. doi: 10.1186/2051-1426-3-S2-P32
254. Zhang W, Liu L, Su H, Liu Q, Shen J, Dai H, et al. Chimeric antigen receptor macrophage therapy for breast tumours mediated by targeting the tumour extracellular matrix. Br J Cancer (2019) 121(10):837–45. doi: 10.1038/s41416-019-0578-3
255. Klichinsky M, Ruella M, Shestova O, Lu XM, Best A, Zeeman M, et al. Human chimeric antigen receptor macrophages for cancer immunotherapy. Nat Biotechnol (2020) 38:947–53. doi: 10.1038/s41587-020-0462-y
Keywords: pediatric sarcoma, tumor-associated macrophage, efferocytosis, tumor microenvironment, immunotherapy
Citation: Koo J, Hayashi M, Verneris MR and Lee-Sherick AB (2020) Targeting Tumor-Associated Macrophages in the Pediatric Sarcoma Tumor Microenvironment. Front. Oncol. 10:581107. doi: 10.3389/fonc.2020.581107
Received: 07 July 2020; Accepted: 09 November 2020;
Published: 14 December 2020.
Edited by:
Alan Wayne, Children’s Hospital of Los Angeles, United StatesReviewed by:
Rosandra Natasha Kaplan, National Cancer Institute (NCI), United StatesCopyright © 2020 Koo, Hayashi, Verneris and Lee-Sherick. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alisa B. Lee-Sherick, YWxpc2EubGVlc2hlcmlja0BjdWFuc2NodXR6LmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.