 Nico Scholz
Nico Scholz Kathreena M. Kurian2
Kathreena M. Kurian2 Florian A. Siebzehnrubl
Florian A. Siebzehnrubl Julien D. F. Licchesi
Julien D. F. Licchesi- 1Department of Biology & Biochemistry, University of Bath, Bath, United Kingdom
- 2Brain Tumour Research Group, Institute of Clinical Neurosciences, University of Bristol, Bristol, United Kingdom
- 3Cardiff University School of Biosciences, European Cancer Stem Cell Research Institute, Cardiff, United Kingdom
Glioblastoma is the most common primary brain tumor in adults with poor overall outcome and 5-year survival of less than 5%. Treatment has not changed much in the last decade or so, with surgical resection and radio/chemotherapy being the main options. Glioblastoma is highly heterogeneous and frequently becomes treatment-resistant due to the ability of glioblastoma cells to adopt stem cell states facilitating tumor recurrence. Therefore, there is an urgent need for novel therapeutic strategies. The ubiquitin system, in particular E3 ubiquitin ligases and deubiquitinating enzymes, have emerged as a promising source of novel drug targets. In addition to conventional small molecule drug discovery approaches aimed at modulating enzyme activity, several new and exciting strategies are also being explored. Among these, PROteolysis TArgeting Chimeras (PROTACs) aim to harness the endogenous protein turnover machinery to direct therapeutically relevant targets, including previously considered “undruggable” ones, for proteasomal degradation. PROTAC and other strategies targeting the ubiquitin proteasome system offer new therapeutic avenues which will expand the drug development toolboxes for glioblastoma. This review will provide a comprehensive overview of E3 ubiquitin ligases and deubiquitinating enzymes in the context of glioblastoma and their involvement in core signaling pathways including EGFR, TGF-β, p53 and stemness-related pathways. Finally, we offer new insights into how these ubiquitin-dependent mechanisms could be exploited therapeutically for glioblastoma.
Glioblastoma
Background
Glioblastoma (GBM) is the most common and aggressive malignant primary brain tumor, categorized as grade IV diffuse glioma by the World Health Organization (WHO) (1). Commonly found in the supratentorial region, GBMs constitute 16% of all primary brain tumors and 54% of all gliomas (2). The CBTRUS Statistical Report (2006–2010) estimated the age-adjusted incidence rate of GBM at 3.19/100,000/year in the United States (2) while data from the National Cancer Registration Service and Hospital Episode Statistics for England (2007–2011) estimated the incidence rate at 4.64/100,000/year in England (3). GBM also has a very poor overall survival rate dropping from 28.4% after one year to 3.4% at five years with a median survival of 6.1 months in the English cohort study (3). When stratified by age, median survival was 16.2 months for 20 to 44-year-olds compared to only 3.2 months for 70+-year-olds. Further, incidences are higher in males compared to females with a relative sex ratio of 1.66:1. Overall GBM has a very poor outlook considering that the median age at diagnosis is 64 (2). In 2016, the WHO published its revised classification of tumors of the central nervous system (CNS) which for the first time used histology as well as molecular parameters to guide appropriate tumor classification (1, 4). Here, GBMs are defined as either isocitrate dehydrogenase (IDH)-wildtype or IDH-mutant, a genotype that in the majority of cases clinically coincides with primary/de novo GBM and secondary GBM, respectively (5). Perhaps unsurprisingly, IDH1 mutations are also very frequent (>80%) in diffuse and anaplastic astrocytomas which are common precursor lesions for recurrent GBM. At the molecular level, IDH mutations result in reduced affinity toward its endogenous substrate, isocitrate, and acquisition of neomorphic enzymatic activity converting α-ketoglutarate into the oncometabolite 2-hydroxyglutarate (6). This gain-of-function has been linked to several oncogenic processes including epigenetic remodeling, which results in the CpG island methylator phenotype (CIMP) (7–9). However, the extent as well as targets of glioma hypermethylation seem to vary considerably when compared to that observed in other IDHmut cancers such as acute myeloid leukemia (AML), possibly explaining why IDH mutational status serves as a favorable prognostic biomarker in GBM only (10). Interestingly, several studies have reported varying methylation patterns between de novo and secondary GBM with, for example, promoter methylation of retinoblastoma protein 1 (RB1) and O6-methylguanine methyltransferase (MGMT) being three-fold and two-fold higher in secondary GBM, respectively (11–15). The epigenetic silencing of the DNA repair enzyme MGMT also serves as IDH-independent prognostic biomarker indicative of increased sensitivity toward temozolomide (TMZ) chemotherapy (16–18). Furthermore, loss of MGMT expression, paired with concomitant TMZ treatment, may select for loss of mismatch repair function resulting in recurrent GBM with hypermutator phenotype (19).
In an attempt to unravel GBM evolution as well as inter- and intra-tumoral heterogeneity, molecular subtyping has been developed as a prognostic strategy. Based on an 840-gene expression profile, GBM samples were grouped into proneural, mesenchymal, classical and neural molecular subtypes (20). Verhaak and colleagues used transcriptomic and genomic profiling to further stratify GBM by identifying patterns of somatic mutations characteristic of individual subtypes. Specifically, EGFR, NF1 and PDGFRA/IDH aberrations were found to define classical, mesenchymal and proneural subtypes, respectively. In recent years, single-cell analysis has become well-established and has offered important insights into tumor complexity. Single-cell RNA-sequencing revealed that tumor bulk transcriptomic profiles do not accurately reflect GBM subtypes (20, 21). Transcriptomic profiles characteristic of the four subgroups (i.e. proneural, mesenchymal, classical and neural), differ at the single-cell level within a tumor, providing further support for the heterogeneity of tumors. In agreement with this, data binning of a proneural tumor according to percentage (%) heterogeneity resulted in patient subsets with diverging overall survival. Hence, higher heterogeneity associated with shorter overall survival. Another single-cell RNA-sequencing study found that infiltrating neoplastic cells from the tumor periphery share a common transcriptomic signature despite having distinct dominant subtypes (22). About 1,000 and 250 genes were found down- and up-regulated, respectively, compared to cells from the tumor core, including genes associated with hypoxia (down) or migration/invasion of the interstitial matrix (up). This indicates that despite intratumoral heterogeneity, some mechanisms such as those driving cell invasion are shared between tumors.
Current Treatments and Future Directions
Treatment of GBM has largely remained unchanged throughout the last decade. A hallmark randomized phase III clinical trial in 2004 by the European Organisation for Research and Treatment of Cancer (EORTC) and the National Cancer Institute of Canada Clinical Trials Group (NCIC), set the following gold standard that is still used today (23, 24). Following maximal safe resection (also referred to as tumor debulking, 84% of patient cohort), patients randomly received radiotherapy alone or radiotherapy with concomitant TMZ chemotherapy followed by six cycles of adjuvant TMZ treatment. The 5-year analysis showed the median survival was 27.2% after two years and 9.8% at five years post treatment commencement. The higher 5-year survival rate compared to previously highlighted epidemiological studies can be explained by the exclusion of 70+-year-olds from the patient cohort. Although surgical debulking followed by radiotherapy and concomitant TMZ chemotherapy remains the current treatment paradigm, several new approaches are being explored including techniques for surgical refinement, immunotherapies and personalized medicine approaches (25–29).
Ongoing efforts in the delineation of the aberrant molecular networks that account for and drive the malignancy and aggressiveness associated with GBM have highlighted key areas that may be exploited therapeutically. In addition to progress made with regards to personalized immunotherapy, the ubiquitin proteasome system has been recognized as one of the most promising fields for novel therapeutics. Proof-of-concept studies have indeed demonstrated that every class of enzymes involved in the ubiquitin-proteasome system can be effectively targeted, including E1-activating, E2-conjugating enzymes, E3 ubiquitin ligases as well as deubiquitinases. With around 1,000 enzymes regulating protein ubiquitination, the number of candidate drug targets is likely to surpass that seen for protein kinases (30). Beyond targeting individual components of the ubiquitin system, new approaches that exploit protein turnover are also being developed and these are bringing new hopes to target the so far “undruggable proteome” (31). In particular, recent developments in proteolysis targeting chimeras (PROTACs) and PROTAC-related molecules such as “molecular glues”, have demonstrated the feasibility of harnessing the endogenous protein turnover machinery for the selective and specific degradation of target proteins. In the next sections, we will discuss key components of the ubiquitin system, in particular E3 ubiquitin ligases and DUBs, in the context of GBM.
The Ubiquitin System
Ubiquitin and UBLs
Post-translational modifications serve a plethora of regulatory functions and thus are integral to cellular homeostasis. In particular, protein ubiquitination plays critical roles by regulating protein fate and function. The small protein modifier ubiquitin (76 aa) is highly conserved in eukaryotes, and it is found expressed in all human tissues (32, 33). More recently, analogous systems in the form of or ubiquitin-like proteins (UBLs) were shown to exist in prokaryotes. UBLs are proteins with shared fold homology to ubiquitin, including a globular β-grasp fold, but for which there is little conservation in the primary protein sequence. As many as 10 UBLs, not including paralogs, have been found in humans with the most well-characterized being interferon-stimulated gene (ISG) 15, autophagy-related genes (Atg) 8 and 12, Nedd8 and small-ubiquitin-related modifiers (SUMO) (34, 35). Furthermore, each post-translational modification has its dedicated conjugation and deconjugation machinery, although some overlapping conjugation systems exists between ubiquitin and UBLs (36, 37). For example, ISG15, a 15 kDa interferon-inducible protein modifier, utilizes its own ubiquitin-like modifier-activating enzyme 7 (UBA7 or UBE1L), ubiquitin/ISG15-conjugating enzyme E2 L6 (UBE2L6 or UbcH8) and the E3 ubiquitin ligases HERC5 or TRIM25 (38–42).
Conjugation of ubiquitin or UBLs to substrate proteins is an ancient, highly conserved protein modification. Even though ubiquitin is absent in prokaryotes, at least two families of post-translational modifications, with analogous function but distinct biochemical pathway to ubiquitin, have been described. Pup (prokaryotic ubiquitin-like protein) which mediates the pupylation of substrate lysine residues was identified in the actinobacteria Mycobacterium tuberculosis/smegmatis and constitutes the first group (43, 44). The second group includes the archaeal SAMPs (small archaeal modifier proteins) and Thermus TtuB (tRNA-two-thiouridine B) which mediate sulfur mobilization (45, 46).
The Ubiquitin Cascade
The pioneering work of Aaron Ciechanover, Avram Hershko and Irwin Rose led to the discovery of ubiquitination as novel post-translational modification that facilitated subsequent degradation in an ATP-dependent manner (47, 48). Subsequently, the Varshavsky lab made seminal contributions to the fundamental understanding of ubiquitin-mediated protein degradation and the regulation of protein half-life. Specifically, delineation of its biological relevance in vivo highlighted ubiquitination as a fundamental requirement for cell viability as well as many cellular pathways including the cell cycle and DNA repair (49, 50).
Substrate protein modification by ubiquitin is mediated by a hierarchical enzymatic cascade (Figure 1). The E1-activating enzyme binds ATP and cofactor Mg2+ and catalyzes ubiquitin C-terminal acyl adenylation, which is then transferred to the sulfhydryl group of the catalytic cysteine residue via acyl substitution forming a thioester bond (51, 52). E1 ubiquitin loading is complete after a second round of ubiquitin adenylate synthesis forming a ternary complex (53). The kinetically charged thioester-conjugate is then transferred to a catalytic cysteine residue in the ubiquitin conjugation (UBC) domain of a cognate E2 via transthioesterification. The thioester bond is susceptible to nucleophilic attack and thus ubiquitin may be transferred to a free substrate lysine (aminolysis) or cysteine residue (transthiolation) (54). Non-canonical transfer may also involve conjugation to serine/threonine residues (oxyester bond) or substrate N-termini (peptide bond) (55–57). The final step of conjugating ubiquitin or UBLs to protein targets is mediated by E3 ubiquitin ligases. This can occur directly by RING E3 ligases which recruit a ubiquitin-loaded E2 and substrate bringing them into close proximity, or indirectly by HECT E3s through an intermediary step where ubiquitin is transthiolated onto a catalytic cysteine residue prior to being transferred to the ϵ-amino group of a substrate lysine residue (58, 59). Single ubiquitin moieties may be attached to a substrate protein at one or multiple sites, monoubiquitination and multi-monoubiquitination respectively, or as ubiquitin chains of varying topology (polyubiquitination), altogether constituting the ubiquitin code (Figure 1) (60).
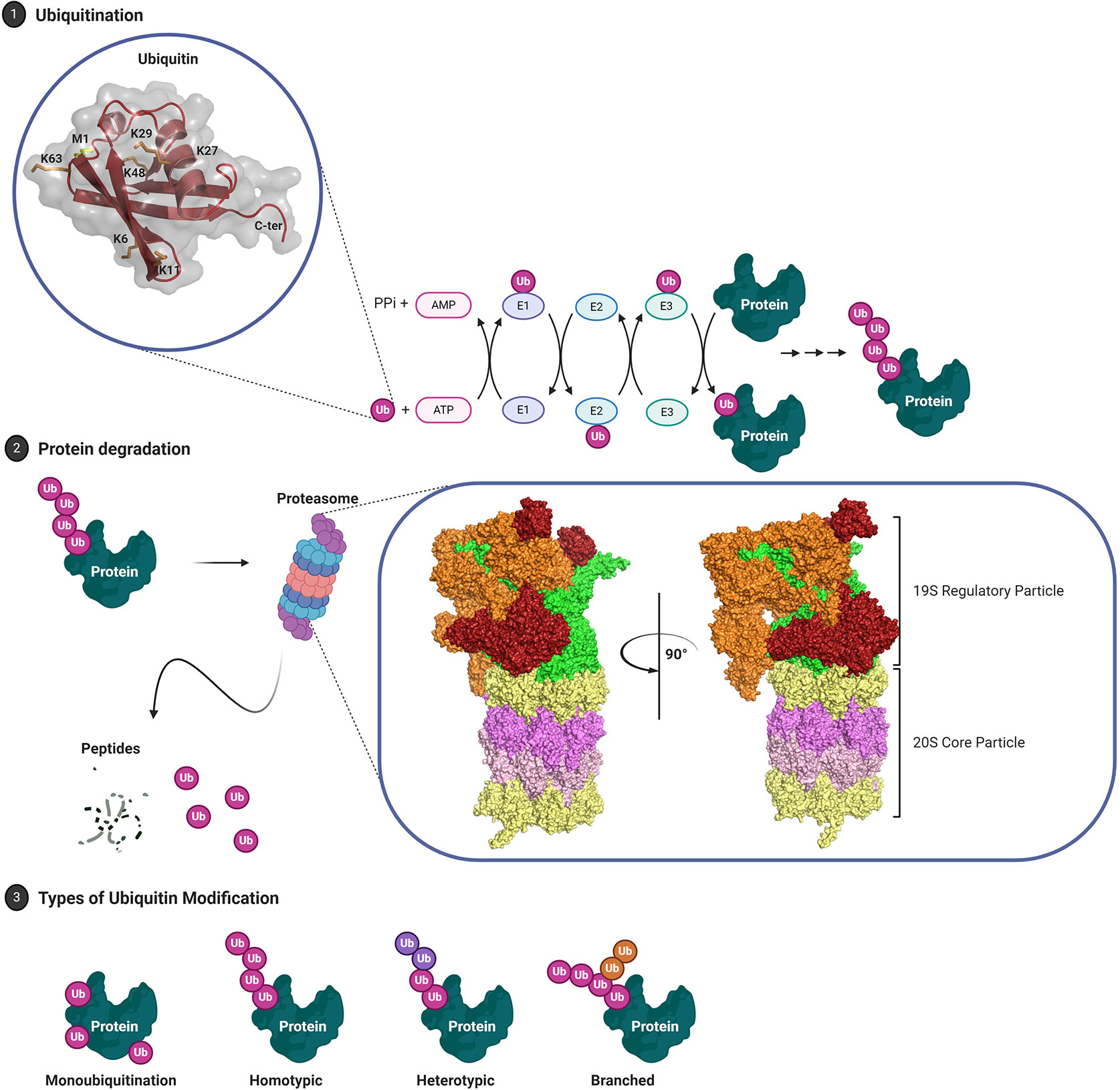
Figure 1 The Ubiquitin Proteasome System. (1) Post-translational modifier ubiquitin, 8.5 kDa, is thiolated to ubiquitin-activating enzyme (E1) and subsequently transthioesterified to a cognate ubiquitin-conjugating enzyme (E2). E3 ubiquitin ligases either serve as scaffold (RING E3 ligases) or catalytic intermediary (HECT E3 ligases) facilitating covalent linkage of ubiquitin C-terminal Gly76 (COOH) to the ϵ-amino group of target lysine residues. (2) Subsequent turnover of ubiquitinated proteins is mediated by the 26S proteasome. Structurally, the proteasome is divided into the 19S regulatory particle, composed of lid and base, and the 20S core particle. Orange: non-ATPase Rpn components (lid); red: ubiquitin receptors Rpn1/10/13 (base); green: AAA+ family ATPases Rpt1–Rpt6 (base); yellow: α heptameric rings, α1–7, that constitute gate/substrate entry portal (20S); magenta: β heptameric rings, β1–7, that constitute the catalytic chamber. (3) Target proteins may either be monoubiquitinated or modified by chains of varying architecture and composition. The complexity of ubiquitin as a signaling molecule, existing as a single moiety or a complex chains, is matched by an impressive diversity in enzymes including 2 E1s, 40 E2s, ~700 E3s and 99 DUBs encoded by the human genome PDB: 1UBQ, Ubiquitin; 6FVW, 26S proteasome.
Below, we will summarize our current understanding of the ubiquitin code in terms of the different types of ubiquitin signals that have been identified, their cellular functions, and the “Writers” (E1, E2, E3) and “Erasers” (i.e. DUBs) that regulate this complex but versatile post-translational modification.
Components of the Ubiquitin System
E3 Ubiquitin Ligases
RING E3 Ligases
E3 ubiquitin ligases catalyze the final step of ubiquitination and impart substrate specificity through protein family diversity. Over 700 E3 ubiquitin ligases are encoded by the human genome and based on their mode of action these have been grouped in four major families: Really Interesting New Gene (RING), Homologous to E6-AP Carboxyl Terminus (HECT), RING-in-between-RING (RBR), and U-box E3 ubiquitin ligases (Figure 2).
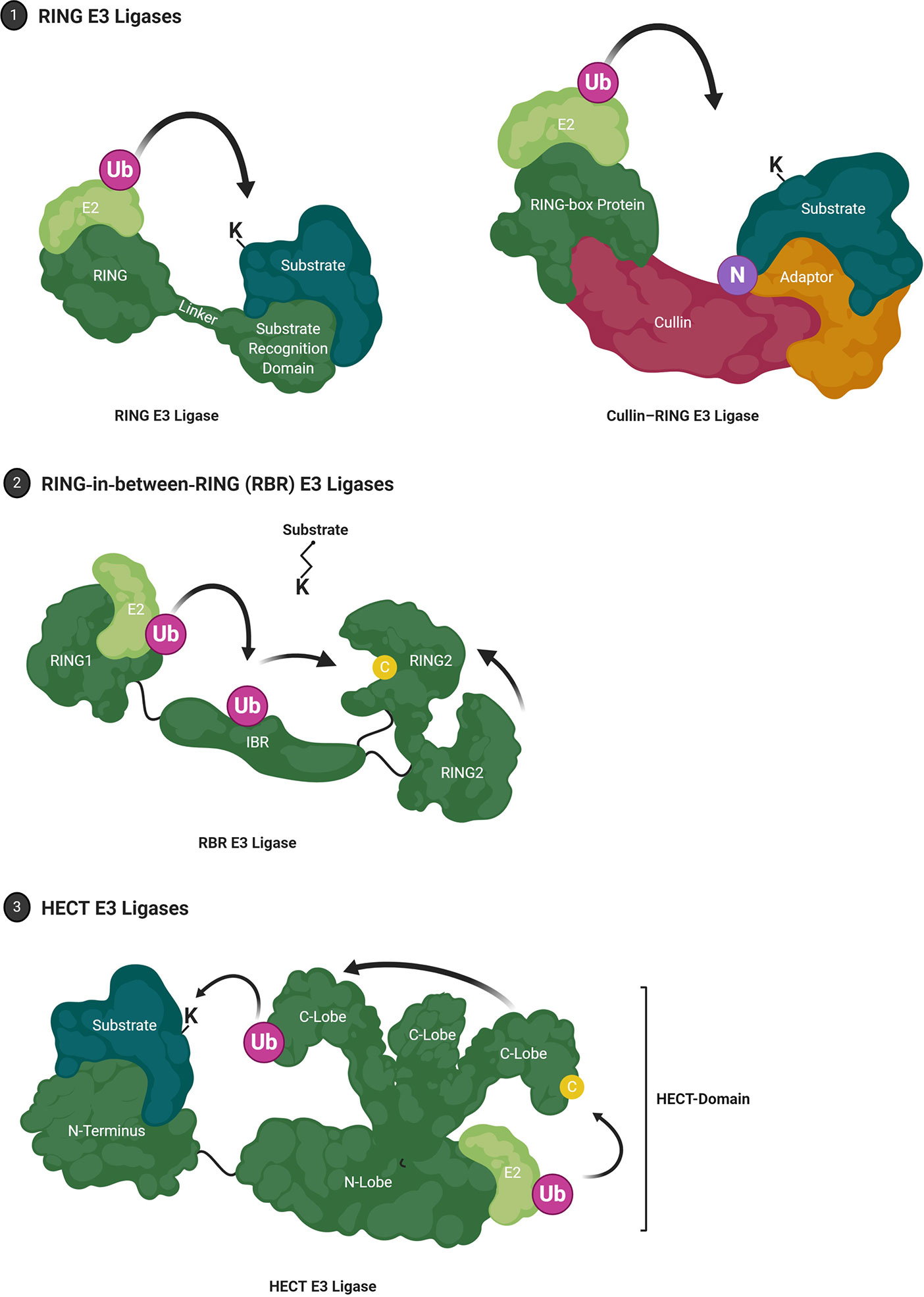
Figure 2 E3 Ubiquitin Ligases. (1) RING (Really Interesting New Gene) E3 ligases facilitate Ub-E2:substrate interaction. RING E3 ligases may also organize into multi-subunit complexes that are commonly composed of Cullins, E2 binding RING-box proteins and an adaptor protein that mediates substrate recognition. Canonically, neddylation (NEDD8) is required to induce the active conformer. (2) RBR (RING-in-between-RING) E3 ligases constitute a hybrid class between RING and HECT E3 ligases where the RING1 domain facilitates E2 interaction while the RING2 domain harbors a catalytic cysteine residue that forms an intermediate thioester. (3) HECT (Homologous to E6-AP Carboxyl Terminus) E3 ligases are characterized by a conserved bi-lobed, catalytic HECT domain. The loaded E2 is bound by the N-lobe where ubiquitin is transferred to the catalytic cysteine residue on the flexible C-lobe. The C-lobe-ubiquitin thioester intermediate rotates toward the substrate which is bound by a substrate-binding domain located N-terminal of the HECT domain.
RING E3s are the largest family with >600 members and are characterized by their zinc finger RING domain (Figure 2) (61). The conserved crossbraced structure of the RING domain is facilitated by two Zn2+ ions and may adopt monomeric or (homo/hetero) dimeric conformations (62–64). Generally, RING domains transiently interact with the E2 UBC domain via a shallow groove constituted by the central α-helix and two adjacent loop regions, thus competing with E1-E2 interaction while other domains are responsible for substrate recruitment (62, 65). Instead of strictly acting as an E2-substrate scaffolding protein, RING E3 ligases also have a passive catalytic capacity and have been shown to facilitate the proximity required for isopeptide bond formation through manifold dynamic conformational rearrangements (66–69). For example, the E2 Ubc13 (Ube2N) and its cofactor Mms2 do not require an E3 to coordinate substrate ubiquitination but exhibit increased reaction kinetics and differential substrate specificity in the presence of E3 ligase TRAF6 (63, 70). A major fraction of RING E3 ligases, the Cullin-RING E3 ligase superfamily, organize into large, multi-subunit proteins (Figure 2) such as the SCF (Skp1, Cullin 1, F-box protein) complex or anaphase-promoting complex/cyclosome (APC/C) [reviewed in (71)].
U-Box E3 Ligases
U-box E3s have a comparable mechanism to RING E3s, utilizing a structurally similar domain they can also operate as monomers or function as subunits of multimeric protein complexes. In contrast to RINGs, U-box E3s lack the conserved cysteine and histidine Zn2+-chelators at the RING domain-binding interface and instead utilize a hydrophobic binding groove constituted by hydrogen bonding networks and polar amino acids (72, 73). A well-researched example is carboxy-terminus of Hsc70 interacting protein (CHIP), a co-chaperone that acts as quality control for misfolded proteins by ubiquitinating Hsp70 and Hsp90 associated substrates (74, 75). The N-terminal tetratricopeptide repeat (TPR) domain mediates Hsp70/Hsp90 interaction, while a coiled-coil domain facilitates CHIP dimerization allowing the homodimer U-box domain to interact with its cognate E2 (76, 77).
RBR E3 Ligases
RING-in-Between RING (RBR) E3 ligases are a comparably small family with the human genome encoding about 12 such proteins [reviewed in (78, 79)] Originally thought to employ a similar mechanism to canonical RING ubiquitination, RBR E3 ligases were rather shown to be hybrids of RING and HECT E3 ligases (80). The RING1 domain mediates interaction with a ubiquitin-loaded E2, while the RING2 domain forms a thioester intermediate via a catalytic cysteine residue (Figure 2). The two-step mechanism is reminiscent of HECT-mediated ubiquitination, although the domain structure differs. Several RBRs have been shown to function in this manner, including HHARI, HOIP and Parkin (81–83). Additionally, Parkin which contributes to neurodegeneration in Parkinson’s disease, but also GBM, can carry out E2-independent monoubiquitination and remains active in the absence of its RING1 domain (84–86). Nevertheless, all RBRs are thought to share stringent intramolecular auto-inhibitory mechanisms. In the case of Parkin, an N-terminal ubiquitin-like domain induces a closed, auto-inhibited conformation by binding to the IBR-RING2 linker region (87). Parkin substrates have been shown to interact with the ubiquitin-like domain, indicating substrate-induced activation (88, 89).
HECT E3 Ligases
HECT E3 ligases can be divided into 16 subfamilies with a total of 28 members encoded by the human genome (90). The HECT family of E3 ligases were discovered during the investigation of E3 ligase E6AP (UBE3A), which then became its first member (91–93). Here, the human papillomavirus (HPV) virulence protein E6 was shown to hijack the mammalian E3 ligase, altering its substrate specificity toward tumor suppressor p53 as well as other regulatory proteins (94, 95). HECT-mediated ubiquitination requires an intermediate step, whereby ubiquitin is first transferred onto the E3 catalytic cysteine residue via a thioester bond, prior to conjugation onto protein substrates. This allows HECT-type E3 ligases to veto any linkage preference conferred by E2 conjugating enzymes (96). The approximately 40 kDa bi-lobed HECT domain is composed of an N-lobe and a C-lobe, with the N- and C-lobes being separated by a hinge glycine residue (Figure 2) (97). While the C-lobe contains the catalytic cysteine residue, the larger N-lobe primarily mediates E2 interaction as evidenced by the crystal structure of E6AP in complex with UBCH7 (97). In the context of at least some HECT E3s such as NEDD4, the N-lobe can also provide a binding interface for ubiquitin itself and this might promote processivity during ubiquitin chain extension (98). These and other structural studies have emphasized that conformational flexibility of the HECT domain is key to bringing the catalytic cysteine of the E3 in close proximity with that of the E2 and thus to enable ubiquitin transfer (97, 99, 100).
Even though HECT E3 ligases share the highly conserved HECT domain, they display considerable diversity in their N-terminal domains which are thought to play important roles in substrate targeting as well as E3 ubiquitin ligase regulation (101). This has been most well-characterized for the NEDD4 HECT E3 ligase family (NEDD4, NEDD4.2, ITCH, SMURF1, SMURF2, WWP1, WWP2, NEDL1 (HECW1) and NEDDL2 (HECW2)) which contain an N-terminal Ca2+-dependent/independent lipid-binding domain (C2 domain) and between two to four WW domains in addition to the C-terminal HECT domain (102, 103). Type I WW domains within NEDD4 family proteins bind a multitude of substrates by engaging PY motifs (PPxY) as well as other proline-rich motifs (104–106). C2 domains mediate targeting of the E3 to the phospholipid bilayer but may also confer substrate specificity (107, 108). Various inter- and intramolecular interactions are prominent in the regulation of HECT E3 ligase activity (109). For example, C2-HECT domain interaction within SMURF2 results in the canonical closed/autoinhibitory conformation that may be outcompeted if a substrate is available (110). Further, NEDD4 forms an autoinhibitory trimer via a conserved α1-helix domain, which is contrasted by E6AP trimerization that constitutes its active conformer (111, 112).
Deubiquitinases
In many aspects, the cellular counterpart to E3 ubiquitin ligases, DUBs remove ubiquitin moieties from substrate proteins ensuring reversibility of the post-translational modification. Around 100 DUBs have been identified in eukaryotes and are divided into seven evolutionary conserved families (USP, JAMM/MPN, OTU, MJD/Josephin, UCH, MINDY, and ZUP1) (113). DUBs are predominantly thiol proteases with a catalytic cysteine residue or in the case of the JAMM family metalloproteases coordinate a Zn2+ ion in the active site (114). DUBs may display substrate specificity and/or ubiquitin linkage specificity. The reversibility of protein ubiquitination was first demonstrated in 1982 by the observation that histone H2A is deubiquitinated during mitosis and re-ubiquitinated during the G1 phase (115). Later, the first DUB, YUH1, was identified in S. cerevisiae and the lack of obvious phenotypic changes suggested the existence of additional DUBs (116). DUBs have since been implicated in most if not all cellular processes including DNA repair, signal transduction and innate immunity (113).
DUBs also play a crucial role in the de novo synthesis of ubiquitin and thus maintenance of the cellular ubiquitin pool. Human ubiquitin is encoded by four genes expressing the ubiquitin precursors UBB, UBC, UBA52 and UBA80. UBB and UBC exist as head-to-tail linked ubiquitin polymers with a C-terminal extension, while UBA52 and UBA80 are ubiquitin monomers fused to the ribosomal proteins L40 and S27A, respectively (117–119). Processing of ubiquitin precursors is carried out by multiple DUBs and likely serves as additional quality control checkpoint. Ribosomal fusion precursors are post-translationally cleaved by UCHL3, USP7 and USP9X, while ubiquitin multimer precursors are processed by USP5 and OTULIN (120).
In addition, DUBs also carry out another important “housekeeping” function by recycling ubiquitin as part of the UPS and the endocytic pathway (121, 122). Upon recognition of ubiquitinated cargoes by ubiquitin receptors on the proteasome lid, including Rpn10, Rpn13 and Rpn1, the polyubiquitin signal is cleaved off by proteasomal DUBs including UCH37 (UCHL5), Usp14 and PSMD14 (Rpn11) (Figure 1) (123). As demonstrated for PSMD14, catalytic activity is in direct competition with ubiquitin unfolding by the proteasomal AAA-ATPases (124). This results in mechanochemical coupling of the two processes, where substrate translocation accelerates conformational switching of PSMD14 into its active β-hairpin conformer. Mechanistically, PSMD14 exists as a dimer with the pseudo-DUB PSMD7 which is subject to steric inhibition by the 20S entry port (125). Therefore, even though PSMD14 may not exert linkage specificity in vitro, it may only catalyze en bloc chain removal, at least in the context of the proteasome (126). The recycling of ubiquitin on the 19S cap is part of a highly orchestrated series of events which leads to cargo unfolding by AAA-ATPases and translocation into the 20S core particle where proteolysis takes place (127).
In other cellular contexts, DUB linkage specificity is of more importance. For example, NFκB signaling relies on the K63-linked polyubiquitination of adaptor proteins for the recruitment of the TAB–TAK1 kinase complex, M1-linked polyubiquitination of NEMO by the linear ubiquitin chain assembly complex (LUBAC), and the K48-linked polyubiquitination of IκBα, the key effector kinase mediating activation of the pathway (128). To regulate NFκB activity, ubiquitin chain disassembly is orchestrated by OTU DUBs OTULIN and CYLD which exert M1 and M1/K63 specificity, respectively (129, 130). Importantly, loss of function of OTULIN drives inflammation and autoimmunity in mice and leads to OTULIN‐related autoinflammatory syndrome (ORAS) in humans (131). Similarly, deficiency in CYLD or A20, a master regulator of NFκB, lead to overt pathway activation and inflammation (132).
The Ubiquitin Code
Ubiquitin can be conjugated to a substrate lysine residue via its C-terminal glycine residue but may also be conjugated to itself. Polyubiquitin chains can thus be assembled through any of its lysine residues (K6, K11, K27, K29, K33, K48, and K63) as well as the N-terminal methionine residue (M1 or linear chains) (Figure 1). Although ubiquitin smears were observed in the initial study by Hershko and colleagues, it took further efforts to confirm the existence of polyubiquitin chains (48). These were first identified as K48-linked polyubiquitin chains attached to lysine residues on short-lived proteins which targeted them for proteolytic degradation by the 26S proteasome in an ATP-dependent manner (133–136). Importantly, each of these linkages has now been identified in yeast and mammalian cells by mass spectrometry (137, 138). Over the last two decades, atypical chains (assembled through linkages other than K48 or K63), as well as more complex polyubiquitin signals such as heterotypic and branched chains, have also been reported, emphasizing the complexity and diversity of ubiquitin as a signaling molecule (60, 139, 140).
Homotypic K48-linked ubiquitin chains canonically signal for proteasomal degradation and also represent the most abundant linkage-type (135, 137, 141). Additional linkage-types that mediate proteasomal targeting include K29, which may also drive lysosomal degradation, and perhaps surprisingly, monoubiquitination and K63 which have been predominantly associated with non-proteasomal functions including endocytosis and autophagy (141–144). Indeed, K63-polyubiquitin chains have been primarily implicated in protein complex assembly which includes TRAF6, Ubc13-Uev1A and TRIKA2 (TAK1, TAB1 and TAB2) that associates with IκB kinase (IKK). Here, K63-polyubiquitination stimulates phosphorylation of IKK by TAD1 which leads to the K48-linked polyubiquitination and degradation of IKK and transcriptional activation of NFκB gene targets (145, 146). Other K63 signaling functions include but are not limited to regulation of DNA repair (also K27) (147–149), protein sorting (150, 151) and mRNA splicing (152) and translation (153). Similarly, K48 chains can also mediate non-proteasomal functions as evidenced by the stabilization of the yeast transcription factor M4 by K48-polyubiquitination (154).
Heterotypic ubiquitin chains may either present with “mixed” or “branched” topology. For example, the E3 ligase complex LUBAC, which assembles linear M1-linked ubiquitin chains, also forms K63/M1-linked hybrid ubiquitin chains. In the context of innate immunity, these hybrid chains mediate activation of the canonical IKK complex, but have also been shown to play a role in the TNFR1 and NOD1 signaling networks through modification of RIP1 and RIP2 kinases, respectively (128, 155). Meyer and Rape showed that APC/C assembles K11/K48-branched ubiquitin chains through its E2s Ube2S and Ube2C resulting in a degradation signal that is superior to homotypic K11/K48 chains (156). To achieve polyubiquitin chains of branched topology, the cooperation of enzymes with differing linkage specificity is key. This is the case with ITCH and Ubr5 which assemble K63-linked chains and K48-linked chains, respectively, resulting in K48/K63-branched ubiquitination of proapoptotic regulator TXNIP (157). Similarly, ubiquitin branching has also been demonstrated in yeast where the E4 enzyme Ufd2p catalyzes K48-linked multi-monoubiquitination of Ufd4p-assembled K29-linked polyubiquitin chains as part of the ubiquitin fusion degradation (UFD) pathway (158, 159). In eukaryotes, K29/K48 branched chains have so far been demonstrated to play a role in targeting substrates to the UPS and ERAD (160, 161). Interestingly, the UPS appears well-equipped for processing these more complex chains, with a recent study showing that the proteasome-associated DUB UCH37 is a debranching DUB with important roles during proteasomal degradation (162). Through continued advancements in mass spectrometry-based techniques including the quantification of polyubiquitin linkage composition (e.g. ubiquitin-AQUA, Middle-Down MS), and the dissection of polyubiquitin architecture (e.g. UBICREST, Ub-Clipping, TUBE, TR-TUBE), new insights into the structure and function of branched ubiquitin chains are now possible (138, 163–167).
The versatility of ubiquitin as a signaling molecule makes it a prime target for cancer cells that seek to escape physiological regulation. Indeed, deregulation of the ubiquitin system is often observed in tumor-suppressing pathways (e.g. overactivation/expression of an E3 ligase leading to the ubiquitin-dependent degradation of a protein with tumor suppressive function) as well as tumor-promoting pathways (e.g. inactivation of an E3 ligase leading to the stabilization of oncoproteins). Thus, E3 ubiquitin ligases and DUBs in particular have emerged as therapeutic candidates, offering the possibility to more accurately control the activity of a given pathway in contrast to targeting protein degradation as a whole through proteasomal inhibition. The contribution of ubiquitin signaling to GBM tumorigenesis is currently not well understood. In the next section, we will highlight ubiquitin-dependent mechanisms relevant to GBM and discuss these in the context of EGFR, TGF-β, p53 and stemness-related pathways. Table 1 and Table 2 provide a comprehensive overview of E3s and DUBs implicated in GBM, respectively.

Table 1 E3 ubiquitin ligases in Glioblastoma (GBM).

Table 2 Deubiquitinases in Glioblastoma (GBM).
The Ubiquitin System in Glioblastoma
Epidermal Growth Factor Receptor
EGFR amplification and mutations rendering the receptor constitutively active are commonly observed in GBM. Most common are deletions of exons 2-7 (EGFRvIIIΔ6-273), which result in constitutive activation of receptor signaling as well as global epigenomic and transcriptomic remodeling with chromatin landscape analysis revealing that activation of 2245 putative enhancers was specific to EGFRvIII (275). Also, EGFR amplification (44%) and point mutations that target the extracellular domain (R108K, A289V/D/T and G598D; 24%) are frequently observed (TCGA, PanCancer Atlas). Likewise, loss of the negative Akt regulator PTEN is associated with poor survival (276). Interestingly, in addition to mutations causing loss of expression or enzymatic activity, L320S and T277A have been found to dysregulate PTEN stability and cellular localization by altering the membrane-binding regulatory interface resulting in increased polyubiquitination (277).
Indeed, EGFR stability and downstream signaling are subject to the ubiquitin regulatory network (Tables 1, 2). In GBM, the DUB CSN6, a subunit of the COP9 signalosome complex (CSN), mediates EGFR stabilization and was also shown to be overexpressed in GBM tumor samples (176). CSN6 may also destabilize EGFR-interacting E3 ligase CHIP by promoting its autoubiquitination (278). Interestingly, in a non-GBM context, it is well-established that the multi-subunit metalloprotease CSN regulates the neddylation of CRLs (279). Here, its CSN6 subunit has been associated with the degradation of tumor suppressor proteins including c-Myc and p53 (280, 281). Another E3 ligase, TRIM11, also regulates EGFR levels and TRIM11 expression correlated closely with glioblastoma stem cell (GSC) markers Nestin and CD133 and promoted tumorsphere formation (223).
TGF-β Signaling
Aberrant rewiring of tumor-suppressing TGF-β signaling that induces potent cell cycle arrest to one that promotes cell growth and EMT is characteristic of tumor progression. It has been shown that in patients with high-grade gliomas TGF-β signaling is highly active and this is associated with poor prognosis (282). The canonical TGF-β pathway signals through receptor-regulated Smads (R-Smads), but the receptor may also directly cross-communicate with non-canonical pathways including MAPK, PI3K or RHO-like GTPases (283). TGF-β signaling is subject to tight regulation by ubiquitination (Figure 3). The inhibitory Smad protein Smad7 functions as a negative feedback loop by complexing with TβR-I and blocking R-Smad phosphorylation, or by binding to the promoter region of PAI1, blocking functional SMAD2/3-DNA complex formation. Further, Smad7 also serves as a docking site for the HECT E3 ligase Smurf2 and DUB USP15 (239, 284). This E3-DUB pair is yet another example of signaling regulation by ubiquitination and a quick and responsive mechanism to regulate pathway activity/output (285). Smurf2 suppresses TGF-β signaling by targeting TβR-I for proteasomal degradation, while USP15 opposes TβR-I polyubiquitination thus stabilizing the receptor complex. Indeed, USP15 knockdown decreased tumorigenic potential in GBM, while more than 2.5 copies of USP15 conferred significantly poorer life expectancy in patients (239).
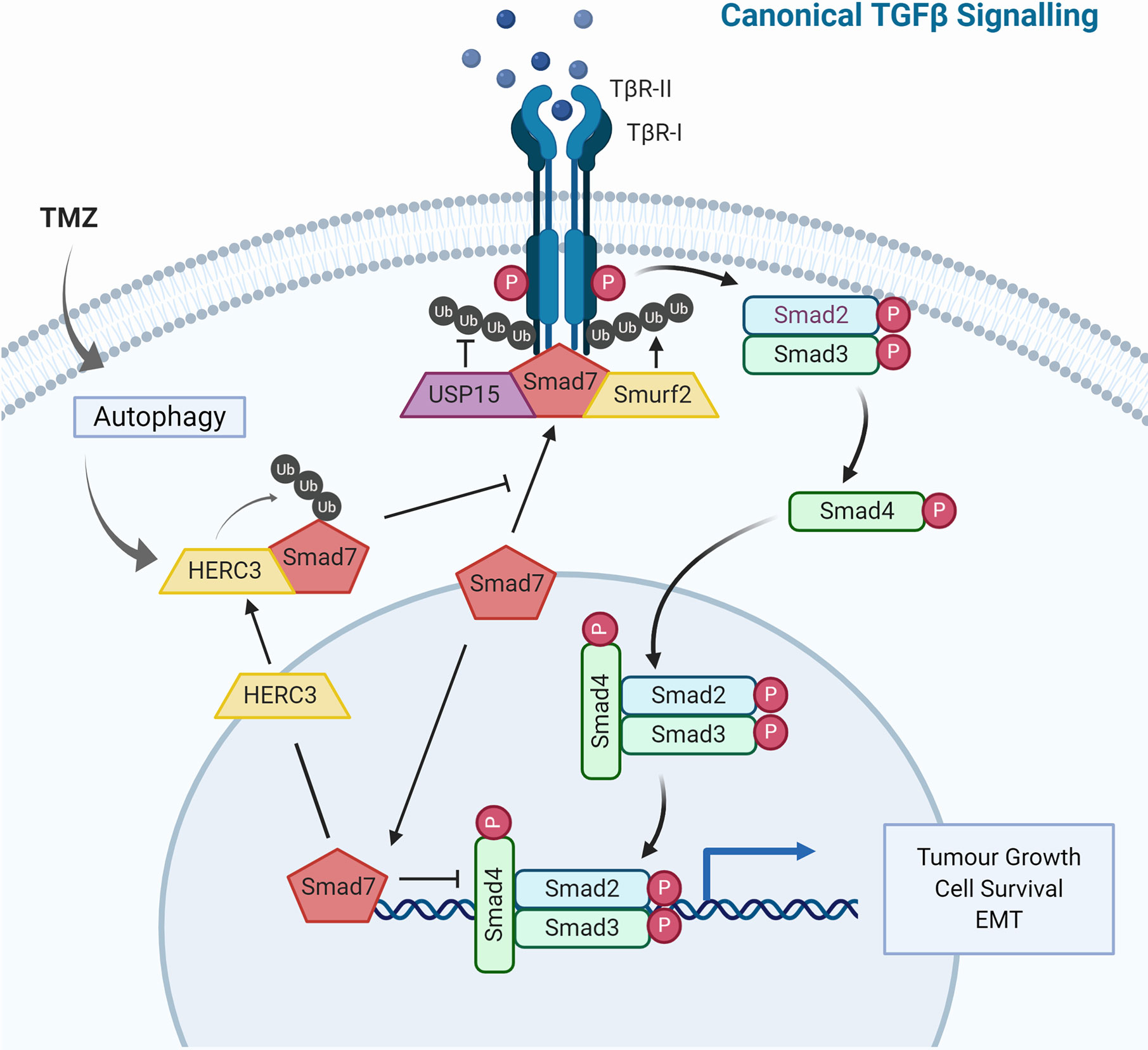
Figure 3 TGF-β Signaling and Ubiquitin in Glioblastoma. The TGF-β signaling cascade is tightly regulated by the ubiquitin-proteasome system. Illustrated are E3 ubiquitin ligases and deubiquitinases that not only regulate TGF-β signaling under physiological conditions but have also been shown to contribute to dysregulation observed in glioblastoma.
The physical and functional interaction between Smad7 and the HECT E3 ligase HERC3 has been shown to play a role in chemoresistance observed in GBM. Concomitant TMZ chemotherapy is a standard treatment for high-grade gliomas and has been shown to induce autophagy-mediated cell death (286). Nonetheless, in a subset of tumor cells, this catabolic process may also have pro-survival effects rendering the tumor chemoresistant (287). HERC3 has also been shown to play a key role in autophagy-induced EMT, a core molecular mechanism for drug resistance in GBM (233, 288). Experiments in GBM cells showed that TMZ-induced autophagy resulted in significant up-regulation of TGF-β signaling and subsequent expression of mesenchymal markers. Specifically, autophagy upregulated HERC3 expression which resulted in HERC3-mediated Smad7 K63-polyubiquitination and subsequent autolysosomal degradation. HERC3 binds Smad7 via its RCC4–7 domains (aa156–366) and in addition to targeting cytoplasmic Smad7, HERC3 also disrupted the inhibitory interaction of nuclear Smad7 with the promoter region of PAI1 (289).
The HECT E3 ligase Smurf1 carries out very similar functions to Smurf2 by also binding to Smad7, however it does not co-precipitate with USP15 indicating a contextually different, USP15/Smurf2-independent role (239, 290). Downstream of receptor complex activation, the DUB USP10 drives TGF-β signaling by stabilizing Smad4 which has been linked to increased metastatic potential in hepatocellular carcinoma (291). Another HECT E3 ligase, NEDD4L, recognizes the phosphorylated PPXY motif of Smad2/3 via its WW domain, resulting in polyubiquitin-mediated turnover and reduced TGF-β signaling output (292). This mechanism is specific to the canonical TGF-β pathway since it requires the phosphorylation of p-Smad2/3 by TGF-β-activated CDK8/9, which does not regulate non-canonical TGF-β pathways. These and additional ubiquitin-dependent mechanisms have been implicated in TGF-β signaling, and their dysregulation is frequently observed in GBM as well as other cancers, therefore opening new avenues for therapeutic intervention (293).
p53 Regulation
The master-regulator p53 integrates various signaling pathways, relaying its tumor suppressive functions through a plethora of target genes. p53 is modified by a large variety of post-translational modifications which regulate its spatial and temporal expression. Ubiquitination of p53 was first discovered in the context of human papillomavirus, which highjacks the HECT E3 ligase E6AP to redirect its E3 ligase activity toward p53, an otherwise non-canonical substrate (92). In addition to oncogenic viruses, p53 is also targeted for ubiquitin-dependent degradation in multiple cancers. In GBM for example (Figure 4), p53 levels are regulated by the RING E3 ligase MDM2 as part of the ARF-MDM2-p53 axis which is dysfunctional in 84% of cases/94% of cell lines (294). Under normal physiological conditions, MDM2-p53 forms a negative feedback loop where p53 activation induces the expression of MDM2 which in turn promotes the ubiquitin-mediated degradation of p53 (295). This equilibrium is disrupted by MDM2 amplification which negates p53 tumor suppressor function such as growth/cell cycle arrest, apoptosis or DNA repair. MDM4 performs a complementary role but lacks intrinsic E3 ligase activity (296). Via protein-protein interactions, MDM4 directly inhibits p53 by binding to its transcription activation and DNA binding domain (297, 298). In contrast to MDM2, MDM4 does not form homodimers but preferentially hetero-oligomerises with MDM2 via their C-terminal RING domains to mediate p53 ubiquitination (299). Indeed, heterodimer formation facilitates increased p53 ubiquitination but also stabilizes MDM2 by reducing its autoubiquitination. In GBM, homozygous deletions of CDKN2A (ARF/56%), gene amplification of MDM2/4 (8.2%/9.4%) and missense mutations in TP53 (31.5%) all lead to loss of p53 tumor suppressive functions, either through reduced activity or through reduced levels (TCGA, PanCancer Atlas).
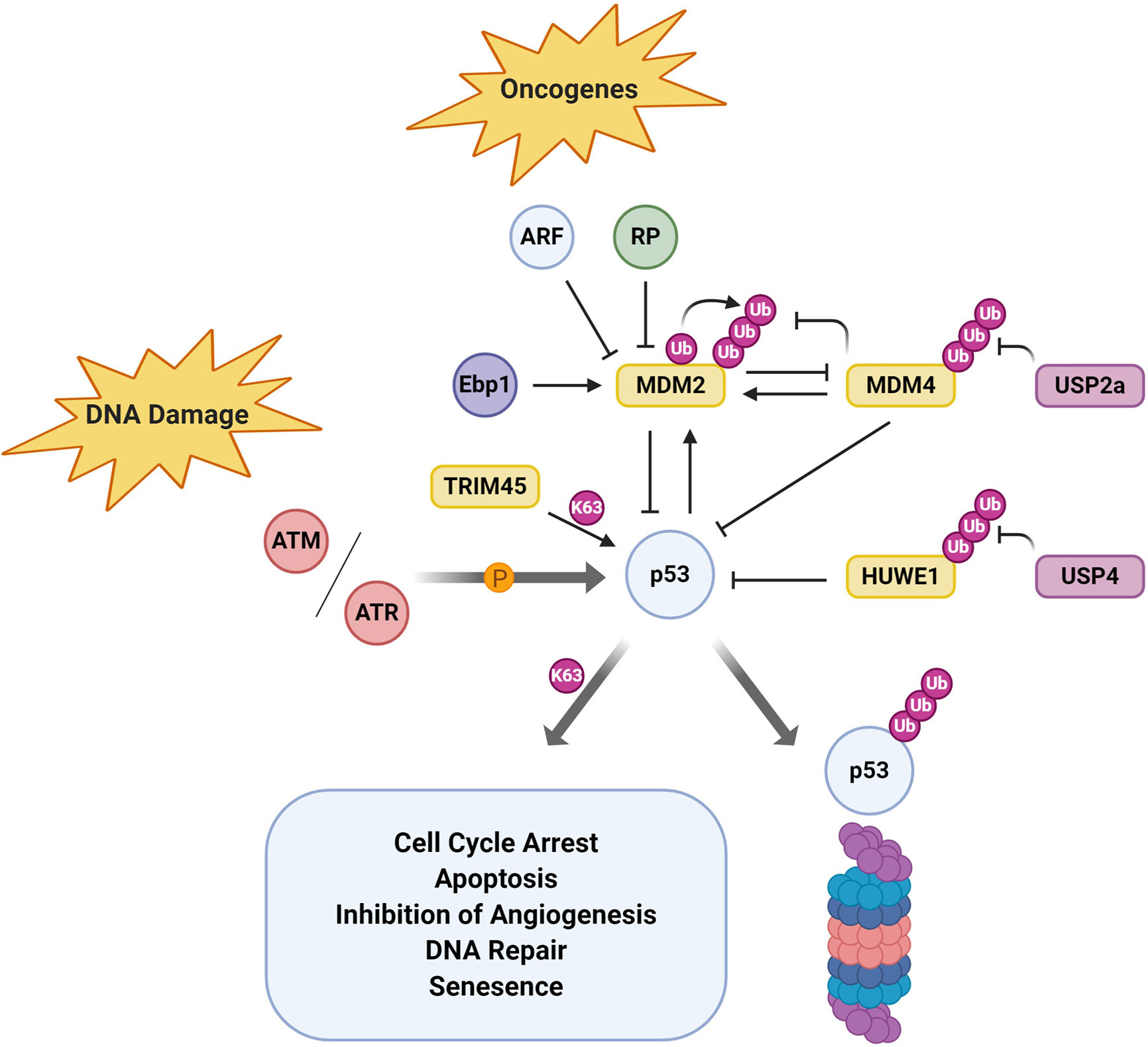
Figure 4 p53 Regulation by the UPS in Glioblastoma. Tumor suppressor p53 is subject to a plethora of upstream regulatory mechanisms including post-translational modification by ubiquitin. Here, E3 ubiquitin ligases and deubiquitinates that have been shown to modulate p53 function/activity in glioblastoma specifically are depicted.
Another interesting regulator of p53 activity in GBM is the DUB USP4 which negatively regulates p53 indirectly by stabilizing the HECT E3 ligase HUWE1/Mule (300). Although USP4 mRNA and protein levels were upregulated in GBM, its transient depletion did not result in changes in cell viability (252). In contrast, cells treated with the chemotherapeutic TMZ were significantly more sensitive to USP4 depletion by siRNA and showed decreased cell viability. This study suggests that USP4 mediates chemoresistance in GBM by preferentially inhibiting p53-mediated apoptosis. However, other E3 ligases such as TRIM45, may also positively regulate p53 activity. The TRIM family of RING E3 ligases are highly expressed in the brain, but TRIM45 mRNA and protein levels have been shown to be significantly downregulated in GBM tissue samples. TRIM45 had been previously shown to negatively regulate the MAPK and NFκB pathway, but its role as tumor suppressor had not been explored in GBM (301, 302). However, in GBM, TRIM45 mediated its tumor suppressor function through direct ubiquitination and stabilization of p53 (226). The authors suggested that K63-ubiquitination of p53 by TRIM45 inhibited subsequent degradative ubiquitination by for example MDM2/4.
Stem Cell Maintenance
The discovery of cells with extensive proliferative and self-renewal capacity in AML gave rise to the cancer stem cell hypothesis (303). Glioblastoma stem cells (here defined as CD133+Nestin+) have since been identified as a distinct subpopulation, critical to tumorigenesis (304). The origin of GSCs may be disputable, but it is evident that the GSC subpopulation is key to the maintenance of tumor growth and invasive capacity, while also providing means for treatment resistance and thus GBM recurrence (305–307). Generally, GSC transcriptomic signature correlates with bulk tumor molecular subtype (excluding neural subtype), thus reflecting clonal heterogeneity and plasticity (308). Indeed, intra tumoral GSC subtype plasticity may allow for adaptation to a particular tumor niche or serves as a survival mechanism in response to microenvironmental cues. Identification of the underlying molecular mechanisms that drive stemness is thus key for successful therapeutic development. Using serial xenotransplantation and DNA barcoding, GSCs have been shown to exhibit a remarkable neutral proliferative hierarchy (309). In this model, a small pool of slow-cycling stem-like cells ensured tumor proliferative capacity by giving rise to rapidly-cycling progenitor cells. Although this model highlights the evolutionary fitness advantage of GSCs over non-GSCs, it does not take into account GSC plasticity. Indeed, multi-lineage plasticity not only extends to molecular subtypes but also exists as a dynamic equilibrium between GSCs and differentiated cancer cells (310). Stemness regulation by the tumor microenvironment results in a bidirectional equilibrium between CSC and non-CSC compartments and therefore GSCs should be regarded as reversible, transient state at the apex of a stem cell hierarchy (311, 312). GSC plasticity itself is now emerging as a key therapeutic target to overcome recurrence and drug resistance.
The underlying molecular mechanisms that contribute to GSC maintenance and plasticity, including the role of ubiquitin signaling, are still being worked out. E3 ligases/DUBs regulate the stability of key mediators of neuronal differentiation, including c-Myc, a core transcriptional regulator of GSCs. c-Myc levels are tightly controlled in a context-dependent manner by several E3 ligases and DUBs. One study showed that USP13 and SCFFBXL14 act as an E3-DUB pair regulating c-Myc ubiquitination in GBM (207). USP13 was found preferentially expressed in GSCs while SCFFBXL14 was predominantly expressed in non-stem glioma cells, enabling preferential stabilization of c-Myc in GSCs. USP28 was previously shown to stabilize c-Myc in HeLa and U2OS cells by antagonizing SCFFBW7α-mediated degradation and a more recent study has now reported its overexpression in GBM (269, 313). It has also been demonstrated that high expression of TRIP13, which stabilized c-Myc by inhibiting FBW7 transcription, correlates with poor patient survival (203). TRIM3 is another E3 ligase that has been shown to suppress c-Myc levels in GBM (219). In Drosophila, TRIM3 is an important regulator of asymmetric cell division, but whether its tumor suppressive effects in GBM are mediated through direct interaction with c-Myc remains to be shown (314).
The gene master regulator REST (repressor element 1-silencing transcription factor) is aberrantly expressed in brain tumors, where it likely maintains stem/progenitor cells through repression of neuronal genes (200, 201, 315). Here, the multi-subunit E3 ligase complex SCFβ-TrCP targets REST for proteasomal degradation via a phospho-degron. Although not in GBM specifically, USP7 has been demonstrated to counterbalance REST ubiquitination by SCFβ-TrCP, facilitating neuronal differentiation in neural stem/progenitor cells. SCFβ-TrCP is a particularly versatile E3 ligase which has been implicated in several pathways including cell cycle regulation, NFκB and Wnt signaling (316). Interestingly, another study reported nuclear mislocalization of SCFβ-TrCP in GBM which led to reduced degradation of its cytosolic targets such as phospho-β-catenin (202). This may lead to increased Wnt signaling which is also commonly observed in GBM (317). Nevertheless, how SCFβ-TrCP regulation of REST and its nuclear mislocalization can be unified remains to be understood.
Another DUB enriched in GBM stem cells is USP1, which stabilizes the DNA damage response and stem cell maintenance regulators ID1/2 and Chk1 (246, 247). Radioresistance in CD133+ cells is conferred by preferential activation of the DNA damage response pathway via phosphorylation of checkpoint proteins ATM, Rad17, Chk1 and Chk2. Loss-of-function experiments on USP1 indeed resulted in impaired GSC survival and radiosensitization (305). Furthermore, CD133+ GSCs drive constitutive activation of the DNA damage response through high levels of replication stress not exhibited by CD133- cells (318). In proneural glioma cells, where the PDGFR gene is frequently amplified, increased PDGF signaling drove expression of members of the E2F transcription factor family (E2F1-3). This in turn promoted E2F interaction with the USP1 promoter and increased USP1 levels which then stabilized the transcriptional regulator ID2 and maintained GSCs stemness (246, 247).
The Ubiquitin-Proteasome System as a Source of Novel Therapeutics in GBM
Drug discovery has largely focused on developing enzyme inhibitors, in particular small molecular kinase inhibitors, with some success (319). Phosphorylation, like ubiquitination, is a reversible post-translational modification and high-throughput screens using small molecule libraries have identified vast numbers of kinase inhibitors that target either the catalytic ATP-binding pocket or adjacent hydrophobic cavities inhibiting substrate phosphorylation (320). The UPS has been dubbed as a new source of therapeutics although the development of small molecules inhibitors has been accompanied by inherent difficulties explaining the slow progress to date (30). E3 ubiquitin ligases may outnumber protein kinases but are inherently more difficult to target. Indeed, components of the UPS are exclusively found intracellularly which in comparison to the extracellular domains of receptor kinases, for example, negates antibody-based approaches. Moreover, E2-E3-substrate interactions are of transient nature and largely independent of well-defined binding pockets making high-throughput screens not readily applicable. Hence, the interface of E2-E3 interaction also does not lend itself to targeting. The identification as well as the fate and function of substrates modified by ubiquitin along with the mechanisms regulating E1, E2, E3 and DUB activity are still being defined. As we learn more about the specificity of enzymes in terms of the ubiquitin-dependent mechanisms they mediate and cellular processes they regulate, the ubiquitin system will offer a diverse therapeutic toolbox. This will be particularly important in the context of complex and heterogeneous pathologies such as GBM, where one “therapeutic magic bullet” might be difficult to achieve. Below we will summarize exciting developments targeting the components of the ubiquitin system and discuss the relevance of these strategies for GBM.
Modulating Proteasomal Activity
Proteasome inhibition marked some of the earliest efforts in targeting the UPS. Since transformed cells exhibit higher proliferative capacity, ability to evade apoptosis and other regulatory mechanisms, these cells were more susceptible to proteasomal inhibition (321, 322). Proteasome inhibitors can be chemically divided into the general categories of peptide aldehydes, peptide vinyl sulfones, peptide epoxyketones, peptide boronates and lactacystin and its derivatives. However, only peptide boronates and epoxyketones bear the appropriate balance of potency, selectivity and metabolic stability required for clinical development (323). Nonetheless, proteasome inhibitors not suitable for the clinic have provided an invaluable understanding of cellular consequences of proteasomal inhibition, with the most prominent example being MG132 (carbobenzoxy-Leu-Leu-leucinal) (324).
Bortezomib (PS-341, Velcade), Carfilzomib (PR-171, Kyprolis) and Ninlaro (Ixazomib, Takeda) are currently the only FDA-approved proteasome inhibitors. All proteasome inhibitors share a similar mechanism of action, they bind active site threonine residues of the proteolytic β-subunits (325, 326). These structural studies identified the hydroxyl group of Thr1 as the catalytic nucleophile, which was confirmed by further crystal structures that demonstrated that only alanine but not serine substitution led to catalytic inactivity (327). The dipeptidyl boronic acid bortezomib (pyrazylcarbonyl-Phe-Leu-boronate) selectively targets the chymotrypsin-like activity of the proteasome with the boron atom forming a tetrahedral adduct with Thr1, exhibiting high potency (EC50 0.6 nM) and a clinical-relevant cytotoxic profile (328). Treatment of various cancer cell lines resulted in cell cycle arrest in G2-M phase and subsequent apoptosis as evidenced by accumulation of cell cycle regulators p21 and p53 as well as other pro-apoptotic proteins (329). Even though the underlying mechanism remains to be fully elucidated, bortezomib is considered to inhibit NFκB activation by blocking the degradation of IκB and also increased sensitivity to chemotherapeutic agents (330). Even though a first phase I clinical trial in solid tumors yielded little success, a second phase I trial for hematologic malignancies showed promising results, ultimately leading to FDA approval of bortezomib in 2003 for multiple myeloma (331, 332).
In contrast to intravenous administration required for both bortezomib and carfilzomib, ixazomib (ninlaro) has become the first FDA-approved oral proteasome inhibitor (333). Ixazomib citrate is metabolized into active ixazomib which selectively and reversibly inhibits the chymotrypsin-like activity of the β5 subunit of the 20S proteasome. Nonetheless, drug delivery, in particular the ability to cross the blood-brain barrier, remains an issue for using proteasome inhibitors to treat brain pathologies such as GBM. Bortezomib was effective in GBM mouse models, but only when administered intracranially but not systemically (334). Proteasome inhibitors do not distinguish between normal and transformed cells which may result in non-specific cytotoxicity. Rather, they rely on the higher proliferative capacity of cancer cells to be more effective in this particular cell pool. However, new delivery strategies such as nanoparticle-derived systems may help overcome specificity issues by directing drugs to specific cellular compartments (335). Indeed, preclinical studies have highlighted the effectiveness and potential of bortezomib nanoparticle delivery, with for example anti-CD38 chitosan nanoparticles improving multiple myeloma cell targeting and resulting in a lower toxicity profile (336–338).
Therapeutic Targeting of E1-Activating and E2-Conjugating Enzymes
Enzymes of the ubiquitination cascade also pose as promising targets for drug discovery. However, given there are only two main mammalian E1-activating enzymes (UBA1 and UBA6), inhibiting their function would also affect ubiquitin-dependent mechanisms as a whole. E1 enzymes carry out the ATP-dependent activation step resulting in the formation of a thiol ester bond between the ubiquitin adenylate and the active site cysteine residue (339). UBA1’s Cys632 has been successfully targeted via covalent modification by pyrazolidine-based inhibitors such as PYR-41 and PYZD-4409 (340, 341). Even though both showed selectivity for malignant cells, with the latter displaying potential for the treatment of hematologic malignancies, its mechanism of action and pharmacological properties are incompletely understood. Currently, the most promising candidate in development is MLN4924 (Pevonedistat) which is being evaluated in several phase I/II/III clinical trials (342). MLN4924 targets NEDD8 Activating Enzyme (NAE), which function as the initiator for the conjugation of ubiquitin-like modifier NEDD8. The small molecule inhibitor induces apoptosis due to accumulation of tumor-suppressive Cullin-RING ligase substrates and S-phase DNA synthesis dysregulation. Structural evidence suggests that MLN4924 inhibits NAE enzymatic activity by forming a NEDD8 adduct via its sulfamate moiety resulting in a NEDD8-AMP mimetic that occupies the adenylation active site (343).
In contrast to targeting the proteasome or E1-activating enzymes, other classes of enzymes in the ubiquitin system are likely to offer more specificity and therefore pose as more desirable therapeutic targets. With 40 E2 conjugating enzymes encoded in the human genome, this class of enzymes play an important role with regards to substrate specificity, in particular for RING E3 ligase-mediated ubiquitination. For example, the small molecule inhibitor CC0651 was originally identified in a screen for SCFSkp2, and exhibited dose-dependent inhibition of CDK inhibitor p27 (344). However, functional studies in budding yeast later revealed that the compound targets human UB2R1 (Cdc34) instead. Structural analysis further confirmed this and also identified CC0651 as an allosteric inhibitor, binding UB2R1 via its biphenyl ring system in a hydrophobic pocket distinct from the active site. Inhibition disrupts ubiquitin chain elongation but also stimulates autoubiquitination.
Another example is the E2 heterodimer UBE2N-UBE2V1 which has been successfully targeted by NSC697923 and BAY 11-7082 (345, 346). NSC697923 blocks ubiquitin transthioesterification by binding the active site cysteine residue of UBE2N (Ubc13), downregulating constitutive NFκB signaling in primary diffuse large B-cell lymphoma cells. BAY 11-7082 was thought to inhibit IκBα phosphorylation but here shown to inhibit K63 polyubiquitin chain formation by forming a covalent adduct with the UBE2N Cyscat. The compound exerts anti-inflammatory effects in primary B cell lymphoma and leukemic cells but is yet to undergo further preclinical evaluation.
Therapeutic Targeting of E3 Ubiquitin Ligases
E3 ubiquitin ligases are at the pinnacle of the ubiquitination cascade, carrying out the final step. This makes them attractive drug targets due to their high degree of specificity and selectivity toward substrates. Nonetheless, the transient and dynamic nature of E3-substrate interaction and their lack of well-defined catalytic cavities makes them inherently difficult to target, especially with small molecule inhibitors. However, GDC-199 (venetoclax) which gained FDA approval for chronic lymphocytic leukemia (CLL) and small lymphocytic lymphoma (SLL) in May 2019 revived interest in the possibility of disrupting protein-protein interactions (347). Venetoclax selectively binds to BCL-2’s BH3-only protein hydrophobic binding groove, leaving the pro-apoptotic protein free to interact with for example BAX and BAK proteins, inducing mitochondrial membrane permeabilization and subsequent cell death (348, 349).
The F-box protein SKP2 is the substrate recognition subunit of the SCFSKP2 E3 ligase complex. Its well-defined role in several human malignancies, as well as availability of structural data, makes it a prime target for high-throughput screens. Indeed, in silico screens identified several hits which selectively target the p27 binding interface, while another screen identified compound 25 which disrupts SKP1 binding (350, 351). The compounds displayed significant effects on cell proliferation through various mechanisms including cell cycle arrest and suppression of Akt-mediated glycolysis in line with their respective targets.
The p53 regulator MDM2 is another well-studied drug target and particularly relevant in GBM where it is frequently amplified. MDM2 binds the transcriptional activation domain of p53 forming an autoregulatory feedback loop, which is complemented by direct binding of MDM2’s hydrophobic cleft to the amphipathic α-helix of p53’s transactivation domain (TAD) resulting in ubiquitination and subsequent proteasomal degradation (352, 353). A screen identified Nutlins, imidazoline analog, as potent and selective inhibitors of TAD binding, inducing p53 stabilization and downstream cell cycle arrest/growth inhibition (354). Crystal structures of imidazoline inhibitors in complex with MDM2 confirmed occupation of the TAD binding cleft as underpinning mechanism. Subsequently, further compounds disrupting MDM2-p53 interaction were identified. However, they suffer from the caveat that canonical ubiquitination of p53 mutants is MDM2-independent (355). An interesting example is AMG-232 (KRT-232) which averaged IC50s in the low nanomolar range in GBM cell lines and patient-derived GBM stem cells (356). More importantly, AMG-232’s suppressive effects seemed to extend selectively to GBM stem cells as the compound displayed efficacious inhibition of stemness-related factors Nestin and ZEB1 in a spheroid culture model. AMG-232 is currently being evaluated in 3 phase I clinical trials, with NCT03107780 probing its ability to penetrate GBM in patients with newly diagnosed or recurrent GBM. Several other imidazoline-based compounds are also currently undergoing early phase clinical trials with however so far modest clinical success (357).
Another class of compounds with several examples currently undergoing phase I clinical trials are inhibitors of apoptosis (IAP) antagonists (358). Proteins of the IAP E3 ligase family are endogenous inhibitors of apoptosis that sequester pro-apoptotic proteins such as caspases via their baculovirus IAP repeat (BIR) domain rendering them inactive (359, 360). Under physiological conditions, activation of the intrinsic apoptotic pathway induces Smac/DIABLO relocalization from the mitochondria to the cytosol where their binding to the hydrophobic BIR domain interface results in IAP dissociation and thus activation of pro-apoptotic proteins (361, 362). Efforts therefore focussed on generating Smac-mimetics, which bind the IAP BIR domain via the characteristic Ala–Val–Pro–Ile interaction motif (363). Smac-mimetics induce dimerization of IAP RING domains, an active conformation, resulting in autoubiquitination and subsequent degradation (364).
The ability of small molecules to alter instead of inhibiting E3 ligase function has been demonstrated in nature as well as experimentally. For example, the plant hormones auxin and jasmonate function as so-called “molecular switches/glues” enhancing E3 ligase substrate affinity (Figure 5) (369, 370). The former is bound by the auxin receptor TIR1, an F-box component of the SCF multi-subunit complex, enhancing degradation of the downstream transcriptional regulators AUX/IAA. In addition, the thalidomide derivative lenalidomide was shown to alter the substrate specificity of Cereblon (CRBN) ubiquitin ligase, inducing the degradation of Ikaros family zinc finger proteins 1 and 3, B cell transcription factors, in multiple B cell malignancies (371, 372). These studies not only elucidate an important mechanism of these immunomodulatory drugs but also provide evidence that small molecules hold the potential to repurpose E3 ubiquitin ligases for targeting “undruggable” targets, in particular GBM-relevant oncoproteins such as c-Myc, β-catenin or MCL1 (373). Database mining and rational screening have been used successfully to identify molecular glue degraders that specifically target cyclin K, and these approaches will have broad applications for drug discovery (374, 375).
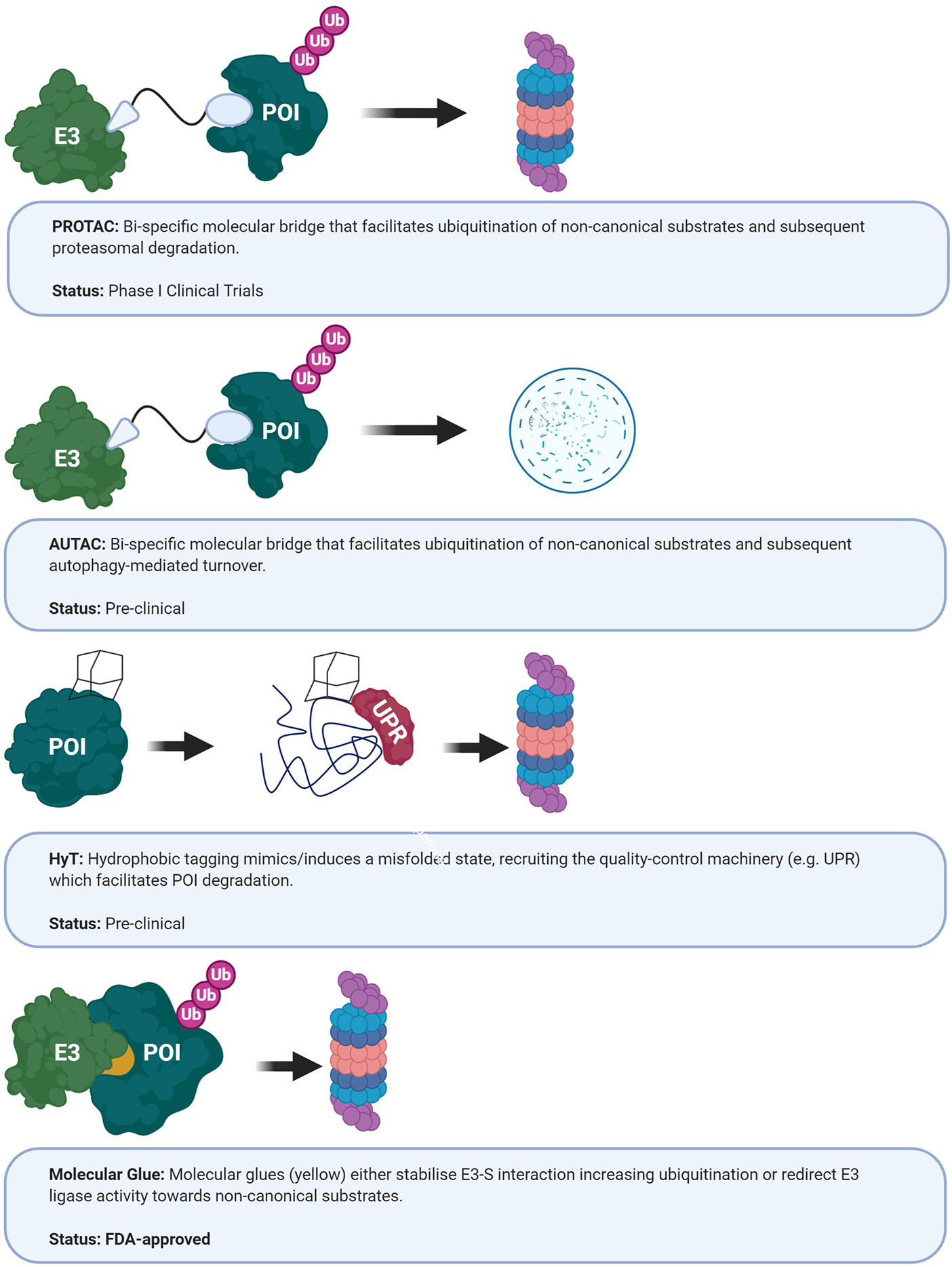
Figure 5 Current approaches targeting E3 ubiquitin ligases. AUTAC, autophagy- targeting chimeric molecules; HyT, hydrophobic tagging; POI, protein of interest; PROTAC, protein-targeting chimeric molecules; UPR, unfolded protein response (365–368).
Therapeutic Targeting of Deubiquitinases
With the previously outlined success of targeting the ubiquitination cascade, it is perhaps not surprising that deubiquitination is also an integral part of current drug discovery efforts. As previously discussed, DUB involvement is frequently observed in various cancers including GBM (Table 2). In an attempt to build on the success of bortezomib but also improve on issues associated with specificity, DUBs associated with ubiquitin hydrolysis at the 26S proteasome are currently evaluated. For example, the 19S subunit-associated DUB USP14, which is involved in ubiquitin recycling, is overexpressed in several diseases such as lung adenocarcinoma and non-small cell lung cancer (376, 377). In this context, the upregulation of USP14 is thought to maintain proteostasis of malignant cells through efficient protein degradation. The growth factor signaling transducer Akt phosphorylates USP14 on Ser432 resulting in an active conformation, thus providing means of globally regulating protein turnover (378). Similarly, UCHL5 is also associated with the 19S cap proteasome complex by binding to ubiquitin receptor RPN13 and functions by editing polyubiquitin degradation signals, cleaving distal ubiquitin moieties (379, 380). However, like USP14, UCHL5 is highly selective, promoting the degradation of certain proteins while guaranteeing the survival of others. For example, it was demonstrated that the RPN13-UCHL5 complex promotes degradation of inducible nitric oxide synthase (iNOS), while stabilizing NFκB suppressor IκBα (381). VLX1570 is a functional analog of the chalcone derivative b-AP15 with a piperidine to azepane ring substitution (382). b-AP15 was previously identified in a screen for lysosomal apoptosis pathway activation and displayed promising in vivo anti-tumor progression activity in several solid tumor models (383). Polyubiquitinated substrate accumulation led to USP14 and UCHL5 target identification and the compound being dubbed second-generation proteasome inhibitor. VLX1570 entered clinical trials in 2015 as a combination study with dexamethasone in myeloma patients, but despite continuous promising preclinical data, the trial had to be suspended in 2017 due to dose-limiting toxicity (NCT02372240). It will be interesting to see how other proteasomal DUB inhibitors fare, with several currently in preclinical development (384).
USP7 is another promising target for the treatment of various cancers as it regulates the stability of a multitude of oncoproteins and tumor suppressors (385). Many of which are also relevant in GBM and add to its previously discussed role in counterbalancing REST ubiquitination by SCFβ-TrCP, facilitating neuronal differentiation in neural stem/progenitor cells (255). These include, for example, stabilization of FOXO (Forkhead box O) transcription factors, regulation of tumor suppressor PTEN nuclear-cytoplasmic partitioning or the p53 pathway (386–388). Several hits are currently investigated but share issues of selectivity and potency. One such compound, amidotetrahydroacridine derivative HBX 19,818, was shown to covalently bind active site Cys223 with an IC50 in the micromolar range (389). Experiments in cancer cell lines confirmed that similar to USP7 knockdown, HBX 19,818 promoted apoptosis and G1 phase cell cycle arrest as well as p53 stabilization. Similarly, P22077, previously identified during an activity-based proteomics screen, showed selective USP7 inhibition in an orthotopic neuroblastoma mouse model (390, 391). Xenograft growth was significantly inhibited via the USP7-MDM2-p53 axis. Recent structures of USP7 in complex with small molecule inhibitors should accelerate informed drug design and development. Importantly, it should be noted that like the previously described Nutlins, USP7 inhibitors are rendered ineffective when faced with p53 mutant malignancies which is the case for ≈32% of GBMs (TCGA, PanCancer Atlas).
USP15 has been implicated in NFκB, Wnt and TGF-β signaling, which are all recognized cancer pathways (392, 393). Building on a previous study that identified USP15 as DUB of receptor-activated SMADs, Eichhorn et al. established USP15 as SMURF2 counterpart (239, 394). USP15 gene amplification is commonly observed in GBM and correlates with aberrant TGF-β signaling. Currently, only weak USP15 inhibitors have been identified, but recent structural insights in its catalytic domain have provided a starting point for a more targeted approach (395). Similarly, USP1 is emerging as a candidate target in GBM, due to its increased expression in GSCs where it contributes to the DNA damage response and stem cell maintenance (247). The FDA-approved antipsychotic pimozide has been identified as USP1 inhibitor and is now being re-evaluated in various preclinical studies for cancer therapy (396). Also, the diphenylbutylpiperidine has CNS activity and was shown to induce radiosensitivity as well as chemosensitivity to TMZ treatment (397).
Future Opportunities for GBM Therapeutics
In addition to small molecule inhibitors, several novel therapeutic avenues that exploit endogenous turnover machinery are being developed (Figure 5). Rather than delineating individual E2/E3-substrate pairings and subsequently subjecting the specific binding interface to a small molecule library screen, these new strategies co-opt endogenous protein degradation machinery – specifically the UPS (i.e. PROTACs, HyT, molecular glues), autophagy (AUTACs) and the endosomal/lysosomal (LYTACs) pathways (398). PROTACs are heterobifunctional molecules which can recruit E3 ubiquitin ligases to the desired protein targets, thereby co-opting the endogenous UPS for targeted protein degradation. Below, we will summarize how PROTACs work and also how some of these strategies provide new opportunities for GBM therapeutics.
Protein-Targeting Chimeric Molecules
PROTACs are an exciting new development in the field. They are bi-specific, artificial molecular bridges that facilitate ubiquitination of non-canonical substrates (Figures 5, 6). Proof-of-concept was demonstrated in 2001, by using the artificial PROTAC-1 to target methionine aminopeptidase-2 (MetAP-2) to SCFβ-TrCP for proteasomal degradation in Xenopus laevis egg extracts (365). β-TrCP is the substrate recognition domain of SCFβ-TrCP, endogenously recognizing a short, phosphorylated peptide stretch within IκBα resulting in subsequent ubiquitination, degradation and thus activation of NFκB signaling (399). MetAP-2 is a primary target of ovalicin (OVA), which covalently binds the active site His231 resulting in a downstream inhibitory effect on endothelial cell proliferation (400). The PROTAC-1 design combines both moieties, the IκBα phosphopeptide and OVA, resulting in the molecular bridging of MetAP-2 and SCFβ-TrCP, two otherwise functionally unrelated proteins. Subsequently, Sakamoto and colleagues designed two similar PROTACs using estradiol and dihydroxytestosterone (DHT) instead of OVA, targeting estrogen receptor (ER) and androgen receptor (AR), respectively (401). The experiments carried out in HEK-293 cells provided important in vitro validation. Issues with poor cell permeability were overcome with PROTAC-4 which was developed by ARIAD Pharmaceuticals. It included a poly-D-arginine tag (-ALAPYIP-(D-Arg)8-NH2) facilitating improved cell permeability and eliminating the previous need for microinjection (402). Nonetheless, complex synthetic chemistry and low efficacy were issues that remained.
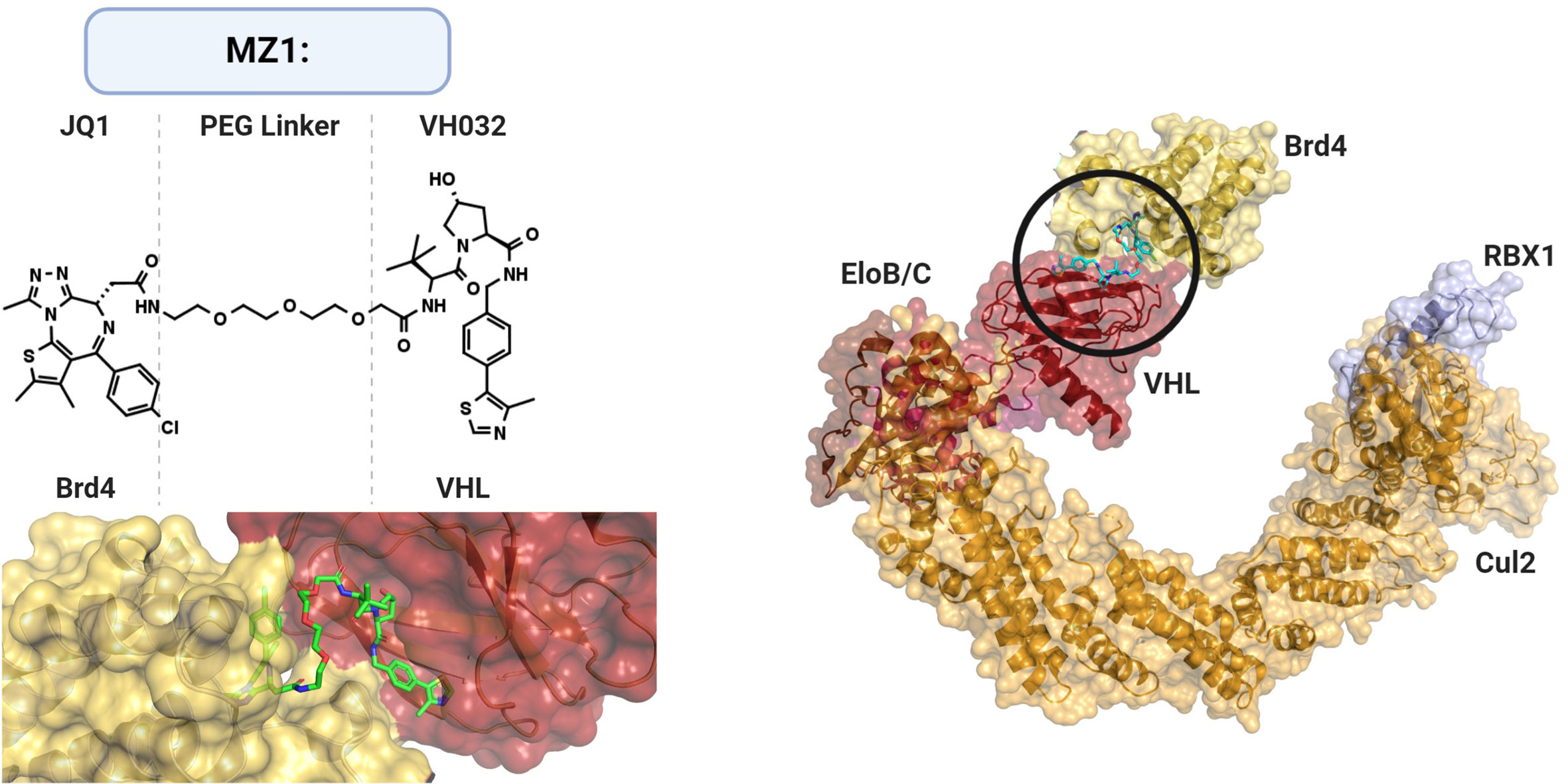
Figure 6 Structural Basis of PROTACs. PROtein-TArgeting Chimeric molecules (PROTACs) are heterobifunctional bridges that link E3 ligase activity to non-canonical substrates. PROTAC MZ1 links bromodomain inhibitor JQ1 to VHL ligand VH032 via a polyethylene glycol (PEG) linker. MZ1 facilitates binding and subsequent ubiquitination of BET (bromodomain and extraterminal) protein family member Brd4 (shown BRD4BD2) to cullin-RING ligase complex CRL2VHL. PDB: 5N4W, Cul2-Rbx1-EloBC-VHL ubiquitin ligase complex; 5T35, PROTAC MZ1 in complex with Brd4BD2 and pVHL : ElonginC:ElonginB.
However, in vivo application and thus revival of the technology became achievable with the development of E3 ligase-specific ligands. These second-generation PROTACs relied on small molecules rather than peptides for E3 ligase recruitment. The first examples included MDM2, cIAP1 and Cullin-Ring ligase (CRL) complex substrate receptors such as CRBN and VHL (403–409). The Crews group who, along with the Deshaies group, first reported PROTAC in 2001, developed its second-generation AR-targeting PROTAC by coupling Nutlin to a selective androgen receptor modulator (SARM) via a polyethylene glycol (PEG) linker (407). Nutlin targets the AR for proteasomal degradation by binding to the p53 interaction interface of MDM2 (354). In 2014, thalidomide and its derivatives lenalidomide and pomalidomide were shown in complex with the E3 ligase DDB1–CRBN, thus validating the E3 ligase as the target of the immunomodulatory drugs (410, 411). The phthalimide ring system found in thalidomide and its derivatives was also utilized for DDB1–CRBN recruitment in a PROTAC design for the degradation of bromodomain and extra-terminal (BET) proteins (403). The other half of the PROTAC consisted of the competitive bromodomain inhibitor JQ1 which binds in the acetyl-lysine binding cavity of BRD4 (Figure 6) (412). The hybrid molecule termed dBET1 displayed in vivo efficacy in a human leukemia xenograft model and induced a more robust apoptotic response in primary human leukemic blast cells compared to BRD inhibition. Similarly, the improved pharmacodynamics could be replicated by another BRD4-targeting PROTAC, ARV-825, in Burkitt’s lymphoma cell lines showing promising results for MYC-driven malignancies (404).
Conceptually similar to PROTAC, hydrophobic tagging (HyT) utilizes a hydrophobic moiety instead of a peptide/small molecule as E3 ligase recruiting domain (Figure 5) (366). Since functional proteins fold in a manner that conceals hydrophobic side chains to assume a lower energy state, the additional hydrophobic surface group is thought to mimic/induce a misfolded state leading to proteasomal degradation (413, 414). The mechanism is not fully elucidated yet, but it is thought that HyT modification initially recruits the chaperone machinery in an attempt to refold the protein, although ultimately targeting HyT-modified proteins to the proteasome (415). Feasibility of the approach was demonstrated by the addition of the cycloalkane adamantane to a bacterial dehalogenase (HaloTag) which resulted in robust degradation of HaloTag fusion proteins in culture and mice (366). Similarly, the pseudo-kinase Her3 which is considered “undruggable” by ATP-competitive small molecules was found to be targetable for degradation by derivatization of the selective ligand TX1-85-1 with the hydrophobic adamantyl moiety (416).
However, many questions remain with regards to clinical application. For example, the large molecular weight of PROTACs may pose challenges to oral bioavailability, pharmacokinetics and tissue specificity. Nonetheless, preclinical evidence are convincing, particularly for the two recently developed BET family protein-targeting PROTACs which exhibit EC50s in the low picomolar range, QCA570 and compound 23 (417, 418). Furthermore, Sun et al. demonstrated that oral, as well as intraperitoneal PROTAC delivery, can mediate robust and global FKBP12 and Bruton’s tyrosine kinase (BTK) degradation in animals from mice to rhesus monkeys (419). Here, the PROTAC RC-32 rendered FKBP12 undetectable after only one day in most organs except the brain, indicating its inability to cross the blood-brain barrier. However, mice treated via intracerebroventricular (i.c.v.) injection displayed localized FKBP12 degradation in the brain, potentially expanding the use of PROTACs to GBM as well as other brain diseases including neurodegenerative disorders such as Alzheimer’s disease. It will be interesting to see how the PROTACs fare in the first clinical trials. The AR-targeting ARV-110 and ER-targeting ARV-471 (Arvinas) are currently undergoing recruitment for phase I clinical trials against prostate and breast cancer, respectively (NCT03888612/NCT04072952). A recent update at the American Association for Clinical Oncology (ASCO) suggests that ARV-110 showed antitumor activity and reduced PSA levels in some patients (J Clin Oncol 38: 2020 (suppl; abstr 3500)).
Conclusions
GBM remains the deadliest cancer with limited therapeutic options. Recent discoveries that are starting to define its heterogeneity indicate that similar to other cancers, personalized therapies will be the way forward. Protein degradation is a ubiquitous feature that is essential to maintain cellular homeostasis, and the small protein modifier ubiquitin plays a key role in regulating protein fate and function, and thereby impacts on most signal transduction pathways and cellular processes.
In this review we have summarized components of the ubiquitin system which are found deregulated in GBM as well as highlighted key molecular mechanisms involved. In just over twenty years or so since the first reports of the discovery of the UPS, PROTACs have shown some exciting potential by being able to control the fate of proteins and trigger their degradation on demand. This has stirred new hopes for effective targeting of the many oncogenic proteins that have been identified as drivers of disease, in particular, those that were dubbed “undruggable”. It has nevertheless taken almost another 20 years to bring PROTACs and targeted protein degradation to the forefront of drug discovery. Recent developments in chemical biology, synthetic biology as well as the first ongoing clinical trials will no doubt accelerate the delivery of new therapies.
The examples included in our review aim to showcase the diversity of ubiquitin-dependent molecular mechanisms that are now being targeted as well as the fast-expanding toolbox of ubiquitin-based therapeutics that are becoming available. PROTACs are prime examples and they are already being adapted to oncoproteins also relevant for GBM including BRD4 (Myc) (Figure 6), ERK1,2 (MAP kinase pathway), EGFR and CDK4/6 (404, 420–423). PROTACs have so far only been designed based on a small number of Cullin-RING E3 ligases, leaving a large number of E3 ligases implicated in GBM still available for investigation (Table 1). Indeed, the tissue-specific expression of E3 ligases used in PROTACs will need to be ascertained as well as the impact that drafting an endogenous E3 ligase for therapeutic help might have on the system. Further exciting developments include combining optogenetics with protein degrader strategies such as Opto-PROTACs, as this could provide added control over the timing and induction of protein degradation (424).
These technological advances will no doubt offer new avenues for GBM where little therapeutic progress has been made throughout the last decades. Ubiquitin-dependent mechanisms have been implicated in the regulation of most if not all hallmarks of GBM, in particular the signal transduction pathways that confer cancer cells properties but also stemness and heterogeneity which have so far hindered the use of potential treatments through mediating drug resistance. Results from the first PROTAC clinical trials are eagerly awaited to inform on pharmacological viability and to outline future hurdles in the field. In the context of GBM and other brain tumors, it will also be important to improve drug delivery systems that could overcome or bypass the blood-brain barrier such as nano-vehicles, strategies for enhancement of brain permeability, active transporter or alternative administration regimens [reviewed in (425, 426)].
Author Contributions
NS and JL designed the content of the review with input from all the co-authors. NS wrote the review with feedback from all the co-authors. All authors contributed to the article and approved the submitted version.
Funding
NS’s PhD studentship is funded by a GW4 BioMed MRC Doctoral Training Partnership. We also acknowledge funding from the University of Bath Alumni for a GBM pump-priming grant for some of the work carried out in the Licchesi laboratory.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We acknowledge GW4CANCER, a cross-disciplinary GW4 network that is building on the substantial cancer research portfolio of Bath, Bristol, Cardiff and Exeter. We acknowledge Zoe Hayes, funded by a Wellcome Trust Biomedical Vacation Scholarship, for her help identifying some E3 ubiquitin ligases presented in Table 1. Figures were created with BioRender.com.
References
1. Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK, et al. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta Neuropathol (2016) 131:803–20. doi: 10.1007/s00401-016-1545-1
2. Ostrom QT, Gittleman H, Farah P, Ondracek A, Chen Y, Wolinsky Y, et al. CBTRUS statistical report: Primary brain and central nervous system tumors diagnosed in the United States in 2006-2010. Neuro Oncol (2013) 15 Suppl 2:ii1–ii56. doi: 10.1093/neuonc/not151
3. Brodbelt A, Greenberg D, Winters T, Williams M, Vernon S, Collins VP. Glioblastoma in England: 2007–2011. Eur J Cancer (2015) 51:533–42. doi: 10.1016/j.ejca.2014.12.014
4. Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A, et al. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol (2007) 114:97–109. doi: 10.1007/s00401-007-0243-4
5. Ohgaki H, Kleihues P. The definition of primary and secondary glioblastoma. Clin Cancer Res (2013) 19:764–72. doi: 10.1158/1078-0432.CCR-12-3002
6. Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature (2009) 462:739–44. doi: 10.1038/nature08617
7. Lu C, Ward PS, Kapoor GS, Rohle D, Turcan S, Abdel-Wahab O, et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature (2012) 483:474–8. doi: 10.1038/nature10860
8. Toyota M, Ahuja N, Ohe-Toyota M, Herman JG, Baylin SB, Issa JP. CpG island methylator phenotype in colorectal cancer. Proc Natl Acad Sci USA (1999) 96:8681–6. doi: 10.1073/pnas.96.15.8681
9. Turcan S, Rohle D, Goenka A, Walsh LA, Fang F, Yilmaz E, et al. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature (2012) 483:479–83. doi: 10.1038/nature10866
10. Unruh D, Zewde M, Buss A, Drumm MR, Tran AN, Scholtens DM, et al. Methylation and transcription patterns are distinct in IDH mutant gliomas compared to other IDH mutant cancers. Sci Rep (2019) 9:8946. doi: 10.1038/s41598-019-45346-1
11. Nakamura M, Yonekawa Y, Kleihues P, Ohgaki H. Promoter hypermethylation of the RB1 gene in glioblastomas. Lab Invest (2001) 81:77–82. doi: 10.1038/labinvest.3780213
12. Martinez R, Martin-Subero JI, Rohde V, Kirsch M, Alaminos M, Fernandez AF, et al. A microarray-based DNA methylation study of glioblastoma multiforme. Epigenetics (2009) 4:255–64. doi: 10.4161/epi.9130
13. Kim TY, Zhong S, Fields CR, Kim JH, Robertson KD. Epigenomic profiling reveals novel and frequent targets of aberrant DNA methylation-mediated silencing in malignant glioma. Cancer Res (2006) 66:7490–501. doi: 10.1158/0008-5472.CAN-05-4552
14. Uhlmann K, Rohde K, Zeller C, Szymas J, Vogel S, Marczinek K, et al. Distinct methylation profiles of glioma subtypes. Int J Cancer (2003) 106:52–9. doi: 10.1002/ijc.11175
15. Nakamura M, Watanabe T, Yonekawa Y, Kleihues P, Ohgaki H. Promoter methylation of the DNA repair gene MGMT in astrocytomas is frequently associated with G:C → A:T mutations of the TP53 tumor suppressor gene. Carcinogenesis (2001) 22:1715–9. doi: 10.1093/carcin/22.10.1715
16. Hegi ME, Diserens AC, Gorlia T, Hamou MF, De Tribolet N, Weller M, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med (2005) 352:997–1003. doi: 10.1056/NEJMoa043331
17. Esteller M, Garcia-Foncillas J, Andion E, Goodman SN, Hidalgo OF, Vanaclocha V, et al. Inactivation of the DNA-repair gene MGMT and the clinical response of gliomas to alkylating agents. N Engl J Med (2000) 343:1350–4. doi: 10.1056/NEJM200011093431901
18. Esteller M, Hamilton SR, Burger PC, Baylin SB, Herman JG. Inactivation of the DNA repair gene O6-methylguanine-DNA methyltransferase by promoter hypermethylation is a common event in primary human neoplasia. Cancer Res (1999) 59:793–7.
19. Cahill DP, Levine KK, Betensky RA, Codd PJ, Romany CA, Reavie LB, et al. Loss of the mismatch repair protein MSH6 in human glioblastomas is associated with tumor progression during temozolomide treatment. Clin Cancer Res (2007) 13:2038–45. doi: 10.1158/1078-0432.CCR-06-2149
20. Verhaak RGW, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, et al. Integrated Genomic Analysis Identifies Clinically Relevant Subtypes of Glioblastoma Characterized by Abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell (2010) 17:98–110. doi: 10.1016/j.ccr.2009.12.020
21. Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science (2014) 344:1396–401. doi: 10.1126/science.1254257
22. Darmanis S, Sloan SA, Croote D, Mignardi M, Chernikova S, Samghababi P, et al. Single-Cell RNA-Seq Analysis of Infiltrating Neoplastic Cells at the Migrating Front of Human Glioblastoma. Cell Rep (2017) 21:1399–410. doi: 10.1016/j.celrep.2017.10.030
23. Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJB, et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N Engl J Med (2005) 352:987–96. doi: 10.1056/NEJMoa043330
24. Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJ, Janzer RC, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol (2009) 10:459–66. doi: 10.1016/S1470-2045(09)70025-7
25. Stummer W, Stocker S, Wagner S, Stepp H, Fritsch C, Goetz C, et al. Intraoperative detection of malignant gliomas by 5-aminolevulinic acid-induced porphyrin fluorescence. Neurosurgery (1998) 42:518–26. doi: 10.1016/S0303-8467(97)81684-8
26. O’Rourke DM, Nasrallah MP, Desai A, Melenhorst JJ, Mansfield K, Morrissette JJD, et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Transl Med (2017) 9:eaaa0984. doi: 10.1126/scitranslmed.aaa0984
27. Dupont C, Vermandel M, Leroy HA, Quidet M, Lecomte F, Delhem N, et al. INtraoperative photoDYnamic Therapy for GliOblastomas (INDYGO): Study Protocol for a Phase I Clinical Trial. Neurosurgery (2019) 84:E414–9. doi: 10.1093/neuros/nyy324
28. Brown CE, Alizadeh D, Starr R, Weng L, Wagner JR, Naranjo A, et al. Regression of Glioblastoma after Chimeric Antigen Receptor T-Cell Therapy. N Engl J Med (2016) 375:2561–9. doi: 10.1056/NEJMoa1610497
29. Hilf N, Kuttruff-Coqui S, Frenzel K, Bukur V, Stevanović S, Gouttefangeas C, et al. Actively personalized vaccination trial for newly diagnosed glioblastoma. Nature (2019) 565:240–5. doi: 10.1038/s41586-018-0810-y
30. Cohen P, Tcherpakov M. Will the ubiquitin system furnish as many drug targets as protein kinases? Cell (2010) 143:686–93. doi: 10.1016/j.cell.2010.11.016
31. Crews CM. Targeting the Undruggable Proteome: The Small Molecules of My Dreams. Chem Biol (2010) 17:551–5. doi: 10.1016/j.chembiol.2010.05.011
32. Goldstein G, Scheid M, Hammerling U, Schlesinger DH, Niall HD, Boyse EA. Isolation of a polypeptide that has lymphocyte-differentiating properties and is probably represented universally in living cells. Proc Natl Acad Sci USA (1975) 72:11–5. doi: 10.1073/pnas.72.1.11
33. Schlesinger DH, Goldstein G, Niall HD. The complete amino acid sequence of ubiquitin, an adenylate cyclase stimulating polypeptide probably universal in living cells. Biochemistry (1975) 14:2214–8. doi: 10.1021/bi00681a026
34. Hochstrasser M. Origin and function of ubiquitin-like proteins. Nature (2009) 458:422–9. doi: 10.1038/nature07958
35. van der Veen AG, Ploegh HL. Ubiquitin-Like Proteins. Annu Rev Biochem (2012) 81:323–57. doi: 10.1146/annurev-biochem-093010-153308
36. Jentsch S, Pyrowolakis G. Ubiquitin and its kin: How close are the family ties? Trends Cell Biol (2000) 10:335–42. doi: 10.1016/S0962-8924(00)01785-2
37. Schulman BA, Wade Harper J. Ubiquitin-like protein activation by E1 enzymes: The apex for downstream signalling pathways. Nat Rev Mol Cell Biol (2009) 10:319–31. doi: 10.1038/nrm2673
38. Yuan W, Krug RM. Influenza B virus NS1 protein inhibits conjugation of the interferon (IFN)-induced ubiquitin-like ISG15 protein. EMBO J (2001) 20:362–71. doi: 10.1093/emboj/20.3.362
39. Zhao C, Beaudenon SL, Kelley ML, Waddell MB, Yuan W, Schulman BA, et al. The UbcH8 ubiquitin E2 enzyme is also the E2 enzyme for ISG15, an. Proc Natl Acad Sci USA (2004) 101:7578–82. doi: 10.1073/pnas.0402528101
40. Dastur A, Beaudenon S, Kelley M, Krug RM, Huibregtse JM. Herc5, an interferon-induced HECT E3 enzyme, is required for conjugation of ISG15 in human cells. J Biol Chem (2006) 281:4334–8. doi: 10.1074/jbc.M512830200
41. Zou W, Zhang D-E. The interferon-inducible ubiquitin-protein isopeptide ligase (E3) EFP also functions as an ISG15 E3 ligase. J Biol Chem (2006) 281:3989–94. doi: 10.1074/jbc.M510787200
42. Haas AL, Ahrens P, Bright PM, Ankel H. Interferon induced a 15-kilodalton protein exhibiting marked homology to ubiquitin. J Biol Chem (1987) 262:11315–23.
43. Pearce MJ, Mintseris J, Ferreyra J, Gygi SP, Darwin KH. Ubiquitin-like protein involved in the proteasome pathway of Mycobacterium tuberculosis. Science (2008) 322:1104–7. doi: 10.1126/science.1163885
44. Burns KE, Liu WT, Boshoff HIM, Dorrestein PC, Barry CE. Proteasomal protein degradation in mycobacteria is dependent upon a prokaryotic ubiquitin-like protein. J Biol Chem (2009) 284:3069–75. doi: 10.1074/jbc.M808032200
45. Maupin-Furlow JA. Prokaryotic Ubiquitin-Like Protein Modification. Annu Rev Microbiol (2014) 68:155–75. doi: 10.1146/annurev-micro-091313-103447
46. Humbard MA, Miranda HV, Lim JM, Krause DJ, Pritz JR, Zhou G, et al. Ubiquitin-like small archaeal modifier proteins (SAMPs) in Haloferax volcanii. Nature (2010) 463:54–60. doi: 10.1038/nature08659
47. Ciechanover A, Heller H, Katz-Etzion R, Hershko A. Activation of the heat-stable polypeptide of the ATP-dependent proteolytic system. Proc Natl Acad Sci USA (1981) 78:761–5. doi: 10.1073/pnas.78.2.761
48. Hershko A, Ciechanover A, Heller H, Haas AL, Rose IA. Proposed role of ATP in protein breakdown: conjugation of protein with multiple chains of the polypeptide of ATP-dependent proteolysis. Proc Natl Acad Sci USA (1980) 77:1783–6. doi: 10.1073/pnas.77.4.1783
49. Finley D, Ciechanover A, Varshavsky A. Thermolability of ubiquitin-activating enzyme from the mammalian cell cycle mutant ts85. Cell (1984) 37:43–55. doi: 10.1016/0092-8674(84)90299-X
50. Ciechanover A, Finley D, Varshavsky A. Ubiquitin dependence of selective protein degradation demonstrated in the mammalian cell cycle mutant ts85. Cell (1984) 37:57–66. doi: 10.1016/0092-8674(84)90300-3
51. Haas AL, Rose IA. The mechanism of ubiquitin activating enzyme. A kinetic and equilibrium analysis. J Biol Chem (1982) 257:10329–37.
52. Haas AL, Warms JV, Hershko A, Rose IA. Ubiquitin-activating enzyme. Mechanism and role in protein-ubiquitin conjugation. J Biol Chem (1982) 257:2543–8.
53. Pickart CM, Kasperek EM, Beal R, Kim A. Substrate properties of site-specific mutant ubiquitin protein (G76a) reveal unexpected mechanistic features of ubiquitin-activating enzyme (E1). J Biol Chem (1994) 269:7115–23.
54. Pickart CM, Rose IA. Functional heterogeneity of ubiquitin carrier proteins. J Biol Chem (1985) 260:1573–81.
55. Wang X, Herr RA, Chua WJ, Lybarger L, Wiertz EJHJ, Hansen TH. Ubiquitination of serine, threonine, or lysine residues on the cytoplasmic tail can induce ERAD of MHC-I by viral E3 ligase mK3. J Cell Biol (2007) 177:613–24. doi: 10.1083/jcb.200611063
56. Scaglione KM, Basrur V, Ashraf NS, Konen JR, Elenitoba-Johnson KSJ, Todi SV, et al. The ubiquitin-conjugating enzyme (E2) ube2w ubiquitinates the N terminus of substrates. J Biol Chem (2013) 288:18784–8. doi: 10.1074/jbc.C113.477596
57. Tatham MH, Plechanovová A, Jaffray EG, Salmen H, Hay RT. Ube2W conjugates ubiquitin to α-amino groups of protein N-termini. Biochem J (2013) 453:137–45. doi: 10.1042/BJ20130244
58. Hershko A, Heller H, Elias S, Ciechanover A. Components of ubiquitin-protein ligase system. Resolution, affinity purification, and role in protein breakdown. J Biol Chem (1983) 258:8206–14.
59. Scheffner M, Nuber U, Huibregtse JM. Protein ubiquitination involving an E1–E2–E3 enzyme ubiquitin thioester cascade. Nature (1995) 373:81–3. doi: 10.1038/373081a0
60. Komander D, Rape M. The Ubiquitin Code. Annu Rev Biochem (2012) 81:203–29. doi: 10.1146/annurev-biochem-060310-170328
61. Lorick KL, Jensen JP, Fang S, Ong AM, Hatakeyama S, Weissman AM. RING fingers mediate ubiquitin-conjugating enzyme (E2)-dependent ubiquitination. Proc Natl Acad Sci USA (1999) 96:11364–9. doi: 10.1073/pnas.96.20.11364
62. Zheng N, Wang P, Jeffrey PD, Pavletich NP. Structure of a c-Cbl-UbcH7 complex: RING domain function in ubiquitin-protein ligases. Cell (2000) 102:533–9. doi: 10.1016/S0092-8674(00)00057-X
63. Yin Q, Lin SC, Lamothe B, Lu M, Lo YC, Hura G, et al. E2 interaction and dimerization in the crystal structure of TRAF6. Nat Struct Mol Biol (2009) 16:658–66. doi: 10.1038/nsmb.1605
64. Brzovic PS, Rajagopal P, Hoyt DW, King MC, Klevit RE. Structure of a BRCA1-BARD1 heterodimeric RING-RING complex. Nat Struct Biol (2001) 8:833–7. doi: 10.1038/nsb1001-833
65. Eletr ZM, Huang DT, Duda DM, Schulman BA, Kuhlman B. E2 conjugating enzymes must disengage from their E1 enzymes before E3-dependent ubiquitin and ubiquitin-like transfer. Nat Struct Mol Biol (2005) 12:933–4. doi: 10.1038/nsmb984
66. Pruneda JN, Littlefield PJ, Soss SE, Nordquist KA, Chazin WJ, Brzovic PS, et al. Structure of an E3:E2∼Ub Complex Reveals an Allosteric Mechanism Shared among RING/U-box Ligases. Mol Cell (2012) 47:933–42. doi: 10.1016/j.molcel.2012.07.001
67. Plechanovov A, Jaffray EG, Tatham MH, Naismith JH, Hay RT. Structure of a RING E3 ligase and ubiquitin-loaded E2 primed for catalysis. Nature (2012) 489:115–20. doi: 10.1038/nature11376
68. Dou H, Buetow L, Sibbet GJ, Cameron K, Huang DT. BIRC7-E2 ubiquitin conjugate structure reveals the mechanism of ubiquitin transfer by a RING dimer. Nat Struct Mol Biol (2012) 19:876–83. doi: 10.1038/nsmb.2379
69. Pruneda JN, Stoll KE, Bolton LJ, Brzovic PS, Klevit RE. Ubiquitin in Motion: Structural Studies of the Ubiquitin-Conjugating Enzyme∼Ubiquitin Conjugate. Biochemistry (2011) 50:1624–33. doi: 10.1021/bi101913m
70. Eddins MJ, Carlile CM, Gomez KM, Pickart CM, Wolberger C. Mms2-Ubc13 covalently bound to ubiquitin reveals the structural basis of linkage-specific polyubiquitin chain formation. Nat Struct Mol Biol (2006) 13:915–20. doi: 10.1038/nsmb1148
71. Petroski MD, Deshaies RJ. Function and regulation of cullin-RING ubiquitin ligases. Nat Rev Mol Cell Biol (2005) 6:9–20. doi: 10.1038/nrm1547
72. Ohi MD, Vander Kooi CW, Rosenberg JA, Chazin WJ, Gould KL. Structural insights into the U-box, a domain associated with multi-ubiquitination. Nat Struct Biol (2003) 10:250–5. doi: 10.1038/nsb906
73. Aravind L, Koonin EV. The U box is a modified RING finger - A common domain in ubiquitination. Curr Biol (2000) 10:R132–4. doi: 10.1016/S0960-9822(00)00398-5
74. Jiang J, Ballinger CA, Wu Y, Dai Q, Cyr DM, Höhfeld J, et al. CHIP is a U-box-dependent E3 ubiquitin ligase: Identification of Hsc70 as a target for ubiquitylation. J Biol Chem (2001) 276:42938–44. doi: 10.1074/jbc.M101968200
75. Murata S, Minami Y, Minami M, Chiba T, Tanaka K. CHIP is a chaperone-dependent E3 ligase that ubiquitylates unfolded protein. EMBO Rep (2001) 2:1133–8. doi: 10.1093/embo-reports/kve246
76. Xu Z, Kohli E, Devlin KI, Bold M, Nix JC, Misra S. Interactions between the quality control ubiquitin ligase CHIP and ubiquitin conjugating enzymes. BMC Struct Biol (2008) 8:26. doi: 10.1186/1472-6807-8-26
77. Nikolay R, Wiederkehr T, Rist W, Kramer G, Mayer MP, Bukau B. Dimerization of the Human E3 Ligase CHIP via a Coiled-coil Domain Is Essential for Its Activity. J Biol Chem (2004) 279:2673–8. doi: 10.1074/jbc.M311112200
78. Marín I, Lucas JI, Gradilla AC, Ferrús A. Parkin and relatives: The RBR family of ubiquitin ligases. Physiol Genomics (2004) 17:253–63. doi: 10.1152/physiolgenomics.00226.2003
79. Spratt DE, Walden H, Shaw GS. RBR E3 Ubiquitin Ligases: New Structures, New Insights, New Questions. Biochem J (2014) 458:421–37. doi: 10.1042/BJ20140006
80. Wenzel DM, Lissounov A, Brzovic PS, Klevit RE. UBCH7 reactivity profile reveals parkin and HHARI to be RING/HECT hybrids. Nature (2011) 474:105–8. doi: 10.1038/nature09966
81. Stieglitz B, Morris-Davies AC, Koliopoulos MG, Christodoulou E, Rittinger K. LUBAC synthesizes linear ubiquitin chains via a thioester intermediate. EMBO Rep (2012) 13:840–6. doi: 10.1038/embor.2012.105
82. Smit JJ, Monteferrario D, Noordermeer SM, van Dijk WJ, van der Reijden BA, Sixma TK. The E3 ligase HOIP specifies linear ubiquitin chain assembly through its RING-IBR-RING domain and the unique LDD extension. EMBO J (2012) 31:3833–44. doi: 10.1038/emboj.2012.217
83. Lazarou M, Narendra DP, Jin SM, Tekle E, Banerjee S, Youle RJ. PINK1 drives parkin self-association and HECT-like E3 activity upstream of mitochondrial binding. J Cell Biol (2013) 200:163–72. doi: 10.1083/jcb.201210111
84. Chew KCM, Matsuda N, Saisho K, Lim GGY, Chai C, Tan H-M, et al. Parkin mediates apparent E2-independent monoubiquitination in vitro and contains an intrinsic activity that catalyzes polyubiquitination. PLoS One (2011) 6:e19720. doi: 10.1371/journal.pone.0019720
85. Veeriah S, Taylor BS, Meng S, Fang F, Yilmaz E, Vivanco I, et al. Somatic Mutations of the Parkinson’s Disease-Associated Gene PARK2 in Glioblastoma and Other Human Malignancies. Nat Genet (2010) 42:77–82. doi: 10.1038/ng.491
86. Matsuda N, Kitami T, Suzuki T, Mizuno Y, Hattori N, Tanaka K. Diverse effects of pathogenic mutations of Parkin that catalyze multiple monoubiquitylation in vitro. J Biol Chem (2006) 281:3204–9. doi: 10.1074/jbc.M510393200
87. Chaugule VK, Burchell L, Barber KR, Sidhu A, Leslie SJ, Shaw GS, et al. Autoregulation of Parkin activity through its ubiquitin-like domain. EMBO J (2011) 30:2853–67. doi: 10.1038/emboj.2011.204
88. Fallon L, Bélanger CML, Corera AT, Kontogiannea M, Regan-Klapisz E, Moreau F, et al. A regulated interaction with the UIM protein Eps15 implicates parkin in EGF receptor trafficking and PI(3)K-Akt signalling. Nat Cell Biol (2006) 8:834–42. doi: 10.1038/ncb1441
89. Tsai YC, Fishman PS, Thakor NV, Oyler GA. Parkin facilitates the elimination of expanded polyglutamine proteins and leads to preservation of proteasome function. J Biol Chem (2003) 278:22044–55. doi: 10.1074/jbc.M212235200
90. Marin I. Animal HECT ubiquitin ligases: Evolution and functional implications. BMC Evol Biol (2010) 10:56. doi: 10.1186/1471-2148-10-56
91. Huibregtse JM, Scheffner M, Beaudenon S, Howley PMA. family of proteins structurally and functionally related to the E6-AP ubiquitin-protein ligase. Proc Natl Acad Sci USA (1995) 92:2563–7. doi: 10.1073/pnas.92.7.2563
92. Scheffner M, Huibregtse JM, Vierstra RD, Howley PM. The HPV-16 E6 and E6-AP complex functions as a ubiquitin-protein ligase in the ubiquitination of p53. Cell (1993) 75:495–505. doi: 10.1016/0092-8674(93)90384-3
93. Huibregtse JM, Scheffner M, Howley PM. A cellular protein mediates association of p53 with the E6 oncoprotein of human papillomavirus types 16 or 18. EMBO J (1991) 10:4129–35. doi: 10.1002/j.1460-2075.1991.tb04990.x
94. Simonson SJS, Difilippantonio MJ, Lambert PF. Two distinct activities contribute to human papillomavirus 16 E6’s oncogenic potential. Cancer Res (2005) 65:8266–73. doi: 10.1158/0008-5472.CAN-05-1651
95. Scheffner M, Werness BA, Huibregtse JM, Levine AJ, Howley PM. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell (1990) 63:1129–36. doi: 10.1016/0092-8674(90)90409-8
96. Kim HC, Huibregtse JM. Polyubiquitination by HECT E3s and the Determinants of Chain Type Specificity. Mol Cell Biol (2009) 29:3307–18. doi: 10.1128/MCB.00240-09
97. Huang L, Kinnucan E, Wang G, Beaudenon S, Howley PM, Huibregtse JM, et al. Structure of an E6AP-UbcH7 complex: Insights into ubiquitination by the E2-E3 enzyme cascade. Science (1999) 286:1321–6. doi: 10.1126/science.286.5443.1321
98. Maspero E, Mari S, Valentini E, Musacchio A, Fish A, Pasqualato S, et al. Structure of the HECT:ubiquitin complex and its role in ubiquitin chain elongation. EMBO Rep (2011) 12:342–9. doi: 10.1038/embor.2011.21
99. Kamadurai HB, Souphron J, Scott DC, Duda DM, Miller DJ, Stringer D, et al. Insights into ubiquitin transfer cascades from a structure of a UbcH5B approximately ubiquitin-HECT(NEDD4L) complex. Mol Cell (2009) 36:1095–102. doi: 10.1016/j.molcel.2009.11.010
100. Verdecia MA, Joazeiro CAP, Wells NJ, Ferrer J-L, Bowman ME, Hunter T, et al. Conformational flexibility underlies ubiquitin ligation mediated by the WWP1 HECT domain E3 ligase. Mol Cell (2003) 11:249–59. doi: 10.1016/S1097-2765(02)00774-8
101. Lorenz S. Structural mechanisms of HECT-type ubiquitin ligases. Biol Chem (2018) 399:127–45. doi: 10.1515/hsz-2017-0184
102. Rotin D, Kumar S. Physiological functions of the HECT family of ubiquitin ligases. Nat Rev Mol Cell Biol (2009) 10:398–409. doi: 10.1038/nrm2690
103. Kumar S, Tomooka Y, Noda M. Identification of a set of genes with developmentally down-regulated expression in the mouse brain. Biochem Biophys Res Commun (1992) 185:1155–61. doi: 10.1016/0006-291X(92)91747-E
104. Ingham RJ, Colwill K, Howard C, Dettwiler S, Lim CSH, Yu J, et al. WW Domains Provide a Platform for the Assembly of Multiprotein Networks. Mol Cell Biol (2005) 25:7092–106. doi: 10.1128/MCB.25.16.7092-7106.2005
105. Kanelis V, Rotin D, Forman-Kay JD. Solution structure of a Nedd4 WW domain-ENaC peptide complex. Nat Struct Biol (2001) 8:407–12. doi: 10.1038/87562
106. Staub O, Dho S, Henry P, Correa J, Ishikawa T, McGlade J, et al. WW domains of Nedd4 bind to the proline-rich PY motifs in the epithelial Na+ channel deleted in Liddle’s syndrome. EMBO J (1996) 15:2371–80. doi: 10.1002/j.1460-2075.1996.tb00593.x
107. Rizo J, Sudhof TC. C2-domains, structure and function of a universal Ca2+-binding domain. J Biol Chem (1998) 273:15879–82. doi: 10.1074/jbc.273.26.15879
108. Tian M, Bai C, Lin Q, Lin H, Liu M, Ding F, et al. Binding of RhoA by the C2 domain of E3 ligase Smurf1 is essential for Smurf1-regulated RhoA ubiquitination and cell protrusive activity. FEBS Lett (2011) 585:2199–204. doi: 10.1016/j.febslet.2011.06.016
109. Weber J, Polo S, Maspero E. HECT E3 ligases: A tale with multiple facets. Front Physiol (2019) 10(10):370. doi: 10.3389/fphys.2019.00370
110. Wiesner S, Ogunjimi AA, Wang H-R, Rotin D, Sicheri F, Wrana JL, et al. Autoinhibition of the HECT-type ubiquitin ligase Smurf2 through its C2 domain. Cell (2007) 130:651–62. doi: 10.1016/j.cell.2007.06.050
111. Attali I, Tobelaim WS, Persaud A, Motamedchaboki K, Simpson-Lavy KJ, Mashahreh B, et al. Ubiquitylation-dependent oligomerization regulates activity of Nedd4 ligases. EMBO J (2017) 36:425–40. doi: 10.15252/embj.201694314
112. Ronchi VP, Klein JM, Edwards DJ, Haas AL. The active form of E6-associated protein (E6AP)/UBE3A ubiquitin ligase is an oligomer. J Biol Chem (2014) 289:1033–48. doi: 10.1074/jbc.M113.517805
113. Clague MJ, Urbé S, Komander D. Breaking the chains: deubiquitylating enzyme specificity begets function. Nat Rev Mol Cell Biol (2019) 20:338–52. doi: 10.1038/s41580-019-0099-1
114. Ambroggio XI, Rees DC, Deshaies RJ. JAMM: A metalloprotease-like zinc site in the proteasome and signalosome. PLoS Biol (2004) 2:e2. doi: 10.1371/journal.pbio.0020002
115. Matsui S-I, Sandberg AA, Negoro S, Seon BK, Goldstein G. Isopeptidase: A novel eukaryotic enzyme that cleaves isopeptide bonds. Proc Natl Acad Sci USA (1982) 79:1535–9. doi: 10.1073/pnas.79.5.1535
116. Miller HI, Henzel WJ, Ridgway JB, Kuang WJ, Chisholm V, Liu CC. Cloning and expression of a yeast ubiquitin protein cleaving activity in escherichia coli. Bio/Technology (1989) 7:689–704. doi: 10.1038/nbt0789-698
117. Redman KL, Rechsteiner M. Identification of the long ubiquitin extension as ribosomal protein S27a. Nature (1989) 338:438–40. doi: 10.1038/338438a0
118. Baker RT, Board PG. The human ubiquitin-52 amino acid fusion protein gene shares several structural features with mammalian ribosomal protein genes. Nucleic Acids Res (1991) 19:1035–40. doi: 10.1093/nar/19.5.1035
119. Finley D, Özkaynak E, Varshavsky A. The yeast polyubiquitin gene is essential for resistance to high temperatures, starvation, and other stresses. Cell (1987) 48:1035–46. doi: 10.1016/0092-8674(87)90711-2
120. Grou CP, Pinto MP, Mendes AV, Domingues P, Azevedo JE. The de novo synthesis of ubiquitin: Identification of deubiquitinases acting on ubiquitin precursors. Sci Rep (2015) 5:1–16. doi: 10.1038/srep12836
121. Verma R, Aravind L, Oania R, McDonald WH, Yates JR, Koonin EV, et al. Role of Rpn11 metalloprotease in deubiquitination and degradation by the 26S proteasome. Science (2002) 298:611–5. doi: 10.1126/science.1075898
122. Williams RL, Urbé S. The emerging shape of the ESCRT machinery. Nat Rev Mol Cell Biol (2007) 8:355–68. doi: 10.1038/nrm2162
123. Finley D. Recognition and Processing of Ubiquitin-Protein Conjugates by the Proteasome. Annu Rev Biochem (2009) 78:477–513. doi: 10.1146/annurev.biochem.78.081507.101607
124. Worden EJ, Dong KC, Martin A. An AAA Motor-Driven Mechanical Switch in Rpn11 Controls Deubiquitination at the 26S Proteasome. Mol Cell (2017) 67:799–811.e8. doi: 10.1016/j.molcel.2017.07.023
125. Worden EJ, Padovani C, Martin A. Structure of the Rpn11-Rpn8 dimer reveals mechanisms of substrate deubiquitination during proteasomal degradation. Nat Struct Mol Biol (2014) 21:220–7. doi: 10.1038/nsmb.2771
126. de Poot SAH, Tian G, Finley D. Meddling with Fate: The Proteasomal Deubiquitinating Enzymes. J Mol Biol (2017) 429:3525–45. doi: 10.1016/j.jmb.2017.09.015
127. Bard JAM, Goodall EA, Greene ER, Jonsson E, Dong KC, Martin A. Structure and Function of the 26S Proteasome. Annu Rev Biochem (2018) 87:697–724. doi: 10.1146/annurev-biochem-062917-011931
128. Emmerich CH, Ordureau A, Strickson S, Arthur JSC, Pedrioli PGA, Komander D, et al. Activation of the canonical IKK complex by K63/M1-linked hybrid ubiquitin chains. Proc Natl Acad Sci USA (2013) 110:15247–52. doi: 10.1073/pnas.1314715110
129. Keusekotten K, Elliott PR, Glockner L, Fiil BK, Damgaard RB, Kulathu Y, et al. OTULIN antagonizes LUBAC signaling by specifically hydrolyzing met1-linked polyubiquitin. Cell (2013) 153:1312–26. doi: 10.1016/j.cell.2013.05.014
130. Hrdinka M, Fiil BK, Zucca M, Leske D, Bagola K, Yabal M, et al. CYLD Limits Lys63- and Met1-Linked Ubiquitin at Receptor Complexes to Regulate Innate Immune Signaling. Cell Rep (2016) 14:2846–58. doi: 10.1016/j.celrep.2016.02.062
131. Damgaard RB, Walker JA, Marco-Casanova P, Morgan NV, Titheradge HL, Elliott PR, et al. The Deubiquitinase OTULIN Is an Essential Negative Regulator of Inflammation and Autoimmunity. Cell (2016) 166:1215–30.e20. doi: 10.1016/j.cell.2016.07.019
132. Wertz IE, O’Rourke KM, Zhou H, Eby M, Aravind L, Seshagiri S, et al. De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-κB signalling. Nature (2004) 430:694–9. doi: 10.1038/nature02794
133. Hershko A, Heller H. Occurrence of a polyubiquitin structure in ubiquitin-protein conjugates. Biochem Biophys Res Commun (1985) 128:1079–86. doi: 10.1016/0006-291X(85)91050-2
134. Dahlmann B, Kopp F, Kuehn L, Niedel B, Pfeifer G, Hegerl R, et al. The multicatalytic proteinase (prosome) is ubiquitous from eukaryotes to archaebacteria. FEBS Lett (1989) 251:125–31. doi: 10.1016/0014-5793(89)81441-3
135. Chau V, Tobias JW, Bachmair A, Marriott D, Ecker DJ, Gonda DK, et al. A multiubiquitin chain is confined to specific lysine in a targeted short-lived protein. Science (1989) 243:1576–83. doi: 10.1126/science.2538923
136. Hershko A, Leshinsky E, Ganoth D, Heller H. ATP-dependent degradation of ubiquitin-protein conjugates. Proc Natl Acad Sci USA (1984) 81:1619–23. doi: 10.1073/pnas.81.6.1619
137. Xu P, Duong DM, Seyfried NT, Cheng D, Xie Y, Robert J, et al. Quantitative Proteomics Reveals the Function of Unconventional Ubiquitin Chains in Proteasomal Degradation. Cell (2009) 137:133–45. doi: 10.1016/j.cell.2009.01.041
138. Phu L, Izrael-Tomasevic A, Matsumoto ML, Bustos D, Dynek JN, Fedorova AV, et al. Improved quantitative mass spectrometry methods for characterizing complex ubiquitin signals. Mol Cell Proteomics (2011) 10:M110.003756. doi: 10.1074/mcp.M110.003756
139. Yau R, Rape M. The increasing complexity of the ubiquitin code. Nat Cell Biol (2016) 18:579–86. doi: 10.1038/ncb3358
140. Deol KK, Lorenz S, Strieter ER. Enzymatic Logic of Ubiquitin Chain Assembly. Front Physiol (2019) 10:835. doi: 10.3389/fphys.2019.00835
141. Pickart CM. Targeting of substrates to the 26S proteasome. FASEB J (1997) 11:1055–66. doi: 10.1096/fasebj.11.13.9367341
142. Saeki Y, Kudo T, Sone T, Kikuchi Y, Yokosawa H, Toh-e A, et al. Lysine 63-linked polyubiquitin chain may serve as a targeting signal for the 26S proteasome. EMBO J (2009) 28:359–71. doi: 10.1038/emboj.2008.305
143. Chastagner P, Israël A, Brou C. Itch/AIP4 mediates Deltex degradation through the formation of K29-linked polyubiquitin chains. EMBO Rep (2006) 7:1147–53. doi: 10.1038/sj.embor.7400822
144. Braten O, Livneh I, Ziv T, Admon A, Kehat I, Caspi LH, et al. Numerous proteins with unique characteristics are degraded by the 26S proteasome following monoubiquitination. Proc Natl Acad Sci USA (2016) 113:E4639–47. doi: 10.1073/pnas.1608644113
145. Wang C, Deng L, Hong M, Akkaraju GR, Inoue J, Chen ZJ. TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature (2001) 412:346–51. doi: 10.1038/35085597
146. Deng L, Wang C, Spencer E, Yang L, Braun A, You J, et al. Activation of the IkappaB kinase complex by TRAF6 requires a dimeric ubiquitin-conjugating enzyme complex and a unique polyubiquitin chain. Cell (2000) 103:351–61. doi: 10.1016/S0092-8674(00)00126-4
147. Stewart GS, Panier S, Townsend K, Al-Hakim AK, Kolas NK, Miller ES, et al. The RIDDLE syndrome protein mediates a ubiquitin-dependent signaling cascade at sites of DNA damage. Cell (2009) 136:420–34. doi: 10.1016/j.cell.2008.12.042
148. Doil C, Mailand N, Bekker-Jensen S, Menard P, Larsen DH, Pepperkok R, et al. RNF168 Binds and Amplifies Ubiquitin Conjugates on Damaged Chromosomes to Allow Accumulation of Repair Proteins. Cell (2009) 136:435–46. doi: 10.1016/j.cell.2008.12.041
149. Gatti M, Pinato S, Maiolica A, Rocchio F, Prato MG, Aebersold R, et al. RNF168 promotes noncanonical K27ubiquitination to signal DNA damage. Cell Rep (2015) 10:226–38. doi: 10.1016/j.celrep.2014.12.021
150. Huang F, Zeng X, Kim W, Balasubramani M, Fortian A, Gygi SP, et al. Lysine 63-linked polyubiquitination is required for EGF receptor degradation. Proc Natl Acad Sci USA (2013) 110:15722–7. doi: 10.1073/pnas.1308014110
151. Lauwers E, Jacob C, Andre B. K63-linked ubiquitin chains as a specific signal for protein sorting into the multivesicular body pathway. J Cell Biol (2009) 185:493–502. doi: 10.1083/jcb.200810114
152. Song EJ, Werner SL, Neubauer J, Stegmeier F, Aspden J, Rio D, et al. The Prp19 complex and the Usp4Sart3 deubiquitinating enzyme control reversible ubiquitination at the spliceosome. Genes Dev (2010) 24:1434–47. doi: 10.1101/gad.1925010
153. Spence J, Gali RR, Dittmar G, Sherman F, Karin M, Finley D. Cell cycle-regulated modification of the ribosome by a variant multiubiquitin chain. Cell (2000) 102:67–76. doi: 10.1016/S0092-8674(00)00011-8
154. Flick K, Ouni I, Wohlschlegel JA, Capati C, McDonald WH, Yates JR, et al. Proteolysis-independent regulation of the transcription factor Met4 by a single Lys 48-linked ubiquitin chain. Nat Cell Biol (2004) 6:634–41. doi: 10.1038/ncb1143
155. Emmerich CH, Bakshi S, Kelsall IR, Ortiz-Guerrero J, Shpiro N, Cohen P. Lys63/Met1-hybrid ubiquitin chains are commonly formed during the activation of innate immune signalling. Biochem Biophys Res Commun (2016) 474:452–61. doi: 10.1016/j.bbrc.2016.04.141
156. Meyer HJ, Rape M. Enhanced protein degradation by branched ubiquitin chains. Cell (2014) 157:910–21. doi: 10.1016/j.cell.2014.03.037
157. Ohtake F, Tsuchiya H, Saeki Y, Tanaka K. K63 ubiquitylation triggers proteasomal degradation by seeding branched ubiquitin chains. Proc Natl Acad Sci USA (2018) 115:E1401–8. doi: 10.1073/pnas.1716673115
158. Liu C, Liu W, Ye Y, Li W. Ufd2p synthesizes branched ubiquitin chains to promote the degradation of substrates modified with atypical chains. Nat Commun (2017) 8:1–15. doi: 10.1038/ncomms14274
159. Johnson ES, Ma PCM, Ota IM, Varshavsky AA. Proteolytic Pathway That Recognizes Ubiquitin as a Degradation Signal. J Biol Chem (1995) 270:17442–56. doi: 10.1074/jbc.270.29.17442
160. Leto DE, Morgens DW, Zhang L, Walczak CP, Elias JE, Bassik MC, et al. Genome-wide CRISPR Analysis Identifies Substrate-Specific Conjugation Modules in ER-Associated Degradation. Mol Cell (2019) 73:377–89. doi: 10.1016/j.molcel.2018.11.015
161. Besche HC, Sha Z, Kukushkin NV, Peth A, Hock E, Kim W, et al. Autoubiquitination of the 26S Proteasome on Rpn13 Regulates Breakdown of Ubiquitin Conjugates. EMBO J (2014) 33:1159–76. doi: 10.1002/embj.201386906
162. Deol KK, Crowe SO, Du J, Bisbee HA, Guenette RG, Strieter ER. Proteasome-Bound UCH37/UCHL5 Debranches Ubiquitin Chains to Promote Degradation. Mol Cell (2020). In Press. doi: 10.1016/j.molcel.2020.10.017
163. Yoshida Y, Saeki Y, Murakami A, Kawawaki J, Tsuchiya H, Yoshihara H, et al. A comprehensive method for detecting ubiquitinated substrates using TR-TUBE. Proc Natl Acad Sci USA (2015) 112:4630–5. doi: 10.1073/pnas.1422313112
164. Valkevich EM, Sanchez NA, Ge Y, Strieter ER. Middle-Down mass spectrometry enables characterization of branched ubiquitin chains. Biochemistry (2014) 53:4979–89. doi: 10.1021/bi5006305
165. Mevissen TET, Hospenthal MK, Geurink PP, Elliott PR, Akutsu M, Arnaudo N, et al. OTU deubiquitinases reveal mechanisms of linkage specificity and enable ubiquitin chain restriction analysis. Cell (2013) 154:169–84. doi: 10.1016/j.cell.2013.05.046
166. Hjerpe R, Aillet F, Lopitz-Otsoa F, Lang V, England P, Rodriguez MS. Efficient protection and isolation of ubiquitylated proteins using tandem ubiquitin-binding entities. EMBO Rep (2009) 10:1250–8. doi: 10.1038/embor.2009.192
167. Swatek KN, Usher JL, Kueck AF, Gladkova C, Mevissen TET, Pruneda JN, et al. Insights into ubiquitin chain architecture using Ub-clipping. Nature (2019) 572:533–7. doi: 10.1038/s41586-019-1482-y
168. Bellail AC, Olson JJ, Yang X, Chen ZJ, Hao C. A20 Ubiquitin Ligase–Mediated Polyubiquitination of RIP1 Inhibits Caspase-8 Cleavage and TRAIL-Induced Apoptosis in Glioblastoma. Cancer Discovery (2012) 2:140–55. doi: 10.1158/2159-8290.CD-11-0172
169. Venere M, Horbinski C, Crish JF, Jin X, Vasanji A, Major J, et al. The mitotic kinesin KIF11 is a driver of invasion, proliferation, and self-renewal in glioblastoma. Sci Transl Med (2015) 7:304ra143. doi: 10.1126/scitranslmed.aac6762
170. De K, Grubb TM, Zalenski AA, Pfaff KE, Pal D, Majumder S, et al. Hyperphosphorylation of CDH1 in glioblastoma cancer stem cells attenuates APC/CCDH1 activity and pharmacologic inhibition of APC/CCDH1/CDC20 compromises viability. Mol Cancer Res (2019) 17:1519–30. doi: 10.1158/1541-7786.MCR-18-1361
171. Mao DD, Gujar AD, Mahlokozera T, Chen I, Pan Y, Luo J, et al. A CDC20-APC/SOX2 Signaling Axis Regulates Human Glioblastoma Stem-like Cells. Cell Rep (2015) 11:1809–21. doi: 10.1016/j.celrep.2015.05.027
172. Wang D, Berglund AE, Kenchappa RS, MacAulay RJ, Mulé JJ, Etame AB. BIRC3 is a biomarker of mesenchymal habitat of glioblastoma, and a mediator of survival adaptation in hypoxia-driven glioblastoma habitats. Sci Rep (2017) 7:9350. doi: 10.1038/s41598-017-09503-8
173. Liu Z, Oh SM, Okada M, Liu X, Cheng D, Peng J, et al. Human BRE1 is an E3 ubiquitin ligase for Ebp1 tumor suppressor. Mol Biol Cell (2009) 20:757–68. doi: 10.1091/mbc.e08-09-0983
174. Lee G-W, Park J.-W.J.B., Park SY, Seo J, Shin S-H, Park J.-W.J.B., et al. The E3 ligase C-CBL inhibits cancer cell migration by neddylating the proto-oncogene c-Src. Oncogene (2018) 37:5552–68. doi: 10.1038/s41388-018-0354-5
175. Seong MW, Park JH, Yoo HM, Yang SW, Oh KH, Ka SH, et al. c-Cbl regulates αPix-mediated cell migration and invasion. Biochem Biophys Res Commun (2014) 455:153–8. doi: 10.1016/j.bbrc.2014.10.129
176. Hou J, Deng Q, Zhou J, Zou J, Zhang Y, Tan P, et al. CSN6 controls the proliferation and metastasis of glioblastoma by CHIP-mediated degradation of EGFR. Oncogene (2017) 36:1134–44. doi: 10.1038/onc.2016.280
177. Xu T, Wang H, Jiang M, Yan Y, Li W, Xu H, et al. The E3 ubiquitin ligase CHIP/miR-92b/PTEN regulatory network contributes to tumorigenesis of glioblastoma. Am J Cancer Res (2017) 7:289–300.
178. Dong J, Wang X-Q, Yao J-J, Li G, Li X-G. Decreased CUL4B expression inhibits malignant proliferation of glioma in vitro and in vivo. Eur Rev Med Pharmacol Sci (2015) 19:1013–21.
179. Queisser MA, Dada LA, Deiss-Yehiely N, Angulo M, Zhou G, Kouri FM, et al. HOIL-1L Functions as the PKCζ Ubiquitin Ligase to Promote Lung Tumor Growth. Am J Respir Crit Care Med (2014) 190:688–98. doi: 10.1164/rccm.201403-0463OC
180. Yang W, Yang X, David G, Dorsey JF. Dissecting the complex regulation of Mad4 in glioblastoma multiforme cells. Cancer Biol Ther (2012) 13:1339–48. doi: 10.4161/cbt.21814
181. Yang W, Cooke M, Duckett C, Yang X, Dorsey JF. Distinctive effects of the cellular inhibitor of apoptosis protein c-IAP2 through stabilization by XIAP in glioblastoma multiforme cells. Cell Cycle (2014) 13:992–1005. doi: 10.4161/cc.27880
182. Abe T, Umeki I, Kanno S II, Inoue S II, Niihori T, Aoki Y. LZTR1 facilitates polyubiquitination and degradation of RAS-GTPases. Cell Death Differ (2020) 27:1023–35. doi: 10.1038/s41418-019-0395-5
183. Kondo S, Kondo Y, Hara H, Kaakaji R, Peterson J, Morimura T, et al. mdm2 gene mediates the expression of mdr1 gene and P-glycoprotein in a human glioblastoma cell line. Br J Cancer (1996) 74:1263–8. doi: 10.1038/bjc.1996.527
184. Biernat W, Kleihues P, Yonekawa Y, Ohgaki H. Amplification and Overexpression of MDM2 in Primary (de novo) Glioblastomas. J Neuropathol Exp Neurol (1997) 56:180–5. doi: 10.1097/00005072-199702000-00009
185. Joshi S, Singh AR, Durden DL. MDM2 Regulates Hypoxic Hypoxia-inducible Factor 1α Stability in an E3 Ligase, Proteasome, and PTEN-Phosphatidylinositol 3-Kinase-AKT-dependent Manner. J Biol Chem (2014) 289:22785–97. doi: 10.1074/jbc.M114.587493
186. Bufalieri F, Caimano M, Severini LL, Basili I, Paglia F, Sampirisi L, et al. The RNA-binding ubiquitin ligase MEX3A affects glioblastoma tumorigenesis by inducing ubiquitylation and degradation of RIG-I. Cancers (Basel) (2020) 12:321. doi: 10.3390/cancers12020321
187. Wald JH, Hatakeyama J, Printsev I, Cuevas A, Fry WHD, Saldana MJ, et al. Suppression of planar cell polarity signaling and migration in glioblastoma by Nrdp1-mediated Dvl polyubiquitination. Oncogene (2017) 36:5158–67. doi: 10.1038/onc.2017.126
188. Dasari VR, Velpula KK, Kaur K, Fassett D, Klopfenstein JD, Dinh DH, et al. Cord Blood Stem Cell-Mediated Induction of Apoptosis in Glioma Downregulates X-Linked Inhibitor of Apoptosis Protein (XIAP). PLoS One (2010) 5:e11813. doi: 10.1371/journal.pone.0011813
189. Bunda S, Heir P, Metcalf J, Li ASC, Agnihotri S, Pusch S, et al. CIC protein instability contributes to tumorigenesis in glioblastoma. Nat Commun (2019) 10:661. doi: 10.1038/s41467-018-08087-9
190. Shin J, Mishra V, Glasgow E, Zaidi S, Ohshiro K, Chitti B, et al. PRAJA is overexpressed in glioblastoma and contributes to neural precursor development. Genes Cancer (2017) 8:640–9. doi: 10.18632/genesandcancer.151
191. Lignitto L, Arcella A, Sepe M, Rinaldi L, Delle Donne R, Gallo A, et al. Proteolysis of MOB1 by the ubiquitin ligase praja2 attenuates Hippo signalling and supports glioblastoma growth. Nat Commun (2013) 4:1822. doi: 10.1038/ncomms2791
192. Xie C, Lu D, Xu M, Qu Z, Zhang W, Wang H. Knockdown of RAD18 inhibits glioblastoma development. J Cell Physiol (2019) 234:21100–12. doi: 10.1002/jcp.28713
193. Jia L, Soengas MS, Sun Y. ROC1/RBX1 E3 Ubiquitin Ligase Silencing Suppresses Tumor Cell Growth via Sequential Induction of G2-M Arrest, Apoptosis, and Senescence. Cancer Res (2009) 69:4974–82. doi: 10.1158/0008-5472.CAN-08-4671
194. Wang X, Bustos M, Zhang X, Ramos R, Tan C, Iida Y, et al. Downregulation of the Ubiquitin-E3 Ligase RNF123 Promotes Upregulation of the NF-κB1 Target SerpinE1 in Aggressive Glioblastoma Tumors. Cancers (Basel) (2020) 12:1081. doi: 10.3390/cancers12051081
195. Liu Y, Wang F, Liu Y, Yao Y, Lv X, Dong B, et al. RNF135, RING finger protein, promotes the proliferation of human glioblastoma cells in vivo and in vitro via the ERK pathway. Sci Rep (2016) 6:20642. doi: 10.1038/srep20642
196. Wu H, Li X, Feng M, Yao L, Deng Z, Zao G, et al. Downregulation of RNF138 inhibits cellular proliferation, migration, invasion and EMT in glioma cells via suppression of the Erk signaling pathway. Oncol Rep (2018) 40:3285–96. doi: 10.3892/or.2018.6744
197. Kim W, Youn H, Lee S, Kim E, Kim D, Sub Lee J, et al. RNF138-mediated ubiquitination of rpS3 is required for resistance of glioblastoma cells to radiation-induced apoptosis. Exp Mol Med (2018) 50:e434–4. doi: 10.1038/emm.2017.247
198. Jin X, Kim LJY, Wu Q, Wallace LC, Prager BC, Sanvoranart T, et al. Targeting Glioma Stem Cells through Combined BMI1 and EZH2 Inhibition. Nat Med (2017) 23:1352–61. doi: 10.1038/nm.4415
199. Du C, Hansen LJ, Singh SX, Wang F, Sun R, Moure CJ, et al. A PRMT5-RNF168-SMURF2 Axis Controls H2AX Proteostasis. Cell Rep (2019) 28:3199–211.e5. doi: 10.1016/j.celrep.2019.08.031
200. Guardavaccaro D, Frescas D, Dorrello NV, Peschiaroli A, Multani AS, Cardozo T, et al. Control of chromosome stability by the β-TrCP-REST-Mad2 axis. Nature (2008) 452:365–9. doi: 10.1038/nature06641
201. Westbrook TF, Hu G, Ang XL, Mulligan P, Pavlova NN, Liang A, et al. SCFβ-TRCP controls oncogenic transformation and neural differentiation through REST degradation. Nature (2008) 452:370–4. doi: 10.1038/nature06780
202. Warfel NA, Niederst M, Stevens MW, Brennan PM, Frame MC, Newton AC. Mislocalization of the E3 Ligase, β-Transducin Repeat-containing Protein 1 (β-TrCP1), in Glioblastoma Uncouples Negative Feedback between the Pleckstrin Homology Domain Leucine-rich Repeat Protein Phosphatase 1 (PHLPP1) and Akt. J Biol Chem (2011) 286:19777–88. doi: 10.1074/jbc.M111.237081
203. Zhang G, Zhu Q, Fu G, Hou J, Hu X, Cao J, et al. TRIP13 promotes the cell proliferation, migration and invasion of glioblastoma through the FBXW7/c-MYC axis. Br J Cancer (2019) 121:1069–78. doi: 10.1038/s41416-019-0633-0
204. Lin J, Ji A, Qiu G, Feng H, Li J, Li S, et al. FBW7 is associated with prognosis, inhibits malignancies and enhances temozolomide sensitivity in glioblastoma cells. Cancer Sci (2018) 109:1001–11. doi: 10.1111/cas.13528
205. Hagedorn M, Delugin M, Abraldes I, Allain N, Belaud-Rotureau M-A, Turmo M, et al. FBXW7/hCDC4 controls glioma cell proliferation in vitro and is a prognostic marker for survival in glioblastoma patients. Cell Div (2007) 2:9. doi: 10.1186/1747-1028-2-9
206. Chen Y, Henson ES, Xiao W, Shome E, Azad MB, Burton TR, et al. Bcl-2 family member Mcl-1 expression is reduced under hypoxia by the E3 ligase FBW7 contributing to BNIP3 induced cell death in glioma cells. Cancer Biol Ther (2016) 17:604–13. doi: 10.1080/15384047.2015.1095399
207. Fang X, Zhou W, Wu Q, Huang Z, Shi Y, Yang K, et al. Deubiquitinase USP13 Maintains Glioblastoma Stem Cells by Antagonizing FBXL14-mediated Myc Ubiquitination. J Exp Med (2017) 214:245–67. doi: 10.1084/jem.20151673
208. Khan M, Muzumdar D, Shiras A. Attenuation of Tumor Suppressive Function of FBXO16 Ubiquitin Ligase Activates Wnt Signaling In Glioblastoma. Neoplasia (2019) 21:106–16. doi: 10.1016/j.neo.2018.11.005
209. Wu J, Su HK, Yu ZH, Xi SY, Guo CC, Hu ZY, et al. Skp2 modulates proliferation, senescence and tumorigenesis of glioma. Cancer Cell Int (2020) 20:71. doi: 10.1186/s12935-020-1144-z
210. Galeano F, Rossetti C, Tomaselli S, Cifaldi L, Lezzerini M, Pezzullo M, et al. ADAR2-editing activity inhibits glioblastoma growth through the modulation of the CDC14B/Skp2/p21/p27 axis. Oncogene (2013) 32:998–1009. doi: 10.1038/onc.2012.125
211. Lee JJ, Lee JS, Cui MN, Yun HH, Kim HY, Lee SH, et al. BIS targeting induces cellular senescence through the regulation of 14-3-3 zeta/STAT3/SKP2/p27 in glioblastoma cells. Cell Death Dis (2014) 5:e1537. doi: 10.1038/cddis.2014.501
212. Mamillapalli R, Gavrilova N, Mihaylova VT, Tsvetkov LM, Wu H, Zhang H, et al. PTEN regulates the ubiquitin-dependent degradation of the CDK inhibitor p27KIP1 through the ubiquitin E3 ligase SCFSKP2. Curr Biol (2001) 11:263–7. doi: 10.1016/S0960-9822(01)00065-3
213. Zhang M, Huang N, Yang X, Luo J, Yan S, Xiao F, et al. A novel protein encoded by the circular form of the SHPRH gene suppresses glioma tumorigenesis. Oncogene (2018) 37:1805–14. doi: 10.1038/s41388-017-0019-9
214. He Y, Roos WP, Wu Q, Hofmann TG, Kaina B. The SIAH1–HIPK2–p53ser46 Damage Response Pathway is Involved in Temozolomide-Induced Glioblastoma Cell Death. Mol Cancer Res (2019) 17:1129–41. doi: 10.1158/1541-7786.MCR-18-1306
215. Yan S, Li A, Liu Y. CacyBP/SIP inhibits the migration and invasion behaviors of glioblastoma cells through activating Siah1 mediated ubiquitination and degradation of cytoplasmic p27. Cell Biol Int (2018) 42:216–26. doi: 10.1002/cbin.10889
216. Fortin Ensign SP, Mathews IT, Eschbacher JM, Loftus JC, Symons MH, Tran NL. The Src homology 3 domain-containing guanine nucleotide exchange factor is overexpressed in high-grade gliomas and promotes tumor necrosis factor-like weak inducer of apoptosis-fibroblast growth factor-inducible 14-induced cell migration and invasion via tumor necrosis factor receptor-associated factor 2. J Biol Chem (2013) 288:21887–97. doi: 10.1074/jbc.M113.468686
217. Kim YH, Joo HS, Kim D-S. Nitric oxide induction of IRE1-α-dependent CREB phosphorylation in human glioma cells. Nitric Oxide (2010) 23:112–20. doi: 10.1016/j.niox.2010.04.009
218. Zheng M, Morgan-Lappe SE, Yang J, Bockbrader KM, Pamarthy D, Thomas D, et al. Growth Inhibition and Radiosensitization of Glioblastoma and Lung Cancer Cells by Small Interfering RNA Silencing of Tumor Necrosis Factor Receptor-Associated Factor 2. Cancer Res (2008) 68:7570–8. doi: 10.1158/0008-5472.CAN-08-0632
219. Chen G, Kong J, Tucker-Burden C, Anand M, Rong Y, Rahman F, et al. Human Brat Ortholog TRIM3 Is a Tumor Suppressor That Regulates Asymmetric Cell Division in Glioblastoma. Cancer Res (2014) 74:4536–48. doi: 10.1158/0008-5472.CAN-13-3703
220. Venuto S, Castellana S, Monti M, Appolloni I, Fusilli C, Fusco C, et al. TRIM8-driven transcriptomic profile of neural stem cells identified glioma-related nodal genes and pathways. Biochim Biophys Acta - Gen Subj (2019) 1863:491–501. doi: 10.1016/j.bbagen.2018.12.001
221. Zhang C, Mukherjee S, Tucker-Burden C, Ross JL, Chau MJ, Kong J, et al. TRIM8 regulates stemness in glioblastoma through PIAS3-STAT3. Mol Oncol (2017) 11:280–94. doi: 10.1002/1878-0261.12034
222. Liu K, Zhang C, Li B, Xie W, Zhang J, Nie X, et al. Mutual Stabilization between TRIM9 Short Isoform and MKK6 Potentiates p38 Signaling to Synergistically Suppress Glioblastoma Progression. Cell Rep (2018) 23:838–51. doi: 10.1016/j.celrep.2018.03.096
223. Di K, Linskey ME, Bota DA. TRIM11 is overexpressed in high-grade gliomas and promotes proliferation, invasion, migration and glial tumor growth. Oncogene (2013) 32:5038–47. doi: 10.1038/onc.2012.531
224. Feng S, Cai X, Li Y, Jian X, Zhang L, Li B. Tripartite motif-containing 14 (TRIM14) promotes epithelial-mesenchymal transition via ZEB2 in glioblastoma cells. J Exp Clin Cancer Res (2019) 38:57. doi: 10.1186/s13046-019-1070-x
225. Xue J, Chen Y, Wu Y, Wang Z, Zhou A, Zhang S, et al. Tumour suppressor TRIM33 targets nuclear β-catenin degradation. Nat Commun (2015) 6:6156. doi: 10.1038/ncomms7156
226. Zhang J, Zhang C, Cui J, Ou J, Han J, Qin Y, et al. TRIM45 functions as a tumor suppressor in the brain via its E3 ligase activity by stabilizing p53 through K63-linked ubiquitination. Cell Death Dis (2017) 8:e2831–1. doi: 10.1038/cddis.2017.149
227. Zhang K-L, Zhou X, Han L, Chen L-Y, Chen L-C, Shi Z-D, et al. MicroRNA-566 activates EGFR signaling and its inhibition sensitizes glioblastoma cells to nimotuzumab. Mol Cancer (2014) 13:63. doi: 10.1186/1476-4598-13-63
228. Xiao B, Zhou X, Ye M, Lv S, Wu M, Liao C, et al. MicroRNA-566 modulates vascular endothelial growth factor by targeting Von Hippel-Landau in human glioblastoma in vitro and in vivo. Mol Med Rep (2016) 13:379–85. doi: 10.3892/mmr.2015.4537
229. Kanno H, Sato H, Yokoyama T-A, Yoshizumi T, Yamada S. The VHL tumor suppressor protein regulates tumorigenicity of U87-derived glioma stem-like cells by inhibiting the JAK/STAT signaling pathway. Int J Oncol (2013) 42:881–6. doi: 10.3892/ijo.2013.1773
230. Wang H, Jiang Z, Na M, Ge H, Tang C, Shen H, et al. PARK2 negatively regulates the metastasis and epithelial-mesenchymal transition of glioblastoma cells via ZEB1. Oncol Lett (2017) 14:2933–9. doi: 10.3892/ol.2017.6488
231. Scott TL, Wicker CA, Suganya R, Dhar B, Pittman T, Horbinski C, et al. Polyubiquitination of apurinic/apyrimidinic endonuclease 1 by Parkin. Mol Carcinog (2017) 56:325–36. doi: 10.1002/mc.22495
232. Oikonomaki M, Bady P, Hegi ME. Ubiquitin Specific Peptidase 15 (USP15) suppresses glioblastoma cell growth via stabilization of HECTD1 E3 ligase attenuating WNT pathway activity. Oncotarget (2017) 8:110490–502. doi: 10.18632/oncotarget.22798
233. Li H, Li J, Chen L, Qi S, Yu S, Weng Z, et al. HERC3-Mediated SMAD7 ubiquitination degradation promotes autophagy-induced EMT and chemoresistance in Glioblastoma. Clin Cancer Res (2019) 25:3602–16. doi: 10.1158/1078-0432.CCR-18-3791
234. Zhao X, D’Arca D, Lim WK, Brahmachary M, Carro MS, Ludwig T, et al. The N-Myc-DLL3 Cascade Is Suppressed by the Ubiquitin Ligase Huwe1 to Inhibit Proliferation and Promote Neurogenesis in the Developing Brain. Dev Cell (2009) 17:210–21. doi: 10.1016/j.devcel.2009.07.009
235. Su C, Wang T, Zhao J, Cheng J, Hou J. Meta-analysis of gene expression alterations and clinical significance of the HECT domain-containing ubiquitin ligase HUWE1 in cancer. Oncol Lett (2019) 18:2292–303. doi: 10.3892/ol.2019.10579
236. Panner A, Crane CA, Changjiang W, Feletti A, Parsa AT, Pieper RO, et al. A Novel PTEN-dependent Link to Ubiquitination Controls FLIPS Stability and TRAIL Sensitivity in Glioblastoma Multiforme. Cancer Res (2009) 69:7911–6. doi: 10.1158/0008-5472.CAN-09-1287
237. Dai B, Pieper RO, Li D, Wei P, Liu M, Woo SY, et al. FoxM1B Regulates NEDD4-1 Expression, Leading to Cellular Transformation and Full Malignant Phenotype in Immortalized Human Astrocytes. Cancer Res (2010) 70:2951–61. doi: 10.1158/0008-5472.CAN-09-3909
238. Chang H, Zhang J, Miao Z, Ding Y, Xu X, Zhao X, et al. Suppression of the Smurf1 Expression Inhibits Tumor Progression in Gliomas. Cell Mol Neurobiol (2018) 38:421–30. doi: 10.1007/s10571-017-0485-1
239. Eichhorn PJA, Rodón L, Gonzàlez-Juncà A, Dirac A, Gili M, Martínez-Sáez E, et al. USP15 stabilizes TGF-β receptor I and promotes oncogenesis through the activation of TGF-β signaling in glioblastoma. Nat Med (2012) 18:429–35. doi: 10.1038/nm.2619
240. Braganza A, Li J, Zeng X, Yates NA, Dey NB, Andrews J, et al. UBE3B is a calmodulin-regulated, mitochondrion-associated E3 ubiquitin ligase. J Biol Chem (2017) 292:2470–84. doi: 10.1074/jbc.M116.766824
241. Svilar D, Dyavaiah M, Brown AR, Tang JB, Li J, McDonald PR, et al. Alkylation sensitivity screens reveal a conserved cross-species functionome. Mol Cancer Res (2012) 10:1580–96. doi: 10.1158/1541-7786.MCR-12-0168
242. Pan S-J, Zhan S-K, Ji W-Z, Pan Y-X, Liu W, Li D-Y, et al. Ubiquitin-protein Ligase E3C Promotes Glioma Progression by Mediating the Ubiquitination and Degrading of Annexin A7. Sci Rep (2015) 5:11066. doi: 10.1038/srep11066
243. Zhao Y, He J, Li Y, Lv S, Cui H. NUSAP1 potentiates chemoresistance in glioblastoma through its SAP domain to stabilize ATR. Signal Transduct Target Ther (2020) 5:44. doi: 10.1038/s41392-020-0137-7
244. Szymura SJ, Bernal GM, Wu L, Zhang Z, Crawley CD, Voce DJ, et al. DDX39B interacts with the pattern recognition receptor pathway to inhibit NF-κB and sensitize to alkylating chemotherapy. BMC Biol (2020) 18:32. doi: 10.1186/s12915-020-0764-z
245. Soares IN, Caetano FA, Pinder J, Rodrigues BR, Beraldo FH, Ostapchenko VG, et al. Regulation of Stress-Inducible Phosphoprotein 1 Nuclear Retention by Protein Inhibitor of Activated STAT PIAS1. Mol Cell Proteomics (2013) 12:3253–70. doi: 10.1074/mcp.M113.031005
246. Rahme GJ, Zhang Z, Young AL, Cheng C, Bivona EJ, Fiering SN, et al. PDGF engages an E2F-USP1 signaling pathway to support ID2-mediated survival of proneural glioma cells. Cancer Res (2016) 76:2964–76. doi: 10.1158/0008-5472.CAN-15-2157
247. Lee J-K, Chang N, Yoon Y, Yang H, Cho H, Kim E, et al. USP1 targeting impedes GBM growth by inhibiting stem cell maintenance and radioresistance. Neuro Oncol (2016) 18:37–47. doi: 10.1093/neuonc/nov091
248. Ma L, Lin K, Chang G, Chen Y, Yue C, Guo Q, et al. Aberrant activation of β-catenin signaling drives glioma tumorigenesis via USP1-mediated stabilization of EZH2. Cancer Res (2019) 79:72–85. doi: 10.1158/0008-5472.CAN-18-1304
249. Wang C-L, Wang J-Y, Liu Z-Y, Ma X-M, Wang X-W, Jin H, et al. Ubiquitin-specific protease 2a stabilizes MDM4 and facilitates the p53-mediated intrinsic apoptotic pathway in glioblastoma. Carcinogenesis (2014) 35:1500–9. doi: 10.1093/carcin/bgu015
250. Tu Y, Chen Z, Zhao P, Sun G, Bao Z, Chao H, et al. Smoothened Promotes Glioblastoma Radiation Resistance Via Activating USP3-Mediated Claspin Deubiquitination. Clin Cancer Res (2020) 26:1749–62. doi: 10.1158/1078-0432.CCR-19-1515
251. Fan L, Chen Z, Wu X, Cai X, Feng S, Lu J, et al. Ubiquitin-specific protease 3 promotes glioblastoma cell invasion and epithelial-mesenchymal transition via stabilizing snail. Mol Cancer Res (2019) 17:1975–84. doi: 10.1158/1541-7786.MCR-19-0197
252. Qin N, Han F, Li L, Ge Y, Lin W, Wang J, et al. Deubiquitinating enzyme 4 facilitates chemoresistance in glioblastoma by inhibiting P53 activity. Oncol Lett (2019) 17:958–64. doi: 10.3892/ol.2018.9654
253. Zhou Y, Liang P, Ji W, Yu Z, Chen H, Jiang L. Ubiquitin-specific protease 4 promotes glioblastoma multiforme via activating ERK pathway. Onco Targets Ther (2019) 12:1825–39. doi: 10.2147/OTT.S176582
254. Izaguirre DI, Zhu W, Hai T, Cheung HC, Krahe R, Cote GJ. PTBP1-dependent regulation of USP5 alternative RNA splicing plays a role in glioblastoma tumorigenesis. Mol Carcinog (2012) 51:895–906. doi: 10.1002/mc.20859
255. Huang Z, Wu Q, Guryanova OA, Cheng L, Shou W, Rich JN, et al. Deubiquitylase HAUSP stabilizes REST and promotes maintenance of neural progenitor cells. Nat Cell Biol (2011) 13:142–52. doi: 10.1038/ncb2153
256. Yi L, Cui Y, Xu Q, Jiang Y. Stabilization of LSD1 by deubiquitinating enzyme USP7 promotes glioblastoma cell tumorigenesis and metastasis through suppression of the p53 signaling pathway. Oncol Rep (2016) 36:2935–45. doi: 10.3892/or.2016.5099
257. Panner A, Crane CA, Weng C, Feletti A, Fang S, Parsa AT, et al. Ubiquitin-specific protease 8 links the PTEN-Akt-AIP4 pathway to the control of FLIPS stability and TRAIL sensitivity in glioblastoma multiforme. Cancer Res (2010) 70:5046–53. doi: 10.1158/0008-5472.CAN-09-3979
258. MacLeod G, Bozek DA, Rajakulendran N, Monteiro V, Ahmadi M, Steinhart Z, et al. Genome-Wide CRISPR-Cas9 Screens Expose Genetic Vulnerabilities and Mechanisms of Temozolomide Sensitivity in Glioblastoma Stem Cells. Cell Rep (2019) 27:971–86. doi: 10.1016/j.celrep.2019.03.047
259. Cox JL, Wilder PJ, Gilmore JM, Wuebben EL, Washburn MP, Rizzino A. The SOX2-Interactome in Brain Cancer Cells Identifies the Requirement of MSI2 and USP9X for the Growth of Brain Tumor Cells. PLoS One (2013) 8:e62857. doi: 10.1371/journal.pone.0062857
260. Wolfsperger F, Hogh-Binder SA, Schittenhelm J, Psaras T, Ritter V, Bornes L, et al. Deubiquitylating enzyme USP9x regulates radiosensitivity in glioblastoma cells by Mcl-1-dependent and -independent mechanisms. Cell Death Dis (2016) 7:e2039. doi: 10.1038/cddis.2015.405
261. Karpel-Massler G, Banu MA, Shu C, Halatsch ME, Westhoff MA, Bruce JN, et al. Inhibition of deubiquitinases primes glioblastoma cells to apoptosis in vitro and in vivo. Oncotarget (2016) 7:12791–805. doi: 10.18632/oncotarget.7302
262. Chen Z, Wang HW, Wang S, Fan L, Feng S, Cai X, et al. USP9X deubiquitinates ALDH1A3 and maintains mesenchymal identity in glioblastoma stem cells. J Clin Invest (2019) 129:2043–55. doi: 10.1172/JCI126414
263. Grunda JM, Nabors LB, Palmer CA, Chhieng DC, Steg A, Mikkelsen T, et al. Increased expression of thymidylate synthetase (TS), ubiquitin specific protease 10 (USP10) and survivin is associated with poor survival in glioblastoma multiforme (GBM). J Neurooncol (2006) 80:261–74. doi: 10.1007/s11060-006-9191-4
264. Zhao H, Wang Y, Yang C, Zhou J, Wang L, Yi K, et al. EGFR-vIII downregulated H2AZK4/7AC though the PI3K/AKT-HDAC2 axis to regulate cell cycle progression. Clin Transl Med (2020) 9:10. doi: 10.1186/s40169-020-0260-7
265. Wu H-C, Lin Y-C, Liu C-H, Chung H-C, Wang Y-T, Lin Y-W, et al. USP11 regulates PML stability to control Notch-induced malignancy in brain tumours. Nat Commun (2014) 5:3214. doi: 10.1038/ncomms4214
266. Xu K, Pei H, Zhang Z, Wang H, Li L, Xia Q. Ubiquitin-specific protease 15 promotes tumor cell invasion and proliferation in glioblastoma. Oncol Lett (2018) 15:3846–51. doi: 10.3892/ol.2018.7747
267. Sgorbissa A, Tomasella A, Potu H, Manini I, Brancolini C. Type I IFNs signaling and apoptosis resistance in glioblastoma cells. Apoptosis (2011) 16:1229–44. doi: 10.1007/s10495-011-0639-4
268. Zhou A, Lin K, Zhang S, Chen Y, Zhang N, Xue J, et al. Nuclear GSK3β promotes tumorigenesis by phosphorylating KDM1A and inducing its deubiquitylation by USP22. Nat Cell Biol (2016) 18:954–66. doi: 10.1038/ncb3396
269. Wang Z, Song Q, Xue J, Zhao Y, Qin S. Ubiquitin-specific protease 28 is overexpressed in human glioblastomas and contributes to glioma tumorigenicity by regulating MYC expression. Exp Biol Med (2016) 241:255–64. doi: 10.1177/1535370215595468
270. Zhou A, Lin K, Zhang S, Ma L, Xue J, Morris S-A, et al. Gli1-induced deubiquitinase USP48 aids glioblastoma tumorigenesis by stabilizing Gli1. EMBO Rep (2017) 18:1318–30. doi: 10.15252/embr.201643124
271. Ding Z, Liu Y, Yao L, Wang D, Zhang J, Cui G, et al. Spy1 induces de-ubiquitinating of RIP1 arrest and confers glioblastoma’s resistance to tumor necrosis factor (TNF-α)-induced apoptosis through suppressing the association of CLIPR-59 and CYLD. Cell Cycle (2015) 14:2149–59. doi: 10.1080/15384101.2015.1041688
272. Guo J, Shinriki S, Su Y, Nakamura T, Hayashi M, Tsuda Y, et al. Hypoxia suppresses cylindromatosis (CYLD) expression to promote inflammation in glioblastoma: Possible link to acquired resistance to anti-VEGF therapy. Oncotarget (2014) 5:6353–64. doi: 10.18632/oncotarget.2216
273. Hjelmeland AB, Wu Q, Wickman S, Eyler C, Heddleston J, Shi Q, et al. Targeting A20 decreases glioma stem cell survival and tumor growth. PLoS Biol (2010) 8:e1000319. doi: 10.1371/journal.pbio.1000319
274. Chai KM, Wang C-Y, Liaw H-J, Fang K-M, Yang C-S, Tzeng S-F. Downregulation of BRCA1-BRCA2-containing complex subunit 3 sensitizes glioma cells to temozolomide. Oncotarget (2014) 5:10901–15. doi: 10.18632/oncotarget.2543
275. Liu F, Hon GC, Villa GR, Turner KM, Ikegami S, Yang H, et al. EGFR Mutation Promotes Glioblastoma through Epigenome and Transcription Factor Network Remodeling. Mol Cell (2015) 60:307–18. doi: 10.1016/j.molcel.2015.09.002
276. Ermoian RP, Furniss CS, Lamborn KR, Basila D, Berger MS, Gottschalk AR, et al. Dysregulation of PTEN and protein kinase B is associated with glioma histology and patient survival. Clin Cancer Res (2002) 8:1100–6.
277. Yang JM, Schiapparelli P, Nguyen HN, Igarashi A, Zhang Q, Abbadi S, et al. Characterization of PTEN mutations in brain cancer reveals that pten mono-ubiquitination promotes protein stability and nuclear localization. Oncogene (2017) 36:3673–85. doi: 10.1038/onc.2016.493
278. Zhou P, Fernandes N, Dodge IL, Lakku Reddi A, Rao N, Safran H, et al. ErbB2 degradation mediated by the co-chaperone protein CHIP. J Biol Chem (2003) 278:13829–37. doi: 10.1074/jbc.M209640200
279. Cope GA, Deshaies RJ. COP9 signalosome: A multifunctional regulator of SCF and other cullin-based ubiquitin ligases. Cell (2003) 114:663–71. doi: 10.1016/S0092-8674(03)00722-0
280. Zhao R, Yeung SCJ, Chen J, Iwakuma T, Su CH, Chen B, et al. Subunit 6 of the COP9 signalosome promotes tumorigenesis in mice through stabilization of MDM2 and is upregulated in human cancers. J Clin Invest (2011) 121:851–65. doi: 10.1172/JCI44111
281. Chen J, Shin JH, Zhao R, Phan L, Wang H, Xue Y, et al. CSN6 drives carcinogenesis by positively regulating Myc stability. Nat Commun (2014) 5:1–15. doi: 10.1038/ncomms6384
282. Bruna A, Darken RS, Rojo F, Ocaña A, Peñuelas S, Arias A, et al. High TGFβ-Smad Activity Confers Poor Prognosis in Glioma Patients and Promotes Cell Proliferation Depending on the Methylation of the PDGF-B Gene. Cancer Cell (2007) 11:147–60. doi: 10.1016/j.ccr.2006.11.023
283. Colak S, ten Dijke P. Targeting TGF-β Signaling in Cancer. Trends Cancer (2017) 3:56–71. doi: 10.1016/j.trecan.2016.11.008
284. Kavsak P, Rasmussen RK, Causing CG, Bonni S, Zhu H, Thomsen GH, et al. Smad7 binds to Smurf2 to form an E3 ubiquitin ligase that targets the TGFβ receptor for degradation. Mol Cell (2000) 6:1365–75. doi: 10.1016/S1097-2765(00)00134-9
286. Hombach-Klonisch S, Mehrpour M, Shojaei S, Harlos C, Pitz M, Hamai A, et al. Glioblastoma and chemoresistance to alkylating agents: Involvement of apoptosis, autophagy, and unfolded protein response. Pharmacol Ther (2018) 184:13–41. doi: 10.1016/j.pharmthera.2017.10.017
287. Sui X, Chen R, Wang Z, Huang Z, Kong N, Zhang M, et al. Autophagy and chemotherapy resistance: A promising therapeutic target for cancer treatment. Cell Death Dis (2013) 4:e838. doi: 10.1038/cddis.2013.350
288. Lu Y, Xiao L, Liu Y, Wang H, Li H, Zhou Q, et al. MIR517C inhibits autophagy and the epithelialto- mesenchymal (-like) transition phenotype in human glioblastoma through KPNA2-dependent disruption of TP53 nuclear translocation. Autophagy (2015) 11:2213–32. doi: 10.1080/15548627.2015.1108507
289. Zhang S, Fei T, Zhang L, Zhang R, Chen F, Ning Y, et al. Smad7 Antagonizes Transforming Growth Factor Signaling in the Nucleus by Interfering with Functional Smad-DNA Complex Formation. Mol Cell Biol (2007) 27:4488–99. doi: 10.1128/MCB.01636-06
290. Ebisawa T, Fukuchi M, Murakami G, Chiba T, Tanaka K, Imamura T, et al. Smurf1 Interacts with Transforming Growth Factor-β Type I Receptor through Smad7 and Induces Receptor Degradation. J Biol Chem (2001) 276:12477–80. doi: 10.1074/jbc.C100008200
291. Yuan T, Chen Z, Yan F, Qian M, Luo H, Ye S, et al. Deubiquitinating enzyme USP10 promotes hepatocellular carcinoma metastasis through deubiquitinating and stabilizing Smad4 protein. Mol Oncol (2020) 14:197–210. doi: 10.1002/1878-0261.12596
292. Gao S, Alarcón C, Sapkota G, Rahman S, Chen PY, Goerner N, et al. Ubiquitin Ligase Nedd4L Targets Activated Smad2/3 to Limit TGF-β Signaling. Mol Cell (2009) 36:457–68. doi: 10.1016/j.molcel.2009.09.043
293. Derynck R, Budi EH. Specificity, versatility, and control of TGF-b family signaling. Sci Signal (2019) 12:eaav5183. doi: 10.1126/scisignal.aav5183
294. Zhang Y, Dube C, Gibert M, Cruickshanks N, Wang B, Coughlan M, et al. The p53 pathway in glioblastoma. Cancers (Basel) (2018) 10:297. doi: 10.3390/cancers10090297
295. Piette J, Neel H, Maréchal V. Mdm2: Keeping p53 under control. Oncogene (1997) 15:1001–10. doi: 10.1038/sj.onc.1201432
296. Iyappan S, Wollscheid HP, Rojas-Fernandez A, Marquardt A, Tang HC, Singh RK, et al. Turning the RING domain protein MdmX into an active ubiquitin-protein ligase. J Biol Chem (2010) 285:33065–72. doi: 10.1074/jbc.M110.115113
297. Huang Q, Chen L, Yang L, Xie X, Gan L, Cleveland JL, et al. MDMX acidic domain inhibits p53 DNA binding in vivo and regulates tumorigenesis. Proc Natl Acad Sci USA (2018) 115:E3368–77. doi: 10.1073/pnas.1719090115
298. Popowicz GM, Czarna A, Holak TA. Structure of the human Mdmx protein bound to the p53 tumor suppressor transactivation domain. Cell Cycle (2008) 7:2441–3. doi: 10.4161/cc.6365
299. Linke K, Mace PD, Smith CA, Vaux DL, Silke J, Day CL, et al. Structure of the MDM2/MDMX RING domain heterodimer reveals dimerization is required for their ubiquitylation in trans. Cell Death Differ (2008) 15:841–8. doi: 10.1038/sj.cdd.4402309
300. Zhang X, Berger FG, Yang J, Lu X. USP4 inhibits p53 through deubiquitinating and stabilizing ARF-BP1. EMBO J (2011) 30:2177–89. doi: 10.1038/emboj.2011.125
301. Sato T, Takahashi H, Hatakeyama S, Iguchi A, Ariga T. The TRIM-FLMN protein TRIM45 directly interacts with RACK1 and negatively regulates PKC-mediated signaling pathway. Oncogene (2015) 34:1280–91. doi: 10.1038/onc.2014.68
302. Shibata M, Sato T, Nukiwa R, Ariga T, Hatakeyama S. TRIM45 negatively regulates NF-κB-mediated transcription and suppresses cell proliferation. Biochem Biophys Res Commun (2012) 423:104–9. doi: 10.1016/j.bbrc.2012.05.090
303. Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med (1997) 3:730–7. doi: 10.1038/nm0797-730
304. Singh SK, Clarke ID, Terasaki M, Bonn VE, Hawkins C, Squire J, et al. Identification of cancer stem cell in human brain tumors. Cancer Res (2003) 63:5821–8.
305. Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature (2006) 444:756–60. doi: 10.1038/nature05236
306. Liu G, Yuan X, Zeng Z, Tunici P, Ng H, Abdulkadir IR, et al. Analysis of gene expression and chemoresistance of CD133+ cancer stem cells in glioblastoma. Mol Cancer (2006) 5:67. doi: 10.1186/1476-4598-5-67
307. Chen J, Li Y, Yu T-S, McKay RM, Burns DK, Kernie SG, et al. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature (2012) 488:522–6. doi: 10.1038/nature11287
308. Joo KM, Kim J, Jin J, Kim M, Seol HJ, Muradov J, et al. Patient-Specific Orthotopic Glioblastoma Xenograft Models Recapitulate the Histopathology and Biology of Human Glioblastomas In Situ. Cell Rep (2013) 3:260–73. doi: 10.1016/j.celrep.2012.12.013
309. Lan X, Jörg DJ, Cavalli FMG, Richards LM, Nguyen LV, Vanner RJ, et al. Fate mapping of human glioblastoma reveals an invariant stem cell hierarchy. Nature (2017) 549:227–32. doi: 10.1038/nature23666
310. Safa AR, Saadatzadeh MR, Cohen-Gadol AA, Pollok KE, Bijangi-Vishehsaraei K. Glioblastoma stem cells (GSCs) epigenetic plasticity and interconversion between differentiated non-GSCs and GSCs. Genes Dis (2015) 2:152–63. doi: 10.1016/j.gendis.2015.02.001
311. Dirkse A, Golebiewska A, Buder T, Nazarov PV, Muller A, Poovathingal S, et al. Stem cell-associated heterogeneity in Glioblastoma results from intrinsic tumor plasticity shaped by the microenvironment. Nat Commun (2019) 10:1787. doi: 10.1038/s41467-019-09853-z
312. Chaffer CL, Brueckmann I, Scheel C, Kaestli AJ, Wiggins PA, Rodrigues LO, et al. Normal and neoplastic nonstem cells can spontaneously convert to a stem-like state. Proc Natl Acad Sci USA (2011) 108:7950–5. doi: 10.1073/pnas.1102454108
313. Popov N, Wanzel M, Madiredjo M, Zhang D, Beijersbergen R, Bernards R, et al. The ubiquitin-specific protease USP28 is required for MYC stability. Nat Cell Biol (2007) 9:765–74. doi: 10.1038/ncb1601
314. Harris RE, Pargett M, Sutcliffe C, Umulis D, Ashe HL. Brat Promotes Stem Cell Differentiation via Control of a Bistable Switch that Restricts BMP Signaling. Dev Cell (2011) 20:72–83. doi: 10.1016/j.devcel.2010.11.019
315. Ballas N, Grunseich C, Lu DD, Speh JC, Mandel G. REST and its corepressors mediate plasticity of neuronal gene chromatin throughout neurogenesis. Cell (2005) 121:645–57. doi: 10.1016/j.cell.2005.03.013
316. Fuchs SY, Spiegelman VS, Kumar KGS. The many faces of β-TrCP E3 ubiquitin ligases: Reflections in the magic mirror of cancer. Oncogene (2004) 23:2028–36. doi: 10.1038/sj.onc.1207389
317. Lee Y, Lee JK, Ahn SH, Lee J, Nam DH. WNT signaling in glioblastoma and therapeutic opportunities. Lab Invest (2016) 96:137–50. doi: 10.1038/labinvest.2015.140
318. Carruthers RD, Ahmed SU, Ramachandran S, Strathdee K, Kurian KM, Hedley A, et al. Replication stress drives constitutive activation of the DNA damage response and radioresistance in glioblastoma stem-like cells. Cancer Res (2018) 78:5060–71. doi: 10.1158/0008-5472.CAN-18-0569
319. Zhang J, Yang PL, Gray NS. Targeting cancer with small molecule kinase inhibitors. Nat Rev Cancer (2009) 9:28–39. doi: 10.1038/nrc2559
320. Goldstein DM, Gray NS, Zarrinkar PP. High-throughput kinase profiling as a platform for drug discovery. Nat Rev Drug Discov (2008) 7:391–7. doi: 10.1038/nrd2541
321. King RW, Deshaies RJ, Peters J-M, Kirschner MW. How Proteolysis Drives the Cell Cycle. Science (1996) 274:1652–9. doi: 10.1126/science.274.5293.1652
322. Adams J. The proteasome: a suitable antineoplastic target. Nat Rev Cancer (2004) 4:349–60. doi: 10.1038/nrc1361
323. Kisselev AF, Goldberg AL. Proteasome inhibitors: from research tools to drug candidates. Chem Biol (2001) 8:739–58. doi: 10.1016/S1074-5521(01)00056-4
324. Rock KL, Gramm C, Rothstein L, Clark K, Stein R, Dick L, et al. Inhibitors of the proteasome block the degradation of most cell proteins and the generation of peptides presented on MHC class I molecules. Cell (1994) 78:761–71. doi: 10.1016/S0092-8674(94)90462-6
325. Groll M, Ditzel L, Löwe J, Stock D, Bochtler M, Bartunik HD, et al. Structure of 20S proteasome from yeast at 2.4Å resolution. Nature (1997) 386:463–71. doi: 10.1038/386463a0
326. Löwe J, Stock D, Jap B, Zwickl P, Baumeister W, Huber R. Crystal structure of the 20S proteasome from the archaeon T. acidophilum at 3.4 A resolution. Science (1995) 268:533–9. doi: 10.1126/science.7725097
327. Seemuller E, Lupas A, Stock D, Lowe J, Huber R, Baumeister W. Proteasome from Thermoplasma acidophilum: a threonine protease. Science (1995) 268:579–82. doi: 10.1126/science.7725107
328. Adams J, Palombella VJ, Sausville EA, Johnson J, Destree A, Lazarus DD, et al. Proteasome inhibitors: a novel class of potent and effective antitumor agents. Cancer Res (1999) 59:2615–22.
329. Hideshima T, Mitsiades C, Akiyama M, Hayashi T, Chauhan D, Richardson P, et al. Molecular mechanisms mediating antimyeloma activity of proteasome inhibitor PS-341. Blood (2003) 101:1530–4. doi: 10.1182/blood-2002-08-2543
330. Ma MH, Yang HH, Parker K, Manyak S, Friedman JM, Altamirano C, et al. The proteasome inhibitor PS-341 markedly enhances sensitivity of multiple myeloma tumor cells to chemotherapeutic agents. Clin Cancer Res (2003) 9:1136–44.
331. Orlowski RZ, Stinchcombe TE, Mitchell BS, Shea TC, Baldwin AS, Stahl S, et al. Phase I Trial of the Proteasome Inhibitor PS-341 in Patients With Refractory Hematologic Malignancies. J Clin Oncol (2002) 20:4420–7. doi: 10.1200/JCO.2002.01.133
332. Aghajanian C, Soignet S, Dizon DS, Pien CS, Adams J, Elliott PJ, et al. A phase I trial of the novel proteasome inhibitor PS341 in advanced solid tumor malignancies. Clin Cancer Res (2002) 8:2505–11.
333. Moreau P, Masszi T, Grzasko N, Bahlis NJ, Hansson M, Pour L, et al. Oral Ixazomib, Lenalidomide, and Dexamethasone for Multiple Myeloma. N Engl J Med (2016) 374:1621–34. doi: 10.1056/NEJMoa1516282
334. Wang W, Cho HY, Rosenstein-Sisson R, Marín Ramos NI, Price R, Hurth K, et al. Intratumoral delivery of bortezomib: Impact on survival in an intracranial glioma tumor model. J Neurosurg (2018) 128:695–700. doi: 10.3171/2016.11.JNS161212
335. Ventola CL. Progress in nanomedicine: Approved and investigational nanodrugs. Pharmacol Ther (2017) 42:742–55.
336. Unsoy G, Yalcin S, Khodadust R, Mutlu P, Onguru O, Gunduz U. Chitosan magnetic nanoparticles for pH responsive Bortezomib release in cancer therapy. BioMed Pharmacother (2014) 68:641–8. doi: 10.1016/j.biopha.2014.04.003
337. Hu X, Chai Z, Lu L, Ruan H, Wang R, Zhan C, et al. Bortezomib Dendrimer Prodrug-Based Nanoparticle System. Adv Funct Mater (2019) 29:1807941. doi: 10.1002/adfm.201807941
338. de la Puente P, Luderer MJ, Federico C, Jin A, Gilson RC, Egbulefu C, et al. Enhancing proteasome-inhibitory activity and specificity of bortezomib by CD38 targeted nanoparticles in multiple myeloma. J Control Release (2018) 270:158–76. doi: 10.1016/j.jconrel.2017.11.045
339. Jin J, Li X, Gygi SP, Harper JW. Dual E1 activation systems for ubiquitin differentially regulate E2 enzyme charging. Nature (2007) 447:1135–8. doi: 10.1038/nature05902
340. Xu GW, Ali M, Wood TE, Wong D, Maclean N, Wang X, et al. The ubiquitin-activating enzyme E1 as a therapeutic target for the treatment of leukemia and multiple myeloma. Blood (2010) 115:2251–9. doi: 10.1182/blood-2009-07-231191
341. Yang Y, Kitagaki J, Dai R-M, Tsai YC, Lorick KL, Ludwig RL, et al. Inhibitors of Ubiquitin-Activating Enzyme (E1), a New Class of Potential Cancer Therapeutics. Cancer Res (2007) 67:9472–81. doi: 10.1158/0008-5472.CAN-07-0568
342. Soucy TA, Smith PG, Milhollen MA, Berger AJ, Gavin JM, Adhikari S, et al. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature (2009) 458:732–6. doi: 10.1038/nature07884
343. Brownell JE, Sintchak MD, Gavin JM, Liao H, Bruzzese FJ, Bump NJ, et al. Substrate-Assisted Inhibition of Ubiquitin-like Protein-Activating Enzymes: The NEDD8 E1 Inhibitor MLN4924 Forms a NEDD8-AMP Mimetic In Situ. Mol Cell (2010) 37:102–11. doi: 10.1016/j.molcel.2009.12.024
344. Ceccarelli DF, Tang X, Pelletier B, Orlicky S, Xie W, Plantevin V, et al. An Allosteric Inhibitor of the Human Cdc34 Ubiquitin-Conjugating Enzyme. Cell (2011) 145:1075–87. doi: 10.1016/j.cell.2011.05.039
345. Strickson S, Campbell DG, Emmerich CH, Knebel A, Plater L, Ritorto MS, et al. The anti-inflammatory drug BAY 11-7082 suppresses the MyD88-dependent signalling network by targeting the ubiquitin system. Biochem J (2013) 451:427–37. doi: 10.1042/BJ20121651
346. Pulvino M, Liang Y, Oleksyn D, DeRan M, Van Pelt E, Shapiro J, et al. Inhibition of proliferation and survival of diffuse large B-cell lymphoma cells by a small-molecule inhibitor of the ubiquitin-conjugating enzyme Ubc13-Uev1A. Blood (2012) 120:1668–77. doi: 10.1182/blood-2012-02-406074
347. Souers AJ, Leverson JD, Boghaert ER, Ackler SL, Catron ND, Chen J, et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat Med (2013) 19:202–8. doi: 10.1038/nm.3048
348. Birkinshaw RW, Gong Jn, Luo CS, Lio D, White CA, Anderson MA, et al. Structures of BCL-2 in complex with venetoclax reveal the molecular basis of resistance mutations. Nat Commun (2019) 10:1–10. doi: 10.1038/s41467-019-10363-1
349. Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA, et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature (2005) 435:677–81. doi: 10.1038/nature03579
350. Wu L, Grigoryan AV, Li Y, Hao B, Pagano M, Cardozo TJ. Specific small molecule inhibitors of skp2-mediated p27 degradation. Chem Biol (2012) 19:1515–24. doi: 10.1016/j.chembiol.2012.09.015
351. Chan C-H, Morrow JK, Li C-F, Gao Y, Jin G, Moten A, et al. Pharmacological Inactivation of Skp2 SCF Ubiquitin Ligase Restricts Cancer Stem Cell Traits and Cancer Progression. Cell (2013) 154:556–68. doi: 10.1016/j.cell.2013.06.048
352. Haupt Y, Maya R, Kazaz A, Oren M. Mdm2 promotes the rapid degradation of p53. Nature (1997) 387:296–9. doi: 10.1038/387296a0
353. Kussie PH, Gorina S, Marechal V, Elenbaas B, Moreau J, Levine AJ, et al. Structure of the MDM2 oncoprotein bound to the p53 tumor suppressor transactivation domain. Science (1996) 274:948–53. doi: 10.1126/science.274.5289.948
354. Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, et al. In Vivo Activation of the p53 Pathway by Small-Molecule Antagonists of MDM2. Science (2004) 303:844–8. doi: 10.1126/science.1092472
355. Lukashchuk N, Vousden KH. Ubiquitination and Degradation of Mutant p53. Mol Cell Biol (2007) 27:8284–95. doi: 10.1128/MCB.00050-07
356. Her NG, Oh JW, Oh YJ, Han S, Cho HJ, Lee Y, et al. Potent effect of the MDM2 inhibitor AMG232 on suppression of glioblastoma stem cells. Cell Death Dis (2018) 9:792. doi: 10.1038/s41419-018-0825-1
357. Burgess A, Chia KM, Haupt S, Thomas D, Haupt Y, Lim E. Clinical Overview of MDM2/X-Targeted Therapies. Front Oncol (2016) 6:7. doi: 10.3389/fonc.2016.00007
358. Derakhshan A, Chen Z, Van Waes C. Therapeutic small molecules target inhibitor of apoptosis proteins in cancers with deregulation of extrinsic and intrinsic cell death pathways. Clin Cancer Res (2017) 23:1379–87. doi: 10.1158/1078-0432.CCR-16-2172
359. Birnbaum MJ, Clem RJ, Miller LK. An apoptosis-inhibiting gene from a nuclear polyhedrosis virus encoding a polypeptide with Cys/His sequence motifs. J Virol (1994) 68:2521–8. doi: 10.1128/JVI.68.4.2521-2528.1994
360. Silke J, Meier P. Inhibitor of Apoptosis (IAP) Proteins–Modulators of Cell Death and Inflammation. Cold Spring Harb Perspect Biol (2013) 5:a008730. doi: 10.1101/cshperspect.a008730
361. Verhagen AM, Ekert PG, Pakusch M, Silke J, Connolly LM, Reid GE, et al. Identification of DIABLO, a mammalian protein that promotes apoptosis by binding to and antagonizing IAP proteins. Cell (2000) 102:43–53. doi: 10.1016/S0092-8674(00)00009-X
362. Du C, Fang M, Li Y, Li L, Wang X. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell (2000) 102:33–42. doi: 10.1016/S0092-8674(00)00008-8
363. Varfolomeev E, Blankenship JW, Wayson SM, Fedorova AV, Kayagaki N, Garg P, et al. IAP Antagonists Induce Autoubiquitination of c-IAPs, NF-κB Activation, and TNFα-Dependent Apoptosis. Cell (2007) 131:669–81. doi: 10.1016/j.cell.2007.10.030
364. Dueber EC, Schoeffler AJ, Lingel A, Elliott JM, Fedorova AV, Giannetti AM, et al. Antagonists induce a conformational change in cIAP1 that promotes autoubiquitination. Science (2011) 334:376–80. doi: 10.1126/science.1207862
365. Sakamoto KM, Kim KB, Kumagai A, Mercurio F, Crews CM, Deshaies RJ. Protacs: chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc Natl Acad Sci USA (2001) 98:8554–9. doi: 10.1073/pnas.141230798
366. Neklesa TK, Tae HS, Schneekloth AR, Stulberg MJ, Corson TW, Sundberg TB, et al. Small-molecule hydrophobic tagging–induced degradation of HaloTag fusion proteins. Nat Chem Biol (2011) 7:538–43. doi: 10.1038/nchembio.597
367. Takahashi D, Moriyama J, Nakamura T, Miki E, Takahashi E, Sato A, et al. AUTACs: Cargo-Specific Degraders Using Selective Autophagy. Mol Cell (2019) 76:797–810.e10. doi: 10.1016/j.molcel.2019.09.009
368. Simonetta KR, Taygerly J, Boyle K, Basham SE, Padovani C, Lou Y, et al. Prospective discovery of small molecule enhancers of an E3 ligase-substrate interaction. Nat Commun (2019) 10:1402. doi: 10.1038/s41467-019-09358-9
369. Sheard LB, Tan X, Mao H, Withers J, Ben-Nissan G, Hinds TR, et al. Jasmonate perception by inositol-phosphate-potentiated COI1–JAZ co-receptor. Nature (2010) 468:400–5. doi: 10.1038/nature09430
370. Gray WM, Kepinski S, Rouse D, Leyser O, Estelle M. Auxin regulates SCFTIR1-dependent degradation of AUX/IAA proteins. Nature (2001) 414:271–6. doi: 10.1038/35104500
371. Lu G, Middleton RE, Sun H, Naniong M, Ott CJ, Mitsiades CS, et al. The Myeloma Drug Lenalidomide Promotes the Cereblon-Dependent Destruction of Ikaros Proteins. Science (2014) 343:305–9. doi: 10.1126/science.1244917
372. Kronke J, Udeshi ND, Narla A, Grauman P, Hurst SN, McConkey M, et al. Lenalidomide Causes Selective Degradation of IKZF1 and IKZF3 in Multiple Myeloma Cells. Science (2014) 343:301–5. doi: 10.1126/science.1244851
373. Nero TL, Morton CJ, Holien JK, Wielens J, Parker MW. Oncogenic protein interfaces: small molecules, big challenges. Nat Rev Cancer (2014) 14:248–62. doi: 10.1038/nrc3690
374. Słabicki M, Kozicka Z, Petzold G, Der Li Y, Manojkumar M, Bunker RD, et al. The CDK inhibitor CR8 acts as a molecular glue degrader that depletes cyclin K. Nature (2020) 585:293–7. doi: 10.1038/s41586-020-2374-x
375. Mayor-Ruiz C, Bauer S, Brand M, Kozicka Z, Siklos M, Imrichova H, et al. Rational discovery of molecular glue degraders via scalable chemical profiling. Nat Chem Biol (2020) 16:1199–207. doi: 10.1038/s41589-020-0594-x
376. Wu N, Liu C, Bai C, Han YP, Cho WCS, Li Q. Over-expression of deubiquitinating enzyme USP14 in lung adenocarcinoma promotes proliferation through the accumulation of β-catenin. Int J Mol Sci (2013) 14:10749–60. doi: 10.3390/ijms140610749
377. Hu M, Li P, Song L, Jeffrey PD, Chenova TA, Wilkinson KD, et al. Structure and mechanisms of the proteasome-associated deubiquitinating enzyme USP14. EMBO J (2005) 24:3747–56. doi: 10.1038/sj.emboj.7600832
378. Xu D, Shan B, Lee B-H, Zhu K, Zhang T, Sun H, et al. Phosphorylation and activation of ubiquitin-specific protease-14 by Akt regulates the ubiquitin-proteasome system. Elife (2015) 4:e10510. doi: 10.7554/eLife.10510
379. Qiu X-B, Ouyang S-Y, Li C-J, Miao S, Wang L, Goldberg AL. hRpn13/ADRM1/GP110 is a novel proteasome subunit that binds the deubiquitinating enzyme, UCH37. EMBO J (2006) 25:5742–53. doi: 10.1038/sj.emboj.7601450
380. Lam YA, Xu W, DeMartino GN, Cohen RE. Editing of ubiquitin conjugates by an isopeptidase in the 26S proteasome. Nature (1997) 385:737–40. doi: 10.1038/385737a0
381. Mazumdar T, Gorgun FM, Sha Y, Tyryshkin A, Zeng S, Hartmann-Petersen R, et al. Regulation of NF-kappaB activity and inducible nitric oxide synthase by regulatory particle non-ATPase subunit 13 (Rpn13). Proc Natl Acad Sci USA (2010) 107:13854–9. doi: 10.1073/pnas.0913495107
382. Wang X, D’Arcy P, Caulfield TR, Paulus A, Chitta K, Mohanty C, et al. Synthesis and Evaluation of Derivatives of the Proteasome Deubiquitinase Inhibitor b-AP15. Chem Biol Drug Des (2015) 86:1036–48. doi: 10.1111/cbdd.12571
383. D’Arcy P, Brnjic S, Olofsson MH, Fryknäs M, Lindsten K, De Cesare M, et al. Inhibition of proteasome deubiquitinating activity as a new cancer therapy. Nat Med (2011) 17:1636–40. doi: 10.1038/nm.2536
384. Manasanch EE, Orlowski RZ. Proteasome inhibitors in cancer therapy. Nat Rev Clin Oncol (2017) 14:417–33. doi: 10.1038/nrclinonc.2016.206
385. Wang Z, Kang W, You Y, Pang J, Ren H, Suo Z, et al. USP7: Novel drug target in cancer therapy. Front Pharmacol (2019) 10:427. doi: 10.3389/fphar.2019.00427
386. Li M, Brooks CL, Kon N, Gu W. A dynamic role of HAUSP in the p53-Mdm2 pathway. Mol Cell (2004) 13:879–86. doi: 10.1016/S1097-2765(04)00157-1
387. van der Horst A, de Vries-Smits AMM, Brenkman AB, van Triest MH, van den Broek N, Colland F, et al. FOXO4 transcriptional activity is regulated by monoubiquitination and USP7/HAUSP. Nat Cell Biol (2006) 8:1064–73. doi: 10.1038/ncb1469
388. Song MS, Salmena L, Carracedo A, Egia A, Lo-Coco F, Teruya-Feldstein J, et al. The deubiquitinylation and localization of PTEN are regulated by a HAUSP-PML network. Nature (2008) 455:813–7. doi: 10.1038/nature07290
389. Reverdy C, Conrath S, Lopez R, Planquette C, Atmanene C, Collura V, et al. Discovery of specific inhibitors of human USP7/HAUSP deubiquitinating enzyme. Chem Biol (2012) 19:467–77. doi: 10.1016/j.chembiol.2012.02.007
390. Fan YH, Cheng J, Vasudevan SA, Dou J, Zhang H, Patel RH, et al. USP7 inhibitor P22077 inhibits neuroblastoma growth via inducing p53-mediated apoptosis. Cell Death Dis (2013) 4:e867. doi: 10.1038/cddis.2013.400
391. Altun M, Kramer HB, Willems LI, McDermott JL, Leach CA, Goldenberg SJ, et al. Activity-based chemical proteomics accelerates inhibitor development for deubiquitylating enzymes. Chem Biol (2011) 18:1401–12. doi: 10.1016/j.chembiol.2011.08.018
392. Schweitzer K, Bozko PM, Dubiel W, Naumann M. CSN controls NF-κB by deubiquitinylation of IκBα. EMBO J (2007) 26:1532–41. doi: 10.1038/sj.emboj.7601600
393. Huang X, Langelotz C, Hetfeld-Pěchoč BKJ, Schwenk W, Dubiel W. The COP9 Signalosome Mediates β-Catenin Degradation by Deneddylation and Blocks Adenomatous Polyposis coli Destruction via USP15. J Mol Biol (2009) 391:691–702. doi: 10.1016/j.jmb.2009.06.066
394. Inui M, Manfrin A, Mamidi A, Martello G, Morsut L, Soligo S, et al. USP15 is a deubiquitylating enzyme for receptor-activated SMADs. Nat Cell Biol (2011) 13:1368–75. doi: 10.1038/ncb2346
395. Ward SJ, Gratton HE, Indrayudha P, Michavila C, Mukhopadhyay R, Maurer SK, et al. The structure of the deubiquitinase USP15 reveals a misaligned catalytic triad and an open ubiquitin-binding channel. J Biol Chem (2018) 293:17362–74. doi: 10.1074/jbc.RA118.003857
396. Chen J, Dexheimer TS, Ai Y, Liang Q, Villamil MA, Inglese J, et al. Selective and cell-active inhibitors of the USP1/ UAF1 deubiquitinase complex reverse cisplatin resistance in non-small cell lung cancer cells. Chem Biol (2011) 18:1390–400. doi: 10.1016/j.chembiol.2011.08.014
397. Roos A, Dhruv HD, Peng S, Inge LJ, Tuncali S, Pineda M, et al. EGFRvIII–Stat5 signaling enhances glioblastoma cell migration and survival. Mol Cancer Res (2018) 16:1185–95. doi: 10.1158/1541-7786.MCR-18-0125
398. Banik SM, Pedram K, Wisnovsky S, Ahn G, Riley NM, Bertozzi CR. Lysosome-targeting chimaeras for degradation of extracellular proteins. Nature (2020) 584:291–7. doi: 10.1038/s41586-020-2545-9
399. Jacobs MD, Harrison SC. Structure of an IkBa/NF-kB Complex. Cell (1998) 95:749–58. doi: 10.1016/S0092-8674(00)81698-0
400. Griffith EC, Su Z, Turk BE, Chen S, Chang YH, Wu Z, et al. Methionine aminopeptidase (type 2) is the common target for angiogenesis inhibitors AGM-1470 and ovalicin. Chem Biol (1997) 4:461–71. doi: 10.1016/S1074-5521(97)90198-8
401. Sakamoto KM, Kim KB, Verma R, Ransick A, Stein B, Crews CM, et al. Development of Protacs to Target Cancer-promoting Proteins for Ubiquitination and Degradation. Mol Cell Proteomics (2003) 2:1350–8. doi: 10.1074/mcp.T300009-MCP200
402. Schneekloth JS, Fonseca FN, Koldobskiy M, Mandal A, Deshaies R, Sakamoto K, et al. Chemical Genetic Control of Protein Levels: Selective in Vivo Targeted Degradation. J Am Chem Soc (2004) 126:3748–54. doi: 10.1021/ja039025z
403. Winter GE, Buckley DL, Paulk J, Roberts JM, Souza A, Dhe-Paganon S, et al. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science (2015) 348:1376–81. doi: 10.1126/science.aab1433
404. Lu J, Qian Y, Altieri M, Dong H, Wang J, Raina K, et al. Hijacking the E3 Ubiquitin Ligase Cereblon to Efficiently Target BRD4. Chem Biol (2015) 22:755–63. doi: 10.1016/j.chembiol.2015.05.009
405. Buckley DL, Raina K, Darricarrere N, Hines J, Gustafson JL, Smith IE, et al. HaloPROTACS: Use of Small Molecule PROTACs to Induce Degradation of HaloTag Fusion Proteins. ACS Chem Biol (2015) 10:1831–7. doi: 10.1021/acschembio.5b00442
406. Bondeson DP, Mares A, Smith IED, Ko E, Campos S, Miah AH, et al. Catalytic in vivo protein knockdown by small-molecule PROTACs. Nat Chem Biol (2015) 11:611–7. doi: 10.1038/nchembio.1858
407. Schneekloth AR, Pucheault M, Tae HS, Crews CM. Targeted intracellular protein degradation induced by a small molecule: En route to chemical proteomics. Bioorg Med Chem Lett (2008) 18:5904–8. doi: 10.1016/j.bmcl.2008.07.114
408. Itoh Y, Ishikawa M, Naito M, Hashimoto Y. Protein knockdown using methyl bestatin-ligand hybrid molecules: Design and synthesis of inducers of ubiquitination-mediated degradation of cellular retinoic acid-binding proteins. J Am Chem Soc (2010) 132:5820–6. doi: 10.1021/ja100691p
409. Buckley DL, Van Molle I, Gareiss PC, Tae HS, Michel J, Noblin DJ, et al. Targeting the von Hippel-Lindau E3 ubiquitin ligase using small molecules to disrupt the VHL/HIF-1α interaction. J Am Chem Soc (2012) 134:4465–8. doi: 10.1021/ja209924v
410. Fischer ES, Böhm K, Lydeard JR, Yang H, Stadler MB, Cavadini S, et al. Structure of the DDB1–CRBN E3 ubiquitin ligase in complex with thalidomide. Nature (2014) 512:49–53. doi: 10.1038/nature13527
411. Chamberlain PP, Lopez-Girona A, Miller K, Carmel G, Pagarigan B, Chie-Leon B, et al. Structure of the human Cereblon–DDB1–lenalidomide complex reveals basis for responsiveness to thalidomide analogs. Nat Struct Mol Biol (2014) 21:803–9. doi: 10.1038/nsmb.2874
412. Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, et al. Selective inhibition of BET bromodomains. Nature (2010) 468:1067–73. doi: 10.1038/nature09504
413. Hershko A, Ciechanover A. The Ubiquitin System. Annu Rev Biochem (1998) 67:425–79. doi: 10.1146/annurev.biochem.67.1.425
414. Lins L, Brasseur R. The hydrophobic effect in protein folding. FASEB J (1995) 9:535–40. doi: 10.1096/fasebj.9.7.7737462
415. McClellan AJ, Tam S, Kaganovich D, Frydman J. Protein quality control: chaperones culling corrupt conformations. Nat Cell Biol (2005) 7:736–41. doi: 10.1038/ncb0805-736
416. Xie T, Lim SM, Westover KD, Dodge ME, Ercan D, Ficarro SB, et al. Pharmacological targeting of the pseudokinase Her3. Nat Chem Biol (2014) 10:1006–12. doi: 10.1038/nchembio.1658
417. Zhou B, Hu J, Xu F, Chen Z, Bai L, Fernandez-Salas E, et al. Discovery of a Small-Molecule Degrader of Bromodomain and Extra-Terminal (BET) Proteins with Picomolar Cellular Potencies and Capable of Achieving Tumor Regression. J Med Chem (2018) 61:462–81. doi: 10.1021/acs.jmedchem.6b01816
418. Qin C, Hu Y, Zhou B, Fernandez-Salas E, Yang CY, Liu L, et al. Discovery of QCA570 as an Exceptionally Potent and Efficacious Proteolysis Targeting Chimera (PROTAC) Degrader of the Bromodomain and Extra-Terminal (BET) Proteins Capable of Inducing Complete and Durable Tumor Regression. J Med Chem (2018) 61:6685–704. doi: 10.1021/acs.jmedchem.8b00506
419. Sun X, Wang J, Yao X, Zheng W, Mao Y, Lan T, et al. A chemical approach for global protein knockdown from mice to non-human primates. Cell Discov (2019) 5:10. doi: 10.1038/s41421-018-0079-1
420. Brand M, Jiang B, Bauer S, Donovan KA, Liang Y, Wang ES, et al. Homolog-Selective Degradation as a Strategy to Probe the Function of CDK6 in AML. Cell Chem Biol (2019) 26:300–6.e9. doi: 10.1016/j.chembiol.2018.11.006
421. Jiang B, Wang ES, Donovan KA, Liang Y, Fischer ES, Zhang T, et al. Development of Dual and Selective Degraders of Cyclin-Dependent Kinases 4 and 6. Angew Chemie Int Ed (2019) 58:6321–6. doi: 10.1002/anie.201901336
422. Burslem GM, Smith BE, Lai AC, Jaime-Figueroa S, McQuaid DC, Bondeson DP, et al. The Advantages of Targeted Protein Degradation Over Inhibition: An RTK Case Study. Cell Chem Biol (2018) 25:67–77.e3. doi: 10.1016/j.chembiol.2017.09.009
423. Lebraud H, Wright DJ, Johnson CN, Heightman TD. Protein degradation by in-cell self-assembly of proteolysis targeting chimeras. ACS Cent Sci (2016) 2:927–34. doi: 10.1021/acscentsci.6b00280
424. Liu J, Chen H, Ma L, He Z, Wang D, Liu Y, et al. Light-induced control of protein destruction by opto-PROTAC. Sci Adv (2020) 6:eaay5154. doi: 10.1126/sciadv.aay5154
425. Banks WA. From blood-brain barrier to blood-brain interface: New opportunities for CNS drug delivery. Nat Rev Drug Discov (2016) 15:275–92. doi: 10.1038/nrd.2015.21
Keywords: glioblastoma, ubiquitin, E3 ubiquitin ligases, deubiquinating enzymes, PROTAC (proteolysis-targeting chimeric molecule), stem cell, cancer, ubiquitin-proteasome system
Citation: Scholz N, Kurian KM, Siebzehnrubl FA and Licchesi JDF (2020) Targeting the Ubiquitin System in Glioblastoma. Front. Oncol. 10:574011. doi: 10.3389/fonc.2020.574011
Received: 18 June 2020; Accepted: 07 October 2020;
Published: 25 November 2020.
Edited by:
Boris Zhivotovsky, Karolinska Institutet (KI), SwedenReviewed by:
Chunming Cheng, The Ohio State University, United StatesDaniele Guardavaccaro, University of Verona, Italy
Copyright © 2020 Scholz, Kurian, Siebzehnrubl and Licchesi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Julien D. F. Licchesi, ai5saWNjaGVzaUBiYXRoLmFjLnVr