 Yinan Wu1,2†
Yinan Wu1,2† Kaimin Hu
Kaimin Hu
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 25 September 2020
Sec. Molecular and Cellular Oncology
Volume 10 - 2020 | https://doi.org/10.3389/fonc.2020.571127
Ferroptosis is a newly described type of programmed cell death and intensively related to both maintaining homeostasis and the development of diseases, especially cancers. Inducing ferroptosis leads to mitochondrial dysfunction and toxic lipid peroxidation in cells, which plays a pivotal role in suppressing cancer growth and progression. Here, we reviewed the existing studies about the molecular mechanisms of ferroptosis involved in different antitumor treatments, such as chemotherapy, targeted therapy, radiotherapy, and immunotherapy. We focused in particular on the distinct combinatorial therapeutic effects such as the synergistic sensitization effect and the drug-resistance reversal achieved when using ferroptosis inducers with conventional cancer therapy. Finally, we discussed the challenges and opportunities in clinical applications of ferroptosis. The application of nanotechnolgy and other novel technologies may provide a new direction in ferroptosis-driven cancer therapies.
The term ferroptosis was coined in 2012 to define a unique iron-dependent form of programmed cell death. In ferroptotic cells, the mitochondria become smaller with increased membrane density, and the mitochondrial cristae usually decrease or disappear (1, 2). Ferroptosis helps maintain the death balance in normal cells and tissues (3). In cancer, some carcinogenic pathways can regulate the key modulatory factors in ferroptosis and induce ferroptosis of cancer cells (4–8).
The existing cancer therapies with unsatisfactory clinical efficacy always meet dilemmas to eradicate cancer cells due to the drug insensitivity or acquired resistance. In the past, inducing apoptosis was considered the main way to cause cancer cell death in conventional treatments. However, increasing studies have reported that inducing ferroptosis can significantly improve the efficacy of killing cancer cells, indicating that ferroptosis is another important way in treatment of cancer, where “all roads lead to Rome,” as the old saying goes. In this review, we summarized the molecular mechanisms and the roles of ferroptosis in different cancer therapies, as well as the challenges and opportunities in clinical applications of ferroptosis such as ferroptosis-driven nanotherapeutics, to draw a conclusion about the recent progress in ferroptosis-inducing cancer therapies and outline the future research and clinical application strategies.
Dysfunctions in iron and lipid metabolism result in ferroptosis, which is characterized by accumulation of reactive oxygen species (ROS) and lipid peroxidation. Free intracellular ferrous ions react with hydrogen peroxide through Fenton reactions and activate the lipoxygenase (LOX) that induces peroxidation of polyunsaturated fatty acids (PUFAs) in cell membranes (1). The toxic free radicals generated by the oxidized lipids produce more oxidized lipids and further oxidative damage. Ferroptosis is inhibited by sequestration of free iron, inhibition of PUFA synthesis, or scavenging of ROS (Figure 1).
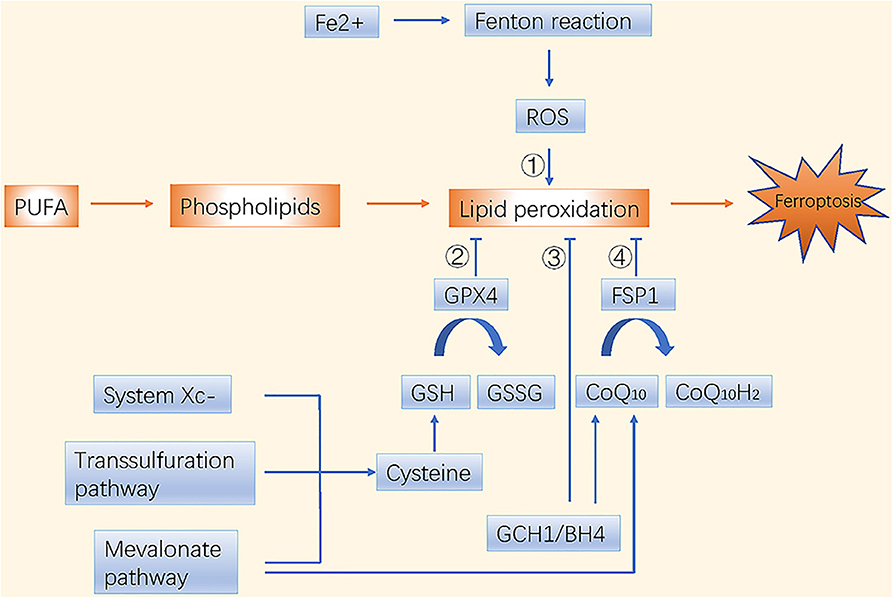
Figure 1. Molecular mechanisms of ferroptosis in brief. There are four main pathways to induce ferroptosis. ➀ Iron metabolism; ➁ GSH/GPX4 pathway; ➂ GCH1/BH4 pathway; ➃ FSP1/CoQ10/NAD(P)H pathway.
Ferroptosis and phospholipid peroxidation are mainly controlled by two parallel systems: glutathione (GSH)/glutathione peroxidase 4 (GPX4) and ferroptosis suppressor protein 1 (FSP1)/ubiquinone (CoQ10)/NAD(P)H axes (9, 10). The GSH/GPX4 pathway includes cystine import via the system (consisting of SLC7A11 and SLC3A2), cysteine production via the transsulfuration pathway, and selenocysteine production via the mevalonate pathway. Interestingly, the mevalonate metabolic pathway is crucial during the synthesis of GPX4 itself and for generation of the CoQ10 backbone. Another highly potent endogenous ferroptosis suppressor, the GTP cyclohydrolase 1 (GCH1)/tetrahydrobiopterin (BH4)/phospholipid axis, was reported recently. It inhibits ferroptosis by selectively preventing depletion of phospholipids with two PUFA tails (11). Nuclear factor erythroid 2-related factor 2 (NRF2), another endogenous antioxidant defense system, is usually maintained at a low level by tumor suppressor Kelch ECH-associated protein 1 (KEAP1)-mediated ubiquitination. Under oxidative stress, NRF2 is stabilized and activated through dissociation from KEAP1 (12); it then suppresses ferroptosis through NRF2/SLC7A11/heme oxygenase-1 (HO-1) and NRF2-FTH1 signaling (13). NRF2 is one of the central factors leading to drug insensitivity or resistance in cancer cells when coping with oxidative stress.
Ferroptosis is a new mechanism of active cancer cell death induced by classic chemotherapeutic drugs discovered in recent years (Tables 1, 2). For example, cisplatin triggers ferroptosis mainly by directly depleting intracellular GSH, which results in the consequent inhibition of GPXs (18). In addition, cisplatin can also induce ferroptosis when ferritinophagy increases free iron levels (17). In consequence, ferroptosis inducers such as erastin or RSL3 always enhance the anticancer effects of cisplatin synergistically by inhibiting system or GPX4 during the treatment of distinct cancer types, such as lung, colorectal, ovarian, and pancreatic ductal adenocarcinoma cancers (17, 18, 30, 31). Suberoylanilide hydroxamic acid (SAHA), a histone deacetylase inhibitor (HDACi), can also sensitize tumors to the effects of cisplatin by inducing ROS (32). Ferritinophagy-mediated ferroptosis is implicated in the therapeutic mechanisms of artemisinin (ART) and its derivatives (19–21). The iron storage protein ferritin binds to NCOA4 in the autophagosome and gets delivered into the lysosomes; ART and its derivative, dihydroartemisinin (DAT), can accumulate in the lysosomes and increase ferritin protein degradation (14), a process that increases the amount of intracellular iron and induces ferroptosis (14, 56). DAT can also regulate iron homeostasis by altering the iron-regulatory proteins (IRPs)/iron-responsive element (IRE) ratio to further increase intracellular free iron (19). Based on this, co-treatment with ART and transferrin (TF), which is responsible for the transport of iron into cells, increased lysosomal free iron and promoted ferroptosis in pancreatic ductal adenocarcinoma (PDAC) (15).
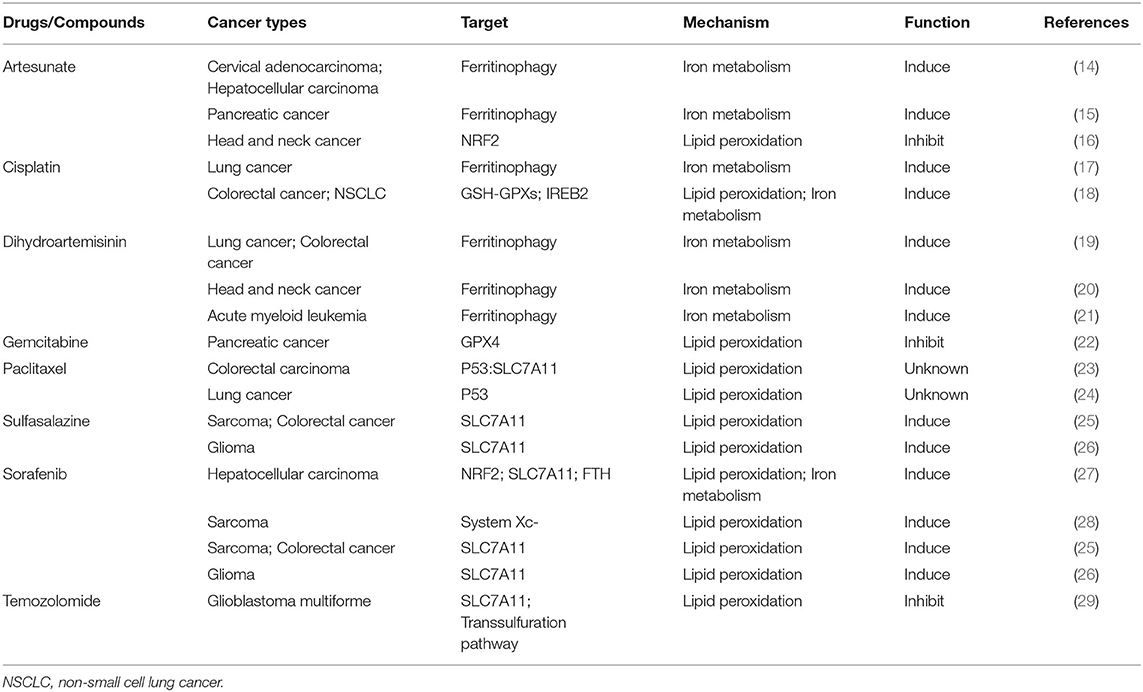
Table 1. Classical anti-tumor drugs and compounds associated with ferroptosis.
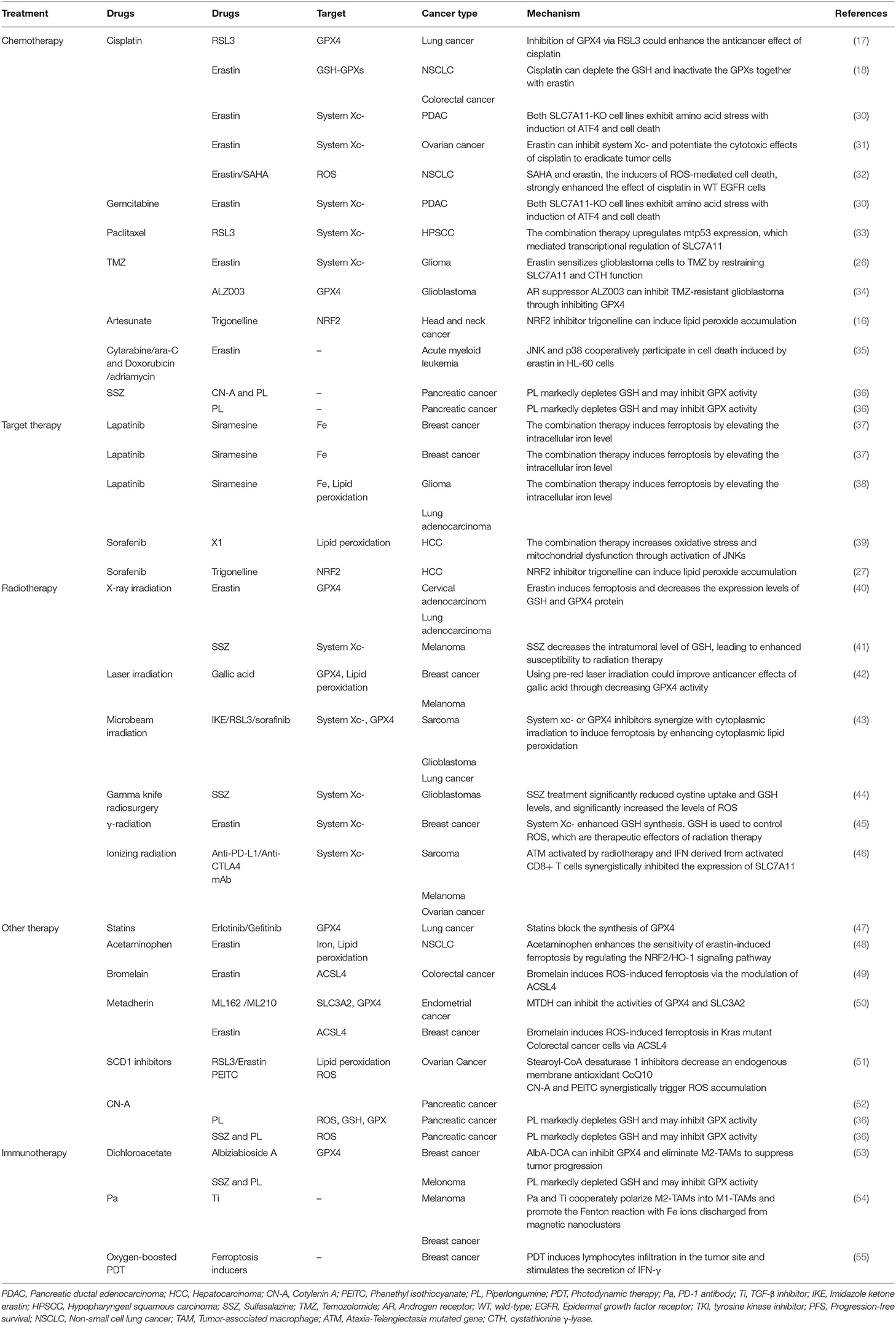
Table 2. The synergistic effect of anti-tumor therapy together with ferroptosis inducers.
Traditional chemotherapy drugs can also inhibit cancerous cell death and lead to ferroptosis resistance through the upregulation of GPX4 and the system Xc−. The heat shock 70-kDa protein 5 (HSPA5)-GPX4 pathway mediated ferroptosis resistance limits the anticancer activity of gemcitabine. Specifically, gemcitabine induces the expression of HSPA5, which in turn prevents GPX4 protein degradation and subsequent lipid peroxidation in pancreatic cancer (22). Co-treatment with gemcitabine and erastin or the genetic inhibition of the HSPA5-GPX4 pathway produced additive effects eradicating pancreatic cancer cells in vitro and in vivo (30). Temozolomide (TMZ) was reported to induce SLC7A11 expression via NRF2 and ATF4 activation pathways both at mRNA and protein levels. Moreover, it can also promote cystathionine γ-lyase (CTH) activity; CTH is a key enzyme in the transsulfuration pathway that secures the cysteine supply and GSH synthesis when SLC7A11 is blocked. This may explain why gliomas with high system expression are more likely to be affected by the erastin-TMZ combination therapy (29, 57). The NRF2-antioxidant response element pathway can also be activated by artesunate, another chemotherapeutic drug used in head and neck cancer (HNC) cells. Inhibition of NRF2 induces lipid lipoxidation and reverses ferroptosis resistance in HNC (16).
The anticancer effects of some other classic chemotherapy drugs are enhanced when combined with ferroptosis inducers. Previous studies showed that paclitaxel (PTX), a classic chemotherapy drug used widely in multiple cancer treatments, can both upregulate the expressions of the p53 and p21 genes and downregulate the expressions of SLC7A11 and SLC1A5 in colorectal carcinoma and lung cancer cells (23, 24). Ye and his colleagues demonstrated that the combination of PTX and RSL3 rather than each agent alone induced ferroptosis in hypopharyngeal squamous carcinoma (HPSCC) cells with mutant p53 (mtp53) through the suppression of SLC7A11. mtp53 expression and, especially, the acetylation of K98 play a key role in PTX and RSL3-induced ferroptosis (33). In addition, a low dose of erastin can dramatically promote the therapeutic effects of cytarabine/ara-C and doxorubicin/adriamycin against leukemic cells in an RAS-independent manner that depends partly on ferroptosis induction. The c-Jun N-terminal kinases (JNKs)/p38-MAPK pathway was shown to play a major role in erastin-induced cell death in leukemic cells (35). The ERK-MAPK pathway is also responsible for RAS-dependent ferroptotic cell death in solid cancer cells (58). These studies suggest that the activation of the MAPK pathway may affect the anticancer efficiency of erastin in different cancers and that exploring the specificity of MAPK substrates may provide a viable option to distinguish diverse erastin-induced types of cell death.
Ferroptosis also plays a role in targeted therapy for cancers by interfering with target molecules involved in the development of cancer (Tables 1, 2). Sorafenib, a multi-kinase inhibitor, can directly inhibit the system function and can also clear the intracellular KEAP1 proteins to activate the NRF2-SLC7A11 signaling as an adaptive response (27, 59, 60). As mentioned previously, cancer cells with high expression of NRF2 can use these protective detoxification systems to prevent lipid peroxide accumulation and survive. Indeed, treatment with the NRF2 inhibitor trigonelline can promote sorafenib-induced ferroptosis in hepatocellular carcinoma cells (27). In addition, sorafenib can also block the formation of BECN1-SLC7A11 complexes and induce ferroptosis through AMP-activated protein kinase activation (25, 26, 61).
VDACs (voltage-dependent anion channels) are crucial mitochondrial membrane proteins, which can be activated to open by the erastin-like compound X1, contributing to lipid peroxidation (58). Sorafenib together with opening of VDACs increases oxidative stress and mitochondrial dysfunction through activation of JNKs in hepatocarcinoma cells (39). Moreover, sorafenib can also induce ferroptosis by triggering the expression of cathepsin B through MEK-ERK signaling phosphorylation, while inhibiting the signal transducer and activator of transcription 3 (STAT3) in human PDAC cell lines (60, 62, 63).
Siramesine combined with lapatinib can induce ferroptosis by elevating the intracellular iron level and subsequently increasing ROS in breast cancer cells (64). This specific cell death was initially induced by ferroptosis, but was regulated by spontaneous ferritin degradation and subsequent upregulation of transferrin (37). The same phenomenon has been observed in glioma and lung adenocarcinoma cell lines; the whole death process was regulated by lysosomal iron release and the degradation of HO-1by the proteasome system (38).
The high-energy ionizing radiation (IR) of radiotherapy always induces direct DNA double-strand breaks (65). Moreover, indirect effects caused by the radiolysis of cellular water and the stimulation of oxidases result in GSH depletion and ROS generation (66–68) (Table 2 and Figure 2). Studies have indicated that the efficacy of radiotherapy increases when GSH is depleted (69, 70).
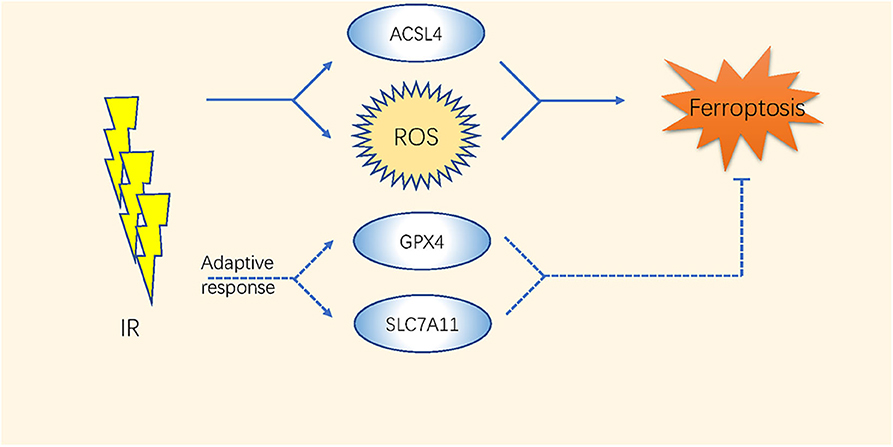
Figure 2. The mechanism of ferroptosis in radiotherapy of cancer.
The expression level of SLC7A11 is higher in either glioblastoma (GBM) patient biopsies or glioma cell lines than in normal brain tissues (44); thus, inhibiting system or GPX4 by ferroptosis inducers enhances the therapeutic effect of radiation in breast cancer, sarcoma, and glioblastoma (40, 41, 43–45). Khorsandi et al. showed that the use of gallic acid (GA, a natural polyhydroxy phenolic compound) after pre-irradiation can significantly reduce the survival rate of breast cancer and melanoma cells mainly through inhibition of GPX4 activity without affecting normal cells (42).
Recently, Lei et al. found that IR also upregulates the expression of ferroptosis marker genes such as PTGS2 in most of the cancer cell lines. IR induces ROS directly by inducing the ACSL4/LPCAT3/ALOX and SLC7A11/GPX4 pathways as an adaptive response. Inactivating SLC7A11 or GPX4 improves the radiosensitivity of cancer cells and xenograft tumors to IR (71). Additionally, ferroptosis has been considered as a direct link to improve tumor control after co-treatments with radiotherapy and CD8+ T cells. The ataxia-telangiectasia mutated gene (ATM) activated by radiotherapy and interferon (IFN) derived from activated CD8+ T cells synergistically inhibited the expression of SLC7A11 (46). These comprehensive studies took a novel direction in improving the therapeutic effects of radiation therapy or fighting against radiotherapy resistance through the regulation of ferroptosis.
Inhibiting GPX4 is a recognized way to induce ferroptosis, while mevalonate blockers are also effective. Statins, such as fluvastatin, lovastatin, and simvastatin, are small molecular inhibitors of 3-hydroxy-3-methyl-glutaryl-CoA reductase (HMGCR); they can inhibit GPX4 by regulating selenoprotein biosynthesis and CoQ10 through the mevalonate pathway, but they cannot be rescued by lipophilic antioxidants (72, 73). After treatment with fluvastatin, the expression of GPX4 decreased in a time-and concentration-dependent manner, and the effect was enhanced by combination with the direct GPX4 inhibitor RSL3 (5). Several previous clinical studies indicate that co-treatment with EGFR-TKIs and statins can improve the therapeutic effect and prolong patients' survival compared with treatment with EGFR-TKIs alone regardless of whether EGFR is mutated (47, 74, 75). It will be interesting to study the role of other mevalonate blockers in ferroptosis-mediated cancer treatments (Table 2).
Moreover, numerous promising therapeutic drugs have emerged for cancer therapy. Acetaminophen enhances the sensitivity of erastin-induced ferroptosis by regulating the NRF2/HO-1 signaling pathway (48). Bromelain induces ROS-induced ferroptosis in Kras mutant colorectal cancer cells via the modulation of ACSL4 (49). Metadherin (MTDH) can inhibit the activities of GPX4 and SLC3A2 (50). Stearoyl-CoA desaturase 1 inhibitors decrease an endogenous membrane antioxidant CoQ10 and make ovarian cancer cells prone to ferroptosis (51). Prominin2 augments the formation of ferritin-containing multivesicular bodies (MVBs) and exosomes to decrease the intracellular iron levels, thereby inhibiting ferroptosis (76). All of these agents combined with ferroptosis inducers (such as RSL3, erastin) can enhance the ferroptosis effect in different cancer cells. Cotylenin A (CN-A, a plant growth regulator) and phenethyl isothiocyanate (PEITC, a dietary anticarcinogenic compound and an inducer of ROS), have been reported to synergistically inhibit the progression of pancreatic cancer via the elevation of ROS (52). The use of CN-A alone did not remarkably affect cell viability, while the use of piperlongumine (PL) alone moderately decreased the viability to 55–70%. When used together, CN-A remarkably enhances PL-induced ferroptosis. In addition, SSZ also promotes PL-induced ferroptosis in pancreatic cancer cells. Moreover, SSZ further enhanced the cytotoxicity of mouse embryonic fibroblasts (MEFs) and cancer cells induced by PL combined with CN-A (36) (Table 2).
The mechanisms of drug resistance in cancer treatment are complex and diverse, restraint of ferroptosis is proved to be an important one.is proved to be an important one (Table 3). When treated with PTX or doxorubicin, activated NRF2 in breast and lung cancers increased drug resistance (89, 90). Improper activation of NRF2 by a mutated KEAP1 or upregulation of P62 was a crucial cytoprotective mechanism against ferroptosis (27, 91). Upregulated NRF2/FTH1 or NRF2/SLC7A11 pathways resulted in ferroptosis resistance (27, 92, 93). Cancer cells with high levels of GSH and system expression always show multidrug and radiation resistance (44, 92–94). Therefore, exploring the effect of small molecule inhibitors targeting the KEAP1/NRF2 pathway in cancer is of therapeutic significance. Ferroptosis agonists (erastin/sorafenib and others) and STAT3 inhibitor BP-1-102 can induce ferroptosis of cisplatin-resistant NSCLC cells via NRF2/SLC7A11 or STAT3/NRF2/GPX4 signals (31, 77, 78). ART was shown to induce ferroptosis in cisplatin-resistant HNC cells when combined with NRF2 inhibitors (16), while its derivative DAT can enhance RSL3-induced ferroptosis by increasing the intracellular free iron level in highly ferroptosis-resistant cancer cells (19). Withaferin A can induce ferroptosis by simultaneously inhibiting both GPX4 and KEAP1 proteins; the consequent upregulation of NRF2 leads to an increase in intracellular labile Fe ions upon excessive activation of HO-1 in chemo-resistant neuroblastoma cells (79).
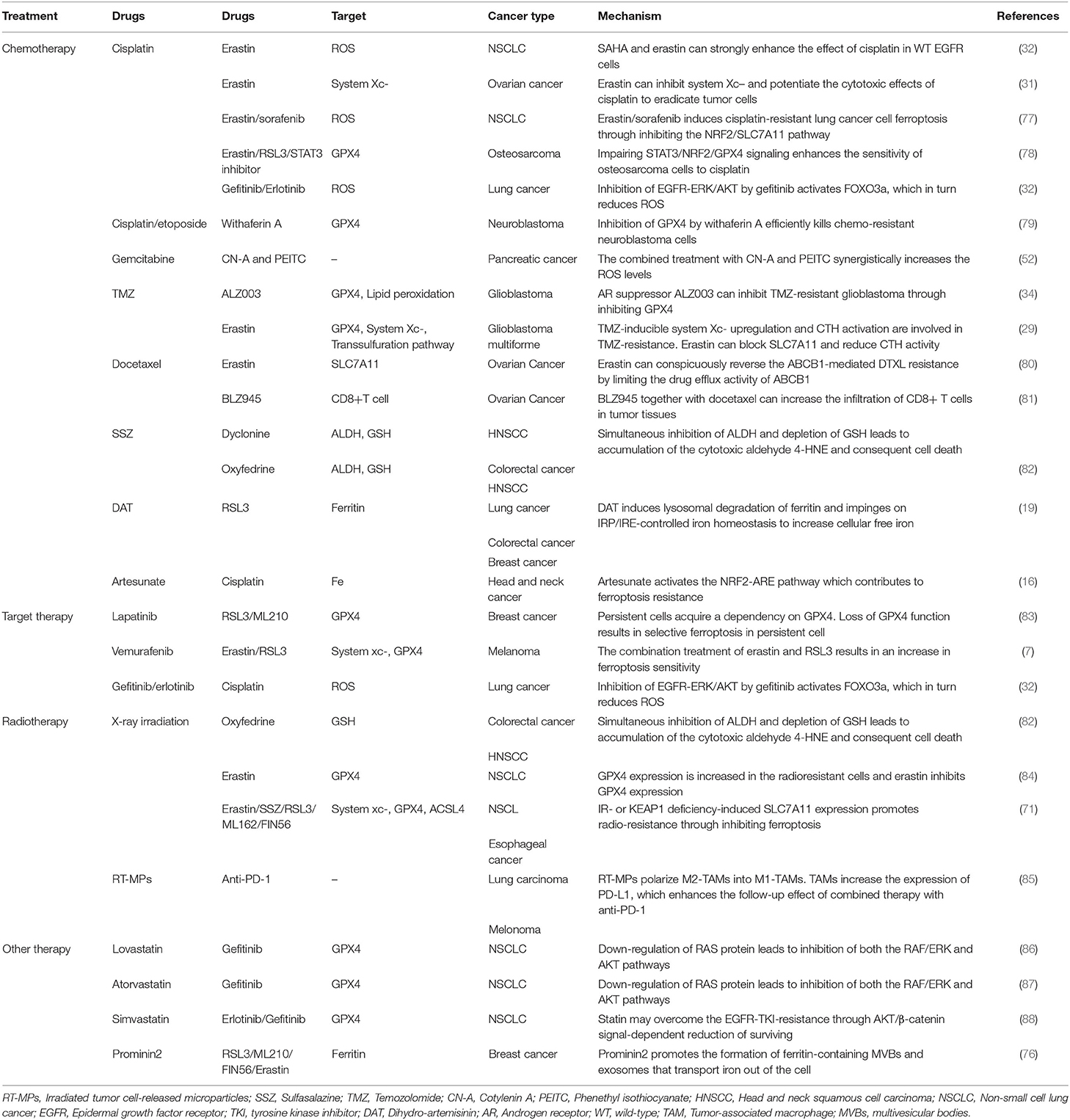
Table 3. Reversing anti-tumor therapy resistance together with ferroptosis inducers.
Drug-resistant cells also tend to express more proteins involved in iron metabolism, such as transferrin receptor (TFR) and ferritin and less ferroportin-1 (FPN, the iron transport protein) than drug-sensitive cells (95–98). In general, increased iron uptake (e.g., due to TFR overexpression), reduced iron storage (e.g., due to the knockdown of ferritin or induction of ferritinophagy), and impaired cellular iron export (e.g., resulting from the knockdown of FPN) enhance the sensitivity to ferroptosis (99). On the other hand, a decrease in intracellular free iron leads to ferroptosis resistance. Targeting TFR, ferritin, or FPN strategies with anticancer therapies are highly effective for overcoming the chemo-resistance of cancer cells. An article published in 1993 showed that downregulation of TFR reversed drug resistance in cancer cells (100). Wu et al. later reported that combination therapy with anticancer drugs and TFR targeting is highly effective for overcoming the chemo-resistance of hematologic malignancy cells (101). Bortezomib prevents the upregulation of ferritin in response to iron, thus limiting the ability to buffer ROS. Reduction of basal ferritin levels or iron supplementation increases cell death and overcomes bortezomib resistance (102). Targeting the downregulation of ferritin L subunit by miR-133a increases the sensitivity of chemo-resistant breast cancer cells to cisplatin and doxorubicin (97). These results suggest that regulating the iron metabolism balance may substantially improve the efficacy of cancer treatments. Elevated levels of aldehyde dehydrogenases (ALDHs) in cancers are a mark of treatment resistance and an effective target for ferroptosis induction. Sulfasalazine (SSZ)-resistant head and neck squamous cell carcinoma (HNSCC) cells often present high expression of ALDH3A1, and genetic or pharmacological inhibition of ALDH3A1 can make these cells regain their sensitivity to SSZ (103). Dyclonine (DYC) and oxyfedrine (OXY), the covalent inhibitors of ALDHs, can cooperate with GSH-depleting agents such as buthionine sulfoximine (BSO) or SSZ to induce intracellular accumulation of the toxic aldehyde 4-hydroxynonenal (4-HNE), a specific lipid peroxidation by-product, and suppress the growth of SSZ-resistant gastric tumors or HNSCCs with high ALDH3A1expression (82, 103).
A recent study showed that drug-resistant cancer cells in a high-mesenchymal cell state were sensitive to ferroptosis induced by GPX4 suppression or statin treatment (5). Similarly, targeting of HER2-amplified breast cancer cell lines with lapatinib leaves a population of persistent cells acting as high-mesenchymal state cells. A remarkable feature of this group of cells is that they depend on GPX4 for survival; thus, GPX4 inhibitors further eradicated this population of residual cells (83). Persistent melanoma cells induced by the BRAF kinase inhibitor vemurafenib also resulted in an increase in ferroptosis sensitivity upon both erastin and RSL3 treatments (7). FSP1, a key component of a non-mitochondrial coenzyme Q10 (CoQ10) antioxidant system, acts in parallel to the GSH/GPX4 pathway. It reduces CoQ10, which acts as a lipophilic radical-trapping antioxidant that halts the propagation of lipid peroxides and inhibits ferroptosis (9). Furthermore, Doll et al. found tumor cells with GPX4KO and FSP1 overexpression that were insensitive to ferroptosis-inducing agents. Treatment with a type of FSP inhibitor (iFSP) selectively induced ferroptosis in these cells (10).
Ferroptosis can also reverse chemo-resistance through other mechanisms. A combined therapy of CN-A and PEITC can trigger ROS accumulation and inhibit the proliferation of gemcitabine-resistant pancreatic cancer cells (52). One of the characteristics of TMZ-resistant cancer cells is the overexpression of androgen receptor (AR); co-treatment with erastin or AR suppressor significantly increased the TMZ-induced cytotoxicity and overcame TMZ resistance through GPX4 inhibition (29, 34). Similarly, overexpression of ABCB1 (P-glycoprotein/MDR1) in ovarian cancers usually leads to chemo-resistance via intracellular drug export (104, 105). Erastin can conspicuously reverse the ABCB1-mediated DTXL resistance by limiting the drug efflux activity of ABCB1 (80).
Inhibiting SLC7A11 or GPX4 sensitizes radio-insensitive cancer cells to IR (71). GPX4 expression is increased in radioresistant NSCLC cells, and knocking down GPX4 or using ferroptosis inducers that target GPX4 can enhance the sensitivity of radiation-resistant lung cancer cells after high-dose hypofractionated irradiation. Erastin and IR exhibited a combined cell killing effect compared with either erastin or IR alone (84). Moreover, co-treatment with oxyfedrine (OXY) can trigger the intracellular accumulation of lipid peroxidation and enhance cell death (82). In conclusion, triggering ferroptosis is a promising method to eradicate treatment-resistant cancer cells.
Ferroptotic cancer cells can continuously release immunostimulatory damage-associated molecular pattern molecules (DAMPs) and activate the adaptive immune system within the tumor microenvironment to enhance anticancer effects (106, 107). The phagocytosis of bone-marrow-derived dendritic cells (BMDCs) is activated by three key DAMPs (CRT, HMGB1, and ATP) from ferroptotic cells (108). Neutrophils are also recruited through a TLR4/TRIF/type 1 IFN or a Wnt signaling pathway induced by ferroptotic cancer cells (109, 110), which are important modulators and potential therapeutic targets against cancer progression (Figure 3).
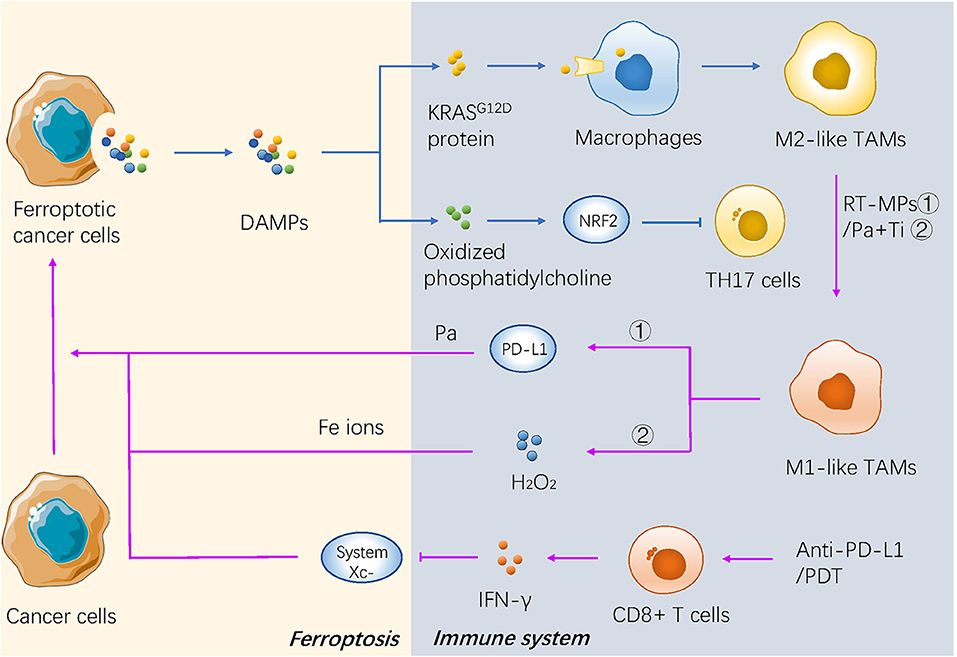
Figure 3. The mechanism of ferroptosis in tumor microenvironment and immunotherapy of cancer. DAMPs secreted by dying cancer cells promotes recruitment of tumor-infiltrating dendritic cells, contributing to immunogenic phagocytosis. And some kind of novel immunotherapies promote the cancerous ferroptosis in turn.
Two major subtypes of tumor-associated macrophages (TAMs) are generated depending on different environmental stresses, including antitumor M1 and procarcinogenic M2 TAMs (111). The KRASG12D protein released from ferroptotic pancreatic cancer cells is further absorbed by macrophages through an advanced glycosylation end product-specific receptor, which then induces STAT3-dependent fatty acid oxidation and drives macrophages to form M2-like TAMs (112). Oxidized phosphatidylcholine from ferroptotic cancer cells also inhibits DC maturation and inhibits the differentiation of T helper 17 (TH17) cells through activation of NRF2 (113). In addition, the pharmacological induction of ferroptosis in vivo may also affect the tumor microenvironment (TME). For example, the activity of GPX4 is positively correlated with the survival of immune cells. GPX4-knockout in the myeloid lineage stimulated the appearance of intestinal tumors in intestinal epithelial cells (114). Antigen-specific CD4+ and CD8+T cells lacking GPX4 are likely to die by ferroptosis (115) (Figure 3).
Taken together, the evidence suggests that the interaction between ferroptosis and the immune system is complicated, and does not just lead to a completely positive or negative effect. Therefore, understanding the mechanisms of the adaptive immune system during ferroptosis in cancer cells is difficult, but necessary to improve the efficacy of clinical anticancer therapy.
A recent study reported that IFN-γ secreted from cytotoxic CD8+ T cells downregulated the expression of system , which makes cancer cells sensitive to ferroptosis (116), indicating that the combination therapy of ferroptosis inducers and immune checkpoint inhibitors could be a promising direction in the future. Some novel treatments also have focused on the interaction between ferroptosis and immunotherapy. Both the irradiated tumor cell-released microparticles and the combined therapy of PD-1 antibody with TGF-β inhibitor can turn M2-polarized TAMs into M1-TAMs to regulate the antitumor interactions between tumor cells and TAMs (85). Nanoparticles with an IO nanocluster core encapsulated in engineered leukocyte membranes containing PD-1 antibody and TGF-β inhibitor were designed; PD-1 antibody and TGF-β inhibitor cooperatively enhanced the immune response in the TME, resulting in an increase in the level of H2O2 in M1 polarized macrophages and triggering Fenton reactions with Fe ions released from the core of the nanoparticles. The generated hydroxyl radicals subsequently induce lethal ferroptosis to tumor cells (54). A novel conjugate AlbA-DCA was developed via inhibition of the GPX4 pathway and elimination of procarcinogenic M2-TAMs to suppress tumor progression in breast cancer (53) (Table 2 and Figure 3). Another study reported that M1 phagocytes exert higher ferroptosis resistance than M2 phagocytes, but that inducible nitric oxide synthase (iNOS)/NO-enrichment of phagocytes can modulate the sensibility of phagocytes to ferroptosis. iNOS depletion/inactivation makes M1 cells sensitive to ferroptosis, while NO donors promote ferroptosis resistance of M2 cells (117). Combining immunotherapy with irradiation can even reverse the chemo-resistance of cancer cells. As previously mentioned, RT-MPs can polarize M2-TAMs into M1-TAMs, which have been reported to release H2O2 (118). When taken up by tumor cells, RT-MPs can cause immunogenic cell death mainly through ferroptosis, thereby making cisplatin-resistant tumor cells more vulnerable to attack by macrophages (85).
Photodynamic therapy is based on photosens (PS-PDT), which are potent inducers of both ferroptosis and phenotypic maturation of BMDCs in two different cancer cell lines (108). After the combination therapy of oxygen-boosted PDT and potent ferroptosis inducers (a 2-in-1 nanoplatform constructed with hemoglobin, photosensitizer chlorin e6, and sorafenib), the level of GSH was decreased while MDA, one of the last-step products of lipid peroxidation, was increased in tumor tissues (corroborating the effects of ferroptosis). In addition, PDT upregulated the level of lymphocyte infiltration in the tumor site and stimulated secretion of IFN-γ (55). As mentioned above, IFN-γ secreted from cytotoxic CD8+ T cells can downregulate the expression of system and induce ferroptosis in cancer cells (116); thus, PDT stimulation could result in both ferroptosis and regulation of the immune TME (Table 2 and Figure 3).
Although researches on ferroptosis in cancer therapy have progressed rapidly in recent years, great problems for its clinical application remain, such as the low solubility and poor membrane permeability of ferroptosis-inducing anticancer drugs or the off-target toxicity to normal cells and tissues (119–121). In view of this plight, development of ferroptosis-driven nanotherapeutics seems to be a promising stratage (122). Ferroptosis-driven nanotherapeutics work in three main ways: First, they can trigger or promote Fenton reactions in tumor cells (123). Nanoparticles are always designed to provide two basic elements of the Fenton response: Fe ions and (or) H2O2. Ferroptosis-combined cancer therapy is mainly based on iron-containing nanomaterials. The hypoxia-induced lower pH inherent to TMEs allows easy release of Fe ions from iron-containing nanomaterials, which triggers the Fenton reaction and leads to ferroptosis in cancer cells. Second, ferroptosis-driven nanotherapeutics are used as carriers of chemotherapeutic drugs or ferroptosis inducers to inhibit the expression of the GSH/GPX4 system in tumor cells (124). For example, sorafenib can be encapsulated in a nanostructure consisting of Fe ions. The suppression of GPX4 by sorafenib combined with the tumor site-specific Fenton reaction induced by Fe ions results in a powerful tumor-specific ferroptosis (125). Nano-carriers can also enhance the internalization of platinum and iron in cancer cells that can bypass the biological barrier of cisplatin (one of the main causes of cisplatin resistance), thereby overcoming this drug resistance by tumor cells (126). Third, nanotherapeutics can provide exogenous regulation of lipid peroxidation to tumor cells, including through oral PUFA supplementation or by delivering PUFAs in nanoparticulate drug delivery systems to regulate lipid peroxidation and directly induce ferroptosis in tumor cells (127, 128).
In addition to nanotherapeutics, Gao and colleagues reported another new method to overcome multidrug resistance in the treatment of cancer through tailored ferroptotic micelles, which consist of ferroptosis inducers with arachidonic acid-conjugated amphiphilic copolymers, opening a new horizon for treating drug-resistant tumors (129). Exosomes have become popular as drug carriers with low immunogenicity, high efficiency, and high biocompatibility. Folate (FA)-vectorized exosomes loaded with erastin (erastin@FA-exo) were developed recently to act on triple-negative breast cancer (TNBC) cells with high expression of FA receptors, and which can overcome the poor water solubility and nephrotoxicity of erastin and result in a better therapeutic effect (130). Moreover, a group of studies are focusing on the relationship between ferroptosis and autophagy (131), the specific association between ferroptosis and other kinds of cell death requires more in-depth research in the future.
Ferroptosis is a novel potential target in cancer treatment, however, precise mechanisms in regulation of ferroptosis in cancers are sometimes complicated and still need to be further explored. For instance, we have mentioned that p53 plays an important role in PTX-induced ferroptosis through downregulation of SLC7A11 and SLC1A5. However, the role of p53 in ferroptosis remains obscure. According to the p53 mutation status and cellular context, p53 can have either pro- or anti-ferroptotic functions during oxidative stress states (132). To be specific, p53 seems to act like a rheostat, suppressing ferroptosis under low oxidative stress, and enhancing it under high ROS stress (133). Similarly, HO-1 also plays contradictory roles promoting or inhibiting ferroptosis depending on the cellular redox status. Downregulated HO-1 in mice renal proximal tubule cells induced ferroptosis, while upregulated HO-1 promoted ferroptosis induced by Bay through NRF2/SLC7A11/HO-1 signaling in breast cancer cells (134). A previous study reported that AMPK-mediated BECN1 phosphorylation promotes ferroptosis by inhibiting SLC7A11-mediated cystine transport (25). However, a recent study proposed the contradictory conclusion that cancer cells with high basal AMPK activation are resistant to ferroptosis and AMPK inactivation sensitizes these cells to ferroptosis. Functional and lipidomic analyses have further revealed that ferroptosis is regulated via AMPK-mediated phosphorylation of acetyl-CoA carboxylase and restrained biosynthesis of PUFAs or other fatty acids (135). It is possible that the function of AMPK in regulating ferroptosis is context dependent, which awaits further exploration in future studies.
In addition to the unclear mechanisms mentioned above, recent studies have also proposed new directions in this field. Previous research indicated that the system /GSH/GPX4 axis was the key pathway in ferroptosis, therefore, the synthesis of GSH is essential. However, inhibition of GSH synthesis did not induce lipid ROS or decrease cell viability in PDAC. Badgley et al. found that cysteine rather than GSH is a central factor in this pathway to induce ferroptosis and that PDAC cells use cysteine to synthesize both coenzyme A and GSH, which cooperatively downregulate cancerous ferroptosis (136). It was once believed that enzymatic lipid peroxidation is primarily catalyzed by arachidonate lipoxygenases (ALOXs), however, the necessity for ALOX involvement in ferroptosis induction was questioned by a recent identification of previously unknown radical-trapping activities of ALOX-inhibiting small molecules used to establish the ALOX-ferroptosis connection (137), but Zou et al. firstly confirmed that cytochrome P450 oxidoreductase (POR), a distinct ferroptosis inducer, is necessary for ferroptosis through peroxidation of polyunsaturated phospholipids on the cell membrane. The pro-ferroptosis effect of POR is applicable to various types of cancer lineages (138).
The complexity of biological system and the difficulty in clinical translation bring challenges and opportunities to the further development of ferroptosis-based cancer therapies concomitantly. The following concerns should be taken into account when applying treatments targeting ferroptosis: (1) Successful experiments in vitro or animal models cannot be directly applied to clinical practice. Clinical trials are needed to be conducted to evaluate the safety and practical efficacy of ferroptosis-inducing therapies. (2) Ferroptosis as a double-edged sword can not only be used as an effective therapeutic target, but also may cause cancers and other diseases. Therefore, special attentions should be paid about the dose, duration and tissue specificity of ferroptosis-inducing drugs to destroy cancer cells and avoid off-target toxicity to normal cells and tissues. (3) Some intrinsic characteristics of available ferroptosis inducers, such as the limited water solubility and toxic effects on kidneys, eyes and the immune systems restricted their clinical applications (119–121). As previously mentioned, antigen-specific CD4+ and CD8+T cells lacking GPX4 are likely to die by ferroptosis (115), and GPX4-knockout in the myeloid lineage can even induce intestinal tumors in intestinal epithelial cells (114), indicating an important role of GPX4 in maintaining a healthy immune system. Once the off-target toxicity to normal cells and tissues occurs, it brings serious aftereffects. Deferoxamine is a classic ferroptosis inhibitor which can be used as a neutralizer to avoid excessive ferroptosis and injury to normal cells and tissues. However, the short half-life of which limits its clinical application, indicating an urgent need to develop an effective and biocompatible novel ferroptosis inhibitor (139). Nanoscale material is another good choice to solve the problem about low efficiency and high side effects of traditional ferroptosis inducers as well as the potential off-target toxicity to normal cells and tissues. The low toxic cisplatin prodrugs can be loaded in iron oxide nanoparticles, which are accumulated at specific site of tumors in the presence of magnetic field, and the prodrugs can be reduced to the toxic cisplatin to induce ferroptosis in cancer cells specifically (126). CY-1-4 (an anti-malaria drug tryptanthrin derivative) carried by nanoparticles can significantly improve the drug's solubility and improve its transportation efficiency in vivo to induce cancerous ferroptosis with a higher efficiency (140). Potential risks also existed in the ferroptosis-driven nanotherapeutics. Nanomaterials themselves have cytotoxic effects, which may lead to a greater risk of side effects in disease treatments. The possibility of cellular protective autophagy induced by some nanomaterials is another great challenge to their further clinical application. Therefore, numerous issues should be taken into consideration in the rational design of efficient and safe ferroptosis-driven nanotherapeutics in the future.
Ferroptosis is an undeniably important manner of cell death in addition to apoptosis in cancer therapies. Here we collated and reviewed how ferroptosis was involved in the cancer treatments according to studies to date. Different therapeutics, including chemicals, targeted agents, radiotherapy and also immunotherapy, induce ferroptosis to kill cancer cells through distinct molecular mechanisms. Insensitivity and acquired resistance are the main obstacles in cancer eradication. Ferroptosis inducers are promising in the field of oncotherapy for their sensitization and synergistic effects. Integrating ferroptosis with burgeoning biotechnologies such as ferroptosis-driven nanotherapeutics may overcome the limits of traditional medicals and ferroptosis inducers with low solubility and poor membrane permeability. Due to the complexity of biological system and the difficulty in clinical translation, further studies and clinical trials are still warranted.
YW, CY, and JQ reviewed the literature and drafted the article. ML, CC, SZ, and KH finalized the paper and provided suggestions to improve it. All authors participated in designing the concept of this manuscript.
This work was funded by National Natural Science Foundation of China (81802628), Zhejiang Provincial Natural Science Foundation of China (LQ19H160045), and Zhejiang Provincial Natural Science Foundation of China (LQ20H060007).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The process of writing this thesis is along with a baby's growth. Welcome to this world, our little baby Seven. We all love you!.
1. Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. (2012) 149:1060–72. doi: 10.1016/j.cell.2012.03.042
2. Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ, et al. Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell. (2017) 171:273–85. doi: 10.1016/j.cell.2017.09.021
3. Conrad M, Kagan VE, Bayir H, Pagnussat GC, Head B, Traber MG, et al. Regulation of lipid peroxidation and ferroptosis in diverse species. Genes Dev. (2018) 32:602–19. doi: 10.1101/gad.314674.118
4. Jiang L, Kon N, Li T, Wang SJ, Su T, Hibshoosh H, et al. Ferroptosis as a p53-mediated activity during tumour suppression. Nature. (2015) 520:57–62. doi: 10.1038/nature14344
5. Viswanathan VS, Ryan MJ, Dhruv HD, Gill S, Eichhoff OM, Seashore-Ludlow B, et al. Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature. (2017) 547:453–7. doi: 10.1038/nature23007
6. Miess H, Dankworth B, Gouw AM, Rosenfeldt M, Schmitz W, Jiang M, et al. The glutathione redox system is essential to prevent ferroptosis caused by impaired lipid metabolism in clear cell renal cell carcinoma. Oncogene. (2018) 37:5435–50. doi: 10.1038/s41388-018-0315-z
7. Tsoi J, Robert L, Paraiso K, Galvan C, Sheu KM, Lay J, et al. Multi-stage differentiation defines melanoma subtypes with differential vulnerability to drug-induced iron-dependent oxidative stress. Cancer Cell. (2018) 33:890–904.e895. doi: 10.1016/j.ccell.2018.03.017
8. Gentric G, Kieffer Y, Mieulet V, Goundiam O, Bonneau C, Nemati F, et al. PML-regulated mitochondrial metabolism enhances chemosensitivity in human ovarian cancers. Cell Metab. (2019) 29:156–173.e110. doi: 10.1016/j.cmet.2018.09.002
9. Bersuker K, Hendricks JM, Li Z, Magtanong L, Ford B, Tang PH, et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature. (2019) 575:688–92. doi: 10.1038/s41586-019-1705-2
10. Doll S, Freitas FP, Shah R, Aldrovandi M, da Silva MC, Ingold I, et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature. (2019) 575:693–8. doi: 10.1038/s41586-019-1707-0
11. Kraft VAN, Bezjian CT, Pfeiffer S, Ringelstetter L, Müller C, Zandkarimi F, et al. GTP Cyclohydrolase 1/tetrahydrobiopterin counteract ferroptosis through lipid remodeling. ACS Cent Sci. (2020) 6:41–53. doi: 10.1021/acscentsci.9b01063
12. Jaramillo MC, Zhang DD. The emerging role of the Nrf2-Keap1 signaling pathway in cancer. Genes Dev. (2013) 27:2179–91. doi: 10.1101/gad.225680.113
13. Zhou B, Liu J, Kang R, Klionsky DJ, Kroemer G, Tang D. Ferroptosis is a type of autophagy-dependent cell death. Semin Cancer Biol. (2019). doi: 10.1016/j.semcancer.2019.03.002. [Epub ahead of print].
14. Yang ND, Tan SH, Ng S, Shi Y, Zhou J, Tan KS, et al. Artesunate induces cell death in human cancer cells via enhancing lysosomal function and lysosomal degradation of ferritin. J Biol Chem. (2014) 289:33425–41. doi: 10.1074/jbc.M114.564567
15. Eling N, Reuter L, Hazin J, Hamacher-Brady A, Brady NR. Identification of artesunate as a specific activator of ferroptosis in pancreatic cancer cells. Oncoscience. (2015) 2:517–32. doi: 10.18632/oncoscience.160
16. Roh JL, Kim EH, Jang H, Shin D. Nrf2 inhibition reverses the resistance of cisplatin-resistant head and neck cancer cells to artesunate-induced ferroptosis. Redox Biol. (2017) 11:254–62. doi: 10.1016/j.redox.2016.12.010
17. Zhang X, Sui S, Wang L, Li H, Zhang L, Xu S, et al. Inhibition of tumor propellant glutathione peroxidase 4 induces ferroptosis in cancer cells and enhances anticancer effect of cisplatin. J Cell Physiol. (2020) 235:3425–37. doi: 10.1002/jcp.29232
18. Guo J, Xu B, Han Q, Zhou H, Xia Y, Gong C, et al. Ferroptosis: a novel anti-tumor action for cisplatin. Cancer Res Treat. (2018) 50:445–60. doi: 10.4143/crt.2016.572
19. Chen GQ, Benthani FA, Wu J, Liang D, Bian ZX, Jiang X. Artemisinin compounds sensitize cancer cells to ferroptosis by regulating iron homeostasis. Cell Death Differ. (2020) 27:242–54. doi: 10.1038/s41418-019-0352-3
20. Lin R, Zhang Z, Chen L, Zhou Y, Zou P, Feng C, et al. Dihydroartemisinin. (DHA) induces ferroptosis and causes cell cycle arrest in head and neck carcinoma cells. Cancer Lett. (2016) 381:165–75. doi: 10.1016/j.canlet.2016.07.033
21. Du J, Wang T, Li Y, Zhou Y, Wang X, Yu X, et al. DHA inhibits proliferation and induces ferroptosis of leukemia cells through autophagy dependent degradation of ferritin. Free Radic Biol Med. (2019) 131:356–69. doi: 10.1016/j.freeradbiomed.2018.12.011
22. Zhu S, Zhang Q, Sun X, Zeh HJ III, Lotze MT, Kang R, et al. HSPA5 regulates ferroptotic cell death in cancer cells. Cancer Res. (2017) 77:2064–77. doi: 10.1158/0008-5472.CAN-16-1979
23. Lv C, Qu H, Zhu W, Xu K, Xu A, Jia B, et al. Low-dose paclitaxel inhibits tumor cell growth by regulating glutaminolysis in colorectal carcinoma cells. Front Pharmacol. (2017) 8:244. doi: 10.3389/fphar.2017.00244
24. Giannakakou P, Robey R, Fojo T, Blagosklonny MV. Low concentrations of paclitaxel induce cell type-dependent p53, p21 and G1/G2 arrest instead of mitotic arrest: molecular determinants of paclitaxel-induced cytotoxicity. Oncogene. (2001) 20:3806–13. doi: 10.1038/sj.onc.1204487
25. Song X, Zhu S, Chen P, Hou W, Wen Q, Liu J, et al. AMPK-mediated BECN1 phosphorylation promotes ferroptosis by directly blocking system Xc(-) activity. Curr Biol. (2018) 28:2388–99.e2385. doi: 10.1016/j.cub.2018.05.094
26. Sehm T, Fan Z, Ghoochani A, Rauh M, Engelhorn T, Minakaki G, et al. Sulfasalazine impacts on ferroptotic cell death and alleviates the tumor microenvironment and glioma-induced brain edema. Oncotarget. (2016) 7:36021–33. doi: 10.18632/oncotarget.8651
27. Sun X, Ou Z, Chen R, Niu X, Chen D, Kang R, et al. Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology. (2016) 63:173–84. doi: 10.1002/hep.28251
28. Dixon SJ, Patel DN, Welsch M, Skouta R, Lee ED, Hayano M, et al. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. Elife. (2014) 3:e02523. doi: 10.7554/eLife.02523.018
29. Chen L, Li X, Liu L, Yu B, Xue Y, Liu Y. Erastin sensitizes glioblastoma cells to temozolomide by restraining xCT and cystathionine-gamma-lyase function. Oncol Rep. (2015) 33:1465–74. doi: 10.3892/or.2015.3712
30. Daher B, Parks SK, Durivault J, Cormerais Y, Baidarjad H, Tambutte E, et al. Genetic ablation of the cystine transporter xCT in PDAC cells inhibits mTORC1, Growth, survival, and tumor formation via nutrient and oxidative stresses. Cancer Res. (2019) 79:3877–90. doi: 10.1158/0008-5472.CAN-18-3855
31. Sato M, Kusumi R, Hamashima S, Kobayashi S, Sasaki S, Komiyama Y, et al. The ferroptosis inducer erastin irreversibly inhibits system xc- and synergizes with cisplatin to increase cisplatin's cytotoxicity in cancer cells. Sci Rep. (2018) 8:968. doi: 10.1038/s41598-018-19213-4
32. Yamaguchi H, Hsu JL, Chen CT, Wang YN, Hsu MC, Chang SS, et al. Caspase-independent cell death is involved in the negative effect of EGF receptor inhibitors on cisplatin in non-small cell lung cancer cells. Clin Cancer Res. (2013) 19:845–54. doi: 10.1158/1078-0432.CCR-12-2621
33. Ye J, Jiang X, Dong Z, Hu S, Xiao M. Low-concentration PTX And RSL3 inhibits tumor cell growth synergistically by inducing ferroptosis in mutant p53 hypopharyngeal squamous carcinoma. Cancer Manag Res. (2019) 11:9783–92. doi: 10.2147/CMAR.S217944
34. Chen TC, Chuang JY, Ko CY, Kao TJ, Yang PY, Yu CH, et al. AR ubiquitination induced by the curcumin analog suppresses growth of temozolomide-resistant glioblastoma through disrupting GPX4-mediated redox homeostasis. Redox Biol. (2020) 30:101413. doi: 10.1016/j.redox.2019.101413
35. Yu Y, Xie Y, Cao L, Yang L, Yang M, Lotze MT, et al. The ferroptosis inducer erastin enhances sensitivity of acute myeloid leukemia cells to chemotherapeutic agents. Mol Cell Oncol. (2015) 2:e1054549. doi: 10.1080/23723556.2015.1054549
36. Yamaguchi Y, Kasukabe T, Kumakura S. Piperlongumine rapidly induces the death of human pancreatic cancer cells mainly through the induction of ferroptosis. Int J Oncol. (2018) 52:1011–22. doi: 10.3892/ijo.2018.4259
37. Ma S, Dielschneider RF, Henson ES, Xiao W, Choquette TR, Blankstein AR, et al. Ferroptosis and autophagy induced cell death occur independently after siramesine and lapatinib treatment in breast cancer cells. PLoS ONE. (2017) 12:e0182921. doi: 10.1371/journal.pone.0182921
38. Villalpando-Rodriguez GE, Blankstein AR, Konzelman C, Gibson SB. Lysosomal destabilizing drug siramesine and the dual tyrosine kinase inhibitor lapatinib induce a synergistic ferroptosis through reduced heme oxygenase-1. (HO-1) levels. Oxid Med Cell Longev. (2019) 2019:9561281. doi: 10.1155/2019/9561281
39. Heslop KA, Rovini A, Hunt EG, Fang D, Morris ME, Christie CF, et al. JNK activation and translocation to mitochondria mediates mitochondrial dysfunction and cell death induced by VDAC opening and sorafenib in hepatocarcinoma cells. Biochem Pharmacol. (2020) 171:113728. doi: 10.1016/j.bcp.2019.113728
40. Shibata Y, Yasui H, Higashikawa K, Miyamoto N, Kuge Y. Erastin, a ferroptosis-inducing agent, sensitized cancer cells to X-ray irradiation via glutathione starvation in vitro and in vivo. PLoS ONE. (2019) 14:e0225931. doi: 10.1371/journal.pone.0225931
41. Nagane M, Kanai E, Shibata Y, Shimizu T, Yoshioka C, Maruo T, et al. Sulfasalazine, an inhibitor of the cystine-glutamate antiporter, reduces DNA damage repair and enhances radiosensitivity in murine B16F10 melanoma. PLoS ONE. (2018) 13:e0195151. doi: 10.1371/journal.pone.0195151
42. Khorsandi K, Kianmehr Z, Hosseinmardi Z, Hosseinzadeh R. Anti-cancer effect of gallic acid in presence of low level laser irradiation: ROS production and induction of apoptosis and ferroptosis. Cancer Cell Int. (2020) 20:18. doi: 10.1186/s12935-020-1100-y
43. Ye LF, Chaudhary KR, Zandkarimi F, Harken AD, Kinslow CJ, Upadhyayula PS, et al. Radiation-induced lipid peroxidation triggers ferroptosis and synergizes with ferroptosis inducers. ACS Chem Biol. (2020) 15:469–84. doi: 10.1021/acschembio.9b00939
44. Sleire L, Skeie BS, Netland IA, Forde HE, Dodoo E, Selheim F, et al. Drug repurposing: sulfasalazine sensitizes gliomas to gamma knife radiosurgery by blocking cystine uptake through system Xc-, leading to glutathione depletion. Oncogene. (2015) 34:5951–9. doi: 10.1038/onc.2015.60
45. Cobler L, Zhang H, Suri P, Park C, Timmerman LA. xCT inhibition sensitizes tumors to gamma-radiation via glutathione reduction. Oncotarget. (2018) 9:32280–97. doi: 10.18632/oncotarget.25794
46. Lang X, Green MD, Wang W, Yu J, Choi JE, Jiang L, et al. Radiotherapy and immunotherapy promote tumoral lipid oxidation and ferroptosis via synergistic repression of SLC7A11. Cancer Discov. (2019) 9:1673–85. doi: 10.1158/2159-8290.CD-19-0338
47. Hung MS, Chen IC, Lee CP, Huang RJ, Chen PC, Tsai YH, et al. Statin improves survival in patients with EGFR-TKI lung cancer: a nationwide population-based study. PLoS ONE. (2017) 12:e0171137. doi: 10.1371/journal.pone.0171137
48. Gai C, Yu M, Li Z, Wang Y, Ding D, Zheng J, et al. Acetaminophen sensitizing erastin-induced ferroptosis via modulation of Nrf2/heme oxygenase-1 signaling pathway in non-small-cell lung cancer. J Cell Physiol. (2020) 235:3329–39. doi: 10.1002/jcp.29221
49. Park S, Oh J, Kim M, Jin EJ. Bromelain effectively suppresses Kras-mutant colorectal cancer by stimulating ferroptosis. Anim Cells Syst. (2018) 22:334–40. doi: 10.1080/19768354.2018.1512521
50. Bi J, Yang S, Li L, Dai Q, Borcherding N, Wagner BA, et al. Metadherin enhances vulnerability of cancer cells to ferroptosis. Cell Death Dis. (2019) 10:682. doi: 10.1038/s41419-019-1897-2
51. Tesfay L, Paul BT, Konstorum A, Deng Z, Cox AO, Lee J, et al. Stearoyl-CoA desaturase 1 protects ovarian cancer cells from ferroptotic cell death. Cancer Res. (2019) 79:5355–66. doi: 10.1158/0008-5472.CAN-19-0369
52. Kasukabe T, Honma Y, Okabe-Kado J, Higuchi Y, Kato N, Kumakura S. Combined treatment with cotylenin A and phenethyl isothiocyanate induces strong antitumor activity mainly through the induction of ferroptotic cell death in human pancreatic cancer cells. Oncol Rep. (2016) 36:968–76. doi: 10.3892/or.2016.4867
53. Wei G, Sun J, Luan W, Hou Z, Wang S, Cui S, et al. Natural product albiziabioside a conjugated with pyruvate dehydrogenase kinase inhibitor dichloroacetate to induce apoptosis-ferroptosis-M2-TAMs polarization for combined cancer therapy. J Med Chem. (2019) 62:8760–72. doi: 10.1021/acs.jmedchem.9b00644
54. Zhang F, Li F, Lu GH, Nie W, Zhang L, Lv Y, et al. Engineering magnetosomes for ferroptosis/immunomodulation synergism in cancer. ACS Nano. (2019) 13:5662–73. doi: 10.1021/acsnano.9b00892
55. Xu T, Ma Y, Yuan Q, Hu H, Hu X, Qian Z, et al. Enhanced ferroptosis by oxygen-boosted phototherapy based on a 2-in-1 nanoplatform of ferrous hemoglobin for tumor synergistic therapy. ACS Nano. (2020) 14:3414–25. doi: 10.1021/acsnano.9b09426
56. Torii S, Shintoku R, Kubota C, Yaegashi M, Torii R, Sasaki M, et al. An essential role for functional lysosomes in ferroptosis of cancer cells. Biochem J. (2016) 473:769–77. doi: 10.1042/BJ20150658
57. Sehm T, Rauh M, Wiendieck K, Buchfelder M, Eyupoglu IY, Savaskan NE. Temozolomide toxicity operates in a xCT/SLC7a11 dependent manner and is fostered by ferroptosis. Oncotarget. (2016) 7:74630–47. doi: 10.18632/oncotarget.11858
58. Yagoda N, von Rechenberg M, Zaganjor E, Bauer AJ, Yang WS, Fridman DJ, et al. RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature. (2007) 447:864–8. doi: 10.1038/nature05859
59. Fan L, Yin S, Zhang E, Hu H. Role of p62 in the regulation of cell death induction. Apoptosis. (2018) 23:187–93. doi: 10.1007/s10495-018-1445-z
60. Wu Y, Zhang S, Gong X, Tam S, Xiao D, Liu S, et al. The epigenetic regulators and metabolic changes in ferroptosis-associated cancer progression. Mol Cancer. (2020) 19:39. doi: 10.1186/s12943-020-01157-x
61. Kang R, Zhu S, Zeh HJ, Klionsky DJ, Tang D. BECN1 is a new driver of ferroptosis. Autophagy. (2018) 14:2173–5. doi: 10.1080/15548627.2018.1513758
62. Gao H, Bai Y, Jia Y, Zhao Y, Kang R, Tang D, et al. Ferroptosis is a lysosomal cell death process. Biochem Biophys Res Commun. (2018) 503:1550–6. doi: 10.1016/j.bbrc.2018.07.078
63. Wu Z, Geng Y, Lu X, Shi Y, Wu G, Zhang M, et al. Chaperone-mediated autophagy is involved in the execution of ferroptosis. Proc Natl Acad Sci USA. (2019) 116:2996–3005. doi: 10.1073/pnas.1819728116
64. Ma S, Henson ES, Chen Y, Gibson SB. Ferroptosis is induced following siramesine and lapatinib treatment of breast cancer cells. Cell Death Dis. (2016) 7:e2307. doi: 10.1038/cddis.2016.208
65. Baidoo KE, Yong K, Brechbiel MW. Molecular pathways: targeted alpha-particle radiation therapy. Clin Cancer Res. (2013) 19:530–7. doi: 10.1158/1078-0432.CCR-12-0298
66. Ashwood-Smith MJ. The effect of whole-body x-irradiation on the glutathione content of rat thymus. Int J Radiat Biol Relat Stud Phys Chem Med. (1961) 3:125–32. doi: 10.1080/09553006114550141
67. Azzam EI, Jay-Gerin JP, Pain D. Ionizing radiation-induced metabolic oxidative stress and prolonged cell injury. Cancer Lett. (2012) 327:48–60. doi: 10.1016/j.canlet.2011.12.012
68. Fierro S, Yoshikawa M, Nagano O, Yoshimi K, Saya H, Einaga Y. In vivo assessment of cancerous tumors using boron doped diamond microelectrode. Sci Rep. (2012) 2:901. doi: 10.1038/srep00901
69. Bump EA, Yu NY, Brown JM. Radiosensitization of hypoxic tumor cells by depletion of intracellular glutathione. Science. (1982) 217:544–5. doi: 10.1126/science.7089580
70. Allalunis-Turner MJ, Day RS III, McKean JD, Petruk KC, Allen PB, Aronyk KE, et al. Glutathione levels and chemosensitizing effects of buthionine sulfoximine in human malignant glioma cells. J Neurooncol. (1991) 11:157–64. doi: 10.1007/BF02390175
71. Lei G, Zhang Y, Koppula P, Liu X, Zhang J, Lin SH, et al. The role of ferroptosis in ionizing radiation-induced cell death and tumor suppression. Cell Res. (2020) 30:146–62. doi: 10.1038/s41422-019-0263-3
72. Warner GJ, Berry MJ, Moustafa ME, Carlson BA, Hatfield DL, Faust JR. Inhibition of selenoprotein synthesis by selenocysteine tRNA[Ser]Sec lacking isopentenyladenosine. J Biol Chem. (2000) 275:28110–9. doi: 10.1074/jbc.M001280200
73. Santoro MM. The antioxidant role of non-mitochondrial CoQ10: mystery solved! Cell Metab 31:13–15. doi: 10.1016/j.cmet.2019.12.007
74. Han JY, Lee SH, Yoo NJ, Hyung LS, Moon YJ, Yun T, et al. A randomized phase II study of gefitinib plus simvastatin versus gefitinib alone in previously treated patients with advanced non-small cell lung cancer. Clin Cancer Res. (2011) 17:1553–60. doi: 10.1158/1078-0432.CCR-10-2525
75. Fiala O, Pesek M, Finek J, Minarik M, Benesova L, Bortlicek Z, et al. Statins augment efficacy of EGFR-TKIs in patients with advanced-stage non-small cell lung cancer harbouring KRAS mutation. Tumour Biol. (2015) 36:5801–5. doi: 10.1007/s13277-015-3249-x
76. Brown CW, Amante JJ, Chhoy P, Elaimy AL, Liu H, Zhu LJ, et al. Prominin2 drives ferroptosis resistance by stimulating iron export. Dev Cell. (2019) 51:575–86.e574. doi: 10.1016/j.devcel.2019.10.007
77. Li Y, Yan H, Xu X, Liu H, Wu C, Zhao L. Erastin/sorafenib induces cisplatin-resistant non-small cell lung cancer cell ferroptosis through inhibition of the Nrf2/xCT pathway. Oncol Lett. (2020) 19:323–33. doi: 10.3892/ol.2019.11066
78. Liu Q, Wang K. The induction of ferroptosis by impairing STAT3/Nrf2/GPx4 signaling enhances the sensitivity of osteosarcoma cells to cisplatin. Cell Biol Int. (2019) 43:1245–56. doi: 10.1002/cbin.11121
79. Hassannia B, Wiernicki B, Ingold I, Qu F, Van Herck S, Tyurina YY, et al. Nano-targeted induction of dual ferroptotic mechanisms eradicates high-risk neuroblastoma. J Clin Invest. (2018) 128:3341–55. doi: 10.1172/JCI99032
80. Zhou HH, Chen X, Cai LY, Nan XW, Chen JH, Chen XX, et al. Erastin reverses ABCB1-mediated docetaxel resistance in ovarian cancer. Front Oncol. (2019) 9:1398. doi: 10.3389/fonc.2019.01398
81. Lu X, Meng T. Depletion of tumor-associated macrophages enhances the anti-tumor effect of docetaxel in a murine epithelial ovarian cancer. Immunobiology. (2019) 224:355–61. doi: 10.1016/j.imbio.2019.03.002
82. Otsuki Y, Yamasaki J, Suina K, Okazaki S, Koike N, Saya H, et al. Vasodilator oxyfedrine inhibits aldehyde metabolism and thereby sensitizes cancer cells to xCT-targeted therapy. Cancer Sci. (2020) 111:127–36. doi: 10.1111/cas.14224
83. Hangauer MJ, Viswanathan VS, Ryan MJ, Bole D, Eaton JK, Matov A, et al. Drug-tolerant persister cancer cells are vulnerable to GPX4 inhibition. Nature. (2017) 551:247–50. doi: 10.1038/nature24297
84. Pan X, Lin Z, Jiang D, Yu Y, Yang D, Zhou H, et al. Erastin decreases radioresistance of NSCLC cells partially by inducing GPX4-mediated ferroptosis. Oncol Lett. (2019) 17:3001–8. doi: 10.3892/ol.2019.9888
85. Wan C, Sun Y, Tian Y, Lu L, Dai X, Meng J, et al. Irradiated tumor cell-derived microparticles mediate tumor eradication via cell killing and immune reprogramming. Sci Adv. (2020) 6:eaay9789. doi: 10.1126/sciadv.aay9789
86. Park IH, Kim JY, Jung JI, Han JY. Lovastatin overcomes gefitinib resistance in human non-small cell lung cancer cells with K-Ras mutations. Invest New Drugs. (2010) 28:791–9. doi: 10.1007/s10637-009-9319-4
87. Chen J, Bi H, Hou J, Zhang X, Zhang C, Yue L, et al. Atorvastatin overcomes gefitinib resistance in KRAS mutant human non-small cell lung carcinoma cells. Cell Death Dis. (2013) 4:e814. doi: 10.1038/cddis.2013.312
88. Hwang KE, Kwon SJ, Kim YS, Park DS, Kim BR, Yoon KH, et al. Effect of simvastatin on the resistance to EGFR tyrosine kinase inhibitors in a non-small cell lung cancer with the T790M mutation of EGFR. Exp Cell Res. (2014) 323:288–96. doi: 10.1016/j.yexcr.2014.02.026
89. Singh A, Misra V, Thimmulappa RK, Lee H, Ames S, Hoque MO, et al. Dysfunctional KEAP1-NRF2 interaction in non-small-cell lung cancer. PLoS Med. (2006) 3:e420. doi: 10.1371/journal.pmed.0030420
90. Loignon M, Miao W, Hu L, Bier A, Bismar TA, Scrivens PJ, et al. Cul3 overexpression depletes Nrf2 in breast cancer and is associated with sensitivity to carcinogens, to oxidative stress, and to chemotherapy. Mol Cancer Ther. (2009) 8:2432–40. doi: 10.1158/1535-7163.MCT-08-1186
91. Padmanabhan B, Tong KI, Ohta T, Nakamura Y, Scharlock M, Ohtsuji M, et al. Structural basis for defects of Keap1 activity provoked by its point mutations in lung cancer. Mol Cell. (2006) 21:689–700. doi: 10.1016/j.molcel.2006.01.013
92. Britten RA, Green JA, Warenius HM. Cellular glutathione. (GSH) and glutathione S-transferase. (GST) activity in human ovarian tumor biopsies following exposure to alkylating agents. Int J Radiat Oncol Biol Phys. (1992) 24:527–31. doi: 10.1016/0360-3016(92)91069-Y
93. Ju HQ, Gocho T, Aguilar M, Wu M, Zhuang ZN, Fu J, et al. Mechanisms of overcoming intrinsic resistance to gemcitabine in pancreatic ductal adenocarcinoma through the redox modulation. Mol Cancer Ther. (2015) 14:788–98. doi: 10.1158/1535-7163.MCT-14-0420
94. Habib E, Linher-Melville K, Lin HX, Singh G. Expression of xCT and activity of system xc(-) are regulated by NRF2 in human breast cancer cells in response to oxidative stress. Redox Biol. (2015) 5:33–42. doi: 10.1016/j.redox.2015.03.003
95. Efferth T, Benakis A, Romero MR, Tomicic M, Rauh R, Steinbach D, et al. Enhancement of cytotoxicity of artemisinins toward cancer cells by ferrous iron. Free Radic Biol Med. (2004) 37:998–1009. doi: 10.1016/j.freeradbiomed.2004.06.023
96. Habashy HO, Powe DG, Staka CM, Rakha EA, Ball G, Green AR, et al. Transferrin receptor. (CD71) is a marker of poor prognosis in breast cancer and can predict response to tamoxifen. Breast Cancer Res Treat. (2010) 119:283–93. doi: 10.1007/s10549-009-0345-x
97. Chekhun VF, Lukyanova NY, Burlaka CA, Bezdenezhnykh NA, Shpyleva SI, Tryndyak VP, et al. Iron metabolism disturbances in the MCF-7 human breast cancer cells with acquired resistance to doxorubicin and cisplatin. Int J Oncol. (2013) 43:1481–6. doi: 10.3892/ijo.2013.2063
98. Basuli D, Tesfay L, Deng Z, Paul B, Yamamoto Y, Ning G, et al. Iron addiction: a novel therapeutic target in ovarian cancer. Oncogene. (2017) 36:4089–99. doi: 10.1038/onc.2017.11
99. Liu J, Kuang F, Kroemer G, Klionsky DJ, Kang R, Tang D. Autophagy-dependent ferroptosis: machinery and regulation. Cell Chem Biol. (2020) 27:420–35. doi: 10.1016/j.chembiol.2020.02.005
100. Barabas K, Faulk WP. Transferrin receptors associate with drug resistance in cancer cells. Biochem Biophys Res Commun. (1993) 197:702–8. doi: 10.1006/bbrc.1993.2536
101. Wu J, Lu Y, Lee A, Pan X, Yang X, Zhao X, et al. Reversal of multidrug resistance by transferrin-conjugated liposomes co-encapsulating doxorubicin and verapamil. J Pharm Pharm Sci. (2007) 10:350–7.
102. Campanella A, Santambrogio P, Fontana F, Frenquelli M, Cenci S, Marcatti M, et al. Iron increases the susceptibility of multiple myeloma cells to bortezomib. Haematologica. (2013) 98:971–9. doi: 10.3324/haematol.2012.074872
103. Okazaki S, Shintani S, Hirata Y, Suina K, Semba T, Yamasaki J, et al. Synthetic lethality of the ALDH3A1 inhibitor dyclonine and xCT inhibitors in glutathione deficiency-resistant cancer cells. Oncotarget. (2018) 9:33832–43. doi: 10.18632/oncotarget.26112
104. Schinkel AH, Jonker JW. Mammalian drug efflux transporters of the ATP binding cassette. (ABC) family: an overview. Adv Drug Deliv Rev. (2003) 55:3–29. doi: 10.1016/S0169-409X(02)00169-2
105. Gottesman MM, Ling V. The molecular basis of multidrug resistance in cancer: the early years of P-glycoprotein research. FEBS Lett. (2006) 580:998–1009. doi: 10.1016/j.febslet.2005.12.060
106. Linkermann A, Skouta R, Himmerkus N, Mulay SR, Dewitz C, De Zen F, et al. Synchronized renal tubular cell death involves ferroptosis. Proc Natl Acad Sci USA. (2014) 111:16836–41. doi: 10.1073/pnas.1415518111
107. Kim SE, Zhang L, Ma K, Riegman M, Chen F, Ingold I, et al. Ultrasmall nanoparticles induce ferroptosis in nutrient-deprived cancer cells and suppress tumour growth. Nat Nanotechnol. (2016) 11:977–85. doi: 10.1038/nnano.2016.164
108. Turubanova VD, Balalaeva IV, Mishchenko TA, Catanzaro E, Alzeibak R, Peskova NN, et al. Immunogenic cell death induced by a new photodynamic therapy based on photosens and photodithazine. J Immunother Cancer. (2019) 7:350. doi: 10.1186/s40425-019-0826-3
109. Del Re DP, Amgalan D, Linkermann A, Liu Q, Kitsis RN. Fundamental mechanisms of regulated cell death and implications for heart disease. Physiol Rev. (2019) 99:1765–817. doi: 10.1152/physrev.00022.2018
110. Zhang Y, Song J, Zhao Z, Yang M, Chen M, Liu C, et al. Single-cell transcriptome analysis reveals tumor immune microenvironment heterogenicity and granulocytes enrichment in colorectal cancer liver metastases. Cancer Lett. (2020) 470:84–94. doi: 10.1016/j.canlet.2019.10.016
111. Mantovani A, Marchesi F, Malesci A, Laghi L, Allavena P. Tumour-associated macrophages as treatment targets in oncology. Nat Rev Clin Oncol. (2017) 14:399–416. doi: 10.1038/nrclinonc.2016.217
112. Dai E, Han L, Liu J, Xie Y, Kroemer G, Klionsky DJ, et al. Autophagy-dependent ferroptosis drives tumor-associated macrophage polarization via release and uptake of oncogenic KRAS protein. Autophagy. (2020) 1–15. doi: 10.1080/15548627.2020.1714209. [Epub ahead of print].
113. Rothe T, Gruber F, Uderhardt S, Ipseiz N, Rossner S, Oskolkova O, et al. 12/15-Lipoxygenase-mediated enzymatic lipid oxidation regulates DC maturation and function. J Clin Invest. (2015) 125:1944–54. doi: 10.1172/JCI78490
114. Canli O, Nicolas AM, Gupta J, Finkelmeier F, Goncharova O, Pesic M, et al. Myeloid cell-derived reactive oxygen species induce epithelial mutagenesis. Cancer Cell. (2017) 32:869–83.e865. doi: 10.1016/j.ccell.2017.11.004
115. Matsushita M, Freigang S, Schneider C, Conrad M, Bornkamm GW, Kopf M. T cell lipid peroxidation induces ferroptosis and prevents immunity to infection. J Exp Med. (2015) 212:555–68. doi: 10.1084/jem.20140857
116. Wang W, Green M, Choi JE, Gijon M, Kennedy PD, Johnson JK, et al. CD8(+) T cells regulate tumour ferroptosis during cancer immunotherapy. Nature. (2019) 569:270–4. doi: 10.1038/s41586-019-1170-y
117. Kapralov AA, Yang Q, Dar HH, Tyurina YY, Anthonymuthu TS, Kim R, et al. Redox lipid reprogramming commands susceptibility of macrophages and microglia to ferroptotic death. Nat Chem Biol. (2020) 16:278–90. doi: 10.1038/s41589-019-0462-8
118. Sindrilaru A, Peters T, Wieschalka S, Baican C, Baican A, Peter H, et al. An unrestrained proinflammatory M1 macrophage population induced by iron impairs wound healing in humans and mice. J Clin Invest. (2011) 121:985–97. doi: 10.1172/JCI44490
119. Rybak LP, Mukherjea D, Jajoo S, Ramkumar V. Cisplatin ototoxicity and protection: clinical and experimental studies. Tohoku J Exp Med. (2009) 219:177–86. doi: 10.1620/tjem.219.177
120. Miller RP, Tadagavadi RK, Ramesh G, Reeves WB. Mechanisms of cisplatin nephrotoxicity. Toxins. (2010) 2:2490–518. doi: 10.3390/toxins2112490
121. Jun KY, Park SE, Liang JL, Jahng Y, Kwon Y. Benzo[b]tryptanthrin inhibits MDR1, topoisomerase activity, and reverses adriamycin resistance in breast cancer cells. ChemMedChem. (2015) 10:827–35. doi: 10.1002/cmdc.201500068
122. Shan X, Li S, Sun B, Chen Q, Sun J, He Z, et al. Ferroptosis-driven nanotherapeutics for cancer treatment. J Control Release. (2020) 319:322–32. doi: 10.1016/j.jconrel.2020.01.008
123. Qian X, Zhang J, Gu Z, Chen Y. Nanocatalysts-augmented Fenton chemical reaction for nanocatalytic tumor therapy. Biomaterials. (2019) 211:1–13. doi: 10.1016/j.biomaterials.2019.04.023
124. Wang S, Li F, Qiao R, Hu X, Liao H, Chen L, et al. Arginine-rich manganese silicate nanobubbles as a ferroptosis-inducing agent for tumor-targeted theranostics. ACS Nano. (2018) 12:12380–92. doi: 10.1021/acsnano.8b06399
125. Liu T, Liu W, Zhang M, Yu W, Gao F, Li C, et al. Ferrous-supply-regeneration nanoengineering for cancer-cell-specific ferroptosis in combination with imaging-guided photodynamic therapy. ACS Nano. (2018) 12:12181–92. doi: 10.1021/acsnano.8b05860
126. Ma P, Xiao H, Yu C, Liu J, Cheng Z, Song H, et al. Enhanced cisplatin chemotherapy by iron oxide nanocarrier-mediated generation of highly toxic reactive oxygen species. Nano Lett. (2017) 17:928–37. doi: 10.1021/acs.nanolett.6b04269
127. Ou W, Mulik RS, Anwar A, McDonald JG, He X, Corbin IR. Low-density lipoprotein docosahexaenoic acid nanoparticles induce ferroptotic cell death in hepatocellular carcinoma. Free Radic Biol Med. (2017) 112:597–607. doi: 10.1016/j.freeradbiomed.2017.09.002
128. Zhou Z, Song J, Tian R, Yang Z, Yu G, Lin L, et al. Activatable singlet oxygen generation from lipid hydroperoxide nanoparticles for cancer therapy. Angew Chem Int Ed Engl. (2017) 56:6492–6. doi: 10.1002/anie.201701181
129. Gao M, Deng J, Liu F, Fan A, Wang Y, Wu H, et al. Triggered ferroptotic polymer micelles for reversing multidrug resistance to chemotherapy. Biomaterials. (2019) 223:119486. doi: 10.1016/j.biomaterials.2019.119486
130. Yu M, Gai C, Li Z, Ding D, Zheng J, Zhang W, et al. Targeted exosome-encapsulated erastin induced ferroptosis in triple negative breast cancer cells. Cancer Sci. (2019) 110:3173–82. doi: 10.1111/cas.14181
131. Hou W, Xie Y, Song X, Sun X, Lotze MT, Zeh HJIII, et al. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy. (2016) 12:1425–8. doi: 10.1080/15548627.2016.1187366
132. Gnanapradeepan K, Basu S, Barnoud T, Budina-Kolomets A, Kung CP, Murphy ME. The p53 tumor suppressor in the control of metabolism and ferroptosis. Front Endocrinol. (2018) 9:124. doi: 10.3389/fendo.2018.00124
133. Kruiswijk F, Labuschagne CF, Vousden KH. p53 in survival, death and metabolic health: a lifeguard with a licence to kill. Nat Rev Mol Cell Biol. (2015) 16:393–405. doi: 10.1038/nrm4007
134. Chang LC, Chiang SK, Chen SE, Yu YL, Chou RH, Chang WC. Heme oxygenase-1 mediates BAY 11-7085 induced ferroptosis. Cancer Lett. (2018) 416:124–37. doi: 10.1016/j.canlet.2017.12.025
135. Lee H, Zandkarimi F, Zhang Y, Meena JK, Kim J, Zhuang L, et al. Energy-stress-mediated AMPK activation inhibits ferroptosis. Nat Cell Biol. (2020) 22:225–34. doi: 10.1038/s41556-020-0461-8
136. Badgley MA, Kremer DM, Maurer HC, DelGiorno KE, Lee HJ, Purohit V, et al. Cysteine depletion induces pancreatic tumor ferroptosis in mice. Science. (2020) 368:85–9. doi: 10.1126/science.aaw9872
137. Shah R, Shchepinov MS, Pratt DA. Resolving the role of lipoxygenases in the initiation and execution of ferroptosis. ACS Cent Sci. (2018) 4:387–96. doi: 10.1021/acscentsci.7b00589
138. Zou Y, Li H, Graham ET, Deik AA, Eaton JK, Wang W, et al. Cytochrome P450 oxidoreductase contributes to phospholipid peroxidation in ferroptosis. Nat Chem Biol. (2020) 16:302–9. doi: 10.1038/s41589-020-0472-6
139. You L, Wang J, Liu T, Zhang Y, Han X, Wang T, et al. Targeted brain delivery of rabies virus glycoprotein 29-modified deferoxamine-loaded nanoparticles reverses functional deficits in parkinsonian mice. ACS Nano. (2018) 12:4123–39. doi: 10.1021/acsnano.7b08172
Keywords: ferroptosis, cancer therapy, drug resistance, immunotherapy, lipid peroxiation
Citation: Wu Y, Yu C, Luo M, Cen C, Qiu J, Zhang S and Hu K (2020) Ferroptosis in Cancer Treatment: Another Way to Rome. Front. Oncol. 10:571127. doi: 10.3389/fonc.2020.571127
Received: 11 June 2020; Accepted: 25 August 2020;
Published: 25 September 2020.
Edited by:
Sergio Giannattasio, Istituto di Biomembrane, Bioenergetica e Biotecnologie Molecolari (IBIOM), ItalyReviewed by:
Gaetano Cairo, University of Milan, ItalyCopyright © 2020 Wu, Yu, Luo, Cen, Qiu, Zhang and Hu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kaimin Hu, aHVrbWJkbkB6anUuZWR1LmNu; Suzhan Zhang, enVjaUB6anUuZWR1LmNu
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.