 Fen Wang
Fen Wang Shubin Wang2
Shubin Wang2 Qing Zhou
Qing Zhou
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 20 October 2020
Sec. Thoracic Oncology
Volume 10 - 2020 | https://doi.org/10.3389/fonc.2020.568059
This article is part of the Research Topic Issues and Challenges in NSCLC Immunotherapy View all 26 articles
Immunotherapy has revolutionized lung cancer treatment in the past decade. By reactivating the host’s immune system, immunotherapy significantly prolongs survival in some advanced lung cancer patients. However, resistance to immunotherapy is frequent, which manifests as a lack of initial response or clinical benefit to therapy (primary resistance) or tumor progression after the initial period of response (acquired resistance). Overcoming immunotherapy resistance is challenging owing to the complex and dynamic interplay among malignant cells and the defense system. This review aims to discuss the mechanisms that drive immunotherapy resistance and the innovative strategies implemented to overcome it in lung cancer.
The discovery of the immune checkpoint inhibitors (ICIs), represented by the monoclonal antibodies that block cytotoxic T−lymphocyte−associated protein 4 (CTLA-4), programmed death protein 1 (PD-1), and programmed death protein ligand 1 (PD-L1), has revolutionized the therapeutic landscape of lung cancer. The significant survival benefit derived from ICI-containing treatment has established it as the mainstay first-line therapy in patients with advanced or locally advanced non-small cell lung cancer (NSCLC) and extensive small-cell lung cancer (SCLC). Unprecedented long-term clinical benefit or even, in some cases, a complete recovery has been witnessed in lung cancer, particularly in patients with high PD-L1-expressing tumors (1–3). Currently, investigations are under way aimed at integrating immunotherapy in the treatment of early-stage lung cancer.
However, most patients with NSCLC develop primary resistance during ICI monotherapy and only 15 to 20% achieve partial or complete response (3). Acquired resistance also occurs in initially responding patients with advanced NSCLC treated with ICIs, after a median progression-free survival (PFS) of 4–10 months (4–9). The mechanisms of resistance to immunotherapy are not yet fully understood, and methods to overcome them must be developed. Herein, we discuss the pathways driving resistance to immunotherapy in lung cancer to help clinicians in their current practice, as well as identify future research priorities and treatment strategies.
Unlike molecular targeted therapy and chemotherapy targeting tumor cells, immunotherapy targets the immune system of the host by mobilizing the immune cells to recognize and eventually eliminate tumor cells. This mechanism of action determines the complexity of the resistance mechanisms in immunotherapy. Different mechanisms of immunotherapy resistance are listed in Table 1.
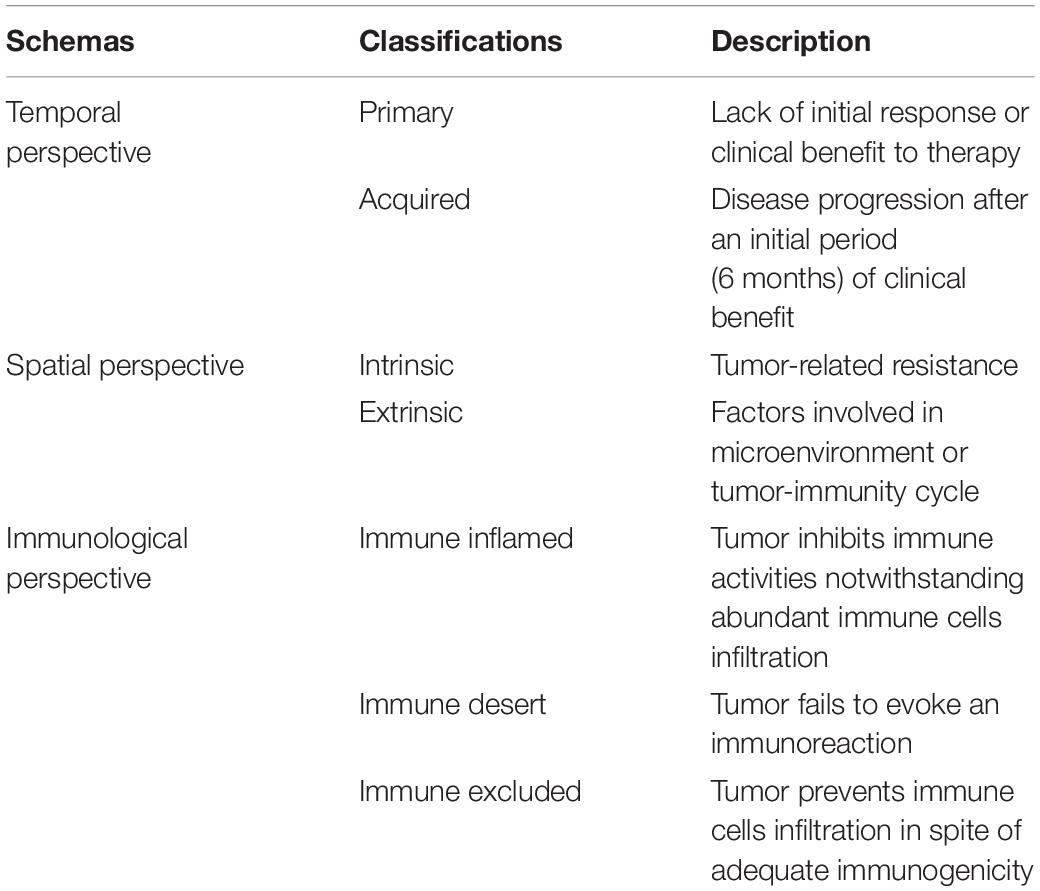
Table 1. Different schemas of resistance to immunotherapy.
In accordance with the timing of development, resistance can be considered as either primary, when no initial response or clinical benefit to the therapy is observed, or acquired, as disease progression occurs after an initial period of clinical benefit (10). Clinically, 6-month treatment duration is adopted as a cutoff value (11). This classification schema correlates with real-time observations by clinicians and contributes to the clinical decision-making process in the absence of other information such as immune characteristics and tumor genetics.
Resistance is additionally classified as intrinsic or extrinsic to cancer cells. The former occurs in the tumor cell itself and encompasses the inherent characteristics related to gene expression, cell signaling, immune recognition, and DNA damage response, whereas the latter is seen in the microenvironment or systemic circulation throughout the T-cell bioactivation process (12, 13).
The cancer−immunity cycle is linked to immunotherapy resistance in another related schema (14). This classification divides resistance from an immunological perspective into immune desert (tumor fails to evoke an immune reaction), immune inflamed (tumor inhibits immune activities notwithstanding abundant immune cells infiltration), or excluded (tumor prevents immune cells infiltration in spite of adequate immunogenicity) (13).
It is noteworthy that the immune response is a continuous and dynamic process rather than categorical (binary). Multiple complex interactions, including immunologic, genomic, and host characteristics and treatment interventions, rather than a single, dominant determinant are involved in the resistance to immunotherapy. The fs can be overlapping or parallel in some cases despite the different timing of occurrence (11).
Underlying mechanisms of primary resistance span an extensive range from tumor factors including genomic features, transcriptomic signatures, and immune landscape, to host factors. The potential mechanisms of acquired resistance at least partly overlap with those involved in primary resistance and mainly include loss of neoantigen and deficiency in presentation, loss of T-cell effector function, and up-regulation of alternate immune checkpoint receptors (10). Here, we will discuss the mechanisms of resistance to immunotherapy from tumor aspects (intrinsic and extrinsic mechanisms) and host-related characteristics in order to avoid confusion and repetition (Figure 1).
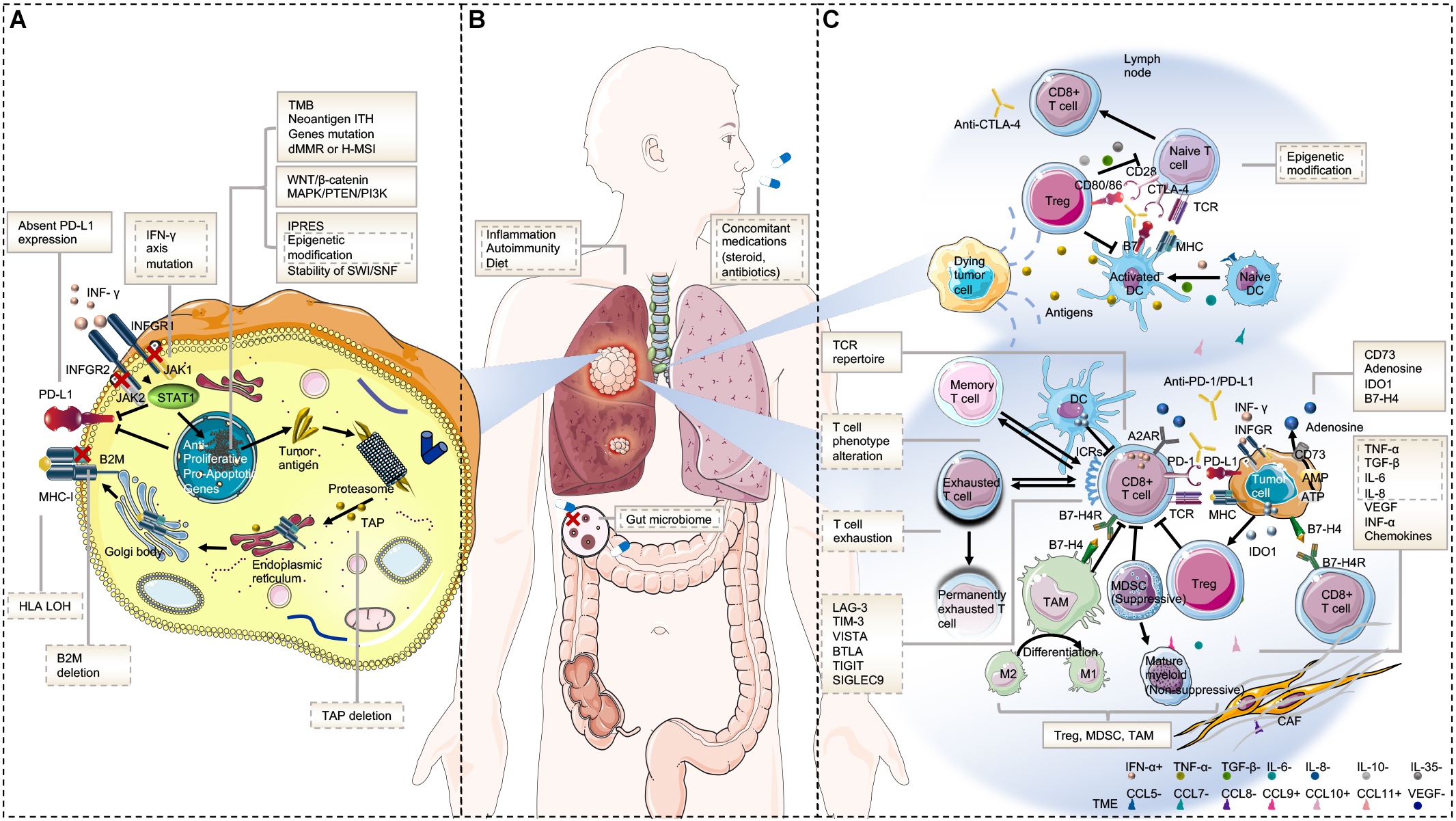
Figure 1. Mechanisms of resistance to immunotherapy. (A) Tumor intrinsic mechanisms that are associated with resistance to immunotherapy include lack of tumor immunogenicity (low TMB, heterogenous antigens, mutation of certain genes, and IPRES transcriptional signatures), deficiency in antigen presentation (alterations in INF-γ signaling pathway, HLA LOH, B2M, and TAP deletion), aberrations in several signaling pathways (MAPK, PI3K, WNT, and IFN), and absent PD-L1 expression. (B) Host-related characteristics that lead to primary or secondary resistance include the gut microbiome, diet, concomitant medications, inflammation state, and autoimmunity. (C) Tumor extrinsic mechanisms involved in resistance to immunotherapy include T cell-related factors (alternative immune checkpoints, T cell exhaustion and phenotype alteration, TCR repertoire, and epigenetic modification), immunosuppressive cells (Treg, MDSC, and M2-TAM), and cytokines and metabolites (e.g., TGF-β, adenosine) released into the tumor microenvironment. Factors in the solid text boxes are involved in primary resistance, whereas those in the dotted text boxes are involved in secondary or acquired resistance. Factors with solid and dotted dual text boxes are involved in both. Cytokines with “+” and “-” represent positive and negative modulators to antitumor immune response, respectively. Abbreviations: TME, tumor microenvironment; MHC, major histocompatibility complex; TCR, T cell receptor; Treg, regulatory T cell; MDSC, myeloid-derived suppressor cell; M2-TAM, type II tumor-associated macrophage; ICR, immune checkpoint receptor; CAF, cancer-associated fibroblast; and IPRES, innate anti-PD-1 resistance.
Tumor-specific antigens are the key to activate T cells to recognize tumor as foreign, which is the first step of tumor-induced adaptive immune responses and immune-mediated tumor killing (15). These neoantigens, interestingly, are derived from somatic mutations and contain new epitopes, and subsequently lead to tumor immunogenicity. Preclinical and clinical studies have revealed that the response of neoantigen-specific effector T cell (Teff) paralleled tumor shrinkage (16–20).
With the improvement of sequencing techniques, it was found that nonsynonymous mutations can generate neoantigens that trigger cytotoxic responses against tumors (21, 22). Nonsynonymous mutation burden, rather than total mutation burden of exons, was demonstrated to be more closely associated with the clinical advantage of anti-PD-1 treatment, validating the importance of neoantigens in dictating response (23). Tumor mutation burden (TMB) is calculated as the total number of nonsynonymous mutations per DNA Megabase (Mb) (21, 24, 25). Low TMB, or low numbers of clonal neoantigens, presenting reduced tumor immunogenicity, is considered as a primary resistance marker to immunotherapy (15, 26).
Clinically, low TMB or neoantigen load has correlated with inferior response and poor PFS to monotherapy of anti−PD1/PD-L1 antibodies in NSCLC (25, 27–30). However, it fails to predict the clinical outcomes, in regard to overall survival (OS) and combination regimens (31, 32). The influence on the OS by subsequent treatments and the additional complexities to the study of immunotherapy resistance added by combinations may partly explain these controversial findings. Recently, a corrected TMB (cTMB) approach based on the adjustment of tumor purity was developed by Anagnostou and colleagues, which was identified on abundant tumor samples mined from The Cancer Genome Atlas (TCGA) and then confirmed in a patient cohort received ICIs therapy. This cTMB more accurately predicted the outcomes of immunotherapy, suggesting that the TMB in samples with low tumor purity was mistakenly underestimated, which was especially important for metastatic NSCLC, because the tumor purity of tissue samples obtained by bronchoscopy or puncture biopsy was often limited (33).
The dilemma of insufficient tissue sample for TMB assessment in a considerable number of patients with NSCLC has given rise to the employment of peripheral blood TMB (bTMB) as a substitute predictor of response or resistance to ICIs in NSCLC (34). In keeping with what was previously reported in tissue, low bTMB evaluated by different plasma sequencing assays was significantly correlated with poor survival or response to immunotherapy in several retrospective and prospective studies (35–37).
In addition to the TMB or the numbers of clonal neoantigens, increased neoantigen intratumor heterogeneity (ITH, defined as relative fraction of subclonal neoantigens) can also impair the sensitivity to ICIs by elevating the likelihood of selection of subclones with poor immunogenicity (25, 38). The considerable variation of neoantigen heterogeneity was demonstrated by McGranahan and colleagues in seven primary NSCLCs (25). On average, 44% of heterogeneous neoantigens were reported only in a subset of tumor regions. They conducted neoantigen and clonality analysis in lung cancer data from TCGA and then validated the approach in a cohort of NSCLC patients treated with ICIs. Compared with high TMB alone, the combination of high TMB with low ITH seems to have a stronger association with clinical benefit to ICIs in this population.
Aberrations in oncogenes and tumor suppressor genes can regulate immune response by amending cytokine profile and immune cell composition and thus render tumor cells resistant or sensitive to ICIs.
Generally, alterations in oncogenic driver genes are characterized as resistant markers to immunotherapy. Although epidermal growth factor receptor (EGFR) mutations and anaplastic lymphoma kinase (ALK) rearrangement tend to have high PD-L1 expression due to the activation of signaling pathways (39, 40), the low mutation or neoantigen load (41), along with the following mechanisms, impairs the immunotherapy sensitivity in this group of patients with lung cancer. First, EGFR mutations have the potential to shape an inert immune environment by up-modulating a series of immune suppressors including inhibitory immune checkpoints (e.g., PD-1 and CTLA-4), immunosuppressive cells (macrophages and regulatory T cells), and cytokines (like TGF-β, IL-6, and IL-10) (42, 43). It has been reported that activated EGFR cascade was associated with elevated T-cell exhaustion and reduced cytotoxic T lymphocytes (CTLs) in a lung adenocarcinoma model (40). Second, downstream pathways of EGFR mutation, such as MAPK, PI3K/AKT, and Janus kinase (JAK)/STAT pathway, negatively affect immunoregulation. Other oncogenic driver-genes that frequently have high PD-L1 expression in lung cancer include ROS1 rearrangements (44), MET exon 14 skipping mutations (45), and BRAF mutations (44, 46). In contrast, RET rearrangements (47) and HER2 mutations (44) have been reported recently to exhibit low PD-L1 expression. None of these oncogenotypes demonstrated favorable clinical responses to ICIs monotherapy except for BRAF mutations, either V600E or non-V600E.
STK11 gene inactivation either by mutational or non-mutational machinery is linked to an indolent immune microenvironment with lower Tumor-infiltrating lymphocyte (TILs; CD3+, CD4+, and CD8+ cells) and PD-L1 expression in spite of the existence of moderate to high TMB (48). Inactivated STK11 gene was recently reported to weaken the innate immune responses by epigenetic inhibition of stimulator of IFN genes (STING), suggesting epigenetic silencing is likely to mediate the promotion of T-cell exclusion by the loss of STK11 (49). In line with these findings, it has been observed in several studies that compared with the wild-type gene, STK11 mutation predicted poorer clinical outcomes of immunotherapy in advanced NSLCL (50, 51). The tumor suppressor TP53 mutation, a well-known negative prognostic factor in lung cancer, is found to be associated with increased PD-L1 expression and higher TMB in non-squamous NSCLC (30) and KRAS-mutated lung adenocarcinoma (51).
KRAS-mutated lung cancer presents distinct immune profiles, biology, and therapeutic vulnerabilities in different subsets classified by co-occurring genetic events (50). Generally, KRAS/TP53 co-mutation predicts sensitivity while KRAS/STK11 co-mutation predicts resistance to immunotherapy in NSCLC. Dong et al. identified TP53/KRAS co-mutated subclass exhibited the highest percentage of PD-L1+/CD8A+ and particular increased PD-L1 expression. They further confirmed a remarkable clinical benefit from pembrolizumab in this population (52). Co-mutation of STK11 was shown to cause the accrual of neutrophils with T-cell-suppressive effects, accompanied with an analogous elevation in the production of T-cell depletion biosignatures and tumor-promoting cytokines (50, 53). TIL numbers and the expression of PD-L1 were also decreased (53). Consistent with these preclinical predictions, patients with KRAS/STK11 co-mutation or single mutation of STK11 had poor response and survival compared with those with wild-type when treated with ICIs (51, 54).
The Kelch-like ECG-associated protein 1 (KEAP1) gene, regulating the cellular antioxidant and cytoprotective transcriptional programs, plays a key role in mediating immune evasion in NSCLC. Depletion of KEAP1 is associated with reduced leukocyte infiltration, increased PD-L1 expression and might also influence other immune cells such as NK cell recruitment and function (55, 56). Co-occurring KEAP1 and phosphatase and tensin homolog (PTEN) inactivation represent an immunologically “cold” tumor while concurrent mutations in KEAP1 and STK11 leads to absence of pro-cancerogenic M2 macrophages (57). However, there are conflicting data on the role of KEAP1 mutation and its co-mutation with STK11 in immunotherapy resistance in NSCLC. KEAP1/STK11 co-mutations were verified to correlate with resistance to ICIs in patients with NSCLC despite high TMB (58). Similarly, STK11, and/or KEAP1 genomic variations posited lack of clinical advantages from combination of immunotherapy with chemotherapy in patients with NSCLC (59). However, inconsistent results were reported recently that clinical benefit from pembrolizumab compared to chemotherapy was poorer in the patients with STK11 and KEAP1 mutation compared with those in wild type in Keynote 042 trial, but the response and survival to immunotherapy were not significantly different between mutant and wild subgroups (60).
The WNT/β-catenin pathway is an additional immunotherapy resistance mechanism. A negative relationship was demonstrated between the level of β-catenin and TILs, which was modulated by deficiency in the recruitment of CD103+ dendritic cells (DCs) essential to T-cell priming and reduced expression of the cytokine CCL4, suggesting WNT/β-catenin signaling pathway is likely to mediate ICIs resistance through T-cell exclusion (61).
Similarly, the MAPK/PTEN/PI3K signaling pathway has been identified to be involved in immunotherapy resistance. Loss of PTEN and the bioactivation of the phosphatidylinositol 3-kinase (PI3K) signaling pathway in tumors decrease the activity of CTLs through the recruitment of inhibitory cells to the microenvironment and Vascular endothelial growth factor (VEGF) expression (62, 63), so that they promote resistance to ICIs (63, 64). The association of PTEN deletions or PIK3CA/AKT mutations with increased PD-L1 expression and immune resistance was also found in glioma (65). It was shown in preclinical models that a PI3K-γ inhibitor decreased myeloid-derived suppressor cells (MDSCs) and improved response to ICIs (66).
Genetic instability caused by alterations in DNA replication and repair genes can augment immunogenicity via a high-TMB neoantigen load (67–69). Correspondingly, deficient DNA mismatch repair (dMMR) or high microsatellite instability (H-MSI) are suggested as sensitive predictors to ICI immunotherapy in many tumor types. Beyond high TMB, increased CD8+ TILs were also reported to be associated with alterations in mismatch repair genes (70), BRCA2 (71), and POLE (72) in different tumors. However, the role of these genes in immunoregulation in NSCLC remains to be elucidated.
The interferon-gamma (INF-γ) signaling cascade is a crucial component of immunotherapy and tends to serve a critical function in primary, adaptive, and acquired resistance to ICI treatment (73–75). IFN-γ is a critical cytokine secreted by activated T cells, natural killer (NK) T cells, in the cancer microenvironment, and it moderates the immune reaction via the downstream enzymes JAK 1/2 and the signal transducer and activators of transcription (STATs) (76). The INF-γ axis exerts both positive and negative impacts on antitumor immune reactions (77). On one hand, it activates an functional antitumor immune reactive via (1) intensifying antigen presentation by up-modulated secretion of MHC-I; (2) recruiting other immune cells by up-regulation of the expression of chemokines (CXCL9, CXCL10, and CXCL11) with effective chemoattractant impacts on T cells (78); and (3) exerting direct anti-proliferative and pro-apoptotic impacts on cancer cells (79). On the other hand, IFN-γ acts in a negative-feedback axis to elevate PD-L1 expression as well as other crucial immune inhibitory components, including IDO1, down-modulating the cytotoxic reaction and adaptive resistance to cancer cells (80, 81) (Figure 1A).
Additionally, copy-number alterations (CNAs) linked to DNA damage response and regulation of DNA editing/repair gene expression were shown to emanate from the malignant exposure to IFN-γ-secreting antigen-specific CTLs in vivo, implying that intensified genetic instability could be among the mechanisms through which CTLs and IFN-γ immunoedits cancers, changing their immune resistance due to genetic evolution (82).
Tumors neutralize the impact of IFN-γ by mutating or down-regulating the molecules involved in the IFN-γ signaling pathway, including IFN-γ receptor chains, regulatory factors, JAK1/2, and STATs upon continuous IFN-γ exposure (73, 83). Multiple studies have demonstrated that mutations of IFN-γ axis and consequent loss of JAK/STAT contribute to immune escape of tumor cells and by that leads to primary or acquired resistance to ICI therapy via incapacity of up-regulating the expression of PD-L1 and MHC-I (73, 78, 84). Any deficiencies in IFN-γ, JAK1/2, or STATs including gene mutations, loss of protein expression, negative regulator presence, or epigenetic silencing would prevent signaling in response to IFN-γ and thereby end up to the up-regulated tumor growth and apoptosis inhibition and down-regulated T-cell infiltration and expression of PD-L1 and MHC-I (74, 78, 85, 86). Correspondingly, genomic changes disturbing IFN-γ pathway genes, including the amplification of suppressor genes PIAS4 and SOCS1 and the deletion of IFNGR1, IFNGR2, IFIT1, IFIT2, IFIT3, IRF1, MTAP, and miR31, have been described as possible machinery of primary resistance to various ICI therapies (73). An IFN-γ-related mRNA profile that contains 10 genes (CCR5, CXCL9, CXCL10, CXCL11, GZMA, HLA-DRA, IDO1, IFNG, PRF1, and STAT1) was additionally identified to predict the response to anti-PD-1 therapy in melanoma (87).
In a recent publication, transcriptional signatures, referred to as innate anti-PD-1 resistance (IPRES) with inflammatory and mesenchymal tumor phenotypes, were shown to manifest poor response to anti-PD-1 therapy in metastatic melanoma (88). Approximately 700 differentially expressed genes (DEGs) were identified between the responsive and non-responsive pretreated tumors. Compared with those of responsive tumors, the transcriptomes of non-responsive tumors were dominated by gene up-regulation events. The up-regulated DEGs in non-responsive tumors, considered as T-cell-suppressive, are involved in mesenchymal transition (TWIST2, TAGLN, FAP, AXL, ROR2, WNT5A, and LOXL2), monocyte/macrophage chemotaxis (CCL2, CCL7, CCL8, and CCL13), immunosuppression (IL10, VEGFA, and VEGFC), and angiogenesis and wound healing (89–91). By contrast, down-regulated gene CDH1 (which is typically down-regulated by mesenchymal tumor cells) was also detected in non-responsive pretreated tumors. Interestingly, there was no difference in the expression of INF-γ pathway signatures, other T-cell-related genes (e.g., CD8A/B, PD-L1, and LAG3), and the genes that presumably modulate immune checkpoint sensitivity between responsive and non-responsive groups, suggesting that T-cell-suppressive inflammatory and mesenchymal phenotypes of tumor are associated with primary resistance to anti-PD-1 therapy.
Emerging evidence has suggested that epigenetic modification may mediate primary resistance and contribute to acquired resistance during ICI therapy through the profound effect on many aspects of antitumor immunity: neoantigen presentation and processing; T-cell functions, differentiation, and proliferation; memory T-cell phenotype acquisition; interfering with T-cell migration; and mediating T-cell exhaustion (10, 92–94).
Epigenetic targeting agents, including those targeting histone deacetylation or methylation as well as targeting DNA methylation, have exhibited encouraging antitumor activity either as monotherapy or in combination with immunotherapy in preclinical studies (94, 95). Clinical trials investigating the performance of these agents combined with adaptive T-cell transfer (ACT) in patients with acquired resistance to prior immunotherapy are ongoing (96).
Stability of chromatin remodeling complexes within tumor cells can also contribute to immunotherapy resistance by multiple mechanisms. It was found that tumor cells were more sensitive to CTL killing, which leads to increased response to anti-PD-1/PD-L1 therapy, due to the deficiency in chromatin remodeling complex SWI/SNF (97, 98). BRG1-associated factor (BAF) and polybromo-associated BAF (PBAF), as the mammalian analogs of the SWI/SNF complex, are essential tumor suppressors and loss of function (LOF) mutations of them were shown to sensitize tumor cells to ICI therapy (98). The inactivated PBAF subunits exhibited elevated CXCL9/CXCL10 expression and TILs recruitment as a result of increase of chromatin accessibility to transcriptional regulators of IFN-γ-inducible genes (97). ARID1A/B subunits are unique to BAF, while other subunits (ARID2, BRD7, and PBRM1) are exclusively contained by PBAF, despite the high similarity of these complexes (99). In another study, loss of ARID1A was found to elevate MSI by defective recruitment of mismatch repair genes and thus increase TMB, which eventual sensitize tumor cells to PD-L1 inhibitor (100).
The PD-1/PD-L1 axis represents one of the foremost mechanisms of modulation of peripheral immune tolerance as well as T-cell activation. Up-regulation of PD-L1 by cancer cells and antigen-presenting cells (APCs) is one approach through which tumors avoid immunosurveillance and constitutes the principle behind PD-1/PD-L1 blockade therapies (101). Absent PD-L expression of tumors has been found to be generally associated with less responses and inferior survival benefits to anti-PD-1/PD-L1 therapies compared with higher expression (102) and may serve as a resistant marker. However, up to 20% of PD−L1−negative malignancies showed responses to PD−1 inhibitors in some cohorts (103), as PD-L1 expression can be up-regulated by other factors including activated IFN-γ cascade (will be discussed in a separated part), suggesting tumor PD-L1 expression alone is not dependable at predicting outcomes of PD-1/PD-L1 inhibitors.
Any factors that affect the PD-L1 expression of tumor cells may lead to resistance to immunotherapy. Beyond encoding genes, PD-L1 expression can be affected by the mutational features of tumor although it is not paralleled with TMB in most of the tumors (104–106). The inherent mechanisms, which have been shown to result in constitutive expression of PD-L1 by tumor cells, consist of alterations in the PTEN/PI3K/AKT pathway (65, 107), MYC overexpression (108), EGFR mutations (40), CDK5 truncation (109), and elevated PD-L1 transcripts stabilized by disruption of the 3-untranslated region (UTR) of this gene (110). Tumor-specific immune response may also be affected by constitutive expression of PD-L1 caused by these oncogenic signaling processes on tumor cell surface. Although it is still unclear whether it causes an increased or decreased possibility of responding to anti-PD-1/PD-L1 therapies, the constitutive PD-L1 expression could result into inadequate response to other immunotherapeutic approaches by suppressing antitumor effect of T cells. The other transcriptional factors constituting HIF1, NFkB, and STAT3, as well as epigenetic factors, additionally participate in the modulation of PD-L1 expression (111).
Inflammatory and hypoxic tumor microenvironment (TME) can also lead to PD-L1 expression on many cell types including tumor cells by Toll-like receptor (TLR) ligands. The recruitment of activated T cells can increase the inflammatory mediators and successively induce the PD-L1 expression on the surface of tumor cells. These tumor cells specifically locate at the invasive periphery where T cells are often abundant (112, 113).
Besides, PD-L1 is stabilized through N-glycosylation and palmitoylation (114, 115). This is crucial for its interaction with PD-1. The resistance to anti-PD-1/PD-L1 treatment could moreover be attributed to the degree of generation and secretion of soluble forms of PD-L1. These variants without the transmembrane domain because of alternative splicing have been reported in recurrent NSCLC incidences that re-occurred after anti-PD-L1 antibody therapy with the ability to act as soluble imitates for anti-PD-L1 antibodies (116).
Loss of neoantigens in the context of immune-mediated pressure is postulated to be another mechanism leading to resistance. In the concept of immunoediting, the constant interactions between tumor cells and the immune system trigger the production of subclones that do not express neoantigens, consequently conferring poor immunogenicity and resistance to ICIs (117). It was demonstrated by Anagnostou and colleagues that seven to eight putative neoantigens were lost in the recurrent NSCLC after ICI treatment, suggesting that immunoediting plays a role in acquired resistance to immunotherapy (118). T-cell-mediated neoantigen immunoediting can be induced by the dynamic interactions between T cells and tumor cells, consequently causing partial or total loss of neoantigen (119). Consistently, deficiency in genes that encode target tumor antigens was demonstrated to be associated with acquired resistance in a murine model treated with adoptive T-cell therapy (ACT) in melanoma (120). However, this relationship between acquired resistance and loss of target neoantigens was not observed in a single patient case who achieved a complete response to ACT in a separate study (121), suggesting that down-regulation/loss of neoantigens may occur during immunotherapy, but should be taken as a canonical mechanism of acquired immune resistance.
Proinflammatory cytokines are likely to contribute to immune escape by inducing loss of antigen expression, resulting in acquired resistance too. The process of Tumor necrosis factor-α (TNF-α)-induced epithelial-to-mesenchymal de-differentiation was shown to lead to a loss of neoantigens causing transformation to a tumor phenotype that is less immunogenic and can more readily evade immune surveillance in the ACT-treated mouse model in melanoma (122). Other TIL generated cytokines, such as IL-6 or TGF-β, are also shown to be involved in the induction of epithelial-to-mesenchymal transition in mouse models across numerous types of tumors, indicating that acquired resistance can be promoted by inflammation.
Defective neoantigen presentation serves a crucial function in ICI acquired resistance. The alterations in this process could happen in beta-2-microglobulin (B2M), transporters associated with antigen processing (TAP), or MHC itself (123, 124) (Figure 1A).
As part of the MHC class I (MHC-I), B2M is crucial during antigen presentation and its genetic deficiency, including loss of heterozygosity (LOH) and deletions or point mutations, was identified to be an important route for primary and acquired resistance to ICIs (125, 126). Other defects that would affect neoantigen presentation include T-cell receptor (TCR) binding domain mutations of MHC-I (127), loss of tapasin (a MHC-I antigen processing molecule), selective epigenetic silencing of the human leukocyte antigen (HLA) A3 antigen, loss of one HLA haplotype (128, 129), and LOH in HLA (130). Homozygosity in one or more of the three highly variable genes (HLA-A, HLA-B, and HLA-C) that encode MHC-I, which are likely to restrict neoantigen presentation to CTLs, was identified to have a significant association between resistance to ICI therapy in a large cohort of cancer patients (131). In contrast to anti-CTLA-4, the expression of MHC-II (but not MHC-I) proteins by tumor and the presence of IFN-γ-mediated gene signatures were found to be associated with the positive responses to anti-PD-1 therapy in melanoma (132).
Defective neoantigen presentation may be mediated by IFN-γ signaling pathway through JAK1/2 and the STATs, by down-regulating the expression of MHC-I (133). Actually, the IFN-γ pathway has both unfavorable and favorable impacts on antitumor immune responses and plays a key role in acquired and primary resistance to ICI therapy (as discussed above).
Tumor-infiltrating lymphocytes constitute a complex group of immune cells with distinct functions and different clinical impacts. Among them, tumor-specific CD8+ T cells can execute anti-cancer activities by killing tumor cells directly and has a strong prognostic effect in NSCLC (134, 135). CD4+ cells are composed of a group of lymphocytes (Tregs CD4+, Th1, Th2, and Th17) secreting diverse cytokine to activate and suppress CD8+ cells. Th1 secretes IFN-γ and IL2, while Th2 secretes IL-4, IL-5, IL-9, IL-10, IL-13, and IL-25 (136, 137). CD45RO+ T cells, also known as memory T lymphocytes, are another subclass of TILs. Regulator and memory T lymphocytes will be discussed in section “Suppressive tumor microenvironment.”
Low CD8+ TIL density was correlated with impaired efficacy and survival in NSCLC patients treated with ICIs (138), suggesting that immunotherapy resistance was mediated by low TILs but was then positively modulated by PD-L1. TILs can be assessed by immunohistochemistry or standard hematoxylin and eosin (H&E) staining; however, no consensus has been reached hitherto in the various scoring models using H&E staining in NSCLC (139–142). A radiomic fingerprint of CD8+ TIL derived via computerized tomography was developed recently and showed promising efficacy in predicting response to ICI therapies but requires further validation (143).
Thus, tumors can be described as three main immune organization profiles (hot, altered, and cold) as per the presence of TILs and correlated proinflammatory cytokines (144). The “cold” immune tumor is characterized as absence of TIL within and at the edges of tumor, manifesting resistance to immunotherapy either due to absent immune stimulation (as with low neoantigen cancer’s poor antigen presentation) or because of failed T-cell priming (as with intrinsic insensitivity to T-cell killing). The “altered” immune tumor is characterized as low TIL within the tumor (“immunosuppressed”) or high TIL at the edges of the tumor (“excluded”), whereas “hot” is high degree of TIL (144). Recently, intratumorally geospatial heterogeneity of TIL was revealed in NSCLC. Tumor subclones from “cold” immune regions were related to mutation space more closely and diversifying more recently compared with those from “hot” immune regions. Higher risk of recurrence was observed in tumors with more than one “cold” immune region (145).
Reduced proliferation and inadequate diversification of T cells possibly contribute to ICI resistance. Impeded priming of naive T cells by blocked DCs recruitment was demonstrated in melanoma to be correlated to the lack of TILs and ICIs resistance (146). The function of DCs can be potentially influenced by the cytokines in the TME through (1) impaired migratory capacity as well as decreased synthesis of costimulatory components (CD86/80) by TGF-β (147, 148); (2) prevented DCs maturation by IL-6-gp130-STAT3 axis; and (3) inhibited activity by Indoleamine 2,3-dioxygenase 1 (IDO, will be discussed in Section “Suppressive tumor microenvironment”). IFN-α signaling pathway is important to the priming of T cells by DCs. It was found that TME with remarkable insufficient IFN-α-producing DCs naturally led to lessened antitumor T-cell priming and thus resistance to ICIs (149, 150). Activated IFN-α stimulated production of the chemokine CXCL10 to recruit TILs to tumor beds and in turn initiate spontaneous antitumor T-cell response (149–151). Preliminary trials combining IFN-α 2b therapy with anti-CTLA-4 inhibitors have indicated clinical activity, which could be caused by diminished populations of MDSC (152, 153). Combinations of other ICIs and IFN-α 2b are currently investigated (154).
Immune resistance also occurs if the tumors evolve the ability to prevent infiltration even if tumor-specific Teffs are formed. Mechanisms that lead to impaired T-cell infiltration involve components in the epigenetic silencing of immune cells (155) and the modification of secreted chemokines (156, 157). Transcriptional program that is associated with T-cell exclusion and thereby predictive resistance to anti-PD-1 therapy was identified in melanoma (158). Stromal cells surrounding tumors within TME can develop the capacity to obstruct effector T-cell entry, and the TGF-β cascade appears to serve a crucial role in promoting T-cell exclusion features in peritumoral fibroblasts (123, 159).
T-cell receptor clonality is emerging as a new biomarker to predict the resistance and immune-related adverse events to ICIs therapy. Since baseline CD8+ T-cell density was found to overlap between respondents and non-respondents to ICI therapy (160–162), it led to the speculation that a constrict TCR arsenal possessed by the baseline T cells concentrated on the antitumor immune reaction and is associated with response to ICI therapies. T-cell clones can be identified by detecting TCR rearrangements constituting genes in the variable (V)-diversity (D)-joining (J) region, which generate the antigen-specific complementarity-determining region 3 (CDR3). The responsivity of TCRs generated by TILs determines their potential to interplay with tumor antigens that are presented on APCs. Thus, the assessment of T-cell clonality divulges the extent of T-cell expansions caused by tumor antigens and contributes to explore the mechanisms underlying T-cell toleration to tumor antigens.
A lower baseline clonal T-cell arsenal has been shown to be linked to worse clinical benefits to ICIs and survival in cancer patients (162, 163). Besides, a remarkable increase in T-cell clones was reported in responders during anti-PD-1 therapy compared to non-responders, implying a cancer-specific reaction to immunotherapy for these patients. Moreover, baseline TCR clonality did not strongly associate with TIL density, implying that low-TIL density tumors could still respond to anti-PD-1 treatment if TIL has a narrow TCR clonality specific to the tumor antigen (164). Inconsistently, it was recently found that T-cell clonality had a positive relationship with T-cell density, PD-L1 expression, and TMB, and a negative relationship with EGFR mutation in NSCLC (165). A corresponding relationship was found between the number of TCR sequences and the number of nonsynonymous mutations, spatial heterogeneity in expanded TCR repertoire, and spatial mutational heterogeneity within tumors in NSCLC, respectively. This intratumorally spatial heterogeneity of TCR repertoire maps the neoantigen landscape, sculptured by focal antigen processing defects or HLA loss (166). Thereby, further investigations to identify the role of TCR clonality in immunotherapy are required.
Compensatory up-regulation of numerous alternate immune checkpoint receptors during ICI therapy as a result of the activation of diverse cellular signals and IFN-γ signaling pathway were observed across multiple studies and have been characterized to be linked to ICI adaptive resistance in NSCLC (84, 167, 168). The expression of CD8+ T cells harboring receptors showed serious flaws in proliferation, migration, and cytokine secretion, indicating their immunosuppressive capacity. In addition, progressive T-cell exhaustion was found in the tumors with highly expressive or co-expressive receptors and different receptor displayed different exhausted phenotype (167).
Among these receptors, lymphocyte activation gene 3 (LAG-3) has great potentiality in cancer immunotherapy because co-expression of PD-1 and LAG-3 was often found in T-cell-depleted immune microenvironment, and PD-1 inhibitors combined with LAG-3 blockades showed strong synergic antitumor responses in preliminary models (169). LAG-3 is a co-inhibitory receptor extensively expressed in TILs in various tumors and serves a crucial function in mediating immune escape by suppressing T-cell antitumor functions. It exerts immunosuppression via binding to MHC-II molecules and other ligands such as galectin-3 and fibrinogen-like protein 1 (FGL1) (170). Thus, blocking LAG-3 can restore antitumor immunity and the combined LAG-3 inhibitors therapy may accordingly overcome immunotherapy resistance. In addition, the expression of these ligands, on the basis of the receptor–ligand interactions, may serve as important biomarkers to predict the efficacy of LAG-3 blockades in lung cancer (167).
The other alternative immune checkpoint receptors, e.g., T-cell immunoglobulin and mucin-3 (TIM-3), V-domain immunoglobulin-containing suppressor of T-cell activation (VISTA), B and T lymphocyte attenuator (BTLA; also referred to as CD272), T-cell immunoreceptor tyrosine-based inhibition motif domain (TIGIT), and sialic acid-binding Ig-like lectin 9 (SIGLEC9), have been discovered (144). Thus, these alternate immune checkpoints are likely to be combined with existing ICI therapy to conquer the resistance. Increased efficacy of PD-1 inhibitors combined with anti-TIM-3 or anti-LAG-3 regimens has been observed in either pre-clinical models or phase I clinical trials (171, 172). Currently, numerous clinical trials evaluating the therapeutic impact of alternate immune checkpoint blockade applied on its own or in combination with PD-1/PD-L1 inhibitors in multiple malignances are ongoing.
T-cell exhaustion is another factor involved in the primary and acquired resistance to ICI therapy (Figure 1C). Exhausted T cells exhibit impaired activity with progressive LOF and antigen persistence compared with Teffs and can be induced by the PD-1/PD-L1 interactions (173). Chronic exposure to cognate antigen triggers increased expression of PD-1, which results in the accumulation of T-cell exhaustion and thus T-cell dysfunction (174). The presence of PD-1 high expression can either exist prior to PD-1 inhibitors, which is associated with primary resistance partially depending on tumor-associated regulatory T cells (Tregs), or develop after the anti-PD-1 therapy, which leads to acquired resistance by severe T-cell exhaustion. In contrast, studies showed that the exhausted T cells with PD-1 low to intermediate phenotype retain the capacity to be reinvigorated by ICIs (158, 175). Epigenetic alterations were found to be associated with T-cell exhaustion too recently. Exhausted T cell displayed a unique chromatin landscape, which alters the transcriptional state, limits its effect function, and determines its capacity to be reprogrammed after therapeutic intervention (176–178).
The formation of memory T cells is crucial to the avoidance of tumor relapse and therapy resistance following drug withdrawal, especially in the long-lasting duration of responses to ICI therapy. Research evidence shows that patients with resistance to anti-PD-1 treatment have fewer tumor-correlated memory T cells relative to sensitive patients (179). Memory T cells remain dormant until antigen re-challenge (180, 181) and if precursor memory T cells are exhausted under chronic antigen exposure, it will lead to memory T-cell deletion and lack of formation (173, 177).
Acquired resistance can be mediated by the alteration from cytotoxic activity to inactivity phenotype of antitumor T cells during TCR-engineered ACT. The original highly cytolytic profile when administrated to patients, which showed strong efficacy initially, was reported to change to a phenotype with impaired cytotoxic functions and Th2-related cytokine release when tumor relapses within months (182, 183).
The TME is a complex net consisting of a variety of immune and stromal cells, cytokines, extracellular matrix, and vasculature, which affect response to immunotherapy. Immune-suppressive cells, including Tregs, MDSCs, M2 macrophages, along with inhibitory cytokines in the TME, can contribute to the inhibition to immune responses (136, 184) (Figure 1C).
Tregs can inhibit Teff reactions by secreting certain inhibitory cytokines (IL-10, IL-35, and TGF-β) or by direct cell contact (185–187). The cytokine IL-10 influences antigen presentation by down-regulating the expression of MHC-II and co-stimulatory components on DCs, thus intercepting the Teff activation (187). The ratio of Teffs to Tregs was shown to be related to the responses to ICIs in mouse models, in that incapacity of either increasing Teffs or decreasing Tregs may cause resistance to immunotherapy (188, 189). Factors that affect Tregs activity, at the same time, are putative biomarkers of resistance. For instance, soluble CD25, an IL-2 receptor whose binding is assumed to stimulate Treg proliferation, was established as a negative predictor of OS for patients treated with anti-CTLA−4 (190). However, tumor-infiltrating Tregs might likely coexist with multiple immune cells, insinuating a potential immunoreactivity. It was reported that a high baseline expression of FoxP3+ Tregs in the tumor is positively associated with better survival in a retrospective study involving patients under the treatment of anti-CTLA-4 antibodies (161).
Myeloid-derived suppressor cells promote immune evasion and tumor growth and have emerged as critical modulators of immune responses in cancer. Studies have suggested the existence of MDSCs in TME correlates with reduced efficacy of immunotherapies, including ICIs therapy (191), ACT (192), and DC vaccination (193). Therefore, reprogramming or eradicating MDSCs might improve clinical response to immunotherapy.
Tumor-associated macrophages (TAMs) can be classified into M1 and M2 macrophages according to disparities in surface molecules, expression of transcription factors, cytokine profiles, and metabolism (194, 195). They promote antitumor immunity effects (mediated by M1) and pro-tumorigenic properties (mediated by M2) that modify the TME (196). The role of TAMs in mediating immunotherapeutic resistance in tumor has been discussed in several reports (197, 198). It was indicated to directly inhibit T-cell responses through PD-L1 in preclinical studies of liver (199) and ovarian cancer (200). The inhibitor of CSF-1R, a receptor for macrophage colony-stimulating growth factor, was investigated in mouse models of pancreatic cancer where it decreased the frequencies of TAMs, while increasing IFN production and delaying tumor progression (201, 202). Similarly, CSF-1R inhibitor was found to synergize ACT therapy in a melanoma model (203). These data suggest that CSF-1R inhibitor may overcome the resistance to immunotherapy.
Specific chemokines, such as CCL5, CCL7, and CXCL8, play an important role in the recruitment of Tregs and MDSCs to the TME, consequently boosting an immunosuppressive climate (204). Alternately, chemokines CXCL9 and CXCL10 recruit CTLs to the TME (205) and the epigenetic silencing of the genes encoding them can reduce TILs and consequently promote resistance to ICIs (205). Epigenetic modulators of these chemokine receptors relieved the suppression of these Th1-cell-type chemokines and increased TILs leading to improved therapeutic efficacy of PD-L1 inhibitor in a model for ovarian cancer (155).
The cytokine milieu is critical to the recruitment, activation, and proliferation of immune cell, performing both immune stimulatory and suppressive effects (206). Transforming growth factor-β (TGF-β) is a cytokine playing key roles in angiogenesis and immunosuppression by stimulating Tregs (207) and excluding T cell in peritumoral fibroblasts (123, 159). Up-regulated TGF-β signaling was correlated with poorly immunogenic tumors and restrained response to ICIs in a colorectal cancer model, indicating resistance to therapy (159). Consequently, enhanced antitumor response to ICIs was observed following application of TGF-β inhibitor either alone or in combination with anti-CTLA-4 or radiation therapy (208, 209). Bintrafusp alfa, a bifunctional fusion protein composed of the extracellular domain of TGF-β receptor II (a TGF-β “trap”) fused to a human immunoglobulin G1 antibody blocking PD-L1, demonstrated favorable efficacy in patients with advanced NSCLC. Ongoing phase III trial is expected to validate the efficacy of bintrafusp alfa vs. pembrolizumab in the first-line setting in advanced NSCLC (NCT03631706).
Tumor necrosis factor-α pathway is postulated to be another immune evasion machinery conferring resistance to PD1 blockade. The expression of TNFα in an inflamed TME positively correlates with the expression of PD-L1 and TIM-3, along with impaired accumulation and increased activation-induced death of CD8+ TILs in melanoma models treated with anti-PD1 therapy. Accordingly, inhibition of TNF-α prevents the expression of PD-L1 and TIM3 and hampers anti-PD1-induced TIL death (210). Therefore, this study offers a rationale for the combination of PD-1/PD-L1 inhibitors with TNFα blockade as a novel immunotherapeutic strategy to overcome resistance in lung cancer, and the phase I clinical trial testing the combination is ongoing (NCT03293784).
Vascular endothelial growth factor has been linked to both decreased T−cell infiltration and immunosuppressive effects in addition to promoting angiogenesis and thus is associated with resistance to ICIs (211). Multiple mechanisms are involved in the interaction of VEGF with antitumor immunity: (1) VEGF prevented the commitment of lymphoid progenitors, decreasing progression to the T-cell lineage (212); (2) VEGF signaling promotes the infiltration of Tregs through a selective endothelium and reduces trafficking and extravasation of CTLs into the TME (213); and (3) VEGF increases expression of inhibitory receptors, contributing to CTL exhaustion (214). Higher levels of VEGF were found in anti-PD-1-resistant patients than sensitive ones (160). Based on these findings and the synergy between angiogenesis blockade and ICI therapies observed in preliminary studies, multiple trials of combination therapy are underway, including bevacizumab and VEGFR-TKI with anti-PD-1 therapy.
Higher levels of interleukin 6 (IL-6) and interleukin 8 (IL-8) have been found recently to be linked to reduced responses and worse clinical outcomes to ICI therapies across multiple types of cancers (215–217). IL-6 is a proinflammatory cytokine generated by T cells and macrophages and is usually involved in the immunoregulation connected to the IFN-γ signaling pathway. It can reduce the expression of PD-L1 and MHC-I, and result in tumor escape and resistance to ICI therapy (218). IL-8 is a proinflammatory chemokine and a chemoattractant for myeloid leukocytes expressed in multiple cancers (219, 220). It potently regulates the chemotaxis of neutrophils (221, 222) and exerts direct pro-tumorigenic effects (223). High levels of IL-8 are regarded to be associated with more neutrophil and monocyte infiltration, defective T-cell functions, and impaired antigen presentation, which subsequently result in resistance to ICI therapy (216, 217).
Contributions from inflammatory processes could participate in quashing the desired impacts of ICIs (Figure 1C). Adenosine can be produced under the condition of hypoxia and ischemia caused by tumor inflammation. It was reported to inhibit the cytotoxic function and proliferation of T cells via the A2A receptor on T cells (224). CD73, which mediates the generation of adenosine through dephosphorylation of adenosine monophosphate, was also demonstrated to suppress immune function (225). CD73 overexpression promotes T-cell exhaustion and is linked to the resistance to ICIs (226, 227).
IDO1 expressed in myeloid cells and cancer cells is a rate-limiting enzyme that converts tryptophan to its immunosuppressive metabolite kynurenine. This enzyme can induce T-cell anergy and apoptosis by gathering kynurenines and consuming the essential amino acid tryptophan and prevent the T-cell clonal expansion (228). It is particularly activated in DCs after binding with CTLA−4 and can be unregulated by CTLA−4 during adaptive immune resistance (229). Reduced expression of IDO at baseline was noted to be associated with poor response to ipilimumab in a phase II study in melanoma (161). Low level of IDO is likely to manifest insufficiency of suppressed TILs feasible to be reactivated by immunotherapy. Correspondingly, IDO-knockout mice exhibited improved OS with ICI compared with wild-type mice, and ICI therapy combined with IDO inhibitors showed both increased numbers and functions of TILs in the TME in an experimental setting (230, 231). However, despite encouraging results observed in preclinical and early-phase clinical studies in different types of tumors, no difference was shown in pembrolizumab combined with IDO1-selective inhibitor vs with placebo in a phase III study in metastatic melanoma (232).
B7-H4 has been proposed as another resistance marker to ICIs recently due to its negative modulation of T cells. B7-H4 constitutes a type I transmembrane protein of the B7 immunoglobulin superfamily and is encoded by the V-set domain containing T-cell activation inhibitor 1 (VTCN 1) gene. It is induced by activated T lymphocytes and down-regulates T-cell function by inhibiting proliferation, cytotoxicity activity, and interleukin secretion, after binding with T cells (233–235). Positive B7-H4 protein expression in patients with advanced NSCLC treated with nivolumab was recently reported to have an enhanced risk of tumor progression and tumor-related death compared with negative expression (236).
Host-related characteristics, including gut microbiome, diet, and antibiotic exposure that adversely affect the gut microbiome, diet, steroid use, vaccine exposure, inflammation state, and autoimmunity, have been shown to relate to primary and acquired resistance to ICI therapy in lung cancer (237, 238) (Figure 1B).
Evidence is arising to support the vigorous impact of the gut microbiome on immunotherapy resistance. Multiple studies have demonstrated that less bacterial diversity and lack of enrichment of specific species showed a significant correlation with resistance to ICI therapies. Relative abundance of Bacteroidales was found in non-responders, while responders were more likely to have Faecalibacterium and Ruminococcaceae (239, 240). Furthermore, transplanting the responder’s feces into aseptic mice exhibited improvement in the treatment of PD-L1 inhibitor (241). Certain species can be altered by the antibiotic’s exposure, which partly explained modified response to ICI therapy either in a good or in a bad way (242–244).
The alteration tendency of gut microbiome structure or dominant bacteria may differentially affect the T-cell immune response. It may be due to the cross-reactivity between intestinal microbiota and tumor-associated antigens, enhancing the inflammatory cytokine production, activation of DCs, and antigen presentation (245, 246). IFN-γ-producing CD8+ T cells were successfully induced from a consortium of 11 bacteria in the intestine, and colonization with this 11-bacterial mixture resulted in enhanced efficacy of ICIs in mouse models (247). Consistently, “good” bacteria introduction was reported to significantly increase IFN-γ production in spleen and tumor-draining lymph nodes (TDLN) (246) and induce DCs to secrete IL-12, resulting in increased recruitment of CCR9+CXCR3+CD4+ T cells into tumor beds (244). The activation of DCs was reported to be modulated by the gut microbiome in animal models and cancer patients. The resistance to anti-CTLA-4 therapy can be reversed by oral administration of Bacteroides fragilis, which induced Th1 immune response in TDLN and promoted DCs maturation (243). Bifidobacterium-feeding mouse presented higher expression of MHC-II in DCs within tumors (248). It remains controversial whether intestinal microbes lead to immunotherapy resistance by affecting the production of Tregs. A higher level of peripheral Tregs was found in patients with “bad” bacteria and was associated with poor response to ipilimumab in metastatic melanoma patients (249). B. fragilis can produce a microbial molecule, polysaccharide A, which can promote the formation of the inducible population of CD4+Foxp3+Tregs (a subset of Tregs), thereby negatively regulating the immune system (250), whereas the other two studies reported no differences in Treg differentiation between pancreatic duct adenosarcoma-bearing mice and control, and the number of Foxp3+ T cells between Bifidobacterium- and PBS-feeding mice (251, 252). The majority of chemokine genes were reported to be up-regulated by specific species of gut microbiome including Fusobacterium nucleatum, B. fragilis, and Escherichia coli in colorectal cancer cells. Additionally, the gut microbiota-derived microbial load was associated with increased chemokine production (251).
The gut microbiome also acts as an instructive modulator of mutant TP53, which ultimately affects tumor proliferation and the immune system. Kadoshi et al. recently found that mutant TP53 presented contrasting effects in different segments of the gut in a mouse model: a remarkable tumor-suppressive effect in proximal gut and the expected oncogenic effect in distal gut (253). The gut microbiome and its single metabolite gallic acid turned mutant TP53 from a tumor-suppressive effect to an oncogenic one, suggesting that the function of mutant TP53 is plastic and under the control of microbiome and microbiota-derived metabolites.
Antibiotics exposure has been reported to be associated with inferior clinical outcomes during ICIs therapy in NSCLC (244, 254). However, it remains debated if antibiotics exposure represents an independent predictive biomarker of ICIs therapy or it is a surrogate for patients with worse prognosis (e.g., poorer performance status, higher comorbidities). The use of antibiotics has an unfavorable impact on the recolonization and subsequent alterations in microbiota composition, which eventually leads to a decline in microbial symbiotic diversity (255). The antibiotics-induced dysbiosis destroys the gut homeostasis and extends from childhood to adulthood, with long-lasting adverse effects on the immune system as well as body metabolism (256–258). The antitumor immune response-induced cyclophosphamide was impaired by antibiotics exposure in the fibrosarcoma model, which was associated with an improvement in the Teff-to-Treg ratio and the loss of Enterococcus hirae-elicited helper T cell in tumor immune infiltrate (259, 260). Correspondingly, the absolute numbers of neutrophils and monocytes decreased after oral administration of imipenem, vancomycin, and neomycin before tumor inoculation in lymphoma and colon models treated with oxaliplatin or IL-10 inhibitor (261). On the contrary, the growth of Fusobacterium spp.-containing tumor was slowed down after the administration of metronidazole in the mouse model of colon cancer, suggesting such bacteria promote tumor progression (262).
Long-term use of steroids adversely impacts the efficacy of ICIs because of their supposed anti-inflammatory and immunosuppressive effects that potentially hamper the mode of ICI action (263), whereas a transient use of steroids aimed at management of ir-AEs did not negatively affect patients’ survival outcomes (264, 265). Beyond affecting gut microbiome, steroids are known to prevent the activation of T lymphocytes, inhibit the amplification of T helper subsets, recruit Tregs, and promote M2 macrophage polarization (266, 267). Hence, early use of steroids on ICI treatment may prevent this phase of T-cell recruitment and thus impair effective antitumor immune response. The increment of neutrophil-to-lymphocyte ratio (NLR), derived NLR (dNLR), and absolute neutrophil count (ANC) after steroids has been shown to be associated with unfavorable clinical outcome in NSCLC patients treated with ICIs (268). Steroids-induced imbalance of immune cells in TME, especially increased MDSCs resulting in elevated ANC and NLR, is the intermediate of immunotherapy resistance in NSCLC (269).
Diet can affect tumor growth within TME through systemic or local effects in multiple ways. First, it may alter the composition and diversity of gut microbiome, which in turn exerts drastically different effects on host immune function. Second, specific ingredients (e.g., vitamins) may be regulated by dietary patterns and then have an impact on immune status. It is well-known that the general metabolic status determining deviations from ideal body weight, as well as the metabolic factors (e.g., low-level arginine and tryptophan, high-level lactate, and the adenosine pathway induced by increased glucose metabolism), highly influences the immune activity (270–272). An appropriate diet can maintain the homeostatic equilibrium between the inflammatory cascade triggered by Th17 cells and the anti-inflammatory pathway mainly based on the activity of Treg (273).
Chronic accumulation and production of inflammatory molecules in the chronic inflammatory status lead to an immunosuppressant state, which is linked to immunotherapy resistance. Proinflammatory and carcinogenic mediators such as IL-6, TNF-α, and chemokines are released in the TME and tend to trigger a variety of molecular signaling cascades including PI3K/MAPK, JAK/STAT, and WNT/B-catenin, which are involved in the resistance to immunotherapy as mentioned previously. The components of immune cells are also altered to be more immunosuppressive with more TAMs, Tregs, and tumor-associated neutrophils within TME (274). Therefore, blocking inflammation might be an effective strategy to improve the outcome of immunotherapy in NSCLC (275).
Tumor development and autoimmunity are two opposite results of imbalanced immune homeostasis in controlling tumor cell growth (low immune responses) and regulating autoreactive responses (immune overreaction). The host autoimmunity affects the efficacy of immunotherapy in bringing more ir-AEs when too strong or incapacity to prime and activate immune cells when too weak (276). In addition, autoimmunity has an inextricable link with host gut microbiome and anti-microbial immunity, as effector responses that lead to inflammatory tissue damage are the same as those that mediate effective host defense (277).
The relationship between smoking and the efficacy of ICIs remains controversial (278–281). Smoking is associated with high TMB, especially nonsynonymous mutations, subsequently enhances the immunogenicity of tumor, and improves the outcome of ICIs therapy (23, 282). Additionally, PD-L1 expression can be up-regulated by smoking through oxidative stress-dependent mechanism (283) and induced by cigarette smoke and the carcinogen benzopyrene (BaP) via aryl hydrocarbon receptor (AhR) (284). Moreover, smoking may also have an impact on the status of TILs (285) and other immune modulators such as B7-H3 (CD276) (286) and in turn affect the efficacy of ICIs therapy.
Research and design of therapies to conquer immunotherapy resistance has been advancing along with mechanistic investigations. Combinatory treatments, either via combinations of diverse immunotherapeutic agents or through combinations with traditional treatments, developed to revitalize the defense system with complementing/synergetic mechanisms, have been introduced to serve as alternative approaches for NSCLC therapy. Diverse targets discussed herein have the potential to serve as both biomarkers of resistance and combination therapy targets. In view of the different resistance mechanisms, the combinatory therapy strategies are mainly manifested in the following aspects (the examples of ongoing studies trying to reverse resistance are summarized in Supplementary Table 1):
1. Emerging evidence has indicated the positive immunologic effects of chemotherapy (287). On one hand, it regulates the composition and function of immune cells such as CTLs (288), MDSCs, and Tregs (289) in the TME and the molecules expressed on tumor cells; on the other hand, it restores the recognition of immune system to tumor through enhancing tumor antigen presentation via up-regulating the expression of MHC-I and through boosting antitumor immune responses via chemo-induced tumor cell apoptosis (290–292). Multiple randomized phase III clinical trials have compared the combination of chemotherapy and ICIs with chemotherapy alone in treatment-naïve advanced lung cancer (4, 9, 293). The results consistently showed that the combination strategies are superior to chemotherapy alone in the first-line setting, regardless of PD-L1 expression, suggesting that the synergistic activity between chemotherapy and ICIs may offset the insensitivity due to low PD-L1 expression.
2. Similar to the effects induced by chemotherapy, radiation therapy combined with ICIs leads to long-lasting tumor regression through escalating antigen exposure secondary to cancer cell apoptosis, enhancing an inflamed TME (294), raising DCs activation and up-regulating proinflammatory cytokines, causing elevated TILs (295), and facilitating cancer relapsing by non-redundant immune mechanisms (296, 297). Consolidative PD-L1 inhibition after concurrent chemo-radiation significantly improved survival in unresectable stage III NSCLC in the PACIFIC study, and this approach has become the standard care of locally advanced NSCLC (298). Clinical trials evaluating the concurrent administration of radiation therapy with ICIs are ongoing.
3. Vaccines using cancer-specific peptides or DCs (237, 299, 300) and oncolytic virus therapy (301, 302) escalate the antigen presentation and priming of T cells.
1. Blocking the MAPK/PTEN/PI3K axis such as BRAF, MEK, and PI3K inhibitors contributes to Teff expansion, avoiding T-cell exhaustion and apoptosis, activating an immune-stimulatory transcriptional program, and promoting the production of proinflammatory cytokines and T-cell cytotoxicity (303, 304). BRAF and MEK inhibitors in combination with PD-1 blockade therapy showed a 73% overall response rate (ORR) and 93% stable disease in BRAF V600-mutated metastatic melanoma (305). However, cobimetinib, a MEK1/2 inhibitor, combined with atezolizumab was evaluated in a phase Ib umbrella platform MORPHEUS. The combination did not show better efficacy compared with the control arm in the NSCLC cohort of this study (306).
2. PARP inhibitors, as synergistic activating CD8+T-cell-mediated antitumor response despite up-regulating PD-L1 expression, which can be complementally inhibited by anti-PD-L1 therapy (307).
3. The combinations of nivolumab with veliparib and pembrolizumab with olaparib were tested in advanced solid tumors (308, 309). No response was observed and PFS and OS were 9.0 and 26.8 weeks, respectively, in the former trial while no results are available in the latter one.
(1) Agonists of TLRs, as contributing to the DC mutations and T-cell priming; (2) STING, as activating inflammatory reactions via IFN-α cascade upon recognition of foreign DNA; (3) dual block CTLA-4 and PD-1, as CTLA-4 inhibitor enhances the T-cell priming, Treg exhaustion, and CTL-mediated immune responses via more antigen recognition (310), while PD-1 inhibitor participates in later reactivation of Teff response; (4) adoptive T-cell transfer either alone or in combination with ICIs therapy, as increasing TILs and T-cell cytotoxicity (311, 312); and (5) bispecific monoclonal antibodies, as redirecting cytotoxic effector cells to the TME, depleting suppressive cells, and activating effector cells by targeting a cancer-specific antigen and either CD3 on CTLs or CD16A on NK cells; or targeting cancer-specific antigen and immune regulators, or targeting dual immunomodulators (313).
(1) Colony-stimulating factor 1 receptor (CSF1R) blockades, as reducing tumor invasion via the MDSCs and M2 macrophages; (2) inhibition of CD73, A2A receptor, as improving TME by targeting suppressive factors; (3) dual blockade of the TGF-β and checkpoint inhibitory receptors, as facilitating tumor penetration with T cells and reversing the immune suppressive TME (208); (4) anti-CXCR2/CXCR4 antibodies, as voiding immune evasion (314); (5) VEGF inhibitors, as normalizing the immune suppressive TME and reversing ICIs resistance (315); and (6) IL-1β inhibitor canakinumab, as targeting tumor inflammatory response and reducing immunosuppression. To date, there are four clinical trials of canakinumab in various settings in the treatment of NSCLC under way, and preliminary results from two of them were released in AACR this year. Pembrolizumab plus chemotherapy combined with canakinumab was safe and well tolerated in the first-line treatment in locally advanced or advanced NSCLC, and the recommended phase III dose of canakinumab was 200 mg s.c. Q3W (316). The efficacy of canakinumab or pembrolizumab monotherapy or in combination as neoadjuvant treatment in resectable NSCLC was assessed in the CANOPY-N study and the results are not reported (317).
(1) Blockade of alternate coinhibitory immune checkpoint receptors, such as LAG-3, TIM-3, TIGIT, BTLA, VISTA, and SIGLEC9; (2) costimulatory agonists, including 4-1BB, OX40, CD40, GITR, and ICOS, as enhancing T-cell expansion and effector functions while controlling Treg cell-suppressive functions (318, 319).
Although not being widely used in clinical practice, the antibodies targeting these immune checkpoints have exhibited promising antitumor activity in early clinical trials in various malignancies. LAG-3 blockades, Relatlimab [humanized anti-LAG-3 monoclonal antibody (mAb)], and Eftilagimod alpha (a soluble LAG-3 protein) combined with PD-1 inhibitors achieved an ORR of 15% in previously treated melanoma (320) and 52.9% in treatment-naïve advanced NSCLC (321), respectively. Based on the positive preclinical results, TIGIT blockade, especially in combination with anti-PD-1/PD-L1 mAb, has been explored in various clinical settings of advanced tumors. MK-7684 (an anti-TIGIT mAb) alone or combined with pembrolizumab showed a disease control rate of 35% and 47%, respectively, in a phase I study (322). Etigilimab (a humanized anti-TIGIT mAb) also presented early signs of efficacy as a monotherapy, with a 0% ORR but 22% stabilized disease in advanced malignancies (322). Tiragolumab is a fully human IgG1/kappa TIGIT monoclonal antibody with an intact Fc region that blocks TIGIT from binding to its PVR ligand and to the co-activating receptor CD226. It improved ORR either alone or combined with atezolizumab compared to historical data in a phase I study (323). The clinically meaningful improvement in ORR and PFS was confirmed recently in the CITYSCAPE study (a phase II study of tiragolumab plus atezolizumab vs placebo plus atezolizumab as first-line treatment in patients with PD-L1-selected NSCLC) (324). Cobolimab is a novel IgG4 anti-TIM-3 mAb and showed clinical benefit with an ORR of 15% and 40% stable disease in combination with anti-PD-1 mAb in a phase I clinical trial (325). Other anti-TIM-3 mAbs including MBG453, Sym023, INCAGN2390, LY3321367, BMS-986258, and SHR1702, as well as a bispecific antibody targeting PD-1 and TIM-3 (RO7121661), have also being evaluated in phase I trials with no clinical results available.
Apart from blocking coinhibitory immune checkpoint receptors mentioned above, several costimulatory agonists are also attractive targets, a few of which have stepped into clinical studies. ATOR-1015, a CTLA-4 × OX40 bispecific antibody, was tested in a phase I study for safety and tolerability in advanced solid tumors (326). GSK998, a humanized IgG1 agonistic OX40 mAb, combined with or without pembrolizumab was also evaluated in a phase I trial of advanced solid tumors including NSCLC (327). A lipid nanoparticle encapsulated mRNA encoding human OX40L, mRNA-2416, showed good tolerability when intratumorally injected as monotherapy in advanced malignancies in a phase I/II trial, and the combination with durvalumab is ongoing (328).
(1) DNA methyltransferase inhibitors, e.g., sensitizing tumors to PD-L1 blockade and elevating the secretion of the immunostimulatory chemokines CXCL10 and CXCL9 (155); (2) histone deacetylase inhibitors, as down-regulating MDSCs, increasing the expression of MHC-I and antigen presentation, and increasing tumor-infiltrating CD8+ T cells (95); and (3) histone methyltransferase Ezh2 inhibitor, as reversing the effects of loss of immunogenicity and antigen presentation (94).
Modifying the composition of gut microbiome might eliminate resistance to ICIs. Dietary modification, probiotics, and fecal microbiota transplantation have been emerging as an adjunct treatment to ICIs.
Of note, combination strategies that have been successful in preclinical models do not necessarily pass safety and performance assessments in clinical trials. In addition to considering the complementarity of immunotherapy resistance mechanisms, the timing and sequence are also important when formulating combination treatment strategies. Therefore, the preclinical model, translational study, and pharmacokinetic study of each of these agents in combination and in isolation are indispensable for the clinical success of combination strategies. Furthermore, multimodal approaches, for example, local therapy for oligo-progression after response to ICIs, should be implemented on therapeutic combinations for better clinical benefits.
It remains challenging to clarify the resistance mechanisms of immunotherapy since they are complex and dynamic, and certain mechanisms alternately overlap. Further understanding of the primary and acquired resistance mechanisms of immunotherapy will help clinicians to make reasonable combination treatment decisions to bring superior survival and avoid additional toxicity for patients with lung cancer.
FW conceptualized and drafted the review. QZ and SW contributed to the significant portions of the manuscript. All authors listed approved it for publication.
The study was supported by the National Key R&D Program of China (Grant No. 2016YFC1303800 to QZ), the High-level Hospital Construction Project (Grant No. DFJH201810 to QZ), the Key Lab System Project of Guangdong Science and Technology Department—Guangdong Provincial Key Lab of Translational Medicine in Lung Cancer (Grant No. 2017B030314120 to Yi-Long Wu), and the Research Foundation of Peking University Shenzhen Hospital (Grant No. JCYJ2018010).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2020.568059/full#supplementary-material
Supplementary Table 1 | Combination therapeutic strategies and examples of ongoing clinical trials to overcome ICIs resistance in advanced lung cancer.
1. Antonia SJ, Borghaei H, Ramalingam SS, Horn L, De Castro Carpeno J, Pluzanski A, et al. Four-year survival with nivolumab in patients with previously treated advanced non-small-cell lung cancer: a pooled analysis. Lancet Oncol. (2019) 20:1395–408. doi: 10.1016/S1470-2045(19)30407-3
2. Garon EB, Hellmann MD, Rizvi NA, Carcereny E, Leighl NB, Ahn MJ, et al. Five-year overall survival for patients with advanced nonsmall-cell lung cancer treated with pembrolizumab: results from the phase I KEYNOTE-001 study. J Clin Oncol. (2019) 37:2518–27. doi: 10.1200/JCO.19.00934
3. Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Five-year survival and correlates among patients with advanced melanoma, renal cell carcinoma, or non-small cell lung cancer treated with nivolumab. JAMA Oncol. (2019). 5:1411–20. doi: 10.1001/jamaoncol.2019.2187
4. West H, McCleod M, Hussein M, Morabito A, Rittmeyer A, Conter HJ, et al. Atezolizumab in combination with carboplatin plus nab-paclitaxel chemotherapy compared with chemotherapy alone as first-line treatment for metastatic non-squamous non-small-cell lung cancer (IMpower130): a multicentre, randomised, open-label, phase 3 trial. Lancet Oncol. (2019) 20:924–37. doi: 10.1016/S1470-2045(19)30167-6
5. Socinski MA, Jotte RM, Cappuzzo F, Orlandi F, Stroyakovskiy D, Nogami N, et al. Atezolizumab for first-line treatment of metastatic nonsquamous NSCLC. N Engl J Med. (2018) 378:2288–301. doi: 10.1056/NEJMoa1716948
6. Reck M, Rodriguez-Abreu D, Robinson AG, Hui R, Csoszi T, Fulop A, et al. Updated analysis of KEYNOTE-024: pembrolizumab versus platinum-based chemotherapy for advanced non-small-cell lung cancer with PD-L1 tumor proportion score of 50% or greater. J Clin Oncol. (2019) 37:537–46. doi: 10.1200/JCO.18.00149
7. Paz-Ares L, Luft A, Vicente D, Tafreshi A, Gumus M, Mazieres J, et al. Pembrolizumab plus chemotherapy for squamous non-small-cell lung cancer. N Engl J Med. (2018) 379:2040–51. doi: 10.1056/NEJMoa1810865
8. Mok TSK, Wu YL, Kudaba I, Kowalski DM, Cho BC, Turna HZ, et al. Pembrolizumab versus chemotherapy for previously untreated, PD-L1-expressing, locally advanced or metastatic non-small-cell lung cancer (KEYNOTE-042): a randomised, open-label, controlled, phase 3 trial. Lancet. (2019) 393:1819–30.
9. Gandhi L, Rodriguez-Abreu D, Gadgeel S, Esteban E, Felip E, De Angelis F, et al. Pembrolizumab plus chemotherapy in metastatic non-small-cell lung cancer. N Engl J Med. (2018) 378:2078–92. doi: 10.1056/NEJMoa1801005
10. Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell. (2017) 168:707–23. doi: 10.1016/j.cell.2017.01.017
11. Remon J, Passiglia F, Ahn MJ, Barlesi F, Forde PM, Garon EB, et al. Immune checkpoint inhibitors in thoracic malignancies: review of the existing evidence by an IASLC expert panel and recommendations. J Thorac Oncol. (2020) 15:914–47. doi: 10.1016/j.jtho.2020.03.006
12. Fares CM, Van Allen EM, Drake CG, Allison JP, Hu-Lieskovan S. Mechanisms of resistance to immune checkpoint blockade: why does checkpoint inhibitor immunotherapy not work for all patients? Am Soc Clin Oncol Educ Book. (2019) 39:147–64. doi: 10.1200/EDBK_240837
13. Chae YK, Oh MS, Giles FJ. Molecular biomarkers of primary and acquired resistance to T-cell-mediated immunotherapy in cancer: landscape, clinical implications, and future directions. Oncologist. (2018) 23:410–21. doi: 10.1634/theoncologist.2017-0354
14. Chen DS, Mellman I. Elements of cancer immunity and the cancer-immune set point. Nature. (2017) 541:321–30. doi: 10.1038/nature21349
15. Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science. (2015) 348:69–74. doi: 10.1126/science.aaa4971
16. Linnemann C, van Buuren MM, Bies L, Verdegaal EM, Schotte R, Calis JJ, et al. High-throughput epitope discovery reveals frequent recognition of neo-antigens by CD4+ T cells in human melanoma. Nat Med. (2015) 21:81–5. doi: 10.1038/nm.3773
17. Matsushita H, Vesely MD, Koboldt DC, Rickert CG, Uppaluri R, Magrini VJ, et al. Cancer exome analysis reveals a T-cell-dependent mechanism of cancer immunoediting. Nature. (2012) 482:400–4. doi: 10.1038/nature10755
18. Matsutake T, Srivastava PK. The immunoprotective MHC II epitope of a chemically induced tumor harbors a unique mutation in a ribosomal protein. Proc Natl Acad Sci USA. (2001) 98:3992–7. doi: 10.1073/pnas.071523398
19. Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med. (2014) 371:2189–99. doi: 10.1056/NEJMoa1406498
20. Tran E, Turcotte S, Gros A, Robbins PF, Lu YC, Dudley ME, et al. Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science. (2014) 344:641–5. doi: 10.1126/science.1251102
21. Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, et al. Signatures of mutational processes in human cancer. Nature. (2013) 500:415–21.
22. Sjoblom T, Jones S, Wood LD, Parsons DW, Lin J, Barber TD, et al. The consensus coding sequences of human breast and colorectal cancers. Science. (2006) 314:268–74.
23. Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, et al. Cancer immunology. mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. (2015) 348:124–8. doi: 10.1126/science.aaa1348
24. Greillier L, Tomasini P, Barlesi F. The clinical utility of tumor mutational burden in non-small cell lung cancer. Transl Lung Cancer Res. (2018) 7:639–46. doi: 10.21037/tlcr.2018.10.08
25. McGranahan N, Furness AJ, Rosenthal R, Ramskov S, Lyngaa R, Saini SK, et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science. (2016) 351:1463–9.
26. Riaz N, Morris L, Havel JJ, Makarov V, Desrichard A, Chan TA. The role of neoantigens in response to immune checkpoint blockade. Int Immunol. (2016) 28:411–9. doi: 10.1093/intimm/dxw019
27. Carbone DP, Reck M, Paz-Ares L, Creelan B, Horn L, Steins M, et al. First-line nivolumab in stage IV or recurrent non-small-cell lung cancer. N Engl J Med. (2017) 376:2415–26. doi: 10.1056/NEJMoa1613493
28. Melendez B, Van Campenhout C, Rorive S, Remmelink M, Salmon I, D’Haene N. Methods of measurement for tumor mutational burden in tumor tissue. Transl Lung Cancer Res. (2018) 7:661–7. doi: 10.21037/tlcr.2018.08.02
29. Owada-Ozaki Y, Muto S, Takagi H, Inoue T, Watanabe Y, Fukuhara M, et al. Prognostic impact of tumor mutation burden in patients with completely resected non-small cell lung cancer: brief report. J Thorac Oncol. (2018) 13:1217–21. doi: 10.1016/j.jtho.2018.04.003
30. Rizvi H, Sanchez-Vega F, La K, Chatila W, Jonsson P, Halpenny D, et al. Molecular determinants of response to anti-programmed cell death (PD)-1 and Anti-programmed death-ligand 1 (PD-L1) blockade in patients with non-small-cell lung cancer profiled with targeted next-generation sequencing. J Clin Oncol. (2018) 36:633–41. doi: 10.1200/JCO.2017.75.3384
31. Hellmann MD, Ciuleanu TE, Pluzanski A, Lee JS, Otterson GA, Audigier-Valette C, et al. Nivolumab plus ipilimumab in lung cancer with a high tumor mutational burden. N Engl J Med. (2018) 378:2093–104. doi: 10.1056/NEJMoa1801946
32. Hellmann MD, Paz-Ares L, Bernabe Caro R, Zurawski B, Kim SW, Carcereny Costa E, et al. Nivolumab plus ipilimumab in advanced non-small-cell lung cancer. N Engl J Med. (2019) 381:2020–31. doi: 10.1056/NEJMoa1910231
33. Anagnostou V, Niknafs N, Marrone KA, Bruhm DC, White JR. Multimodal genomic features predict outcome of immune checkpoint blockade in non-small-cell lung cancer. Nat Cancer. (2020) 1:99–111. doi: 10.1038/s43018-019-0008-8
34. Russo A, De Miguel Perez D, Gunasekaran M, Scilla K, Lapidus R, Cooper B, et al. Liquid biopsy tracking of lung tumor evolutions over time. Expert Rev Mol Diagn. (2019) 19:1099–108. doi: 10.1080/14737159.2020.1680287
35. Gandara DR, Paul SM, Kowanetz M, Schleifman E, Zou W, Li Y, et al. Blood-based tumor mutational burden as a predictor of clinical benefit in non-small-cell lung cancer patients treated with atezolizumab. Nat Med. (2018) 24:1441–8. doi: 10.1038/s41591-018-0134-3
36. Georgiadis A, Durham JN, Keefer LA, Bartlett BR, Zielonka M, Murphy D, et al. Noninvasive detection of microsatellite instability and high tumor mutation burden in cancer patients treated with PD-1 blockade. Clin Cancer Res. (2019) 25:7024–34. doi: 10.1158/1078-0432.CCR-19-1372
37. Wang Z, Duan J, Cai S, Han M, Dong H, Zhao J, et al. Assessment of blood tumor mutational burden as a potential biomarker for immunotherapy in patients with non-small cell lung cancer with use of a next-generation sequencing cancer gene panel. JAMA Oncol. (2019) 5:696–702. doi: 10.1001/jamaoncol.2018.7098
38. Riaz N, Havel JJ, Makarov V, Desrichard A, Urba WJ, Sims JS, et al. Tumor and microenvironment evolution during immunotherapy with nivolumab. Cell. (2017) 171:934–49.e16. doi: 10.1016/j.cell.2017.09.028
39. Koh J, Jang JY, Keam B, Kim S, Kim MY, Go H, et al. EML4-ALK enhances programmed cell death-ligand 1 expression in pulmonary adenocarcinoma via hypoxia-inducible factor (HIF)-1alpha and STAT3. Oncoimmunology. (2016) 5:e1108514. doi: 10.1080/2162402X.2015.1108514
40. Akbay EA, Koyama S, Carretero J, Altabef A, Tchaicha JH, Christensen CL, et al. Activation of the PD-1 pathway contributes to immune escape in EGFR-driven lung tumors. Cancer Discov. (2013) 3:1355–63. doi: 10.1158/1535-7163.TARG-13-B290
41. Offin M, Rizvi H, Tenet M, Ni A, Sanchez-Vega F, Li BT, et al. Tumor mutation burden and efficacy of EGFR-tyrosine kinase inhibitors in patients with EGFR-mutant lung cancers. Clin Cancer Res. (2019) 25:1063–9. doi: 10.1158/1078-0432.CCR-18-1102
42. Gainor JF, Shaw AT, Sequist LV, Fu X, Azzoli CG, Piotrowska Z, et al. EGFR mutations and ALK rearrangements are associated with low response rates to PD-1 pathway blockade in non-small cell lung cancer: a retrospective analysis. Clin Cancer Res. (2016) 22:4585–93. doi: 10.1158/1078-0432.CCR-15-3101
43. Busch SE, Hanke ML, Kargl J, Metz HE, MacPherson D, Houghton AM. Lung cancer subtypes generate unique immune responses. J Immunol. (2016) 197:4493–503. doi: 10.4049/jimmunol.1600576
44. Mazieres J, Drilon A, Lusque A, Mhanna L, Cortot AB, Mezquita L, et al. Immune checkpoint inhibitors for patients with advanced lung cancer and oncogenic driver alterations: results from the IMMUNOTARGET registry. Ann Oncol. (2019) 30:1321–8. doi: 10.1093/annonc/mdz167
45. Sabari JK, Leonardi GC, Shu CA, Umeton R, Montecalvo J, Ni A, et al. PD-L1 expression, tumor mutational burden, and response to immunotherapy in patients with MET exon 14 altered lung cancers. Ann Oncol. (2018) 29:2085–91. doi: 10.1093/annonc/mdy334
46. Dudnik E, Peled N, Nechushtan H, Wollner M, Onn A, Agbarya A, et al. BRAF mutant lung cancer: programmed death ligand 1 expression, tumor mutational burden, microsatellite instability status, and response to immune check-point inhibitors. J Thorac Oncol. (2018) 13:1128–37. doi: 10.1016/j.jtho.2018.04.024
47. Offin M, Guo R, Wu SL, Sabari J, Land JD, Ni A, et al. Immunophenotype and response to immunotherapy of RET-rearranged lung cancers. JCO Precis Oncol. (2019) 3:PO.18.00386. doi: 10.1200/PO.18.00386
48. Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. (2012) 2:401–4. doi: 10.1158/2159-8290.CD-12-0095
49. Kitajima S, Ivanova E, Guo S, Yoshida R, Campisi M, Sundararaman SK, et al. Suppression of STING associated with LKB1 loss in KRAS-driven lung cancer. Cancer Discov. (2019) 9:34–45. doi: 10.1158/2159-8290.CD-18-0689
50. Skoulidis F, Byers LA, Diao L, Papadimitrakopoulou VA, Tong P, Izzo J, et al. Co-occurring genomic alterations define major subsets of KRAS-mutant lung adenocarcinoma with distinct biology, immune profiles, and therapeutic vulnerabilities. Cancer Discov. (2015) 5:860–77. doi: 10.1158/2159-8290.CD-14-1236
51. Skoulidis F, Goldberg ME, Greenawalt DM, Hellmann MD, Awad MM, Gainor JF, et al. STK11/LKB1 mutations and PD-1 inhibitor resistance in KRAS-mutant lung adenocarcinoma. Cancer Discov. (2018) 8:822–35.
52. Dong ZY, Zhong WZ, Zhang XC, Su J, Xie Z, Liu SY, et al. Potential predictive value of TP53 and KRAS mutation status for response to PD-1 blockade immunotherapy in lung adenocarcinoma. Clin Cancer Res. (2017) 23:3012–24. doi: 10.1016/j.jtho.2016.11.504
53. Koyama S, Akbay EA, Li YY, Aref AR, Skoulidis F, Herter-Sprie GS, et al. STK11/LKB1 deficiency promotes neutrophil recruitment and proinflammatory cytokine production to suppress T-cell activity in the lung tumor microenvironment. Cancer Res. (2016) 76:999–1008. doi: 10.1158/0008-5472.CAN-15-1439
54. Bange E, Marmarelis ME, Hwang WT, Yang YX, Thompson JC, Rosenbaum J, et al. Impact of KRAS and TP53 co-mutations on outcomes after first-line systemic therapy among patients with STK11-mutated advanced non-small-cell lung cancer. JCO Precis Oncol. (2019) 3:10.1200/PO.18.00326. doi: 10.1200/PO.18.00326
55. Best SA, Ding S, Kersbergen A, Dong X, Song JY, Xie Y, et al. Distinct initiating events underpin the immune and metabolic heterogeneity of KRAS-mutant lung adenocarcinoma. Nat Commun. (2019) 10:4190. doi: 10.1038/s41467-019-12164-y
56. Best SA, Sutherland KD. “Keaping” a lid on lung cancer: the Keap1-Nrf2 pathway. Cell Cycle. (2018) 17:1696–707. doi: 10.1080/15384101.2018.1496756
57. Kadara H, Choi M, Zhang J, Parra ER, Rodriguez-Canales J, Gaffney SG, et al. Whole-exome sequencing and immune profiling of early-stage lung adenocarcinoma with fully annotated clinical follow-up. Ann Oncol. (2018) 29:1072. doi: 10.1093/annonc/mdx062
58. Arbour K, Shen R, Plodkowski A, Rizvi H, Ni A, Long N, et al. Concurrent mutations in STK11 and KEAP1 is associated with resistance to PD-(L)1 blockade in patients with NSCLC despite high TMB. J Thorac Oncol. (2018) 13:S424. doi: 10.1016/j.jtho.2018.08.480
59. Skoulidis F, Arbour KC, Hellmann MD, Patil PD, Marmarelis ME, Awad MM, et al. STK11 and/or KEAP1 genomic alterations showed lack of clinical benefits from addition of immunotherapy to chemotherapy in NSCLC patients. Proceedings of the ASCO Annual Meeting 2019, Chicago. (2019).
60. Cho BC, Lopes G, Kowalski DM, Kasahara K, Wu YL, Castro G, et al. Relationship between STK11 and KAEP1 Mutational Status and Efficacy in KEYNOTE-042: Pembrolizumab Monotherapy versus Platinum-based Chemotherapy as First-line Therapy for PD-L1-Positive advanced NSCLC. AACR 2020, Abstract 084. (2020). doi: 10.1158/1538-7445.AM2020-CT084
61. Spranger S, Bao R, Gajewski TF. Melanoma-intrinsic beta-catenin signalling prevents anti-tumour immunity. Nature. (2015) 523:231–5. doi: 10.1038/nature14404
62. George S, Miao D, Demetri GD, Adeegbe D, Rodig SJ, Shukla S, et al. Loss of PTEN is associated with resistance to anti-PD-1 checkpoint blockade therapy in metastatic uterine leiomyosarcoma. Immunity. (2017) 46:197–204. doi: 10.1016/j.immuni.2017.02.001
63. Peng W, Chen JQ, Liu C, Malu S, Creasy C, Tetzlaff MT, et al. Loss of PTEN promotes resistance to T cell-mediated immunotherapy. Cancer Discov. (2016) 6:202–16. doi: 10.1158/1538-7445.AM2016-4363
64. Toso A, Revandkar A, Di Mitri D, Guccini I, Proietti M, Sarti M, et al. Enhancing chemotherapy efficacy in Pten-deficient prostate tumors by activating the senescence-associated antitumor immunity. Cell Rep. (2014) 9:75–89. doi: 10.1016/j.celrep.2014.08.044
65. Parsa AT, Waldron JS, Panner A, Crane CA, Parney IF, Barry JJ, et al. Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nat Med. (2007) 13:84–8. doi: 10.1038/nm1517
66. De Henau O, Rausch M, Winkler D, Campesato LF, Liu C, Cymerman DH, et al. Overcoming resistance to checkpoint blockade therapy by targeting PI3Kgamma in myeloid cells. Nature. (2016) 539:443–7. doi: 10.1038/nature20554
67. Howitt BE, Shukla SA, Sholl LM, Ritterhouse LL, Watkins JC, Rodig S, et al. Association of polymerase e-mutated and microsatellite-instable endometrial cancers with neoantigen load, number of tumor-infiltrating lymphocytes, and expression of PD-1 and PD-L1. JAMA Oncol. (2015) 1:1319–23. doi: 10.1001/jamaoncol.2015.2151
68. Timmermann B, Kerick M, Roehr C, Fischer A, Isau M, Boerno ST, et al. Somatic mutation profiles of MSI and MSS colorectal cancer identified by whole exome next generation sequencing and bioinformatics analysis. PLoS One. (2010) 5:e15661. doi: 10.1371/journal.pone.0015661
69. Tomlinson IP, Novelli MR, Bodmer WF. The mutation rate and cancer. Proc Natl Acad Sci USA. (1996) 93:14800–3. doi: 10.1073/pnas.93.25.14800
70. Maby P, Tougeron D, Hamieh M, Mlecnik B, Kora H, Bindea G, et al. Correlation between density of CD8+ T-cell infiltrate in microsatellite unstable colorectal cancers and frameshift mutations: a rationale for personalized immunotherapy. Cancer Res. (2015) 75:3446–55. doi: 10.1158/0008-5472.CAN-14-3051
71. Wen WX, Leong CO. Association of BRCA1- and BRCA2-deficiency with mutation burden, expression of PD-L1/PD-1, immune infiltrates, and T cell-inflamed signature in breast cancer. PLoS One. (2019) 14:e0215381. doi: 10.1371/journal.pone.0215381
72. Mehnert JM, Panda A, Zhong H, Hirshfield K, Damare S, Lane K, et al. Immune activation and response to pembrolizumab in POLE-mutant endometrial cancer. J Clin Invest. (2016) 126:2334–40. doi: 10.1172/JCI84940
73. Gao J, Shi LZ, Zhao H, Chen J, Xiong L, He Q, et al. Loss of IFN-gamma pathway genes in tumor cells as a mechanism of resistance to anti-CTLA-4 therapy. Cell. (2016) 167:397–404.e9. doi: 10.1016/j.cell.2016.08.069
74. Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, et al. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N Engl J Med. (2016) 375:819–29. doi: 10.1056/NEJMoa1604958
75. Sharma P, Allison JP. The future of immune checkpoint therapy. Science. (2015) 348:56–61. doi: 10.1126/science.aaa8172
76. Taube JM, Anders RA, Young GD, Xu H, Sharma R, McMiller TL, et al. Colocalization of inflammatory response with B7-h1 expression in human melanocytic lesions supports an adaptive resistance mechanism of immune escape. Sci Transl Med. (2012) 4:127ra37. doi: 10.1126/scitranslmed.3003689
77. Ikeda H, Old LJ, Schreiber RD. The roles of IFN gamma in protection against tumor development and cancer immunoediting. Cytokine Growth Factor Rev. (2002) 13:95–109. doi: 10.1016/S1359-6101(01)00038-7
78. Shin DS, Zaretsky JM, Escuin-Ordinas H, Garcia-Diaz A, Hu-Lieskovan S, Kalbasi A, et al. Primary resistance to PD-1 blockade mediated by JAK1/2 mutations. Cancer Discov. (2017) 7:188–201. doi: 10.1158/2159-8290.CD-16-1223
79. Platanias LC. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat Rev Immunol. (2005) 5:375–86. doi: 10.1038/nri1604
80. Prendergast GC, Smith C, Thomas S, Mandik-Nayak L, Laury-Kleintop L, Metz R, et al. Indoleamine 2,3-dioxygenase pathways of pathogenic inflammation and immune escape in cancer. Cancer Immunol Immunother. (2014) 63:721–35. doi: 10.1007/s00262-014-1549-4
81. Toulmonde M, Penel N, Adam J, Chevreau C, Blay JY, Le Cesne A, et al. Use of PD-1 targeting, macrophage infiltration, and IDO pathway activation in sarcomas: a phase 2 clinical trial. JAMA Oncol. (2018) 4:93–7. doi: 10.1001/jamaoncol.2017.1617
82. Takeda K, Nakayama M, Hayakawa Y, Kojima Y, Ikeda H, Imai N, et al. IFN-gamma is required for cytotoxic T cell-dependent cancer genome immunoediting. Nat Commun. (2017) 8:14607. doi: 10.1038/ncomms14607
83. Darnell JE Jr., Kerr IM, Stark GR. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science. (1994) 264:1415–21. doi: 10.1126/science.8197455
84. Benci JL, Xu B, Qiu Y, Wu TJ, Dada H, Twyman-Saint Victor C, et al. Tumor interferon signaling regulates a multigenic resistance program to immune checkpoint blockade. Cell. (2016) 167:1540–54.e12. doi: 10.1016/j.cell.2016.11.022
85. Sucker A, Zhao F, Pieper N, Heeke C, Maltaner R, Stadtler N, et al. Acquired IFNgamma resistance impairs anti-tumor immunity and gives rise to T-cell-resistant melanoma lesions. Nat Commun. (2017) 8:15440. doi: 10.1038/ncomms15440
86. Bach EA, Aguet M, Schreiber RD. The IFN gamma receptor: a paradigm for cytokine receptor signaling. Annu Rev Immunol. (1997) 15:563–91. doi: 10.1146/annurev.immunol.15.1.563
87. Ayers M, Lunceford J, Nebozhyn M, Murphy E, Loboda A, Kaufman DR, et al. IFN-gamma-related mRNA profile predicts clinical response to PD-1 blockade. J Clin Invest. (2017) 127:2930–40. doi: 10.1172/JCI91190
88. Hugo W, Zaretsky JM, Sun L, Song C, Moreno BH, Hu-Lieskovan S, et al. Genomic and transcriptomic features of response to anti-PD-1 therapy in metastatic melanoma. Cell. (2016) 165:35–44. doi: 10.1016/j.cell.2016.02.065
89. Motz GT, Coukos G. The parallel lives of angiogenesis and immunosuppression: cancer and other tales. Nat Rev Immunol. (2011) 11:702–11. doi: 10.1038/nri3064
90. Schafer M, Werner S. Cancer as an overhealing wound: an old hypothesis revisited. Nat Rev Mol Cell Biol. (2008) 9:628–38. doi: 10.1038/nrm2455
91. Voron T, Marcheteau E, Pernot S, Colussi O, Tartour E, Taieb J, et al. Control of the immune response by pro-angiogenic factors. Front Oncol. (2014) 4:70. doi: 10.3389/fonc.2014.00070
92. Gallagher SJ, Shklovskaya E, Hersey P. Epigenetic modulation in cancer immunotherapy. Curr Opin Pharmacol. (2017) 35:48–56. doi: 10.1016/j.coph.2017.05.006
93. Nagarsheth N, Peng D, Kryczek I, Wu K, Li W, Zhao E, et al. PRC2 epigenetically silences Th1-type chemokines to suppress effector T-cell trafficking in colon cancer. Cancer Res. (2016) 76:275–82. doi: 10.1158/0008-5472.CAN-15-1938
94. Zingg D, Arenas-Ramirez N, Sahin D, Rosalia RA, Antunes AT, Haeusel J, et al. The histone methyltransferase Ezh2 controls mechanisms of adaptive resistance to tumor immunotherapy. Cell Rep. (2017) 20:854–67. doi: 10.1016/j.celrep.2017.07.007
95. Wang LX, Mei ZY, Zhou JH, Yao YS, Li YH, Xu YH, et al. Low dose decitabine treatment induces CD80 expression in cancer cells and stimulates tumor specific cytotoxic T lymphocyte responses. PLoS One. (2013) 8:e62924. doi: 10.1371/journal.pone.0062924
96. Vo DD, Prins RM, Begley JL, Donahue TR, Morris LF, Bruhn KW, et al. Enhanced antitumor activity induced by adoptive T-cell transfer and adjunctive use of the histone deacetylase inhibitor LAQ824. Cancer Res. (2009) 69:8693–9. doi: 10.1158/0008-5472.CAN-09-1456
97. Pan D, Kobayashi A, Jiang P, de Andrade L, Tay RE, Luoma AM, et al. A major chromatin regulator determines resistance of tumor cells to T cell-mediated killing. Science. (2018) 359:770–5. doi: 10.1126/science.aao1710
98. Miao D, Margolis CA, Gao W, Voss MH, Li W, Martini DJ, et al. Genomic correlates of response to immune checkpoint therapies in clear cell renal cell carcinoma. Science. (2018) 359:801–6. doi: 10.1126/science.aan5951
99. Kadoch C, Crabtree GR. Mammalian SWI/SNF chromatin remodeling complexes and cancer: mechanistic insights gained from human genomics. Sci Adv. (2015) 1:e1500447. doi: 10.1126/sciadv.1500447
100. Shen J, Ju Z, Zhao W, Wang L, Peng Y, Ge Z, et al. ARID1A deficiency promotes mutability and potentiates therapeutic antitumor immunity unleashed by immune checkpoint blockade. Nat Med. (2018) 24:556–62. doi: 10.1038/s41591-018-0012-z
101. Zou W, Wolchok JD, Chen L. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: mechanisms, response biomarkers, and combinations. Sci Transl Med. (2016) 8:328rv4. doi: 10.1126/scitranslmed.aad7118
102. Martin AM, Nirschl TR, Nirschl CJ, Francica BJ, Kochel CM, van Bokhoven A, et al. Paucity of PD-L1 expression in prostate cancer: innate and adaptive immune resistance. Prostate Cancer Prostatic Dis. (2015) 18:325–32. doi: 10.1038/pcan.2015.39
103. Lipson EJ, Forde PM, Hammers HJ, Emens LA, Taube JM, Topalian SL. Antagonists of PD-1 and PD-L1 in cancer treatment. Semin Oncol. (2015) 42:587–600. doi: 10.1053/j.seminoncol.2015.05.013
104. Yarchoan M, Albacker LA, Hopkins AC, Montesion M, Murugesan K, Vithayathil TT, et al. PD-L1 expression and tumor mutational burden are independent biomarkers in most cancers. JCI Insight. (2019) 4:e126908. doi: 10.1172/jci.insight.126908
105. Prestipino A, Zeiser R. Clinical implications of tumor-intrinsic mechanisms regulating PD-L1. Sci Transl Med. (2019) 11:eaav4810. doi: 10.1126/scitranslmed.aav4810
106. Sun C, Mezzadra R, Schumacher TN. Regulation and function of the PD-L1 checkpoint. Immunity. (2018) 48:434–52. doi: 10.1016/j.immuni.2018.03.014
107. Lastwika KJ, Wilson W III, Li QK, Norris J, Xu H, Ghazarian SR, et al. Control of PD-L1 expression by oncogenic activation of the AKT-mTOR pathway in non-small cell lung cancer. Cancer Res. (2016) 76:227–38. doi: 10.1158/0008-5472.CAN-14-3362
108. Casey SC, Tong L, Li Y, Do R, Walz S, Fitzgerald KN, et al. MYC regulates the antitumor immune response through CD47 and PD-L1. Science. (2016) 352:227–31. doi: 10.1126/science.aac9935
109. Dorand RD, Nthale J, Myers JT, Barkauskas DS, Avril S, Chirieleison SM, et al. Cdk5 disruption attenuates tumor PD-L1 expression and promotes antitumor immunity. Science. (2016) 353:399–403. doi: 10.1126/science.aae0477
110. Kataoka K, Shiraishi Y, Takeda Y, Sakata S, Matsumoto M, Nagano S, et al. Aberrant PD-L1 expression through 3’-UTR disruption in multiple cancers. Nature. (2016) 534:402–6.
111. Chen J, Jiang CC, Jin L, Zhang XD. Regulation of PD-L1: a novel role of pro-survival signalling in cancer. Ann Oncol. (2016) 27:409–16. doi: 10.1093/annonc/mdv615
112. Deschoolmeester V, Baay M, Van Marck E, Weyler J, Vermeulen P, Lardon F, et al. Tumor infiltrating lymphocytes: an intriguing player in the survival of colorectal cancer patients. BMC Immunol. (2010) 11:19. doi: 10.1186/1471-2172-11-19
113. He J, Hu Y, Hu M, Li B. Development of PD-1/PD-L1 pathway in tumor immune microenvironment and treatment for non-small cell lung cancer. Sci Rep. (2015) 5:13110. doi: 10.1038/srep13110
114. Li CW, Lim SO, Chung EM, Kim YS, Park AH, Yao J, et al. Eradication of triple-negative breast cancer cells by targeting Glycosylated PD-L1. Cancer Cell. (2018) 33:187–201.e10. doi: 10.1016/j.ccell.2018.01.009
115. Yao H, Lan J, Li C, Shi H, Brosseau JP, Wang H, et al. Inhibiting PD-L1 palmitoylation enhances T-cell immune responses against tumours. Nat Biomed Eng. (2019) 3:306–17. doi: 10.1038/s41551-019-0375-6
116. Gong B, Kiyotani K, Sakata S, Nagano S, Kumehara S, Baba S, et al. Secreted PD-L1 variants mediate resistance to PD-L1 blockade therapy in non-small cell lung cancer. J Exp Med. (2019) 216:982–1000. doi: 10.1084/jem.20180870
117. Dunn GP, Old LJ, Schreiber RD. The three Es of cancer immunoediting. Annu Rev Immunol. (2004) 22:329–60. doi: 10.1146/annurev.immunol.22.012703.104803
118. Anagnostou V, Smith KN, Forde PM, Niknafs N, Bhattacharya R, White J, et al. Evolution of neoantigen landscape during immune checkpoint blockade in non-small cell lung cancer. Cancer Discov. (2017) 7:264–76. doi: 10.1158/1538-7445.AM2017-NG01
119. Verdegaal EM, de Miranda NF, Visser M, Harryvan T, van Buuren MM, Andersen RS, et al. Neoantigen landscape dynamics during human melanoma-T cell interactions. Nature. (2016) 536:91–5. doi: 10.1038/nature18945
120. Ribas A, Hamid O, Daud A, Hodi FS, Wolchok JD, Kefford R, et al. Association of pembrolizumab with tumor response and survival among patients with advanced melanoma. JAMA. (2016) 315:1600–9. doi: 10.1001/jama.2016.4059
121. Kaluza KM, Thompson JM, Kottke TJ, Flynn Gilmer HC, Knutson DL, Vile RG. Adoptive T cell therapy promotes the emergence of genomically altered tumor escape variants. Int J Cancer. (2012) 131:844–54. doi: 10.1002/ijc.26447
122. Landsberg J, Kohlmeyer J, Renn M, Bald T, Rogava M, Cron M, et al. Melanomas resist T-cell therapy through inflammation-induced reversible dedifferentiation. Nature. (2012) 490:412–6. doi: 10.1038/nature11538
123. Mariathasan S, Turley SJ, Nickles D, Castiglioni A, Yuen K, Wang Y, et al. TGFbeta attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature. (2018) 554:544–8.
124. Sucker A, Zhao F, Real B, Heeke C, Bielefeld N, Mabetaen S, et al. Genetic evolution of T-cell resistance in the course of melanoma progression. Clin Cancer Res. (2014) 20:6593–604. doi: 10.1158/1078-0432.CCR-14-0567
125. Pereira C, Gimenez-Xavier P, Pros E, Pajares MJ, Moro M, Gomez A, et al. Genomic profiling of patient-derived xenografts for lung cancer identifies B2M inactivation impairing immunorecognition. Clin Cancer Res. (2017) 23:3203–13. doi: 10.1158/1078-0432.CCR-16-1946
126. Gettinger S, Choi J, Hastings K, Truini A, Datar I, Sowell R, et al. Impaired HLA Class I antigen processing and presentation as a mechanism of acquired resistance to immune checkpoint inhibitors in lung cancer. Cancer Discov. (2017) 7:1420–35. doi: 10.1158/2159-8290.CD-17-0593
127. Giannakis M, Mu XJ, Shukla SA, Qian ZR, Cohen O, Nishihara R, et al. Genomic correlates of immune-cell infiltrates in colorectal carcinoma. Cell Rep. (2016) 15:857–65. doi: 10.1016/j.celrep.2016.03.075
128. Chang CC, Pirozzi G, Wen SH, Chung IH, Chiu BL, Errico S, et al. Multiple structural and epigenetic defects in the human leukocyte antigen class I antigen presentation pathway in a recurrent metastatic melanoma following immunotherapy. J Biol Chem. (2015) 290:26562–75. doi: 10.1074/jbc.M115.676130
129. Donia M, Harbst K, van Buuren M, Kvistborg P, Lindberg MF, Andersen R, et al. Acquired immune resistance follows complete tumor regression without loss of target antigens or IFNgamma signaling. Cancer Res. (2017) 77:4562–6. doi: 10.1158/0008-5472.CAN-16-3172
130. McGranahan N, Rosenthal R, Hiley CT, Rowan AJ, Watkins TBK, Wilson GA, et al. Allele-specific HLA loss and immune escape in lung cancer evolution. Cell. (2017) 171:1259–71.e11.
131. Chowell D, Morris LGT, Grigg CM, Weber JK, Samstein RM, Makarov V, et al. Patient HLA class I genotype influences cancer response to checkpoint blockade immunotherapy. Science. (2018) 359:582–7. doi: 10.1126/science.aao4572
132. Rodig SJ, Gusenleitner D, Jackson DG, Gjini E, Giobbie-Hurder A, Jin C, et al. MHC proteins confer differential sensitivity to CTLA-4 and PD-1 blockade in untreated metastatic melanoma. Sci Transl Med. (2018) 10:eaar3342. doi: 10.1126/scitranslmed.aar3342
133. Boehm U, Klamp T, Groot M, Howard JC. Cellular responses to interferon-gamma. Annu Rev Immunol. (1997) 15:749–95. doi: 10.1146/annurev.immunol.15.1.749
134. Donnem T, Kilvaer TK, Andersen S, Richardsen E, Paulsen EE, Hald SM, et al. Strategies for clinical implementation of TNM-Immunoscore in resected nonsmall-cell lung cancer. Ann Oncol. (2016) 27:225–32. doi: 10.1093/annonc/mdv560
135. Geng Y, Shao Y, He W, Hu W, Xu Y, Chen J, et al. Prognostic role of tumor-infiltrating lymphocytes in lung cancer: a meta-analysis. Cell Physiol Biochem. (2015) 37:1560–71. doi: 10.1159/000438523
136. Gajewski TF, Schreiber H, Fu YX. Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol. (2013) 14:1014–22. doi: 10.1038/ni.2703
137. Kim HJ, Cantor H. CD4 T-cell subsets and tumor immunity: the helpful and the not-so-helpful. Cancer Immunol Res. (2014) 2:91–8. doi: 10.1158/2326-6066.CIR-13-0216
138. Rittmeyer A, Barlesi F, Waterkamp D, Park K, Ciardiello F, von Pawel J, et al. Atezolizumab versus docetaxel in patients with previously treated non-small-cell lung cancer (OAK): a phase 3, open-label, multicentre randomised controlled trial. Lancet. (2017) 389:255–65. doi: 10.1016/S0140-6736(16)32517-X
139. Brambilla E, Le Teuff G, Marguet S, Lantuejoul S, Dunant A, Graziano S, et al. Prognostic effect of tumor lymphocytic infiltration in resectable non-small-cell lung cancer. J Clin Oncol. (2016) 34:1223–30. doi: 10.1200/JCO.2015.63.0970
140. Feng W, Li Y, Shen L, Cai XW, Zhu ZF, Chang JH, et al. Prognostic value of tumor-infiltrating lymphocytes for patients with completely resected stage IIIA(N2) non-small cell lung cancer. Oncotarget. (2016) 7:7227–40. doi: 10.18632/oncotarget.6979
141. Kilic A, Landreneau RJ, Luketich JD, Pennathur A, Schuchert MJ. Density of tumor-infiltrating lymphocytes correlates with disease recurrence and survival in patients with large non-small-cell lung cancer tumors. J Surg Res. (2011) 167:207–10. doi: 10.1016/j.jss.2009.08.029
142. Ruffini E, Asioli S, Filosso PL, Lyberis P, Bruna MC, Macri L, et al. Clinical significance of tumor-infiltrating lymphocytes in lung neoplasms. Ann Thorac Surg. (2009) 87:365–71. doi: 10.1016/j.athoracsur.2008.10.067
143. Sun R, Limkin EJ, Vakalopoulou M, Dercle L, Champiat S, Han SR, et al. A radiomics approach to assess tumour-infiltrating CD8 cells and response to anti-PD-1 or anti-PD-L1 immunotherapy: an imaging biomarker, retrospective multicohort study. Lancet Oncol. (2018) 19:1180–91. doi: 10.1016/S1470-2045(18)30413-3
144. Galon J, Bruni D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat Rev Drug Discov. (2019) 18:197–218. doi: 10.1038/s41573-018-0007-y
145. AbdulJabbar K, Raza SEA, Rosenthal R, Jamal-Hanjani M, Veeriah S, Akarca A, et al. Geospatial immune variability illuminates differential evolution of lung adenocarcinoma. Nat Med. (2020) 26:1054–62. doi: 10.1038/s41591-020-0900-x
146. Spranger S, Luke JJ, Bao R, Zha Y, Hernandez KM, Li Y, et al. Density of immunogenic antigens does not explain the presence or absence of the T-cell-inflamed tumor microenvironment in melanoma. Proc Natl Acad Sci USA. (2016) 113:E7759–68. doi: 10.1073/pnas.1609376113
147. Flavell RA, Sanjabi S, Wrzesinski SH, Licona-Limon P. The polarization of immune cells in the tumour environment by TGFbeta. Nat Rev Immunol. (2010) 10:554–67. doi: 10.1038/nri2808
148. MartIn-Fontecha A, Sebastiani S, Hopken UE, Uguccioni M, Lipp M, Lanzavecchia A, et al. Regulation of dendritic cell migration to the draining lymph node: impact on T lymphocyte traffic and priming. J Exp Med. (2003) 198:615–21. doi: 10.1084/jem.20030448
149. Diamond MS, Kinder M, Matsushita H, Mashayekhi M, Dunn GP, Archambault JM, et al. Type I interferon is selectively required by dendritic cells for immune rejection of tumors. J Exp Med. (2011) 208:1989–2003. doi: 10.1084/jem.20101158
150. Fuertes MB, Kacha AK, Kline J, Woo SR, Kranz DM, Murphy KM, et al. Host type I IFN signals are required for antitumor CD8+ T cell responses through CD8{alpha}+ dendritic cells. J Exp Med. (2011) 208:2005–16. doi: 10.1084/jem.20101159
151. Sistigu A, Yamazaki T, Vacchelli E, Chaba K, Enot DP, Adam J, et al. Cancer cell-autonomous contribution of type I interferon signaling to the efficacy of chemotherapy. Nat Med. (2014) 20:1301–9. doi: 10.1038/nm.3708
152. Tarhini AA, Butterfield LH, Shuai Y, Gooding WE, Kalinski P, Kirkwood JM. Differing patterns of circulating regulatory T cells and myeloid-derived suppressor cells in metastatic melanoma patients receiving anti-CTLA4 antibody and interferon-alpha or TLR-9 agonist and GM-CSF with peptide vaccination. J Immunother. (2012) 35:702–10. doi: 10.1097/CJI.0b013e318272569b
153. Tarhini AA, Cherian J, Moschos SJ, Tawbi HA, Shuai Y, Gooding WE, et al. Safety and efficacy of combination immunotherapy with interferon alfa-2b and tremelimumab in patients with stage IV melanoma. J Clin Oncol. (2012) 30:322–8. doi: 10.1200/JCO.2011.37.5394
154. Buque A, Bloy N, Aranda F, Castoldi F, Eggermont A, Cremer I, et al. Trial watch: immunomodulatory monoclonal antibodies for oncological indications. Oncoimmunology. (2015) 4:e1008814. doi: 10.1080/2162402X.2015.1008814
155. Peng D, Kryczek I, Nagarsheth N, Zhao L, Wei S, Wang W, et al. Epigenetic silencing of TH1-type chemokines shapes tumour immunity and immunotherapy. Nature. (2015) 527:249–53. doi: 10.1038/nature15520
156. Molon B, Ugel S, Del Pozzo F, Soldani C, Zilio S, Avella D, et al. Chemokine nitration prevents intratumoral infiltration of antigen-specific T cells. J Exp Med. (2011) 208:1949–62. doi: 10.1084/jem.20101956
157. Proost P, Mortier A, Loos T, Vandercappellen J, Gouwy M, Ronsse I, et al. Proteolytic processing of CXCL11 by CD13/aminopeptidase N impairs CXCR3 and CXCR7 binding and signaling and reduces lymphocyte and endothelial cell migration. Blood. (2007) 110:37–44. doi: 10.1182/blood-2006-10-049072
158. Blackburn SD, Shin H, Freeman GJ, Wherry EJ. Selective expansion of a subset of exhausted CD8 T cells by alphaPD-L1 blockade. Proc Natl Acad Sci USA. (2008) 105:15016–21. doi: 10.1073/pnas.0801497105
159. Tauriello DVF, Palomo-Ponce S, Stork D, Berenguer-Llergo A, Badia-Ramentol J, Iglesias M, et al. TGFbeta drives immune evasion in genetically reconstituted colon cancer metastasis. Nature. (2018) 554:538–43. doi: 10.1038/nature25492
160. Chen PL, Roh W, Reuben A, Cooper ZA, Spencer CN, Prieto PA, et al. Analysis of immune signatures in longitudinal tumor samples yields insight into biomarkers of response and mechanisms of resistance to immune checkpoint blockade. Cancer Discov. (2016) 6:827–37. doi: 10.1158/2159-8290.CD-15-1545
161. Hamid O, Schmidt H, Nissan A, Ridolfi L, Aamdal S, Hansson J, et al. A prospective phase II trial exploring the association between tumor microenvironment biomarkers and clinical activity of ipilimumab in advanced melanoma. J Transl Med. (2011) 9:204. doi: 10.1186/1479-5876-9-204
162. Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. (2014) 515:568–71.
163. Cha E, Klinger M, Hou Y, Cummings C, Ribas A, Faham M, et al. Improved survival with T cell clonotype stability after anti-CTLA-4 treatment in cancer patients. Sci Transl Med. (2014) 6:238ra70. doi: 10.1126/scitranslmed.3008211
164. Hirsch FR, Scagliotti GV, Mulshine JL, Kwon R, Curran WJ Jr., Wu YL, et al. Lung cancer: current therapies and new targeted treatments. Lancet. (2017) 389:299–311. doi: 10.1016/S0140-6736(16)30958-8
165. Reuben A, Zhang J, Chiou SH, Gittelman RM, Li J, Lee WC, et al. Comprehensive T cell repertoire characterization of non-small cell lung cancer. Nat Commun. (2020) 11:603. doi: 10.1038/s41467-019-14273-0
166. Joshi K, de Massy MR, Ismail M, Reading JL, Uddin I, Woolston A, et al. Spatial heterogeneity of the T cell receptor repertoire reflects the mutational landscape in lung cancer. Nat Med. (2019) 25:1549–59. doi: 10.1038/s41591-019-0592-2
167. Thommen DS, Schreiner J, Muller P, Herzig P, Roller A, Belousov A, et al. Progression of lung cancer is associated with increased dysfunction of T cells defined by coexpression of multiple inhibitory receptors. Cancer Immunol Res. (2015) 3:1344–55. doi: 10.1158/2326-6066.CIR-15-0097
168. Topalian SL, Drake CG, Pardoll DM. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell. (2015) 27:450–61. doi: 10.1016/j.ccell.2015.03.001
169. Hellmann MD, Friedman CF, Wolchok JD. Combinatorial Cancer Immunotherapies. Adv Immunol. (2016) 130:251–77. doi: 10.1016/bs.ai.2015.12.005
170. Balko JJ, Wang D, Ericsson-Gonzalez P, Nixon M, Salgado R, Sanchez V, et al. MHC-II expression to drive a unique pattern of adaptive resistance to antitumor immunity through receptor checkpoint engagement. J Clin Oncol. (2018) 36:180. doi: 10.1200/JCO.2018.36.5_suppl.180
171. Du W, Yang M, Turner A, Xu C, Ferris RL, Huang J, et al. TIM-3 as a target for cancer immunotherapy and mechanisms of action. Int J Mol Sci. (2017) 18:645. doi: 10.3390/ijms18030645
172. Manieri NA, Chiang EY, Grogan JL. TIGIT: a key inhibitor of the cancer immunity cycle. Trends Immunol. (2017) 38:20–8. doi: 10.1016/j.it.2016.10.002
173. Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol. (2015) 15:486–99. doi: 10.1038/nri3862
174. Wei F, Zhong S, Ma Z, Kong H, Medvec A, Ahmed R, et al. Strength of PD-1 signaling differentially affects T-cell effector functions. Proc Natl Acad Sci USA. (2013) 110:E2480–9. doi: 10.1073/pnas.1305394110
175. Ngiow SF, Young A, Jacquelot N, Yamazaki T, Enot D, Zitvogel L, et al. A Threshold Level of Intratumor CD8+ T-cell PD1 Expression Dictates Therapeutic Response to Anti-PD1. Cancer Res. (2015) 75:3800–11. doi: 10.1158/0008-5472.CAN-15-1082
176. Pauken KE, Sammons MA, Odorizzi PM, Manne S, Godec J, Khan O, et al. Epigenetic stability of exhausted T cells limits durability of reinvigoration by PD-1 blockade. Science. (2016) 354:1160–5. doi: 10.1126/science.aaf2807
177. Sen DR, Kaminski J, Barnitz RA, Kurachi M, Gerdemann U, Yates KB, et al. The epigenetic landscape of T cell exhaustion. Science. (2016) 354:1165–9. doi: 10.1126/science.aae0491
178. Mognol GP, Spreafico R, Wong V, Scott-Browne JP, Togher S, Hoffmann A, et al. Exhaustion-associated regulatory regions in CD8(+) tumor-infiltrating T cells. Proc Natl Acad Sci USA. (2017) 114:E2776–85. doi: 10.1073/pnas.1620498114
179. Ribas A, Shin DS, Zaretsky J, Frederiksen J, Cornish A, Avramis E, et al. PD-1 blockade expands intratumoral memory T cells. Cancer Immunol Res. (2016) 4:194–203. doi: 10.1158/2326-6066.CIR-15-0210
180. Farber DL, Yudanin NA, Restifo NP. Human memory T cells: generation, compartmentalization and homeostasis. Nat Rev Immunol. (2014) 14:24–35. doi: 10.1038/nri3567
181. Woodland DL, Kohlmeier JE. Migration, maintenance and recall of memory T cells in peripheral tissues. Nat Rev Immunol. (2009) 9:153–61. doi: 10.1038/nri2496
182. Ma C, Cheung AF, Chodon T, Koya RC, Wu Z, Ng C, et al. Multifunctional T-cell analyses to study response and progression in adoptive cell transfer immunotherapy. Cancer Discov. (2013) 3:418–29. doi: 10.1158/2159-8290.CD-12-0383
183. Ma C, Fan R, Ahmad H, Shi Q, Comin-Anduix B, Chodon T, et al. A clinical microchip for evaluation of single immune cells reveals high functional heterogeneity in phenotypically similar T cells. Nat Med. (2011) 17:738–43. doi: 10.1038/nm.2375
184. Fridman WH, Zitvogel L, Sautes-Fridman C, Kroemer G. The immune contexture in cancer prognosis and treatment. Nat Rev Clin Oncol. (2017) 14:717–34. doi: 10.1038/nrclinonc.2017.101
185. Oida T, Zhang X, Goto M, Hachimura S, Totsuka M, Kaminogawa S, et al. CD4+CD25- T cells that express latency-associated peptide on the surface suppress CD4+CD45RBhigh-induced colitis by a TGF-beta-dependent mechanism. J Immunol. (2003) 170:2516–22. doi: 10.4049/jimmunol.170.5.2516
186. Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell. (2008) 133:775–87. doi: 10.1016/j.cell.2008.05.009
187. Sundstedt A, O’Neill EJ, Nicolson KS, Wraith DC. Role for IL-10 in suppression mediated by peptide-induced regulatory T cells in vivo. J Immunol. (2003) 170:1240–8. doi: 10.4049/jimmunol.170.3.1240
188. Simpson TR, Li F, Montalvo-Ortiz W, Sepulveda MA, Bergerhoff K, Arce F, et al. Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti-CTLA-4 therapy against melanoma. J Exp Med. (2013) 210:1695–710. doi: 10.1084/jem.20130579
189. Viehl CT, Moore TT, Liyanage UK, Frey DM, Ehlers JP, Eberlein TJ, et al. Depletion of CD4+CD25+ regulatory T cells promotes a tumor-specific immune response in pancreas cancer-bearing mice. Ann Surg Oncol. (2006) 13:1252–8. doi: 10.1245/s10434-006-9015-y
190. Hannani D, Vetizou M, Enot D, Rusakiewicz S, Chaput N, Klatzmann D, et al. Anticancer immunotherapy by CTLA-4 blockade: obligatory contribution of IL-2 receptors and negative prognostic impact of soluble CD25. Cell Res. (2015) 25:208–24. doi: 10.1038/cr.2015.3
191. Meyer C, Cagnon L, Costa-Nunes CM, Baumgaertner P, Montandon N, Leyvraz L, et al. Frequencies of circulating MDSC correlate with clinical outcome of melanoma patients treated with ipilimumab. Cancer Immunol Immunother. (2014) 63:247–57. doi: 10.1007/s00262-013-1508-5
192. Kodumudi KN, Weber A, Sarnaik AA, Pilon-Thomas S. Blockade of myeloid-derived suppressor cells after induction of lymphopenia improves adoptive T cell therapy in a murine model of melanoma. J Immunol. (2012) 189:5147–54. doi: 10.4049/jimmunol.1200274
193. Laborde RR, Lin Y, Gustafson MP, Bulur PA, Dietz AB. Cancer vaccines in the world of immune suppressive monocytes (CD14(+)HLA-DR(lo/neg) Cells): the gateway to improved responses. Front Immunol. (2014) 5:147. doi: 10.3389/fimmu.2014.00147
194. Biswas SK, Mantovani A. Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nat Immunol. (2010) 11:889–96. doi: 10.1038/ni.1937
195. Hu W, Li X, Zhang C, Yang Y, Jiang J, Wu C. Tumor-associated macrophages in cancers. Clin Transl Oncol. (2016) 18:251–8. doi: 10.1007/s12094-015-1373-0
196. Chanmee T, Ontong P, Konno K, Itano N. Tumor-associated macrophages as major players in the tumor microenvironment. Cancers (Basel). (2014) 6:1670–90. doi: 10.3390/cancers6031670
197. De Palma M, Lewis CE. Macrophage regulation of tumor responses to anticancer therapies. Cancer Cell. (2013) 23:277–86. doi: 10.1016/j.ccr.2013.02.013
198. Ruffell B, Coussens LM. Macrophages and therapeutic resistance in cancer. Cancer Cell. (2015) 27:462–72. doi: 10.1016/j.ccell.2015.02.015
199. Kuang DM, Zhao Q, Peng C, Xu J, Zhang JP, Wu C, et al. Activated monocytes in peritumoral stroma of hepatocellular carcinoma foster immune privilege and disease progression through PD-L1. J Exp Med. (2009) 206:1327–37. doi: 10.1084/jem.20082173
200. Kryczek I, Zou L, Rodriguez P, Zhu G, Wei S, Mottram P, et al. B7-H4 expression identifies a novel suppressive macrophage population in human ovarian carcinoma. J Exp Med. (2006) 203:871–81. doi: 10.1084/jem.20050930
201. Le DT, Lutz E, Uram JN, Sugar EA, Onners B, Solt S, et al. Evaluation of ipilimumab in combination with allogeneic pancreatic tumor cells transfected with a GM-CSF gene in previously treated pancreatic cancer. J Immunother. (2013) 36:382–9. doi: 10.1097/CJI.0b013e31829fb7a2
202. Zhu Y, Knolhoff BL, Meyer MA, Nywening TM, West BL, Luo J, et al. CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res. (2014) 74:5057–69. doi: 10.1158/0008-5472.CAN-13-3723
203. Mok S, Koya RC, Tsui C, Xu J, Robert L, Wu L, et al. Inhibition of CSF-1 receptor improves the antitumor efficacy of adoptive cell transfer immunotherapy. Cancer Res. (2014) 74:153–61. doi: 10.1158/0008-5472.CAN-13-1816
204. Highfill SL, Cui Y, Giles AJ, Smith JP, Zhang H, Morse E, et al. Disruption of CXCR2-mediated MDSC tumor trafficking enhances anti-PD1 efficacy. Sci Transl Med. (2014) 6:237ra67. doi: 10.1126/scitranslmed.3007974
205. Rabin RL, Park MK, Liao F, Swofford R, Stephany D, Farber JM. Chemokine receptor responses on T cells are achieved through regulation of both receptor expression and signaling. J Immunol. (1999) 162:3840–50.
206. Joyce JA, Fearon DT. T cell exclusion, immune privilege, and the tumor microenvironment. Science. (2015) 348:74–80. doi: 10.1126/science.aaa6204
207. Lebrun JJ. The dual role of TGFbeta in human cancer: from tumor suppression to cancer metastasis. ISRN Mol Biol. (2012) 2012:381428. doi: 10.5402/2012/381428
208. Vanpouille-Box C, Diamond JM, Pilones KA, Zavadil J, Babb JS, Formenti SC, et al. TGFbeta is a master regulator of radiation therapy-induced antitumor immunity. Cancer Res. (2015) 75:2232–42. doi: 10.1158/0008-5472.CAN-14-3511
209. Hanks B, Holtzhausen A, Evans K, Heid M, Blobe G. Combinatorial TGF-{beta} signaling blockade and anti-CTLA-4 antibody immunotherapy in a murine BRAFV600E-PTEN-/-transgenic model of melanoma. Paper Presented at: ASCO Annual Meeting Proceedings 2014, Chicago. (2014). doi: 10.1200/jco.2014.32.15_suppl.3011
210. Bertrand F, Montfort A, Marcheteau E, Imbert C, Gilhodes J, Filleron T, et al. TNFalpha blockade overcomes resistance to anti-PD-1 in experimental melanoma. Nat Commun. (2017) 8:2256. doi: 10.1038/s41467-017-02358-7
211. Ott PA, Hodi FS, Buchbinder EI. Inhibition of immune checkpoints and vascular endothelial growth factor as combination therapy for metastatic melanoma: an overview of rationale, preclinical evidence, and initial clinical data. Front Oncol. (2015) 5:202. doi: 10.3389/fonc.2015.00202
212. Ohm JE, Gabrilovich DI, Sempowski GD, Kisseleva E, Parman KS, Nadaf S, et al. VEGF inhibits T-cell development and may contribute to tumor-induced immune suppression. Blood. (2003) 101:4878–86. doi: 10.1182/blood-2002-07-1956
213. Motz GT, Santoro SP, Wang LP, Garrabrant T, Lastra RR, Hagemann IS, et al. Tumor endothelium FasL establishes a selective immune barrier promoting tolerance in tumors. Nat Med. (2014) 20:607–15. doi: 10.1038/nm.3541
214. Voron T, Colussi O, Marcheteau E, Pernot S, Nizard M, Pointet AL, et al. VEGF-A modulates expression of inhibitory checkpoints on CD8+ T cells in tumors. J Exp Med. (2015) 212:139–48. doi: 10.1084/jem.20140559
215. Bjoern J, Juul Nitschke N, Zeeberg Iversen T, Schmidt H, Fode K, Svane IM. Immunological correlates of treatment and response in stage IV malignant melanoma patients treated with Ipilimumab. Oncoimmunology. (2016) 5:e1100788. doi: 10.1080/2162402X.2015.1100788
216. Schalper KA, Carleton M, Zhou M, Chen T, Feng Y, Huang SP, et al. Elevated serum interleukin-8 is associated with enhanced intratumor neutrophils and reduced clinical benefit of immune-checkpoint inhibitors. Nat Med. (2020) 26:688–92. doi: 10.1038/s41591-020-0856-x
217. Yuen KC, Liu LF, Gupta V, Madireddi S, Keerthivasan S, Li C, et al. High systemic and tumor-associated IL-8 correlates with reduced clinical benefit of PD-L1 blockade. Nat Med. (2020) 26:693–8. doi: 10.1158/1538-7445.AM2020-2000
218. Garcia-Diaz A, Shin DS, Moreno BH, Saco J, Escuin-Ordinas H, Rodriguez GA, et al. Interferon receptor signaling pathways regulating PD-L1 and PD-L2 expression. Cell Rep. (2017) 19:1189–201. doi: 10.1016/j.celrep.2017.04.031
219. Moore BB, Arenberg DA, Stoy K, Morgan T, Addison CL, Morris SB, et al. Distinct CXC chemokines mediate tumorigenicity of prostate cancer cells. Am J Pathol. (1999) 154:1503–12. doi: 10.1016/S0002-9440(10)65404-1
220. Najjar YG, Rayman P, Jia X, Pavicic PG Jr., Rini BI, Tannenbaum C, et al. Myeloid-derived suppressor cell subset accumulation in renal cell carcinoma parenchyma is associated with intratumoral expression of IL1beta, IL8, CXCL5, and Mip-1alpha. Clin Cancer Res. (2017) 23:2346–55. doi: 10.1158/1078-0432.CCR-15-1823
221. Baggiolini M. CXCL8–the first chemokine. Front Immunol. (2015) 6:285. doi: 10.3389/fimmu.2015.00285
222. Baggiolini M, Walz A, Kunkel SL. Neutrophil-activating peptide-1/interleukin 8, a novel cytokine that activates neutrophils. J Clin Invest. (1989) 84:1045–9. doi: 10.1172/JCI114265
223. Alfaro C, Teijeira A, Onate C, Perez G, Sanmamed MF, Andueza MP, et al. Tumor-produced interleukin-8 attracts human myeloid-derived suppressor cells and elicits extrusion of neutrophil extracellular traps (NETs). Clin Cancer Res. (2016) 22:3924–36. doi: 10.1158/1078-0432.CCR-15-2463
224. Zhang H, Conrad DM, Butler JJ, Zhao C, Blay J, Hoskin DW. Adenosine acts through A2 receptors to inhibit IL-2-induced tyrosine phosphorylation of STAT5 in T lymphocytes: role of cyclic adenosine 3’,5’-monophosphate and phosphatases. J Immunol. (2004) 173:932–44. doi: 10.4049/jimmunol.173.2.932
225. Stagg J, Divisekera U, McLaughlin N, Sharkey J, Pommey S, Denoyer D, et al. Anti-CD73 antibody therapy inhibits breast tumor growth and metastasis. Proc Natl Acad Sci USA. (2010) 107:1547–52. doi: 10.1073/pnas.0908801107
226. Allard B, Pommey S, Smyth MJ, Stagg J. Targeting CD73 enhances the antitumor activity of anti-PD-1 and anti-CTLA-4 mAbs. Clin Cancer Res. (2013) 19:5626–35. doi: 10.1158/1078-0432.CCR-13-0545
227. Beavis PA, Slaney CY, Milenkovski N, Henderson MA, Loi S, Stagg J, et al. CD73: a potential biomarker for anti-PD-1 therapy. Oncoimmunology. (2015) 4:e1046675. doi: 10.1080/2162402X.2015.1046675
228. Platten M, von Knebel Doeberitz N, Oezen I, Wick W, Ochs K. Cancer immunotherapy by targeting IDO1/TDO and their downstream effectors. Front Immunol. (2014) 5:673. doi: 10.3389/fimmu.2014.00673
229. Munn DH, Sharma MD, Mellor AL. Ligation of B7-1/B7-2 by human CD4+ T cells triggers indoleamine 2,3-dioxygenase activity in dendritic cells. J Immunol. (2004) 172:4100–10. doi: 10.4049/jimmunol.172.7.4100
230. Holmgaard RB, Zamarin D, Munn DH, Wolchok JD, Allison JP. Indoleamine 2,3-dioxygenase is a critical resistance mechanism in antitumor T cell immunotherapy targeting CTLA-4. J Exp Med. (2013) 210:1389–402. doi: 10.1084/jem.20130066
231. Spranger S, Koblish HK, Horton B, Scherle PA, Newton R, Gajewski TF. Mechanism of tumor rejection with doublets of CTLA-4, PD-1/PD-L1, or IDO blockade involves restored IL-2 production and proliferation of CD8(+) T cells directly within the tumor microenvironment. J Immunother Cancer. (2014) 2:3. doi: 10.1186/2051-1426-2-3
232. Long GV, Dummer R, Hamid O, Gajewski TF, Caglevic C, Dalle S, et al. Epacadostat plus pembrolizumab versus placebo plus pembrolizumab in patients with unresectable or metastatic melanoma (ECHO-301/KEYNOTE-252): a phase 3, randomised, double-blind study. Lancet Oncol. (2019) 20:1083–97. doi: 10.1016/S1470-2045(19)30274-8
233. Mott KR, Gate D, Matundan HH, Ghiasi YN, Town T, Ghiasi H. CD8+ T cells play a bystander role in mice latently infected with herpes simplex virus 1. J Virol. (2016) 90:5059–67. doi: 10.1128/JVI.00255-16
234. Zang X, Loke P, Kim J, Murphy K, Waitz R, Allison JP. B7x: a widely expressed B7 family member that inhibits T cell activation. Proc Natl Acad Sci USA. (2003) 100:10388–92. doi: 10.1073/pnas.1434299100
235. Zou W, Chen L. Inhibitory B7-family molecules in the tumour microenvironment. Nat Rev Immunol. (2008) 8:467–77. doi: 10.1038/nri2326
236. Genova C, Boccardo S, Mora M, Rijavec E, Biello F, Rossi G, et al. Correlation between B7-H4 and survival of non-small-cell lung cancer patients treated with nivolumab. J Clin Med. (2019) 8:1566. doi: 10.3390/jcm8101566
237. Ott PA, Hu Z, Keskin DB, Shukla SA, Sun J, Bozym DJ, et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature. (2017) 547:217–21.
238. Son CH, Bae JH, Shin DY, Lee HR, Choi YJ, Jo WS, et al. CTLA-4 blockade enhances antitumor immunity of intratumoral injection of immature dendritic cells into irradiated tumor in a mouse colon cancer model. J Immunother. (2014) 37:1–7. doi: 10.1097/CJI.0000000000000007
239. Chaput N, Lepage P, Coutzac C, Soularue E, Le Roux K, Monot C, et al. Baseline gut microbiota predicts clinical response and colitis in metastatic melanoma patients treated with ipilimumab. Ann Oncol. (2019) 30:2012. doi: 10.1093/annonc/mdz224
240. Gopalakrishnan V, Spencer CN, Nezi L, Reuben A, Andrews MC, Karpinets TV, et al. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science. (2018) 359:97–103.
241. Matson V, Fessler J, Bao R, Chongsuwat T, Zha Y, Alegre ML, et al. The commensal microbiome is associated with anti-PD-1 efficacy in metastatic melanoma patients. Science. (2018) 359:104–8. doi: 10.1126/science.aao3290
242. Derosa L, Routy B, Fidelle M, Iebba V, Alla L, Pasolli E, et al. Gut bacteria composition drives primary resistance to cancer immunotherapy in renal cell carcinoma patients. Eur Urol. (2020) 78:195–206. doi: 10.1016/j.eururo.2020.04.044
243. Vetizou M, Pitt JM, Daillere R, Lepage P, Waldschmitt N, Flament C, et al. Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science. (2015) 350:1079–84. doi: 10.1126/science.aad1329
244. Routy B, Le Chatelier E, Derosa L, Duong CPM, Alou MT, Daillere R, et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science. (2018) 359:91–7.
245. Pitt JM, Vetizou M, Daillere R, Roberti MP, Yamazaki T, Routy B, et al. Resistance mechanisms to immune-checkpoint blockade in cancer: tumor-intrinsic and -extrinsic factors. Immunity. (2016) 44:1255–69. doi: 10.1016/j.immuni.2016.06.001
246. Sivan A, Corrales L, Hubert N, Williams JB, Aquino-Michaels K, Earley ZM, et al. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science. (2015) 350:1084–9. doi: 10.1126/science.aac4255
247. Tanoue T, Morita S, Plichta DR, Skelly AN, Suda W, Sugiura Y, et al. A defined commensal consortium elicits CD8 T cells and anti-cancer immunity. Nature. (2019) 565:600–5. doi: 10.1038/s41586-019-0878-z
248. Kim YG, Udayanga KG, Totsuka N, Weinberg JB, Nunez G, Shibuya A. Gut dysbiosis promotes M2 macrophage polarization and allergic airway inflammation via fungi-induced PGE(2). Cell Host Microbe. (2014) 15:95–102. doi: 10.1016/j.chom.2013.12.010
249. Chaput N, Lepage P, Coutzac C, Soularue E, Le Roux K, Monot C, et al. Baseline gut microbiota predicts clinical response and colitis in metastatic melanoma patients treated with ipilimumab. Ann Oncol. (2017) 28:1368–79. doi: 10.1093/annonc/mdx108
250. Round JL, Mazmanian SK. Inducible Foxp3+ regulatory T-cell development by a commensal bacterium of the intestinal microbiota. Proc Natl Acad Sci USA. (2010) 107:12204–9. doi: 10.1073/pnas.0909122107
251. Cremonesi E, Governa V, Garzon JFG, Mele V, Amicarella F, Muraro MG, et al. Gut microbiota modulate T cell trafficking into human colorectal cancer. Gut. (2018) 67:1984–94. doi: 10.1158/1538-7445.AM2018-1001
252. Pushalkar S, Hundeyin M, Daley D, Zambirinis CP, Kurz E, Mishra A, et al. The pancreatic cancer microbiome promotes oncogenesis by induction of innate and adaptive immune suppression. Cancer Discov. (2018) 8:403–16. doi: 10.1158/2159-8290.CD-17-1134
253. Kadosh E, Snir-Alkalay I, Venkatachalam A, May S, Lasry A, Elyada E, et al. The gut microbiome switches mutant p53 from tumour-suppressive to oncogenic. Nature. (2020) 586:133–38. doi: 10.1038/s41586-020-2541-0
254. Derosa L, Hellmann MD, Spaziano M, Halpenny D, Fidelle M, Rizvi H, et al. Negative association of antibiotics on clinical activity of immune checkpoint inhibitors in patients with advanced renal cell and non-small-cell lung cancer. Ann Oncol. (2018) 29:1437–44. doi: 10.1093/annonc/mdy103
255. Suez J, Zmora N, Zilberman-Schapira G, Mor U, Dori-Bachash M, Bashiardes S, et al. Post-antibiotic gut mucosal microbiome reconstitution is impaired by probiotics and improved by autologous FMT. Cell. (2018) 174:1406–23.e16. doi: 10.1016/j.cell.2018.08.047
256. Vangay P, Ward T, Gerber JS, Knights D. Antibiotics, pediatric dysbiosis, and disease. Cell Host Microbe. (2015) 17:553–64. doi: 10.1016/j.chom.2015.04.006
257. Tamburini S, Shen N, Wu HC, Clemente JC. The microbiome in early life: implications for health outcomes. Nat Med. (2016) 22:713–22. doi: 10.1038/nm.4142
258. Routy B, Gopalakrishnan V, Daillere R, Zitvogel L, Wargo JA, Kroemer G. The gut microbiota influences anticancer immunosurveillance and general health. Nat Rev Clin Oncol. (2018) 15:382–96. doi: 10.1038/s41571-018-0006-2
259. Viaud S, Saccheri F, Mignot G, Yamazaki T, Daillere R, Hannani D, et al. The intestinal microbiota modulates the anticancer immune effects of cyclophosphamide. Science. (2013) 342:971–6. doi: 10.1126/science.1240537
260. Daillere R, Vetizou M, Waldschmitt N, Yamazaki T, Isnard C, Poirier-Colame V, et al. Enterococcus hirae and barnesiella intestinihominis facilitate cyclophosphamide-induced therapeutic immunomodulatory effects. Immunity. (2016) 45:931–43. doi: 10.1016/j.immuni.2016.09.009
261. Iida N, Dzutsev A, Stewart CA, Smith L, Bouladoux N, Weingarten RA, et al. Commensal bacteria control cancer response to therapy by modulating the tumor microenvironment. Science. (2013) 342:967–70. doi: 10.1126/science.1240527
262. Bullman S, Pedamallu CS, Sicinska E, Clancy TE, Zhang X, Cai D, et al. Analysis of Fusobacterium persistence and antibiotic response in colorectal cancer. Science. (2017) 358:1443–8. doi: 10.1126/science.aal5240
263. Coutinho AE, Chapman KE. The anti-inflammatory and immunosuppressive effects of glucocorticoids, recent developments and mechanistic insights. Mol Cell Endocrinol. (2011) 335:2–13. doi: 10.1016/j.mce.2010.04.005
264. Schadendorf D, Wolchok JD, Hodi FS, Chiarion-Sileni V, Gonzalez R, Rutkowski P, et al. Efficacy and safety outcomes in patients with advanced melanoma who discontinued treatment with nivolumab and ipilimumab because of adverse events: a pooled analysis of randomized phase II and III trials. J Clin Oncol. (2017) 35:3807–14. doi: 10.1200/JCO.2017.73.2289
265. Horvat TZ, Adel NG, Dang TO, Momtaz P, Postow MA, Callahan MK, et al. Immune-related adverse events, need for systemic immunosuppression, and effects on survival and time to treatment failure in patients with melanoma treated with ipilimumab at memorial sloan kettering cancer center. J Clin Oncol. (2015) 33:3193–8. doi: 10.1200/JCO.2015.60.8448
266. Mahata B, Zhang X, Kolodziejczyk AA, Proserpio V, Haim-Vilmovsky L, Taylor AE, et al. Single-cell RNA sequencing reveals T helper cells synthesizing steroids de novo to contribute to immune homeostasis. Cell Rep. (2014) 7:1130–42. doi: 10.1016/j.celrep.2014.04.011
267. Planchard D, Popat S, Kerr K, Novello S, Smit EF, Faivre-Finn C, et al. Metastatic non-small cell lung cancer: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol. (2018) 29(Suppl. 4):iv192–237. doi: 10.1093/annonc/mdy275
268. Fuca G, Galli G, Poggi M, Lo Russo G, Proto C, Imbimbo M, et al. Modulation of peripheral blood immune cells by early use of steroids and its association with clinical outcomes in patients with metastatic non-small cell lung cancer treated with immune checkpoint inhibitors. ESMO Open. (2019) 4:e000457. doi: 10.1136/esmoopen-2018-000457
269. Della Corte CM, Morgillo F. Early use of steroids affects immune cells and impairs immunotherapy efficacy. ESMO Open. (2019) 4:e000477. doi: 10.1136/esmoopen-2018-000477
270. Vijayan D, Young A, Teng MWL, Smyth MJ. Targeting immunosuppressive adenosine in cancer. Nat Rev Cancer. (2017) 17:709–24. doi: 10.1038/nrc.2017.86
271. Renner K, Singer K, Koehl GE, Geissler EK, Peter K, Siska PJ, et al. Metabolic hallmarks of tumor and immune cells in the tumor microenvironment. Front Immunol. (2017) 8:248. doi: 10.3389/fimmu.2017.00248
272. De Rosa V, Di Rella F, Di Giacomo A, Matarese G. Regulatory T cells as suppressors of anti-tumor immunity: role of metabolism. Cytokine Growth Factor Rev. (2017) 35:15–25. doi: 10.1016/j.cytogfr.2017.04.001
273. Soldati L, Di Renzo L, Jirillo E, Ascierto PA, Marincola FM, De Lorenzo A. The influence of diet on anti-cancer immune responsiveness. J Transl Med. (2018) 16:75. doi: 10.1186/s12967-018-1448-0
274. Qu X, Tang Y, Hua S. Immunological approaches towards cancer and inflammation: a cross talk. Front Immunol. (2018) 9:563. doi: 10.3389/fimmu.2018.00563
275. Maccio A, Madeddu C. Blocking inflammation to improve immunotherapy of advanced cancer. Immunology. (2020) 159:357–64. doi: 10.1111/imm.13164
276. June CH, Warshauer JT, Bluestone JA. Is autoimmunity the Achilles’ heel of cancer immunotherapy? Nat Med. (2017) 23:540–7. doi: 10.1038/nm.4321
277. Caspi RR. Immunotherapy of autoimmunity and cancer: the penalty for success. Nat Rev Immunol. (2008) 8:970–6. doi: 10.1038/nri2438
278. Mo J, Hu X, Gu L, Chen B, Khadaroo PA, Shen Z, et al. Smokers or non-smokers: who benefits more from immune checkpoint inhibitors in treatment of malignancies? An up-to-date meta-analysis. World J Surg Oncol. (2020) 18:15. doi: 10.1186/s12957-020-1792-4
279. Li B, Huang X, Fu L. Impact of smoking on efficacy of PD-1/PD-L1 inhibitors in non-small cell lung cancer patients: a meta-analysis. Onco Targets Ther. (2018) 11:3691–6. doi: 10.2147/OTT.S156421
280. Lee CK, Man J, Lord S, Cooper W, Links M, Gebski V, et al. Clinical and molecular characteristics associated with survival among patients treated with checkpoint inhibitors for advanced non-small cell lung carcinoma: a systematic review and meta-analysis. JAMA Oncol. (2018) 4:210–6. doi: 10.1001/jamaoncol.2017.4427
281. Abdel-Rahman O. Smoking and EGFR status may predict outcomes of advanced NSCLC treated with PD-(L)1 inhibitors beyond first line: a meta-analysis. Clin Respir J. (2018) 12:1809–19. doi: 10.1111/crj.12742
282. Ng TL, Liu Y, Dimou A, Patil T, Aisner DL, Dong Z, et al. Predictive value of oncogenic driver subtype, programmed death-1 ligand (PD-L1) score, and smoking status on the efficacy of PD-1/PD-L1 inhibitors in patients with oncogene-driven non-small cell lung cancer. Cancer. (2019) 125:1038–49. doi: 10.1002/cncr.31871
283. Kerdidani D, Magkouta S, Chouvardas P, Karavana V, Glynos K, Roumelioti F, et al. Cigarette smoke-induced emphysema exhausts early Cytotoxic CD8(+) T cell responses against nascent lung cancer cells. J Immunol. (2018) 201:1558–69. doi: 10.4049/jimmunol.1700700
284. Wang GZ, Zhang L, Zhao XC, Gao SH, Qu LW, Yu H, et al. The Aryl hydrocarbon receptor mediates tobacco-induced PD-L1 expression and is associated with response to immunotherapy. Nat Commun. (2019) 10:1125. doi: 10.1038/s41467-019-08887-7
285. Gettinger SN, Choi J, Mani N, Sanmamed MF, Datar I, Sowell R, et al. A dormant TIL phenotype defines non-small cell lung carcinomas sensitive to immune checkpoint blockers. Nat Commun. (2018) 9:3196. doi: 10.1038/s41467-018-05032-8
286. Inamura K, Yokouchi Y, Kobayashi M, Sakakibara R, Ninomiya H, Subat S, et al. Tumor B7-H3 (CD276) expression and smoking history in relation to lung adenocarcinoma prognosis. Lung Cancer. (2017) 103:44–51. doi: 10.1016/j.lungcan.2016.11.013
287. Hato SV, Khong A, de Vries IJ, Lesterhuis WJ. Molecular pathways: the immunogenic effects of platinum-based chemotherapeutics. Clin Cancer Res. (2014) 20:2831–7. doi: 10.1158/1078-0432.CCR-13-3141
288. Zitvogel L, Kepp O, Kroemer G. Immune parameters affecting the efficacy of chemotherapeutic regimens. Nat Rev Clin Oncol. (2011) 8:151–60. doi: 10.1038/nrclinonc.2010.223
289. van der Most RG, Currie AJ, Mahendran S, Prosser A, Darabi A, Robinson BW, et al. Tumor eradication after cyclophosphamide depends on concurrent depletion of regulatory T cells: a role for cycling TNFR2-expressing effector-suppressor T cells in limiting effective chemotherapy. Cancer Immunol Immunother. (2009) 58:1219–28. doi: 10.1007/s00262-008-0628-9
290. Kepp O, Galluzzi L, Martins I, Schlemmer F, Adjemian S, Michaud M, et al. Molecular determinants of immunogenic cell death elicited by anticancer chemotherapy. Cancer Metastasis Rev. (2011) 30:61–9. doi: 10.1007/s10555-011-9273-4
291. Emens LA, Middleton G. The interplay of immunotherapy and chemotherapy: harnessing potential synergies. Cancer Immunol Res. (2015) 3:436–43. doi: 10.1158/2326-6066.CIR-15-0064
292. Chen G, Emens LA. Chemoimmunotherapy: reengineering tumor immunity. Cancer Immunol Immunother. (2013) 62:203–16. doi: 10.1007/s00262-012-1388-0
293. Horn L, Mansfield AS, Szczesna A, Havel L, Krzakowski M, Hochmair MJ, et al. First-line atezolizumab plus chemotherapy in extensive-stage small-cell lung cancer. N Engl J Med. (2018) 379:2220–9. doi: 10.1056/NEJMoa1809064
294. Wang Y, Deng W, Li N, Neri S, Sharma A, Jiang W, et al. Combining immunotherapy and radiotherapy for cancer treatment: current challenges and future directions. Front Pharmacol. (2018) 9:185. doi: 10.3389/fphar.2018.00185
295. Demaria S, Golden EB, Formenti SC. Role of local radiation therapy in cancer immunotherapy. JAMA Oncol. (2015) 1:1325–32. doi: 10.1001/jamaoncol.2015.2756
296. Deng L, Liang H, Burnette B, Beckett M, Darga T, Weichselbaum RR, et al. Irradiation and anti-PD-L1 treatment synergistically promote antitumor immunity in mice. J Clin Invest. (2014) 124:687–95. doi: 10.1172/JCI67313
297. Twyman-Saint Victor C, Rech AJ, Maity A, Rengan R, Pauken KE, Stelekati E, et al. Radiation and dual checkpoint blockade activate non-redundant immune mechanisms in cancer. Nature. (2015) 520:373–7. doi: 10.1038/nature14292
298. Antonia SJ, Villegas A, Daniel D, Vicente D, Murakami S, Hui R, et al. Overall survival with durvalumab after chemoradiotherapy in stage III NSCLC. N Engl J Med. (2018) 379:2342–50. doi: 10.1056/NEJMoa1809697
299. Saha A, Chatterjee SK. Combination of CTL-associated antigen-4 blockade and depletion of CD25 regulatory T cells enhance tumour immunity of dendritic cell-based vaccine in a mouse model of colon cancer. Scand J Immunol. (2010) 71:70–82. doi: 10.1111/j.1365-3083.2009.02355.x
300. Gibney GT, Kudchadkar RR, DeConti RC, Thebeau MS, Czupryn MP, Tetteh L, et al. Safety, correlative markers, and clinical results of adjuvant nivolumab in combination with vaccine in resected high-risk metastatic melanoma. Clin Cancer Res. (2015) 21:712–20. doi: 10.1158/1078-0432.CCR-14-2468
301. Guo ZS, Liu Z, Bartlett DL. Oncolytic immunotherapy: dying the right way is a key to eliciting potent antitumor immunity. Front Oncol. (2014) 4:74. doi: 10.3389/fonc.2014.00074
302. Puzanov I, Milhem MM, Minor D, Hamid O, Li A, Chen L, et al. Talimogene laherparepvec in combination with ipilimumab in previously untreated, unresectable stage IIIB-IV melanoma. J Clin Oncol. (2016) 34:2619–26. doi: 10.1200/JCO.2016.67.1529
303. O’Donnell JS, Massi D, Teng MWL, Mandala M. PI3K-AKT-mTOR inhibition in cancer immunotherapy, redux. Semin Cancer Biol. (2018) 48:91–103. doi: 10.1016/j.semcancer.2017.04.015
304. Ebert PJR, Cheung J, Yang Y, McNamara E, Hong R, Moskalenko M, et al. MAP kinase inhibition promotes T cell and anti-tumor activity in combination with PD-L1 checkpoint blockade. Immunity. (2016) 44:609–21. doi: 10.1016/j.immuni.2016.01.024
305. Ribas A, Lawrence D, Atkinson V, Agarwal S, Miller WH Jr., Carlino MS, et al. Combined BRAF and MEK inhibition with PD-1 blockade immunotherapy in BRAF-mutant melanoma. Nat Med. (2019) 25:936–40. doi: 10.1038/s41591-019-0476-5
306. Cho BC, Bahary N, Bendell J, Felip E, Johnson M, Kang Y-K, et al. Abstract CT201: phase Ib/II open-label, randomized evaluation of atezolizumab + cobimetinib vs control in MORPHEUS-NSCLC (non-small cell lung cancer), MORPHEUS-PDAC (pancreatic ductal adenocarcinoma) and MORPHEUS-GC (gastric cancer). American Association for Cancer Research (AACR) Virtual Annual Meeting 2020. (2020). doi: 10.1158/1538-7445.AM2020-CT201
307. Vikas P, Borcherding N, Chennamadhavuni A, Garje R. Therapeutic potential of combining PARP inhibitor and immunotherapy in solid tumors. Front Oncol. (2020) 10:570. doi: 10.3389/fonc.2020.00570
308. Vagia E, Viveiros P, Kang C-Y, Nelson V, Kalyan A, Kircher S, et al. Abstract CT157: phase I/Ib study of nivolumab and veliparib in patients with advanced solid tumors and lymphoma harboring mutations in selected DNA repair genes. American Association for Cancer Research (AACR) Virtual Annual Meeting 2020. (2020). doi: 10.1158/1538-7445.AM2020-CT157
309. Yap TA, Dhawan M, Hendifar AE, Miao M, Owonikoko TK, Quintela-Fandino M, et al. Abstract CT269: a phase 2 study of olaparib in combination with pembrolizumab in patients with previously treated advanced solid tumors with homologous recombination repair mutation (HRRm) and/or Homologous recombination repair deficiency (HDR): KEYLYNK-007. American Association for Cancer Research (AACR) Virtual Annual Meeting 2020. (2020). doi: 10.1158/1538-7445.AM2020-CT269
310. Wolchok JD, Saenger Y. The mechanism of anti-CTLA-4 activity and the negative regulation of T-cell activation. Oncologist. (2008) 13(Suppl. 4):2–9. doi: 10.1634/theoncologist.13-S4-2
311. Blake SJ, Ching AL, Kenna TJ, Galea R, Large J, Yagita H, et al. Blockade of PD-1/PD-L1 promotes adoptive T-cell immunotherapy in a tolerogenic environment. PLoS One. (2015) 10:e0119483. doi: 10.1371/journal.pone.0119483
312. Houot R, Schultz LM, Marabelle A, Kohrt H. T-cell-based Immunotherapy: adoptive cell transfer and checkpoint inhibition. Cancer Immunol Res. (2015) 3:1115–22. doi: 10.1158/2326-6066.CIR-15-0190
313. Dahlen E, Veitonmaki N, Norlen P. Bispecific antibodies in cancer immunotherapy. Ther Adv Vaccines Immunother. (2018) 6:3–17. doi: 10.1177/2515135518763280
314. Chen Y, Ramjiawan RR, Reiberger T, Ng MR, Hato T, Huang Y, et al. CXCR4 inhibition in tumor microenvironment facilitates anti-programmed death receptor-1 immunotherapy in sorafenib-treated hepatocellular carcinoma in mice. Hepatology. (2015) 61:1591–602. doi: 10.1002/hep.27665
315. Reck M, Mok TSK, Nishio M, Jotte RM, Cappuzzo F, Orlandi F, et al. Atezolizumab plus bevacizumab and chemotherapy in non-small-cell lung cancer (IMpower150): key subgroup analyses of patients with EGFR mutations or baseline liver metastases in a randomised, open-label phase 3 trial. Lancet Respir Med. (2019) 7:387–401. doi: 10.1016/S2213-2600(19)30084-0
316. Johnson BE, Kim TM, Hiltermann TJN, Barlesi F, Grohe C, Goto Y, et al. Abstract CT214: CANOPY-1: safety run-in results from phase (ph) 3 study of canakinumab (CAN) or placebo (PBO) in combination (comb) with pembrolizumab (PEM) plus platinum-based doublet chemotherapy (Ctx) as 1st line therapy in patients (pts) with advanced or metastatic NSCLC. American Association for Cancer Research (AACR) Virtual Annual Meeting 2020. (2020). doi: 10.1158/1538-7445.AM2020-CT214
317. Lee JM, Garrido P, Kim ES, Arslan C, Pujol J-L, Song Y, et al. Abstract CT192: randomized phase II study of canakinumab (CAN) or pembrolizumab (PEM) alone or in combination as neoadjuvant therapy in patients (Pts) with surgically resected (Stage IB-IIIA) non-small cell lung cancer (NSCLC): CANOPY-N. American Association for Cancer Research (AACR) Virtual Annual Meeting 2020. (2020). doi: 10.1158/1538-7445.AM2020-CT192
318. Mahoney KM, Rennert PD, Freeman GJ. Combination cancer immunotherapy and new immunomodulatory targets. Nat Rev Drug Discov. (2015) 14:561–84. doi: 10.1038/nrd4591
319. Hu-Lieskovan S, Ribas A. New combination strategies using programmed cell death 1/programmed cell death ligand 1 checkpoint inhibitors as a backbone. Cancer J. (2017) 23:10–22. doi: 10.1097/PPO.0000000000000246
320. Kwiatkowska D, Kluska P, Reich A. Beyond PD-1 immunotherapy in malignant melanoma. Dermatol Ther (Heidelb). (2019) 9:243–57. doi: 10.1007/s13555-019-0292-3
321. Forster M, Felip E, Doger B, Majem M, Carcereny E, Peguero J, et al. Abstract CT202: initial results from a phase II study (TACTI-002) in metastatic non-small cell lung or head and neck carcinoma patients receiving eftilagimod alpha (soluble lag-3 protein) and pembrolizumab. American Association for Cancer Research (AACR) Virtual Annual Meeting 2020. (2020). doi: 10.1158/1538-7445.AM2020-CT202
322. FIERCE Biotech SITC18: Merck Highlights New LAG-3 and TIGIT Data. [Press Release]. (2019). Available online at: https://www.fiercebiotech.com/biotech/sitc-merck-highlights-new-lag-3-and-tigit-data (accessed September 21, 2019).
323. Bendell JC, Bedard P, Bang Y-J, LoRusso P, Hodi S, Gordon M, et al. Abstract CT302: phase Ia/Ib dose-escalation study of the anti-TIGIT antibody tiragolumab as a single agent and in combination with atezolizumab in patients with advanced solid tumors. American Association for Cancer Research (AACR) Virtual Annual Meeting 2020. (2020). doi: 10.1158/1538-7445.AM2020-CT302
324. Rodriguez-Abreu D, Johnson ML, Hussein MA, Cobo M, Patel AJ, Secen NM, et al. Abstract 9503: primary analysis of a randomized, double-blind, phase II study of the anti-TIGIT antibody tiragolumab (tira) plus atezolizumab (atezo) versus placebo plus atezo as first-line (1L) treatment in patients with PD-L1-selected NSCLC (CITYSCAPE). American Society of Clinical Oncology (ASCO) Virtual Annual Meeting 2020: Journal of Clincal Oncology. (2020). doi: 10.1200/JCO.2020.38.15_suppl.9503
325. Chen X, Song X, Li K, Zhang T. FcgammaR-binding is an important functional attribute for immune checkpoint antibodies in cancer immunotherapy. Front Immunol. (2019) 10:292. doi: 10.3389/fimmu.2019.00292
326. Yachnin J, Ullenhag GJ, Carneiro A, Nielsen DL, Rohrberg KS, Kvarnhammar AM, et al. Abstract CT145: a first-in-human phase 1 study in patients with advanced and/or refractory solid malignancies to evaluate the safety of ATOR-1015, a CTLA-4 x OX40 bispecific antibody. American Association for Cancer Research (AACR) Virtual Annual Meeting 2020. (2020). doi: 10.1158/1538-7445.AM2020-CT145
327. Postel-Vinay S, Lam VK, Ros W, Bauer TM, Hansen AR, Cho DC, et al. Abstract CT150: a first-in-human phase I study of the OX40 agonist GSK3174998 (GSK998) +/- pembrolizumab in patients (Pts) with selected advanced solid tumors (ENGAGE-1). American Association for Cancer Research (AACR) Virtual Annual Meeting 2020. (2020). doi: 10.1158/1538-7445.AM2020-CT150
328. Jimeno A, Gupta S, Sullivan R, Do KT, Akerley WL, Wang D, et al. Abstract CT032: a phase 1/2, open-label, multicenter, dose escalation and efficacy study of mRNA-2416, a lipid nanoparticle encapsulated mRNA encoding human OX40L, for intratumoral injection alone or in combination with durvalumab for patients with advanced malignancies. American Association for Cancer Research (AACR) Virtual Annual Meeting 2020. (2020). doi: 10.1158/1538-7445.AM2020-CT032
Keywords: resistance mechanism, immunotherapy, PD-1/PD-L1, Immune check inhibitor, lung cance
Citation: Wang F, Wang S and Zhou Q (2020) The Resistance Mechanisms of Lung Cancer Immunotherapy. Front. Oncol. 10:568059. doi: 10.3389/fonc.2020.568059
Received: 31 May 2020; Accepted: 14 September 2020;
Published: 20 October 2020.
Edited by:
Laura Mezquita, Hospital Clínic de Barcelona, SpainReviewed by:
Ignacio Matos, University College London, United KingdomCopyright © 2020 Wang, Wang and Zhou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qing Zhou, Z3p6aG91cWluZ0AxMjYuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.