 Yibing Bai
Yibing Bai Jiani Yang
Jiani Yang Ying Cui
Ying Cui Yuanfei Yao1,2
Yuanfei Yao1,2 Xiaona Fan
Xiaona Fan Yanqiao Zhang
Yanqiao Zhang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 08 January 2021
Sec. Cancer Metabolism
Volume 10 - 2020 | https://doi.org/10.3389/fonc.2020.562950
Sirtuins (SIRTs) are members of the silent information regulator-2 family. They are a conserved family of nicotinamide adenine dinucleotide-dependent protein lysine deacylases. SIRTS are involved in intricate cellular processes. There are seven subtypes of SIRTs (1–7) in mammals. SIRT4 is located mainly in mitochondria and has various catalytic activities. These enzyme activities give it a diverse range of important biologic functions, such as energy metabolism, oxidative stress, and aging. Cancer is characterized as reprogramming of energy metabolism and redox imbalance, and SIRT4 can affect tumorigenesis. Here, we review the structure, localization, and enzyme activity of SIRT4 and its role in various neoplasms.
Cancer has become the first or second most prevalent cause of premature death in ~100 countries (1). Cancer occurrence is closely related to genomic instability, which enables cells to maintain proliferation signals, escape growth inhibitors, avoid immune damage, resist cell death and, eventually, develop into a life-threatening malignant tumor. Hanahan et al. described the “hotspots” and new advances in oncology during the last decade and proposed new tumor features: avoidance of immune destruction, promotion of tumor inflammation, abnormal cell energy, genomic instability, and mutations (2). In recent years, a new feature of malignant tumors has attracted increasing attention: the reprogramming of energy metabolism (3).
Sirtuin4 (SIRT4) belongs to sirtuins (SIRTs). The latter is a family of highly conserved nicotinamide adenine dinucleotide (NAD+)-dependent enzymes that regulate lifespan, ageing and metabolism (4). Mammalian SIRTs are considered class-III histone deacetylases (HDACs). According to sequence homology, HDAC enzymes can be divided into three types (5, 6). Class I contains HDAC1, 2, 3, and 8, all of which are homologs of the RPD3 protein in yeast. Class II contains HDAC4, 5, 6, 7, 9, and 10, all of which are homologs of the yeast Hda1 protein. While class III contains seven SIRT homologs of the silent information regulator 2 (Sir2) protein of yeast (7).
SIRT4 is located mainly in the mitochondria of cells. SIRT4 has adenosine diphosphate (ADP)-ribosyltransferase activity and can deacetylate (8, 9). In recent years, it has been found that SIRT4 catalyzes removal of lipoyl- and biotinyl-lysine modifications more efficiently than deacetylate (10). SIRT4 also has lysine-deacylase activity for removing 3,3-dimethylsuccinyl (DMS), 3-hydroxy-3-methylpentadienyl (HMG), 3-methylglutaryl (MG) and 3-methylglutamyl (MGc) (11, 12). These enzyme activities enable SIRT4 to regulate the targets of glutamate dehydrogenase (GDH), malonyl-CoA decarboxylase and the pyruvate dehydrogenase complex (PDHC) directly. In this way, SIRT4 plays an irreplaceable part in the metabolic regulation of glutamine, lipids, and glucose in malignant tumors (8–10).
In this review, we summarize the structure and localization, enzyme activity, and role of SIRT4 in malignant tumors.
SIRT4 is a protein of size 35.2 kDa located on chromosome 12q24.31 (13). According to sequencing and analyses of its primary structure, SIRT4 has essential amino acids that are involved in the deacylase reaction. SIRTs have a conserved catalytic core of ~275 amino acids flanked by N-terminal and C-terminal sequences of variable length. The small zinc-binding domain and large Rossmann-folding domain enable all SIRTs to bind specifically to NAD+ as a cofactor (14). SIRT4 contains a homologous SIRTs deacylase domain, a conserved catalytic histidine (H161), substrate specificity, as well as a Zn2+-binding and a Rossmann fold/NAD+-binding motif (amino acids 62–82, 143–146, 260–262, 286–288). The main sequencing analysis and alignment of three types of mitochondrial SIRTs (SIRT3, SIRT4, and SIRT5) have shown that the position of amino acids is very important for mitochondrial localization, NAD+ binding, catalysis, substrate specificity, and Zn2+ binding (12).
The secondary structure of SIRT4 contains α-helices and β-sheets, which are at virtually the same location as those in other mitochondrial SIRTs (SIRT3 and SIRT5). The evolutionarily conserved α-helices of the SIRT4 catalytic pocket can confer specific interactions with negatively charged lysine molecules. Comparison of the crystal structure of SIRT4 with that of other SIRTs has revealed that the most significant difference is an extended ~12-residue SIRT4 loop (residues 195–206) between α8 and α9 in the Zn2+-binding module. This loop is oriented deep into the catalytic core and contributes to the lining of the active site. Specifically, this loop is not present in any other SIRT isotypes, so it is called the “SIRT4 loop”. The latter folds into active sites to potentially regulate catalysis. Another unusual feature of the catalytic core of SIRT4 is the channel that branches from the acyl-lysine binding channel to the protein surface. It can accommodate longer substrate acyl groups, and may act as a binding site that regulates metabolites, which rationalizes its activities. This channel is conservative in SIRT4, but is different in other isotypes. However, the specific function of this SIRT4 channel has yet to be studied deeply (11).
Phylogenetic analyses can reveal the patterns of evolutionary stress of specific amino acids, which may be important for protein function. Phylogenetic analyses of 5869 SIRTs domains of proteins from 3,562 species are consistent with studies which divided human SIRTs into four categories: SIRT1–3 occupy class I, SIRT4 occupies class II, SIRT5 occupies class III, and SIRT6 and SIRT7 occupy class IV (12).
SIRT4 is distributed in humans in the heart, brain, liver, kidney, muscle, pancreas, thymus, lung, spleen, bone marrow, uterus, ovary, and testis (9, 15–18). The wide distribution of SIRT4 may indicate the importance of its biologic functions.
Upon fusion of SIRTs with green fluorescent protein (GFP), SIRT4-GFP co-localizes with the mitochondrial-specific probe MitoFluor™ as well as the mitochondrial labeler MitoTracker™. SIRT4 has a N-terminal mitochondrial targeting sequence (MTS) of length of 28 amino acids. This MTS is responsible for localization of this enzyme in the mitochondrial cavity (15). Studies have shown that SIRT4-EGFP and SIRT4-FLAG are co-located with mitochondrial markers, and that SIRT4 is closely related to the metabolic activity and significant biologic functions of mitochondria (9, 10, 15, 19). Moreover, mitochondria are responsible for oxidative metabolism through the Krebs cycle, β-oxidation of fatty acids (FAs) and oxidative phosphorylation, which result in production of most cellular adenosine 5′ triphosphate (ATP) (20). Therefore, there is an association between SIRT4 and energy metabolism.
Recently, it was shown that SIRT4 exists not only in mitochondria but also in the cytoplasm and nucleus (21). Fusion of SIRT4 with a very mature fluorescent protein (FP) variant to form a “super folder GFP” (sfGFP) can enable localization and visualization of de novo protein synthesis dynamically (22, 23). Even at very low expression, SIRT4-sfGFP was not fully introduced into mitochondria and accumulated in the cytoplasm and nucleus. Moreover, SIRT4-sfGFP accumulated in the outer mitochondrial membrane (OMM) but did not enter the mitochondrial matrix. However, the fast-folding sfGFP fused with mature MTS to be introduced into the mitochondrial cavity. Those findings suggest that SIRT4 enters mitochondria slowly. Moreover, SIRT4-sfGFP accumulation in the OMM is related to the swelling of mitochondria, which indicates the toxicity of this specific fusion construct (21).
Another maneuver is based on “split FP technology” for studying introduction of proteins into the mitochondrial matrix (24). To avoid the mis-localization and mitochondrial swelling of “clunky” SIRT4-FP fusion constructors, FP splitting based on sfCherry2 self-complementarity was used. Using this established method, SIRT4-sfCherry211 was introduced and located in the mitochondrial matrix (25). Furthermore, the complementary sfCherry21–10 constructors targeting the mitochondrial cavity and nucleus were co-expressed. This new trigonal probe was named “mitochondrial sirtuin 4 tripartite abundance reporter” (mito-STAR), which realized the visualization of SIRT4 introduction into the mitochondrial matrix and nucleus. Using mito-STAR, it was found that, although the mitochondrial targeting of SIRT4-sfCherry211 had been improved significantly compared with that using SIRT4-sfGFP, many cells showing nuclear fluorescence signals could be detected, which suggested the nuclear abundance of SIRT4-sfCherry211 under a dormant condition (21).
During the mitochondrial unfolded protein response (mtUPR), the stress state caused by accumulation of unfolded proteins in mitochondria and accumulation of mitochondrial-targeted proteins in the cytoplasm and nucleus contributes to a retrograde signaling pathway (26, 27). It has been reported that SIRT3 participates in mtUPR (28, 29). When mito-STAR was applied under mitochondrial stress, SIRT4 expression in the nucleus was increased significantly (21). Once again, the idea that SIRT4 promotes retrograde signals from mitochondria to the nucleus was supported by data.
Consistently, it has been found that a fraction of endogenous or ectopically expressed SIRT4 is present in the cytosol and localizes predominantly to centrosomes. Confocal spinning disk microscopy has revealed that SIRT4 is found during the cell cycle dynamically at centrosomes with an intensity peak in the G2 phase and early mitosis. Moreover, SIRT4 precipitates with microtubules and interacts with structural (α, β-tubulin, γ-tubulin, TUBGCP2, TUBGCP3) and regulatory (HDAC6) microtubule components as detected by co-immunoprecipitation experiments and mass spectrometry of the mitotic SIRT4 interactome. SIRT4 overexpression resulted in a pronounced decrease in the level of acetylated α-tubulin (K40) associated with altered microtubule dynamics in mitotic cells. Importantly, the N-terminally truncated variant SIRT4 (ΔN28), which cannot translocate into mitochondria, delayed mitotic progression and reduced cell proliferation. Hence, SIRT4 may exert its anti-proliferative role through mitochondrial/metabolism-dependent and mitochondria-independent functions (30).
All SIRTs have conserved deacetylase domains. Hence, these enzymes are usually called “lysine deacetylases” which, in contrast to acetyltransferases, can deacetylate from lysine residues (31). However, SIRTs show different catalytic efficiency for this modification. Initial studies revealed scarce deacetylase activity in SIRT4 using histone substrates or substrates of bovine serum albumin (9, 18, 32). Nevertheless, the lack of deacetylase activity in SIRT4 may be due to the absence of a suitable substrate, German et al. showed, for the first time, that SIRT4 has substrate-specific deacetylase activity. SIRT4 controls lipid catabolism through deacetylation of malonyl-CoA decarboxylase (8). In addition, SIRT4 has been shown to have significant deacetylation activity on acetylome peptide microarrays, thereby revealing the physiologic candidates for deacetylation. MtHsp60 (Lys31) and Stress-70 (Lys135, 595) have the highest hit rates, which are related to the proliferation and senescence of cells, and also involve NAD+ transhydrogenase (Lys394, 331, 397), which is important for to responses redox stress (33). Recently, it has been shown that in pancreatic ductal adenocarcinoma (PDAC), SIRT4 could deacetylate branched-chain amino acid transaminase 2 (BCAT2) at lysine 44 (K44), and that BCAT2 was stabilized by SIRT4 deacetylation, which promoted catabolism of branched-chain amino acids, cell proliferation and the growth of pancreatic tumors (34).
SIRT4 ADP-ribosylates GDH to repress its activity using NAD in pancreatic β-cell mitochondria. In this way, it prevents glutamine being an insulin secretagogue and down-regulating insulin secretion in response to amino acids. This effect has been shown to be alleviated during calorie restriction. During calorie restriction, the ratio of NAD/NADH decreased, SIRT4 activity decreased, and GDH activity increased, thereby up-regulating insulin secretion in response to glutamine and leucine (9). Ahuja and colleagues showed that SIRT4 has a highly efficient ADP-ribosyltransferase function for histone and bovine serum albumin (18). However, some scholars believe that the ADP-ribosyltransferase activity of SIRT4 may be due to the inefficient side-effects of deacetylase activity, and to be unrelated to physiology (35). Therefore, the role of the ADP-ribosyltransferase activity of SIRT4 merits further study.
SIRT4 can inhibit the activity of PDHC components by hydrolyzing lipoamide cofactors from dihydrolipoyllysine-residue acetyltransferase (DLAT) E2 component of the PDHC. SIRT4 shows enzymatic activity only in the presence of NAD+. To compare the relative preference of SIRT4 for different acyl lysine peptides, ion chromatography was used to quantify the percentage of unmodified peptides produced by each substrate. SIRT4 showed the highest efficacy for removing the lipoyl modification. The relative amount of unmodified product formed after reacting with SIRT4 was 11% for lipoyl, 3% for biotinyl, and 0.3% for acetyl. Moreover, compared with other mitochondrial SIRTs, SIRT4 had the highest catalytic efficiency for lipamide modification (10).
To screen the activity of recombinant SIRT4 against various acyl histone peptides, SIRT4 displayed dedecanoylase and deoctanoylase activity, as well as exhibited low (but measurable) activity against lipoyl, succinyl, and dodecanoyl substrates (36).
Testing SIRT4 from an acyl library of carbamoyl phosphate synthetase 1 (CPS1)-Lys527 peptides showed a specific profile in monitoring of nicotinamide (NAM) release from NAD+ by conjugated enzyme assays. Testing revealed that use of lipoylated and biotinylated substrates could enhance activity further, but the highest activity was observed with DMS substrates (eight-fold stronger than deacetylation). DMS was compared with the physiologically occurring acyl part of activated coenzyme A (CoA)-thioesters to potentially modify the sidechains of lysine (37, 38), and the HMG group was found to be the most relevant. HMG-modified CPS1-Lys527 peptides elicited SIRT4 activity that was similar to that of the DMS-CPS1 substrate, and approximately three-fold higher than that of the acetyl peptide (11).
Anderson and colleagues used a combination of phylogeny, structural biology, and enzymology to demonstrate that SIRT4 catalyzes removal of HMG, as well as the associated modified MG and MGc, from the lysine residues of the target protein. Moreover, they found that the metabolites responsible for these post-translational modifications were intermediates of leucine oxidation, and that SIRT4 removed these modifications to restore leucine catabolism. In addition, disorders in leucine metabolism in SIRT4 knockout (KO) mice led to increased basal and stimulated insulin secretion, which developed gradually into glucose tolerance and insulin resistance. Those findings confirmed the strong enzyme activity of SIRT4, revealed the mechanism of controlling fluxes in branched-chain amino acids, and positioned SIRT4 as a key factor in maintaining glucose homeostasis during insulin secretion and senescence (12) (Figure 1).
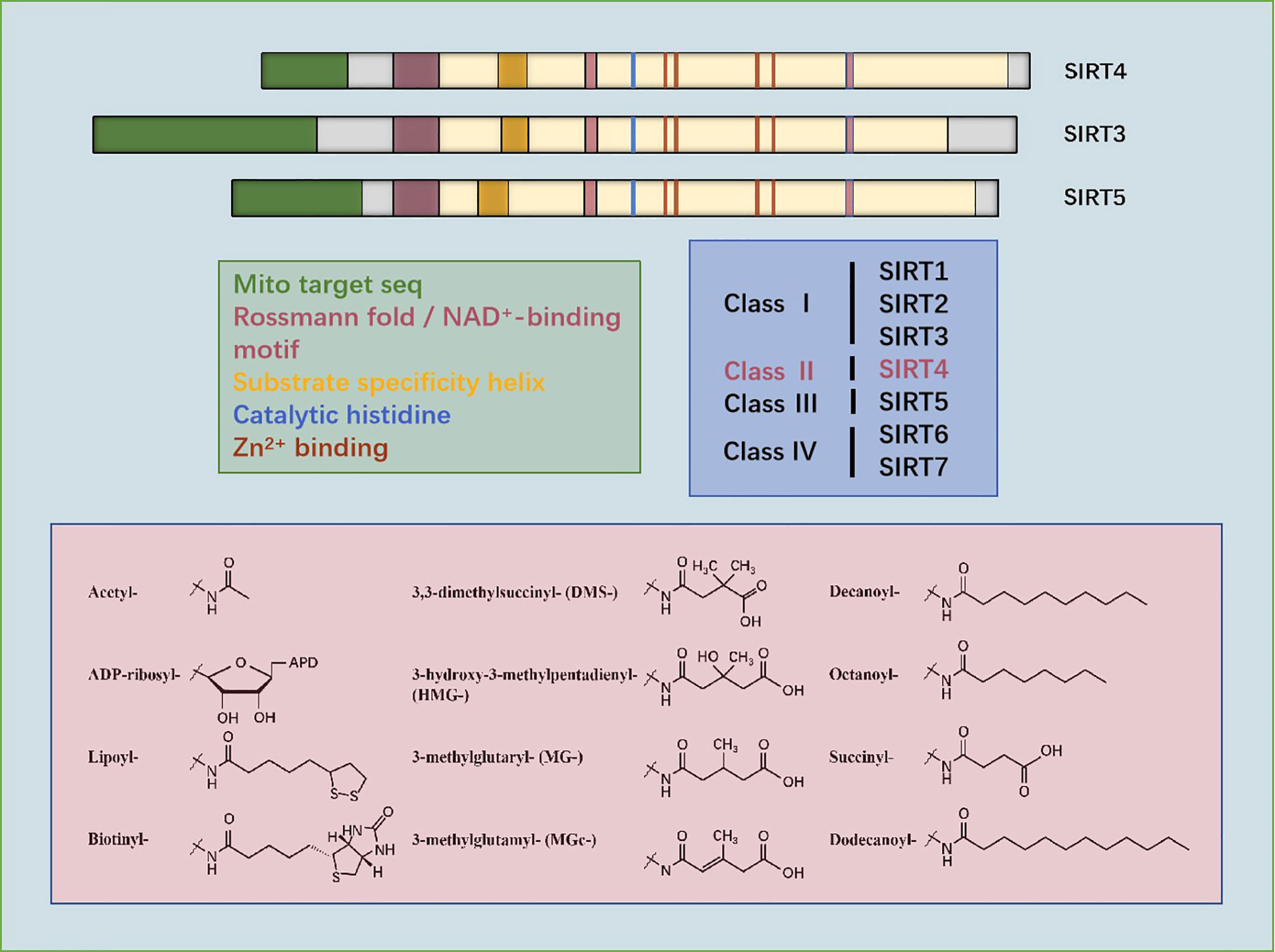
Figure 1 Sequencing of amino acids and mitochondrial localization, NAD+-binding, substrate specificity, catalytic, histidine- and Zn2+-binding domains of SIRT3, SIRT4, and SIRT5. Phylogenetic analyses show that SIRT4 occupies class II. Structures of the substrates catalyzed by SIRT4 are shown.
Analyses of data from The Cancer Genome Atlas (TCGA) database demonstrated that SIRT4 expression was downregulated in 30 cancer types. Similarly, compared with samples of normal tissue, 19,120 human-cancer tissues also showed reduced expression of SIRT4, which were analyzed from three datasets (GTEX, TARGET, TCGA). Wang and colleagues studied the copy number of SIRT4 in various cancer types using the Tumorscape database, and found that SIRT4 was located in the region in chromosome 12 that was deleted significantly in the whole dataset (39).
Through a comprehensive meta-analysis of SIRT4 expression in human tumors, it was found that expression of SIRT4 mRNA in tissues with bladder, breast, colon, gastric, ovarian, or thyroid cancer was significantly lower than that in corresponding normal tissues. Moreover, the loss of SIRT4 expression showed a strong association with short-term metastasis in breast-cancer patients. In addition, SIRT4 overexpression led to tumorigenesis and inhibited tumor development in a xenotransplantation model using Tsc2−/− mouse embryonic fibroblasts (MEFs) (40).
Fu and colleagues employed immunohistochemical (IHC) staining to measure SIRT4 expression in lung, ovarian, pancreatic, hepatocellular, bladder, prostate, or renal cancer tissues. Compared with the positive staining of the corresponding normal adjacent tissues, SIRT4-negative or weak staining was observed in most tumor types (41).
A lack of SIRT4 accelerates the morbidity of spontaneous tumors throughout life. Jeong and colleagues found that several types of neoplasms were incurred in ~63% (22/35) of SIRT4-KO animals, whereas 24% (6/25) of wild-type (WT) littermates had tumors. At the age of 18−26 months, more SIRT4-KO mice (45.5%) developed lung tumors than WT mice (8.3%). Also, 41% (7/17) of these lung tumors were adenomas and 59% (10/17) were carcinomas, from SIRT4-KO mice. In addition, 60% of female SIRT4-KO mice had cystic endometrial hyperplasia, whereas 15% of WT mice showed mild endometrial hyperplasia (42).
The evidence provided above suggests that SIRT4 is a tumor suppressor. Two mechanisms have been proposed by which SIRT4 inhibits tumorigenesis and tumor development, as studied in MEFs. The first hypothesis is based on the observation that genomic instability and metabolic changes are key features of many cancer cells (43, 44). DNA damage triggers a closely coordinated signal-transduction response to maintain genomic integrity by promoting cell-cycle arrest and DNA repair (45, 46). It has been discovered that DNA damage triggers a key barrier to glutamine metabolism, which is necessary for an appropriate response to DNA damage. This blockade requires mitochondrial SIRT4, which is induced by various genotoxic proxies and inhibits glutamine metabolism into the tricarboxylic acid (TCA) cycle. SIRT4 can inhibit glutamine metabolism in mitochondria after DNA damage and regulate cell-cycle progression and genomic fidelity in response to DNA damage. Loss of SIRT4 leads to increased glutamine-dependent proliferation and stress-induced genomic instability, which results in a carcinogenic phenotype. Therefore, SIRT4 plays an important part in regulating glutamine metabolism and genomic instability (42). The second hypothesis is based on the observation that atypical activation of serine/threonine kinase in mammalian target of rapamycin complex 1 (mTORC1) controls GDH activity by inhibiting SIRT4 expression, thereby stimulating glutamine uptake, not only in MEFs, but also in human epithelial tumor cell lines [colon cancer (DLD1) and prostate cancer (DU14)]. The mTORC1 pathway controls SIRT4 negatively by promoting proteasome-mediated degradation of cyclic adenosine monophosphate response element binding (CREB)-2. The latter is recognized as a transcription factor of SIRT4. mTORC1 activation promotes the binding of CREB2 to E3 ubiquitin ligase βTrCP and regulates the ubiquitin-based degradation of CREB2 (40) (Figure 2).
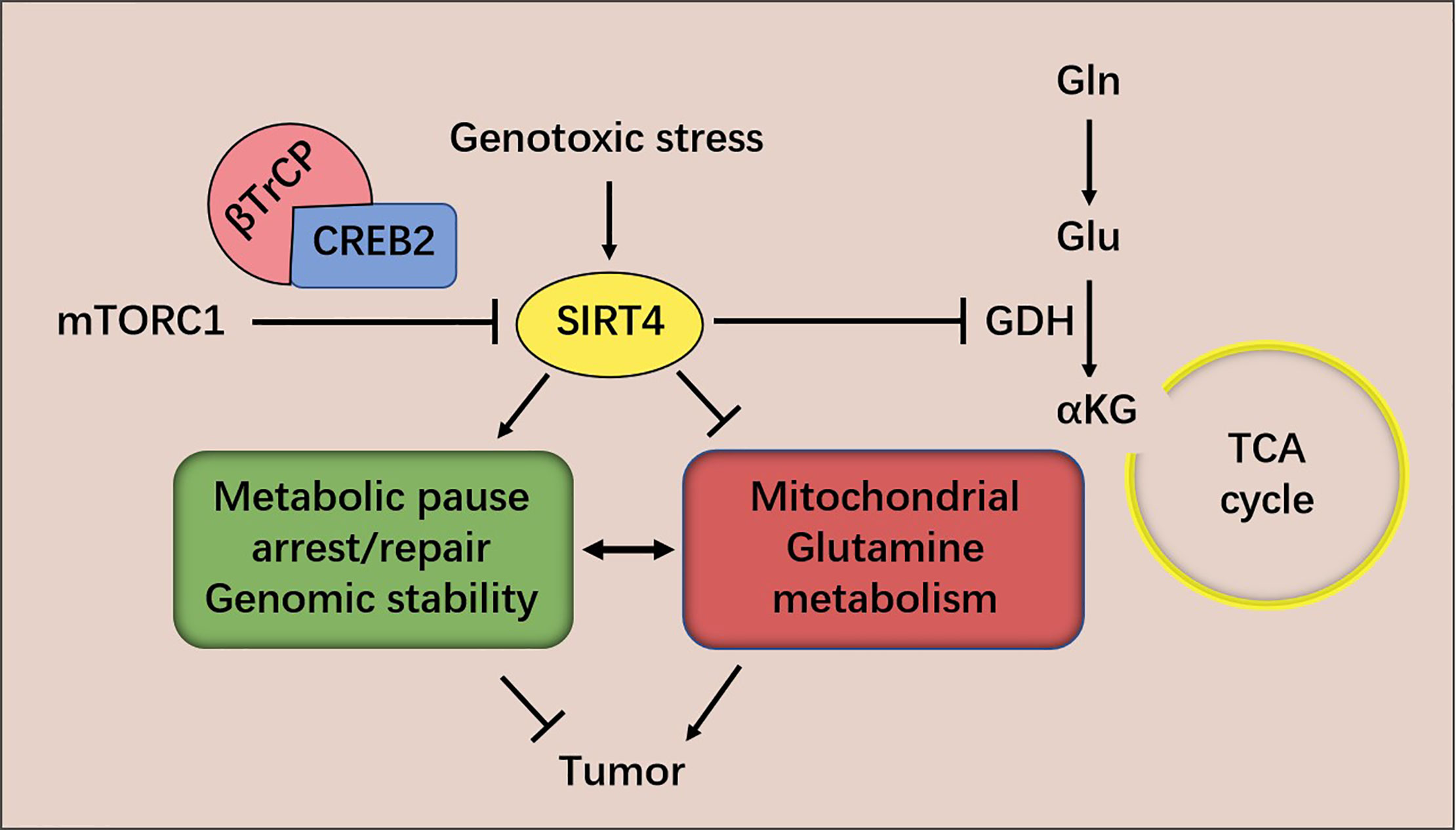
Figure 2 mTORC1 regulates SIRT4 expression and glutamine metabolism. SIRT4 inhibits glutamine metabolism in mitochondria after DNA damage and regulates cell-cycle progression and genomic fidelity in response to DNA damage.
SIRT4 expression is reduced in human HCC. The genomic mutation, amplification, and mRNA expression of SIRT4 in HCC were analyzed using the cBioPortal tool of cancer genomics (www.cbioportal.org/). As a result, there was almost no mutation or amplification of SIRT4, and down-regulation of mRNA expression was found in 5.23% of human HCC samples, which could be explained by frequent and deep deletions of the locus. Also, down-regulation of SIRT4 expression was detected in 83 individual HCC datasets compared with that in matched normal tissues. SIRT4 expression was decreased in early stage (I–II) HCC, especially during cancer progression, indicating that downregulation of SIRT4 expression may be necessary for the initiation and maintenance of tumors. By measuring SIRT4 expression in 364 HCC samples, it was found that low expression of SIRT4 was associated with shorter recurrence time. Moreover, SIRT4 deficiency promoted the proliferation and migration of HCC cells in vitro and promoted tumorigenesis and lung metastasis of HCC in vivo (39).
Unbiased gene set enrichment analysis revealed that SIRT4 defection was related to activation of mTOR signal transduction. SIRT4 deletion increased expression of the downstream genes of mTOR (MYC, CCND1, HIF1A, and SREBP1) rather than expression of mTOR itself. Consistently, expression of the upstream inhibitors of mTOR (ULK1, TP53, and CDKN1A) was decreased. In addition, IHC analyses and western blotting exhibited that SIRT4-KO increased mTOR phosphorylation, further implying that SIRT4 ablation mediates the activity of mTOR rather than its transcription. Rapamycin (classical inhibitor of mTOR) diminishes mTOR activation intensely, as well as the consequent hyper-proliferative phenotypes aroused by SIRT4 ablation in vitro and in vivo. More importantly, rapamycin is almost undetectable in control HCC cells with sufficient SIRT4 expression, which may help to explain the resistance to mTOR-targeted therapy for HCC (39).
Tumor cells are characterized by rapid proliferation which, in some scenarios, is driven by increased utilization of glutamine (47). As mentioned above, SIRT4 restricts glutamine utilization to inhibit TCA circulation in response to DNA damage (42). In HCC, SIRT4 depletion also leads to a remarkable increase in glutamine uptake and NH4+ production and, importantly, glucose uptake and production of lactic acid are not affected. Hence, SIRT4 loss fortifies the ability of cells to utilize glutamine. As a metabolic sensor, adenosine monophosphate-activated protein kinase (AMPK) inhibits metabolic reprogramming of glycolysis and biosynthesis pathways in cancer by regulating expression of hypoxia-inducible factor 1α (HIF1α) or mTOR (48, 49). Studies have shown that SIRT4 ablation reduces the phosphorylation of AMPKα and its target acetyl-CoA carboxylase (ACC) significantly. As a result, it was observed that mTOR activation increased through phosphorylation of the downstream effectors of TORC1: S6K and 4E-BP1. In addition, SIRT4 status had no effect on the gene expression of AMPK, ACC, S6K, or 4E-BP1. Interestingly, AMPK deficiency restored the glutamate reduction caused by SIRT4 overexpression. Taken together, those results suggest that SIRT4 deletion enhances the metabolic flux of glutamyl conversion, which is combined with AMPK inactivation in HCC cells. Moreover, AMPK is activated by the tumor suppressor liver kinase B1 (LKB1), a serine/threonine kinase that phosphorylates AMPK at high levels of AMP (50). Silencing expression of LKB1 or AMPKα counteracts the effects of SIRT4 depletion or overexpression, respectively, which demonstrates that SIRT4 inhibits the mTOR pathway through a mechanism that is dependent upon LKB1 and AMPKα (39).
The anti-hyperglycemia drug metformin has been shown to inhibit expression of mitochondrial complex I (51). This action leads to an increase in expression of intracellular AMP and ADP, which bind to the gamma regulatory subunit of AMPK and promote LKB1-mediated AMPK phosphorylation (52). After treatment with the AMPKα agonist metformin, activation of AMPKα and subsequent activation of mTOR were not observed in SIRT4-deficient cells. Consistently, activation of AMPKα expression by metformin significantly eliminated the hyperproliferative phenotype in SIRT4-negative cells, a result that was consistent with the data in vivo. Therefore, SIRT4 deletion-dependent tumor biology is dependent mainly on activation of AMPKα stimulated by AMP/ADP. Metformin-activated AMPKα expression completely reversed the carcinogenic characteristics of SIRT4-negative cells. Interestingly, metformin had almost no effect on SIRT4-control HCC cells, which may explain the resistance of metformin-treated HCC patients (39). However, more clinical evidence is needed to ascertain if SIRT4 expression in human HCC can predict patient responses to metformin itself or in combination with sorafenib.
Zhi and colleagues used tissue microarrays to measure SIRT4 expression in 90 HCC samples. IHC analyses showed that SIRT4 expression was down-regulated significantly in tumor tissues compared with that in paracancerous tissues. In addition, total proteins and RNA were extracted from 16 pairs of fresh HCC tissues and matched paracancerous tissues, and revealed identical results (53).
SIRT4 expression in peritumoral tissue of HCC patients was negatively correlated with tumor size, pathology grade, T stage, and clinical stage. In addition, Kaplan–Meier analysis and logarithmic rank tests showed that SIRT4 expression in the tissue around the HCC was positively correlated with the survival rate of HCC patients. However, SIRT4 expression in tumor tissues was not related to the factors mentioned above. Therefore, those results suggested that SIRT4 may have a tumor-inhibitory role in pericarcinomatous tissues of HCC by inhibiting the development and migration of tumor cells and, thus, improving the prognosis of patients. Therefore, according to SIRT4 expression in the paracancerous tissues of HCC, all HCC cases were divided into two categories (high SIRT4 expression and low SIRT4 expression). Using IHC analyses and flow cytometry (FCM), two interesting phenomena were discovered: (i) increased infiltration of tumor-associated macrophages (TAMs); (ii) a higher proportion of M2/M1 macrophages in the tissue around the HCC in the group of low SIRT4 expression. Macrophage infiltration may be caused by several monocyte/macrophage chemokines. Increased expression of monocyte chemotactic protein-1 (MCP-1) was caused by down-regulation of SIRT4 expression in HCC, and it was the reason for the increase in TAM infiltration. Down-regulation of SIRT4 expression can activate the nuclear factor-kappa B (NF-κB) pathway, which leads to upregulation of MCP-1 expression downstream (53). Furthermore, whether SIRT4 can catalyze the ADP ribosylation of mitochondrial NF-κB, and thereby increase nuclear translocation of p65, remains to be studied.
Another phenomenon observed was an increase in the proportion of M2/M1 macrophages in the tissues around HCC (53). Tao and colleagues reported that SIRT4 can reprogram endotoxin tolerance and promote acute inflammation of monocytes (54), thereby revealing the role of SIRT4 in immunoregulation. HCC-conditioned medium (HCM) was found to inhibit SIRT4 expression, and SIRT4 silencing stimulated macrophages to M2 polarization (53). The products of lipid metabolism play an important part in changing the function of macrophages and in inflammation (55).
M2 polarization of macrophages is related to activation of oxidative metabolism, including the functional TCA cycle promoted by glutamine and glucose catabolism, the increase of mitochondrial oxidative phosphorylation and fortification of fatty-acid oxidation (FAO) (56). SIRT4 is also involved in lipid metabolism and glutamine metabolism. It was shown that SIRT4 inhibits peroxisome proliferator-activated receptor α (PPARα) activity, thereby inhibiting FAO in the liver. More explicitly, SIRT4 disrupted the interaction of SIRT1 and PPARα, hence the activation of PPARα transcriptional activity by SIRT1 was inhibited (9, 57, 58).
In SIRT4-silenced TAMs, expression of FAO genes, including MCAD, PDK4, CPT1, PGC1-α, PPARδ, and PPARα, was increased, as was that of phosphorylated signal transducer and activator of transcription 3 (p-STAT3), which is necessary for differentiation into the M2 phenotype. Moreover, PPARδ suppressed by retroviruses reversed the effect of SIRT4-KO on expression of the phenotypic markers of TAMs. Therefore, SIRT4 silencing regulates TAMs to M2-like polarization through a FAO-PPARδ-STAT3 signaling pathway. Moreover, in the paracancerous tissues of HCC with low expression of SIRT4, the apoptosis rate of M1-like TAMs was high. Furthermore, the high expression of interleukin (IL)-10 induced by SIRT4 silencing in M2-like TAMs can prompt the apoptosis of M1 macrophages (53). Consistent with data from other studies, a high proportion of M2/M1 macrophages in pericarcinomatous tissue is associated with a poor prognosis in patients after tumor resection (59).
In HCM-stimulated TAMs, SIRT4 silencing: (i) promotes IL-10 expression but also promotes the production of cytokines, including IL-6 and vascular endothelial growth factor, which have been reported to prompt tumor progression; (ii) inhibits IL-12 production, enhances anti-tumor immunity, and prevents cancer development. Therefore, SIRT4 silencing in macrophages prominently prompts macrophage-induced tumor development in vitro and in vivo (53). The potential influence of SIRT4 on the STAT3-IL-6 axis needs to be studied further.
AMPK may also be a crucial moiety for FAO to regulate macrophages (60). AMPK can activate FAO at the expense of FA synthesis through at least two mechanisms: (i) phosphorylation and inactivation of ACC (the first step in FA synthesis); (ii) activation of PGC1-α which, in turn, stimulates the biogenesis and function of mitochondria. As mentioned above, SIRT4 can also affect the AMPK pathway, so whether the M2 polarization of TAMs is related to the SIRT4-AMPK-FAO axis merits further study.
Most tumor cells (including HCC cells) use aerobic glycolysis to maintain anabolic growth, and the preference for glycolysis often leads to adverse clinical outcomes. As mentioned above, SIRT4 can inhibit the PDHC to participate in aerobic glycolysis. HCC cells that overexpress SIRT4 show a reduced ability for glycolysis, and the mRNA and protein expression of key enzymes in glycolysis is also decreased (61).
Researchers have discovered that upregulation of the histone methyltransferase SET8 is positively correlated with poor survival in HCC patients, and aggravates glycolysis changes and tumor progression in vitro and in vivo. Notably, enhanced expression of SIRT4 is sufficient to reverse the increased expression of key glycolytic genes in SET8-overexpressed cells. The enhanced aerobic glycolysis observed in cells with SET8 overexpression is the result of SIRT4 inactivation (61). SET8 is the only known monomethyltransferase during monomethylation of histone H4 lysine 20 (H4K20me1) (62). Expression of SIRT4 mRNA and protein is decreased in HCC cells stably expressing SET8, whereas that of H4K20me1 is increased prominently. Chromatin immunoprecipitation (ChIP) studies have demonstrated that H4K20me1 is enriched in the SIRT4 promoter region. Those results suggest that SET8 inhibits the transcription of SIRT4 through H4K20me1. Krüppel-like factor 4 (KLF4) has a repressive role in the glycolysis of HCC cells, whereas mitochondrial oxygen consumption is enhanced. ChIP studies have shown that KLF4 binds directly to the SIRT4 promoter to aid its transcription. Also, SET8 and KLF4 are co-precipitated and co-localized, and SET8 overexpression leads to prominent reductions in expression of KLF4 mRNA and protein. However, the mechanism is not clear and may be related to DNA methylation (61).
Researchers have established a senescence model of the SMCC7721 liver-cancer cell line induced by doxorubicin. Use of the β-galactosidase method to measure cell senescence revealed that doxorubicin-induced SMCC7721 senescent cells had increased expression of the senescence-related proteins p53 and P16, increased expression of SIRT4, and blocked the Janus kinase 2 (JAK2) signaling pathway. In this model, SIRT4 overexpression could increase expression of the senescence-related protein p53, whereas the JAK2 signaling pathway was inhibited. CP-690550 (inhibitor of the JAK2/STAT3 signaling pathway) enhanced the cell senescence induced by doxorubicin significantly. Hence, SIRT4 promoted doxorubicin-induced senescence of SMCC7721 cells by inhibiting the JAK2/STAT3 signaling pathway (63).
SIRT4 deacetylates glycerol O-acyltransferase (GNPAT) (64), expression of which is upregulated and which promotes FA synthesis in HCC cells at the K128 residue, while acetyl-CoA acetyltransferase 1 (ACAT1) (65), which is a mitochondrial enzyme, acetylates GNPAT at K128. Moreover, SIRT4 can antagonize ACAT1-mediated GNPAT acetylation. That is, SIRT4 deacetylates GNPAT at K128 with dissociation of ACAT1. In addition, ectopic expression of SIRT4 WT leads to a reduction in expression of GNPAT protein without a change in mRNA expression. This phenomenon occurs because deacetylation can promote TRIM21-mediated ubiquitination and degradation of GNPAT (66). Menendez and colleagues found that GNPAT interacts closely with fatty acid synthase (FASN) (67) which is a key enzyme in malonyl-CoA synthesis of long-chain FAs. FASN expression is up-regulated in many types of cancer, and is closely related to tumor invasiveness and a poor prognosis (68). GNPAT acetylation can inhibit TRIM21-mediated FASN degradation and promote lipid synthesis and tumor metabolism. In brief, SIRT4 deacetylates and reduces expression of GNPAT, which can promote lipid metabolism and hepatocarcinogenesis by stabilizing FASN (66) (Figure 3).
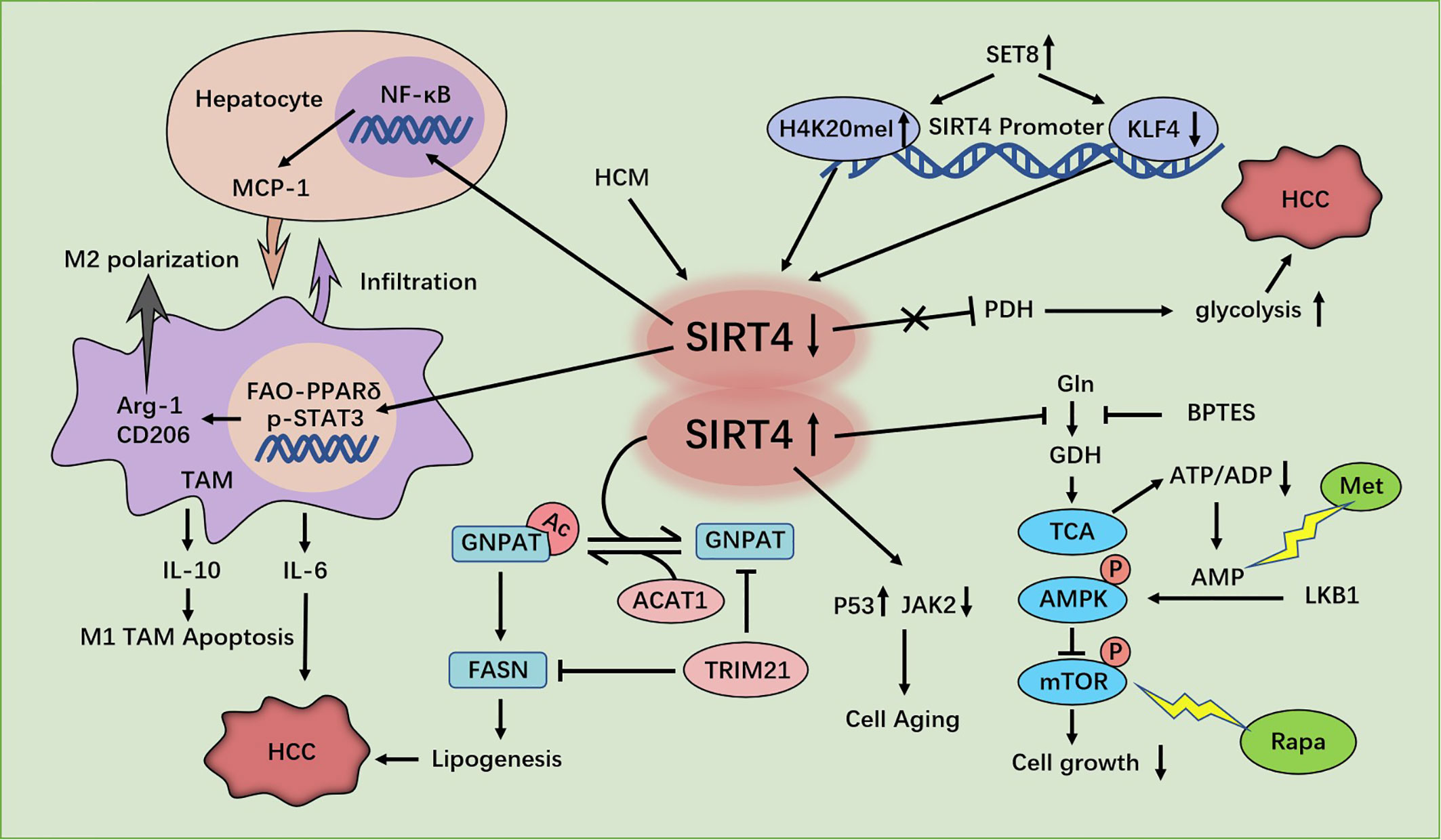
Figure 3 Regulation of HCC cells by SIRT4. SIRT4 depletion promotes hepatocellular carcinoma (HCC) genesis by regulating the AMPKα-mTOR axis. SIRT4 silencing in tumor-associated macrophages (TAMs) promotes HCC development through PPAR signaling-mediated macrophage activation. SET8 inhibits sirt4 transcription by regulating H4K20me1 and KLF4, thereby enhancing aerobic glycolysis and promoting the growth of HCC cells. SIRT4 deficiency promotes GNPAT acetylation, inhibits FASN degradation, promotes lipid synthesis, and promotes HCC. SIRT4 promotes the senescence of SMCC7721 cells in HCC by inhibiting the JAK2-STAT3 signaling pathway.
In PC cell lines, SIRT4 overexpression strongly inhibits cell proliferation and increases apoptosis. It has been shown that SIRT4 is a negative regulator of aerobic glycolysis. SIRT4 mediates the effects of ubiquitin-like plant homologous domain and ring finger domain 1 (UHRF1) (69) on the proliferation and glycolysis metabolism in PC cells (70). Using an extracellular flux analyzer in seahorses to measure glycolysis, Hu and colleagues observed that SIRT4 overexpression inhibited the extracellular acidification rate, thereby indicating its negative regulation of aerobic glycolysis. In addition, if SIRT4 is overexpressed, the oxygen-consumption rate is increased accordingly, thereby supporting the positive role of SIRT4 in mitochondrial respiration (70). Conversely, UHRF1 (an epigenetic modifier (71), overexpressed in PC cells) predicts a poor prognosis and prompts PC-cell proliferation by promoting aerobic glycolysis, and positively regulates the transcriptional activity of HIF1 α in a dose-dependent manner. Whether using TCGA database, or employing IHC analyses of 70 PC patients, or experimenting with PC cell lines, a negative correlation between UHRF1 and SIRT4 has been confirmed. ChIP analyses have shown that UHRF1 combines specifically with the SIRT4 promoter to inhibit SIRT4 expression in PC cells. SIRT4 overexpression reduces the protein expression of HIF1α, as well as the expression of glycolysis genes (GLUT1, HK2, and LDHA) at mRNA and protein levels. In vitro studies have also shown a significant negative correlation between SIRT4 and glycolysis genes (GLUT1, HK2 and LDHA) in PC patients in a cohort in TCGA database (70). In brief, UHRF1 promotes aerobic glycolysis and proliferation of PC cells via suppression of SIRT4 expression.
However, Lei and colleagues claimed that SIRT4 is involved in catabolism of branched-chain amino acids and promotes cell proliferation and tumor growth in PDAC by regulating BCAT2. They found that BCAT2 could be deacetylated and stabilized by SIRT4 at K44. Conversely, cAMP-responsive element-binding (CREB)-binding protein (CBP) is the acetyltransferase of BCAT2. BCAT2 acetylation leads to its degradation through a ubiquitin-proteasome pathway, and is stimulated by deprivation of branched-chain amino acids (34) (Figure 4).
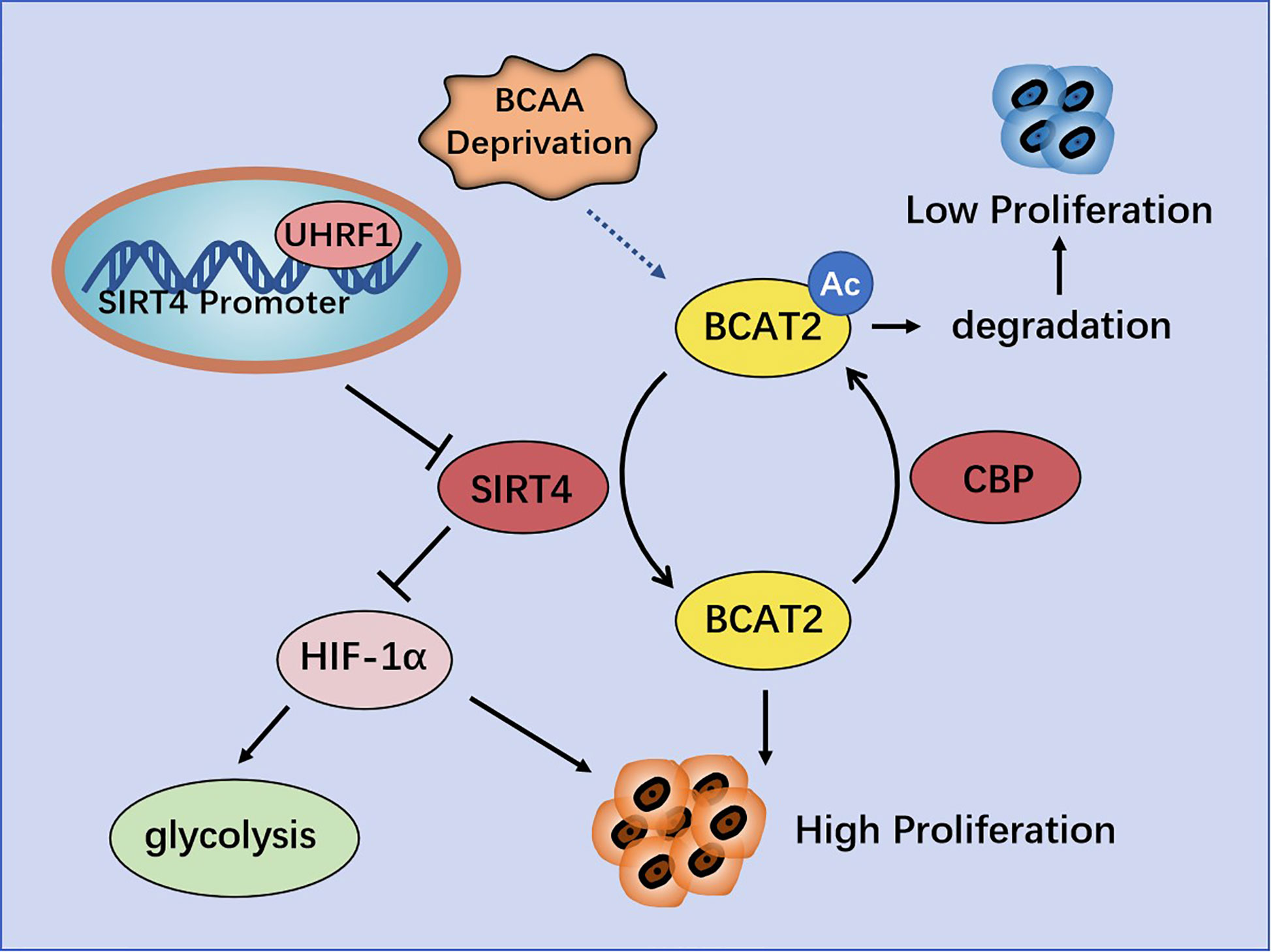
Figure 4 Regulation of pancreatic cancer (PC) by SIRT4. UHRF1 promotes aerobic glycolysis and proliferation of PC cells by inhibiting SIRT4 expression. BCAT2 is deacetylated and stabilized by SIRT4, and is acetylated and degenerated by CBP.
In a study of 75 patients with gastric adenocarcinoma in 2015, SIRT4 expression in tumor tissue was significantly lower than that in corresponding normal tissues. In addition, SIRT4 expression was negatively correlated with the pathologic grade, depth of tumor invasion, number of positive lymph nodes, and International Union Against Cancer stage (72).
Sun and colleagues collected 86 pairs of GC tissues and paracancerous normal tissues. They also found that SIRT4 expression was down-regulated in GC tissues and negatively correlated with tumor size, pathologic grade, and lymph-node metastasis. Moreover, the survival rate of patients with low expression of SIRT4 was decreased (73).
In GC cell lines, SIRT4 expression is decreased, and functional experiments have shown that SIRT4 inhibits the proliferation, migration, and invasion of GC cells. Enzyme-linked immunosorbent assays have shown that ectopic expression of SIRT4 inhibits secretion of matrix metallopeptidase (MMP)2 and MMP9 (73), which are closely related to tumor invasiveness in humans and a poor prognosis (74). SIRT4 regulates epithelial–mesenchymal transition (EMT) to control the migration and invasion of cells. SIRT4 overexpression significantly upregulates expression of the mRNA and protein of E-cadherin, down-regulates expression of the protein of N-cadherin and vimentin, but has no effect on their mRNA. It has been postulated that SIRT4 may inhibit EMT by regulating E-cadherin expression. Measurement of the RNA expression of EMT-related transcription factors (slug, snail, and twist) has revealed that ectopic expression of SIRT4 reduces expression of slug but has no effect on snail or twist (73).
Researchers have shown that SIRT4 overexpression induced arrest of the G1 phase of the cell cycle by inhibiting expression of phosphorylated extracellular signal-regulated kinase (p-ERK), cyclin D and cyclin E (75). FCM and staining (propidium iodide) revealed that SIRT4 overexpression prominently increased the proportion of cells in the G1 phase and decreased the number of cells in the S phase of the cell cycle. SIRT4 inhibited expression of cyclin D and cyclin E prominently. In addition, SIRT4 predominantly decreased p-ERK expression, but did not affect ERK expression (75). It has been reported that ERK controls the G1 phase of the cell cycle by regulating expression of cyclin D (76). In contrast, SIRT4 overexpression does not affect the apoptosis of GC cells (75).
Nakahara and co-workers evaluated 172 patients with esophageal squamous cell carcinoma (ESCC) who underwent resection. They measured expression of SIRT4 mRNA in cohort 1 (n = 79) and measured expression of SIRT4 protein in cohort 2 (n = 93). They found that SIRT4 expression was not correlated with any clinicopathologic parameter in either cohort. However, in cohort 1, the overall survival rate of patients with low expression of SIRT4 was lower than that of cases with high SIRT4 expression. In cohort 2, the rate of overall survival and disease-free survival of SIRT4-negative patients was lower than that of their SIRT4-positive counterparts. Multivariate analysis showed that SIRT4 expression was an independent prognostic factor for overall survival. Also, the rate of distant recurrence of SIRT4-negative patients was notably higher than that of SIRT4-positive cases. Moreover, SIRT4-KO can predominantly increase GDH activity and promote the proliferation and migration of esophageal-cancer cells in vitro (77). Recently, our team witnessed that SIRT4 is a specific target gene for micro (mi)R-424-5p in ESCC and negatively modulated by miR-424-5p. miR-424-5p expression was up-regulated in ESCC and dramatically facilitated the proliferative and migratory capacity of ESCC cells. SIRT4 overexpression strongly rescued the promotive influence of miR-424-5p on the proliferative and migratory capacity of ESCC cells. Hence, manipulation of the miR-424-5p/SIRT4 axis could provide a novel strategy for further ESCC treatment (78).
Miyo and colleagues collected 142 samples of CRC tissue and part of its paracancerous tissue, as well as 38 samples of colorectal adenomas tissue, from patients after resection. They found that the proportion of patients with high expression of SIRT4 in normal tissues (61.1%) was greater than that for patients with adenomas (52.6%) or CRC (52.5%). SIRT4 expression was negatively correlated with tumor–node–metastasis (TNM) stage, but not with sex, age, venous invasion or histology type. The recurrence rate of patients with low expression of SIRT4 was increased, and 5-year overall survival rate tended to decrease. It was also found that SIRT4 inhibited the proliferation, invasion, and migration of CRC cell lines. SIRT4 inhibited the enzyme activity of GDH, and E-cadherin gene (CDH1) expression was up-regulated notably by SIRT4. Immunofluorescence staining also demonstrated that SIRT4 overexpression could increase E-cadherin expression. In addition, it was shown that vimentin expression was predominantly down-regulated by SIRT4 overexpression (79). Shen and colleagues showed that E-cadherin expression was inhibited by the zinc finger E-box binding homeobox (ZEB) family, whereas expression of the ZEB family was inhibited by miRNA-200c (80). MiRNA-200c expression was inhibited by SIRT4, but SIRT4 had an inconsistent effect on ZEB1 expression, suggesting that the mechanism by which SIRT4 regulates E-cadherin expression may be different. Further studies showed that inhibition of glutamine metabolism by SIRT4 led to positive regulation of E-cadherin expression. In addition, α-ketoglutarate (α-KG) eliminated the induction of E-cadherin expression by SIRT4, which suggested that SIRT4 inhibited EMT by inactivating GDH and reducing a-KG expression in cells. Those data revealed other molecular mechanisms of glutamine-metabolism regulation, which affected E-cadherin expression through SIRT4 and, in turn, affected cell mobility. In brief, glutamine metabolism may be important for the mobility of cancer cells (79).
Huang and colleagues used tissue microarrays in 89 cases of CRC. They found that SIRT4 expression in CRC tissues was notably lower than that in normal tissues. The lower the SIRT4 expression, the worse was the pathologic differentiation and the lower was the overall survival rate. mRNA expression of SIRT2, SIRT4 and SIRT5 in 16 pairs of CRC tissues and adjacent normal tissues showed that SIRT4 expression was reduced significantly, whereas that of SIRT2 and SIRT5 showed no significant changes. Analyses of 236 cases of CRC samples and 22 normal colorectal-tissue samples from TCGA database provided data that were consistent with the results stated above. Moreover, SIRT4 expression was down-regulated in the early stage and, importantly, its low expression was maintained during cancer progression, suggesting that down-regulation of SIRT4 expression may be necessary for tumorigenesis and maintenance of tumor cells. Further studies showed that SIRT4 overexpression inhibited proliferation of CRC cells in vivo and in vitro. SIRT4 inhibited glutamine metabolism and cooperated with glycolysis inhibition to induce the death of CRC cells. In addition, SIRT4 overexpression significantly reduced the rates of the S phase and G2/M phases of two CRC cell lines after 5-fluorouracil treatment, but had no effect on the apoptosis rate, indicating that SIRT4 increased the sensitivity of CRC cells to 5-fluorouracil by delaying mitosis (81) (Figure 5).
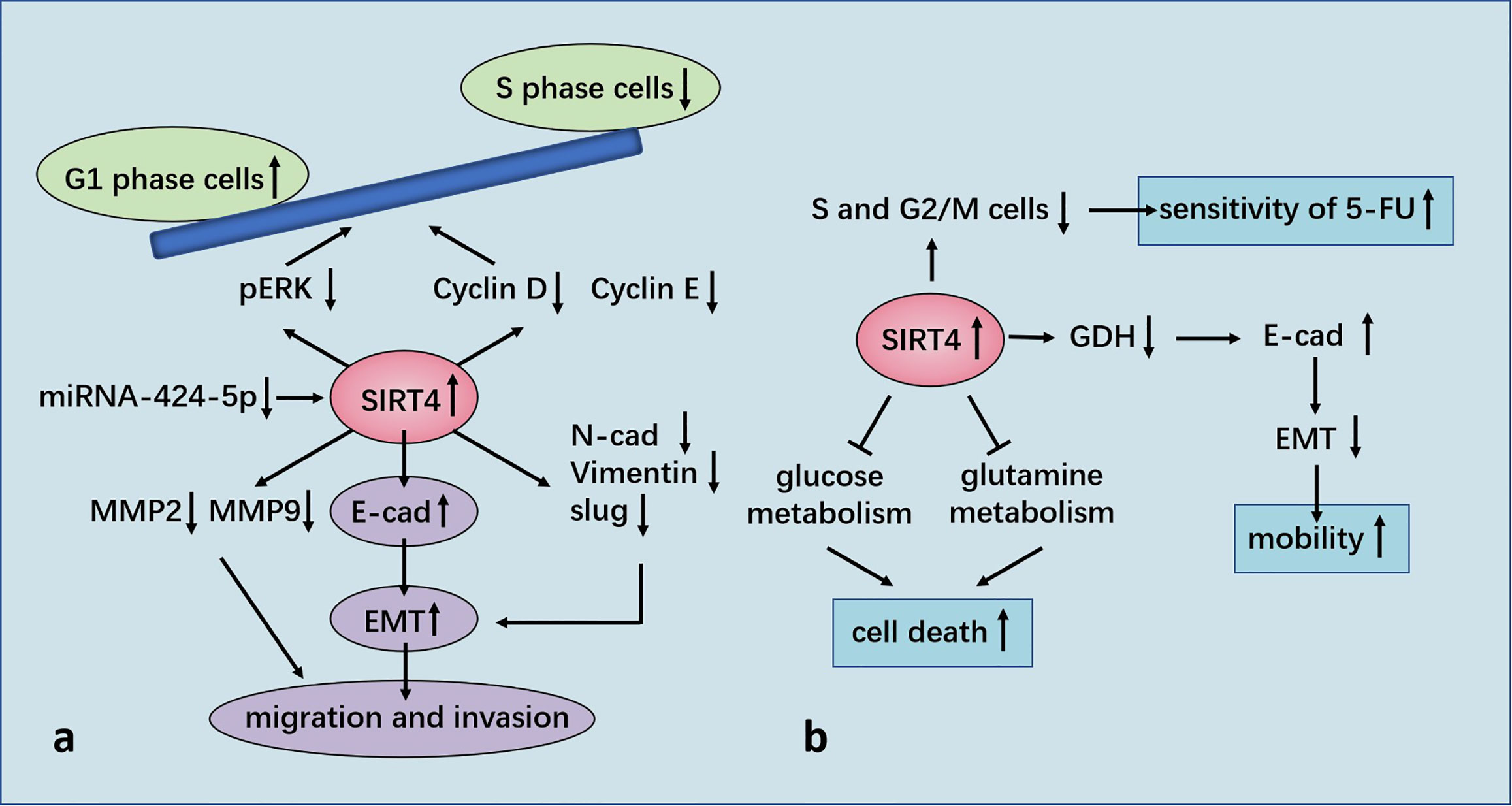
Figure 5 (A) Regulation of GC by SIRT4. SIRT4 inhibits the proliferation, migration, and invasion of GC cells by regulating epithelial–mesenchymal transition (EMT). SIRT4 overexpression induces stagnation of the G1 phase of the cell cycle by inhibiting expression of p-ERK, cyclin D, and cyclin E. SIRT4 is a specific target gene for miR-424-5p in ESCC and negatively modulated by miR-424-5p. (B) Regulation of CRC by SIRT4. SIRT4 expression leads to positive regulation of E-cadherin expression by inhibition of glutamate dehydrogenase (GDH) expression, which inhibits EMT and influences cell mobility. SIRT4 inhibits glutamine metabolism and induces death of colorectral cancer (CRC) cells in conjunction with glycolysis inhibition. SIRT4 increases the sensitivity of CRC cells to 5-FU by delaying mitosis.
High-throughput real-time polymerase chain reaction analyses have shown that SIRT4 expression in breast-cancer tissue is predominantly lower than that in paracancerous tissue (82). The analysis of TCGA and 3 Gene Expression Omnibus (GEO) datasets showed that decreased SIRT4 expression correlated with increased risk of disease progression and poor clinical outcome. Besides, immunoblotting and IHC staining also showed a significant decrease in SIRT4 protein levels in breast tumor tissues (83). Wang and colleagues found that down-regulation of SIRT4 expression promoted glutamine decomposition, promoted the growth of breast-cancer cells and inhibited their apoptosis. ChIP analyses demonstrated that C-terminal-binding protein (CtBP) was bound to the SIRT4 promoter and inhibited its expression directly. Inhibition of SIRT4 expression by CtBP helped to maintain the pH homeostasis of breast-cancer cells and was beneficial to their growth. CtBP also down-regulated SIRT4 expression to promote the growth of breast-cancer cells and inhibited their apoptosis (84).
Glucose and glutamine are essential components for cell growth. The maximum yield from the original nutrients can be achieved by coordinating the metabolic pathways of different substrates. A mechanism for coordinating consumption of glucose and glutamine in cancer cells has been proposed: the CtBP-SIRT4-GDH axis mediates crosstalk between glycolysis and glutamine decomposition. Synergistic consumption of glucose and glutamine among different cell lines was observed. That is, glutamine consumption was delayed under low and high levels of glucose. Application of glycolysis inhibitors showed that, with an increase in the drug dose, the decrease in glutamine consumption was consistent with a decrease in glucose consumption (85).
Researchers found that when cells were cultured in low-glucose medium, SIRT4 expression increased in a time-dependent manner. Application of a glycolysis inhibitor showed that restricted glycolysis upregulated SIRT4 expression and eroded GDH activity. Accordingly, treatment with a glycolysis inhibitor also up-regulated p21 expression, indicating that cell growth stopped, which was similar to the effect of DNA damage. This synergistic effect was related to the inhibition of SIRT4 expression by CtBP at high glucose levels, and GDH activity also indicated that CtBP needed a high glucose supply to inhibit SIRT4 expression. However, low glucose abolished the binding of CtBP to the SIRT4 promoter and inhibition of SIRT4 expression. Dimerized CtBP has great potential to inhibit expression of target genes. Use of the bimolecular fluorescence complementation method revealed that a low glucose supply reduced the binding activity of CtBP as a co-inhibitor by inhibiting CtBP dimerization (85).
Xing and collaborators showed that SIRT4 increased the sensitivity of breast-cancer cells to tamoxifen. SIRT4 overexpression decreased the half-maximal inhibitory concentration of tamoxifen in estrogen receptor (ER)-negative MCF7 and T47D cells, whereas its depletion increased it. Glutamine is an essential nutrient for the growth and survival of cells. Tamoxifen inhibited the proliferation of ER-negative cells by inhibiting glutamine uptake. Cells treated with tamoxifen after SIRT4 overexpression consumed less glutamine than cells treated with tamoxifen alone. This observation suggested that SIRT4 and tamoxifen work together to reduce glutamine absorption by ER-positive breast-cancer cells. In addition, SIRT4 enhanced inhibition of the proliferation and apoptosis of ER-positive breast-cancer cells induced by tamoxifen (86).
STAT3 is activated in breast cancer, and increased phosphorylation of the tyrosine 705 (Y705) site in STAT3 has been observed in tamoxifen-resistant MCF7/TAM cells (87). STAT3 mediates expression of many genes in response to cellular stimuli, and overactivation of STAT3 is led by phosphorylation of the Y705 site (88). Importantly, SIRT4 inhibits the phosphorylation and nuclear translocation of STAT3, thereby inhibiting the STAT3 signaling pathway, and amplification of the target genes MYC and CCND1 of the STAT3 signaling pathway provide tamoxifen resistance in ER-positive breast-cancer cells (89, 90). The transcription and translation of MYC and CCND1 are also blocked by SIRT4. In addition, activation or inhibition of STAT3 expression can counteract the effect of SIRT4 overexpression/depletion on cell proliferation. STAT3 signal transduction is stimulated by cytokines such as IL-6. If SIRT4 is overexpressed, IL-6 release in ER-positive breast-cancer cells decrease, and inhibition of STAT3 expression induced by SIRT4 is reversed after IL-6 treatment. Those data suggest that SIRT4 enhances the sensitivity of ER-positive breast-cancer cells to tamoxifen by inhibiting the IL-6/STAT3 pathway (86).
Recent studies have shown that SIRT4 plays a novel part in the negative regulation of mammary-gland development and stemness. SIRT4 can inhibit the self-renewal and expansion of breast tumor-initiating cells and may protect mice from breast tumors and lung metastasis by preventing the formation of mammary stem cells. In this mechanism, SIRT4 negatively regulates SIRT1 expression by inhibiting glutamine metabolism, ensuring inhibition of breast stem cells. H4K16ac and breast cancer susceptibility gene 1 (BRCA1) are new major targets for SIRT4 in breast cancer. SIRT4 deficiency increases the expression of acetyl-histone H3 at lys9 (H3K9ac), acetyl-histone H4 at lys16 (H4K16ac) and stem-cell markers, and down-regulates BRCA1 expression by regulating H4K16ac rather than well-defined H3K9ac. These processes cannot be separated from SIRT1, which regulates the acetylation pattern of histone H4 and regulates stemness through BRCA1 (83) (Figure 6).
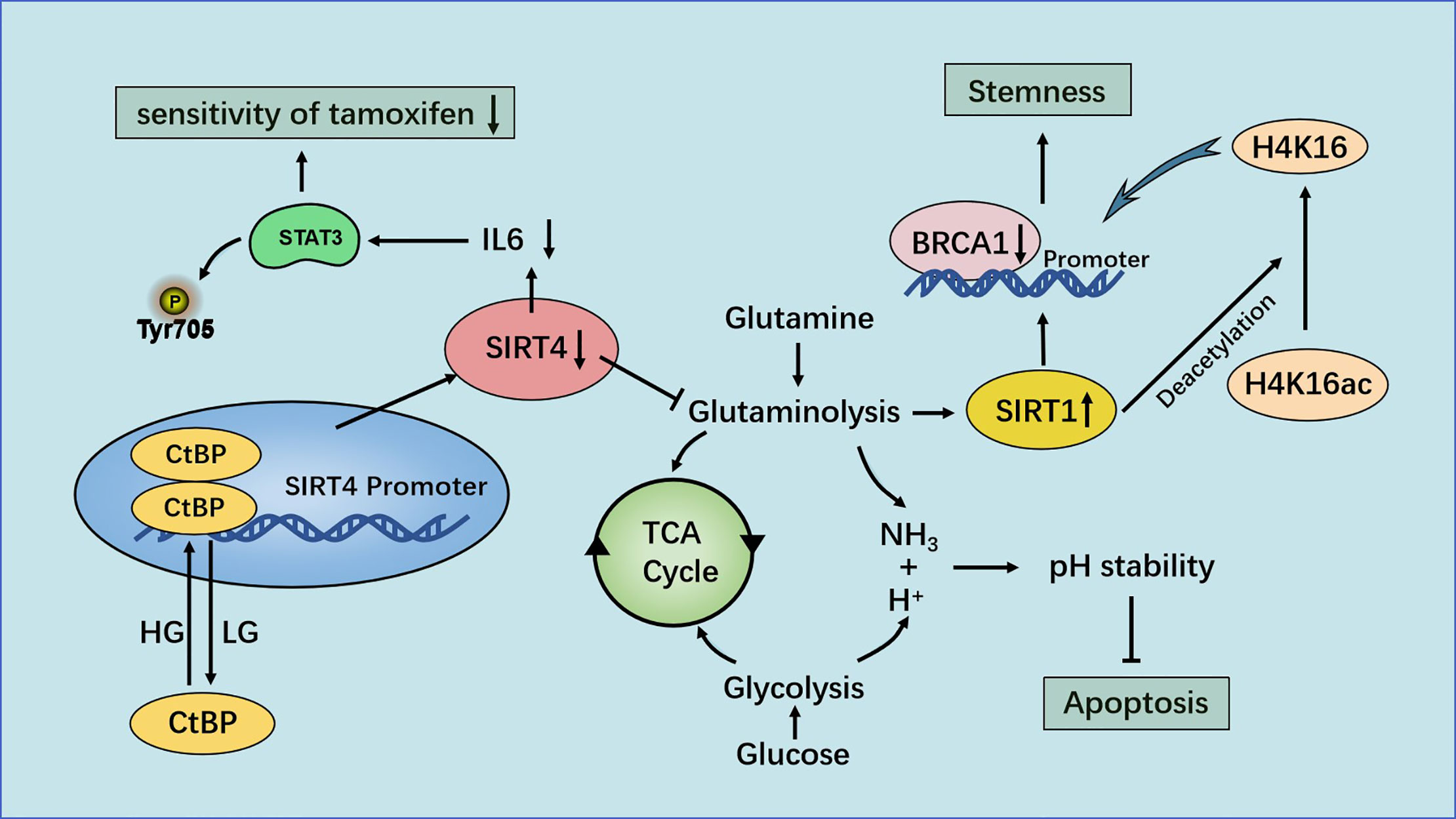
Figure 6 Regulation of breast cancer by SIRT4. Under the condition of high glucose levels, CtBP dimerization helps CtBP to promote glutamine metabolism by inhibiting SIRT4 expression. This action can help maintain the pH homeostasis of cells, promote the growth of breast-cancer cells and inhibit their apoptosis. SIRT4 enhances the sensitivity of ER-positive breast cancer cells to tamoxifen by inhibiting glutamine metabolism and the IL-6-STAT3 signaling pathway.
Use of an online database of Kaplan–Meier plots ascertained the prognostic value of expression of SIRT4 mRNA in patients with ovarian cancer. High expression of SIRT4 mRNA indicated the poor survival of patients with ovarian cancer. In pathologic grade-III carcinoma and all clinical stages, overexpression of SIRT4 mRNA was correlated with poor overall survival. In addition, SIRT4 expression was negatively correlated with the overall survival of patients treated with drugs (91).
IHC analyses were undertaken to measure expression of SIRT4 protein in 65 samples of endometrioid-adenocarcinoma tissue using microarrays. SIRT4 expression was significantly lower in endometrioid-adenocarcinoma tissue than that in non-neoplastic tissue. In addition, SIRT4 expression was lower in tissues from patients with an advanced stage of endometrioid-adenocarcinoma according to American Joint Committee on Cancer classification (92).
Li and colleagues found that, based on IHC analyses, SIRT4 expression in prostate-cancer tissues was significantly lower than that in matched paracancerous tissues, and that expression of SIRT4 protein was low in 9/12 frozen prostate-cancer tissues. They discovered that the P21 activated kinase 6-SIRT4-adenine nucleotide translocase-2 (PAK6-SIRT4-ANT2) complex could inhibit the apoptosis of prostate-cancer cells (93). Previously, it had been reported that ANT2 (94), in the inner membrane of mitochondria, is the regulatory substrate of SIRT4. SIRT4 can regulate expression of ANT2 protein through deacetylation, and maintain the energy balance in mitochondria by coupling with ANT2 (18, 95). Furthermore, they found that SIRT4 regulates the acetylation level of ANT2 on the K105 site and degrades ANT2 by promoting its ubiquitination, which is dependent upon the deacetylation activity of SIRT4. Notably, regulation of acetylation activity of ANT2 by SIRT4 was dependent upon the presence of glutamine in the medium (93). In addition, P21 activated kinase 6 (PAK6) (96) is a conserved family of serine/threonine kinases, which is also located mainly in the mitochondrial inner membrane. PAK6 was found to promote SIRT4 ubiquitin-mediated proteolysis. Moreover, PAK6 phosphorylated ANT2 directly at the T107 site, and directly/indirectly affected ANT2 acetylation through SIRT4. The interaction between phosphorylation and deacetylation of ANT2 regulated the apoptosis of prostate-cancer cells. In summary, multiple post-translational modifications between proteins in the PAK6-SIRT4-ANT2 complex (including the promotion of ubiquitin degradation through deacetylation and the interaction between acetylation at K105 and phosphorylation at T107) lead to various modifications that interact with the target protein ANT2. These modifications, in turn, eventually regulate the apoptosis of prostate-cancer cells, and lead to tumor growth in the body (93).
Using publicly available TCGA pan-cancer datasets, as well as western blotting and real-time reverse transcription-quantitative polymerase chain reaction, the expression and prognostic value of SIRTs in ccRCC was evaluated. SIRT4 expression in ccRCC was lower than that in normal tissues, and low SIRT4 expression was obviously involved with poor overall survival and advanced tumor stage in ccRCC patients (97, 98). Wang and coworkers also indicated that SIRT4 overexpression significantly reduced the proliferation, migration and invasion ability of ccRCC cells in vitro (98).
Consistent with the findings in ESCC, Yu and colleagues found that miR-424 could regulate SIRT4 expression in bladder cancer. They found that high expression of miR-424 was closely related to the poor clinical prognosis of patients with bladder cancer. In this mechanism, cisplatin induced miR-424 expression in bladder cancer cells in a HIF-1 α-dependent manner, and miR-424 conferred cisplatin resistance on bladder cancer cells in vitro and in vivo. Importantly, miR-424 mediated this resistance by down-regulation of expression of SIRT4 and pro-apoptotic UNC5B. SIRT4 knockout prohibited cisplatin-mediated PARP proteolysis and reduced the sensitivity of these cells to cisplatin (99).
Low expression of SIRT4 in non-small cell lung cancer (NSCLC) patients is related to clinicopathologic factors and a poor prognosis. Carrying out IHC analyses on 133 lung-cancer tissues, it was found that SIRT4 expression was decreased dramatically in 52.6% of lung-cancer tissues. There was no significant difference between SIRT4 status and age, sex or histology differentiation, but it was negatively correlated with TNM stage. In addition, the proportion of SIRT4 down-regulation in adenocarcinoma (52/84, 61.9%) was higher than that in squamous cell carcinoma (18/49, 36.73%). Patients with low expression of SIRT4 showed stronger expression of Ki-67 and shorter survival than those with high SIRT4 expression (41).
Upregulation of SIRT4 expression inhibited the proliferation, invasion and migration of lung-cancer cell lines. FCM showed that the percentage of cells in the S phase decreased in cell lines with SIRT4 transfection, suggesting that SIRT4 may inhibit cell growth by inhibiting the transition from the S phase to the G1 phase in mice. SIRT4 damaged mitochondrial fission and inhibited the activity of the mitochondrial fission-related protein Drp1. Mitochondria in cells overexpressing SIRT4 found it easier to fuse and had longer shapes. SIRT4 overexpression resulted in a significant down-regulation of Drp1 expression (41). The active form of Drp1, phosphate Drp1 (S616), promoted the entrapment of Drp1 around the mitochondrial membrane and induced an increase in mitochondrial fission (100). SIRT4 attenuated mitochondrial fission by inhibiting Drp1S616 phosphorylation. SIRT4 inhibited the migration and invasion of lung-cancer cells by inhibiting the mitochondrial fission induced by Drp1. SIRT4 overexpression inhibited the activity of mitogen-activated protein kinase (MEK) and ERK significantly, whereas inhibition of MEK/ERK expression reversed the upregulation of p-Drp1 expression and enhanced the invasive ability and mitochondrial fission induced by SIRT4 depletion.
To evaluate further the function of SIRT4 in lung-cancer metastasis, 60 samples of primary NSCLC tissues and their corresponding lymph-node metastatic tumors were analyzed. SIRT4 expression in the latter was lower than that in corresponding primary NSCLC tissues. Also, there was a negative correlation between expression of SIRT4 and Drp1 in primary NSCLC tissues and lymph-node metastatic tumors. In vivo experiments also showed that SIRT4 overexpression delayed tumor growth notably, and that SIRT4 expression was negatively correlated with p-Drp1 expression in lung-cancer samples (41).
In Myc-driven human Burkitt’s lymphoma cells, SIRT4 overexpression inhibits mitochondrial glutamine metabolism which, in turn, inhibits cell proliferation (101). Glutaminase is the first enzyme in the mitochondrial metabolism of glutamine, and its inhibition limits the entry of glutamine into the TCA cycle (102). It has been shown that if the glutaminase level is reduced using short-hairpin RNAs, SIRT4 overexpression no longer inhibits glutamine uptake (42). BPTES (glutaminase inhibitor) treatment can inhibit cell proliferation much more than that observed with SIRT4 overexpression, but the precise relationship between SIRT4 and glutaminase is not clear. The synergistic effect of SIRT4 and glycolysis inhibition leads to the death of Burkitt’s lymphoma cells. However, SIRT4 can regulate glutamine metabolism in cells independent of Myc. SIRT4 expression in Burkitt’s lymphoma cells does not affect expression of Myc and its target genes, which is incompatible with the effects seen in HCC cells and PC cells. This phenomenon may be caused by specificity to tumor tissues or organ functions. In a mouse model, SIRT4 deletion accelerated the formation of lymphoma and mortality in Em-Myc transgenic mice (101).
Bradbury and colleagues characterized expression of all 18 members of the HDAC family in primary acute myeloid leukemia blasts, and in four types of control cell (CD34+ progenitors from the umbilical cord, either quiescent or cycling (post-culture), cycling CD34+ progenitors from granulocyte-colony stimulating factor-stimulated adult donors, and peripheral blood mononuclear cells). Among them, SIRT4 expression was decreased in acute myeloid leukemia cells compared with that in control cells (103).
Analyses of tissue samples from 89 patients with thyroid cancer by Chen and colleagues showed that expression of SIRT4 protein decreased significantly compared with that in normal tissue, but there was no significant correlation with clinicopathologic features. In vitro experiments showed that SIRT4 inhibited the proliferation, migration, and invasion of thyroid-cancer cells. SIRT4 overexpression blocked the G0/G1 phase of the cell cycle, and induced apoptosis. SIRT4 inhibited the migration and invasion of thyroid-cancer cells by inhibiting glutamine metabolism (104).
Expression of mitochondrial tumor-suppressor genes (SIRT3, SIRT4, and MTUS1), a mitochondrial DNA repair gene (OGG1) and a proliferation marker (Ki-67) was measured by reverse transcription-quantitative polymerase chain reaction and real-time fluorescence quantitative polymerase chain reaction in HNSCC patients and controls by Mahjabeen and colleagues. Compared with the control group, expression of SIRT3, SIRT4, MTUS1, and OGG1 in HNSCC was down-regulated significantly, whereas Ki-67 expression was up-regulated. SIRT4 expression in the advanced clinical stage (III–IV) was significantly lower than that in early clinical stage (I–II), and SIRT4 expression was negatively correlated with TNM stage. A positive correlation between expression of SIRT4 and SIRT3, SIRT4 and OGG1 was observed (105). In conclusion, down-regulation of expression of certain mitochondrial tumor-suppressor genes, such as SIRT3, SIRT4, and MTUS1, in HNSCC cells indicates aggressive tumor behaviors, and may predict an unfavorable clinical outcome.
Analyses of human neuroblastoma tissues and adjacent normal tissues were undertaken by Wang and colleagues. They showed that SIRT4 expression was decreased dramatically in neuroblastoma tissues. SIRT4 expression was negatively correlated with the stage (according to the International Neuroblastoma Staging System) and lymph-node metastasis. The duration of survival of neuroblastoma patients with low expression of SIRT4 was shortened significantly. In vitro experiments showed that SIRT4 overexpression reduced the proliferation, invasion, and migration of neuroblastoma cells significantly. Up-regulation of SIRT4 expression decreased the basic oxygen consumption rate of SH-SY5Y cells significantly, suggesting that up-regulation of SIRT4 expression inhibited respiration and decreased energy production in mitochondria. Simultaneously, up-regulation of SIRT4 expression diminished the value of spare respiratory capacity dramatically, indicating that up-regulation of SIRT4 expression may reduce maximum energy production in mitochondria and increase mitochondrial damage. SIRT4 overexpression reduced expression of SIRT1 protein (106). The role of SIRT1 in regulating tumors through energy metabolism has been demonstrated by several scholars. For example, SIRT1 was shown by Rodgers and colleagues to promote tumor development by interfering with mitochondrial biosynthesis (107). SIRT1 also interferes with glycolysis in tumor cells by activating HIF-2α, thereby regulating energy metabolism in tumors (108). In brief, SIRT4 exhibited a tumor-suppressor function in human neuroblastoma, and reduced the energy metabolism of tumor cells by inhibiting mitochondrial metabolism and SIRT1 expression (106) (Table 1).
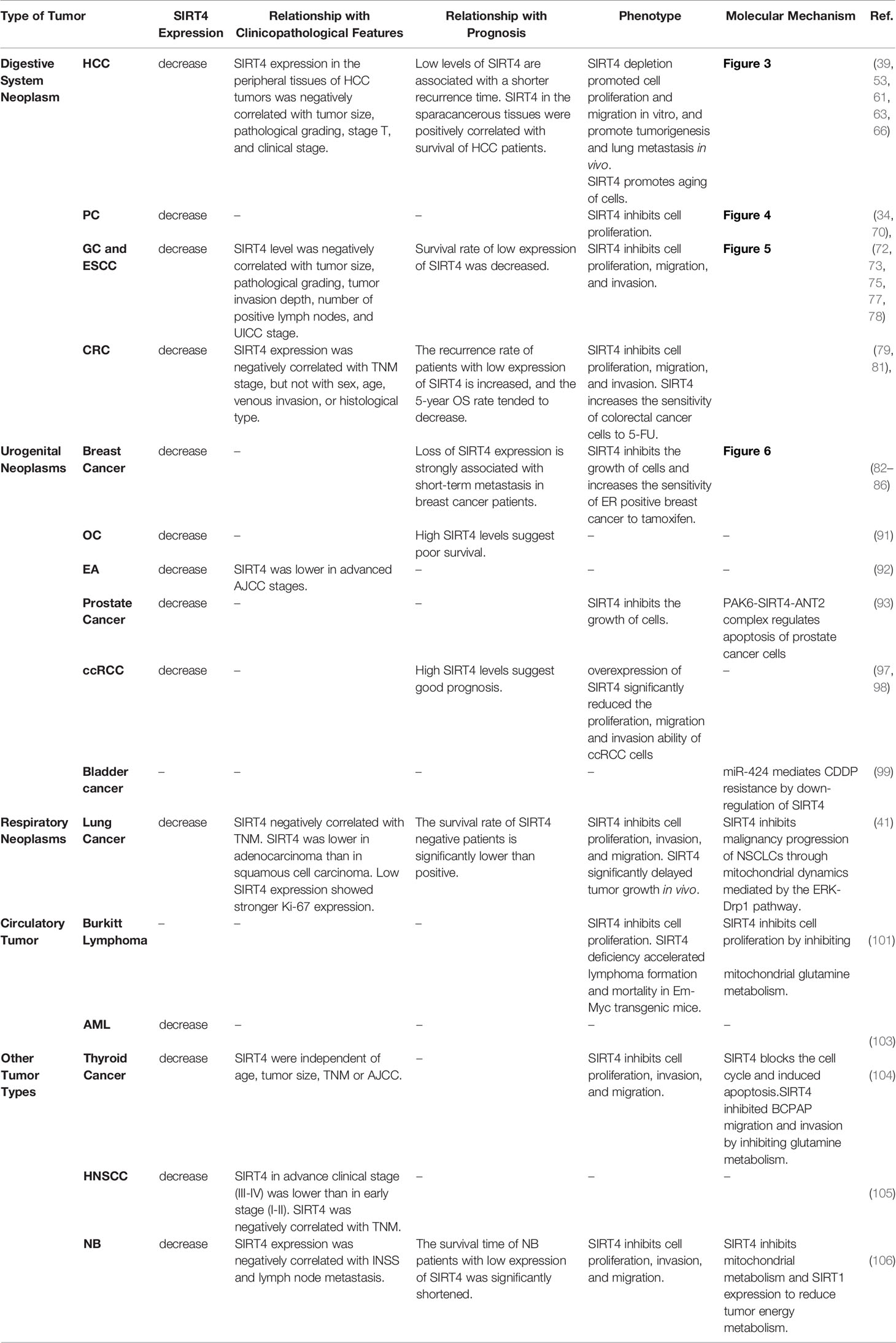
Table 1 SIRT4 in various types of tumor.
Substantial studies have focused on SIRT4 from structure to location, and from enzyme activity to function. Structure determines the localization and enzyme activity, which determines function. SIRT4 with a N-terminal MTS is responsible for localization in the mitochondrial cavity. Recent studies have demonstrated that SIRT4 is also present in the nucleus and cytoplasm, which may involve new crosstalk. SIRT4 has various enzyme activities. SIRT5 has distinct desuccinylase activity, and SIRT1–3 have remarkable deacetylase activity (109). The main enzyme activity of SIRT4 is not known because the identity of its specific substrates is not known.
However, various physiologic functions regulated by SIRT4 have been identified. SIRT4 has important roles in energy metabolism and oxidative stress, metabolic diseases and cancer, physical fitness, health, and ageing (110).
We reviewed mainly the function of SIRT4 in various malignant neoplasms. SIRT4 expression is down-regulated in most cancer types, and loss of SIRT4 increases the risk of spontaneous tumors throughout life. SIRT4 participates in regulation of glutamine metabolism by inhibiting GDH expression, and glucose metabolism by inhibiting PDHC expression. SIRT4 is induced by various genetic toxicants, which can regulate cell-cycle progression and genomic fidelity in response to DNA damage. Moreover, oncogenes, tumor-suppressor genes, cancer stem cells, inflammation, and metabolism are the main foci of cancer research. In particular, “metabolic change” has become a new direction for cancer research in recent years. SIRT4 has an important role in suppressing cancer, so SIRT4 could be used as a new biomarker for cancer diagnosis, and to develop targets for new strategies for cancer treatment. Therefore, it is necessary to develop specific pharmacologic activators and inhibitors of SIRT4 that can be used as tools to understand the biology of SIRT4, and which may also be employed as a novel cancer treatment.
YB conceived and designed the content of this review. YB wrote the manuscript with the help of JY, YC, FW, CL, and XF. YZ and YY contributed to the final version of the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by the National Natural Science Foundation of China (81672428, 81872427, 81703000), the Applied Technology Research and Development Program of Heilongjiang Province (GA19C002), the Nn10 Excellent Discipline Construction Program (Hepatobiliary and Pancreatic Tumor 2017), and the Fundamental Research Funds for the Provincial Universities of Heilongjiang Province.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
I am deeply appreciative of my mentors and friends for guidance and help. Also I thank The Charlesworth Group (www.cwauthors.com/), for providing medical-editing assistance during preparation of this manuscript.
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin (2018) 68(6):394–424. doi: 10.3322/caac.21492
2. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144(5):646–74. doi: 10.1016/j.cell.2011.02.013
3. Yoo HC, Park SJ, Nam M, Kang J, Kim K, Yeo JH, et al. A Variant of SLC1A5 Is a Mitochondrial Glutamine Transporter for Metabolic Reprogramming in Cancer Cells. Cell Metab (2020) 31(2):267–83.e12. doi: 10.1016/j.cmet.2019.11.020
4. Finkel T, Deng CX, Mostoslavsky R. Recent progress in the biology and physiology of sirtuins. Nature (2009) 460(7255):587–91. doi: 10.1038/nature08197
5. de Ruijter AJ, van Gennip AH, Caron HN, Kemp S, van Kuilenburg AB. Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem J (2003) 370(Pt 3):737–49. doi: 10.1042/bj20021321
6. Gregoretti IV, Lee YM, Goodson HV. Molecular evolution of the histone deacetylase family: functional implications of phylogenetic analysis. J Mol Biol (2004) 338(1):17–31. doi: 10.1016/j.jmb.2004.02.006
7. Blander G, Guarente L. The Sir2 family of protein deacetylases. Annu Rev Biochem (2004) 73:417–35. doi: 10.1146/annurev.biochem.73.011303.073651
8. Laurent G, German NJ, Saha AK, de Boer VC, Davies M, Koves TR, et al. SIRT4 coordinates the balance between lipid synthesis and catabolism by repressing malonyl CoA decarboxylase. Mol Cell (2013) 50(5):686–98. doi: 10.1016/j.molcel.2013.05.012
9. Haigis MC, Mostoslavsky R, Haigis KM, Fahie K, Christodoulou DC, Murphy AJ, et al. SIRT4 inhibits glutamate dehydrogenase and opposes the effects of calorie restriction in pancreatic beta cells. Cell (2006) 126(5):941–54. doi: 10.1016/j.cell.2006.06.057
10. Mathias RA, Greco TM, Oberstein A, Budayeva HG, Chakrabarti R, Rowland EA, et al. Sirtuin 4 is a lipoamidase regulating pyruvate dehydrogenase complex activity. Cell (2014) 159(7):1615–25. doi: 10.1016/j.cell.2014.11.046
11. Pannek M, Simic Z, Fuszard M, Meleshin M, Rotili D, Mai A, et al. Crystal structures of the mitochondrial deacylase Sirtuin 4 reveal isoform-specific acyl recognition and regulation features. Nat Commun (2017) 8(1):1513. doi: 10.1038/s41467-017-01701-2
12. Anderson KA, Huynh FK, Fisher-Wellman K, Stuart JD, Peterson BS, Douros JD, et al. SIRT4 Is a Lysine Deacylase that Controls Leucine Metabolism and Insulin Secretion. Cell Metab (2017) 25(4)838–55.e15. doi: 10.1016/j.cmet.2017.03.003
13. Frye RA. Characterization of five human cDNAs with homology to the yeast SIR2 gene: Sir2-like proteins (sirtuins) metabolize NAD and may have protein ADP-ribosyltransferase activity. Biochem Biophys Res Commun (1999) 260(1):273–9. doi: 10.1006/bbrc.1999.0897
14. Michan S, Sinclair D. Sirtuins in mammals: insights into their biological function. Biochem J (2007) 404(1):1–13. doi: 10.1042/BJ20070140
15. Michishita E, Park JY, Burneskis JM, Barrett JC, Horikawa I. Evolutionarily conserved and nonconserved cellular localizations and functions of human SIRT proteins. Mol Biol Cell (2005) 16(10):4623–35. doi: 10.1091/mbc.e05-01-0033
16. Osborne B, Bentley NL, Montgomery MK, Turner N. The role of mitochondrial sirtuins in health and disease. Free Radical Biol Med (2016) 100:164–74. doi: 10.1016/j.freeradbiomed.2016.04.197
17. Komlos D, Mann KD, Zhuo Y, Ricupero CL, Hart RP, Liu AY, et al. Glutamate dehydrogenase 1 and SIRT4 regulate glial development. Glia (2013) 61(3):394–408. doi: 10.1002/glia.22442
18. Ahuja N, Schwer B, Carobbio S, Waltregny D, North BJ, Castronovo V, et al. Regulation of insulin secretion by SIRT4, a mitochondrial ADP-ribosyltransferase. J Biol Chem (2007) 282(46):33583–92. doi: 10.1074/jbc.M705488200
19. Lang A, Anand R, Altinoluk-Hambüchen S, Ezzahoini H, Stefanski A, Iram A, et al. SIRT4 interacts with OPA1 and regulates mitochondrial quality control and mitophagy. Aging (2017) 9(10):2163–89. doi: 10.18632/aging.101307
20. Lombard DB, Tishkoff DX, Bao J. Mitochondrial sirtuins in the regulation of mitochondrial activity and metabolic adaptation. Handb Exp Pharmacol (2011) 206:163–88. doi: 10.1007/978-3-642-21631-2_8
21. Ramadani-Muja J, Gottschalk B, Pfeil K, Burgstaller S, Rauter T, Bischof H, et al. Visualization of Sirtuin 4 Distribution between Mitochondria and the Nucleus, Based on Bimolecular Fluorescence Self-Complementation. Cells (2019) 8(12):1583. doi: 10.3390/cells8121583
22. Khmelinskii A, Keller PJ, Bartosik A, Meurer M, Barry JD, Mardin BR, et al. Tandem fluorescent protein timers for in vivo analysis of protein dynamics. Nat Biotechnol (2012) 30(7):708–14. doi: 10.1038/nbt.2281
23. Pédelacq JD, Cabantous S, Tran T, Terwilliger TC, Waldo GS. Engineering and characterization of a superfolder green fluorescent protein. Nat Biotechnol (2006) 24(1):79–88. doi: 10.1038/nbt1172
24. Cabantous S, Terwilliger TC, Waldo GS. Protein tagging and detection with engineered self-assembling fragments of green fluorescent protein. Nat Biotechnol (2005) 23(1):102–7. doi: 10.1038/nbt1044
25. Ruan L, Zhou C, Jin E, Kucharavy A, Zhang Y, Wen Z, et al. Cytosolic proteostasis through importing of misfolded proteins into mitochondria. Nature (2017) 543(7645):443–6. doi: 10.1038/nature21695
26. Melber A, Haynes CM. UPR(mt) regulation and output: a stress response mediated by mitochondrial-nuclear communication. Cell Res (2018) 28(3):281–95. doi: 10.1038/cr.2018.16
27. Desai R, Campanella M. MitoCPR: Meticulous Monitoring of Mitochondrial Proteostasis. Mol Cell (2018) 71(1):8–9. doi: 10.1016/j.molcel.2018.06.027
28. Kenny TC, Hart P, Ragazzi M, Sersinghe M, Chipuk J, Sagar MAK, et al. Selected mitochondrial DNA landscapes activate the SIRT3 axis of the UPR(mt) to promote metastasis. Oncogene (2017) 36(31):4393–404. doi: 10.1038/onc.2017.52
29. Kenny TC, Manfredi G, Germain D. The Mitochondrial Unfolded Protein Response as a Non-Oncogene Addiction to Support Adaptation to Stress during Transformation in Cancer and Beyond. Front Oncol (2017) 7:159. doi: 10.3389/fonc.2017.00159
30. Bergmann L, Lang A, Bross C, Altinoluk-Hambüchen S, Fey I, Overbeck N, et al. Subcellular Localization and Mitotic Interactome Analyses Identify SIRT4 as a Centrosomally Localized and Microtubule Associated Protein. Cells (2020) 9(9):1950. doi: 10.3390/cells9091950
31. Imai S, Armstrong CM, Kaeberlein M, Guarente L. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature (2000) 403(6771):795–800. doi: 10.1038/35001622
32. Schwer B, North BJ, Frye RA, Ott M, Verdin E. The human silent information regulator (Sir)2 homologue hSIRT3 is a mitochondrial nicotinamide adenine dinucleotide-dependent deacetylase. J Cell Biol (2002) 158(4):647–57. doi: 10.1083/jcb.200205057
33. Rauh D, Fischer F, Gertz M, Lakshminarasimhan M, Bergbrede T, Aladini F, et al. An acetylome peptide microarray reveals specificities and deacetylation substrates for all human sirtuin isoforms. Nat Commun (2013) 4:2327. doi: 10.1038/ncomms3327
34. Lei MZ, Li XX, Zhang Y, Li JT, Zhang F, Wang YP, et al. Acetylation promotes BCAT2 degradation to suppress BCAA catabolism and pancreatic cancer growth. Signal Transduct Targeted Ther (2020) 5(1):70. doi: 10.1038/s41392-020-0168-0
35. Du J, Jiang H, Lin H. Investigating the ADP-ribosyltransferase activity of sirtuins with NAD analogues and 32P-NAD. Biochemistry (2009) 48(13):2878–90. doi: 10.1021/bi802093g
36. Feldman JL, Baeza J, Denu JM. Activation of the protein deacetylase SIRT6 by long-chain fatty acids and widespread deacylation by mammalian sirtuins. J Biol Chem (2013) 288(43):31350–6. doi: 10.1074/jbc.C113.511261
37. Wagner GR, Bhatt DP, O’Connell TM, Thompson JW, Dubois LG, Backos DS, et al. A Class of Reactive Acyl-CoA Species Reveals the Non-enzymatic Origins of Protein Acylation. Cell Metab (2017) 25(4):823–37.e8. doi: 10.1016/j.cmet.2017.03.006
38. Simic Z, Weiwad M, Schierhorn A, Steegborn C, Schutkowski M. The ε-Amino Group of Protein Lysine Residues Is Highly Susceptible to Nonenzymatic Acylation by Several Physiological Acyl-CoA Thioesters. Chembiochem Eur J Chem Biol (2015) 16(16):2337–47. doi: 10.1002/cbic.201500364
39. Wang YS, Du L, Liang X, Meng P, Bi L, Wang YL, et al. Sirtuin 4 Depletion Promotes Hepatocellular Carcinoma Tumorigenesis Through Regulating Adenosine-Monophosphate-Activated Protein Kinase Alpha/Mammalian Target of Rapamycin Axis in Mice. Hepatology (2019) 69(4):1614–31. doi: 10.1002/hep.30421
40. Csibi A, Fendt SM, Li C, Poulogiannis G, Choo AY, Chapski DJ, et al. The mTORC1 pathway stimulates glutamine metabolism and cell proliferation by repressing SIRT4. Cell (2013) 153(4):840–54. doi: 10.1016/j.cell.2013.04.023
41. Fu L, Dong Q, He J, Wang X, Xing J, Wang E, et al. SIRT4 inhibits malignancy progression of NSCLCs, through mitochondrial dynamics mediated by the ERK-Drp1 pathway. Oncogene (2017) 36(19):2724–36. doi: 10.1038/onc.2016.425
42. Jeong SM, Xiao C, Finley LW, Lahusen T, Souza AL, Pierce K, et al. SIRT4 has tumor-suppressive activity and regulates the cellular metabolic response to DNA damage by inhibiting mitochondrial glutamine metabolism. Cancer Cell (2013) 23(4):450–63. doi: 10.1016/j.ccr.2013.02.024
43. Jones RG, Thompson CB. Tumor suppressors and cell metabolism: a recipe for cancer growth. Genes Dev (2009) 23(5):537–48. doi: 10.1101/gad.1756509
44. Wise DR, Thompson CB. Glutamine addiction: a new therapeutic target in cancer. Trends Biochem Sci (2010) 35(8):427–33. doi: 10.1016/j.tibs.2010.05.003
45. Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol Cell (2010) 40(2):179–204. doi: 10.1016/j.molcel.2010.09.019
46. Cosentino C, Grieco D, Costanzo V. ATM activates the pentose phosphate pathway promoting anti-oxidant defence and DNA repair. EMBO J (2011) 30(3):546–55. doi: 10.1038/emboj.2010.330
47. Jin L, Alesi GN, Kang S. Glutaminolysis as a target for cancer therapy. Oncogene (2016) 35(28):3619–25. doi: 10.1038/onc.2015.447
48. Faubert B, Boily G, Izreig S, Griss T, Samborska B, Dong Z, et al. AMPK is a negative regulator of the Warburg effect and suppresses tumor growth in vivo. Cell Metab (2013) 17(1):113–24. doi: 10.1016/j.cmet.2012.12.001
49. Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, et al. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell (2008) 30(2):214–26. doi: 10.1016/j.molcel.2008.03.003
50. Shaw RJ, Kosmatka M, Bardeesy N, Hurley RL, Witters LA, DePinho RA, et al. The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc Natl Acad Sci U States America (2004) 101(10):3329–35. doi: 10.1073/pnas.0308061100
51. Dykens JA, Jamieson J, Marroquin L, Nadanaciva S, Billis PA, Will Y. Biguanide-induced mitochondrial dysfunction yields increased lactate production and cytotoxicity of aerobically-poised HepG2 cells and human hepatocytes in vitro. Toxicol Appl Pharmacol (2008) 233(2):203–10. doi: 10.1016/j.taap.2008.08.013
52. Hawley SA, Ross FA, Chevtzoff C, Green KA, Evans A, Fogarty S, et al. Use of cells expressing gamma subunit variants to identify diverse mechanisms of AMPK activation. Cell Metab (2010) 11(6):554–65. doi: 10.1016/j.cmet.2010.04.001
53. Li Z, Li H, Zhao ZB, Zhu W, Feng PP, Zhu XW, et al. SIRT4 silencing in tumor-associated macrophages promotes HCC development via PPARδ signalling-mediated alternative activation of macrophages. J Exp Clin Cancer Res CR (2019) 38(1):469. doi: 10.1186/s13046-019-1456-9
54. Tao J, Zhang J, Ling Y, McCall CE, Liu TF. Mitochondrial Sirtuin 4 Resolves Immune Tolerance in Monocytes by Rebalancing Glycolysis and Glucose Oxidation Homeostasis. Front Immunol (2018) 9:419. doi: 10.3389/fimmu.2018.00419
55. Saha S, Shalova IN, Biswas SK. Metabolic regulation of macrophage phenotype and function. Immunol Rev (2017) 280(1):102–11. doi: 10.1111/imr.12603
56. Huang SC, Everts B, Ivanova Y, O’Sullivan D, Nascimento M, Smith AM, et al. Cell-intrinsic lysosomal lipolysis is essential for alternative activation of macrophages. Nat Immunol (2014) 15(9):846–55. doi: 10.1038/ni.2956
57. Laurent G, de Boer VC, Finley LW, Sweeney M, Lu H, Schug TT, et al. SIRT4 represses peroxisome proliferator-activated receptor α activity to suppress hepatic fat oxidation. Mol Cell Biol (2013) 33(22):4552–61. doi: 10.1128/mcb.00087-13
58. Hershberger KA, Martin AS, Hirschey MD. Role of NAD(+) and mitochondrial sirtuins in cardiac and renal diseases. Nat Rev Nephrol (2017) 13(4):213–25. doi: 10.1038/nrneph.2017.5
59. Zhang H, Wang X, Shen Z, Xu J, Qin J, Sun Y. Infiltration of diametrically polarized macrophages predicts overall survival of patients with gastric cancer after surgical resection. Gastric Cancer Off J Int Gastric Cancer Assoc Japan Gastric Cancer Assoc (2015) 18(4):740–50. doi: 10.1007/s10120-014-0422-7
60. Ji J, Xue TF, Guo XD, Yang J, Guo RB, Wang J, et al. Antagonizing peroxisome proliferator-activated receptor γ facilitates M1-to-M2 shift of microglia by enhancing autophagy via the LKB1-AMPK signaling pathway. Aging Cell (2018) 17(4):e12774. doi: 10.1111/acel.12774
61. Chen X, Ding X, Wu Q, Qi J, Zhu M, Miao C. Monomethyltransferase SET8 facilitates hepatocellular carcinoma growth by enhancing aerobic glycolysis. Cell Death Dis (2019) 10(4):312. doi: 10.1038/s41419-019-1541-1
62. Milite C, Feoli A, Viviano M, Rescigno D, Cianciulli A, Balzano AL, et al. The emerging role of lysine methyltransferase SETD8 in human diseases. Clin Epigenet (2016) 8:102. doi: 10.1186/s13148-016-0268-4
63. Xia XH, Xiao CJ, Shan H. Facilitation of liver cancer SMCC7721 cell aging by sirtuin 4 via inhibiting JAK2/STAT3 signal pathway. Eur Rev Med Pharmacol Sci (2017) 21(6):1248–53.
64. Gu L, Zhu Y, Lin X, Li Y, Cui K, Prochownik EV, et al. Amplification of Glyceronephosphate O-Acyltransferase and Recruitment of USP30 Stabilize DRP1 to Promote Hepatocarcinogenesis. Cancer Res (2018) 78(20):5808–19. doi: 10.1158/0008-5472.Can-18-0340
65. Fan J, Lin R, Xia S, Chen D, Elf SE, Liu S, et al. Tetrameric Acetyl-CoA Acetyltransferase 1 Is Important for Tumor Growth. Mol Cell (2016) 64(5):859–74. doi: 10.1016/j.molcel.2016.10.014
66. Gu L, Zhu Y, Lin X, Tan X, Lu B, Li Y. Stabilization of FASN by ACAT1-mediated GNPAT acetylation promotes lipid metabolism and hepatocarcinogenesis. Oncogene (2020) 39(11):2437–49. doi: 10.1038/s41388-020-1156-0
67. Menendez JA, Lupu R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat Rev Cancer (2007) 7(10):763–77. doi: 10.1038/nrc2222
68. Che L, Chi W, Qiao Y, Zhang J, Song X, Liu Y, et al. Cholesterol biosynthesis supports the growth of hepatocarcinoma lesions depleted of fatty acid synthase in mice and humans. Gut (2020) 69(1):177–86. doi: 10.1136/gutjnl-2018-317581
69. Kong X, Chen J, Xie W, Brown SM, Cai Y, Wu K, et al. Defining UHRF1 Domains that Support Maintenance of Human Colon Cancer DNA Methylation and Oncogenic Properties. Cancer Cell (2019) 35(4):633–48.e7. doi: 10.1016/j.ccell.2019.03.003
70. Hu Q, Qin Y, Ji S, Xu W, Liu W, Sun Q, et al. UHRF1 promotes aerobic glycolysis and proliferation via suppression of SIRT4 in pancreatic cancer. Cancer Lett (2019) 452:226–36. doi: 10.1016/j.canlet.2019.03.024
71. Abu-Alainin W, Gana T, Liloglou T, Olayanju A, Barrera LN, Ferguson R, et al. UHRF1 regulation of the Keap1-Nrf2 pathway in pancreatic cancer contributes to oncogenesis. J Pathol (2016) 238(3):423–33. doi: 10.1002/path.4665
72. Huang G, Cui F, Yu F, Lu H, Zhang M, Tang H, et al. Sirtuin-4 (SIRT4) is downregulated and associated with some clinicopathological features in gastric adenocarcinoma. BioMed Pharmacother (2015) 72:135–9. doi: 10.1016/j.biopha.2015.04.013
73. Sun H, Huang D, Liu G, Jian F, Zhu J, Zhang L. SIRT4 acts as a tumor suppressor in gastric cancer by inhibiting cell proliferation, migration, and invasion. Onco Targets Ther (2018) 11:3959–68. doi: 10.2147/OTT.S156143
74. Cai X, Zhu H, Li Y. PKCζ, MMP−2 and MMP−9 expression in lung adenocarcinoma and association with a metastatic phenotype. Mol Med Rep (2017) 16(6):8301–6. doi: 10.3892/mmr.2017.7634
75. Hu Y, Lin J, Lin Y, Chen X, Zhu G, Huang G. Overexpression of SIRT4 inhibits the proliferation of gastric cancer cells through cell cycle arrest. Oncol Lett (2019) 17(2):2171–6. doi: 10.3892/ol.2018.9877
76. Chambard JC, Lefloch R, Pouysségur J, Lenormand P. ERK implication in cell cycle regulation. Biochim Biophys Acta (2007) 1773(8):1299–310. doi: 10.1016/j.bbamcr.2006.11.010
77. Nakahara Y, Yamasaki M, Sawada G, Miyazaki Y, Makino T, Takahashi T, et al. Downregulation of SIRT4 Expression Is Associated with Poor Prognosis in Esophageal Squamous Cell Carcinoma. Oncology (2016) 90(6):347–55. doi: 10.1159/000445323
78. Cui Y, Yang J, Bai Y, Zhang Y, Yao Y, Zheng T, et al. miR-424-5p regulates cell proliferation and migration of esophageal squamous cell carcinoma by targeting SIRT4. J Cancer (2020) 11(21):6337–47. doi: 10.7150/jca.50587
79. Miyo M, Yamamoto H, Konno M, Colvin H, Nishida N, Koseki J, et al. Tumour-suppressive function of SIRT4 in human colorectal cancer. Br J Cancer (2015) 113(3):492–9. doi: 10.1038/bjc.2015.226
80. Shen A, Lin W, Chen Y, Liu L, Chen H, Zhuang Q, et al. Pien Tze Huang inhibits metastasis of human colorectal carcinoma cells via modulation of TGF-β1/ZEB/miR-200 signaling network. Int J Oncol (2015) 46(2):685–90. doi: 10.3892/ijo.2014.2772
81. Huang G, Cheng J, Yu F, Liu X, Yuan C, Liu C, et al. Clinical and therapeutic significance of sirtuin-4 expression in colorectal cancer. Oncol Rep (2016) 35(5):2801–10. doi: 10.3892/or.2016.4685
82. Igci M, Kalender ME, Borazan E, Bozgeyik I, Bayraktar R, Bozgeyik E, et al. High-throughput screening of Sirtuin family of genes in breast cancer. Gene (2016) 586(1):123–8. doi: 10.1016/j.gene.2016.04.023
83. Du L, Liu X, Ren Y, Li J, Li P, Jiao Q, et al. Loss of SIRT4 promotes the self-renewal of Breast Cancer Stem Cells. Theranostics (2020) 10(21):9458–76. doi: 10.7150/thno.44688
84. Wang L, Zhou H, Wang Y, Cui G, Di LJ. CtBP maintains cancer cell growth and metabolic homeostasis via regulating SIRT4. Cell Death Dis (2015) 6:e1620. doi: 10.1038/cddis.2014.587
85. Wang L, Li JJ, Guo LY, Li P, Zhao Z, Zhou H, et al. Molecular link between glucose and glutamine consumption in cancer cells mediated by CtBP and SIRT4. Oncogenesis (2018) 7(3):26. doi: 10.1038/s41389-018-0036-8
86. Xing J, Li J, Fu L, Gai J, Guan J, Li Q. SIRT4 enhances the sensitivity of ER-positive breast cancer to tamoxifen by inhibiting the IL-6/STAT3 signal pathway. Cancer Med (2019) 8(16):7086–97. doi: 10.1002/cam4.2557
87. Yi EH, Lee CS, Lee JK, Lee YJ, Shin MK, Cho CH, et al. STAT3-RANTES autocrine signaling is essential for tamoxifen resistance in human breast cancer cells. Mol Cancer Res MCR (2013) 11(1):31–42. doi: 10.1158/1541-7786.Mcr-12-0217
88. Ball DP, Lewis AM, Williams D, Resetca D, Wilson DJ, Gunning PT. Signal transducer and activator of transcription 3 (STAT3) inhibitor, S3I-201, acts as a potent and non-selective alkylating agent. Oncotarget (2016) 7(15):20669–79. doi: 10.18632/oncotarget.7838
89. Cheng R, Liu YJ, Cui JW, Yang M, Liu XL, Li P, et al. Aspirin regulation of c-myc and cyclinD1 proteins to overcome tamoxifen resistance in estrogen receptor-positive breast cancer cells. Oncotarget (2017) 8(18):30252–64. doi: 10.18632/oncotarget.16325
90. Li Z, Cui J, Yu Q, Wu X, Pan A, Li L. Evaluation of CCND1 amplification and CyclinD1 expression: diffuse and strong staining of CyclinD1 could have same predictive roles as CCND1 amplification in ER positive breast cancers. Am J Trans Res (2016) 8(1):142–53.
91. He Q, Chen K, Ye R, Dai N, Guo P, Wang L. Associations of sirtuins with clinicopathological variables and prognosis in human ovarian cancer. Oncol Lett (2020) 19(4):3278–88. doi: 10.3892/ol.2020.11432
92. Chen X, Lai X, Wu C, Tian Q, Lei T, Pan J, et al. Decreased SIRT4 protein levels in endometrioid adenocarcinoma tissues are associated with advanced AJCC stage. Cancer Biomarkers Section A Dis Markers (2017) 19(4):419–24. doi: 10.3233/cbm-160419
93. Li T, Li Y, Liu T, Hu B, Li J, Liu C, et al. Mitochondrial PAK6 inhibits prostate cancer cell apoptosis via the PAK6-SIRT4-ANT2 complex. Theranostics (2020) 10(6):2571–86. doi: 10.7150/thno.42874
94. Hubackova S, Davidova E, Rohlenova K, Stursa J, Werner L, Andera L, et al. Selective elimination of senescent cells by mitochondrial targeting is regulated by ANT2. Cell Death Diff (2019) 26(2):276–90. doi: 10.1038/s41418-018-0118-3
95. Ho L, Titus AS, Banerjee KK, George S, Lin W, Deota S, et al. SIRT4 regulates ATP homeostasis and mediates a retrograde signaling via AMPK. Aging (2013) 5(11):835–49. doi: 10.18632/aging.100616
96. Kumar R, Sanawar R, Li X, Li F. Structure, biochemistry, and biology of PAK kinases. Gene (2017) 605:20–31. doi: 10.1016/j.gene.2016.12.014
97. Tan Y, Li B, Peng F, Gong G, Li N. Integrative Analysis of Sirtuins and Their Prognostic Significance in Clear Cell Renal Cell Carcinoma. Front Oncol (2020) 10:218. doi: 10.3389/fonc.2020.00218
98. Wang C, Piao C, Liu J, Zhang Z, Zhu Y, Kong C. Mammalian SIRT4 is a tumor suppressor of clear cell renal cell carcinoma by inhibiting cancer proliferation, migration and invasion. Cancer Biomarkers Section A Dis Markers (2020) 29(4):453–62. doi: 10.3233/cbm-191253
99. Yu M, Ozaki T, Sun D, Xing H, Wei B, An J, et al. HIF-1α-dependent miR-424 induction confers cisplatin resistance on bladder cancer cells through down-regulation of pro-apoptotic UNC5B and SIRT4. J Exp Clin Cancer Res CR (2020) 39(1):108. doi: 10.1186/s13046-020-01613-y
100. Taguchi N, Ishihara N, Jofuku A, Oka T, Mihara K. Mitotic phosphorylation of dynamin-related GTPase Drp1 participates in mitochondrial fission. J Biol Chem (2007) 282(15):11521–9. doi: 10.1074/jbc.M607279200
101. Jeong SM, Lee A, Lee J, Haigis MC. SIRT4 protein suppresses tumor formation in genetic models of Myc-induced B cell lymphoma. J Biol Chem (2014) 289(7):4135–44. doi: 10.1074/jbc.M113.525949
102. Le A, Lane AN, Hamaker M, Bose S, Gouw A, Barbi J, et al. Glucose-independent glutamine metabolism via TCA cycling for proliferation and survival in B cells. Cell Metab (2012) 15(1):110–21. doi: 10.1016/j.cmet.2011.12.009
103. Bradbury CA, Khanim FL, Hayden R, Bunce CM, White DA, Drayson MT, et al. Histone deacetylases in acute myeloid leukaemia show a distinctive pattern of expression that changes selectively in response to deacetylase inhibitors. Leukemia (2005) 19(10):1751–9. doi: 10.1038/sj.leu.2403910
104. Chen Z, Lin J, Feng S, Chen X, Huang H, Wang C, et al. SIRT4 inhibits the proliferation, migration, and invasion abilities of thyroid cancer cells by inhibiting glutamine metabolism. Onco Targets Ther (2019) 12:2397–408. doi: 10.2147/OTT.S189536
105. Mahjabeen I, Kayani MA. Loss of Mitochondrial Tumor Suppressor Genes Expression Is Associated with Unfavorable Clinical Outcome in Head and Neck Squamous Cell Carcinoma: Data from Retrospective Study. PloS One (2016) 11(1):e0146948. doi: 10.1371/journal.pone.0146948
106. Wang Y, Guo Y, Gao J, Yuan X. Tumor-suppressive function of SIRT4 in neuroblastoma through mitochondrial damage. Cancer Manag Res (2018) 10:5591–603. doi: 10.2147/CMAR.S172509
107. Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM, Puigserver P. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature (2005) 434(7029):113–8. doi: 10.1038/nature03354
108. Dioum EM, Chen R, Alexander MS, Zhang Q, Hogg RT, Gerard RD, et al. Regulation of hypoxia-inducible factor 2alpha signaling by the stress-responsive deacetylase sirtuin 1. Sci (N Y NY) (2009) 324(5932):1289–93. doi: 10.1126/science.1169956
109. Gertz M, Steegborn C. Using mitochondrial sirtuins as drug targets: disease implications and available compounds. Cell Mol Life Sci (2016) 73(15):2871–96. doi: 10.1007/s00018-016-2180-7
Keywords: sirtuin4, cancer, structure, localization, enzymatic activities, metabolism, mitochondria
Citation: Bai Y, Yang J, Cui Y, Yao Y, Wu F, Liu C, Fan X and Zhang Y (2021) Research Progress of Sirtuin4 in Cancer. Front. Oncol. 10:562950. doi: 10.3389/fonc.2020.562950
Received: 08 September 2020; Accepted: 03 November 2020;
Published: 08 January 2021.
Edited by:
Stefano Falone, University of L’Aquila, ItalyReviewed by:
Athanasios Vasilopoulos, Northwestern University, United StatesCopyright © 2021 Bai, Yang, Cui, Yao, Wu, Liu, Fan and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yanqiao Zhang, eWFucWlhb3poYW5nQGVtcy5ocmJtdS5lZHUuY24=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.