
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Oncol. , 22 December 2020
Sec. Cancer Metabolism
Volume 10 - 2020 | https://doi.org/10.3389/fonc.2020.537930
This article is part of the Research Topic Using New Metabolic Approaches to Target and Eradicate Cancer Stem Cells View all 14 articles
 Anna Mukha1,2*†
Anna Mukha1,2*† Anna Dubrovska1,2,3,4*
Anna Dubrovska1,2,3,4*Most human tumors possess a high heterogeneity resulting from both clonal evolution and cell differentiation program. The process of cell differentiation is initiated from a population of cancer stem cells (CSCs), which are enriched in tumor‐regenerating and tumor‐propagating activities and responsible for tumor maintenance and regrowth after treatment. Intrinsic resistance to conventional therapies, as well as a high degree of phenotypic plasticity, makes CSCs hard-to-target tumor cell population. Reprogramming of CSC metabolic pathways plays an essential role in tumor progression and metastatic spread. Many of these pathways confer cell adaptation to the microenvironmental stresses, including a shortage of nutrients and anti-cancer therapies. A better understanding of CSC metabolic dependences as well as metabolic communication between CSCs and the tumor microenvironment are of utmost importance for efficient cancer treatment. In this mini-review, we discuss the general characteristics of CSC metabolism and potential metabolic targeting of CSC populations as a potent strategy to enhance the efficacy of conventional treatment approaches.
According to the world health organization (WHO), cancer is responsible for one in six deaths worldwide, and global cancer rates continue to grow (1, 2). Although the mono-therapy such as surgery, chemotherapy and radiotherapy is a commonly accepted treatment modality for different types of cancers, the combination of two or more types of treatment targeting the key cancer mechanisms in synergistic or additive manners is currently a cornerstone of anticancer therapy especially for advanced and aggressive cancers (3, 4). Recent innovations in treatment technologies as well as in precision of radiation and drug delivery substantially increased efficiency and quality of treatment. However, treatment-related toxicities and tumor therapy resistance still constitute a fundamental clinical and scientific challenge (5–7).
The difficulty of cancer treatment has its roots in the nature of this disease. Tumors are highly heterogeneous, consisting of different types of cells. Intratumoral heterogeneity is evidenced at the multiple levels, including genetic and epigenetic landscapes, histological and molecular specificities as well as functional differences between tumor cells including their abilities to propagate tumor growth and give rise to other types of cancer cells by the process of differentiation (8).
The process of cell differentiation is initiated from a population of cancer stem cells (CSCs) that possess unique properties such as the unlimited capacity of self-renewal and asymmetric division, which leads to the production of different cell types within tumors. These properties of CSCs make them equipped with tumor‐regenerating and tumor‐propagating activities and, therefore, responsible for the tumor maintenance and regrowth after treatment. The density of CSCs substantially varies between individual tumors, and its analysis is proven to have prognostic significance for different types of cancers (9, 10). Several CSC-specific markers have been described, among them the expression of CD133, CD44, CD117 (c-kit), Oct4, high aldehyde dehydrogenase (ALDH) activity, etc. as discussed elsewhere (11–13). However, some of these markers can be found in normal stem cells, which make identification and targeting of CSCs more challenging (14). A high plasticity of CSC populations is an additional obstacle on the way of clinical translation as tumor cells possess the ability of shifting their state from the CSC- to non-CSC populations and vice versa that is regulated by multiple genetic, epigenetic and microenvironmental stimuli (15–18). Although tumor stemness is described as a highly dynamic state, eradication of all CSC populations during tumor treatment is of high clinical importance as remaining CSCs might re-initiate local tumor growth and lead to metastatic dissemination.
Many preclinical and clinical studies suggested that some CSC populations can be equipped with intrinsic and extrinsic mechanisms providing them with high radioresistance and chemoresistance compared to the bulk of tumor cells. This relatively high therapy resistance of CSCs is attributed to the efficient DNA repair, low proliferative rate, protective tumor microenvironment, maintenance of cellular redox homeostasis, and immune escape. Altered metabolism of CSCs substantially contributes to their treatment resistance. A deep understanding of the CSC metabolic features and their molecular background will help to develop novel therapeutic strategies that precisely target CSCs and improve the efficiency of cancer control.
Reprogramming of cellular metabolism plays a crucial role in tumor initiation, progression, resistance to conventional therapy, and immunosuppression. Unique features of tumor metabolism were noticed almost one hundred years ago. At the beginning of the XX century, Otto Warburg and co-workers described aerobic glycolysis, accompanied by excessive production of lactate, as one of the distinct characteristics of tumor cells and tissue slices (19). Since then, many other alterations of biochemical pathways have been described for cancer cells (11, 20, 21). Studying the metabolism of CSCs is a challenging task due to the small size and high plasticity of these cell populations. Nevertheless, current experimental data shows that the metabolic features of CSCs are highly heterogeneous, and tumor type-dependent (Table 1).
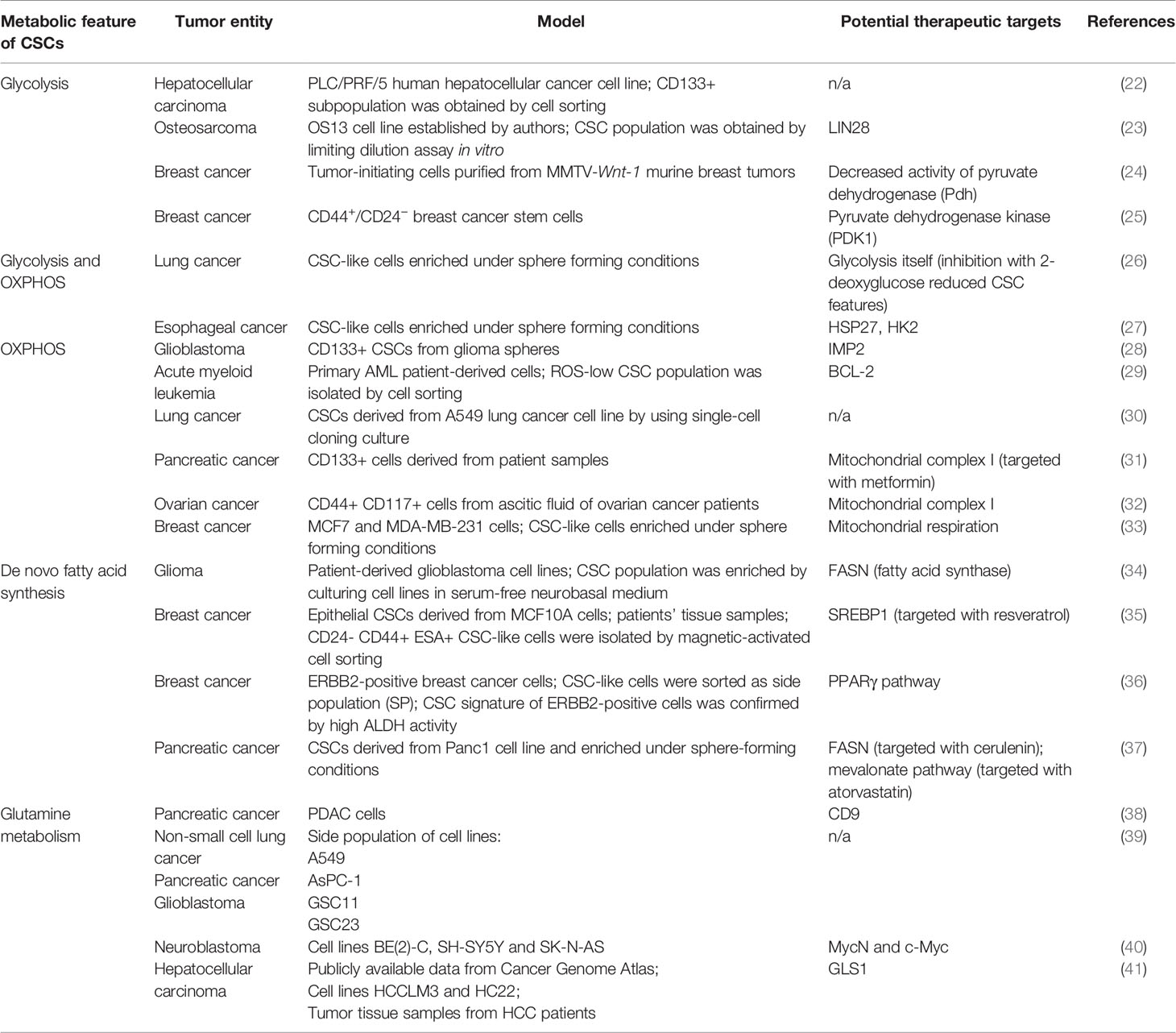
Table 1 Examples of the metabolic features of CSCs described for the different tumor models.
Glycolysis is one of the major and best-studied metabolic characteristics of cancer cells. Fast-growing tissues, such as the most malignant tumors, demand more energy. In differentiated cells, energy in the form of adenosine triphosphate (ATP) is produced via oxidative phosphorylation (OXPHOS) that occurs in mitochondria. Complete oxidation of glucose molecule leads to the production of about 30 molecules of ATP, whereas about 26 out of these 30 ATP molecules are generated by OXPHOS (42). Fast-proliferating cancer cells switch from OXPHOS to glycolysis that requires the consumption of a high amount of glucose since only two molecules of ATP per one molecule of consumed glucose can be produced via this pathway. Lactate, a byproduct of aerobic glycolysis, is shuttled to the extracellular space and was shown to support stemness by upregulation of the expression of genes related to stem cell properties, such as transcription factor SP1, sterol regulatory element-binding protein 1 (SREBP1) which is a transcriptional activator required for regulation of lipid homeostasis, etc., to increase aggressiveness and invasive properties of cancer cells as well as to promote immunosuppression (43–48). Glycolytic CSCs were described for several tumor entities. Song et al. showed that CD133+ liver carcinoma cells had enhanced glycolysis (22). Osteosarcoma-initiating cells also showed a highly glycolytic phenotype (49). Breast CSCs demonstrated the upregulated glycolysis and simultaneously decreased OXPHOS (24). Heterogeneous results were showed for glioblastoma stem cells: Zhou et al. described highly glycolytic glioblastoma cells which were enriched for CSC populations by cell growth conditions (50) while Janiszewska et al. showed the importance of OXPHOS for CD133+ glioblastoma CSCs (28). OXPHOS, as the primary energy production pathway was also shown for leukemic (29), pancreatic (51) and ovarian (32) CSCs.
Many cancer cells demonstrate altered amino acid metabolism. For the majority of cancer cells, glutamine—usually a non-essential amino acid—becomes critically essential as they consume high amounts of it to cover their biosynthetic and energetic needs (52). The rewiring of glutamine metabolism in tumor cells is associated with specific genetic alterations including mitochondrial DNA (mtDNA) mutations (53), oncogenic KRAS (54, 55) and c-Myc overexpression (56). Glutamine enters cells via specific transporters (most of them belong to the alanine/serine/cysteine transporter (ASCT) family) and is used in various biochemical pathways. Bi-directional transporters of amino acids export glutamine in exchange for other amino acids (for example, cysteine). In the cytoplasm, glutamine is converted into glutamate and, subsequently, α-ketoglutarate (α-KG). Glutamate is a building block of glutathione—one of the main scavenges of reactive oxygen species (ROS), which protects the cells from oxidative injury and lethal DNA damage (57, 58). In glutamine metabolism, α-KG is an essential intermediate fueling tricarboxylic acid (TCA) cycle in mitochondria. Metabolites of the TCA cycle are, in turn, used for various other pathways, for example, nucleotide and fatty acid biosynthesis. Moreover, α-KG is a co-factor of the ten-eleven translocation (TET) family DNA demethylases and Jumonji-C (JMJ-C) family histone demethylases—enzymes that play a role in epigenetic regulation of gene transcription. Some pieces of evidence suggest that elevated α-KG to succinate ratio is a marker of stemness (59).
Another critical metabolic characteristic of cancer cells is their lipid metabolism. De novo lipid biosynthesis, enhanced lipid oxidation, and increased storage of lipids are unique characteristics of many cancers. For some of them, such as prostate cancer, lipid content was proposed as a potential biomarker, since the accumulation of lipids in prostate tissue of mice correlated with tumor stage (60). Increased lipid droplet content was shown for colorectal CSCs (61).
De novo lipid biosynthesis and fatty acid oxidation are among the most targetable features of CSCs (62, 63). CSCs from glioma (34) and pancreatic cancer (37) demonstrated upregulated lipogenesis; interesting that pancreatic CSCs fuelled their lipogenesis via enhanced glycolysis. Fatty acid synthase (FASN) is the critical enzyme in de novo lipid synthesis. Its expression is upregulated in many cancers, including lung, colon, breast, and ovarian cancer (64–67). SREBP-2, a transcription factor associated with de novo lipid synthesis, was shown to activate transcription of c-Myc in prostate cancer, therefore contributing to the increase of CSC properties (68). Increased fatty acid oxidation is critical for maintaining the stemness of breast cancer (69, 70) and leukemic cells (71).
Interaction of tumor microenvironment with cancer stem cells can support the survival and phenotype of CSCs. The tumor microenvironment consists of cancer-associated fibroblasts, endothelial cells, immune cells, extracellular matrix. Several factors are critically important for the sustaining of CSC metabolism, and hypoxia is one of them. Hypoxia is one of the major hallmarks of tumor microenvironment playing a critical role in CSC maintenance, quiescence, and therapy resistance (72). Hypoxia can affect CSCs in different ways, including activation of the hypoxia-inducible factor (HIF) mediated signaling that controls the tumorigenicity of CSCs (73). HIF-mediated signaling can interfere with the metabolism of cancer cells by upregulation of many glycolysis-associated genes, including glucose transporters from GLUT family (74). Pharmacological inhibition of GLUT-1 was shown to decrease the self-renewal properties of CSCs in vitro (75). Acidic microenvironment associated with hypoxic tumor areas is shown to promote CSC features by activation of the HIF-dependent transcription program (76). Interesting that cervical cancer cells located in hypoxic areas can produce lactate that is scavenged by cancer cells of oxygenated regions, fueling their proliferation (77). Cancer-associated fibroblasts (CAFs) can support the metabolic needs of cancer cells by feeding them via production of alanine (78), lactate, fatty acids or ketone bodies (79). CSCs from certain cancers (e.g., hepatocellular carcinoma and breast cancer) can promote angiogenesis and, therefore, increase nutrient supply, by releasing pro-angiogenic factors (such as VEGF) (80, 81). Tumor-associated immune cells contribute to the cancer progression and survival of CSCs via different mechanisms. Thus, cancer-associated macrophages can secrete various cytokines (e.g., TGFβ, IL-6) that induce the conversion of cancer cells to cells with CSC phenotype and contribute to chronic inflammation in tumor region (82, 83). Lactate produced by cancer cells in the hypoxic environment is known to induce conversion of tumor-associated macrophages into their pro-tumorigenic phenotype (84, 85). To survive under nutrient shortage conditions, CSCs may activate autophagy, the process of recycling their own nutrients by degrading organelles and large molecules. Enhanced autophagy as a pro-survival and pro-tumorigenic mechanism was demonstrated for breast (86), liver (87), osteosarcoma (88), and ovarian CSCs (89). Many of the above-described metabolic pathways confer CSC adaptation to the microenvironmental stresses, including a shortage of nutrients and anti-cancer therapies. These pathways are attractive targets for the eradication of CSC populations and better treatment outcomes (Figure 1, Table 2).
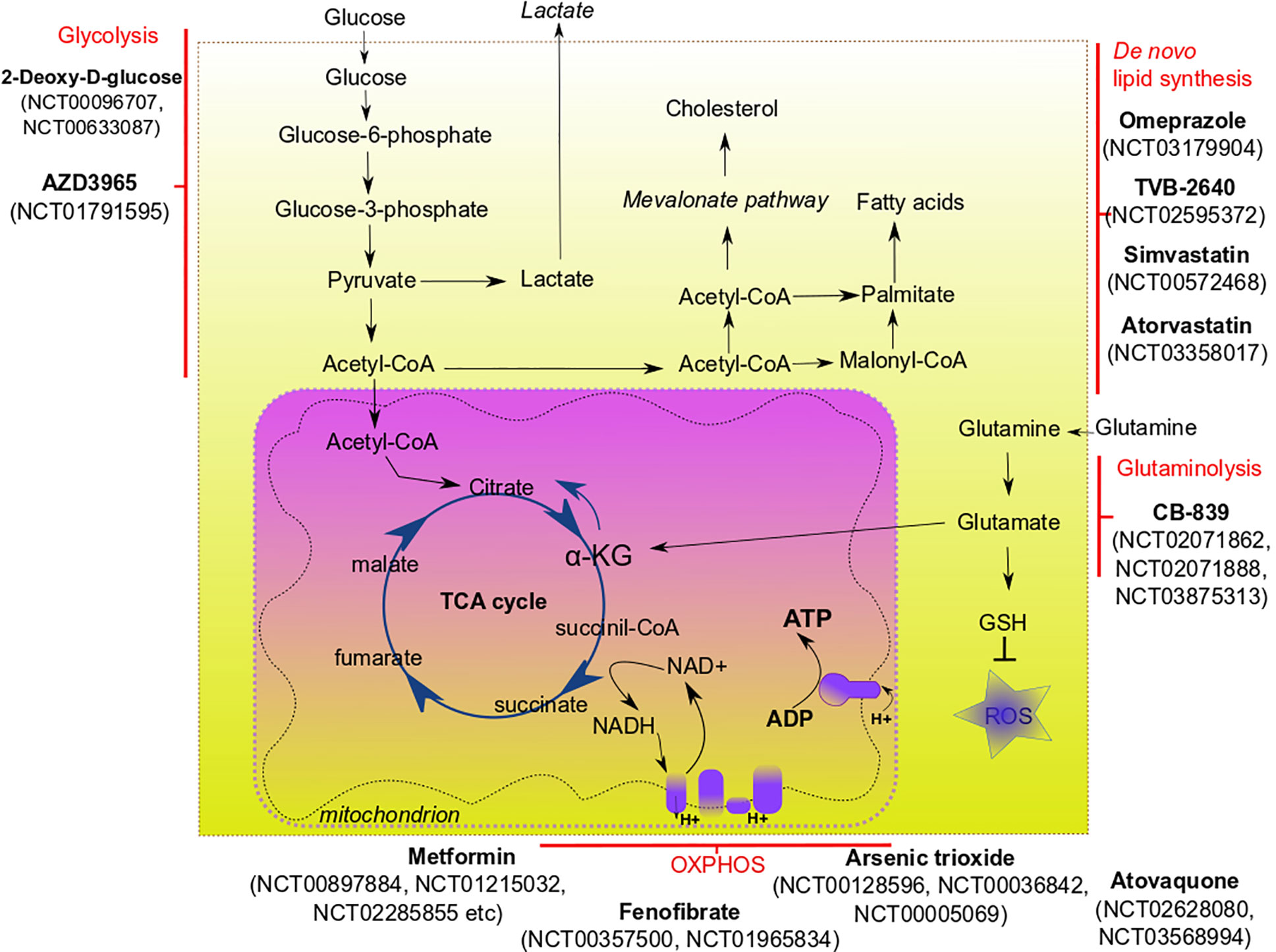
Figure 1 Main metabolic pathways of CSCs and their potential targeting in clinical trials.

Table 2 Compounds for metabolic targeting of cancer stem cells.
The most straightforward approach to inhibit glycolysis is to starve tumors for glucose. The effect on patients can be achieved by subjecting them to a ketogenic diet, containing low amounts of carbohydrates and balanced amounts of proteins and fat. Ketogenic diet-mimicking treatment in vitro effectively reduced CSC-signature in glioma cells (109). Experimental evidence showing the benefit of a ketogenic diet for cancer patients, especially those with glioblastoma and pancreatic cancer, prompted to investigate the potency of this approach as adjuvant therapy for these types of malignancy. However, current clinical data demonstrates mixed results (110). Although the ketogenic diet is usually well-tolerated, compliance with its strict regimes is generally challenging for patients; therefore, it is not considered as monotherapy, and even its usage as adjuvant therapy is discussable (111).
Compound-mediated targeting of glycolysis demonstrated better results in many preclinical studies. Metformin—an antidiabetic drug—has drawn recent attention in cancer research due to its ability to inhibit various molecular pathways leading to the elimination of cancer cells (112). Metformin attenuates glycolysis in a variety of tumor entities. Interesting that metformin can either downregulate glycolytic flux in hepatocellular carcinoma cells (90) or increase glycolysis in breast cancer cells (113). Moreover, it can also inhibit mitochondrial complex I, therefore impairing OXPHOS (114). Altering cancer cell respiration by metformin treatment led to a significant improvement in radiotherapy response in tumor xenograft models of prostate and colon cancer (91). Epigallocatechin gallate (EGCG) was tested as an inhibitor of glycolysis together with conventional chemotherapeutic drugs, and shown as a potent enhancer of chemotherapy (94). A synthetic analog of glucose, 2-deoxy-D-glucose, was tested in vitro and showed the ability to inhibit glycolysis and decrease the CSC phenotype of triple-negative breast cancer cells (93). Experiments on colon cancer cells demonstrated that a combination of 2-deoxyglucose with biguanides (such as 3-bromopyruvate) substantially reduced their proliferation (92). Deoxyglucose is now evaluated in clinical trials as a treatment agent for different cancers, such as lung, breast, and pancreatic cancer (clinicaltrials.gov numbers NCT00096707, NCT00633087).
OXPHOS is another promising metabolic target for CSCs. To date, many compounds have been designed to precisely target OXPHOS. Each compound targets a specific protein element of the electron transport chain blocking the transport of electrons and production of ATP. Most compounds that have shown their efficacy in vitro, in vivo, and in clinical trials, are directed towards mitochondrial complex I (115). The list of these compounds includes, but is not limited to metformin, phenofibrate, pyrvinium, rosiglitazone, pioglitazone, etc. Molecular mechanisms and efficacy of many OXPHOS-targeting compounds are described in reviews by Ashton and co-authors (115) and Sica et al. (116). Such OXPHOS-targeting compounds as atovaquone (clinicaltrials.gov No NCT02628080, NCT03568994), phenformin (NCT03026517) and arsenic trioxide (NCT00128596, NCT00036842, NCT00005069) are now under clinical trials for various solid tumors and leukemias. A combination of OXPHOS inhibition with other treatment modalities (particularly, radiotherapy) shows promising results in vitro and in vivo (117).
As an essential amino acid for most cancer cells, glutamine represents an attractive anticancer target: depriving cells for glutamine seems to be an effective therapeutic option. However, in reality, targeting glutamine metabolism is a challenging task. Systemic approaches to direct glutamine deprivation may be inefficient as glutamine can be synthesized de novo by non-cancerous tissues, such as muscles (118). Other amino acids, such as asparagine and arginine, may also contribute to cancer cell survival under gluatamine deprivation conditions (119, 120). Moreover, some components of tumor microenvironment (e.g. cancer-associated fibroblasts) are able to supplement cancer cells with de novo synthesized glutamine, supporting their proliferation (121).
Glutamine metabolism can be precisely targeted via blocking critical steps of glutamine utilization. One of the most potent targets is glutaminase 1 (GLS1)—the enzyme that converts glutamine to glutamate. Numerous in vitro studies showed that GLS1 was associated with cancer progression, metastasis and CSCs for hepatocellular carcinoma (41), triple-negative breast cancer (122) and pancreatic cancer (123). Inhibition of GLS1 disrupts redox balance in CSCs and can sensitize them to other types of therapy (e.g., radiotherapy) (97, 123). Several inhibitors of GLS1 have been developed, among them BPTES (124) and CB-839. After showing high efficacy in vitro (125) and in vivo, CB-839 entered clinical trials. Currently, CB-839 is tested in Phase I and II clinical trials alone or in combination with other chemotherapeutic drugs for such malignancies as leukemia, breast cancer, colorectal cancer, and lung cancer (NCT02071862, NCT02071888, NCT03875313).
As discussed above, the metabolism of fatty acids is substantially altered in many cancers. Cancer cells can be deprived of exogenous fatty acids or precursors for de novo fatty acid synthesis (such as glucose), which may be a promising strategy to slow tumor growth. Indeed, de novo fatty acid synthesis, which occurs in CSCs, but not healthy cells, seems to be one of the most promising targetable processes to eliminate the CSC population. Fatty acid synthase (FASN) is a target that received the most attention among all enzymes involved in the lipid metabolism of CSCs. Overexpression of FASN has been shown for a number of cancers, such as lung, prostate, ovarian and colon (66, 67, 126, 127). Inhibitors of FASN have pleiotropic effects on tumor cells, mostly because of the different pathways they can target. Cerulenin, a classical inhibitor of FASN, demonstrated high efficacy in reducing stem cell markers in glioblastoma and colon cells in vitro (34, 104). Chemical modifications of cerulenin, such as C75, were developed as the more stable analog of this drug, and C75 showed good results in inhibiting breast cancer cell proliferation (105). Such inhibitors of FASN as omeprazole and TVB-2640 are now evaluated in clinical trials for the treatment of breast cancer (NCT03179904, NCT02595372).
Not only FASN can be inhibited to target de novo lipid synthesis in cancer cells. Sterol regulatory element-binding proteins (SREBPs) are essential components of de novo lipid synthesis. A few compounds have been synthesized to target their functions. One of the most potent ones is fatostatin (128). It had a remarkable anti-tumor activity for prostate cancer; however, experiments with breast cancer cells showed mixed results, as fatostatin induced accumulation of both pro- and antiapoptotic lipids (108).
Altered tumor metabolism is of utmost clinical importance as it mediates tumor resistance toward conventional anticancer agents, and metabolic co-targeting emerges as a novel, highly promising concept to enhance the efficacy of conventional treatment approaches. Metabolic inhibition of tumor growth by targeting CSCs is of specific interest as these cell populations are responsible for tumor maintenance and regrowth after treatment. Limitations of the current CSC assays and lack of the experimental models representing complex tumor microenvironments are a severe challenge to the development of the metabolic CSC-targeting approaches and their clinical translation. Many pitfalls also arise from the intratumoral heterogeneity of CSC metabolic features as well as the high plasticity of CSC nutritional demand during tumor progression and treatment. Future studies on heterogeneous CSC metabolic states at the level of single-cell resolution and employment advanced computational approaches to merge multi-omics data might yield clues for the development of novel metabolic targeting approaches and their implementation in current treatment regimens.
AM and AD contributed to the conception and design of the figure, tables, and manuscript. AM and AD wrote and edited the manuscript. All authors contributed to the article and approved the submitted version.
Work in AD lab is partially supported by grants from Deutsche Forschungsgemeinschaft (DFG) (273676790, 401326337 SPP 2084: µBONE, and 416001651), from Wilhelm Sander-Stiftung (2017.106.1), BMBF (Grant-No. 03Z1NN11) and DLR Project Management Agency (01DK17047). This work was in part supported by Sächsischen Landesstipendiums for AM (Tyutyunnykova).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Ferlay J, Colombet M, Soerjomataram I, Mathers C, Parkin DM, Piñeros M, et al. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int J Cancer (2019) 144:1941–53. doi: 10.1002/ijc.31937
2. Lin L, Yan L, Liu Y, Yuan F, Li H, Ni J. Incidence and death in 29 cancer groups in 2017 and trend analysis from 1990 to 2017 from the Global Burden of Disease Study. J Hematol Oncol (2019) 12(1):96. doi: 10.1186/s13045-019-0783-9
3. Mokhtari RB, Homayouni TS, Baluch N, Morgatskaya E, Kumar S, Das B, et al. Combination therapy in combating cancer. Oncotarget (2017) 8:38022–43. doi: 10.18632/oncotarget.16723
4. Palmer AC, Sorger PK. Combination Cancer Therapy Can Confer Benefit via Patient-to-Patient Variability without Drug Additivity or Synergy. Cell (2017) 171(7):1678–91.e13. doi: 10.1016/j.cell.2017.11.009
5. Bristow RG, Alexander B, Baumann M, Bratman SV, Brown JM, Camphausen K, et al. Combining precision radiotherapy with molecular targeting and immunomodulatory agents: a guideline by the American Society for Radiation Oncology. Lancet Oncol (2018) 19:e240–51. doi: 10.1016/S1470-2045(18)30096-2
6. Hwang WL, Pike LRG, Royce TJ, Mahal BA, Loeffler JS. Safety of combining radiotherapy with immune-checkpoint inhibition. Nat Rev Clin Oncol (2018) 15:477–94. doi: 10.1038/s41571-018-0046-7
7. Cleeland CS, Allen JD, Roberts SA, Brell JM, Giralt SA, Khakoo AY, et al. Reducing the toxicity of cancer therapy: Recognizing needs, taking action. Nat Rev Clin Oncol (2012) 9:471–8. doi: 10.1038/nrclinonc.2012.99
8. Gerdes MJ, Sood A, Sevinsky C, Pris AD, Zavodszky MI, Ginty F. Emerging understanding of multiscale tumor heterogeneity. Front Oncol (2014) 4:1–12. doi: 10.3389/fonc.2014.00366
9. Krause M, Dubrovska A, Linge A, Baumann M. Cancer stem cells: Radioresistance, prediction of radiotherapy outcome and specific targets for combined treatments. Adv Drug Delivery Rev (2017) 109:63–73. doi: 10.1016/j.addr.2016.02.002
10. Bütof R, Dubrovska A, Baumann M. Clinical perspectives of cancer stem cell research in radiation oncology. Radiother Oncol (2013) 108:388–96. doi: 10.1016/j.radonc.2013.06.002
11. Bezuidenhout N, Shoshan M. A shifty target: Tumor-initiating cells and their metabolism. Int J Mol Sci (2019) 20:1–19. doi: 10.3390/ijms20215370
12. Cojoc M, Peitzsch C, Kurth I, Trautmann F, Kunz-Schughart LA, Telegeev GD, et al. Aldehyde Dehydrogenase Is Regulated by β-Catenin/TCF and Promotes Radioresistance in Prostate Cancer Progenitor Cells. Cancer Res (2015) 75(7):1482–94. doi: 10.1158/0008-5472.CAN-14-1924
13. Peitzsch C, Tyutyunnykova A, Pantel K, Dubrovska A. Cancer stem cells: The root of tumor recurrence and metastases. Semin Cancer Biol (2017) 44:10–24. doi: 10.1016/j.semcancer.2017.02.011
14. Kim W-T, Ryu CJ. Cancer stem cell surface markers on normal stem cells. BMB Rep (2017) 50(6):285–98. doi: 10.5483/BMBRep.2017.50.6.039
15. Linge A, Lock S, Gudziol V, Nowak A, Lohaus F, Von Neubeck C, et al. Low cancer stem cell marker expression and low hypoxia identify good prognosis subgroups in HPV(-) HNSCC after postoperative radiochemotherapy: A multicenter study of the DKTK-ROG. Clin Cancer Res (2016) 22(11):2639–49. doi: 10.1158/1078-0432.CCR-15-1990
16. Prasetyanti PR, Medema JP. Intra-tumor heterogeneity from a cancer stem cell perspective. Mol Cancer (2017) 16:1–9. doi: 10.1186/s12943-017-0600-4
17. Wang KJ, Wang C, Dai LH, Yang J, Huang H, Ma XJ, et al. Targeting an autocrine regulatory loop in cancer stem-like cells impairs the progression and chemotherapy resistance of bladder cancer. Clin Cancer Res (2019) 25(3):1070–86. doi: 10.1158/1078-0432.CCR-18-0586
18. Kreso A, Dick JE. Evolution of the cancer stem cell model. Cell Stem Cell (2014) 14:275–91. doi: 10.1016/j.stem.2014.02.006
19. Warburg O. On the origin of cancer cells. Sci (80- ) (1956) 123(3191):309–14. doi: 10.1126/science.123.3191.309
20. Intlekofer AM, Finley LWS. Metabolic signatures of cancer cells and stem cells. Nat Metab (2019) 1(2):177–88. doi: 10.1038/s42255-019-0032-0
21. DeBerardinis RJ, Chandel NS. Fundamentals of cancer metabolism. Sci Adv (2016) 2(5):e1600200. doi: 10.1126/sciadv.1600200
22. Song K, Kwon H, Han C, Zhang J, Dash S, Lim K, et al. Active glycolytic metabolism in CD133(+) hepatocellular cancer stem cells: Regulation by MIR-122. Oncotarget (2015) 6(38):40822–35. doi: 10.18632/oncotarget.5812
23. Mizushima E, Tsukahara T, Emori M, Murata K, Akamatsu A, Shibayama Y, et al. Osteosarcoma-initiating cells show high aerobic glycolysis and attenuation of oxidative phosphorylation mediated by LIN28B. Cancer Sci.
24. Feng W, Gentles A, Nair RV, Huang M, Lin Y, Lee CY, et al. Targeting unique metabolic properties of breast tumor initiating cells. Stem Cells (2014) 32(7):1734–45. doi: 10.1002/stem.1662
25. Peng F, Wang JH, Fan WJ, Meng YT, Li MM, Li TT, et al. Glycolysis gatekeeper PDK1 reprograms breast cancer stem cells under hypoxia. Oncogene (2018) 37(8):1062–74. doi: 10.1038/onc.2017.368
26. Hsu HS, Liu CC, Lin JH, Hsu TW, Hsu JW, Li AFY, et al. Involvement of collagen XVII in pluripotency gene expression and metabolic reprogramming of lung cancer stem cells. J BioMed Sci (2020) 27(1):115–20. doi: 10.1186/s12929-019-0593-y
27. Liu C, Chou K, Hsu J, Lin J, Hsu T, Yen DH, et al. High metabolic rate and stem cell characteristics of esophageal cancer stem-like cells depend on the Hsp27–AKT–HK2 pathway. Int J Cancer (2019) 145(8):2144–56. doi: 10.1002/ijc.32301
28. Janiszewska M, Suvà ML, Riggi N, Houtkooper RH, Auwerx J, Clément-Schatlo V, et al. Imp2 controls oxidative phosphorylation and is crucial for preservin glioblastoma cancer stem cells. Genes Dev (2012) 26(17):1926–44. doi: 10.1101/gad.188292.112
29. Lagadinou ED, Sach A, Callahan K, Rossi RM, Neering SJ, Minhajuddin M, et al. BCL-2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells. Cell Stem Cell (2013) 12(3):329–41. doi: 10.1016/j.stem.2012.12.013
30. Ye X-Q, Li Q, Wang G-H, Sun F-F, Huang G-J, Bian X-W, et al. Mitochondrial and energy metabolism-related properties as novel indicators of lung cancer stem cells. Int J Cancer (2011) 129(4):820–31. doi: 10.1002/ijc.25944
31. Sancho P, Burgos-Ramos E, Tavera A, Bou Kheir T, Jagust P, Schoenhals M, et al. MYC/PGC-1α balance determines the metabolic phenotype and plasticity of pancreatic cancer stem cells. Cell Metab (2015) 22(4):590–605. doi: 10.1016/j.cmet.2015.08.015
32. Pastò A, Bellio C, Pilotto G, Ciminale V, Silic-Benussi M, Guzzo G, et al. Cancer stem cells from epithelial ovarian cancer patients privilege oxidative phosphorylation, and resist glucose deprivation. Oncotarget (2014) 5(12):4305–19. doi: 10.18632/oncotarget.2010
33. De Francesco EM, Ózsvári B, Sotgia F, Lisanti MP. Dodecyl-TPP Targets Mitochondria and Potently Eradicates Cancer Stem Cells (CSCs): Synergy With FDA-Approved Drugs and Natural Compounds (Vitamin C and Berberine). Front Oncol (2019) 9::615. doi: 10.3389/fonc.2019.00615/full
34. Yasumoto Y, Miyazaki H, Vaidyan LK, Kagawa Y, Ebrahimi M, Yamamoto Y, et al. Inhibition of fatty acid synthase decreases expression of stemness markers in glioma stem cells. PloS One (2016) 11(1):329–41. doi: 10.1371/journal.pone.0147717
35. Pandey PR, Xing F, Sharma S, Watabe M, Pai SK, Iiizumi-Gairani M, et al. Elevated lipogenesis in epithelial stem-like cell confers survival advantage in ductal carcinoma in situ of breast cancer. Oncogene (2013) 32(42):5111–22. doi: 10.1038/onc.2012.519
36. Wang X, Sun Y, Wong J, Conklin DS. PPARγ maintains ERBB2-positive breast cancer stem cells. Oncogene (2013) 32(49):5512–21. doi: 10.1038/onc.2013.217
37. Brandi J, Dando I, Pozza ED, Biondani G, Jenkins R, Elliott V, et al. Proteomic analysis of pancreatic cancer stem cells: Functional role of fatty acid synthesis and mevalonate pathways. J Proteomics (2017) 150:310–22. doi: 10.1016/j.jprot.2016.10.002
38. Wang VMY, Ferreira RMM, Almagro J, Evan T, Legrave N, Zaw Thin M, et al. CD9 identifies pancreatic cancer stem cells and modulates glutamine metabolism to fuel tumour growth. Nat Cell Biol (2019) 21(11):1425–35. doi: 10.1038/s41556-019-0407-1
39. Liao J, Liu P-P, Hou G, Shao J, Yang J, Liu K, et al. Regulation of stem-like cancer cells by glutamine through β-catenin pathway mediated by redox signaling. Mol Cancer (2017) 16(1):51. doi: 10.1186/s12943-017-0623-x
40. Le Grand M, Mukha A, Püschel J, Valli E, Kamili A, Vittorio O, et al. Interplay between MycN and c-Myc regulates radioresistance and cancer stem cell phenotype in neuroblastoma upon glutamine deprivation. Theranostics (2020) 10(14):6411–29. doi: 10.7150/thno.42602
41. Li B, Cao Y, Meng G, Qian L, Xu T, Yan C, et al. Targeting glutaminase 1 attenuates stemness properties in hepatocellular carcinoma by increasing reactive oxygen species and suppressing Wnt/beta-catenin pathway. EBioMedicine (2019) 39:239–54. doi: 10.1016/j.ebiom.2018.11.063
42. Koopman WJH, Distelmaier F, Smeitink JA, Willems PH. OXPHOS mutations and neurodegeneration. EMBO J (2012) 32(1):9–29. doi: 10.1038/emboj.2012.300
43. Estrella V, Chen T, Lloyd M, Wojtkowiak J, Cornnell HH, Ibrahim-Hashim A, et al. Acidity generated by the tumor microenvironment drives local invasion. Cancer Res (2013) 73(5):1524–35. doi: 10.1158/0008-5472.CAN-12-2796
44. Martinez-Outschoorn UE, Prisco M, Ertel A, Tsirigos A, Lin Z, Pavlides S, et al. Ketones and lactate increase cancer cell “stemness”, driving recurrence, metastasis and poor clinical outcome in breast cancer: Achieving personalized medicine via metabolo-genomics. Cell Cycle (2011) 10(8):1271–86. doi: 10.4161/cc.10.8.15330
45. Lin S, Sun L, Lyu X, Ai X, Du D, Su N, et al. Lactate-activated macrophages induced aerobic glycolysis and epithelial-mesenchymal transition in breast cancer by regulation of CCL5-CCR5 axis: A positive metabolic feedback loop. Oncotarget (2017) 8(66):110426–43. doi: 10.18632/oncotarget.22786
46. Tasdogan A, Faubert B, Ramesh V, Ubellacker JM, Shen B, Solmonson A, et al. Metabolic heterogeneity confers differences in melanoma metastatic potential. Nature (2020) 577(7788):115–20. doi: 10.1038/s41586-019-1847-2
47. Brand A, Singer K, Koehl GE, Kolitzus M, Schoenhammer G, Thiel A, et al. LDHA-Associated Lactic Acid Production Blunts Tumor Immunosurveillance by T and NK Cells. Cell Metab (2016) 24(5):657–71. doi: 10.1016/j.cmet.2016.08.011
48. Daneshmandi S, Wegiel B, Seth P. Blockade of lactate dehydrogenase-A (LDH-A) improves efficacy of anti-programmed cell death-1 (PD-1) therapy in melanoma. Cancers (Basel) (2019) 11(4):2367–78. doi: 10.3390/cancers11040450
49. Mizushima E, Tsukahara T, Emori M, Murata K, Akamatsu A, Shibayama Y, et al. Osteosarcoma-initiating cells show high aerobic glycolysis and attenuation of oxidative phosphorylation mediated by LIN28B. Cancer Sci (2020) 111(1):36–46. doi: 10.1111/cas.14229
50. Zhou Y, Zhou Y, Shingu T, Feng L, Chen Z, Ogasawara M, et al. Metabolic alterations in highly tumorigenic glioblastoma cells: Preference for hypoxia and high dependency on glycolysis. J Biol Chem (2011) 286(37):32843–53. doi: 10.1074/jbc.M111.260935
51. Sancho P, Burgos-Ramos E, Tavera A, Bou Kheir T, Jagust P, Schoenhals M, et al. MYC/PGC-1α Balance Determines the Metabolic Phenotype and Plasticity of Pancreatic Cancer Stem Cells. Cell Metab (2015) 22(4):590–605. doi: 10.1016/j.cmet.2015.08.015
52. Choi Y-K, Park K-G. Targeting Glutamine Metabolism for Cancer Treatment. Biomol Ther (Seoul) (2018) 26(1):19–28. doi: 10.4062/biomolther.2017.178
53. Chen Q, Kirk K, Shurubor YI, Zhao D, Arreguin AJ, Shahi I, et al. Rewiring of Glutamine Metabolism Is a Bioenergetic Adaptation of Human Cells with Mitochondrial DNA Mutations. Cell Metab (2018) 27(5):1007–25.e5. doi: 10.1016/j.cmet.2018.03.002
54. Mukhopadhyay S, Goswami D, Adiseshaiah PP, Burgan W, Yi M, Guerin TM, et al. Undermining glutaminolysis bolsters chemotherapy while NRF2 promotes chemoresistance in KRAS-driven pancreatic cancers. Cancer Res (2020) 41(6):405–9. doi: 10.1158/0008-5472.CAN-19-1363
55. Romero R, Sayin VI, Davidson SM, Bauer MR, Singh SX, Leboeuf SE, et al. Keap1 loss promotes Kras-driven lung cancer and results in dependence on glutaminolysis. Nat Med (2017) 23(11):1362–8. doi: 10.1038/nm.4407
56. Wise DR, Deberardinis RJ, Mancuso A, Sayed N, Zhang XY, Pfeiffer HK, et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc Natl Acad Sci USA (2008) 105(48):18782–7. doi: 10.1073/pnas.0810199105
57. Alvarez-Idaboy JR, Galano A. On the Chemical Repair of DNA Radicals by Glutathione: Hydrogen vs Electron Transfer. J Phys Chem B (2012) 116(31):9316–25. doi: 10.1021/jp303116n
58. Evans JW, Taylor YC, Brown JM. The role of glutathione and DNA strand break repair in determining the shoulder of the radiation survival curve. Br J Cancer Suppl (1984) 6:49–53. doi: 10.1038/nm.2882
59. Carey BW, Finley LWS, Cross JR, Allis CD, Thompson CB. Intracellular α-ketoglutarate maintains the pluripotency of embryonic stem cells. Nature (2015) 518(7539):413–6. doi: 10.1038/nature13981
60. O’Malley J, Kumar R, Kuzmin AN, Pliss A, Yadav N, Balachandar S, et al. Lipid quantification by Raman microspectroscopy as a potential biomarker in prostate cancer. Cancer Lett (2017) 397:52–60. doi: 10.1016/j.canlet.2017.03.025
61. Tirinato L, Liberale C, Di Franco S, Candeloro P, Benfante A, La Rocca R, et al. Lipid droplets: A new player in colorectal cancer stem cells unveiled by spectroscopic imaging. Stem Cells (2015) 33(1):35–44. doi: 10.1002/stem.1837
62. Yi M, Li J, Chen S, Cai J, Ban Y, Peng Q, et al. Emerging role of lipid metabolism alterations in Cancer stem cells. J Exp Clin Cancer Res (2018) 37(1):118. doi: 10.1186/s13046-018-0784-5
63. Mancini R, Noto A, Pisanu ME, De Vitis C, Maugeri-Saccà M, Ciliberto G. Metabolic features of cancer stem cells: the emerging role of lipid metabolism. Oncogene (2018) 37(18):2367–78. doi: 10.1038/s41388-018-0141-3
64. Bartolacci C, Padanad M, Andreani C, Melegari M, Rindhe S, George K, et al. Fatty Acid Synthase Is a Therapeutic Target in Mutant KRAS Lung Cancer. J Thorac Oncol (2017) 12(8):S1538. doi: 10.1016/j.jtho.2017.06.030
65. Lupu R, Menendez JA. Targeting Fatty Acid Synthase in Breast and Endometrial Cancer: An Alternative to Selective Estrogen Receptor Modulators? Endocrinology (2006) 147(9):4056–66. doi: 10.1210/en.2006-0486
66. Cai Y, Wang J, Zhang L, Wu D, Yu D, Tian X, et al. Expressions of fatty acid synthase and HER2 are correlated with poor prognosis of ovarian cancer. Med Oncol (2015) 32(1):1–6. doi: 10.1007/s12032-014-0391-z
67. Cerne D, Prodan Zitnik I, Sok M. Increased Fatty Acid Synthase Activity in Non-small Cell Lung Cancer Tissue Is a Weaker Predictor of Shorter Patient Survival than Increased Lipoprotein Lipase Activity. Arch Med Res (2010) 41(6):405–9. doi: 10.1016/j.arcmed.2010.08.007
68. Li X, Wu JB, Li Q, Shigemura K, Chung LWK, Huang WC. SREBP-2 promotes stem cell-like properties and metastasis by transcriptional activation of c-Myc in prostate cancer. Oncotarget (2016) 7(11):12869–84. doi: 10.18632/oncotarget.7331
69. Ann D, Somlo G, Fahrmann JF, Yuan Y, Tripathi SC, Li Y-J, et al. JAK/STAT3-Regulated Fatty Acid β-Oxidation Is Critical for Breast Cancer Stem Cell Self-Renewal and Chemoresistance. Cell Metab (2018) 27(6):1357. doi: 10.1016/j.cmet.2017.11.001
70. Wang T, Fahrmann JF, Lee H, Li YJ, Tripathi SC, Yue C, et al. JAK/STAT3-Regulated Fatty Acid β-Oxidation Is Critical for Breast Cancer Stem Cell Self-Renewal and Chemoresistance. Cell Metab (2018) 27(1):136–50.e5. doi: 10.1016/j.cmet.2017.11.001
71. Ito K, Carracedo A, Weiss D, Arai F, Ala U, Avigan DE, et al. A PML-PPAR-δ pathway for fatty acid oxidation regulates hematopoietic stem cell maintenance. Nat Med (2012) 18(9):1350–8. doi: 10.1038/nm.2882
72. Qiu GZ, Jin MZ, Dai JX, Sun W, Feng JH, Jin WL. Reprogramming of the Tumor in the Hypoxic Niche: The Emerging Concept and Associated Therapeutic Strategies. Trends Pharmacol Sci (2017) 38:669–86. doi: 10.1016/j.tips.2017.05.002
73. Li Z, Bao S, Wu Q, Wang H, Eyler C, Sathornsumetee S, et al. Hypoxia-Inducible Factors Regulate Tumorigenic Capacity of Glioma Stem Cells. Cancer Cell (2009) 15(6):501–13. doi: 10.1016/j.ccr.2009.03.018
74. Singh D, Arora R, Kaur P, Singh B, Mannan R, Arora S. Overexpression of hypoxia-inducible factor and metabolic pathways: possible targets of cancer. Cell Biosci (2017) 7(1):62. doi: 10.1186/s13578-017-0190-2
75. Shibuya K, Okada M, Suzuki S, Seino M, Seino S, Takeda H, et al. Targeting the facilitative glucose transporter GLUT1 inhibits the self-renewal and tumor-initiating capacity of cancer stem cells. Oncotarget (2015) 6(2):651–61. doi: 10.18632/oncotarget.2892
76. Filatova A, Seidel S, Böǧürcü N, Gräf S, Garvalov BK, Acker T. Acidosis acts through HSP90 in a PHD/ VHL-independent manner to promote HIF function and stem cell maintenance in glioma. Cancer Res (2016) 76(19):5845–56. doi: 10.1158/0008-5472.CAN-15-2630
77. Sonveaux P, Végran F, Schroeder T, Wergin MC, Verrax J, Rabbani ZN, et al. Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J Clin Invest (2008) 118(12):3930–42. doi: 10.1172/JCI36843
78. Sousa CM, Biancur DE, Wang X, Halbrook CJ, Sherman MH, Zhang L, et al. Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature (2016) 536(7617):479–83. doi: 10.1038/nature19084
79. Fu Y, Liu S, Yin S, Niu W, Xiong W, Tan M, et al. The reverse Warburg effect is likely to be an Achilles’ heel of cancer that can be exploited for cancer therapy. Oncotarget (2017) 8(34):57813–25. doi: 10.18632/oncotarget.18175
80. Yao H, Liu N, Lin MC, Zheng J. Positive feedback loop between cancer stem cells and angiogenesis in hepatocellular carcinoma. Cancer Lett (2017) 379:213–9. doi: 10.1016/j.canlet.2016.03.014
81. Sun H, Jia J, Wang X, Ma B, Di L, Song G, et al. CD44+/CD24- breast cancer cells isolated from MCF-7 cultures exhibit enhanced angiogenic properties. Clin Transl Oncol (2013) 15(1):46–54. doi: 10.1007/s12094-012-0891-2
82. Chen Y, Tan W, Wang C. Tumor-associated macrophage-derived cytokines enhance cancer stem-like characteristics through epithelial–mesenchymal transition. OncoTargets Ther (2018) 11:3817–26. doi: 10.2147/OTT.S168317
83. Yang J, Liao D, Chen C, Liu Y, Chuang TH, Xiang R, et al. Tumor-associated macrophages regulate murine breast cancer stem cells through a novel paracrine egfr/stat3/sox-2 signaling pathway. Stem Cells (2013) 31(2):248–58. doi: 10.1002/stem.1281
84. Ohashi T, Aoki M, Tomita H, Akazawa T, Sato K, Kuze B, et al. M2-like macrophage polarization in high lactic acid-producing head and neck cancer. Cancer Sci (2017) 108(6):1128–34. doi: 10.1111/cas.13244
85. Mu X, Shi W, Xu Y, Xu C, Zhao T, Geng B, et al. Tumor-derived lactate induces M2 macrophage polarization via the activation of the ERK/STAT3 signaling pathway in breast cancer. Cell Cycle (2018) 17(4):428–38. doi: 10.1080/15384101.2018.1444305
86. Gong C, Bauvy C, Tonelli G, Yue W, Deloménie C, Nicolas V, et al. Beclin 1 and autophagy are required for the tumorigenicity of breast cancer stem-like/progenitor cells. Oncogene (2013) 32(18):2261–72. doi: 10.1038/onc.2012.252
87. Song YJ, Zhang SS, Guo XL, Sun K, Han ZP, Li R, et al. Autophagy contributes to the survival of CD133+ liver cancer stem cells in the hypoxic and nutrient-deprived tumor microenvironment. Cancer Lett (2013) 339(1):70–81. doi: 10.1016/j.canlet.2013.07.021
88. Zhang D, Zhao Q, Sun H, Yin L, Wu J, Xu J, et al. Defective autophagy leads to the suppression of stem-like features of CD271+ osteosarcoma cells. J BioMed Sci (2016) 23(1):1–12. doi: 10.1186/s12929-016-0297-5
89. Peng Q, Qin J, Zhang Y, Cheng X, Wang X, Lu W, et al. Autophagy maintains the stemness of ovarian cancer stem cells by FOXA2. J Exp Clin Cancer Res (2017) 36(1):171. doi: 10.1186/s13046-017-0644-8
90. Hu L, Zeng Z, Xia Q, Liu Z, Feng X, Chen J, et al. Metformin attenuates hepatoma cell proliferation by decreasing glycolytic flux through the HIF-1α/PFKFB3/PFK1 pathway. Life Sci (2019) 239:116966. doi: 10.1016/j.lfs.2019.116966
91. Zannella VE, Pra AD, Muaddi H, McKee TD, Stapleton S, Sykes J, et al. Reprogramming metabolism with metformin improves tumor oxygenation and radiotherapy response. Clin Cancer Res (2013) 19(24):6741–50. doi: 10.1158/1078-0432.CCR-13-1787
92. Lea MA, Qureshi MS, Buxhoeveden M, Gengel N, Kleinschmit J, Desbordes C. Regulation of the proliferation of colon cancer cells by compounds that affect glycolysis, including 3-bromopyruvate, 2-deoxyglucose and biguanides. Anticancer Res (2013) 33(2):401–7. doi: 10.1158/1538-7445.AM2013-1855
93. O’Neill S, Porter RK, McNamee N, Martinez VG, O’Driscoll L. 2-Deoxy-D-Glucose inhibits aggressive triple-negative breast cancer cells by targeting glycolysis and the cancer stem cell phenotype. Sci Rep (2019) 9(1):706–20. doi: 10.1038/s41598-019-39789-9
94. Wei R, Hackman RM, Wang Y, Mackenzie GG. Targeting glycolysis with epigallocatechin-3-gallate enhances the efficacy of chemotherapeutics in pancreatic cancer cells and xenografts. Cancers (Basel) (2019) 11(10):685–700. doi: 10.3390/cancers11101496
95. Gross MI, Demo SD, Dennison JB, Chen L, Chernov-Rogan T, Goyal B, et al. Antitumor activity of the glutaminase inhibitor CB-839 in triple-negative breast cancer. Mol Cancer Ther (2014) 13(4):890–901. doi: 10.1158/1535-7163.MCT-13-0870
96. Ciombor KK, Whisenant J, Cardin DB, Goff LW, Das S, Schulte M, et al. CB-839, panitumumab, and irinotecan in RAS wildtype (WT) metastatic colorectal cancer (mCRC): Phase I results. J Clin Oncol (2019) 37(4_suppl):574–4. doi: 10.1200/JCO.2019.37.4_suppl.574
97. Boysen G, Jamshidi-Parsian A, Davis MA, Siegel ER, Simecka CM, Kore RA, et al. Glutaminase inhibitor CB-839 increases radiation sensitivity of lung tumor cells and human lung tumor xenografts in mice. Int J Radiat Biol (2019) 95(4):436–42. doi: 10.1080/09553002.2018.1558299
98. Lian X, Wang G, Zhou H, Zheng Z, Fu Y, Cai L. Anticancer properties of fenofibrate: A repurposing use. J Cancer (2018) 9(9):1527–37. doi: 10.7150/jca.24488
99. Alimoghaddam K. A review of arsenic trioxide and acute promyelocytic Leukemia. Int J Hematol Stem Cell Res (2014) 8(3):44–54. doi: 10.1042/BJ20070039
100. Gao X, Liu X, Shan W, Liu Q, Wang C, Zheng J, et al. Anti-malarial atovaquone exhibits anti-tumor effects by inducing DNA damage in hepatocellular carcinoma. Am J Cancer Res (2018) 8(9):1697–711. doi: 10.18632/oncotarget.12944
101. Fiorillo M, Lamb R, Tanowitz HB, Mutti L, Krstic-Demonacos M, Cappello AR, et al. Repurposing atovaquone: Targeting mitochondrial complex III and OXPHOS to eradicate cancer stem cells. Oncotarget (2016) 7(23):34084–99. doi: 10.18632/oncotarget.9122
102. Mody M, Dharker N, Bloomston M, Wang PS, Chou FS, Glickman TS, et al. Rosiglitazone sensitizes MDA-MB-231 breast cancer cells to anti-tumour effects of tumour necrosis factor-α, CH11 and CYC202. Endocr Relat Cancer (2007) 14(2):305–15. doi: 10.1677/ERC-06-0003
103. Liu Y, Hu X, Shan X, Chen K, Tang H. Rosiglitazone metformin adduct inhibits hepatocellular carcinoma proliferation via activation of AMPK/p21 pathway. Cancer Cell Int (2019) 19(1):13. doi: 10.1186/s12935-019-0732-2
104. Shiragami R, Murata S, Kosugi C, Tezuka T, Yamazaki M, Hirano A, et al. Enhanced antitumor activity of cerulenin combined with oxaliplatin in human colon cancer cells. Int J Oncol (2013) 43(2):431–8. doi: 10.3892/ijo.2013.1978
105. Kridel SJ, Lowther WT, Pemble IV CW. Fatty acid synthase inhibitors: new directions for oncology. Expert Opin Invest Drugs (2007) 16(11):1817–29. doi: 10.1517/13543784.16.11.1817
106. Jin UH, Lee SO, Pfent C, Safe S. The aryl hydrocarbon receptor ligand omeprazole inhibits breast cancer cell invasion and metastasis. BMC Cancer (2014) 14(1):1–14. doi: 10.1186/1471-2407-14-498
107. Li X, Chen YT, Hu P, Huang WC. Fatostatin displays high antitumor activity in prostate cancer by blocking SREBP-regulated metabolic pathways and androgen receptor signaling. Mol Cancer Ther (2014) 13(4):855–66. doi: 10.1158/1535-7163.MCT-13-0797
108. Brovkovych V, Izhar Y, Danes JM, Dubrovskyi O, Sakallioglu IT, Morrow LM, et al. Fatostatin induces pro- and anti-apoptotic lipid accumulation in breast cancer. Oncogenesis (2018) 7(8):66. doi: 10.1038/s41389-018-0076-0
109. Ji C, Hu Y, Cheng G, Liang L, Gao B, Ren Y, et al. A ketogenic diet attenuates proliferation and stemness of glioma stem−like cells by altering metabolism resulting in increased ROS production. Int J Oncol (2019) 27(1):1–16. doi: 10.3892/ijo.2019.4942
110. Weber DD, Aminzadeh-Gohari S, Tulipan J, Catalano L, Feichtinger RG, Kofler B. Ketogenic diet in the treatment of cancer – Where do we stand? Mol Metab (2019) 145(8):2144–56. doi: 10.1016/j.molmet.2019.06.026
111. Erickson N, Boscheri A, Linke B, Huebner J. Systematic review: isocaloric ketogenic dietary regimes for cancer patients. Med Oncol (2017) 34:72. doi: 10.1007/s12032-017-0930-5
112. Yu X, Mao W, Zhai Y, Tong C, Liu M, Ma L, et al. Anti-tumor activity of metformin: From metabolic and epigenetic perspectives. Oncotarget (2017) 8:5619–28. doi: 10.18632/oncotarget.13639
113. Andrzejewski S, Gravel S-P, Pollak M, St-Pierre J. Metformin directly acts on mitochondria to alter cellular bioenergetics. Cancer Metab (2014) 2(1):12. doi: 10.1186/2049-3002-2-12
114. Viollet B, Guigas B, Sanz Garcia N, Leclerc J, Foretz M, Andreelli F. Cellular and molecular mechanisms of metformin: An overview. Clin Sci (2012) 122:253–70. doi: 10.1042/CS20110386
115. Ashton TM, Gillies McKenna W, Kunz-Schughart LA, Higgins GS. Oxidative phosphorylation as an emerging target in cancer therapy. Clin Cancer Res (2018) 24:2482–90. doi: 10.1158/1078-0432.CCR-17-3070
116. Sica V, Bravo-San Pedro JM, Stoll G, Kroemer G. Oxidative phosphorylation as a potential therapeutic target for cancer therapy. Int J Cancer (2020) 146(1):10–7. doi: 10.1002/ijc.32616
117. Chen D, Barsoumian HB, Fischer G, Yang L, Verma V, Younes AI, et al. Combination treatment with radiotherapy and a novel oxidative phosphorylation inhibitor overcomes PD-1 resistance and enhances antitumor immunity. J Immunother Cancer (2020) 8(1):289. doi: 10.1136/jitc-2019-000289
118. Curthoys NP, Watford M. Regulation of glutaminase activity and glutamine metabolism. Annu Rev Nutr (1995) 15:133–59. doi: 10.1146/annurev.nu.15.070195.001025
119. Zhang J, Fan J, Venneti S, Cross JR, Takagi T, Bhinder B, et al. Asparagine plays a critical role in regulating cellular adaptation to glutamine depletion. Mol Cell (2014) 56(2):205–18. doi: 10.1016/j.molcel.2014.08.018
120. Alkan HF, Walter KE, Luengo A, Madreiter-Sokolowski CT, Stryeck S, Lau AN, et al. Cytosolic Aspartate Availability Determines Cell Survival When Glutamine Is Limiting. Cell Metab (2018) 28(5):706–20.e6. doi: 10.1016/j.cmet.2018.07.021
121. Yang L, Achreja A, Yeung T-L, Mangala LS, Jiang D, Han C, et al. Targeting Stromal Glutamine Synthetase in Tumors Disrupts Tumor Microenvironment-Regulated Cancer Cell Growth. Cell Metab (2016) 24(5):685–700. doi: 10.1016/j.cmet.2016.10.011
122. Gross MI, Demo SD, Dennison JB, Chen L, Chernov-Rogan T, Goyal B, et al. Antitumor activity of the glutaminase inhibitor CB-839 in triple-negative breast cancer. Mol Cancer Ther (2014) 13(4):890–901. doi: 10.1158/1535-7163.MCT-13-0870
123. Li D, Fu Z, Chen R, Zhao X, Zhou Y, Zeng B, et al. Inhibition of glutamine metabolism counteracts pancreatic cancer stem cell features and sensitizes cells to radiotherapy. Oncotarget (2015) 6(31):31151–63. doi: 10.18632/oncotarget.5150
124. Robinson MM, McBryant SJ, Tsukamoto T, Rojas C, Ferraris DV, Hamilton SK, et al. Novel mechanism of inhibition of rat kidney-type glutaminase by BPTES. Biochem J (2007) 7(23):34084–99. doi: 10.18632/oncotarget.9122
125. Matre P, Velez J, Jacamo R, Qi Y, Su X, Cai T, et al. Inhibiting glutaminase in acute myeloid leukemia: Metabolic dependency of selected AML subtypes. Oncotarget (2016) 7(48):79722–35. doi: 10.18632/oncotarget.12944
126. Migita T, Ruiz S, Fornari A, Fiorentino M, Priolo C, Zadra G, et al. Fatty Acid Synthase: A Metabolic Enzyme and Candidate Oncogene in Prostate Cancer. JNCI J Natl Cancer Inst (2009) 101(7):519–32. doi: 10.1093/jnci/djp030
127. Zadra G, Ribeiro CF, Chetta P, Ho Y, Cacciatore S, Gao X, et al. Inhibition of de novo lipogenesis targets androgen receptor signaling in castration-resistant prostate cancer. Proc Natl Acad Sci USA (2019) 116(2):631–40. doi: 10.1186/1471-2407-14-498
Keywords: cancer stem cells, therapy resistance, metabolic targeting, OXPHOS, glycolysis, glutamine metabolism, fatty acid metabolism, tumor microenvironment
Citation: Mukha A and Dubrovska A (2020) Metabolic Targeting of Cancer Stem Cells. Front. Oncol. 10:537930. doi: 10.3389/fonc.2020.537930
Received: 25 February 2020; Accepted: 05 November 2020;
Published: 22 December 2020.
Edited by:
Federica Sotgia, University of Salford, United KingdomReviewed by:
Toshihiko Torigoe, Sapporo Medical University, JapanCopyright © 2020 Mukha and Dubrovska. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anna Mukha, YW11a2hhQHVtY3V0cmVjaHQubmw=; Anna Dubrovska, YS5kdWJyb3Zza2FAaHpkci5kZQ==
†Present address: Anna Mukha, Center for Molecular Medicine, UMC Utrecht, Utrecht University, Utrecht, Netherlands
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.