 Yining Jiang
Yining Jiang Liyan Zhao2†
Liyan Zhao2† Yubo Wang
Yubo Wang- 1Department of Neurosurgery, First Hospital of Jilin University, Changchun, China
- 2Department of Medical Laboratory, Second Hospital, Jilin University, Changchun, China
Background: Primary intracranial Ewing sarcoma (ES)/peripheral primitive neuroectodermal tumors (pPNETs) are extremely rare malignancies, which arise in children and adolescents, with only 9 cases reported in patients over 30 years of age. Due to its rarity, MRI features and treatment strategies for primary intracranial ES/pPNETs remain unclear. The purpose of this study was to explore the clinical features, imaging findings, pathological characteristics, different diagnoses, treatment, and prognosis of cerebellar liponeurocytoma in adults.
Case Description: A 55-year-old female was admitted to the hospital with memory decline over 1 month, which aggravated in the last 2 weeks. MRI showed a 4.3 × 6.5 × 3.5 cm heterogeneous large mass in the left frontal lobe with mild peritumoral edema. The mass was successfully removed under neuronavigation and electrophysiological monitoring. The entire mass was removed, and postoperative pathology indicated an ES pPNET diagnosis, with an EWSR1 gene rearrangement. Subsequently, the patient underwent disciplinary radiotherapy.
Conclusion: The diagnosis of primary intracranial ES/pPNETs depends on the comprehensive consideration of histological examination, immunohistochemical analysis, and genetic detection. Gross tumor resection combined with radiotherapy and chemotherapy might be the most beneficial treatment.
Introduction
Primary intracranial ES/pPNETs are a group of poorly differentiated, highly malignant, and aggressive small round cell neoplasms, generally originating from bone and soft tissue, which are common among children and adolescents (1–4). ES/pPNETs that occur intracranially are extremely rare, with only 57 cases reported so far. For patients over 30 years of age, only 9 cases have been reported (4–12). Fusion of the EWSR1 gene with a member of the ETS gene family is thought to be the primary cause of the ES/pPNETs (3, 4, 13–15). Herein, we present a case of ES/pPNET located in the left frontal lobe of a 55-year-old female, who was tested through histological examination, immunohistochemical analysis and fluorescence in situ hybridization (FISH). Moreover, we have concluded the patients over 30 years of age, and summarized the typical pathological and radiological features of this rare tumor entity, with surgery, adjuvant therapy, prognosis, and important differential diagnoses discussed in detail.
Case Report
History and Examination
A 55-year-old female patient was admitted to the hospital due to progressive memory decline for over 1 month, which aggravated in the last 2 weeks. Physical examination showed normal higher mental functions. Left limb muscle strength was normal; right limbs were slightly weaker than left limbs.
Neuroimaging Finding
Magnetic resonance imaging (MRI) showed a 4.3 × 6.5 × 3.5 cm large irregular mass located in the left frontal lobe with mixed isointense-to-hypointense signals on T1-weighted imaging (T1WI), uneven hypointense-to-hyperintense signals on T2-weighted imaging (T2WI), and T2 dark-fluid. After gadolinium administration, obvious heterogenous enhancement was observed (Figures 1A–C). Computerized tomography (CT) showed a heterogeneous hypo-and isoindense mass in the left frontal lobe with a CT value of 15–37 HU (Figure 1D). Magnetic resonance venography indicated that the forehead sagittal sinus was not visible as it was compressed by the tumor (Figure 1E). Diffusion tensor imaging showed that the nerve fibers in the lesion area were compressed, displaced, and partially interrupted (Figure 1F).
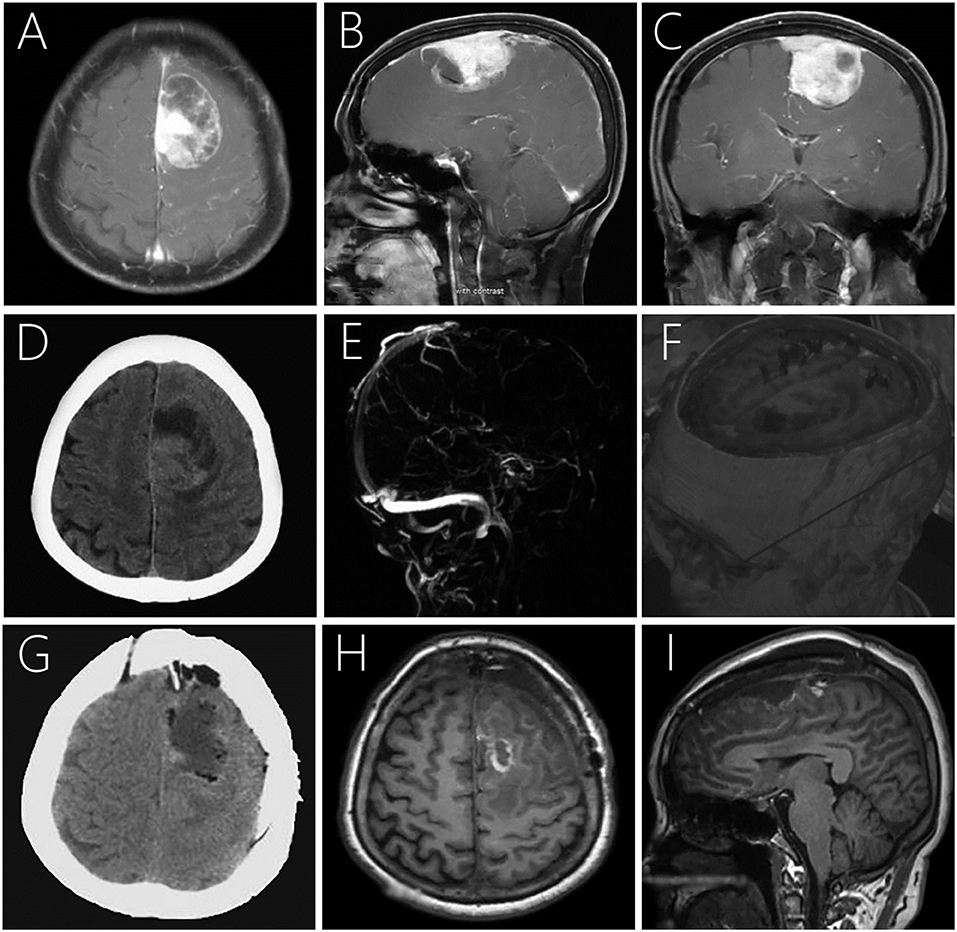
Figure 1. Post-contrast MRI (A–C) showing a large irregular mass with heterogeneous enhancement in the left frontal lobe with mild peritumoral edema. Plain CT demonstrating a large mass mixed hypo- and isodensity (D). Magnetic resonance venography showing no development was seen in the superior sagittal sinus of the frontal area (E). Diffusion tensor imaging showing compression, displacement, and partial interruption of nerve fibers in the lesion area (F). Postoperative CT (G) and MRI 7-days postoperatively (H,I) revealed that the lesion was completely removed with no signs of recurrence.
Surgery
A preoperative clinical diagnosis of meningioma was made, and surgery was performed under preoperative neuronavigation and intraoperative electrophysiological monitoring of somatosensory, and muscle-evoked potentials. The tumor mass was soft, rich in blood supply, and invaded into the sagittal sinus from the left side. The tumor and eroded dura were completely removed.
Pathological and Genetic Finding
Pathology was suggestive of ES/pPNET. Hematoxylin and eosin-stained paraffin sections predominantly showed closely packed small, round to oval, undifferentiated cells with hyperchromatic nuclei, increased mitotic activity, and little basophilic cytoplasm (Figure 2A). Immunohistochemical analysis indicated that the Ki-67 index was ± 50%. Moreover, the neoplasm was positive for CD99 (Figure 2B), FLI-1, MAP-2, vimentin, synaptophysin, and the progesterone receptor, and was negative for TTF-1, CK-pan, CK5/6, CD56, CgA, LCA, S-100, glial fibrillary acidic protein, myeloperoxidase, MyoD1, NeuN, Olig-2, STAT6, and epithelial membrane antigen. Moreover, fluorescence in situ hybridization (FISH) with a EWSR1 break apart probe showed 41.0% of split signals, thereby confirming the diagnosis of ES/pPNETs (Figures 2C,D).
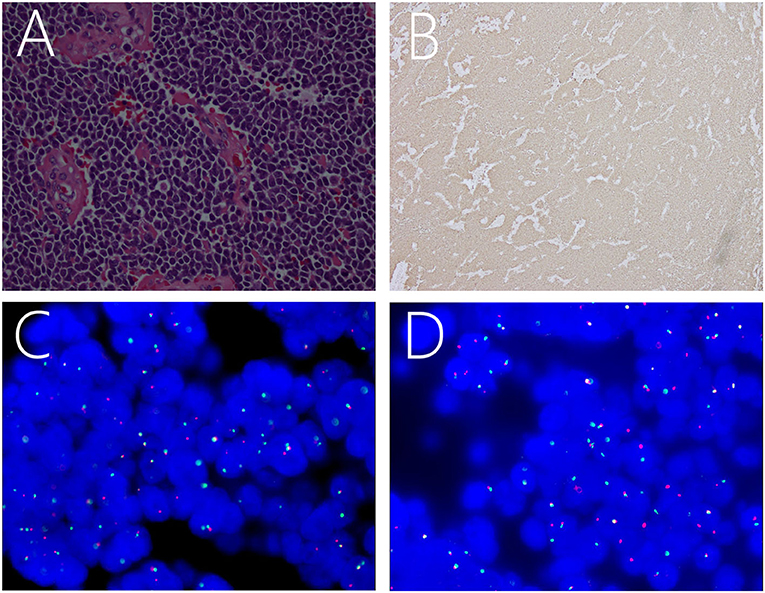
Figure 2. Light microscopic image showing a number of small, round, oval, undifferentiated cells, with intense staining of the nuclei and basophilic cytoplasm (A). Immunohistochemical staining showing positivity for CD99 (B). FISH revealing positive results for EWSR1 rearrangement as indicated by the separation of red and green signals (C,D).
Postoperative Course
The patient's postoperative course was uneventful, and positron emission tomography was performed to identify potential extracranial primary sites, and serum tumor markers were measured, but both were negative. Postoperative CT (Figure 1G) and MRI 1 week postoperatively (Figures 1H,I) demonstrated that the lesion was completely removed, and no signs of recurrence were observed. Subsequently, the patient underwent disciplinary radiotherapy for 1 month. During the last telephone follow-up in August 2020, 18 months after surgery, the patient reported to be living a normal daily life with no apparent symptoms. We believe the patient's condition is stable and will continue to follow-up.
Discussion
Epidemiology
Intracranial ES/pPNETs are very rare malignant tumors, accounting for 0.03% of the total number of intracranial tumors (3, 4), with only 57 cases reported until now. ES/pPNETs mostly occur in children and adolescents with a median age at first onset of disease being 15 years of age, and the peak of disease is prominent in the second decade, ranging from 5 months to 67 years of age, with a slight male predisposition (14, 16–18). The case presented here is an ES/pPNET in the left frontal lobe of a 55-year-old female patient. Notably, the incidence of ES/pPNET in adults is exceedingly rare. Here, we reviewed all reported cases in patients older than 30 years of age, only 9 cases have been reported. Our study is the first to report a systematic review for primary intracranial ES/pPNET in adult patients. In our study, the mean patient age was 50.7 years (range: 34–67 years), and there were seven females and four males (Table 1). Moreover, supratentorial cerebral hemispheres are predilection sites for ES/pPNET (14). In our study, the presenting sites include the hemispheric surface (6 cases), cerebellopontine angle (2 cases), cavernous sinus (1 case), and posterior fossa (1 case). Moreover, the origin of primary intracranial ES/pPNETs has not been clearly elucidated, however, the presumptive precursor cells are speculated to stem from the neural crest or mesenchymal stem cells (19, 20).
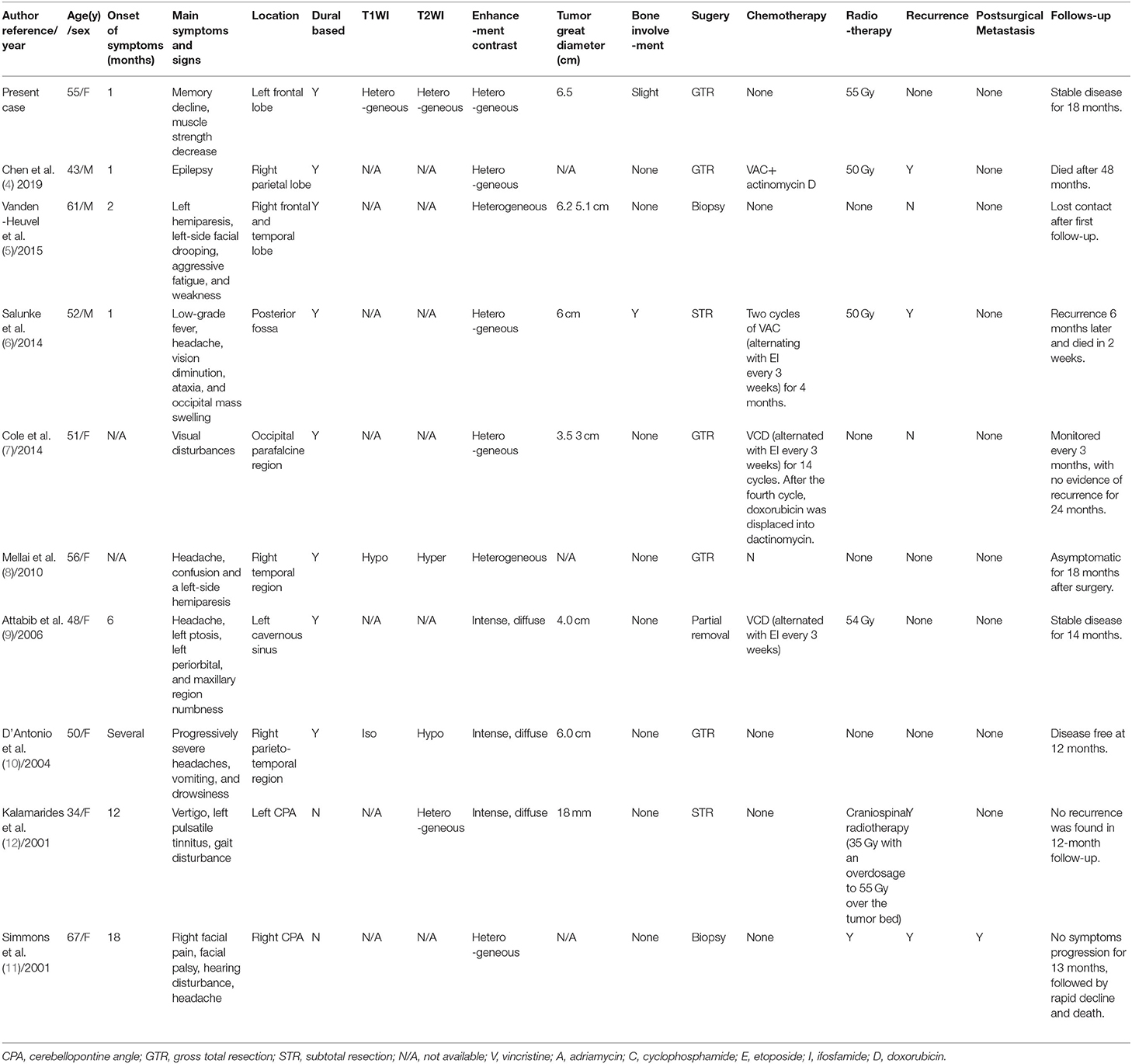
Table 1. Summary of Cases of Primary Intracranial ES/ pPNETs over 30 years of age Reported in the Literature.
Clinical Presentation
The clinical manifestations of primary intracranial ES/pPNETs are diverse and highly variable, considering tumor location, size, and invasion site. Our study showed that clinical symptoms include intracranial pressure, increasing presenting headache, and vomiting (5 cases, [50.0%]), hemiplegia and muscle strength decrease (3 cases, [30.0%]), facial palsy (3 cases, [30.0%]), deafness and hearing disturbance (2 cases, [20.0%]), drowsiness and fatigue (2 cases, [20.0%]), epilepsy (1 case, [10.0%]), memory decline (1 case, [10.0%]), and ataxia (1 case, [10.0%]). The mean duration of clinical symptoms was 5.9 months (range: 1–18 months), and the duration in our results is longer than what has been described in the literature (with a median of 9 weeks) (14). This might associate with the compensation of unclosed or not firmly integrated cranial sutures in children. Some highly uncommon clinical signs of ES/pPNETs have been reported, including the formation of large intra-cerebral hematomas and elevated serum levels of carcinoembryonic antigens (1), which are important to be noticed during clinical evaluation. In this case, the patient's major complaint was memory decline, suggesting functional damage of the dominant side frontal lobe.
Radiologic Features
We extensively reviewed previous reports and found that primary intracranial ES/pPNETs mostly showed mixed isointense-to-hypointense signals on T1WI, and isointense-to-hyperintense signals on T2WI (4). Our case was consistent with these findings. As for post-contrast MRI, Cherif et al. reviewed all reported ES/pPNETs in 2018, and presented a heterogeneous enhancement of about 40.0%, intense enhancement of about 52.5%, and moderate enhancement of 7.5% of these lesions (14). In our study, we showed heterogeneous enhancement of about 60.0% (6 cases), and intense enhancement of 40.0% (4 cases) of these lesions. Jing et al. regarded the reason for such heterogeneous enhancement as a characteristic of high density small round cells under the microscope, and a large amount of protein-rich mucus in some areas, which was accompanied by hemorrhage and necrosis (16). In our study, skull involvement was observed in two cases. In addition, tumors with a dural base were another characteristic of primary intracranial ES/pPNET, which was observed in 80.0% in patients included in our study. This was one of the key aspects for ES/pPNET to be confused with hemangiopericytoma or meningioma among others (5, 21). In our case, the tumor adhered to the sagittal sinus with a broad base and grew in a semi-circular shape, with a preoperative diagnosis of hemangiopericytoma. This was probably due to the ES/pPNET undermining surrounding normal meninges. Furthermore, the meninges, blood-brain barrier, and skull affected the growth pattern of the tumor. Therefore, to some degree, imaging in general and MRI examinations have limited value in the differential diagnoses of ES/pPNET (22).
Diagnosis
ES/pPNETs are highly aggressive, malignant tumors with focal necrosis (1). They are mainly composed of small, round or oval, undifferentiated cells, with hyperchromatic nuclei, increased mitotic activity, and a slightly basophilic cytoplasm (1, 3, 4). Moreover, the tumor cells are markedly fibrotic, highly mitotic, and separated by groups of cells containing collagen bands (1, 3, 4, 14, 18). Membranous expression of CD99 is a highly reliable and sensitive diagnostic biomarker for primary intracranial ES/pPNETs (3, 4, 13–15, 18), and was detected in nearly all patients. However, CD99 is not recommended as a specific immunohistochemical marker for diagnosing ES/pPNETs, because CD99 can also be detected in other small, blue round cell tumors, including lymphoblastic lymphomas, ependymomas, and rhabdomyosarcomas (2, 4, 14, 21, 23). Nevertheless, the staining pattern in these cases is often cytoplasmic, rather than the distinct membranous staining typical of ES/pPNETs (1, 17, 22, 24). In general, the membrane protein FLI-1 is also expressed in ES/pPNETs (1, 4).
At present, molecular testing depicting EWSR1 gene rearrangement is a golden standard for diagnosing ES/pPNET (13, 14), which can be detected by reverse transcription polymerase chain reaction and FISH methods with a sensitivity and specificity about 91–100% (5). Chromosomal translocation t(11, 22)(q24;q12) is the most common genetic aberration in ES/pPNETs. This translocation results in formation of a chimeric transcription factor EWS-FLI-1 (friend leukemia integration 1 transcription factor) in 85–90% of ES/pPNETs, being the in-frame fusion of the 5' end of EWSR1 gene with the 3' portion of the FLI-1 gene with abnormal transcription regulator properties (1, 4, 13–15, 18). The downstream effects of the EWS-FLI-1 fusion gene include dysregulation of cell proliferation, differentiation, apoptosis, angiogenesis, invasion, and metastasis (25). There are also other chromosomal translocations, including ESW/ERG t(21, 22)(q22;q12), which is the second most common translocation, accounting 10% of ES/pPNETs (18). Less than 1% of chromosome translocations lead to EWSR1 gene fusion with other ETS family transcription factor genes, including t(2, 22) (q36;q12) (EWS-FEV), t(7, 22) (q22;q12) (EWS-ETV1), t(17, 22) (q21;q12) (EWS-EIAF) (1, 26–28). Nonetheless, it is worth noting that these translocations are not only associated with ES/pPNETs. Thorner et al. demonstrated that two rhabdomyosarcomas and two polyphenotypic tumors with t(11, 22)(q24;q12) translocations were negative for CD99 (29). Thus, the diagnosis of ES/pPNETs is highly dependent on the comprehensive consideration of histological examination, immunohistochemical analysis, and molecular genetic analyses (18). The pseudoautosomal gene, MIC2 gene, is another important gene, which has been demonstrated to be present in ES/pPNETs (17, 18). The cell surface glycoprotein CD99 is the product of the MIC2 gene, which is observed in essentially all cases of ES/pPNETs (15, 17).
In differential diagnosis, due to histological similarities, primary intracranial ES/pPNETs can be misdiagnosed as central nervous system embryonal tumors, cPNETs (such as medulloblastoma, central neuroblastoma, and other neuroepithelial tumors), malignant meningioma, and melanoma, among others (3, 18, 20, 30). Central nervous system embryonal tumors do not express the MIC2 gene, and are negative for CD99 (13). As for cPNETs, they can be confirmed by the absence of CD99 expression and t(11, 22) translocation (1, 2, 14, 22). In our clinical case, diffuse membranous positivity for CD99 and FLI-1 were observed, combined with the expression of EWSR1 in FISH. Furthermore, whole-body CT, bone marrow aspirates, bone scans, lumbar punctures, and positron emission tomography scans are recommended to confirm the tumor to be primarily intracranial.
Treatment Modalities
Due to the rarity of ES/pPNETs, the standard treatment approach for this type of malignancy has not yet been established. To date, surgical resection, with the aim of total tumor resection, is the main therapy cornerstone (3, 4). Wide surgical resection margins at the time of primary surgery have markedly reduced local recurrences (13, 15). Chen et al. found the lifetime of gross total resection (GTR) to be significantly longer when compared to partial resection (38 months vs. 20 months, respectively) (4). In our study cohort, GTR was performed in five out ten patients, subtotal resection two patients, biopsy in two patients, and partial excision in one patient. The mean follow-up period for patients with GTR was 24 months (range: 12–48 months), with 1-year follow-up for 100.0% (5/5), 1.5-year for 80.0% (4/5), and 2-year for 40.0% (2/5). However, the mean follow-up period for patients with incomplete tumor resection (ITR) was 11.3 months (range: 6–14 months), with 1-year follow-up for 60.0% (3/5) and 1.5-year follow-up for none.
Due to the rarity of this disease, the standard first-line adjuvant treatment remains unclear. Some studies showed that the use of chemotherapy significantly improved the long-term survival rate, from 5–10 to 70–80% (31, 32), and presented lower recurrence rates (4, 14, 31, 32). In our study cohort, no recurrence was found in patients who underwent ITR and chemotherapy. However, for three patients who only underwent ITR, recurrence was found in two patients. Moreover, the mean follow-up period for patients with chemotherapy (23 months) was longer than for patients who underwent surgery only (11 months). The chemotherapeutics included vincristine, cyclophosphamide, doxorubicin, ifosfamide, etoposide, Adriamycin, and actinomycin D (3, 14). The chemotherapeutic combinations in our study include vincristine-cyclophosphamide-doxorubicin (VCD), vincristine-adriamycin-cyclophosphamide (VAC), and VAC-actinomycin D (VACA) alternating with etoposide-ifosfamide (EI), which was consistent with the findings presented in previous studies (4, 14). Moreover, the European Cooperative Group proposed vincristine-ifosfamide-doxorubicin-etoposide (VIDE) as the intensive induction chemotherapy for Ewing sarcoma (33). In addition, Jain et al. proposed neoadjuvant chemotherapy, which improved cytoreduction and achieved local control of tumors prior to surgical resection (4, 34). Moreover, radiotherapy is known to be an important adjuvant treatment option for ES/pPNETs (14). Chen et al. (4) suggested that the 1- and 2-year survival rates of patients receiving adjuvant radiotherapy (88.9 and 66.7%) were significantly higher than those of patients who did not receive radiotherapy (60.0 and 0%). In addition, the median survival time was significantly extended (38 months vs. 13 months). However, in our study, due to the small sample size, there was no significant survival discrepancy in patients with or without radiotherapy.
After extensive literature review, we observed that GTR combined with adjuvant chemotherapy and radiotherapy was the most beneficial treatment protocol (3, 4, 14, 21). Moreover, close follow-up is recommended and essentially necessary. Chen et al. presented that patients who underwent GTR, chemotherapy, and radiotherapy had a longest 2-year survival rate and the longest median survival time (4). In our study cohort, only one patient underwent GTR, adjuvant chemotherapy and radiotherapy, and he showed the longest survival period of 4 years. Notably, for patients who underwent ITR, radiotherapy combined with chemotherapy is highly recommended (3, 14).
Prognosis
Primary intracranial ES/ pPNETs are aggressive malignant tumors with a poor prognosis, and ITR is one of the most important reasons for its recurrence (14). Therefore, GTR should be the surgical aim. However, even when treated with total resection, recurrence may be unavoidable. Moreover, postoperative metastasis along cerebrospinal fluid was observed in one case in our study (11). Tumors located in the infratentorial region, with skull involvement, metastasis, and postoperative radio- and chemotherapy are highly associated with a poor prognosis (14). Adversely, age, sex, onset of symptoms, and presence of hemorrhagic or cystic component, do not appear to influence the prognosis of ES/pPNETs (14, 18, 33).
Furthermore, with an emerging understanding of the molecular underpinnings of ES, a prognosis based on the biological profile of tumors is possible. For example, a better prognosis has been associated with tumors containing the EWS-FLI-1 chimeric-type gene, when compared with other types of gene translocation (35).
Conclusion
In summary, primary intracranial ES/pPNETs are rarely reported in the literature and, with only 9 known cases, is extremely rare in patients over 30 years of age. Thus, more cases describing ES/pPNETs as well as long-term follow-up studies are warranted to fully understand ES/pPNETs in the adult population. Therefore, because of these limitations, our present case report might represent an additional reference among the few available that might serve as a potential guide for clinicians and radiologists.
Data Availability Statement
All datasets generated for this study are included in the article/supplementary material.
Ethics Statement
The studies involving human participants were reviewed and approved by the First Hospital of Jilin University Ethics Committee. The patients/participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author Contributions
YJ, LZ, and YW made study design, data collection, data analysis and interpretation, and composed the manuscript and literature review. YL and XL was the surgeon that performed the surgery and did data collection, data analysis, and interpretation. XW made English and grammar corrections, critical revisions, and approved final version. YL had the acquisition, analysis or interpretation of data for the work, revising it critically for important intellectual content, final approval of the version to be published, and agreement to be accountable for all aspects of the work in ensuring that questions related to the accuracy, or integrity of any part of the work are appropriately investigated and resolved. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
CT, Computerized tomography; ES/pPNETs, Ewing sarcoma/ peripheral primitive neuroectodermal tumors; FISH, Fluorescence in situ hybridization; GTR, Gross total resection; ITR, Incomplete tumor resection; MRI, Magnetic resonance imaging; T1WI, T1-weighted imaging; T2WI: T2-weighted imaging.
References
1. Ke C, Duan Q, Yang H, Zhu F, Yan M, Xu SP, et al. Meningeal Ewing sarcoma/peripheral PNET: clinicopathological, immunohistochemical and FISH study of four cases. Neuropathology. (2017) 37:35–44. doi: 10.1111/neup.12325
2. Mattogno PP, Nasi D, Iaccarino C, Oretti G, Santoro L, Romano A. First case of primary sellar/suprasellar-intraventricular Ewing sarcoma: case report and review of the literature. World Neurosurg. (2017) 98:869.e1–5. doi: 10.1016/j.wneu.2016.12.045
3. Yim J, Lee WS, Kim SK, Kang HJ, Bae J, Park SH. Intracranial Ewing sarcoma with whole genome study. Childs Nerv Syst. (2019) 35:547–52. doi: 10.1007/s00381-018-3997-1
4. Chen J, Jiang Q, Zhang Y, Yu Y, Zheng Y, Chen J, et al. Clinical features and long-term outcome of primary intracranial Ewing sarcoma/peripheral primitive neuroectodermal tumors: 14 cases from a single institution. World Neurosurg. (2019) 122:e1606–14. doi: 10.1016/j.wneu.2018.11.151
5. vandenHeuvel KA, Al-Rohil RN, Stevenson ME, Qian J, Gross NL, McNall-Knapp R, et al. Primary intracranial Ewing's sarcoma with unusual features. Int J Clin Exp Pathol. (2015) 8:260–74.
6. Salunke P, Sharma M, Gupta K. Ewing sarcoma of the occipital bone in an elderly patient. World Neurosurg. (2014) 81:e10–2. doi: 10.1016/j.wneu.2010.12.050
7. Cole M, Parajuli S, Laske D, Goldstein L, Morrison T, Mukherjee A, et al. Peripheral primitive neuroectodermal tumor of the dura in a 51-year-old woman following intensive treatment for breast cancer. Am J Case Rep. (2014) 15:294–9. doi: 10.12659/AJCR.890656
8. Mellai M, Caldera V, Comino A, Fortunato M, Bernucci C, Schiffer D. PNET/ESFT of the cranial vault: a case report. Clin Neuropathol. (2010) 29:372–7. doi: 10.5414/NPP29372
9. Attabib NA, West M, Rhodes RH. Peripheral primitive neuroectodermal tumor of the cavernous sinus: case report. Neurosurgery. (2006) 58:E992. doi: 10.1227/01.NEU.0000210215.73374.EE
10. D'Antonio A, Caleo A, Garcia JF, Marsilia GM, De Dominicis G, Boscaino A. Primary peripheral PNET/Ewing's sarcoma of the dura with FISH analysis. Histopathology. (2004) 45:651–4. doi: 10.1111/j.1365-2559.2004.01961.x
11. Simmons MA, Luff DA, Banerjee SS, Ramsden RT. Peripheral primitive neuroectodermal tumour (pPNET) of the cerebellopontine angle presenting in adult life. J Laryngol Otol. (2001) 115:848–52. doi: 10.1258/0022215011909161
12. Kalamarides M, Dewolf E, Shahidi A, Couvelard A, Bouccara D, Cyna-Gorse F, et al. Extraaxial primitive neuroectodermal tumor mimicking a vestibular schwannoma: diagnostic and therapeutic difficulties. Report of two cases. J Neurosurg. (2001) 94:612–6. doi: 10.3171/jns.2001.94.4.0612
13. Yang MJ, Whelan R, Madden J, Mulcahy Levy JM, Kleinschmidt-DeMasters BK, Hankinson TC, et al. Intracranial Ewing sarcoma: four pediatric examples. Childs Nerv Syst. (2018) 34:441–8. doi: 10.1007/s00381-017-3684-7
14. Cherif El Asri A, Benzagmout M, Chakour K, Chaoui MF, Laaguili J, Chahdi H, et al. Primary intracranial pPNET/Ewing sarcoma: diagnosis, management, and prognostic factors dilemma-a systematic review of the literature. World Neurosurg. (2018) 115:346–56. doi: 10.1016/j.wneu.2018.04.164
15. Choi SW, Ko H. Primary Ewing sarcoma of the squamous temporal bone with intracranial and extracranial extension: a rare cause of sudden sensorineural hearing loss. Head Neck. (2019) 41:E38–41. doi: 10.1002/hed.25449
16. Jing Z, Wen-Yi L, Jian-Li L, Jun-Lin Z, Chi D. The imaging features of meningeal Ewing sarcoma/peripheral primitive neuroectodermal tumours (pPNETs). Br J Radiol. (2014) 87:20130631. doi: 10.1259/bjr.20130631
17. Mobley BC, Roulston D, Shah GV, Bijwaard KE, McKeever PE. Peripheral primitive neuroectodermal tumor/Ewing's sarcoma of the craniospinal vault: case reports and review. Hum Pathol. (2006) 37:845–53. doi: 10.1016/j.humpath.2006.02.011
18. Idrees M, Gandhi C, Betchen S, Strauchen J, King W, Wolfe D. Intracranial peripheral primitive neuroectodermal tumors of the cavernous sinus: a diagnostic peculiarity. Arch Pathol Lab Med. (2005) 129:e11–5. doi: 10.1043/1543-2165(2005)129<e11:IPPNTO>2.0.CO;2
19. Torchia EC, Jaishankar S, Baker SJ. Ewing tumor fusion proteins block the differentiation of pluripotent marrow stromal cells. Cancer Res. (2003) 63:3464–8.
20. Mazur MA, Gururangan S, Bridge JA, Cummings TJ, Mukundan S, Fuchs H, et al. Intracranial Ewing sarcoma. Pediatr Blood Cancer. (2005) 45:850–6. doi: 10.1002/pbc.20430
21. Kumar V, Singh A, Sharma V, Kumar M. Primary intracranial dural-based Ewing sarcoma/peripheral primitive neuroectodermal tumor mimicking a meningioma: a rare tumor with review of literature. Asian J Neurosurg. (2017) 12:351–7. doi: 10.4103/1793-5482.185060
22. Velivela K, Rajesh A, Uppin MS, Purohit AK. Primary intracranial peripheral PNET"–a case report and review. Neurology India. (2014) 62:669–73. doi: 10.4103/0028-3886.149400
23. Singh AK, Srivastava AK, Pal L, Sardhara J, Yadav R, Singh S, et al. Extraosseous primary intracranial Ewing Sarcoma/peripheral primitive neuroectodermal tumor: series of seven cases and review of literature. Asian J Neurosurg. (2018) 13:288–96. doi: 10.4103/1793-5482.228570
24. Srivastava G, Jallo GI, Miller NR. Primary Ewing sarcoma of the cavernous sinus. Childs Nerv Syst. (2015) 31:1583–8. doi: 10.1007/s00381-015-2743-1
25. Desmaze C, Brizard F, Turc-Carel C, Melot T, Delattre O, Thomas G, et al. Multiple chromosomal mechanisms generate an EWS/FLI1 or an EWS/ERG fusion gene in Ewing tumors. Cancer Genet Cytogenet. (1997) 97:12–9. doi: 10.1016/S0165-4608(96)00326-3
26. Smith SC, Palanisamy N, Martin E, Almenara J, McHugh JB, Choi EK, et al. The utility of ETV1, ETV4 and ETV5 RNA in-situ hybridization in the diagnosis of CIC-DUX sarcomas. Histopathology. (2017) 70:657–63. doi: 10.1111/his.13112
27. Charville GW, Wang WL, Ingram DR, Roy A, Thomas D, Patel RM, et al. EWSR1 fusion proteins mediate PAX7 expression in Ewing sarcoma. Modern Pathol. (2017) 30:1312–20. doi: 10.1038/modpathol.2017.49
28. Maire G, Brown CW, Bayani J, Pereira C, Gravel DH, Bell JC, et al. Complex rearrangement of chromosomes 19, 21, and 22 in Ewing sarcoma involving a novel reciprocal inversion-insertion mechanism of EWS-ERG fusion gene formation: a case analysis and literature review. Cancer Genet Cytogenet. (2008) 181:81–92. doi: 10.1016/j.cancergencyto.2007.11.002
29. Thorner P, Squire J, Chilton-MacNeil S, Marrano P, Bayani J, Malkin D, et al. Is the EWS/FLI-1 fusion transcript specific for Ewing sarcoma and peripheral primitive neuroectodermal tumor? A report of four cases showing this transcript in a wider range of tumor types. Am J Pathol. (1996) 148:1125–38.
30. Navarro R, Laguna A, de Torres C, Cigudosa JC, Sunol M, Cruz O, et al. Primary Ewing sarcoma of the tentorium presenting with intracranial hemorrhage in a child. J Neurosurg. (2007) 107(Suppl. 5):411–5. doi: 10.3171/PED-07/11/411
31. Craft AW, Cotterill SJ, Bullimore JA, Pearson D. Long-term results from the first UKCCSG Ewing's Tumour Study (ET-1). United Kingdom children's cancer study group (UKCCSG) and the medical research council bone sarcoma working party. Eur J Cancer. (1997) 33:1061–9. doi: 10.1016/S0959-8049(97)00043-9
32. Craft A, Cotterill S, Malcolm A, Spooner D, Grimer R, Souhami R, et al. Ifosfamide-containing chemotherapy in Ewing's sarcoma: the second United Kingdom children's cancer study group and the medical research council Ewing's tumor study. J Clin Oncol. (1998) 16:3628–33. doi: 10.1200/JCO.1998.16.11.3628
33. Juergens C, Weston C, Lewis I, Whelan J, Paulussen M, Oberlin O, et al. Safety assessment of intensive induction with vincristine, ifosfamide, doxorubicin, and etoposide (VIDE) in the treatment of Ewing tumors in the EURO-Ewing. 99 clinical trial. Pediatr Blood Cancer. (2006) 47:22–9. doi: 10.1002/pbc.20820
34. Jain S, Kapoor G. Chemotherapy in Ewing's sarcoma. Ind J Orthop. (2010) 44:369–77. doi: 10.4103/0019-5413.69305
Keywords: ewing sarcoma, peripheral primitive neuroectodermal tumor, primary intracranial, diagnosis, central nervous system, treatment
Citation: Jiang Y, Zhao L, Wang Y, Liu X, Wu X and Li Y (2020) Primary Intracranial Ewing Sarcoma/Peripheral Primitive Neuroectodermal Tumor Mimicking Meningioma: A Case Report and Literature Review. Front. Oncol. 10:528073. doi: 10.3389/fonc.2020.528073
Received: 18 January 2020; Accepted: 28 August 2020;
Published: 06 October 2020.
Edited by:
German Torres, New York Institute of Technology, United StatesReviewed by:
Matthew Tate, Northwestern University, United StatesMagimairajan Issai Vanan, CancerCare Manitoba, Canada
Copyright © 2020 Jiang, Zhao, Wang, Liu, Wu and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yunqian Li, eXVucWlhbkBqbHUuZWR1LmNu
†These authors have contributed equally to this work