 Eivind Salmorin Birkeland
Eivind Salmorin Birkeland Lisa Maria Koch1,2†
Lisa Maria Koch1,2† Reinhard Dechant
Reinhard Dechant
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Oncol., 18 August 2020
Sec. Cancer Metabolism
Volume 10 - 2020 | https://doi.org/10.3389/fonc.2020.01561
This article is part of the Research TopicAnnual Meeting of the International Society of Cancer Metabolism (ISCaM)- Cancer Metabolic Rewiring: Mapping the Road to Clinical TranslationView all 12 articles
To adjust cell growth and proliferation to changing environmental conditions or developmental requirements, cells have evolved a remarkable network of signaling cascades that integrates cues from cellular metabolism, growth factor availability and a large variety of stresses. In these networks, cellular information flow is mostly mediated by posttranslational modifications, most notably phosphorylation, or signaling molecules such as GTPases. Yet, a large body of evidence also implicates cytosolic pH (pHc) as a highly conserved cellular signal driving cell growth and proliferation, suggesting that pH-dependent protonation of specific proteins also regulates cellular signaling. In mammalian cells, pHc is regulated by growth factor derived signals and responds to metabolic cues in response to glucose stimulation. Importantly, high pHc has also been identified as a hall mark of cancer, but mechanisms of pH regulation in cancer are only poorly understood. Here, we discuss potential mechanisms of pH regulation with emphasis on metabolic signals regulating pHc by Na+/H+-exchangers. We hypothesize that elevated NHE activity and pHc in cancer are a direct consequence of the metabolic adaptations in tumor cells including enhanced aerobic glycolysis, generally referred to as the Warburg effect. This hypothesis not only provides an explanation for the growth advantage conferred by a switch to aerobic glycolysis beyond providing precursors for accumulation of biomass, but also suggests that treatments targeting pH regulation as a potential anti-cancer therapy may effectively target the result of altered tumor cell metabolism.
Evidence for pH-dependent cell growth is largely based on experiments modulating the activity of Na+/H+-exchangers (NHE) of the SLC9A family of transport proteins. These proteins regulate intracellular pH by using the Na+ gradient to transport protons across their target membranes. NHE1-5 (SLC9A1-5) localize to the plasma-membrane and regulate pHc (1). In contrast, NHE6-9 are found in organelles of the endomembrane system to regulate luminal pH, but can also affect pHc (1, 2). NHE proteins form homo-dimers with 12 transmembrane domains located in the N-terminal part of the protein and a large C-terminal cytoplasmic domain, which is target of several kinases. The C-terminal domain also interacts with phospholipids and the actin cytoskeleton to regulate cell migration and contribute to metastasis [Figure 1A and Ref. (3)].
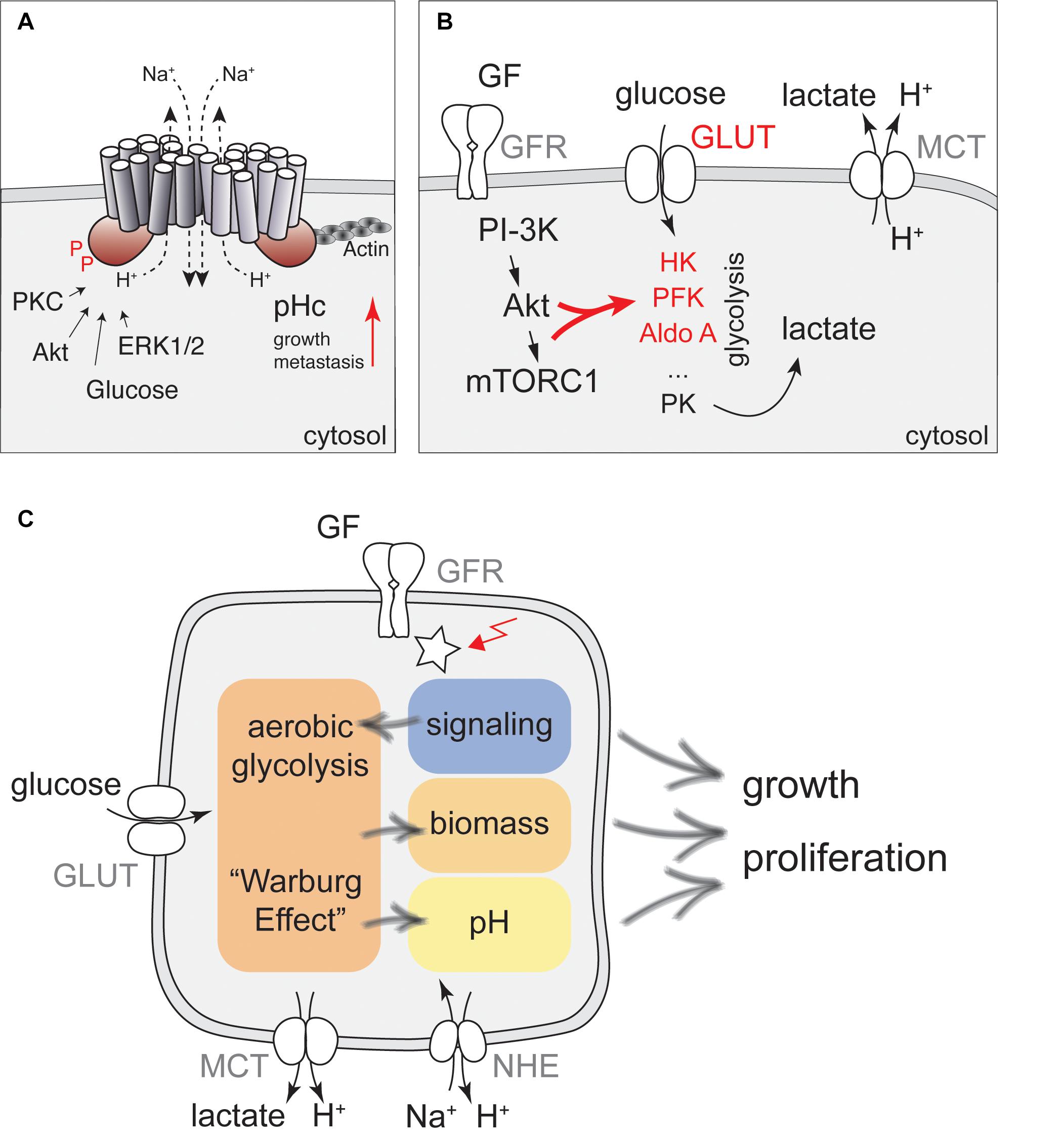
Figure 1. A network of cellular metabolism and cellular signaling governs cell growth and regulation of pHc. (A) NHE1 is a key regulator of pHc in mammalian cells. A schematic representation of the NHE1 structure together with key regulatory inputs and potential functions for cancer development is shown. (B) Regulation of glycolytic activity by PI3K/Akt signaling. Glucose transporters (GLUT) and the metabolic enzymes hexokinase (HK), phosphofructokinase (PFK), aldolase A (Aldo A) indicated in red are all direct or indirect targets of growth factor (GF) dependent regulation, leading to enhanced aerobic glycolysis upon stimulation with GFs. Glycolytic activity is also directly coupled to pHc regulation by lactate export using Monocarboxylate transporters. (C) Proposed model for how signaling, metabolism, and pH interact to regulate cell growth and proliferation. Glucose metabolism can be stimulated by glucose, growth factors and oncogenic activation (red arrow) to produce precursors for biomass production, but may also increase pHc through activation of NHE activity, further contributing to enhanced cell growth and proliferation.
In addition to NHEs, several other regulators of pHc have been identified. These include Na+/Bicarbonate transporters and monocarboxylate transporters (MCTs), ATP-driven proton pumps, as well as carbonic anhydrases (4). All these pH regulators have been linked to a growing number of physiological activities including regulation of cellular signaling, transcription and cell growth, and have been associated with cancer (5–9). While NHE proteins are ubiquitously expressed to regulate intracellular pH (10), other pH regulators become critical only under specific conditions. For example, MCTs, which transport lactate and protons across the plasma-membrane, are critical to maintain pHc in rapidly proliferating cells and primary tumors, but are less important in differentiated cells (11–14).
A large body of evidence suggests that activation of NHE proteins upon growth factor stimulation is a critical step in promoting cell growth and proliferation (15). Growth factor stimulation triggers an increase in pHc of about 0.2–0.3 pH units (16–24). As this increase in pH is tightly correlated with increased Na+ influx and blocked by amiloride, it was concluded that pH regulation depends on Na+/H+-exchangers. Similar conclusions were drawn from overexpression or injection of activated Ras into quiescent cells (25, 26), or overexpression of v-Mos (26). Importantly, increased pHc is also necessary for initiation of cell-cycle progression under these conditions (25, 27). Although the increase in pHc is maximal several minutes following injection of active Ras, addition of amiloride as late as 6 h after the injection of the activated protein effectively suppresses DNA replication (25), suggesting that high pHc may act at a specific step during early G1. Interestingly, increased pHc in response to growth factor stimulation is also necessary for high translation efficiency and correlates with phosphorylation of the ribosomal protein S6 (28), a critical target of the mTORC1 pathway (29). mTORC1 activity was subsequently confirmed to be sensitive to pH (30) and inhibition of NHE activity (31). The underlying mechanisms remain to be fully established, but may involve regulation of amino-acid uptake by macropinocytosis (31).
In addition, elevated pHc may further promote cell growth by regulating metabolism through the intrinsic pH-sensitivity of metabolic enzymes. In particular, phosphofructokinase and lactate dehydrogenase have received special attention due to a sharp pH optimum at slightly alkaline pH, which may lead to enhanced glycolysis and glucose uptake upon NHE activation (32–34).
Increased NHE activity has also been linked to multiple aspects of cancer and causes a reversed proton gradient across the plasma-membrane due to elevated pHc, enhanced tumor growth and invasion (3, 35–38). Accordingly, pharmacological inhibitors of NHE activity have been proposed as anti-cancer therapies (39–42). Indeed, the FDA approved drug amiloride, a broad specificity NHE inhibitor and potassium sparing diuretic, or derivatives thereof, are highly effective in mouse models for multiple myeloma (43) and pancreatic cancer (2). High NHE expression correlates with poor survival in several cancer types, further underscoring the importance of pH regulation in these diseases (2, 44). Mechanisms for increasing NHE activity may vary in different cancers and might include increasing the specific activity of NHE proteins. Therefore, detailed understanding of NHE regulation is needed to better understand their contribution to the disease.
Regulation of NHE activity in response mitogenic stimuli has been mostly attributed to direct phosphorylation of NHE proteins by growth factor activated kinases. Indeed, activation of Protein Kinase C (PKC) by phorbol esters (15–17, 45–47), or its endogenous activator diacyl-glyceride triggers an NHE-dependent increase in pHc (16, 48, 49), while inhibition of PKC by trifluoperazine can abolish the rise in pHc upon growth factor stimulation (50). Thus, it was suggested that PKC is the primary target downstream of growth factors for pH regulation. Yet, as no PKC-dependent phosphorylation sites on NHE proteins have been identified, the effect of PKC on NHE activation may be indirect.
In contrast, both Akt and Erk1/2 have been suggested to directly phosphorylate NHE1 (Figure 1A). In vitro, Akt phosphorylates NHE1 at Ser-648, while Erk1/2 phosphorylates NHE1 at Ser-770 and Ser-771 (51–55). Akt is required to reestablish physiological pH following an acidification stress due to acid loading and mutating S648 to a non-phosphorylatable residue impairs NHE1 function (52), strongly suggesting that Akt-dependent phosphorylation of NHE1 is key for increased pHc and cell proliferation. Yet, evidence for in vivo phosphorylation of this site has remained limiting. Nevertheless, these phosphorylation sites are conserved in NHE2 and NHE4, and other NHE proteins harbor MAPK or CDK consensus sites (SP) at the corresponding position. Thus, phosphorylation might be a general mechanism for the regulation of Na+/H+-exchangers.
Excellent reviews are available that summarize NHE regulation by phosphorylation (10, 51, 53). Here, we rather focus on the interaction between cellular metabolism and pHc. We hypothesize that an increased pHc and increased glycolytic activity, both commonly found in cancers, are two sides of the same coin, which contribute to enhanced cell growth and proliferation. Specifically, we discuss potential mechanisms of regulation of Na+/H+-exchange by cellular metabolism as a mechanism to control cell growth.
Several reports have shown that glucose stimulation of cells is sufficient to increase pHc in an NHE1-dependent manner. Specifically, glucose availability stimulates pHc via NHEs in pancreatic beta-cells, which may contribute to glucose stimulated insulin release (56–60). NHE-dependent regulation of pHc by glucose was also observed in liver or muscle cells (61–63), but the underlying mechanisms are only poorly understood. Importantly, glucose-dependent regulation of pHc can be observed in the absence of growth-factors (59, 60), and inhibition of glycolysis by 2-deoxy-glucose strongly decreased NHE activity (64–66), suggesting that Na+/H+-exchange is regulated by a metabolic signal derived from glycolysis, or might be coupled to energy metabolism.
Increased glucose uptake and metabolism to fuel aerobic glycolysis is also a general feature of mitogenic stimulation under physiological conditions and oncogenic transformation [Figure 1B and Refs. (67–69)] and is generally referred to as the Warburg effect. Although it was originally assumed that tumor cells upregulate glycolysis even in the presence of oxygen due to defective mitochondria, it is now clear that increased rates of glycolysis allow the redirection of metabolic fluxes toward more efficient biomass production (69, 70) in actively proliferating cells and form the molecular basis of using PET scans for tumor detection (71).
However, if NHE activity is regulated by a metabolic signal in response to increased glucose concentration, the same mechanisms activating NHE activity should also be in place when glucose uptake and glycolysis are activated by growth factors or oncogenic transformation. Thus, understanding the regulation of NHE activity by metabolic cues might identify mechanisms how altered tumor cell metabolism contributes to elevated pHc and enhanced cell growth (Figure 1C).
Activation of aerobic glycolysis is best understood in response to PI3-kinase signaling and Akt activation, which has been suggested to be the Warburg kinase (72) (Figure 1B). Akt is required for growth factor-stimulated glucose uptake by triggering translocation of glucose transporters (GLUTs) to the plasma-membrane (73–75), via phosphorylation of the GTPase activating protein (GAP) for Rab10, AS160 (76). mTORC1 activation downstream of PI3-kinase/Akt signaling also promotes expression of Hexokinase II (77–79). In addition, Akt phosphorylates Hexokinase to promote its association with mitochondria, which protects cells from apoptosis, but may not contribute to enhanced enzymatic activity (80). Akt also indirectly stimulates phosphofructokinase 1 (PFK-1), the major control point of glycolysis. Specifically, Akt promotes the accumulation of the glycolytic side-product and most potent allosteric activator of PFK-1, Fructose-2,6-bisphosphate by phosphorylation of PFK-2 (81, 82). Finally, PI3-kinase signaling increases Aldolase A activity (83). Collectively, these processes lead to enhanced glycolytic activity and ATP production (84).
The PI3-Kinase/Akt pathway is probably the most frequently activated pathway in cancer, therefore explaining the shift toward high aerobic glycolysis in a large number of tumors. However, at least on a transcriptional level, upregulation of glucose uptake and glycolysis can also be observed upon activation of c-myc, mTORC1, and K-Ras, or upon loss of p53 (85–91), further underscoring the importance of enhanced aerobic glycolysis to sustain, or even signal enhanced cell growth.
How could glucose metabolism be linked to NHE-dependent pH regulation? In principle, glucose could regulate the specific activity of NHE proteins, or affect the Na+ gradient by the Na+/K+-ATPase (Figure 2A). Similar to NHE proteins, the Na+/K+-ATPase is also subject to growth factor-dependent activation and the same kinases regulating NHEs have been linked to activation of the Na+/K+-ATPase (92–94). In particular, PKC directly phosphorylates Na+/K+-ATPase, possibly explaining PKC-dependent activation of Na+/H+-exchange (95). Indeed, Na+/K+-ATPase activity rapidly increases upon glucose stimulation in different cell types (96, 97), which could translate into changes of pHc.
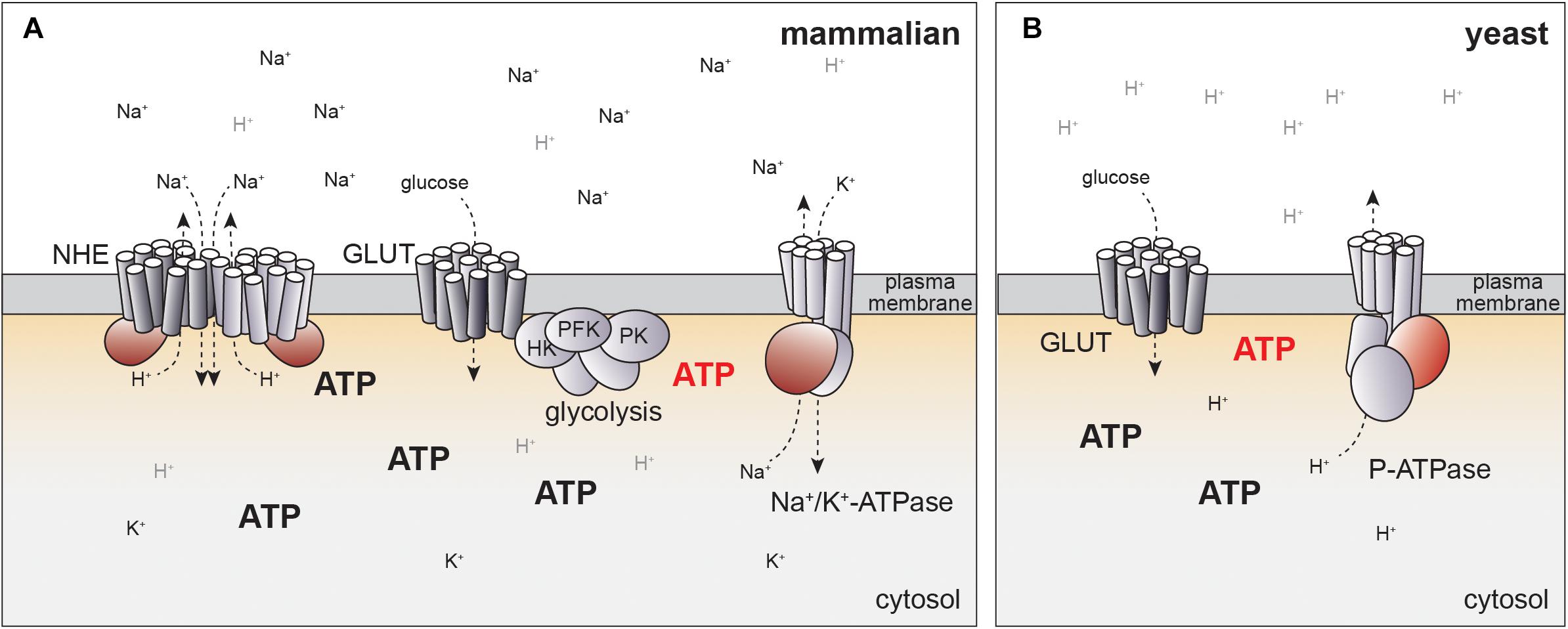
Figure 2. Potential mechanisms for how NHE activity can be linked to glucose metabolism. (A) In mammalian cells, pHc is regulated by NHE activity, which may be linked to ATP production directly or indirectly via Na+/K+-ATPase. Localized production of ATP by glycolysis at the plasma membrane may generate distinct pools of ATP (red) that might help to explain coupling of cellular metabolism to pH regulation. Potential regulatory or catalytic ATP binding sites in ion pumps and exchangers are indicated in red. See text for details. (B) Evolutionary conservation of glucose-dependent regulation of pHc. In yeast, pHc is regulated by an ATP-dependent proton pump (P-ATPase) independent of Na+/H+ exchange. P-ATPase activity is coupled to glycolytic activity, but molecular mechanisms remain to be fully established. Color code for ATP regulated domains and hypothetical localized ATP pools same as in panel (A).
At least two key kinases regulating NHEs respond to changes in cellular metabolism. PKC is regulated by glucose availability through modulating the levels of its activator DAG (98). Similarly, Erk1/2 activity is subject to glucose regulation (99, 100). Thus, both kinases could directly or indirectly contribute to NHE activation in response to glucose (101). Yet, careful assessment of the basal activity of these kinases and NHE phosphorylation in the absence of growth factors would be required to study a potential role in glucose-dependent regulation of pHc, before potential molecular mechanisms can be addressed.
Conceptually, direct coupling of metabolism to NHE activity may be more appealing. In particular, coupling of glycolytic activity, or flux, to pH regulation would readily explain the observed increase in pHc by glucose availability, stimulation of glucose metabolism by mitogenic activation, and metabolic reprogramming in cancer. Interestingly, in all highly glycolytic cells, glycolysis is directly coupled to pHc via MCTs, which secrete lactic acid, the end-product of fermentation (102, 103). This is also the basis of using extracellular acidification rates as a means of estimating glycolytic flux (104, 105). Yet, it is less clear how glycolysis could be coupled to NHE activity.
It has been hypothesized that sensing of metabolic flux may depend on the accumulation of metabolites, which tightly correlate with pathway activity and trigger the appropriate cellular response (106). For example, the abundance of fructose-1,6-bisphosphate (FBP) tightly correlates with glycolytic flux in yeast and bacteria, and binding of FBP to a transcription factor allows for coupling of glycolytic flux to gene expression (107, 108). Similarly, in pancreatic beta-cells, ATP correlates with glucose concentration and glycolytic activity and triggers insulin secretion by binding to ATP-sensitive K+-channels (109).
ATP concentration may also link glycolytic flux to the regulation of pHc by NHE activity. As discussed above, Akt activation results in increased ATP concentrations (84). Moreover, the establishment of the Na+ gradient necessary for NHE activity consumes a large fraction of cellular ATP production (110), suggesting tight linkage to energy metabolism.
Indeed, NHE1 activity directly depends on the presence of ATP. Basal Na+/H+-exchange can occur in the absence of ATP based on the concentration gradient of the transported cations (111, 112). Yet, depletion of cells from ATP abolishes NHE activity (65, 66), while readdition of ATP restores NHE activity in patch-clamp experiments (65). Half maximal activation of NHE1 was achieved at 5 mM ATP, suggesting that NHE activity could be modulated by changes in the ATP concentration in vivo. Interestingly, ATP depletion reduces the affinity of NHE proteins to protons by 0.5 pH units, readily explaining ATP-dependent pH regulation (66, 113).
Surprisingly, NHE activity could also be triggered by the poorly hydrolysable ATP analog ATPγS and did not depend on the presence of Mg2+ (65). Thus, ATP-dependency is unlikely to be mediated by associated kinases or ATPases. Instead, Na+/H+-exchange may depend on direct binding of ATP to NHE proteins or a membrane associated activator. Consistently, ATP dependence of NHE1 requires the presence of its cytoplasmic C-terminal domain (114, 115). Although no consensus sequences from known ATP-binding motives can be identified in the primary sequence, cross-linking experiments have revealed evidence for direct binding of ATP to the cytoplasmic domain of NHE1 (116). While mapping of the potential ATP binding site will be required to generate mutants to directly test the significance of ATP binding in vivo, this model offers an attractive mechanism of coupling metabolic activity to NHE activity (Figure 2A).
An alternative model for coupling of energy metabolism to regulation of pHc through ATP-dependent ion pumps may be suggested by evolutionary considerations of pH-dependent cell growth. In yeast, pHc is regulated by a P-type ATPase, PMA1, that directly pumps protons across the plasma-membrane in an ATP-dependent manner (117, 118), but does not require NHE activity at the plasma-membrane (Figure 2B). PMA1 activity and pHc increase with the quality and quantity of the available carbon source (119). As in mammalian cells, high pHc drives cell growth and proliferation, at least in part, by activating TORC1 (99, 119).
The differences in pH regulation in yeast and mammals are readily explained by the different environmental constraints for single cellular organisms and cells embedded within a complex organism. In their natural environment yeast cells are constantly exposed to changes in osmolarity and thus may rely on ATP-dependent proton pumps rather than a Na+ gradient. In contrast, establishing a proton gradient with a similar concentration profile as the Na+ gradient in mammalian cells (120, 121) would yield pH differences of more than 1 pH unit across the plasma-membrane and may thus require indirect regulation of pH via Na+-H+-exchange. Yet, the conservation of pHc as a glucose-dependent signal regulating cell growth indicates that regulation of pH in mammalian cells may also be mediated by coupling of Na+/K+-ATPase to glucose metabolism (Figure 2A).
Indeed, several ATP-dependent ion pumps have been suggested to be regulated by energy metabolism in mammalian cells. In particular, direct modulation by ATP has been proposed for ATP-dependent cation exchangers and ion channels (122–130) despite the fact that the Km of these pumps for ATP hydrolysis have been consistently found to be significantly lower than physiological ATP concentrations (131–133), making a direct coupling of their activity to changing ATP concentrations in cells unlikely. To resolve this contradiction, it has been suggested that these pumps may be coupled to glycolytic ATP production by concentrating ATP producing enzymes close to the ATP-dependent pumps, thereby allowing physical or kinetic coupling of ATP synthesis to hydrolysis (124, 134). For example, glycolytic enzymes including hexokinase, phosphofructokinase and pyruvate kinase co-purify with the plasma-membrane in pancreatic cancer cells, which may allow direct regulation of an ATP-dependent Ca2+-pump (124). In cardiomyocytes and erythrocytes, glycolytic enzymes localize at the plasma-membrane (130, 135) and have been suggested to regulate Na+/K+-ATPase (122) as well as ATP-sensitive K+-channels (130, 134), possibly by direct interaction and localized ATP production (125).
Theoretical considerations argue against the formation of localized pools of metabolites based on local enrichment of metabolic enzymes, as rapid intracellular diffusion of metabolites outcompetes even the fastest enzymes, leading to rapid dissipation of local concentration differences (136). Yet, it remains a possibility that in vitro determination of enzymatic parameters fall short of accurately replicating the specific conditions in the microenvironment at the target membranes. Interestingly, indirect measurements of local ATP concentrations by targeting luciferase, which emits light in an ATP-dependent manner, to different cellular locations clearly support the existence of separated, local pools of ATP. Specifically, this method allows to follow the dynamics of ATP concentration in different cellular compartments following a glucose pulse. While ATP only very transiently accumulates in the cytosol upon glucose stimulation, ATP stabilizes at elevated levels at the plasma-membrane upon glucose stimulation (137). More detailed measurements of localized metabolite distributions, for example using FRET reporters would be key to further support localized pools of metabolites regulating ATP-dependent processes at the plasma-membrane. Similarly, to discriminate between direct and indirect mechanisms to couple metabolism to NHE activity, dynamic measurements of Na+/K+-ATPase and/or the resulting Na+ gradient would be required.
While the molecular mechanisms linking glucose or energy metabolism to increased pHc await further clarification, the existing data strongly argue for a tight coupling of glucose metabolism to NHE activity. As glucose metabolism can be stimulated by increasing glucose concentration or induction of the Warburg effect, this coupling readily explains the elevated pHc found in cancer cells. In turn, an elevated pHc may further enhance aerobic glycolysis due to the pH-sensitivity of phosphofructokinase (33) as part of a positive feedback loop to regulate cell growth. Thus, an elevated pHc may act as a signal that relays changes in cellular metabolism to cell growth and proliferation, but further identification of pH-sensitive steps governing cell growth and proliferation will be required to better understand the functional importance of pHc in normal and cancer cells.
Targeting cellular metabolism for cancer therapy has triggered great interest as potential treatments could be widely applicable to a range of tumors with similar metabolic alterations. Indeed, several glycolytic inhibitors have been tested as potential anti-cancer treatments, but have largely failed due to high toxicity at the effective dose (138, 139). Targeting pH regulation by NHE proteins may act as a treatment of the consequences of altered tumor metabolism and may help to devise novel treatment strategies. While similar considerations of toxicity by targeting NHE proteins may apply, a better understanding of NHE regulation in normal and cancer cells will help to further dissect the interplay between cellular metabolism and signaling, and to define windows of opportunities to treat cancer by targeting pHc regulation.
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
The work in the Dechant laboratory was supported by the SNF and ETHZ (ETH Zurich Research Grant 28 17-2).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors thank Sung Sik Lee and Alfredo J. Ibanez for helpful comments on the manuscript.
1. Fuster DG, Alexander RT. Traditional and emerging roles for the SLC9 Na+/H+ exchangers. Pflugers Arch. (2014) 466:61–76. doi: 10.1007/s00424-013-1408-8
2. Galenkamp KMO, Sosicka P, Jung M, Recouvreux MV, Zhang Y, Moldenhauer MR, et al. Golgi acidification by NHE7 regulates cytosolic pH homeostasis in pancreatic cancer cells. Cancer Discov. (2020) 10:822–35. doi: 10.1158/2159-8290.cd-19-1007
3. Cardone RA, Casavola V, Reshkin SJ. The role of disturbed pH dynamics and the Na+/H+ exchanger in metastasis. Nat Rev Cancer. (2005) 5:786–95. doi: 10.1038/nrc1713
4. Casey JR, Grinstein S, Orlowski J. Sensors and regulators of intracellular pH. Nat Rev Mol Cell Biol. (2009) 11:50–61. doi: 10.1038/nrm2820
5. Raja DA, Gotherwal V, Burse SA, Subramaniam YJ, Sultan F, Vats A, et al. pH-controlled histone acetylation amplifies melanocyte differentiation downstream of MITF. EMBO Rep. (2020) 21:e48333.
6. Pamarthy S, Kulshrestha A, Katara GK, Beaman KD. The curious case of vacuolar ATPase: regulation of signaling pathways. Mol Cancer. (2018) 17:41.
7. Gorbatenko A, Olesen CW, Boedtkjer E, Pedersen SF. Regulation and roles of bicarbonate transporters in cancer. Front Physiol. (2014) 5:130.
8. Supuran CT, Winum JY. Carbonic anhydrase IX inhibitors in cancer therapy: an update. Future Med Chem. (2015) 7:1407–14. doi: 10.4155/fmc.15.71
9. Tozzi M, Sorensen CE, Magni L, Christensen NM, Bouazzi R, Buch CM, et al. proton pump inhibitors reduce pancreatic adenocarcinoma progression by selectively targeting H(+), K(+)-ATPases in pancreatic cancer and stellate cells. Cancers (Basel). (2020) 12:640. doi: 10.3390/cancers12030640
10. Baumgartner M, Patel H, Barber DL. Na(+)/H(+) exchanger NHE1 as plasma membrane scaffold in the assembly of signaling complexes. Am J Physiol Cell Physiol. (2004) 287:C844–50.
11. Tasdogan A, Faubert B, Ramesh V, Ubellacker JM, Shen B, Solmonson A, et al. Metabolic heterogeneity confers differences in melanoma metastatic potential. Nature. (2020) 577:115–20. doi: 10.1038/s41586-019-1847-2
12. Flores A, Schell J, Krall AS, Jelinek D, Miranda M, Grigorian M, et al. Lactate dehydrogenase activity drives hair follicle stem cell activation. Nat Cell Biol. (2017) 19:1017–26. doi: 10.1038/ncb3575
13. Counillon L, Bouret Y, Marchiq I, Pouyssegur J. Na(+)/H(+) antiporter (NHE1) and lactate/H(+) symporters (MCTs) in pH homeostasis and cancer metabolism. Biochim Biophys Acta. (2016) 1863:2465–80. doi: 10.1016/j.bbamcr.2016.02.018
14. Marchiq I, Le Floch R, Roux D, Simon MP, Pouyssegur J. Genetic disruption of lactate/H+ symporters (MCTs) and their subunit CD147/BASIGIN sensitizes glycolytic tumor cells to phenformin. Cancer Res. (2015) 75:171–80. doi: 10.1158/0008-5472.can-14-2260
15. Moolenaar WH. Effects of growth factors on intracellular pH regulation. Annu Rev Physiol. (1986) 48:363–76. doi: 10.1146/annurev.ph.48.030186.002051
16. Moolenaar WH, Tertoolen LG, De Laat SW. Phorbol ester and diacylglycerol mimic growth factors in raising cytoplasmic pH. Nature. (1984) 312:371–4. doi: 10.1038/312371a0
17. Burns CP, Rozengurt E. Serum, platelet-derived growth factor, vasopressin and phorbol esters increase intracellular pH in Swiss 3T3 cells. Biochem Biophys Res Commun. (1983) 116:931–8. doi: 10.1016/s0006-291x(83)80231-9
18. Moolenaar WH, Tsien RY, Van Der Saag PT, De Laat SW. Na+/H+ exchange and cytoplasmic pH in the action of growth factors in human fibroblasts. Nature. (1983) 304:645–8. doi: 10.1038/304645a0
19. Cassel D, Rothenberg P, Zhuang YX, Deuel TF, Glaser L. Platelet-derived growth factor stimulates Na+/H+ exchange and induces cytoplasmic alkalinization in NR6 cells. Proc Natl Acad Sci USA. (1983) 80:6224–8. doi: 10.1073/pnas.80.20.6224
20. Rothenberg P, Glaser L, Schlesinger P, Cassel D. Activation of Na+/H+ exchange by epidermal growth factor elevates intracellular pH in A431 cells. J Biol Chem. (1983) 258:12644–53.
21. Hesketh TR, Moore JP, Morris JD, Taylor MV, Rogers J, Smith GA, et al. A common sequence of calcium and pH signals in the mitogenic stimulation of eukaryotic cells. Nature. (1985) 313:481–4. doi: 10.1038/313481a0
22. Lopez-Rivas A, Stroobant P, Waterfield MD, Rozengurt E. Ionic responses rapidly elicited by porcine platelet-derived growth factor in Swiss 3T3 cells. EMBO J. (1984) 3:939–44. doi: 10.1002/j.1460-2075.1984.tb01911.x
23. Moore RD. Stimulation of Na:H exchange by insulin. Biophys J. (1981) 33:203–10. doi: 10.1016/s0006-3495(81)84881-3
24. Putnam RW. Effect of insulin on intracellular pH in frog skeletal muscle fibers. Am J Physiol. (1985) 248(3 Pt 1):C330–6.
25. Hagag N, Lacal JC, Graber M, Aaronson S, Viola MV. Microinjection of ras p21 induces a rapid rise in intracellular pH. Mol Cell Biol. (1987) 7:1984–8. doi: 10.1128/mcb.7.5.1984
26. Doppler W, Jaggi R, Groner B. Induction of v-mos and activated Ha-ras oncogene expression in quiescent NIH 3T3 cells causes intracellular alkalinisation and cell-cycle progression. Gene. (1987) 54:147–53. doi: 10.1016/0378-1119(87)90357-x
27. Pouyssegur J, Franchi A, L’Allemain G, Paris S. Cytoplasmic pH, a key determinant of growth factor-induced DNA synthesis in quiescent fibroblasts. FEBS Lett. (1985) 190:115–9. doi: 10.1016/0014-5793(85)80439-7
28. Chambard JC, Pouyssegur J. Intracellular pH controls growth factor-induced ribosomal protein S6 phosphorylation and protein synthesis in the G0—-G1 transition of fibroblasts. Exp Cell Res. (1986) 164:282–94. doi: 10.1016/0014-4827(86)90029-7
29. Fingar DC, Blenis J. Target of rapamycin (TOR): an integrator of nutrient and growth factor signals and coordinator of cell growth and cell cycle progression. Oncogene. (2004) 23:3151–71. doi: 10.1038/sj.onc.1207542
30. Balgi AD, Diering GH, Donohue E, Lam KK, Fonseca BD, Zimmerman C, et al. Regulation of mTORC1 Signaling by pH. PLoS One. (2011) 6:e21549.
31. Charpentier JC, Chen D, Lapinski PE, Turner J, Grigorova I, Swanson JA, et al. Macropinocytosis drives T cell growth by sustaining the activation of mTORC1. Nat Commun. (2020) 11:180.
32. Goto Y, Kida K, Kaino Y, Ito T, Matsuda H. Inhibitory effect of amiloride on glucose transport in isolated rat adipocytes. Diabetes Res Clin Pract. (1993) 20:1–5. doi: 10.1016/0168-8227(93)90015-w
33. White KA, Grillo-Hill BK, Barber DL. Cancer cell behaviors mediated by dysregulated pH dynamics at a glance. J Cell Sci. (2017) 130:663–9. doi: 10.1242/jcs.195297
34. Flinck M, Kramer SH, Pedersen SF. Roles of pH in control of cell proliferation. Acta Physiol (Oxf). (2018) 223:e13068. doi: 10.1111/apha.13068
35. Webb BA, Chimenti M, Jacobson MP, Barber DL. Dysregulated pH: a perfect storm for cancer progression. Nat Rev Cancer. (2011) 11:671–7. doi: 10.1038/nrc3110
36. Huber V, De Milito A, Harguindey S, Reshkin SJ, Wahl ML, Rauch C, et al. Proton dynamics in cancer. J Transl Med. (2010) 8:57.
37. Orive G, Reshkin SJ, Harguindey S, Pedraz JL. Hydrogen ion dynamics and the Na+/H+ exchanger in cancer angiogenesis and antiangiogenesis. Br J Cancer. (2003) 89:1395–9. doi: 10.1038/sj.bjc.6601286
38. Reshkin SJ, Cardone RA, Harguindey S. Na+-H+ exchanger, pH regulation and cancer. Recent Patents Anti Cancer Drug discover. (2013) 8:85–99. doi: 10.2174/1574892811308010085
39. Harguindey S, Orive G, Luis Pedraz J, Paradiso A, Reshkin SJ. The role of pH dynamics and the Na+/H+ antiporter in the etiopathogenesis and treatment of cancer. Two faces of the same coin–one single nature. Biochim Biophys Acta. (2005) 1756:1–24. doi: 10.1016/j.bbcan.2005.06.004
40. Harguindey S, Arranz JL, Wahl ML, Orive G, Reshkin SJ. Proton transport inhibitors as potentially selective anticancer drugs. Anticancer Res. (2009) 29:2127–36.
41. Harguindey S, Arranz JL, Polo Orozco JD, Rauch C, Fais S, Cardone RA, et al. Cariporide and other new and powerful NHE1 inhibitors as potentially selective anticancer drugs–an integral molecular/biochemical/metabolic/clinical approach after one hundred years of cancer research. J Transl Med. (2013) 11:282. doi: 10.1186/1479-5876-11-282
42. Harguindey S, Koltai T, Reshkin SJ. Curing cancer? Further along the new pH-centric road and paradigm. Oncoscience. (2018) 5:132–3. doi: 10.18632/oncoscience.422
43. Rojas EA, Corchete LA, San-Segundo L, Martinez-Blanch JF, Codoner FM, Paino T, et al. Amiloride, an old diuretic drug, is a potential therapeutic agent for multiple myeloma. Clin Cancer Res. (2017) 23:6602–15. doi: 10.1158/1078-0432.ccr-17-0678
44. Uhlen M, Zhang C, Lee S, Sjostedt E, Fagerberg L, Bidkhori G, et al. A pathology atlas of the human cancer transcriptome. Science (2017) 357:6352.
45. Nishizuka Y. The role of protein kinase C in cell surface signal transduction and tumour promotion. Nature. (1984) 308:693–8. doi: 10.1038/308693a0
46. Whiteley B, Cassel D, Zhuang YX, Glaser L. Tumor promoter phorbol 12-myristate 13-acetate inhibits mitogen-stimulated Na+/H+ exchange in human epidermoid carcinoma A431 cells. J Cell Biol. (1984) 99:1162–6. doi: 10.1083/jcb.99.3.1162
47. Grinstein S, Cohen S, Goetz JD, Rothstein A, Gelfand EW. Characterization of the activation of Na+/H+ exchange in lymphocytes by phorbol esters: change in cytoplasmic pH dependence of the antiport. Proc Natl Acad Sci USA. (1985) 82:1429–33. doi: 10.1073/pnas.82.5.1429
48. Grinstein S, Cohen S, Goetz JD, Rothstein A. Osmotic and phorbol ester-induced activation of Na+/H+ exchange: possible role of protein phosphorylation in lymphocyte volume regulation. J Cell Biol. (1985) 101:269–76. doi: 10.1083/jcb.101.1.269
49. Vara F, Schneider JA, Rozengurt E. Ionic responses rapidly elicited by activation of protein kinase C in quiescent Swiss 3T3 cells. Proc Natl Acad Sci USA. (1985) 82:2384–8. doi: 10.1073/pnas.82.8.2384
50. Grinstein S, Cohen S, Goetz JD, Rothstein A, Mellors A, Gelfand EW. Activation of the Na+-H+ antiport by changes in cell volume and by pnorbol esters; possible role of protein kinase. In: Arnost KFB, Peter S A, Walter F B editors. Current Topics in Membranes and Transport. Cambridge, MA: Academic Press (1986). p.26.
51. Amith SR, Fliegel L. Regulation of the Na+/H+ Exchanger (NHE1) in breast cancer metastasis. Cancer Res. (2013) 73:1259–64. doi: 10.1158/0008-5472.can-12-4031
52. Meima ME, Webb BA, Witkowska HE, Barber DL. The sodium-hydrogen exchanger NHE1 is an Akt substrate necessary for actin filament reorganization by growth factors. J Biol Chem. (2009) 284:26666–75. doi: 10.1074/jbc.m109.019448
53. Fliegel L. Structural and functional changes in the Na(+)/H(+) exchanger isoform 1, Induced by Erk1/2 phosphorylation. Int J Mol Sci. (2019) 20:2378. doi: 10.3390/ijms20102378
54. Malo ME, Li L, Fliegel L. Mitogen-activated protein kinase-dependent activation of the Na+/H+ exchanger is mediated through phosphorylation of amino acids Ser770 and Ser771. J Biol Chem. (2007) 282:6292–9. doi: 10.1074/jbc.m611073200
55. Wang H, Silva NL, Lucchesi PA, Haworth R, Wang K, Michalak M, et al. Phosphorylation and regulation of the Na+/H+ exchanger through mitogen-activated protein kinase. Biochemistry. (1997) 36:9151–8. doi: 10.1021/bi970802f
56. Stiernet P, Nenquin M, Moulin P, Jonas JC, Henquin JC. Glucose-induced cytosolic pH changes in beta-cells and insulin secretion are not causally related: studies in islets lacking the Na+/H+ exchangeR NHE1. J Biol Chem. (2007) 282:24538–46. doi: 10.1074/jbc.m702862200
57. Gunawardana SC, Rocheleau JV, Head WS, Piston DW. Nutrient-stimulated insulin secretion in mouse islets is critically dependent on intracellular pH. BMC Endocr Disord. (2004) 4:1.
58. Lindstrom P, Sehlin J. Effect of glucose on the intracellular pH of pancreatic islet cells. Biochem J. (1984) 218:887–92. doi: 10.1042/bj2180887
59. Juntti-Berggren L, Arkhammar P, Nilsson T, Rorsman P, Berggren PO. Glucose-induced increase in cytoplasmic pH in pancreatic beta-cells is mediated by Na+/H+ exchange, an effect not dependent on protein kinase C. J Biol Chem. (1991) 266:23537–41.
60. Trebilcock R, Lynch A, Tomlinson S, Best L. Na+/H+ exchange is responsible for intracellular pH regulation in insulin-secreting HIT-T15 cells. Mol Cell Endocrinol. (1990) 71:21–5. doi: 10.1016/0303-7207(90)90070-o
61. Comolli R, Casale A, Mariotti D. Amiloride and glucose effects on the intracellular pH of Yoshida rat ascites hepatoma AH-130 cells grown in vivo. Cell Biol Int Rep. (1984) 8:297–307. doi: 10.1016/0309-1651(84)90156-5
62. Siczkowski M, Ng LL. Glucose-induced changes in activity and phosphorylation of the Na+/H+ exchanger, NHE-1, in vascular myocytes from Wistar-Kyoto and spontaneously hypertensive rats. Metabolism. (1996) 45:114–9. doi: 10.1016/s0026-0495(96)90208-5
63. Williams B, Howard RL. Glucose-induced changes in Na+/H+ antiport activity and gene expression in cultured vascular smooth muscle cells. Role of protein kinase C. J Clin Invest. (1994) 93:2623–31. doi: 10.1172/jci117275
64. Demaurex N, Grinstein S. Na+/H+ antiport: modulation by ATP and role in cell volume regulation. J Exp Biol. (1994) 196:389–404.
65. Demaurex N, Romanek RR, Orlowski J, Grinstein SATP. dependence of Na+/H+ exchange. Nucleotide specificity and assessment of the role of phospholipids. J Gen Physiol. (1997) 109:117–28.
66. Cassel D, Katz M, Rotman M. Depletion of cellular ATP inhibits Na+/H+ antiport in cultured human cells. Modulation of the regulatory effect of intracellular protons on the antiporter activity. J Biol Chem. (1986) 261:5460–6.
67. Baltazar F, Afonso J, Costa M, Granja S. Lactate beyond a waste metabolite: metabolic affairs and signaling in malignancy. Front Oncol. (2020) 10:231.
68. Lunt SY, Vander Heiden MG. Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu Rev Cell Dev Biol. (2011) 27:441–64. doi: 10.1146/annurev-cellbio-092910-154237
69. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. (2009) 324:1029–33. doi: 10.1126/science.1160809
70. DeBerardinis RJ, Chandel NS. We need to talk about the Warburg effect. Nat Metab. (2020) 2:127–9. doi: 10.1038/s42255-020-0172-2
71. Bensinger SJ, Christofk HR. New aspects of the Warburg effect in cancer cell biology. Semin Cell Dev Biol. (2012) 23:352–61. doi: 10.1016/j.semcdb.2012.02.003
72. Robey RB, Hay N. Is Akt the “Warburg kinase”?-Akt-energy metabolism interactions and oncogenesis. Sem. Cancer Biol. (2009) 19:25–31. doi: 10.1016/j.semcancer.2008.11.010
73. Makinoshima H, Takita M, Saruwatari K, Umemura S, Obata Y, Ishii G, et al. Signaling through the phosphatidylinositol 3-kinase (PI3K)/mammalian target of rapamycin (mTOR) axis is responsible for aerobic glycolysis mediated by glucose transporter in epidermal growth factor receptor (EGFR)-mutated Lung adenocarcinoma. J Biol Chem. (2015) 290:17495–504. doi: 10.1074/jbc.m115.660498
74. Yu Y, Maguire TG, Alwine JC. Human cytomegalovirus activates glucose transporter 4 expression to increase glucose uptake during infection. J Virol. (2011) 85:1573–80. doi: 10.1128/jvi.01967-10
75. Dasgupta S, Rai RC. PPAR-gamma and Akt regulate GLUT1 and GLUT3 surface localization during Mycobacterium tuberculosis infection. Mol Cell Biochem. (2018) 440:127–38. doi: 10.1007/s11010-017-3161-3
76. Sano H, Eguez L, Teruel MN, Fukuda M, Chuang TD, Chavez JA, et al. Rab10, a target of the AS160 Rab GAP, is required for insulin-stimulated translocation of GLUT4 to the adipocyte plasma membrane. Cell Metab. (2007) 5:293–303. doi: 10.1016/j.cmet.2007.03.001
77. Roberts DJ, Miyamoto S. Hexokinase II integrates energy metabolism and cellular protection: Akting on mitochondria and TORCing to autophagy. Cell Death Differ. (2015) 22:364. doi: 10.1038/cdd.2014.208
78. Osawa H, Sutherland C, Robey RB, Printz RL, Granner DK. Analysis of the signaling pathway involved in the regulation of hexokinase II gene transcription by insulin. J Biol Chem. (1996) 271:16690–4. doi: 10.1074/jbc.271.28.16690
79. Zhuo B, Li Y, Li Z, Qin H, Sun Q, Zhang F, et al. PI3K/Akt signaling mediated Hexokinase-2 expression inhibits cell apoptosis and promotes tumor growth in pediatric osteosarcoma. Biochem Biophys Res Commun. (2015) 464:401–6. doi: 10.1016/j.bbrc.2015.06.092
80. Robey RB, Hay N. Mitochondrial hexokinases, novel mediators of the antiapoptotic effects of growth factors and Akt. Oncogene. (2006) 25:4683–96. doi: 10.1038/sj.onc.1209595
81. Deprez J, Vertommen D, Alessi DR, Hue L, Rider MH. Phosphorylation and activation of heart 6-phosphofructo-2-kinase by protein kinase B and other protein kinases of the insulin signaling cascades. J Biol Chem. (1997) 272:17269–75. doi: 10.1074/jbc.272.28.17269
82. Mouton V, Toussaint L, Vertommen D, Gueuning MA, Maisin L, Havaux X, et al. Heart 6-phosphofructo-2-kinase activation by insulin requires PKB (protein kinase B), but not SGK3 (serum- and glucocorticoid-induced protein kinase 3). Biochem J. (2010) 431:267–75. doi: 10.1042/bj20101089
83. Hu H, Juvekar A, Lyssiotis CA, Lien EC, Albeck JG, Oh D, et al. Phosphoinositide 3-kinase regulates glycolysis through mobilization of aldolase from the actin cytoskeleton. Cell. (2016) 164:433–46. doi: 10.1016/j.cell.2015.12.042
84. Hahn-Windgassen A, Nogueira V, Chen CC, Skeen JE, Sonenberg N, Hay N. Akt activates the mammalian target of rapamycin by regulating cellular ATP level and AMPK activity. J Biol Chem. (2005) 280:32081–9. doi: 10.1074/jbc.m502876200
85. Yu L, Chen X, Sun X, Wang L, Chen S. The glycolytic switch in tumors: how many players are involved? J Cancer. (2017) 8:3430–40. doi: 10.7150/jca.21125
86. Yun J, Rago C, Cheong I, Pagliarini R, Angenendt P, Rajagopalan H, et al. Glucose deprivation contributes to the development of KRAS pathway mutations in tumor cells. Science. (2009) 325:1555–9. doi: 10.1126/science.1174229
87. Sun Q, Chen X, Ma J, Peng H, Wang F, Zha X, et al. Mammalian target of rapamycin up-regulation of pyruvate kinase isoenzyme type M2 is critical for aerobic glycolysis and tumor growth. Proc Natl Acad Sci USA. (2011) 108:4129–34. doi: 10.1073/pnas.1014769108
88. Shukla SK, Gunda V, Abrego J, Haridas D, Mishra A, Souchek J, et al. MUC16-mediated activation of mTOR and c-Myc reprograms pancreatic cancer metabolism. Oncotarget. (2015) 6:19118–31. doi: 10.18632/oncotarget.4078
89. Valera A, Pujol A, Gregori X, Riu E, Visa J, Bosch F. Evidence from transgenic mice that myc regulates hepatic glycolysis. FASEB J. (1995) 9:1067–78. doi: 10.1096/fasebj.9.11.7649406
90. Bensaad K, Tsuruta A, Selak MA, Vidal MN, Nakano K, Bartrons R, et al. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell. (2006) 126:107–20. doi: 10.1016/j.cell.2006.05.036
91. Schwartzenberg-Bar-Yoseph F, Armoni M, Karnieli E. The tumor suppressor p53 down-regulates glucose transporters GLUT1 and GLUT4 gene expression. Cancer Res. (2004) 64:2627–33. doi: 10.1158/0008-5472.can-03-0846
92. Chibalin AV, Kovalenko MV, Ryder JW, Feraille E, Wallberg-Henriksson H, Zierath JR. Insulin- and glucose-induced phosphorylation of the Na(+),K(+)-adenosine triphosphatase alpha-subunits in rat skeletal muscle. Endocrinology. (2001) 142:3474–82. doi: 10.1210/endo.142.8.8294
93. Al-Khalili L, Kotova O, Tsuchida H, Ehren I, Feraille E, Krook A, et al. ERK1/2 mediates insulin stimulation of Na(+),K(+)-ATPase by phosphorylation of the alpha-subunit in human skeletal muscle cells. J Biol Chem. (2004) 279:25211–8. doi: 10.1074/jbc.m402152200
94. Serhan MF, Kreydiyyeh SI. Insulin targets the Na(+)/K(+) ATPase in enterocytes via PI3K, PKC, and MAPKS. J Recept Signal Transduct Res. (2011) 31:299–306. doi: 10.3109/10799893.2011.587821
95. Pedemonte CH, Pressley TA, Lokhandwala MF, Cinelli AR. Regulation of Na, K-ATPase transport activity by protein kinase C. J Membr Biol. (1997) 155:219–27. doi: 10.1007/s002329900174
96. Balaban RS, Bader JP. Studies on the relationship between glycolysis and (Na+ + K+)-ATPase in cultured cells. Biochim Biophys Acta. (1984) 804:419–26. doi: 10.1016/0167-4889(84)90069-7
97. Elmi A, Idahl LA, Sandstrom PE, Sehlin J. D-glucose stimulates the Na+/K+ pump in mouse pancreatic islet cells. Int J Exp Diabetes Res. (2000) 1:155–64. doi: 10.1155/edr.2000.155
98. Miele C, Paturzo F, Teperino R, Sakane F, Fiory F, Oriente F, et al. Glucose regulates diacylglycerol intracellular levels and protein kinase C activity by modulating diacylglycerol kinase subcellular localization. J Biol Chem. (2007) 282:31835–43. doi: 10.1074/jbc.m702481200
99. Dechant R, Binda M, Lee SS, Pelet S, Winderickx J, Peter M. Cytosolic pH is a second messenger for glucose and regulates the PKA pathway through V-ATPase. EMBO J. (2010) 29:2515–26. doi: 10.1038/emboj.2010.138
100. Costes S, Longuet C, Broca C, Faruque O, Hani EH, Bataille D, et al. Cooperative effects between protein kinase A and p44/p42 mitogen-activated protein kinase to promote cAMP-responsive element binding protein activation after beta cell stimulation by glucose and its alteration due to glucotoxicity. Ann N Y Acad Sci. (2004) 1030:230–42. doi: 10.1196/annals.1329.029
101. Da Costa-Pessoa JM, Damasceno RS, Machado UF, Beloto-Silva O, Oliveira-Souza M. High glucose concentration stimulates NHE-1 activity in distal nephron cells: the role of the Mek/Erk1/2/p90RSK and p38MAPK signaling pathways. Cell Physiol Biochem. (2014) 33:333–43. doi: 10.1159/000356673
102. Payen VL, Mina E, Van Hee VF, Porporato PE, Sonveaux P. Monocarboxylate transporters in cancer. Mol Metab. (2020) 33:48–66.
103. Benjamin D, Robay D, Hindupur SK, Pohlmann J, Colombi M, El-Shemerly MY, et al. Dual Inhibition of the lactate transporters MCT1 and MCT4 Is synthetic lethal with metformin due to NAD+ depletion in cancer cells. Cell Rep (2018) 25, 3047–58.e4.
104. Divakaruni AS, Paradyse A, Ferrick DA, Murphy AN, Jastroch M. Analysis and interpretation of microplate-based oxygen consumption and pH data. Methods Enzymol. (2014) 547:309–54. doi: 10.1016/b978-0-12-801415-8.00016-3
105. TeSlaa T, Teitell MA. Techniques to monitor glycolysis. Methods Enzymol. (2014) 542:91–114. doi: 10.1016/b978-0-12-416618-9.00005-4
106. Kotte O, Zaugg JB, Heinemann M. Bacterial adaptation through distributed sensing of metabolic fluxes. Mol Syst Biol. (2010) 6:355. doi: 10.1038/msb.2010.10
107. Monteiro F, Hubmann G, Takhaveev V, Vedelaar SR, Norder J, Hekelaar J, et al. Measuring glycolytic flux in single yeast cells with an orthogonal synthetic biosensor. Mol Syst Biol. (2019) 15:e9071.
108. Kochanowski K, Volkmer B, Gerosa L. Haverkorn van Rijsewijk BR, Schmidt A, Heinemann M. Functioning of a metabolic flux sensor in Escherichia coli. Proc Natl Acad Sci USA. (2013) 110:1130–5. doi: 10.1073/pnas.1202582110
109. MacDonald PE, Joseph JW, Rorsman P. Glucose-sensing mechanisms in pancreatic beta-cells. Philos Trans R Soc Lond B Biol Sci. (2005) 360:2211–25. doi: 10.1098/rstb.2005.1762
110. Berg J, Tymoczko JL, Stryer L. Biochemistry. New York, NY: W H Freeman (2002). doi: 10.1098/rstb.2005.1762
111. Murer H, Hopfer U, Kinne R. Sodium/proton antiport in brush-border-membrane vesicles isolated from rat small intestine and kidney. Biochem J. (1976) 154:597–604. doi: 10.1042/bj1540597
112. Kinsella JL, Aronson PS. Properties of the Na+-H+ exchanger in renal microvillus membrane vesicles. Am J Physiol. (1980) 238:F461–9.
113. Little PJ, Weissberg PL, Cragoe EJ Jr., Bobik A. Dependence of Na+/H+ antiport activation in cultured rat aortic smooth muscle on calmodulin, calcium, and ATP. Evidence for the involvement of calmodulin-dependent kinases. J Biol Chem. (1988) 263:16780–6.
114. Wakabayashi S, Fafournoux P, Sardet C, Pouyssegur J. The Na+/H+ antiporter cytoplasmic domain mediates growth factor signals and controls “H(+)-sensing”. Proc Natl Acad Sci USA. (1992) 89:2424–8. doi: 10.1073/pnas.89.6.2424
115. Goss GG, Woodside M, Wakabayashi S, Pouyssegur J, Waddell T, Downey GP, et al. ATP dependence of NHE-1, the ubiquitous isoform of the Na+/H+ antiporter. Analysis of phosphorylation and subcellular localization. J Biol Chem. (1994) 269:8741–8.
116. Shimada-Shimizu N, Hisamitsu T, Nakamura TY, Wakabayashi S. Evidence that Na+/H+ exchanger 1 is an ATP-binding protein. FEBS J. (2013) 280:1430–42. doi: 10.1111/febs.12138
117. Rao R, Nakamoto RK, Verjovski-Almeida S, Slayman CW. Structure and function of the yeast plasma-membrane H(+)-ATPase. Ann N Y Acad Sci. (1992) 671:195–203.
118. Rao R, Drummond-Barbosa D, Slayman CW. Transcriptional regulation by glucose of the yeast PMA1 gene encoding the plasma membrane H(+)-ATPase. Yeast. (1993) 9:1075–84. doi: 10.1002/yea.320091006
119. Dechant R, Saad S, Ibáñez AJ, Peter M. Cytosolic pH regulates cell growth through distinct gtpases, Arf1 and Gtr1, to promote ras/PKA and TORC1 activity. Molecular Cell. (2014) 55:409–21. doi: 10.1016/j.molcel.2014.06.002
120. Karmazyn M, Avkiran M, Fliegel L. The Sodium-Hydrogen Exchanger, From Molecule to its Role in Disease. New York, NY: Springer Science+Business Media (2003). doi: 10.1016/j.molcel.2014.06.002
121. Milosavljevic N, Monet M, Lena I, Brau F, Lacas-Gervais S, Feliciangeli S, et al. The intracellular Na(+)/H(+) exchanger NHE7 effects a Na(+)-coupled, but not K(+)-coupled proton-loading mechanism in endocytosis. Cell Rep. (2014) 7:689–96. doi: 10.1016/j.celrep.2014.03.054
122. Sepp M, Sokolova N, Jugai S, Mandel M, Peterson P, Vendelin M. Tight coupling of Na+/K+-ATPase with glycolysis demonstrated in permeabilized rat cardiomyocytes. PLoS One. (2014) 9:e99413. doi: 10.1371/journal.pone.0099413
123. James AD, Patel W, Butt Z, Adiamah M, Dakhel R, Latif A, et al. The plasma membrane calcium pump in pancreatic cancer cells exhibiting the warburg effect relies on glycolytic ATP. J Biol Chem. (2015) 290:24760–71. doi: 10.1074/jbc.m115.668707
124. James AD, Richardson DA, Oh IW, Sritangos P, Attard T, Barrett L, et al. Cutting off the fuel supply to calcium pumps in pancreatic cancer cells: role of pyruvate kinase-M2 (PKM2). Br J Cancer. (2020) 122:266–78. doi: 10.1038/s41416-019-0675-3
125. Dhar-Chowdhury P, Harrell MD, Han SY, Jankowska D, Parachuru L, Morrissey A, et al. The glycolytic enzymes, glyceraldehyde-3-phosphate dehydrogenase, triose-phosphate isomerase, and pyruvate kinase are components of the K(ATP) channel macromolecular complex and regulate its function. J Biol Chem. (2005) 280:38464–70. doi: 10.1074/jbc.m508744200
126. Dubinsky WP, Mayorga-Wark O, Schultz SG. Colocalization of glycolytic enzyme activity and KATP channels in basolateral membrane of Necturus enterocytes. Am J Physiol. (1998) 275(6 Pt 1):C1653–9.
127. Dutka TL, Lamb GD. Na+-K+ pumps in the transverse tubular system of skeletal muscle fibers preferentially use ATP from glycolysis. Am J Physiol Cell Physiol. (2007) 293:C967–77.
128. Chu H, Puchulu-Campanella E, Galan JA, Tao WA, Low PS, Hoffman JF. Identification of cytoskeletal elements enclosing the ATP pools that fuel human red blood cell membrane cation pumps. Proc Natl Acad Sci USA. (2012) 109:12794–9. doi: 10.1073/pnas.1209014109
129. Hoffman JF, Dodson A, Proverbio F. On the functional use of the membrane compartmentalized pool of ATP by the Na+ and Ca++ pumps in human red blood cell ghosts. J Gen Physiol. (2009) 134:351–61. doi: 10.1085/jgp.200910270
130. Hong M, Kefaloyianni E, Bao L, Malester B, Delaroche D, Neubert TA, et al. Cardiac ATP-sensitive K+ channel associates with the glycolytic enzyme complex. FASEB J. (2011) 25:2456–67. doi: 10.1096/fj.10-176669
131. Bruce J. Plasma membrane calcium pump regulation by metabolic stress. World J Biol Chem. (2010) 1:221–8. doi: 10.4331/wjbc.v1.i7.221
132. Bruce JIE. Metabolic regulation of the PMCA: role in cell death and survival. Cell Calcium. (2018) 69:28–36. doi: 10.1016/j.ceca.2017.06.001
133. Soltoff SP, Mandel LJ. Active ion transport in the renal proximal tubule. III. The ATP dependence of the Na pump. J Gen Physiol. (1984) 84:643–62. doi: 10.1085/jgp.84.4.643
134. Weiss JN, Lamp ST. Glycolysis preferentially inhibits ATP-sensitive K+ channels in isolated guinea pig cardiac myocytes. Science. (1987) 238:67–9. doi: 10.1126/science.2443972
135. Campanella ME, Chu H, Low PS. Assembly and regulation of a glycolytic enzyme complex on the human erythrocyte membrane. Proc Natl Acad Sci USA. (2005) 102:2402–7. doi: 10.1073/pnas.0409741102
136. Barros LF, Martinez C. An enquiry into metabolite domains. Biophys J. (2007) 92:3878–84. doi: 10.1529/biophysj.106.100925
137. Kennedy HJ, Pouli AE, Ainscow EK, Jouaville LS, Rizzuto R, Rutter GA. Glucose generates sub-plasma membrane ATP microdomains in single islet beta-cells. Potential role for strategically located mitochondria. J Biol Chem. (1999) 274:13281–91. doi: 10.1074/jbc.274.19.13281
138. Pelicano H, Martin DS, Xu RH, Huang P. Glycolysis inhibition for anticancer treatment. Oncogene. (2006) 25:4633–46. doi: 10.1038/sj.onc.1209597
Keywords: Na+/H+-exchanger, cytosolic pH, growth and proliferation, metabolism, aerobic glycolysis
Citation: Birkeland ES, Koch LM and Dechant R (2020) Another Consequence of the Warburg Effect? Metabolic Regulation of Na+/H+ Exchangers May Link Aerobic Glycolysis to Cell Growth. Front. Oncol. 10:1561. doi: 10.3389/fonc.2020.01561
Received: 17 June 2020; Accepted: 20 July 2020;
Published: 18 August 2020.
Edited by:
Silvia Pastorekova, Slovak Academy of Sciences, SlovakiaReviewed by:
Cesare Indiveri, University of Calabria, ItalyCopyright © 2020 Birkeland, Koch and Dechant. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Reinhard Dechant, cmVpbmhhcmQuZGVjaGFudEBiYy5iaW9sLmV0aHouY2g=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.