
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Oncol. , 31 August 2020
Sec. Cancer Molecular Targets and Therapeutics
Volume 10 - 2020 | https://doi.org/10.3389/fonc.2020.01475
 Katia Khoury1
Katia Khoury1 Antoinette R. Tan2
Antoinette R. Tan2 Andrew Elliott3Joanne Xiu3
Andrew Elliott3Joanne Xiu3 Zoran Gatalica4Arielle L. Heeke2,3Claudine Isaacs1
Zoran Gatalica4Arielle L. Heeke2,3Claudine Isaacs1 Paula R. Pohlmann1Lee S. Schwartzberg5Michael Simon6W. Michael Korn3Sandra M. Swain1
Paula R. Pohlmann1Lee S. Schwartzberg5Michael Simon6W. Michael Korn3Sandra M. Swain1 Filipa Lynce1*†
Filipa Lynce1*†Background: PI3K/AKT signaling pathway is activated in breast cancer and associated with cell survival. We explored the prevalence of PI3K pathway alterations and co-expression with other markers in breast cancer subtypes.
Methods: Samples of non-matched primary and metastatic breast cancer submitted to a CLIA-certified genomics laboratory were molecularly profiled to identify pathogenic or presumed pathogenic mutations in the PIK3CA-AKT1-PTEN pathway using next generation sequencing. Cases with loss of PTEN by IHC were also included. The frequency of co-alterations was examined, including DNA damage response pathways and markers of response to immuno-oncology agents.
Results: Of 4,895 tumors profiled, 3,558 (72.7%) had at least one alteration in the PIK3CA-AKT1-PTEN pathway: 1,472 (30.1%) harbored a PIK3CA mutation, 174 (3.6%) an AKT1 mutation, 2,682 (54.8%) had PTEN alterations (PTEN mutation in 7.0% and/or PTEN loss by IHC in 51.4% of cases), 81 (1.7%) harbored a PIK3R1 mutation, and 4 (0.08%) a PIK3R2 mutation. Most of the cohort consisted of metastatic sites (n = 2974, 60.8%), with PIK3CA mutation frequency increased in metastatic (32.1%) compared to primary sites (26.9%), p < 0.001. Other PIK3CA mutations were identified in 388 (7.9%) specimens, classified as “off-label,” as they were not included in the FDA-approved companion test for PIK3CA mutations. Notable co-alterations included increased PD-L1 expression and high tumor mutational burden in PIK3CA-AKT1-PTEN mutated cohorts. Novel concurrent mutations were identified including CDH1 mutations.
Conclusions: Findings from this cohort support further exploration of the clinical benefit of PI3K inhibitors for “off-label” PIK3CA mutations and combination strategies with potential clinical benefit for patients with breast cancer.
Phosphatidylinositol-3-kinase/protein kinase B (PI3K/AKT) signaling pathway is dysregulated in various carcinomas including breast cancer, through several genomic abnormalities. Gain-of-function mutations of PI3K subunits including PIK3CA encoding the catalytic subunit p110α, PIK3CB encoding subunit p110β, and genes like PIK3R1 encoding regulatory subunit p85α have been described. PIK3CA involves two main “hotspots” for activating mutations: E545K of the helical domain on exon 9, and H1047R of the kinase domain on exon 20 (1–9). Mutations in AKT1, and loss-of-function mutations and alterations in PTEN also affect the PI3K pathway, thus promoting cell survival and resistance (8, 10, 11).
About 40% of HR-positive breast cancers harbor PIK3CA mutations. Alpelisib (PIQRAY, Novartis Pharmaceuticals Corporation), a PI3K inhibitor, received FDA approval in combination with fulvestrant for patients with hormone receptor (HR)-positive, human epidermal growth factor receptor 2 (HER2)-negative PIK3CA-mutated advanced breast cancer. Approval was based on SOLAR-1, a phase 3 randomized trial that showed a benefit of 5.3 months in progression-free survival with the addition of alpelisib in the cohort of patients with PIK3CA-mutated breast cancer (12). PIK3CA mutations that were considered for trial enrollment in SOLAR-1 included C420R, E542K, E545A, E545D (1635G > T only), E545G, E545K, Q546E, Q546R, H1047L, H1047R, and H1047Y. The FDA also approved the therascreen® PIK3CA RGQ PCR Kit, (QIAGEN Manchester, Ltd.), a companion test able to select patients who have these specific mutations. For the purpose of the current report, mutations detectable by the companion test were considered alpelisib “on-label” (12).
The use of PI3K and AKT inhibitors in combination with chemotherapy, endocrine therapy, HER2-targeted therapies, cyclin-dependent kinase 4/6 (CDK4/6) and poly ADP ribose polymerase (PARP) inhibitors, has been explored in pre-clinical and clinical models across multiple tumor types (13–27). Based on emerging data that loss of PTEN may represent a potential mechanism for resistance to checkpoint inhibitors, combinations of a triplet regimen of AKT inhibitor, programmed death-ligand 1 (PD-L1) inhibitor and chemotherapy are also being explored in the setting of advanced breast cancer (28).
In this study, we report the prevalence of PI3K pathway alterations and co-expression with other markers of clinical interest in different breast cancer subtypes, based on somatic molecular profiling. This approach can lead to the identification of novel drug combinations with potential synergy that could be further evaluated in the clinical trial setting.
A retrospective review of molecular profiles was performed for 4,845 female and 50 male breast cancer cases submitted to Caris Life Sciences, a Clinical Laboratory Improvement Amendments (CLIA)/College of American Pathologists (CAP)/ISO15189/New York State Department of Health (NYSDOH)-certified clinical laboratory (Phoenix, AZ), between January 2015 and June 2019. Specimens were obtained from more than 500 centers, primarily within the United States, and patient demographics were de-identified (29–31).
IHC was performed on formalin-fixed paraffin-embedded (FFPE) sections of glass slides. Slides were stained using automated staining techniques (Benchmark XT, Ventana, Tucson, AZ; and Autostainer Link 48, Dako, Carpenteria, CA), per the manufacturer's instructions, and were optimized and validated per CLIA/CAO and International Organization for Standardization (ISO) requirements. Staining was scored for intensity (0 = no staining; 1+ = weak staining; 2+ = moderate staining; 3+ = strong staining) and cancer cell staining percentage (0–100%). Results were categorized as positive or negative by defined thresholds specific to each marker based on published clinical literature. The cutoff for ER and PR positivity was ≥1% staining according to the American Society of Clinical Oncology/College of American Pathologists (ASCO/CAP) guidelines (29–31) (Table S1). A board-certified pathologist evaluated all IHC results independently. For PD-L1, a laboratory developed test (LDT) Ventana SP142 assay was initially used to test for PD-L1 expression of tumor cells, and after FDA approval in March 2019, the FDA CDx Ventana PD-L1 (SP142) assay was used to test for PD-L1 expression of immune cells.
HER2/neu amplification was examined by CISH (INFORM HER2 Dual ISH DNA Probe Cocktail, Ventana, Tucson, AZ). All HER2 test results were classified according to the 2018 ASCO/CAP HER2 testing recommendations (31, 32).
NGS was performed on genomic DNA isolated from FFPE tumor samples using the NextSeq platform (Illumina, Inc., San Diego, CA). Matched normal tissue was not sequenced. A custom-designed SureSelect XT assay was used to enrich 592 whole-gene targets (Agilent Technologies, Santa Clara, CA). All variants were detected with > 99% confidence based on allele frequency and amplicon coverage, with an average sequencing depth of coverage of >500 and an analytic sensitivity of 5%. Prior to molecular testing, tumor enrichment was achieved by harvesting targeted tissue using manual microdissection techniques. Genetic variants identified were interpreted by board-certified molecular geneticists and categorized as “pathogenic,” “presumed pathogenic,” “variant of unknown significance (VUS),” “presumed benign,” or “benign,” according to the American College of Medical Genetics and Genomics (ACMG) standards. When assessing mutation frequencies of individual genes, “pathogenic,” and “presumed pathogenic” were counted as mutations while “benign,” “presumed benign” variants, and “VUS” were excluded.
Up to three different testing methods were used to determine MSI/MMR status of tumors profiled, including Fragment Analysis (FA), IHC, and NGS. FA was tested with Microsatellite Instability Analysis (Promega, Madison, WI), which included fluorescently labeled primers for co-amplification of seven markers including five mononucleotide repeat markers (BAT-25, BAT26, NR-21, NR24, and MONO-27) and two pentanucleotide repeat markers (Penta C and D). The mononucleotide markers were used for MSI determination, while the pentanucleotide markers were used to detect either sample mix-ups or contamination. A tumor sample was considered MSI if two or more mononucleotide repeats were abnormal; if one mononucleotide repeat was abnormal or repeats were identical between the tumor and adjacent normal tissue, then the tumor sample was considered microsatellite stable (MSS). MMR protein expression was tested by IHC (using the following antibody clones: MLH1, M1 antibody; MSH2, G2191129 antibody; MSH6, 44 antibody, and PMS2, EPR3947 antibody [Ventana Medical Systems, Inc., Tucson, AZ, USA]). The complete absence of protein expression of any of the four MMR proteins tested (0 intensity in 100% of cells) was considered MMR deficient (dMMR). NGS method for measuring MSI (MSI-NGS) used over 7,000 target microsatellite loci and compared to the reference genome hg19 from the University of California, Santa Cruz (UCSC) Genome Browser database. The number of microsatellite loci that were altered by somatic insertion or deletion was counted for each sample. Only insertions or deletions that increased or decreased the number of repeats were considered. Genomic variants in the microsatellite loci were detected using the same depth and frequency criteria as used for mutation detection. MSI-NGS results were compared with results from over 2,000 matching clinical cases analyzed with traditional PCR-based methods. The threshold to determine MSI by NGS was determined to be 46 or more loci with insertions or deletions to generate a sensitivity of >95% and specificity of >99%. The three platforms generated highly concordant results as previously reported, and in the rare cases of discordant results, the MSI or MMR status of the tumor was determined in the order of FA, IHC, and NGS (33).
TMB was measured (592 genes and 1.4 megabases [MB] sequenced per tumor) by counting all non-synonymous missense mutations found per tumor that had not been previously described as germline alterations. The threshold to define TMB-high was ≥10 mutations/MB (34).
The proportion of pathogenic or presumed pathogenic co-alterations (mutation and/or expression) identified from all tumor specimens tested for each specific mutation were calculated and compared between mutated (MT) and wild type (WT) breast tumors, defined based on the presence of PIK3CA-AKT-PTEN alterations, and among the breast cancer subtypes. Sequencing tests with indeterminate results due to low depth of coverage were excluded from the total number for percentage calculation. The total frequency of PIK3CA-AKT-PTEN-MT cases in the complete cohort and per subtype was calculated by dividing the number of tumors with at least one alteration in PIK3CA, AKT1, or PTEN by the total number of tumors tested. Statistical analysis was performed using Chi-square tests. P < 0.05 were considered statistically significant. The log2 odds ratio was calculated for biomarker pairs to assess the tendency of mutual exclusivity (value ≤ 0) or co-occurrence (value > 0), with p-values derived from a one-sided Fisher's exact test and q-values derived from a Benjamini-Hochberg correction procedure to decrease the false discovery rate.
A total of 4,895 breast cancer cases from 2015 to 2019 with available PIK3CA, AKT1, and PTEN NGS results were reviewed. Four thousand eight hundred and forty-five (99%) were female, and fifty (1%) were male. The median age at the time of testing was 58 years (17–90, SD = 12.8). One thousand nine hundred and twenty-one (39.2%) tissue samples were from primary sites, and 2,974 (60.8%) were from metastatic sites. Metastatic samples included 776 (26.1%) from liver, 320 (10.8%) from bone, 319 (10.7%) from lung, and 135 (4.5%) from brain. Two hundred and fifty cases (5.1%) were lobular. Two thousand five hundred and forty-nine (52.1%) cases were HR-positive HER2-negative, 1,863 (38.1%) were triple negative breast cancer (TNBC), 263 (5.4%) were HR-positive, HER2-positive, and 220 (4.5%) were HR-negative, HER2-positive (Table 1). One thousand eight hundred and fifty-five of 1863 TNBC samples (99.6%) had IHC Androgen Receptor (AR) results available, with 376 (20.3%) samples positive for AR expression.
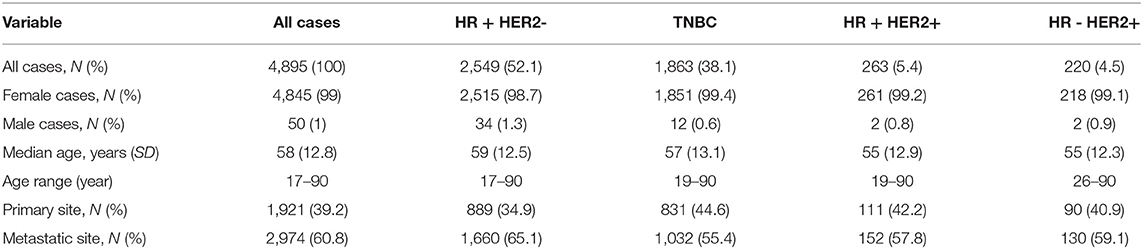
Table 1. Patient and tumor characteristics.
Three thousand five hundred and fifty-eight (72.7%) cases had at least one alteration in the PIK3CA-AKT1-PTEN pathway: 1,472 (30.1%) of cases harbored a PIK3CA mutation, 174 (3.6%) harbored an AKT1 mutation, 2,682 (54.8%) had PTEN alterations including 344 (7.0%) PTEN mutations, and/or PTEN loss by IHC in 2,516 (51.4%) cases. Eighty-one (1.7%) tumors harbored a PIK3R1 mutation, with 66 unique alterations identified (including 4 pathogenic, 55 presumed pathogenic), and 4 (0.08%) cases harbored a PIK3R2 mutation (G373R mutation). The most common hotspot mutations in PIK3CA were in the kinase domain (H1047R in 567 (38.5%) of all PIK3CA alterations) and in the helical domain (E545K in 304 (20.7%) of all PIK3CA alterations) (4–8, 35). Importantly, alpelisib “off-label” activating PIK3CA mutations (those not detected by therascreen® PIK3CA RGQ PCR Kit in SOLAR-1 trial) were seen in 388 (7.9%) of all breast tumors (12). The most common off-label mutations included: N345K (n = 74), E726K (n = 45), G1049R (n = 19), and Q546K (n = 15) (35, 36). The most common AKT1 mutation was in the single hotspot mutation E17K in 164 (94.3%) of all AKT1 mutations. Table S2 describes the most common PIK3CA-AKT1-PTEN pathway-associated genes and their frequencies in our cohort.
With respect to breast cancer subtypes, 100/263 (38.0%) HR-positive HER2-positive, 85/220 (38.6%) HR-negative HER2-positive, 1,857/2,549 (72.9%) HR-positive HER2-negative, and 1,516/18,63 (81.4%) TNBC samples had at least one alteration in the PIK3CA-AKT1-PTEN pathway (Figure 1). Among TNBC samples with a PIK3CA-AKT1-PTEN pathway alteration, 1,192 (78.6%) were negative for AR expression by IHC (i.e., quadruple-negative breast cancer). PIK3CA was the most frequent alteration in HER2-positive breast cancer, in 178/185 (96.2%) cases. Within HER2-negative subtypes, PTEN was the most frequent alteration with PTEN mutation or PTEN loss by IHC present in 79.3% of mutated cases. TNBC was the subtype with the lowest frequency of PIK3CA mutations (18.0% in TNBC vs. 37% in other subtypes). No AKT1 mutations were noted in HER2-positive tumors, and they were found in low frequencies in HER2-negative tumors (6.2%). PIK3CA mutation frequency increased with age, with mutations identified in 26.1% of tumors from patients younger than 60 years and in 35.0% of patients 60 years or older (p < 0.0001). Specifically, in HR-positive HER2-negative, PIK3CA mutations were found in 42.0% of tumors in age ≥60 vs. 33.5% in age <60 (p < 0.0001), and in TNBC at 23.5% in age ≥60 vs. 14.1% in age <60 (p < 0.0001). PIK3CA mutations were increased overall in metastatic vs. primary sites (32.1 vs. 26.9%, p < 0.0001), specifically in HR-positive HER2-positive breast cancer (44.1 vs. 25.2%, respectively, p = 0.002) and TNBC (21 vs. 14.3%, respectively, p = 0.0002). Table S3 also describes the patient and tumor characteristics by PIK3CA, AKT1, and PTEN mutation status.
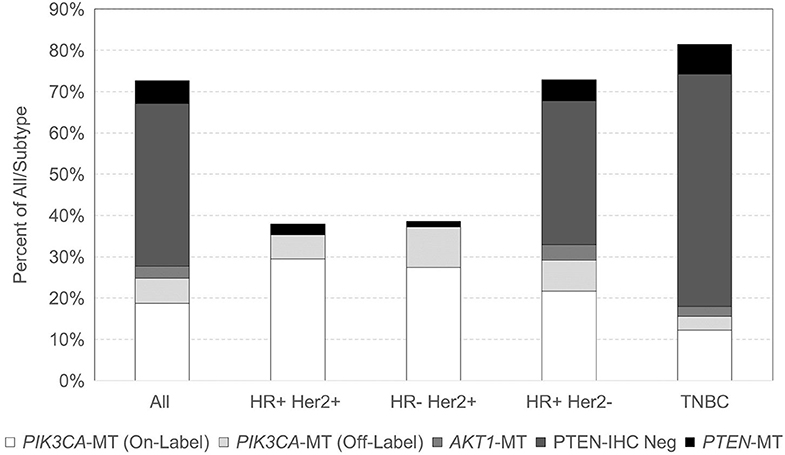
Figure 1. Distribution of PIK3CA, AKT1, and PTEN mutations across breast cancer subtypes.
Figure S1 illustrates the prevalence and co-occurrence of PIK3CA, AKT1, and PTEN alterations in breast tumors. Table 2 summarizes the biomarker subgroup co-alterations for all subtypes. Of the PIK3CA-AKT1-PTEN-MT cases (n = 3,558), only 5 (0.1%) cases had co-occurring alterations in PIK3CA, AKT1, and PTEN.
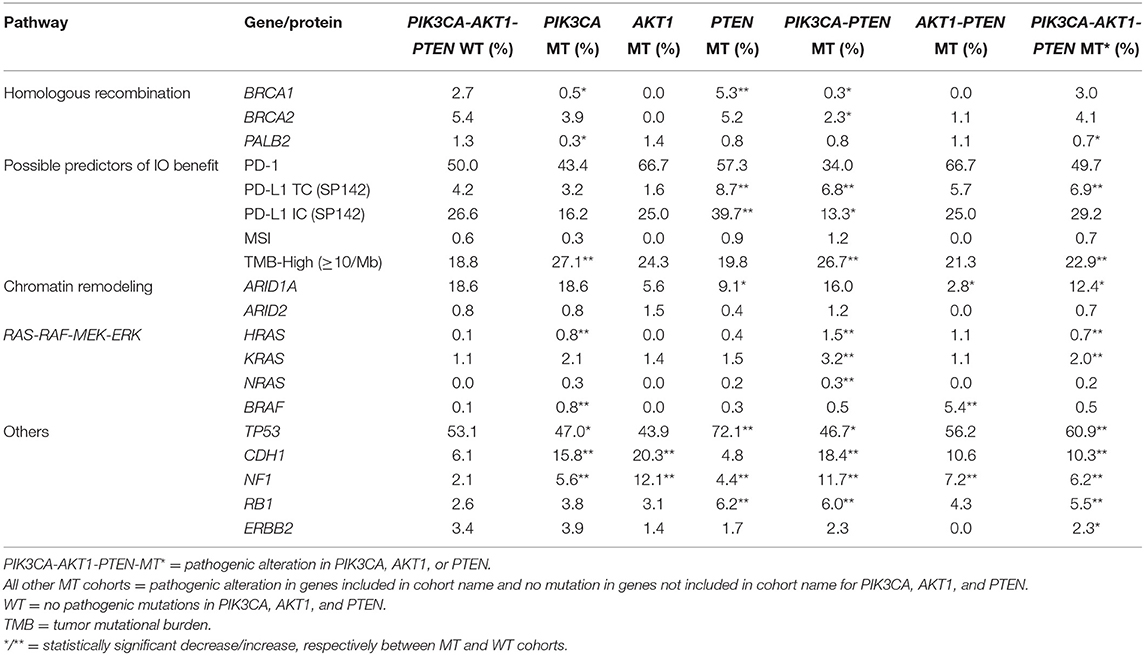
Table 2. Summary of biomarker subgroup co-alterations for all subtypes.
The co-existence of PIK3CA-AKT1-PTEN alterations with other alterations in pathways of clinical relevance was explored, including genes involved in homologous recombination (HR) and DNA damage sensors, chromatin remodeling, RAS-RAF-MEK-ERK pathway, and potential predictors of benefit to immunotherapy. The frequency of selected co-mutations with PIK3CA-AKT1-PTEN alterations is illustrated in Table 3. There was overall low co-alteration frequency for the HR deficiency (HRD)-related genes across all subtypes. CDH1, NF1, and RB1 mutations were significantly increased in the PIK3CA-AKT1-PTEN-MT cohort, particularly in HR-positive and TNBC subtypes.
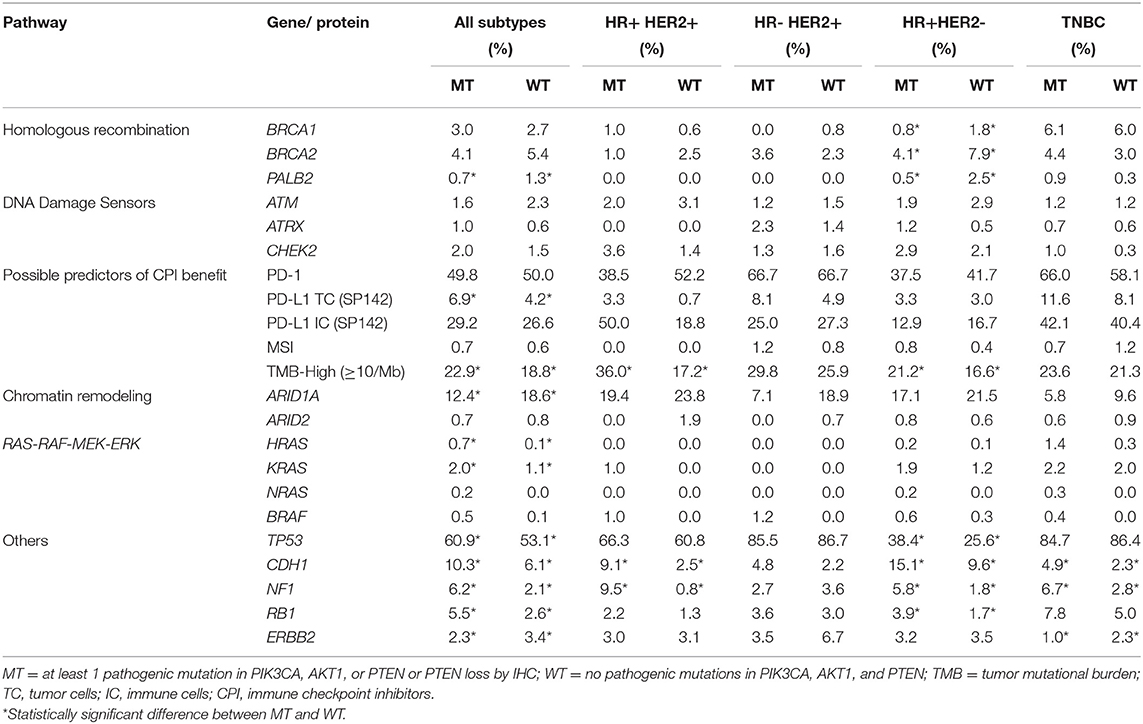
Table 3. Selected co-alterations based on statistical significance.
LDT PD-L1 tumor cells (TC) antibody was used in 4,276/4,895 samples (87.4%), 501/4,895 (10.2%) were tested with FDA CDx PD-L1 immune cell (IC) stain and 21/4,985 (0.4%) were tested with both methods. This difference reflects the change in practice after the approval of the FDA CDx Ventana PD-L1 (SP142) assay. TMB-high and LDT PD-L1 (TC) expression frequency was increased in the PIK3CA-AKT1-PTEN-MT cohort (p < 0.05), whereas the frequency of FDA CDx PD-L1 (IC) expression was not significantly different between MT and WT cohorts (Table 3). In the RAS signaling pathway, there was an increased HRAS, KRAS, and NRAS co-mutation frequency in the MT cohort across all subtypes, with no HRAS or NRAS mutations identified in the HER2-positive subtypes, and no KRAS mutations identified in the HR-negative HER2-positive subtype. BRAF co-mutation frequency was increased in PIK3CA-AKT1-PTEN-MT cohort across all subtypes (p ns); however, BRAF co-mutation frequency was very low for both MT and WT cohorts. Other statistically significant increased co-alterations between PIK3CA-AKT1-PTEN-WT and PIK3CA-AKT1-PTEN-MT cohorts were seen with TP53 (53.1 vs. 60.9%), CDH1 (6.1 vs. 10.3%), NF1 (2.1 vs. 6.2%), and RB1 (2.6 vs. 5.5%). The frequency of CDH1 mutations in PIK3CA-AKT1-PTEN-MT was higher in lobular than in non-lobular carcinoma (73.5 vs. 6.8%), although frequency of CDH1 mutations remained positively associated with PIK3CA-AKT-PTEN-MT after lobular cases were excluded from the analysis. We evaluated the co-occurrence of possible driver events and events associated with mutual exclusivity using an Oncoprint plot (Figure 2). Genomic features were selected based on mutual exclusivity analysis that identified TMB, CDH1, NF1, PD-L1, CHEK2, and BRCA1/2 as significant tendencies of co-occurrence.
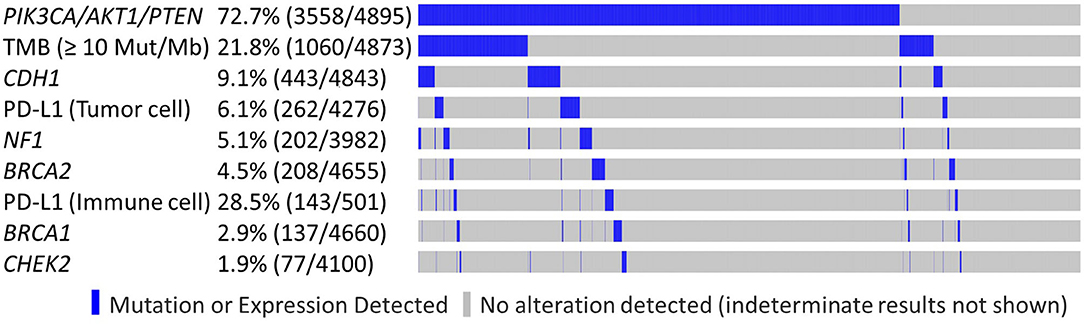
Figure 2. Genomic features observed in PIK3CA/AKT1/PTEN altered and non-altered tumors. Oncoprint plot illustrates co-occurrence of possible driver events and events associated with mutual exclusivity.
In this cohort, PIK3CA pathogenic/presumed pathogenic mutations (n = 1616) were classified as on-label (if included in the SOLAR-1 trial) or off-label. There were 1,204 (74.5%) PIK3CA on-label mutations, and 412 (25.5%) PIK3CA off-label mutations. Of all PIK3CA mutations identified, 57/1,616 (3.5%) were off-label mutations (56 pathogenic/presumed pathogenic, 1 VUS) at amino acid positions that correspond to those of on-label mutations. Some of these off-label mutations have been described as activating in preclinical studies, including N345K, Q546K, and G1049R. In our study, N345K (n = 74), Q546K (n = 15), and G1049R (n = 19) comprised 108/412 (~26%) of the off-label pathogenic/presumed pathogenic mutations (35, 36). Other novel off-label pathogenic/presumed pathogenic mutations have not been defined as activating vs. deleterious. The prevalence of alpelisib on/off-label PIK3CA mutations was similar across breast cancer subtypes, with the lowest frequency seen in TNBC. More than 60% of alpelisib on/off-label mutations occurred in HR-positive HER2-negative subtype (Figure 3). The alpelisib on-label (n = 1,040) and off-label (n = 264) cohorts included cases with exclusively on- or off-label mutations, respectively. Cases with PIK3CA VUS mutations (n =1 14) and cases with both alpelisib on- and off-label mutations in the same tumor (n = 115) were excluded from the analysis.
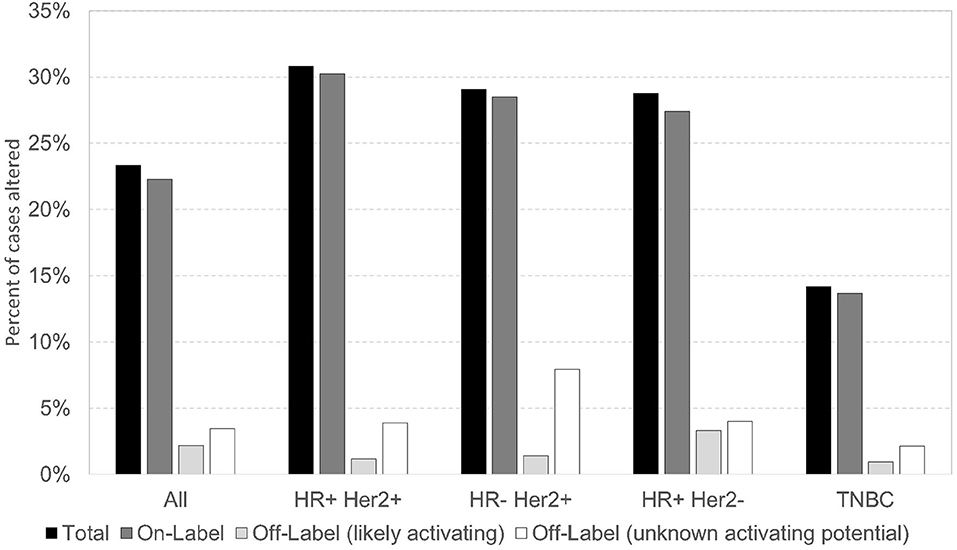
Figure 3. Prevalence of alpelisib On/Off Label PIK3CA mutations across breast cancer subtypes.
Few co-alterations were significantly different between alpelisib on-label and off-label PIK3CA-mutant cohorts, illustrated in Table S4. In all breast cancer subtypes, there was an increased co-mutation frequency in alpelisib off-label cohort compared to on-label cohort in CHEK2 and ERBB2, and increased co-mutation frequency in alpelisib on- and off-label cohorts compared to PIK3CA-WT in CHEK2, HRAS, TP53, CDH1, and NF1.
In this large cohort of 4,895 NGS molecularly profiled breast tumors, we observed a high prevalence of mutations in the PIK3CA-AKT1-PTEN pathway, in up to 72.7% of tumors across all breast cancer subtypes. Other studies have described distinct prevalence of mutations in the PIK3CA-AKT-PTEN pathway, varying from 38.9% PIK3CA mutations in the METABRIC database, 25% pathway mutations in the PAKT trial and 41% pathway mutations in the LOTUS trial (37–39). This variation likely reflects different criteria defining an altered pathway, distinct assays and variant calling or differences in patient populations. The majority of our tumor samples were from metastatic sites (60.8%), while the entire METABRIC database and 82% of the PAKT samples were obtained from primary sites (38, 39). The PAKT and the LOTUS trials enrolled patients with TNBC only (37, 38). In addition, there may also be racial and ethnic differences in the prevalence of PIK3CA-AKT1-PTEN alterations. It has been previously reported that there were differences on the location of PIK3CA mutations and were overall less prevalent in African Americans compared to Caucasians (40). In the LOTUS trial, nearly half of the patients were of Asian ethnicity (37).
As previously reported, PIK3CA was commonly mutated in HR-positive subtypes (37.6%), in a higher percentage of cases than in SOLAR-1 (29%) which can be explained by a broader number of PIK3CA alterations included in our analysis (12, 41). PIK3CA was also the most frequent alteration in HER2-positive breast cancer. PTEN alterations mostly occurred in HER2-negative subtypes and were present in more than half of tumors tested (54.8%), by mutation and/or PTEN loss by IHC. AKT1 mutations were rare, with none identified in HER2-positive tumors. This type of information may be relevant for clinical trial design. Recent phase II and III trials using AKT inhibitors have not used a specific biomarker selected population for trial participation, however, trials conducted with alpelisib enrolled a biomarker selected population (38, 42).
The use of immunotherapy in combination with chemotherapy has been established as the new standard of care in advanced PD-L1 positive TNBC with improved outcomes seen in IMpassion 130 trial (43). In our cohort, the most notable co-alteration identified was a significant increase in PD-L1 expression in tumor cells and high TMB in PIK3CA-AKT1-PTEN mutated cohorts, especially in HR-positive subtypes. This finding could form a basis for further development of drug combinations that affect the PIK3CA-AKT1-PTEN pathway in combination with agents that target the immune system. Such studies are underway, and include for example, a Phase Ib trial evaluating the safety and efficacy for ipatasertib, an AKT inhibitor, combined with atezolizumab and paclitaxel or nab-paclitaxel in patients with advanced TNBC, which showed an objective response rate of 73% for the combination in 26 patients at a median follow up of 6.1 months, regardless of PD-L1 or PIK3CA-AKT1-PTEN status (28). A phase III trial is currently underway for patients with advanced TNBC, evaluating the use of paclitaxel with ipatasertib vs. placebo, and atezolizumab vs. placebo for non-PD-L1 positive patients, and paclitaxel with atezolizumab and ipatasertib vs. placebo for PD-L1 positive patients (44).
Of interest, most cases of CDH1 mutations also demonstrated concurrent mutations in the PIK3CA-AKT1-PTEN pathway and/or high TMB, regardless of histology. Of the 443 total CDH1-MT cases included in the cohort, none had a ROS1 mutation or fusion, indicating that CDH1 and ROS1 result in synthetic lethality, as has been previously described in breast cancer (45). In vivo, inhibition of ROS1 has been shown to produce significant antitumor effects in different models of E-cadherin-defective breast cancer. Therefore, ROS1 inhibitors may be of benefit for patients with CDH1 mutated breast cancers, in combination with PI3K or AKT inhibitors, with or without immunotherapy, and warrant further investigation in early clinical trials.
In this report, the most common hotspot mutations in PIK3CA were in the kinase domain and helical domain, considered on-label mutations. However, off-label activating PIK3CA mutations were also seen, and we identified pathogenic and presumed pathogenic mutations not previously defined, with more than half (293/412, 71.1%) of cases occurring in HR-positive, HER2-negative subtype. At this point, not all novel off-label mutations have data regarding their functional consequences (i.e., activating vs. deleterious), and only a few off-label mutations have been previously reported as deleterious in preclinical studies (35, 36). Therefore, it remains difficult to interpret the functional consequences of new genetic mutations, and the efficacy of PI3K inhibitors in tumors with alpelisib off-label mutations remains unknown. Notable co-alterations seen in off-label mutations include CHEK2 mutations and MSI-High/TMB-High, which may lead to novel targets and drug combinations, and wider use of NGS molecular profiling, given that the current FDA-approved companion test includes only on-label mutations tested in SOLAR-1 (12).
The major limitations of this study are the lack of matched clinical data and outcomes, and therefore the clinical implications of our findings remain to be determined. In addition, the use of molecular profiling in this dataset was determined by clinicians and may have been influenced by patient and tumor characteristics. Therefore, the actual incidence of alterations in the PIK3CA-AKT1-PTEN pathway here described may not fully represent the general population. Lastly, given the lack of known activating potential of most “off-label” PIK3CA mutations, the clinical implications of our findings remain to be determined.
In conclusion, we showed that the prevalence of alterations in the PIK3CA-AKT1-PTEN pathway is elevated across all tumor subtypes and that a considerable number of tumors harbor off-label mutations. This is a very large and comprehensive dataset that characterized the PIK3CA-AKT1-PTEN pathway beyond PIK3CA mutations and included both primary and metastatic breast cancer cases. Although our dataset lacks outcome data, we believe that our results are hypothesis-generating and it is worth exploring the clinical implications of the “off-label” PIK3CA mutations. Finally, our data identified potential targets of interest for combination strategies and support the continuous investigation of the use of agents targeting the PIK3CA-AKT1-PTEN pathway in combination with immunotherapy.
The datasets presented in this article are not readily available because the raw data is protected proprietary information. Requests to access the datasets should be directed to aelliott@carisls.com of Caris Life Sciences.
Ethical review and approval was not required for the study on human participants in accordance with the local legislation and institutional requirements. Written informed consent from the patients was not required to participate in this study in accordance with the national legislation and the institutional requirements.
KK, AT, AE, JX, ZG, AH, CI, PP, LS, MS, WK, SS, and FL contributed to the design, implementation of the research, to the analysis of the results, and to the writing of the manuscript. All authors contributed to the article and approved the submitted version.
AT reports consulting/advisory role from Celgene, Immunomedics, Genentech, Novartis, Pfizer, and Merck, institution-associated research funding from Merck, Pfizer, Tesaro, Genentech/Roche, G1 Therapeutics, and Daiichi Sankyo, and travel, accommodations, and expenses from Caris Life Sciences. AE, JX, and AH are employees of Caris Life Sciences. ZG reports stock/ownership in Caris Life Sciences. CI reports consulting/advisory role from Pfizer, Genentech/Roche, Novartis, AstraZeneca, Puma Biotechnology, Context Therapeutics, and Seattle Genetics, association with the speakers' bureau for Genentech, Pfizer, and AstraZeneca, received honoraria from Genentech/Roche, AstraZeneca, and Pfizer, and institution-associated research funding from Novartis, Pfizer, Genentech, and Tesaro. PP reports consulting/advisory role from Personalized Cancer Therapy, OncoPlex Diagnostics, Immunonet BioSciences, Pfizer, HERON, Puma Biotechnology, Sirtex Medical, Caris Life Sciences, and Juniper, association with the speakers' bureau for Genentech/Roche, patents: United States Patent Nos. 8,486,413, 8,501,417, 9,023,362, and 9,745,377, stock/ownership and leadership in Immunonet BioSciences, received honoraria from ASCO and Dava Oncology, and institution-associated research funding from Genentech/Roche, Fabre-Kramer, Advanced Cancer Therapeutics, Caris Centers of Excellence, Pfizer, Pieris Pharmaceuticals, and Cascadian Therapeutics. LS reports consulting/advisory role from Helsinn, Pfizer, Amgen, Genentech/Roche, BMS, Genomic Health, Myriad, AstraZeneca, Spectrum, Caris Life Sciences, and Lilly, association with the speakers' bureau for Coherus and PUMA, and has received research funding from Amgen, Spectrum, Genentech, Novartis, and AstraZeneca. MS reports association with the speakers' bureau for Astra Zeneca. WK reports stock/ownership interests and employment from Caris Life Sciences, consulting/advisory role from Merck. SS reports consulting/advisory role from Pieris Pharmaceuticals, Inivata, Tocagen, Genomic Health, Genentech/Roche, Lilly, Daiichi Sankyo, Molecular Templates, Athenex, and Silverback Therapeutics, travel, accommodations, and expenses from Inivata, Caris Centers of Excellence, Genentech/Roche, Lilly, Daiichi Sankyo, NanoString Technologies, Bristol-Myers Squibb, Novartis, and Caris Life Sciences, professional services agreement for IDMC from AstraZeneca, support for third party writing assistance from Roche, and institution-associated research funding from Genentech and Pfizer. FL reports consulting/advisory role from Bristol-Myers Squibb (unpaid), Jounce Therapeutics (unpaid), AstraZeneca (unpaid), and Pfizer/EMD Serono; received honoraria from ASCO; received travel, accommodations and expenses from Bristol-Myers Squibb and Genentech, has received research funding from Pfizer, Bristol-Myers Squibb, Jounce Therapeutics and Inivata, and institution-associated research funding from Calithera Biosciences, Immunomedics, Chugai Pharma USA, Regeneron, Inivata, and Tesaro.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2020.01475/full#supplementary-material
1. Avivar-Valderas A, McEwen R, Taheri-Ghahfarokhi A, Carnevalli LS, Hardaker EL, Maresca M, et al. Functional significance of co-occurring mutations in PIK3CA and MAP3K1 in breast cancer. Oncotarget. (2018) 9:21444–58. doi: 10.18632/oncotarget.25118
2. Chow LM, Baker SJ. PTEN function in normal and neoplastic growth. Cancer Lett. (2006) 241:184–96. doi: 10.1016/j.canlet.2005.11.042
3. Cully M, You H, Levine AJ, Mak TW. Beyond PTEN mutations: the PI3K pathway as an integrator of multiple inputs during tumorigenesis. Nat Rev Cancer. (2006) 6:184–92. doi: 10.1038/nrc1819
4. Katso R, Okkenhaug K, Ahmadi K, White S, Timms J, Waterfield MD. Cellular function of phosphoinositide 3-kinases: implications for development, immunity, homeostasis, and cancer. Annu Rev Cell Dev Biol. (2001) 17:615–75. doi: 10.1146/annurev.cellbio.17.1.615
5. Mankoo PK, Sukumar S, Karchin R. PIK3CA somatic mutations in breast cancer: mechanistic insights from langevin dynamics simulations. Proteins. (2009) 75:499–508. doi: 10.1002/prot.22265
6. Zhao L, Vogt PK. Helical domain and kinase domain mutations in p110alpha of phosphatidylinositol 3-kinase induce gain of function by different mechanisms. Proc Natl Acad Sci USA. (2008) 105:2652–7. doi: 10.1073/pnas.0712169105
7. Liu P, Cheng H, Roberts TM, Zhao JJ. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat Rev Drug Discover. (2009) 8:627–44. doi: 10.1038/nrd2926
8. McCubrey JA, Steelman LS, Abrams SL, Lee JT, Chang F, Bertrand FE, et al. Roles of the RAF/MEK/ERK and PI3K/PTEN/AKT pathways in malignant transformation and drug resistance. Adv Enzyme Regul. (2006) 46:249–79. doi: 10.1016/j.advenzreg.2006.01.004
9. Liu S, Chen S, Yuan W, Wang H, Chen K, Li D, et al. PD-1/PD-L1 interaction up-regulates MDR1/P-gp expression in breast cancer cells via PI3K/AKT and MAPK/ERK pathways. Oncotarget. (2017) 8:99901–12. doi: 10.18632/oncotarget.21914
10. Elmenier FM, Lasheen DS, Abouzid KAM. Phosphatidylinositol 3 kinase (PI3K) inhibitors as new weapon to combat cancer. Eur J Med Chem. (2019) 183:111718. doi: 10.1016/j.ejmech.2019.111718
11. Juric D, Castel P, Griffith M, Griffith OL, Won HH, Ellis H, et al. Convergent loss of PTEN leads to clinical resistance to a PI(3)Kalpha inhibitor. Nature. (2015) 518:240–4. doi: 10.1038/nature13948
12. André F, Ciruelos E, Rubovszky G, Campone M, Loibl S, Rugo HS, et al. Alpelisib for PIK3CA-mutated, hormone receptor–positive advanced breast cancer. N Engl J Med. (2019) 380:1929–40. doi: 10.1056/NEJMoa1813904
13. De P, Sun Y, Carlson JH, Friedman LS, Leyland-Jones BR, Dey N. Doubling down on the PI3K-AKT-mTOR pathway enhances the antitumor efficacy of PARP inhibitor in triple negative breast cancer model beyond BRCA-ness. Neoplasia. (2014) 16:43–72. doi: 10.1593/neo.131694
14. Costa R, Han H, Gradishar W. Targeting the PI3K/AKT/mTOR pathway in triple-negative breast cancer: a review. Breast Cancer Res Treat. (2018) 169:397–406. doi: 10.1007/s10549-018-4697-y
15. Almozyan S, Colak D, Mansour F, Alaiya A, Al-Harazi O, Qattan A, et al. PD-L1 promotes OCT4 and Nanog expression in breast cancer stem cells by sustaining PI3K/AKT pathway activation. Int J Cancer. (2017) 141:1402–12. doi: 10.1002/ijc.30834
16. Banerji U, Dean EJ, Perez-Fidalgo JA, Batist G, Bedard PL, You B, et al. A phase I open-label study to identify a dosing regimen of the pan-AKT inhibitor AZD5363 for evaluation in solid tumors and in PIK3CA-mutated breast and gynecologic cancers. Clin Cancer Res. (2018) 24:2050–9. doi: 10.1158/1078-0432.CCR-17-2260
17. Crabb SJ, Birtle AJ, Martin K, Downs N, Ratcliffe I, Maishman T, et al. ProCAID: a phase I clinical trial to combine the AKT inhibitor AZD5363 with docetaxel and prednisolone chemotherapy for metastatic castration resistant prostate cancer. Invest New Drugs. (2017) 35:599–607. doi: 10.1007/s10637-017-0433-4
18. Crafter C, Vincent JP, Tang E, Dudley P, James NH, Klinowska T, et al. Combining AZD8931, a novel EGFR/HER2/HER3 signalling inhibitor, with AZD5363 limits AKT inhibitor induced feedback and enhances antitumour efficacy in HER2-amplified breast cancer models. Int J Oncol. (2015) 47:446–54. doi: 10.3892/ijo.2015.3062
19. Ibrahim YH, Garcia-Garcia C, Serra V, He L, Torres-Lockhart K, Prat A, et al. PI3K inhibition impairs BRCA1/2 expression and sensitizes BRCA-proficient triple-negative breast cancer to PARP inhibition. Cancer Discov. (2012) 2:1036–47. doi: 10.1158/2159-8290.CD-11-0348
20. Matulonis UA, Wulf GM, Barry WT, Birrer M, Westin SN, Farooq S, et al. Phase I dose escalation study of the PI3kinase pathway inhibitor BKM120 and the oral poly (ADP ribose) polymerase (PARP) inhibitor olaparib for the treatment of high-grade serous ovarian and breast cancer. Ann Oncol. (2017) 28:512–8. doi: 10.1093/annonc/mdw672
21. Mayer IA, Abramson VG, Formisano L, Balko JM, Estrada MV, Sanders ME, et al. A phase Ib study of alpelisib (BYL719), a PI3Kα-specific inhibitor, with letrozole in ER + /HER2 – metastatic breast cancer. Clin Cancer Res. (2017) 23:26–34. doi: 10.1158/1078-0432.CCR-16-0134
22. Ribas R, Pancholi S, Guest SK, Marangoni E, Gao Q, Thuleau A, et al. AKT antagonist AZD5363 influences estrogen receptor function in endocrine-resistant breast cancer and synergizes with fulvestrant (ICI182780) in vivo. Mol Cancer Ther. (2015) 14:2035–48. doi: 10.1158/1535-7163.MCT-15-0143
23. Toren P, Kim S, Cordonnier T, Crafter C, Davies BR, Fazli L, et al. Combination AZD5363 with enzalutamide significantly delays enzalutamide-resistant prostate cancer in preclinical models. Eur Urol. (2015) 67:986–90. doi: 10.1016/j.eururo.2014.08.006
24. Vora SR, Juric D, Kim N, Mino-Kenudson M, Huynh T, Costa C, et al. CDK 4/6 inhibitors sensitize PIK3CA mutant breast cancer to PI3K inhibitors. Cancer Cell. (2014) 26:136–49. doi: 10.1016/j.ccr.2014.05.020
25. Spathas N, Economopoulou P, Kotsantis I, Oikonomopoulos N, Psyrri A. Exceptional response to copanlisib in a heavily pretreated patient with PIK3CA-mutated metastatic breast cancer. JCO Precision Oncol. (2020) 4:335–40. doi: 10.1200/PO.19.00049
26. Franzese E, Centonze S, Diana A, Lombardi A, Carlino F, Guerrera LP, et al. Genomic profile and BRCA-1 promoter methylation status in BRCA mutated ovarian cancer: new insights in predictive biomarkers of olaparib response. Front Oncol. (2019) 9:1289. doi: 10.3389/fonc.2019.01289
27. Orditura M, Della Corte CM, Diana A, Ciaramella V, Franzese E, Famiglietti V, et al. Three dimensional primary cultures for selecting human breast cancers that are sensitive to the anti-tumor activity of ipatasertib or taselisib in combination with anti-microtubule cytotoxic drugs. Breast. (2018) 41:165–71. doi: 10.1016/j.breast.2018.08.002
28. Schmid P, Loirat D, Savas P, Espinosa E, Boni V, Italiano A, et al. Abstract CT049: phase Ib study evaluating a triplet combination of ipatasertib (IPAT), atezolizumab (atezo), and paclitaxel (PAC) or nab-PAC as first-line (1L) therapy for locally advanced/metastatic triple-negative breast cancer (TNBC). Cancer Res. (2019) 79:CT049. doi: 10.1158/1538-7445.AM2019-CT049
29. Hammond ME, Hayes DF, Dowsett M, Allred DC, Hagerty KL, Badve S, et al. American society of clinical oncology/college of American pathologists guideline recommendations for immunohistochemical testing of estrogen and progesterone receptors in breast cancer. Arch Pathol Lab Med. (2010) 134:907–22. doi: 10.1043/1543-2165-134.7.e48
30. Hammond ME, Hayes DF, Dowsett M, Allred DC, Hagerty KL, Badve S, et al. American society of clinical oncology/college of american pathologists guideline recommendations for immunohistochemical testing of estrogen and progesterone receptors in breast cancer. J Clin Oncol. (2010) 28:2784–95. doi: 10.1200/JCO.2009.25.6529
31. Wolff AC, Hammond M, Allison KH, Harvey BE, Mangu PB, Bartlett J, et al. Human epidermal growth factor receptor 2 testing in breast cancer: American society of clinical oncology/college of American pathologists clinical practice guideline focused update. Arch Pathol Lab Med. (2018) 142:1364–82. doi: 10.1200/JOP.18.00206
32. Wolff AC, Hammond ME, Hicks DG, Dowsett M, McShane LM, Allison KH, et al. Recommendations for human epidermal growth factor receptor 2 testing in breast cancer: American society of clinical oncology/college of American pathologists clinical practice guideline update. J Clin Oncol. (2013) 31:3997–4013. doi: 10.1200/JCO.2013.50.9984
33. Vanderwalde A, Spetzler D, Xiao N, Gatalica Z, Marshall J. Microsatellite instability status determined by next-generation sequencing and compared with PD-L1 and tumor mutational burden in 11,348 patients. Cancer Med. (2018) 7:746–56. doi: 10.1002/cam4.1372
34. Hellmann MD, Ciuleanu TE, Pluzanski A, Lee JS, Otterson GA, Audigier-Valette C, et al. Nivolumab plus ipilimumab in lung cancer with a high tumor mutational burden. N Engl J Med. (2018) 378:2093–104. doi: 10.1056/NEJMoa1801946
35. Martínez-Sáez O, Chic N, Pascual T, Adamo B, Vidal M, González-Farré B, et al. Frequency and spectrum of PIK3CA somatic mutations in breast cancer. Breast Cancer Res. (2020) 22:45–9. doi: 10.1186/s13058-020-01284-9
36. Dogruluk T, Tsang YH, Espitia M, Chen F, Chen T, Chong Z, et al. Identification of variant-specific functions of PIK3CA by rapid phenotyping of rare mutations. Cancer Res. (2015) 75:5341–54. doi: 10.1158/0008-5472.CAN-15-1654
37. Kim S, Dent R, Im S, Espié M, Blau S, Tan AR, et al. Ipatasertib plus paclitaxel versus placebo plus paclitaxel as first-line therapy for metastatic triple-negative breast cancer (LOTUS): a multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Oncol. (2017) 18:1360–72. doi: 10.1016/S1470-2045(17)30450-3
38. Schmid P, Abraham J, Chan S, Wheatley D, Brunt AM, Nemsadze G, et al. Capivasertib plus paclitaxel versus placebo plus paclitaxel as first-line therapy for metastatic triple-negative breast cancer: the PAKT trial. J Clin Oncol. (2020) 38:423–33. doi: 10.1200/JCO.19.00368
39. Pereira B, Chin S, Rueda OM, Vollan HM, Provenzano E, Bardwell HA, et al. The somatic mutation profiles of 2,433 breast cancers refines their genomic and transcriptomic landscapes. Nat Commun. (2016) 7:11479. doi: 10.1038/ncomms11908
40. Lynce F, Xiu J, Nunes MR, Swain SM, Gatalica Z, Isaacs C, et al. Abstract PD8-04: Racial differences in the molecular landscape of breast cancer. Cancer Res. (2017) 77:PD8–04. doi: 10.1158/1538-7445.SABCS16-PD8-04
41. Bareche Y, Venet D, Ignatiadis M, Aftimos P, Piccart M, Rothe F, et al. Unravelling triple-negative breast cancer molecular heterogeneity using an integrative multiomic analysis. Ann Oncol. (2018) 29:895–902. doi: 10.1093/annonc/mdy024
42. ClinicalTrials.gov. Capivasertib+Paclitaxel as First Line Treatment for Patients With Locally Advanced or Metastatic TNBC Status: Recruiting. Available online at: https://ClinicalTrials.gov/show/NCT03997123 (accessed May 10, 2020).
43. Schmid P, Rugo HS, Adams S, Schneeweiss A, Barrios CH, Iwata H, et al. Atezolizumab plus nab-paclitaxel as first-line treatment for unresectable, locally advanced or metastatic triple-negative breast cancer (IMpassion130): updated efficacy results from a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. (2020) 21:44–59. doi: 10.1016/S1470-2045(19)30689-8
44. ClinicalTrials.gov. A Study of Ipatasertib in Combination WITH atezolizumab and Paclitaxel as a Treatment for Participants With Locally Advanced or Metastatic Triple-Negative Breast Cancer. Status: Recruiting. Available online at: https://ClinicalTrials.gov/show/NCT04177108 (accessed May 10, 2020).
Keywords: breast cancer, molecular profiling, PIK3CA-AKT1-PTEN pathway, PI3K inhibitor, PD-L1, CDH1
Citation: Khoury K, Tan AR, Elliott A, Xiu J, Gatalica Z, Heeke AL, Isaacs C, Pohlmann PR, Schwartzberg LS, Simon M, Korn WM, Swain SM and Lynce F (2020) Prevalence of Phosphatidylinositol-3-Kinase (PI3K) Pathway Alterations and Co-alteration of Other Molecular Markers in Breast Cancer. Front. Oncol. 10:1475. doi: 10.3389/fonc.2020.01475
Received: 12 May 2020; Accepted: 10 July 2020;
Published: 31 August 2020.
Edited by:
Erica Golemis, Fox Chase Cancer Center, United StatesReviewed by:
Vladislav Korobeynikov, Columbia University Irving Medical Center, United StatesCopyright © 2020 Khoury, Tan, Elliott, Xiu, Gatalica, Heeke, Isaacs, Pohlmann, Schwartzberg, Simon, Korn, Swain and Lynce. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Filipa Lynce, ZmlsaXBhX2x5bmNlQGRmY2kuaGFydmFyZC5lZHU=
†Present address: Filipa Lynce, Dana Farber Cancer Institute, Harvard Medical School, Boston, MA, United States
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.