 Qing Li
Qing Li Zhen-Yu Ding
Zhen-Yu Ding- Department of Biotherapy, Cancer Center, National Clinical Research Center for Geriatrics, West China Hospital, Sichuan University, Chengdu, China
Immunotherapy has revolutionized the standard of care for a range of malignancies. Accumulating evidence suggests that the success of immunotherapy is likely attributable to neoantigen-specific T cells. Thus, adoptive cell therapy with these neoantigen-specific T cells is highly promising. This strategy has proven to successfully elicit tumor regression or even complete remission in metastatic cancer patients. However, a fundamental challenge is to effectively identify and isolate neoantigen-specific T cells or their T cell receptors (TCRs), from either tumor-infiltrating lymphocytes (TILs) or peripheral blood lymphocytes (PBLs), and many methods have been developed to this end. In this review, we focus on the current proposed strategies for identifying and isolating neoantigen-specific T cells.
Introduction
Several immunotherapeutic strategies that harness the exquisite specificity of the immune system to eliminate tumors have emerged during the past decade; these include cancer vaccines, immune checkpoint blockade, and adoptive cell therapy (ACT) (1) with the potential to revolutionize the standard of care for a range of malignancies.
To a large extent, the specificity of immunotherapy is dependent on the recognition of specific tumor antigens, especially neoantigens. Neoantigens are a kind of tumor antigen derived from tumor-specific somatic mutations and are highly restricted to tumor cells with minimal established immune tolerance (1). Neoantigen-based cancer vaccines have shown promising therapeutic effects in the clinic (2–8). In addition, a growing body of evidence indicates that neoantigen-specific T cells underlie the success of the recently emergent immune checkpoint inhibitor therapy (9–13). Adoptive transfer of autologous, in vitro expanded, tumor-infiltrating lymphocytes (TILs) was reported to achieve dramatic clinical responses in some metastatic cancer patients, especially in those with melanoma and cervical cancer (14–19). In-depth studies have revealed the critical roles of neoantigen-specific T cells in maintaining durable responses following ACT (20–26). In support of these findings, the adoptive transfer of selected TILs targeting neoantigens led to significant tumor regression (27–29). Increasing research attention has been shifted to identifying and selecting neoantigen-specific T cells (30–34). However, such a “precise targeting” strategy poses a great challenge in terms of the identification and isolation of neoantigen-specific T cells. Methods have been proposed and developed for this purpose. Here, we attempt to summarize the known strategies for isolating neoantigen-specific T cells.
Identification and Isolation of Neoantigen-Specific T Cells From TILs
Researchers have long attempted to isolate neoantigen-specific subpopulations from the background of transferred TILs. In early studies, an autologous tumor cell cDNA library was constructed and used as a pool to screen for neoantigen-specific T cells (20, 21). In a study of a melanoma patient who experienced a complete response going beyond 7 years following adoptive TIL transfer, one T cell clone specific for a mutated antigen PPP1R3B was identified and shown to be responsible for the antitumor effects (22).
However, the time-consuming and laborious process required to identify neoepitope-responsive T cells has hindered their extensive functional assessment (32). Advances in next-generation sequencing have enabled the rapid assessment of the mutational landscape of human cancers and made it possible to identify immunogenic mutated tumor antigens through in silico analysis. Rosenberg's group first employed predicted neo-peptides, obtained by whole-exome sequencing and human leucocyte antigen (HLA) class I–binding algorithms, for TIL screening. Using this approach, they identified 7 neoantigens recognized by 3 therapeutic bulk TIL cultures that mediated objective tumor regressions in three individuals with melanoma (23). Using a similar method, neoantigen-specific CD8+ TILs could also be identified in hematological malignancies, such as acute lymphoblastic leukemia (ALL) (35). Prickett et al. (25) and Stevanovic et al. (26) also demonstrated that neoantigen-specific T cells could be identified from therapeutic TILs by screening tandem minigene (TMG) libraries encoding cancer mutations identified from patients' tumors by whole-exome sequencing. This finding might further facilitate the recognition of neoantigen-specific T cells because it circumvents the need for prediction of HLA–peptide binding and synthesis of a large number of peptides.
With the advent of these techniques, the field of ACT took a great leap from bulk TILs to neoantigen-specific T cells. A concise flowchart showing the steps involved in identifying and isolating neoantigen-specific T cells for ACT is summarized in Figure 1. Tran et al. (27) successfully performed neoantigen-specific T cell therapy in a 43-year-old woman with extensively metastatic and intensively treated cholangiocarcinoma. After administration of a bulk lymphocyte population containing a high percentage of neoantigen ERBB2IP-specific CD4+T cells, the patient showed a long-lasting objective clinical response without obvious toxicity. Subsequently, neoantigen-specific T cells were identified in one colon cancer patient and another breast cancer patient, and reinfusion of these specific T cells led to a partial response in one patient and a durable complete response in another (28, 29). Currently, ACT with neoantigen-specific T cells is being tested in clinical trials in both solid and hematological tumors (Supplementary Table 1).
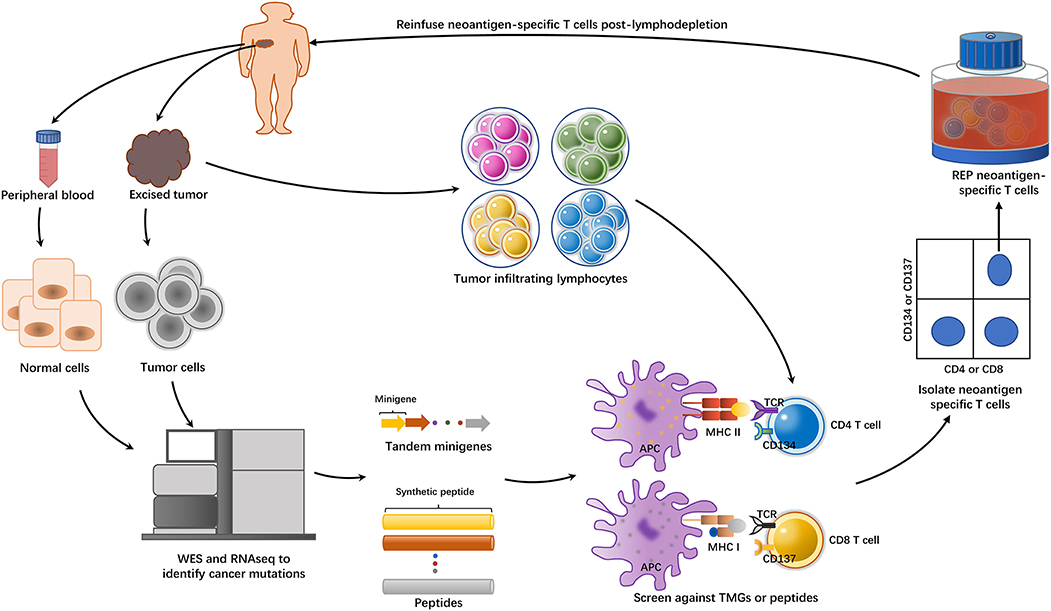
Figure 1. The general approach of identifying and isolating neoantigen-specific TILs for ACT. The tumor cells from excised tumor tissue and matched normal cells underwent whole-exome sequencing (WES) and RNA sequencing to identify non-synonymous mutations. Based on the information, either tandem minigenes (TMGs) or peptides were then synthesized. These TMGs or peptides were pulsed into autologous antigen presenting cells (APCs), such as dendritic cells (DCs) or B cells, and they were processed and presented in the context of major histocompatibility complex (MHC). On the other side, the excised tumors were minced into ~1–2 mm3 fragments and placed in 24-well plates stimulated with IL-2. Then, the TILs were cocultured with these pulsed APCs. The identification of the individual neoantigen-specific T subpopulation was dependent on the IFN-γ enzyme-linked immunospot (ELISPOT) assay and the activation of the markers such as CD137(41BB) or CD134(OX40) on the T cell surfaces when recognizing their cognate target antigen. T cells with these activation surface markers would be purified by flow cytometry. Then, the sorted T cells were subject to rapid expansion in vitro and reinfusion to the tumor-bearing patient.
However, the extensive expansion of neoantigen-specific T cells during preparation compromises their proliferation potential (36). In addition, the method involved requires sophisticated equipment and a time period of several months. For most metastatic patients, this time frame is unacceptable. To address these issues, additional attempts have been made, using either surface markers or T cell receptor (TCR) redundancy.
Approaches Based on Surface Markers
CD137 belongs to the tumor necrosis factor receptor superfamily (37, 38). It functions as a costimulatory molecule to promote the proliferation and survival of activated T cells (39, 40). CD137 expression is highly restricted to transiently activated CD8+ T cells but almost undetectable in resting cells. Upregulated CD137 can be detected on stimulated CD8+ T cells of all phenotypes (e.g., naïve T cells as well as early and late memory effector T cells) (41). Naturally occurring tumor-reactive T cells stimulated by tumor antigens also express CD137 as proven by Ye et al. (42). In a clinical trial (Trial registration ID: NCT02111863) among 6 patients with melanoma who underwent adoptive transfer with CD137-selected TILs, only 1 patient achieved partial response, and the remaining 5 progressed. The study was terminated.
This approach has its pitfalls: Because CD137 is an activation marker, CD137+ T cells obtained by large-scale production are generally overactivated and highly differentiated with limited proliferative potential. A potential solution is to obtain TCRs from these CD137+T cells instead. This strategy was reported by Parkhurst et al. (43). Briefly, CD8+ T cells were stimulated overnight with immunogenic mutated TMG RNAs. Subsequently, the CD8+ T cell population with the highest CD137 expression was sorted by fluorescent-activated cell sorting (FACS), and expanded in vitro. Then, dominant TCR α and β chains were sequenced in the enriched populations. Twenty-seven TCRs from 6 patients that recognized 14 neoantigens expressed by autologous tumor cells were identified. However, this process was time-consuming (2–3 months).
A simplified protocol was proposed by Seliktar-Ofir et al. (44). Here, TILs, but not CD8+ T cells, were cocultured with autologous tumor cells; CD137+ T cells were isolated by magnetic bead separation and expanded. No further TCR sequencing was performed. The entire process took only 35 days. T cells stimulated with neoantigens or other tumor-associated antigens exhibit upregulated CD137 expression (25, 42, 43, 45). Therefore, a CD137-based selection protocol was advocated for its broad antigen coverage including both neoantigen and shared tumor antigens without prior knowledge of epitope specificity. However, the prerequisite of the establishment of autologous tumor cell lines poses a challenge.
Direct and indirect evidence shows that the interaction between PD-1 and PD-L1 inhibits T lymphocyte function, leading to evasion of persistent inflammatory or autoimmune reactions (46–48). However, this protective mechanism is hijacked by tumors to escape immune surveillance, PD-1 has been characterized as an inhibitory receptor on chronically stimulated T-cells in the tumor microenvironment (46). At the tumor site, TILs are exposed to tumor antigens; the binding of TCR and antigen upregulates either costimulatory or coinhibitory receptors to promote or inhibit T cell activation and function, respectively (49). Therefore, PD-1+ T cell populations among TILs may contain a large proportion of tumor-specific T cells. The findings of Inozume et al. (50) and Ahmadzadeh et al. (51), that tumor-responsive T cells are enriched among CD8+PD1+ lymphocytes from fresh melanoma specimens, provide direct support for this notion.
In another study, Gros et al. (49) demonstrated that PD-1 expression on CD8+ TILs in fresh melanoma tumor specimens enabled identification of a diverse patient-specific repertoire of clonally expanded tumor-reactive cells, including mutated neoantigen-specific CD8+ lymphocytes. Although PD-1 is an inhibitory receptor expressed on T cells, studies have shown that IL-2 restored the antitumor function of T cells in vitro (49, 50). However, on antigen-experienced terminally differentiated effector memory (TEMRA) cells, PD-1 is either not expressed or expressed at very low levels (49, 52). Therefore, a PD-1-based enrichment strategy may not be suitable for these cells.
Screening strategies based on CD137 or PD-1 expression are suitable for CD8+ T cells, mainly in melanoma. Epithelial cancers, which account for more than 80% of all human malignancies, harbor fewer mutations than melanoma (53). They exhibit compromised capability to induce mutation-specific T cell responses, together with a limited number of infiltrating neoantigen-specific TILs (32). In addition, CD4+T cells have been shown to play an important role in mediating tumor regression in animal models and patients (27, 36, 54–56). However, CD137 or PD-1 is expressed on CD8+ cells as a sole marker; therefore, it may not be reliably used to enrich activated CD4+ cells (42, 43, 57). CD134 is transiently expressed on CD4 + T cells stimulated by antigens and can be used as a marker for the classification of mutant reactive T cells (58).
Recently, Yossef et al. (36) reported an approach in which the TILs that expressed CD134 or CD137and/or PD-1 were isolated by FACS. Thus, both CD4+ T and TEMRA cells were rescued, which would otherwise be missed if a single marker were used. Sorted cells underwent limiting-dilution in microwell plates to avoid the overgrowth of non-specific T cells. Cultures were tested for the ability to recognize a 25-mer peptide pool encompassing possible neoantigens. Notably, the highly oligoclonal nature of these T cells makes possible the convenient application of single cell sequencing of their TCRs. In 6 patients with metastatic epithelial cancer, this high-throughput approach led to the detection of CD4+ and CD8+ T cells targeting 18 and 1 neoantigens, respectively, whereas only 6 and 2 neoantigens were identified by using the TIL fragment screening approach. In 2 patients in which no neoantigen was found by traditional screening, the novel approach identified 5 distinct neoantigen-specific TCR clones for one patient and a highly potent MHC class II–restricted KRASG12V-reactive TCR for the other. In a metastatic tumor sample from a patient with serous ovarian cancer, 3 MHC class II–restricted TCRs targeting the TP53G245S hot-spot mutation were identified.
TCR Frequency
TCR sequence analysis is used as a tool to monitor T cell responses to specific antigens by measuring the abundance of T cell clonotypes (49, 59, 60). The advent of next-generation sequencing has enabled identification of the full TCR repertoire of TILs (61, 62). This valuable data for TCRs from tumor-reactive TILs could be used to modify T cells (TCR-T). However, the lengthy expansion process and excessive stimulation would result in TCR repertoire switching (63). To avoid this problem, Pasetto et al. (63) directly performed TCR sequencing of the fresh enzymatically digested melanoma tissues prior to in vitro expansion. As described earlier, tumor-reactive clonotypes were enriched in CD8+PD-1+ TIL subsets in melanoma (49, 50). The authors analyzed the TCR repertoire of TILs in CD8+, CD8–, CD8+PD-1–, or CD8+PD-1+ subsets, respectively, and found that many of the most frequently occurring TCR clonotypes present in the CD8+PD-1+ TIL subset recognized the autologous tumor and tumor antigens, included neoantigens. This report provided a much more convenient approach to efficiently identify tumor-reactive T cells based solely on the frequency of TCR and PD-1 expression, without prior knowledge of the specific neoantigen. However, this strategy must be applied with caution because the isolated TCR clones may be self-reactive and result in deleterious on-target, off-tumor toxicities (43).
Isolation of Neoantigen-Specific T Cells From Peripheral Blood Lymphocytes (PBLs)
In some situations, neoantigen-specific T cells were undetectable in the TIL compartment, possibly owing to the following factors: presentation of neoantigens in a non-inflammatory context (64), impaired T cell infiltration because of the sparse distribution of adhesion molecules on these cells (65, 66), and presence of immunosuppressive cytokines and cells (e.g., regulatory T cells) in the tumor microenvironment (67). Furthermore, the tissue from which TILs may be obtained poses a challenge. In this regard, peripheral blood is an alternative and reliable source for neoantigen-specific T cells.
The first attempt is considered to have been made by a group led by Lennerz et al. (68). In this study, a system of “mixed lymphocyte-tumor cells” (MLTC) was established, wherein peripheral blood mononuclear cells (PBMCs) from a patient with metastatic melanoma were cocultured with autologous tumor cells. The MTLC system could be viewed as a simplified in vitro simulation of the tumor microenvironment. Furthermore, cytotoxic T lymphocyte (CTLs) clone derived by limiting dilution from the MLTC system or MLTC were subject to autologous tumor cell cDNA library screening. T cell clones reactive to 5 mutated epitopes were obtained.
The use of MHC-peptide tetramers is a canonical method to identify and study a certain antigen-specific T cell subset (69–71). For ACT, tetramers were used to isolate and expand tumor antigen-specific T cells (72). Moreover, in immune checkpoint inhibitor (ICI)-treated cancer patients, MHC-peptide tetramers have been successfully used to monitor neoantigen-specific T cells (9, 12). Cohen et al. (73) used this method to sort neoantigen-specific T cells from the PBLs of patients with metastatic melanoma. In brief, a panel of MHC-peptide tetramers consisting of predicted neo-epitopes was synthesized and used to screen PBLs. Neoantigen-specific T cells targeting 8 of the 9 mutated epitopes identified from TILs could be isolated from autologous peripheral blood with frequencies ranging between 0.4 and 0.002%. In cancers with intermediate mutational loads, such as multiple myeloma, the use of MHC-peptide tetramers could also isolate neoantigen-specific T cells from the PBLs (74). However, this method was only applied to CD8+ T cells and required HLA-binding prediction algorithms to guide the synthesis of HLA-peptide tetramers.
A previous study has shown that PD-1 expression could guide the identification of neoantigen-specific CD8+ T cells from the tumor microenvironment (49). The same strategy could be adopted for isolation from PBLs (75). In one study, 4 patients with metastatic melanoma were enrolled. CD8+ PBLs were expanded in vitro and cocultured with autologous DCs, which were electroporated with in vitro transcribed TMG RNA for mutant epitopes. In 3 out of 4 patients, neoantigen-specific lymphocytes could be isolated from the CD8+PD-1+ lymphocyte subset, but not the CD8+PD-1– lymphocyte subset (75).
The isolation of neoantigen-specific cells from the PBLs of patients with epithelial cancer is even more challenging. Preexisting antigen-specific memory T cells may represent a potential solution. Memory T cells, including central memory T cells (TCM), effector memory T cells (TEM), and TEMRA from PBLs were cocultured with DCs loaded with candidate neoantigens in the TMG or peptide form (76). After coculturing, memory cells were restimulated with DCs loaded with all TMGs and then sorted by the expression of CD134 and CD137 to enrich for neoantigen-reactive T cells. The resulting cells were then expanded and screened against all TMGs to test for neoantigen recognition. With this highly sensitive “in vitro stimulation (IVS)” method, T cells targeting KRASG12D and KRASG12V were successfully isolated from 3 out of 6 epithelial cancer patients. This new method enabled identification and isolation of neoantigen-reactive T cells from the blood circulation at very low frequencies.
The identification of neoantigen-specific T cells from naïve T cells is also of interest. A previous report showed that both naïve and activated neoantigen-specific T cells could be expanded from the peripheral blood of follicular lymphoma patients by priming with peptide-pulsed DCs (77). Using the same method, neoantigen-specific T cells were successfully expanded from the peripheral blood of HLA-matched healthy donors (30, 78). These preliminary results support the use of naïve T cells as an alternative source for ACT; however, their exceptionally low frequencies in peripheral blood and requirement for repeated stimulation pose hurdles (79).
Recently, a large, library-based “mini-lines” screening approach was proposed, which aimed to identify naïve antigen-reactive T cells from small volumes of blood (80–82). This system began with a small-scale culture in 96-well plates with 2,000 initial T cells in each well. The small-scale culture underwent a rapid 1,000- to 5,000-fold expansion (mini-line). Thousands of such well-scaled cultures were conducted simultaneously. Each T cell clone was maintained at a frequency of 1 in 2,000 but amplified to an absolute number of 1,000–5,000 cells, which is a sufficient number for routine detection. Applying this high-throughput parallel T cell culture system, neoantigen-specific T cells were identified and expanded 3–9 months prior to the first tumor recurrence in a patient with high-grade serous ovarian cancer. However, the long duration of culture possibly rendered this method more suitable as a preemptive therapeutic strategy (83).
Discussion
After decades of efforts, the adoptive transfer of neoantigen-specific T cells is finally close to readiness for clinical application. High efficacy of this immunotherapeutic strategy has been achieved in a number of cancer patients and the prospects are promising. However, these approaches are also quite costly and hard to apply to large numbers of patients. The current methods of identifying neoantigen-specific T cells are summarized in Figure 2A and Supplementary Table 2. More convenient and effective screening methods for neoantigen-specific T cells remain necessary, some strategies to improve neoantigen-specific T cells identification are shown in Figure 2A.
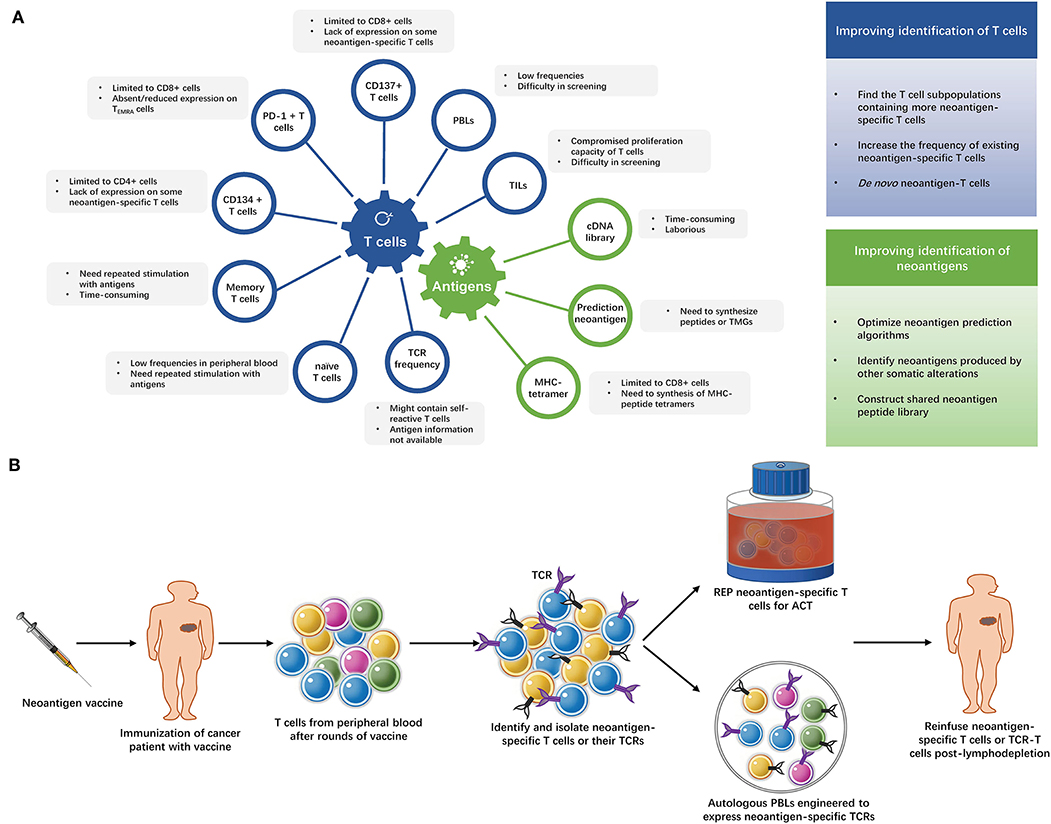
Figure 2. (A) Strategies of identifying neoantigen-specific T cells. The limitations of current methods of identifying neoantigen-specific T cells and strategies to improve neoantigen-specific T cells identification. TILs, tumor infiltrating lymphocytes; PBLs, peripheral blood lymphocytes; PD-1, programmed cell death-1; TEMRA cells, terminally differentiated effector memory cells; TCR, T cell receptor; TMG, tandem minigene; MHC, major histocompatibility complex. (B) The “blueprint” of isolating neoantigen-specific T cells from peripheral blood after neoantigen-targeting vaccine. After several rounds of immunization with neoantigen vaccines, T cells are collected from the patient's peripheral blood and neoantigen-specific T cells are identified and isolated from these T cells. Then, the neoantigen-specific T cells undergo rapid expansion (REP), or their TCRs are exploited to modify autologous lymphocytes. The expanded neoantigen-specific T cells or modified TCR-T cells are reinfused to the patient.
It is feasible to obtain neoantigen-targeting T cells from PBLs although their frequencies are generally much lower than TILs (73, 75, 76). However, increasing the frequencies of these valuable neoantigen-specific T cells in peripheral blood remains a challenge.
Vaccination with neo-peptides has been shown to prime CD4+ and CD8+ T-cell responses in mouse models (54, 84). Patients treated with vaccines generated neoantigen-specific T cells (2–8). It could be reasonably inferred that the isolation of neoantigen-reactive T cells from the peripheral blood would be more easily achieved following neoantigen-specific vaccination. This neoantigen-based combo immunotherapy has its advantages: first, isolation and expansion of TILs in vitro is not necessary. Second, cancer vaccines not only elicit neoantigen-specific T cell responses and amplify existing tumor-specific T cells responses, but they also increase the breadth and diversity of the tumor-specific T cell response (1, 7). Multiclonal T cells may, thus, be obtained. Third, the relatively easy preparation of cancer vaccines would buy time for the isolation of neoantigen-specific T cells in maintaining the performance of patients. The “blueprint” is shown in Figure 2B.
Conclusion
The previous decade has witnessed the emergence of immunotherapy for cancer. Accumulating evidence suggests that neoantigen-specific T cells underlie successful immunotherapy. Therefore, the isolation of neoantigen-specific T lymphocytes represents the “holy grail” for cancer immunotherapy. However, a fundamental challenge is to effectively identify and isolate neoantigen-specific T cells. The developments summarized in this review and future breakthroughs are anticipated to translate the adoptive transfer of neoantigen-specific T cells into a powerful weapon in our armamentarium against cancer.
Author Contributions
QL prepared the manuscript draft. Z-YD revised it critically for important intellectual content and approved the final version. QL and Z-YD contributed to the conception and design of the review. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Clinical Research Center for Geriatrics, West China Hospital, Sichuan University (Z2018B18).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2020.01347/full#supplementary-material
References
1. Hu Z, Ott PA, Wu CJ. Towards personalized, tumour-specific, therapeutic vaccines for cancer. Nat Rev Immunol. (2018) 18:168–82. doi: 10.1038/nri.2017.131
2. Chen FJ, Zou ZY, Du J, Su S, Shao J, Meng FY, et al. Neoantigen identification strategies enable personalized immunotherapy in refractory solid tumors. J Clin Invest. (2019) 129:2056–70. doi: 10.1172/JCI99538
3. Keskin DB, Anandappa AJ, Sun J, Tirosh I, Mathewson ND, Li SQ, et al. Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial. Nature. (2019) 565:234–9.
4. Hilf N, Kuttruff-Coqui S, Frenzel K, Bukur V, Stevanovic S, Gouttefangeas C, et al. Actively personalized vaccination trial for newly diagnosed glioblastoma. Nature. (2019) 565:240–5. doi: 10.1038/s41586-018-0810-y
5. Ott PA, Hu ZT, Keskin DB, Shukla SA, Sun J, Bozym DJ, et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature. (2017) 547:217–21. doi: 10.1038/nature22991
6. Sahin U, Derhovanessian E, Miller M, Kloke BP, Simon P, Lower M, et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature. (2017) 547:222–6. doi: 10.1038/nature23003
7. Carreno BM, Magrini V, Becker-Hapak M, Kaabinejadian S, Hundal J, Petti AA, et al. A dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specific T cells. Science. (2015) 348:803–8. doi: 10.1126/science.aaa3828
8. Tanyi JL, Bobisse S, Ophir E, Tuyaerts S, Roberti A, Genolet R, et al. Personalized cancer vaccine effectively mobilizes antitumor T cell immunity in ovarian cancer. Sci Transl Med. (2018) 10:eaao5931. doi: 10.1126/scitranslmed.aao5931
9. van Rooij N, van Buuren MM, Philips D, Velds A, Toebes M, Heemskerk B, et al. Tumor exome analysis reveals neoantigen-specific T-cell reactivity in an ipilimumab-responsive melanoma. J Clin Oncol. (2013) 31:E439–42. doi: 10.1200/JCO.2012.47.7521
10. Gubin MM, Zhang XL, Schuster H, Caron E, Ward JP, Noguchi T, et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature. (2014) 515:577–81. doi: 10.1038/nature13988
11. Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, et al. PD-1 blockade in tumors with mismatch-repair deficiency. New Engl J Med. (2015) 372:2509–20. doi: 10.1056/NEJMc1510353
12. Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, et al. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. (2015) 348:124–8. doi: 10.1126/science.aaa1348
13. Van Allen EM, Miao D, Schilling B, Shukla SA, Blank C, Zimmer L, et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science. (2015) 350:207–11. doi: 10.1126/science.aad0095
14. Dudley ME, Wunderlich JR, Robbins PF, Yang JC, Hwu P, Schwartzentruber DJ, et al. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science. (2002) 298:850–4. doi: 10.1126/science.1076514
15. Dudley ME, Yang JC, Sherry R, Hughes MS, Royal R, Kammula U, et al. Adoptive cell therapy for patients with metastatic melanoma: evaluation of intensive myeloablative chemoradiation preparative regimens. J Clin Oncol. (2008) 26:5233–9. doi: 10.1200/JCO.2008.16.5449
16. Rosenberg SA, Yang JC, Sherry RM, Kammula US, Hughes MS, Phan GQ, et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin Cancer Res. (2011) 17:4550–7. doi: 10.1158/1078-0432.CCR-11-0116
17. Besser MJ, Shapira-Frommer R, Itzhaki O, Treves AJ, Zippel DB, Levy D, et al. Adoptive transfer of tumor-infiltrating lymphocytes in patients with metastatic melanoma: intent-to-treat analysis and efficacy after failure to prior immunotherapies. Clin Cancer Res. (2013) 19:4792–800. doi: 10.1158/1078-0432.CCR-13-0380
18. Andersen R, Donia M, Ellebaek E, Borch TH, Kongsted P, Iversen TZ, et al. Long-lasting complete responses in patients with metastatic melanoma after adoptive cell therapy with tumor-infiltrating lymphocytes and an attenuated IL2 regimen. Clin Cancer Res. (2016) 22:3734–45. doi: 10.1158/1078-0432.CCR-15-1879
19. Stevanovic S, Draper LM, Langhan MM, Campbell TE, Kwong ML, Wunderlich JR, et al. Complete regression of metastatic cervical cancer after treatment with human papillomavirus-targeted tumor-infiltrating T cells. J Clin Oncol. (2015) 33:1543–50. doi: 10.1200/JCO.2014.58.9093
20. Huang J, El-Gamil M, Dudley ME, Li YF, Rosenberg SA, Robbins PF. T cells associated with tumor regression recognize frameshifted products of the CDKN2A tumor suppressor gene locus and a mutated HLA class I gene product. J Immunol. (2004) 172:6057–64. doi: 10.4049/jimmunol.172.10.6057
21. Zhou JH, Dudley ME, Rosenberg SA, Robbins PF. Persistence of multiple tumor-specific T-Cell clones is associated with complete tumor regression in a melanoma patient receiving adoptive cell transfer therapy. J Immunother. (2005) 28:53–62. doi: 10.1097/00002371-200501000-00007
22. Lu YC, Yao X, Li YF, El-Gamil M, Dudley ME, Yang JC, et al. Mutated PPP1R3B is recognized by T cells used to treat a melanoma patient who experienced a durable complete tumor regression. J Immunol. (2013) 190:6034–42. doi: 10.4049/jimmunol.1202830
23. Robbins PF, Lu YC, El-Gamil M, Li YF, Gross C, Gartner J, et al. Mining exomic sequencing data to identify mutated antigens recognized by adoptively transferred tumor-reactive T cells. Nat Med. (2013) 19:747–52. doi: 10.1038/nm.3161
24. Lu YC, Yao X, Crystal JS, Li YF, El-Gamil M, Gross C, et al. Efficient identification of mutated cancer antigens recognized by T cells associated with durable tumor regressions. Clin Cancer Res. (2014) 20:3401–10. doi: 10.1158/1078-0432.CCR-14-0433
25. Prickett TD, Crystal JS, Cohen CJ, Pasetto A, Parkhurst MR, Gartner JJ, et al. Durable complete response from metastatic melanoma after transfer of autologous T cells recognizing 10 mutated tumor antigens. Cancer Immunol Res. (2016) 4:669–78. doi: 10.1158/2326-6066.CIR-15-0215
26. Stevanovic S, Pasetto A, Helman SR, Gartner JJ, Prickett TD, Howie B, et al. Landscape of immunogenic tumor antigens in successful immunotherapy of virally induced epithelial cancer. Science. (2017) 356:200–5. doi: 10.1126/science.aak9510
27. Tran E, Turcotte S, Gros A, Robbins PF, Lu YC, Dudley ME, et al. Cancer immunotherapy based on mutation-specific CD4+T cells in a patient with epithelial cancer. Science. (2014) 344:641–5. doi: 10.1126/science.1251102
28. Tran E, Robbins PF, Lu YC, Prickett TD, Gartner JJ, Jia L, et al. T-cell transfer therapy targeting mutant KRAS in cancer. New Engl J Med. (2016) 375:2255–62. doi: 10.1056/NEJMoa1609279
29. Zacharakis N, Chinnasamy H, Black M, Xu H, Lu YC, Zheng ZL, et al. Immune recognition of somatic mutations leading to complete durable regression in metastatic breast cancer. Nat Med. (2018) 24:724–30. doi: 10.1038/s41591-018-0040-8
30. Stronen E, Toebes M, Kelderman S, van Buuren MM, Yang WW, van Rooij N, et al. Targeting of cancer neoantigens with donor-derived T cell receptor repertoires. Science. (2016) 352:1337–41. doi: 10.1126/science.aaf2288
31. Yarchoan M, Johnson BA, Lutz ER, Laheru DA, Jaffee EM. Targeting neoantigens to augment antitumour immunity. Nat Rev Cancer. (2017) 17:209–22. doi: 10.1038/nrc.2016.154
32. Tran E, Robbins PF, Rosenberg SA. 'Final common pathway' of human cancer immunotherapy: targeting random somatic mutations. Nat Immunol. (2017) 18:255–62. doi: 10.1038/ni.3682
33. Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science. (2015) 348:69–74. doi: 10.1126/science.aaa4971
34. Wirth TC, Kuhnel F. Neoantigen targeting-dawn of a new era in cancer immunotherapy? Front Immunol. (2017) 8:1848. doi: 10.3389/fimmu.2017.01848
35. Zamora AE, Crawford JC, Allen EK, Guo XJ, Bakke J, Carter RA, et al. Pediatric patients with acute lymphoblastic leukemia generate abundant and functional neoantigen-specific CD8(+) T cell responses. Sci Transl Med. (2019) 11: eaat8549. doi: 10.1126/scitranslmed.aat8549
36. Yossef R, Tran E, Deniger DC, Gros A, Pasetto A, Parkhurst MR, et al. Enhanced detection of neoantigen-reactive T cells targeting unique and shared oncogenes for personalized cancer immunotherapy. JCI Insight. (2018) 3:122467. doi: 10.1172/jci.insight.122467
37. Vinay DS, Kwon BS. Role of 4-1BB in immune responses. Semin Immunol. (1998) 10:481–9. doi: 10.1006/smim.1998.0157
38. Watts TH. Tnf/tnfr family members in costimulation of T cell responses. Annu Rev Immunol. (2005) 23:23–68. doi: 10.1146/annurev.immunol.23.021704.115839
39. Cannons JL, Lau P, Ghumman B, de Benedette MA, Yagita H, Okumura K, et al. 4-1BB ligand induces cell division, sustains survival, and enhances effector function of CD4 and CD8 T cells with similar efficacy. J Immunol. (2001) 167:1313–24. doi: 10.4049/jimmunol.167.3.1313
40. Halstead ES, Mueller YM, Altman JD, Katsikis PD. In vivo stimulation of CD137 broadens primary antiviral CD8(+) T cell responses. Nat Immunol. (2002) 3:536–41. doi: 10.1038/ni798
41. Wolfl M, Kuball J, Ho WY, Nguyen H, Manley TJ, Bleakley M, et al. Activation-induced expression of CD137 permits detection, isolation, and expansion of the full repertoire of CD8(+) T cells responding to antigen without requiring knowledge of epitope specificities. Blood. (2007) 110:201–10. doi: 10.1182/blood-2006-11-056168
42. Ye QR, Song DG, Poussin M, Yamamoto T, Best A, Li CS, et al. CD137 accurately identifies and enriches for naturally occurring tumor-reactive T cells in tumor. Clin Cancer Res. (2014) 20:44–55. doi: 10.1158/1078-0432.CCR-13-0945
43. Parkhurst M, Gros A, Pasetto A, Prickett T, Crystal JS, Robbins P, et al. Isolation of T-cell receptors specifically reactive with mutated tumor-associated antigens from tumor-infiltrating lymphocytes based on CD137 expression. Clin Cancer Res. (2017) 23:2491–505. doi: 10.1158/1078-0432.CCR-16-2680
44. Seliktar-Ofir S, Merhavi-Shoham E, Itzhaki O, Yunger S, Markel G, Schachter J, et al. Selection of shared and neoantigen-reactive T cells for adoptive cell therapy based on CD137 separation. Front Immunol. (2017) 8:1211 doi: 10.3389/fimmu.2017.01211
45. Makkouk A, Chester C, Kohrt HE. Rationale for anti-CD137 cancer immunotherapy. Eur J Cancer. (2016) 54:112–9. doi: 10.1016/j.ejca.2015.09.026
46. Chen L. Co-inhibitory molecules of the B7-CD28 family in the control of T-cell immunity. Nat Rev Immunol. (2004) 4:336–47. doi: 10.1038/nri1349
47. Greenwald RJ, Freeman GJ, Sharpe AH. The B7 family revisited. Annu Rev Immunol. (2005) 23:515–48. doi: 10.1146/annurev.immunol.23.021704.115611
48. Sharpe AH, Wherry EJ, Ahmed R, Freeman GJ. The function of programmed cell death 1 and its ligands in regulating autoimmunity and infection. Nat Immunol. (2007) 8:239–45. doi: 10.1038/ni1443
49. Gros A, Robbins PF, Yao X, Li YF, Turcotte S, Tran E, et al. PD-1 identifies the patient-specific CD8(+) tumor-reactive repertoire infiltrating human tumors. J Clin Invest. (2014) 124:2246–59. doi: 10.1172/JCI73639
50. Inozume T, Hanada KI, Wang QJ, Ahmadzadeh M, Wunderlich JR, Rosenberg SA, et al. Selection of CD8(+)PD-1(+) lymphocytes in fresh human melanomas enriches for tumor-reactive T cells. J Immunother. (2010) 33:956–64. doi: 10.1097/CJI.0b013e3181fad2b0
51. Ahmadzadeh M, Johnson LA, Heemskerk B, Wunderlich JR, Dudley ME, White DE, et al. Tumor antigen-specific CD8 T cells infiltrating the tumor express high levels of PD-1 and are functionally impaired. Blood. (2009) 114:1537–44. doi: 10.1182/blood-2008-12-195792
52. Larbi A, Fulop T. From “Truly Naive” to “Exhausted Senescent” T cells: when markers predict functionality. Cytom A. (2014) 85a:25–35. doi: 10.1002/cyto.a.22351
53. Vogelstein B, Papadopoulos N, Velculescu VE, Zhou SB, Diaz LA, Kinzler KW. Cancer genome landscapes. Science. (2013) 339:1546–58. doi: 10.1126/science.1235122
54. Kreiter S, Vormehr M, van de Roemer N, Diken M, Lower M, Diekmann J, et al. Mutant MHC class II epitopes drive therapeutic immune responses to cancer. Nature. (2015) 520:692–6. doi: 10.1038/nature14426
55. Hunder NN, Wallen H, Cao JH, Hendricks DW, Reilly JZ, Rodmyre R, et al. Treatment of metastatic melanoma with autologous CD4+T cells against NY-ESO-1. New Engl J Med. (2008) 358:2698–703. doi: 10.1056/NEJMoa0800251
56. Linnemann C, van Buuren MM, Bies L, Verdegaal EME, Schotte R, Calis JJA, et al. High-throughput epitope discovery reveals frequent recognition of neo-antigens by CD4(+) T cells in human melanoma. Nat Med. (2015) 21:81–5. doi: 10.1038/nm.3773
57. Watanabe K, Suzuki S, Kamei M, Toji S, Kawase T, Takahashi T, et al. CD137-guided isolation and expansion of antigen-specific CD8 cells for potential use in adoptive immunotherapy. Int J Hematol. (2008) 88:311–20. doi: 10.1007/s12185-008-0134-z
58. Tran E, Ahmadzadeh M, Lu YC, Gros A, Turcotte S, Robbins PF, et al. Immunogenicity of somatic mutations in human gastrointestinal cancers. Science. (2015) 350:1387–90. doi: 10.1126/science.aad1253
59. Dziubianau M, Hecht J, Kuchenbecker L, Sattler A, Stervbo U, Rodelsperger C, et al. TCR repertoire analysis by next generation sequencing allows complex differential diagnosis of t cell-related pathology. Am J Transplant. (2013) 13:2842–54. doi: 10.1111/ajt.12431
60. Hohn H, Neukirch C, Freitag K, Necker A, Hitzler W, Seliger B, et al. Longitudinal analysis of the T-cell receptor (TCR)-VA and -VB repertoire in CD8(+) T cells from individuals immunized with recombinant hepatitis B surface antigen. Clin Exp Immunol. (2002) 129:309–17. doi: 10.1046/j.1365-2249.2002.01841.x
61. Sherwood AM, Emerson RO, Scherer D, Habermann N, Buck K, Staffa J, et al. Tumor-infiltrating lymphocytes in colorectal tumors display a diversity of T cell receptor sequences that differ from the T cells in adjacent mucosal tissue. Cancer Immunol Immunother. (2013) 62:1453–61. doi: 10.1007/s00262-013-1446-2
62. Emerson RO, Sherwood AM, Rieder MJ, Guenthoer J, Williamson DW, Carlson CS, et al. High-throughput sequencing of T-cell receptors reveals a homogeneous repertoire of tumour-infiltrating lymphocytes in ovarian cancer. J Pathol. (2013) 231:433–40. doi: 10.1002/path.4260
63. Pasetto A, Gros A, Robbins PF, Deniger DC, Prickett TD, Matus-Nicodemos R, et al. Tumor- and neoantigen-reactive T-cell receptors can be identified based on their frequency in fresh tumor. Cancer Immunol Res. (2016) 4:734–43. doi: 10.1158/2326-6066.CIR-16-0001
64. Willimsky G, Blankenstein T. Sporadic immunogenic tumours avoid destruction by inducing T-cell tolerance. Nature. (2005) 437:141–6. doi: 10.1038/nature03954
65. Lanitis E, Irving M, Coukos G. Targeting the tumor vasculature to enhance T cell activity. Curr Opin Immunol. (2015) 33:55–63. doi: 10.1016/j.coi.2015.01.011
66. Motz GT, Santoro SP, Wang LP, Garrabrant T, Lastra RR, Hagemann IS, etal. Tumor endothelium FasL establishes a selective immune barrier promoting tolerance in tumors. Nat Med. (2014) 20:607–15. doi: 10.1038/nm.3541
67. Munn DH, Bronte V. Immune suppressive mechanisms in the tumor microenvironment. Curr Opin Immunol. (2016) 39:1–6. doi: 10.1016/j.coi.2015.10.009
68. Lennerz V, Fatho M, Gentilini C, Frye RA, Lifke A, Ferel D, et al. The response of autologous T cells to a human melanoma is dominated by mutated neoantigens. Proc Natl Acad Sci USA. (2005) 102:16013–8. doi: 10.1073/pnas.0500090102
69. Lee PP, Yee C, Savage PA, Fong L, Brockstedt D, Weber JS, et al. Characterization of circulating T cells specific for tumor-associated antigens in melanoma patients. Nat Med. (1999) 5:677–85. doi: 10.1038/9525
70. Rubio V, Stuge TB, Singh N, Betts MR, Weber JS, Roederer M, et al. Ex vivo identification, isolation and analysis of tumor-cytolytic T cells. Nat Med. (2003) 9:1377–82. doi: 10.1038/nm942
71. Yee C, Savage PA, Lee PP, Davis MM, Greenberg PD. Isolation of high avidity melanoma-reactive CTL from heterogeneous populations using peptide-MHC tetramers. J Immunol. (1999) 162:2227–34.
72. Yee C, Thompson JA, Byrd D, Riddell SR, Roche P, Celis E, et al. Adoptive T cell therapy using antigen-specific CD8(+) T cell clones for the treatment of patients with metastatic melanoma: in vivo persistence, migration, and antitumor effect of transferred T cells. Proc Natl Acad Sci USA. (2002) 99:16168–73. doi: 10.1073/pnas.242600099
73. Cohen CJ, Gartner JJ, Horovitz-Fried M, Shamalov K, Trebska-McGowan K, Bliskovsky VV, et al. Isolation of neoantigen-specific T cells from tumor and peripheral lymphocytes. J Clin Invest. (2015) 125:3981–91. doi: 10.1172/JCI82416
74. Perumal D, Imai N, Lagana A, Finnigan J, Melnekoff D, Leshchenko VV, et al. Mutation-derived neoantigen-specific T-cell responses in multiple myeloma. Clin Cancer Res. (2020) 26:450–64. doi: 10.1158/1078-0432.CCR-19-2309
75. Gros A, Parkhurst MR, Tran E, Pasetto A, Robbins PF, Ilyas S, et al. Prospective identification of neoantigen-specific lymphocytes in the peripheral blood of melanoma patients. Nat Med. (2016) 22:433–8. doi: 10.1038/nm.4051
76. Cafri G, Yossef R, Pasetto A, Deniger DC, Lu YC, Parkhurst M, et al. Memory T cells targeting oncogenic mutations detected in peripheral blood of epithelial cancer patients. Nat Commun. (2019) 10:449. doi: 10.1038/s41467-019-08304-z
77. Nielsen JS, Sedgwick CG, Shahid A, Zong ZS, Brumme ZL, Yu S, et al. Toward personalized lymphoma immunotherapy: identification of common driver mutations recognized by patient CD8(+) T cells. Clin Cancer Res. (2016) 22:2226–36. doi: 10.1158/1078-0432.CCR-15-2023
78. Nielsen JS, Chang AR, Wick DA, Sedgwick CG, Zong ZS, Mungall AJ, et al. Mapping the human T cell repertoire to recurrent driver mutations in MYD88 and EZH2 in lymphoma. Oncoimmunology. (2017) 6:e1321184. doi: 10.1080/2162402X.2017.1321184
79. Wolfl M, Greenberg PD. Antigen-specific activation and cytokine-facilitated expansion of naive, human CD8+ T cells. Nat Protoc. (2014) 9:950–66. doi: 10.1038/nprot.2014.064
80. Geiger R, Duhen T, Lanzavecchia A, Sallusto F. Human naive and memory CD4(+) T cell repertoires specific for naturally processed antigens analyzed using libraries of amplified T cells. J Exp Med. (2009) 206:1525–34. doi: 10.1084/jem.20090504
81. Theaker SM, Rius C, Greenshields-Watson A, Lloyd A, Trimby A, Fuller A, et al. T-cell libraries allow simple parallel generation of multiple peptide-specific human T-cell clones. J Immunol Methods. (2016) 430:43–50. doi: 10.1016/j.jim.2016.01.014
82. Costa-Nunes C, Cachot A, Bobisse S, Arnaud M, Genolet R, Baumgaertner P, et al. High-throughput screening of human tumor antigen-specific CD4 T cells, including neoantigen-reactive T cells. Clin Cancer Res. (2019) 25:4320–31. doi: 10.1158/1078-0432.CCR-18-1356
83. Martin SD, Wick DA, Nielsen JS, Little N, Holt RA, Nelson BH. A library-based screening method identifies neoantigen-reactive T cells in peripheral blood prior to relapse of ovarian cancer. Oncoimmunology. (2018) 7:e1371895. doi: 10.1080/2162402X.2017.1371895
Keywords: adoptive cell therapy (ACT), neoantigen-specific T cells, T cell receptor (TCR), tumor infiltrating lymphocytes (TILs), peripheral blood lymphocytes (PBLs)
Citation: Li Q and Ding Z-Y (2020) The Ways of Isolating Neoantigen-Specific T Cells. Front. Oncol. 10:1347. doi: 10.3389/fonc.2020.01347
Received: 12 December 2019; Accepted: 26 June 2020;
Published: 11 August 2020.
Edited by:
Cyrille J. Cohen, Bar-Ilan University, IsraelReviewed by:
Manel Juan, Hospital Clínic de Barcelona, SpainRodabe N. Amaria, University of Texas MD Anderson Cancer Center, United States
Copyright © 2020 Li and Ding. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhen-Yu Ding, ZGluZ3poZW55dUBzY3UuZWR1LmNu