 Yina Yu
Yina Yu Liang Gong2†
Liang Gong2† Jun Ye
Jun Ye- 1Department of Gastroenterology, The Second Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, China
- 2Department of Otolaryngology, Cixi People's Hospital, Ningbo, China
Metabolic syndrome (MetS) is characterized by hyperglycemia, hypertension, dyslipidemia and abdominal obesity. Patients with MetS or other metabolic disorders are more susceptible to cancer development and recurrence and have a worse long-term prognosis. Moreover, the metabolic reprogramming observed in cancer cells has also been described as one of the new hallmarks of cancer. Thus, aberrant metabolism has been proposed as an important risk factor for cancer. Chronic inflammation, reactive oxygen species (ROS), and oncogenic signaling pathways are considered as main potential triggers. Considering the strong association between metabolism and cancer, metabolism-modulating drugs, including metformin and statins, as well as adopting a healthy lifestyle, have been extensively investigated as strategies to combat cancer. Furthermore, strategies that interfere with the metabolic rewiring of cells may also have potent anti-cancer effects. In this article, we provide a comprehensive review of current knowledge on the relationship between aberrant metabolism and cancer and discuss the potential use of metabolism-targeting strategy for the treatment of cancer.
Introduction
Metabolic Syndrome (MetS) is a constellation of metabolic risk factors and a significant cause of morbidity and mortality. The five main components of MetS are abdominal obesity, hyperglycemia, high blood pressure, hypertriglyceridemia, and low high-density lipoprotein (HDL)-cholesterol levels (1, 2). Although the reported prevalence of MetS varies among different studies, its prevalence is increasing at an alarming rate worldwide.In Western countries, one in five adults are diagnosed with MetS (3). Cancer is the leading cause of death and, despite advances in cancer prevention, its incidence remains exceptionally high. Given the high prevalence of metabolic disorders and cancer, as well as the fact that many patients suffer from both, the potential association between MetS and cancer has been extensively investigated.
Several studies have suggested that people who suffer from MetS have higher chances of developing cancer; the rate of cancer recurrence and mortality are also higher. A recent study has shown that people diagnosed with MetS have a 33% higher cancer mortality rate compared to patients who have no metabolic disorders (4). Furthermore, the number of MetS components was directly proportional to the cancer-related mortality rate (4). Among the MetS components, obesity and hyperglycemia have been suggested as the determinants of tumor-associated clinicopathology. Furthermore, mounting evidence highlights the impact of certain metabolic disorders on the risk for several types of cancer, including colorectal, prostate, pancreatic, renal, liver, post-menopausal breast, and endometrial cancer (5–8). Moreover, malignant cells acquire changes in anabolic and catabolic pathways to meet their high metabolic and energy demands, a phenomenon known as metabolic reprogramming (9). Importantly, metabolic reprogramming has emerged as a hallmark of cancer and has been shown to be involved in cancer initiation, progression, and metastasis, as well as the survival of cancer cells and the development of resistance to antitumor therapies.
The identification of metabolic reprogramming and metabolic syndrome as important regulators of cancer development and progression has provided a rationale for the development of metabolism-targeting therapies as a promising therapeutic approach for cancer. Herein, we provide a comprehensive review of the current understanding related to the association between metabolism and cancer, the potential underlying mechanisms, and emerging metabolism-targeting anticancer therapies.
Cancer Cell Metabolic Reprogramming
Rewiring of Cancer Cell Metabolism
The rapidly dividing cancer cells have high demands for energy and nutrients to meet their high metabolic needs. Consequently, the cellular metabolism undergoes a rewiring during malignant transformation. Importantly, cancer cells switch from oxidative phosphorylation to aerobic glycolysis to generate ATP (adenosine triphosphate), a phenomenon commonly referred to as the Warburg effect (10). Several mechanisms have been identified to mediate the metabolic rewiring of cancer cells. Genome instability play a vital part in the alternation of energy metabolism (11). The aberrant activation of certain oncogenes such as K-ras (12), MYC (13), mTOR (14), and P53 (15), have been identified as cell-autonomous mechanisms regulating various aspects of the Warburg effect. Somatic mutations in the mitochondrial genome (mtDNA) or changes in the mtDNA content leading to mitochondrial dysfunction have been associated with an increased glycolytic rate in malignant cells (16).
Apart from cell-autonomous mechanisms, extrinsic factors have also been identified as driving metabolic reprogramming in cancer cells. The tumor microenvironment is often hypoxic in solid cancers, leading to the activation of HIF-1α. The transcriptional activity of HIF-1α inhibits mitochondrial respiratory chains and induces glycolysis, among other things. Several types of stromal cells, including tumor-associated macrophages (TAM), have also been implicated in establishing a hypoxic tumor microenvironment and promoting aerobic glycolysis (17), subsequently leading to metabolic reprogramming in cancer cells (18).
Increased uptake of glutamine and enhanced glutaminolysis have been identified as a hallmark of cancer cells. Glutamine is essential in cancer cells and has been implicated in cancer progression, as it serves as a substrate for oxidative metabolism, which generates more than half of the ATP that is required by malignant cells (19). As well as an important source of energy, glutamine also acts as a biosynthetic precursor for numerous molecules that are essential for rapidly proliferating cells, including fatty acids, pyrimidines, purines, and amino acids (19). Therefore, glutaminolysis is indispensable for metabolic reprogramming in cancer cells.
Mounting evidence suggests that lipid metabolism is also rewired in rapidly proliferating cells (20). The up-regulation of fatty acid synthase (FASN) and subsequent enhanced de novo synthesis of fatty acids have been described as a frequent event in cancer cells (21). The elevated levels of fatty acids will act as signaling molecules, energy storage, and cell membrane components, which enable tumor cells to meet the increased demands in energy and cellular components (20). Oncogenic mutations and loss of tumor suppressor genes have been shown to contribute to alterations in glutamine and lipid metabolism (11).
Metabolic Reprogramming and Metastatic Potential of Tumor Cells
Metabolic rewiring has been implicated in the enhanced ability of cancer cells to survive and proliferate, enabling them to survive under stressful conditions and resist anticancer therapies (22, 23). Importantly, increasing evidence suggests a role for metabolic reprogramming in enhancing the metastatic ability and development of cancer cells (Figure 1). Promoting the epithelial to mesenchymal transition (EMT) and subsequent detachment of cancer cells from the extracellular matrix early during metastasis are vital potential mechanisms (24). Lactate accumulation and extracellular matrix acidification due to the enhanced glycolytic rate can promote EMT by regulating the expression of EMT-related proteins (25). Moreover, extracellular acidosis can activate matrix-metalloproteinases (MMPs), which play a crucial role in the degradation of extracellular matrix and subsequent invasion of cancer cells into the vascular wall (26). The increase in the levels of the glycolysis by-product methylglyoxal, as well as the overexpression of glucose transporter 1 (GLUT1) and hexokinase 2 (HK2), have also been linked to enhanced metastatic potential (27–29). Aerobic glycolysis suppresses mitochondrial oxidative metabolism and induces anoikis resistance, promoting cancer cell migration and invasion (30).
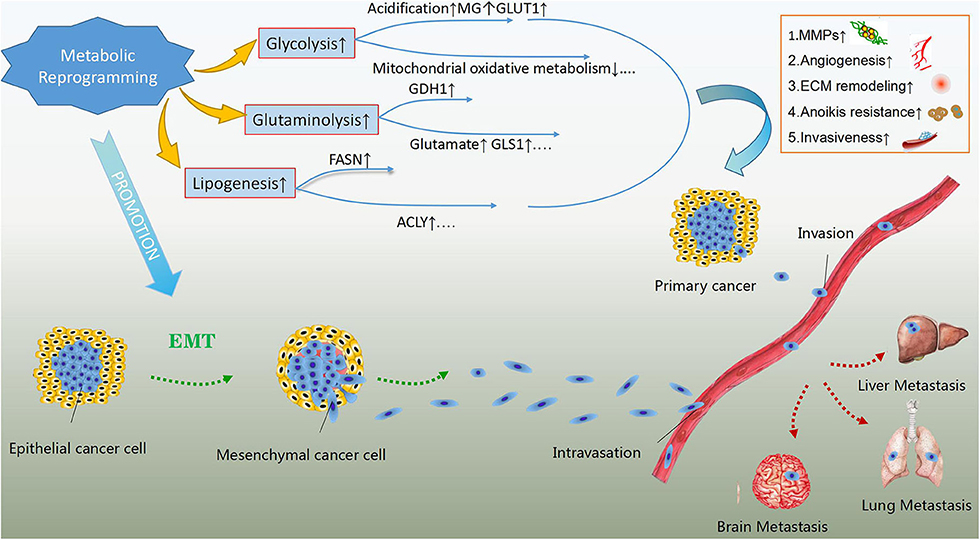
Figure 1. The effects of metabolic reprogramming in cancer development and metastasis. Metabolic reprogramming in cancer cells, mainly including glycolysis, glutaminolysis and lipogenesis can enhance their metastatic abilities. The products of rewired metabolism like lactate, methylglyoxal (MG) and glutamate as well as up-regulated key enzymes or concerning transporters in energy metabolism such as glutamate dehydrogenase 1(GDH1), glutaminase 1(GLS1), fatty acid synthase (FASN), ATP-citrate lyse (ACLY) and glucose transporter 1(GLUT1) are the potential regulatory mechanisms in addition to suppressed mitochondrial oxidative metabolism. These regulators can give rise to activation of matrix-metalloproteinases (MMPs), extracellular matrix (ECM) remodeling, angiogenesis, anoikis resistance, and enhanced invasiveness, which assist primary cancer cells in invading blood vessels and metastasizing to distant organs. In addition, rewired metabolism also promotes epithelial to mesenchymal transition, contributing to the metastasis of malignant cells.
Similarly, glutaminolysis contributes to EMT and metastasis by inducing the expression of glutamate dehydrogenase 1 (GDH1) (31). The serum levels of glutamate, the end product of glutaminolysis, have been linked to enhanced invasiveness and proliferation of prostate cancer cells (32), while in breast cancer cells, glutamate has been shown to induce migration by up-regulating the expression of transmembrane matrix metalloproteinases (33). Additionally, glutaminase 1 (GLS1), a key enzyme of glutamine metabolism, has also been implicated in cancer metastasis (34). FASN can also promote tumor cell growth and metastasis by activating the AMP-activated protein kinase (AMPK)/mTOR signaling pathway (35). The overexpression of another key enzyme of de novo lipogenesis, ATP-citrate lyase (ACLY), has been linked to enhanced metastatic ability and resistance to radiotherapy (36).
Potential Mechanisms Linking Metabolic Syndrome to Cancer
Inflammation
Inflammatory immune responses protect organisms from external pathological insults, including mechanical trauma and infectious microorganisms. In contrast to acute inflammation that has protective roles, chronic inflammation has been linked to numerous diseases, including cancer (37). Conditions caused by the disruption in metabolic homeostasis, such as central obesity and hyperglycemia, have been linked to chronic inflammation (38, 39). Anti-inflammation roles have been attributed to HDL, and a decrease in HDL levels combined with hyperglycemia can have synergistic effects in the establishment of chronic inflammation (40), linking the disruption in metabolic homeostasis to cancer development and progression.
Epidemiological studies have shown that approximately 15–20% of all cancers can be attributed to chronic inflammation or chronic infections (37). Inflammatory bowel disease (IBD), chronic hepatitis, and gastritis, respectively, elevating the risk of colorectal cancer, liver cancer, and gastric cancer are most notable examples. Inflammatory response makes a significant contribution to different stages of cancer progression (Table 1). Genetic mutations or alternations are recognized to account for most carcinogenic initiation. The long-term infiltration of inflammatory immune cells and chronic secretion of inflammatory cytokines, including tumor necrosis factor-alpha (TNF-α) and interleukins (ILs), may promote mutagenesis and abnormal gene expression (41). The increased generation of ROS by immune cells like macrophages and neutrophils during inflammation may also give rise to genetic alterations in normal cells (42). Herein, persistent inflammation can potentially predispose cells to undergo malignant transformation via inducing accumulated gene mutations. Furthermore, creating a favorable niche for transformed cells to reproduce and grow also makes a big difference in cancer development. Inflammatory cells and cytokines have been shown to induce EMT and promote cancer cell invasion (43, 44), as well as enhance cancer stemness (45), augmenting metastatic property of malignant cells. STAT3 and nuclear factor kappa B (NF-κB) signaling pathway can be activated by inflammatory responses which may mediate survival-favoring signaling and induce metabolic reprogramming, driving cancer cell growth, migration, and invasion (46, 47). The crosstalk between infiltrating inflammatory cells and malignant cells and recruitment of tumor-promoting auxiliary cells like fibroblasts confirm the decisive effect that inflammation exerts in modulating tumor microenvironment (TME) (48). Additionally, the relationship between inflammation and carcinogenesis is extremely complex, as tumors or anti-cancer therapy can also trigger inflammation via several pathways, further promoting cancer progression (62).
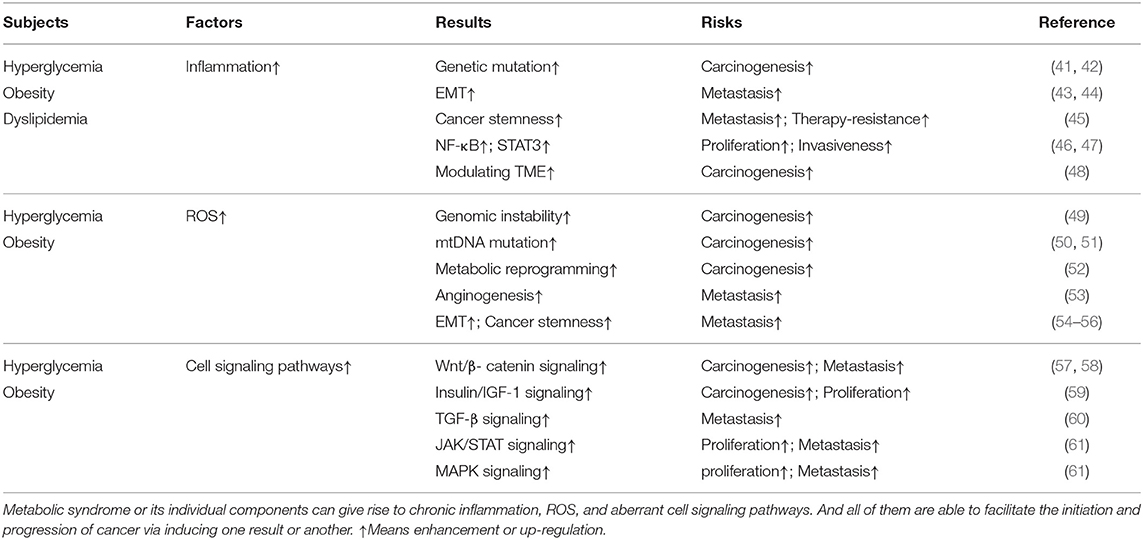
Table 1. The mechanism linking MetS to cancer.
ROS and Oxidative Stress
Reactive oxygen species (ROS) are chemically reactive molecules that are produced during oxygen metabolism. Superoxide anion (), hydrogen peroxide (H2O2), and hydroxyl radical (HO∙) are the most important ROS and are primarily produced in mitochondria by nicotinamide adenine dinucleotide phosphate (NADPH) oxidases (NOXs) (63). ROS serve as singling molecules and, in normal cells, the homeostasis in ROS levels is maintained by several cellular antioxidant mechanisms, protecting from oxidative damage. Abnormal metabolism and metabolic disorders, including hyperglycemia and adipose tissue expansion, can lead to enhanced ROS generation (64, 65).
Detection of a high ROS level in many cancer cells suggests that ROS play an essential role in cancer initiation and progression (66) (Table 1). An important mechanism by which ROS can drive carcinogenesis is the induction of DNA damage and genomic instability, contributing to malignant transformation (49). Additionally, ROS have been shown to induce mutagenesis in mitochondrial DNA (mtDNA), which has been linked to the development of certain malignancies, such as colon (50) and prostate (51) cancer. Promoting metabolic shift to glycolysis and doing damage to electron transfer may serve as underlying reasons why mutations in mtDNA can lead to tumorgenesis (52). However, only when cancer cells own the abilities to adapt to ROS stress can they survive and progress. The redox adaption including activation of redox-sensitive transcription factors like NF-κB and subsequent elevated expression of ROS-scavenging enzymes is not only responsible for cell survival but for progression as well (67). Some researchers already found ROS stress can promote angiogenesis and metastasis by inducing VEGF-A secretion and subsequent endothelial cell migration and proliferation (53). Additionally, the Wnt/β-catenin signaling pathway, which is known to drive EMT, cancer stem cell (CSC) development, and chemotherapy resistance, can be activated by ROS (54–56). Intriguingly, oncogene activation can also result in elevated ROS levels, thereby promoting cancer progression (68).
Cell Signaling Pathways Associated With Cancer
Mutagenic signaling pathways like Mitogen-activated protein kinase (MAPK), Wnt, TGF-β, and JAK/STAT have been well-demonstrated to be critical for cancer initiation and progression (Table 1). Accumulating evidence suggests that metabolic disorders are involved in the dysregulation of these and other oncogenic pathways. In accordance with previous studies demonstrating the role of hyperglycemia in carcinogenesis, a recent study has shown that hyperglycemia can induce the Wnt/β-catenin signaling pathway, promoting cancer cell survival and the progression of hyperglycemia-related cancer (57). Obesity has also been reported to activate the Wnt/β-catenin signaling pathway in a TNF-α-dependent way (58). Moreover, aberrant insulin/insulin-like growth factor-1 (IGF-1) signaling, which has a long-standing role in cancer, is activated by hyperglycemia or obesity and is believed to be an essential mediator of the oncogenic effects of these metabolic disorders (59). Hyperinsulinemia and insulin resistance augment the activation of the MAPK pathway, promoting cancer cell proliferative survival (69). Moreover, insulin has been shown to enhance TGF-β signaling (60). In an animal model, the AMPK/mTORC1/Nox4 signaling axis was highlighted as a critical molecular mechanism linking hyperglycemia to colorectal cancer (CRC) (70). The expansion of adipocytes and subsequent disruption of adipokine signaling homeostasis has also been suggested to be an important mechanism driving the progression of cancers associated with metabolic disorders. The enhanced secretion of leptin has been linked to cancer cell growth and migration by inducing the activation of STAT3, MAPK, JAK/STAT signaling pathways (61). On the other hand, adiponectin counteracting the carcinogenic effects of leptin by interfering with the activation of oncogenic signaling pathways is remarkably reduced in obese individuals (71). In summary, the aberrant activation of oncogenic signaling pathways observed in patients with metabolic disorders represents a link between metabolic syndrome and cancer.
Therapeutic Approaches to Target Metabolic Disorders
Metformin
Several epidemiological studies have suggested the potential anti-cancer effects of the antidiabetic agent metformin, especially in colorectal cancer (72) and endometrial cancer (73). The mechanisms underlying the anti-cancer effects of metformin have been extensively investigated (74). Metformin administration can reduce the levels of glucose in the blood, potentially contributing to its anti-cancer effects. In a study in hepatocellular carcinoma, metformin has been demonstrated to reduce the glycolytic flux by inhibiting phosphofructokinase-1 (PFK-1), suppressing the proliferation of cancer cells (75). Additionally, metformin suppresses the expression of HIF-1α and elevation of PDH levels under hypoxic conditions, interfering with aerobic glycolysis in cancer cells (76). A study by Kitson et al. showed that metformin was capable of interfering with the function of cancer stem cells and repressing the expression of CSC-related genes (77); the inhibitory effect of metformin on CSCs is likely to be dependent on AMPK-mTOR and glutamine metabolism (78). Furthermore, a recent study suggested that the vascular effects of metformin are potential mechanisms by which metformin suppresses metastasis and sensitizes to chemotherapy (79). Metformin has also been shown to suppress cancer cell growth, as well as induce apoptosis and autophagy (80). Despite mounting evidence suggesting the potential clinical benefit of metformin for cancer prevention and treatment, Farmer et al. pointed out that different types of bias existed in many observational clinical studies on metformin and cancer (81). Notably, latest findings show that when the studies are better designed, the protective effect of metformin is attenuated (82). Accordingly, the exact efficacy of metformin in cancer and clinical application need further exploration.
Statins
Statins are lipid-lowering agents commonly prescribed in patients with hyperlipidemia. Numerous epidemiological studies have suggested that statins may be useful in lowering the risk of cancer development. A prospective cohort study has demonstrated that statins can improve cancer patient survival and decrease the rate of cancer-related mortality in postmenopausal women (83). Patients with colorectal cancer (84) and liver cancer (85) may benefit from statins according to clinical or preclinical studies. The mechanisms underlying the anti-tumor effects of statins remain elusive. Statins are known to interfere with cholesterol synthesis by inhibiting 3-hydroxy-3methylglutary-coenzyme A(HMG-CoA) reductase. Interfering with cellular lipid biogenesis, mitochondrial metabolism, and ROS generation have been suggested as potential mechanisms by which statins may suppress cancer initiation and progression (86, 87). A study by Sadaria et al. has shown that intracellular adhesion molecule-1 (ICAM-1), an essential mediator of cancer cell metastasis, was suppressed by statins (88). Statins could also inhibit the transcription of MACC1 in colon cancer cells, suppressing cancer cell growth and metastasis (89). Importantly, statins have also been reported to have radiosensitizing effects in cancer cells (90). Autophagy evasion has been described as a hallmark of cancer, and statins have recently been shown to exert anti-cancer effects by inducing autophagy (91). A study in prostate cancer suggested that the combination of metformin and statins has synergistic effects in inducing apoptosis in chemotherapy-resistant cancer cells (92). Nevertheless, some meta-analyses demonstrated that more evidence is needed which supports the preventing role stains play in cancer (93, 94). Risk of bias must be attached more significance and be avoided in observational studies before evaluating links between statins and cancer (95). Taking all aforementioned findings into consideration, while statins seem to emerge as potential anti-cancer agents, especially for cancer patients with obesity or hypercholesterolemia, the application of statins to cancer needs further investigation.
Lifestyle Interventions
Extensive multicenter cohort studies have demonstrated that an unhealthy lifestyle is a strong risk factor for all-cause mortality (96), including death related to MetS and MetS-associated cancer. Therefore, adopting a healthy lifestyle has been proposed as a promising approach to prevent MetS and MetS-associated malignancies. Exercise is strongly recommended, not only for reducing the risk of obesity and other MetS components but also for lowering the risk of cancer development and cancer recurrence, as well as improving the quality of life of cancer patients (97). Additionally, exercise can improve insulin resistance and modulate chronic inflammation in obese individuals (98). Undoubtedly, regular exercise is extremely important in preventing MetS and tumor development (99). Keeping a disturbed circadian rhythms at a distance is also vigorously proposed due to its intimate relation with cancer and metabolism (100). Moreover, eating too fast has been associated with hypertriglyceridemia and other MetS components (101). According to the American Dietary Guidelines, its recommended food intake patterns, moderate alcohol consumption, and a diet high in vegetables, fruits, and whole grains and low in sugar or fat, can provide a benefit to numerous chronic diseases (102). Notably, a strong link between dietary patterns and microbiota composition has been described, suggesting that a healthy diet can promote gut microbiota diversity (103), reducing the risk of MetS and cancer. In summary, increasing evidence indicates that adopting a healthy lifestyle is a potent, easy, and low economic burden mechanism for preventing MetS and cancer (104, 105).
Discussion
As indicated by numerous epidemiological studies, along with changes in the pace and patterns of modern lifestyles, an increasing number of people are suffering from metabolic syndrome and cancer. A positive association between metabolic syndrome and cancer has been established (8). Metabolic disorders are widely accepted to be involved in the development and progression of several types of human cancers. Increased ROS production, chronic inflammation, and aberrant activation of oncogenic signaling pathways represent important links between metabolic disorders and cancer (41, 106). However, additional underlying mechanisms potentially exist, which are yet to be elucidated. For example, elevated estrogen levels in adipose tissue, as well as the increased synthesis of extracellular matrix proteins and hyaluronan (HA) in hyperglycemia, are believed to be implicated in cancer development and progression (107, 108). Cancer is strongly linked not only to alterations in metabolism at the organismal level but also at the cellular level. Cancer cells undergo metabolic reprogramming, which facilitates their malignant properties, including rapid growth, migration, and invasion (109). Given the essential role of metabolic rewiring in tumor development, drugs interfering with glycolysis, glutaminolysis, or lipogenesis, may provide a clinical benefit in cancer patients (110). Furthermore, considering the strong links between the disruption of organismal metabolic homeostasis and malignancy, therapeutic interventions already in use for patients with metabolic disorders, including metformin and statins, may serve as promising strategies to inhibit cancer development and progression. It has also become evident that a healthy lifestyle can prevent not only the development of MetS but also the development of cancer (104, 105).
In conclusion, metabolic syndrome has been established as an important risk factor for cancer development; hence, cancer patients or individuals who are at high risk of cancer development are likely to benefit from the prevention and treatment of metabolic diseases. Further efforts are required for the development of therapeutic interventions that target both aberrant metabolism and cancer.
Author Contributions
YY and LG wrote the review, were responsible for figure and legend and final editing. JY contributes to defining the topic, analyzing article and preparing for submission. All the authors have approved for the final version of manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (No. 81773065); Natural Science Foundation of Zhejiang Province (No. LR19H160001); the Health and Family Planning Commission of Zhejiang Province (No. 2015RCB026).
Abbreviations
MetS, metabolic syndrome; ROS, reactive oxygen species; ATP, adenosine triphosphate; TRIM59, tripartite motif-containing59; PI3K, phosphatidylinositol 3-kinase; mtDNA, mitochondrial genome; TAM, tumor-associated macrophages; MG, methylglyoxal; GLUT1, glucose-transporter1; HK2, hexokinase 2; GLS1, glutaminase 1; MMPs, matrix-metalloproteinases; ECM, extracellular matrix; EMT, epithelial to mesenchymal transition; GDH1, glutamate dehydrogenase1; FASN, fattyacid synthase; ACLY, ATP-citrate lyase; TNF-α, tumor necrosis factor-alpha; IL, interleukin; NF-κB, nuclear factor kappa B; TME, tumor microenvironment; NADPH, nicotinamide adenine dinucleotide phosphate; CSC, cancer stem cell; MAPK, Mitogen-activated protein kinase; CRC, colorectal cancer; PFK-1, phosphofructokinase-1; HMG-CoA, 3-hydroxy-3methylglutary-coenzyme A; ICAM-1, intracellular adhesion molecule-1; HA, hyaluronan.
References
1. Alberti KG, Eckel RH, Grundy SM, Zimmet PZ, Cleeman JI, Donato KA, et al. Harmonizing the metabolic syndrome: a joint interim statement of the International diabetes federation task force on epidemiology and prevention; national heart, lung, and blood institute; American heart association; world heart federation; International atherosclerosis society; and International association for the study of obesity. Circulation. (2009) 120:1640–5. doi: 10.1161/CIRCULATIONAHA.109.192644
2. Eckel RH, Grundy SM, Zimmet PZ. The metabolic syndrome. Lancet. (2005) 365:1415–28. doi: 10.1016/S0140-6736(05)66378-7
3. Beltrán-Sánchez H, Harhay MO, Harhay MM, McElligott S. Prevalence and trends of metabolic syndrome in the adult U.S. population, 1999-2010. J Am Coll Cardiol. (2013) 62:697–703. doi: 10.1016/j.jacc.2013.05.064
4. Gathirua-Mwangi WG, Monahan PO, Murage MJ, Zhang J. Metabolic syndrome and total cancer mortality in the third National health and nutrition examination survey. Cancer Causes Control. (2017) 28:127–36. doi: 10.1007/s10552-016-0843-1
5. Harlid S, Myte R, Van Guelpen B. The metabolic syndrome, inflammation, and colorectal cancer risk: an evaluation of large panels of plasma protein markers using repeated, prediagnostic samples. Mediators Inflamm. (2017). 2017:4803156. doi: 10.1155/2017/4803156
6. Guo M, Liu T, Li P, Wang T, Zeng C, Yang M, et al. Association between metabolic syndrome and breast cancer risk: an updated meta-analysis of follow-up studies. Front Oncol. (2019) 9:1290. doi: 10.3389/fonc.2019.01290
7. Dickerman BA, Torfadottir JE, Valdimarsdottir UA, Wilson KM, Steingrimsdottir L, Aspelund T, et al. Midlife metabolic factors and prostate cancer risk in later life. Int J Cancer. (2018) 142:1166–73. doi: 10.1002/ijc.31142
8. Stocks T, Bjørge T, Ulmer H, Manjer J, Häggström C, Nagel G, et al. Metabolic risk score and cancer risk: pooled analysis of seven cohorts. Int J Epidemiol. (2015) 44:1353–63. doi: 10.1093/ije/dyv001
9. Pavlova NN, Thompson CB. The emerging hallmarks of cancer metabolism. Cell Metabol. (2016) 23:27–47. doi: 10.1016/j.cmet.2015.12.006
10. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the warburg effect: the metabolic requirements of cell proliferation. Science. (2009) 324:1029–33. doi: 10.1126/science.1160809
11. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
12. Hu Y, Lu W, Chen G, Wang P, Chen Z, Zhou Y, et al. K-ras(G12V) transformation leads to mitochondrial dysfunction and a metabolic switch from oxidative phosphorylation to glycolysis. Cell Res. (2012) 22:399–412. doi: 10.1038/cr.2011.145
13. Stine ZE, Walton ZE, Altman BJ, Hsieh AL, Dang CV. MYC, metabolism, and cancer. Cancer Discov. (2015) 5:1024–39. doi: 10.1158/2159-8290.CD-15-0507
14. Kobliakov VA. The mechanisms of regulation of aerobic glycolysis (warburg effect) by oncoproteins in carcinogenesis. Biochemistry. (2019) 84:1117–28. doi: 10.1134/S0006297919100018
15. Hernández-Reséndiz I, Gallardo-Pérez JC, López-Macay A, Robledo-Cadena DX, García-Villa E, Gariglio P, et al. Mutant p53 downregulates oxidative phosphorylation and upregulates glycolysis under normoxia and hypoxia in human cervix cancer cells. J Cell Physiol. (2019) 234:5524–36. doi: 10.1002/jcp.27354
16. Guerra F, Arbini AA, Moro L. Mitochondria and cancer chemoresistance. Biochim Biophys Acta Bioenerg. (2017) 1858:686–99. doi: 10.1016/j.bbabio.2017.01.012
17. Jeong H, Kim S, Hong BJ, Lee CJ, Kim YE, Bok S, et al. Tumor-associated macrophages enhance tumor hypoxia and aerobic glycolysis. Cancer Res. (2019) 79:795–806. doi: 10.1158/0008-5472.CAN-18-2545
18. Muir A, Danai LV, Vander Heiden MG. Microenvironmental regulation of cancer cell metabolism: implications for experimental design and translational studies. Dis Model Mech. (2018) 11:dmm035758. doi: 10.1242/dmm.035758
19. Matés JM, Di Paola FJ, Campos-Sandoval JA, Mazurek S, Márquez J. Therapeutic targeting of glutaminolysis as an essential strategy to combat cancer. Semin Cell Dev Biol. (2020). 98:34–43. doi: 10.1016/j.semcdb.2019.05.012
20. Currie E, Schulze A, Zechner R, Walther TC, Farese RV. Cellular fatty acid metabolism and cancer. Cell Metab. (2013) 18:153–61. doi: 10.1016/j.cmet.2013.05.017
21. Kuhajda FP. Fatty acid synthase and cancer: new application of an old pathway. Cancer Res. (2006) 66:5977–80. doi: 10.1158/0008-5472.CAN-05-4673
22. Hirschey MD, DeBerardinis RJ, Diehl AME, Drew JE, Frezza C, Green MF, et al. Dysregulated metabolism contributes to oncogenesis. Semin Cancer Biol. (2015) 35:S129–50. doi: 10.1016/j.semcancer.2015.10.002
23. Boroughs LK, DeBerardinis RJ. Metabolic pathways promoting cancer cell survival and growth. Nat Cell Biol. (2015) 17:351–9. doi: 10.1038/ncb3124
24. Kang H, Kim H, Lee S, Youn H, Youn B. Role of metabolic reprogramming in epithelial−mesenchymal transition (EMT). Int J Mol Sci. (2019) 20:2042. doi: 10.3390/ijms20082042
25. Riemann A, Rauschner M, Gießelmann M, Reime S, Haupt V, Thews O. Extracellular acidosis modulates the expression of Epithelial-Mesenchymal Transition (EMT) markers and adhesion of epithelial and tumor cells. Neoplasia. (2019) 21:450–8. doi: 10.1016/j.neo.2019.03.004
26. Thews O, Riemann A. Tumor pH and metastasis: a malignant process beyond hypoxia. Cancer Metastasis Rev. (2019) 38:113–29. doi: 10.1007/s10555-018-09777-y
27. Botzer LE, Maman S, Sagi-Assif O, Meshel T, Nevo I, Yron I, et al. Hexokinase 2 is a determinant of neuroblastoma metastasis. Br J Cancer. (2016) 114:759–66. doi: 10.1038/bjc.2016.26
28. Nokin MJ, Bellier J, Durieux F, Peulen O, Rademaker G, Gabriel M, et al. Methylglyoxal, a glycolysis metabolite, triggers metastasis through MEK/ERK/SMAD1 pathway activation in breast cancer. Breast Cancer Res. (2019) 21:11. doi: 10.1186/s13058-018-1095-7
29. Xi J, Wang Y, Liu H. GLUT-1 participates in the promotion of LncRNA CASC9 in proliferation and metastasis of laryngeal carcinoma cells. Gene. (2020) 726:144194. doi: 10.1016/j.gene.2019.144194
30. Lu J, Tan M, Cai Q. The warburg effect in tumor progression: mitochondrial oxidative metabolism as an anti-metastasis mechanism. Cancer Lett. (2015) 356:156–64. doi: 10.1016/j.canlet.2014.04.001
31. Jin L, Chun J, Pan C, Kumar A, Zhang G, Ha Y, et al. The PLAG1-GDH1 axis promotes anoikis resistance and tumor metastasis through CamKK2-AMPK signaling in LKB1-deficient lung cancer. Mol Cell. (2018) 69:87–99.e7. doi: 10.1016/j.molcel.2017.11.025
32. Koochekpour S, Majumdar S, Azabdaftari G, Attwood K, Scioneaux R, Subramani D, et al. Serum glutamate levels correlate with gleason score and glutamate blockade decreases proliferation, migration, and invasion and induces apoptosis in prostate cancer cells. Clin Cancer Res. (2012) 18:5888–901. doi: 10.1158/1078-0432.CCR-12-1308
33. Dornier E, Rabas N, Mitchell L, Novo D, Dhayade S, Marco S, et al. Glutaminolysis drives membrane trafficking to promote invasiveness of breast cancer cells. Nat Commun. (2017) 8:2255. doi: 10.1038/s41467-017-02101-2
34. Xiang L, Mou J, Shao B, Wei Y, Liang H, Takano N, et al. Glutaminase 1 expression in colorectal cancer cells is induced by hypoxia and required for tumor growth, invasion, and metastatic colonization. Cell Death Dis. (2019) 10:40. doi: 10.1038/s41419-018-1291-5
35. Lu T, Sun L, Wang Z, Zhang Y, He Z, Xu C. Fatty acid synthase enhances colorectal cancer cell proliferation and metastasis via regulating AMPK/mTOR pathway. OncoTargets Ther. (2019) 12:3339–47. doi: 10.2147/OTT.S199369
36. Wen J, Min X, Shen M, Hua Q, Han Y, Zhao L, et al. ACLY facilitates colon cancer cell metastasis by CTNNB1. J Exp Clin Cancer Res. (2019) 38:401. doi: 10.1186/s13046-019-1391-9
37. Greten FR, Grivennikov SI. Inflammation and cancer: triggers, mechanisms, and consequences. Immunity. (2019) 51:27–41. doi: 10.1016/j.immuni.2019.06.025
38. Magnuson AM, Regan DP, Booth AD, Fouts JK, Solt CM, Hill JL, et al. High-fat diet induced central adiposity (visceral fat) is associated with increased fibrosis and decreased immune cellularity of the mesenteric lymph node in mice. Eur J Nutr. (2019) 59:1641–54. doi: 10.1007/s00394-019-02019-z
39. Kumar P, Raman T, Swain MM, Mishra R, Pal A. Hyperglycemia-induced oxidative-nitrosative stress induces inflammation and neurodegeneration via augmented tuberous sclerosis complex-2 (TSC-2) activation in neuronal cells. Mol Neurobiol. (2017) 54:238–54. doi: 10.1007/s12035-015-9667-3
40. Ebtehaj S, Gruppen EG, Parvizi M, Tietge UJF, Dullaart RPF. The anti-inflammatory function of HDL is impaired in type 2 diabetes: role of hyperglycemia, paraoxonase-1 and low grade inflammation. Cardiovasc Diabetol. (2017) 16:132. doi: 10.1186/s12933-017-0613-8
41. Ikwegbue PC, Masamba P, Mbatha LS, Oyinloye BE, Kappo AP. Interplay between heat shock proteins, inflammation and cancer: a potential cancer therapeutic target. Am J Cancer Res. (2019) 9:242–9.
42. Canli Ö, Nicolas AM, Gupta J, Finkelmeier F, Goncharova O, Pesic M, et al. Myeloid cell-derived reactive oxygen species induce epithelial mutagenesis. Cancer Cell. (2017) 32:869–83.e5. doi: 10.1016/j.ccell.2017.11.004
43. Che D, Zhang S, Jing Z, Shang L, Jin S, Liu F, et al. Macrophages induce EMT to promote invasion of lung cancer cells through the IL-6-mediated COX-2/PGE/β-catenin signalling pathway. Mol Immunol. (2017) 90:197–210. doi: 10.1016/j.molimm.2017.06.018
44. Suarez-Carmona M, Lesage J, Cataldo D, Gilles C. EMT and inflammation: inseparable actors of cancer progression. Mol Oncol. (2017) 11:805–23. doi: 10.1002/1878-0261.12095
45. Wang T, Song P, Zhong T, Wang X, Xiang X, Liu Q, et al. The inflammatory cytokine IL-6 induces FRA1 deacetylation promoting colorectal cancer stem-like properties. Oncogene. (2019) 38:4932–47. doi: 10.1038/s41388-019-0763-0
46. Qu D, Shen L, Liu S, Li H, Ma Y, Zhang R, et al. Chronic inflammation confers to the metabolic reprogramming associated with tumorigenesis of colorectal cancer. Cancer Biol Ther. (2017) 18:237–44. doi: 10.1080/15384047.2017.1294292
47. Taniguchi K, Karin M. NF-κB, inflammation, immunity and cancer: coming of age. Nat Rev Immunol. (2018) 18:309–24. doi: 10.1038/nri.2017.142
48. Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. (2010) 140:883–99. doi: 10.1016/j.cell.2010.01.025
49. Sallmyr A, Fan J, Rassool FV. Genomic instability in myeloid malignancies: increased reactive oxygen species (ROS), DNA double strand breaks (DSBs) and error-prone repair. Cancer Lett. (2008) 270:1–9. doi: 10.1016/j.canlet.2008.03.036
50. Koshikawa N, Akimoto M, Hayashi JI, Nagase H, Takenaga K. Association of predicted pathogenic mutations in mitochondrial ND genes with distant metastasis in NSCLC and colon cancer. Sci Rep. (2017) 7:15535. doi: 10.1038/s41598-017-15592-2
51. Ray AM, Zuhlke KA, Levin AM, Douglas JA, Cooney KA, Petros JA. Sequence variation in the mitochondrial gene cytochrome c oxidase subunit I and prostate cancer in African American men. Prostate. (2009) 69:956–60. doi: 10.1002/pros.20943
52. Hart PC, Mao M, de Abreu AL, Ansenberger-Fricano K, Ekoue DN, Ganini D, et al. MnSOD upregulation sustains the warburg effect via mitochondrial ROS and AMPK-dependent signalling in cancer. Nat Commun. (2015) 6:6053. doi: 10.1038/ncomms7053
53. Cai H. Hydrogen peroxide regulation of endothelial function: origins, mechanisms, and consequences. Cardiovasc Res. (2005) 68:26–36. doi: 10.1016/j.cardiores.2005.06.021
54. Shenoy AK, Fisher RC, Butterworth EA, Pi L, Chang LJ, Appelman HD, et al. Transition from colitis to cancer: high Wnt activity sustains the tumor-initiating potential of colon cancer stem cell precursors. Cancer Res. (2012) 72:5091–100. doi: 10.1158/0008-5472.CAN-12-1806
55. El Ayachi I, Fatima I, Wend P, Alva-Ornelas JA, Runke S, Kuenzinger WL, et al. The WNT10B network is associated with survival and metastases in chemoresistant triple-negative breast cancer. Cancer Res. (2019) 79:982–93. doi: 10.1158/0008-5472.CAN-18-1069
56. Peluffo G, Subedee A, Harper NW, Kingston N, Jovanović B, Flores F, et al. EN1 is a transcriptional dependency in triple-negative breast cancer associated with brain metastasis. Cancer Res. (2019) 79:4173–83. doi: 10.1158/0008-5472.CAN-18-3264
57. Chouhan S, Singh S, Athavale D, Ramteke P, Pandey V, Joseph J, et al. Glucose induced activation of canonical Wnt signaling pathway in hepatocellular carcinoma is regulated by DKK4. Sci Rep. (2016) 6:27558. doi: 10.1038/srep27558
58. Guo C, Kim SJ, Frederick AM, Li J, Jin Y, Zeng H, et al. Genetic ablation of tumor necrosis factor-alpha attenuates the promoted colonic Wnt signaling in high fat diet-induced obese mice. J Nutr Biochem. (2019) 77:108302. doi: 10.1016/j.jnutbio.2019.108302
59. Arcidiacono D, Dedja A, Giacometti C, Fassan M, Nucci D, Francia S, et al. Hyperinsulinemia promotes esophageal cancer development in a surgically-induced duodeno-esophageal reflux murine model. Int J Mol Sci. (2018) 19:1198. doi: 10.3390/ijms19041198
60. Budi EH, Muthusamy BP, Derynck R. The insulin response integrates increased TGF-β signaling through Akt-induced enhancement of cell surface delivery of TGF-β receptors. Sci Signal. (2015) 8:ra96. doi: 10.1126/scisignal.aaa9432
61. Ghasemi A, Saeidi J, Azimi-Nejad M, Hashemy SI. Leptin-induced signaling pathways in cancer cell migration and invasion. Cell Oncol. (2019) 42:243–60. doi: 10.1007/s13402-019-00428-0
62. Takahashi K, Nagai N, Ogura K, Tsuneyama K, Saiki I, Irimura T, et al. Mammary tissue microenvironment determines T cell-dependent breast cancer-associated inflammation. Cancer Sci. (2015) 106:867–74. doi: 10.1111/cas.12685
63. Jiang H, Zhang XW, Liao QL, Wu WT, Liu YL, Huang WH. Electrochemical monitoring of paclitaxel-induced ROS release from mitochondria inside single cells. Small. (2019) 15:e1901787. doi: 10.1002/smll.201901787
64. Sedlic F, Muravyeva MY, Sepac A, Sedlic M, Williams AM, Yang M, et al. Targeted modification of mitochondrial ROS production converts high glucose-induced cytotoxicity to cytoprotection: effects on anesthetic preconditioning. J Cell Physiol. (2017) 232:216–24. doi: 10.1002/jcp.25413
65. Mahbouli S, Der Vartanian A, Ortega S, Rougé S, Vasson MP, Rossary A. Leptin induces ROS via NOX5 in healthy and neoplastic mammary epithelial cells. Oncol Rep. (2017) 38:3254–64. doi: 10.3892/or.2017.6009
66. Sabharwal SS, Schumacker PT. Mitochondrial ROS in cancer: initiators, amplifiers or an achilles' heel? Nat Rev Cancer. (2014) 14:709–21. doi: 10.1038/nrc3803
67. Trachootham D, Alexandre J, Huang P. Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach? Nat RevDrug Discov. (2009) 8:579–91. doi: 10.1038/nrd2803
68. Ogrunc M, Di Micco R, Liontos M, Bombardelli L, Mione M, Fumagalli M, et al. Oncogene-induced reactive oxygen species fuel hyperproliferation and DNA damage response activation. Cell Death Differ. (2014) 21:998–1012. doi: 10.1038/cdd.2014.16
69. Muntoni S, Muntoni S. Insulin resistance: pathophysiology and rationale for treatment. Ann Nutr Metab. (2011) 58:25–36. doi: 10.1159/000323395
70. Mroueh FM, Noureldein M, Zeidan YH, Boutary S, Irani SAM, Eid S, et al. Unmasking the interplay between mTOR and Nox4: novel insights into the mechanism connecting diabetes and cancer. FASEB J. (2019) 33:14051–66. doi: 10.1096/fj.201900396RR
71. Parida S, Siddharth S, Sharma D. Adiponectin, obesity, and cancer: clash of the bigwigs in health and disease. Int J Mol Sci. (2019) 20:2519. doi: 10.3390/ijms20102519
72. Amable G, Martínez-León E, Picco ME, Di Siervi N, Davio C, Rozengurt E, et al. Metformin inhibits β-catenin phosphorylation on Ser-552 through an AMPK/PI3K/Akt pathway in colorectal cancer cells. Int J Biochem Cell Biol. (2019) 112:88–94. doi: 10.1016/j.biocel.2019.05.004
73. Bai M, Yang L, Liao H, Liang X, Xie B, Xiong J, et al. Metformin sensitizes endometrial cancer cells to chemotherapy through IDH1-induced Nrf2 expression via an epigenetic mechanism. Oncogene. (2018) 37:5666–81. doi: 10.1038/s41388-018-0360-7
74. Pernicova I, Korbonits M. Metformin–mode of action and clinical implications for diabetes and cancer. Nat Rev Endocrinol. (2014) 10:143–56. doi: 10.1038/nrendo.2013.256
75. Hu L, Zeng Z, Xia Q, Liu Z, Feng X, Chen J, et al. Metformin attenuates hepatoma cell proliferation by decreasing glycolytic flux through the HIF-1α/PFKFB3/PFK1 pathway. Life Sci. (2019) 239:116966. doi: 10.1016/j.lfs.2019.116966
76. Guimarães TA, Farias LC, Santos ES, de Carvalho Fraga CA, Orsini LA, de Freitas Teles L, et al. Metformin increases PDH and suppresses HIF-1α under hypoxic conditions and induces cell death in oral squamous cell carcinoma. Oncotarget. (2016) 7:55057–68. doi: 10.18632/oncotarget.10842
77. Kitson SJ, Rosser M, Fischer DP, Marshall KM, Clarke RB, Crosbie EJ. Targeting endometrial cancer stem cell activity with metformin is inhibited by patient-derived adipocyte-secreted factors. Cancers. (2019) 11:653. doi: 10.3390/cancers11050653
78. Kim JH, Lee KJ, Seo Y, Kwon JH, Yoon JP, Kang JY, et al. Effects of metformin on colorectal cancer stem cells depend on alterations in glutamine metabolism. Sci Rep. (2018) 8:409. doi: 10.1038/s41598-018-29895-5
79. Wang JC, Li GY, Wang B, Han SX, Sun X, Jiang YN, et al. Metformin inhibits metastatic breast cancer progression and improves chemosensitivity by inducing vessel normalization via PDGF-downregulation B. J Exp Clin Cancer Res. (2019) 38:235. doi: 10.1186/s13046-019-1211-2
80. Sharma P, Kumar S. Metformin inhibits human breast cancer cell growth by promoting apoptosis via a ROS-independent pathway involving mitochondrial dysfunction: pivotal role of superoxide dismutase (SOD). Cell Oncology. (2018) 41:637–50. doi: 10.1007/s13402-018-0398-0
81. Farmer RE, Ford D, Forbes HJ, Chaturvedi N, Kaplan R, Smeeth L, et al. Metformin and cancer in type 2 diabetes: a systematic review and comprehensive bias evaluation. Int J Epidemiol. (2017) 46:728–44. doi: 10.1093/ije/dyx046
82. Dankner R, Roth J. More recent, better designed studies have weakened links between antidiabetes medications and cancer risk. Diabetic Med. (2020) 37:194–202. doi: 10.1111/dme.14179
83. Wang A, Aragaki AK, Tang JY, Kurian AW, Manson JE, Chlebowski RT, et al. Statin use and all-cancer survival: prospective results from the Women's health initiative. Br J Cancer. (2016) 115:129–35. doi: 10.1038/bjc.2016.149
84. Li Y, He X, Ding Y, Chen H, Sun L. Statin uses and mortality in colorectal cancer patients: an updated systematic review and meta-analysis. Cancer Med. (2019) 8:3305–13. doi: 10.1002/cam4.2151
85. Tran KT, McMenamin ÚC, Coleman HG, Cardwell CR, Murchie P, Iversen L, et al. Statin use and risk of liver cancer: evidence from two population-based studies. Int J Cancer. (2020) 146:1250–60. doi: 10.1002/ijc.32426
86. Christie CF, Fang D, Hunt EG, Morris ME, Rovini A, Heslop KA, et al. Statin-dependent modulation of mitochondrial metabolism in cancer cells is independent of cholesterol content. FASEB J. (2019) 33:8186–201. doi: 10.1096/fj.201802723R
87. Chen J, Liu B, Yuan J, Yang J, Zhang J, An Y, et al. Atorvastatin reduces vascular endothelial growth factor (VEGF) expression in human non-small cell lung carcinomas (NSCLCs) via inhibition of reactive oxygen species (ROS) production. Mol Oncol. (2012) 6:62–72. doi: 10.1016/j.molonc.2011.11.003
88. Sadaria MR, Reppert AE, Yu JA, Meng X, Fullerton DA, Reece TB, et al. Statin therapy attenuates growth and malignant potential of human esophageal adenocarcinoma cells. J Thorac Cardiovasc Surg. (2011) 142:1152–60. doi: 10.1016/j.jtcvs.2011.08.004
89. Juneja M, Kobelt D, Walther W, Voss C, Smith J, Specker E, et al. Statin and rottlerin small-molecule inhibitors restrict colon cancer progression and metastasis via MACC1. PLoS Biol. (2017) 15:e2000784. doi: 10.1371/journal.pbio.2000784
90. Chen YA, Shih HW, Lin YC, Hsu HY, Wu TF, Tsai CH, et al. Simvastatin sensitizes radioresistant prostate cancer cells by compromising DNA double-strand break repair. Front Pharmacol. (2018) 9:600. doi: 10.3389/fphar.2018.00600
91. Yang Z, Su Z, DeWitt JP, Xie L, Chen Y, Li X, et al. Fluvastatin prevents lung adenocarcinoma bone metastasis by triggering autophagy. EBioMedicine. (2017) 19:49–59. doi: 10.1016/j.ebiom.2017.04.017
92. Babcook MA, Sramkoski RM, Fujioka H, Daneshgari F, Almasan A, Shukla S, et al. Combination simvastatin and metformin induces G1-phase cell cycle arrest and Ripk1- and Ripk3-dependent necrosis in C4-2B osseous metastatic castration-resistant prostate cancer cells. Cell Death Dis. (2014) 5:e1536. doi: 10.1038/cddis.2014.500
93. Jeong GH, Lee KH, Kim JY, Eisenhut M, Kronbichler A, van der Vliet HJ, et al. Statin and cancer mortality and survival: an umbrella systematic review and meta-analysis. J Clin Med. (2020) 9:326. doi: 10.3390/jcm9020326
94. Dale KM, Coleman CI, Henyan NN, Kluger J, White CM. Statins and cancer risk: a meta-analysis. JAMA. (2006) 295:74–80. doi: 10.1001/jama.295.1.74
95. Dickerman BA, García-Albéniz X, Logan RW, Denaxas S, Hernán MA. Avoidable flaws in observational analyses: an application to statins and cancer. Nat Med. (2019). 25:1601–6. doi: 10.1038/s41591-019-0597-x
96. Ding D, Rogers K, van der Ploeg H, Stamatakis E, Bauman AE. Traditional and emerging lifestyle risk behaviors and all-cause mortality in middle-aged and older adults: evidence from a large population-based Australian cohort. PLoS Med. (2015) 12:e1001917. doi: 10.1371/journal.pmed.1001917
97. The Oncology Lancet. Exercise and cancer treatment: balancing patient needs. Lancet Oncol. (2018). 19:715. doi: 10.1016/S1470-2045(18)30376-0
98. Mendelson M, Michallet AS, Monneret D, Perrin C, Estève F, Lombard PR, et al. Impact of exercise training without caloric restriction on inflammation, insulin resistance and visceral fat mass in obese adolescents. Pediatric Obesity. (2015) 10:311–9. doi: 10.1111/ijpo.255
99. Fuhr L, El-Athman R, Scrima R, Cela O, Carbone A, Knoop H, et al. The circadian clock regulates metabolic phenotype rewiring Via HKDC1 and modulates tumor progression and drug response in colorectal cancer. EBioMedicine. (2018) 33:105–21. doi: 10.1016/j.ebiom.2018.07.002
100. Sahar S, Sassone-Corsi P. Metabolism and cancer: the circadian clock connection. Nat Rev Cancer. (2009) 9:886–96. doi: 10.1038/nrc2747
101. Paz-Graniel I, Babio N, Mendez I, Salas-Salvadó J. Association between eating speed and classical cardiovascular risk factors: a cross-sectional study. Nutrients. (2019) 11:83. doi: 10.3390/nu11010083
102. McGuire S. Scientific Report of the 2015 Dietary guidelines advisory committee. Washington, DC: US departments of agriculture and health and human services 2015. Adv Nutr. (2016) 7:202–4. doi: 10.3945/an.115.011684
103. Laitinen K, Mokkala K. Overall dietary quality relates to gut microbiota diversity and abundance. Int J Mol Sci. (2019) 20:1835. doi: 10.3390/ijms20081835
104. Wang Q, Chair SY, Wong EM. The effects of a lifestyle intervention program on physical outcomes, depression, and quality of life in adults with metabolic syndrome: a randomized clinical trial. Int J Cardiol. (2017) 230:461–7. doi: 10.1016/j.ijcard.2016.12.084
105. Kushi LH, Doyle C, McCullough M, Rock CL, Demark-Wahnefried W, Bandera EV, et al. American cancer society guidelines on nutrition and physical activity for cancer prevention: reducing the risk of cancer with healthy food choices and physical activity. CA Cancer J Clin. (2012) 62:30–67. doi: 10.3322/caac.20140
106. Saikolappan S, Kumar B, Shishodia G, Koul S, Koul HK. Reactive oxygen species and cancer: a complex interaction. Cancer Lett. (2019) 452:132–43. doi: 10.1016/j.canlet.2019.03.020
107. Moore SC, Matthews CE, Shu OX, Yu K, Gail MH, Xu X, et al. Endogenous estrogens, estrogen metabolites, and breast cancer risk in postmenopausal Chinese women. J Natl Cancer Inst. (2016) 108:djw103. doi: 10.1093/jnci/djw103
108. Twarock S, Reichert C, Peters U, Gorski DJ, Röck K, Fischer JW. Hyperglycaemia and aberrated insulin signalling stimulate tumour progression via induction of the extracellular matrix component hyaluronan. Int J Cancer. (2017) 141:791–804. doi: 10.1002/ijc.30776
109. DeBerardinis RJ, Chandel NS. Fundamentals of cancer metabolism. Sci Adv. (2016) 2:e1600200. doi: 10.1126/sciadv.1600200
Keywords: metabolic syndrome, metabolic reprogramming, cancer, metastasis, metabolism-targeting therapy
Citation: Yu Y, Gong L and Ye J (2020) The Role of Aberrant Metabolism in Cancer: Insights Into the Interplay Between Cell Metabolic Reprogramming, Metabolic Syndrome, and Cancer. Front. Oncol. 10:942. doi: 10.3389/fonc.2020.00942
Received: 04 March 2020; Accepted: 13 May 2020;
Published: 11 June 2020.
Edited by:
Federica Sotgia, University of Salford, United KingdomReviewed by:
Elisabet Cuyàs, Institute of Biomedical Research of Girona, SpainPatrizia Pasanisi, Istituto Nazionale dei Tumori (IRCCS), Italy
Copyright © 2020 Yu, Gong and Ye. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jun Ye, d3ptY3llanVuQHpqdS5lZHUuY24=
†These authors have contributed equally to this work