 Hirohito Yamaguchi
Hirohito Yamaguchi Ghina M. Taouk
Ghina M. Taouk
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 11 June 2020
Sec. Cancer Metabolism
Volume 10 - 2020 | https://doi.org/10.3389/fonc.2020.00928
This article is part of the Research Topic Cancer Cell Metabolism and Immunomodulation in the Context of Tumor Metastasis View all 30 articles
Yes-Associated Protein (YAP) and Transcriptional Co-activator with PDZ-binding Motif (TAZ) are the downstream effectors of the Hippo signaling pathway that play a crucial role in various aspects of cancer progression including metastasis. Metastasis is the multistep process of disseminating cancer cells in a body and responsible for the majority of cancer-related death. Emerging evidence has shown that cancer cells reprogram their metabolism to gain proliferation, invasion, migration, and anti-apoptotic abilities and adapt to various environment during metastasis. Moreover, it has increasingly been recognized that YAP/TAZ regulates cellular metabolism that is associated with the phenotypic changes, and recent studies suggest that the YAP/TAZ-mediated metabolic alterations contribute to metastasis. In this review, we will introduce the latest knowledge of YAP/TAZ regulation and function in cancer metastasis and metabolism, and discuss possible links between the YAP/TAZ-mediated metabolic reprogramming and metastasis.
Metastasis is the spread of cancer cells from primary tumors to distinct organs and a major cause of cancer-related death. Therefore, targeting metastasis is expected to be an effective therapeutic strategy to improve the survival of cancer patients. Metastasis is a complex process including various distinct steps, and cancer cells need to gain diverse abilities in each step that are regulated by a variety of cellular signaling pathways (1). Thus, the understanding of regulatory mechanisms of metastasis is particularly crucial for targeting it.
During tumor development, cancer cells are exposed to dynamic changes of tumor microenvironment such as availability of nutrient and oxygen. Hence, cancer cells need to adapt their metabolism to these changes to develop tumors (2). Emerging evidence has suggested that metabolic reprogramming also plays a significant role in metastasis, during which cancer cells are exposed to different environment from primary tumors to distant metastatic sites (3).
Yes-Associated Protein (YAP) and Transcriptional Co-activator with PDZ-binding Motif (TAZ) are the transcriptional co-regulators and downstream effectors of the Hippo signaling pathway (4). Numerous studies using mouse models have demonstrated that YAP/TAZ promotes tumor development, progression, and metastasis (4). At cellular level, YAP/TAZ regulates cell cycles, migration, invasion, anchorage-independent growth, epithelial-mesenchymal transition (EMT), and stemness. Because all these functions of YAP/TAZ are crucial for various steps of metastasis, YAP/TAZ serves as a central regulator of metastasis. Besides, it has gradually been recognized that YAP/TAZ regulates cellular metabolic reprograming to adapt the various tumor microenvironment (5).
Based on the fact that YAP/TAZ has the ability to regulate both metastasis and metabolism, it is expected that YAP/TAZ functions as a central hub for metastasis by regulating both metabolic adaptation and phenotypic changes required for metastasis. Thus, in this review, we will introduce the latest knowledge of YAP/TAZ regulation and function in cancer metastasis and metabolism, and discuss possible links between the YAP/TAZ-mediated metabolic reprogramming and metastasis.
YAP/TAZ is the downstream effector of the Hippo signaling pathway, which is an evolutionally conserved kinase cascade (Figure 1) (6, 7). The key regulators of the Hippo pathway include two kinases mammalian sterile 20-like kinase 1 and 2 (MST1/2) and large tumor suppressor kinase 1 and 2 (LATS1/2) and their regulatory subunits SAV1 and MOB1, respectively (Figure 1) (6). MST1/2 forms a complex with SAV1, which phosphorylates and activates the downstream LATS1/2-MOB1 complex (6). Activated LATS1/2 phosphorylates YAP/TAZ at multiple serine/threonine residues. Upon phosphorylation, YAP/TAZ interacts with 14-3-3, resulting in its cytoplasmic sequestration (8). Alternatively, phosphorylated YAP/TAZ is ubiquitinated by E3 ligase β-TrCP and undergoes proteasome-dependent degradation (9, 10). Thus, MST1/2 and LATS1/2 are the negative regulators of YAP/TAZ. Unphosphorylated YAP/TAZ moves to the nucleus, where YAP/TAZ interacts with (TEA domain) TEAD family transcription factors and functions as a transcription co-activator (11, 12). Major YAP/TAZ target genes include molecules that are related to cell growth, proliferation, and migration such as CTGF, CYR61, MYC, AXL, BIRC5, and CCND1 (13, 14).
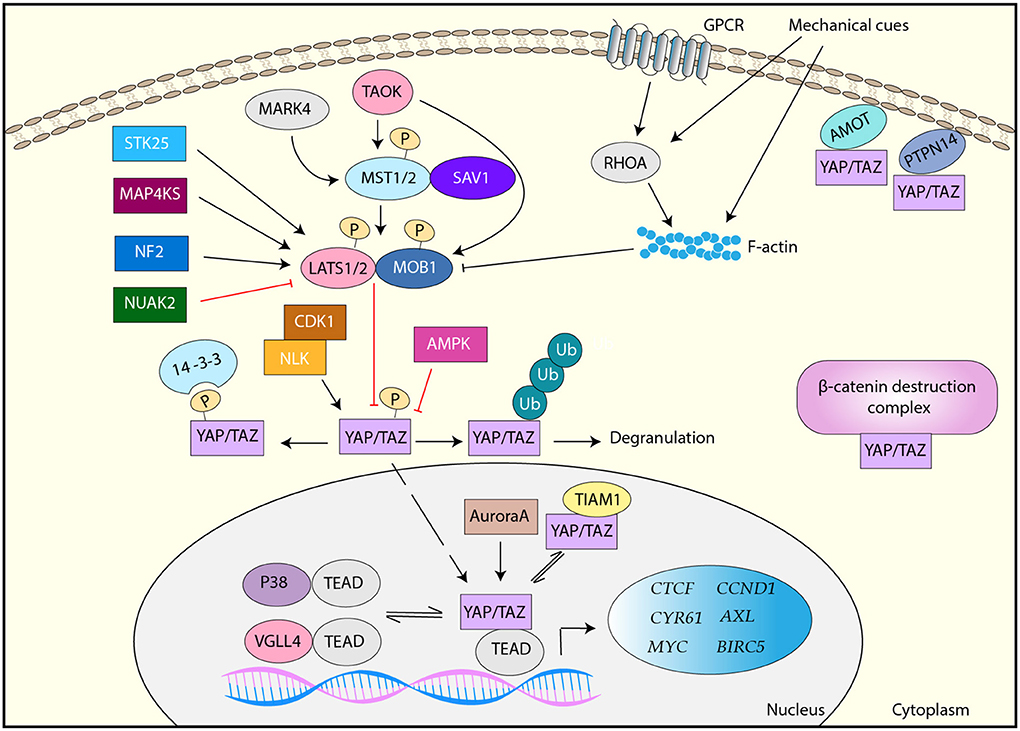
Figure 1. Regulation of YAP/TAZ. YAP/TAZ is primarily regulated by the canonical Hippo pathway, MST1/2-SAV1 > LATS1/2-MOB1. LATS1/2 phosphorylates YAP/TAZ and inactivate it via either 14-3-3-mediated cytoplasmic sequestration or ubiquitination and proteasome-mediated degradation. Unphosphorylated YAP/TAZ translocates to the nucleus, where it interacts with TEAD transcription factors and induces target genes. LATS1/2 is activated by TAOK, STK25, MAP4KS, and NF2, but inactivated by mechanical cues- and GPCR-RHOA-mediated F-actin and NUAK2. MST1/2 is activated by MARK4 and TAOK. YAP/TAZ is also regulated by LATS-independently. AMOT and PTPN14 interact with YAP/TAZ and sequester it in the plasma membrane. YAP/TAZ is inhibited by the β-catenin destruction complex or TIAM1 via a direct interaction. YAP/TAZ is also directly phosphorylated and regulated by AMPK, CDK1, NLK, Aurora A, and several RTKs. In addition, VGLL4 and p38 interact TEAD and inhibit YAP/TAZ activity.
Various extracellular stimuli regulate YAP/TAZ through the Hippo signaling pathway-dependent and independent mechanisms. In particular, YAP/TAZ plays a central role in cellular mechanotransduction, which is a molecular process to convert various extracellular mechanical forces to cellular responses (15). Namely, mechanical cues from surrounding extracellular matrix (ECM) and cells are converted to biochemical signals to control gene expression and protein functions, resulting in cell proliferation, survival, differentiation, and migration (16). YAP/TAZ nuclear localization and activity are regulated by various mechanical cues, and activated YAP/TAZ is required for biological function induced by mechanical cues (17–20). Increased polymerized actin (F-actin) induced by mechanical cues contributes to YAP/TAZ activation through Hippo-dependent and independent mechanism, although the detail mechanism is still uncertain (21). Moreover, various G-protein-coupled receptors (GPCR) induces F-actin through RHOA activation, resulting in LATS1/2 inactivation and subsequent YAP/TAZ activation (22).
Besides, several upstream regulators of MST/LATS have been identified. Tao kinase (TAOK) phosphorylates and activates MST1 and LATS1/2 (23), and several mitogen-activated protein kinase kinase kinase kinase (MAP4K) family members directly phosphorylate and activate LATS1/2 (24, 25). A recent study has also shown that STK25 phosphorylates LATS1/2 and contribute to the activation of LATS1/2 in response to contact inhibition and cell detachment (26). In addition, tumor suppressor Neurofibromin 2 (NF2) is a critical activator of LATS1/2 (27). Therefore, these molecules ultimately inactivate YAP/TAZ. In contrast, NUAK2 phosphorylates LATS1 and inhibits it, thereby activating YAP/TAZ (28), while MAP/microtubule affinity-regulating kinase 4 (MARK4) can phosphorylate MST2 and SAV1, resulting in the disruption of MST2/SAV1/LATS1 complex and subsequent YAP/TAZ activation (29).
In addition to LATS-mediated phosphorylation, YAP/TAZ is regulated by a variety of molecules. Angiomotin (AMOT) and protein tyrosine phosphatase non-receptor type 14 (PTPN14) directly bind to YAP to sequester YAP in the plasma membrane (30, 31). Besides, it has been shown that YAP/TAZ is found in the β-catenin destruction complex and it is released from the complex upon Wnt ligand and translocates to the nucleus (32). Moreover, multiple kinases directly phosphorylate YAP/TAZ at the various sites that are different from LATS-mediated phosphorylation sites, and activate or inactivate it. For example, in response to cellular energy stress, AMP-activated protein kinase (AMPK) phosphorylates YAP and inactivates it (33, 34). CDK1 phosphorylates YAP during the G2–M phase of the cell cycle, and the CDK1-mediated YAP phosphorylation has been implicated to be linked to mitotic defects (35). In response to osmotic stress, NLK phosphorylates YAP and inhibits the interaction between YAP and 14-3-3, thereby enhancing its nuclear localization (36). Aurora A also interacts with and phosphorylates YAP in the nucleus, and contributes to YAP transcriptional activity (37). Furthermore, PYK2 is implicated to phosphorylate TAZ at its tyrosine residues and regulate its protein stability (38). Several receptor tyrosine kinases (RTKs) including FGFR, RET, and MERTK are also shown to phosphorylate YAP/TAZ and activate it (39).
A major function of YAP/TAZ is transcriptional regulation through DNA binding protein TEAD family proteins (40, 41). Several regulatory mechanisms for the YAP/TAZ-TEAD complex have been identified. VGLL4 inhibits YAP by competing with YAP for its TEAD-binding (42). TIAM1 interacts with YAP/TAZ and impairs the YAP/TAZ-TEAD interaction (43). Moreover, osmotic stress induces the interaction between TEAD and p38 MAP kinase, resulting in TEAD translocation to the cytoplasm and YAP/TAZ inactivation (44). In addition to these factors, YAP/TAZ has been reported to interact with other transcription regulators such as RUNX2 and AP-1, cooperate to regulate the expression of target genes (45). Therefore, YAP/TAZ is regulated by a variety of stress and stimuli through multiple mechanisms (Figure 1).
YAP/TAZ functions as an oncogenic transcription factor in majority of solid tumors. Numerous studies have demonstrated that high expression and/or nuclear localization of YAP/TAZ in cancer cells are correlated with poor clinical outcomes in many cancer types such as lung, colorectal, breast, liver, gastric, pancreatic, prostate, endometrial, esophageal, bladder, and ovarian cancer (4, 46, 47). Moreover, it has been shown that the signatures of YAP/TAZ regulated gene expression are correlated with poor prognosis of lung and breast cancers (48, 49). Moreover, gene inactivation by mutations or deletions in multiple upstream regulators of the Hippo pathway, such as LATS1/2, PTPN14, and NF2 has been identified in several cancers including mesothelioma, skin basal cell carcinoma, and bladder and colon cancer (50–52).
In addition to retrospective analysis of clinical samples, numerous animal studies have also verified the function of YAP/TAZ as a bona-fide oncogenic factor. For example, conditional knockout of MST1/2 in liver or liver-specific YAP overexpression induces spontaneous liver cancer (53–55). Ectopic expression of YAP promotes tumor formation and progression in the genetically engineered mouse (GEM) model of lung cancer (56). Moreover, genetic inactivation of YAP reduces or suppresses tumor formation in various GEM models of lung, breast, colon, and pancreatic cancer (56–59).
At the cellular level, YAP/TAZ is involved in diverse functions required for tumor progression. For example, YAP/TAZ promotes cell proliferation by upregulating many genes involved in cell cycle control such as CCND1, CDK1, CDC25, and MCMs (40, 60, 61). YAP/TAZ also promotes anchorage-independent growth in various cancer cells (37, 62, 63).
To establish tumors, cancer cells need to escape from immune surveillance. Recently, it has been demonstrated that YAP/TAZ is involved in immune evasion as well as the resistance to immune checkpoint inhibitors (64). For instance, YAP/TAZ has been shown to upregulate chemokine CXCL5 and CCL2, resulting in the recruitment of immune suppressive myeloid-derived suppressor cells (MDSC) and M2 macrophages, respectively (65, 66). YAP/TAZ also upregulates PD-L1 in cancer cells, which is a critical regulator for the immune checkpoints (67, 68).
Furthermore, some studies have indicated that YAP/TAZ is essential for the maintenance of cancer stem cells. For example, TAZ is required for maintenance of breast cancer stem cells (49), while in osteosarcoma, YAP functions downstream of SOX2 to maintain cancer stem cells (69). In basal like breast cancer cells, serum response factor (SRF) interacts with YAP and induces IL-6 expression, which promotes stemness of basal like breast cancer cells (70). In esophageal cancer cells, YAP induces SOX9, which is engaged in cancer stem cell properties (71, 72).
During cancer development, cancer cells require high energy and a large amount of building blocks to maintain their growth and proliferation. However, because of limited oxygen and nutrients in tumor microenvironment, cancer cells are exposed to various metabolic stress and need to adapt their metabolism to it (73). Recent studies have shown that YAP/TAZ is not only regulated by diverse metabolic signals, but also serves as a key regulator of various metabolic pathways (5).
Therefore, YAP/TAZ obviously plays an essential role in cancer progression (Figure 2). In addition to the functions that were discussed earlier, YAP/TAZ regulates various cellular functions that are essential for metastasis, such as cell migration, invasion, survival, and EMT. We will discuss these functions in detail in the next section.
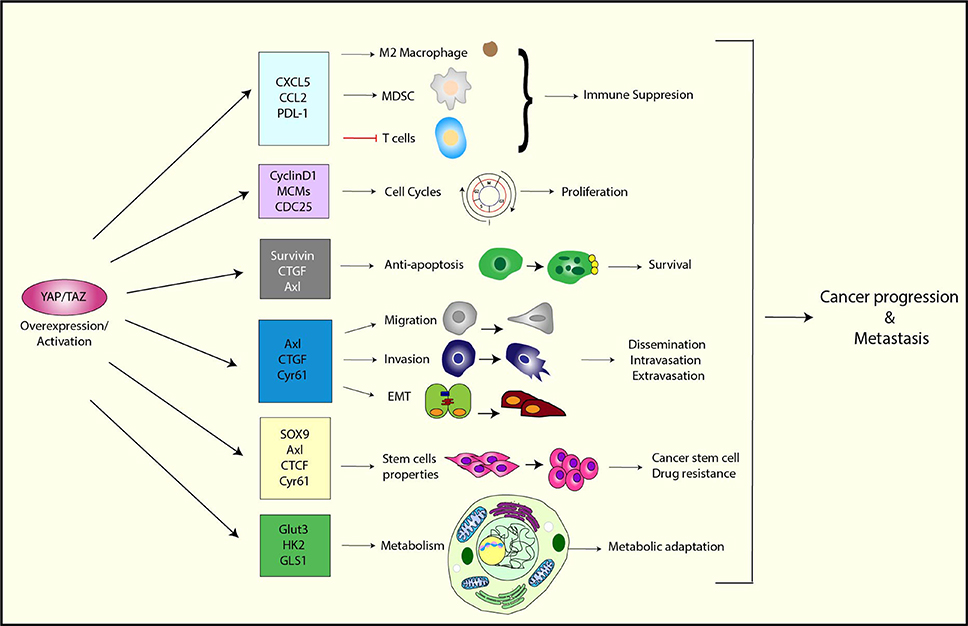
Figure 2. The function of YAP/TAZ in cancer progression and metastasis. YAP/TAZ promotes tumor progression and metastasis by upregulating a variety of its target genes that are involved in immune suppression, cell cycle control, anti-apoptosis, migration/invasion, EMT, stem cell properties, and metabolism.
It has been recognized that metastasis occurs through multiple steps known as the metastatic cascade. First, cancer cells gain the ability of migration and invasion, and these cells are dissociated from primary tumors to enter blood and lymphatic vessels. Then, the cells circulates in the blood stream, and some surviving cells extravasates at distant tissues and organs to form secondary tumors (1, 74).
Multiple retrospective analyses of clinical samples have shown that the upregulation or nuclear localization of YAP/TAZ is correlated with metastasis in a variety of cancer such as breast, colorectal, gastric, lung, liver, and pancreatic cancer (75–80). Similarly, the downregulation of upstream negative regulators of the Hippo pathway is also associated with metastasis in the various cancer (81–84).
Moreover, a number of in vivo studies have demonstrated the role of YAP/TAZ in metastasis. Overexpression or activation of YAP/TAZ promotes the ability of cancer cells to metastasize in xenograft mouse models of several cancer types (85–87). In contrast, inactivation of YAP/TAZ suppresses metastasis in several mouse models (87, 88).
At the cellular level, YAP/TAZ has been demonstrated to regulate multiple essential steps of the metastatic cascade (Figure 2). Cell migration and invasion abilities are crucial for cancer cell metastasis, and many studies have shown that overexpression and activation of YAP/TAZ promotes cell migration and invasion (85, 89–92), while the inhibition of YAP/TAZ reduces the migration and invasion abilities of cancer cells (91–93). Mechanistically, YAP/TAZ has been shown to control cell motility by limiting cytoskeletal and focal adhesion maturation (94). Moreover, YAP/TAZ promotes cell migration and invasion through the upregulation of various downstream targets, such as Axl, Cyr61, and receptor for hyaluronan-mediated motility (RHAMM) (89, 95, 96).
EMT is the process that epithelial cells lose their characteristics of cell polarity and cell-cell adhesion and gain mesenchymal cell properties, and associated with cell motility (97, 98). Because the initial step of metastasis is the detachment of cells from a tumor mass, EMT is considered to be the first event for metastasis (97, 99). Numerous studies have shown that YAP/TAZ is a key regulator of EMT. Overexpression of YAP/TAZ induce EMT (11, 12, 93, 100, 101), while the inhibition of YAP/ TAZ reverses EMT (93, 102, 103). Mechanistically, several YAP/TAZ targets such as Axl, Cyr61, and CTGF have been shown to have the ability to induce EMT and stemness (104–106). Moreover, YAP has been shown to interact with Smad2/3/4 and regulate the mRNA expression of EMT-inducing transcription factors, Snail, Twist1, and Slug (107). EMT-inducing transcription factor ZEB1 also interacts with YAP and enhances its transcription activity (108).
During metastasis, cancer cells get into blood vessels and circulate in a body. For cancer cells to metastasize, they need to evolve a mechanism, which helps them to survive during their circulation in blood vessels. Anoikis is the type of apoptotic cell death that is induced by cell detachment, and gaining an ability of resistance toward anoikis is critical for cancer cells to metastasize (109). It has been shown that YAP/TAZ is able to inhibit anoikis (84). Interestingly, platelets interacts with cancer cells, in which YAP is activated through RHOA-MYPT1-PP1-mediated YAP dephosphorylation (110). The platelets-mediated YAP activation contributes to resistance to anoikis and metastasis (110). Moreover, it has been shown that fluid shared stress activates YAP/TAZ, suggesting that YAP/TAZ is activated during circulation and the activated YAP/TAZ may contribute to cancer cell survival during metastasis (111, 112).
Glucose is an essential source of energy and building blocks for cells. It is metabolized through glycolysis, TCA cycle, and oxidative phosphorylation (OXPHOS) to produce ATP. In the presence of oxygen, normal cells primarily generate ATP through oxidative phosphorylation and exhibit reduced glucose consumption. However, cancer cells uptake high levels of glucose and convert it to lactate via glycolysis regardless of the presence of oxygen (aerobic glycolysis). The enhanced glycolysis, pentose phosphate, and hexosamine biosynthetic pathways support the energy and building blocks required for high replication of cancer cells (113).
It has been demonstrated that YAP/TAZ is inactivated under the low glucose condition or by the inhibition of glycolysis while it is activated in response to high levels of glucose (33, 34, 114, 115). Several distinct mechanisms of glucose-mediated YAP/TAZ activation have been identified (Figure 3).
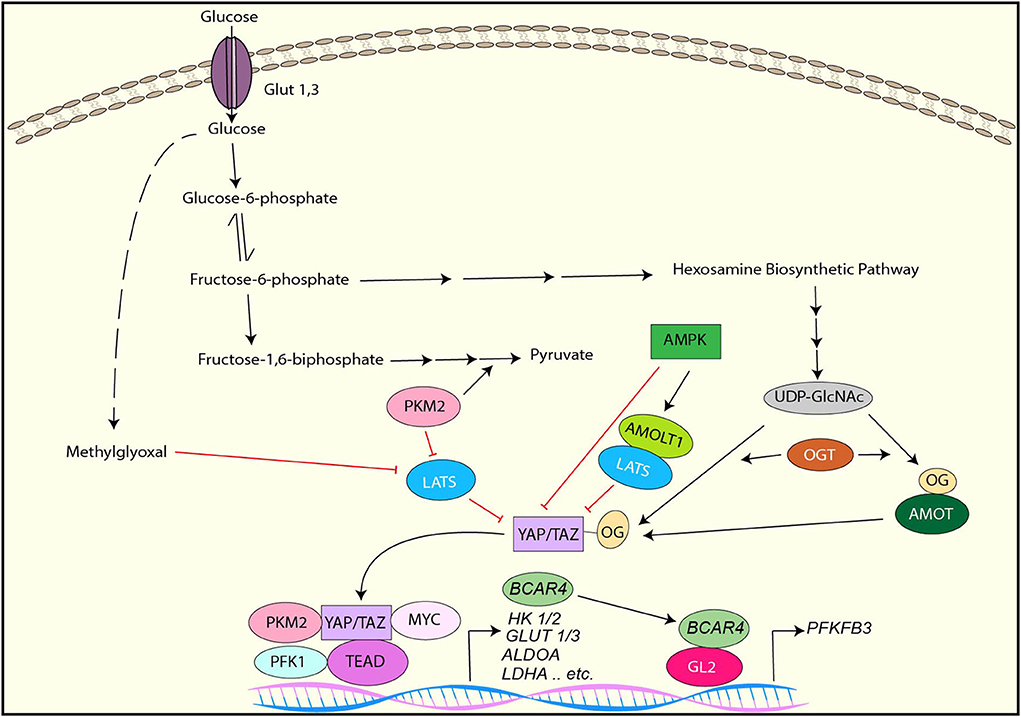
Figure 3. YAP/TAZ regulation by glucose metabolism and YAP/TAZ-targets for glucose metabolism. YAP/TAZ is inhibited by AMPK directly or indirectly through AMOTL1-mediated LATS activation. LATS is inhibited by PKM2 or Methylglyoxal, resulting in YAP/TAZ activation. YAP activation is also induced by direct O-GlcNAcylation or indirect O-GlcNAcylation of AMOT. In the nucleus, PKM2, PFK1, or Myc interacts with the YAP/TAZ-TEAD complex and enhances its transcriptional activity toward to the genes involved in glucose metabolism. YAP/TAZ induces lncRNA BCAR4, which interacts with GLI2 and induces PFKFB3 transcription.
AMPK is a key cellular metabolic sensor that is activated by a high AMP to ATP ratio (116). As described earlier section, studies have shown that AMPK directly phosphorylates YAP and inhibits its transcriptional activity (33, 34). Also, these studies have indicated that LATS is also activated by glucose deprivation and contributes to YAP inactivation independently of AMPK, although the underlying mechanism is uncertain (33, 34). Another study has revealed that AMPK phosphorylates angiomotin-like 1 (AMOTL1), resulting in the stabilization of AMOTL1, which contributes to LATS-mediated YAP phosphorylation and inactivation (114).
O-GlcNAcylation is a posttranslational modification of proteins that is catalyzed by O-GlcNAc transferase (OGT) (117, 118). Uridine diphosphate N-acetylglucosamine (UDP-GlcNAc) is a donor sugar of O-GlcNAcylation and transferred to serine/threonine residues of myriad target proteins by OGT (117, 118). UDP-GlcNAc is synthesized from glucose through the hexosamine biosynthetic pathway, which links to cellular O-GlcNAcylation levels (118). Moreover, it has been shown that aberrant protein O-GlcNAcylation is associated with metabolic reprogramming in cancer (119). It has been reported that YAP is also O-GlcNAcylated in response to high levels of glucose (120, 121). One study has shown that YAP is O-GlcNAcylated at Serine 109 (S109) by OGT under high glucose, and S109-O-GlcNAcylation of YAP prevents YAP-LATS interaction, thereby activating YAP (121). In contrast, another study has revealed that O-GlcNAcylation of YAP at threonine 214 (T214), which is also induced by high levels of glucose, enhances its stability by inhibiting the β-TrCP-YAP interaction. Besides, T214 O-GlcNAcylation of YAP is associated with YAP tumorigenic activity (120). Moreover, it has been reported that AMOT is also O-GlcNAcylated, and AMOT expression and O-GlcNAcylation is enhanced by high levels of glucose (122). Interestingly, AMOT enhances YAP nuclear localization and transcriptional activity under the high glucose condition while AMOT inhibits them in normal levels of glucose, suggesting that AMOT O-GlcNAcylation is another mechanism of YAP/TAZ regulation by high level of glucose (122).
In addition to the above mentioned mechanism, phosphofructokinase (PFK1), which is one of the enzymes regulating glycolysis, has been implicated to regulate YAP/TAZ activity (115). Inhibition of glycolysis with 2-deoxy-glucose (2DG) reduces TEAD-YAP complex formation, thereby reducing YAP-mediated transcription. Activated PFK1 interacts with TEAD and form a complex with TEAD and YAP, resulting in stabilization of the YAP-TEAD complex and YAP activation (115).
Methylglyoxal (MG) is a byproduct of glycolysis that induces protein glycation and formation of advanced glycation end products (AGEs). A study has revealed that high MG-adducts and nuclear YAP are correlated in breast cancer, and MG induces YAP activation in breast cancer cells (123). Mechanistically, MG induces HSP90 glycation, which impairs its chaperon activity on LATS1, resulting in destabilization of LATS1 protein and subsequent YAP activation (123).
Pyruvate kinase M2 (PKM2) is a splice isoform of pyruvate kinase that plays an critical role in aerobic glycolysis in cancer (124). It has been reported that tyrosine 105-phosphorylated PKM2, which is induced by several tyrosine kinases, reduces LATS1 protein stability and downregulates it, thereby activating YAP (125). Furthermore, another report has shown that PKM2 directly interacts with YAP and enhances YAP transcriptional activity (126).
Because YAP/TAZ promotes cell growth and proliferation that require high nutrients, it is rational that YAP/TAZ is activated by high cellular glucose and inhibited under a limited energy source. However, in this context, it is also reasonable that YAP/TAZ controls glycolysis to support cell growth and proliferation. Indeed, emerging evidence has shown that YAP/TAZ regulates glycolysis by upregulating the key enzymes for the pathway as well as glucose transporters (Figure 3).
It has been demonstrated that knockdown of YAP/TAZ downregulates various genes involved in glycolysis including GLUT3, HK1,HK2, PFKFB4, PFKP, GAPDH, PGK1, PGAM1, LDHA, PDHA1, and PDHB, resulting in the downregulation of glycolysis and upregulation of mitochondrial oxidative phosphorylation in renal cell carcinoma cells (127). In cancer cells from a KRAS mutant pancreatic cancer GEM, deletion of YAP downregulates the expression of HK2, ALDOA, GPADH, PGK1, PGAM1, and LDHA genes (128). Interestingly, YAP cooperates with other transcription regulators to control many genes related to glycolysis. For example, YAP interacts with HIF1α and regulates the transcription of various genes involved in aerobic glycolysis such as GLUT1, HK2, ALDOA, LDHA, and PKM2 (129, 130). The YAP-TEAD complex interacts with p65 and upregulates the transcription of HK2 (131). YAP associates with c-Myc and regulates PGAM1 mRNA expression (132). Besides, YAP directly regulates transcription of GLUT3, and PKM2 enhances GLUT3 expression by interacting with YAP (126). In addition to direct transcriptional regulation, YAP also indirectly upregulates HK2 and PFKFB3 expression and glycolysis via the lncRNA BCAR4-GLI2 axis (133).
Another key metabolic reprograming that is critical for cancer progression is adaptation of lipid metabolism (134). Cancer cells require high levels of lipid to maintain their growth and proliferation, and exhibit elevated de novo synthesis or uptake of lipid compared to normal cells (134). A growing body of evidence has shown that YAP/TAZ is not only activated by the dysregulated lipid metabolism pathways but also plays a role in the adaptation of lipid metabolism in cancer (Figure 4)
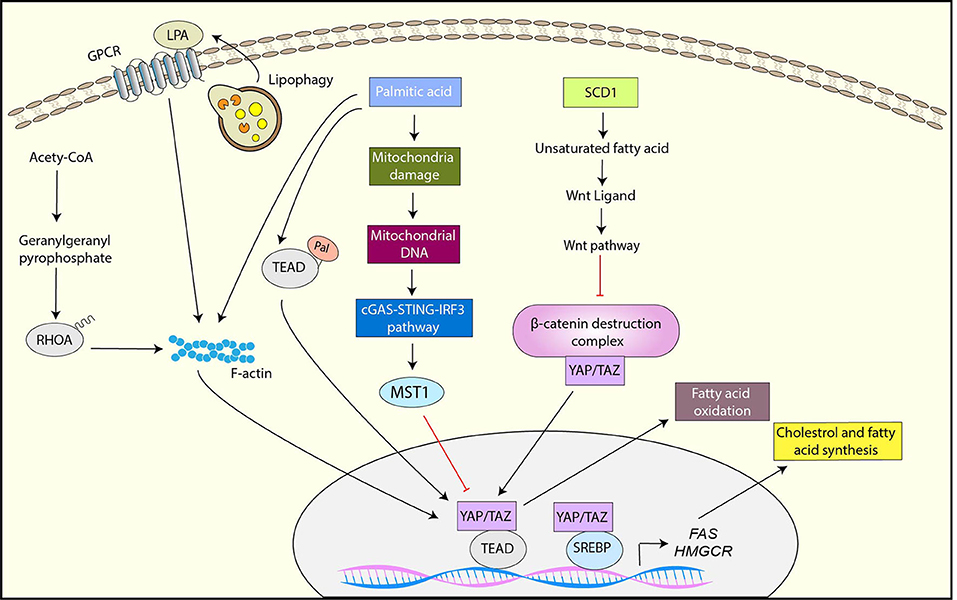
Figure 4. YAP/TAZ regulation by lipid metabolism and YAP/TAZ-targets for Lipid metabolism. SCD1-mediated unsaturated fatty acid activates YAP/TAZ by inhibiting the β-catenin destruction complex via Wnt activation. Palmitic acid inhibits YAP through the mitochondrial damage-mediated cGAS-STING-IRF3-MST1 pathway while it activates YAP-TAZ via palmitoylation of TEAD or F-actin rearrangement. The mevalonate pathway activates YAP/TAZ through geranylgeranylation of RHOA. Lipophagy is also associated with YAP activation likely through the upregulation of LPA, a ligand of GPCR. YAP interacts with SREBP1 and SREBP2 and enhances their transcriptional activity toward FAS and HMGCR. YAP also enhances fatty acid oxidation.
De novo fatty acid synthesis is activated in cancer cells for the maintenance of membrane synthesis and generation of signaling molecules (135). Stearoyl-CoA-desaturase 1 (SCD1) is an enzyme that synthesizes monounsaturated fatty acids, and a study has shown that the inhibition of SCD1 downregulates YAP/TAZ expression, nuclear localization, and activity, suggesting that monounsaturated fatty acids are crucial for YAP function (136). The study has also indicated that SCD1 activates YAP/TAZ through the inhibition of the β-catenin destruction complex, which is likely induced by the upregulation of lipid modified Wnt proteins (136). Palmitic acid is a common saturated fatty acid in organisms. It has been reported that palmitic acid inhibits YAP by upregulating MST1, thereby inhibiting endothelial cell proliferation, migration, and angiogenesis (137). Mechanistically, palmitic acid induces mitochondrial damage, which results in releasing mitochondrial DNA (mtDNA) to the cytoplasm. The cytoplasmic mtDNA further activates cGAS–STING–IRF3 signaling, which subsequently induces MST1 transcription (137). In contrast to endothelial cells, free fatty acids (FFA) including palmitic acid activate YAP via F-actin rearrangement in pancreatic β-cells (138). Notably, CTGF induced by YAP plays a role in protection of β-cells from FFA-induced apoptosis (138).
Protein S-palmitoylation is the post-translational modification by which palmitate is attached to cysteine residues of proteins, leading to protein association with membrane, localization changes, and/or functional alterations. Several studies have demonstrated that TEAD is palmitoylated and TEAD palmitylation is critical for YAP/TAZ function (139–141). Although TEAD palmitolaylation does not affect TEAD localization or membrane binding, it is important for the TEAD-YAP/TAZ interaction or proper TEAD folding and stability (139–141). Interestingly, TEAD is autopalmitoylated under physiological conditions (139). Moreover, a recent study has shown that TEAD palmitoylation is regulated by cell density (140). Mechanistically, high cell density induces NF2-dependent downregulation of fatty acid synthase (FASN) and acetyl-CoA carboxylase (ACC), which are the crucial enzymes for de novo biosynthesis of palmitate (140). Moreover, high cell density upregulates the expression of depalmitoylases including APT2 and ABHD17A, which may contribute to TEAD depalmitoylation and subsequent TEAD downregulation (140). Therefore, these results suggest that TEAD palmitoylation serves as the potential regulatory mechanism of YAP/TAZ in response to cellular fatty acid metabolism.
The mevalonate pathway is another lipid metabolic pathway to synthesize sterols and isoprenoids from acetyl-CoA. The intermediate products of the mevalonate pathway including farnesyl pyrophosphate and geranylgeranyl pyrophosphate serve as substrates for protein farnesylation and geranylgeranylation, which plays a significant role in protein localization and function (142). Several studies have revealed that YAP/TAZ is regulated by the mevalonate pathway (96, 143, 144). These studies have further identified the underlying mechanism, in which the activation of YAP/TAZ by the mevalonate pathway is mediated by geranylgeranylation of RHOA and subsequent RHOA activation (96, 143, 144).
Lipophagy is the autophagic degradation process of lipid droplets, and a recent study has shown that oxidized low-density lipoprotein (oxLDL) lipophagy is associated with YAP activation and hepatocellular carcinoma (HCC) progression (145). In brief, endoplasmic reticulum-residential protein, Nogo-B is upregulated by the CD36-mediated oxLDL uptake, and Nogo-B interacts with ATG5 to promote oxLDL lipophagy, resulting in YAP activation likely through the upregulation of lysophosphatidic acid (LPA), which is a ligand of GPCR (145).
In addition to YAP/TAZ regulation by lipid metabolism, recent studies have shown that YAP/TAZ regulates lipid metabolism. The sterol regulatory element-binding proteins (SREBPs) are transcription factors that control the expression of enzymes involved in fatty acid and cholesterol biosynthesis (146). It has been reported that YAP interacts with the nuclear forms of SREBP1 and SREBP2 and enhances their transcriptional activities toward fatty acids synthase (FAS) and 30-hydroxylmethyl glutaryl coenzyme A reductase (HMGCR) (147). Also, the study has shown that the activation of LATS1 or inhibition of YAP reduced hepatic steatosis and hyperlipidaemia in diet-induced diabetic mice (147).
Fatty acids oxidation (FAO) is the metabolic pathway that degrade fatty acid to supply ATP and acetyl-CoA and the activity of FAO is promoted in various cancer types (148). Recently, it has been proved that knocking-down YAP reduces FAO while YAP overexpression activates it (149). In addition, the study has indicated that YAP-mediated metabolic shift to FAO is critical for lymph nodes metastasis in a melanoma mouse model (149).
In cancer, amino acid metabolism is also dysregulated. In particular, because glutamine is a key amino acid involved in energy generation in TCA cycle, biosynthesis of essential molecules such as amino acid, nucleotide, and fatty acid, control of redox homeostasis, and regulation of cell signaling (150), enhanced glutamine uptake and glutaminolysis are essential for cancer cell proliferation and survival (151). YAP/TAZ has been shown to be involved in glutamine metabolism by upregulating various genes involved in it (Figure 5).
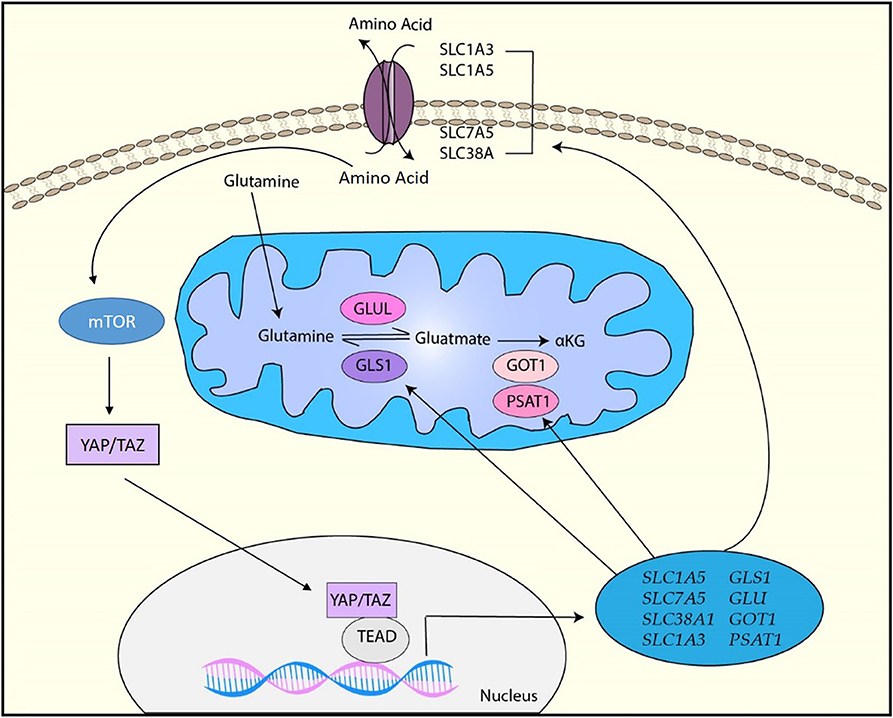
Figure 5. YAP/TAZ-targets for amino acid metabolism. YAP/TAZ regulates amino acid metabolism by upregulating various factors involved in amino acid transportation such as SLC1A3, SLC1A5, SCL7A5, and SLC38A1 as well as glutamine metabolism by upregulating GLS1, GLUL, GOT1, and PSAT1. Moreover, upregulation of amino acids induces the activation of mTOR, which also activates YAP/TAZ through various mechanisms.
It has been reported that YAP/TAZ directly regulates the mRNA expression of glutamine transporters SLC38A1 and SLC7A5 in HCC and these transporters are required for YAP/TAZ-mediated proliferation of HCC cells (152). In a zebra fish model, YAP has been shown to reprogram glutamine metabolism and enhance liver tumor formation (153). Briefly, YAP upregulates glutamine synthetase (GLUL) expression and activity, resulting in the upregulation of glutamine and nucleotide biosynthesis (153). In pulmonary vascular cells, YAP/TAZ upregulates the expression of glutaminase 1 (GLS1) that is the enzyme regulating the first step in the glutaminolysis by generating glutamate from glutamine (154). Physiologically, endothelial cell proliferation is enhanced by vascular stiffness in pulmonary hypertension. YAP/TAZ is activated by mechanical cues from stiff ECM and results in endothelial cell proliferation by upregulating GLS1-mediated glutaminolysis (154). It has been revealed that EphA2 induces YAP/TAZ activation through RHOA activation, and activated YAP/TAZ promotes glutaminolysis through upregulation of GLS1 and SLC1A5 in HER2-positive breast cancer cells (155). This study has also showed that YAP/TAZ expression positively correlates with GLS1 and SLC1A5 expression in human breast cancer patient samples (155). Another study with breast cancer cell lines has indicated that YAP/TAZ reprograms metabolism of breast cancer cells to glutamine-dependent one by inducing glutamic-oxaloacetic transaminase (GOT1) and phosphoserine aminotransferase (PSAT1), which are the key enzymes to convert glutamate to α-ketoglutarate (α-KG) (156). Thus, these studies demonstrated that the YAP/TAZ-mediated glutamine metabolism plays a role in breast cancer progression. Moreover, a recent study has demonstrated that mechanical cues from stiff ECM activates glutamine metabolism though YAP/TAZ activation in both cancer cells and cancer-associated fibroblasts (CAFs) (157). YAP/TAZ upregulates not only GLS1 to increase glutamine metabolism, but also SLC1A3 to exchange amino acids between cancer cells and CAFs (157). Cancer cells utilize CAFs-derived aspartate for the nucleotide biosynthesis pathway to maintain their proliferation while CAFs use cancer cell-derived glutamate for the glutathione pathway to maintain redox homeostasis (157). Thus, the study has indicated that YAP/TAZ regulates tumor growth by coordinating non-essential amino acids flux within the tumor niche.
In addition to glutamine metabolism, YAP/TAZ is also associated with the regulation of other amino acids. For example, YAP/TAZ increases leucine uptake by upregulating high-affinity leucine transporter LAT1 (158). Moreover, a study has suggested that YAP/TAZ activates serine metabolism (159). Upregulation of amino acids leads to the activation of the mammalian target of rapamycin (mTOR) signaling pathway, which is crucial for cell growth and survival (160). Numerous studies have shown that YAP/TAZ is closely associated with mTOR signaling through amino acids. For example, YAP/TAZ upregulates mTOR activity through the upregulation of amino acid transporters (152, 158). In addition, YAP activates mTOR by downregulating PTEN (161). Alternatively, the mTOR pathway has been shown to control YAP/TAZ activity through various mechanisms such as inhibition of autophagy, MST1, and AMOTL2, suggesting that cellular amino acids can regulate YAP/TAZ via activation of mTOR (162–164).
Growing evidence has indicated that metabolic reprograming plays a critical role in metastasis (165). As mentioned in the earlier section, metastasis consists of multiple steps, in which cancer cells need to reprogram cellular metabolism to adapt to the drastic changes of environment. In addition, it has been revealed that metabolic reprogramming is associated with the phonotypic changes of cancer cells to gain specific ability required for metastasis (3).
Glycolysis is upregulated in metastatic cancer cells compared to non-metastatic ones (166), and the inhibition of glycolysis attenuates metastasis (167). EMT is a critical step for cancer cells to initiate metastasis and associated with cell migration and invasion ability, and glycolysis has been shown to be closely associated with EMT (168, 169). Induction of EMT in pancreatic ductal adenocarcinoma by tumor necrosis factor-α (TNF-α) and transforming growth factor-β (TGF-β) simultaneously reprograms glucose metabolism to enhance glycolysis (168). Moreover, EMT regulator Snail silences the expression of fructose-1,6-biphosphatase (FBP1), which is a rate-limiting enzyme in gluconeogenesis (170). Loss of FBP1 enhances glycolysis and reduces OXPHOS, while, FBP1 inhibits tumorigenesis in vivo (170). Because YAP/TAZ is activated by high levels of glucose and can regulate both EMT and glycolysis, it may coordinate glycolysis and EMT to initiate metastasis.
Indeed, one report have demonstrated that YAP/TAZ-mediated glucose metabolism is vital for metastasis. In brief, glycolysis and glucose transporter Glut3 are upregulated in metastatic colorectal cancer cells, and Glut3 expression is correlated with poor survival in colorectal cancer patients (126). Interestingly, Glut3 overexpression increases YAP/TAZ expression while Glut3-induced cell migration, invasion, and tumor sphere formation activity are abrogated by YAP knockdown (126). Moreover, YAP induces Glut3 expression in colorectal cancer cells, and both YAP and Glut3 are required for high-fat/sucrose diet-mediated metastasis in a xenograft mouse model (126). Thus, YAP/TAZ and Glut3 regulates each other to control glycolysis and metastasis in colorectal cancer (Figure 6). In addition to glycolysis, OXHOS and activated mitochondria have been shown to play a role in metastasis (171, 172). Moreover, a study has revealed that specific metabolic adaptations are required for breast cancer cells to metastasize to particular organs (173). Liver-metastatic cancer cells show enhanced glycolysis and reduced OXPHOS while bone- or brain-metastatic cancer cells display enhanced OXPHOS (173). So far, it is uncertain whether YAP/TAZ is involved in organ-specific metastasis. However, considering YAP/TAZ promotes glycolysis, it may contribute to organ-specific metastasis.
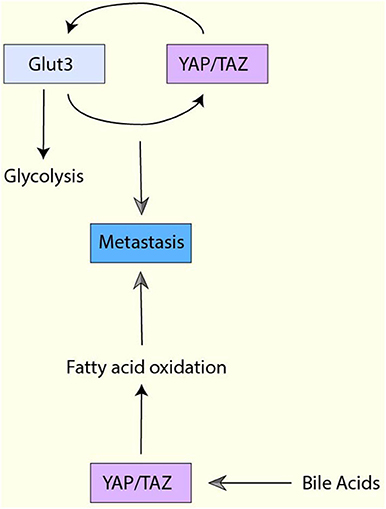
Figure 6. The interplay between YAP/TAZ-mediated metabolism and metastasis. Glut3 overexpression increases YAP/TAZ expression, and Glut3 promotes cell migration, invasion, and tumor sphere formation YAP/TAZ-dependently. Moreover, YAP/TAZ also induces Glut3 expression, and both YAP/TAZ and Glut3 are required for metastasis. YAP activation enhances fatty acid oxidation (FAO) and the enhanced FAO is required for lymph node metastasis. Moreover, YAP in the cancer cells is activated by bile acids, which is accumulated to high levels in the metastatic lymph nodes.
Several reports have indicated that the lipid metabolic pathways are also associated with metastasis. For example, in ovarian cancer, fatty acid incorporation via CD36 is required for cell migration and invasion in vitro and metastasis in vivo (174). In contrast, in A549 non-small lung cancer cells, lipogenesis plays a negative role in TGF-β-induced EMT and metastasis (175). Therefore, the role of lipid metabolism may be context-dependent. In mevalonate-mediated YAP activation, YAP regulates RHAMM, which upregulates breast cancer invasion and migration activity (96). Moreover, the recent study using a melanoma mouse model has also showed that YAP-mediated FAO is critical for metastasis (149) (Figure 6). In this model, YAP activation enhances FAO and the enhanced FAO is required for lymph node metastasis. Additionally, YAP in the cancer cells is activated by bile acids, which is accumulated to high levels in the metastatic lymph nodes (149).
Glutamine metabolism has also been reported to be engaged in metastasis. For example, glutaminolytic enzyme, glutamate dehydrogenase 1 (GDH1), has been shown to contribute to anoikis resistance and promote metastasis (176). Mechanistically, GDH1 enhances α-KG, which further regulates energy balance through the CamKK2-AMPK pathway after cell detachment (176). In colorectal cancer tissues, GLS1 expression is positively correlated with lymph node metastasis and advanced clinical stage (177). Moreover, GLS1 is induced by hypoxia via HIF1α in colorectal cancer cell lines, and knockdown of GLS1 reduces metastasis in a xenograft mouse model with colorectal cancer cells (177). So far, there is no direct evidence that YAP/TAZ-mediated gluaminolysis contributes to metastasis. However, because YAP/TAZ can regulate GLS1 expression and glutaminolysis, it is likely that YAP/TAZ-mediated glutaminolysis contributes to metastasis. Moreover, although the GDH1-CamKK2-AMPK pathway confers resistance to anoikis, YAP/TAZ is known to be negatively regulated by AMPK. Thus, the glutamine metabolism-mediated anti-apoptotic function may be context-dependent. Obviously further studies are necessary to address the role of YAP/TAZ in glutaminolysis-mediated metastasis.
Recent advances in targeted therapy and immunotherapy have significantly improved survival rates of cancer patients. However, majority of metastatic cancer is still challenging to be cured. Therefore, it is important to understand the pathways that are involved in metastasis and identify the potential druggable targets. As we described here, YAP/TAZ is activated by high nutrient conditions, and regulates multiple aspects of metastasis as well as cancer metabolism. Thus, YAP/TAZ would be a promising drug target for metastatic cancer. Although no effective clinical drugs targeting YAP/TAZ are available, multiple direct and indirect inhibitors for YAP/TAZ have been developed (178). Verteporfin, which is a photosensitizer for photodynamic therapy, has been known to inhibit YAP/TAZ activity by blocking YAP/TAZ-TEAD interaction and downregulating YAP/TAZ expression (179, 180). Indeed, multiple studies have demonstrated that verteporfin inhibits YAP/TAZ-dependent expression of the genes related to glycolysis and glutaminolysis and subsequent glycolysis and glutaminoslysis in vitro and in vivo (126, 129, 153, 154, 157, 181). In addition to verteporfin, a peptide mimicking VGLL4 has also been shown to inhibit YAP-TEAD interaction and suppress tumor growth in vitro and in vivo (42). Moreover, several specific small molecule inhibitors for YAP-TEAD interaction have been developed (182–184). These inhibitors may have better therapeutic effects with less toxicity than verteporfin because of their specificity. As an alternative approach, YAP/TAZ upstream regulators, in particular protein kinases and other enzymes, would serve as potential drug targets for attenuating YAP/TAZ activity (178). However, YAP/TAZ is regulated by many upstream regulators and the regulatory mechanism may be dependent on cell context. Therefore, it is critical to identify key YAP/TAZ regulators in each tumor for targeting it. Moreover, the expression of YAP/TAZ-target genes would serve as a biomarker to monitor efficacy of therapy.
In recent years, numerous studies have demonstrated the role of YAP/TAZ in metabolism and metastasis. However, there are several critical questions that need to be answered to understand YAP/TAZ-mediated metabolism and metastasis.
Because YAP/TAZ is a multifunctional transcriptional regulator, it is expected to be involved in various steps of metastasis. However, it is still uncertain exactly how YAP/TAZ is regulated during metastasis. For example, EMT is considered as a first step for metastasis, but mesenchymal to epithelial transition (MET) also plays a role in it (185). Moreover, cancer cells reprogram their metabolism between high glycolysis and high OXPHOS during metastasis (3). Therefore, YAP/TAZ activity needs to be controlled strictly during metastasis. Cancer cells are exposed to diverse types of mechanical cues, and changes of nutrient and oxygen during metastasis, which may contribute to the regulation of YAP/TAZ and coordinate the cancer progression and metastasis. Indeed, some mechanical cues from ECM have been shown to promote EMT and metastasis as well as alter cellular metabolism (154, 157, 186, 187). In addition to LATS-mediated phosphorylation, YAP/TAZ is regulated by the interaction with other transcription factors and other proteins. Thus, during the metastasis and metabolic alterations, YAP/TAZ may change its binding partners so that it alters its function according to the need of microenvironment. In addition, various extracellular metabolites may affect YAP/TAZ binding partners and signaling, and contribute to metabolic adaptation and metastasis. Moreover, several reports have suggested that YAP also exhibits context-dependent tumor suppressor function (188, 189). The complicated metabolic regulation by YAP/TAZ may be associated with its tumor suppressor function. Thus, the understanding of the diverse role of YAP/TAZ in different cellular contexts may lead to a better therapeutic strategy to target metastatic cancer.
HY wrote the manuscript. GT made the figures and contributed to editing the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by a startup grant from Qatar Biomedical Research Institute, Qatar Foundation.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Qatar Biomedical Research Institute and Qatar Foundation for their support.
1. Valastyan S, Weinberg RA. Tumor metastasis: molecular insights and evolving paradigms. Cell. (2011) 147:275–92. doi: 10.1016/j.cell.2011.09.024
2. Pavlova NN, Thompson CB. The emerging hallmarks of cancer metabolism. Cell Metab. (2016) 23:27–47. doi: 10.1016/j.cmet.2015.12.006
3. Lehuede C, Dupuy F, Rabinovitch R, Jones RG, Siegel PM. Metabolic plasticity as a determinant of tumor growth and metastasis. Cancer Res. (2016) 76:5201–8. doi: 10.1158/0008-5472.CAN-16-0266
4. Zanconato F, Cordenonsi M, Piccolo S. YAP/TAZ at the roots of cancer. Cancer Cell. (2016) 29:783–803. doi: 10.1016/j.ccell.2016.05.005
5. Koo JH, Guan KL. Interplay between YAP/TAZ and metabolism. Cell Metab. (2018) 28:196–206. doi: 10.1016/j.cmet.2018.07.010
6. Yu FX, Guan KL. The hippo pathway: regulators and regulations. Genes Dev. (2013) 27:355–71. doi: 10.1101/gad.210773.112
7. Zhao B, Tumaneng K, Guan KL. The hippo pathway in organ size control, tissue regeneration and stem cell self-renewal. Nat Cell Biol. (2011) 13:877–83. doi: 10.1038/ncb2303
8. Zhao B, Wei X, Li W, Udan RS, Yang Q, Kim J, et al. Inactivation of YAP oncoprotein by the hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev. (2007) 21:2747–61. doi: 10.1101/gad.1602907
9. Liu CY, Zha ZY, Zhou X, Zhang H, Huang W, Zhao D, et al. The hippo tumor pathway promotes TAZ degradation by phosphorylating a phosphodegron and recruiting the SCF{beta}-TrCP E3 ligase. J Biol Chem. (2010) 285:37159–69. doi: 10.1074/jbc.M110.152942
10. Zhao B, Li L, Tumaneng K, Wang CY, Guan KL. A coordinated phosphorylation by Lats and CK1 regulates YAP stability through SCF(beta-TRCP). Genes Dev. (2010) 24:72–85. doi: 10.1101/gad.1843810
11. Zhao B, Ye X, Yu J, Li L, Li W, Li S, et al. TEAD mediates YAP-dependent gene induction and growth control. Genes Dev. (2008) 22:1962–71. doi: 10.1101/gad.1664408
12. Zhang H, Liu CY, Zha ZY, Zhao B, Yao J, Zhao S, et al. TEAD transcription factors mediate the function of TAZ in cell growth and epithelial-mesenchymal transition. J Biol Chem. (2009) 284:13355–62. doi: 10.1074/jbc.M900843200
13. Mizuno T, Murakami H, Fujii M, Ishiguro F, Tanaka I, Kondo Y, et al. YAP induces malignant mesothelioma cell proliferation by upregulating transcription of cell cycle-promoting genes. Oncogene. (2012) 31:5117–22. doi: 10.1038/onc.2012.5
14. Hong W, Guan KL. The YAP and TAZ transcription co-activators: key downstream effectors of the mammalian Hippo pathway. Semin Cell Dev Biol. (2012) 23:785–93. doi: 10.1016/j.semcdb.2012.05.004
15. Panciera T, Azzolin L, Cordenonsi M, Piccolo S. Mechanobiology of YAP and TAZ in physiology and disease. Nat Rev Mol Cell Biol. (2017) 18:758–770. doi: 10.1038/nrm.2017.87
16. Jaalouk DE, Lammerding J. Mechanotransduction gone awry. Nat Rev Mol Cell Biol. (2009) 10:63–73. doi: 10.1038/nrm2597
17. Dupont S, Morsut L, Aragona M, Enzo E, Giulitti S, Cordenonsi M, et al. Role of YAP/TAZ in mechanotransduction. Nature. (2011) 474:179–83. doi: 10.1038/nature10137
18. Aragona M, Panciera T, Manfrin A, Giulitti S, Michielin F, Elvassore N, et al. A mechanical checkpoint controls multicellular growth through YAP/TAZ regulation by actin-processing factors. Cell. (2013) 154:1047–59. doi: 10.1016/j.cell.2013.07.042
19. Meng Z, Qiu Y, Lin KC, Kumar A, Placone JK, Fang C, et al. RAP2 mediates mechanoresponses of the Hippo pathway. Nature. (2018) 560:655–60. doi: 10.1038/s41586-018-0444-0
20. Chang L, Azzolin L, Di Biagio D, Zanconato F, Battilana G, Lucon Xiccato R, et al. The SWI/SNF complex is a mechanoregulated inhibitor of YAP and TAZ. Nature. (2018) 563:265–9. doi: 10.1038/s41586-018-0658-1
21. Dasgupta I, McCollum D. Control of cellular responses to mechanical cues through YAP/TAZ regulation. J Biol Chem. (2019) 294:17693–706. doi: 10.1074/jbc.REV119.007963
22. Yu FX, Zhao B, Panupinthu N, Jewell JL, Lian I, Wang LH, et al. Regulation of the hippo-YAP pathway by G-protein-coupled receptor signaling. Cell. (2012) 150:780–91. doi: 10.1016/j.cell.2012.06.037
23. Plouffe SW, Meng Z, Lin KC, Lin B, Hong AW, Chun JV, et al. Characterization of hippo pathway components by gene inactivation. Mol Cell. (2016) 64:993–1008. doi: 10.1016/j.molcel.2016.10.034
24. Meng Z, Moroishi T, Mottier-Pavie V, Plouffe SW, Hansen CG, Hong AW, et al. MAP4K family kinases act in parallel to MST1/2 to activate LATS1/2 in the hippo pathway. Nat Commun. (2015) 6:8357. doi: 10.1038/ncomms9357
25. Zheng Y, Wang W, Liu B, Deng H, Uster E, Pan D. Identification of happyhour/MAP4K as alternative Hpo/Mst-like kinases in the hippo kinase cascade. Dev Cell. (2015) 34:642–55. doi: 10.1016/j.devcel.2015.08.014
26. Lim S, Hermance N, Mudianto T, Mustaly HM, Mauricio IPM, Vittoria MA, et al. Identification of the kinase STK25 as an upstream activator of LATS signaling. Nat Commun. (2019) 10:1547. doi: 10.1038/s41467-019-09597-w
27. Yin F, Yu J, Zheng Y, Chen Q, Zhang N, Pan D. Spatial organization of hippo signaling at the plasma membrane mediated by the tumor suppressor Merlin/NF2. Cell. (2013) 154:1342–55. doi: 10.1016/j.cell.2013.08.025
28. Gill MK, Christova T, Zhang YY, Gregorieff A, Zhang L, Narimatsu M, et al. A feed forward loop enforces YAP/TAZ signaling during tumorigenesis. Nat Commun. (2018) 9:3510. doi: 10.1038/s41467-018-05939-2
29. Arash EH, Shiban A, Song S, Attisano L. MARK4 inhibits Hippo signaling to promote proliferation and migration of breast cancer cells. EMBO Rep. (2017) 18:420–36. doi: 10.15252/embr.201642455
30. Zhao B, Li L, Lu Q, Wang LH, Liu CY, Lei Q, et al. Angiomotin is a novel hippo pathway component that inhibits YAP oncoprotein. Genes Dev. (2011) 25:51–63. doi: 10.1101/gad.2000111
31. Michaloglou C, Lehmann W, Martin T, Delaunay C, Hueber A, Barys L, et al. The tyrosine phosphatase PTPN14 is a negative regulator of YAP activity. PLoS ONE. (2013) 8:e61916. doi: 10.1371/journal.pone.0061916
32. Azzolin L, Panciera T, Soligo S, Enzo E, Bicciato S, Dupont S, et al. YAP/TAZ incorporation in the beta-catenin destruction complex orchestrates the Wnt response. Cell. (2014) 158:157–70. doi: 10.1016/j.cell.2014.06.013
33. Mo JS, Meng Z, Kim YC, Park HW, Hansen CG, Kim S, et al. Cellular energy stress induces AMPK-mediated regulation of YAP and the hippo pathway. Nat Cell Biol. (2015) 17:500–10. doi: 10.1038/ncb3111
34. Wang W, Xiao ZD, Li X, Aziz KE, Gan B, Johnson RL, et al. AMPK modulates Hippo pathway activity to regulate energy homeostasis. Nat Cell Biol. (2015) 17:490–9. doi: 10.1038/ncb3113
35. Yang S, Zhang L, Liu M, Chong R, Ding SJ, Chen Y, et al. CDK1 phosphorylation of YAP promotes mitotic defects and cell motility and is essential for neoplastic transformation. Cancer Res. (2013) 73:6722–33. doi: 10.1158/0008-5472.CAN-13-2049
36. Hong AW, Meng Z, Yuan HX, Plouffe SW, Moon S, Kim W, et al. Osmotic stress-induced phosphorylation by NLK at Ser128 activates YAP. EMBO Rep. (2017) 18:72–86. doi: 10.15252/embr.201642681
37. Chang SS, Yamaguchi H, Xia W, Lim SO, Khotskaya Y, Wu y, et al. Aurora A kinase activates YAP signaling in triple-negative breast cancer. Oncogene. (2017) 36:1265–75. doi: 10.1038/onc.2016.292
38. Kedan A, Verma N, Saroha A, Shreberk-Shaked M, Muller AK, Nair NU, et al. PYK2 negatively regulates the hippo pathway in TNBC by stabilizing TAZ protein. Cell Death Dis. (2018) 9:985. doi: 10.1038/s41419-018-1005-z
39. Azad T, Nouri K, Janse van Rensburg HJ, Maritan SM, Wu L, Hao Y, et al. A gain-of-functional screen identifies the hippo pathway as a central mediator of receptor tyrosine kinases during tumorigenesis. Oncogene. (2019) 39:334–55. doi: 10.1038/s41388-019-0988-y
40. Zanconato F, Forcato M, Battilana G, Azzolin L, Quaranta E, Bodega B, et al. Genome-wide association between YAP/TAZ/TEAD and AP-1 at enhancers drives oncogenic growth. Nat Cell Biol. (2015) 17:1218–27. doi: 10.1038/ncb3216
41. Stein C, Bardet AF, Roma G, Bergling S, Clay I, Ruchti A, et al. YAP1 Exerts its transcriptional control via TEAD-mediated activation of enhancers. PLoS Genet. (2015) 11:e1005465. doi: 10.1371/journal.pgen.1005465
42. Jiao S, Wang H, Shi Z, Dong A, Zhang W, Song X, et al. A peptide mimicking VGLL4 function acts as a YAP antagonist therapy against gastric cancer. Cancer Cell. (2014) 25:166–80. doi: 10.1016/j.ccr.2014.01.010
43. Diamantopoulou Z, White G, Fadlullah MZH, Dreger M, Pickering K, Maltas J, et al. TIAM1 antagonizes TAZ/YAP both in the destruction complex in the cytoplasm and in the nucleus to inhibit invasion of intestinal epithelial cells. Cancer Cell. (2017) 31:621–34.e6. doi: 10.1016/j.ccell.2017.03.007
44. Lin KC, Moroishi T, Meng Z, Jeong HS, Plouffe SW, Sekido Y, et al. Regulation of hippo pathway transcription factor TEAD by p38 MAPK-induced cytoplasmic translocation. Nat Cell Biol. (2017) 19:996–1002. doi: 10.1038/ncb3581
45. Totaro A, Panciera T, Piccolo S. YAP/TAZ upstream signals and downstream responses. Nat Cell Biol. (2018) 20:888–99. doi: 10.1038/s41556-018-0142-z
46. Moroishi T, Hansen CG, Guan KL. The emerging roles of YAP and TAZ in cancer. Nat Rev Cancer. (2015) 15:73–9. doi: 10.1038/nrc3876
47. Lo Sardo F, Strano S, Blandino G. YAP and TAZ in lung cancer: oncogenic role and clinical targeting. Cancers. (2018) 10:137. doi: 10.3390/cancers10050137
48. Noguchi S, Saito A, Horie M, Mikami Y, Suzuki HI, Morishita Y, et al. An integrative analysis of the tumorigenic role of TAZ in human non-small cell lung cancer. Clin Cancer Res. (2014) 20:4660–72. doi: 10.1158/1078-0432.CCR-13-3328
49. Cordenonsi M, Zanconato F, Azzolin L, Forcato M, Rosato A, Frasson C, et al. The hippo transducer TAZ confers cancer stem cell-related traits on breast cancer cells. Cell. (2011) 147:759–72. doi: 10.1016/j.cell.2011.09.048
50. Bonilla X, Parmentier L, King B, Bezrukov F, Kaya G, Zoete V, et al. Genomic analysis identifies new drivers and progression pathways in skin basal cell carcinoma. Nat Genet. (2016) 48:398–406. doi: 10.1038/ng.3525
51. Miyanaga A, Masuda M, Tsuta K, Kawasaki K, Nakamura Y, Sakuma T, et al. Hippo pathway gene mutations in malignant mesothelioma: revealed by RNA and targeted exon sequencing. J Thorac Oncol. (2015) 10:844–51. doi: 10.1097/JTO.0000000000000493
52. Saadeldin MK, Shawer H, Mostafa A, Kassem NM, Amleh A, Siam R. New genetic variants of LATS1 detected in urinary bladder and colon cancer. Front Genet. (2014) 5:425. doi: 10.3389/fgene.2014.00425
53. Zhou D, Conrad C, Xia F, Park JS, Payer B, Yin Y, et al. Mst1 and Mst2 maintain hepatocyte quiescence and suppress hepatocellular carcinoma development through inactivation of the Yap1 oncogene. Cancer Cell. (2009) 16:425–38. doi: 10.1016/j.ccr.2009.09.026
54. Lu L, Li Y, Kim SM, Bossuyt W, Liu P, Qiu Q, et al. Hippo signaling is a potent in vivo growth and tumor suppressor pathway in the mammalian liver. Proc Natl Acad Sci USA. (2010) 107:1437–42. doi: 10.1073/pnas.0911427107
55. Dong J, Feldmann G, Huang J, Wu S, Zhang N, Comerford S, et al. Elucidation of a universal size-control mechanism in Drosophila and mammals. Cell. (2007) 130:1120–33. doi: 10.1016/j.cell.2007.07.019
56. Zhang W, Gao Y, Li F, Tong X, Ren Y, Han X, et al. YAP promotes malignant progression of Lkb1-deficient lung adenocarcinoma through downstream regulation of survivin. Cancer Res. (2015) 75:4450–7. doi: 10.1158/0008-5472.CAN-14-3396
57. Chen Q, Zhang N, Gray RS, Li H, Ewald AJ, Zahnow CA, et al. A temporal requirement for hippo signaling in mammary gland differentiation, growth, and tumorigenesis. Genes Dev. (2014) 28:432–7. doi: 10.1101/gad.233676.113
58. Cai J, Maitra A, Anders RA, Taketo MM, Pan D. β-Catenin destruction complex-independent regulation of Hippo-YAP signaling by APC in intestinal tumorigenesis. Genes Dev. (2015) 29:1493–506. doi: 10.1101/gad.264515.115
59. Zhang W, Nandakumar N, Shi Y, Manzano M, Smith A, Graham G, et al. Downstream of mutant KRAS, the transcription regulator YAP is essential for neoplastic progression to pancreatic ductal adenocarcinoma. Sci Signal. (2014) 7:ra42. doi: 10.1126/scisignal.2005049
60. Kapoor A, Yao W, Ying H, Hua S, Liewen A, Wang Q, et al. Yap1 activation enables bypass of oncogenic Kras addiction in pancreatic cancer. Cell. (2014) 158:185–97. doi: 10.1016/j.cell.2014.06.003
61. Kim W, Cho YS, Wang X, Park O, Ma X, Kim H, et al. Hippo signaling is intrinsically regulated during cell cycle progression by APC/C(Cdh1). Proc Natl Acad Sci USA. (2019) 116:9423–32. doi: 10.1073/pnas.1821370116
62. Yang N, Morrison CD, Liu P, Miecznikowski J, Bshara W, Han S, et al. TAZ induces growth factor-independent proliferation through activation of EGFR ligand amphiregulin. Cell Cycle. (2012) 11:2922–30. doi: 10.4161/cc.21386
63. Matsushita A, Sato T, Mukai S, Fujishita T, Mishiro-Sato E, Okuda M, et al. TAZ activation by hippo pathway dysregulation induces cytokine gene expression and promotes mesothelial cell transformation. Oncogene. (2019) 38:1966–78. doi: 10.1038/s41388-018-0417-7
64. Pan Z, Tian Y, Cao C, Niu G. The emerging role of YAP/TAZ in tumor immunity. Mol Cancer Res. (2019) 17:1777–86. doi: 10.1158/1541-7786.MCR-19-0375
65. Guo X, Zhao Y, Yan H, Yang Y, Shen S, Dai X, et al. Single tumor-initiating cells evade immune clearance by recruiting type II macrophages. Genes Dev. (2017) 31:247–59. doi: 10.1101/gad.294348.116
66. Wang G, Lu X, Dey P, Deng P, Wu CC, Jiang S, et al. Targeting YAP-dependent MDSC infiltration impairs tumor progression. Cancer Discov. (2016) 6:80–95. doi: 10.1158/2159-8290.CD-15-0224
67. Janse van Rensburg HJ, Azad T, Ling M, Hao Y, Snetsinger B, Khanal P, et al. The hippo pathway component TAZ promotes immune evasion in human cancer through PD-L1. Cancer Res. (2018) 78:1457–70. doi: 10.1158/0008-5472.CAN-17-3139
68. Kim MH, Kim CG, Kim SK, Shin SJ, Choe EA, Park SH, et al. YAP-induced PD-L1 expression drives immune evasion in BRAFi-resistant melanoma. Cancer Immunol Res. (2018) 6:255–66. doi: 10.1158/2326-6066.CIR-17-0320
69. Basu-Roy U, Bayin NS, Rattanakorn K, Han E, Placantonakis DG, Mansukhani A, et al. Sox2 antagonizes the hippo pathway to maintain stemness in cancer cells. Nat Commun. (2015) 6:6411. doi: 10.1038/ncomms7411
70. Kim T, Yang SJ, Hwang D, Song J, Kim M, Kyum Kim S, et al. Lim: a basal-like breast cancer-specific role for SRF-IL6 in YAP-induced cancer stemness. Nat Commun. (2015) 6:10186. doi: 10.1038/ncomms10186
71. Wang L, Zhang Z, Yu X, Huang X, Liu Z, Chai Y, et al. Unbalanced YAP-SOX9 circuit drives stemness and malignant progression in esophageal squamous cell carcinoma. Oncogene. (2019) 38:2042–55. doi: 10.1038/s41388-018-0476-9
72. Song S, Ajani JA, Honjo S, Maru DM, Chen Q, Scott AW, et al. Johnson: hippo coactivator YAP1 upregulates SOX9 and endows esophageal cancer cells with stem-like properties. Cancer Res. (2014) 74:4170–82. doi: 10.1158/0008-5472.CAN-13-3569
73. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
74. Chaffer CL, Weinberg RA. A perspective on cancer cell metastasis. Science. (2011) 331:1559–64. doi: 10.1126/science.1203543
75. Xie M, Zhang L, He CS, Hou JH, Lin SX, Hu ZH, et al. Zhao: prognostic significance of TAZ expression in resected non-small cell lung cancer. J Thorac Oncol. (2012) 7:799–807. doi: 10.1097/JTO.0b013e318248240b
76. Bartucci M, Dattilo R, Moriconi C, Pagliuca A, Mottolese M, Federici G, et al. TAZ is required for metastatic activity and chemoresistance of breast cancer stem cells. Oncogene. (2015) 34:681–90. doi: 10.1038/onc.2014.5
77. Wang L, Shi S, Guo Z, Zhang X, Han S, Yang A, et al. Overexpression of YAP and TAZ is an independent predictor of prognosis in colorectal cancer and related to the proliferation and metastasis of colon cancer cells. PLoS ONE. (2013) 8:e65539. doi: 10.1371/journal.pone.0065539
78. Guo Y, Pan Q, Zhang J, Xu X, Liu X, Wang Q, et al. Functional and clinical evidence that TAZ is a candidate oncogene in hepatocellular carcinoma. J Cell Biochem. (2015) 116:2465–75. doi: 10.1002/jcb.25117
79. Yang S, Zhang L, Purohit V, Shukla SK, Chen X, Yu F, et al. Active YAP promotes pancreatic cancer cell motility, invasion and tumorigenesis in a mitotic phosphorylation-dependent manner through LPAR3. Oncotarget. (2015) 6:36019–31. doi: 10.18632/oncotarget.5935
80. Hu X, Xin Y, Xiao Y, Zhao J. Overexpression of YAP1 is correlated with progression, metastasis and poor prognosis in patients with gastric carcinoma. Pathol Oncol Res. (2014) 20:805–11. doi: 10.1007/s12253-014-9757-y
81. Takahashi Y, Miyoshi Y, Takahata C, Irahara N, Taguchi T, Tamaki Y, et al. Down-regulation of LATS1 and LATS2 mRNA expression by promoter hypermethylation and its association with biologically aggressive phenotype in human breast cancers. Clin Cancer Res. (2005) 11:1380–5. doi: 10.1158/1078-0432.CCR-04-1773
82. Zhang J, Wang G, Chu SJ, Zhu JS, Zhang R, Lu WW, et al. Loss of large tumor suppressor 1 promotes growth and metastasis of gastric cancer cells through upregulation of the YAP signaling. Oncotarget. (2016) 7:16180–93. doi: 10.18632/oncotarget.7568
83. Yamashita S, Yamamoto H, Mimori K, Nishida N, Takahashi H, Haraguchi N, et al. MicroRNA-372 is associated with poor prognosis in colorectal cancer. Oncology. (2012) 82:205–12. doi: 10.1159/000336809
84. Zhao B, Li L, Wang L, Wang CY, Yu J, Guan KL. Cell detachment activates the hippo pathway via cytoskeleton reorganization to induce anoikis. Genes Dev. (2012) 26:54–68. doi: 10.1101/gad.173435.111
85. Lamar JM, Stern P, Liu H, Schindler JW, Jiang ZG, Hynes RO. The hippo pathway target, YAP, promotes metastasis through its TEAD-interaction domain. Proc Natl Acad Sci USA. (2012) 109:E2441–50. doi: 10.1073/pnas.1212021109
86. Gu JJ, Rouse C, Xu X, Wang J, Onaitis MW, Pendergast AM. Inactivation of ABL kinases suppresses non-small cell lung cancer metastasis. JCI Insight. (2016) 1:e89647. doi: 10.1172/jci.insight.89647
87. Wang J, Rouse C, Jasper JS, Pendergast AM. ABL kinases promote breast cancer osteolytic metastasis by modulating tumor-bone interactions through TAZ and STAT5 signaling. Sci Signal. (2016) 9:ra12. doi: 10.1126/scisignal.aad3210
88. Zhou Z, Zhu JS, Gao CP, Li LP, Zhou C, Wang H, et al. siRNA targeting YAP gene inhibits gastric carcinoma growth and tumor metastasis in SCID mice. Oncol Lett. (2016) 11:2806–14. doi: 10.3892/ol.2016.4319
89. Xu MZ, Chan SW, Liu AM, Wong KF, Fan ST, Chen J, et al. AXL receptor kinase is a mediator of YAP-dependent oncogenic functions in hepatocellular carcinoma. Oncogene. (2011) 30:1229–40. doi: 10.1038/onc.2010.504
90. Chan SW, Lim CJ, Guo K, Ng CP, Lee I, Hunziker W, et al. A role for TAZ in migration, invasion, and tumorigenesis of breast cancer cells. Cancer Res. (2008) 68:2592–8. doi: 10.1158/0008-5472.CAN-07-2696
91. Zhang L, Yang S, Chen X, Stauffer S, Yu F, Lele SM, et al. The hippo pathway effector YAP regulates motility, invasion, and castration-resistant growth of prostate cancer cells. Mol Cell Biol. (2015) 35:1350–62. doi: 10.1128/MCB.00102-15
92. Wang Q, Xu Z, An Q, Jiang D, Wang L, Liang B, et al. TAZ promotes epithelial to mesenchymal transition via the upregulation of connective tissue growth factor expression in neuroblastoma cells. Mol Med Rep. (2015) 11:982–8. doi: 10.3892/mmr.2014.2818
93. Xiao H, Jiang N, Zhou B, Liu Q, Du C. TAZ regulates cell proliferation and epithelial-mesenchymal transition of human hepatocellular carcinoma. Cancer Sci. (2015) 106:151–9. doi: 10.1111/cas.12587
94. Mason DE, Collins JM, Dawahare JH, Nguyen TD, Lin Y, Voytik-Harbin SL, et al. YAP and TAZ limit cytoskeletal and focal adhesion maturation to enable persistent cell motility. J Cell Biol. (2019) 218:1369–89. doi: 10.1083/jcb.201806065
95. Hsu YL, Hung JY, Chou SH, Huang MS, Tsai MJ, Lin YS, et al. Angiomotin decreases lung cancer progression by sequestering oncogenic YAP/TAZ and decreasing Cyr61 expression. Oncogene. (2015) 34:4056–68. doi: 10.1038/onc.2014.333
96. Wang Z, Wu Y, Wang H, Zhang Y, Mei L, Fang X, et al. Interplay of mevalonate and hippo pathways regulates RHAMM transcription via YAP to modulate breast cancer cell motility. Proc Natl Acad Sci USA. (2014) 111:E89–98. doi: 10.1073/pnas.1319190110
97. Shibue T, Weinberg RA. EMT, CSCs, and drug resistance: the mechanistic link and clinical implications. Nat Rev Clin Oncol. (2017) 14:611–29. doi: 10.1038/nrclinonc.2017.44
98. Ye X, Weinberg RA. Epithelial-mesenchymal plasticity: a central regulator of cancer progression. Trends Cell Biol. (2015) 25:675–86. doi: 10.1016/j.tcb.2015.07.012
99. Dongre A, Weinberg RA. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat Rev Mol Cell Biol. (2019) 20:69–84. doi: 10.1038/s41580-018-0080-4
100. Overholtzer M, Zhang J, Smolen GA, Muir B, Li W, Sgroi DC, et al. Transforming properties of YAP, a candidate oncogene on the chromosome 11q22 amplicon. Proc Natl Acad Sci USA. (2006) 103:12405–10. doi: 10.1073/pnas.0605579103
101. Lei QY, Zhang H, Zhao B, Zha ZY, Bai F, Pei XH, et al. TAZ promotes cell proliferation and epithelial-mesenchymal transition and is inhibited by the hippo pathway. Mol Cell Biol. (2008) 28:2426–36. doi: 10.1128/MCB.01874-07
102. Xie D, Cui J, Xia T, Jia Z, Wang L, Wei W, et al. Hippo transducer TAZ promotes epithelial mesenchymal transition and supports pancreatic cancer progression. Oncotarget. (2015) 6:35949–63. doi: 10.18632/oncotarget.5772
103. Pan Z, Tian Y, Zhang B, Zhang X, Shi H, Liang Z, et al. YAP signaling in gastric cancer-derived mesenchymal stem cells is critical for its promoting role in cancer progression. Int J Oncol. (2017) 51:1055–66. doi: 10.3892/ijo.2017.4101
104. Asiedu MK, Beauchamp-Perez FD, Ingle JN, Behrens MD, Radisky DC, Knutson KL. AXL induces epithelial-to-mesenchymal transition and regulates the function of breast cancer stem cells. Oncogene. (2014) 33:1316–24. doi: 10.1038/onc.2013.57
105. Liu BC, Zhang JD, Zhang XL, Wu GQ, Li MX. Role of connective tissue growth factor (CTGF) module 4 in regulating epithelial mesenchymal transition (EMT) in HK-2 cells. Clin Chim Acta. (2006) 373:144–50. doi: 10.1016/j.cca.2006.05.029
106. Hou CH, Lin FL, Hou SM, Liu JF. Cyr61 promotes epithelial-mesenchymal transition and tumor metastasis of osteosarcoma by Raf-1/MEK/ERK/Elk-1/TWIST-1 signaling pathway. Mol Cancer. (2014) 13:236. doi: 10.1186/1476-4598-13-236
107. Zhang H, von Gise A, Liu Q, Hu T, Tian X, He L, et al. Yap1 is required for endothelial to mesenchymal transition of the atrioventricular cushion. J Biol Chem. (2014) 289:18681–92. doi: 10.1074/jbc.M114.554584
108. Lehmann W, Mossmann D, Kleemann J, Mock K, Meisinger C, Brummer T, et al. ZEB1 turns into a transcriptional activator by interacting with YAP1 in aggressive cancer types. Nat Commun. (2016) 7:10498. doi: 10.1038/ncomms10498
109. Paoli P, Giannoni E, Chiarugi P. Anoikis molecular pathways and its role in cancer progression. Biochim Biophys Acta. (2013) 1833:3481–98. doi: 10.1016/j.bbamcr.2013.06.026
110. Haemmerle M, Taylor ML, Gutschner T, Pradeep S, Cho MS, Sheng J, et al. Platelets reduce anoikis and promote metastasis by activating YAP1 signaling. Nat Commun. (2017) 8:310. doi: 10.1038/s41467-017-00411-z
111. Lee HJ, Diaz MF, Price KM, Ozuna JA, Zhang S, Sevick-Muraca EM, et al. Fluid shear stress activates YAP1 to promote cancer cell motility. Nat Commun. (2017) 8:14122. doi: 10.1038/ncomms14122
112. Lee HJ, Ewere A, Diaz MF, Wenzel PL. TAZ responds to fluid shear stress to regulate the cell cycle. Cell Cycle. (2018) 17:147–53. doi: 10.1080/15384101.2017.1404209
113. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the warburg effect: the metabolic requirements of cell proliferation. Science. (2009) 324:1029–33. doi: 10.1126/science.1160809
114. DeRan M, Yang J, Shen CH, Peters EC, Fitamant J, Chan P, et al. Energy stress regulates hippo-YAP signaling involving AMPK-mediated regulation of angiomotin-like 1 protein. Cell Rep. (2014) 9:495–503. doi: 10.1016/j.celrep.2014.09.036
115. Enzo E, Santinon G, Pocaterra A, Aragona M, Bresolin S, Forcato M, et al. Aerobic glycolysis tunes YAP/TAZ transcriptional activity. EMBO J. (2015) 34:1349–70. doi: 10.15252/embj.201490379
116. Hardie DG, Carling D, Carlson M. The AMP-activated/SNF1 protein kinase subfamily: metabolic sensors of the eukaryotic cell? Annu Rev Biochem. (1998) 67:821–55. doi: 10.1146/annurev.biochem.67.1.821
117. Hanover JA, Krause MW, Love DC. Bittersweet memories: linking metabolism to epigenetics through O-GlcNAcylation. Nat Rev Mol Cell Biol. (2012) 13:312–21. doi: 10.1038/nrm3334
118. Butkinaree C, Park K, Hart GW. O-linked beta-N-acetylglucosamine (O-GlcNAc): extensive crosstalk with phosphorylation to regulate signaling and transcription in response to nutrients and stress. Biochim Biophys Acta. (2010) 1800:96–106. doi: 10.1016/j.bbagen.2009.07.018
119. Singh JP, Zhang K, Wu J, Yang X. O-GlcNAc signaling in cancer metabolism and epigenetics. Cancer Lett. (2015) 356:244–50. doi: 10.1016/j.canlet.2014.04.014
120. Zhang X, Qiao Y, Wu Q, Chen Y, Zou S, Liu X, et al. The essential role of YAP O-GlcNAcylation in high-glucose-stimulated liver tumorigenesis. Nat Commun. (2017) 8:15280. doi: 10.1038/ncomms15280
121. Peng C, Zhu Y, Zhang W, Liao Q, Chen Y, Zhao X, et al. Regulation of the hippo-YAP pathway by glucose sensor O-GlcNAcylation. Mol Cell. (2017) 68:591–604.e5. doi: 10.1016/j.molcel.2017.10.010
122. Liu Y, Lu Z, Shi Y, Sun F. AMOT is required for YAP function in high glucose induced liver malignancy. Biochem Biophys Res Commun. (2018) 495:1555–61. doi: 10.1016/j.bbrc.2017.12.010
123. Nokin MJ, Durieux F, Peixoto P, Chiavarina B, Peulen O, Blomme A, et al. Methylglyoxal, a glycolysis side-product, induces Hsp90 glycation and YAP-mediated tumor growth and metastasis. Elife. (2016) 5:e19375. doi: 10.7554/eLife.19375.034
124. Zhang Z, Deng X, Liu Y, Liu Y, Sun L, Chen F. PKM2, function and expression and regulation. Cell Biosci. (2019) 9:52. doi: 10.1186/s13578-019-0317-8
125. Zhou Z, Li M, Zhang L, Zhao H, Sahin O, Chen J, et al. Oncogenic kinase-induced PKM2 tyrosine 105 phosphorylation converts nononcogenic PKM2 to a tumor promoter and induces cancer stem-like cells. Cancer Res. (2018) 78:2248–61. doi: 10.1158/0008-5472.CAN-17-2726
126. Kuo CC, Ling HH, Chiang MC, Chung CH, Lee WY, Chu CY, et al. Metastatic colorectal cancer rewrites metabolic program through a Glut3-YAP-dependent signaling circuit. Theranostics. (2019) 9:2526–40. doi: 10.7150/thno.32915
127. White SM, Avantaggiati ML, Nemazanyy I, Di Poto C, Yang Y, Pende M, et al. YAP/TAZ inhibition induces metabolic and signaling rewiring resulting in targetable vulnerabilities in NF2-deficient tumor cells. Dev Cell. (2019) 49:425–43.e9. doi: 10.1016/j.devcel.2019.04.014
128. Murakami S, Nemazanyy I, White SM, Chen H, Nguyen CDK, Graham GT, et al. A Yap-Myc-Sox2-p53 regulatory network dictates metabolic homeostasis and differentiation in kras-driven pancreatic ductal adenocarcinomas. Dev Cell. (2019) 51:113–28.e9. doi: 10.1016/j.devcel.2019.07.022
129. Chen R, Zhu S, Fan XG, Wang H, Lotze MT, Zeh HJ, et al. High mobility group protein B1 controls liver cancer initiation through yes-associated protein -dependent aerobic glycolysis. Hepatology. (2018) 67:1823–41. doi: 10.1002/hep.29663
130. Jia Y, Li HY, Wang J, Wang Y, Zhang P, Ma N, Mo SJ. Phosphorylation of 14-3-3zeta links YAP transcriptional activation to hypoxic glycolysis for tumorigenesis. Oncogenesis. (2019) 8:31. doi: 10.1038/s41389-019-0143-1
131. Gao Y, Yang Y, Yuan F, Huang J, Xu W, Mao B, et al. TNFalpha-YAP/p65-HK2 axis mediates breast cancer cell migration. Oncogenesis. (2017) 6:e383. doi: 10.1038/oncsis.2017.83
132. Shen Y, Zhao S, Wang S, Pan X, Zhang Y, Xu J, et al. S1P/S1PR3 axis promotes aerobic glycolysis by YAP/c-MYC/PGAM1 axis in osteosarcoma. EBioMedicine. (2019) 40:210–23. doi: 10.1016/j.ebiom.2018.12.038
133. Zheng X, Han H, Liu GP, Ma YX, Pan RL, Sang LJ, et al. LncRNA wires up hippo and Hedgehog signaling to reprogramme glucose metabolism. EMBO J. (2017) 36:3325–35. doi: 10.15252/embj.201797609
134. Santos CR, Schulze A. Lipid metabolism in cancer. FEBS J. (2012) 279:2610–23. doi: 10.1111/j.1742-4658.2012.08644.x
135. Rohrig F, Schulze A. The multifaceted roles of fatty acid synthesis in cancer. Nat Rev Cancer. (2016) 16:732–49. doi: 10.1038/nrc.2016.89
136. Noto A, De Vitis C, Pisanu ME, Roscilli G, Ricci G, Catizone A, et al. Stearoyl-CoA-desaturase 1 regulates lung cancer stemness via stabilization and nuclear localization of YAP/TAZ. Oncogene. (2017) 36:4573–84. doi: 10.1038/onc.2017.75
137. Yuan L, Mao Y, Luo W, Wu W, Xu H, Wang XL YH. Palmitic acid dysregulates the hippo-YAP pathway and inhibits angiogenesis by inducing mitochondrial damage and activating the cytosolic DNA sensor cGAS-STING-IRF3 signaling mechanism. J Biol Chem. (2017) 292:15002–15. doi: 10.1074/jbc.M117.804005
138. Deng Y, Matsui Y, Pan W, Li Q, Lai ZC. Yap1 plays a protective role in suppressing free fatty acid-induced apoptosis and promoting beta-cell survival. Protein Cell. (2016) 7:362–72. doi: 10.1007/s13238-016-0258-5
139. Chan P, Han X, Zheng B, DeRan M, Yu J, Jarugumilli GK, et al. Autopalmitoylation of TEAD proteins regulates transcriptional output of the Hippo pathway. Nat Chem Biol. (2016) 12:282–9. doi: 10.1038/nchembio.2036
140. Kim NG, Gumbiner BM. Cell contact and Nf2/Merlin-dependent regulation of TEAD palmitoylation and activity. Proc Natl Acad Sci USA. (2019) 116:9877–82. doi: 10.1073/pnas.1819400116
141. Noland CL, Gierke S, Schnier PD, Murray J, Sandoval WN, Sagolla M, et al. Cunningham: palmitoylation of TEAD transcription factors is required for their stability and function in hippo pathway signaling. Structure. (2016) 24:179–86. doi: 10.1016/j.str.2015.11.005
142. Palsuledesai CC, Distefano MD. Protein prenylation: enzymes, therapeutics, and biotechnology applications. ACS Chem Biol. (2015) 10:51–62. doi: 10.1021/cb500791f
143. Mi W, Lin Q, Childress C, Sudol M, Robishaw J, Berlot CH, et al. Geranylgeranylation signals to the hippo pathway for breast cancer cell proliferation and migration. Oncogene. (2015) 34:3095–106. doi: 10.1038/onc.2014.251
144. Sorrentino G, Ruggeri N, Specchia V, Cordenonsi M, Mano M, Dupont S, et al. Metabolic control of YAP and TAZ by the mevalonate pathway. Nat Cell Biol. (2014) 16:357–66. doi: 10.1038/ncb2936
145. Tian Y, Yang B, Qiu W, Hao Y, Zhang Z, Yang B, et al. ER-residential nogo-B accelerates NAFLD-associated HCC mediated by metabolic reprogramming of oxLDL lipophagy. Nat Commun. (2019) 10:3391. doi: 10.1038/s41467-019-11274-x
146. Horton JD. Sterol regulatory element-binding proteins: transcriptional activators of lipid synthesis. Biochem Soc Trans. (2002) 30:1091–5. doi: 10.1042/bst0301091
147. Shu Z, Gao Y, Zhang G, Zhou Y, Cao J, Wan D, et al. A functional interaction between hippo-YAP signalling and SREBPs mediates hepatic steatosis in diabetic mice. J Cell Mol Med. (2019) 23:3616–28. doi: 10.1111/jcmm.14262
148. Ma Y, Temkin SM, Hawkridge AM, Guo C, Wang W, Wang XY, et al. Fatty acid oxidation: an emerging facet of metabolic transformation in cancer. Cancer Lett. (2018) 435:92–100. doi: 10.1016/j.canlet.2018.08.006
149. Lee CK, Jeong SH, Jang C, Bae H, Kim YH, Park I, et al. Tumor metastasis to lymph nodes requires YAP-dependent metabolic adaptation. Science. (2019) 363:644–49. doi: 10.1126/science.aav0173
150. Altman BJ, Stine ZE, Dang CV. From krebs to clinic: glutamine metabolism to cancer therapy. Nat Rev Cancer. (2016) 16:619–34. doi: 10.1038/nrc.2016.114
151. Mates JM, Di Paola FJ, Campos-Sandoval JA, Mazurek S, Marquez J. Therapeutic targeting of glutaminolysis as an essential strategy to combat cancer. Semin Cell Dev Biol. (2019) 98:34–43. doi: 10.1016/j.semcdb.2019.05.012
152. Park YY, Sohn BH, Johnson RL, Kang MH, Kim SB, Shim JJ, et al. Yes-associated protein 1 and transcriptional coactivator with PDZ-binding motif activate the mammalian target of rapamycin complex 1 pathway by regulating amino acid transporters in hepatocellular carcinoma. Hepatology. (2016) 63:159–72. doi: 10.1002/hep.28223
153. Cox AG, Hwang KL, Brown KK, Evason K, Beltz S, Tsomides A, et al. Yap reprograms glutamine metabolism to increase nucleotide biosynthesis and enable liver growth. Nat Cell Biol. (2016) 18:886–96. doi: 10.1038/ncb3389
154. Bertero T, Oldham WM, Cottrill KA, Pisano S, Vanderpool RR, Yu Q, et al. Vascular stiffness mechanoactivates YAP/TAZ-dependent glutaminolysis to drive pulmonary hypertension. J Clin Invest. (2016) 126:3313–35. doi: 10.1172/JCI86387
155. Edwards DN, Ngwa VM, Wang S, Shiuan E, Brantley-Sieders DM, Kim LC, et al. The receptor tyrosine kinase EphA2 promotes glutamine metabolism in tumors by activating the transcriptional coactivators YAP and TAZ. Sci Signal. (2017) 10:eaan4667. doi: 10.1126/scisignal.aan4667
156. Yang CS, Stampouloglou E, Kingston NM, Zhang L, Monti S, Varelas X. Glutamine-utilizing transaminases are a metabolic vulnerability of TAZ/YAP-activated cancer cells. EMBO Rep. (2018) 19:e43577. doi: 10.15252/embr.201643577
157. Bertero T, Oldham WM, Grasset EM, Bourget I, Boulter E, Pisano S, et al. Tumor-stroma mechanics coordinate amino acid availability to sustain tumor growth and malignancy. Cell Metab. (2019) 29:124–40.e10. doi: 10.1016/j.cmet.2018.09.012
158. Hansen CG, Ng YL, Lam WL, Plouffe SW, Guan KL. The hippo pathway effectors YAP and TAZ promote cell growth by modulating amino acid signaling to mTORC1. Cell Res. (2015) 25:1299–313. doi: 10.1038/cr.2015.140
159. Wu Q, Li J, Sun S, Chen X, Zhang H, Li B, et al. YAP/TAZ-mediated activation of serine metabolism and methylation regulation is critical for LKB1-deficient breast cancer progression. Biosci Rep. (2017) 37:BSR20171072. doi: 10.1042/BSR20171072
160. Bar-Peled L, Sabatini DM. Regulation of mTORC1 by amino acids. Trends Cell Biol. (2014) 24:400–6. doi: 10.1016/j.tcb.2014.03.003
161. Tumaneng K, Schlegelmilch K, Russell RC, Yimlamai D, Basnet H, Mahadevan N, et al. YAP mediates crosstalk between the hippo and PI(3)K-TOR pathways by suppressing PTEN via miR-29. Nat Cell Biol. (2012) 14:1322–9. doi: 10.1038/ncb2615
162. Artinian N, Cloninger C, Holmes B, Benavides-Serrato A, Bashir T, Gera J. Phosphorylation of the hippo pathway component AMOTL2 by the mTORC2 kinase promotes YAP signaling, resulting in enhanced glioblastoma growth and invasiveness. J Biol Chem. (2015) 290:19387–401. doi: 10.1074/jbc.M115.656587
163. Liang N, Zhang C, Dill P, Panasyuk G, Pion D, Koka V, et al. Regulation of YAP by mTOR and autophagy reveals a therapeutic target of tuberous sclerosis complex. J Exp Med. (2014) 211:2249–63. doi: 10.1084/jem.20140341
164. Collak FK, Yagiz K, Luthringer DJ, Erkaya B, Cinar B. Threonine-120 phosphorylation regulated by phosphoinositide-3-kinase/Akt and mammalian target of rapamycin pathway signaling limits the antitumor activity of mammalian sterile 20-like kinase 1. J Biol Chem. (2012) 287:23698–709. doi: 10.1074/jbc.M112.358713
165. Schild T, Low V, Blenis J, Gomes AP. Unique metabolic adaptations dictate distal organ-specific metastatic colonization. Cancer Cell. (2018) 33:347–54. doi: 10.1016/j.ccell.2018.02.001
166. Lu X, Bennet B, Mu E, Rabinowitz J, Kang Y. Metabolomic changes accompanying transformation and acquisition of metastatic potential in a syngeneic mouse mammary tumor model. J Biol Chem. (2010) 285:9317–21. doi: 10.1074/jbc.C110.104448
167. Sottnik JL, Lori JC, Rose BJ, Thamm DH. Glycolysis inhibition by 2-deoxy-D-glucose reverts the metastatic phenotype in vitro and in vivo. Clin Exp Metastasis. (2011) 28:865–75. doi: 10.1007/s10585-011-9417-5
168. Liu M, Quek LE, Sultani G, Turner N. Epithelial-mesenchymal transition induction is associated with augmented glucose uptake and lactate production in pancreatic ductal adenocarcinoma. Cancer Metab. (2016) 4:19. doi: 10.1186/s40170-016-0160-x
169. Kondaveeti Y, Guttilla Reed IK, White BA. Epithelial-mesenchymal transition induces similar metabolic alterations in two independent breast cancer cell lines. Cancer Lett. (2015) 364:44–58. doi: 10.1016/j.canlet.2015.04.025
170. Dong C, Yuan T, Wu Y, Wang Y, Fan TW, Miriyala S, et al. Loss of FBP1 by Snail-mediated repression provides metabolic advantages in basal-like breast cancer. Cancer Cell. (2013) 23:316–31. doi: 10.1016/j.ccr.2013.01.022
171. LeBleu VS, O'Connell JT, Gonzalez Herrera KN, Wikman H, Pantel K, Haigis MC, et al. PGC-1α mediates mitochondrial biogenesis and oxidative phosphorylation in cancer cells to promote metastasis. Nat Cell Biol. (2014) 16:992–1003. doi: 10.1038/ncb3039
172. Porporato PE, Payen VL, Perez-Escuredo J, De Saedeleer CJ, Danhier P, Copetti T, et al. A mitochondrial switch promotes tumor metastasis. Cell Rep. (2014) 8:754–66. doi: 10.1016/j.celrep.2014.06.043
173. Dupuy F, Tabaries S, Andrzejewski S, Dong Z, Blagih J, Annis MG, et al. PDK1-dependent metabolic reprogramming dictates metastatic potential in breast cancer. Cell Metab. (2015) 22:577–89. doi: 10.1016/j.cmet.2015.08.007
174. Ladanyi A, Mukherjee A, Kenny HA, Johnson A, Mitra AK, Sundaresan S, et al. Adipocyte-induced CD36 expression drives ovarian cancer progression and metastasis. Oncogene. (2018) 37:2285–301. doi: 10.1038/s41388-017-0093-z
175. Jiang L, Xiao L, Sugiura H, Huang X, Ali A, Kuro-o M, et al. Metabolic reprogramming during TGFβ1-induced epithelial-to-mesenchymal transition. Oncogene. (2015) 34:3908–16. doi: 10.1038/onc.2014.321
176. Jin L, Chun J, Pan C, Kumar A, Zhang G, Ha Y, et al. The PLAG1-GDH1 axis promotes anoikis resistance and tumor metastasis through CamKK2-AMPK signaling in LKB1-deficient lung cancer. Mol Cell. (2018) 69:87–99.e7. doi: 10.1016/j.molcel.2017.11.025
177. Xiang L, Mou J, Shao B, Wei Y, Liang H, Takano N, et al. Glutaminase 1 expression in colorectal cancer cells is induced by hypoxia and required for tumor growth, invasion, and metastatic colonization. Cell Death Dis. (2019) 10:40. doi: 10.1038/s41419-018-1291-5
178. Elisi GM, Santucci M, D'Arca D, Lauriola A, Marverti G, Losi L, et al. Repurposing of drugs targeting YAP-TEAD functions. Cancers. (2018) 10:329. doi: 10.3390/cancers10090329
179. Brodowska K, Al-Moujahed A, Marmalidou A, Meyer Zu Horste M, Cichy J, Miller JW, et al. The clinically used photosensitizer verteporfin (VP) inhibits YAP-TEAD and human retinoblastoma cell growth in vitro without light activation. Exp Eye Res. (2014) 124:67–73. doi: 10.1016/j.exer.2014.04.011
180. Liu-Chittenden Y, Huang B, Shim JS, Chen Q, Lee SJ, Anders RA, et al. Genetic and pharmacological disruption of the TEAD-YAP complex suppresses the oncogenic activity of YAP. Genes Dev. (2012) 26:1300–5. doi: 10.1101/gad.192856.112
181. Du K, Hyun J, Premont RT, Choi SS, Michelotti GA, Swiderska-Syn M, et al. Hedgehog-YAP signaling pathway regulates glutaminolysis to control activation of hepatic stellate cells. Gastroenterology. (2018) 154:1465–79.e13. doi: 10.1053/j.gastro.2017.12.022
182. Kurppa KJ, Liu Y, To C, Zhang T, Fan M, Vajdi A, et al. Treatment-induced tumor dormancy through YAP-mediated transcriptional reprogramming of the apoptotic pathway. Cancer Cell. (2020) 37:104–22.e12. doi: 10.1016/j.ccell.2019.12.006
183. Bum-Erdene K, Zhou D, Gonzalez-Gutierrez G, Ghozayel MK, Si Y, Xu D, et al. Small-molecule covalent modification of conserved cysteine leads to allosteric inhibition of the TEADYap protein-protein interaction. Cell Chem Biol. (2019) 26:378–89.e13. doi: 10.1016/j.chembiol.2018.11.010
184. Nouri K, Azad T, Ling M, van Rensburg HJJ, Pipchuk A, Shen H, et al. Identification of celastrol as a novel YAP-TEAD inhibitor for cancer therapy by high throughput screening with ultrasensitive YAP/TAZ-TEAD biosensors. Cancers. (2019) 11:1596. doi: 10.3390/cancers11101596
185. Jolly MK, Ware KE, Gilja S, Somarelli JA, Levine H. EMT and MET: necessary or permissive for metastasis? Mol Oncol. (2017) 11:755–69. doi: 10.1002/1878-0261.12083
186. Wei SC, Fattet L, Tsai JH, Guo Y, Pai VH, Majeski HE, et al. Matrix stiffness drives epithelial-mesenchymal transition and tumour metastasis through a TWIST1-G3BP2 mechanotransduction pathway. Nat Cell Biol. (2015) 17:678–88. doi: 10.1038/ncb3157
187. Romani P, Brian I, Santinon G, Pocaterra A, Audano M, Pedretti S, et al. Extracellular matrix mechanical cues regulate lipid metabolism through Lipin-1 and SREBP. Nat Cell Biol. (2019) 21:338–47. doi: 10.1038/s41556-018-0270-5
188. Jho E. Dual role of YAP: oncoprotein and tumor suppressor. J Thorac Dis. (2018) 10(Suppl. 33):S3895–8. doi: 10.21037/jtd.2018.10.70
Keywords: YAP, TAZ, cancer, metabolism, metastasis
Citation: Yamaguchi H and Taouk GM (2020) A Potential Role of YAP/TAZ in the Interplay Between Metastasis and Metabolic Alterations. Front. Oncol. 10:928. doi: 10.3389/fonc.2020.00928
Received: 19 December 2019; Accepted: 12 May 2020;
Published: 11 June 2020.
Edited by:
Yue Zhao, University of Cologne, GermanyReviewed by:
Lei Chang, Soochow University Medical College (SUMC), ChinaCopyright © 2020 Yamaguchi and Taouk. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hirohito Yamaguchi, aHlhbWFndWNoaUBoYmt1LmVkdS5xYQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.